User login
Mylan issues voluntary recall of certain valsartan-containing products
Mylan has announced that it is voluntarily recalling 15 lots of products containing valsartan because of the detection of trace amounts of N-nitrosodiethylamine within the active ingredient, according to a company announcement posted on the website of the Food and Drug Administration.

The affected products include six lots of amlodipine/valsartan tablets (5-mg/160-mg, 10-mg/160-mg, and 10-mg/320-mg strengths), seven lots of valsartan tablets (40-mg, 80-mg, 160-mg, and 320-mg strengths), and two lots of valsartan/hydrochlorothiazide tablets (320-mg/25-mg strength). All products were distributed between March 2017 and November 2018.
“Patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication. Patients who are on valsartan should continue taking their medication, as the risk of harm to the patient’s health may be higher if the treatment is stopped immediately without any alternative treatment,” the company statement said.
N-nitrosodiethylamine is a naturally occurring substance that has been identified as a human carcinogen by the International Agency for Research on Cancer.
The lot and NDC (National Drug Code) numbers of the affected products can be found in the full press release on the FDA website.
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers, and consumers in possession of recalled product should contact Stericycle at 1-888-406-9305 for the return of the recalled product. Normal business hours are Monday through Friday, 8 a.m. to 5 p.m. EST, according to the statement.
Mylan has announced that it is voluntarily recalling 15 lots of products containing valsartan because of the detection of trace amounts of N-nitrosodiethylamine within the active ingredient, according to a company announcement posted on the website of the Food and Drug Administration.
The affected products include six lots of amlodipine/valsartan tablets (5-mg/160-mg, 10-mg/160-mg, and 10-mg/320-mg strengths), seven lots of valsartan tablets (40-mg, 80-mg, 160-mg, and 320-mg strengths), and two lots of valsartan/hydrochlorothiazide tablets (320-mg/25-mg strength). All products were distributed between March 2017 and November 2018.
“Patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication. Patients who are on valsartan should continue taking their medication, as the risk of harm to the patient’s health may be higher if the treatment is stopped immediately without any alternative treatment,” the company statement said.
N-nitrosodiethylamine is a naturally occurring substance that has been identified as a human carcinogen by the International Agency for Research on Cancer.
The lot and NDC (National Drug Code) numbers of the affected products can be found in the full press release on the FDA website.
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers, and consumers in possession of recalled product should contact Stericycle at 1-888-406-9305 for the return of the recalled product. Normal business hours are Monday through Friday, 8 a.m. to 5 p.m. EST, according to the statement.
Mylan has announced that it is voluntarily recalling 15 lots of products containing valsartan because of the detection of trace amounts of N-nitrosodiethylamine within the active ingredient, according to a company announcement posted on the website of the Food and Drug Administration.
The affected products include six lots of amlodipine/valsartan tablets (5-mg/160-mg, 10-mg/160-mg, and 10-mg/320-mg strengths), seven lots of valsartan tablets (40-mg, 80-mg, 160-mg, and 320-mg strengths), and two lots of valsartan/hydrochlorothiazide tablets (320-mg/25-mg strength). All products were distributed between March 2017 and November 2018.
“Patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication. Patients who are on valsartan should continue taking their medication, as the risk of harm to the patient’s health may be higher if the treatment is stopped immediately without any alternative treatment,” the company statement said.
N-nitrosodiethylamine is a naturally occurring substance that has been identified as a human carcinogen by the International Agency for Research on Cancer.
The lot and NDC (National Drug Code) numbers of the affected products can be found in the full press release on the FDA website.
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers, and consumers in possession of recalled product should contact Stericycle at 1-888-406-9305 for the return of the recalled product. Normal business hours are Monday through Friday, 8 a.m. to 5 p.m. EST, according to the statement.
FDA warns stopping fingolimod linked to severe MS worsening
Patients with multiple sclerosis (MS) who stop taking the MS medication Gilenya (fingolimod) may experience severe disease worsening, according to a safety announcement from the Food and Drug Administration. The disease may become worse than it was before patients started the medication or while patients were taking the drug. Severe worsening is rare but can result in permanent disability, according to the FDA statement.

“The severe increase in disability in these patients was more severe than typical MS relapses, and in cases where baseline disability was known, appeared unrelated to the patients’ prior disease state,” according to the announcement. “Several patients who were able to walk without assistance prior to discontinuing Gilenya progressed to needing wheelchairs or becoming totally bed bound.” Seventeen of the patients partially recovered, eight had permanent disability, and six returned to the level of disability that they had before or during Gilenya treatment.
The patients most often discontinued treatment because they intended to or had become pregnant. Other reasons for halting therapy included lack of efficacy, lymphopenia, infections, or cancer.
The best approach to discontinuing treatment and the best way to treat a severe increase in disability if it occurs has not been determined, according to the FDA statement.
The drug’s safety labeling has been updated to include warnings about the potential for a severe increase in disability after stopping Gilenya. Health care professionals should tell patients about the risk of a severe increase in disability after stopping Gilenya and should monitor patients carefully if they do stop treatment. Patients should seek immediate medical attention after stopping the therapy if they have new or worsened MS symptoms, including trouble using their arms or legs, or changes in thinking, vision, or balance.
Patients with multiple sclerosis (MS) who stop taking the MS medication Gilenya (fingolimod) may experience severe disease worsening, according to a safety announcement from the Food and Drug Administration. The disease may become worse than it was before patients started the medication or while patients were taking the drug. Severe worsening is rare but can result in permanent disability, according to the FDA statement.
“The severe increase in disability in these patients was more severe than typical MS relapses, and in cases where baseline disability was known, appeared unrelated to the patients’ prior disease state,” according to the announcement. “Several patients who were able to walk without assistance prior to discontinuing Gilenya progressed to needing wheelchairs or becoming totally bed bound.” Seventeen of the patients partially recovered, eight had permanent disability, and six returned to the level of disability that they had before or during Gilenya treatment.
The patients most often discontinued treatment because they intended to or had become pregnant. Other reasons for halting therapy included lack of efficacy, lymphopenia, infections, or cancer.
The best approach to discontinuing treatment and the best way to treat a severe increase in disability if it occurs has not been determined, according to the FDA statement.
The drug’s safety labeling has been updated to include warnings about the potential for a severe increase in disability after stopping Gilenya. Health care professionals should tell patients about the risk of a severe increase in disability after stopping Gilenya and should monitor patients carefully if they do stop treatment. Patients should seek immediate medical attention after stopping the therapy if they have new or worsened MS symptoms, including trouble using their arms or legs, or changes in thinking, vision, or balance.
Patients with multiple sclerosis (MS) who stop taking the MS medication Gilenya (fingolimod) may experience severe disease worsening, according to a safety announcement from the Food and Drug Administration. The disease may become worse than it was before patients started the medication or while patients were taking the drug. Severe worsening is rare but can result in permanent disability, according to the FDA statement.
“The severe increase in disability in these patients was more severe than typical MS relapses, and in cases where baseline disability was known, appeared unrelated to the patients’ prior disease state,” according to the announcement. “Several patients who were able to walk without assistance prior to discontinuing Gilenya progressed to needing wheelchairs or becoming totally bed bound.” Seventeen of the patients partially recovered, eight had permanent disability, and six returned to the level of disability that they had before or during Gilenya treatment.
The patients most often discontinued treatment because they intended to or had become pregnant. Other reasons for halting therapy included lack of efficacy, lymphopenia, infections, or cancer.
The best approach to discontinuing treatment and the best way to treat a severe increase in disability if it occurs has not been determined, according to the FDA statement.
