User login
Captopril questioned for diabetes patients in COVID-19 setting
Captopril appears to be associated with a higher rate of pulmonary adverse reactions in patients with diabetes than that of other ACE inhibitors or angiotensin receptor blockers (ARBs) and therefore may not be the best choice for patients with diabetes and COVID-19, a new study suggests.
The study was published online in the Journal of the American Pharmacists Association.
The authors, led by Emma G. Stafford, PharmD, University of Missouri-Kansas City School of Pharmacy, note that diabetes seems to confer a higher risk of adverse outcomes in COVID-19 infection and there is conflicting data on the contribution of ACE inhibitors and ARBs, commonly used medications in diabetes, on the mortality and morbidity of COVID-19.
“In light of the recent COVID-19 outbreak, more research is needed to understand the effects that diabetes (and its medications) may have on the respiratory system and how that could affect the management of diseases such as COVID-19,” they say.
“Although ACE inhibitors and ARBs are generally considered to have similar adverse event profiles, evaluation of postmarketing adverse events may shed light on minute differences that could have important clinical impacts,” they add.
For the current study, the researchers analyzed data from multiple publicly available data sources on adverse drug reactions in patients with diabetes taking ACE inhibitors or ARBs. The data included all adverse drug events (ADEs) reported nationally to the US Food and Drug Administration and internationally to the Medical Dictionary for Regulatory Activities (MedDRA).
Results showed that captopril, the first ACE inhibitor approved back in 1981, has a higher incidence of pulmonary ADEs in patients with diabetes as compared with other ACE-inhibitor drugs (P = .005) as well as a statistically significant difference in pulmonary events compared with ARBs (P = .012).
“These analyses suggest that pharmacists and clinicians will need to consider the specific medication’s adverse event profile, particularly captopril, on how it may affect infections and other acute disease states that alter pulmonary function, such as COVID-19,” the authors conclude.
They say that the high incidence of pulmonary adverse drug effects with captopril “highlights the fact that the drugs belonging in one class are not identical and that its pharmacokinetics and pharmacodynamics can affect the patients’ health especially during acute processes like COVID-19.”
“This is especially important as current observational studies of COVID-19 patients tend to group drugs within a class and are not analyzing the potential differences within each class,” they add.
They note that ACE inhibitors can be broadly classified into 3 structural classes: sulfhydryl-, dicarboxyl-, and phosphorous- containing molecules. Notably, captopril is the only currently available ACE inhibitor belonging to the sulfhydryl-containing class and may explain the higher incidence of adverse drug effects observed, they comment.
“Health care providers have been left with many questions when treating patients with COVID-19, including how ACE inhibitors or ARBs may affect their clinical course. Results from this study may be helpful when prescribing or continuing ACE inhibitors or ARBs for patients with diabetes and infections or illnesses that may affect pulmonary function, such as COVID-19,” they conclude.
Questioning safety in COVID-19 an “overreach”
Commenting for Medscape Medical News, Michael A. Weber, MD, professor of medicine at State University of New York, said he thought the current article appears to overreach in questioning captopril’s safety in the COVID-19 setting.
“Captopril was the first ACE inhibitor available for clinical use. In early prescribing its dosage was not well understood and it might have been administered in excessive amounts,” Weber notes.
“There were some renal and other adverse effects reported that at first were attributed to the fact that captopril, unlike any other popular ACE inhibitors, contained a sulfhydryl (SH) group in its molecule,” he said. “It is not clear whether this feature could be responsible for the increased pulmonary side effects and potential danger to COVID-19 patients now reported with captopril in this new pharmacy article.”
But he adds: “The article contains no evidence that the effect of captopril or any other ACE inhibitor on the pulmonary ACE-2 enzyme has a deleterious effect on outcomes of COVID-19 disease. In any case, captopril — which should be prescribed in a twice-daily dose — is not frequently prescribed these days since newer ACE inhibitors are effective with just once-daily dosing.”
This article first appeared on Medscape.com.
Captopril appears to be associated with a higher rate of pulmonary adverse reactions in patients with diabetes than that of other ACE inhibitors or angiotensin receptor blockers (ARBs) and therefore may not be the best choice for patients with diabetes and COVID-19, a new study suggests.
The study was published online in the Journal of the American Pharmacists Association.
The authors, led by Emma G. Stafford, PharmD, University of Missouri-Kansas City School of Pharmacy, note that diabetes seems to confer a higher risk of adverse outcomes in COVID-19 infection and there is conflicting data on the contribution of ACE inhibitors and ARBs, commonly used medications in diabetes, on the mortality and morbidity of COVID-19.
“In light of the recent COVID-19 outbreak, more research is needed to understand the effects that diabetes (and its medications) may have on the respiratory system and how that could affect the management of diseases such as COVID-19,” they say.
“Although ACE inhibitors and ARBs are generally considered to have similar adverse event profiles, evaluation of postmarketing adverse events may shed light on minute differences that could have important clinical impacts,” they add.
For the current study, the researchers analyzed data from multiple publicly available data sources on adverse drug reactions in patients with diabetes taking ACE inhibitors or ARBs. The data included all adverse drug events (ADEs) reported nationally to the US Food and Drug Administration and internationally to the Medical Dictionary for Regulatory Activities (MedDRA).
Results showed that captopril, the first ACE inhibitor approved back in 1981, has a higher incidence of pulmonary ADEs in patients with diabetes as compared with other ACE-inhibitor drugs (P = .005) as well as a statistically significant difference in pulmonary events compared with ARBs (P = .012).
“These analyses suggest that pharmacists and clinicians will need to consider the specific medication’s adverse event profile, particularly captopril, on how it may affect infections and other acute disease states that alter pulmonary function, such as COVID-19,” the authors conclude.
They say that the high incidence of pulmonary adverse drug effects with captopril “highlights the fact that the drugs belonging in one class are not identical and that its pharmacokinetics and pharmacodynamics can affect the patients’ health especially during acute processes like COVID-19.”
“This is especially important as current observational studies of COVID-19 patients tend to group drugs within a class and are not analyzing the potential differences within each class,” they add.
They note that ACE inhibitors can be broadly classified into 3 structural classes: sulfhydryl-, dicarboxyl-, and phosphorous- containing molecules. Notably, captopril is the only currently available ACE inhibitor belonging to the sulfhydryl-containing class and may explain the higher incidence of adverse drug effects observed, they comment.
“Health care providers have been left with many questions when treating patients with COVID-19, including how ACE inhibitors or ARBs may affect their clinical course. Results from this study may be helpful when prescribing or continuing ACE inhibitors or ARBs for patients with diabetes and infections or illnesses that may affect pulmonary function, such as COVID-19,” they conclude.
Questioning safety in COVID-19 an “overreach”
Commenting for Medscape Medical News, Michael A. Weber, MD, professor of medicine at State University of New York, said he thought the current article appears to overreach in questioning captopril’s safety in the COVID-19 setting.
“Captopril was the first ACE inhibitor available for clinical use. In early prescribing its dosage was not well understood and it might have been administered in excessive amounts,” Weber notes.
“There were some renal and other adverse effects reported that at first were attributed to the fact that captopril, unlike any other popular ACE inhibitors, contained a sulfhydryl (SH) group in its molecule,” he said. “It is not clear whether this feature could be responsible for the increased pulmonary side effects and potential danger to COVID-19 patients now reported with captopril in this new pharmacy article.”
But he adds: “The article contains no evidence that the effect of captopril or any other ACE inhibitor on the pulmonary ACE-2 enzyme has a deleterious effect on outcomes of COVID-19 disease. In any case, captopril — which should be prescribed in a twice-daily dose — is not frequently prescribed these days since newer ACE inhibitors are effective with just once-daily dosing.”
This article first appeared on Medscape.com.
Captopril appears to be associated with a higher rate of pulmonary adverse reactions in patients with diabetes than that of other ACE inhibitors or angiotensin receptor blockers (ARBs) and therefore may not be the best choice for patients with diabetes and COVID-19, a new study suggests.
The study was published online in the Journal of the American Pharmacists Association.
The authors, led by Emma G. Stafford, PharmD, University of Missouri-Kansas City School of Pharmacy, note that diabetes seems to confer a higher risk of adverse outcomes in COVID-19 infection and there is conflicting data on the contribution of ACE inhibitors and ARBs, commonly used medications in diabetes, on the mortality and morbidity of COVID-19.
“In light of the recent COVID-19 outbreak, more research is needed to understand the effects that diabetes (and its medications) may have on the respiratory system and how that could affect the management of diseases such as COVID-19,” they say.
“Although ACE inhibitors and ARBs are generally considered to have similar adverse event profiles, evaluation of postmarketing adverse events may shed light on minute differences that could have important clinical impacts,” they add.
For the current study, the researchers analyzed data from multiple publicly available data sources on adverse drug reactions in patients with diabetes taking ACE inhibitors or ARBs. The data included all adverse drug events (ADEs) reported nationally to the US Food and Drug Administration and internationally to the Medical Dictionary for Regulatory Activities (MedDRA).
Results showed that captopril, the first ACE inhibitor approved back in 1981, has a higher incidence of pulmonary ADEs in patients with diabetes as compared with other ACE-inhibitor drugs (P = .005) as well as a statistically significant difference in pulmonary events compared with ARBs (P = .012).
“These analyses suggest that pharmacists and clinicians will need to consider the specific medication’s adverse event profile, particularly captopril, on how it may affect infections and other acute disease states that alter pulmonary function, such as COVID-19,” the authors conclude.
They say that the high incidence of pulmonary adverse drug effects with captopril “highlights the fact that the drugs belonging in one class are not identical and that its pharmacokinetics and pharmacodynamics can affect the patients’ health especially during acute processes like COVID-19.”
“This is especially important as current observational studies of COVID-19 patients tend to group drugs within a class and are not analyzing the potential differences within each class,” they add.
They note that ACE inhibitors can be broadly classified into 3 structural classes: sulfhydryl-, dicarboxyl-, and phosphorous- containing molecules. Notably, captopril is the only currently available ACE inhibitor belonging to the sulfhydryl-containing class and may explain the higher incidence of adverse drug effects observed, they comment.
“Health care providers have been left with many questions when treating patients with COVID-19, including how ACE inhibitors or ARBs may affect their clinical course. Results from this study may be helpful when prescribing or continuing ACE inhibitors or ARBs for patients with diabetes and infections or illnesses that may affect pulmonary function, such as COVID-19,” they conclude.
Questioning safety in COVID-19 an “overreach”
Commenting for Medscape Medical News, Michael A. Weber, MD, professor of medicine at State University of New York, said he thought the current article appears to overreach in questioning captopril’s safety in the COVID-19 setting.
“Captopril was the first ACE inhibitor available for clinical use. In early prescribing its dosage was not well understood and it might have been administered in excessive amounts,” Weber notes.
“There were some renal and other adverse effects reported that at first were attributed to the fact that captopril, unlike any other popular ACE inhibitors, contained a sulfhydryl (SH) group in its molecule,” he said. “It is not clear whether this feature could be responsible for the increased pulmonary side effects and potential danger to COVID-19 patients now reported with captopril in this new pharmacy article.”
But he adds: “The article contains no evidence that the effect of captopril or any other ACE inhibitor on the pulmonary ACE-2 enzyme has a deleterious effect on outcomes of COVID-19 disease. In any case, captopril — which should be prescribed in a twice-daily dose — is not frequently prescribed these days since newer ACE inhibitors are effective with just once-daily dosing.”
This article first appeared on Medscape.com.
FDA approves in-home breast cancer treatment
Advantageous for infusion centers?
The Food and Drug Administration approved a combination of pertuzumab (Perjeta, Genentech/Roche), trastuzumab (Herceptin, Genentech/Roche) and hyaluronidase (Phesgo, Genentech/Roche) that is administered subcutaneously – rather than intravenously – for the treatment of early and metastatic HER2-positive breast cancers.
Phesgo is initially used in combination with chemotherapy at an infusion center but could continue to be administered in a patient’s home by a qualified health care professional once chemotherapy is complete, according to the FDA.
Administration takes approximately 8 minutes for the initial loading dose and approximately 5 minutes for maintenance doses, according to a Genentech press statement. This compares favorably with the 150 minutes needed for the combined loading dose of intravenous pertuzumab and trastuzumab, and the 60-150 minutes for intravenous maintenance infusions, the company said.
“Currently, most patients with HER2-positive breast cancer receive trastuzumab and pertuzumab at infusion centers. With a new administration route, Phesgo offers an outpatient option for patients to receive trastuzumab and pertuzumab,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in an agency press release.
“The fixed-dose combination of trastuzumab and pertuzumab offers a simpler, faster, and easier treatment experience for patients with HER2-positive breast cancer,” said Antoinette Tan, MD, MHSc, chief of breast medical oncology at Levine Cancer Institute, Charlotte, N.C., in the company statement.
Dr. Tan also said that home administration “can be advantageous for patients and infusion centers.”
However, in April, the Community Oncology Alliance strenuously objected to this type of treatment in a patient’s home, as reported by Medscape Medical News.
The group, which represents U.S. community-based practices, said it “fundamentally opposes home infusion of chemotherapy, cancer immunotherapy, and cancer treatment supportive drugs because of serious patient safety concerns.”
The FDA’s approval was based on the results of the pivotal phase 3 FeDeriCa trial, a noninferiority study in patients with HER2-positive early breast cancer, which demonstrated that the new product had comparable efficacy and safety as intravenous pertuzumab and intravenous trastuzumab.
In terms of efficacy, the subcutaneous product demonstrated noninferior plasma levels of pertuzumab, which was the primary endpoint, when compared with IV administration of pertuzumab.
Safety was comparable between the two approaches, with no new safety signals using the subcutaneous delivery method, including no “meaningful difference” in cardiac toxicity, according to Genentech. However, there were more administration-related reactions with the new product. The most common adverse events in both groups were alopecia, nausea, diarrhea, and anemia.
The new product uses a drug delivery technology (Enhanze, Halozyme Therapeutics) that employs a proprietary enzyme that temporarily degrades hyaluronan, a glycosaminoglycan or chain of natural sugars in the body, to facilitate the dispersion and absorption of injected therapeutic drugs, according to Genentech.
In May, at the European Society for Medical Oncology Breast Cancer Virtual Meeting 2020, investigators of the phase 2 PHranceSCa study reported that “more than 80%” of patients preferred subcutaneous to intravenous administration of pertuzumab and trastuzumab.
This article first appeared on Medscape.com.
Advantageous for infusion centers?
Advantageous for infusion centers?
The Food and Drug Administration approved a combination of pertuzumab (Perjeta, Genentech/Roche), trastuzumab (Herceptin, Genentech/Roche) and hyaluronidase (Phesgo, Genentech/Roche) that is administered subcutaneously – rather than intravenously – for the treatment of early and metastatic HER2-positive breast cancers.
Phesgo is initially used in combination with chemotherapy at an infusion center but could continue to be administered in a patient’s home by a qualified health care professional once chemotherapy is complete, according to the FDA.
Administration takes approximately 8 minutes for the initial loading dose and approximately 5 minutes for maintenance doses, according to a Genentech press statement. This compares favorably with the 150 minutes needed for the combined loading dose of intravenous pertuzumab and trastuzumab, and the 60-150 minutes for intravenous maintenance infusions, the company said.
“Currently, most patients with HER2-positive breast cancer receive trastuzumab and pertuzumab at infusion centers. With a new administration route, Phesgo offers an outpatient option for patients to receive trastuzumab and pertuzumab,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in an agency press release.
“The fixed-dose combination of trastuzumab and pertuzumab offers a simpler, faster, and easier treatment experience for patients with HER2-positive breast cancer,” said Antoinette Tan, MD, MHSc, chief of breast medical oncology at Levine Cancer Institute, Charlotte, N.C., in the company statement.
Dr. Tan also said that home administration “can be advantageous for patients and infusion centers.”
However, in April, the Community Oncology Alliance strenuously objected to this type of treatment in a patient’s home, as reported by Medscape Medical News.
The group, which represents U.S. community-based practices, said it “fundamentally opposes home infusion of chemotherapy, cancer immunotherapy, and cancer treatment supportive drugs because of serious patient safety concerns.”
The FDA’s approval was based on the results of the pivotal phase 3 FeDeriCa trial, a noninferiority study in patients with HER2-positive early breast cancer, which demonstrated that the new product had comparable efficacy and safety as intravenous pertuzumab and intravenous trastuzumab.
In terms of efficacy, the subcutaneous product demonstrated noninferior plasma levels of pertuzumab, which was the primary endpoint, when compared with IV administration of pertuzumab.
Safety was comparable between the two approaches, with no new safety signals using the subcutaneous delivery method, including no “meaningful difference” in cardiac toxicity, according to Genentech. However, there were more administration-related reactions with the new product. The most common adverse events in both groups were alopecia, nausea, diarrhea, and anemia.
The new product uses a drug delivery technology (Enhanze, Halozyme Therapeutics) that employs a proprietary enzyme that temporarily degrades hyaluronan, a glycosaminoglycan or chain of natural sugars in the body, to facilitate the dispersion and absorption of injected therapeutic drugs, according to Genentech.
In May, at the European Society for Medical Oncology Breast Cancer Virtual Meeting 2020, investigators of the phase 2 PHranceSCa study reported that “more than 80%” of patients preferred subcutaneous to intravenous administration of pertuzumab and trastuzumab.
This article first appeared on Medscape.com.
The Food and Drug Administration approved a combination of pertuzumab (Perjeta, Genentech/Roche), trastuzumab (Herceptin, Genentech/Roche) and hyaluronidase (Phesgo, Genentech/Roche) that is administered subcutaneously – rather than intravenously – for the treatment of early and metastatic HER2-positive breast cancers.
Phesgo is initially used in combination with chemotherapy at an infusion center but could continue to be administered in a patient’s home by a qualified health care professional once chemotherapy is complete, according to the FDA.
Administration takes approximately 8 minutes for the initial loading dose and approximately 5 minutes for maintenance doses, according to a Genentech press statement. This compares favorably with the 150 minutes needed for the combined loading dose of intravenous pertuzumab and trastuzumab, and the 60-150 minutes for intravenous maintenance infusions, the company said.
“Currently, most patients with HER2-positive breast cancer receive trastuzumab and pertuzumab at infusion centers. With a new administration route, Phesgo offers an outpatient option for patients to receive trastuzumab and pertuzumab,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in an agency press release.
“The fixed-dose combination of trastuzumab and pertuzumab offers a simpler, faster, and easier treatment experience for patients with HER2-positive breast cancer,” said Antoinette Tan, MD, MHSc, chief of breast medical oncology at Levine Cancer Institute, Charlotte, N.C., in the company statement.
Dr. Tan also said that home administration “can be advantageous for patients and infusion centers.”
However, in April, the Community Oncology Alliance strenuously objected to this type of treatment in a patient’s home, as reported by Medscape Medical News.
The group, which represents U.S. community-based practices, said it “fundamentally opposes home infusion of chemotherapy, cancer immunotherapy, and cancer treatment supportive drugs because of serious patient safety concerns.”
The FDA’s approval was based on the results of the pivotal phase 3 FeDeriCa trial, a noninferiority study in patients with HER2-positive early breast cancer, which demonstrated that the new product had comparable efficacy and safety as intravenous pertuzumab and intravenous trastuzumab.
In terms of efficacy, the subcutaneous product demonstrated noninferior plasma levels of pertuzumab, which was the primary endpoint, when compared with IV administration of pertuzumab.
Safety was comparable between the two approaches, with no new safety signals using the subcutaneous delivery method, including no “meaningful difference” in cardiac toxicity, according to Genentech. However, there were more administration-related reactions with the new product. The most common adverse events in both groups were alopecia, nausea, diarrhea, and anemia.
The new product uses a drug delivery technology (Enhanze, Halozyme Therapeutics) that employs a proprietary enzyme that temporarily degrades hyaluronan, a glycosaminoglycan or chain of natural sugars in the body, to facilitate the dispersion and absorption of injected therapeutic drugs, according to Genentech.
In May, at the European Society for Medical Oncology Breast Cancer Virtual Meeting 2020, investigators of the phase 2 PHranceSCa study reported that “more than 80%” of patients preferred subcutaneous to intravenous administration of pertuzumab and trastuzumab.
This article first appeared on Medscape.com.
Where does dexamethasone fit in with diabetic ketoacidosis in COVID-19?
A new article in the Journal of Clinical Endocrinology & Metabolism (JCEM) addresses unique concerns and considerations regarding diabetic ketoacidosis (DKA) in the setting of COVID-19.
Corresponding author Marie E. McDonnell, MD, director of the diabetes program at Brigham and Women’s Hospital, Boston, Massachusetts, discussed the recommendations with Medscape Medical News and also spoke about the news this week that the corticosteroid dexamethasone reduced death rates in severely ill patients with COVID-19.
The full JCEM article, by lead author Nadine E. Palermo, DO, Division of Endocrinology, Diabetes, and Hypertension, also at Brigham and Women’s Hospital, covers DKA diagnosis and triage, and emphasizes that usual hospital protocols for DKA management may need to be adjusted during COVID-19 to help preserve personal protective equipment and ICU beds.
“Hospitals and clinicians need to be able to quickly identify and manage DKA in COVID patients to save lives. This involves determining the options for management, including when less intensive subcutaneous insulin is indicated, and understanding how to guide patients on avoiding this serious complication,” McDonnell said in an Endocrine Society statement.
What about dexamethasone for severe COVID-19 in diabetes?
