User login
Heart disease remains the leading cause of death in U.S.
The 10 leading causes of death in the United States remained unchanged over the past year, according to a new report from the Centers for Disease Control (CDC). Though life expectancy at birth decreased to 78.6 years in 2017, down from 78.7 years in 2016, that change was driven primarily by suicide and drug overdose.
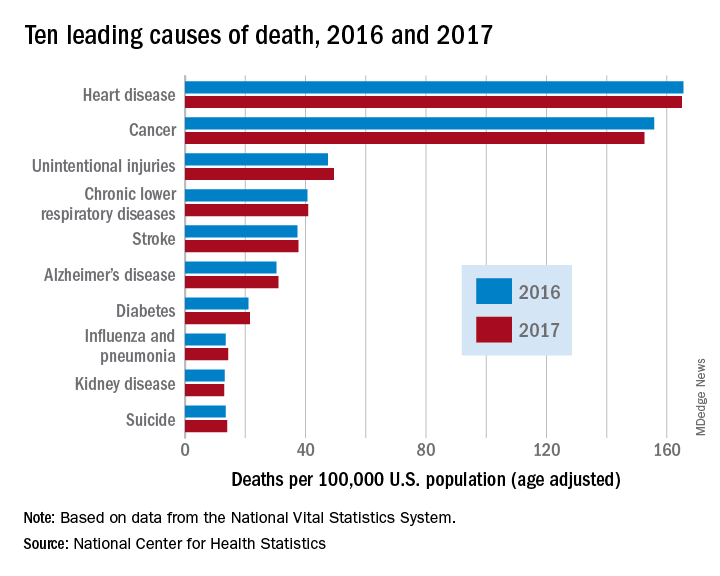
However, heart disease remains the leading cause of death in the United States, at 165 deaths per 100,000 individuals in 2017. This represents a slight, statistically nonsignificant, decrease from the 165.5 deaths per 100,000 caused by heart disease in the previous year.
Other diseases related to cardiometabolic health saw increases. Stroke and diabetes each caused a small but significant increase in deaths in 2017, which saw a 1-year increase to 37.6 from 37.3 stroke deaths per 100,000 people. Diabetes deaths increased to 21.5 from 21 per 100,000 the previous year. Stroke was the fifth and diabetes the seventh most common cause of death, according to the data brief published by the CDC’s National Center for Health Statistics (NCHS).
Alzheimer’s disease deaths also increased significantly, from 30.3 per 100,000 in 2016 to 31 per 100,000 in 2017. Although Alzheimer’s exact etiology remains under study, cardiovascular disease factors and Alzheimer’s disease share many risk factors and are often comorbid .
“With a slight decrease in deaths from heart disease in 2017 and a slight increase in deaths from stroke, this lack of any major movement in these areas has been a trend we’ve seen the last couple of years,” said Ivor Benjamin, MD, president of the American Heart Association, in a press release. “It is discouraging after experiencing decades when heart disease and stroke death rates both dropped more dramatically.”
Infant deaths from congenital malformations decreased from 2016 to 2017, from 122.1 to 118.8 deaths per 100,000 live births. “While the report doesn’t specify death rates for specific types of congenital malformations, this is heartening news as it could reflect fewer deaths from congenital heart defects,” said the AHA in its release.
According to the CDC, the 10 leading causes of death together account for about three quarters of United States deaths. Cancer caused nearly as many deaths as heart disease – 152.5 per 100,000. This represented a significant decrease from the 155.8 cancer deaths per 100,000 seen in 2016. The remaining top 10 causes of death, in decreasing order, were unintentional injuries, chronic lower respiratory diseases, influenza and pneumonia, kidney disease, and suicide.
The 10 leading causes of death in the United States remained unchanged over the past year, according to a new report from the Centers for Disease Control (CDC). Though life expectancy at birth decreased to 78.6 years in 2017, down from 78.7 years in 2016, that change was driven primarily by suicide and drug overdose.
However, heart disease remains the leading cause of death in the United States, at 165 deaths per 100,000 individuals in 2017. This represents a slight, statistically nonsignificant, decrease from the 165.5 deaths per 100,000 caused by heart disease in the previous year.
Other diseases related to cardiometabolic health saw increases. Stroke and diabetes each caused a small but significant increase in deaths in 2017, which saw a 1-year increase to 37.6 from 37.3 stroke deaths per 100,000 people. Diabetes deaths increased to 21.5 from 21 per 100,000 the previous year. Stroke was the fifth and diabetes the seventh most common cause of death, according to the data brief published by the CDC’s National Center for Health Statistics (NCHS).
Alzheimer’s disease deaths also increased significantly, from 30.3 per 100,000 in 2016 to 31 per 100,000 in 2017. Although Alzheimer’s exact etiology remains under study, cardiovascular disease factors and Alzheimer’s disease share many risk factors and are often comorbid .
“With a slight decrease in deaths from heart disease in 2017 and a slight increase in deaths from stroke, this lack of any major movement in these areas has been a trend we’ve seen the last couple of years,” said Ivor Benjamin, MD, president of the American Heart Association, in a press release. “It is discouraging after experiencing decades when heart disease and stroke death rates both dropped more dramatically.”
Infant deaths from congenital malformations decreased from 2016 to 2017, from 122.1 to 118.8 deaths per 100,000 live births. “While the report doesn’t specify death rates for specific types of congenital malformations, this is heartening news as it could reflect fewer deaths from congenital heart defects,” said the AHA in its release.
According to the CDC, the 10 leading causes of death together account for about three quarters of United States deaths. Cancer caused nearly as many deaths as heart disease – 152.5 per 100,000. This represented a significant decrease from the 155.8 cancer deaths per 100,000 seen in 2016. The remaining top 10 causes of death, in decreasing order, were unintentional injuries, chronic lower respiratory diseases, influenza and pneumonia, kidney disease, and suicide.