The drug’s safety labeling has been updated to include warnings about the potential for a severe increase in disability after stopping Gilenya. Health care professionals should tell patients about the risk of a severe increase in disability after stopping Gilenya and should monitor patients carefully if they do stop treatment. Patients should seek immediate medical attention after stopping the therapy if they have new or worsened MS symptoms, including trouble using their arms or legs, or changes in thinking, vision, or balance.
FDA approves emapalumab for primary HLH
The (HLH).
Emapalumab, an interferon gamma-blocking antibody, is approved to treat patients of all ages (newborn and older) with primary HLH who have refractory, recurrent, or progressive disease or who cannot tolerate conventional HLH therapy. Emapalumab is the first treatment to be FDA approved for primary HLH, and it is expected to be available in the United States in the first quarter of 2019. The FDA previously granted emapalumab priority review, breakthrough therapy designation, orphan drug designation, and rare pediatric disease designation. The FDA’s approval of emapalumab is based on results from a phase 2/3 trial (NCT01818492).
The trial included 34 patients, 27 of whom had refractory, recurrent, or progressive disease or could not tolerate conventional HLH therapy. Patients received emapalumab in combination with dexamethasone. At the end of treatment, 63% (17/27) of patients had achieved a response, which was defined as complete response (n = 7), partial response (n=8), or HLH improvement (n = 2). A total of 70% (n=19) of patients went on to hematopoietic stem cell transplant. The most common adverse events were infections (56%), hypertension (41%), infusion-related reactions (27%), and pyrexia (24%).
Results also are scheduled to be presented at the 2018 annual meeting of the American Society of Hematology in the late-breaker abstract session (Abstract LBA-6).
Emapalumab was developed by Novimmune SA. Sobi acquired global rights to the drug in August 2018.
The (HLH).
Emapalumab, an interferon gamma-blocking antibody, is approved to treat patients of all ages (newborn and older) with primary HLH who have refractory, recurrent, or progressive disease or who cannot tolerate conventional HLH therapy. Emapalumab is the first treatment to be FDA approved for primary HLH, and it is expected to be available in the United States in the first quarter of 2019. The FDA previously granted emapalumab priority review, breakthrough therapy designation, orphan drug designation, and rare pediatric disease designation. The FDA’s approval of emapalumab is based on results from a phase 2/3 trial (NCT01818492).
The trial included 34 patients, 27 of whom had refractory, recurrent, or progressive disease or could not tolerate conventional HLH therapy. Patients received emapalumab in combination with dexamethasone. At the end of treatment, 63% (17/27) of patients had achieved a response, which was defined as complete response (n = 7), partial response (n=8), or HLH improvement (n = 2). A total of 70% (n=19) of patients went on to hematopoietic stem cell transplant. The most common adverse events were infections (56%), hypertension (41%), infusion-related reactions (27%), and pyrexia (24%).
Results also are scheduled to be presented at the 2018 annual meeting of the American Society of Hematology in the late-breaker abstract session (Abstract LBA-6).
Emapalumab was developed by Novimmune SA. Sobi acquired global rights to the drug in August 2018.
The (HLH).
Emapalumab, an interferon gamma-blocking antibody, is approved to treat patients of all ages (newborn and older) with primary HLH who have refractory, recurrent, or progressive disease or who cannot tolerate conventional HLH therapy. Emapalumab is the first treatment to be FDA approved for primary HLH, and it is expected to be available in the United States in the first quarter of 2019. The FDA previously granted emapalumab priority review, breakthrough therapy designation, orphan drug designation, and rare pediatric disease designation. The FDA’s approval of emapalumab is based on results from a phase 2/3 trial (NCT01818492).
The trial included 34 patients, 27 of whom had refractory, recurrent, or progressive disease or could not tolerate conventional HLH therapy. Patients received emapalumab in combination with dexamethasone. At the end of treatment, 63% (17/27) of patients had achieved a response, which was defined as complete response (n = 7), partial response (n=8), or HLH improvement (n = 2). A total of 70% (n=19) of patients went on to hematopoietic stem cell transplant. The most common adverse events were infections (56%), hypertension (41%), infusion-related reactions (27%), and pyrexia (24%).
Results also are scheduled to be presented at the 2018 annual meeting of the American Society of Hematology in the late-breaker abstract session (Abstract LBA-6).
Emapalumab was developed by Novimmune SA. Sobi acquired global rights to the drug in August 2018.
Draft guidelines advise HIV screening for most teens and adults
Individuals aged 15-65 years, including pregnant women, should be screened for HIV infection, and those at risk should be given prophylaxis, according to draft recommendations issued by the U.S. Preventive Services Task Force. The screening recommendation extends to younger adolescents and older adults at increased risk for HIV infection. The recommendations are level A.
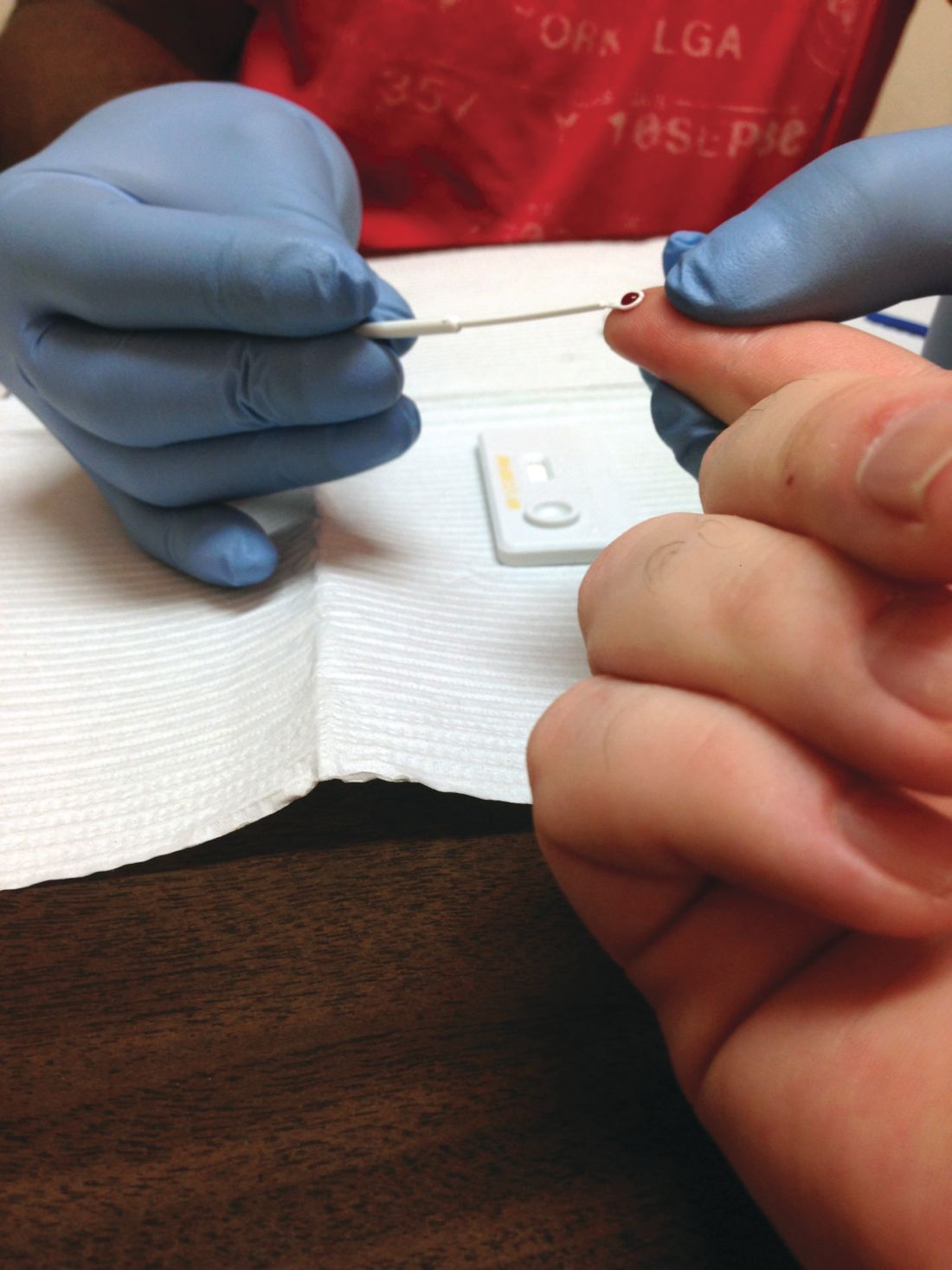
HIV remains a significant public health issue in the United States, with rates rising among individuals aged 25-29 years, although the overall number of cases has dropped slightly, according to the USPSTF report.