The new article briefly touches on the fact that upward adjustments to intensive intravenous insulin therapy for DKA may be necessary in patients with COVID-19 who are receiving concomitant corticosteroids or vasopressors.
But it was written prior to the June 16 announcement of the “RECOVERY” trial results with dexamethasone. The UK National Health Service immediately approved the drug’s use in the COVID-19 setting, despite the fact that there has been no published article on the findings yet.
McDonnell told Medscape Medical News that she would need to see formal results to better understand exactly which patients were studied and which ones benefited.
“The peer review will be critical. It looks as if it only benefits people who need respiratory support, but I want to understand that in much more detail,” she said. “If they all had acute respiratory distress syndrome [ARDS],” that’s different.
“There are already some data supporting steroid use in ARDS,” she noted, but added that not all of it suggests benefit.
She pointed to one of several studies now showing that diabetes, and hyperglycemia among people without a prior diabetes diagnosis, are both strong predictors of mortality in hospitalized patients with COVID-19.
“There was a very clear relationship between hyperglycemia and outcomes. We really shouldn’t put people at risk until we have clear data,” she said.
If, once the data are reviewed and appropriate dexamethasone becomes an established treatment for severe COVID-19, hyperglycemia would be a concern among all patients, not just those with previously diagnosed diabetes, she noted.
“We know a good number of people with prediabetes develop hyperglycemia when put on steroids. They can push people over the edge. We’re not going to miss anybody, but treating steroid-induced hyperglycemia is really hard,” McDonnell explained.
She also recommended 2014 guidance from Diabetes UK and the Association of British Clinical Diabetologists, which addresses management of inpatient steroid-induced DKA in patients with and without pre-existing diabetes.
Another major concern, she said, is “patients trying to get dexamethasone when they start to get sick” because this is not the right population to use this agent.
“We worry about people who do not need this drug. If they have diabetes, they put themselves at risk of hyperglycemia, which then increases the risk of severe COVID-19. And then they’re also putting themselves at risk of DKA. It would just be bad medicine,” she said.
Managing DKA in the face of COVID-19: Flexibility is key
In the JCEM article, Palermo and colleagues emphasize that the usual hospital protocols for DKA management may need to be adjusted during COVID-19 in the interest of reducing transmission risk and preserving scare resources.
They provide evidence for alternative treatment strategies, such as the use of subcutaneous rather than intravenous insulin when appropriate.
“We wanted to outline when exactly you should consider nonintensive management strategies for DKA,” McDonnell further explained to Medscape Medical News.
“That would include those with mild or some with moderate DKA. ... The idea is to remind our colleagues about that because hospitals tend to operate on a protocol-driven algorithmic methodology, they can forget to step off the usual care pathway even if evidence supports that,” she said.
But on the other hand, she also said that, in some very complex or severely ill patients with COVID-19, classical intravenous insulin therapy makes the most sense even if their DKA is mild.
The outpatient setting: Prevention and preparation
The new article also addresses several concerns regarding DKA prevention in the outpatient setting.
As with other guidelines, it includes a reminder that patients with diabetes should be advised to discontinue sodium-glucose cotransporter 2 (SGLT2) inhibitors if they become ill with COVID-19, especially if they’re not eating or drinking normally, because they raise the risk for DKA.
Also, for patients with type 1 diabetes, particularly those with a history of repeated DKA, “this is the time to make sure we reach out to patients to refill their insulin prescriptions and address issues related to cost and other access difficulties,” McDonnell said.
The authors also emphasize that insulin starts and education should not be postponed during the pandemic. “Patients identified as meeting criteria to start insulin should be referred for urgent education, either in person or, whenever possible and practical, via video teleconferencing,” they urge.
McDonnell has reported receiving research funding from Novo Nordisk. The other two authors have reported no relevant financial relationships.
This article first appeared on Medscape.com.
A new article in the Journal of Clinical Endocrinology & Metabolism (JCEM) addresses unique concerns and considerations regarding diabetic ketoacidosis (DKA) in the setting of COVID-19.
Corresponding author Marie E. McDonnell, MD, director of the diabetes program at Brigham and Women’s Hospital, Boston, Massachusetts, discussed the recommendations with Medscape Medical News and also spoke about the news this week that the corticosteroid dexamethasone reduced death rates in severely ill patients with COVID-19.
The full JCEM article, by lead author Nadine E. Palermo, DO, Division of Endocrinology, Diabetes, and Hypertension, also at Brigham and Women’s Hospital, covers DKA diagnosis and triage, and emphasizes that usual hospital protocols for DKA management may need to be adjusted during COVID-19 to help preserve personal protective equipment and ICU beds.
“Hospitals and clinicians need to be able to quickly identify and manage DKA in COVID patients to save lives. This involves determining the options for management, including when less intensive subcutaneous insulin is indicated, and understanding how to guide patients on avoiding this serious complication,” McDonnell said in an Endocrine Society statement.
What about dexamethasone for severe COVID-19 in diabetes?
The new article briefly touches on the fact that upward adjustments to intensive intravenous insulin therapy for DKA may be necessary in patients with COVID-19 who are receiving concomitant corticosteroids or vasopressors.
But it was written prior to the June 16 announcement of the “RECOVERY” trial results with dexamethasone. The UK National Health Service immediately approved the drug’s use in the COVID-19 setting, despite the fact that there has been no published article on the findings yet.
McDonnell told Medscape Medical News that she would need to see formal results to better understand exactly which patients were studied and which ones benefited.
“The peer review will be critical. It looks as if it only benefits people who need respiratory support, but I want to understand that in much more detail,” she said. “If they all had acute respiratory distress syndrome [ARDS],” that’s different.
“There are already some data supporting steroid use in ARDS,” she noted, but added that not all of it suggests benefit.
She pointed to one of several studies now showing that diabetes, and hyperglycemia among people without a prior diabetes diagnosis, are both strong predictors of mortality in hospitalized patients with COVID-19.
“There was a very clear relationship between hyperglycemia and outcomes. We really shouldn’t put people at risk until we have clear data,” she said.
If, once the data are reviewed and appropriate dexamethasone becomes an established treatment for severe COVID-19, hyperglycemia would be a concern among all patients, not just those with previously diagnosed diabetes, she noted.
“We know a good number of people with prediabetes develop hyperglycemia when put on steroids. They can push people over the edge. We’re not going to miss anybody, but treating steroid-induced hyperglycemia is really hard,” McDonnell explained.
She also recommended 2014 guidance from Diabetes UK and the Association of British Clinical Diabetologists, which addresses management of inpatient steroid-induced DKA in patients with and without pre-existing diabetes.
Another major concern, she said, is “patients trying to get dexamethasone when they start to get sick” because this is not the right population to use this agent.
“We worry about people who do not need this drug. If they have diabetes, they put themselves at risk of hyperglycemia, which then increases the risk of severe COVID-19. And then they’re also putting themselves at risk of DKA. It would just be bad medicine,” she said.
Managing DKA in the face of COVID-19: Flexibility is key
In the JCEM article, Palermo and colleagues emphasize that the usual hospital protocols for DKA management may need to be adjusted during COVID-19 in the interest of reducing transmission risk and preserving scare resources.
They provide evidence for alternative treatment strategies, such as the use of subcutaneous rather than intravenous insulin when appropriate.
“We wanted to outline when exactly you should consider nonintensive management strategies for DKA,” McDonnell further explained to Medscape Medical News.
“That would include those with mild or some with moderate DKA. ... The idea is to remind our colleagues about that because hospitals tend to operate on a protocol-driven algorithmic methodology, they can forget to step off the usual care pathway even if evidence supports that,” she said.
But on the other hand, she also said that, in some very complex or severely ill patients with COVID-19, classical intravenous insulin therapy makes the most sense even if their DKA is mild.
The outpatient setting: Prevention and preparation
The new article also addresses several concerns regarding DKA prevention in the outpatient setting.
As with other guidelines, it includes a reminder that patients with diabetes should be advised to discontinue sodium-glucose cotransporter 2 (SGLT2) inhibitors if they become ill with COVID-19, especially if they’re not eating or drinking normally, because they raise the risk for DKA.
Also, for patients with type 1 diabetes, particularly those with a history of repeated DKA, “this is the time to make sure we reach out to patients to refill their insulin prescriptions and address issues related to cost and other access difficulties,” McDonnell said.
The authors also emphasize that insulin starts and education should not be postponed during the pandemic. “Patients identified as meeting criteria to start insulin should be referred for urgent education, either in person or, whenever possible and practical, via video teleconferencing,” they urge.
McDonnell has reported receiving research funding from Novo Nordisk. The other two authors have reported no relevant financial relationships.
This article first appeared on Medscape.com.
A new article in the Journal of Clinical Endocrinology & Metabolism (JCEM) addresses unique concerns and considerations regarding diabetic ketoacidosis (DKA) in the setting of COVID-19.
Corresponding author Marie E. McDonnell, MD, director of the diabetes program at Brigham and Women’s Hospital, Boston, Massachusetts, discussed the recommendations with Medscape Medical News and also spoke about the news this week that the corticosteroid dexamethasone reduced death rates in severely ill patients with COVID-19.
The full JCEM article, by lead author Nadine E. Palermo, DO, Division of Endocrinology, Diabetes, and Hypertension, also at Brigham and Women’s Hospital, covers DKA diagnosis and triage, and emphasizes that usual hospital protocols for DKA management may need to be adjusted during COVID-19 to help preserve personal protective equipment and ICU beds.
“Hospitals and clinicians need to be able to quickly identify and manage DKA in COVID patients to save lives. This involves determining the options for management, including when less intensive subcutaneous insulin is indicated, and understanding how to guide patients on avoiding this serious complication,” McDonnell said in an Endocrine Society statement.
What about dexamethasone for severe COVID-19 in diabetes?
The new article briefly touches on the fact that upward adjustments to intensive intravenous insulin therapy for DKA may be necessary in patients with COVID-19 who are receiving concomitant corticosteroids or vasopressors.
But it was written prior to the June 16 announcement of the “RECOVERY” trial results with dexamethasone. The UK National Health Service immediately approved the drug’s use in the COVID-19 setting, despite the fact that there has been no published article on the findings yet.
McDonnell told Medscape Medical News that she would need to see formal results to better understand exactly which patients were studied and which ones benefited.
“The peer review will be critical. It looks as if it only benefits people who need respiratory support, but I want to understand that in much more detail,” she said. “If they all had acute respiratory distress syndrome [ARDS],” that’s different.
“There are already some data supporting steroid use in ARDS,” she noted, but added that not all of it suggests benefit.
She pointed to one of several studies now showing that diabetes, and hyperglycemia among people without a prior diabetes diagnosis, are both strong predictors of mortality in hospitalized patients with COVID-19.
“There was a very clear relationship between hyperglycemia and outcomes. We really shouldn’t put people at risk until we have clear data,” she said.
If, once the data are reviewed and appropriate dexamethasone becomes an established treatment for severe COVID-19, hyperglycemia would be a concern among all patients, not just those with previously diagnosed diabetes, she noted.
“We know a good number of people with prediabetes develop hyperglycemia when put on steroids. They can push people over the edge. We’re not going to miss anybody, but treating steroid-induced hyperglycemia is really hard,” McDonnell explained.
She also recommended 2014 guidance from Diabetes UK and the Association of British Clinical Diabetologists, which addresses management of inpatient steroid-induced DKA in patients with and without pre-existing diabetes.
Another major concern, she said, is “patients trying to get dexamethasone when they start to get sick” because this is not the right population to use this agent.
“We worry about people who do not need this drug. If they have diabetes, they put themselves at risk of hyperglycemia, which then increases the risk of severe COVID-19. And then they’re also putting themselves at risk of DKA. It would just be bad medicine,” she said.
Managing DKA in the face of COVID-19: Flexibility is key
In the JCEM article, Palermo and colleagues emphasize that the usual hospital protocols for DKA management may need to be adjusted during COVID-19 in the interest of reducing transmission risk and preserving scare resources.
They provide evidence for alternative treatment strategies, such as the use of subcutaneous rather than intravenous insulin when appropriate.
“We wanted to outline when exactly you should consider nonintensive management strategies for DKA,” McDonnell further explained to Medscape Medical News.
“That would include those with mild or some with moderate DKA. ... The idea is to remind our colleagues about that because hospitals tend to operate on a protocol-driven algorithmic methodology, they can forget to step off the usual care pathway even if evidence supports that,” she said.
But on the other hand, she also said that, in some very complex or severely ill patients with COVID-19, classical intravenous insulin therapy makes the most sense even if their DKA is mild.
The outpatient setting: Prevention and preparation
The new article also addresses several concerns regarding DKA prevention in the outpatient setting.
As with other guidelines, it includes a reminder that patients with diabetes should be advised to discontinue sodium-glucose cotransporter 2 (SGLT2) inhibitors if they become ill with COVID-19, especially if they’re not eating or drinking normally, because they raise the risk for DKA.
Also, for patients with type 1 diabetes, particularly those with a history of repeated DKA, “this is the time to make sure we reach out to patients to refill their insulin prescriptions and address issues related to cost and other access difficulties,” McDonnell said.
The authors also emphasize that insulin starts and education should not be postponed during the pandemic. “Patients identified as meeting criteria to start insulin should be referred for urgent education, either in person or, whenever possible and practical, via video teleconferencing,” they urge.
McDonnell has reported receiving research funding from Novo Nordisk. The other two authors have reported no relevant financial relationships.
This article first appeared on Medscape.com.
First-in-kind anti-CD47 antibody shows promise for MDS and AML treatment
Magrolimab plus azacitidine (AZA) improved outcomes in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients according to the results of a phase 1b study (NCT03248479) presented at the virtual ASCO meeting. The combo especially was promising for the underserved patient population that have the TP53 (p53) mutation.
Magrolimab is a first-in-kind IgG anti-CD47 monoclonal antibody that promotes the elimination of tumor cells through macrophage phagocytosis. CD47 is a “do not eat me” signal on cancer cells that allows the cells to evade macrophages. Its increased expression is predictive of a worse outcome in AML patients, according to David A. Sallman, MD, of the Moffitt Cancer Center, Tampa, Fla., and colleagues.
Dr. Sallman presented the results of a study examining whether magrolimab would provide a synergistic benefit when combined with AZA (which induces other prophagocytic “eat me” signals such as calreticulin on cancer cells). The primary objectives of the study were to examine the safety of magrolimab alone or with AZA, and to assess the efficacy of the magrolimab/AZA combo in 29 untreated AML patients and 39 untreated MDS patients. The majority of both the MDS and AML patients were poor cytogenetic risk at 64% and 72%, respectively. Mutant p53 was present in 13% of the MDS patients and 45% of the AML patients.
No deaths occurred in the first 60 days of the study among either the MDS or AML patients and discontinuation of treatment because of drug-related adverse events was seen in only one of the patients (1.5%) treated with magrolimab/AZA. There was no significant neutropenia or thrombocytopenia caused by the therapy seen, and the majority of the patients improved their neutrophil and platelet counts while on therapy.
Anemia from CD47 blockade was mitigated by the use of a priming dose of magrolimab coupled to a maintenance-dose regimen, resulting in a mild hemoglobin drop on the first dose, which returned to baseline with a majority of patients experiencing significant hemoglobin improvement and a decrease in transfusion frequency over time, according to Dr. Sallman and his colleagues.
The results showed that magrolimab/AZA induced a 91% overall response rate (ORR), with a 42% complete remission (CR) that increased to 56% at 6 months, in the MDS patients. AML patients experienced a 64% ORR (56% CR/CRi [CR with incomplete hematological remission]). These results compare favorably with the CR rate of 6%-17% rate seen for AZA monotherapy, according to Dr. Sallman.
Red blood cell transfusion independence was achieved in 58% of the MDS patients and 64% of the AML patients, and a complete cytogenetic response was seen in 35% and 50% of the MDS and AML patients, respectively.
The combined treatment was especially effective in the patients with p53 mutations, with an overall response rate of 75% for both MDS and AML, and a complete response of 42% and 50%, respectively. During the reported time of the study, the median survival was not reached, which compares favorably with current therapies, according to Dr. Sallman.
“Specifically looking at a very-high-risk p53-mutant subset, complete remissions have been observed in the majority of patients. And again, these have been durable. Based on all of these data, expansion cohorts both in MDS and p53 and AML continue to accrue with registrational studies in progress for MDS and planned for p53-mutant AML,” Dr. Sallman concluded.
The trial was sponsored by Gilead Sciences, and funding was obtained from the California Institute for Regenerative Medicine. Dr. Sallman disclosed that he received research funding from Celgene and has acted in a consulting or advisory role for Agios, argenx, and Celyad. He was also on the speaker’s bureau for a variety of pharmaceutical/biotech companies.
SOURCE: Sallman DA et al. ASCO 2020, Abstract 7507.
Magrolimab plus azacitidine (AZA) improved outcomes in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients according to the results of a phase 1b study (NCT03248479) presented at the virtual ASCO meeting. The combo especially was promising for the underserved patient population that have the TP53 (p53) mutation.
Magrolimab is a first-in-kind IgG anti-CD47 monoclonal antibody that promotes the elimination of tumor cells through macrophage phagocytosis. CD47 is a “do not eat me” signal on cancer cells that allows the cells to evade macrophages. Its increased expression is predictive of a worse outcome in AML patients, according to David A. Sallman, MD, of the Moffitt Cancer Center, Tampa, Fla., and colleagues.
Dr. Sallman presented the results of a study examining whether magrolimab would provide a synergistic benefit when combined with AZA (which induces other prophagocytic “eat me” signals such as calreticulin on cancer cells). The primary objectives of the study were to examine the safety of magrolimab alone or with AZA, and to assess the efficacy of the magrolimab/AZA combo in 29 untreated AML patients and 39 untreated MDS patients. The majority of both the MDS and AML patients were poor cytogenetic risk at 64% and 72%, respectively. Mutant p53 was present in 13% of the MDS patients and 45% of the AML patients.
No deaths occurred in the first 60 days of the study among either the MDS or AML patients and discontinuation of treatment because of drug-related adverse events was seen in only one of the patients (1.5%) treated with magrolimab/AZA. There was no significant neutropenia or thrombocytopenia caused by the therapy seen, and the majority of the patients improved their neutrophil and platelet counts while on therapy.
Anemia from CD47 blockade was mitigated by the use of a priming dose of magrolimab coupled to a maintenance-dose regimen, resulting in a mild hemoglobin drop on the first dose, which returned to baseline with a majority of patients experiencing significant hemoglobin improvement and a decrease in transfusion frequency over time, according to Dr. Sallman and his colleagues.
The results showed that magrolimab/AZA induced a 91% overall response rate (ORR), with a 42% complete remission (CR) that increased to 56% at 6 months, in the MDS patients. AML patients experienced a 64% ORR (56% CR/CRi [CR with incomplete hematological remission]). These results compare favorably with the CR rate of 6%-17% rate seen for AZA monotherapy, according to Dr. Sallman.
Red blood cell transfusion independence was achieved in 58% of the MDS patients and 64% of the AML patients, and a complete cytogenetic response was seen in 35% and 50% of the MDS and AML patients, respectively.
The combined treatment was especially effective in the patients with p53 mutations, with an overall response rate of 75% for both MDS and AML, and a complete response of 42% and 50%, respectively. During the reported time of the study, the median survival was not reached, which compares favorably with current therapies, according to Dr. Sallman.
“Specifically looking at a very-high-risk p53-mutant subset, complete remissions have been observed in the majority of patients. And again, these have been durable. Based on all of these data, expansion cohorts both in MDS and p53 and AML continue to accrue with registrational studies in progress for MDS and planned for p53-mutant AML,” Dr. Sallman concluded.
The trial was sponsored by Gilead Sciences, and funding was obtained from the California Institute for Regenerative Medicine. Dr. Sallman disclosed that he received research funding from Celgene and has acted in a consulting or advisory role for Agios, argenx, and Celyad. He was also on the speaker’s bureau for a variety of pharmaceutical/biotech companies.
SOURCE: Sallman DA et al. ASCO 2020, Abstract 7507.
Magrolimab plus azacitidine (AZA) improved outcomes in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients according to the results of a phase 1b study (NCT03248479) presented at the virtual ASCO meeting. The combo especially was promising for the underserved patient population that have the TP53 (p53) mutation.
Magrolimab is a first-in-kind IgG anti-CD47 monoclonal antibody that promotes the elimination of tumor cells through macrophage phagocytosis. CD47 is a “do not eat me” signal on cancer cells that allows the cells to evade macrophages. Its increased expression is predictive of a worse outcome in AML patients, according to David A. Sallman, MD, of the Moffitt Cancer Center, Tampa, Fla., and colleagues.
Dr. Sallman presented the results of a study examining whether magrolimab would provide a synergistic benefit when combined with AZA (which induces other prophagocytic “eat me” signals such as calreticulin on cancer cells). The primary objectives of the study were to examine the safety of magrolimab alone or with AZA, and to assess the efficacy of the magrolimab/AZA combo in 29 untreated AML patients and 39 untreated MDS patients. The majority of both the MDS and AML patients were poor cytogenetic risk at 64% and 72%, respectively. Mutant p53 was present in 13% of the MDS patients and 45% of the AML patients.
No deaths occurred in the first 60 days of the study among either the MDS or AML patients and discontinuation of treatment because of drug-related adverse events was seen in only one of the patients (1.5%) treated with magrolimab/AZA. There was no significant neutropenia or thrombocytopenia caused by the therapy seen, and the majority of the patients improved their neutrophil and platelet counts while on therapy.
