User login
Understanding, Diagnosing, and Treating Long COVID
As the pandemic wanes, the public is clamoring for a return to normal. But individuals with long COVID face a challenging journey to get back to their baseline. Here’s what clinicians need to know to help patients with long COVID.
The COVID-19 pandemic is waning. The official federal public health emergency ended on May 11, 2023. Moreover, the public is ready to move on 3 years after the beginning of a pandemic that resulted in over a million deaths in the United States.
But not everyone can go back to normal. The Centers for Disease Control and Prevention (CDC) estimates that 1 in 13 US adults (7.5%) have long COVID symptoms. Many of these people feel as if they are the forgotten patients. While everyone else is moving on, a significant number of people have not returned to their baseline.
A group from Yale School of Medicine, myself included, reviewed a number of studies to gain a better understanding of 1) how long COVID manifests and 2) potential treatment options. Highlights of our evaluation are presented here.
Long COVID: The Basics
What exactly is long COVID-19, and how is it thought to develop?
The World Health Organization (WHO) defines long COVID as symptoms that persist 3 months postinfection, last for ≥ 2 months, and are not attributable to another cause.
Hypotheses about the mechanisms of long COVID include the presence of a persistent viral reservoir, an imbalance in the viral and microbial ecosystems, reactivation of latent DNA viruses, and endothelial dysfunction.
Who is most at risk?
Females
Older individuals
Individuals with preexisting conditions, including hypertension, diabetes, obesity, and lung disease
Individuals who experienced > 5 symptoms within the first week of COVID-19 illness
Individuals with breakthrough infections after vaccination against COVID-19 appear to be at increased risk of at least 1 postacute condition
Additionally, as the risk of contracting COVID-19 is demonstrably higher in certain racial and ethnic populations, it stands to reason that more of these individuals will experience long COVID.
Long COVID symptoms: how long is long?
Long COVID symptoms may persist for 2 years after initial infection. One analysis from China showed that nearly 7 in 10 patients experienced at least 1 ongoing symptom 6 months following infection, with more than half reporting symptoms at 24 months. Dyspnea, anxiety, and depression are especially persistent.
In another analysis, 90% of individuals reported symptoms 35 weeks postinfection. Symptoms did not only occur in people who were hospitalized; they also occurred in people who had a “mild case.”
Clinical Manifestations of Long COVID
More than 50 symptoms have been identified as potentially associated with long COVID. The most common manifestations involve pulmonary, cardiac, and neuropsychiatric sequelae. There is no single test to determine if symptoms are due to long COVID.
Pulmonary
How it manifests
Chronic cough
Shortness of breath
Interstitial lung disease
Treatment options
Treatment options are variable and depend on predominant symptoms. Chronic cough should be managed based on primary etiology. Treatment for interstitial lung disease depends on whether the process continues to evolve or stabilizes. The role of antifibrotics in these patients is being investigated. Lung transplantation has largely been reserved for unresolved acute injury.
Cardiac
How it manifests
Postacute sequelae cardiovascular disease, where cardiovascular disorders are uncovered during diagnostic testing
Postacute sequelae cardiovascular syndrome, such as exercise intolerance, tachycardia and chest pain, and dyspnea
Other important considerations:
Cardiac symptoms can occur independent of preexisting conditions, severity, course of acute illness, and time from original diagnosis
Cardiac involvement can occur in any age group
One analysis revealed increased risk of stroke, arrythmias, pericarditis, myocarditis, and ischemic heart disease 1 year after COVID-19 infection
Postural orthostatic tachycardia syndrome (POTS) and neurogenic orthostatic hypotension have also been observed
Treatment options
Treatment options are dictated by clinical manifestations and course. Patients who have autonomic dysfunction can be advised to increase salt and water intake since hypovolemia can worsen symptoms. Consider fludrocortisone and midodrine along with recumbent and semirecumbent exercises as tolerated, as exercise can sometimes worsen symptoms.
Neuropsychiatric
How it manifests
Patients can present with fatigue, memory disorders, headache, vertigo, myalgia, neuropathy, and smell and taste disorders, and there have been reports of cognitive decline postinfection.
Other important considerations:
A retrospective cohort study revealed that 34% of individuals had a new neurological or psychiatric diagnosis in the first 6 months after infection, including intracranial hemorrhage, ischemic stroke, parkinsonism, and dementia. Many COVID survivors experienced critical illness requiring mechanical ventilation, sedation, and paralytics, increasing the odds of developing postintensive care syndrome
Treatment options
Use of standard of care treatments, as well as neurocognitive rehabilitation and psychosocial support, is recommended for specific neuropsychiatric conditions. Patients with headache may benefit from treatment with amitriptyline or similar medications. Olfactory training and intranasal treatments can benefit those with loss of smell.
Future Directions
Two medications that may hold promise for treating individuals long COVID symptoms are currently undergoing early investigation.
Pyridostigmine may help improve peak exercise capacity
Pyridostigmine improved peak exercise oxygen uptake in patients with chronic fatigue syndrome in a randomized, double-blind, placebo-controlled trial involving 45 individuals. Participants were assigned to receive either pyridostigmine 60 mg orally or placebo, and the pyridostigmine group showed an improved peak exercise uptake via increased cardiac output and right ventricular filling pressures.
An investigational compound may improve fatigue-based symptoms
A 4-week protocol using the compound AXA1125 improved fatigue-based symptoms in patients with long COVID in a double-blind, randomized, controlled phase 2a pilot study involving 41 individuals. Investigators looked at average change in postexertional skeletal muscle phosphocreatine (PCr) recovery rate from baseline to day 28 after moderate exercise as well as fatigue levels. Although PCr recovery rate did not differ significantly between groups, use of the compound was linked with significant reduction in fatigue-based symptoms.
Summary
It is important to exercise caution when interpreting data involving individuals with long COVID. Most studies to date are retrospective and observational, definitions and assessments are not yet standardized, and long-term follow-up is lacking, among other factors.
Clinicians should remain vigilant, keeping the following in mind as they see patients who may be experiencing long COVID:
Those most at risk include females, older individuals, those with obesity, people with preexisting conditions, individuals who experienced multiple symptoms early in their COVID-19 illness, and those who had breakthrough infections after COVID-19 vaccination
Symptoms may persist up to 2 years after acute infection
The most common manifestations of long COVID involve pulmonary, cardiac, and neuropsychiatric complications
Two medications, pyridostigmine and the compound AXA1125, are under investigation and may hold promise in treating some symptoms
As the pandemic wanes, the public is clamoring for a return to normal. But individuals with long COVID face a challenging journey to get back to their baseline. Here’s what clinicians need to know to help patients with long COVID.
The COVID-19 pandemic is waning. The official federal public health emergency ended on May 11, 2023. Moreover, the public is ready to move on 3 years after the beginning of a pandemic that resulted in over a million deaths in the United States.
But not everyone can go back to normal. The Centers for Disease Control and Prevention (CDC) estimates that 1 in 13 US adults (7.5%) have long COVID symptoms. Many of these people feel as if they are the forgotten patients. While everyone else is moving on, a significant number of people have not returned to their baseline.
A group from Yale School of Medicine, myself included, reviewed a number of studies to gain a better understanding of 1) how long COVID manifests and 2) potential treatment options. Highlights of our evaluation are presented here.
Long COVID: The Basics
What exactly is long COVID-19, and how is it thought to develop?
The World Health Organization (WHO) defines long COVID as symptoms that persist 3 months postinfection, last for ≥ 2 months, and are not attributable to another cause.
Hypotheses about the mechanisms of long COVID include the presence of a persistent viral reservoir, an imbalance in the viral and microbial ecosystems, reactivation of latent DNA viruses, and endothelial dysfunction.
Who is most at risk?
Females
Older individuals
Individuals with preexisting conditions, including hypertension, diabetes, obesity, and lung disease
Individuals who experienced > 5 symptoms within the first week of COVID-19 illness
Individuals with breakthrough infections after vaccination against COVID-19 appear to be at increased risk of at least 1 postacute condition
Additionally, as the risk of contracting COVID-19 is demonstrably higher in certain racial and ethnic populations, it stands to reason that more of these individuals will experience long COVID.
Long COVID symptoms: how long is long?
Long COVID symptoms may persist for 2 years after initial infection. One analysis from China showed that nearly 7 in 10 patients experienced at least 1 ongoing symptom 6 months following infection, with more than half reporting symptoms at 24 months. Dyspnea, anxiety, and depression are especially persistent.
In another analysis, 90% of individuals reported symptoms 35 weeks postinfection. Symptoms did not only occur in people who were hospitalized; they also occurred in people who had a “mild case.”
Clinical Manifestations of Long COVID
More than 50 symptoms have been identified as potentially associated with long COVID. The most common manifestations involve pulmonary, cardiac, and neuropsychiatric sequelae. There is no single test to determine if symptoms are due to long COVID.
Pulmonary
How it manifests
Chronic cough
Shortness of breath
Interstitial lung disease
Treatment options
Treatment options are variable and depend on predominant symptoms. Chronic cough should be managed based on primary etiology. Treatment for interstitial lung disease depends on whether the process continues to evolve or stabilizes. The role of antifibrotics in these patients is being investigated. Lung transplantation has largely been reserved for unresolved acute injury.
Cardiac
How it manifests
Postacute sequelae cardiovascular disease, where cardiovascular disorders are uncovered during diagnostic testing
Postacute sequelae cardiovascular syndrome, such as exercise intolerance, tachycardia and chest pain, and dyspnea
Other important considerations:
Cardiac symptoms can occur independent of preexisting conditions, severity, course of acute illness, and time from original diagnosis
Cardiac involvement can occur in any age group
One analysis revealed increased risk of stroke, arrythmias, pericarditis, myocarditis, and ischemic heart disease 1 year after COVID-19 infection
Postural orthostatic tachycardia syndrome (POTS) and neurogenic orthostatic hypotension have also been observed
Treatment options
Treatment options are dictated by clinical manifestations and course. Patients who have autonomic dysfunction can be advised to increase salt and water intake since hypovolemia can worsen symptoms. Consider fludrocortisone and midodrine along with recumbent and semirecumbent exercises as tolerated, as exercise can sometimes worsen symptoms.
Neuropsychiatric
How it manifests
Patients can present with fatigue, memory disorders, headache, vertigo, myalgia, neuropathy, and smell and taste disorders, and there have been reports of cognitive decline postinfection.
Other important considerations:
A retrospective cohort study revealed that 34% of individuals had a new neurological or psychiatric diagnosis in the first 6 months after infection, including intracranial hemorrhage, ischemic stroke, parkinsonism, and dementia. Many COVID survivors experienced critical illness requiring mechanical ventilation, sedation, and paralytics, increasing the odds of developing postintensive care syndrome
Treatment options
Use of standard of care treatments, as well as neurocognitive rehabilitation and psychosocial support, is recommended for specific neuropsychiatric conditions. Patients with headache may benefit from treatment with amitriptyline or similar medications. Olfactory training and intranasal treatments can benefit those with loss of smell.
Future Directions
Two medications that may hold promise for treating individuals long COVID symptoms are currently undergoing early investigation.
Pyridostigmine may help improve peak exercise capacity
Pyridostigmine improved peak exercise oxygen uptake in patients with chronic fatigue syndrome in a randomized, double-blind, placebo-controlled trial involving 45 individuals. Participants were assigned to receive either pyridostigmine 60 mg orally or placebo, and the pyridostigmine group showed an improved peak exercise uptake via increased cardiac output and right ventricular filling pressures.
An investigational compound may improve fatigue-based symptoms
A 4-week protocol using the compound AXA1125 improved fatigue-based symptoms in patients with long COVID in a double-blind, randomized, controlled phase 2a pilot study involving 41 individuals. Investigators looked at average change in postexertional skeletal muscle phosphocreatine (PCr) recovery rate from baseline to day 28 after moderate exercise as well as fatigue levels. Although PCr recovery rate did not differ significantly between groups, use of the compound was linked with significant reduction in fatigue-based symptoms.
Summary
It is important to exercise caution when interpreting data involving individuals with long COVID. Most studies to date are retrospective and observational, definitions and assessments are not yet standardized, and long-term follow-up is lacking, among other factors.
Clinicians should remain vigilant, keeping the following in mind as they see patients who may be experiencing long COVID:
Those most at risk include females, older individuals, those with obesity, people with preexisting conditions, individuals who experienced multiple symptoms early in their COVID-19 illness, and those who had breakthrough infections after COVID-19 vaccination
Symptoms may persist up to 2 years after acute infection
The most common manifestations of long COVID involve pulmonary, cardiac, and neuropsychiatric complications
Two medications, pyridostigmine and the compound AXA1125, are under investigation and may hold promise in treating some symptoms
As the pandemic wanes, the public is clamoring for a return to normal. But individuals with long COVID face a challenging journey to get back to their baseline. Here’s what clinicians need to know to help patients with long COVID.
The COVID-19 pandemic is waning. The official federal public health emergency ended on May 11, 2023. Moreover, the public is ready to move on 3 years after the beginning of a pandemic that resulted in over a million deaths in the United States.
But not everyone can go back to normal. The Centers for Disease Control and Prevention (CDC) estimates that 1 in 13 US adults (7.5%) have long COVID symptoms. Many of these people feel as if they are the forgotten patients. While everyone else is moving on, a significant number of people have not returned to their baseline.
A group from Yale School of Medicine, myself included, reviewed a number of studies to gain a better understanding of 1) how long COVID manifests and 2) potential treatment options. Highlights of our evaluation are presented here.
Long COVID: The Basics
What exactly is long COVID-19, and how is it thought to develop?
The World Health Organization (WHO) defines long COVID as symptoms that persist 3 months postinfection, last for ≥ 2 months, and are not attributable to another cause.
Hypotheses about the mechanisms of long COVID include the presence of a persistent viral reservoir, an imbalance in the viral and microbial ecosystems, reactivation of latent DNA viruses, and endothelial dysfunction.
Who is most at risk?
Females
Older individuals
Individuals with preexisting conditions, including hypertension, diabetes, obesity, and lung disease
Individuals who experienced > 5 symptoms within the first week of COVID-19 illness
Individuals with breakthrough infections after vaccination against COVID-19 appear to be at increased risk of at least 1 postacute condition
Additionally, as the risk of contracting COVID-19 is demonstrably higher in certain racial and ethnic populations, it stands to reason that more of these individuals will experience long COVID.
Long COVID symptoms: how long is long?
Long COVID symptoms may persist for 2 years after initial infection. One analysis from China showed that nearly 7 in 10 patients experienced at least 1 ongoing symptom 6 months following infection, with more than half reporting symptoms at 24 months. Dyspnea, anxiety, and depression are especially persistent.
In another analysis, 90% of individuals reported symptoms 35 weeks postinfection. Symptoms did not only occur in people who were hospitalized; they also occurred in people who had a “mild case.”
Clinical Manifestations of Long COVID
More than 50 symptoms have been identified as potentially associated with long COVID. The most common manifestations involve pulmonary, cardiac, and neuropsychiatric sequelae. There is no single test to determine if symptoms are due to long COVID.
Pulmonary
How it manifests
Chronic cough
Shortness of breath
Interstitial lung disease
Treatment options
Treatment options are variable and depend on predominant symptoms. Chronic cough should be managed based on primary etiology. Treatment for interstitial lung disease depends on whether the process continues to evolve or stabilizes. The role of antifibrotics in these patients is being investigated. Lung transplantation has largely been reserved for unresolved acute injury.
Cardiac
How it manifests
Postacute sequelae cardiovascular disease, where cardiovascular disorders are uncovered during diagnostic testing
Postacute sequelae cardiovascular syndrome, such as exercise intolerance, tachycardia and chest pain, and dyspnea
Other important considerations:
Cardiac symptoms can occur independent of preexisting conditions, severity, course of acute illness, and time from original diagnosis
Cardiac involvement can occur in any age group
One analysis revealed increased risk of stroke, arrythmias, pericarditis, myocarditis, and ischemic heart disease 1 year after COVID-19 infection
Postural orthostatic tachycardia syndrome (POTS) and neurogenic orthostatic hypotension have also been observed
Treatment options
Treatment options are dictated by clinical manifestations and course. Patients who have autonomic dysfunction can be advised to increase salt and water intake since hypovolemia can worsen symptoms. Consider fludrocortisone and midodrine along with recumbent and semirecumbent exercises as tolerated, as exercise can sometimes worsen symptoms.
Neuropsychiatric
How it manifests
Patients can present with fatigue, memory disorders, headache, vertigo, myalgia, neuropathy, and smell and taste disorders, and there have been reports of cognitive decline postinfection.
Other important considerations:
A retrospective cohort study revealed that 34% of individuals had a new neurological or psychiatric diagnosis in the first 6 months after infection, including intracranial hemorrhage, ischemic stroke, parkinsonism, and dementia. Many COVID survivors experienced critical illness requiring mechanical ventilation, sedation, and paralytics, increasing the odds of developing postintensive care syndrome
Treatment options
Use of standard of care treatments, as well as neurocognitive rehabilitation and psychosocial support, is recommended for specific neuropsychiatric conditions. Patients with headache may benefit from treatment with amitriptyline or similar medications. Olfactory training and intranasal treatments can benefit those with loss of smell.
Future Directions
Two medications that may hold promise for treating individuals long COVID symptoms are currently undergoing early investigation.
Pyridostigmine may help improve peak exercise capacity
Pyridostigmine improved peak exercise oxygen uptake in patients with chronic fatigue syndrome in a randomized, double-blind, placebo-controlled trial involving 45 individuals. Participants were assigned to receive either pyridostigmine 60 mg orally or placebo, and the pyridostigmine group showed an improved peak exercise uptake via increased cardiac output and right ventricular filling pressures.
An investigational compound may improve fatigue-based symptoms
A 4-week protocol using the compound AXA1125 improved fatigue-based symptoms in patients with long COVID in a double-blind, randomized, controlled phase 2a pilot study involving 41 individuals. Investigators looked at average change in postexertional skeletal muscle phosphocreatine (PCr) recovery rate from baseline to day 28 after moderate exercise as well as fatigue levels. Although PCr recovery rate did not differ significantly between groups, use of the compound was linked with significant reduction in fatigue-based symptoms.
Summary
It is important to exercise caution when interpreting data involving individuals with long COVID. Most studies to date are retrospective and observational, definitions and assessments are not yet standardized, and long-term follow-up is lacking, among other factors.
Clinicians should remain vigilant, keeping the following in mind as they see patients who may be experiencing long COVID:
Those most at risk include females, older individuals, those with obesity, people with preexisting conditions, individuals who experienced multiple symptoms early in their COVID-19 illness, and those who had breakthrough infections after COVID-19 vaccination
Symptoms may persist up to 2 years after acute infection
The most common manifestations of long COVID involve pulmonary, cardiac, and neuropsychiatric complications
Two medications, pyridostigmine and the compound AXA1125, are under investigation and may hold promise in treating some symptoms
COVID boosters effective, but not for long
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study.
I am here today to talk about the effectiveness of COVID vaccine boosters in the midst of 2023. The reason I want to talk about this isn’t necessarily to dig into exactly how effective vaccines are. This is an area that’s been trod upon multiple times. But it does give me an opportunity to talk about a neat study design called the “test-negative case-control” design, which has some unique properties when you’re trying to evaluate the effect of something outside of the context of a randomized trial.
So, just a little bit of background to remind everyone where we are. These are the number of doses of COVID vaccines administered over time throughout the pandemic.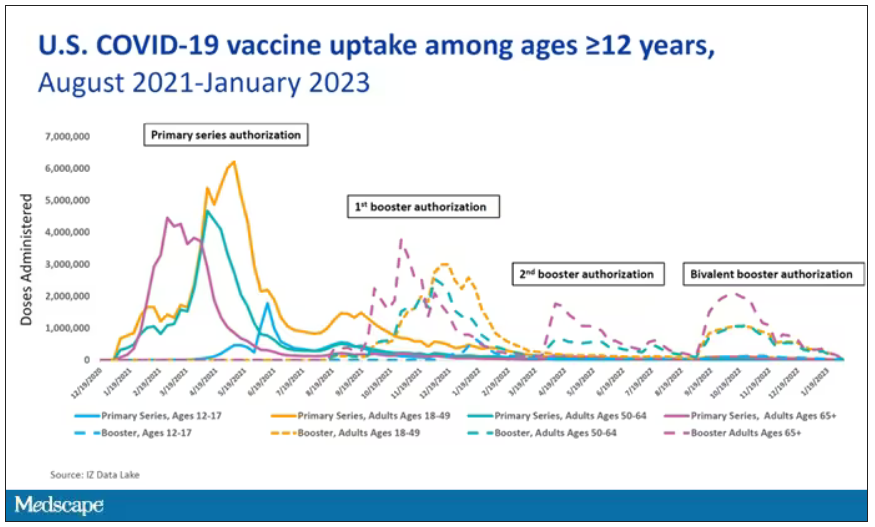
You can see that it’s stratified by age. The orange lines are adults ages 18-49, for example. You can see a big wave of vaccination when the vaccine first came out at the start of 2021. Then subsequently, you can see smaller waves after the first and second booster authorizations, and maybe a bit of a pickup, particularly among older adults, when the bivalent boosters were authorized. But still very little overall pickup of the bivalent booster, compared with the monovalent vaccines, which might suggest vaccine fatigue going on this far into the pandemic. But it’s important to try to understand exactly how effective those new boosters are, at least at this point in time.
I’m talking about Early Estimates of Bivalent mRNA Booster Dose Vaccine Effectiveness in Preventing Symptomatic SARS-CoV-2 Infection Attributable to Omicron BA.5– and XBB/XBB.1.5–Related Sublineages Among Immunocompetent Adults – Increasing Community Access to Testing Program, United States, December 2022–January 2023, which came out in the Morbidity and Mortality Weekly Report very recently, which uses this test-negative case-control design to evaluate the ability of bivalent mRNA vaccines to prevent hospitalization.
The question is: Does receipt of a bivalent COVID vaccine booster prevent hospitalizations, ICU stay, or death? That may not be the question that is of interest to everyone. I know people are interested in symptoms, missed work, and transmission, but this paper was looking at hospitalization, ICU stay, and death.
What’s kind of tricky here is that the data they’re using are in people who are hospitalized with various diseases. You might look at that on the surface and say: “Well, you can’t – that’s impossible.” But you can, actually, with this cool test-negative case-control design.
Here’s basically how it works. You take a population of people who are hospitalized and confirmed to have COVID. Some of them will be vaccinated and some of them will be unvaccinated. And the proportion of vaccinated and unvaccinated people doesn’t tell you very much because it depends on how that compares with the rates in the general population, for instance. Let me clarify this. If 100% of the population were vaccinated, then 100% of the people hospitalized with COVID would be vaccinated. That doesn’t mean vaccines are bad. Put another way, if 90% of the population were vaccinated and 60% of people hospitalized with COVID were vaccinated, that would actually show that the vaccines were working to some extent, all else being equal. So it’s not just the raw percentages that tell you anything. Some people are vaccinated, some people aren’t. You need to understand what the baseline rate is.
The test-negative case-control design looks at people who are hospitalized without COVID. Now who those people are (who the controls are, in this case) is something you really need to think about. In the case of this CDC study, they used people who were hospitalized with COVID-like illnesses – flu-like illnesses, respiratory illnesses, pneumonia, influenza, etc. This is a pretty good idea because it standardizes a little bit for people who have access to healthcare. They can get to a hospital and they’re the type of person who would go to a hospital when they’re feeling sick. That’s a better control than the general population overall, which is something I like about this design.