The 10 leading causes of death in the United States remained unchanged over the past year, according to a new report from the Centers for Disease Control (CDC). Though life expectancy at birth decreased to 78.6 years in 2017, down from 78.7 years in 2016, that change was driven primarily by suicide and drug overdose.
However, heart disease remains the leading cause of death in the United States, at 165 deaths per 100,000 individuals in 2017. This represents a slight, statistically nonsignificant, decrease from the 165.5 deaths per 100,000 caused by heart disease in the previous year.
Other diseases related to cardiometabolic health saw increases. Stroke and diabetes each caused a small but significant increase in deaths in 2017, which saw a 1-year increase to 37.6 from 37.3 stroke deaths per 100,000 people. Diabetes deaths increased to 21.5 from 21 per 100,000 the previous year. Stroke was the fifth and diabetes the seventh most common cause of death, according to the data brief published by the CDC’s National Center for Health Statistics (NCHS).
Alzheimer’s disease deaths also increased significantly, from 30.3 per 100,000 in 2016 to 31 per 100,000 in 2017. Although Alzheimer’s exact etiology remains under study, cardiovascular disease factors and Alzheimer’s disease share many risk factors and are often comorbid .
“With a slight decrease in deaths from heart disease in 2017 and a slight increase in deaths from stroke, this lack of any major movement in these areas has been a trend we’ve seen the last couple of years,” said Ivor Benjamin, MD, president of the American Heart Association, in a press release. “It is discouraging after experiencing decades when heart disease and stroke death rates both dropped more dramatically.”
Infant deaths from congenital malformations decreased from 2016 to 2017, from 122.1 to 118.8 deaths per 100,000 live births. “While the report doesn’t specify death rates for specific types of congenital malformations, this is heartening news as it could reflect fewer deaths from congenital heart defects,” said the AHA in its release.
According to the CDC, the 10 leading causes of death together account for about three quarters of United States deaths. Cancer caused nearly as many deaths as heart disease – 152.5 per 100,000. This represented a significant decrease from the 155.8 cancer deaths per 100,000 seen in 2016. The remaining top 10 causes of death, in decreasing order, were unintentional injuries, chronic lower respiratory diseases, influenza and pneumonia, kidney disease, and suicide.
FROM A CDC DATA BRIEF
U.S. life expectancy down; drug overdose, suicide up sharply
Average life expectancy fell in the United States fell from 78.7 years to 78.6 years from 2016 to 2017, according to a new report on the nation’s health. The decrease is primarily attributable to increases in suicide and drug overdose rates, according to new data from the Centers for Disease Control (CDC).
“The latest CDC data show that the U.S. life expectancy has declined over the past few years. Tragically, this troubling trend is largely driven by deaths from drug overdose and suicide,” said CDC Director Robert Redfield, MD, in a statement.
Two subreports that looked specifically at suicide mortality and drug overdose deaths mapped out where, when, and for whom the sharpest increases in mortality are being seen.
For suicide, though rates have increased by 33% overall for both men and women since 1999, the greatest annual increases in suicide rates have happened since 2006, according to a new report from the CDC’s National Center for Health Statistics (NCHS).
Overall, suicide rates have climbed from 10.5 to 14.0 per 100,000 individuals, with statistically significant increases in suicide rates among all age groups except those aged 75 years and older.
Suicide rates rose more steeply in the most rural counties. The age-adjusted increase in the most rural counties was 53%, compared with an increase of 16% in suicide rates for the nation’s most urban counties over the 1999-2017 time period.
Over the entire period studied, men were more likely than women to experience suicide, as rates rose among most age groups. For example, the rates of suicide for men aged 15-24 years rose from 16.8 to 22.7 per 100,000; for women in that age group, suicide rates went from 3.0 to 5.8 per 100,000.
Though suicide has remained the 10th leading cause of death overall in the United States, suicide was the second leading cause of death for adolescents and young adults (aged 10-34) in 2016, and the fourth leading cause of death for those aged 35-54 in that year.
These increases come despite a goal set by the CDC and a national coalition of health partners to reduce suicide rates to 10.2 per 100,000 by 2020, as part of the Healthy People 2020 initiative, noted Molly Hedegaard, MD, of NCHS, and her coauthors, in the suicide mortality data briefing.
Drug overdoses increased by nearly 10% in one year, with the highest rates seen in adults aged 25-54 years, according to a second CDC data briefing.
The number of people who died of drug overdoses in the United States in 2017 was 70,237. This represents a year-over-year age-adjusted increase of 9.6%, from 19.8 to 21.7 per 100,000 individuals, said Dr. Hedegaard and the coauthors of the drug overdose mortality report.
Reflecting known national trends in opioid use disorder, age-adjusted drug overdose deaths were highest in the states of West Virginia, Ohio, and Pennsylvania, where rates were 57.8, 46.3, and 44.3 per 100,000 residents, respectively. The District of Columbia had the fourth-highest age adjusted drug overdose death rate, at 44 per 100,000.
Twenty states, clustered primarily in the Eastern half of the United States, “had age-adjusted drug overdose death rate that were statistically higher than the national rate,” wrote Dr. Hedegaard and her coauthors.
Compared with 1999, more than six times as many adults in older midlife (aged 55-64 years) died from drug overdoses in 2017 (4.2 versus 28 per 100,000).
Adults aged 25-34 years, 35-44 years, and 45-54 years also had significant increases in drug overdose rates; in 2017, rates were 38.4, 29, and 37.7 per 100,000, respectively. Adolescent and young adults died from drug overdoses at a rate of 12.6 per 100,000, and those over 65 years old had a death rate of 6.9 per 100,000.
Deaths attributable to synthetic opioid use, excluding methadone, rose by 45% in just one year, going from 6.2 to 9.0 per 100,000 nationally. In 1999, synthetic opioids other than methadone were implicated in just 0.3 per 100,000 deaths. Synthetic opioids include fentanyl and fentanyl analogs, such as carfentanyl.