HIV prevention is a multistep process that includes not only screening but also wearing condoms during sex and using clean needles and syringes if injecting drugs, the researchers noted.
However, those at high risk for HIV, such as intravenous drug users, can help reduce their risk by taking a daily pill, the researchers wrote.
In an evidence report submitted to the Agency for Healthcare Research and Quality, researchers reviewed the Cochrane databases, MEDLINE, and Embase for studies up to June 2018. Based on data from 11 trials, pre-exposure prophylaxis (PrEP) consisting of antiretroviral therapy was associated with decreased risk of HIV infection, compared with placebo or no PrEP, with consistent effects across risk categories, the investigators noted.
The most common HIV risk factors include man-to-man sexual contact, injection drug use, having sex without a condom, exchanging sex for drugs or money, and having sex with an HIV-infected partner, according to the USPSTF report.
Although PrEP was associated with renal and gastrointestinal adverse effects, most were mild and resolved when the therapy either ended or continued long term. The use of PrEP does not absolve high-risk individuals from observing safety in sex activity and intravenous drug use, the researchers noted.
The Task Force’s draft recommendation statements and draft evidence reviews are available for public comment and are posted on the Task Force website at www.uspreventiveservicestaskforce.org. Comments can be submitted from Nov. 20, 2018, to Dec. 26, 2018, at www.uspreventiveservicestaskforce.org/tfcomment.htm.
Individuals aged 15-65 years, including pregnant women, should be screened for HIV infection, and those at risk should be given prophylaxis, according to draft recommendations issued by the U.S. Preventive Services Task Force. The screening recommendation extends to younger adolescents and older adults at increased risk for HIV infection. The recommendations are level A.
HIV remains a significant public health issue in the United States, with rates rising among individuals aged 25-29 years, although the overall number of cases has dropped slightly, according to the USPSTF report.
HIV prevention is a multistep process that includes not only screening but also wearing condoms during sex and using clean needles and syringes if injecting drugs, the researchers noted.
However, those at high risk for HIV, such as intravenous drug users, can help reduce their risk by taking a daily pill, the researchers wrote.
In an evidence report submitted to the Agency for Healthcare Research and Quality, researchers reviewed the Cochrane databases, MEDLINE, and Embase for studies up to June 2018. Based on data from 11 trials, pre-exposure prophylaxis (PrEP) consisting of antiretroviral therapy was associated with decreased risk of HIV infection, compared with placebo or no PrEP, with consistent effects across risk categories, the investigators noted.
The most common HIV risk factors include man-to-man sexual contact, injection drug use, having sex without a condom, exchanging sex for drugs or money, and having sex with an HIV-infected partner, according to the USPSTF report.
Although PrEP was associated with renal and gastrointestinal adverse effects, most were mild and resolved when the therapy either ended or continued long term. The use of PrEP does not absolve high-risk individuals from observing safety in sex activity and intravenous drug use, the researchers noted.
The Task Force’s draft recommendation statements and draft evidence reviews are available for public comment and are posted on the Task Force website at www.uspreventiveservicestaskforce.org. Comments can be submitted from Nov. 20, 2018, to Dec. 26, 2018, at www.uspreventiveservicestaskforce.org/tfcomment.htm.
Individuals aged 15-65 years, including pregnant women, should be screened for HIV infection, and those at risk should be given prophylaxis, according to draft recommendations issued by the U.S. Preventive Services Task Force. The screening recommendation extends to younger adolescents and older adults at increased risk for HIV infection. The recommendations are level A.
HIV remains a significant public health issue in the United States, with rates rising among individuals aged 25-29 years, although the overall number of cases has dropped slightly, according to the USPSTF report.
HIV prevention is a multistep process that includes not only screening but also wearing condoms during sex and using clean needles and syringes if injecting drugs, the researchers noted.
However, those at high risk for HIV, such as intravenous drug users, can help reduce their risk by taking a daily pill, the researchers wrote.
In an evidence report submitted to the Agency for Healthcare Research and Quality, researchers reviewed the Cochrane databases, MEDLINE, and Embase for studies up to June 2018. Based on data from 11 trials, pre-exposure prophylaxis (PrEP) consisting of antiretroviral therapy was associated with decreased risk of HIV infection, compared with placebo or no PrEP, with consistent effects across risk categories, the investigators noted.
The most common HIV risk factors include man-to-man sexual contact, injection drug use, having sex without a condom, exchanging sex for drugs or money, and having sex with an HIV-infected partner, according to the USPSTF report.
Although PrEP was associated with renal and gastrointestinal adverse effects, most were mild and resolved when the therapy either ended or continued long term. The use of PrEP does not absolve high-risk individuals from observing safety in sex activity and intravenous drug use, the researchers noted.
The Task Force’s draft recommendation statements and draft evidence reviews are available for public comment and are posted on the Task Force website at www.uspreventiveservicestaskforce.org. Comments can be submitted from Nov. 20, 2018, to Dec. 26, 2018, at www.uspreventiveservicestaskforce.org/tfcomment.htm.
FDA approves rifamycin for treatment of traveler’s diarrhea
The Food and Drug Administration has approved rifamycin (Aemcolo) for the treatment of traveler’s diarrhea caused by noninvasive strains of Escherichia coli.
FDA approval was based on results of three clinical trials. The efficacy of rifamycin was shown in a trial of 264 adults with traveler’s diarrhea in Guatemala and Mexico. Compared with placebo, rifamycin significantly reduced symptoms of the condition. The safety of rifamycin was illustrated in a pair of studies including 619 adults with traveler’s diarrhea who took rifamycin orally for 3-4 days. The most common adverse events were headache and constipation.
Traveler’s diarrhea is the most common travel-related illness, affecting 10%-40% of travelers. It can be caused by a multitude of pathogens, but bacteria from food or water is the most common source. High-risk areas include much of Asia, the Middle East, Mexico, Central and South America, and Africa.
Rifamycin was not effective in patients with diarrhea complicated by fever and/or bloody stool or in diarrhea caused by a pathogen other than E. coli.
“Travelers’ diarrhea affects millions of people each year, and having treatment options for this condition can help reduce symptoms of the condition,” Edward Cox, MD, MPH, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved rifamycin (Aemcolo) for the treatment of traveler’s diarrhea caused by noninvasive strains of Escherichia coli.
FDA approval was based on results of three clinical trials. The efficacy of rifamycin was shown in a trial of 264 adults with traveler’s diarrhea in Guatemala and Mexico. Compared with placebo, rifamycin significantly reduced symptoms of the condition. The safety of rifamycin was illustrated in a pair of studies including 619 adults with traveler’s diarrhea who took rifamycin orally for 3-4 days. The most common adverse events were headache and constipation.
