User login
FDA approves Nplate for pediatric ITP
The Food and Drug Administration has approved romiplostim (Nplate) for pediatric patients aged 1 year and older who have had immune thrombocytopenia (ITP) for at least 6 months and have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.

The FDA based the approval on two trials in pediatric patients 1 year and older with ITP for at least 6 months duration.
In the first trial, 62 patients were randomized 2:1 to receive romiplostim or placebo; differences in durable platelet response, overall platelet response, and duration of response were all statistically significant, with P values less than .05.
Durable platelet response (at least 6 weekly platelet counts greater than or equal to 50 × 109/L during weeks 18 through 25 of treatment) was achieved in 22 patients (52%) who received romiplostim and 2 (10%) who received placebo. Overall platelet response was achieved in 30 (71%) and 4 (20%) patients, respectively. Patients who received romiplostim had platelet counts greater than or equal to 50 x 109/L for a median of 12 weeks, compared with 1 week in patients who received placebo, the FDA said in a statement.
In the second randomized trial, 22 patients were randomized 3:1 to receive romiplostim or placebo; 15 patients in the romiplostim arm achieved a platelet count greater than or equal to 50 x 109/L for 2 consecutive weeks and an increase in platelet count of greater than or equal to 20 × 109/L above baseline for 2 consecutive weeks during the treatment period (88%; 95% confidence interval, 64%-99%), compared with 0 patients in the placebo arm.
The most common adverse reactions observed in children receiving romiplostim include contusion, upper respiratory tract infection, and oropharyngeal pain.
The recommended initial romiplostim dose for pediatric patients is 1 mcg/kg based on actual body weight and administered as a weekly subcutaneous injection. Dose should be adjusted in increments of 1 mcg/kg until the patient achieves a platelet count greater than or equal to 50 x 109/L. Body weight should be reassessed every 12 weeks, according to the FDA announcement.
The Food and Drug Administration has approved romiplostim (Nplate) for pediatric patients aged 1 year and older who have had immune thrombocytopenia (ITP) for at least 6 months and have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
The FDA based the approval on two trials in pediatric patients 1 year and older with ITP for at least 6 months duration.
In the first trial, 62 patients were randomized 2:1 to receive romiplostim or placebo; differences in durable platelet response, overall platelet response, and duration of response were all statistically significant, with P values less than .05.
Durable platelet response (at least 6 weekly platelet counts greater than or equal to 50 × 109/L during weeks 18 through 25 of treatment) was achieved in 22 patients (52%) who received romiplostim and 2 (10%) who received placebo. Overall platelet response was achieved in 30 (71%) and 4 (20%) patients, respectively. Patients who received romiplostim had platelet counts greater than or equal to 50 x 109/L for a median of 12 weeks, compared with 1 week in patients who received placebo, the FDA said in a statement.
In the second randomized trial, 22 patients were randomized 3:1 to receive romiplostim or placebo; 15 patients in the romiplostim arm achieved a platelet count greater than or equal to 50 x 109/L for 2 consecutive weeks and an increase in platelet count of greater than or equal to 20 × 109/L above baseline for 2 consecutive weeks during the treatment period (88%; 95% confidence interval, 64%-99%), compared with 0 patients in the placebo arm.
The most common adverse reactions observed in children receiving romiplostim include contusion, upper respiratory tract infection, and oropharyngeal pain.
The recommended initial romiplostim dose for pediatric patients is 1 mcg/kg based on actual body weight and administered as a weekly subcutaneous injection. Dose should be adjusted in increments of 1 mcg/kg until the patient achieves a platelet count greater than or equal to 50 x 109/L. Body weight should be reassessed every 12 weeks, according to the FDA announcement.
The Food and Drug Administration has approved romiplostim (Nplate) for pediatric patients aged 1 year and older who have had immune thrombocytopenia (ITP) for at least 6 months and have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
The FDA based the approval on two trials in pediatric patients 1 year and older with ITP for at least 6 months duration.
In the first trial, 62 patients were randomized 2:1 to receive romiplostim or placebo; differences in durable platelet response, overall platelet response, and duration of response were all statistically significant, with P values less than .05.
Durable platelet response (at least 6 weekly platelet counts greater than or equal to 50 × 109/L during weeks 18 through 25 of treatment) was achieved in 22 patients (52%) who received romiplostim and 2 (10%) who received placebo. Overall platelet response was achieved in 30 (71%) and 4 (20%) patients, respectively. Patients who received romiplostim had platelet counts greater than or equal to 50 x 109/L for a median of 12 weeks, compared with 1 week in patients who received placebo, the FDA said in a statement.