Deaths involving heroin remained stable from 2016 to 2017, at 4.9 per 100,000. Deaths attributable to natural and semisynthetic prescription opioids, such as oxycodone and hydrocodone, also were the same in 2017 as 2016, at 4.4 per 100,000.
Looking at trends over time since 1999, the rate of increase in drug overdose deaths had risen slowly since 1999 and stabilized in the mid-2000s. However, beginning in 2012, rates have increased steeply, particularly for males.
“Male rates were significantly higher than female rates for all years,” reported Dr. Hedegaard and her coauthors (P less than .05). Though female drug overdose death rates have climbed from 3.9 to 14.4 per 100,000 since 1999, the male death rate has gone from 8.2 to 29.1 per 100,000 during the study period.
“Life expectancy gives us a snapshot of the nation’s overall health and these sobering statistics are a wakeup call that we are losing too many Americans, too early and too often, to conditions that are preventable. CDC is committed to putting science into action to protect U.S. health, but we must all work together to reverse this trend and help ensure that all Americans live longer and healthier lives,” said Dr. Redfield.
Average life expectancy fell in the United States fell from 78.7 years to 78.6 years from 2016 to 2017, according to a new report on the nation’s health. The decrease is primarily attributable to increases in suicide and drug overdose rates, according to new data from the Centers for Disease Control (CDC).
“The latest CDC data show that the U.S. life expectancy has declined over the past few years. Tragically, this troubling trend is largely driven by deaths from drug overdose and suicide,” said CDC Director Robert Redfield, MD, in a statement.
Two subreports that looked specifically at suicide mortality and drug overdose deaths mapped out where, when, and for whom the sharpest increases in mortality are being seen.
For suicide, though rates have increased by 33% overall for both men and women since 1999, the greatest annual increases in suicide rates have happened since 2006, according to a new report from the CDC’s National Center for Health Statistics (NCHS).
Overall, suicide rates have climbed from 10.5 to 14.0 per 100,000 individuals, with statistically significant increases in suicide rates among all age groups except those aged 75 years and older.
Suicide rates rose more steeply in the most rural counties. The age-adjusted increase in the most rural counties was 53%, compared with an increase of 16% in suicide rates for the nation’s most urban counties over the 1999-2017 time period.
Over the entire period studied, men were more likely than women to experience suicide, as rates rose among most age groups. For example, the rates of suicide for men aged 15-24 years rose from 16.8 to 22.7 per 100,000; for women in that age group, suicide rates went from 3.0 to 5.8 per 100,000.
Though suicide has remained the 10th leading cause of death overall in the United States, suicide was the second leading cause of death for adolescents and young adults (aged 10-34) in 2016, and the fourth leading cause of death for those aged 35-54 in that year.
These increases come despite a goal set by the CDC and a national coalition of health partners to reduce suicide rates to 10.2 per 100,000 by 2020, as part of the Healthy People 2020 initiative, noted Molly Hedegaard, MD, of NCHS, and her coauthors, in the suicide mortality data briefing.
Drug overdoses increased by nearly 10% in one year, with the highest rates seen in adults aged 25-54 years, according to a second CDC data briefing.
The number of people who died of drug overdoses in the United States in 2017 was 70,237. This represents a year-over-year age-adjusted increase of 9.6%, from 19.8 to 21.7 per 100,000 individuals, said Dr. Hedegaard and the coauthors of the drug overdose mortality report.
Reflecting known national trends in opioid use disorder, age-adjusted drug overdose deaths were highest in the states of West Virginia, Ohio, and Pennsylvania, where rates were 57.8, 46.3, and 44.3 per 100,000 residents, respectively. The District of Columbia had the fourth-highest age adjusted drug overdose death rate, at 44 per 100,000.
Twenty states, clustered primarily in the Eastern half of the United States, “had age-adjusted drug overdose death rate that were statistically higher than the national rate,” wrote Dr. Hedegaard and her coauthors.
Compared with 1999, more than six times as many adults in older midlife (aged 55-64 years) died from drug overdoses in 2017 (4.2 versus 28 per 100,000).
Adults aged 25-34 years, 35-44 years, and 45-54 years also had significant increases in drug overdose rates; in 2017, rates were 38.4, 29, and 37.7 per 100,000, respectively. Adolescent and young adults died from drug overdoses at a rate of 12.6 per 100,000, and those over 65 years old had a death rate of 6.9 per 100,000.
Deaths attributable to synthetic opioid use, excluding methadone, rose by 45% in just one year, going from 6.2 to 9.0 per 100,000 nationally. In 1999, synthetic opioids other than methadone were implicated in just 0.3 per 100,000 deaths. Synthetic opioids include fentanyl and fentanyl analogs, such as carfentanyl.
Deaths involving heroin remained stable from 2016 to 2017, at 4.9 per 100,000. Deaths attributable to natural and semisynthetic prescription opioids, such as oxycodone and hydrocodone, also were the same in 2017 as 2016, at 4.4 per 100,000.
Looking at trends over time since 1999, the rate of increase in drug overdose deaths had risen slowly since 1999 and stabilized in the mid-2000s. However, beginning in 2012, rates have increased steeply, particularly for males.
“Male rates were significantly higher than female rates for all years,” reported Dr. Hedegaard and her coauthors (P less than .05). Though female drug overdose death rates have climbed from 3.9 to 14.4 per 100,000 since 1999, the male death rate has gone from 8.2 to 29.1 per 100,000 during the study period.
“Life expectancy gives us a snapshot of the nation’s overall health and these sobering statistics are a wakeup call that we are losing too many Americans, too early and too often, to conditions that are preventable. CDC is committed to putting science into action to protect U.S. health, but we must all work together to reverse this trend and help ensure that all Americans live longer and healthier lives,” said Dr. Redfield.
