User login
Atypical Skin Bronzing in Response to Belumosudil for Graft-vs-Host Disease
Atypical Skin Bronzing in Response to Belumosudil for Graft-vs-Host Disease
To the Editor:
Drug-induced hyperpigmentation is a common cause of an acquired increase in pigmentation. Belumosudil is an oral selective inhibitor of Rho-associated coiled-coil containing protein kinase (ROCK2) that is approved for the treatment of chronic graft-vs-host disease (GVHD). We describe a patient who developed diffuse skin bronzing 3 weeks after initiation of belumosudil treatment.
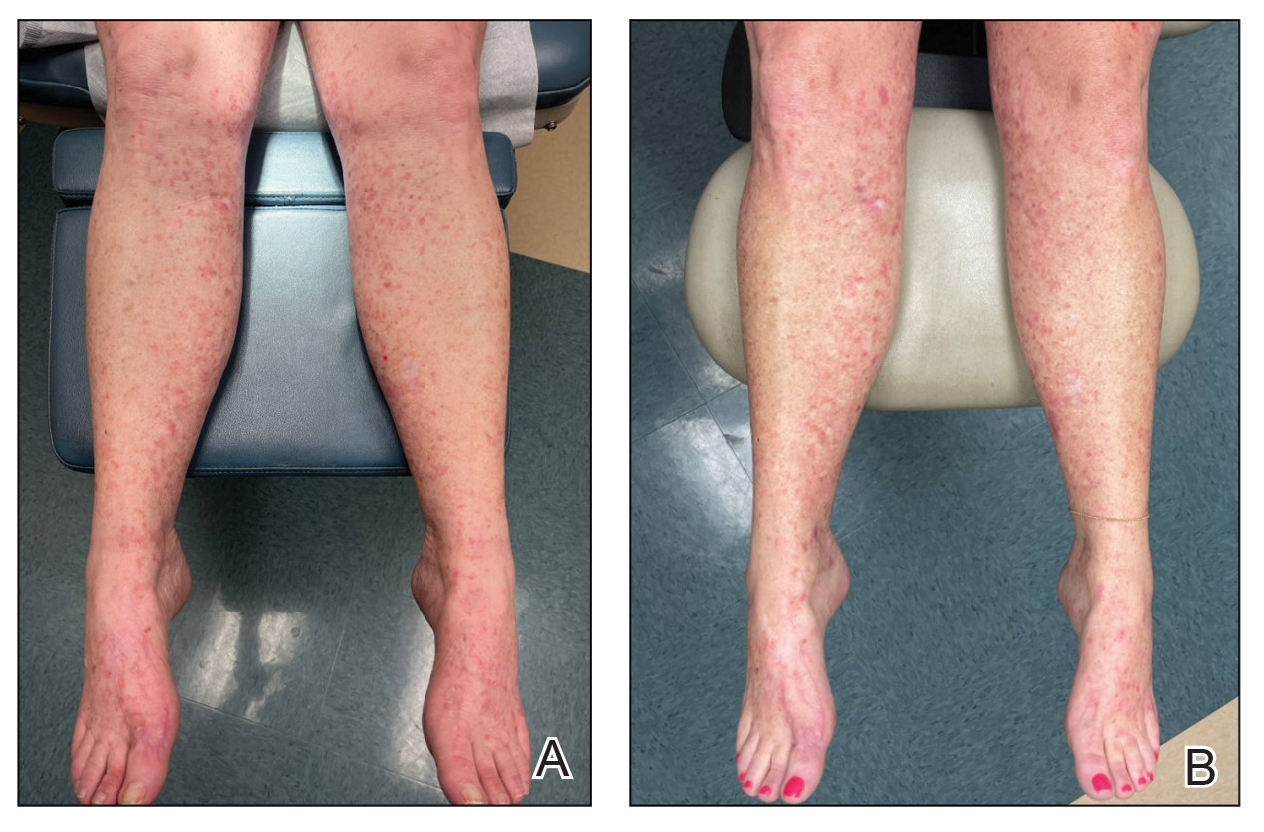
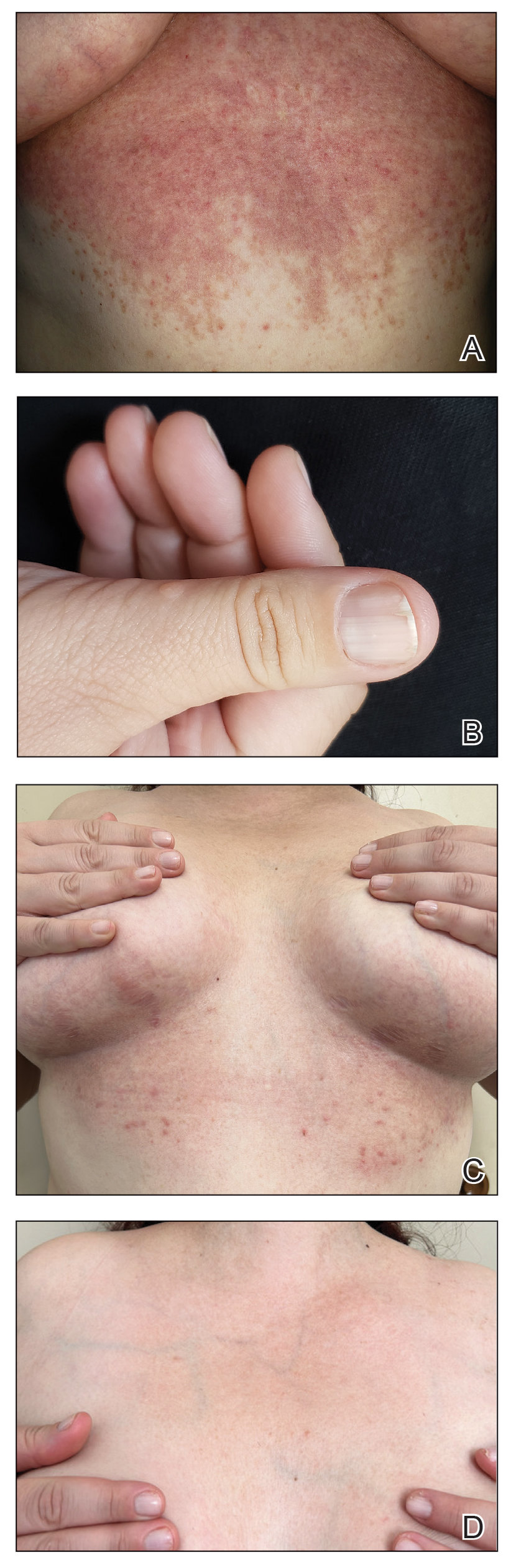