In the second randomized trial, 22 patients were randomized 3:1 to receive romiplostim or placebo; 15 patients in the romiplostim arm achieved a platelet count greater than or equal to 50 x 109/L for 2 consecutive weeks and an increase in platelet count of greater than or equal to 20 × 109/L above baseline for 2 consecutive weeks during the treatment period (88%; 95% confidence interval, 64%-99%), compared with 0 patients in the placebo arm.
The most common adverse reactions observed in children receiving romiplostim include contusion, upper respiratory tract infection, and oropharyngeal pain.
The recommended initial romiplostim dose for pediatric patients is 1 mcg/kg based on actual body weight and administered as a weekly subcutaneous injection. Dose should be adjusted in increments of 1 mcg/kg until the patient achieves a platelet count greater than or equal to 50 x 109/L. Body weight should be reassessed every 12 weeks, according to the FDA announcement.
Duodenoscopes contain more bacteria than expected
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
FDA issues alert over e-liquids with undeclared drugs
The Food and Drug Administration has issued an alert regarding two . E-liquid is the flavored mixture used in electronic cigarettes.
In a laboratory analysis, the FDA found that “E-Cialis HelloCig E-Liquid” contained both sildenafil and tadalafil, and that “E-Rimonabant HelloCig E-Liquid” contained sildenafil. Sildenafil and tadalafil are approved for the treatment of erectile dysfunction. Unapproved usage of these drugs in over-the-counter e-liquids is therefore illegal.
Both sildenafil and tadalafil can interact with nitrates found in some prescription drugs and can cause a dangerous lowering of blood pressure. Conditions commonly treated with nitrates include diabetes, high blood pressure, high cholesterol, or heart disease.
“FDA recently warned HelloCig of these issues and contacted the company several times to recommend they recall these products due to the risks to consumers. However, HelloCig has not responded to the agency’s recommendation. Therefore, FDA urges consumers to stop using these products and to contact their health care professional with any concerns associated with their use,” the FDA said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an alert regarding two . E-liquid is the flavored mixture used in electronic cigarettes.
In a laboratory analysis, the FDA found that “E-Cialis HelloCig E-Liquid” contained both sildenafil and tadalafil, and that “E-Rimonabant HelloCig E-Liquid” contained sildenafil. Sildenafil and tadalafil are approved for the treatment of erectile dysfunction. Unapproved usage of these drugs in over-the-counter e-liquids is therefore illegal.
Both sildenafil and tadalafil can interact with nitrates found in some prescription drugs and can cause a dangerous lowering of blood pressure. Conditions commonly treated with nitrates include diabetes, high blood pressure, high cholesterol, or heart disease.
“FDA recently warned HelloCig of these issues and contacted the company several times to recommend they recall these products due to the risks to consumers. However, HelloCig has not responded to the agency’s recommendation. Therefore, FDA urges consumers to stop using these products and to contact their health care professional with any concerns associated with their use,” the FDA said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an alert regarding two . E-liquid is the flavored mixture used in electronic cigarettes.
In a laboratory analysis, the FDA found that “E-Cialis HelloCig E-Liquid” contained both sildenafil and tadalafil, and that “E-Rimonabant HelloCig E-Liquid” contained sildenafil. Sildenafil and tadalafil are approved for the treatment of erectile dysfunction. Unapproved usage of these drugs in over-the-counter e-liquids is therefore illegal.
Both sildenafil and tadalafil can interact with nitrates found in some prescription drugs and can cause a dangerous lowering of blood pressure. Conditions commonly treated with nitrates include diabetes, high blood pressure, high cholesterol, or heart disease.
“FDA recently warned HelloCig of these issues and contacted the company several times to recommend they recall these products due to the risks to consumers. However, HelloCig has not responded to the agency’s recommendation. Therefore, FDA urges consumers to stop using these products and to contact their health care professional with any concerns associated with their use,” the FDA said in the press release.
Find the full press release on the FDA website.
Bacterial contamination behind most cosmetics recalls
Most of the 313 cosmetic and personal care products recalled from 2002 to 2016 had problems with bacterial contamination, according to data obtained from the Food and Drug Administration.