Average life expectancy fell in the United States fell from 78.7 years to 78.6 years from 2016 to 2017, according to a new report on the nation’s health. The decrease is primarily attributable to increases in suicide and drug overdose rates, according to new data from the Centers for Disease Control (CDC).
“The latest CDC data show that the U.S. life expectancy has declined over the past few years. Tragically, this troubling trend is largely driven by deaths from drug overdose and suicide,” said CDC Director Robert Redfield, MD, in a statement.
Two subreports that looked specifically at suicide mortality and drug overdose deaths mapped out where, when, and for whom the sharpest increases in mortality are being seen.
For suicide, though rates have increased by 33% overall for both men and women since 1999, the greatest annual increases in suicide rates have happened since 2006, according to a new report from the CDC’s National Center for Health Statistics (NCHS).
Overall, suicide rates have climbed from 10.5 to 14.0 per 100,000 individuals, with statistically significant increases in suicide rates among all age groups except those aged 75 years and older.
Suicide rates rose more steeply in the most rural counties. The age-adjusted increase in the most rural counties was 53%, compared with an increase of 16% in suicide rates for the nation’s most urban counties over the 1999-2017 time period.
Over the entire period studied, men were more likely than women to experience suicide, as rates rose among most age groups. For example, the rates of suicide for men aged 15-24 years rose from 16.8 to 22.7 per 100,000; for women in that age group, suicide rates went from 3.0 to 5.8 per 100,000.
Though suicide has remained the 10th leading cause of death overall in the United States, suicide was the second leading cause of death for adolescents and young adults (aged 10-34) in 2016, and the fourth leading cause of death for those aged 35-54 in that year.
These increases come despite a goal set by the CDC and a national coalition of health partners to reduce suicide rates to 10.2 per 100,000 by 2020, as part of the Healthy People 2020 initiative, noted Molly Hedegaard, MD, of NCHS, and her coauthors, in the suicide mortality data briefing.
Drug overdoses increased by nearly 10% in one year, with the highest rates seen in adults aged 25-54 years, according to a second CDC data briefing.
The number of people who died of drug overdoses in the United States in 2017 was 70,237. This represents a year-over-year age-adjusted increase of 9.6%, from 19.8 to 21.7 per 100,000 individuals, said Dr. Hedegaard and the coauthors of the drug overdose mortality report.
Reflecting known national trends in opioid use disorder, age-adjusted drug overdose deaths were highest in the states of West Virginia, Ohio, and Pennsylvania, where rates were 57.8, 46.3, and 44.3 per 100,000 residents, respectively. The District of Columbia had the fourth-highest age adjusted drug overdose death rate, at 44 per 100,000.
Twenty states, clustered primarily in the Eastern half of the United States, “had age-adjusted drug overdose death rate that were statistically higher than the national rate,” wrote Dr. Hedegaard and her coauthors.
Compared with 1999, more than six times as many adults in older midlife (aged 55-64 years) died from drug overdoses in 2017 (4.2 versus 28 per 100,000).
Adults aged 25-34 years, 35-44 years, and 45-54 years also had significant increases in drug overdose rates; in 2017, rates were 38.4, 29, and 37.7 per 100,000, respectively. Adolescent and young adults died from drug overdoses at a rate of 12.6 per 100,000, and those over 65 years old had a death rate of 6.9 per 100,000.
Deaths attributable to synthetic opioid use, excluding methadone, rose by 45% in just one year, going from 6.2 to 9.0 per 100,000 nationally. In 1999, synthetic opioids other than methadone were implicated in just 0.3 per 100,000 deaths. Synthetic opioids include fentanyl and fentanyl analogs, such as carfentanyl.
Deaths involving heroin remained stable from 2016 to 2017, at 4.9 per 100,000. Deaths attributable to natural and semisynthetic prescription opioids, such as oxycodone and hydrocodone, also were the same in 2017 as 2016, at 4.4 per 100,000.
Looking at trends over time since 1999, the rate of increase in drug overdose deaths had risen slowly since 1999 and stabilized in the mid-2000s. However, beginning in 2012, rates have increased steeply, particularly for males.
“Male rates were significantly higher than female rates for all years,” reported Dr. Hedegaard and her coauthors (P less than .05). Though female drug overdose death rates have climbed from 3.9 to 14.4 per 100,000 since 1999, the male death rate has gone from 8.2 to 29.1 per 100,000 during the study period.
“Life expectancy gives us a snapshot of the nation’s overall health and these sobering statistics are a wakeup call that we are losing too many Americans, too early and too often, to conditions that are preventable. CDC is committed to putting science into action to protect U.S. health, but we must all work together to reverse this trend and help ensure that all Americans live longer and healthier lives,” said Dr. Redfield.
FDA approves gilteritinib for AML with FLT3 mutation
as detected by an FDA-approved test.

The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
FDA approves rituximab biosimilar for lymphoma
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
Teva recalls valsartan-containing combo pills
Teva Pharmaceuticals has issued a voluntary recall of all unexpired lots of amlodipine/valsartan and amlodipine/valsartan /hydrochlorothiazide combination tablets because of contamination with N-nitrosodiethylamine (NDEA), according to a company announcement posted on the website of the Food and Drug Administration.
The contaminant was detected at above acceptable limits in the valsartan component of the pills, which were manufactured by Mylan India. Mylan has already recalled its own valsartan-containing products. Teva has recalled other valsartan-containing products in recent months because of the presence of N-nitrosodimethylamine (NDMA).
Teva has now recalled all of its unexpired valsartan-containing products from the U.S. market. The Teva recall affects almost 50 lots, all distributed nationwide to Teva’s direct accounts, which include wholesalers, retailers, repackagers, Veterans Affairs pharmacies, and others. The lot and National Drug Code numbers of the affected products, as well as the company’s announcement, are available on the Food and Drug Administration website.