Traveler’s diarrhea is the most common travel-related illness, affecting 10%-40% of travelers. It can be caused by a multitude of pathogens, but bacteria from food or water is the most common source. High-risk areas include much of Asia, the Middle East, Mexico, Central and South America, and Africa.
Rifamycin was not effective in patients with diarrhea complicated by fever and/or bloody stool or in diarrhea caused by a pathogen other than E. coli.
“Travelers’ diarrhea affects millions of people each year, and having treatment options for this condition can help reduce symptoms of the condition,” Edward Cox, MD, MPH, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved rifamycin (Aemcolo) for the treatment of traveler’s diarrhea caused by noninvasive strains of Escherichia coli.
FDA approval was based on results of three clinical trials. The efficacy of rifamycin was shown in a trial of 264 adults with traveler’s diarrhea in Guatemala and Mexico. Compared with placebo, rifamycin significantly reduced symptoms of the condition. The safety of rifamycin was illustrated in a pair of studies including 619 adults with traveler’s diarrhea who took rifamycin orally for 3-4 days. The most common adverse events were headache and constipation.
Traveler’s diarrhea is the most common travel-related illness, affecting 10%-40% of travelers. It can be caused by a multitude of pathogens, but bacteria from food or water is the most common source. High-risk areas include much of Asia, the Middle East, Mexico, Central and South America, and Africa.
Rifamycin was not effective in patients with diarrhea complicated by fever and/or bloody stool or in diarrhea caused by a pathogen other than E. coli.
“Travelers’ diarrhea affects millions of people each year, and having treatment options for this condition can help reduce symptoms of the condition,” Edward Cox, MD, MPH, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
Adjunctive Promacta approved for first-line SAA
The Food and Drug Administration has approved use of eltrombopag (Promacta) for patients severe aplastic anemia (SAA), Novartis announced Nov. 16.
The agency’s move means that eltrombopag, a synthetic thrombopoietin-receptor agonist, is now approved for use in combination with standard immunosuppressive therapy as first-line treatment for adults, and pediatric patients aged 2 and older with SAA. The drug received breakthrough therapy designation and priority review for this indication, the company said in a press release.
In addition, eltrombopag is FDA-approved for SAA patients who have had an insufficient response to immunosuppressive therapy, those with chronic immune thrombocytopenia who have had an insufficient response to other treatments, and those with thrombocytopenia and with chronic hepatitis C infection.
. The trial included 153 previously untreated SAA patients aged 2 and older. The patients received eltrombopag in combination with horse antithymocyte globulin and cyclosporine.
The starting dose of eltrombopag in the trial was 150 mg once daily for patients aged 12 and older (75 mg for East and Southeast Asians), 75 mg once daily for patients aged 6 to 11 (37.5 mg for East and Southeast Asians), and 2.5 mg/kg once daily for patients aged 2 to 5 (1.25 mg/kg for East and Southeast Asians).
Patients were divided into three cohorts with different dosing schedules. The recommended schedule from the third cohort (n = 92) was eltrombopag from day 1 to month 6, plus horse antithymocyte globulin and cyclosporine. All patients in this cohort were eligible to receive a low dose of cyclosporine for an additional 18 months if they achieved a hematologic response at 6 months.
Among the patients treated at the recommended dosing schedule, the 6-month overall response rate was 79%, and the complete response rate was 44%. The median duration of both overall and complete response was 24.3 months.
The most common adverse events in these patients were increases in ALT (29%), AST (17%), and blood bilirubin (17%). Also, rash (8%), and skin discoloration, including hyperpigmentation (5%), were cited as adverse events.
Updated results from the trial are available in the prescribing information for eltrombopag. In most countries outside of the United States, the drug is marketed as Revolade.
The Food and Drug Administration has approved use of eltrombopag (Promacta) for patients severe aplastic anemia (SAA), Novartis announced Nov. 16.
The agency’s move means that eltrombopag, a synthetic thrombopoietin-receptor agonist, is now approved for use in combination with standard immunosuppressive therapy as first-line treatment for adults, and pediatric patients aged 2 and older with SAA. The drug received breakthrough therapy designation and priority review for this indication, the company said in a press release.
In addition, eltrombopag is FDA-approved for SAA patients who have had an insufficient response to immunosuppressive therapy, those with chronic immune thrombocytopenia who have had an insufficient response to other treatments, and those with thrombocytopenia and with chronic hepatitis C infection.
. The trial included 153 previously untreated SAA patients aged 2 and older. The patients received eltrombopag in combination with horse antithymocyte globulin and cyclosporine.
The starting dose of eltrombopag in the trial was 150 mg once daily for patients aged 12 and older (75 mg for East and Southeast Asians), 75 mg once daily for patients aged 6 to 11 (37.5 mg for East and Southeast Asians), and 2.5 mg/kg once daily for patients aged 2 to 5 (1.25 mg/kg for East and Southeast Asians).
Patients were divided into three cohorts with different dosing schedules. The recommended schedule from the third cohort (n = 92) was eltrombopag from day 1 to month 6, plus horse antithymocyte globulin and cyclosporine. All patients in this cohort were eligible to receive a low dose of cyclosporine for an additional 18 months if they achieved a hematologic response at 6 months.
Among the patients treated at the recommended dosing schedule, the 6-month overall response rate was 79%, and the complete response rate was 44%. The median duration of both overall and complete response was 24.3 months.
The most common adverse events in these patients were increases in ALT (29%), AST (17%), and blood bilirubin (17%). Also, rash (8%), and skin discoloration, including hyperpigmentation (5%), were cited as adverse events.
Updated results from the trial are available in the prescribing information for eltrombopag. In most countries outside of the United States, the drug is marketed as Revolade.
The Food and Drug Administration has approved use of eltrombopag (Promacta) for patients severe aplastic anemia (SAA), Novartis announced Nov. 16.
The agency’s move means that eltrombopag, a synthetic thrombopoietin-receptor agonist, is now approved for use in combination with standard immunosuppressive therapy as first-line treatment for adults, and pediatric patients aged 2 and older with SAA. The drug received breakthrough therapy designation and priority review for this indication, the company said in a press release.
In addition, eltrombopag is FDA-approved for SAA patients who have had an insufficient response to immunosuppressive therapy, those with chronic immune thrombocytopenia who have had an insufficient response to other treatments, and those with thrombocytopenia and with chronic hepatitis C infection.
. The trial included 153 previously untreated SAA patients aged 2 and older. The patients received eltrombopag in combination with horse antithymocyte globulin and cyclosporine.
The starting dose of eltrombopag in the trial was 150 mg once daily for patients aged 12 and older (75 mg for East and Southeast Asians), 75 mg once daily for patients aged 6 to 11 (37.5 mg for East and Southeast Asians), and 2.5 mg/kg once daily for patients aged 2 to 5 (1.25 mg/kg for East and Southeast Asians).
Patients were divided into three cohorts with different dosing schedules. The recommended schedule from the third cohort (n = 92) was eltrombopag from day 1 to month 6, plus horse antithymocyte globulin and cyclosporine. All patients in this cohort were eligible to receive a low dose of cyclosporine for an additional 18 months if they achieved a hematologic response at 6 months.
Among the patients treated at the recommended dosing schedule, the 6-month overall response rate was 79%, and the complete response rate was 44%. The median duration of both overall and complete response was 24.3 months.
The most common adverse events in these patients were increases in ALT (29%), AST (17%), and blood bilirubin (17%). Also, rash (8%), and skin discoloration, including hyperpigmentation (5%), were cited as adverse events.