Some of those people who don’t have COVID (they’re in the hospital for flu or whatever) will have been vaccinated for COVID, and some will not have been vaccinated for COVID. And of course, we don’t expect COVID vaccines necessarily to protect against the flu or pneumonia, but that gives us a way to standardize.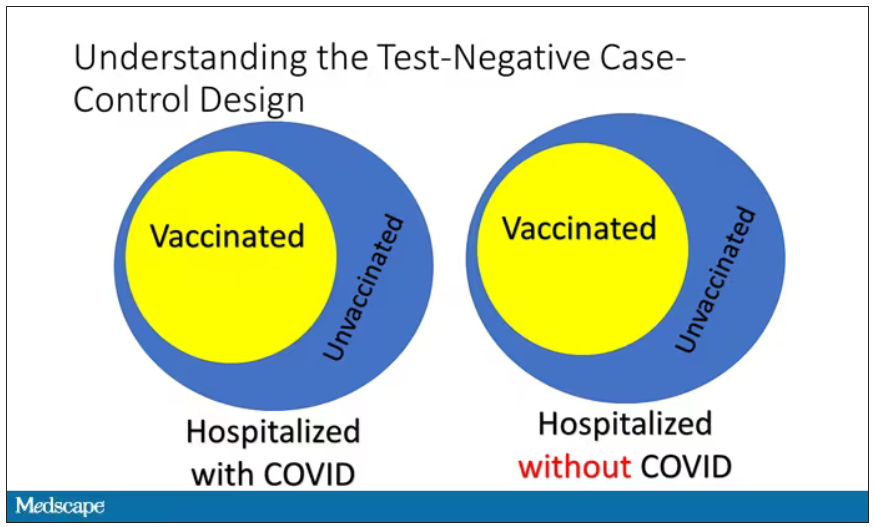
If you look at these Venn diagrams, I’ve got vaccinated/unvaccinated being exactly the same proportion, which would suggest that you’re just as likely to be hospitalized with COVID if you’re vaccinated as you are to be hospitalized with some other respiratory illness, which suggests that the vaccine isn’t particularly effective.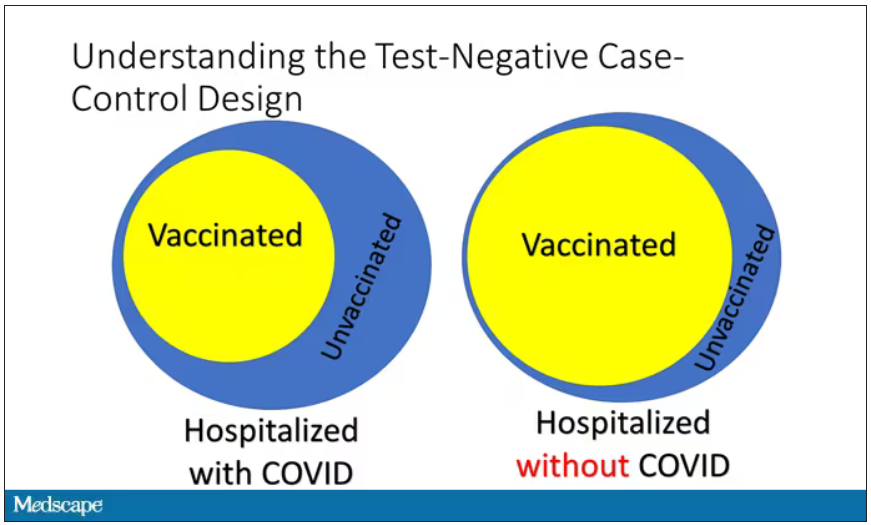
However, if you saw something like this, looking at all those patients with flu and other non-COVID illnesses, a lot more of them had been vaccinated for COVID. What that tells you is that we’re seeing fewer vaccinated people hospitalized with COVID than we would expect because we have this standardization from other respiratory infections. We expect this many vaccinated people because that’s how many vaccinated people there are who show up with flu. But in the COVID population, there are fewer, and that would suggest that the vaccines are effective. So that is the test-negative case-control design. You can do the same thing with ICU stays and death.
There are some assumptions here which you might already be thinking about. The most important one is that vaccination status is not associated with the risk for the disease. I always think of older people in this context. During the pandemic, at least in the United States, older people were much more likely to be vaccinated but were also much more likely to contract COVID and be hospitalized with COVID. The test-negative design actually accounts for this in some sense, because older people are also more likely to be hospitalized for things like flu and pneumonia. So there’s some control there.
But to the extent that older people are uniquely susceptible to COVID compared with other respiratory illnesses, that would bias your results to make the vaccines look worse. So the standard approach here is to adjust for these things. I think the CDC adjusted for age, sex, race, ethnicity, and a few other things to settle down and see how effective the vaccines were.
Let’s get to a worked example.
This is the actual data from the CDC paper. They had 6,907 individuals who were hospitalized with COVID, and 26% of them were unvaccinated. What’s the baseline rate that we would expect to be unvaccinated? A total of 59,234 individuals were hospitalized with a non-COVID respiratory illness, and 23% of them were unvaccinated. So you can see that there were more unvaccinated people than you would think in the COVID group. In other words, fewer vaccinated people, which suggests that the vaccine works to some degree because it’s keeping some people out of the hospital.
Now, 26% versus 23% is not a very impressive difference. But it gets more interesting when you break it down by the type of vaccine and how long ago the individual was vaccinated.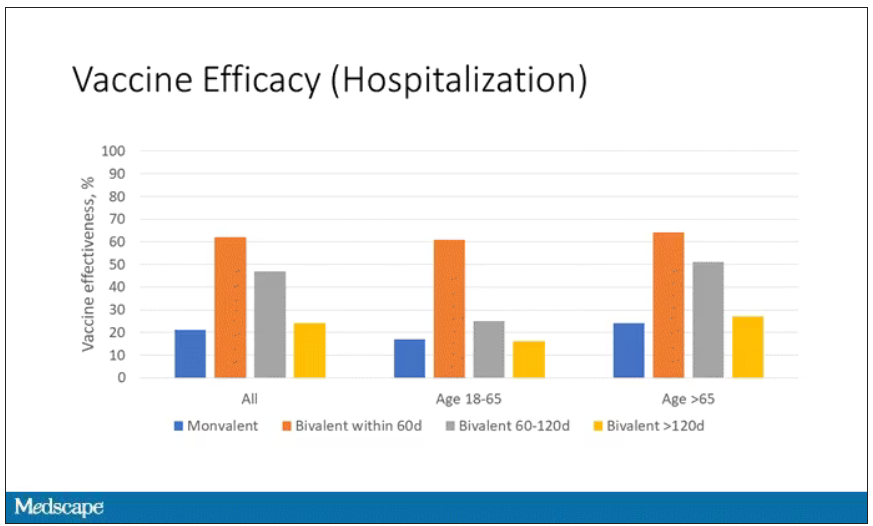
Let’s walk through the “all” group on this figure. What you can see is the calculated vaccine effectiveness. If you look at just the monovalent vaccine here, we see a 20% vaccine effectiveness. This means that you’re preventing 20% of hospitalizations basically due to COVID by people getting vaccinated. That’s okay but it’s certainly not anything to write home about. But we see much better vaccine effectiveness with the bivalent vaccine if it had been received within 60 days.
This compares people who received the bivalent vaccine within 60 days in the COVID group and the non-COVID group. The concern that the vaccine was given very recently affects both groups equally so it shouldn’t result in bias there. You see a step-off in vaccine effectiveness from 60 days, 60-120 days, and greater than 120 days. This is 4 months, and you’ve gone from 60% to 20%. When you break that down by age, you can see a similar pattern in the 18-to-65 group and potentially some more protection the greater than 65 age group.
Why is vaccine efficacy going down? The study doesn’t tell us, but we can hypothesize that this might be an immunologic effect – the antibodies or the protective T cells are waning over time. This could also reflect changes in the virus in the environment as the virus seeks to evade certain immune responses. But overall, this suggests that waiting a year between booster doses may leave you exposed for quite some time, although the take-home here is that bivalent vaccines in general are probably a good idea for the proportion of people who haven’t gotten them.
When we look at critical illness and death, the numbers look a little bit better.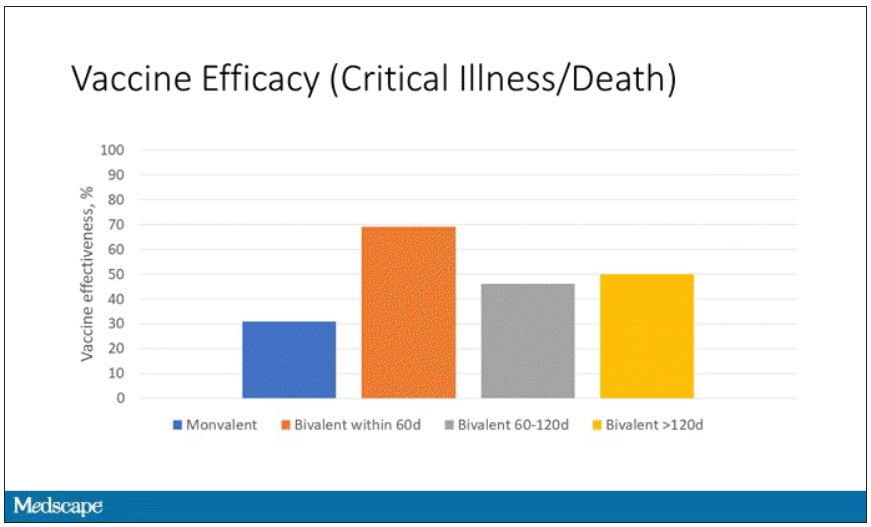
You can see that bivalent is better than monovalent – certainly pretty good if you’ve received it within 60 days. It does tend to wane a little bit, but not nearly as much. You’ve still got about 50% vaccine efficacy beyond 120 days when we’re looking at critical illness, which is stays in the ICU and death.
The overriding thing to think about when we think about vaccine policy is that the way you get immunized against COVID is either by vaccine or by getting infected with COVID, or both.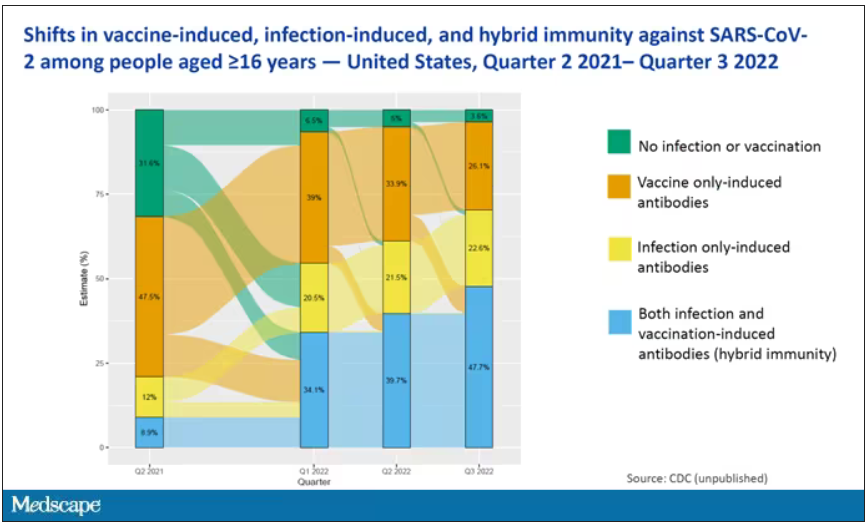
This really interesting graph from the CDC (although it’s updated only through quarter three of 2022) shows the proportion of Americans, based on routine lab tests, who have varying degrees of protection against COVID. What you can see is that, by quarter three of 2022, just 3.6% of people who had blood drawn at a commercial laboratory had no evidence of infection or vaccination. In other words, almost no one was totally naive. Then 26% of people had never been infected – they only have vaccine antibodies – plus 22% of people had only been infected but had never been vaccinated. And then 50% of people had both. So there’s a tremendous amount of existing immunity out there.
The really interesting question about future vaccination and future booster doses is, how does it work on the background of this pattern? The CDC study doesn’t tell us, and I don’t think they have the data to tell us the vaccine efficacy in these different groups. Is it more effective in people who have only had an infection, for example? Is it more effective in people who have only had vaccination versus people who had both, or people who have no protection whatsoever? Those are the really interesting questions that need to be answered going forward as vaccine policy gets developed in the future.
I hope this was a helpful primer on how the test-negative case-control design can answer questions that seem a little bit unanswerable.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and director of Yale’s Clinical and Translational Research Accelerator. He disclosed no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study.
I am here today to talk about the effectiveness of COVID vaccine boosters in the midst of 2023. The reason I want to talk about this isn’t necessarily to dig into exactly how effective vaccines are. This is an area that’s been trod upon multiple times. But it does give me an opportunity to talk about a neat study design called the “test-negative case-control” design, which has some unique properties when you’re trying to evaluate the effect of something outside of the context of a randomized trial.
So, just a little bit of background to remind everyone where we are. These are the number of doses of COVID vaccines administered over time throughout the pandemic.
You can see that it’s stratified by age. The orange lines are adults ages 18-49, for example. You can see a big wave of vaccination when the vaccine first came out at the start of 2021. Then subsequently, you can see smaller waves after the first and second booster authorizations, and maybe a bit of a pickup, particularly among older adults, when the bivalent boosters were authorized. But still very little overall pickup of the bivalent booster, compared with the monovalent vaccines, which might suggest vaccine fatigue going on this far into the pandemic. But it’s important to try to understand exactly how effective those new boosters are, at least at this point in time.
I’m talking about Early Estimates of Bivalent mRNA Booster Dose Vaccine Effectiveness in Preventing Symptomatic SARS-CoV-2 Infection Attributable to Omicron BA.5– and XBB/XBB.1.5–Related Sublineages Among Immunocompetent Adults – Increasing Community Access to Testing Program, United States, December 2022–January 2023, which came out in the Morbidity and Mortality Weekly Report very recently, which uses this test-negative case-control design to evaluate the ability of bivalent mRNA vaccines to prevent hospitalization.
The question is: Does receipt of a bivalent COVID vaccine booster prevent hospitalizations, ICU stay, or death? That may not be the question that is of interest to everyone. I know people are interested in symptoms, missed work, and transmission, but this paper was looking at hospitalization, ICU stay, and death.
What’s kind of tricky here is that the data they’re using are in people who are hospitalized with various diseases. You might look at that on the surface and say: “Well, you can’t – that’s impossible.” But you can, actually, with this cool test-negative case-control design.
Here’s basically how it works. You take a population of people who are hospitalized and confirmed to have COVID. Some of them will be vaccinated and some of them will be unvaccinated. And the proportion of vaccinated and unvaccinated people doesn’t tell you very much because it depends on how that compares with the rates in the general population, for instance. Let me clarify this. If 100% of the population were vaccinated, then 100% of the people hospitalized with COVID would be vaccinated. That doesn’t mean vaccines are bad. Put another way, if 90% of the population were vaccinated and 60% of people hospitalized with COVID were vaccinated, that would actually show that the vaccines were working to some extent, all else being equal. So it’s not just the raw percentages that tell you anything. Some people are vaccinated, some people aren’t. You need to understand what the baseline rate is.
The test-negative case-control design looks at people who are hospitalized without COVID. Now who those people are (who the controls are, in this case) is something you really need to think about. In the case of this CDC study, they used people who were hospitalized with COVID-like illnesses – flu-like illnesses, respiratory illnesses, pneumonia, influenza, etc. This is a pretty good idea because it standardizes a little bit for people who have access to healthcare. They can get to a hospital and they’re the type of person who would go to a hospital when they’re feeling sick. That’s a better control than the general population overall, which is something I like about this design.
Some of those people who don’t have COVID (they’re in the hospital for flu or whatever) will have been vaccinated for COVID, and some will not have been vaccinated for COVID. And of course, we don’t expect COVID vaccines necessarily to protect against the flu or pneumonia, but that gives us a way to standardize.
If you look at these Venn diagrams, I’ve got vaccinated/unvaccinated being exactly the same proportion, which would suggest that you’re just as likely to be hospitalized with COVID if you’re vaccinated as you are to be hospitalized with some other respiratory illness, which suggests that the vaccine isn’t particularly effective.
However, if you saw something like this, looking at all those patients with flu and other non-COVID illnesses, a lot more of them had been vaccinated for COVID. What that tells you is that we’re seeing fewer vaccinated people hospitalized with COVID than we would expect because we have this standardization from other respiratory infections. We expect this many vaccinated people because that’s how many vaccinated people there are who show up with flu. But in the COVID population, there are fewer, and that would suggest that the vaccines are effective. So that is the test-negative case-control design. You can do the same thing with ICU stays and death.
There are some assumptions here which you might already be thinking about. The most important one is that vaccination status is not associated with the risk for the disease. I always think of older people in this context. During the pandemic, at least in the United States, older people were much more likely to be vaccinated but were also much more likely to contract COVID and be hospitalized with COVID. The test-negative design actually accounts for this in some sense, because older people are also more likely to be hospitalized for things like flu and pneumonia. So there’s some control there.
But to the extent that older people are uniquely susceptible to COVID compared with other respiratory illnesses, that would bias your results to make the vaccines look worse. So the standard approach here is to adjust for these things. I think the CDC adjusted for age, sex, race, ethnicity, and a few other things to settle down and see how effective the vaccines were.
Let’s get to a worked example.
This is the actual data from the CDC paper. They had 6,907 individuals who were hospitalized with COVID, and 26% of them were unvaccinated. What’s the baseline rate that we would expect to be unvaccinated? A total of 59,234 individuals were hospitalized with a non-COVID respiratory illness, and 23% of them were unvaccinated. So you can see that there were more unvaccinated people than you would think in the COVID group. In other words, fewer vaccinated people, which suggests that the vaccine works to some degree because it’s keeping some people out of the hospital.
Now, 26% versus 23% is not a very impressive difference. But it gets more interesting when you break it down by the type of vaccine and how long ago the individual was vaccinated.
Let’s walk through the “all” group on this figure. What you can see is the calculated vaccine effectiveness. If you look at just the monovalent vaccine here, we see a 20% vaccine effectiveness. This means that you’re preventing 20% of hospitalizations basically due to COVID by people getting vaccinated. That’s okay but it’s certainly not anything to write home about. But we see much better vaccine effectiveness with the bivalent vaccine if it had been received within 60 days.
This compares people who received the bivalent vaccine within 60 days in the COVID group and the non-COVID group. The concern that the vaccine was given very recently affects both groups equally so it shouldn’t result in bias there. You see a step-off in vaccine effectiveness from 60 days, 60-120 days, and greater than 120 days. This is 4 months, and you’ve gone from 60% to 20%. When you break that down by age, you can see a similar pattern in the 18-to-65 group and potentially some more protection the greater than 65 age group.
Why is vaccine efficacy going down? The study doesn’t tell us, but we can hypothesize that this might be an immunologic effect – the antibodies or the protective T cells are waning over time. This could also reflect changes in the virus in the environment as the virus seeks to evade certain immune responses. But overall, this suggests that waiting a year between booster doses may leave you exposed for quite some time, although the take-home here is that bivalent vaccines in general are probably a good idea for the proportion of people who haven’t gotten them.
When we look at critical illness and death, the numbers look a little bit better.
You can see that bivalent is better than monovalent – certainly pretty good if you’ve received it within 60 days. It does tend to wane a little bit, but not nearly as much. You’ve still got about 50% vaccine efficacy beyond 120 days when we’re looking at critical illness, which is stays in the ICU and death.
The overriding thing to think about when we think about vaccine policy is that the way you get immunized against COVID is either by vaccine or by getting infected with COVID, or both.
This really interesting graph from the CDC (although it’s updated only through quarter three of 2022) shows the proportion of Americans, based on routine lab tests, who have varying degrees of protection against COVID. What you can see is that, by quarter three of 2022, just 3.6% of people who had blood drawn at a commercial laboratory had no evidence of infection or vaccination. In other words, almost no one was totally naive. Then 26% of people had never been infected – they only have vaccine antibodies – plus 22% of people had only been infected but had never been vaccinated. And then 50% of people had both. So there’s a tremendous amount of existing immunity out there.
The really interesting question about future vaccination and future booster doses is, how does it work on the background of this pattern? The CDC study doesn’t tell us, and I don’t think they have the data to tell us the vaccine efficacy in these different groups. Is it more effective in people who have only had an infection, for example? Is it more effective in people who have only had vaccination versus people who had both, or people who have no protection whatsoever? Those are the really interesting questions that need to be answered going forward as vaccine policy gets developed in the future.
I hope this was a helpful primer on how the test-negative case-control design can answer questions that seem a little bit unanswerable.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and director of Yale’s Clinical and Translational Research Accelerator. He disclosed no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study.
I am here today to talk about the effectiveness of COVID vaccine boosters in the midst of 2023. The reason I want to talk about this isn’t necessarily to dig into exactly how effective vaccines are. This is an area that’s been trod upon multiple times. But it does give me an opportunity to talk about a neat study design called the “test-negative case-control” design, which has some unique properties when you’re trying to evaluate the effect of something outside of the context of a randomized trial.
So, just a little bit of background to remind everyone where we are. These are the number of doses of COVID vaccines administered over time throughout the pandemic.
You can see that it’s stratified by age. The orange lines are adults ages 18-49, for example. You can see a big wave of vaccination when the vaccine first came out at the start of 2021. Then subsequently, you can see smaller waves after the first and second booster authorizations, and maybe a bit of a pickup, particularly among older adults, when the bivalent boosters were authorized. But still very little overall pickup of the bivalent booster, compared with the monovalent vaccines, which might suggest vaccine fatigue going on this far into the pandemic. But it’s important to try to understand exactly how effective those new boosters are, at least at this point in time.
I’m talking about Early Estimates of Bivalent mRNA Booster Dose Vaccine Effectiveness in Preventing Symptomatic SARS-CoV-2 Infection Attributable to Omicron BA.5– and XBB/XBB.1.5–Related Sublineages Among Immunocompetent Adults – Increasing Community Access to Testing Program, United States, December 2022–January 2023, which came out in the Morbidity and Mortality Weekly Report very recently, which uses this test-negative case-control design to evaluate the ability of bivalent mRNA vaccines to prevent hospitalization.
The question is: Does receipt of a bivalent COVID vaccine booster prevent hospitalizations, ICU stay, or death? That may not be the question that is of interest to everyone. I know people are interested in symptoms, missed work, and transmission, but this paper was looking at hospitalization, ICU stay, and death.
What’s kind of tricky here is that the data they’re using are in people who are hospitalized with various diseases. You might look at that on the surface and say: “Well, you can’t – that’s impossible.” But you can, actually, with this cool test-negative case-control design.
Here’s basically how it works. You take a population of people who are hospitalized and confirmed to have COVID. Some of them will be vaccinated and some of them will be unvaccinated. And the proportion of vaccinated and unvaccinated people doesn’t tell you very much because it depends on how that compares with the rates in the general population, for instance. Let me clarify this. If 100% of the population were vaccinated, then 100% of the people hospitalized with COVID would be vaccinated. That doesn’t mean vaccines are bad. Put another way, if 90% of the population were vaccinated and 60% of people hospitalized with COVID were vaccinated, that would actually show that the vaccines were working to some extent, all else being equal. So it’s not just the raw percentages that tell you anything. Some people are vaccinated, some people aren’t. You need to understand what the baseline rate is.
The test-negative case-control design looks at people who are hospitalized without COVID. Now who those people are (who the controls are, in this case) is something you really need to think about. In the case of this CDC study, they used people who were hospitalized with COVID-like illnesses – flu-like illnesses, respiratory illnesses, pneumonia, influenza, etc. This is a pretty good idea because it standardizes a little bit for people who have access to healthcare. They can get to a hospital and they’re the type of person who would go to a hospital when they’re feeling sick. That’s a better control than the general population overall, which is something I like about this design.
Some of those people who don’t have COVID (they’re in the hospital for flu or whatever) will have been vaccinated for COVID, and some will not have been vaccinated for COVID. And of course, we don’t expect COVID vaccines necessarily to protect against the flu or pneumonia, but that gives us a way to standardize.
If you look at these Venn diagrams, I’ve got vaccinated/unvaccinated being exactly the same proportion, which would suggest that you’re just as likely to be hospitalized with COVID if you’re vaccinated as you are to be hospitalized with some other respiratory illness, which suggests that the vaccine isn’t particularly effective.
However, if you saw something like this, looking at all those patients with flu and other non-COVID illnesses, a lot more of them had been vaccinated for COVID. What that tells you is that we’re seeing fewer vaccinated people hospitalized with COVID than we would expect because we have this standardization from other respiratory infections. We expect this many vaccinated people because that’s how many vaccinated people there are who show up with flu. But in the COVID population, there are fewer, and that would suggest that the vaccines are effective. So that is the test-negative case-control design. You can do the same thing with ICU stays and death.
There are some assumptions here which you might already be thinking about. The most important one is that vaccination status is not associated with the risk for the disease. I always think of older people in this context. During the pandemic, at least in the United States, older people were much more likely to be vaccinated but were also much more likely to contract COVID and be hospitalized with COVID. The test-negative design actually accounts for this in some sense, because older people are also more likely to be hospitalized for things like flu and pneumonia. So there’s some control there.
But to the extent that older people are uniquely susceptible to COVID compared with other respiratory illnesses, that would bias your results to make the vaccines look worse. So the standard approach here is to adjust for these things. I think the CDC adjusted for age, sex, race, ethnicity, and a few other things to settle down and see how effective the vaccines were.
Let’s get to a worked example.
This is the actual data from the CDC paper. They had 6,907 individuals who were hospitalized with COVID, and 26% of them were unvaccinated. What’s the baseline rate that we would expect to be unvaccinated? A total of 59,234 individuals were hospitalized with a non-COVID respiratory illness, and 23% of them were unvaccinated. So you can see that there were more unvaccinated people than you would think in the COVID group. In other words, fewer vaccinated people, which suggests that the vaccine works to some degree because it’s keeping some people out of the hospital.
Now, 26% versus 23% is not a very impressive difference. But it gets more interesting when you break it down by the type of vaccine and how long ago the individual was vaccinated.
Let’s walk through the “all” group on this figure. What you can see is the calculated vaccine effectiveness. If you look at just the monovalent vaccine here, we see a 20% vaccine effectiveness. This means that you’re preventing 20% of hospitalizations basically due to COVID by people getting vaccinated. That’s okay but it’s certainly not anything to write home about. But we see much better vaccine effectiveness with the bivalent vaccine if it had been received within 60 days.