Teva is notifying its U.S. customers by certified mail and arranging for returns and reimbursements. Distribution of recalled products should stop immediately, the company said.
Patients, however, should contact their pharmacists or physicians for alternative options before stopping treatment. “The risk of harm to a patient’s health may be higher if the treatment is stopped immediately without any comparable alternative treatment,” Teva said in its announcement.
The company plans regular updates as it works through the problem. NDEA is typically found in very small amounts in certain foods, drinking water, air pollution, and certain industrial processes.
Questions and other concerns can be directed to Teva directly at 888-838-2872 or via email to druginfo@tevapharm.com.
Teva Pharmaceuticals has issued a voluntary recall of all unexpired lots of amlodipine/valsartan and amlodipine/valsartan /hydrochlorothiazide combination tablets because of contamination with N-nitrosodiethylamine (NDEA), according to a company announcement posted on the website of the Food and Drug Administration.
The contaminant was detected at above acceptable limits in the valsartan component of the pills, which were manufactured by Mylan India. Mylan has already recalled its own valsartan-containing products. Teva has recalled other valsartan-containing products in recent months because of the presence of N-nitrosodimethylamine (NDMA).
Teva has now recalled all of its unexpired valsartan-containing products from the U.S. market. The Teva recall affects almost 50 lots, all distributed nationwide to Teva’s direct accounts, which include wholesalers, retailers, repackagers, Veterans Affairs pharmacies, and others. The lot and National Drug Code numbers of the affected products, as well as the company’s announcement, are available on the Food and Drug Administration website.
Teva is notifying its U.S. customers by certified mail and arranging for returns and reimbursements. Distribution of recalled products should stop immediately, the company said.
Patients, however, should contact their pharmacists or physicians for alternative options before stopping treatment. “The risk of harm to a patient’s health may be higher if the treatment is stopped immediately without any comparable alternative treatment,” Teva said in its announcement.
The company plans regular updates as it works through the problem. NDEA is typically found in very small amounts in certain foods, drinking water, air pollution, and certain industrial processes.
Questions and other concerns can be directed to Teva directly at 888-838-2872 or via email to druginfo@tevapharm.com.
Teva Pharmaceuticals has issued a voluntary recall of all unexpired lots of amlodipine/valsartan and amlodipine/valsartan /hydrochlorothiazide combination tablets because of contamination with N-nitrosodiethylamine (NDEA), according to a company announcement posted on the website of the Food and Drug Administration.
The contaminant was detected at above acceptable limits in the valsartan component of the pills, which were manufactured by Mylan India. Mylan has already recalled its own valsartan-containing products. Teva has recalled other valsartan-containing products in recent months because of the presence of N-nitrosodimethylamine (NDMA).
Teva has now recalled all of its unexpired valsartan-containing products from the U.S. market. The Teva recall affects almost 50 lots, all distributed nationwide to Teva’s direct accounts, which include wholesalers, retailers, repackagers, Veterans Affairs pharmacies, and others. The lot and National Drug Code numbers of the affected products, as well as the company’s announcement, are available on the Food and Drug Administration website.
Teva is notifying its U.S. customers by certified mail and arranging for returns and reimbursements. Distribution of recalled products should stop immediately, the company said.
Patients, however, should contact their pharmacists or physicians for alternative options before stopping treatment. “The risk of harm to a patient’s health may be higher if the treatment is stopped immediately without any comparable alternative treatment,” Teva said in its announcement.
The company plans regular updates as it works through the problem. NDEA is typically found in very small amounts in certain foods, drinking water, air pollution, and certain industrial processes.
Questions and other concerns can be directed to Teva directly at 888-838-2872 or via email to druginfo@tevapharm.com.
Temixys plus other antiretrovirals approved for HIV-1
The Food and Drug Administration has approved the combination of lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) known as Temixys for treatment of HIV-1 when used with other antiretrovirals. The approval is for adult and pediatric patients with HIV-1 who weigh at least 35 kg.
The approval is based on data through 144 weeks in a double-blind, active-controlled, multicenter trial in 600 antiretroviral-naive patients. The trial compared TDF/3TC plus efavirenz (EFV) with 3TC/EFV plus stavudine (d4T). The results showed similar responses at 144 weeks between both groups: 62% of patients taking TDF/3TC/EFV and 58% of patients taking d4T/3TC/EFV achieved and maintained fewer than 50 copies/mL of HIV-1 RNA.
The most common adverse events include headache, pain, depression, rash, and diarrhea. Prior to initiating treatment, patients should be tested for hepatitis B virus because there have been reports of 3TC-resistant strains of hepatitis B virus associated with treatment of HIV-1 with 3TC-containing regimens in coinfected patients. Patients should also be tested for estimated creatinine clearance, urine glucose, and urine protein because TDF/3TC is not recommended for patients with renal impairment.
The full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved the combination of lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) known as Temixys for treatment of HIV-1 when used with other antiretrovirals. The approval is for adult and pediatric patients with HIV-1 who weigh at least 35 kg.
The approval is based on data through 144 weeks in a double-blind, active-controlled, multicenter trial in 600 antiretroviral-naive patients. The trial compared TDF/3TC plus efavirenz (EFV) with 3TC/EFV plus stavudine (d4T). The results showed similar responses at 144 weeks between both groups: 62% of patients taking TDF/3TC/EFV and 58% of patients taking d4T/3TC/EFV achieved and maintained fewer than 50 copies/mL of HIV-1 RNA.
The most common adverse events include headache, pain, depression, rash, and diarrhea. Prior to initiating treatment, patients should be tested for hepatitis B virus because there have been reports of 3TC-resistant strains of hepatitis B virus associated with treatment of HIV-1 with 3TC-containing regimens in coinfected patients. Patients should also be tested for estimated creatinine clearance, urine glucose, and urine protein because TDF/3TC is not recommended for patients with renal impairment.