Updated results from the trial are available in the prescribing information for eltrombopag. In most countries outside of the United States, the drug is marketed as Revolade.
FDA expands approval of brentuximab vedotin to PTCL
The , marking the first FDA approval of a treatment for newly-diagnosed PTCL.
The drug, which is marketed by Seattle Genetics as Adcetris, is a monoclonal antibody that binds to CD30 protein found on some cancer cells.
It was previously approved for adult patients with untreated stage III or IV classical Hodgkin lymphoma (cHL), cHL after relapse, cHL after stem cell transplant in patients at high risk for relapse or progression, systemic anaplastic large cell lymphoma (ALCL) after other treatments fail, and primary cutaneous ALCL or CD30-expressing mycosis fungoides after other treatments fail.
The expanded approval, which followed the granting of Priority Review and Breakthrough Therapy designations for the supplemental Biologic License Application, was made using the FDA’s new Real-Time Oncology Review pilot program (RTOR). This program allows for data review and communication with a sponsor prior to official application submission with the goal of speeding up the review process.
The brentuximab vedotin approval now extends to previously untreated systemic ALCL and other CD30-expressing PTCLs in combination with chemotherapy.
Approval was based on the ECHELON-2 clinical trial involving 452 patients, which demonstrated improved progression-free survival (PFS) in patients with certain types of PTCL who were treated first-line with either brentuximab vedotin plus chemotherapy with cyclophosphamide, doxorubicin, prednisone (CHP), or standard chemotherapy with CHP and vincristine (CHOP). Median PFS was 48 months vs. 21 months in the groups, respectively (hazard ratio, 0.71).

The FDA advises health care providers to “monitor patients for infusion reactions, life-threatening allergic reactions (anaphylaxis), neuropathy, fever, gastrointestinal complications, and infections,” according to a press release announcing the approval, which also states that patients should be monitored for tumor lysis syndrome, serious skin reactions, pulmonary toxicity, and hepatotoxicity.
The drug may cause harm to a developing fetus or newborn and should not be used in women who are pregnant or breastfeeding. A Boxed Warning regarding risk of progressive multifocal leukoencephalopathy is also included in the prescribing information.
The current standard of care for initial treatment of PTCL is multiagent chemotherapy – a treatment that “has not significantly changed in decades and is too often unsuccessful in leading to long-term remissions, underscoring the need for new treatments, ” Steven Horwitz, MD, of Memorial Sloan Kettering Cancer Center, New York, said in a statement issued by Seattle Genetics.
“With this approval, clinicians have the opportunity to transform the way newly diagnosed CD30-expressing PTCL patients are treated,” Dr. Horwitz said.
The ECHELON-2 data will be presented at the American Society of Hematology annual meeting in San Diego on Monday, Dec. 3, 2018.
The , marking the first FDA approval of a treatment for newly-diagnosed PTCL.
The drug, which is marketed by Seattle Genetics as Adcetris, is a monoclonal antibody that binds to CD30 protein found on some cancer cells.
It was previously approved for adult patients with untreated stage III or IV classical Hodgkin lymphoma (cHL), cHL after relapse, cHL after stem cell transplant in patients at high risk for relapse or progression, systemic anaplastic large cell lymphoma (ALCL) after other treatments fail, and primary cutaneous ALCL or CD30-expressing mycosis fungoides after other treatments fail.
The expanded approval, which followed the granting of Priority Review and Breakthrough Therapy designations for the supplemental Biologic License Application, was made using the FDA’s new Real-Time Oncology Review pilot program (RTOR). This program allows for data review and communication with a sponsor prior to official application submission with the goal of speeding up the review process.
The brentuximab vedotin approval now extends to previously untreated systemic ALCL and other CD30-expressing PTCLs in combination with chemotherapy.
Approval was based on the ECHELON-2 clinical trial involving 452 patients, which demonstrated improved progression-free survival (PFS) in patients with certain types of PTCL who were treated first-line with either brentuximab vedotin plus chemotherapy with cyclophosphamide, doxorubicin, prednisone (CHP), or standard chemotherapy with CHP and vincristine (CHOP). Median PFS was 48 months vs. 21 months in the groups, respectively (hazard ratio, 0.71).
The FDA advises health care providers to “monitor patients for infusion reactions, life-threatening allergic reactions (anaphylaxis), neuropathy, fever, gastrointestinal complications, and infections,” according to a press release announcing the approval, which also states that patients should be monitored for tumor lysis syndrome, serious skin reactions, pulmonary toxicity, and hepatotoxicity.
The drug may cause harm to a developing fetus or newborn and should not be used in women who are pregnant or breastfeeding. A Boxed Warning regarding risk of progressive multifocal leukoencephalopathy is also included in the prescribing information.
The current standard of care for initial treatment of PTCL is multiagent chemotherapy – a treatment that “has not significantly changed in decades and is too often unsuccessful in leading to long-term remissions, underscoring the need for new treatments, ” Steven Horwitz, MD, of Memorial Sloan Kettering Cancer Center, New York, said in a statement issued by Seattle Genetics.
“With this approval, clinicians have the opportunity to transform the way newly diagnosed CD30-expressing PTCL patients are treated,” Dr. Horwitz said.
The ECHELON-2 data will be presented at the American Society of Hematology annual meeting in San Diego on Monday, Dec. 3, 2018.
The , marking the first FDA approval of a treatment for newly-diagnosed PTCL.
The drug, which is marketed by Seattle Genetics as Adcetris, is a monoclonal antibody that binds to CD30 protein found on some cancer cells.
It was previously approved for adult patients with untreated stage III or IV classical Hodgkin lymphoma (cHL), cHL after relapse, cHL after stem cell transplant in patients at high risk for relapse or progression, systemic anaplastic large cell lymphoma (ALCL) after other treatments fail, and primary cutaneous ALCL or CD30-expressing mycosis fungoides after other treatments fail.
The expanded approval, which followed the granting of Priority Review and Breakthrough Therapy designations for the supplemental Biologic License Application, was made using the FDA’s new Real-Time Oncology Review pilot program (RTOR). This program allows for data review and communication with a sponsor prior to official application submission with the goal of speeding up the review process.
The brentuximab vedotin approval now extends to previously untreated systemic ALCL and other CD30-expressing PTCLs in combination with chemotherapy.
Approval was based on the ECHELON-2 clinical trial involving 452 patients, which demonstrated improved progression-free survival (PFS) in patients with certain types of PTCL who were treated first-line with either brentuximab vedotin plus chemotherapy with cyclophosphamide, doxorubicin, prednisone (CHP), or standard chemotherapy with CHP and vincristine (CHOP). Median PFS was 48 months vs. 21 months in the groups, respectively (hazard ratio, 0.71).
The FDA advises health care providers to “monitor patients for infusion reactions, life-threatening allergic reactions (anaphylaxis), neuropathy, fever, gastrointestinal complications, and infections,” according to a press release announcing the approval, which also states that patients should be monitored for tumor lysis syndrome, serious skin reactions, pulmonary toxicity, and hepatotoxicity.
The drug may cause harm to a developing fetus or newborn and should not be used in women who are pregnant or breastfeeding. A Boxed Warning regarding risk of progressive multifocal leukoencephalopathy is also included in the prescribing information.
The current standard of care for initial treatment of PTCL is multiagent chemotherapy – a treatment that “has not significantly changed in decades and is too often unsuccessful in leading to long-term remissions, underscoring the need for new treatments, ” Steven Horwitz, MD, of Memorial Sloan Kettering Cancer Center, New York, said in a statement issued by Seattle Genetics.
“With this approval, clinicians have the opportunity to transform the way newly diagnosed CD30-expressing PTCL patients are treated,” Dr. Horwitz said.