This compares people who received the bivalent vaccine within 60 days in the COVID group and the non-COVID group. The concern that the vaccine was given very recently affects both groups equally so it shouldn’t result in bias there. You see a step-off in vaccine effectiveness from 60 days, 60-120 days, and greater than 120 days. This is 4 months, and you’ve gone from 60% to 20%. When you break that down by age, you can see a similar pattern in the 18-to-65 group and potentially some more protection the greater than 65 age group.
Why is vaccine efficacy going down? The study doesn’t tell us, but we can hypothesize that this might be an immunologic effect – the antibodies or the protective T cells are waning over time. This could also reflect changes in the virus in the environment as the virus seeks to evade certain immune responses. But overall, this suggests that waiting a year between booster doses may leave you exposed for quite some time, although the take-home here is that bivalent vaccines in general are probably a good idea for the proportion of people who haven’t gotten them.
When we look at critical illness and death, the numbers look a little bit better.
You can see that bivalent is better than monovalent – certainly pretty good if you’ve received it within 60 days. It does tend to wane a little bit, but not nearly as much. You’ve still got about 50% vaccine efficacy beyond 120 days when we’re looking at critical illness, which is stays in the ICU and death.
The overriding thing to think about when we think about vaccine policy is that the way you get immunized against COVID is either by vaccine or by getting infected with COVID, or both.
This really interesting graph from the CDC (although it’s updated only through quarter three of 2022) shows the proportion of Americans, based on routine lab tests, who have varying degrees of protection against COVID. What you can see is that, by quarter three of 2022, just 3.6% of people who had blood drawn at a commercial laboratory had no evidence of infection or vaccination. In other words, almost no one was totally naive. Then 26% of people had never been infected – they only have vaccine antibodies – plus 22% of people had only been infected but had never been vaccinated. And then 50% of people had both. So there’s a tremendous amount of existing immunity out there.
The really interesting question about future vaccination and future booster doses is, how does it work on the background of this pattern? The CDC study doesn’t tell us, and I don’t think they have the data to tell us the vaccine efficacy in these different groups. Is it more effective in people who have only had an infection, for example? Is it more effective in people who have only had vaccination versus people who had both, or people who have no protection whatsoever? Those are the really interesting questions that need to be answered going forward as vaccine policy gets developed in the future.
I hope this was a helpful primer on how the test-negative case-control design can answer questions that seem a little bit unanswerable.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and director of Yale’s Clinical and Translational Research Accelerator. He disclosed no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Emerging Treatment Options for Mantle Cell Lymphoma

Mantle cell lymphoma (MCL) is a rare, B-cell non-Hodgkin lymphoma whose biological heterogeneity has long challenged researchers and clinicians. There are no firmly-established therapies, and many individuals experience relapse even after successful treatment. There is a clear unmet need in MCL in the relapsed setting. In recent years, researchers have worked to address this need, demonstrating efficacy with covalent Bruton tyrosine kinase (BTK) inhibitors, led by ibrutinib, and anti-CD19 chimeric antigen receptor T-cell therapy. While these are helpful additions, relapse remains a challenge.
Fortunately, progress continues. Owing to encouraging results in recent trials, individuals with relapsed/refractory MCL are now experiencing clinical benefit from the noncovalent BTK inhibitor pirtrobrutinib. Investigational bispecific antibody (bsAb) therapy awaits in the wings.
Similarly, both younger and older patients with treatment naïve MCL could soon see improvement from the addition of BTK inhibitors to each age group’s standard treatment option. The following is a description of recent developments and their potential implications for practice.
One of the most exciting developments is the US Food and Drug Administration’s accelerated approval of pirtrobrutinib. A noncovalent BTK inhibitor, pirtrobrutinib has been found to have activity in individuals with MCL who have failed on multiple therapies, including standard BTK inhibitors. Pirtrobrutinib targets certain mutations in the BTK protein that are associated with resistance to covalent BTK inhibitors. In addition to resistance, some patients discontinue treatment with non-reversible BTK inhibitors because of intolerable toxicity.
Approval of pirtrobrutinib was based on an evaluation involving 120 individuals (median age 71) who were previously treated with a non-reversible BTK inhibitor. Two-thirds were previously treated with ibrutinib; 30% with acalabrutinib, and 8% zanubrutinib (some received more than one BTK inhibitor previously). The vast majority (83%) discontinued treatment due to refractory or progressive disease; 10% stopped due to toxicity; and the remainder halted treatment for other reasons.
Six in every 10 of the participants were classified on the MCL International Prognostic Index as intermediate; one-fourth were classified as high; and the remainder low. Patients received 200 mg of pirtrobrutinib once a day until disease either progressed or they experienced intolerable toxicity. Among the results:
Overall response rate was 50%; 13% responded completely
Median duration of response was 8.3 months
Duration of response rate at 6 months was 65%
Grade 3 or 4 abnormalities experienced by 10% or more of participants included decreased neutrophil counts, lymphocyte counts, and platelet counts
Further, bsAb therapy targeting CD20-CD3 is not yet approved but is showing promise as a potential therapy following BTK inhibitor failure. The treatment consists of an antibody containing two prongs. One is a CD20 protein that attaches to the lymphoma cell. The other is an anti-CD3 antibody that attaches to the T cell to bring the patient’s own T cells closer to the lymphoma to increase the cell kill.
Preliminary studies evaluating bsAbs in individuals with MCL, many of whom have failed on multiple other types of therapies, show a remarkably high response rate. In one such investigation, the bsAb glofitamab was given to 21 individuals as monotherapy following pretreatment with obinutuzumab. The regimen produced an overall response rate of 81% (n = 17) and a complete response rate of 68% (n = 14). Response was similar in participants who had and had not received prior BTK therapy. Among those who achieved a complete response, median duration was 2.4 months, and 12 of those who reached a complete response were still in remission at the study’s data cutoff point.
For younger individuals with treatment-naïve MCL, the current standard is chemotherapy and autologous stem-cell transplant (ASCT). For older individuals the standard is chemoimmunotherapy. The replacement or addition of the BTK inhibitor ibrutinib to these regimens is showing the promise of added clinical benefit in both age contingents.
Investigators presented results of the three-arm TRIANGLE trial at the 64th ASH Annual Meeting in December 2022. The study compared 1) chemotherapy followed by ASCT; 2) ibrutinib plus chemotherapy followed by ASCT and ibrutinib maintenance; and 3) ibrutinib plus chemotherapy followed by ibrutinib maintenance. Participants (n = 870) ≤ 65 years of age (median age 57) with previously untreated advanced-stage MCL were randomized to 1 of the 3 regimens. Investigators looked at overall response, complete response, and failure-free survival rates (FFS). Among the results:
Overall response rates were 98% in the 2 groups whose treatments included ibrutinib, versus 94% in the chemotherapy followed by ASCT group.
Complete response rates were 45% and 36%, respectively.
The non-ibrutinib regimen did not attain FFS superiority over ibrutinib plus chemotherapy, with a 3-year FFS rate of 72% and 86%, respectively (p=0.9979, hazard ratio [HR]: 1.77).
Ibrutinib plus chemotherapy was shown to be superior to chemotherapy/ASCT, with a 3-year FFS rate of 88% and 72%, respectively (p=0.0008, HR: 0.52).
The only adverse event differences of note occurred during maintenance treatment; there were significantly more grade 3-5 adverse events in the ibrutinib/chemotherapy/ASCT group, compared with the other 2 contingents.
Researchers noted in materials accompanying their presentation that, “It has been clearly demonstrated that the current standard high-dose regimen is not superior to the new ibrutinib-containing regimen without ASCT. More follow-up is needed to clarify the role of ASCT in the context of ibrutinib-containing treatment. However, the current results already support the use of ibrutinib in the first-line treatment of younger MCL patients.”
It also appears that ibrutinib added to standard chemoimmunotherapy can improve outcomes in older individuals with treatment-naïve MCL. In 2022, researchers published results from the international, randomized, double-blind, phase 3 SHINE trial. Participants (n = 523) were ≥ 65 years of age with previously untreated MCL and were randomized to receive either ibrutinib 560 mg daily or placebo added to chemoimmunotherapy consisting of bendamustine and rituximab every 4 weeks for 6 cycles. Individuals with a partial or complete response continued treatment every 8 weeks for up to 12 more doses. Investigators looked primarily at progression-free survival (PFS), as well as complete response, undetectable minimal residual disease, and time to worsening. Among the results:
116 participants (44%) in the ibrutinib group experienced disease progression or died, compared with 152 (58%) in the placebo contingent.
Median PFS was 80.6 months and 52.9 months, respectively.
PFS benefit was seen across most subgroups (patients categorized as high risk, and those with TP53 mutations did not benefit).
Complete response was seen in 66% and 58% of participants, respectively.
Undetectable minimal residual disease was observed in 62% and 57%, respectively.
Deaths attributed to disease progression or adverse events occurred in 22% and 28%, respectively.
Grade 3 or 4 adverse event incident rates were 82% and 77%, respectively.
Researchers noted that, “Given the shorter progression-free survival with current standard-care chemoimmunotherapy options, a prolongation of progression-free survival in response to primary therapy may provide patients with an improved opportunity for durable disease control in order to prevent or delay relapse.”
Data on the use of other BTK inhibitors as first-line treatment for MCL are forthcoming, including:
ECHO, a phase 3 trial assessing the efficacy of acalabrutinib versus placebo added to bendamustine and rituximab.
MANGROVE, a phase 3 study comparing zanubrutinib plus rituximab versus bendamustine plus rituximab.
ENRICH, a phase 2 study evaluating a chemotherapy-free option–ibrutinib and rituximab in older individuals.
OASIS, a randomized, phase 2 trial comparing ibrutinib/anti-CD20 antibodies (Ab) and Ibrutinib/anti-CD20 Ab/venetoclax given as fixed duration combinations.
The evolution of BTK inhibitors for relapsed MCL has great potential; further benefits continue to be explored.
Mantle cell lymphoma (MCL) is a rare, B-cell non-Hodgkin lymphoma whose biological heterogeneity has long challenged researchers and clinicians. There are no firmly-established therapies, and many individuals experience relapse even after successful treatment. There is a clear unmet need in MCL in the relapsed setting. In recent years, researchers have worked to address this need, demonstrating efficacy with covalent Bruton tyrosine kinase (BTK) inhibitors, led by ibrutinib, and anti-CD19 chimeric antigen receptor T-cell therapy. While these are helpful additions, relapse remains a challenge.
Fortunately, progress continues. Owing to encouraging results in recent trials, individuals with relapsed/refractory MCL are now experiencing clinical benefit from the noncovalent BTK inhibitor pirtrobrutinib. Investigational bispecific antibody (bsAb) therapy awaits in the wings.
Similarly, both younger and older patients with treatment naïve MCL could soon see improvement from the addition of BTK inhibitors to each age group’s standard treatment option. The following is a description of recent developments and their potential implications for practice.
One of the most exciting developments is the US Food and Drug Administration’s accelerated approval of pirtrobrutinib. A noncovalent BTK inhibitor, pirtrobrutinib has been found to have activity in individuals with MCL who have failed on multiple therapies, including standard BTK inhibitors. Pirtrobrutinib targets certain mutations in the BTK protein that are associated with resistance to covalent BTK inhibitors. In addition to resistance, some patients discontinue treatment with non-reversible BTK inhibitors because of intolerable toxicity.
Approval of pirtrobrutinib was based on an evaluation involving 120 individuals (median age 71) who were previously treated with a non-reversible BTK inhibitor. Two-thirds were previously treated with ibrutinib; 30% with acalabrutinib, and 8% zanubrutinib (some received more than one BTK inhibitor previously). The vast majority (83%) discontinued treatment due to refractory or progressive disease; 10% stopped due to toxicity; and the remainder halted treatment for other reasons.
Six in every 10 of the participants were classified on the MCL International Prognostic Index as intermediate; one-fourth were classified as high; and the remainder low. Patients received 200 mg of pirtrobrutinib once a day until disease either progressed or they experienced intolerable toxicity. Among the results:
Overall response rate was 50%; 13% responded completely
Median duration of response was 8.3 months
Duration of response rate at 6 months was 65%
Grade 3 or 4 abnormalities experienced by 10% or more of participants included decreased neutrophil counts, lymphocyte counts, and platelet counts
Further, bsAb therapy targeting CD20-CD3 is not yet approved but is showing promise as a potential therapy following BTK inhibitor failure. The treatment consists of an antibody containing two prongs. One is a CD20 protein that attaches to the lymphoma cell. The other is an anti-CD3 antibody that attaches to the T cell to bring the patient’s own T cells closer to the lymphoma to increase the cell kill.
Preliminary studies evaluating bsAbs in individuals with MCL, many of whom have failed on multiple other types of therapies, show a remarkably high response rate. In one such investigation, the bsAb glofitamab was given to 21 individuals as monotherapy following pretreatment with obinutuzumab. The regimen produced an overall response rate of 81% (n = 17) and a complete response rate of 68% (n = 14). Response was similar in participants who had and had not received prior BTK therapy. Among those who achieved a complete response, median duration was 2.4 months, and 12 of those who reached a complete response were still in remission at the study’s data cutoff point.
For younger individuals with treatment-naïve MCL, the current standard is chemotherapy and autologous stem-cell transplant (ASCT). For older individuals the standard is chemoimmunotherapy. The replacement or addition of the BTK inhibitor ibrutinib to these regimens is showing the promise of added clinical benefit in both age contingents.
Investigators presented results of the three-arm TRIANGLE trial at the 64th ASH Annual Meeting in December 2022. The study compared 1) chemotherapy followed by ASCT; 2) ibrutinib plus chemotherapy followed by ASCT and ibrutinib maintenance; and 3) ibrutinib plus chemotherapy followed by ibrutinib maintenance. Participants (n = 870) ≤ 65 years of age (median age 57) with previously untreated advanced-stage MCL were randomized to 1 of the 3 regimens. Investigators looked at overall response, complete response, and failure-free survival rates (FFS). Among the results:
Overall response rates were 98% in the 2 groups whose treatments included ibrutinib, versus 94% in the chemotherapy followed by ASCT group.
Complete response rates were 45% and 36%, respectively.
The non-ibrutinib regimen did not attain FFS superiority over ibrutinib plus chemotherapy, with a 3-year FFS rate of 72% and 86%, respectively (p=0.9979, hazard ratio [HR]: 1.77).
Ibrutinib plus chemotherapy was shown to be superior to chemotherapy/ASCT, with a 3-year FFS rate of 88% and 72%, respectively (p=0.0008, HR: 0.52).
The only adverse event differences of note occurred during maintenance treatment; there were significantly more grade 3-5 adverse events in the ibrutinib/chemotherapy/ASCT group, compared with the other 2 contingents.
Researchers noted in materials accompanying their presentation that, “It has been clearly demonstrated that the current standard high-dose regimen is not superior to the new ibrutinib-containing regimen without ASCT. More follow-up is needed to clarify the role of ASCT in the context of ibrutinib-containing treatment. However, the current results already support the use of ibrutinib in the first-line treatment of younger MCL patients.”
It also appears that ibrutinib added to standard chemoimmunotherapy can improve outcomes in older individuals with treatment-naïve MCL. In 2022, researchers published results from the international, randomized, double-blind, phase 3 SHINE trial. Participants (n = 523) were ≥ 65 years of age with previously untreated MCL and were randomized to receive either ibrutinib 560 mg daily or placebo added to chemoimmunotherapy consisting of bendamustine and rituximab every 4 weeks for 6 cycles. Individuals with a partial or complete response continued treatment every 8 weeks for up to 12 more doses. Investigators looked primarily at progression-free survival (PFS), as well as complete response, undetectable minimal residual disease, and time to worsening. Among the results:
116 participants (44%) in the ibrutinib group experienced disease progression or died, compared with 152 (58%) in the placebo contingent.
Median PFS was 80.6 months and 52.9 months, respectively.
PFS benefit was seen across most subgroups (patients categorized as high risk, and those with TP53 mutations did not benefit).
Complete response was seen in 66% and 58% of participants, respectively.
Undetectable minimal residual disease was observed in 62% and 57%, respectively.
Deaths attributed to disease progression or adverse events occurred in 22% and 28%, respectively.
Grade 3 or 4 adverse event incident rates were 82% and 77%, respectively.
Researchers noted that, “Given the shorter progression-free survival with current standard-care chemoimmunotherapy options, a prolongation of progression-free survival in response to primary therapy may provide patients with an improved opportunity for durable disease control in order to prevent or delay relapse.”
Data on the use of other BTK inhibitors as first-line treatment for MCL are forthcoming, including:
ECHO, a phase 3 trial assessing the efficacy of acalabrutinib versus placebo added to bendamustine and rituximab.
MANGROVE, a phase 3 study comparing zanubrutinib plus rituximab versus bendamustine plus rituximab.
ENRICH, a phase 2 study evaluating a chemotherapy-free option–ibrutinib and rituximab in older individuals.
OASIS, a randomized, phase 2 trial comparing ibrutinib/anti-CD20 antibodies (Ab) and Ibrutinib/anti-CD20 Ab/venetoclax given as fixed duration combinations.
The evolution of BTK inhibitors for relapsed MCL has great potential; further benefits continue to be explored.
Mantle cell lymphoma (MCL) is a rare, B-cell non-Hodgkin lymphoma whose biological heterogeneity has long challenged researchers and clinicians. There are no firmly-established therapies, and many individuals experience relapse even after successful treatment. There is a clear unmet need in MCL in the relapsed setting. In recent years, researchers have worked to address this need, demonstrating efficacy with covalent Bruton tyrosine kinase (BTK) inhibitors, led by ibrutinib, and anti-CD19 chimeric antigen receptor T-cell therapy. While these are helpful additions, relapse remains a challenge.
Fortunately, progress continues. Owing to encouraging results in recent trials, individuals with relapsed/refractory MCL are now experiencing clinical benefit from the noncovalent BTK inhibitor pirtrobrutinib. Investigational bispecific antibody (bsAb) therapy awaits in the wings.
Similarly, both younger and older patients with treatment naïve MCL could soon see improvement from the addition of BTK inhibitors to each age group’s standard treatment option. The following is a description of recent developments and their potential implications for practice.
One of the most exciting developments is the US Food and Drug Administration’s accelerated approval of pirtrobrutinib. A noncovalent BTK inhibitor, pirtrobrutinib has been found to have activity in individuals with MCL who have failed on multiple therapies, including standard BTK inhibitors. Pirtrobrutinib targets certain mutations in the BTK protein that are associated with resistance to covalent BTK inhibitors. In addition to resistance, some patients discontinue treatment with non-reversible BTK inhibitors because of intolerable toxicity.
Approval of pirtrobrutinib was based on an evaluation involving 120 individuals (median age 71) who were previously treated with a non-reversible BTK inhibitor. Two-thirds were previously treated with ibrutinib; 30% with acalabrutinib, and 8% zanubrutinib (some received more than one BTK inhibitor previously). The vast majority (83%) discontinued treatment due to refractory or progressive disease; 10% stopped due to toxicity; and the remainder halted treatment for other reasons.
Six in every 10 of the participants were classified on the MCL International Prognostic Index as intermediate; one-fourth were classified as high; and the remainder low. Patients received 200 mg of pirtrobrutinib once a day until disease either progressed or they experienced intolerable toxicity. Among the results:
Overall response rate was 50%; 13% responded completely
Median duration of response was 8.3 months
Duration of response rate at 6 months was 65%
Grade 3 or 4 abnormalities experienced by 10% or more of participants included decreased neutrophil counts, lymphocyte counts, and platelet counts
Further, bsAb therapy targeting CD20-CD3 is not yet approved but is showing promise as a potential therapy following BTK inhibitor failure. The treatment consists of an antibody containing two prongs. One is a CD20 protein that attaches to the lymphoma cell. The other is an anti-CD3 antibody that attaches to the T cell to bring the patient’s own T cells closer to the lymphoma to increase the cell kill.
Preliminary studies evaluating bsAbs in individuals with MCL, many of whom have failed on multiple other types of therapies, show a remarkably high response rate. In one such investigation, the bsAb glofitamab was given to 21 individuals as monotherapy following pretreatment with obinutuzumab. The regimen produced an overall response rate of 81% (n = 17) and a complete response rate of 68% (n = 14). Response was similar in participants who had and had not received prior BTK therapy. Among those who achieved a complete response, median duration was 2.4 months, and 12 of those who reached a complete response were still in remission at the study’s data cutoff point.
For younger individuals with treatment-naïve MCL, the current standard is chemotherapy and autologous stem-cell transplant (ASCT). For older individuals the standard is chemoimmunotherapy. The replacement or addition of the BTK inhibitor ibrutinib to these regimens is showing the promise of added clinical benefit in both age contingents.
Investigators presented results of the three-arm TRIANGLE trial at the 64th ASH Annual Meeting in December 2022. The study compared 1) chemotherapy followed by ASCT; 2) ibrutinib plus chemotherapy followed by ASCT and ibrutinib maintenance; and 3) ibrutinib plus chemotherapy followed by ibrutinib maintenance. Participants (n = 870) ≤ 65 years of age (median age 57) with previously untreated advanced-stage MCL were randomized to 1 of the 3 regimens. Investigators looked at overall response, complete response, and failure-free survival rates (FFS). Among the results:
Overall response rates were 98% in the 2 groups whose treatments included ibrutinib, versus 94% in the chemotherapy followed by ASCT group.
Complete response rates were 45% and 36%, respectively.
The non-ibrutinib regimen did not attain FFS superiority over ibrutinib plus chemotherapy, with a 3-year FFS rate of 72% and 86%, respectively (p=0.9979, hazard ratio [HR]: 1.77).
Ibrutinib plus chemotherapy was shown to be superior to chemotherapy/ASCT, with a 3-year FFS rate of 88% and 72%, respectively (p=0.0008, HR: 0.52).
The only adverse event differences of note occurred during maintenance treatment; there were significantly more grade 3-5 adverse events in the ibrutinib/chemotherapy/ASCT group, compared with the other 2 contingents.
Researchers noted in materials accompanying their presentation that, “It has been clearly demonstrated that the current standard high-dose regimen is not superior to the new ibrutinib-containing regimen without ASCT. More follow-up is needed to clarify the role of ASCT in the context of ibrutinib-containing treatment. However, the current results already support the use of ibrutinib in the first-line treatment of younger MCL patients.”
It also appears that ibrutinib added to standard chemoimmunotherapy can improve outcomes in older individuals with treatment-naïve MCL. In 2022, researchers published results from the international, randomized, double-blind, phase 3 SHINE trial. Participants (n = 523) were ≥ 65 years of age with previously untreated MCL and were randomized to receive either ibrutinib 560 mg daily or placebo added to chemoimmunotherapy consisting of bendamustine and rituximab every 4 weeks for 6 cycles. Individuals with a partial or complete response continued treatment every 8 weeks for up to 12 more doses. Investigators looked primarily at progression-free survival (PFS), as well as complete response, undetectable minimal residual disease, and time to worsening. Among the results:
116 participants (44%) in the ibrutinib group experienced disease progression or died, compared with 152 (58%) in the placebo contingent.
Median PFS was 80.6 months and 52.9 months, respectively.
PFS benefit was seen across most subgroups (patients categorized as high risk, and those with TP53 mutations did not benefit).
Complete response was seen in 66% and 58% of participants, respectively.
Undetectable minimal residual disease was observed in 62% and 57%, respectively.
Deaths attributed to disease progression or adverse events occurred in 22% and 28%, respectively.
Grade 3 or 4 adverse event incident rates were 82% and 77%, respectively.
Researchers noted that, “Given the shorter progression-free survival with current standard-care chemoimmunotherapy options, a prolongation of progression-free survival in response to primary therapy may provide patients with an improved opportunity for durable disease control in order to prevent or delay relapse.”
Data on the use of other BTK inhibitors as first-line treatment for MCL are forthcoming, including:
ECHO, a phase 3 trial assessing the efficacy of acalabrutinib versus placebo added to bendamustine and rituximab.
MANGROVE, a phase 3 study comparing zanubrutinib plus rituximab versus bendamustine plus rituximab.
ENRICH, a phase 2 study evaluating a chemotherapy-free option–ibrutinib and rituximab in older individuals.
OASIS, a randomized, phase 2 trial comparing ibrutinib/anti-CD20 antibodies (Ab) and Ibrutinib/anti-CD20 Ab/venetoclax given as fixed duration combinations.
The evolution of BTK inhibitors for relapsed MCL has great potential; further benefits continue to be explored.
Update on Migraine Prevention 2023

What is your experience with prescribing preventive medication for your patients with migraine?
Roughly 40% of patients living with migraine should be on preventive medication or other treatment, but probably fewer than 15% of patients with migraine are currently receiving therapy. There are several reasons for this: General physicians rarely put patients on preventive medication unless they are interested in or knowledgeable about headache, and the older preventive medicines that neurologists and headache specialists have used for many years have a lot of potential side effects and do not begin to work quickly.