The full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved the combination of lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) known as Temixys for treatment of HIV-1 when used with other antiretrovirals. The approval is for adult and pediatric patients with HIV-1 who weigh at least 35 kg.
The approval is based on data through 144 weeks in a double-blind, active-controlled, multicenter trial in 600 antiretroviral-naive patients. The trial compared TDF/3TC plus efavirenz (EFV) with 3TC/EFV plus stavudine (d4T). The results showed similar responses at 144 weeks between both groups: 62% of patients taking TDF/3TC/EFV and 58% of patients taking d4T/3TC/EFV achieved and maintained fewer than 50 copies/mL of HIV-1 RNA.
The most common adverse events include headache, pain, depression, rash, and diarrhea. Prior to initiating treatment, patients should be tested for hepatitis B virus because there have been reports of 3TC-resistant strains of hepatitis B virus associated with treatment of HIV-1 with 3TC-containing regimens in coinfected patients. Patients should also be tested for estimated creatinine clearance, urine glucose, and urine protein because TDF/3TC is not recommended for patients with renal impairment.
The full prescribing information can be found on the FDA website.
FDA approves larotrectinib for cancers with NTRK gene fusions
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
CDC: No medical therapy can yet be recommended for acute flaccid myelitis
The updated guidance on managing acute flaccid myelitis is unlikely to relieve the frustrations of physicians struggling to treat the condition.
![]()
After reviewing the extant data on the baffling disorder, the Centers for Disease Control and Prevention found no evidence that corticosteroids, interferon, antivirals, or any other immunologic or biologic therapy is an effective treatment.
All of the treatments mentioned in the guidance have been used anecdotally, and often for cases proven to be associated with enterovirus-related cases. However, there are no well validated studies confirming benefit for any of these approaches, the agency said in its clinical management document.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the CDC. The number of confirmed cases is triple that seen in 2017. Whether the disease is an infectious or autoimmune process, or something else entirely, remains unknown.
In response to the outbreak – the largest since 2014 – an expert panel of 4 CDC staff physicians reviewed the literature to find what, if any, treatments were effective; another 14 external experts provided input on the recommendations. At this point, nothing can be officially recommended, the agency said.
Corticosteroids
Corticosteroids should not be administered to most patients with AFM. In addition to “a theoretical concern” about the potential adverse effects of these drugs in acute infections, there is some hard evidence that they are associated with worse outcomes in enteroviral neuroinvasive diseases, particularly those caused by EV-71.
This observation, following a 2012 outbreak in Cambodia, led a World Health Organization commission to conclude that corticosteroids were contraindicated in the management of EV-71–associated neuroinvasive disease. This year, there has been an uptick in EV-A71-associated neurologic disease.
The CDC did hedge its advice on corticosteroids a bit in the setting of AFM, however. “There may be theoretical benefit for steroids in the setting of severe cord swelling or long tract signs suggesting white matter involvement, where steroids may salvage tissue that may be harmed due to an ongoing immune/inflammatory response. While AFM is clinically and radiographically defined by the predominance of gray matter damage in the spinal cord, some patients may have some white matter involvement. It is not clear if these different patterns are important relative to therapeutic considerations.”
Nevertheless, the agency does not recommend corticosteroid use for these patients. “The possible benefits of the use of corticosteroids to manage spinal cord edema or white matter involvement in AFM should be balanced with the possible harm due to immunosuppression in the setting of possible viral infection.”
IVIG
While IVIG holds some theoretical benefit for AFM, there are no high-level human data, the guidelines state. The treatment is generally safe and well tolerated, but the few reports of its use in AFM did not show clear benefit. These include two case series. One suggested an acute improvement of neurologic status, but no long-term resolution of deficits. The other indicated neither significant improvement nor deterioration.
However, current practice at Children’s Hospital of Philadelphia is to initiate IVIG therapy at AFM diagnosis in hopes of boosting humoral immunity.
Nevertheless, the CDC said, “For IVIG to modify disease in an active viral infectious process, early administration is likely required, and possibly prior to exposure,” and the treatment cannot be recommended.
Plasma exchange
Plasma exchange in combination with IVIG and corticosteroids was ineffective in a case series of four Argentinian children, although a single case published last year found that the combination was associated with significant improvement. However, there are not enough data to recommend this approach.
Fluoxetine
Fluoxetine’s antiviral potential turned up in a high-throughput screening project to identify novel compounds with antiviral efficacy against enteroviruses. In 2012, researchers from the University of California, Los Angeles, tested more than 1,000 compounds and found that the SSRI is a potent inhibitor of coxsackievirus. A later project at the National Institutes of Health replicated this finding, and determined that fluoxetine inhibited several enteroviruses, including the AFM suspect, EV-D68.
Fluoxetine concentrates more highly in the central nervous system than it does in plasma, but its antiviral properties have nothing to do with neurotransmitter activity. Rather, it appears to inhibit protein 2C, a highly conserved nonstructural protein that’s crucial to the assembly of RNA into virion particles.
In early November, a retrospective study examined fluoxetine’s use in 30 AFM patients, compared with 26 who did not receive it. The primary outcome was change in summative limb strength score. The study did little to clarify any benefit, however. The authors concluded that fluoxetine was preferentially given to patients with EDV-68 infections. They had more severe impairment at nadir, and at the last follow-up of about 1 year, they had worse outcomes.
“There is no clear human evidence for efficacy of fluoxetine in the treatment of AFM based on a single retrospective evaluation conducted in patients with AFM, and data from a mouse model also did not support efficacy,” the CDC said.
Antiviral medications
The CDC is quite clear on its recommendation that these drugs are not indicated in AFM, since it is not yet proven to be an infectious process.