, said Timothy M. Janetos, MD, and his associates at Northwestern University in Chicago. Bacterial contamination was by far the most common reason – 76% of the recalls over that period (11 class I, 217 class II, and 9 class II) – with unapproved ingredients and labeling problems well behind at 6%.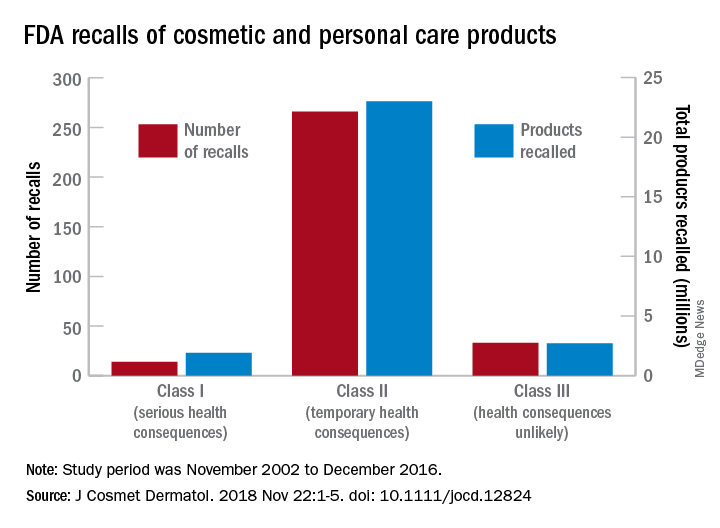
Recalls are classified by the FDA according to risk to patient safety: Class I means there is “reasonable probability of causing serious adverse health outcomes or death,” class II defines the risk as “temporary or reversible,” and class III recalls are “unlikely to cause an adverse health consequence,” they explained.
“While the number of total recalls per year was low in the context of the industry’s size and the ubiquity of cosmetic use by consumers (median: 17/year), these events involved millions of products distributed worldwide,” Dr. Janetos and his associates wrote. The class I recalls covered over 1.9 million products in distribution, the class II recalls accounted for 23 million products, and class II recalls involved over 2.7 million products.
Baby products were the category most likely to be affected, accounting for 76 (24%) of all recalls, the investigators said, with 30 involving one manufacturer of cleansing kits intended for hospital use. All but 3 of the 76 recalls resulted from bacterial contamination.
“The FDA currently has no authority to order a cosmetics manufacturer to recall a product,” they wrote, and “inspectors are only capable of inspecting 0.3% of foreign-imported products yearly,” so underreporting of such problems is likely. “Dermatologists are often the first to encounter [adverse events] related to cosmetic products and can help strengthen public safety by actively reporting these events and advocating for recalls,” said Dr. Janetos and his associates, who did not declare any conflicts of interest.
Information on reporting cosmetic-related complaints to the FDA is available on the FDA website at: https://www.fda.gov/Cosmetics/ComplianceEnforcement/AdverseEventReporting/default.htm.
rfranki@mdedge.com
SOURCE: Janetos TM et al. J Cosmet Dermatol. 2018 Nov 22:1-5. doi: 10.1111/jocd.12824.
Most of the 313 cosmetic and personal care products recalled from 2002 to 2016 had problems with bacterial contamination, according to data obtained from the Food and Drug Administration.
, said Timothy M. Janetos, MD, and his associates at Northwestern University in Chicago. Bacterial contamination was by far the most common reason – 76% of the recalls over that period (11 class I, 217 class II, and 9 class II) – with unapproved ingredients and labeling problems well behind at 6%.
Recalls are classified by the FDA according to risk to patient safety: Class I means there is “reasonable probability of causing serious adverse health outcomes or death,” class II defines the risk as “temporary or reversible,” and class III recalls are “unlikely to cause an adverse health consequence,” they explained.
“While the number of total recalls per year was low in the context of the industry’s size and the ubiquity of cosmetic use by consumers (median: 17/year), these events involved millions of products distributed worldwide,” Dr. Janetos and his associates wrote. The class I recalls covered over 1.9 million products in distribution, the class II recalls accounted for 23 million products, and class II recalls involved over 2.7 million products.
Baby products were the category most likely to be affected, accounting for 76 (24%) of all recalls, the investigators said, with 30 involving one manufacturer of cleansing kits intended for hospital use. All but 3 of the 76 recalls resulted from bacterial contamination.