Anemia from CD47 blockade was mitigated by the use of a priming dose of magrolimab coupled to a maintenance-dose regimen, resulting in a mild hemoglobin drop on the first dose, which returned to baseline with a majority of patients experiencing significant hemoglobin improvement and a decrease in transfusion frequency over time, according to Dr. Sallman and his colleagues.
The results showed that magrolimab/AZA induced a 91% overall response rate (ORR), with a 42% complete remission (CR) that increased to 56% at 6 months, in the MDS patients. AML patients experienced a 64% ORR (56% CR/CRi [CR with incomplete hematological remission]). These results compare favorably with the CR rate of 6%-17% rate seen for AZA monotherapy, according to Dr. Sallman.
Red blood cell transfusion independence was achieved in 58% of the MDS patients and 64% of the AML patients, and a complete cytogenetic response was seen in 35% and 50% of the MDS and AML patients, respectively.
The combined treatment was especially effective in the patients with p53 mutations, with an overall response rate of 75% for both MDS and AML, and a complete response of 42% and 50%, respectively. During the reported time of the study, the median survival was not reached, which compares favorably with current therapies, according to Dr. Sallman.
“Specifically looking at a very-high-risk p53-mutant subset, complete remissions have been observed in the majority of patients. And again, these have been durable. Based on all of these data, expansion cohorts both in MDS and p53 and AML continue to accrue with registrational studies in progress for MDS and planned for p53-mutant AML,” Dr. Sallman concluded.
The trial was sponsored by Gilead Sciences, and funding was obtained from the California Institute for Regenerative Medicine. Dr. Sallman disclosed that he received research funding from Celgene and has acted in a consulting or advisory role for Agios, argenx, and Celyad. He was also on the speaker’s bureau for a variety of pharmaceutical/biotech companies.
SOURCE: Sallman DA et al. ASCO 2020, Abstract 7507.
FROM ASCO 2020
DAPA-HF: Dapagliflozin slows T2D onset in heart failure patients
Dapagliflozin treatment of patients with heart failure but without diabetes in the DAPA-HF trial led to a one-third cut in the relative incidence of new-onset diabetes over a median follow-up of 18 months in a prespecified analysis from the multicenter trial that included 2,605 heart failure patients without diabetes at baseline.

The findings represented the first evidence that a drug from dapagliflozin’s class, the sodium-glucose cotransporter 2 (SGLT2) inhibitors, could prevent or slow the onset of type 2 diabetes. It represents “an additional benefit” that dapagliflozin (Farxiga) offers to patients with heart failure with reduced ejection fraction (HFrEF) like those enrolled in the DAPA-HF trial, Silvio E. Inzucchi, MD, said at the virtual annual scientific sessions of the American Diabetes Association. DAPA-HF had previously proved that treatment with this drug significantly reduced the study’s primary endpoint of cardiovascular death or heart failure worsening.
During 18 months of follow-up, 7.1% of patients in the placebo arm developed type 2 diabetes, compared with 4.9% in those who received dapagliflozin, a 2.2% absolute difference and a 32% relative risk reduction that was statistically significant for this prespecified but “exploratory” endpoint, reported Dr. Inzucchi, an endocrinologist and professor of medicine at Yale University, New Haven, Conn.
For this analysis, a hemoglobin A1c level of at least 6.5% measured in two consecutive assessments was the criterion for diagnosing incident diabetes. The 2,605 enrolled patients without diabetes in the DAPA-HF trial represented 55% of the entire trial cohort of 4,744 patients with HFrEF.
The 32% relative risk reduction for incident diabetes was primarily relevant to enrolled patients with prediabetes at entry, who constituted 67% of the enrolled cohort based on the usual definition of prediabetes, an A1c of 5.7%-6.4%.
Among all 157 (6%) of the DAPA-HF patients who developed diabetes during the trial, 150 (96%) occurred in patients with prediabetes by the usual definition; 136 of the incident cases (87%) had prediabetes by a more stringent criterion of an A1c of 6.0%-6.4%.
To put the preventive efficacy of dapagliflozin into more context, Dr. Inzucchi cited the 31% relative protection rate exerted by metformin in the Diabetes Prevention Program study (N Engl J Med. 2002 Feb 7;346[6]:393-403).
The findings showed that “dapagliflozin is the first medication demonstrated to reduce both incident type 2 diabetes and mortality in a single trial,” as well as the first agent from the SGLT2 inhibitor class to show a diabetes prevention effect, Dr. Inzucchi noted. Patients with both heart failure and diabetes are known to have a substantially increased mortality risk, compared with patients with just one of these diseases, and the potent risk posed by the confluence of both was confirmed in the results Dr. Inzucchi reported.
The 157 HFrEF patients in the trial who developed diabetes had a statistically significant 70% increased incidence of all-cause mortality during the trial’s follow-up, compared with similar HFrEF patients who remained free from a diabetes diagnosis, and they also had a significant 77% relative increase in their incidence of cardiovascular death. This analysis failed to show that incident diabetes had a significant impact on hospitalizations for heart failure coupled with cardiovascular death, another endpoint of the trial.

“This is a tremendously important analysis. We recognize that diabetes is an important factor that can forecast heart failure risk, even over relatively short follow-up. A drug that targets both diseases can be quite beneficial,” commented Muthiah Vaduganathan, MD, a cardiologist at Brigham and Women’s Hospital in Boston.
The impact of dapagliflozin on average A1c levels during the DAPA-HF trial was minimal, reducing levels by an average of 0.04% among those who entered with prediabetes and by 0.05% among the other patients. This suggests that the mechanisms by which dapagliflozin reduced incident diabetes was by routes that did not involve simply reducing hyperglycemia, and the observed decrease in incident diabetes was not apparently caused by “masking” of hyperglycemia by dapagliflozin, said Dr. Inzucchi.
One possibility is that dapagliflozin, which also improved quality of life and reduced hospitalizations in the DAPA-HF trial, led to improved function and mobility among patients that had beneficial effects on their insulin sensitivity, Dr. Vaduganathan speculated in an interview.

The new finding of dapagliflozin’s benefit “is great news,” commented Yehuda Handelsman, MD, an endocrinologist and diabetes specialist who is medical director of the Metabolic Institute of America in Tarzana, Calif. “It’s an impressive and important result, and another reason to use dapagliflozin in patients with HFrEF, a group of patients whom you want to prevent from having worse outcomes” by developing diabetes.
The DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial enrolled HFrEF patients at 410 centers in 20 countries during February 2017–August 2018. The study’s primary endpoint was the composite incidence of cardiovascular death or worsening heart failure, which occurred in 16.3% of patients randomized to receive dapagliflozin and in 21.2% of control patients on standard care but on placebo instead of the study drug, a statistically significant relative risk reduction of 26% (N Engl J Med. 2019 Nov 21;381[21]:1995-2008). In the 2,605-patient subgroup without type 2 diabetes at baseline the primary endpoint fell by a statistically significant 27% with dapagliflozin treatment, the first time an SGLT2 inhibitor drug was shown effective for reducing this endpoint in patients with HFrEF but without diabetes. DAPA-HF did not enroll any patients with type 1 diabetes.
DAPA-HF was sponsored by AstraZeneca, the company that markets dapagliflozin (Farxiga). Dr. Inzucchi has been a consultant to AstraZeneca and to Abbott, Boehringer Ingelheim, Merck, Novo Nordisk, Sanofi/Lexicon, and vTv Therapeutics. Dr. Vaduganathan has been an adviser to AstraZeneca and to Amgen, Baxter, Bayer, Boehringer Ingelheim, Cytokinetics, and Relypsa. Dr. Handelsman has been a consultant to several drug companies including AstraZeneca.
SOURCE: Inzucchi SE et al. ADA 2020, abstract 271-OR.
Dapagliflozin treatment of patients with heart failure but without diabetes in the DAPA-HF trial led to a one-third cut in the relative incidence of new-onset diabetes over a median follow-up of 18 months in a prespecified analysis from the multicenter trial that included 2,605 heart failure patients without diabetes at baseline.
The findings represented the first evidence that a drug from dapagliflozin’s class, the sodium-glucose cotransporter 2 (SGLT2) inhibitors, could prevent or slow the onset of type 2 diabetes. It represents “an additional benefit” that dapagliflozin (Farxiga) offers to patients with heart failure with reduced ejection fraction (HFrEF) like those enrolled in the DAPA-HF trial, Silvio E. Inzucchi, MD, said at the virtual annual scientific sessions of the American Diabetes Association. DAPA-HF had previously proved that treatment with this drug significantly reduced the study’s primary endpoint of cardiovascular death or heart failure worsening.
During 18 months of follow-up, 7.1% of patients in the placebo arm developed type 2 diabetes, compared with 4.9% in those who received dapagliflozin, a 2.2% absolute difference and a 32% relative risk reduction that was statistically significant for this prespecified but “exploratory” endpoint, reported Dr. Inzucchi, an endocrinologist and professor of medicine at Yale University, New Haven, Conn.
For this analysis, a hemoglobin A1c level of at least 6.5% measured in two consecutive assessments was the criterion for diagnosing incident diabetes. The 2,605 enrolled patients without diabetes in the DAPA-HF trial represented 55% of the entire trial cohort of 4,744 patients with HFrEF.
The 32% relative risk reduction for incident diabetes was primarily relevant to enrolled patients with prediabetes at entry, who constituted 67% of the enrolled cohort based on the usual definition of prediabetes, an A1c of 5.7%-6.4%.
Among all 157 (6%) of the DAPA-HF patients who developed diabetes during the trial, 150 (96%) occurred in patients with prediabetes by the usual definition; 136 of the incident cases (87%) had prediabetes by a more stringent criterion of an A1c of 6.0%-6.4%.
To put the preventive efficacy of dapagliflozin into more context, Dr. Inzucchi cited the 31% relative protection rate exerted by metformin in the Diabetes Prevention Program study (N Engl J Med. 2002 Feb 7;346[6]:393-403).
The findings showed that “dapagliflozin is the first medication demonstrated to reduce both incident type 2 diabetes and mortality in a single trial,” as well as the first agent from the SGLT2 inhibitor class to show a diabetes prevention effect, Dr. Inzucchi noted. Patients with both heart failure and diabetes are known to have a substantially increased mortality risk, compared with patients with just one of these diseases, and the potent risk posed by the confluence of both was confirmed in the results Dr. Inzucchi reported.
The 157 HFrEF patients in the trial who developed diabetes had a statistically significant 70% increased incidence of all-cause mortality during the trial’s follow-up, compared with similar HFrEF patients who remained free from a diabetes diagnosis, and they also had a significant 77% relative increase in their incidence of cardiovascular death. This analysis failed to show that incident diabetes had a significant impact on hospitalizations for heart failure coupled with cardiovascular death, another endpoint of the trial.
“This is a tremendously important analysis. We recognize that diabetes is an important factor that can forecast heart failure risk, even over relatively short follow-up. A drug that targets both diseases can be quite beneficial,” commented Muthiah Vaduganathan, MD, a cardiologist at Brigham and Women’s Hospital in Boston.
The impact of dapagliflozin on average A1c levels during the DAPA-HF trial was minimal, reducing levels by an average of 0.04% among those who entered with prediabetes and by 0.05% among the other patients. This suggests that the mechanisms by which dapagliflozin reduced incident diabetes was by routes that did not involve simply reducing hyperglycemia, and the observed decrease in incident diabetes was not apparently caused by “masking” of hyperglycemia by dapagliflozin, said Dr. Inzucchi.
One possibility is that dapagliflozin, which also improved quality of life and reduced hospitalizations in the DAPA-HF trial, led to improved function and mobility among patients that had beneficial effects on their insulin sensitivity, Dr. Vaduganathan speculated in an interview.
The new finding of dapagliflozin’s benefit “is great news,” commented Yehuda Handelsman, MD, an endocrinologist and diabetes specialist who is medical director of the Metabolic Institute of America in Tarzana, Calif. “It’s an impressive and important result, and another reason to use dapagliflozin in patients with HFrEF, a group of patients whom you want to prevent from having worse outcomes” by developing diabetes.
The DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial enrolled HFrEF patients at 410 centers in 20 countries during February 2017–August 2018. The study’s primary endpoint was the composite incidence of cardiovascular death or worsening heart failure, which occurred in 16.3% of patients randomized to receive dapagliflozin and in 21.2% of control patients on standard care but on placebo instead of the study drug, a statistically significant relative risk reduction of 26% (N Engl J Med. 2019 Nov 21;381[21]:1995-2008). In the 2,605-patient subgroup without type 2 diabetes at baseline the primary endpoint fell by a statistically significant 27% with dapagliflozin treatment, the first time an SGLT2 inhibitor drug was shown effective for reducing this endpoint in patients with HFrEF but without diabetes. DAPA-HF did not enroll any patients with type 1 diabetes.
DAPA-HF was sponsored by AstraZeneca, the company that markets dapagliflozin (Farxiga). Dr. Inzucchi has been a consultant to AstraZeneca and to Abbott, Boehringer Ingelheim, Merck, Novo Nordisk, Sanofi/Lexicon, and vTv Therapeutics. Dr. Vaduganathan has been an adviser to AstraZeneca and to Amgen, Baxter, Bayer, Boehringer Ingelheim, Cytokinetics, and Relypsa. Dr. Handelsman has been a consultant to several drug companies including AstraZeneca.
SOURCE: Inzucchi SE et al. ADA 2020, abstract 271-OR.
Dapagliflozin treatment of patients with heart failure but without diabetes in the DAPA-HF trial led to a one-third cut in the relative incidence of new-onset diabetes over a median follow-up of 18 months in a prespecified analysis from the multicenter trial that included 2,605 heart failure patients without diabetes at baseline.
The findings represented the first evidence that a drug from dapagliflozin’s class, the sodium-glucose cotransporter 2 (SGLT2) inhibitors, could prevent or slow the onset of type 2 diabetes. It represents “an additional benefit” that dapagliflozin (Farxiga) offers to patients with heart failure with reduced ejection fraction (HFrEF) like those enrolled in the DAPA-HF trial, Silvio E. Inzucchi, MD, said at the virtual annual scientific sessions of the American Diabetes Association. DAPA-HF had previously proved that treatment with this drug significantly reduced the study’s primary endpoint of cardiovascular death or heart failure worsening.
During 18 months of follow-up, 7.1% of patients in the placebo arm developed type 2 diabetes, compared with 4.9% in those who received dapagliflozin, a 2.2% absolute difference and a 32% relative risk reduction that was statistically significant for this prespecified but “exploratory” endpoint, reported Dr. Inzucchi, an endocrinologist and professor of medicine at Yale University, New Haven, Conn.
For this analysis, a hemoglobin A1c level of at least 6.5% measured in two consecutive assessments was the criterion for diagnosing incident diabetes. The 2,605 enrolled patients without diabetes in the DAPA-HF trial represented 55% of the entire trial cohort of 4,744 patients with HFrEF.
The 32% relative risk reduction for incident diabetes was primarily relevant to enrolled patients with prediabetes at entry, who constituted 67% of the enrolled cohort based on the usual definition of prediabetes, an A1c of 5.7%-6.4%.
Among all 157 (6%) of the DAPA-HF patients who developed diabetes during the trial, 150 (96%) occurred in patients with prediabetes by the usual definition; 136 of the incident cases (87%) had prediabetes by a more stringent criterion of an A1c of 6.0%-6.4%.
To put the preventive efficacy of dapagliflozin into more context, Dr. Inzucchi cited the 31% relative protection rate exerted by metformin in the Diabetes Prevention Program study (N Engl J Med. 2002 Feb 7;346[6]:393-403).
The findings showed that “dapagliflozin is the first medication demonstrated to reduce both incident type 2 diabetes and mortality in a single trial,” as well as the first agent from the SGLT2 inhibitor class to show a diabetes prevention effect, Dr. Inzucchi noted. Patients with both heart failure and diabetes are known to have a substantially increased mortality risk, compared with patients with just one of these diseases, and the potent risk posed by the confluence of both was confirmed in the results Dr. Inzucchi reported.
The 157 HFrEF patients in the trial who developed diabetes had a statistically significant 70% increased incidence of all-cause mortality during the trial’s follow-up, compared with similar HFrEF patients who remained free from a diabetes diagnosis, and they also had a significant 77% relative increase in their incidence of cardiovascular death. This analysis failed to show that incident diabetes had a significant impact on hospitalizations for heart failure coupled with cardiovascular death, another endpoint of the trial.
“This is a tremendously important analysis. We recognize that diabetes is an important factor that can forecast heart failure risk, even over relatively short follow-up. A drug that targets both diseases can be quite beneficial,” commented Muthiah Vaduganathan, MD, a cardiologist at Brigham and Women’s Hospital in Boston.
The impact of dapagliflozin on average A1c levels during the DAPA-HF trial was minimal, reducing levels by an average of 0.04% among those who entered with prediabetes and by 0.05% among the other patients. This suggests that the mechanisms by which dapagliflozin reduced incident diabetes was by routes that did not involve simply reducing hyperglycemia, and the observed decrease in incident diabetes was not apparently caused by “masking” of hyperglycemia by dapagliflozin, said Dr. Inzucchi.
One possibility is that dapagliflozin, which also improved quality of life and reduced hospitalizations in the DAPA-HF trial, led to improved function and mobility among patients that had beneficial effects on their insulin sensitivity, Dr. Vaduganathan speculated in an interview.
The new finding of dapagliflozin’s benefit “is great news,” commented Yehuda Handelsman, MD, an endocrinologist and diabetes specialist who is medical director of the Metabolic Institute of America in Tarzana, Calif. “It’s an impressive and important result, and another reason to use dapagliflozin in patients with HFrEF, a group of patients whom you want to prevent from having worse outcomes” by developing diabetes.
The DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial enrolled HFrEF patients at 410 centers in 20 countries during February 2017–August 2018. The study’s primary endpoint was the composite incidence of cardiovascular death or worsening heart failure, which occurred in 16.3% of patients randomized to receive dapagliflozin and in 21.2% of control patients on standard care but on placebo instead of the study drug, a statistically significant relative risk reduction of 26% (N Engl J Med. 2019 Nov 21;381[21]:1995-2008). In the 2,605-patient subgroup without type 2 diabetes at baseline the primary endpoint fell by a statistically significant 27% with dapagliflozin treatment, the first time an SGLT2 inhibitor drug was shown effective for reducing this endpoint in patients with HFrEF but without diabetes. DAPA-HF did not enroll any patients with type 1 diabetes.
DAPA-HF was sponsored by AstraZeneca, the company that markets dapagliflozin (Farxiga). Dr. Inzucchi has been a consultant to AstraZeneca and to Abbott, Boehringer Ingelheim, Merck, Novo Nordisk, Sanofi/Lexicon, and vTv Therapeutics. Dr. Vaduganathan has been an adviser to AstraZeneca and to Amgen, Baxter, Bayer, Boehringer Ingelheim, Cytokinetics, and Relypsa. Dr. Handelsman has been a consultant to several drug companies including AstraZeneca.
SOURCE: Inzucchi SE et al. ADA 2020, abstract 271-OR.
FROM ADA 2020
FDA revokes emergency use of hydroxychloroquine
The U.S. Food and Drug Administration revoked its decision from March 28 allowing use of hydroxychloroquine and chloroquine to treat people hospitalized with COVID-19 under an emergency use authorization (EUA).
“Based on its ongoing analysis of the EUA and emerging scientific data, the FDA determined that chloroquine and hydroxychloroquine are unlikely to be effective in treating COVID-19 for the authorized uses in the EUA,” the agency announced in a June 15 statement.
The FDA also warned today that the use of hydroxychloroquine or chloroquine may have a potential drug interaction with the investigational antiviral drug remdesivir that limits its effectiveness against COVID-19.
Remdesivir was granted emergency use authorization by the FDA on May 1.
“Based on a recently completed nonclinical laboratory study, the FDA is revising the fact sheet for healthcare providers that accompanies the drug to state that coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulfate is not recommended as it may result in reduced antiviral activity of remdesivir. The agency is not aware of instances of this reduced activity occurring in the clinical setting but is continuing to evaluate all data related to remdesivir,” the FDA said in a news release.
Controversy over hydroxychloroquine
Even with such federal permission, since late March the use of these two agents has been mired in controversy.
President Donald J. Trump promoted the use of hydroxychloroquine and chloroquine to treat Americans with COVID-19, while scientific studies raised questions about their safety and effectiveness. Recent research, for example, pointed to elevated cardiovascular risks, as reported by Medscape Medical News.
The FDA acknowledged this recent evidence. “Additionally, in light of ongoing serious cardiac adverse events and other potential serious side effects, the known and potential benefits of chloroquine and hydroxychloroquine no longer outweigh the known and potential risks for the authorized use.”
The full suspension of the EUA follows a warning the agency issued on April 24. The FDA’s Safety Communication cautioned against use of the two agents outside of a hospital setting, citing an increase in outpatient prescriptions and “reports of serious heart rhythm problems.”
“While additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19, we determined the emergency use authorization was no longer appropriate,” based on a rigorous assessment by scientists in our Center for Drug Evaluation and Research,” Patrizia Cavazzoni, MD, acting director of CDER, noted in the FDA statement.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration revoked its decision from March 28 allowing use of hydroxychloroquine and chloroquine to treat people hospitalized with COVID-19 under an emergency use authorization (EUA).
“Based on its ongoing analysis of the EUA and emerging scientific data, the FDA determined that chloroquine and hydroxychloroquine are unlikely to be effective in treating COVID-19 for the authorized uses in the EUA,” the agency announced in a June 15 statement.