It takes approximately 2 to 3 months for preventive medication to become effective, and many patients need to be slowly titrated up to an effective dose. By the time patients reach a steady state over a few weeks, if it is still not working well, they must slowly taper it and try something else. This is what often occurs with older preventive migraine medications—especially one of the most commonly used preventives, topiramate (Topamax). This drug was first indicated for epilepsy and then later for mood stabilization. Though it has good efficacy in reducing migraine attacks, it has many possible side effects, some of them troublesome. I often had multiple calls from patients in their first month of taking it complain of memory or word-finding issues and tingling in the extremities. More serious adverse events can be increased pressure in the eyes, such as glaucoma, and kidney stones. I often get referrals from other neurologists and headache specialists regarding patients who have failed multiple preventive medicines; 90% percent of these referrals need to be switched to the newer, more costly calcitonin gene-related peptide (CGRP)-blocking preventative medications, if insurance companies will cover them.
What categories of migraine preventive drugs do you generally prescribe your patients?
Of the older medications, most are epilepsy medicines, beta blockers, antidepressants, or cardiac medications such as angiotensin receptor blockers (candesartan). Of the newer medications, I use 1 of the 4 injectable monoclonal antibodies (mAbs), or 1 of the 2 gepants.
Older migraine preventive medication
Anticonvulsants (epilepsy medications)
Anticonvulsants are used for the treatment of several conditions, including epilepsy and pain control, but some can help reduce migraine attacks. These medicines, like all drugs, have the potential to cause side effects, especially topiramate; this medicine often causes paresthesia or tingling in the extremities as well as trouble with speech and memory, kidney stones, pancreatitis, and weight loss. The weight loss side effect of this drug has made it more appealing for some patients who had previously gained 10 to 15 pounds taking antidepressant medication to treat their migraine. I personally thought it was the most effective of all the preventive migraine medications if the patient could tolerate it.
Beta Blockers
Beta blockers cause the heart rate to decrease and also lower blood pressure. Most of my migraine patients are healthy females in their 20s and 30s and, when taking a beta blocker, can get short of breath when they exercise. These medications can also cause some depression and gastrointestinal issues and raise cholesterol levels.
Antidepressants
The type of antidepressants that I normally prescribe for migraine prevention are the tricyclic antidepressants. The one that has the best data in the literature and is often prescribed is amitriptyline (Elavil); I prefer a cousin to this medicine, nortriptyline. I prescribe tricyclics because many of my migraine patients have 2 other comorbid problems: depression and trouble staying asleep at night. Amitriptyline tends to cause drowsiness and can help patients sleep. It can also cause dry mouth, trouble urinating (especially in men), constipation, weight gain, and can slow patients down mentally, so it should not be prescribed to elderly patients. These antidepressants should be prescribed in very low doses and taken an hour before bedtime. The dose should be increased gradually over several weeks to help reduce adverse events. The best dose for migraine is often lower than the antidepressant dose, so sometimes a depressed patient needs 2 types of antidepressants. The typical dose for migraine prevention is about 50 to 75 mg. For depression, it is about 150 mg.
The patient would then need to increase their dose gradually for a month and remain on the target dose for at least another month. At the end of 2 months, they would have some idea whether it was working for them. If it was not, I might increase the dose even further. It is important to set expectations with patients at the beginning of treatment and tell them it is going to take 2 to 3 months to see if it works. If it does not work, I tell them, we will have to try another one, and that is going to take 2 or 3 months as well, until we can switch to the newer medications, which start to work in the first month, often in the first few days.
Why wouldn’t we just start with the newer preventives? Insurance companies require patients to fail, on average, 2 categories of the older medications before they will pay for the newer ones. Medicare usually only covers the older generic medications.
New migraine preventive medications
Monoclonal Antibodies
mAbs that block CGRP for the prevention of migraine, such as erenumab, fremanezumab, galcanezumab, and eptinezumab, target either the CGRP ligand itself or block the receptor to CGRP. This class of medication became available about 5 years ago. The first one approved was erenumab (Aimovig). It was tried by a lot of headache specialists, many neurologists, and then some general physicians once it came to market. It is the only one in its class that grabs the ligand CGRP and prevents it from docking on its receptor. Recently, 5-year safety data indicated it is extremely safe with only a few side effects, (it has been shown to cause some constipation and hypertension). It does, however, tend to lower the number of migraine days per month by about 40% to 50%. At the beginning of erenumab’s availability, researchers took patients that had 8 to 22 days of migraine per month and put them in double-blind, placebo-controlled, randomized trials. They found that some patients' migraine days went down gradually to 10 to 12 days from 20 migraine days per month. Erenumab works quickly, and most patients improve within 2 weeks.
Fremanezumab (AJOVY™) was the second mAb approved, followed pretty quickly by the third, galcanezumab (Emgality™). All 3 of these mAbs are administered once a month by a subcutaneous injection from an autoinjector. If a patient takes 3 fremanezumab injections in 1 day, they do not have to repeat that dose for 3 months. The upside of these 3 treatments is that the patient can self-administer the medication at home with few, if any, adverse events; the downside is they are expensive medications, costing about $600 per month.
Shortly thereafter, a fourth mAb, eptinezumab (VYEPTI™), was brought to market. Unlike the other 3 mAbs, it is administered as an intravenous infusion. The patient must come to an office or infusion center for a 30-minute intravenous infusion, which is not as convenient as treating themselves with an autoinjector at home. Eptinezumab is a strong medication that is often prescribed when other treatments are not effective. Each of the 4 mAbs has its own possible adverse events, but these are few and usually mild. The mAbs have a half-life of about 28 to 32 days; it takes 5 to 6 months after an injection for these mAbs to be metabolized by the reticuloendothelial system.
Gepants
The gepants are small molecule CGRP receptor blockers with much shorter half-lives than mAbs. They work by blocking the CGRP receptor so the CGRP ligand cannot dock there and cause vasodilation and increased pain transmission. Gepants have half-lives of 6 to 12 hours and can be used to treat a migraine acutely. Several drug companies studied the effects of taking a gepant every day or every other day, showing it can also be used as a migraine preventive medication. Ubrogepant (Ubrelvy®) was the first gepant to receive approval from the US Food and Drug Administration (FDA), but it was authorized only for acute care. Rimegepant (Nurtec®) was the second gepant approved, initially for acute treatment and later becoming the first gepant approved for migraine prevention. The same tablet can be used for acute care or for prevention. Preventive treatment consists of one 75 mg oral disintegrating tablet taken every second day. It works quite well as a preventive and has very few side effects. Nausea and abdominal discomfort occur in < 3% of patients. Some patients prefer to take a pill every other day over having an injection once per month or once every 3 months. It makes more sense for a woman of childbearing potential to take a drug with very short half-life vs one that lasts for 5 to 6 months in case she decides to become pregnant (or unexpectedly becomes pregnant).
A third gepant, atogepant (Qulipta™), was later approved, but only for prevention. It is available in 3 different strengths: 10 mg, 30 mg, and 60 mg. I tend to prescribe the 60-mg strength, and the dose is 1 pill every day.
If you compare rimegepant, which is taken once every other day, and atogepant, taken once daily, the latter tends to have slightly more side effects of nausea, drowsiness, and constipation, whereas rimegepant has been shown to have fewer side effects in double-blind, randomized studies. Like all gepants, it is quite effective and fast acting.
The goal of preventive medications is to decrease the frequency, severity, and duration of migraine attacks. Effective treatment can increase responsiveness to acute migraine therapy and improve the quality of life in patients suffering from migraine. Every patient is different and thus the side effects they experience vary. With time and patience, most patients find the relief from migraine they have been desperately seeking through the preventive medicines discussed above. This is a good time to have migraine, if you can get in to see a knowledgeable doctor and your insurance company cooperates. When I started my neurology practice 51 years ago, we had few preventives, and none approved by the FDA. Now we have several older, approved preventives—4 newer mAbs, and 2 newer gepants—as well as several devices, which we will discuss in the future.
What is your experience with prescribing preventive medication for your patients with migraine?
Roughly 40% of patients living with migraine should be on preventive medication or other treatment, but probably fewer than 15% of patients with migraine are currently receiving therapy. There are several reasons for this: General physicians rarely put patients on preventive medication unless they are interested in or knowledgeable about headache, and the older preventive medicines that neurologists and headache specialists have used for many years have a lot of potential side effects and do not begin to work quickly.
It takes approximately 2 to 3 months for preventive medication to become effective, and many patients need to be slowly titrated up to an effective dose. By the time patients reach a steady state over a few weeks, if it is still not working well, they must slowly taper it and try something else. This is what often occurs with older preventive migraine medications—especially one of the most commonly used preventives, topiramate (Topamax). This drug was first indicated for epilepsy and then later for mood stabilization. Though it has good efficacy in reducing migraine attacks, it has many possible side effects, some of them troublesome. I often had multiple calls from patients in their first month of taking it complain of memory or word-finding issues and tingling in the extremities. More serious adverse events can be increased pressure in the eyes, such as glaucoma, and kidney stones. I often get referrals from other neurologists and headache specialists regarding patients who have failed multiple preventive medicines; 90% percent of these referrals need to be switched to the newer, more costly calcitonin gene-related peptide (CGRP)-blocking preventative medications, if insurance companies will cover them.
What categories of migraine preventive drugs do you generally prescribe your patients?
Of the older medications, most are epilepsy medicines, beta blockers, antidepressants, or cardiac medications such as angiotensin receptor blockers (candesartan). Of the newer medications, I use 1 of the 4 injectable monoclonal antibodies (mAbs), or 1 of the 2 gepants.
Older migraine preventive medication
Anticonvulsants (epilepsy medications)
Anticonvulsants are used for the treatment of several conditions, including epilepsy and pain control, but some can help reduce migraine attacks. These medicines, like all drugs, have the potential to cause side effects, especially topiramate; this medicine often causes paresthesia or tingling in the extremities as well as trouble with speech and memory, kidney stones, pancreatitis, and weight loss. The weight loss side effect of this drug has made it more appealing for some patients who had previously gained 10 to 15 pounds taking antidepressant medication to treat their migraine. I personally thought it was the most effective of all the preventive migraine medications if the patient could tolerate it.
Beta Blockers
Beta blockers cause the heart rate to decrease and also lower blood pressure. Most of my migraine patients are healthy females in their 20s and 30s and, when taking a beta blocker, can get short of breath when they exercise. These medications can also cause some depression and gastrointestinal issues and raise cholesterol levels.
Antidepressants
The type of antidepressants that I normally prescribe for migraine prevention are the tricyclic antidepressants. The one that has the best data in the literature and is often prescribed is amitriptyline (Elavil); I prefer a cousin to this medicine, nortriptyline. I prescribe tricyclics because many of my migraine patients have 2 other comorbid problems: depression and trouble staying asleep at night. Amitriptyline tends to cause drowsiness and can help patients sleep. It can also cause dry mouth, trouble urinating (especially in men), constipation, weight gain, and can slow patients down mentally, so it should not be prescribed to elderly patients. These antidepressants should be prescribed in very low doses and taken an hour before bedtime. The dose should be increased gradually over several weeks to help reduce adverse events. The best dose for migraine is often lower than the antidepressant dose, so sometimes a depressed patient needs 2 types of antidepressants. The typical dose for migraine prevention is about 50 to 75 mg. For depression, it is about 150 mg.
The patient would then need to increase their dose gradually for a month and remain on the target dose for at least another month. At the end of 2 months, they would have some idea whether it was working for them. If it was not, I might increase the dose even further. It is important to set expectations with patients at the beginning of treatment and tell them it is going to take 2 to 3 months to see if it works. If it does not work, I tell them, we will have to try another one, and that is going to take 2 or 3 months as well, until we can switch to the newer medications, which start to work in the first month, often in the first few days.
Why wouldn’t we just start with the newer preventives? Insurance companies require patients to fail, on average, 2 categories of the older medications before they will pay for the newer ones. Medicare usually only covers the older generic medications.
New migraine preventive medications
Monoclonal Antibodies
mAbs that block CGRP for the prevention of migraine, such as erenumab, fremanezumab, galcanezumab, and eptinezumab, target either the CGRP ligand itself or block the receptor to CGRP. This class of medication became available about 5 years ago. The first one approved was erenumab (Aimovig). It was tried by a lot of headache specialists, many neurologists, and then some general physicians once it came to market. It is the only one in its class that grabs the ligand CGRP and prevents it from docking on its receptor. Recently, 5-year safety data indicated it is extremely safe with only a few side effects, (it has been shown to cause some constipation and hypertension). It does, however, tend to lower the number of migraine days per month by about 40% to 50%. At the beginning of erenumab’s availability, researchers took patients that had 8 to 22 days of migraine per month and put them in double-blind, placebo-controlled, randomized trials. They found that some patients' migraine days went down gradually to 10 to 12 days from 20 migraine days per month. Erenumab works quickly, and most patients improve within 2 weeks.
Fremanezumab (AJOVY™) was the second mAb approved, followed pretty quickly by the third, galcanezumab (Emgality™). All 3 of these mAbs are administered once a month by a subcutaneous injection from an autoinjector. If a patient takes 3 fremanezumab injections in 1 day, they do not have to repeat that dose for 3 months. The upside of these 3 treatments is that the patient can self-administer the medication at home with few, if any, adverse events; the downside is they are expensive medications, costing about $600 per month.
Shortly thereafter, a fourth mAb, eptinezumab (VYEPTI™), was brought to market. Unlike the other 3 mAbs, it is administered as an intravenous infusion. The patient must come to an office or infusion center for a 30-minute intravenous infusion, which is not as convenient as treating themselves with an autoinjector at home. Eptinezumab is a strong medication that is often prescribed when other treatments are not effective. Each of the 4 mAbs has its own possible adverse events, but these are few and usually mild. The mAbs have a half-life of about 28 to 32 days; it takes 5 to 6 months after an injection for these mAbs to be metabolized by the reticuloendothelial system.
Gepants
The gepants are small molecule CGRP receptor blockers with much shorter half-lives than mAbs. They work by blocking the CGRP receptor so the CGRP ligand cannot dock there and cause vasodilation and increased pain transmission. Gepants have half-lives of 6 to 12 hours and can be used to treat a migraine acutely. Several drug companies studied the effects of taking a gepant every day or every other day, showing it can also be used as a migraine preventive medication. Ubrogepant (Ubrelvy®) was the first gepant to receive approval from the US Food and Drug Administration (FDA), but it was authorized only for acute care. Rimegepant (Nurtec®) was the second gepant approved, initially for acute treatment and later becoming the first gepant approved for migraine prevention. The same tablet can be used for acute care or for prevention. Preventive treatment consists of one 75 mg oral disintegrating tablet taken every second day. It works quite well as a preventive and has very few side effects. Nausea and abdominal discomfort occur in < 3% of patients. Some patients prefer to take a pill every other day over having an injection once per month or once every 3 months. It makes more sense for a woman of childbearing potential to take a drug with very short half-life vs one that lasts for 5 to 6 months in case she decides to become pregnant (or unexpectedly becomes pregnant).
A third gepant, atogepant (Qulipta™), was later approved, but only for prevention. It is available in 3 different strengths: 10 mg, 30 mg, and 60 mg. I tend to prescribe the 60-mg strength, and the dose is 1 pill every day.
If you compare rimegepant, which is taken once every other day, and atogepant, taken once daily, the latter tends to have slightly more side effects of nausea, drowsiness, and constipation, whereas rimegepant has been shown to have fewer side effects in double-blind, randomized studies. Like all gepants, it is quite effective and fast acting.
The goal of preventive medications is to decrease the frequency, severity, and duration of migraine attacks. Effective treatment can increase responsiveness to acute migraine therapy and improve the quality of life in patients suffering from migraine. Every patient is different and thus the side effects they experience vary. With time and patience, most patients find the relief from migraine they have been desperately seeking through the preventive medicines discussed above. This is a good time to have migraine, if you can get in to see a knowledgeable doctor and your insurance company cooperates. When I started my neurology practice 51 years ago, we had few preventives, and none approved by the FDA. Now we have several older, approved preventives—4 newer mAbs, and 2 newer gepants—as well as several devices, which we will discuss in the future.
What is your experience with prescribing preventive medication for your patients with migraine?
Roughly 40% of patients living with migraine should be on preventive medication or other treatment, but probably fewer than 15% of patients with migraine are currently receiving therapy. There are several reasons for this: General physicians rarely put patients on preventive medication unless they are interested in or knowledgeable about headache, and the older preventive medicines that neurologists and headache specialists have used for many years have a lot of potential side effects and do not begin to work quickly.
It takes approximately 2 to 3 months for preventive medication to become effective, and many patients need to be slowly titrated up to an effective dose. By the time patients reach a steady state over a few weeks, if it is still not working well, they must slowly taper it and try something else. This is what often occurs with older preventive migraine medications—especially one of the most commonly used preventives, topiramate (Topamax). This drug was first indicated for epilepsy and then later for mood stabilization. Though it has good efficacy in reducing migraine attacks, it has many possible side effects, some of them troublesome. I often had multiple calls from patients in their first month of taking it complain of memory or word-finding issues and tingling in the extremities. More serious adverse events can be increased pressure in the eyes, such as glaucoma, and kidney stones. I often get referrals from other neurologists and headache specialists regarding patients who have failed multiple preventive medicines; 90% percent of these referrals need to be switched to the newer, more costly calcitonin gene-related peptide (CGRP)-blocking preventative medications, if insurance companies will cover them.
What categories of migraine preventive drugs do you generally prescribe your patients?
Of the older medications, most are epilepsy medicines, beta blockers, antidepressants, or cardiac medications such as angiotensin receptor blockers (candesartan). Of the newer medications, I use 1 of the 4 injectable monoclonal antibodies (mAbs), or 1 of the 2 gepants.
Older migraine preventive medication
Anticonvulsants (epilepsy medications)
Anticonvulsants are used for the treatment of several conditions, including epilepsy and pain control, but some can help reduce migraine attacks. These medicines, like all drugs, have the potential to cause side effects, especially topiramate; this medicine often causes paresthesia or tingling in the extremities as well as trouble with speech and memory, kidney stones, pancreatitis, and weight loss. The weight loss side effect of this drug has made it more appealing for some patients who had previously gained 10 to 15 pounds taking antidepressant medication to treat their migraine. I personally thought it was the most effective of all the preventive migraine medications if the patient could tolerate it.
Beta Blockers
Beta blockers cause the heart rate to decrease and also lower blood pressure. Most of my migraine patients are healthy females in their 20s and 30s and, when taking a beta blocker, can get short of breath when they exercise. These medications can also cause some depression and gastrointestinal issues and raise cholesterol levels.
Antidepressants
The type of antidepressants that I normally prescribe for migraine prevention are the tricyclic antidepressants. The one that has the best data in the literature and is often prescribed is amitriptyline (Elavil); I prefer a cousin to this medicine, nortriptyline. I prescribe tricyclics because many of my migraine patients have 2 other comorbid problems: depression and trouble staying asleep at night. Amitriptyline tends to cause drowsiness and can help patients sleep. It can also cause dry mouth, trouble urinating (especially in men), constipation, weight gain, and can slow patients down mentally, so it should not be prescribed to elderly patients. These antidepressants should be prescribed in very low doses and taken an hour before bedtime. The dose should be increased gradually over several weeks to help reduce adverse events. The best dose for migraine is often lower than the antidepressant dose, so sometimes a depressed patient needs 2 types of antidepressants. The typical dose for migraine prevention is about 50 to 75 mg. For depression, it is about 150 mg.
The patient would then need to increase their dose gradually for a month and remain on the target dose for at least another month. At the end of 2 months, they would have some idea whether it was working for them. If it was not, I might increase the dose even further. It is important to set expectations with patients at the beginning of treatment and tell them it is going to take 2 to 3 months to see if it works. If it does not work, I tell them, we will have to try another one, and that is going to take 2 or 3 months as well, until we can switch to the newer medications, which start to work in the first month, often in the first few days.
Why wouldn’t we just start with the newer preventives? Insurance companies require patients to fail, on average, 2 categories of the older medications before they will pay for the newer ones. Medicare usually only covers the older generic medications.
New migraine preventive medications
Monoclonal Antibodies
mAbs that block CGRP for the prevention of migraine, such as erenumab, fremanezumab, galcanezumab, and eptinezumab, target either the CGRP ligand itself or block the receptor to CGRP. This class of medication became available about 5 years ago. The first one approved was erenumab (Aimovig). It was tried by a lot of headache specialists, many neurologists, and then some general physicians once it came to market. It is the only one in its class that grabs the ligand CGRP and prevents it from docking on its receptor. Recently, 5-year safety data indicated it is extremely safe with only a few side effects, (it has been shown to cause some constipation and hypertension). It does, however, tend to lower the number of migraine days per month by about 40% to 50%. At the beginning of erenumab’s availability, researchers took patients that had 8 to 22 days of migraine per month and put them in double-blind, placebo-controlled, randomized trials. They found that some patients' migraine days went down gradually to 10 to 12 days from 20 migraine days per month. Erenumab works quickly, and most patients improve within 2 weeks.
Fremanezumab (AJOVY™) was the second mAb approved, followed pretty quickly by the third, galcanezumab (Emgality™). All 3 of these mAbs are administered once a month by a subcutaneous injection from an autoinjector. If a patient takes 3 fremanezumab injections in 1 day, they do not have to repeat that dose for 3 months. The upside of these 3 treatments is that the patient can self-administer the medication at home with few, if any, adverse events; the downside is they are expensive medications, costing about $600 per month.
Shortly thereafter, a fourth mAb, eptinezumab (VYEPTI™), was brought to market. Unlike the other 3 mAbs, it is administered as an intravenous infusion. The patient must come to an office or infusion center for a 30-minute intravenous infusion, which is not as convenient as treating themselves with an autoinjector at home. Eptinezumab is a strong medication that is often prescribed when other treatments are not effective. Each of the 4 mAbs has its own possible adverse events, but these are few and usually mild. The mAbs have a half-life of about 28 to 32 days; it takes 5 to 6 months after an injection for these mAbs to be metabolized by the reticuloendothelial system.
Gepants
The gepants are small molecule CGRP receptor blockers with much shorter half-lives than mAbs. They work by blocking the CGRP receptor so the CGRP ligand cannot dock there and cause vasodilation and increased pain transmission. Gepants have half-lives of 6 to 12 hours and can be used to treat a migraine acutely. Several drug companies studied the effects of taking a gepant every day or every other day, showing it can also be used as a migraine preventive medication. Ubrogepant (Ubrelvy®) was the first gepant to receive approval from the US Food and Drug Administration (FDA), but it was authorized only for acute care. Rimegepant (Nurtec®) was the second gepant approved, initially for acute treatment and later becoming the first gepant approved for migraine prevention. The same tablet can be used for acute care or for prevention. Preventive treatment consists of one 75 mg oral disintegrating tablet taken every second day. It works quite well as a preventive and has very few side effects. Nausea and abdominal discomfort occur in < 3% of patients. Some patients prefer to take a pill every other day over having an injection once per month or once every 3 months. It makes more sense for a woman of childbearing potential to take a drug with very short half-life vs one that lasts for 5 to 6 months in case she decides to become pregnant (or unexpectedly becomes pregnant).
A third gepant, atogepant (Qulipta™), was later approved, but only for prevention. It is available in 3 different strengths: 10 mg, 30 mg, and 60 mg. I tend to prescribe the 60-mg strength, and the dose is 1 pill every day.
If you compare rimegepant, which is taken once every other day, and atogepant, taken once daily, the latter tends to have slightly more side effects of nausea, drowsiness, and constipation, whereas rimegepant has been shown to have fewer side effects in double-blind, randomized studies. Like all gepants, it is quite effective and fast acting.
The goal of preventive medications is to decrease the frequency, severity, and duration of migraine attacks. Effective treatment can increase responsiveness to acute migraine therapy and improve the quality of life in patients suffering from migraine. Every patient is different and thus the side effects they experience vary. With time and patience, most patients find the relief from migraine they have been desperately seeking through the preventive medicines discussed above. This is a good time to have migraine, if you can get in to see a knowledgeable doctor and your insurance company cooperates. When I started my neurology practice 51 years ago, we had few preventives, and none approved by the FDA. Now we have several older, approved preventives—4 newer mAbs, and 2 newer gepants—as well as several devices, which we will discuss in the future.
The Current and Future Role of JAK Inhibitors for Psoriatic Arthritis

Introduction
The first Janus kinase (JAK) inhibitor received regulatory approval for the treatment of psoriatic arthritis (PsA) more than 5 years ago. Although there are limited comparative data between this and other JAK inhibitors approved or in development for the treatment of PsA, it is reasonable to anticipate variability in therapeutic effect and the risk of adverse events between different JAK inhibitors. So far, there have been considerable differences in the relative selectivity of each agent on the 4 JAK isoform enzymes, JAK1, JAK2, JAK3, and TYK2. This selectivity determines the downstream signal transducers and activators of transcription proteins (JAK-STAT [signal transducer and activator of transcription] pathway) that ultimately mediate both anti-inflammatory and off-target effects. In this review of JAK inhibitors in PsA, differences between JAK inhibitors will be explored for their potential impact on benefit-to-risk ratio while treating PsA.