“The FDA currently has no authority to order a cosmetics manufacturer to recall a product,” they wrote, and “inspectors are only capable of inspecting 0.3% of foreign-imported products yearly,” so underreporting of such problems is likely. “Dermatologists are often the first to encounter [adverse events] related to cosmetic products and can help strengthen public safety by actively reporting these events and advocating for recalls,” said Dr. Janetos and his associates, who did not declare any conflicts of interest.
Information on reporting cosmetic-related complaints to the FDA is available on the FDA website at: https://www.fda.gov/Cosmetics/ComplianceEnforcement/AdverseEventReporting/default.htm.
rfranki@mdedge.com
SOURCE: Janetos TM et al. J Cosmet Dermatol. 2018 Nov 22:1-5. doi: 10.1111/jocd.12824.
Most of the 313 cosmetic and personal care products recalled from 2002 to 2016 had problems with bacterial contamination, according to data obtained from the Food and Drug Administration.
, said Timothy M. Janetos, MD, and his associates at Northwestern University in Chicago. Bacterial contamination was by far the most common reason – 76% of the recalls over that period (11 class I, 217 class II, and 9 class II) – with unapproved ingredients and labeling problems well behind at 6%.
Recalls are classified by the FDA according to risk to patient safety: Class I means there is “reasonable probability of causing serious adverse health outcomes or death,” class II defines the risk as “temporary or reversible,” and class III recalls are “unlikely to cause an adverse health consequence,” they explained.
“While the number of total recalls per year was low in the context of the industry’s size and the ubiquity of cosmetic use by consumers (median: 17/year), these events involved millions of products distributed worldwide,” Dr. Janetos and his associates wrote. The class I recalls covered over 1.9 million products in distribution, the class II recalls accounted for 23 million products, and class II recalls involved over 2.7 million products.
Baby products were the category most likely to be affected, accounting for 76 (24%) of all recalls, the investigators said, with 30 involving one manufacturer of cleansing kits intended for hospital use. All but 3 of the 76 recalls resulted from bacterial contamination.
“The FDA currently has no authority to order a cosmetics manufacturer to recall a product,” they wrote, and “inspectors are only capable of inspecting 0.3% of foreign-imported products yearly,” so underreporting of such problems is likely. “Dermatologists are often the first to encounter [adverse events] related to cosmetic products and can help strengthen public safety by actively reporting these events and advocating for recalls,” said Dr. Janetos and his associates, who did not declare any conflicts of interest.
Information on reporting cosmetic-related complaints to the FDA is available on the FDA website at: https://www.fda.gov/Cosmetics/ComplianceEnforcement/AdverseEventReporting/default.htm.
rfranki@mdedge.com
SOURCE: Janetos TM et al. J Cosmet Dermatol. 2018 Nov 22:1-5. doi: 10.1111/jocd.12824.
FROM THE JOURNAL OF COSMETIC DERMATOLOGY
Atezolizumab combination regimen approved for advanced non-squamous NSCLC
The Food and Drug Administration has approved atezolizumab (Tecentriq) in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations.
Approval was based on greater overall survival (OS) among patients receiving the four drug combination, compared with patients who did not receive the checkpoint inhibitor but received the other three drugs in the randomized IMpower150 trial.
For the trial, 1,202 patients with metastatic NSq NSCLC were randomized to three arms for first-line treatment:
• atezolizumab, carboplatin, paclitaxel, and bevacizumab (4-drug regimen);
• atezolizumab, carboplatin and paclitaxel (3-drug regimen); or
• carboplatin, paclitaxel, and bevacizumab (control arm).
Among patients with NSq NSCLC without an EGFR or ALK mutation (87%), the estimated median OS was 19.2 months for patients receiving the 4-drug regimen and 14.7 months for those in the control arm (hazard ratio [HR] 0.78; 95% CI: 0.64, 0.96; P = .016), the FDA said in a press statement announcing the approval.
The median progression-free survival was 8.5 months for patients receiving the 4-drug regimen and 7.0 months for those in the control arm (HR 0.71; 95% CI 0.59, 0.85; P = .0002). The overall response rates were 55% in the 4-drug arm and 42% in the control arm. There were no significant differences in OS or final progression-free survival between the 3-drug arm containing atezolizumab and the control arm.
The most common adverse reactions with atezolizumab were fatigue/asthenia, alopecia, nausea, diarrhea, constipation, decreased appetite, arthralgia, hypertension, and neuropathy. Treatment with atezolizumab was discontinued in 15% of patients due to adverse reactions, the most common reason being pneumonitis.