The ECHELON-2 data will be presented at the American Society of Hematology annual meeting in San Diego on Monday, Dec. 3, 2018.
FDA aims to squash youth vaping, smoking
The Food and Drug Administration once again has upped the ante in its war on youth smoking and vaping.

“Today, I’m pursuing actions aimed at addressing the disturbing trend of youth nicotine use and continuing to advance the historic declines we’ve achieved in recent years in the rates of combustible cigarette use among kids,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
First and foremost, the FDA wants to reduce the lure of e-cigarettes by limiting the variety of flavored products for sale in retail outlets. Under the proposal unveiled Nov. 15, only electronic nicotine delivery systems (ENDS) that are unflavored or have tobacco, mint, or menthol flavors would be widely available. Flavored products – think cherry, cotton candy, and mango – would be sold in age-restricted environments, such as stand-alone tobacco retailers like vape shops. The FDA also seeks more stringent enforcement of age verification on ENDS products sold online.
The proposal also would reexamine regulations governing flavored cigars, with the possible aim of banning them.

“These efforts to address flavors and protect youth would dramatically impact the ability of American kids to access tobacco products that we know are both appealing and addicting,” Dr. Gottlieb said in a statement. “This policy framework reflects a redoubling of the FDA’s efforts to protect kids from all nicotine-containing products.”
In a move that seems to be aimed at youth-oriented products like Juul, the FDA will be seeking to remove from the market any ENDS product that is marketed specifically to young people.
Finally, the FDA intends to pursue regulation that would ban menthol from combustible tobacco products.
“I believe these menthol-flavored products represent one of the most common and pernicious routes by which kids initiate on combustible cigarettes,” Dr. Gottlieb said. “The menthol serves to mask some of the unattractive features of smoking that might otherwise discourage a child from smoking. Moreover, I believe that menthol products disproportionately and adversely affect underserved communities. And as a matter of public health, they exacerbate troubling disparities in health related to race and socioeconomic status.”
The policy shift comes as the Centers for Disease Control and Prevention released data from the 2018 National Youth Tobacco Survey showing that use of e-cigarettes among high schoolers is on the rise, growing from 1.5% in 2011 to 20.8% in 2018. Middle schoolers saw use over the same time period increase from 0.6% to 4.9%.
The rise of current use of e-cigarettes was enough to reverse a declining trend in overall tobacco use in recent years between 2015 and 2017.
“FDA’s enforcement efforts and policy framework would restrict access to most flavored e-cigarettes and limit the chances of youth beginning to use these products, while ensuring the products are available to adult smokers as an alternative to combustible cigarettes,” Alex M. Azar II, secretary of the Department of Health & Human Services, said in a statement supporting the FDA’s efforts. “Our obligation at HHS is always to the public health, and we believe FDA’s goals strike the right public health balance in addressing the multifaceted challenge we have before us today.”
Under Dr. Gottlieb, the FDA has been aggressively pursuing ways to reduce tobacco consumption, targeting both ENDS and combustible tobacco regulations in an effort to limit nicotine exposure and reduce the number of people addicted to nicotine and the health issues that come with it.
The American College of Cardiology voiced its support of the FDA’s actions.
“The FDA’s announcement restricting the sale of flavored e-cigarettes and other tobacco products shows they are ready to do their part in making tobacco products less available to our children,” ACC President C. Michael Valentine, MD, said in a statement, adding that the medical community needs to continue to do its part to make sure tobacco use continues to decline, especially in the nonadult population.
The FDA proposals were published as part of an advance notice of proposed rulemaking in the Federal Register. Comments can be made at www.regulations.gov through June 19.
The Food and Drug Administration once again has upped the ante in its war on youth smoking and vaping.
“Today, I’m pursuing actions aimed at addressing the disturbing trend of youth nicotine use and continuing to advance the historic declines we’ve achieved in recent years in the rates of combustible cigarette use among kids,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
First and foremost, the FDA wants to reduce the lure of e-cigarettes by limiting the variety of flavored products for sale in retail outlets. Under the proposal unveiled Nov. 15, only electronic nicotine delivery systems (ENDS) that are unflavored or have tobacco, mint, or menthol flavors would be widely available. Flavored products – think cherry, cotton candy, and mango – would be sold in age-restricted environments, such as stand-alone tobacco retailers like vape shops. The FDA also seeks more stringent enforcement of age verification on ENDS products sold online.
The proposal also would reexamine regulations governing flavored cigars, with the possible aim of banning them.
“These efforts to address flavors and protect youth would dramatically impact the ability of American kids to access tobacco products that we know are both appealing and addicting,” Dr. Gottlieb said in a statement. “This policy framework reflects a redoubling of the FDA’s efforts to protect kids from all nicotine-containing products.”
In a move that seems to be aimed at youth-oriented products like Juul, the FDA will be seeking to remove from the market any ENDS product that is marketed specifically to young people.
Finally, the FDA intends to pursue regulation that would ban menthol from combustible tobacco products.
“I believe these menthol-flavored products represent one of the most common and pernicious routes by which kids initiate on combustible cigarettes,” Dr. Gottlieb said. “The menthol serves to mask some of the unattractive features of smoking that might otherwise discourage a child from smoking. Moreover, I believe that menthol products disproportionately and adversely affect underserved communities. And as a matter of public health, they exacerbate troubling disparities in health related to race and socioeconomic status.”
The policy shift comes as the Centers for Disease Control and Prevention released data from the 2018 National Youth Tobacco Survey showing that use of e-cigarettes among high schoolers is on the rise, growing from 1.5% in 2011 to 20.8% in 2018. Middle schoolers saw use over the same time period increase from 0.6% to 4.9%.
The rise of current use of e-cigarettes was enough to reverse a declining trend in overall tobacco use in recent years between 2015 and 2017.
“FDA’s enforcement efforts and policy framework would restrict access to most flavored e-cigarettes and limit the chances of youth beginning to use these products, while ensuring the products are available to adult smokers as an alternative to combustible cigarettes,” Alex M. Azar II, secretary of the Department of Health & Human Services, said in a statement supporting the FDA’s efforts. “Our obligation at HHS is always to the public health, and we believe FDA’s goals strike the right public health balance in addressing the multifaceted challenge we have before us today.”
Under Dr. Gottlieb, the FDA has been aggressively pursuing ways to reduce tobacco consumption, targeting both ENDS and combustible tobacco regulations in an effort to limit nicotine exposure and reduce the number of people addicted to nicotine and the health issues that come with it.
The American College of Cardiology voiced its support of the FDA’s actions.
“The FDA’s announcement restricting the sale of flavored e-cigarettes and other tobacco products shows they are ready to do their part in making tobacco products less available to our children,” ACC President C. Michael Valentine, MD, said in a statement, adding that the medical community needs to continue to do its part to make sure tobacco use continues to decline, especially in the nonadult population.
The FDA proposals were published as part of an advance notice of proposed rulemaking in the Federal Register. Comments can be made at www.regulations.gov through June 19.
The Food and Drug Administration once again has upped the ante in its war on youth smoking and vaping.
“Today, I’m pursuing actions aimed at addressing the disturbing trend of youth nicotine use and continuing to advance the historic declines we’ve achieved in recent years in the rates of combustible cigarette use among kids,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
First and foremost, the FDA wants to reduce the lure of e-cigarettes by limiting the variety of flavored products for sale in retail outlets. Under the proposal unveiled Nov. 15, only electronic nicotine delivery systems (ENDS) that are unflavored or have tobacco, mint, or menthol flavors would be widely available. Flavored products – think cherry, cotton candy, and mango – would be sold in age-restricted environments, such as stand-alone tobacco retailers like vape shops. The FDA also seeks more stringent enforcement of age verification on ENDS products sold online.