Background
Data from the National Psoriasis Foundation (NPF) estimates that 8 million individuals in the United States have psoriasis.1 PsA, an inflammatory spondyloarthritis associated with psoriasis, develops in about 30% of these individuals, but precise epidemiology on this subset of psoriasis patients is complicated by missed and delayed diagnoses. Of patients with psoriasis, only about 15% of patients with PsA have joint inflammation at the time or in advance of skin lesions.2 This might explain delays in diagnosis. In one study, 15% of patients treated for psoriasis were found to have concomitant but unrecognized PsA.3
PsA was first classified as a distinct pathologic condition only about 50 years ago, even though skeletal remains indicate that this disease existed in early civilizations.2 Based on consensus that PsA deserved definition as a distinct entity, the Classification Criteria for Psoriatic Arthritis (CASPAR) were published in 2006.4 By these criteria, cumulative points are allotted for clinical signs of skin, nail, and joint involvement, as well as radiographic signs in patients judged to have inflammatory disease in the joints, spine, or entheses to classify them as having PsA.
There are numerous recommendations for the treatment of PsA, including those issued by the American College of Rheumatology (ACR),5 the European Alliance of Associations for Rheumatology (EULAR),6 and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA).7 Although generally compatible with the others, the GRAPPA recommendations, which are the most recent, have addressed the heterogeneity of PsA by recommending therapies for specific disease domains, such as the skin, nail, and joint manifestations.
For treatment of PsA, the available drug classes for moderate-to-severe disease include immunomodulators, such as methotrexate, biologics that inhibit cytokines, such as tumor necrosis factor (TNF) and the interleukin (IL) cytokines IL-17, 1L-23, and IL12/IL-23, phosphodiesterase-4 (PDE4) inhibitors, and JAK inhibitors. In the GRAPPA recommendations, JAK inhibitors are listed along with other targeted therapies as first-line choices for peripheral arthritis, axial disease, enthesitis, dactylitis, and plaque psoriasis.
JAK Inhibitors and PsA
There are multiple ways to classify JAK inhibitors. Tofacitinib, the first JAK inhibitor approved for PsA, is labeled a first-generation agent because it is relatively nonselective for the 4 JAK isoforms.8 Second-generation agents, such as upadacitinib, have been distinguished from tofacitinib, baricitinib, and other first-generation drugs by greater relative selectivity on the JAK1 enzyme. Other drugs in development for PsA target different JAK isoforms. Deucravacitinib, for example, which was approved for psoriasis after a favorable phase 3 trial9 and has shown promise for PsA in a phase 2 trial, is selective for the TYK2 isoform.10 A rapidly growing list of JAK inhibitors with different selectivity profiles, including dual JAK inhibitory effects, are being explored in a host of inflammatory diseases.
The relationship between selectivity on specific JAK isoforms, anti-inflammatory effects, and off-target effects is not fully understood.8 In addition, characteristics beyond JAK selectivity have potential pharmacologic importance. For example, JAK inhibitors can be classified as ATP competitive inhibitors and allosteric inhibitors, both of which are reversible binding modes.8 Within each of these subcategories, the site of kinase binding has the potential to influence clinical activity.8
JAK Inhibitors: Clinical Experience in PsA
Tofacitinib, a first-generation JAK inhibitor, initially licensed for use in the treatment of rheumatoid arthritis (RA), received regulatory approval for PsA on the basis of the OPAL Beyond trial.11 Approval of upadacitinib for PsA followed about 4 years later on the basis of the SELECT PsA-1 trial.12 The primary endpoint in both of these studies was proportion of patients with an ACR response, signifying degree of improvement from baseline, of ≥20%. For the JAK inhibitors, the ACR20 rates were about 50% and 70% in the tofacitinib and upadacitinib phase 3 trials, respectively. Other JAK inhibitors have been evaluated in PsA but none so far are approved in the United States.
Despite experimental evidence supporting the hypothesis that JAK1 selectivity is clinically relevant to the treatment of PsA and other spondyloarthritides,13 there is no level 1 evidence of an efficacy or safety advantage for second- relative to first-generation JAK inhibitors. A small number of indirect comparisons, such as one employing a network Bayesian analysis to compare these drugs for the treatment of RA,14 have supported a clinical advantage for JAK1 selectivity, but head-to-head comparisons are needed to confirm differences.
Prescribing information for both tofacitinib and upadacitinib in PsA and other indications include a black box warning for risk of serious adverse events, including major adverse cardiac events (MACE) and thromboembolism. The warning is based on the placebo-controlled ORAL trial with tofacitinib in RA.15 The study population was enhanced for risk with eligibility that required older age and the presence of cardiovascular risk factors. In this high-risk RA population, tofacitinib was associated with modest increases in serious adverse events, including MACE and thromboembolism, relative to placebo over several years of follow-up. A similar trial has not been conducted with upadacitinib or in patients with PsA.
In a phase 3 trial with the TYK2-selective deucravacitinib in psoriasis, there was no increase in the rate of MACE or thromboembolism.9 When granted regulatory approval for psoriasis, the product information did not include a black box warning, differentiating it from other currently available JAK inhibitors. It has not yet been proven whether the absence of serious adverse events in the phase 3 psoriasis and phase 2 PsA trials with deucravacitinib are related to TYK2 JAK enzyme selectivity.
Although TYK2 is closely associated with upregulation of IL-23 and other inflammatory cytokines implicated in the pathophysiology of PSA, the JAK-STAT signaling pathway is incompletely understood.8 Moreover, all of the JAK inhibitors synthesized so far have relative rather than absolute selectivity for any specific JAK isoform. This complicates the ability to attribute benefits and risks to the inhibition of any single JAK enzyme isoform and amplifies the need for comparative studies.
While other JAK inhibitors have reached late stages of development for the treatment of PsA, such as filgotinib (a JAK1 selective drug) and brepocitinib (which is selective for both JAK1 and TYK2),16,17 it is appropriate to emphasize that currently available JAK inhibitors are effective and acceptably safe for PsA. The goal of continued drug development is the potential to develop agents with even greater efficacy but with a lower risk of off-target effects. Currently, the black box warnings included in the labeling of tofacitinib and upadacitinib give pause, leading many clinicians to move to these agents after an inadequate response to biologics. Newer therapies in the JAK inhibitor class free of serious adverse effects might reverse the order, given the preference of many patients for oral agents.
The JAK inhibitor development program is rich not just for inflammatory diseases and autoimmune diseases, but for myeloproliferative diseases and neoplasms. JAK inhibitors are already identified in the GRAPPA recommendations as appropriate first-line options for most manifestations of PsA, including joint and skin involvement, but newer drugs with a more favorable JAK selectivity or other pharmacologic characteristics and decreased adverse risks might make these a more dominant treatment choice.
Summary
Relative selectivity for JAK isoforms promises therapies that are both more effective and safer for PsA as well as other inflammatory diseases. This promise is now being explored in experimental trials testing therapies with variable degrees of selectivity in the context of other characteristics, such as kinase binding, with the potential to influence clinical effects. However, the promise will not be fulfilled until large clinical trials, particularly comparative trials, can confirm the importance of JAK isoform selectivity. If specific types of selectivity prove relevant to the benefit-to-risk ratio of JAK inhibitors in PsA, it may alter the current order of treatment preferences for this disease.
Introduction
The first Janus kinase (JAK) inhibitor received regulatory approval for the treatment of psoriatic arthritis (PsA) more than 5 years ago. Although there are limited comparative data between this and other JAK inhibitors approved or in development for the treatment of PsA, it is reasonable to anticipate variability in therapeutic effect and the risk of adverse events between different JAK inhibitors. So far, there have been considerable differences in the relative selectivity of each agent on the 4 JAK isoform enzymes, JAK1, JAK2, JAK3, and TYK2. This selectivity determines the downstream signal transducers and activators of transcription proteins (JAK-STAT [signal transducer and activator of transcription] pathway) that ultimately mediate both anti-inflammatory and off-target effects. In this review of JAK inhibitors in PsA, differences between JAK inhibitors will be explored for their potential impact on benefit-to-risk ratio while treating PsA.
Background
Data from the National Psoriasis Foundation (NPF) estimates that 8 million individuals in the United States have psoriasis.1 PsA, an inflammatory spondyloarthritis associated with psoriasis, develops in about 30% of these individuals, but precise epidemiology on this subset of psoriasis patients is complicated by missed and delayed diagnoses. Of patients with psoriasis, only about 15% of patients with PsA have joint inflammation at the time or in advance of skin lesions.2 This might explain delays in diagnosis. In one study, 15% of patients treated for psoriasis were found to have concomitant but unrecognized PsA.3
PsA was first classified as a distinct pathologic condition only about 50 years ago, even though skeletal remains indicate that this disease existed in early civilizations.2 Based on consensus that PsA deserved definition as a distinct entity, the Classification Criteria for Psoriatic Arthritis (CASPAR) were published in 2006.4 By these criteria, cumulative points are allotted for clinical signs of skin, nail, and joint involvement, as well as radiographic signs in patients judged to have inflammatory disease in the joints, spine, or entheses to classify them as having PsA.
There are numerous recommendations for the treatment of PsA, including those issued by the American College of Rheumatology (ACR),5 the European Alliance of Associations for Rheumatology (EULAR),6 and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA).7 Although generally compatible with the others, the GRAPPA recommendations, which are the most recent, have addressed the heterogeneity of PsA by recommending therapies for specific disease domains, such as the skin, nail, and joint manifestations.
For treatment of PsA, the available drug classes for moderate-to-severe disease include immunomodulators, such as methotrexate, biologics that inhibit cytokines, such as tumor necrosis factor (TNF) and the interleukin (IL) cytokines IL-17, 1L-23, and IL12/IL-23, phosphodiesterase-4 (PDE4) inhibitors, and JAK inhibitors. In the GRAPPA recommendations, JAK inhibitors are listed along with other targeted therapies as first-line choices for peripheral arthritis, axial disease, enthesitis, dactylitis, and plaque psoriasis.
JAK Inhibitors and PsA
There are multiple ways to classify JAK inhibitors. Tofacitinib, the first JAK inhibitor approved for PsA, is labeled a first-generation agent because it is relatively nonselective for the 4 JAK isoforms.8 Second-generation agents, such as upadacitinib, have been distinguished from tofacitinib, baricitinib, and other first-generation drugs by greater relative selectivity on the JAK1 enzyme. Other drugs in development for PsA target different JAK isoforms. Deucravacitinib, for example, which was approved for psoriasis after a favorable phase 3 trial9 and has shown promise for PsA in a phase 2 trial, is selective for the TYK2 isoform.10 A rapidly growing list of JAK inhibitors with different selectivity profiles, including dual JAK inhibitory effects, are being explored in a host of inflammatory diseases.
The relationship between selectivity on specific JAK isoforms, anti-inflammatory effects, and off-target effects is not fully understood.8 In addition, characteristics beyond JAK selectivity have potential pharmacologic importance. For example, JAK inhibitors can be classified as ATP competitive inhibitors and allosteric inhibitors, both of which are reversible binding modes.8 Within each of these subcategories, the site of kinase binding has the potential to influence clinical activity.8
JAK Inhibitors: Clinical Experience in PsA
Tofacitinib, a first-generation JAK inhibitor, initially licensed for use in the treatment of rheumatoid arthritis (RA), received regulatory approval for PsA on the basis of the OPAL Beyond trial.11 Approval of upadacitinib for PsA followed about 4 years later on the basis of the SELECT PsA-1 trial.12 The primary endpoint in both of these studies was proportion of patients with an ACR response, signifying degree of improvement from baseline, of ≥20%. For the JAK inhibitors, the ACR20 rates were about 50% and 70% in the tofacitinib and upadacitinib phase 3 trials, respectively. Other JAK inhibitors have been evaluated in PsA but none so far are approved in the United States.
Despite experimental evidence supporting the hypothesis that JAK1 selectivity is clinically relevant to the treatment of PsA and other spondyloarthritides,13 there is no level 1 evidence of an efficacy or safety advantage for second- relative to first-generation JAK inhibitors. A small number of indirect comparisons, such as one employing a network Bayesian analysis to compare these drugs for the treatment of RA,14 have supported a clinical advantage for JAK1 selectivity, but head-to-head comparisons are needed to confirm differences.
Prescribing information for both tofacitinib and upadacitinib in PsA and other indications include a black box warning for risk of serious adverse events, including major adverse cardiac events (MACE) and thromboembolism. The warning is based on the placebo-controlled ORAL trial with tofacitinib in RA.15 The study population was enhanced for risk with eligibility that required older age and the presence of cardiovascular risk factors. In this high-risk RA population, tofacitinib was associated with modest increases in serious adverse events, including MACE and thromboembolism, relative to placebo over several years of follow-up. A similar trial has not been conducted with upadacitinib or in patients with PsA.
In a phase 3 trial with the TYK2-selective deucravacitinib in psoriasis, there was no increase in the rate of MACE or thromboembolism.9 When granted regulatory approval for psoriasis, the product information did not include a black box warning, differentiating it from other currently available JAK inhibitors. It has not yet been proven whether the absence of serious adverse events in the phase 3 psoriasis and phase 2 PsA trials with deucravacitinib are related to TYK2 JAK enzyme selectivity.
Although TYK2 is closely associated with upregulation of IL-23 and other inflammatory cytokines implicated in the pathophysiology of PSA, the JAK-STAT signaling pathway is incompletely understood.8 Moreover, all of the JAK inhibitors synthesized so far have relative rather than absolute selectivity for any specific JAK isoform. This complicates the ability to attribute benefits and risks to the inhibition of any single JAK enzyme isoform and amplifies the need for comparative studies.
While other JAK inhibitors have reached late stages of development for the treatment of PsA, such as filgotinib (a JAK1 selective drug) and brepocitinib (which is selective for both JAK1 and TYK2),16,17 it is appropriate to emphasize that currently available JAK inhibitors are effective and acceptably safe for PsA. The goal of continued drug development is the potential to develop agents with even greater efficacy but with a lower risk of off-target effects. Currently, the black box warnings included in the labeling of tofacitinib and upadacitinib give pause, leading many clinicians to move to these agents after an inadequate response to biologics. Newer therapies in the JAK inhibitor class free of serious adverse effects might reverse the order, given the preference of many patients for oral agents.
The JAK inhibitor development program is rich not just for inflammatory diseases and autoimmune diseases, but for myeloproliferative diseases and neoplasms. JAK inhibitors are already identified in the GRAPPA recommendations as appropriate first-line options for most manifestations of PsA, including joint and skin involvement, but newer drugs with a more favorable JAK selectivity or other pharmacologic characteristics and decreased adverse risks might make these a more dominant treatment choice.
Summary
Relative selectivity for JAK isoforms promises therapies that are both more effective and safer for PsA as well as other inflammatory diseases. This promise is now being explored in experimental trials testing therapies with variable degrees of selectivity in the context of other characteristics, such as kinase binding, with the potential to influence clinical effects. However, the promise will not be fulfilled until large clinical trials, particularly comparative trials, can confirm the importance of JAK isoform selectivity. If specific types of selectivity prove relevant to the benefit-to-risk ratio of JAK inhibitors in PsA, it may alter the current order of treatment preferences for this disease.
Introduction
The first Janus kinase (JAK) inhibitor received regulatory approval for the treatment of psoriatic arthritis (PsA) more than 5 years ago. Although there are limited comparative data between this and other JAK inhibitors approved or in development for the treatment of PsA, it is reasonable to anticipate variability in therapeutic effect and the risk of adverse events between different JAK inhibitors. So far, there have been considerable differences in the relative selectivity of each agent on the 4 JAK isoform enzymes, JAK1, JAK2, JAK3, and TYK2. This selectivity determines the downstream signal transducers and activators of transcription proteins (JAK-STAT [signal transducer and activator of transcription] pathway) that ultimately mediate both anti-inflammatory and off-target effects. In this review of JAK inhibitors in PsA, differences between JAK inhibitors will be explored for their potential impact on benefit-to-risk ratio while treating PsA.
Background
Data from the National Psoriasis Foundation (NPF) estimates that 8 million individuals in the United States have psoriasis.1 PsA, an inflammatory spondyloarthritis associated with psoriasis, develops in about 30% of these individuals, but precise epidemiology on this subset of psoriasis patients is complicated by missed and delayed diagnoses. Of patients with psoriasis, only about 15% of patients with PsA have joint inflammation at the time or in advance of skin lesions.2 This might explain delays in diagnosis. In one study, 15% of patients treated for psoriasis were found to have concomitant but unrecognized PsA.3
PsA was first classified as a distinct pathologic condition only about 50 years ago, even though skeletal remains indicate that this disease existed in early civilizations.2 Based on consensus that PsA deserved definition as a distinct entity, the Classification Criteria for Psoriatic Arthritis (CASPAR) were published in 2006.4 By these criteria, cumulative points are allotted for clinical signs of skin, nail, and joint involvement, as well as radiographic signs in patients judged to have inflammatory disease in the joints, spine, or entheses to classify them as having PsA.
There are numerous recommendations for the treatment of PsA, including those issued by the American College of Rheumatology (ACR),5 the European Alliance of Associations for Rheumatology (EULAR),6 and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA).7 Although generally compatible with the others, the GRAPPA recommendations, which are the most recent, have addressed the heterogeneity of PsA by recommending therapies for specific disease domains, such as the skin, nail, and joint manifestations.
For treatment of PsA, the available drug classes for moderate-to-severe disease include immunomodulators, such as methotrexate, biologics that inhibit cytokines, such as tumor necrosis factor (TNF) and the interleukin (IL) cytokines IL-17, 1L-23, and IL12/IL-23, phosphodiesterase-4 (PDE4) inhibitors, and JAK inhibitors. In the GRAPPA recommendations, JAK inhibitors are listed along with other targeted therapies as first-line choices for peripheral arthritis, axial disease, enthesitis, dactylitis, and plaque psoriasis.
JAK Inhibitors and PsA
There are multiple ways to classify JAK inhibitors. Tofacitinib, the first JAK inhibitor approved for PsA, is labeled a first-generation agent because it is relatively nonselective for the 4 JAK isoforms.8 Second-generation agents, such as upadacitinib, have been distinguished from tofacitinib, baricitinib, and other first-generation drugs by greater relative selectivity on the JAK1 enzyme. Other drugs in development for PsA target different JAK isoforms. Deucravacitinib, for example, which was approved for psoriasis after a favorable phase 3 trial9 and has shown promise for PsA in a phase 2 trial, is selective for the TYK2 isoform.10 A rapidly growing list of JAK inhibitors with different selectivity profiles, including dual JAK inhibitory effects, are being explored in a host of inflammatory diseases.
The relationship between selectivity on specific JAK isoforms, anti-inflammatory effects, and off-target effects is not fully understood.8 In addition, characteristics beyond JAK selectivity have potential pharmacologic importance. For example, JAK inhibitors can be classified as ATP competitive inhibitors and allosteric inhibitors, both of which are reversible binding modes.8 Within each of these subcategories, the site of kinase binding has the potential to influence clinical activity.8
JAK Inhibitors: Clinical Experience in PsA
Tofacitinib, a first-generation JAK inhibitor, initially licensed for use in the treatment of rheumatoid arthritis (RA), received regulatory approval for PsA on the basis of the OPAL Beyond trial.11 Approval of upadacitinib for PsA followed about 4 years later on the basis of the SELECT PsA-1 trial.12 The primary endpoint in both of these studies was proportion of patients with an ACR response, signifying degree of improvement from baseline, of ≥20%. For the JAK inhibitors, the ACR20 rates were about 50% and 70% in the tofacitinib and upadacitinib phase 3 trials, respectively. Other JAK inhibitors have been evaluated in PsA but none so far are approved in the United States.
Despite experimental evidence supporting the hypothesis that JAK1 selectivity is clinically relevant to the treatment of PsA and other spondyloarthritides,13 there is no level 1 evidence of an efficacy or safety advantage for second- relative to first-generation JAK inhibitors. A small number of indirect comparisons, such as one employing a network Bayesian analysis to compare these drugs for the treatment of RA,14 have supported a clinical advantage for JAK1 selectivity, but head-to-head comparisons are needed to confirm differences.
Prescribing information for both tofacitinib and upadacitinib in PsA and other indications include a black box warning for risk of serious adverse events, including major adverse cardiac events (MACE) and thromboembolism. The warning is based on the placebo-controlled ORAL trial with tofacitinib in RA.15 The study population was enhanced for risk with eligibility that required older age and the presence of cardiovascular risk factors. In this high-risk RA population, tofacitinib was associated with modest increases in serious adverse events, including MACE and thromboembolism, relative to placebo over several years of follow-up. A similar trial has not been conducted with upadacitinib or in patients with PsA.
In a phase 3 trial with the TYK2-selective deucravacitinib in psoriasis, there was no increase in the rate of MACE or thromboembolism.9 When granted regulatory approval for psoriasis, the product information did not include a black box warning, differentiating it from other currently available JAK inhibitors. It has not yet been proven whether the absence of serious adverse events in the phase 3 psoriasis and phase 2 PsA trials with deucravacitinib are related to TYK2 JAK enzyme selectivity.
Although TYK2 is closely associated with upregulation of IL-23 and other inflammatory cytokines implicated in the pathophysiology of PSA, the JAK-STAT signaling pathway is incompletely understood.8 Moreover, all of the JAK inhibitors synthesized so far have relative rather than absolute selectivity for any specific JAK isoform. This complicates the ability to attribute benefits and risks to the inhibition of any single JAK enzyme isoform and amplifies the need for comparative studies.
While other JAK inhibitors have reached late stages of development for the treatment of PsA, such as filgotinib (a JAK1 selective drug) and brepocitinib (which is selective for both JAK1 and TYK2),16,17 it is appropriate to emphasize that currently available JAK inhibitors are effective and acceptably safe for PsA. The goal of continued drug development is the potential to develop agents with even greater efficacy but with a lower risk of off-target effects. Currently, the black box warnings included in the labeling of tofacitinib and upadacitinib give pause, leading many clinicians to move to these agents after an inadequate response to biologics. Newer therapies in the JAK inhibitor class free of serious adverse effects might reverse the order, given the preference of many patients for oral agents.
The JAK inhibitor development program is rich not just for inflammatory diseases and autoimmune diseases, but for myeloproliferative diseases and neoplasms. JAK inhibitors are already identified in the GRAPPA recommendations as appropriate first-line options for most manifestations of PsA, including joint and skin involvement, but newer drugs with a more favorable JAK selectivity or other pharmacologic characteristics and decreased adverse risks might make these a more dominant treatment choice.
Summary
Relative selectivity for JAK isoforms promises therapies that are both more effective and safer for PsA as well as other inflammatory diseases. This promise is now being explored in experimental trials testing therapies with variable degrees of selectivity in the context of other characteristics, such as kinase binding, with the potential to influence clinical effects. However, the promise will not be fulfilled until large clinical trials, particularly comparative trials, can confirm the importance of JAK isoform selectivity. If specific types of selectivity prove relevant to the benefit-to-risk ratio of JAK inhibitors in PsA, it may alter the current order of treatment preferences for this disease.
Clinical trials: Top priority for long COVID
The Centers for Disease Control and Prevention and the U.S. Census Bureau estimate that 6.1% of the U.S. adult population is living with long COVID, with millions more debilitated worldwide. The demand for substantial treatment is enormous, but the urgency to fund and begin the necessary range of clinical trials has not met the severity of the problem.
While trials are slowly beginning to happen, the treatment choices and trial design require crucial nuances and understanding of viral-onset illnesses, and few research groups are creating strong trials that fully reflect the complexities of this landscape.
These recommendations recognize that roughly half of long COVID patients have new-onset myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and dysautonomia from COVID, which must be at the forefront of how trials are designed and conducted, and are additionally based on the current hypotheses about long COVID’s pathophysiologies.
1: Drugs proposed by experts in postviral fields should be prioritized
Upward of 50 drugs for viral-onset conditions like ME/CFS, dysautonomia, AIDS, and others have been waiting for years to go to trial, but have not had the funding to do so.
Treatments proposed by experts in viral-onset illnesses (such as ME/CFS and dysautonomia) should be prioritized (PM R. 2022 Oct;14[10]:1270-91), as outside researchers are not familiar with these fields and their potential treatment options.
2: Drugs targeting a wide range of mechanisms should be trialed
Treatments that should be trialed include anticoagulants/antiplatelets for clotting and vascular functioning, immunomodulators including JAK-STAT inhibitors, COVID-specific antivirals and antivirals against reactivated herpesviruses (Valcyte, Valacyclovir, EBV vaccine).
Other options include prescription mast cell stabilizers (ketotifen, cromolyn sodium), drugs that regulate microglial activation (low-dose naltrexone, low-dose aripiprazole), anti-CGRP medications, beta-blockers, and intravenous immunoglobulin.
Others include medications that target mitochondrial dysfunction; ivabradine; pyridostigmine;, DRP1 inhibitors; supplements showing success in patient communities including lactoferrin, ubiquinone, and nattokinase; and therapies targeting glymphatic/lymphatic dysfunction, microbiome therapies, and therapeutic peptides.