The recommended atezolizumab dose is 1,200 mg intravenously over 60 minutes every 3 weeks, the FDA said.
The Food and Drug Administration has approved atezolizumab (Tecentriq) in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations.
Approval was based on greater overall survival (OS) among patients receiving the four drug combination, compared with patients who did not receive the checkpoint inhibitor but received the other three drugs in the randomized IMpower150 trial.
For the trial, 1,202 patients with metastatic NSq NSCLC were randomized to three arms for first-line treatment:
• atezolizumab, carboplatin, paclitaxel, and bevacizumab (4-drug regimen);
• atezolizumab, carboplatin and paclitaxel (3-drug regimen); or
• carboplatin, paclitaxel, and bevacizumab (control arm).
Among patients with NSq NSCLC without an EGFR or ALK mutation (87%), the estimated median OS was 19.2 months for patients receiving the 4-drug regimen and 14.7 months for those in the control arm (hazard ratio [HR] 0.78; 95% CI: 0.64, 0.96; P = .016), the FDA said in a press statement announcing the approval.
The median progression-free survival was 8.5 months for patients receiving the 4-drug regimen and 7.0 months for those in the control arm (HR 0.71; 95% CI 0.59, 0.85; P = .0002). The overall response rates were 55% in the 4-drug arm and 42% in the control arm. There were no significant differences in OS or final progression-free survival between the 3-drug arm containing atezolizumab and the control arm.
The most common adverse reactions with atezolizumab were fatigue/asthenia, alopecia, nausea, diarrhea, constipation, decreased appetite, arthralgia, hypertension, and neuropathy. Treatment with atezolizumab was discontinued in 15% of patients due to adverse reactions, the most common reason being pneumonitis.
The recommended atezolizumab dose is 1,200 mg intravenously over 60 minutes every 3 weeks, the FDA said.
The Food and Drug Administration has approved atezolizumab (Tecentriq) in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations.
Approval was based on greater overall survival (OS) among patients receiving the four drug combination, compared with patients who did not receive the checkpoint inhibitor but received the other three drugs in the randomized IMpower150 trial.
For the trial, 1,202 patients with metastatic NSq NSCLC were randomized to three arms for first-line treatment:
• atezolizumab, carboplatin, paclitaxel, and bevacizumab (4-drug regimen);
• atezolizumab, carboplatin and paclitaxel (3-drug regimen); or
• carboplatin, paclitaxel, and bevacizumab (control arm).
Among patients with NSq NSCLC without an EGFR or ALK mutation (87%), the estimated median OS was 19.2 months for patients receiving the 4-drug regimen and 14.7 months for those in the control arm (hazard ratio [HR] 0.78; 95% CI: 0.64, 0.96; P = .016), the FDA said in a press statement announcing the approval.
The median progression-free survival was 8.5 months for patients receiving the 4-drug regimen and 7.0 months for those in the control arm (HR 0.71; 95% CI 0.59, 0.85; P = .0002). The overall response rates were 55% in the 4-drug arm and 42% in the control arm. There were no significant differences in OS or final progression-free survival between the 3-drug arm containing atezolizumab and the control arm.
The most common adverse reactions with atezolizumab were fatigue/asthenia, alopecia, nausea, diarrhea, constipation, decreased appetite, arthralgia, hypertension, and neuropathy. Treatment with atezolizumab was discontinued in 15% of patients due to adverse reactions, the most common reason being pneumonitis.
The recommended atezolizumab dose is 1,200 mg intravenously over 60 minutes every 3 weeks, the FDA said.
Phase 3 study of novel pemphigus treatment is initiated
and will enroll about 120 patients with moderate to severe disease, according to Principia Biopharma, which is developing the drug.
In a press release, the company said that the randomized, double-blind PEGASYS study will compare PRN1008 with placebo, in about 120 patients with newly diagnosed or relapsing moderate to severe pemphigus.
The company also reported the results of an open label phase 2 study of patients with newly diagnosed or relapsing mild or moderate pemphigus, including pemphigus vulgaris and pemphigus foliaceus, which found that control of disease activity within 4 weeks of starting treatment – the primary efficacy endpoint – was achieved by more than 50% of patients taking PRN1008. Principia has extended the trial’s active treatment period from 12 to 24 weeks. The results also led the company to initiate the phase 3 trial.