The proposal also would reexamine regulations governing flavored cigars, with the possible aim of banning them.
“These efforts to address flavors and protect youth would dramatically impact the ability of American kids to access tobacco products that we know are both appealing and addicting,” Dr. Gottlieb said in a statement. “This policy framework reflects a redoubling of the FDA’s efforts to protect kids from all nicotine-containing products.”
In a move that seems to be aimed at youth-oriented products like Juul, the FDA will be seeking to remove from the market any ENDS product that is marketed specifically to young people.
Finally, the FDA intends to pursue regulation that would ban menthol from combustible tobacco products.
“I believe these menthol-flavored products represent one of the most common and pernicious routes by which kids initiate on combustible cigarettes,” Dr. Gottlieb said. “The menthol serves to mask some of the unattractive features of smoking that might otherwise discourage a child from smoking. Moreover, I believe that menthol products disproportionately and adversely affect underserved communities. And as a matter of public health, they exacerbate troubling disparities in health related to race and socioeconomic status.”
The policy shift comes as the Centers for Disease Control and Prevention released data from the 2018 National Youth Tobacco Survey showing that use of e-cigarettes among high schoolers is on the rise, growing from 1.5% in 2011 to 20.8% in 2018. Middle schoolers saw use over the same time period increase from 0.6% to 4.9%.
The rise of current use of e-cigarettes was enough to reverse a declining trend in overall tobacco use in recent years between 2015 and 2017.
“FDA’s enforcement efforts and policy framework would restrict access to most flavored e-cigarettes and limit the chances of youth beginning to use these products, while ensuring the products are available to adult smokers as an alternative to combustible cigarettes,” Alex M. Azar II, secretary of the Department of Health & Human Services, said in a statement supporting the FDA’s efforts. “Our obligation at HHS is always to the public health, and we believe FDA’s goals strike the right public health balance in addressing the multifaceted challenge we have before us today.”
Under Dr. Gottlieb, the FDA has been aggressively pursuing ways to reduce tobacco consumption, targeting both ENDS and combustible tobacco regulations in an effort to limit nicotine exposure and reduce the number of people addicted to nicotine and the health issues that come with it.
The American College of Cardiology voiced its support of the FDA’s actions.
“The FDA’s announcement restricting the sale of flavored e-cigarettes and other tobacco products shows they are ready to do their part in making tobacco products less available to our children,” ACC President C. Michael Valentine, MD, said in a statement, adding that the medical community needs to continue to do its part to make sure tobacco use continues to decline, especially in the nonadult population.
The FDA proposals were published as part of an advance notice of proposed rulemaking in the Federal Register. Comments can be made at www.regulations.gov through June 19.
More acute flaccid myelitis cases confirmed by CDC
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the Centers for Disease Control and Prevention.

The number of confirmed cases is triple that seen in 2017.
Nearly all of the patients (90%) were children aged 2-8 years, and 99% experienced a fever and /or respiratory illness 7-10 days before the onset of symptoms. But although the prodrome and seasonality of AFM suggest an infective process, only 54% of the patients tested positive for the virus, Nancy Messonnier, MD, said during a briefing held by CDC officials. The most common findings were the enteroviruses EV-A71 (29%) and EV-D68 (37%); other viruses were recovered in the remaining pathogen-positive cases.
It’s not at all clear that these were causative agents, said Dr. Messonnier, director of the National Center for Immunization and Respiratory Diseases.
“At this time of year lots of children have a fever and respiratory infections,” she said. AFM may be caused by one of the identified viruses, a still-undetected pathogen, or a pathogen hiding in untested tissue. “Or, it could be an infection that’s kicking off an immune process,” attacking gray matter in the spinal cord.
The reported increase in cases must be viewed cautiously, Dr. Messonnier said. Physicians are becoming more aware of AFM, so the spike could represent an increase in reporting as well as actual incidence.
It’s not clear why the disease manifests almost exclusively in children, Dr. Messonnier said. Nor do health officials have much of a grasp on AFM’s long-term sequelae.
“We know that patients can recover fully, but at least half don’t, and some of those have serious sequelae. Unfortunately, we have not been following every patient, so this is a gap in our knowledge.”
A newly created national task force will examine AFM’s long-term effects, Dr. Messonnier said. The task force will also look at mortality; health departments across the country will examine mortality records to identify any past deaths preceded by AFM-like symptoms.
“One of the reasons we have convened this task force is to think about this hypothesis [of an autoimmune syndrome]. We have not backed off on the idea of an infectious organism causing it, but we are thinking more broadly,” Dr. Messonnier said.
Some anti-immunization groups are blaming vaccines for the disease, noting that several childhood vaccines list encephalomyelitis and transverse myelitis as possible adverse events.
“We are investigating every one of the cases in this and prior years and have a list of hypotheses based on the epidemiology,” Dr. Messonnier said. “I would say toxins are low on that list. Many of the children may have been vaccinated [before developing AFM] and that is something we will look at, but for now we recommend that all children should be vaccinated” according to the recommended schedule.
Additional details were published on 80 of the cases. Patients’ mean age was 4 years; 59% were male. Symptoms suggesting a viral illness occurred in 99%; these included fever (81%), cough, rhinorrhea, and congestion (78%), and vomiting and diarrhea (38%).
AFM symptoms varied; 47% had only upper limb involvement, 9% only lower limb, 15% two or three upper, and 29% all four limbs. All the patients with confirmed AFM were hospitalized, and 59% treated in intensive care units. There were no deaths (MMWR. 2018;ePub:13 November. DOI: http://dx.doi.org/10.15585/mmwr.mm6745e1).
AFM remains extremely rare, Dr. Messonnier said. But physicians should be alert for any signs of sudden limb weakness in children and report those immediately. The workup should include questions about recent fever with or without respiratory or gastrointestinal symptoms. Prompt collection of viral testing samples (cerebrospinal fluid, serum, respiratory, and stool specimens) is critical.
Additional information for health care professionals is available on the CDC AFM web page.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the Centers for Disease Control and Prevention.
The number of confirmed cases is triple that seen in 2017.
Nearly all of the patients (90%) were children aged 2-8 years, and 99% experienced a fever and /or respiratory illness 7-10 days before the onset of symptoms. But although the prodrome and seasonality of AFM suggest an infective process, only 54% of the patients tested positive for the virus, Nancy Messonnier, MD, said during a briefing held by CDC officials. The most common findings were the enteroviruses EV-A71 (29%) and EV-D68 (37%); other viruses were recovered in the remaining pathogen-positive cases.
It’s not at all clear that these were causative agents, said Dr. Messonnier, director of the National Center for Immunization and Respiratory Diseases.
“At this time of year lots of children have a fever and respiratory infections,” she said. AFM may be caused by one of the identified viruses, a still-undetected pathogen, or a pathogen hiding in untested tissue. “Or, it could be an infection that’s kicking off an immune process,” attacking gray matter in the spinal cord.
The reported increase in cases must be viewed cautiously, Dr. Messonnier said. Physicians are becoming more aware of AFM, so the spike could represent an increase in reporting as well as actual incidence.
It’s not clear why the disease manifests almost exclusively in children, Dr. Messonnier said. Nor do health officials have much of a grasp on AFM’s long-term sequelae.
“We know that patients can recover fully, but at least half don’t, and some of those have serious sequelae. Unfortunately, we have not been following every patient, so this is a gap in our knowledge.”
A newly created national task force will examine AFM’s long-term effects, Dr. Messonnier said. The task force will also look at mortality; health departments across the country will examine mortality records to identify any past deaths preceded by AFM-like symptoms.