3: Use appropriate long COVID subtypes
Long COVID is an umbrella term that encompasses multiple new-onset and worsened conditions and symptoms after COVID. Roughly half of long COVID patients likely meet the criteria for ME/CFS and/or dysautonomia. Others may have new-onset diabetes, major clotting events, lung damage, neurological disorders, loss of smell or taste, and other manifestations.
Patients in different categories likely have different responses to treatments. It’s critical to identify appropriate subtypes for each trial, ideally performing detailed analyses to identify the treatments that work best, and don’t, for each subtype.
4: Behavioral treatments, especially those that have harmed similar populations, should not be trialed
Behavioral treatments including exercise, graded exercise therapy (GET), and cognitive-behavioral therapy (CBT) should not be trialed, let alone prioritized, for long COVID.
In patients with postexertional malaise (PEM), one of the most common long COVID symptoms, exercise is actively harmful and causes dysfunctional metabolic patterns, cardiac preload failure, impaired systemic oxygen extraction, and more. GET and CBT have failed similar populations , and exercise is explicitly contraindicated by the World Health Organization, the British National Institute for Health and Care Excellence, the CDC, and other organizations.
Resources should instead be put toward the wide range of medications that have not yet adequately undergone clinical trials.
5: PCR and antibody tests should not be used as inclusion criteria for trial participants
Only an estimated 1%-3% of cases in the first wave of COVID were documented, and the CDC estimates that only 25% of cases through September 2021 were documented. Similarly, antibody tests are unreliable to determine past infection, as roughly a third of patients don’t seroconvert, and a similar proportion serorevert within a few months. Using polymerase chain reaction (PCR) and antibody testing to determine who should be included in clinical trials limits who is eligible to participate in research, particularly those who have been ill for longer. Additionally, the majority of those who serorevert are women, so using antibody tests for inclusion introduces a selection bias and may miss mechanisms of immune system functioning that are part of long COVID.
PCR tests also have high false-negative rates and requiring them in research excludes people with lower viral loads with long COVID, which would confound findings.
These issues with testing also lead to COVID-infected people accidentally being included in control groups, which ruins the credibility of the research findings completely.
6: Include comparator groups
There are several common diagnoses that occur in people with long COVID, including ME/CFS, postural orthostatic tachycardia syndrome, small-fiber neuropathy, mast cell activation syndrome, and Ehlers-Danlos syndrome.
Identifying people with these conditions within the trial cohort improves research across all fields, benefiting all groups, and helps clarify what types of patients benefit most from certain medications.
7: Identify the right endpoints; avoid the wrong ones
Even though our understanding of the pathophysiology of long COVID is still evolving, it’s still possible to do clinical trials by identifying strong endpoints and outcome measures.
Several tools have been designed for viral-onset conditions and should be used alongside other endpoints. Postexertional malaise and autonomic symptoms, which are some of the most common symptoms of long COVID, can be measured with the validated DSQ-PEM and COMPASS-31, respectively. Tools for cognitive dysfunction trials should capture specific and common types of impairment, like processing speed.
Endpoints should be high-impact and aim for large improvements that have clinical significance over small improvements that do not have clinical significance.
Objective tests should be incorporated where possible; some to consider include natural killer cell functioning, cerebral blood flow, T-cell functioning, levels of reactivated herpesviruses, blood lactate levels, and microclots, as testing becomes available.
Mental health outcomes shouldn’t be primary endpoints, except where a trial is targeting a specific mental health condition because of COVID (for example, premenstrual dysphoric disorder).
If mental health conditions are tracked secondarily, it’s vital not to use questionnaires that include physical symptoms like fatigue, difficulty concentrating, difficulty sleeping, or palpitations, as these artificially increase depression and anxiety scores in chronically ill respondents. Tools that include physical symptoms (Patient Health Questionnaire–9, Beck Anxiety Inventory, Beck Depression Inventory) can be replaced with scales like the PHQ-2, General Anxiety Disorder–7, Hospital Anxiety and Depression Scale, or PROMIS-29 subscales.
Because certain cytokines and other inflammatory markers may naturally decrease over time without corresponding improvement in the ME/CFS subtype, caution should be taken when using cytokines as endpoints.
8: Consider enrollment and objectives carefully
A proportion of people with long COVID will recover in the early months after infection. Ideally, clinical trials will primarily study treatments in patients who have been ill 6 months or longer, as some natural recovery will happen before that can bias studies.
But where resources are abundant, it is ideal for trials to additionally look at whether the treatments can help patients in the early months recover and prevent progression to the later stage.
9: Tracking illness duration is crucial
Research from ME/CFS shows that there may be an immune change in the first few years of the illness, where cytokines decrease without any corresponding change in symptom improvement.
Because of this and the possibility that other markers follow the same pattern, disease duration should be a core feature of all analyses and trial designs. Trial outcomes should be designed to answer the question of whether the medication helps patients at different durations of illness.
10: Prioritize patient populations less likely to recover without intervention
Some long COVID phenotypes seem less likely to recover without intervention. Trials should take care to focus on these patient populations, which include those with neurologic symptoms and those meeting ME/CFS criteria.
11: Account for the relapsing/remitting nature
Outcome measures need to be assessed in a way that can distinguish a temporary remission, which is part of the natural course of the disease, from a permanent cure.
Factors that can contribute to the relapsing/remitting nature include physical and cognitive postexertional malaise, menstrual cycle changes, and seasonal changes.
12: Trial participants should reflect the diversity of the long COVID population
Certain demographics are more likely to be affected by acute and long COVID and need to be appropriately recruited and reflected in research, including in patient engagement.
Trials must include high numbers of Hispanic/Latinx, Black, and indigenous communities, queer and transgender populations, and women. Trial materials and design need to incorporate linguistic diversity in addition to racial/ethnic diversity.
Upward of 75% of long COVID cases happen after mild acute cases; clinical researchers should ensure that nonhospitalized patients make up the bulk of trial participants.
13: Utilize meaningful engagement of patients, especially in treatment selection and study design
Meaningful patient engagement means engaging multiple patients at every step of the trial process, from treatment selection to study design to analysis to communication of the results.
Patient experiences are extremely valuable and contain information that researchers may not be familiar with, including the nature and patterns of the illness, insights into possible treatments, and barriers to documentation and care that may also impact research. Tapping into those patient experiences will make trials stronger.
Overall, the landscape of long COVID clinical trials is ripe for discovery, and researchers choosing to go down this path will be deeply appreciated by the patient community.
Hannah Davis is a long COVID patient-researcher and cofounder of the Patient-Led Research Collaborative, an organization studying the long-term effects of COVID.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention and the U.S. Census Bureau estimate that 6.1% of the U.S. adult population is living with long COVID, with millions more debilitated worldwide. The demand for substantial treatment is enormous, but the urgency to fund and begin the necessary range of clinical trials has not met the severity of the problem.
While trials are slowly beginning to happen, the treatment choices and trial design require crucial nuances and understanding of viral-onset illnesses, and few research groups are creating strong trials that fully reflect the complexities of this landscape.
These recommendations recognize that roughly half of long COVID patients have new-onset myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and dysautonomia from COVID, which must be at the forefront of how trials are designed and conducted, and are additionally based on the current hypotheses about long COVID’s pathophysiologies.
1: Drugs proposed by experts in postviral fields should be prioritized
Upward of 50 drugs for viral-onset conditions like ME/CFS, dysautonomia, AIDS, and others have been waiting for years to go to trial, but have not had the funding to do so.
Treatments proposed by experts in viral-onset illnesses (such as ME/CFS and dysautonomia) should be prioritized (PM R. 2022 Oct;14[10]:1270-91), as outside researchers are not familiar with these fields and their potential treatment options.
2: Drugs targeting a wide range of mechanisms should be trialed
Treatments that should be trialed include anticoagulants/antiplatelets for clotting and vascular functioning, immunomodulators including JAK-STAT inhibitors, COVID-specific antivirals and antivirals against reactivated herpesviruses (Valcyte, Valacyclovir, EBV vaccine).
Other options include prescription mast cell stabilizers (ketotifen, cromolyn sodium), drugs that regulate microglial activation (low-dose naltrexone, low-dose aripiprazole), anti-CGRP medications, beta-blockers, and intravenous immunoglobulin.
Others include medications that target mitochondrial dysfunction; ivabradine; pyridostigmine;, DRP1 inhibitors; supplements showing success in patient communities including lactoferrin, ubiquinone, and nattokinase; and therapies targeting glymphatic/lymphatic dysfunction, microbiome therapies, and therapeutic peptides.
3: Use appropriate long COVID subtypes
Long COVID is an umbrella term that encompasses multiple new-onset and worsened conditions and symptoms after COVID. Roughly half of long COVID patients likely meet the criteria for ME/CFS and/or dysautonomia. Others may have new-onset diabetes, major clotting events, lung damage, neurological disorders, loss of smell or taste, and other manifestations.
Patients in different categories likely have different responses to treatments. It’s critical to identify appropriate subtypes for each trial, ideally performing detailed analyses to identify the treatments that work best, and don’t, for each subtype.
4: Behavioral treatments, especially those that have harmed similar populations, should not be trialed
Behavioral treatments including exercise, graded exercise therapy (GET), and cognitive-behavioral therapy (CBT) should not be trialed, let alone prioritized, for long COVID.
In patients with postexertional malaise (PEM), one of the most common long COVID symptoms, exercise is actively harmful and causes dysfunctional metabolic patterns, cardiac preload failure, impaired systemic oxygen extraction, and more. GET and CBT have failed similar populations , and exercise is explicitly contraindicated by the World Health Organization, the British National Institute for Health and Care Excellence, the CDC, and other organizations.
Resources should instead be put toward the wide range of medications that have not yet adequately undergone clinical trials.
5: PCR and antibody tests should not be used as inclusion criteria for trial participants
Only an estimated 1%-3% of cases in the first wave of COVID were documented, and the CDC estimates that only 25% of cases through September 2021 were documented. Similarly, antibody tests are unreliable to determine past infection, as roughly a third of patients don’t seroconvert, and a similar proportion serorevert within a few months. Using polymerase chain reaction (PCR) and antibody testing to determine who should be included in clinical trials limits who is eligible to participate in research, particularly those who have been ill for longer. Additionally, the majority of those who serorevert are women, so using antibody tests for inclusion introduces a selection bias and may miss mechanisms of immune system functioning that are part of long COVID.
PCR tests also have high false-negative rates and requiring them in research excludes people with lower viral loads with long COVID, which would confound findings.
These issues with testing also lead to COVID-infected people accidentally being included in control groups, which ruins the credibility of the research findings completely.
6: Include comparator groups
There are several common diagnoses that occur in people with long COVID, including ME/CFS, postural orthostatic tachycardia syndrome, small-fiber neuropathy, mast cell activation syndrome, and Ehlers-Danlos syndrome.
Identifying people with these conditions within the trial cohort improves research across all fields, benefiting all groups, and helps clarify what types of patients benefit most from certain medications.
7: Identify the right endpoints; avoid the wrong ones
Even though our understanding of the pathophysiology of long COVID is still evolving, it’s still possible to do clinical trials by identifying strong endpoints and outcome measures.
Several tools have been designed for viral-onset conditions and should be used alongside other endpoints. Postexertional malaise and autonomic symptoms, which are some of the most common symptoms of long COVID, can be measured with the validated DSQ-PEM and COMPASS-31, respectively. Tools for cognitive dysfunction trials should capture specific and common types of impairment, like processing speed.
Endpoints should be high-impact and aim for large improvements that have clinical significance over small improvements that do not have clinical significance.
Objective tests should be incorporated where possible; some to consider include natural killer cell functioning, cerebral blood flow, T-cell functioning, levels of reactivated herpesviruses, blood lactate levels, and microclots, as testing becomes available.
Mental health outcomes shouldn’t be primary endpoints, except where a trial is targeting a specific mental health condition because of COVID (for example, premenstrual dysphoric disorder).
If mental health conditions are tracked secondarily, it’s vital not to use questionnaires that include physical symptoms like fatigue, difficulty concentrating, difficulty sleeping, or palpitations, as these artificially increase depression and anxiety scores in chronically ill respondents. Tools that include physical symptoms (Patient Health Questionnaire–9, Beck Anxiety Inventory, Beck Depression Inventory) can be replaced with scales like the PHQ-2, General Anxiety Disorder–7, Hospital Anxiety and Depression Scale, or PROMIS-29 subscales.
Because certain cytokines and other inflammatory markers may naturally decrease over time without corresponding improvement in the ME/CFS subtype, caution should be taken when using cytokines as endpoints.
8: Consider enrollment and objectives carefully
A proportion of people with long COVID will recover in the early months after infection. Ideally, clinical trials will primarily study treatments in patients who have been ill 6 months or longer, as some natural recovery will happen before that can bias studies.
But where resources are abundant, it is ideal for trials to additionally look at whether the treatments can help patients in the early months recover and prevent progression to the later stage.
9: Tracking illness duration is crucial
Research from ME/CFS shows that there may be an immune change in the first few years of the illness, where cytokines decrease without any corresponding change in symptom improvement.
Because of this and the possibility that other markers follow the same pattern, disease duration should be a core feature of all analyses and trial designs. Trial outcomes should be designed to answer the question of whether the medication helps patients at different durations of illness.
10: Prioritize patient populations less likely to recover without intervention
Some long COVID phenotypes seem less likely to recover without intervention. Trials should take care to focus on these patient populations, which include those with neurologic symptoms and those meeting ME/CFS criteria.
11: Account for the relapsing/remitting nature
Outcome measures need to be assessed in a way that can distinguish a temporary remission, which is part of the natural course of the disease, from a permanent cure.
Factors that can contribute to the relapsing/remitting nature include physical and cognitive postexertional malaise, menstrual cycle changes, and seasonal changes.
12: Trial participants should reflect the diversity of the long COVID population
Certain demographics are more likely to be affected by acute and long COVID and need to be appropriately recruited and reflected in research, including in patient engagement.
Trials must include high numbers of Hispanic/Latinx, Black, and indigenous communities, queer and transgender populations, and women. Trial materials and design need to incorporate linguistic diversity in addition to racial/ethnic diversity.
Upward of 75% of long COVID cases happen after mild acute cases; clinical researchers should ensure that nonhospitalized patients make up the bulk of trial participants.
13: Utilize meaningful engagement of patients, especially in treatment selection and study design
Meaningful patient engagement means engaging multiple patients at every step of the trial process, from treatment selection to study design to analysis to communication of the results.
Patient experiences are extremely valuable and contain information that researchers may not be familiar with, including the nature and patterns of the illness, insights into possible treatments, and barriers to documentation and care that may also impact research. Tapping into those patient experiences will make trials stronger.
Overall, the landscape of long COVID clinical trials is ripe for discovery, and researchers choosing to go down this path will be deeply appreciated by the patient community.
Hannah Davis is a long COVID patient-researcher and cofounder of the Patient-Led Research Collaborative, an organization studying the long-term effects of COVID.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention and the U.S. Census Bureau estimate that 6.1% of the U.S. adult population is living with long COVID, with millions more debilitated worldwide. The demand for substantial treatment is enormous, but the urgency to fund and begin the necessary range of clinical trials has not met the severity of the problem.
While trials are slowly beginning to happen, the treatment choices and trial design require crucial nuances and understanding of viral-onset illnesses, and few research groups are creating strong trials that fully reflect the complexities of this landscape.
These recommendations recognize that roughly half of long COVID patients have new-onset myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and dysautonomia from COVID, which must be at the forefront of how trials are designed and conducted, and are additionally based on the current hypotheses about long COVID’s pathophysiologies.
1: Drugs proposed by experts in postviral fields should be prioritized
Upward of 50 drugs for viral-onset conditions like ME/CFS, dysautonomia, AIDS, and others have been waiting for years to go to trial, but have not had the funding to do so.
Treatments proposed by experts in viral-onset illnesses (such as ME/CFS and dysautonomia) should be prioritized (PM R. 2022 Oct;14[10]:1270-91), as outside researchers are not familiar with these fields and their potential treatment options.
2: Drugs targeting a wide range of mechanisms should be trialed
Treatments that should be trialed include anticoagulants/antiplatelets for clotting and vascular functioning, immunomodulators including JAK-STAT inhibitors, COVID-specific antivirals and antivirals against reactivated herpesviruses (Valcyte, Valacyclovir, EBV vaccine).
Other options include prescription mast cell stabilizers (ketotifen, cromolyn sodium), drugs that regulate microglial activation (low-dose naltrexone, low-dose aripiprazole), anti-CGRP medications, beta-blockers, and intravenous immunoglobulin.
Others include medications that target mitochondrial dysfunction; ivabradine; pyridostigmine;, DRP1 inhibitors; supplements showing success in patient communities including lactoferrin, ubiquinone, and nattokinase; and therapies targeting glymphatic/lymphatic dysfunction, microbiome therapies, and therapeutic peptides.
3: Use appropriate long COVID subtypes
Long COVID is an umbrella term that encompasses multiple new-onset and worsened conditions and symptoms after COVID. Roughly half of long COVID patients likely meet the criteria for ME/CFS and/or dysautonomia. Others may have new-onset diabetes, major clotting events, lung damage, neurological disorders, loss of smell or taste, and other manifestations.
Patients in different categories likely have different responses to treatments. It’s critical to identify appropriate subtypes for each trial, ideally performing detailed analyses to identify the treatments that work best, and don’t, for each subtype.
4: Behavioral treatments, especially those that have harmed similar populations, should not be trialed
Behavioral treatments including exercise, graded exercise therapy (GET), and cognitive-behavioral therapy (CBT) should not be trialed, let alone prioritized, for long COVID.
In patients with postexertional malaise (PEM), one of the most common long COVID symptoms, exercise is actively harmful and causes dysfunctional metabolic patterns, cardiac preload failure, impaired systemic oxygen extraction, and more. GET and CBT have failed similar populations , and exercise is explicitly contraindicated by the World Health Organization, the British National Institute for Health and Care Excellence, the CDC, and other organizations.
Resources should instead be put toward the wide range of medications that have not yet adequately undergone clinical trials.
5: PCR and antibody tests should not be used as inclusion criteria for trial participants
Only an estimated 1%-3% of cases in the first wave of COVID were documented, and the CDC estimates that only 25% of cases through September 2021 were documented. Similarly, antibody tests are unreliable to determine past infection, as roughly a third of patients don’t seroconvert, and a similar proportion serorevert within a few months. Using polymerase chain reaction (PCR) and antibody testing to determine who should be included in clinical trials limits who is eligible to participate in research, particularly those who have been ill for longer. Additionally, the majority of those who serorevert are women, so using antibody tests for inclusion introduces a selection bias and may miss mechanisms of immune system functioning that are part of long COVID.
PCR tests also have high false-negative rates and requiring them in research excludes people with lower viral loads with long COVID, which would confound findings.
These issues with testing also lead to COVID-infected people accidentally being included in control groups, which ruins the credibility of the research findings completely.
6: Include comparator groups
There are several common diagnoses that occur in people with long COVID, including ME/CFS, postural orthostatic tachycardia syndrome, small-fiber neuropathy, mast cell activation syndrome, and Ehlers-Danlos syndrome.
Identifying people with these conditions within the trial cohort improves research across all fields, benefiting all groups, and helps clarify what types of patients benefit most from certain medications.
7: Identify the right endpoints; avoid the wrong ones
Even though our understanding of the pathophysiology of long COVID is still evolving, it’s still possible to do clinical trials by identifying strong endpoints and outcome measures.
Several tools have been designed for viral-onset conditions and should be used alongside other endpoints. Postexertional malaise and autonomic symptoms, which are some of the most common symptoms of long COVID, can be measured with the validated DSQ-PEM and COMPASS-31, respectively. Tools for cognitive dysfunction trials should capture specific and common types of impairment, like processing speed.
Endpoints should be high-impact and aim for large improvements that have clinical significance over small improvements that do not have clinical significance.
Objective tests should be incorporated where possible; some to consider include natural killer cell functioning, cerebral blood flow, T-cell functioning, levels of reactivated herpesviruses, blood lactate levels, and microclots, as testing becomes available.
Mental health outcomes shouldn’t be primary endpoints, except where a trial is targeting a specific mental health condition because of COVID (for example, premenstrual dysphoric disorder).
If mental health conditions are tracked secondarily, it’s vital not to use questionnaires that include physical symptoms like fatigue, difficulty concentrating, difficulty sleeping, or palpitations, as these artificially increase depression and anxiety scores in chronically ill respondents. Tools that include physical symptoms (Patient Health Questionnaire–9, Beck Anxiety Inventory, Beck Depression Inventory) can be replaced with scales like the PHQ-2, General Anxiety Disorder–7, Hospital Anxiety and Depression Scale, or PROMIS-29 subscales.
Because certain cytokines and other inflammatory markers may naturally decrease over time without corresponding improvement in the ME/CFS subtype, caution should be taken when using cytokines as endpoints.
8: Consider enrollment and objectives carefully
A proportion of people with long COVID will recover in the early months after infection. Ideally, clinical trials will primarily study treatments in patients who have been ill 6 months or longer, as some natural recovery will happen before that can bias studies.
But where resources are abundant, it is ideal for trials to additionally look at whether the treatments can help patients in the early months recover and prevent progression to the later stage.
9: Tracking illness duration is crucial
Research from ME/CFS shows that there may be an immune change in the first few years of the illness, where cytokines decrease without any corresponding change in symptom improvement.
Because of this and the possibility that other markers follow the same pattern, disease duration should be a core feature of all analyses and trial designs. Trial outcomes should be designed to answer the question of whether the medication helps patients at different durations of illness.
10: Prioritize patient populations less likely to recover without intervention
Some long COVID phenotypes seem less likely to recover without intervention. Trials should take care to focus on these patient populations, which include those with neurologic symptoms and those meeting ME/CFS criteria.
11: Account for the relapsing/remitting nature
Outcome measures need to be assessed in a way that can distinguish a temporary remission, which is part of the natural course of the disease, from a permanent cure.
Factors that can contribute to the relapsing/remitting nature include physical and cognitive postexertional malaise, menstrual cycle changes, and seasonal changes.
12: Trial participants should reflect the diversity of the long COVID population
Certain demographics are more likely to be affected by acute and long COVID and need to be appropriately recruited and reflected in research, including in patient engagement.
Trials must include high numbers of Hispanic/Latinx, Black, and indigenous communities, queer and transgender populations, and women. Trial materials and design need to incorporate linguistic diversity in addition to racial/ethnic diversity.
Upward of 75% of long COVID cases happen after mild acute cases; clinical researchers should ensure that nonhospitalized patients make up the bulk of trial participants.
13: Utilize meaningful engagement of patients, especially in treatment selection and study design
Meaningful patient engagement means engaging multiple patients at every step of the trial process, from treatment selection to study design to analysis to communication of the results.
Patient experiences are extremely valuable and contain information that researchers may not be familiar with, including the nature and patterns of the illness, insights into possible treatments, and barriers to documentation and care that may also impact research. Tapping into those patient experiences will make trials stronger.
Overall, the landscape of long COVID clinical trials is ripe for discovery, and researchers choosing to go down this path will be deeply appreciated by the patient community.
Hannah Davis is a long COVID patient-researcher and cofounder of the Patient-Led Research Collaborative, an organization studying the long-term effects of COVID.
A version of this article first appeared on Medscape.com.
Antibody Drug Conjugates: a growing field of targeted therapy for breast cancer

The landscape of breast cancer care and how we're working on pushing the targeted treatment movement forward is rapidly changing, especially with antibody drug conjugates (ADCs).
I like to think of ADCs as targeted missiles. They're essentially composed of antibodies against specific antigens, or targets of interest, and then they're combined with a linker to a chemotherapy payload. It's a way to deliver the chemotherapy in a more targeted manner than traditional chemotherapy, which is an exciting opportunity to allow us to target those patients who otherwise prefer agents that were more difficult to tolerate before this technology was invented.
This field has grown exponentially in the last 5 to 10 years and has presented multiple new opportunities for research. What is most exciting is that we have new targets for these treatments—new antigens that we can target with novel ADCs.
The NeoSTAR trial evaluated the ADC sacituzumab govitecan (SG), which is used for patients who have earlier stage triple negative breast cancer before surgery. The idea of this is to hopefully spare patients from many of the more toxic effects of traditional chemotherapy, while still providing them precision in terms of the treatment that we're targeting in the body.
I was involved in the ASCENT trial, which is also notable for precise treatment. It demonstrated the superiority of SG in metastatic triple negative breast cancer, and more recently the US Food and Drug Administration label has been expanded to the metastatic hormone-positive space as well. As we're developing these ADCs and broadening their use, we're able to reach larger patient populations. It's really exciting because we know there's such an appetite among our patients to use these agents, given how effective they can be and, in some situations, less toxic than the standard chemotherapy they would have otherwise gotten.