PRN1008 is an inhibitor of BTK, an enzyme that “is present in the signaling pathways of most types of white blood cells except for T cells and plasma cells,” according to the company’s press release.
and will enroll about 120 patients with moderate to severe disease, according to Principia Biopharma, which is developing the drug.
In a press release, the company said that the randomized, double-blind PEGASYS study will compare PRN1008 with placebo, in about 120 patients with newly diagnosed or relapsing moderate to severe pemphigus.
The company also reported the results of an open label phase 2 study of patients with newly diagnosed or relapsing mild or moderate pemphigus, including pemphigus vulgaris and pemphigus foliaceus, which found that control of disease activity within 4 weeks of starting treatment – the primary efficacy endpoint – was achieved by more than 50% of patients taking PRN1008. Principia has extended the trial’s active treatment period from 12 to 24 weeks. The results also led the company to initiate the phase 3 trial.
PRN1008 is an inhibitor of BTK, an enzyme that “is present in the signaling pathways of most types of white blood cells except for T cells and plasma cells,” according to the company’s press release.
and will enroll about 120 patients with moderate to severe disease, according to Principia Biopharma, which is developing the drug.
In a press release, the company said that the randomized, double-blind PEGASYS study will compare PRN1008 with placebo, in about 120 patients with newly diagnosed or relapsing moderate to severe pemphigus.
The company also reported the results of an open label phase 2 study of patients with newly diagnosed or relapsing mild or moderate pemphigus, including pemphigus vulgaris and pemphigus foliaceus, which found that control of disease activity within 4 weeks of starting treatment – the primary efficacy endpoint – was achieved by more than 50% of patients taking PRN1008. Principia has extended the trial’s active treatment period from 12 to 24 weeks. The results also led the company to initiate the phase 3 trial.
PRN1008 is an inhibitor of BTK, an enzyme that “is present in the signaling pathways of most types of white blood cells except for T cells and plasma cells,” according to the company’s press release.
FDA approves congenital CMV diagnostic test
, a new test to be used as an aid in the diagnosis of congenital cytomegalovirus (CMV) in newborns less than 21 days of age.
The Alethia CMV Assay Test System detects CMV DNA from a saliva swab. Results from the test should be used only in conjunction with the results of other diagnostic tests and clinical information, according to an FDA statement.
“This test for detecting the virus, when used in conjunction with the results of other diagnostic tests, may help health care providers more quickly identify the virus in newborns,” said Timothy Stenzel, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health.
In a prospective clinical study, 1,472 saliva samples out of 1,475 samples collected from newborns were correctly identified by the device as negative for the presence of CMV DNA. Three samples were incorrectly identified as positive when they were negative. Five collected saliva specimens were correctly identified as positive for the presence of CMV DNA.
In a testing of 34 samples of archived specimens from babies known to be infected with CMV, all of the archived specimens were correctly identified by the device as positive for the presence of CMV DNA.
The FDA reviewed the Alethia CMV Assay Test System through a regulatory pathway established for novel, low- to moderate-risk devices. Along with this authorization, the FDA is establishing criteria, called special controls, which determine the requirements for demonstrating accuracy, reliability, and effectiveness of tests intended to be used as an aid in the diagnosis of congenital CMV infection.
With this new regulatory classification, subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) process, whereby devices can obtain marketing authorization by demonstrating substantial equivalence to a previously approved device.
The FDA granted marketing authorization of the Alethia CMV Assay Test System to Meridian Bioscience.
, a new test to be used as an aid in the diagnosis of congenital cytomegalovirus (CMV) in newborns less than 21 days of age.
The Alethia CMV Assay Test System detects CMV DNA from a saliva swab. Results from the test should be used only in conjunction with the results of other diagnostic tests and clinical information, according to an FDA statement.
“This test for detecting the virus, when used in conjunction with the results of other diagnostic tests, may help health care providers more quickly identify the virus in newborns,” said Timothy Stenzel, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health.
In a prospective clinical study, 1,472 saliva samples out of 1,475 samples collected from newborns were correctly identified by the device as negative for the presence of CMV DNA. Three samples were incorrectly identified as positive when they were negative. Five collected saliva specimens were correctly identified as positive for the presence of CMV DNA.
In a testing of 34 samples of archived specimens from babies known to be infected with CMV, all of the archived specimens were correctly identified by the device as positive for the presence of CMV DNA.
The FDA reviewed the Alethia CMV Assay Test System through a regulatory pathway established for novel, low- to moderate-risk devices. Along with this authorization, the FDA is establishing criteria, called special controls, which determine the requirements for demonstrating accuracy, reliability, and effectiveness of tests intended to be used as an aid in the diagnosis of congenital CMV infection.