“Any guidance regarding antiviral medications should be interpreted with great caution, given the unknowns about the pathogenesis of this illness at present ... Testing has been conducted at CDC for antiviral activity of compounds pleconaril, pocapavir, and vapendavir and none have significant activity against currently circulating strains of EV-D68 at clinically relevant concentrations.”
Interferon
There is some anecdotal evidence that interferon alpha-2b was beneficial in treating a polio-like syndrome associated with West Nile virus and Saint Louis encephalitis. “Although there are limited in vitro, animal, and anecdotal human data suggesting activity of some interferons against viral infections, sufficient data are lacking in the setting of AFM,” the agency said. “There is no indication that interferon should be used for the treatment of AFM, and there is concern about the potential for harm from the use of interferon given the immunomodulatory effects in the setting of possible ongoing viral replication.”
SOURCE: CDC Acute Flaccid Myelitis: Interim Considerations for Clinical Management
The updated guidance on managing acute flaccid myelitis is unlikely to relieve the frustrations of physicians struggling to treat the condition.
![]()
After reviewing the extant data on the baffling disorder, the Centers for Disease Control and Prevention found no evidence that corticosteroids, interferon, antivirals, or any other immunologic or biologic therapy is an effective treatment.
All of the treatments mentioned in the guidance have been used anecdotally, and often for cases proven to be associated with enterovirus-related cases. However, there are no well validated studies confirming benefit for any of these approaches, the agency said in its clinical management document.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the CDC. The number of confirmed cases is triple that seen in 2017. Whether the disease is an infectious or autoimmune process, or something else entirely, remains unknown.
In response to the outbreak – the largest since 2014 – an expert panel of 4 CDC staff physicians reviewed the literature to find what, if any, treatments were effective; another 14 external experts provided input on the recommendations. At this point, nothing can be officially recommended, the agency said.
Corticosteroids
Corticosteroids should not be administered to most patients with AFM. In addition to “a theoretical concern” about the potential adverse effects of these drugs in acute infections, there is some hard evidence that they are associated with worse outcomes in enteroviral neuroinvasive diseases, particularly those caused by EV-71.
This observation, following a 2012 outbreak in Cambodia, led a World Health Organization commission to conclude that corticosteroids were contraindicated in the management of EV-71–associated neuroinvasive disease. This year, there has been an uptick in EV-A71-associated neurologic disease.
The CDC did hedge its advice on corticosteroids a bit in the setting of AFM, however. “There may be theoretical benefit for steroids in the setting of severe cord swelling or long tract signs suggesting white matter involvement, where steroids may salvage tissue that may be harmed due to an ongoing immune/inflammatory response. While AFM is clinically and radiographically defined by the predominance of gray matter damage in the spinal cord, some patients may have some white matter involvement. It is not clear if these different patterns are important relative to therapeutic considerations.”
Nevertheless, the agency does not recommend corticosteroid use for these patients. “The possible benefits of the use of corticosteroids to manage spinal cord edema or white matter involvement in AFM should be balanced with the possible harm due to immunosuppression in the setting of possible viral infection.”
IVIG
While IVIG holds some theoretical benefit for AFM, there are no high-level human data, the guidelines state. The treatment is generally safe and well tolerated, but the few reports of its use in AFM did not show clear benefit. These include two case series. One suggested an acute improvement of neurologic status, but no long-term resolution of deficits. The other indicated neither significant improvement nor deterioration.
However, current practice at Children’s Hospital of Philadelphia is to initiate IVIG therapy at AFM diagnosis in hopes of boosting humoral immunity.
Nevertheless, the CDC said, “For IVIG to modify disease in an active viral infectious process, early administration is likely required, and possibly prior to exposure,” and the treatment cannot be recommended.
Plasma exchange
Plasma exchange in combination with IVIG and corticosteroids was ineffective in a case series of four Argentinian children, although a single case published last year found that the combination was associated with significant improvement. However, there are not enough data to recommend this approach.
Fluoxetine
Fluoxetine’s antiviral potential turned up in a high-throughput screening project to identify novel compounds with antiviral efficacy against enteroviruses. In 2012, researchers from the University of California, Los Angeles, tested more than 1,000 compounds and found that the SSRI is a potent inhibitor of coxsackievirus. A later project at the National Institutes of Health replicated this finding, and determined that fluoxetine inhibited several enteroviruses, including the AFM suspect, EV-D68.
Fluoxetine concentrates more highly in the central nervous system than it does in plasma, but its antiviral properties have nothing to do with neurotransmitter activity. Rather, it appears to inhibit protein 2C, a highly conserved nonstructural protein that’s crucial to the assembly of RNA into virion particles.
In early November, a retrospective study examined fluoxetine’s use in 30 AFM patients, compared with 26 who did not receive it. The primary outcome was change in summative limb strength score. The study did little to clarify any benefit, however. The authors concluded that fluoxetine was preferentially given to patients with EDV-68 infections. They had more severe impairment at nadir, and at the last follow-up of about 1 year, they had worse outcomes.
“There is no clear human evidence for efficacy of fluoxetine in the treatment of AFM based on a single retrospective evaluation conducted in patients with AFM, and data from a mouse model also did not support efficacy,” the CDC said.
Antiviral medications
The CDC is quite clear on its recommendation that these drugs are not indicated in AFM, since it is not yet proven to be an infectious process.
“Any guidance regarding antiviral medications should be interpreted with great caution, given the unknowns about the pathogenesis of this illness at present ... Testing has been conducted at CDC for antiviral activity of compounds pleconaril, pocapavir, and vapendavir and none have significant activity against currently circulating strains of EV-D68 at clinically relevant concentrations.”