The other big category of trials involves another ADC called trastuzumab deruxtecan (TD). Trastuzumab is conjugated against HER2, a breast cancer specific agent, and is combined with the linker, deruxtecan—a very potent chemotherapy payload. TD was initially used in patients who had HER2-positive breast cancer. In fact, trastuzumab, the first half of the drug, was used as an antibody in and of itself in a lot of earlier stage and metastatic cancer for years. We've known about that for a long time. But more recently, with the series of DESTINY trials, we have seen the major impact that TD in HER2 can have compared to other chemotherapy agents and against other ADCs as well.
What was so exciting about the trials presented in 2022 is that they created a new category of patients called HER2-low. Before, we had always considered patients as HER2-positive or HER2-negative. We now know it's not that binary. We had already known by the way we do the pathology that people can have levels of HER2 expression. HER2-low patients are people who would have been considered HER2-negative before this—but have some HER2 expression. They have what we consider low on a scale of 0 to 3+, typically. Therefore, they're 1+ or 2+, not 0, and not 3+, because that would be considered HER2-positive. It's a little more complicated because when it's triple 2-positive, they could also do a back-up test, if a patient is positive on that, they are considered as HER2-positive.
There is now a new category of HER2-low patients who were also shown to have a tremendous improvement benefit with TD; this new category of patients could be candidates for this treatment, although they would never have been used for HER2 targeting before. These agents can be so effective that it's even causing us to rethink our classifications of disease.
Some of what we're working on right now is specifically looking at how patients are resistant to these agents; that's one of our major focuses as Mass General. We know these treatments are highly effective, but unfortunately, they don't last forever. Very few of these cancer treatments do, because as we know, cancer has this remarkable ability to evolve resistance to agents that we use. The amazing thing about ADCs is they've extended (in some cases) overall survival for patients, which is fantastic. But as I said, we know that they are not lasting forever. Part of what we're involved in with that clinical and research setting is to look at patients who've had success with these agents and ultimately progressed, and then figure out what changed before and after treatment (down to the single cell or genetic level) in order to continue expanding use of these treatments, extending use of the treatments, and making them more effective for more patients.
The landscape of breast cancer care and how we're working on pushing the targeted treatment movement forward is rapidly changing, especially with antibody drug conjugates (ADCs).
I like to think of ADCs as targeted missiles. They're essentially composed of antibodies against specific antigens, or targets of interest, and then they're combined with a linker to a chemotherapy payload. It's a way to deliver the chemotherapy in a more targeted manner than traditional chemotherapy, which is an exciting opportunity to allow us to target those patients who otherwise prefer agents that were more difficult to tolerate before this technology was invented.
This field has grown exponentially in the last 5 to 10 years and has presented multiple new opportunities for research. What is most exciting is that we have new targets for these treatments—new antigens that we can target with novel ADCs.
The NeoSTAR trial evaluated the ADC sacituzumab govitecan (SG), which is used for patients who have earlier stage triple negative breast cancer before surgery. The idea of this is to hopefully spare patients from many of the more toxic effects of traditional chemotherapy, while still providing them precision in terms of the treatment that we're targeting in the body.
I was involved in the ASCENT trial, which is also notable for precise treatment. It demonstrated the superiority of SG in metastatic triple negative breast cancer, and more recently the US Food and Drug Administration label has been expanded to the metastatic hormone-positive space as well. As we're developing these ADCs and broadening their use, we're able to reach larger patient populations. It's really exciting because we know there's such an appetite among our patients to use these agents, given how effective they can be and, in some situations, less toxic than the standard chemotherapy they would have otherwise gotten.
The other big category of trials involves another ADC called trastuzumab deruxtecan (TD). Trastuzumab is conjugated against HER2, a breast cancer specific agent, and is combined with the linker, deruxtecan—a very potent chemotherapy payload. TD was initially used in patients who had HER2-positive breast cancer. In fact, trastuzumab, the first half of the drug, was used as an antibody in and of itself in a lot of earlier stage and metastatic cancer for years. We've known about that for a long time. But more recently, with the series of DESTINY trials, we have seen the major impact that TD in HER2 can have compared to other chemotherapy agents and against other ADCs as well.
What was so exciting about the trials presented in 2022 is that they created a new category of patients called HER2-low. Before, we had always considered patients as HER2-positive or HER2-negative. We now know it's not that binary. We had already known by the way we do the pathology that people can have levels of HER2 expression. HER2-low patients are people who would have been considered HER2-negative before this—but have some HER2 expression. They have what we consider low on a scale of 0 to 3+, typically. Therefore, they're 1+ or 2+, not 0, and not 3+, because that would be considered HER2-positive. It's a little more complicated because when it's triple 2-positive, they could also do a back-up test, if a patient is positive on that, they are considered as HER2-positive.
There is now a new category of HER2-low patients who were also shown to have a tremendous improvement benefit with TD; this new category of patients could be candidates for this treatment, although they would never have been used for HER2 targeting before. These agents can be so effective that it's even causing us to rethink our classifications of disease.
Some of what we're working on right now is specifically looking at how patients are resistant to these agents; that's one of our major focuses as Mass General. We know these treatments are highly effective, but unfortunately, they don't last forever. Very few of these cancer treatments do, because as we know, cancer has this remarkable ability to evolve resistance to agents that we use. The amazing thing about ADCs is they've extended (in some cases) overall survival for patients, which is fantastic. But as I said, we know that they are not lasting forever. Part of what we're involved in with that clinical and research setting is to look at patients who've had success with these agents and ultimately progressed, and then figure out what changed before and after treatment (down to the single cell or genetic level) in order to continue expanding use of these treatments, extending use of the treatments, and making them more effective for more patients.
The landscape of breast cancer care and how we're working on pushing the targeted treatment movement forward is rapidly changing, especially with antibody drug conjugates (ADCs).
I like to think of ADCs as targeted missiles. They're essentially composed of antibodies against specific antigens, or targets of interest, and then they're combined with a linker to a chemotherapy payload. It's a way to deliver the chemotherapy in a more targeted manner than traditional chemotherapy, which is an exciting opportunity to allow us to target those patients who otherwise prefer agents that were more difficult to tolerate before this technology was invented.
This field has grown exponentially in the last 5 to 10 years and has presented multiple new opportunities for research. What is most exciting is that we have new targets for these treatments—new antigens that we can target with novel ADCs.
The NeoSTAR trial evaluated the ADC sacituzumab govitecan (SG), which is used for patients who have earlier stage triple negative breast cancer before surgery. The idea of this is to hopefully spare patients from many of the more toxic effects of traditional chemotherapy, while still providing them precision in terms of the treatment that we're targeting in the body.
I was involved in the ASCENT trial, which is also notable for precise treatment. It demonstrated the superiority of SG in metastatic triple negative breast cancer, and more recently the US Food and Drug Administration label has been expanded to the metastatic hormone-positive space as well. As we're developing these ADCs and broadening their use, we're able to reach larger patient populations. It's really exciting because we know there's such an appetite among our patients to use these agents, given how effective they can be and, in some situations, less toxic than the standard chemotherapy they would have otherwise gotten.
The other big category of trials involves another ADC called trastuzumab deruxtecan (TD). Trastuzumab is conjugated against HER2, a breast cancer specific agent, and is combined with the linker, deruxtecan—a very potent chemotherapy payload. TD was initially used in patients who had HER2-positive breast cancer. In fact, trastuzumab, the first half of the drug, was used as an antibody in and of itself in a lot of earlier stage and metastatic cancer for years. We've known about that for a long time. But more recently, with the series of DESTINY trials, we have seen the major impact that TD in HER2 can have compared to other chemotherapy agents and against other ADCs as well.
What was so exciting about the trials presented in 2022 is that they created a new category of patients called HER2-low. Before, we had always considered patients as HER2-positive or HER2-negative. We now know it's not that binary. We had already known by the way we do the pathology that people can have levels of HER2 expression. HER2-low patients are people who would have been considered HER2-negative before this—but have some HER2 expression. They have what we consider low on a scale of 0 to 3+, typically. Therefore, they're 1+ or 2+, not 0, and not 3+, because that would be considered HER2-positive. It's a little more complicated because when it's triple 2-positive, they could also do a back-up test, if a patient is positive on that, they are considered as HER2-positive.
There is now a new category of HER2-low patients who were also shown to have a tremendous improvement benefit with TD; this new category of patients could be candidates for this treatment, although they would never have been used for HER2 targeting before. These agents can be so effective that it's even causing us to rethink our classifications of disease.
Some of what we're working on right now is specifically looking at how patients are resistant to these agents; that's one of our major focuses as Mass General. We know these treatments are highly effective, but unfortunately, they don't last forever. Very few of these cancer treatments do, because as we know, cancer has this remarkable ability to evolve resistance to agents that we use. The amazing thing about ADCs is they've extended (in some cases) overall survival for patients, which is fantastic. But as I said, we know that they are not lasting forever. Part of what we're involved in with that clinical and research setting is to look at patients who've had success with these agents and ultimately progressed, and then figure out what changed before and after treatment (down to the single cell or genetic level) in order to continue expanding use of these treatments, extending use of the treatments, and making them more effective for more patients.
Raising Awareness for Managing Disease-Modifying Therapies in Aging Persons With Multiple Sclerosis

Multiple sclerosis (MS) is a chronic, inflammatory demyelinating and neurodegenerative disease that affects the central nervous system. While there is no cure for MS, significant progress has been made during the last 2 decades, with over 25 medications developed, including disease modifying therapies (DMTs) that have shown benefit in reducing the number of acute events (relapses), curbing the development of new lesions seen on magnetic resonance imaging (MRI), and slowing disease progression/worsening. However, the benefit of available DMTs is seen primarily during the inflammatory stage of the disease (relapsing remitting) and is less clear in the later stages (secondary-progressive disease). Age was shown to be one of the most consistent contributing factors linked to disease worsening, and most studies suggest limited benefit of available DMTs in patients older than 50 years.
Meanwhile, the aging MS population is increasing worldwide, with most patients being between ages 55 and 65 years—a trend considered related to a general extended life expectancy, better diagnosis, early initiation of efficient DMTs, and improved general medical care. As persons with MS (pwMS) age, there is a clear change in the clinical presentation, with reduced risk for relapses and/or development of new MRI lesions but increased risk for disease worsening, with physical and cognitive decline. Systematic analysis–gathered data from clinical trials suggest an inadequate benefit of available DMTs in patients over 50 years, although the data have limitations, as most studies in relapsing MS did not enroll patients over the age of 55 years. In progressive MS trials, the median age of participants is 47 years; therefore, available data on aging populations are currently limited and cannot fully justify whether the medication is actually beneficial.
Another challenge in treating aging pwMS besides the limited benefit of DMTs is concern regarding safety and tolerability, especially as the most potent medications, which are now considered the most efficacious interventions for MS, are immunosuppressive agents. Aging populations with known weaker immune systems (immunosenescence) that are exposed to immunosuppressive interventions can be more susceptible to infections, have a decreased response to vaccinations, and face an increased risk of cancer.
The aging population is also known to have other health issues (comorbidities) and, therefore, may become more vulnerable to side effects from DMTs, making it necessary to consider a different management approach. Until more effective and safe therapeutic interventions become available for aging pwMS, discontinuation or de-escalation are the most frequently used approaches. Choosing between continuing, discontinuing, or de-escalating DMTs when treating aging MS patients is a complex process that requires careful consideration as well as active patient and patient family engagement in the final management decision.
The 2022 DISCO-MS trial was the first randomized discontinuation trial of MS drugs in older pwMS. The trial was designed to investigate the effect of discontinuing DMTs in patients aged 55 years and older who had not had recent relapses for at least 5 years and had no recent or new MRI lesions for at least 3 years. This multicenter study was conducted by the University of Colorado (supported/funded by a Patient-Centered Outcomes Research Institute grant) and included 259 participants with a median age of 63 years. Participants were randomly assigned to either continue or discontinue treatment and were followed for up to 22 months. The results of the DISCO-MS trial showed that 1 of 128 participants who stayed on medication had a relapse, and 3 of 131 people who discontinued medication had a relapse. There were no significant differences between the groups in progression of disability, cognition, quality of life, or adverse events. However, more participants who discontinued DMTs had new MRI lesions (16 vs 6), although there was no relationship to relapses or disability progression. Based on a noninferiority study design, the primary outcome (combined relapses and/or new MRI lesions) was not reached in this study. Other retrospective studies, such as a large study conducted in 2018, showed that most patients over age 60 years who discontinued DMTs remained off DMTs. These studies provide preliminary data that may guide clinicians who are considering discontinuing DMTs in their aging patients.
The second approach is de-escalation, which aims primarily to minimize the risk of side effects and complications while maintaining efficacy. Therefore, de-escalating MS medication in aging pwMS should always be done with great care. Some factors that should be considered when de-escalating treatment include the patient's age, their overall health, and the severity of their MS symptoms. Some approaches to de-escalating MS medication include gradually reducing the dosage of the medication over time or increasing the interval between the administration of infusible medications. This can help minimize the risk of side effects and complications, while still monitoring for maintained efficacy. These changes require shared decision-making between practitioners and patients after discussing the potential risks of MS relapse, new MRI lesions, or disease progression, along with the potential benefits of reducing medication-related side effects. Another approach is to switch to a different type of medication that is less immunosuppressive (ie, immunomodulatory) and that may be better suited to the patient's needs; these medications are less likely to cause side effects in older patients or may be better tolerated by patients with certain health conditions.
DMTs may cause side effects in patients of any age, but aging patients may be more susceptible to certain side effects due to changes in their physiology and increased vulnerability due to other health issues. Some side effects of DMTs in aging pwMS that should be considered include:
Cardiovascular issues: some DMTs may increase the risk of cardiovascular complications such as hypertension, hyperlipidemia, and heart failure, which may be more concerning in aging patients who may already have cardiovascular risk factors.
Infections: aging patients may be more vulnerable to more severe infections, which often require hospitalization. Such patients are also at higher risk for opportunistic infections, such as zoster infections, or progressive multifocal leukoencephalopathy due to changes in the weakening of their immune system function and higher prevalence of other health issues.
Skin reactions/change to skin pathology: sphingosine-1-phosphate receptor modulators are oral DMTs for MS that were associated with cases of basal cell carcinoma in clinical trials.
Ultimately, the decision to continue, discontinue, or de-escalate DMTs in aging pwMS should be based on the individual patient's needs and circumstances. It is important for clinicians to work closely with their patients to develop a personalized treatment plan that considers all the relevant benefits and risks. In the meantime, more research is needed on this topic to provide better outcomes for our growing population of aging patients who are living with MS.
Multiple sclerosis (MS) is a chronic, inflammatory demyelinating and neurodegenerative disease that affects the central nervous system. While there is no cure for MS, significant progress has been made during the last 2 decades, with over 25 medications developed, including disease modifying therapies (DMTs) that have shown benefit in reducing the number of acute events (relapses), curbing the development of new lesions seen on magnetic resonance imaging (MRI), and slowing disease progression/worsening. However, the benefit of available DMTs is seen primarily during the inflammatory stage of the disease (relapsing remitting) and is less clear in the later stages (secondary-progressive disease). Age was shown to be one of the most consistent contributing factors linked to disease worsening, and most studies suggest limited benefit of available DMTs in patients older than 50 years.
Meanwhile, the aging MS population is increasing worldwide, with most patients being between ages 55 and 65 years—a trend considered related to a general extended life expectancy, better diagnosis, early initiation of efficient DMTs, and improved general medical care. As persons with MS (pwMS) age, there is a clear change in the clinical presentation, with reduced risk for relapses and/or development of new MRI lesions but increased risk for disease worsening, with physical and cognitive decline. Systematic analysis–gathered data from clinical trials suggest an inadequate benefit of available DMTs in patients over 50 years, although the data have limitations, as most studies in relapsing MS did not enroll patients over the age of 55 years. In progressive MS trials, the median age of participants is 47 years; therefore, available data on aging populations are currently limited and cannot fully justify whether the medication is actually beneficial.
Another challenge in treating aging pwMS besides the limited benefit of DMTs is concern regarding safety and tolerability, especially as the most potent medications, which are now considered the most efficacious interventions for MS, are immunosuppressive agents. Aging populations with known weaker immune systems (immunosenescence) that are exposed to immunosuppressive interventions can be more susceptible to infections, have a decreased response to vaccinations, and face an increased risk of cancer.
The aging population is also known to have other health issues (comorbidities) and, therefore, may become more vulnerable to side effects from DMTs, making it necessary to consider a different management approach. Until more effective and safe therapeutic interventions become available for aging pwMS, discontinuation or de-escalation are the most frequently used approaches. Choosing between continuing, discontinuing, or de-escalating DMTs when treating aging MS patients is a complex process that requires careful consideration as well as active patient and patient family engagement in the final management decision.
The 2022 DISCO-MS trial was the first randomized discontinuation trial of MS drugs in older pwMS. The trial was designed to investigate the effect of discontinuing DMTs in patients aged 55 years and older who had not had recent relapses for at least 5 years and had no recent or new MRI lesions for at least 3 years. This multicenter study was conducted by the University of Colorado (supported/funded by a Patient-Centered Outcomes Research Institute grant) and included 259 participants with a median age of 63 years. Participants were randomly assigned to either continue or discontinue treatment and were followed for up to 22 months. The results of the DISCO-MS trial showed that 1 of 128 participants who stayed on medication had a relapse, and 3 of 131 people who discontinued medication had a relapse. There were no significant differences between the groups in progression of disability, cognition, quality of life, or adverse events. However, more participants who discontinued DMTs had new MRI lesions (16 vs 6), although there was no relationship to relapses or disability progression. Based on a noninferiority study design, the primary outcome (combined relapses and/or new MRI lesions) was not reached in this study. Other retrospective studies, such as a large study conducted in 2018, showed that most patients over age 60 years who discontinued DMTs remained off DMTs. These studies provide preliminary data that may guide clinicians who are considering discontinuing DMTs in their aging patients.
The second approach is de-escalation, which aims primarily to minimize the risk of side effects and complications while maintaining efficacy. Therefore, de-escalating MS medication in aging pwMS should always be done with great care. Some factors that should be considered when de-escalating treatment include the patient's age, their overall health, and the severity of their MS symptoms. Some approaches to de-escalating MS medication include gradually reducing the dosage of the medication over time or increasing the interval between the administration of infusible medications. This can help minimize the risk of side effects and complications, while still monitoring for maintained efficacy. These changes require shared decision-making between practitioners and patients after discussing the potential risks of MS relapse, new MRI lesions, or disease progression, along with the potential benefits of reducing medication-related side effects. Another approach is to switch to a different type of medication that is less immunosuppressive (ie, immunomodulatory) and that may be better suited to the patient's needs; these medications are less likely to cause side effects in older patients or may be better tolerated by patients with certain health conditions.
DMTs may cause side effects in patients of any age, but aging patients may be more susceptible to certain side effects due to changes in their physiology and increased vulnerability due to other health issues. Some side effects of DMTs in aging pwMS that should be considered include:
Cardiovascular issues: some DMTs may increase the risk of cardiovascular complications such as hypertension, hyperlipidemia, and heart failure, which may be more concerning in aging patients who may already have cardiovascular risk factors.
Infections: aging patients may be more vulnerable to more severe infections, which often require hospitalization. Such patients are also at higher risk for opportunistic infections, such as zoster infections, or progressive multifocal leukoencephalopathy due to changes in the weakening of their immune system function and higher prevalence of other health issues.
Skin reactions/change to skin pathology: sphingosine-1-phosphate receptor modulators are oral DMTs for MS that were associated with cases of basal cell carcinoma in clinical trials.
Ultimately, the decision to continue, discontinue, or de-escalate DMTs in aging pwMS should be based on the individual patient's needs and circumstances. It is important for clinicians to work closely with their patients to develop a personalized treatment plan that considers all the relevant benefits and risks. In the meantime, more research is needed on this topic to provide better outcomes for our growing population of aging patients who are living with MS.
Multiple sclerosis (MS) is a chronic, inflammatory demyelinating and neurodegenerative disease that affects the central nervous system. While there is no cure for MS, significant progress has been made during the last 2 decades, with over 25 medications developed, including disease modifying therapies (DMTs) that have shown benefit in reducing the number of acute events (relapses), curbing the development of new lesions seen on magnetic resonance imaging (MRI), and slowing disease progression/worsening. However, the benefit of available DMTs is seen primarily during the inflammatory stage of the disease (relapsing remitting) and is less clear in the later stages (secondary-progressive disease). Age was shown to be one of the most consistent contributing factors linked to disease worsening, and most studies suggest limited benefit of available DMTs in patients older than 50 years.
Meanwhile, the aging MS population is increasing worldwide, with most patients being between ages 55 and 65 years—a trend considered related to a general extended life expectancy, better diagnosis, early initiation of efficient DMTs, and improved general medical care. As persons with MS (pwMS) age, there is a clear change in the clinical presentation, with reduced risk for relapses and/or development of new MRI lesions but increased risk for disease worsening, with physical and cognitive decline. Systematic analysis–gathered data from clinical trials suggest an inadequate benefit of available DMTs in patients over 50 years, although the data have limitations, as most studies in relapsing MS did not enroll patients over the age of 55 years. In progressive MS trials, the median age of participants is 47 years; therefore, available data on aging populations are currently limited and cannot fully justify whether the medication is actually beneficial.
Another challenge in treating aging pwMS besides the limited benefit of DMTs is concern regarding safety and tolerability, especially as the most potent medications, which are now considered the most efficacious interventions for MS, are immunosuppressive agents. Aging populations with known weaker immune systems (immunosenescence) that are exposed to immunosuppressive interventions can be more susceptible to infections, have a decreased response to vaccinations, and face an increased risk of cancer.
The aging population is also known to have other health issues (comorbidities) and, therefore, may become more vulnerable to side effects from DMTs, making it necessary to consider a different management approach. Until more effective and safe therapeutic interventions become available for aging pwMS, discontinuation or de-escalation are the most frequently used approaches. Choosing between continuing, discontinuing, or de-escalating DMTs when treating aging MS patients is a complex process that requires careful consideration as well as active patient and patient family engagement in the final management decision.
The 2022 DISCO-MS trial was the first randomized discontinuation trial of MS drugs in older pwMS. The trial was designed to investigate the effect of discontinuing DMTs in patients aged 55 years and older who had not had recent relapses for at least 5 years and had no recent or new MRI lesions for at least 3 years. This multicenter study was conducted by the University of Colorado (supported/funded by a Patient-Centered Outcomes Research Institute grant) and included 259 participants with a median age of 63 years. Participants were randomly assigned to either continue or discontinue treatment and were followed for up to 22 months. The results of the DISCO-MS trial showed that 1 of 128 participants who stayed on medication had a relapse, and 3 of 131 people who discontinued medication had a relapse. There were no significant differences between the groups in progression of disability, cognition, quality of life, or adverse events. However, more participants who discontinued DMTs had new MRI lesions (16 vs 6), although there was no relationship to relapses or disability progression. Based on a noninferiority study design, the primary outcome (combined relapses and/or new MRI lesions) was not reached in this study. Other retrospective studies, such as a large study conducted in 2018, showed that most patients over age 60 years who discontinued DMTs remained off DMTs. These studies provide preliminary data that may guide clinicians who are considering discontinuing DMTs in their aging patients.
The second approach is de-escalation, which aims primarily to minimize the risk of side effects and complications while maintaining efficacy. Therefore, de-escalating MS medication in aging pwMS should always be done with great care. Some factors that should be considered when de-escalating treatment include the patient's age, their overall health, and the severity of their MS symptoms. Some approaches to de-escalating MS medication include gradually reducing the dosage of the medication over time or increasing the interval between the administration of infusible medications. This can help minimize the risk of side effects and complications, while still monitoring for maintained efficacy. These changes require shared decision-making between practitioners and patients after discussing the potential risks of MS relapse, new MRI lesions, or disease progression, along with the potential benefits of reducing medication-related side effects. Another approach is to switch to a different type of medication that is less immunosuppressive (ie, immunomodulatory) and that may be better suited to the patient's needs; these medications are less likely to cause side effects in older patients or may be better tolerated by patients with certain health conditions.
DMTs may cause side effects in patients of any age, but aging patients may be more susceptible to certain side effects due to changes in their physiology and increased vulnerability due to other health issues. Some side effects of DMTs in aging pwMS that should be considered include:
Cardiovascular issues: some DMTs may increase the risk of cardiovascular complications such as hypertension, hyperlipidemia, and heart failure, which may be more concerning in aging patients who may already have cardiovascular risk factors.
Infections: aging patients may be more vulnerable to more severe infections, which often require hospitalization. Such patients are also at higher risk for opportunistic infections, such as zoster infections, or progressive multifocal leukoencephalopathy due to changes in the weakening of their immune system function and higher prevalence of other health issues.
Skin reactions/change to skin pathology: sphingosine-1-phosphate receptor modulators are oral DMTs for MS that were associated with cases of basal cell carcinoma in clinical trials.
Ultimately, the decision to continue, discontinue, or de-escalate DMTs in aging pwMS should be based on the individual patient's needs and circumstances. It is important for clinicians to work closely with their patients to develop a personalized treatment plan that considers all the relevant benefits and risks. In the meantime, more research is needed on this topic to provide better outcomes for our growing population of aging patients who are living with MS.