With this new regulatory classification, subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) process, whereby devices can obtain marketing authorization by demonstrating substantial equivalence to a previously approved device.
The FDA granted marketing authorization of the Alethia CMV Assay Test System to Meridian Bioscience.
, a new test to be used as an aid in the diagnosis of congenital cytomegalovirus (CMV) in newborns less than 21 days of age.
The Alethia CMV Assay Test System detects CMV DNA from a saliva swab. Results from the test should be used only in conjunction with the results of other diagnostic tests and clinical information, according to an FDA statement.
“This test for detecting the virus, when used in conjunction with the results of other diagnostic tests, may help health care providers more quickly identify the virus in newborns,” said Timothy Stenzel, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health.
In a prospective clinical study, 1,472 saliva samples out of 1,475 samples collected from newborns were correctly identified by the device as negative for the presence of CMV DNA. Three samples were incorrectly identified as positive when they were negative. Five collected saliva specimens were correctly identified as positive for the presence of CMV DNA.
In a testing of 34 samples of archived specimens from babies known to be infected with CMV, all of the archived specimens were correctly identified by the device as positive for the presence of CMV DNA.
The FDA reviewed the Alethia CMV Assay Test System through a regulatory pathway established for novel, low- to moderate-risk devices. Along with this authorization, the FDA is establishing criteria, called special controls, which determine the requirements for demonstrating accuracy, reliability, and effectiveness of tests intended to be used as an aid in the diagnosis of congenital CMV infection.
With this new regulatory classification, subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) process, whereby devices can obtain marketing authorization by demonstrating substantial equivalence to a previously approved device.
The FDA granted marketing authorization of the Alethia CMV Assay Test System to Meridian Bioscience.
Firdapse approved: First treatment for rare autoimmune disorder
The Food and Drug Administration has approved amifampridine (Firdapse) as the first treatment for the rare autoimmune disorder known as Lambert-Eaton myasthenic syndrome, which causes the immune system to attack the neuromuscular junction and thereby disrupts the nerves’ ability to send signals to muscle cells. This causes fatigue and weakness in those affected, so they can experience difficulties with activities of daily living as a result.
The most common side effects included prickling sensation, upper respiratory tract infection, abdominal pain, and muscle spasms.
More information can be found in the FDA’s press announcement.
cpalmer@mdedge.com
The Food and Drug Administration has approved amifampridine (Firdapse) as the first treatment for the rare autoimmune disorder known as Lambert-Eaton myasthenic syndrome, which causes the immune system to attack the neuromuscular junction and thereby disrupts the nerves’ ability to send signals to muscle cells. This causes fatigue and weakness in those affected, so they can experience difficulties with activities of daily living as a result.
The most common side effects included prickling sensation, upper respiratory tract infection, abdominal pain, and muscle spasms.
More information can be found in the FDA’s press announcement.
cpalmer@mdedge.com
The Food and Drug Administration has approved amifampridine (Firdapse) as the first treatment for the rare autoimmune disorder known as Lambert-Eaton myasthenic syndrome, which causes the immune system to attack the neuromuscular junction and thereby disrupts the nerves’ ability to send signals to muscle cells. This causes fatigue and weakness in those affected, so they can experience difficulties with activities of daily living as a result.
The most common side effects included prickling sensation, upper respiratory tract infection, abdominal pain, and muscle spasms.
More information can be found in the FDA’s press announcement.
cpalmer@mdedge.com
FDA warns of serious side effect of AML treatment
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
Stroke, arterial dissection events reported with Lemtrada, FDA says
Instances of stroke and arterial dissection in the head and neck have been reported in some multiple sclerosis patients soon after an infusion of alemtuzumab (Lemtrada), according to a safety announcement issued by the Food and Drug Administration on Nov. 29.

Since the FDA approved alemtuzumab in 2014 for relapsing forms of MS, 13 cases of ischemic and hemorrhagic stroke or arterial dissection have been reported worldwide via the FDA Adverse Event Reporting System, but “additional cases we are unaware of may have occurred,” the FDA said in the announcement.
Most of the patients who developed stroke or arterial lining tears showed symptoms within a day of taking the medication, although one patient reported symptoms three days after treatment. The drug is given via intravenous infusion and is generally reserved for patients with relapsing MS who have not responded adequately to other approved MS medications, according to the FDA.