The FDA also warned today that the use of hydroxychloroquine or chloroquine may have a potential drug interaction with the investigational antiviral drug remdesivir that limits its effectiveness against COVID-19.
Remdesivir was granted emergency use authorization by the FDA on May 1.
“Based on a recently completed nonclinical laboratory study, the FDA is revising the fact sheet for healthcare providers that accompanies the drug to state that coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulfate is not recommended as it may result in reduced antiviral activity of remdesivir. The agency is not aware of instances of this reduced activity occurring in the clinical setting but is continuing to evaluate all data related to remdesivir,” the FDA said in a news release.
Controversy over hydroxychloroquine
Even with such federal permission, since late March the use of these two agents has been mired in controversy.
President Donald J. Trump promoted the use of hydroxychloroquine and chloroquine to treat Americans with COVID-19, while scientific studies raised questions about their safety and effectiveness. Recent research, for example, pointed to elevated cardiovascular risks, as reported by Medscape Medical News.
The FDA acknowledged this recent evidence. “Additionally, in light of ongoing serious cardiac adverse events and other potential serious side effects, the known and potential benefits of chloroquine and hydroxychloroquine no longer outweigh the known and potential risks for the authorized use.”
The full suspension of the EUA follows a warning the agency issued on April 24. The FDA’s Safety Communication cautioned against use of the two agents outside of a hospital setting, citing an increase in outpatient prescriptions and “reports of serious heart rhythm problems.”
“While additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19, we determined the emergency use authorization was no longer appropriate,” based on a rigorous assessment by scientists in our Center for Drug Evaluation and Research,” Patrizia Cavazzoni, MD, acting director of CDER, noted in the FDA statement.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration revoked its decision from March 28 allowing use of hydroxychloroquine and chloroquine to treat people hospitalized with COVID-19 under an emergency use authorization (EUA).
“Based on its ongoing analysis of the EUA and emerging scientific data, the FDA determined that chloroquine and hydroxychloroquine are unlikely to be effective in treating COVID-19 for the authorized uses in the EUA,” the agency announced in a June 15 statement.
The FDA also warned today that the use of hydroxychloroquine or chloroquine may have a potential drug interaction with the investigational antiviral drug remdesivir that limits its effectiveness against COVID-19.
Remdesivir was granted emergency use authorization by the FDA on May 1.
“Based on a recently completed nonclinical laboratory study, the FDA is revising the fact sheet for healthcare providers that accompanies the drug to state that coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulfate is not recommended as it may result in reduced antiviral activity of remdesivir. The agency is not aware of instances of this reduced activity occurring in the clinical setting but is continuing to evaluate all data related to remdesivir,” the FDA said in a news release.
Controversy over hydroxychloroquine
Even with such federal permission, since late March the use of these two agents has been mired in controversy.
President Donald J. Trump promoted the use of hydroxychloroquine and chloroquine to treat Americans with COVID-19, while scientific studies raised questions about their safety and effectiveness. Recent research, for example, pointed to elevated cardiovascular risks, as reported by Medscape Medical News.
The FDA acknowledged this recent evidence. “Additionally, in light of ongoing serious cardiac adverse events and other potential serious side effects, the known and potential benefits of chloroquine and hydroxychloroquine no longer outweigh the known and potential risks for the authorized use.”
The full suspension of the EUA follows a warning the agency issued on April 24. The FDA’s Safety Communication cautioned against use of the two agents outside of a hospital setting, citing an increase in outpatient prescriptions and “reports of serious heart rhythm problems.”
“While additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19, we determined the emergency use authorization was no longer appropriate,” based on a rigorous assessment by scientists in our Center for Drug Evaluation and Research,” Patrizia Cavazzoni, MD, acting director of CDER, noted in the FDA statement.
This article first appeared on Medscape.com.
No OS benefit with gefitinib vs. chemo for EGFR+ NSCLC
The median OS was 75.5 months in patients randomized to adjuvant gefitinib and 62.8 months in patients randomized to vinorelbine plus cisplatin.
Yi-Long Wu, MD, of Guangdong Lung Cancer Institute in Guangzhou, China, reported these results as part of the American Society of Clinical Oncology virtual scientific program.
Prior results from this trial had shown a disease-free survival (DFS) benefit with gefitinib, but this did not translate to an OS benefit at the final analysis, Dr. Wu said.
He noted, however, that the median OS of 75.5 months in the gefitinib arm “was one of the best in resected EGFR-mutant non–small cell lung cancer, compared with historical data.”
The findings also suggest a possible benefit with at least 18 months of gefitinib and show that adjuvant EGFR tyrosine kinase inhibitors (TKIs) should be considered the optimal therapy to improve DFS and achieve potentially better OS in this setting, Dr. Wu said.
Study details and DFS
The ADJUVANT trial (NCT01405079) randomized 222 patients, aged 18-75 years, with EGFR-mutant, stage II-IIIA (N1-N2) NSCLC who had undergone complete resection. Patients were enrolled at 27 sites between September 2011 and April 2014.
The patients were randomized 1:1 to receive 250 mg of gefitinib once daily for 24 months, or 25 mg/m2 of vinorelbine on days 1 and 8 plus 75 mg/m2 of cisplatin on day 1 every 3 weeks for 4 cycles.
The intent-to-treat (ITT) population included 111 patients in each arm. The per-protocol population included 106 patients in the gefitinib arm and 87 patients in the chemotherapy arm.
Primary results from this trial showed a significant improvement in DFS with gefitinib (Lancet Oncol. 2018 Jan;19[1]:139-48). That improvement was maintained in the final analysis.
The median DFS was 30.8 months in the gefitinib arm and 19.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The hazard ratio (HR) was 0.56 (P = .001) in the ITT population and 0.51 (P < .001) in the per-protocol population.
In the ITT population, the 5-year DFS rates were 22.6% in the gefitinib arm and 23.2% in the chemotherapy arm. In the per-protocol population, the 5-year DFS rates were 22.6% and 22.8%, respectively.
OS results
The median OS was 75.5 months in the gefitinib arm and 62.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The HR was 0.92 in both the ITT (P = .674) and per-protocol populations (P = .686).
In the ITT population, the 5-year OS rates were 53.2% in the gefitinib arm and 51.2% in the chemotherapy arm. In the per-protocol population, the 5-year OS rates were 53.2% and 50.7%, respectively.
Subgroup analyses by age, gender, lymph node status, and EGFR mutation showed trends toward improved OS with gefitinib, but the differences were not statistically significant.
The researchers conducted a post hoc analysis to assess the effect of subsequent treatment on patient outcomes. The analysis showed that patients who received gefitinib with subsequent EGFR-TKIs had the best responses and OS.
The median OS was not reached among patients who received gefitinib and subsequent EGFR-TKIs, whereas the median OS ranged from 15.6 months to 62.8 months in other groups. The shortest OS was observed in patients who received adjuvant chemotherapy without subsequent therapy.
The duration of gefitinib treatment also appeared to affect OS. The median OS was 35.7 months in patients who received gefitinib for less than 18 months, and the median OS was not reached in patients who received gefitinib for 18 months or longer (HR, 0.38; P < .001).
Implications and potential next steps
Despite the lack of OS improvement with gefitinib, “all of the patients on this study did much, much better than historical non–small cell lung cancer not specified by the EGFR mutation, with 70 months median survival compared to 35 months median survival for N2-positive disease,” said invited discussant Christopher G. Azzoli, MD, director of thoracic oncology at Lifespan Cancer Institute at Brown University in Providence, R.I.
“But you can’t avoid noticing how the curves come back together in terms of disease-free survival when your effective treatment is limited to 24 months,” he added.
An apparent risk of late brain recurrence in the gefitinib arm is also a concern, Dr. Azzoli said. “So ... longer duration of treatment with a drug that has better control of CNS [central nervous system] disease, such as osimertinib, may improve both DFS and OS,” he added.
Only about 50% of patients in the chemotherapy arm received a TKI at recurrence. The post hoc analysis showing that TKI recipients had the best outcomes raises the question of whether “the survival benefit could be conferred by delivering a superior drug merely at recurrence, or is there benefit to earlier delivery of an effective drug,” Dr. Azzoli said.
Given the high cost of continuous therapy, biomarker refinement could help improve treatment decision-making, he said, noting that “early testing of blood DNA to detect cancer in the body as minimal residual disease is showing promise,” and that many phase 3 studies of EGFR-TKIs are ongoing.
The current trial was sponsored by the Guangdong Association of Clinical Trials. Dr. Wu disclosed relationships with AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/China, Lilly, MSD Oncology, Pfizer, and Roche. Dr. Azzoli reported having no disclosures.
SOURCE: Wu Y et al. ASCO 2020, Abstract 9005.
The median OS was 75.5 months in patients randomized to adjuvant gefitinib and 62.8 months in patients randomized to vinorelbine plus cisplatin.
Yi-Long Wu, MD, of Guangdong Lung Cancer Institute in Guangzhou, China, reported these results as part of the American Society of Clinical Oncology virtual scientific program.
Prior results from this trial had shown a disease-free survival (DFS) benefit with gefitinib, but this did not translate to an OS benefit at the final analysis, Dr. Wu said.
He noted, however, that the median OS of 75.5 months in the gefitinib arm “was one of the best in resected EGFR-mutant non–small cell lung cancer, compared with historical data.”
The findings also suggest a possible benefit with at least 18 months of gefitinib and show that adjuvant EGFR tyrosine kinase inhibitors (TKIs) should be considered the optimal therapy to improve DFS and achieve potentially better OS in this setting, Dr. Wu said.
Study details and DFS
The ADJUVANT trial (NCT01405079) randomized 222 patients, aged 18-75 years, with EGFR-mutant, stage II-IIIA (N1-N2) NSCLC who had undergone complete resection. Patients were enrolled at 27 sites between September 2011 and April 2014.
The patients were randomized 1:1 to receive 250 mg of gefitinib once daily for 24 months, or 25 mg/m2 of vinorelbine on days 1 and 8 plus 75 mg/m2 of cisplatin on day 1 every 3 weeks for 4 cycles.
The intent-to-treat (ITT) population included 111 patients in each arm. The per-protocol population included 106 patients in the gefitinib arm and 87 patients in the chemotherapy arm.
Primary results from this trial showed a significant improvement in DFS with gefitinib (Lancet Oncol. 2018 Jan;19[1]:139-48). That improvement was maintained in the final analysis.
The median DFS was 30.8 months in the gefitinib arm and 19.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The hazard ratio (HR) was 0.56 (P = .001) in the ITT population and 0.51 (P < .001) in the per-protocol population.
In the ITT population, the 5-year DFS rates were 22.6% in the gefitinib arm and 23.2% in the chemotherapy arm. In the per-protocol population, the 5-year DFS rates were 22.6% and 22.8%, respectively.
OS results
The median OS was 75.5 months in the gefitinib arm and 62.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The HR was 0.92 in both the ITT (P = .674) and per-protocol populations (P = .686).
In the ITT population, the 5-year OS rates were 53.2% in the gefitinib arm and 51.2% in the chemotherapy arm. In the per-protocol population, the 5-year OS rates were 53.2% and 50.7%, respectively.
Subgroup analyses by age, gender, lymph node status, and EGFR mutation showed trends toward improved OS with gefitinib, but the differences were not statistically significant.
The researchers conducted a post hoc analysis to assess the effect of subsequent treatment on patient outcomes. The analysis showed that patients who received gefitinib with subsequent EGFR-TKIs had the best responses and OS.
The median OS was not reached among patients who received gefitinib and subsequent EGFR-TKIs, whereas the median OS ranged from 15.6 months to 62.8 months in other groups. The shortest OS was observed in patients who received adjuvant chemotherapy without subsequent therapy.
The duration of gefitinib treatment also appeared to affect OS. The median OS was 35.7 months in patients who received gefitinib for less than 18 months, and the median OS was not reached in patients who received gefitinib for 18 months or longer (HR, 0.38; P < .001).
Implications and potential next steps
Despite the lack of OS improvement with gefitinib, “all of the patients on this study did much, much better than historical non–small cell lung cancer not specified by the EGFR mutation, with 70 months median survival compared to 35 months median survival for N2-positive disease,” said invited discussant Christopher G. Azzoli, MD, director of thoracic oncology at Lifespan Cancer Institute at Brown University in Providence, R.I.
“But you can’t avoid noticing how the curves come back together in terms of disease-free survival when your effective treatment is limited to 24 months,” he added.
An apparent risk of late brain recurrence in the gefitinib arm is also a concern, Dr. Azzoli said. “So ... longer duration of treatment with a drug that has better control of CNS [central nervous system] disease, such as osimertinib, may improve both DFS and OS,” he added.
Only about 50% of patients in the chemotherapy arm received a TKI at recurrence. The post hoc analysis showing that TKI recipients had the best outcomes raises the question of whether “the survival benefit could be conferred by delivering a superior drug merely at recurrence, or is there benefit to earlier delivery of an effective drug,” Dr. Azzoli said.
Given the high cost of continuous therapy, biomarker refinement could help improve treatment decision-making, he said, noting that “early testing of blood DNA to detect cancer in the body as minimal residual disease is showing promise,” and that many phase 3 studies of EGFR-TKIs are ongoing.
The current trial was sponsored by the Guangdong Association of Clinical Trials. Dr. Wu disclosed relationships with AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/China, Lilly, MSD Oncology, Pfizer, and Roche. Dr. Azzoli reported having no disclosures.
SOURCE: Wu Y et al. ASCO 2020, Abstract 9005.
The median OS was 75.5 months in patients randomized to adjuvant gefitinib and 62.8 months in patients randomized to vinorelbine plus cisplatin.
Yi-Long Wu, MD, of Guangdong Lung Cancer Institute in Guangzhou, China, reported these results as part of the American Society of Clinical Oncology virtual scientific program.
Prior results from this trial had shown a disease-free survival (DFS) benefit with gefitinib, but this did not translate to an OS benefit at the final analysis, Dr. Wu said.
He noted, however, that the median OS of 75.5 months in the gefitinib arm “was one of the best in resected EGFR-mutant non–small cell lung cancer, compared with historical data.”
The findings also suggest a possible benefit with at least 18 months of gefitinib and show that adjuvant EGFR tyrosine kinase inhibitors (TKIs) should be considered the optimal therapy to improve DFS and achieve potentially better OS in this setting, Dr. Wu said.
Study details and DFS
The ADJUVANT trial (NCT01405079) randomized 222 patients, aged 18-75 years, with EGFR-mutant, stage II-IIIA (N1-N2) NSCLC who had undergone complete resection. Patients were enrolled at 27 sites between September 2011 and April 2014.
The patients were randomized 1:1 to receive 250 mg of gefitinib once daily for 24 months, or 25 mg/m2 of vinorelbine on days 1 and 8 plus 75 mg/m2 of cisplatin on day 1 every 3 weeks for 4 cycles.
The intent-to-treat (ITT) population included 111 patients in each arm. The per-protocol population included 106 patients in the gefitinib arm and 87 patients in the chemotherapy arm.
Primary results from this trial showed a significant improvement in DFS with gefitinib (Lancet Oncol. 2018 Jan;19[1]:139-48). That improvement was maintained in the final analysis.
The median DFS was 30.8 months in the gefitinib arm and 19.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The hazard ratio (HR) was 0.56 (P = .001) in the ITT population and 0.51 (P < .001) in the per-protocol population.
In the ITT population, the 5-year DFS rates were 22.6% in the gefitinib arm and 23.2% in the chemotherapy arm. In the per-protocol population, the 5-year DFS rates were 22.6% and 22.8%, respectively.
OS results
The median OS was 75.5 months in the gefitinib arm and 62.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The HR was 0.92 in both the ITT (P = .674) and per-protocol populations (P = .686).
In the ITT population, the 5-year OS rates were 53.2% in the gefitinib arm and 51.2% in the chemotherapy arm. In the per-protocol population, the 5-year OS rates were 53.2% and 50.7%, respectively.
Subgroup analyses by age, gender, lymph node status, and EGFR mutation showed trends toward improved OS with gefitinib, but the differences were not statistically significant.
The researchers conducted a post hoc analysis to assess the effect of subsequent treatment on patient outcomes. The analysis showed that patients who received gefitinib with subsequent EGFR-TKIs had the best responses and OS.
The median OS was not reached among patients who received gefitinib and subsequent EGFR-TKIs, whereas the median OS ranged from 15.6 months to 62.8 months in other groups. The shortest OS was observed in patients who received adjuvant chemotherapy without subsequent therapy.
The duration of gefitinib treatment also appeared to affect OS. The median OS was 35.7 months in patients who received gefitinib for less than 18 months, and the median OS was not reached in patients who received gefitinib for 18 months or longer (HR, 0.38; P < .001).
Implications and potential next steps
Despite the lack of OS improvement with gefitinib, “all of the patients on this study did much, much better than historical non–small cell lung cancer not specified by the EGFR mutation, with 70 months median survival compared to 35 months median survival for N2-positive disease,” said invited discussant Christopher G. Azzoli, MD, director of thoracic oncology at Lifespan Cancer Institute at Brown University in Providence, R.I.
“But you can’t avoid noticing how the curves come back together in terms of disease-free survival when your effective treatment is limited to 24 months,” he added.
An apparent risk of late brain recurrence in the gefitinib arm is also a concern, Dr. Azzoli said. “So ... longer duration of treatment with a drug that has better control of CNS [central nervous system] disease, such as osimertinib, may improve both DFS and OS,” he added.
Only about 50% of patients in the chemotherapy arm received a TKI at recurrence. The post hoc analysis showing that TKI recipients had the best outcomes raises the question of whether “the survival benefit could be conferred by delivering a superior drug merely at recurrence, or is there benefit to earlier delivery of an effective drug,” Dr. Azzoli said.
Given the high cost of continuous therapy, biomarker refinement could help improve treatment decision-making, he said, noting that “early testing of blood DNA to detect cancer in the body as minimal residual disease is showing promise,” and that many phase 3 studies of EGFR-TKIs are ongoing.
The current trial was sponsored by the Guangdong Association of Clinical Trials. Dr. Wu disclosed relationships with AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/China, Lilly, MSD Oncology, Pfizer, and Roche. Dr. Azzoli reported having no disclosures.
SOURCE: Wu Y et al. ASCO 2020, Abstract 9005.
FROM ASCO 2020
Preliminary evidence indicates famotidine might improve COVID-19 symptoms
High-dose oral famotidine might improve cardinal symptoms of COVID-19 infection, according to the findings of a small outpatient case series and a subsequent retrospective study.
After developing COVID-19 symptoms, the 10 patients in the case series began self-medicating with 60-240 mg famotidine daily over a median of 11 days. “All patients reported marked improvements of disease-related symptoms after starting famotidine,” first author Tobias Janowitz, MD, PhD, of Cold Spring Harbor Laboratory, N.Y., and associates wrote in Gut.
Improvements began within 24-48 hours of starting on the histamine-2 receptor antagonist. By 14 days after treatment initiation, all patients reported near-normalization of both respiratory and systemic symptoms, the researchers reported.
The patients were 23-71 years old. Seven tested positive for COVID-19, two had antibodies to COVID-19, and one had a clinical diagnosis of COVID-19 without laboratory confirmation. Over a median of 11 days (range, 5-21 days), six patients self-administered 80 mg famotidine three times daily and four self-administered lower amounts – from 60 to 150 mg of famotidine daily, divided into two or three doses. Patients started on famotidine between 2 and 26 days after symptom onset.
Through phone interviews and questionnaires, the researchers ascertained changes in cough, dyspnea, fatigue, headache, anosmia, and general unwellness by using a modified four-point Eastern Cooperative Oncology Group (ECOG) performance status scale. Improvements were seen across all symptom categories, and respiratory symptoms improved faster than systemic symptoms. Apart from two cases of persistent anosmia, symptoms resolved completely within 14 days of starting famotidine.
Seven patients reported no side effects of famotidine; one reported grade 1 dizziness and infrequent perceptions of tachycardia; one reported grade 1 dizziness, dry skin, and insomnia; and one reported grade 1 gastrointestinal symptoms and temporary forgetfulness. “Other than forgetfulness, all of these side effects are listed in the prescription information for famotidine, and all side effects resolved on discontinuation of famotidine,” the investigators wrote.
While the findings are intriguing, Dr. Janowitz and associates cautioned against overinterpretation of them. Another expert agreed: “This is a preliminary study based on a hypothesized antiviral effect. It’s important to know that it doesn’t really prove it works,” said Amesh Adalja, MD, senior scholar at the Johns Hopkins University Center for Health Security, Baltimore, and a spokesperson for the Infectious Diseases Society of America, during an interview with MDedge.
These patients might have improved anyway, without self-administering famotidine, said Dr. Adalja, who was not involved in the study.
Furthermore, the mechanism by which famotidine might act on COVID-19 remains unclear. The drug “could have a viral target, for example, one of the viral proteases, or a host target, resulting, for example, in modulation of the immunological response to the virus,” Dr. Janowitz and associates wrote.
Dr. Adalja noted that many compounds show effects against COVID-19 that are not well understood. He called for randomized trials to evaluate the biological plausibility of famotidine use, and its potential efficacy.
“This is a cheap, over-the-counter drug, but no drug is without side effects,” he added. “We need to know whether it works.”
Based on the case series findings, researchers conducted another retrospective study of patients hospitalized with COVID-19 infection. Those who were incidentally taking famotidine before or at hospitalization had a significantly reduced risk of intubation or death, with a hazard ratio of 0.43 (Gastroenterology. 2020 May 22. doi: 10.1053/j.gastro.2020.05.053)
The National Institutes of Health provided partial support. The investigators reported having no conflicts of interest.