“One of the reasons we have convened this task force is to think about this hypothesis [of an autoimmune syndrome]. We have not backed off on the idea of an infectious organism causing it, but we are thinking more broadly,” Dr. Messonnier said.
Some anti-immunization groups are blaming vaccines for the disease, noting that several childhood vaccines list encephalomyelitis and transverse myelitis as possible adverse events.
“We are investigating every one of the cases in this and prior years and have a list of hypotheses based on the epidemiology,” Dr. Messonnier said. “I would say toxins are low on that list. Many of the children may have been vaccinated [before developing AFM] and that is something we will look at, but for now we recommend that all children should be vaccinated” according to the recommended schedule.
Additional details were published on 80 of the cases. Patients’ mean age was 4 years; 59% were male. Symptoms suggesting a viral illness occurred in 99%; these included fever (81%), cough, rhinorrhea, and congestion (78%), and vomiting and diarrhea (38%).
AFM symptoms varied; 47% had only upper limb involvement, 9% only lower limb, 15% two or three upper, and 29% all four limbs. All the patients with confirmed AFM were hospitalized, and 59% treated in intensive care units. There were no deaths (MMWR. 2018;ePub:13 November. DOI: http://dx.doi.org/10.15585/mmwr.mm6745e1).
AFM remains extremely rare, Dr. Messonnier said. But physicians should be alert for any signs of sudden limb weakness in children and report those immediately. The workup should include questions about recent fever with or without respiratory or gastrointestinal symptoms. Prompt collection of viral testing samples (cerebrospinal fluid, serum, respiratory, and stool specimens) is critical.
Additional information for health care professionals is available on the CDC AFM web page.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the Centers for Disease Control and Prevention.
The number of confirmed cases is triple that seen in 2017.
Nearly all of the patients (90%) were children aged 2-8 years, and 99% experienced a fever and /or respiratory illness 7-10 days before the onset of symptoms. But although the prodrome and seasonality of AFM suggest an infective process, only 54% of the patients tested positive for the virus, Nancy Messonnier, MD, said during a briefing held by CDC officials. The most common findings were the enteroviruses EV-A71 (29%) and EV-D68 (37%); other viruses were recovered in the remaining pathogen-positive cases.
It’s not at all clear that these were causative agents, said Dr. Messonnier, director of the National Center for Immunization and Respiratory Diseases.
“At this time of year lots of children have a fever and respiratory infections,” she said. AFM may be caused by one of the identified viruses, a still-undetected pathogen, or a pathogen hiding in untested tissue. “Or, it could be an infection that’s kicking off an immune process,” attacking gray matter in the spinal cord.
The reported increase in cases must be viewed cautiously, Dr. Messonnier said. Physicians are becoming more aware of AFM, so the spike could represent an increase in reporting as well as actual incidence.
It’s not clear why the disease manifests almost exclusively in children, Dr. Messonnier said. Nor do health officials have much of a grasp on AFM’s long-term sequelae.
“We know that patients can recover fully, but at least half don’t, and some of those have serious sequelae. Unfortunately, we have not been following every patient, so this is a gap in our knowledge.”
A newly created national task force will examine AFM’s long-term effects, Dr. Messonnier said. The task force will also look at mortality; health departments across the country will examine mortality records to identify any past deaths preceded by AFM-like symptoms.
“One of the reasons we have convened this task force is to think about this hypothesis [of an autoimmune syndrome]. We have not backed off on the idea of an infectious organism causing it, but we are thinking more broadly,” Dr. Messonnier said.
Some anti-immunization groups are blaming vaccines for the disease, noting that several childhood vaccines list encephalomyelitis and transverse myelitis as possible adverse events.
“We are investigating every one of the cases in this and prior years and have a list of hypotheses based on the epidemiology,” Dr. Messonnier said. “I would say toxins are low on that list. Many of the children may have been vaccinated [before developing AFM] and that is something we will look at, but for now we recommend that all children should be vaccinated” according to the recommended schedule.
Additional details were published on 80 of the cases. Patients’ mean age was 4 years; 59% were male. Symptoms suggesting a viral illness occurred in 99%; these included fever (81%), cough, rhinorrhea, and congestion (78%), and vomiting and diarrhea (38%).
AFM symptoms varied; 47% had only upper limb involvement, 9% only lower limb, 15% two or three upper, and 29% all four limbs. All the patients with confirmed AFM were hospitalized, and 59% treated in intensive care units. There were no deaths (MMWR. 2018;ePub:13 November. DOI: http://dx.doi.org/10.15585/mmwr.mm6745e1).
AFM remains extremely rare, Dr. Messonnier said. But physicians should be alert for any signs of sudden limb weakness in children and report those immediately. The workup should include questions about recent fever with or without respiratory or gastrointestinal symptoms. Prompt collection of viral testing samples (cerebrospinal fluid, serum, respiratory, and stool specimens) is critical.
Additional information for health care professionals is available on the CDC AFM web page.
FROM A CDC BRIEFING
FDA clears new blood typing, screening instrument
The Food and Drug Administration has granted marketing clearance for the immunohematology instrument NEO Iris.
NEO Iris is a fully automated blood bank instrument designed for the mid- to high-volume laboratory, according to Immucor, the company marketing the device. Immucor says NEO Iris provides the highest type and screen throughput on the market – up to 60 types and screens per hour.
NEO Iris performs ABO/Rh D typing, weak D testing, donor confirmation, cytomegalovirus screening, immunoglobulin G direct antiglobulin test and crossmatch, and antibody identification and screening.
The workflow management tool on Neo Iris has STAT priority and allows operators to run tests in any order at any time, according to Immucor.
The company says NEO Iris can hold up to 224 samples, and “modules can pipette, incubate, centrifuge, and read simultaneously.” NEO Iris integrates with Immucor’s data management software, ImmuLINK, to aggregate test results and produce reports with complete testing history.
The Food and Drug Administration has granted marketing clearance for the immunohematology instrument NEO Iris.
NEO Iris is a fully automated blood bank instrument designed for the mid- to high-volume laboratory, according to Immucor, the company marketing the device. Immucor says NEO Iris provides the highest type and screen throughput on the market – up to 60 types and screens per hour.
NEO Iris performs ABO/Rh D typing, weak D testing, donor confirmation, cytomegalovirus screening, immunoglobulin G direct antiglobulin test and crossmatch, and antibody identification and screening.
The workflow management tool on Neo Iris has STAT priority and allows operators to run tests in any order at any time, according to Immucor.
The company says NEO Iris can hold up to 224 samples, and “modules can pipette, incubate, centrifuge, and read simultaneously.” NEO Iris integrates with Immucor’s data management software, ImmuLINK, to aggregate test results and produce reports with complete testing history.
The Food and Drug Administration has granted marketing clearance for the immunohematology instrument NEO Iris.
NEO Iris is a fully automated blood bank instrument designed for the mid- to high-volume laboratory, according to Immucor, the company marketing the device. Immucor says NEO Iris provides the highest type and screen throughput on the market – up to 60 types and screens per hour.
NEO Iris performs ABO/Rh D typing, weak D testing, donor confirmation, cytomegalovirus screening, immunoglobulin G direct antiglobulin test and crossmatch, and antibody identification and screening.
The workflow management tool on Neo Iris has STAT priority and allows operators to run tests in any order at any time, according to Immucor.
The company says NEO Iris can hold up to 224 samples, and “modules can pipette, incubate, centrifuge, and read simultaneously.” NEO Iris integrates with Immucor’s data management software, ImmuLINK, to aggregate test results and produce reports with complete testing history.