The Prediabetes Debate: Should the Diabetes Diagnostic Threshold Be Lowered, Allowing Clinicians to Intervene Earlier?

I believe that the diagnosis of type 2 diabetes mellitus (T2DM) should be broadened to match the glycemic thresholds currently used for prediabetes. This would eliminate the need for a separate category, the “prediabetes” nomenclature, and allow for earlier therapeutic intervention in patients. Current diabetes diagnostic thresholds do not reflect the latest advancements in T2DM understanding. The latest in T2DM research suggests that intervening and treating prediabetes earlier could potentially offer better clinical outcomes and enhance patients’ quality of life, as cell and tissue damage occurs early and leads to dysfunction prior to a diabetes diagnosis.1 T2DM is a highly complex disease with multifactorial causes beyond hyperglycemia that should be considered, such as hyperlipidemia, insulin resistance, hyperinsulinemia, and autoimmune inflammatory mechanisms that lead to β-cell dysfunction or failure, which results in hyperglycemia.
Prediabetes is associated with micro- and macrovascular complications that can occur early in the progression to frank disease state.1,2 This phase of diabetes also includes insulin resistance, impaired incretin action, insulin hypersecretion, increased lipolysis, and ectopic lipid storage—all of which damage β cells. These dysfunctions are also present in the frank diabetic disease state.3,4 Furthermore, diabetic retinopathy occurs in 8% to 12% of patients with prediabetes, and retinopathy begins earlier than previously thought, with neuroinflammation occurring even before vascular damage.5,6 Unfortunately, these neuro-inflammatory lesions cannot be detected with the typical instruments used in an ophthalmologist’s office.
It is believed that, through the principle of metabolic memory, even a moderate increase or episodic spikes in blood glucose can lead to negative effects in prediabetic patients who are susceptible to T2DM.1,5 Therefore, a lower diabetes diagnostic threshold could allow for earlier, more precise, and personalized therapies based on each patient’s individual risk factors and biomarkers. With a diagnosis of T2DM at the current prediabetes threshold, patients could receive treatment covered by health insurance while in the “prediabetic” state—treatment that would not have been previously approved. Patients should be treated earlier and on an individual basis with counseling on diet and lifestyle changes and antidiabetic agents to reduce glycemic levels, preserve β cells, and reduce cardiovascular [CV] or renal risk, among other complications.6,7
Moreover, the newer agents for treating diabetes such as glucagon-like pepetide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors, which are also associated with reduction in adverse CV and/or renal outcomes, could be beneficial if administered early in the prediabetic stage of disease.8,9
Early lifestyle and pharmacologic interventions can reduce the rate of progression from prediabetes to diabetes as well as complications and associated conditions, and even potentially result in remission or a full reversal of diabetes. These “side benefits” of lowering the diabetes diagnostic threshold (over and above glycemic control) make any cost-effectiveness calculations all the more advantageous to individual patients as well as to society.
Given the numerous benefits of earlier intervention for diabetes treatment, I believe a call to re-evaluate the current diabetes diagnostic threshold is in order, as it will do a great service for all patients who are currently at risk for developing T2DM.
Armato JP, DeFronzo RA, Abdul-Ghani M, Ruby RJ. Successful treatment of prediabetes in clinical practice using physiological assessment (STOP DIABETES). Lancet Diabetes Endocrinol. 2018;6:781-789.
Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28:645-655.
American Diabetes Association. Standards of Medical Care in Diabetes – 2021. Diabetes Care. 2021;44(S15–S39):S111-S124.
Brannick B, Wynn A, Dagogo-Jack S. Prediabetes as a toxic environment for the initiation of microvascular and macrovascular complications. Exp Biol Med (Maywood). 2016;241:1323-1331.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183-1197.
Sinclair SH, Schwartz SS. Diabetic retinopathy–an underdiagnosed and undertreated inflammatory, neuro-vascular complication of diabetes. Front Endocrinol (Lausanne). 2019;10:843.
Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin N Am. 2016;45:751-764.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World J Diabetes. 2015;6:1207-1222.
Dankner R, Roth J. The personalized approach for detecting prediabetes and diabetes. Curr Diabetes Rev. 2016;12:58-65.
I believe that the diagnosis of type 2 diabetes mellitus (T2DM) should be broadened to match the glycemic thresholds currently used for prediabetes. This would eliminate the need for a separate category, the “prediabetes” nomenclature, and allow for earlier therapeutic intervention in patients. Current diabetes diagnostic thresholds do not reflect the latest advancements in T2DM understanding. The latest in T2DM research suggests that intervening and treating prediabetes earlier could potentially offer better clinical outcomes and enhance patients’ quality of life, as cell and tissue damage occurs early and leads to dysfunction prior to a diabetes diagnosis.1 T2DM is a highly complex disease with multifactorial causes beyond hyperglycemia that should be considered, such as hyperlipidemia, insulin resistance, hyperinsulinemia, and autoimmune inflammatory mechanisms that lead to β-cell dysfunction or failure, which results in hyperglycemia.
Prediabetes is associated with micro- and macrovascular complications that can occur early in the progression to frank disease state.1,2 This phase of diabetes also includes insulin resistance, impaired incretin action, insulin hypersecretion, increased lipolysis, and ectopic lipid storage—all of which damage β cells. These dysfunctions are also present in the frank diabetic disease state.3,4 Furthermore, diabetic retinopathy occurs in 8% to 12% of patients with prediabetes, and retinopathy begins earlier than previously thought, with neuroinflammation occurring even before vascular damage.5,6 Unfortunately, these neuro-inflammatory lesions cannot be detected with the typical instruments used in an ophthalmologist’s office.
It is believed that, through the principle of metabolic memory, even a moderate increase or episodic spikes in blood glucose can lead to negative effects in prediabetic patients who are susceptible to T2DM.1,5 Therefore, a lower diabetes diagnostic threshold could allow for earlier, more precise, and personalized therapies based on each patient’s individual risk factors and biomarkers. With a diagnosis of T2DM at the current prediabetes threshold, patients could receive treatment covered by health insurance while in the “prediabetic” state—treatment that would not have been previously approved. Patients should be treated earlier and on an individual basis with counseling on diet and lifestyle changes and antidiabetic agents to reduce glycemic levels, preserve β cells, and reduce cardiovascular [CV] or renal risk, among other complications.6,7
Moreover, the newer agents for treating diabetes such as glucagon-like pepetide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors, which are also associated with reduction in adverse CV and/or renal outcomes, could be beneficial if administered early in the prediabetic stage of disease.8,9
Early lifestyle and pharmacologic interventions can reduce the rate of progression from prediabetes to diabetes as well as complications and associated conditions, and even potentially result in remission or a full reversal of diabetes. These “side benefits” of lowering the diabetes diagnostic threshold (over and above glycemic control) make any cost-effectiveness calculations all the more advantageous to individual patients as well as to society.
Given the numerous benefits of earlier intervention for diabetes treatment, I believe a call to re-evaluate the current diabetes diagnostic threshold is in order, as it will do a great service for all patients who are currently at risk for developing T2DM.
I believe that the diagnosis of type 2 diabetes mellitus (T2DM) should be broadened to match the glycemic thresholds currently used for prediabetes. This would eliminate the need for a separate category, the “prediabetes” nomenclature, and allow for earlier therapeutic intervention in patients. Current diabetes diagnostic thresholds do not reflect the latest advancements in T2DM understanding. The latest in T2DM research suggests that intervening and treating prediabetes earlier could potentially offer better clinical outcomes and enhance patients’ quality of life, as cell and tissue damage occurs early and leads to dysfunction prior to a diabetes diagnosis.1 T2DM is a highly complex disease with multifactorial causes beyond hyperglycemia that should be considered, such as hyperlipidemia, insulin resistance, hyperinsulinemia, and autoimmune inflammatory mechanisms that lead to β-cell dysfunction or failure, which results in hyperglycemia.
Prediabetes is associated with micro- and macrovascular complications that can occur early in the progression to frank disease state.1,2 This phase of diabetes also includes insulin resistance, impaired incretin action, insulin hypersecretion, increased lipolysis, and ectopic lipid storage—all of which damage β cells. These dysfunctions are also present in the frank diabetic disease state.3,4 Furthermore, diabetic retinopathy occurs in 8% to 12% of patients with prediabetes, and retinopathy begins earlier than previously thought, with neuroinflammation occurring even before vascular damage.5,6 Unfortunately, these neuro-inflammatory lesions cannot be detected with the typical instruments used in an ophthalmologist’s office.
It is believed that, through the principle of metabolic memory, even a moderate increase or episodic spikes in blood glucose can lead to negative effects in prediabetic patients who are susceptible to T2DM.1,5 Therefore, a lower diabetes diagnostic threshold could allow for earlier, more precise, and personalized therapies based on each patient’s individual risk factors and biomarkers. With a diagnosis of T2DM at the current prediabetes threshold, patients could receive treatment covered by health insurance while in the “prediabetic” state—treatment that would not have been previously approved. Patients should be treated earlier and on an individual basis with counseling on diet and lifestyle changes and antidiabetic agents to reduce glycemic levels, preserve β cells, and reduce cardiovascular [CV] or renal risk, among other complications.6,7
Moreover, the newer agents for treating diabetes such as glucagon-like pepetide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors, which are also associated with reduction in adverse CV and/or renal outcomes, could be beneficial if administered early in the prediabetic stage of disease.8,9
Early lifestyle and pharmacologic interventions can reduce the rate of progression from prediabetes to diabetes as well as complications and associated conditions, and even potentially result in remission or a full reversal of diabetes. These “side benefits” of lowering the diabetes diagnostic threshold (over and above glycemic control) make any cost-effectiveness calculations all the more advantageous to individual patients as well as to society.
Given the numerous benefits of earlier intervention for diabetes treatment, I believe a call to re-evaluate the current diabetes diagnostic threshold is in order, as it will do a great service for all patients who are currently at risk for developing T2DM.
Armato JP, DeFronzo RA, Abdul-Ghani M, Ruby RJ. Successful treatment of prediabetes in clinical practice using physiological assessment (STOP DIABETES). Lancet Diabetes Endocrinol. 2018;6:781-789.
Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28:645-655.
American Diabetes Association. Standards of Medical Care in Diabetes – 2021. Diabetes Care. 2021;44(S15–S39):S111-S124.
Brannick B, Wynn A, Dagogo-Jack S. Prediabetes as a toxic environment for the initiation of microvascular and macrovascular complications. Exp Biol Med (Maywood). 2016;241:1323-1331.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183-1197.
Sinclair SH, Schwartz SS. Diabetic retinopathy–an underdiagnosed and undertreated inflammatory, neuro-vascular complication of diabetes. Front Endocrinol (Lausanne). 2019;10:843.
Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin N Am. 2016;45:751-764.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World J Diabetes. 2015;6:1207-1222.
Dankner R, Roth J. The personalized approach for detecting prediabetes and diabetes. Curr Diabetes Rev. 2016;12:58-65.
Armato JP, DeFronzo RA, Abdul-Ghani M, Ruby RJ. Successful treatment of prediabetes in clinical practice using physiological assessment (STOP DIABETES). Lancet Diabetes Endocrinol. 2018;6:781-789.
Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28:645-655.
American Diabetes Association. Standards of Medical Care in Diabetes – 2021. Diabetes Care. 2021;44(S15–S39):S111-S124.
Brannick B, Wynn A, Dagogo-Jack S. Prediabetes as a toxic environment for the initiation of microvascular and macrovascular complications. Exp Biol Med (Maywood). 2016;241:1323-1331.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183-1197.
Sinclair SH, Schwartz SS. Diabetic retinopathy–an underdiagnosed and undertreated inflammatory, neuro-vascular complication of diabetes. Front Endocrinol (Lausanne). 2019;10:843.
Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin N Am. 2016;45:751-764.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World J Diabetes. 2015;6:1207-1222.
Dankner R, Roth J. The personalized approach for detecting prediabetes and diabetes. Curr Diabetes Rev. 2016;12:58-65.
Antibody-Drug Conjugates: Targeted Treatments Providing Hope for Patients With Breast Cancer

The restrictive therapeutic index of chemotherapy has led to the emergence of antibody-drug conjugates (ADCs), medicines that combine the specificity of monoclonal antibodies (mAbs) with the cytotoxic effects of chemotherapy to deliver cytotoxic payloads to cancer cells. This targeted approach can reduce the side effects of chemotherapy and improve the effectiveness of treatment. Several ADCs, including ado-trastuzumab emtansine (Kadcyla), sacituzumab govitecan-hziy (Trodelvy), and fam-trastuzumab deruxtecan-nxki (Enhertu), are currently approved for treating some types of breast cancer (BC).
The ADC trastuzumab emtansine was approved specifically for treating human epidermal growth factor receptor 2 positive (HER2+) metastatic breast cancer (mBC) in patients who have previously been treated with trastuzumab and a taxane (paclitaxel or docetaxel) and who have already been treated for mBC or have developed tumor recurrence within 6 months of receiving adjuvant therapy. The US Food and Drug Administration (FDA) approval was based on the results of the EMILIA study, a phase 3 clinical trial that compared treatment with trastuzumab emtansine vs capecitabine + lapatinib in participants with HER2+, locally advanced, or metastatic BC. This trial emerged from the need for well-tolerated, HER2-directed therapies for patients with this type of cancer. Trastuzumab emtansine consists of trastuzumab, a mAb that targets HER2 (which is overexpressed in about 20% of BCs), linked to emtansine, a cytotoxic payload that inhibits cell division. The trastuzumab emtansine group had a median overall survival (OS) of 29.9 months vs 25.9 months in the capecitabine + lapatinib group, for a hazard ratio (HR) of 0.75 (95% CI: 0.64, 0.88).
Another ADC, sacituzumab govitecan, targets the Trop-2 protein, which is overexpressed in BC. This ADC includes a mAb that is linked to SN-38, a cytotoxic payload that inhibits DNA replication. Triple-negative breast cancer (TNBC) is a subtype of BC that does not have receptors for estrogen, progesterone, or HER2—making it more difficult to treat than other forms of BC. Sacituzumab govitecan is used to treat patients with metastatic TNBC who have received at least 2 prior therapies for metastatic disease. Sacituzumab govitecan is also approved for the treatment of patients with unresectable locally advanced or metastatic hormone-receptor–positive (HR+), and HER2-negative (HER2−) BC who have received endocrine-based therapy and at least 2 additional systemic therapies in the metastatic setting. Sacituzumab govitecan was the first Trop-2–directed ADC to demonstrate OS benefit in patients with HR+/HER2− mBC who had received prior endocrine-based therapy and at least 2 chemotherapies. It is now also recommended as a Category 1 preferred treatment for metastatic HR+/HER2− BC by the National Comprehensive Cancer Network.
The results of the TROPiCS-02 study, which led to the FDA approval of sacituzumab govitecan, demonstrated a median OS of 14.4 months with sacituzumab govitecan vs 11.2 months with treatment of physician’s choice (HR, 0.79; 95% CI: 0.65, 0.96; P = 0.02). This represents a 3.2-month improvement in survival and a 21% reduction in the risk for patient death. Before this medicine was approved, there were limited options to offer patients with BC after endocrine-based therapy and chemotherapy.
A third ADC, trastuzumab deruxtecan, targets the HER2 protein, like trastuzumab emtansine, but with a different cytotoxic payload. It consists of trastuzumab linked to deruxtecan, whose cytotoxicity inhibits DNA replication. It is approved for the treatment of HER2+ mBC. Its FDA approval was based on the results of the DESTINY-Breast04 phase 3 clinical trial, which demonstrated that treatment with trastuzumab deruxtecan, when compared with standard-of-care chemotherapy, doubles progression-free survival among patients with mBC that expresses low levels of HER2. The median OS for patients in the HR+ group who received trastuzumab deruxtecan was 23.9 months vs 17.5 months for those who received chemotherapy. In the hormone receptor-negative (HR−) group, the median OS for those who took trastuzumab deruxtecan was 16.6 months vs 10.3 months for those treated with chemotherapy.
The emergence of ADCs have demonstrated promising advancements in the treatment of BC, particularly in patients with HER2+ or triple-negative disease. ADCs have given new hope to and prolonged life for patients living with pretreated HR+/HER2− mBC. ADCs also have the potential to provide a more effective and less toxic treatment option for patients with BC. However, further research is needed to fully understand their long-term effects and to develop new ADCs that target other types of BC.
The restrictive therapeutic index of chemotherapy has led to the emergence of antibody-drug conjugates (ADCs), medicines that combine the specificity of monoclonal antibodies (mAbs) with the cytotoxic effects of chemotherapy to deliver cytotoxic payloads to cancer cells. This targeted approach can reduce the side effects of chemotherapy and improve the effectiveness of treatment. Several ADCs, including ado-trastuzumab emtansine (Kadcyla), sacituzumab govitecan-hziy (Trodelvy), and fam-trastuzumab deruxtecan-nxki (Enhertu), are currently approved for treating some types of breast cancer (BC).
The ADC trastuzumab emtansine was approved specifically for treating human epidermal growth factor receptor 2 positive (HER2+) metastatic breast cancer (mBC) in patients who have previously been treated with trastuzumab and a taxane (paclitaxel or docetaxel) and who have already been treated for mBC or have developed tumor recurrence within 6 months of receiving adjuvant therapy. The US Food and Drug Administration (FDA) approval was based on the results of the EMILIA study, a phase 3 clinical trial that compared treatment with trastuzumab emtansine vs capecitabine + lapatinib in participants with HER2+, locally advanced, or metastatic BC. This trial emerged from the need for well-tolerated, HER2-directed therapies for patients with this type of cancer. Trastuzumab emtansine consists of trastuzumab, a mAb that targets HER2 (which is overexpressed in about 20% of BCs), linked to emtansine, a cytotoxic payload that inhibits cell division. The trastuzumab emtansine group had a median overall survival (OS) of 29.9 months vs 25.9 months in the capecitabine + lapatinib group, for a hazard ratio (HR) of 0.75 (95% CI: 0.64, 0.88).
Another ADC, sacituzumab govitecan, targets the Trop-2 protein, which is overexpressed in BC. This ADC includes a mAb that is linked to SN-38, a cytotoxic payload that inhibits DNA replication. Triple-negative breast cancer (TNBC) is a subtype of BC that does not have receptors for estrogen, progesterone, or HER2—making it more difficult to treat than other forms of BC. Sacituzumab govitecan is used to treat patients with metastatic TNBC who have received at least 2 prior therapies for metastatic disease. Sacituzumab govitecan is also approved for the treatment of patients with unresectable locally advanced or metastatic hormone-receptor–positive (HR+), and HER2-negative (HER2−) BC who have received endocrine-based therapy and at least 2 additional systemic therapies in the metastatic setting. Sacituzumab govitecan was the first Trop-2–directed ADC to demonstrate OS benefit in patients with HR+/HER2− mBC who had received prior endocrine-based therapy and at least 2 chemotherapies. It is now also recommended as a Category 1 preferred treatment for metastatic HR+/HER2− BC by the National Comprehensive Cancer Network.
The results of the TROPiCS-02 study, which led to the FDA approval of sacituzumab govitecan, demonstrated a median OS of 14.4 months with sacituzumab govitecan vs 11.2 months with treatment of physician’s choice (HR, 0.79; 95% CI: 0.65, 0.96; P = 0.02). This represents a 3.2-month improvement in survival and a 21% reduction in the risk for patient death. Before this medicine was approved, there were limited options to offer patients with BC after endocrine-based therapy and chemotherapy.
A third ADC, trastuzumab deruxtecan, targets the HER2 protein, like trastuzumab emtansine, but with a different cytotoxic payload. It consists of trastuzumab linked to deruxtecan, whose cytotoxicity inhibits DNA replication. It is approved for the treatment of HER2+ mBC. Its FDA approval was based on the results of the DESTINY-Breast04 phase 3 clinical trial, which demonstrated that treatment with trastuzumab deruxtecan, when compared with standard-of-care chemotherapy, doubles progression-free survival among patients with mBC that expresses low levels of HER2. The median OS for patients in the HR+ group who received trastuzumab deruxtecan was 23.9 months vs 17.5 months for those who received chemotherapy. In the hormone receptor-negative (HR−) group, the median OS for those who took trastuzumab deruxtecan was 16.6 months vs 10.3 months for those treated with chemotherapy.
The emergence of ADCs have demonstrated promising advancements in the treatment of BC, particularly in patients with HER2+ or triple-negative disease. ADCs have given new hope to and prolonged life for patients living with pretreated HR+/HER2− mBC. ADCs also have the potential to provide a more effective and less toxic treatment option for patients with BC. However, further research is needed to fully understand their long-term effects and to develop new ADCs that target other types of BC.
The restrictive therapeutic index of chemotherapy has led to the emergence of antibody-drug conjugates (ADCs), medicines that combine the specificity of monoclonal antibodies (mAbs) with the cytotoxic effects of chemotherapy to deliver cytotoxic payloads to cancer cells. This targeted approach can reduce the side effects of chemotherapy and improve the effectiveness of treatment. Several ADCs, including ado-trastuzumab emtansine (Kadcyla), sacituzumab govitecan-hziy (Trodelvy), and fam-trastuzumab deruxtecan-nxki (Enhertu), are currently approved for treating some types of breast cancer (BC).
The ADC trastuzumab emtansine was approved specifically for treating human epidermal growth factor receptor 2 positive (HER2+) metastatic breast cancer (mBC) in patients who have previously been treated with trastuzumab and a taxane (paclitaxel or docetaxel) and who have already been treated for mBC or have developed tumor recurrence within 6 months of receiving adjuvant therapy. The US Food and Drug Administration (FDA) approval was based on the results of the EMILIA study, a phase 3 clinical trial that compared treatment with trastuzumab emtansine vs capecitabine + lapatinib in participants with HER2+, locally advanced, or metastatic BC. This trial emerged from the need for well-tolerated, HER2-directed therapies for patients with this type of cancer. Trastuzumab emtansine consists of trastuzumab, a mAb that targets HER2 (which is overexpressed in about 20% of BCs), linked to emtansine, a cytotoxic payload that inhibits cell division. The trastuzumab emtansine group had a median overall survival (OS) of 29.9 months vs 25.9 months in the capecitabine + lapatinib group, for a hazard ratio (HR) of 0.75 (95% CI: 0.64, 0.88).
Another ADC, sacituzumab govitecan, targets the Trop-2 protein, which is overexpressed in BC. This ADC includes a mAb that is linked to SN-38, a cytotoxic payload that inhibits DNA replication. Triple-negative breast cancer (TNBC) is a subtype of BC that does not have receptors for estrogen, progesterone, or HER2—making it more difficult to treat than other forms of BC. Sacituzumab govitecan is used to treat patients with metastatic TNBC who have received at least 2 prior therapies for metastatic disease. Sacituzumab govitecan is also approved for the treatment of patients with unresectable locally advanced or metastatic hormone-receptor–positive (HR+), and HER2-negative (HER2−) BC who have received endocrine-based therapy and at least 2 additional systemic therapies in the metastatic setting. Sacituzumab govitecan was the first Trop-2–directed ADC to demonstrate OS benefit in patients with HR+/HER2− mBC who had received prior endocrine-based therapy and at least 2 chemotherapies. It is now also recommended as a Category 1 preferred treatment for metastatic HR+/HER2− BC by the National Comprehensive Cancer Network.
The results of the TROPiCS-02 study, which led to the FDA approval of sacituzumab govitecan, demonstrated a median OS of 14.4 months with sacituzumab govitecan vs 11.2 months with treatment of physician’s choice (HR, 0.79; 95% CI: 0.65, 0.96; P = 0.02). This represents a 3.2-month improvement in survival and a 21% reduction in the risk for patient death. Before this medicine was approved, there were limited options to offer patients with BC after endocrine-based therapy and chemotherapy.
A third ADC, trastuzumab deruxtecan, targets the HER2 protein, like trastuzumab emtansine, but with a different cytotoxic payload. It consists of trastuzumab linked to deruxtecan, whose cytotoxicity inhibits DNA replication. It is approved for the treatment of HER2+ mBC. Its FDA approval was based on the results of the DESTINY-Breast04 phase 3 clinical trial, which demonstrated that treatment with trastuzumab deruxtecan, when compared with standard-of-care chemotherapy, doubles progression-free survival among patients with mBC that expresses low levels of HER2. The median OS for patients in the HR+ group who received trastuzumab deruxtecan was 23.9 months vs 17.5 months for those who received chemotherapy. In the hormone receptor-negative (HR−) group, the median OS for those who took trastuzumab deruxtecan was 16.6 months vs 10.3 months for those treated with chemotherapy.
The emergence of ADCs have demonstrated promising advancements in the treatment of BC, particularly in patients with HER2+ or triple-negative disease. ADCs have given new hope to and prolonged life for patients living with pretreated HR+/HER2− mBC. ADCs also have the potential to provide a more effective and less toxic treatment option for patients with BC. However, further research is needed to fully understand their long-term effects and to develop new ADCs that target other types of BC.