SOURCE: Janowitz T et al. Gut. 2020 Jun 4. doi: 10.1136/gutjnl-2020-321852.
High-dose oral famotidine might improve cardinal symptoms of COVID-19 infection, according to the findings of a small outpatient case series and a subsequent retrospective study.
After developing COVID-19 symptoms, the 10 patients in the case series began self-medicating with 60-240 mg famotidine daily over a median of 11 days. “All patients reported marked improvements of disease-related symptoms after starting famotidine,” first author Tobias Janowitz, MD, PhD, of Cold Spring Harbor Laboratory, N.Y., and associates wrote in Gut.
Improvements began within 24-48 hours of starting on the histamine-2 receptor antagonist. By 14 days after treatment initiation, all patients reported near-normalization of both respiratory and systemic symptoms, the researchers reported.
The patients were 23-71 years old. Seven tested positive for COVID-19, two had antibodies to COVID-19, and one had a clinical diagnosis of COVID-19 without laboratory confirmation. Over a median of 11 days (range, 5-21 days), six patients self-administered 80 mg famotidine three times daily and four self-administered lower amounts – from 60 to 150 mg of famotidine daily, divided into two or three doses. Patients started on famotidine between 2 and 26 days after symptom onset.
Through phone interviews and questionnaires, the researchers ascertained changes in cough, dyspnea, fatigue, headache, anosmia, and general unwellness by using a modified four-point Eastern Cooperative Oncology Group (ECOG) performance status scale. Improvements were seen across all symptom categories, and respiratory symptoms improved faster than systemic symptoms. Apart from two cases of persistent anosmia, symptoms resolved completely within 14 days of starting famotidine.
Seven patients reported no side effects of famotidine; one reported grade 1 dizziness and infrequent perceptions of tachycardia; one reported grade 1 dizziness, dry skin, and insomnia; and one reported grade 1 gastrointestinal symptoms and temporary forgetfulness. “Other than forgetfulness, all of these side effects are listed in the prescription information for famotidine, and all side effects resolved on discontinuation of famotidine,” the investigators wrote.
While the findings are intriguing, Dr. Janowitz and associates cautioned against overinterpretation of them. Another expert agreed: “This is a preliminary study based on a hypothesized antiviral effect. It’s important to know that it doesn’t really prove it works,” said Amesh Adalja, MD, senior scholar at the Johns Hopkins University Center for Health Security, Baltimore, and a spokesperson for the Infectious Diseases Society of America, during an interview with MDedge.
These patients might have improved anyway, without self-administering famotidine, said Dr. Adalja, who was not involved in the study.
Furthermore, the mechanism by which famotidine might act on COVID-19 remains unclear. The drug “could have a viral target, for example, one of the viral proteases, or a host target, resulting, for example, in modulation of the immunological response to the virus,” Dr. Janowitz and associates wrote.
Dr. Adalja noted that many compounds show effects against COVID-19 that are not well understood. He called for randomized trials to evaluate the biological plausibility of famotidine use, and its potential efficacy.
“This is a cheap, over-the-counter drug, but no drug is without side effects,” he added. “We need to know whether it works.”
Based on the case series findings, researchers conducted another retrospective study of patients hospitalized with COVID-19 infection. Those who were incidentally taking famotidine before or at hospitalization had a significantly reduced risk of intubation or death, with a hazard ratio of 0.43 (Gastroenterology. 2020 May 22. doi: 10.1053/j.gastro.2020.05.053)
The National Institutes of Health provided partial support. The investigators reported having no conflicts of interest.
SOURCE: Janowitz T et al. Gut. 2020 Jun 4. doi: 10.1136/gutjnl-2020-321852.
High-dose oral famotidine might improve cardinal symptoms of COVID-19 infection, according to the findings of a small outpatient case series and a subsequent retrospective study.
After developing COVID-19 symptoms, the 10 patients in the case series began self-medicating with 60-240 mg famotidine daily over a median of 11 days. “All patients reported marked improvements of disease-related symptoms after starting famotidine,” first author Tobias Janowitz, MD, PhD, of Cold Spring Harbor Laboratory, N.Y., and associates wrote in Gut.
Improvements began within 24-48 hours of starting on the histamine-2 receptor antagonist. By 14 days after treatment initiation, all patients reported near-normalization of both respiratory and systemic symptoms, the researchers reported.
The patients were 23-71 years old. Seven tested positive for COVID-19, two had antibodies to COVID-19, and one had a clinical diagnosis of COVID-19 without laboratory confirmation. Over a median of 11 days (range, 5-21 days), six patients self-administered 80 mg famotidine three times daily and four self-administered lower amounts – from 60 to 150 mg of famotidine daily, divided into two or three doses. Patients started on famotidine between 2 and 26 days after symptom onset.
Through phone interviews and questionnaires, the researchers ascertained changes in cough, dyspnea, fatigue, headache, anosmia, and general unwellness by using a modified four-point Eastern Cooperative Oncology Group (ECOG) performance status scale. Improvements were seen across all symptom categories, and respiratory symptoms improved faster than systemic symptoms. Apart from two cases of persistent anosmia, symptoms resolved completely within 14 days of starting famotidine.
Seven patients reported no side effects of famotidine; one reported grade 1 dizziness and infrequent perceptions of tachycardia; one reported grade 1 dizziness, dry skin, and insomnia; and one reported grade 1 gastrointestinal symptoms and temporary forgetfulness. “Other than forgetfulness, all of these side effects are listed in the prescription information for famotidine, and all side effects resolved on discontinuation of famotidine,” the investigators wrote.
While the findings are intriguing, Dr. Janowitz and associates cautioned against overinterpretation of them. Another expert agreed: “This is a preliminary study based on a hypothesized antiviral effect. It’s important to know that it doesn’t really prove it works,” said Amesh Adalja, MD, senior scholar at the Johns Hopkins University Center for Health Security, Baltimore, and a spokesperson for the Infectious Diseases Society of America, during an interview with MDedge.
These patients might have improved anyway, without self-administering famotidine, said Dr. Adalja, who was not involved in the study.
Furthermore, the mechanism by which famotidine might act on COVID-19 remains unclear. The drug “could have a viral target, for example, one of the viral proteases, or a host target, resulting, for example, in modulation of the immunological response to the virus,” Dr. Janowitz and associates wrote.
Dr. Adalja noted that many compounds show effects against COVID-19 that are not well understood. He called for randomized trials to evaluate the biological plausibility of famotidine use, and its potential efficacy.
“This is a cheap, over-the-counter drug, but no drug is without side effects,” he added. “We need to know whether it works.”
Based on the case series findings, researchers conducted another retrospective study of patients hospitalized with COVID-19 infection. Those who were incidentally taking famotidine before or at hospitalization had a significantly reduced risk of intubation or death, with a hazard ratio of 0.43 (Gastroenterology. 2020 May 22. doi: 10.1053/j.gastro.2020.05.053)
The National Institutes of Health provided partial support. The investigators reported having no conflicts of interest.
SOURCE: Janowitz T et al. Gut. 2020 Jun 4. doi: 10.1136/gutjnl-2020-321852.
FROM GUT
Analysis of Pharmacist Interventions Used to Resolve Safety Target of Polypharmacy (STOP) Drug Interactions
Statins are one of the most common medications dispensed in the US and are associated with clinically significant drug interactions.1,2 The most common adverse drug reaction (ADR) of statin drug interactions is muscle-related toxicities.2 Despite technology advances to alert clinicians to drug interactions, updated statin manufacturer labeling, and guideline recommendations, inappropriate prescribing and dispensing of statin drug interactions continues to occur in health care systems.2-10
The medical literature has demonstrated many opportunities for pharmacists to prevent and mitigate drug interactions. At the points of prescribing and dispensing, pharmacists can reduce the number of potential drug interactions for the patient.11-13 Pharmacists also have identified and resolved drug interactions through quality assurance review after dispensing to a patient.7,8
Regardless of the time point of an intervention, the most common method pharmacists used to resolve drug interactions was through recommendations to a prescriber. The recommendations were generated through academic detailing, clinical decision support algorithms, drug conversions, or the pharmacist’s expertise. Regardless of the method the pharmacist used, the prescriber had the final authority to accept or decline the recommendation.7,8,11-13 Although these interventions were effective, pharmacists could further streamline the process by autonomously resolving drug interactions. However, these types of interventions are not well described in the medical literature.
Background
The US Department of Veterans Affairs (VA) Veterans Integrated Service Network (VISN), established the Safety Target of Polypharmacy (STOP) report in 2015. At each facility in the network, the report identified patients who were dispensed medications known to have drug interactions. The interactions were chosen by the VISN, and the severity of the interactions was based on coding parameters within the VA computerized order entry system, which uses a severity score based on First Databank data. At the Harry S. Truman Memorial Veterans’ Hospital (Truman VA) in Columbia, Missouri, > 500 drug interactions were initially active on the STOP report. The most common drug interactions were statins with gemfibrozil and statins with niacin.14-18 The Truman VA Pharmacy Service was charged with resolving the interactions for the facility.
The Truman VA employs 3 Patient Aligned Care Team (PACT) Clinical Pharmacy Specialists (CPS) practicing within primary care clinics. PACT is the patientcentered medical home model used by the VA. PACT CPS are ambulatory care pharmacists who assist providers in managing diseases using a scope of practice. Having a scope of practice would have allowed the PACT CPS to manage drug interactions with independent prescribing authority. However, due to the high volume of STOP report interactions and limited PACT CPS resources, the Pharmacy Service needed to develop an efficient, patient-centered method to resolve them. The intervention also needed to allow pharmacists, both with and without a scope of practice, to address the interactions.
Methods
The Truman VA Pharmacy Service developed protocols, approved by the Pharmacy and Therapeutics (P&T) Committee, to manage the specific gemfibrozil-statin and niacinstatin interactions chosen for the VISN 15 STOP report (Figures 1 and 2). The protocols were designed to identify patients who did not have a clear indication for gemfibrozil or niacin, were likely to maintain triglycerides (TGs) < 500 mg/dL without these medications, and would not likely require close monitoring after discontinuation.19 The protocols allowed pharmacists to autonomously discontinue gemfibrozil or niacin if patients did not have a history of pancreatitis, TGs ≥ 400 mg/dL or a nonlipid indication for niacin (eg, pellagra) after establishing care at Truman VA. Additionally, both interacting medications had to be dispensed by the VA. When pharmacists discontinued a medication, it was documented in a note in the patient electronic health record. The prescriber was notified through the note and the patient received a notification letter. Follow-up laboratory monitoring was not required as part of the protocol.
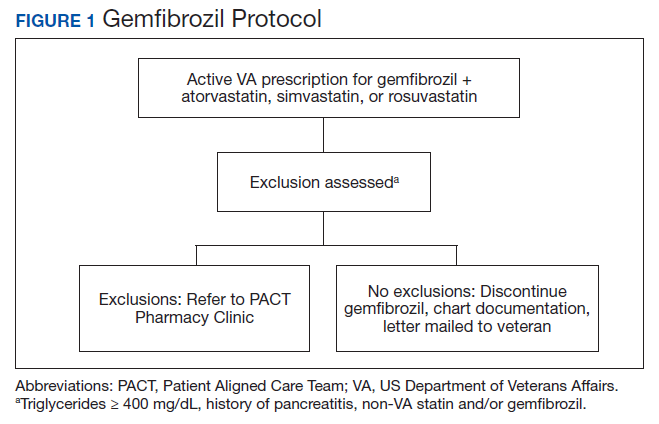
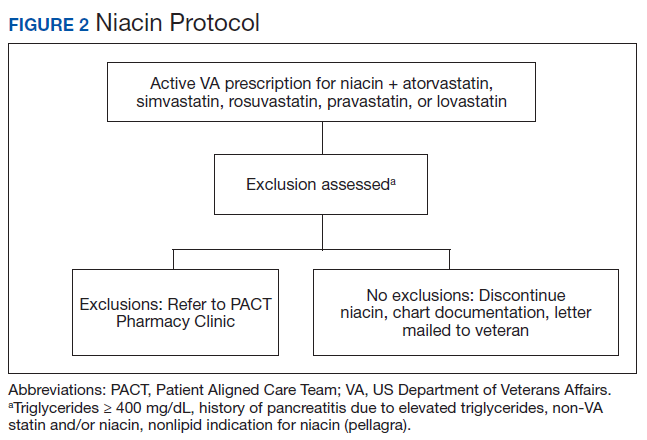
If patients met any of the exclusion criteria for discontinuation, the primary care provider (PCP) was notified to place a consult to the PACT Pharmacy Clinic for individualized interventions and close monitoring. Patients prescribed niacin for nonlipid indications were allowed to continue with their current drug regimen. At each encounter, the PACT CPS assessed for ADRs, made individualized medication changes, and arranged follow-up appointments. Once the interaction was resolved and treatment goals met, the PCP resumed monitoring of the patient’s lipid therapy.
Following all pharmacist interventions, a retrospective quality improvement analysis was conducted. The primary outcome was to evaluate the impact of discontinuing gemfibrozil and niacin by protocol on patients’ laboratory results. The coprimary endpoints were to describe the change in TG levels and the percentage of patients with TGs ≥ 500 mg/dL at least 5 weeks following the pharmacist-directed discontinuation by protocol. Secondary outcomes included the time required to resolve the interactions and a description of the PACT CPS pharmacologic interventions. Additionally, a quality assurance peer review was used to ensure the pharmacists appropriately utilized the protocols.
Data were collected from August 2016 to September 2017 for patients prescribed gemfibrozil and from May 2017 to January 2018 for patients prescribed niacin. The time spent resolving interactions was quantified based on encounter data. Descriptive statistics were used to analyze demographic information and the endpoints associated with each outcome. The project was reviewed by the University of Missouri Institutional Review Board, Truman VA privacy and information security officers, and was determined to meet guidelines for quality improvement.
Results
The original STOP report included 397 drug interactions involving statins with gemfibrozil or niacin (Table 1). The majority of patients were white and male aged 60 to 79 years. Gemfibrozil was the most common drug involved in all interactions (79.8%). The most common statins were atorvastatin (40%) and simvastatin (36.5%).
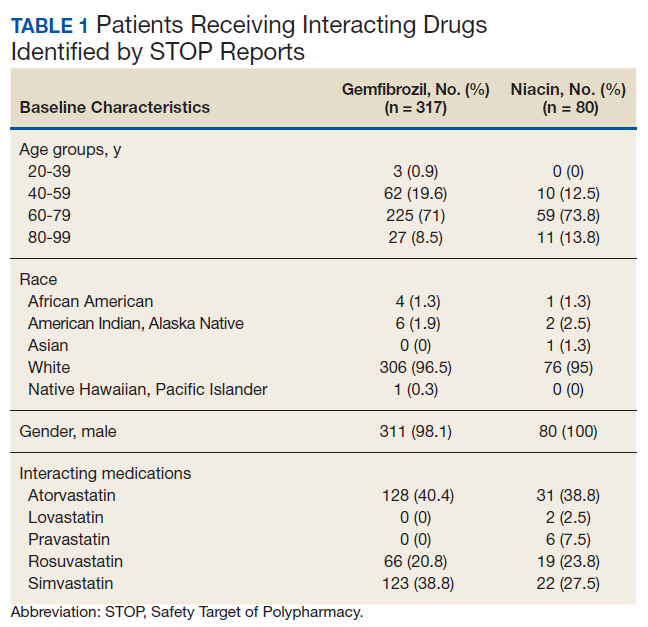
Gemfibrozil-Statin Interactions
Pharmacists discontinued gemfibrozil by protocol for 94 patients (29.6%), and 107 patients (33.8%) were referred to the PACT Pharmacy Clinic (Figure 3). For the remaining 116 patients (36.6%), the drug interaction was addressed outside of the protocol for the following reasons: the drug interaction was resolved prior to pharmacist review; an interacting prescription was expired and not to be continued; the patient self-discontinued ≥ 1 interacting medications; the patient was deceased; the patient moved; the patient was receiving ≥ 1 interacting medications outside of the VA; or the prescriber resolved the interaction following notification by the pharmacist.
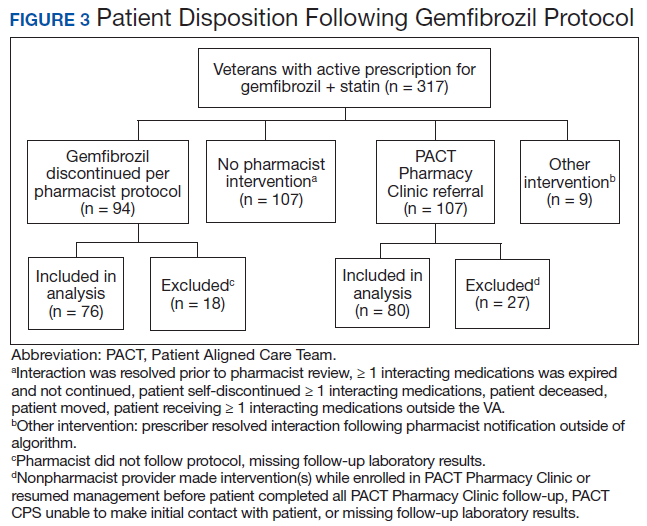
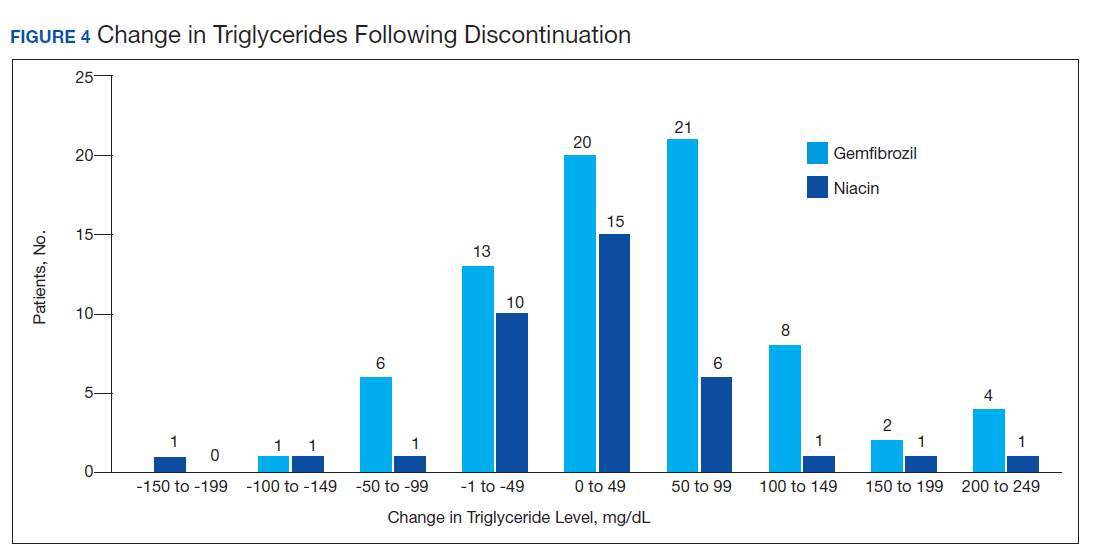
Ultimately, the interaction was resolved for all patients with a gemfibrozil-statin interaction on the STOP report. Following gemfibrozil discontinuation by protocol, 76 patients (80.9%) had TG laboratory results available and were included in the analysis. Sixty-two patients’ (82%) TG levels decreased or increased by < 100 mg/dL (Figure 4), and the TG levels of 1 patient (1.3%) increased above the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter was 6.5 (3.6) months (range, 1-17). The pharmacists spent a mean of 16 minutes per patient resolving each interaction.
Of the 107 patients referred to the PACT Pharmacy Clinic, 80 (74.8%) had TG laboratory results available and were included in the analysis. These patients were followed by the PACT CPS until the drug interaction was resolved and confirmed to have TG levels at goal (< 500 mg/dL). Gemfibrozil doses ranged from 300 mg daily to 600 mg twice daily, with 70% (n = 56) of patients taking 600 mg twice daily. The PACT CPS made 148 interventions (Table 2). Twenty-three (29%) patients required only gemfibrozil discontinuation. The remaining 57 patients (71%) required at least 2 medication interventions. The PACT CPS generated 213 encounters for resolving drug interactions with a median of 2 encounters per patient.
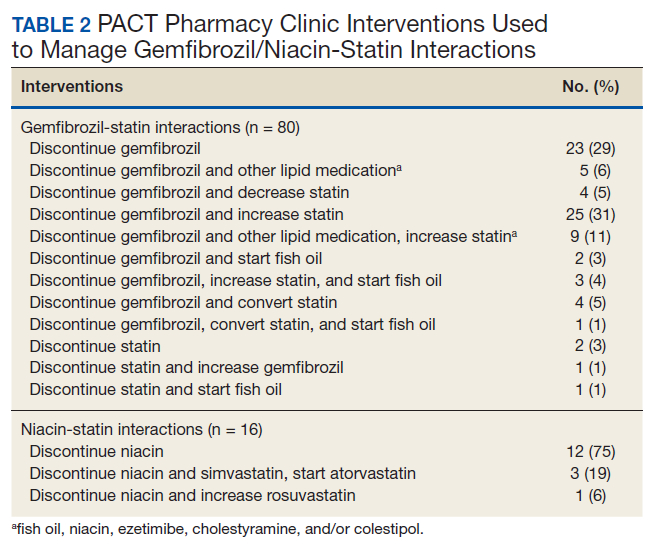
Quality assurance review identified 5 patients (5.3%) who underwent gemfibrozil discontinuation by protocol, despite having criteria that would have recommended against discontinuation. In accordance with the protocol criteria, these patients were later referred to the PACT Pharmacy Clinic. None of these patients experienced a TG increase at or above the threshold of 500 mg/dL after gemfibrozil was initially discontinued but were excluded from the earlier analysis.