Interferon
There is some anecdotal evidence that interferon alpha-2b was beneficial in treating a polio-like syndrome associated with West Nile virus and Saint Louis encephalitis. “Although there are limited in vitro, animal, and anecdotal human data suggesting activity of some interferons against viral infections, sufficient data are lacking in the setting of AFM,” the agency said. “There is no indication that interferon should be used for the treatment of AFM, and there is concern about the potential for harm from the use of interferon given the immunomodulatory effects in the setting of possible ongoing viral replication.”
SOURCE: CDC Acute Flaccid Myelitis: Interim Considerations for Clinical Management
The updated guidance on managing acute flaccid myelitis is unlikely to relieve the frustrations of physicians struggling to treat the condition.
![]()
After reviewing the extant data on the baffling disorder, the Centers for Disease Control and Prevention found no evidence that corticosteroids, interferon, antivirals, or any other immunologic or biologic therapy is an effective treatment.
All of the treatments mentioned in the guidance have been used anecdotally, and often for cases proven to be associated with enterovirus-related cases. However, there are no well validated studies confirming benefit for any of these approaches, the agency said in its clinical management document.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the CDC. The number of confirmed cases is triple that seen in 2017. Whether the disease is an infectious or autoimmune process, or something else entirely, remains unknown.
In response to the outbreak – the largest since 2014 – an expert panel of 4 CDC staff physicians reviewed the literature to find what, if any, treatments were effective; another 14 external experts provided input on the recommendations. At this point, nothing can be officially recommended, the agency said.
Corticosteroids
Corticosteroids should not be administered to most patients with AFM. In addition to “a theoretical concern” about the potential adverse effects of these drugs in acute infections, there is some hard evidence that they are associated with worse outcomes in enteroviral neuroinvasive diseases, particularly those caused by EV-71.
This observation, following a 2012 outbreak in Cambodia, led a World Health Organization commission to conclude that corticosteroids were contraindicated in the management of EV-71–associated neuroinvasive disease. This year, there has been an uptick in EV-A71-associated neurologic disease.
The CDC did hedge its advice on corticosteroids a bit in the setting of AFM, however. “There may be theoretical benefit for steroids in the setting of severe cord swelling or long tract signs suggesting white matter involvement, where steroids may salvage tissue that may be harmed due to an ongoing immune/inflammatory response. While AFM is clinically and radiographically defined by the predominance of gray matter damage in the spinal cord, some patients may have some white matter involvement. It is not clear if these different patterns are important relative to therapeutic considerations.”
Nevertheless, the agency does not recommend corticosteroid use for these patients. “The possible benefits of the use of corticosteroids to manage spinal cord edema or white matter involvement in AFM should be balanced with the possible harm due to immunosuppression in the setting of possible viral infection.”
IVIG
While IVIG holds some theoretical benefit for AFM, there are no high-level human data, the guidelines state. The treatment is generally safe and well tolerated, but the few reports of its use in AFM did not show clear benefit. These include two case series. One suggested an acute improvement of neurologic status, but no long-term resolution of deficits. The other indicated neither significant improvement nor deterioration.
However, current practice at Children’s Hospital of Philadelphia is to initiate IVIG therapy at AFM diagnosis in hopes of boosting humoral immunity.
Nevertheless, the CDC said, “For IVIG to modify disease in an active viral infectious process, early administration is likely required, and possibly prior to exposure,” and the treatment cannot be recommended.
Plasma exchange
Plasma exchange in combination with IVIG and corticosteroids was ineffective in a case series of four Argentinian children, although a single case published last year found that the combination was associated with significant improvement. However, there are not enough data to recommend this approach.
Fluoxetine
Fluoxetine’s antiviral potential turned up in a high-throughput screening project to identify novel compounds with antiviral efficacy against enteroviruses. In 2012, researchers from the University of California, Los Angeles, tested more than 1,000 compounds and found that the SSRI is a potent inhibitor of coxsackievirus. A later project at the National Institutes of Health replicated this finding, and determined that fluoxetine inhibited several enteroviruses, including the AFM suspect, EV-D68.
Fluoxetine concentrates more highly in the central nervous system than it does in plasma, but its antiviral properties have nothing to do with neurotransmitter activity. Rather, it appears to inhibit protein 2C, a highly conserved nonstructural protein that’s crucial to the assembly of RNA into virion particles.
In early November, a retrospective study examined fluoxetine’s use in 30 AFM patients, compared with 26 who did not receive it. The primary outcome was change in summative limb strength score. The study did little to clarify any benefit, however. The authors concluded that fluoxetine was preferentially given to patients with EDV-68 infections. They had more severe impairment at nadir, and at the last follow-up of about 1 year, they had worse outcomes.
“There is no clear human evidence for efficacy of fluoxetine in the treatment of AFM based on a single retrospective evaluation conducted in patients with AFM, and data from a mouse model also did not support efficacy,” the CDC said.
Antiviral medications
The CDC is quite clear on its recommendation that these drugs are not indicated in AFM, since it is not yet proven to be an infectious process.
“Any guidance regarding antiviral medications should be interpreted with great caution, given the unknowns about the pathogenesis of this illness at present ... Testing has been conducted at CDC for antiviral activity of compounds pleconaril, pocapavir, and vapendavir and none have significant activity against currently circulating strains of EV-D68 at clinically relevant concentrations.”
Interferon
There is some anecdotal evidence that interferon alpha-2b was beneficial in treating a polio-like syndrome associated with West Nile virus and Saint Louis encephalitis. “Although there are limited in vitro, animal, and anecdotal human data suggesting activity of some interferons against viral infections, sufficient data are lacking in the setting of AFM,” the agency said. “There is no indication that interferon should be used for the treatment of AFM, and there is concern about the potential for harm from the use of interferon given the immunomodulatory effects in the setting of possible ongoing viral replication.”
SOURCE: CDC Acute Flaccid Myelitis: Interim Considerations for Clinical Management
FDA approves glasdegib for AML
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
Venetoclax gets accelerated approval in older AML patients
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.