Symptoms include sudden onset of the following: severe headache or neck pain; numbness or weakness in the arms or legs, especially on only one side of the body; confusion or trouble speaking or understanding speech; vision problems in one or both eyes; and dizziness, loss of balance, or difficulty walking.
As a result of the reports, the FDA has updated the drug label prescribing information and the patient Medication Guide to reflect these risks, and added the risk of stroke to the medication’s existing boxed warning.
Health care providers should remind patients of the potential for stroke and arterial dissection at each treatment visit and advise them to seek immediate medical attention if they experience any of the symptoms reported in previous cases. “The diagnosis is often complicated because early symptoms such as headache and neck pain are not specific,” according to the agency, but patients complaining of such symptoms should be evaluated immediately.
Alemtuzumab was also approved in May 2001 for treating B-cell chronic lymphocytic leukemia (B-CLL) under the brand name Campath. The FDA will update the Campath label to reflect the new warnings and risks.
Instances of stroke and arterial dissection in the head and neck have been reported in some multiple sclerosis patients soon after an infusion of alemtuzumab (Lemtrada), according to a safety announcement issued by the Food and Drug Administration on Nov. 29.
Since the FDA approved alemtuzumab in 2014 for relapsing forms of MS, 13 cases of ischemic and hemorrhagic stroke or arterial dissection have been reported worldwide via the FDA Adverse Event Reporting System, but “additional cases we are unaware of may have occurred,” the FDA said in the announcement.
Most of the patients who developed stroke or arterial lining tears showed symptoms within a day of taking the medication, although one patient reported symptoms three days after treatment. The drug is given via intravenous infusion and is generally reserved for patients with relapsing MS who have not responded adequately to other approved MS medications, according to the FDA.
Symptoms include sudden onset of the following: severe headache or neck pain; numbness or weakness in the arms or legs, especially on only one side of the body; confusion or trouble speaking or understanding speech; vision problems in one or both eyes; and dizziness, loss of balance, or difficulty walking.
As a result of the reports, the FDA has updated the drug label prescribing information and the patient Medication Guide to reflect these risks, and added the risk of stroke to the medication’s existing boxed warning.
Health care providers should remind patients of the potential for stroke and arterial dissection at each treatment visit and advise them to seek immediate medical attention if they experience any of the symptoms reported in previous cases. “The diagnosis is often complicated because early symptoms such as headache and neck pain are not specific,” according to the agency, but patients complaining of such symptoms should be evaluated immediately.
Alemtuzumab was also approved in May 2001 for treating B-cell chronic lymphocytic leukemia (B-CLL) under the brand name Campath. The FDA will update the Campath label to reflect the new warnings and risks.
Instances of stroke and arterial dissection in the head and neck have been reported in some multiple sclerosis patients soon after an infusion of alemtuzumab (Lemtrada), according to a safety announcement issued by the Food and Drug Administration on Nov. 29.
Since the FDA approved alemtuzumab in 2014 for relapsing forms of MS, 13 cases of ischemic and hemorrhagic stroke or arterial dissection have been reported worldwide via the FDA Adverse Event Reporting System, but “additional cases we are unaware of may have occurred,” the FDA said in the announcement.
Most of the patients who developed stroke or arterial lining tears showed symptoms within a day of taking the medication, although one patient reported symptoms three days after treatment. The drug is given via intravenous infusion and is generally reserved for patients with relapsing MS who have not responded adequately to other approved MS medications, according to the FDA.
Symptoms include sudden onset of the following: severe headache or neck pain; numbness or weakness in the arms or legs, especially on only one side of the body; confusion or trouble speaking or understanding speech; vision problems in one or both eyes; and dizziness, loss of balance, or difficulty walking.
As a result of the reports, the FDA has updated the drug label prescribing information and the patient Medication Guide to reflect these risks, and added the risk of stroke to the medication’s existing boxed warning.
Health care providers should remind patients of the potential for stroke and arterial dissection at each treatment visit and advise them to seek immediate medical attention if they experience any of the symptoms reported in previous cases. “The diagnosis is often complicated because early symptoms such as headache and neck pain are not specific,” according to the agency, but patients complaining of such symptoms should be evaluated immediately.
Alemtuzumab was also approved in May 2001 for treating B-cell chronic lymphocytic leukemia (B-CLL) under the brand name Campath. The FDA will update the Campath label to reflect the new warnings and risks.