Niacin-Statin Interactions
Pharmacists discontinued niacin by protocol for 48 patients (60.0%), and 22 patients (27.5%) were referred to the PACT Pharmacy Clinic (Figure 5). For the remaining 5 patients (6.3%), the interaction was either addressed outside the protocol prior to pharmacist review, or an interacting prescription was expired and not to be continued. Additionally, niacin was continued per prescriber preference in 5 patients (6.3%).
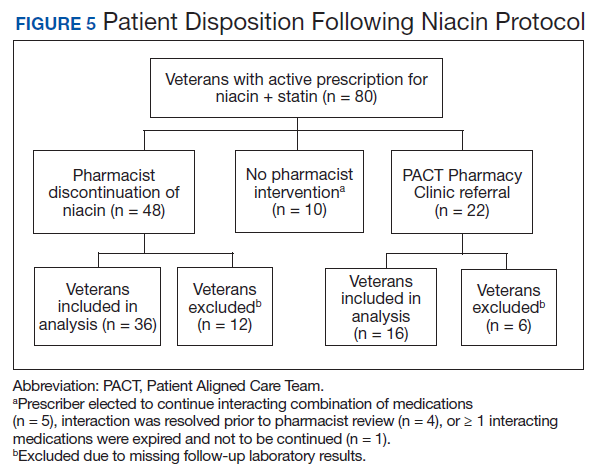
Thirty-six patients (75%) had TG laboratory results available following niacin discontinuation by protocol and were included in the analysis. Most patients’ (n = 33, 91.7%) TG levels decreased or increased by < 100 mg/dL. No patient had a TG level that increased higher than the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter, was 5.3 (2.5) months (range, 1.2-9.8). The pharmacists spent a mean of 15 minutes per patient resolving each interaction. The quality assurance review found no discrepancies in the pharmacists’ application of the protocol.
Of the 22 patients referred to the PACT Pharmacy Clinic, 16 (72.7%) patients had TG laboratory results available and were included in the analysis. As with the gemfibrozil interactions, these patients were followed by the PACT Pharmacy Clinic until the drug interaction was resolved and confirmed to have TGs at goal (< 500 mg/dL). Niacin doses ranged from 500 mg daily to 2,000 mg daily, with the majority of patients taking 1,000 mg daily. The PACT CPS made 23 interventions. The PACT CPS generated 46 encounters for resolving drug interactions with a median of 2 encounters per patient.
Discussion
Following gemfibrozil or niacin discontinuation by protocol, most patients with available laboratory results experienced either a decrease or modest TG elevation. The proportion of patients experiencing a decrease in TGs was unexpected but potentially multifactorial. Individual causes for the decrease in TGs were beyond the scope of this analysis. The retrospective design limited the ability to identify variables that could have impacted TG levels when gemfibrozil or niacin were started and discontinued. Although the treatment of TG levels is not indicated until it is ≥ 500 mg/dL, due to an increased risk of pancreatitis, both protocols excluded patients with a history of TGs ≥ 400 mg/dL.19 The lower threshold was set to compensate for anticipated increase in TG levels, following gemfibrozil or niacin discontinuation, and to minimize the number of patients with TG levels ≥ 500 mg/dL. The actual impact on patients’ TG levels supports the use of this lower threshold in the protocol.
When TG levels increased by 200 to 249 mg/dL after gemfibrozil or niacin discontinuation, patients were evaluated for possible underlying causes, which occurred for 4 gemfibrozil and 1 niacin patient. One patient started a β-blocker after gemfibrozil was initiated, and 3 patients were taking gemfibrozil prior to establishing care at the VA. The TG levels of the patient taking niacin correlated with an increased hemoglobin A1c. The TG level for only 1 patient taking gemfibrozil increased above the 500 mg/dL threshold. The patient had several comorbidities known to increase TG levels, but the comorbidities were previously well controlled. No additional medication changes were made at that time, and the TG levels on the next fasting lipid panel decreased to goal. The patient did not experience any negative clinical sequelae from the elevated TG levels.
Thirty-five patients (36%) who were referred to the PACT Pharmacy Clinic required only either gemfibrozil or niacin discontinuation. These patients were evaluated to identify whether adjustments to the protocols would have allowed for pharmacist discontinuation without referral to the PACT Pharmacy Clinic. Twenty-four of these patients (69%) had repeated TG levels ≥ 400 mg/dL prior to referral to the PACT Pharmacy Clinic. Additionally, there was no correlation between the gemfibrozil or niacin doses and the change in TG levels following discontinuation. These data indicate the protocols appropriately identified patients who did not have an indication for gemfibrozil or niacin.
In addition to drug interactions identified on the STOP report, the PACT CPS resolved 12 additional interactions involving simvastatin and gemfibrozil. Additionally, unnecessary lipid medications were deprescribed. The PACT CPS identified 13 patients who experienced myalgias, an ADR attributed to the gemfibrozil- statin interaction. Of those, 9 patients’ ADRs resolved after discontinuing gemfibrozil alone. For the remaining 4 patients, additional interventions to convert the patient to another statin were required to resolve the ADR.
Using pharmacists to address the drug interactions shifted workload from the prescribers and other primary care team members. The mean time spent to resolve both gemfibrozil and niacin interactions by protocol was 15.5 minutes. One hundred fortytwo patients (35.8%) had drug interactions resolved by protocol, saving the PACT CPS’ expertise for patients requiring individualized interventions. Drug interactions were resolved within 4 PACT CPS encounters for 93.8% of the patients taking gemfibrozil and within 3 PACT CPS encounters for 93.8% of the patients taking niacin.
The protocols allowed 12 additional pharmacists who did not have an ambulatory care scope of practice to assist the PACT CPS in mitigating the STOP drug interactions. These pharmacists otherwise would have been limited to making consultative recommendations. Simultaneously, the design allowed for the PACT pharmacists’ expertise to be allocated for patients most likely to require interventions beyond the protocols. This type of intraprofessional referral process is not well described in the medical literature. To the authors’ knowledge, the only studies described referrals from hospital pharmacists to community pharmacists during transitions of care on hospital discharge.20,21
Limitations
The results of this study are derived from a retrospective chart review at a single VA facility. The autonomous nature of PACT CPS interventions may be difficult to replicate in other settings that do not permit pharmacists the same prescriptive authority. This analysis was designed to demonstrate the impact of the pharmacist in resolving major drug interactions. Patients referred to the PACT Pharmacy Clinic who also had their lipid medications adjusted by a nonpharmacist provider were excluded. However, this may have minimized the impact of the PACT CPS on the patient care provided. As postintervention laboratory results were not available for all patients, some patients’ TG levels could have increased above the 500 mg/dL threshold but were not identified. The time investment was extensive and likely underestimates the true cost of implementing the interventions.
Because notification letters were used to instruct patients to stop gemfibrozil or niacin, several considerations need to be addressed when interpreting the follow-up laboratory results. First, we cannot confirm whether the patients received the letter or the exact date the letter was received. Additionally, we cannot confirm whether the patients followed the instructions to stop the interacting medications or the date the medications were stopped. It is possible some patients were still taking the interacting medication when the first laboratory was drawn. Should a patient have continued the interacting medication, most would have run out and been unable to obtain a refill within 90 days of receiving the letter, as this is the maximum amount dispensed at one time. The mean time to the first laboratory result for both gemfibrozil and niacin was 6.5 and 5.3 months, respectively. Approximately 85% of patients completed the first laboratory test at least 3 months after the letter was mailed.
The protocols were designed to assess whether gemfibrozil or niacin was indicated and did not assess whether the statin was indicated. Therefore, discontinuing the statin also could have resolved the interaction appropriately. However, due to characteristics of the patient population and recommendations in current lipid guidelines, it was more likely the statin would be indicated.22,23 The protocols also assumed that patients eligible for gemfibrozil or niacin discontinuation would not need additional changes to their lipid medications. The medication changes made by the PACT CPS may have gone beyond those minimally necessary to resolve the drug interaction and maintain TG goals. Patients who had gemfibrozil or niacin discontinued by protocol also may have benefited from additional optimization of their lipid medications.
Conclusions
This quality improvement analysis supports further evaluation of the complementary use of protocols and PACT CPS prescriptive authority to resolve statin drug interactions. The gemfibrozil and niacin protocols appropriately identified patients who were less likely to experience an adverse change in TG laboratory results. Patients more likely to require additional medication interventions were appropriately referred to the PACT Pharmacy Clinics for individualized care. These data support expanded roles for pharmacists, across various settings, to mitigate select drug interactions at the Truman VA.
Acknowledgments
This quality improvement project is the result of work supported with resources and use of the Harry S. Truman Memorial Veterans’ Hospital in Columbia, Missouri.
1. The top 200 drugs of 2020 Provided by the ClinCalc DrugStats Database. http://clincalc.com/DrugStats /Top200Drugs.aspx. Updated February 11, 2017. Accessed May 12, 2020.
2. Wiggins BS, Saseen JJ, Page RL 2nd, et al; American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134(21):e468‐e495. doi:10.1161/CIR.0000000000000456
3. Smithburger PL, Buckley MS, Bejian S, Burenheide K, Kane-Gill SL. A critical evaluation of clinical decision support for the detection of drug-drug interactions. Expert Opin Drug Saf. 2011;10(6):871‐882. doi:10.1517/14740338.2011.583916
4. US Food and Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/Drugs/DrugSafety /ucm256581.htm. Updated December 15, 2017. Accessed May 12, 2020.
5. US Food and Drug Administration. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. https://www.fda.gov /Drugs/DrugSafety/ucm293101.htm. Updated January 19, 2016. Accessed May 12, 2020.
6. US Food and Drug Administration Federal Register. AbbVie Inc. et al; withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://www.federalregister .gov/documents/2016/04/18/2016-08887/abbvie-inc -et-al-withdrawal-of-approval-of-indications-related -to-the-coadministration-with-statins. Published April 18, 2016. Accessed May 12, 2020.
7. Lamprecht DG Jr, Todd BA, Denham AM, Ruppe LK, Stadler SL. Clinical pharmacist patient-safety initiative to reduce against-label prescribing of statins with cyclosporine. Ann Pharmacother. 2017;51(2):140‐145. doi:10.1177/1060028016675352
8. Roblek T, Deticek A, Leskovar B, et al. Clinical-pharmacist intervention reduces clinically relevant drugdrug interactions in patients with heart failure: A randomized, double-blind, controlled trial. Int J Cardiol. 2016;203:647‐652. doi:10.1016/j.ijcard.2015.10.206
9. Tuchscherer RM, Nair K, Ghushchyan V, Saseen JJ. Simvastatin prescribing patterns before and after FDA dosing restrictions: a retrospective analysis of a large healthcare claims database. Am J Cardiovasc Drugs. 2015;15(1):27‐34. doi:10.1007/s40256-014-0096-x
10. Alford JC, Saseen JJ, Allen RR, Nair KV. Persistent use of against-label statin-fibrate combinations from 2003-2009 despite United States Food and Drug Administration dose restrictions. Pharmacotherapy. 2012;32(7):623‐630. doi:10.1002/j.1875-9114.2011.01090.x
11. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit [published correction appears in JAMA 2000 Mar 8;283(10):1293]. JAMA. 1999;282(3):267‐270. doi:10.1001/jama.282.3.267
12. Kucukarslan SN, Peters M, Mlynarek M, Nafziger DA. Pharmacists on rounding teams reduce preventable adverse drug events in hospital general medicine units. Arch Intern Med. 2003;163(17):2014‐2018. doi:10.1001/archinte.163.17.2014
13. Humphries TL, Carroll N, Chester EA, Magid D, Rocho B. Evaluation of an electronic critical drug interaction program coupled with active pharmacist intervention. Ann Pharmacother. 2007;41(12):1979‐1985. doi:10.1345/aph.1K349
14. Zocor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2018.
15. Lipitor [package insert]. New York, NY: Pfizer; 2017.
16. Crestor [package insert]. Wilmington, DE: AstraZeneca; 2018.
17. Mevacor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2012.
18. Wolters Kluwer Health, Lexi-Drugs, Lexicomp. Pravastatin. www.online.lexi.com. [Source not verified.]
19. Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333. doi: 10.1161/CIR.0b013e3182160726
20. Ferguson J, Seston L, Ashcroft DM. Refer-to-pharmacy: a qualitative study exploring the implementation of an electronic transfer of care initiative to improve medicines optimisation following hospital discharge. BMC Health Serv Res. 2018;18(1):424. doi:10.1186/s12913-018-3262-z
21. Ensing HT, Koster ES, Dubero DJ, van Dooren AA, Bouvy ML. Collaboration between hospital and community pharmacists to address drug-related problems: the HomeCoMe-program. Res Social Adm Pharm. 2019;15(3):267‐278. doi:10.1016/j.sapharm.2018.05.001
22. US Department of Defense, US Department of Veterans Affairs. VA/DoD clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction guideline summary. https://www.healthquality.va.gov /guidelines/CD/lipids/LipidSumOptSinglePg31Aug15.pdf. Published 2014. Accessed May 14, 2020.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation. 2014 Jun 24;129(25) (suppl 2):S46-48] [published correction appears in Circulation. 2015 Dec 22;132(25):e396]. Circulation. 2014;129(25)(suppl 2): S1‐S45. doi:10.1161/01.cir.0000437738.63853.7a
Statins are one of the most common medications dispensed in the US and are associated with clinically significant drug interactions.1,2 The most common adverse drug reaction (ADR) of statin drug interactions is muscle-related toxicities.2 Despite technology advances to alert clinicians to drug interactions, updated statin manufacturer labeling, and guideline recommendations, inappropriate prescribing and dispensing of statin drug interactions continues to occur in health care systems.2-10
The medical literature has demonstrated many opportunities for pharmacists to prevent and mitigate drug interactions. At the points of prescribing and dispensing, pharmacists can reduce the number of potential drug interactions for the patient.11-13 Pharmacists also have identified and resolved drug interactions through quality assurance review after dispensing to a patient.7,8
Regardless of the time point of an intervention, the most common method pharmacists used to resolve drug interactions was through recommendations to a prescriber. The recommendations were generated through academic detailing, clinical decision support algorithms, drug conversions, or the pharmacist’s expertise. Regardless of the method the pharmacist used, the prescriber had the final authority to accept or decline the recommendation.7,8,11-13 Although these interventions were effective, pharmacists could further streamline the process by autonomously resolving drug interactions. However, these types of interventions are not well described in the medical literature.
Background
The US Department of Veterans Affairs (VA) Veterans Integrated Service Network (VISN), established the Safety Target of Polypharmacy (STOP) report in 2015. At each facility in the network, the report identified patients who were dispensed medications known to have drug interactions. The interactions were chosen by the VISN, and the severity of the interactions was based on coding parameters within the VA computerized order entry system, which uses a severity score based on First Databank data. At the Harry S. Truman Memorial Veterans’ Hospital (Truman VA) in Columbia, Missouri, > 500 drug interactions were initially active on the STOP report. The most common drug interactions were statins with gemfibrozil and statins with niacin.14-18 The Truman VA Pharmacy Service was charged with resolving the interactions for the facility.
The Truman VA employs 3 Patient Aligned Care Team (PACT) Clinical Pharmacy Specialists (CPS) practicing within primary care clinics. PACT is the patientcentered medical home model used by the VA. PACT CPS are ambulatory care pharmacists who assist providers in managing diseases using a scope of practice. Having a scope of practice would have allowed the PACT CPS to manage drug interactions with independent prescribing authority. However, due to the high volume of STOP report interactions and limited PACT CPS resources, the Pharmacy Service needed to develop an efficient, patient-centered method to resolve them. The intervention also needed to allow pharmacists, both with and without a scope of practice, to address the interactions.
Methods
The Truman VA Pharmacy Service developed protocols, approved by the Pharmacy and Therapeutics (P&T) Committee, to manage the specific gemfibrozil-statin and niacinstatin interactions chosen for the VISN 15 STOP report (Figures 1 and 2). The protocols were designed to identify patients who did not have a clear indication for gemfibrozil or niacin, were likely to maintain triglycerides (TGs) < 500 mg/dL without these medications, and would not likely require close monitoring after discontinuation.19 The protocols allowed pharmacists to autonomously discontinue gemfibrozil or niacin if patients did not have a history of pancreatitis, TGs ≥ 400 mg/dL or a nonlipid indication for niacin (eg, pellagra) after establishing care at Truman VA. Additionally, both interacting medications had to be dispensed by the VA. When pharmacists discontinued a medication, it was documented in a note in the patient electronic health record. The prescriber was notified through the note and the patient received a notification letter. Follow-up laboratory monitoring was not required as part of the protocol.
If patients met any of the exclusion criteria for discontinuation, the primary care provider (PCP) was notified to place a consult to the PACT Pharmacy Clinic for individualized interventions and close monitoring. Patients prescribed niacin for nonlipid indications were allowed to continue with their current drug regimen. At each encounter, the PACT CPS assessed for ADRs, made individualized medication changes, and arranged follow-up appointments. Once the interaction was resolved and treatment goals met, the PCP resumed monitoring of the patient’s lipid therapy.
Following all pharmacist interventions, a retrospective quality improvement analysis was conducted. The primary outcome was to evaluate the impact of discontinuing gemfibrozil and niacin by protocol on patients’ laboratory results. The coprimary endpoints were to describe the change in TG levels and the percentage of patients with TGs ≥ 500 mg/dL at least 5 weeks following the pharmacist-directed discontinuation by protocol. Secondary outcomes included the time required to resolve the interactions and a description of the PACT CPS pharmacologic interventions. Additionally, a quality assurance peer review was used to ensure the pharmacists appropriately utilized the protocols.
Data were collected from August 2016 to September 2017 for patients prescribed gemfibrozil and from May 2017 to January 2018 for patients prescribed niacin. The time spent resolving interactions was quantified based on encounter data. Descriptive statistics were used to analyze demographic information and the endpoints associated with each outcome. The project was reviewed by the University of Missouri Institutional Review Board, Truman VA privacy and information security officers, and was determined to meet guidelines for quality improvement.
Results
The original STOP report included 397 drug interactions involving statins with gemfibrozil or niacin (Table 1). The majority of patients were white and male aged 60 to 79 years. Gemfibrozil was the most common drug involved in all interactions (79.8%). The most common statins were atorvastatin (40%) and simvastatin (36.5%).
Gemfibrozil-Statin Interactions
Pharmacists discontinued gemfibrozil by protocol for 94 patients (29.6%), and 107 patients (33.8%) were referred to the PACT Pharmacy Clinic (Figure 3). For the remaining 116 patients (36.6%), the drug interaction was addressed outside of the protocol for the following reasons: the drug interaction was resolved prior to pharmacist review; an interacting prescription was expired and not to be continued; the patient self-discontinued ≥ 1 interacting medications; the patient was deceased; the patient moved; the patient was receiving ≥ 1 interacting medications outside of the VA; or the prescriber resolved the interaction following notification by the pharmacist.
Ultimately, the interaction was resolved for all patients with a gemfibrozil-statin interaction on the STOP report. Following gemfibrozil discontinuation by protocol, 76 patients (80.9%) had TG laboratory results available and were included in the analysis. Sixty-two patients’ (82%) TG levels decreased or increased by < 100 mg/dL (Figure 4), and the TG levels of 1 patient (1.3%) increased above the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter was 6.5 (3.6) months (range, 1-17). The pharmacists spent a mean of 16 minutes per patient resolving each interaction.
Of the 107 patients referred to the PACT Pharmacy Clinic, 80 (74.8%) had TG laboratory results available and were included in the analysis. These patients were followed by the PACT CPS until the drug interaction was resolved and confirmed to have TG levels at goal (< 500 mg/dL). Gemfibrozil doses ranged from 300 mg daily to 600 mg twice daily, with 70% (n = 56) of patients taking 600 mg twice daily. The PACT CPS made 148 interventions (Table 2). Twenty-three (29%) patients required only gemfibrozil discontinuation. The remaining 57 patients (71%) required at least 2 medication interventions. The PACT CPS generated 213 encounters for resolving drug interactions with a median of 2 encounters per patient.
Quality assurance review identified 5 patients (5.3%) who underwent gemfibrozil discontinuation by protocol, despite having criteria that would have recommended against discontinuation. In accordance with the protocol criteria, these patients were later referred to the PACT Pharmacy Clinic. None of these patients experienced a TG increase at or above the threshold of 500 mg/dL after gemfibrozil was initially discontinued but were excluded from the earlier analysis.
Niacin-Statin Interactions
Pharmacists discontinued niacin by protocol for 48 patients (60.0%), and 22 patients (27.5%) were referred to the PACT Pharmacy Clinic (Figure 5). For the remaining 5 patients (6.3%), the interaction was either addressed outside the protocol prior to pharmacist review, or an interacting prescription was expired and not to be continued. Additionally, niacin was continued per prescriber preference in 5 patients (6.3%).
Thirty-six patients (75%) had TG laboratory results available following niacin discontinuation by protocol and were included in the analysis. Most patients’ (n = 33, 91.7%) TG levels decreased or increased by < 100 mg/dL. No patient had a TG level that increased higher than the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter, was 5.3 (2.5) months (range, 1.2-9.8). The pharmacists spent a mean of 15 minutes per patient resolving each interaction. The quality assurance review found no discrepancies in the pharmacists’ application of the protocol.
Of the 22 patients referred to the PACT Pharmacy Clinic, 16 (72.7%) patients had TG laboratory results available and were included in the analysis. As with the gemfibrozil interactions, these patients were followed by the PACT Pharmacy Clinic until the drug interaction was resolved and confirmed to have TGs at goal (< 500 mg/dL). Niacin doses ranged from 500 mg daily to 2,000 mg daily, with the majority of patients taking 1,000 mg daily. The PACT CPS made 23 interventions. The PACT CPS generated 46 encounters for resolving drug interactions with a median of 2 encounters per patient.
Discussion
Following gemfibrozil or niacin discontinuation by protocol, most patients with available laboratory results experienced either a decrease or modest TG elevation. The proportion of patients experiencing a decrease in TGs was unexpected but potentially multifactorial. Individual causes for the decrease in TGs were beyond the scope of this analysis. The retrospective design limited the ability to identify variables that could have impacted TG levels when gemfibrozil or niacin were started and discontinued. Although the treatment of TG levels is not indicated until it is ≥ 500 mg/dL, due to an increased risk of pancreatitis, both protocols excluded patients with a history of TGs ≥ 400 mg/dL.19 The lower threshold was set to compensate for anticipated increase in TG levels, following gemfibrozil or niacin discontinuation, and to minimize the number of patients with TG levels ≥ 500 mg/dL. The actual impact on patients’ TG levels supports the use of this lower threshold in the protocol.
When TG levels increased by 200 to 249 mg/dL after gemfibrozil or niacin discontinuation, patients were evaluated for possible underlying causes, which occurred for 4 gemfibrozil and 1 niacin patient. One patient started a β-blocker after gemfibrozil was initiated, and 3 patients were taking gemfibrozil prior to establishing care at the VA. The TG levels of the patient taking niacin correlated with an increased hemoglobin A1c. The TG level for only 1 patient taking gemfibrozil increased above the 500 mg/dL threshold. The patient had several comorbidities known to increase TG levels, but the comorbidities were previously well controlled. No additional medication changes were made at that time, and the TG levels on the next fasting lipid panel decreased to goal. The patient did not experience any negative clinical sequelae from the elevated TG levels.
Thirty-five patients (36%) who were referred to the PACT Pharmacy Clinic required only either gemfibrozil or niacin discontinuation. These patients were evaluated to identify whether adjustments to the protocols would have allowed for pharmacist discontinuation without referral to the PACT Pharmacy Clinic. Twenty-four of these patients (69%) had repeated TG levels ≥ 400 mg/dL prior to referral to the PACT Pharmacy Clinic. Additionally, there was no correlation between the gemfibrozil or niacin doses and the change in TG levels following discontinuation. These data indicate the protocols appropriately identified patients who did not have an indication for gemfibrozil or niacin.
In addition to drug interactions identified on the STOP report, the PACT CPS resolved 12 additional interactions involving simvastatin and gemfibrozil. Additionally, unnecessary lipid medications were deprescribed. The PACT CPS identified 13 patients who experienced myalgias, an ADR attributed to the gemfibrozil- statin interaction. Of those, 9 patients’ ADRs resolved after discontinuing gemfibrozil alone. For the remaining 4 patients, additional interventions to convert the patient to another statin were required to resolve the ADR.
Using pharmacists to address the drug interactions shifted workload from the prescribers and other primary care team members. The mean time spent to resolve both gemfibrozil and niacin interactions by protocol was 15.5 minutes. One hundred fortytwo patients (35.8%) had drug interactions resolved by protocol, saving the PACT CPS’ expertise for patients requiring individualized interventions. Drug interactions were resolved within 4 PACT CPS encounters for 93.8% of the patients taking gemfibrozil and within 3 PACT CPS encounters for 93.8% of the patients taking niacin.
The protocols allowed 12 additional pharmacists who did not have an ambulatory care scope of practice to assist the PACT CPS in mitigating the STOP drug interactions. These pharmacists otherwise would have been limited to making consultative recommendations. Simultaneously, the design allowed for the PACT pharmacists’ expertise to be allocated for patients most likely to require interventions beyond the protocols. This type of intraprofessional referral process is not well described in the medical literature. To the authors’ knowledge, the only studies described referrals from hospital pharmacists to community pharmacists during transitions of care on hospital discharge.20,21
Limitations
The results of this study are derived from a retrospective chart review at a single VA facility. The autonomous nature of PACT CPS interventions may be difficult to replicate in other settings that do not permit pharmacists the same prescriptive authority. This analysis was designed to demonstrate the impact of the pharmacist in resolving major drug interactions. Patients referred to the PACT Pharmacy Clinic who also had their lipid medications adjusted by a nonpharmacist provider were excluded. However, this may have minimized the impact of the PACT CPS on the patient care provided. As postintervention laboratory results were not available for all patients, some patients’ TG levels could have increased above the 500 mg/dL threshold but were not identified. The time investment was extensive and likely underestimates the true cost of implementing the interventions.
Because notification letters were used to instruct patients to stop gemfibrozil or niacin, several considerations need to be addressed when interpreting the follow-up laboratory results. First, we cannot confirm whether the patients received the letter or the exact date the letter was received. Additionally, we cannot confirm whether the patients followed the instructions to stop the interacting medications or the date the medications were stopped. It is possible some patients were still taking the interacting medication when the first laboratory was drawn. Should a patient have continued the interacting medication, most would have run out and been unable to obtain a refill within 90 days of receiving the letter, as this is the maximum amount dispensed at one time. The mean time to the first laboratory result for both gemfibrozil and niacin was 6.5 and 5.3 months, respectively. Approximately 85% of patients completed the first laboratory test at least 3 months after the letter was mailed.
The protocols were designed to assess whether gemfibrozil or niacin was indicated and did not assess whether the statin was indicated. Therefore, discontinuing the statin also could have resolved the interaction appropriately. However, due to characteristics of the patient population and recommendations in current lipid guidelines, it was more likely the statin would be indicated.22,23 The protocols also assumed that patients eligible for gemfibrozil or niacin discontinuation would not need additional changes to their lipid medications. The medication changes made by the PACT CPS may have gone beyond those minimally necessary to resolve the drug interaction and maintain TG goals. Patients who had gemfibrozil or niacin discontinued by protocol also may have benefited from additional optimization of their lipid medications.
Conclusions
This quality improvement analysis supports further evaluation of the complementary use of protocols and PACT CPS prescriptive authority to resolve statin drug interactions. The gemfibrozil and niacin protocols appropriately identified patients who were less likely to experience an adverse change in TG laboratory results. Patients more likely to require additional medication interventions were appropriately referred to the PACT Pharmacy Clinics for individualized care. These data support expanded roles for pharmacists, across various settings, to mitigate select drug interactions at the Truman VA.
Acknowledgments
This quality improvement project is the result of work supported with resources and use of the Harry S. Truman Memorial Veterans’ Hospital in Columbia, Missouri.
Statins are one of the most common medications dispensed in the US and are associated with clinically significant drug interactions.1,2 The most common adverse drug reaction (ADR) of statin drug interactions is muscle-related toxicities.2 Despite technology advances to alert clinicians to drug interactions, updated statin manufacturer labeling, and guideline recommendations, inappropriate prescribing and dispensing of statin drug interactions continues to occur in health care systems.2-10
The medical literature has demonstrated many opportunities for pharmacists to prevent and mitigate drug interactions. At the points of prescribing and dispensing, pharmacists can reduce the number of potential drug interactions for the patient.11-13 Pharmacists also have identified and resolved drug interactions through quality assurance review after dispensing to a patient.7,8
Regardless of the time point of an intervention, the most common method pharmacists used to resolve drug interactions was through recommendations to a prescriber. The recommendations were generated through academic detailing, clinical decision support algorithms, drug conversions, or the pharmacist’s expertise. Regardless of the method the pharmacist used, the prescriber had the final authority to accept or decline the recommendation.7,8,11-13 Although these interventions were effective, pharmacists could further streamline the process by autonomously resolving drug interactions. However, these types of interventions are not well described in the medical literature.
Background
The US Department of Veterans Affairs (VA) Veterans Integrated Service Network (VISN), established the Safety Target of Polypharmacy (STOP) report in 2015. At each facility in the network, the report identified patients who were dispensed medications known to have drug interactions. The interactions were chosen by the VISN, and the severity of the interactions was based on coding parameters within the VA computerized order entry system, which uses a severity score based on First Databank data. At the Harry S. Truman Memorial Veterans’ Hospital (Truman VA) in Columbia, Missouri, > 500 drug interactions were initially active on the STOP report. The most common drug interactions were statins with gemfibrozil and statins with niacin.14-18 The Truman VA Pharmacy Service was charged with resolving the interactions for the facility.
The Truman VA employs 3 Patient Aligned Care Team (PACT) Clinical Pharmacy Specialists (CPS) practicing within primary care clinics. PACT is the patientcentered medical home model used by the VA. PACT CPS are ambulatory care pharmacists who assist providers in managing diseases using a scope of practice. Having a scope of practice would have allowed the PACT CPS to manage drug interactions with independent prescribing authority. However, due to the high volume of STOP report interactions and limited PACT CPS resources, the Pharmacy Service needed to develop an efficient, patient-centered method to resolve them. The intervention also needed to allow pharmacists, both with and without a scope of practice, to address the interactions.
Methods
The Truman VA Pharmacy Service developed protocols, approved by the Pharmacy and Therapeutics (P&T) Committee, to manage the specific gemfibrozil-statin and niacinstatin interactions chosen for the VISN 15 STOP report (Figures 1 and 2). The protocols were designed to identify patients who did not have a clear indication for gemfibrozil or niacin, were likely to maintain triglycerides (TGs) < 500 mg/dL without these medications, and would not likely require close monitoring after discontinuation.19 The protocols allowed pharmacists to autonomously discontinue gemfibrozil or niacin if patients did not have a history of pancreatitis, TGs ≥ 400 mg/dL or a nonlipid indication for niacin (eg, pellagra) after establishing care at Truman VA. Additionally, both interacting medications had to be dispensed by the VA. When pharmacists discontinued a medication, it was documented in a note in the patient electronic health record. The prescriber was notified through the note and the patient received a notification letter. Follow-up laboratory monitoring was not required as part of the protocol.
If patients met any of the exclusion criteria for discontinuation, the primary care provider (PCP) was notified to place a consult to the PACT Pharmacy Clinic for individualized interventions and close monitoring. Patients prescribed niacin for nonlipid indications were allowed to continue with their current drug regimen. At each encounter, the PACT CPS assessed for ADRs, made individualized medication changes, and arranged follow-up appointments. Once the interaction was resolved and treatment goals met, the PCP resumed monitoring of the patient’s lipid therapy.
Following all pharmacist interventions, a retrospective quality improvement analysis was conducted. The primary outcome was to evaluate the impact of discontinuing gemfibrozil and niacin by protocol on patients’ laboratory results. The coprimary endpoints were to describe the change in TG levels and the percentage of patients with TGs ≥ 500 mg/dL at least 5 weeks following the pharmacist-directed discontinuation by protocol. Secondary outcomes included the time required to resolve the interactions and a description of the PACT CPS pharmacologic interventions. Additionally, a quality assurance peer review was used to ensure the pharmacists appropriately utilized the protocols.
Data were collected from August 2016 to September 2017 for patients prescribed gemfibrozil and from May 2017 to January 2018 for patients prescribed niacin. The time spent resolving interactions was quantified based on encounter data. Descriptive statistics were used to analyze demographic information and the endpoints associated with each outcome. The project was reviewed by the University of Missouri Institutional Review Board, Truman VA privacy and information security officers, and was determined to meet guidelines for quality improvement.
Results
The original STOP report included 397 drug interactions involving statins with gemfibrozil or niacin (Table 1). The majority of patients were white and male aged 60 to 79 years. Gemfibrozil was the most common drug involved in all interactions (79.8%). The most common statins were atorvastatin (40%) and simvastatin (36.5%).
Gemfibrozil-Statin Interactions
Pharmacists discontinued gemfibrozil by protocol for 94 patients (29.6%), and 107 patients (33.8%) were referred to the PACT Pharmacy Clinic (Figure 3). For the remaining 116 patients (36.6%), the drug interaction was addressed outside of the protocol for the following reasons: the drug interaction was resolved prior to pharmacist review; an interacting prescription was expired and not to be continued; the patient self-discontinued ≥ 1 interacting medications; the patient was deceased; the patient moved; the patient was receiving ≥ 1 interacting medications outside of the VA; or the prescriber resolved the interaction following notification by the pharmacist.
Ultimately, the interaction was resolved for all patients with a gemfibrozil-statin interaction on the STOP report. Following gemfibrozil discontinuation by protocol, 76 patients (80.9%) had TG laboratory results available and were included in the analysis. Sixty-two patients’ (82%) TG levels decreased or increased by < 100 mg/dL (Figure 4), and the TG levels of 1 patient (1.3%) increased above the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter was 6.5 (3.6) months (range, 1-17). The pharmacists spent a mean of 16 minutes per patient resolving each interaction.
Of the 107 patients referred to the PACT Pharmacy Clinic, 80 (74.8%) had TG laboratory results available and were included in the analysis. These patients were followed by the PACT CPS until the drug interaction was resolved and confirmed to have TG levels at goal (< 500 mg/dL). Gemfibrozil doses ranged from 300 mg daily to 600 mg twice daily, with 70% (n = 56) of patients taking 600 mg twice daily. The PACT CPS made 148 interventions (Table 2). Twenty-three (29%) patients required only gemfibrozil discontinuation. The remaining 57 patients (71%) required at least 2 medication interventions. The PACT CPS generated 213 encounters for resolving drug interactions with a median of 2 encounters per patient.
Quality assurance review identified 5 patients (5.3%) who underwent gemfibrozil discontinuation by protocol, despite having criteria that would have recommended against discontinuation. In accordance with the protocol criteria, these patients were later referred to the PACT Pharmacy Clinic. None of these patients experienced a TG increase at or above the threshold of 500 mg/dL after gemfibrozil was initially discontinued but were excluded from the earlier analysis.
Niacin-Statin Interactions
Pharmacists discontinued niacin by protocol for 48 patients (60.0%), and 22 patients (27.5%) were referred to the PACT Pharmacy Clinic (Figure 5). For the remaining 5 patients (6.3%), the interaction was either addressed outside the protocol prior to pharmacist review, or an interacting prescription was expired and not to be continued. Additionally, niacin was continued per prescriber preference in 5 patients (6.3%).
Thirty-six patients (75%) had TG laboratory results available following niacin discontinuation by protocol and were included in the analysis. Most patients’ (n = 33, 91.7%) TG levels decreased or increased by < 100 mg/dL. No patient had a TG level that increased higher than the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter, was 5.3 (2.5) months (range, 1.2-9.8). The pharmacists spent a mean of 15 minutes per patient resolving each interaction. The quality assurance review found no discrepancies in the pharmacists’ application of the protocol.
Of the 22 patients referred to the PACT Pharmacy Clinic, 16 (72.7%) patients had TG laboratory results available and were included in the analysis. As with the gemfibrozil interactions, these patients were followed by the PACT Pharmacy Clinic until the drug interaction was resolved and confirmed to have TGs at goal (< 500 mg/dL). Niacin doses ranged from 500 mg daily to 2,000 mg daily, with the majority of patients taking 1,000 mg daily. The PACT CPS made 23 interventions. The PACT CPS generated 46 encounters for resolving drug interactions with a median of 2 encounters per patient.
Discussion
Following gemfibrozil or niacin discontinuation by protocol, most patients with available laboratory results experienced either a decrease or modest TG elevation. The proportion of patients experiencing a decrease in TGs was unexpected but potentially multifactorial. Individual causes for the decrease in TGs were beyond the scope of this analysis. The retrospective design limited the ability to identify variables that could have impacted TG levels when gemfibrozil or niacin were started and discontinued. Although the treatment of TG levels is not indicated until it is ≥ 500 mg/dL, due to an increased risk of pancreatitis, both protocols excluded patients with a history of TGs ≥ 400 mg/dL.19 The lower threshold was set to compensate for anticipated increase in TG levels, following gemfibrozil or niacin discontinuation, and to minimize the number of patients with TG levels ≥ 500 mg/dL. The actual impact on patients’ TG levels supports the use of this lower threshold in the protocol.
When TG levels increased by 200 to 249 mg/dL after gemfibrozil or niacin discontinuation, patients were evaluated for possible underlying causes, which occurred for 4 gemfibrozil and 1 niacin patient. One patient started a β-blocker after gemfibrozil was initiated, and 3 patients were taking gemfibrozil prior to establishing care at the VA. The TG levels of the patient taking niacin correlated with an increased hemoglobin A1c. The TG level for only 1 patient taking gemfibrozil increased above the 500 mg/dL threshold. The patient had several comorbidities known to increase TG levels, but the comorbidities were previously well controlled. No additional medication changes were made at that time, and the TG levels on the next fasting lipid panel decreased to goal. The patient did not experience any negative clinical sequelae from the elevated TG levels.
Thirty-five patients (36%) who were referred to the PACT Pharmacy Clinic required only either gemfibrozil or niacin discontinuation. These patients were evaluated to identify whether adjustments to the protocols would have allowed for pharmacist discontinuation without referral to the PACT Pharmacy Clinic. Twenty-four of these patients (69%) had repeated TG levels ≥ 400 mg/dL prior to referral to the PACT Pharmacy Clinic. Additionally, there was no correlation between the gemfibrozil or niacin doses and the change in TG levels following discontinuation. These data indicate the protocols appropriately identified patients who did not have an indication for gemfibrozil or niacin.
In addition to drug interactions identified on the STOP report, the PACT CPS resolved 12 additional interactions involving simvastatin and gemfibrozil. Additionally, unnecessary lipid medications were deprescribed. The PACT CPS identified 13 patients who experienced myalgias, an ADR attributed to the gemfibrozil- statin interaction. Of those, 9 patients’ ADRs resolved after discontinuing gemfibrozil alone. For the remaining 4 patients, additional interventions to convert the patient to another statin were required to resolve the ADR.
Using pharmacists to address the drug interactions shifted workload from the prescribers and other primary care team members. The mean time spent to resolve both gemfibrozil and niacin interactions by protocol was 15.5 minutes. One hundred fortytwo patients (35.8%) had drug interactions resolved by protocol, saving the PACT CPS’ expertise for patients requiring individualized interventions. Drug interactions were resolved within 4 PACT CPS encounters for 93.8% of the patients taking gemfibrozil and within 3 PACT CPS encounters for 93.8% of the patients taking niacin.
The protocols allowed 12 additional pharmacists who did not have an ambulatory care scope of practice to assist the PACT CPS in mitigating the STOP drug interactions. These pharmacists otherwise would have been limited to making consultative recommendations. Simultaneously, the design allowed for the PACT pharmacists’ expertise to be allocated for patients most likely to require interventions beyond the protocols. This type of intraprofessional referral process is not well described in the medical literature. To the authors’ knowledge, the only studies described referrals from hospital pharmacists to community pharmacists during transitions of care on hospital discharge.20,21
Limitations
The results of this study are derived from a retrospective chart review at a single VA facility. The autonomous nature of PACT CPS interventions may be difficult to replicate in other settings that do not permit pharmacists the same prescriptive authority. This analysis was designed to demonstrate the impact of the pharmacist in resolving major drug interactions. Patients referred to the PACT Pharmacy Clinic who also had their lipid medications adjusted by a nonpharmacist provider were excluded. However, this may have minimized the impact of the PACT CPS on the patient care provided. As postintervention laboratory results were not available for all patients, some patients’ TG levels could have increased above the 500 mg/dL threshold but were not identified. The time investment was extensive and likely underestimates the true cost of implementing the interventions.
Because notification letters were used to instruct patients to stop gemfibrozil or niacin, several considerations need to be addressed when interpreting the follow-up laboratory results. First, we cannot confirm whether the patients received the letter or the exact date the letter was received. Additionally, we cannot confirm whether the patients followed the instructions to stop the interacting medications or the date the medications were stopped. It is possible some patients were still taking the interacting medication when the first laboratory was drawn. Should a patient have continued the interacting medication, most would have run out and been unable to obtain a refill within 90 days of receiving the letter, as this is the maximum amount dispensed at one time. The mean time to the first laboratory result for both gemfibrozil and niacin was 6.5 and 5.3 months, respectively. Approximately 85% of patients completed the first laboratory test at least 3 months after the letter was mailed.
The protocols were designed to assess whether gemfibrozil or niacin was indicated and did not assess whether the statin was indicated. Therefore, discontinuing the statin also could have resolved the interaction appropriately. However, due to characteristics of the patient population and recommendations in current lipid guidelines, it was more likely the statin would be indicated.22,23 The protocols also assumed that patients eligible for gemfibrozil or niacin discontinuation would not need additional changes to their lipid medications. The medication changes made by the PACT CPS may have gone beyond those minimally necessary to resolve the drug interaction and maintain TG goals. Patients who had gemfibrozil or niacin discontinued by protocol also may have benefited from additional optimization of their lipid medications.
Conclusions
This quality improvement analysis supports further evaluation of the complementary use of protocols and PACT CPS prescriptive authority to resolve statin drug interactions. The gemfibrozil and niacin protocols appropriately identified patients who were less likely to experience an adverse change in TG laboratory results. Patients more likely to require additional medication interventions were appropriately referred to the PACT Pharmacy Clinics for individualized care. These data support expanded roles for pharmacists, across various settings, to mitigate select drug interactions at the Truman VA.
Acknowledgments
This quality improvement project is the result of work supported with resources and use of the Harry S. Truman Memorial Veterans’ Hospital in Columbia, Missouri.
1. The top 200 drugs of 2020 Provided by the ClinCalc DrugStats Database. http://clincalc.com/DrugStats /Top200Drugs.aspx. Updated February 11, 2017. Accessed May 12, 2020.
2. Wiggins BS, Saseen JJ, Page RL 2nd, et al; American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134(21):e468‐e495. doi:10.1161/CIR.0000000000000456
3. Smithburger PL, Buckley MS, Bejian S, Burenheide K, Kane-Gill SL. A critical evaluation of clinical decision support for the detection of drug-drug interactions. Expert Opin Drug Saf. 2011;10(6):871‐882. doi:10.1517/14740338.2011.583916
4. US Food and Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/Drugs/DrugSafety /ucm256581.htm. Updated December 15, 2017. Accessed May 12, 2020.
5. US Food and Drug Administration. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. https://www.fda.gov /Drugs/DrugSafety/ucm293101.htm. Updated January 19, 2016. Accessed May 12, 2020.
6. US Food and Drug Administration Federal Register. AbbVie Inc. et al; withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://www.federalregister .gov/documents/2016/04/18/2016-08887/abbvie-inc -et-al-withdrawal-of-approval-of-indications-related -to-the-coadministration-with-statins. Published April 18, 2016. Accessed May 12, 2020.
7. Lamprecht DG Jr, Todd BA, Denham AM, Ruppe LK, Stadler SL. Clinical pharmacist patient-safety initiative to reduce against-label prescribing of statins with cyclosporine. Ann Pharmacother. 2017;51(2):140‐145. doi:10.1177/1060028016675352
8. Roblek T, Deticek A, Leskovar B, et al. Clinical-pharmacist intervention reduces clinically relevant drugdrug interactions in patients with heart failure: A randomized, double-blind, controlled trial. Int J Cardiol. 2016;203:647‐652. doi:10.1016/j.ijcard.2015.10.206
9. Tuchscherer RM, Nair K, Ghushchyan V, Saseen JJ. Simvastatin prescribing patterns before and after FDA dosing restrictions: a retrospective analysis of a large healthcare claims database. Am J Cardiovasc Drugs. 2015;15(1):27‐34. doi:10.1007/s40256-014-0096-x
10. Alford JC, Saseen JJ, Allen RR, Nair KV. Persistent use of against-label statin-fibrate combinations from 2003-2009 despite United States Food and Drug Administration dose restrictions. Pharmacotherapy. 2012;32(7):623‐630. doi:10.1002/j.1875-9114.2011.01090.x
11. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit [published correction appears in JAMA 2000 Mar 8;283(10):1293]. JAMA. 1999;282(3):267‐270. doi:10.1001/jama.282.3.267
12. Kucukarslan SN, Peters M, Mlynarek M, Nafziger DA. Pharmacists on rounding teams reduce preventable adverse drug events in hospital general medicine units. Arch Intern Med. 2003;163(17):2014‐2018. doi:10.1001/archinte.163.17.2014
13. Humphries TL, Carroll N, Chester EA, Magid D, Rocho B. Evaluation of an electronic critical drug interaction program coupled with active pharmacist intervention. Ann Pharmacother. 2007;41(12):1979‐1985. doi:10.1345/aph.1K349
14. Zocor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2018.
15. Lipitor [package insert]. New York, NY: Pfizer; 2017.
16. Crestor [package insert]. Wilmington, DE: AstraZeneca; 2018.
17. Mevacor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2012.
18. Wolters Kluwer Health, Lexi-Drugs, Lexicomp. Pravastatin. www.online.lexi.com. [Source not verified.]
19. Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333. doi: 10.1161/CIR.0b013e3182160726
20. Ferguson J, Seston L, Ashcroft DM. Refer-to-pharmacy: a qualitative study exploring the implementation of an electronic transfer of care initiative to improve medicines optimisation following hospital discharge. BMC Health Serv Res. 2018;18(1):424. doi:10.1186/s12913-018-3262-z
21. Ensing HT, Koster ES, Dubero DJ, van Dooren AA, Bouvy ML. Collaboration between hospital and community pharmacists to address drug-related problems: the HomeCoMe-program. Res Social Adm Pharm. 2019;15(3):267‐278. doi:10.1016/j.sapharm.2018.05.001
22. US Department of Defense, US Department of Veterans Affairs. VA/DoD clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction guideline summary. https://www.healthquality.va.gov /guidelines/CD/lipids/LipidSumOptSinglePg31Aug15.pdf. Published 2014. Accessed May 14, 2020.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation. 2014 Jun 24;129(25) (suppl 2):S46-48] [published correction appears in Circulation. 2015 Dec 22;132(25):e396]. Circulation. 2014;129(25)(suppl 2): S1‐S45. doi:10.1161/01.cir.0000437738.63853.7a
1. The top 200 drugs of 2020 Provided by the ClinCalc DrugStats Database. http://clincalc.com/DrugStats /Top200Drugs.aspx. Updated February 11, 2017. Accessed May 12, 2020.
2. Wiggins BS, Saseen JJ, Page RL 2nd, et al; American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134(21):e468‐e495. doi:10.1161/CIR.0000000000000456
3. Smithburger PL, Buckley MS, Bejian S, Burenheide K, Kane-Gill SL. A critical evaluation of clinical decision support for the detection of drug-drug interactions. Expert Opin Drug Saf. 2011;10(6):871‐882. doi:10.1517/14740338.2011.583916
4. US Food and Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/Drugs/DrugSafety /ucm256581.htm. Updated December 15, 2017. Accessed May 12, 2020.
5. US Food and Drug Administration. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. https://www.fda.gov /Drugs/DrugSafety/ucm293101.htm. Updated January 19, 2016. Accessed May 12, 2020.
6. US Food and Drug Administration Federal Register. AbbVie Inc. et al; withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://www.federalregister .gov/documents/2016/04/18/2016-08887/abbvie-inc -et-al-withdrawal-of-approval-of-indications-related -to-the-coadministration-with-statins. Published April 18, 2016. Accessed May 12, 2020.
7. Lamprecht DG Jr, Todd BA, Denham AM, Ruppe LK, Stadler SL. Clinical pharmacist patient-safety initiative to reduce against-label prescribing of statins with cyclosporine. Ann Pharmacother. 2017;51(2):140‐145. doi:10.1177/1060028016675352
8. Roblek T, Deticek A, Leskovar B, et al. Clinical-pharmacist intervention reduces clinically relevant drugdrug interactions in patients with heart failure: A randomized, double-blind, controlled trial. Int J Cardiol. 2016;203:647‐652. doi:10.1016/j.ijcard.2015.10.206
9. Tuchscherer RM, Nair K, Ghushchyan V, Saseen JJ. Simvastatin prescribing patterns before and after FDA dosing restrictions: a retrospective analysis of a large healthcare claims database. Am J Cardiovasc Drugs. 2015;15(1):27‐34. doi:10.1007/s40256-014-0096-x
10. Alford JC, Saseen JJ, Allen RR, Nair KV. Persistent use of against-label statin-fibrate combinations from 2003-2009 despite United States Food and Drug Administration dose restrictions. Pharmacotherapy. 2012;32(7):623‐630. doi:10.1002/j.1875-9114.2011.01090.x
11. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit [published correction appears in JAMA 2000 Mar 8;283(10):1293]. JAMA. 1999;282(3):267‐270. doi:10.1001/jama.282.3.267
12. Kucukarslan SN, Peters M, Mlynarek M, Nafziger DA. Pharmacists on rounding teams reduce preventable adverse drug events in hospital general medicine units. Arch Intern Med. 2003;163(17):2014‐2018. doi:10.1001/archinte.163.17.2014
13. Humphries TL, Carroll N, Chester EA, Magid D, Rocho B. Evaluation of an electronic critical drug interaction program coupled with active pharmacist intervention. Ann Pharmacother. 2007;41(12):1979‐1985. doi:10.1345/aph.1K349
14. Zocor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2018.
15. Lipitor [package insert]. New York, NY: Pfizer; 2017.
16. Crestor [package insert]. Wilmington, DE: AstraZeneca; 2018.
17. Mevacor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2012.
18. Wolters Kluwer Health, Lexi-Drugs, Lexicomp. Pravastatin. www.online.lexi.com. [Source not verified.]
19. Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333. doi: 10.1161/CIR.0b013e3182160726
20. Ferguson J, Seston L, Ashcroft DM. Refer-to-pharmacy: a qualitative study exploring the implementation of an electronic transfer of care initiative to improve medicines optimisation following hospital discharge. BMC Health Serv Res. 2018;18(1):424. doi:10.1186/s12913-018-3262-z
21. Ensing HT, Koster ES, Dubero DJ, van Dooren AA, Bouvy ML. Collaboration between hospital and community pharmacists to address drug-related problems: the HomeCoMe-program. Res Social Adm Pharm. 2019;15(3):267‐278. doi:10.1016/j.sapharm.2018.05.001
22. US Department of Defense, US Department of Veterans Affairs. VA/DoD clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction guideline summary. https://www.healthquality.va.gov /guidelines/CD/lipids/LipidSumOptSinglePg31Aug15.pdf. Published 2014. Accessed May 14, 2020.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation. 2014 Jun 24;129(25) (suppl 2):S46-48] [published correction appears in Circulation. 2015 Dec 22;132(25):e396]. Circulation. 2014;129(25)(suppl 2): S1‐S45. doi:10.1161/01.cir.0000437738.63853.7a
Biologics may carry melanoma risk for patients with immune-mediated inflammatory diseases
The in a systematic review and meta-analysis published in JAMA Dermatology.
The studies included in the analysis, however, had limitations, including a lack of those comparing biologic and conventional systemic therapy in psoriasis and inflammatory bowel disease (IBD), according to Shamarke Esse, MRes, of the division of musculoskeletal and dermatological sciences at the University of Manchester (England) and colleagues. “We advocate for more large, well-designed studies of this issue to be performed to help improve certainty” regarding this association, they wrote.
Previous studies that have found an increased risk of melanoma in patients on biologics for psoriasis, rheumatoid arthritis, and IBD have “typically used the general population as the comparator,” they noted. There is a large amount of evidence that has established short-term efficacy and safety of biologics, compared with conventional systemic treatments, but concerns about longer-term cancer risk associated with biologics remains a concern. Moreover, they added, “melanoma is a highly immunogenic skin cancer and therefore of concern to patients treated with TNFIs [tumor necrosis factor inhibitors] because melanoma risk increases with suppression of the immune system and TNF-alpha plays an important role in the immune surveillance of tumors.12,13
In their review, the researchers identified seven cohort studies from MEDLINE, Embase, and Cochrane Central Register of Controlled Trials (CENTRAL) databases published between January 1995 and February 2019 that evaluated melanoma risk in about 34,000 patients receiving biologics and 135,370 patients who had never been treated with biologics, and were receiving conventional systemic therapy for psoriasis, RA, or IBD. Of these, four studies were in patients with RA, two studies were in patients with IBD, and a single study was in patients with psoriasis. Six studies examined patients taking TNF inhibitors, but only one of six studies had information on specific TNF inhibitors (adalimumab, etanercept, and infliximab) in patients with RA. One study evaluated abatacept and rituximab in RA patients.
The researchers analyzed the pooled relative risk across all studies. Compared with patients who received conventional systemic therapy, there was a nonsignificant association with risk of melanoma in patients with psoriasis (hazard ratio, 1.57; 95% confidence interval, 0.61-4.09), RA (pooled relative risk, 1.20; 95% CI, 0.83-1.74), and IBD (pRR, 1.20; 95% CI, 0.60-2.40).
Among RA patients who received TNF inhibitors only, there was a slightly elevated nonsignificant risk of melanoma (pRR, 1.08; 95% CI, 0.81-1.43). Patients receiving rituximab had a pRR of 0.73 (95% CI, 0.38-1.39), and patients taking abatacept had a pRR of 1.43 (95% CI, 0.66-3.09), compared with RA patients receiving conventional systemic therapy. When excluding two major studies in the RA subgroup of patients in a sensitivity analysis, pooled risk estimates varied from 0.91 (95% CI, 0.69-1.18) to 1.95 (95% CI, 1.16- 3.30). There were no significant between-study heterogeneity or publication bias among the IBD and RA studies.
Mr. Esse and colleagues acknowledged the small number of IBD and psoriasis studies in the meta-analysis, which could affect pooled risk estimates. “Any future update of our study through the inclusion of newly published studies may produce significantly different pooled risk estimates than those reported in our meta-analysis,” they said. In addition, the use of health insurance databases, lack of risk factors for melanoma, and inconsistent information about treatment duration for patients receiving conventional systemic therapy were also limitations.
“Prospective cohort studies using an active comparator, new-user study design providing detailed information on treatment history, concomitant treatments, biologic and conventional systemic treatment duration, recreational and treatment-related UV exposure, skin color, and date of melanoma diagnosis are required to help improve certainty. These studies would also need to account for key risk factors and the latency period of melanoma,” the researchers said.
Mr. Esse disclosed being funded by a PhD studentship from the Psoriasis Association. One author disclosed receiving personal fees from Janssen, LEO Pharma, Lilly, and Novartis outside the study; another disclosed receiving grants and personal fees from those and several other pharmaceutical companies during the study, and personal fees from several pharmaceutical companies outside of the submitted work; the fourth author had no disclosures.
SOURCE: Esse S et al. JAMA Dermatol. 2020 May 20;e201300.
The in a systematic review and meta-analysis published in JAMA Dermatology.
The studies included in the analysis, however, had limitations, including a lack of those comparing biologic and conventional systemic therapy in psoriasis and inflammatory bowel disease (IBD), according to Shamarke Esse, MRes, of the division of musculoskeletal and dermatological sciences at the University of Manchester (England) and colleagues. “We advocate for more large, well-designed studies of this issue to be performed to help improve certainty” regarding this association, they wrote.
Previous studies that have found an increased risk of melanoma in patients on biologics for psoriasis, rheumatoid arthritis, and IBD have “typically used the general population as the comparator,” they noted. There is a large amount of evidence that has established short-term efficacy and safety of biologics, compared with conventional systemic treatments, but concerns about longer-term cancer risk associated with biologics remains a concern. Moreover, they added, “melanoma is a highly immunogenic skin cancer and therefore of concern to patients treated with TNFIs [tumor necrosis factor inhibitors] because melanoma risk increases with suppression of the immune system and TNF-alpha plays an important role in the immune surveillance of tumors.12,13
In their review, the researchers identified seven cohort studies from MEDLINE, Embase, and Cochrane Central Register of Controlled Trials (CENTRAL) databases published between January 1995 and February 2019 that evaluated melanoma risk in about 34,000 patients receiving biologics and 135,370 patients who had never been treated with biologics, and were receiving conventional systemic therapy for psoriasis, RA, or IBD. Of these, four studies were in patients with RA, two studies were in patients with IBD, and a single study was in patients with psoriasis. Six studies examined patients taking TNF inhibitors, but only one of six studies had information on specific TNF inhibitors (adalimumab, etanercept, and infliximab) in patients with RA. One study evaluated abatacept and rituximab in RA patients.
The researchers analyzed the pooled relative risk across all studies. Compared with patients who received conventional systemic therapy, there was a nonsignificant association with risk of melanoma in patients with psoriasis (hazard ratio, 1.57; 95% confidence interval, 0.61-4.09), RA (pooled relative risk, 1.20; 95% CI, 0.83-1.74), and IBD (pRR, 1.20; 95% CI, 0.60-2.40).
Among RA patients who received TNF inhibitors only, there was a slightly elevated nonsignificant risk of melanoma (pRR, 1.08; 95% CI, 0.81-1.43). Patients receiving rituximab had a pRR of 0.73 (95% CI, 0.38-1.39), and patients taking abatacept had a pRR of 1.43 (95% CI, 0.66-3.09), compared with RA patients receiving conventional systemic therapy. When excluding two major studies in the RA subgroup of patients in a sensitivity analysis, pooled risk estimates varied from 0.91 (95% CI, 0.69-1.18) to 1.95 (95% CI, 1.16- 3.30). There were no significant between-study heterogeneity or publication bias among the IBD and RA studies.
Mr. Esse and colleagues acknowledged the small number of IBD and psoriasis studies in the meta-analysis, which could affect pooled risk estimates. “Any future update of our study through the inclusion of newly published studies may produce significantly different pooled risk estimates than those reported in our meta-analysis,” they said. In addition, the use of health insurance databases, lack of risk factors for melanoma, and inconsistent information about treatment duration for patients receiving conventional systemic therapy were also limitations.
“Prospective cohort studies using an active comparator, new-user study design providing detailed information on treatment history, concomitant treatments, biologic and conventional systemic treatment duration, recreational and treatment-related UV exposure, skin color, and date of melanoma diagnosis are required to help improve certainty. These studies would also need to account for key risk factors and the latency period of melanoma,” the researchers said.
Mr. Esse disclosed being funded by a PhD studentship from the Psoriasis Association. One author disclosed receiving personal fees from Janssen, LEO Pharma, Lilly, and Novartis outside the study; another disclosed receiving grants and personal fees from those and several other pharmaceutical companies during the study, and personal fees from several pharmaceutical companies outside of the submitted work; the fourth author had no disclosures.
SOURCE: Esse S et al. JAMA Dermatol. 2020 May 20;e201300.
The in a systematic review and meta-analysis published in JAMA Dermatology.
The studies included in the analysis, however, had limitations, including a lack of those comparing biologic and conventional systemic therapy in psoriasis and inflammatory bowel disease (IBD), according to Shamarke Esse, MRes, of the division of musculoskeletal and dermatological sciences at the University of Manchester (England) and colleagues. “We advocate for more large, well-designed studies of this issue to be performed to help improve certainty” regarding this association, they wrote.
Previous studies that have found an increased risk of melanoma in patients on biologics for psoriasis, rheumatoid arthritis, and IBD have “typically used the general population as the comparator,” they noted. There is a large amount of evidence that has established short-term efficacy and safety of biologics, compared with conventional systemic treatments, but concerns about longer-term cancer risk associated with biologics remains a concern. Moreover, they added, “melanoma is a highly immunogenic skin cancer and therefore of concern to patients treated with TNFIs [tumor necrosis factor inhibitors] because melanoma risk increases with suppression of the immune system and TNF-alpha plays an important role in the immune surveillance of tumors.12,13
In their review, the researchers identified seven cohort studies from MEDLINE, Embase, and Cochrane Central Register of Controlled Trials (CENTRAL) databases published between January 1995 and February 2019 that evaluated melanoma risk in about 34,000 patients receiving biologics and 135,370 patients who had never been treated with biologics, and were receiving conventional systemic therapy for psoriasis, RA, or IBD. Of these, four studies were in patients with RA, two studies were in patients with IBD, and a single study was in patients with psoriasis. Six studies examined patients taking TNF inhibitors, but only one of six studies had information on specific TNF inhibitors (adalimumab, etanercept, and infliximab) in patients with RA. One study evaluated abatacept and rituximab in RA patients.
The researchers analyzed the pooled relative risk across all studies. Compared with patients who received conventional systemic therapy, there was a nonsignificant association with risk of melanoma in patients with psoriasis (hazard ratio, 1.57; 95% confidence interval, 0.61-4.09), RA (pooled relative risk, 1.20; 95% CI, 0.83-1.74), and IBD (pRR, 1.20; 95% CI, 0.60-2.40).
Among RA patients who received TNF inhibitors only, there was a slightly elevated nonsignificant risk of melanoma (pRR, 1.08; 95% CI, 0.81-1.43). Patients receiving rituximab had a pRR of 0.73 (95% CI, 0.38-1.39), and patients taking abatacept had a pRR of 1.43 (95% CI, 0.66-3.09), compared with RA patients receiving conventional systemic therapy. When excluding two major studies in the RA subgroup of patients in a sensitivity analysis, pooled risk estimates varied from 0.91 (95% CI, 0.69-1.18) to 1.95 (95% CI, 1.16- 3.30). There were no significant between-study heterogeneity or publication bias among the IBD and RA studies.
Mr. Esse and colleagues acknowledged the small number of IBD and psoriasis studies in the meta-analysis, which could affect pooled risk estimates. “Any future update of our study through the inclusion of newly published studies may produce significantly different pooled risk estimates than those reported in our meta-analysis,” they said. In addition, the use of health insurance databases, lack of risk factors for melanoma, and inconsistent information about treatment duration for patients receiving conventional systemic therapy were also limitations.
“Prospective cohort studies using an active comparator, new-user study design providing detailed information on treatment history, concomitant treatments, biologic and conventional systemic treatment duration, recreational and treatment-related UV exposure, skin color, and date of melanoma diagnosis are required to help improve certainty. These studies would also need to account for key risk factors and the latency period of melanoma,” the researchers said.
Mr. Esse disclosed being funded by a PhD studentship from the Psoriasis Association. One author disclosed receiving personal fees from Janssen, LEO Pharma, Lilly, and Novartis outside the study; another disclosed receiving grants and personal fees from those and several other pharmaceutical companies during the study, and personal fees from several pharmaceutical companies outside of the submitted work; the fourth author had no disclosures.
SOURCE: Esse S et al. JAMA Dermatol. 2020 May 20;e201300.
FROM JAMA DERMATOLOGY