A 64-year-old fair-skinned woman presented to the dermatology clinic with bronzing of the skin and dystrophic nails 3 weeks after starting belumosudil for treatment of chronic GVHD. Six months prior to presentation, the patient had received a bone marrow transplant for chronic lymphoid leukemia. She presented to dermatology 6 months after the transplant with a new-onset rash that was suspicious for GVHD. Physical examination revealed pruritic pink papules diffusely scattered on the legs and forearms (Figure 1). The patient declined biopsy at that time and later followed up with oncology. The patient’s oncologist supported a diagnosis of GVHD, and the patient began treatment with belumosudil 200 mg/d which was intended to be taken until treatment failure due to progression of chronic GVHD.
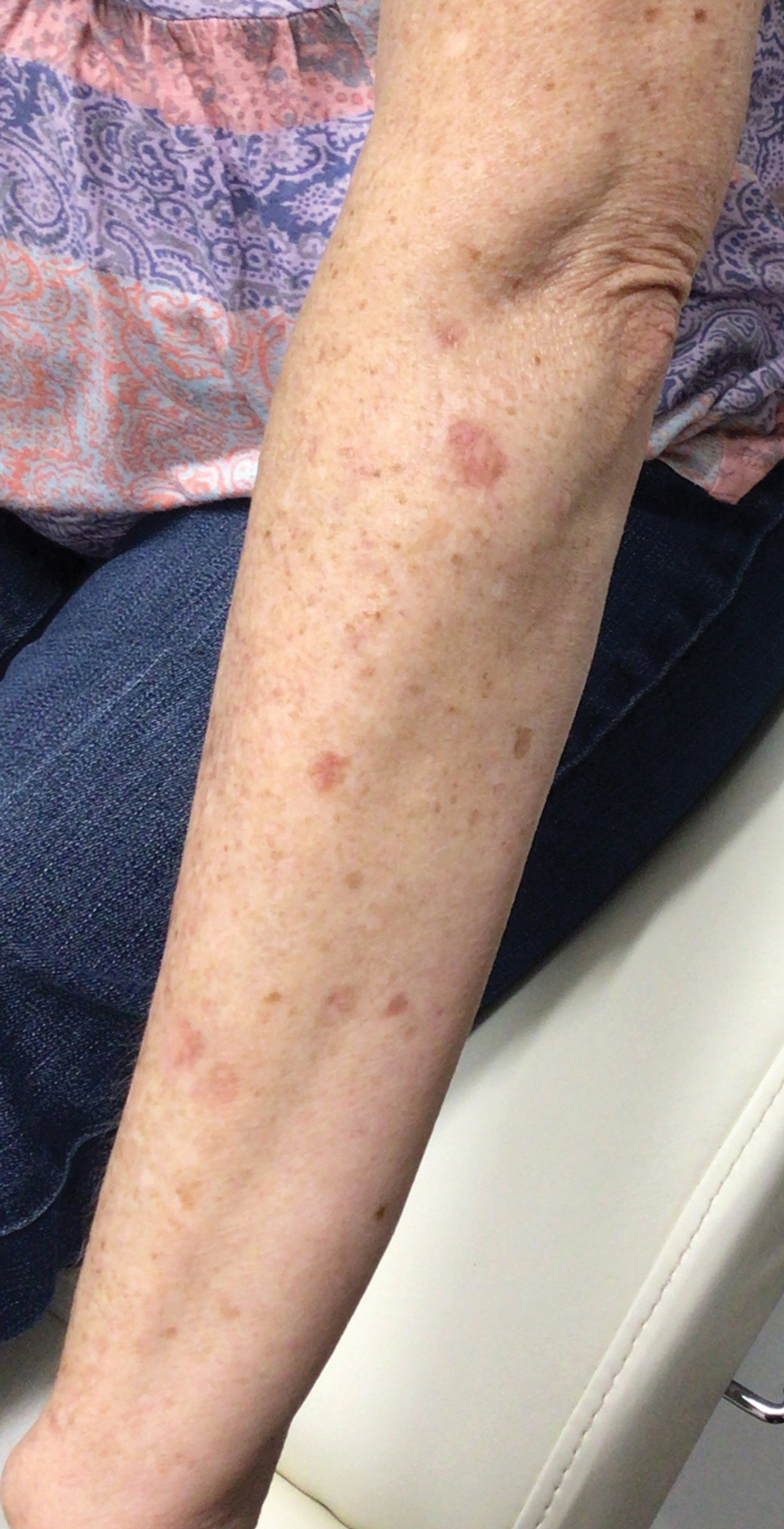
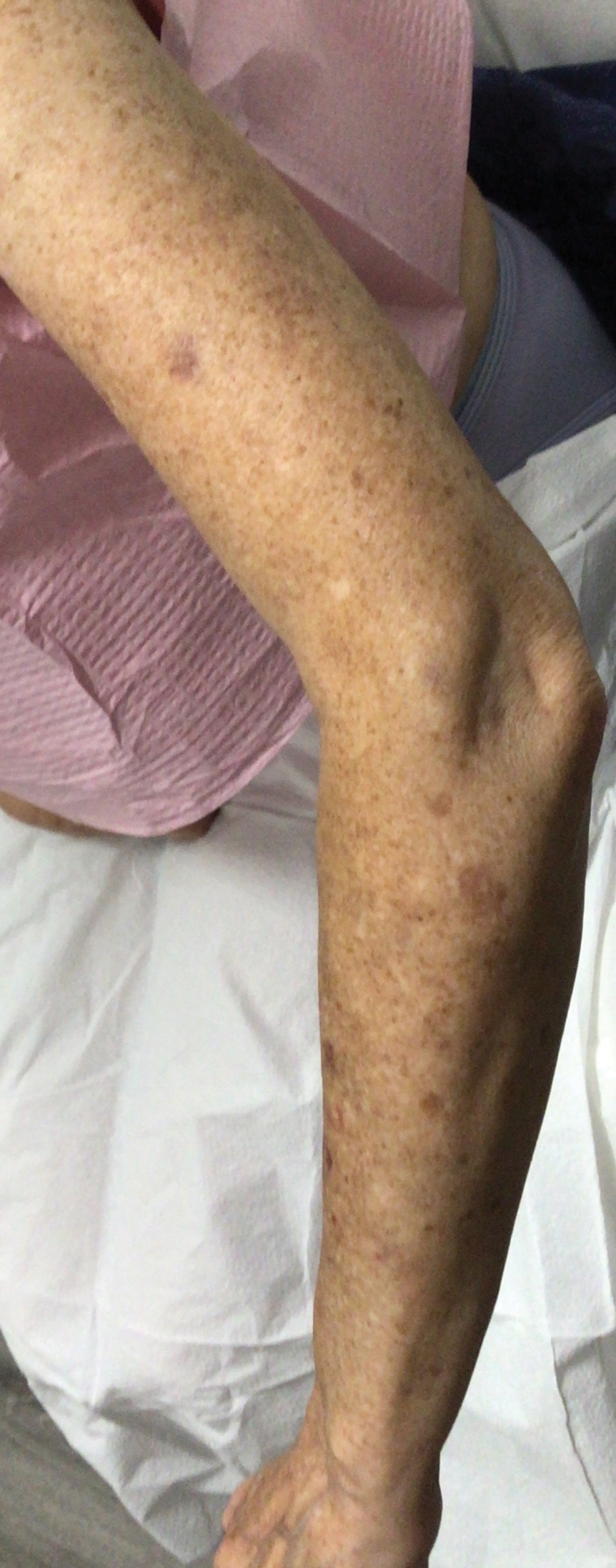
Three weeks after starting belumosudil, the patient developed diffuse bronzing of the skin and brown, evenly colored patches scattered on the trunk, back, and upper and lower extremities on a background of the presumed GVHD rash (Figure 2). The hyperpigmentation was abrupt, starting on the chest and spreading to the abdomen, extremities, and back (Figure 3).
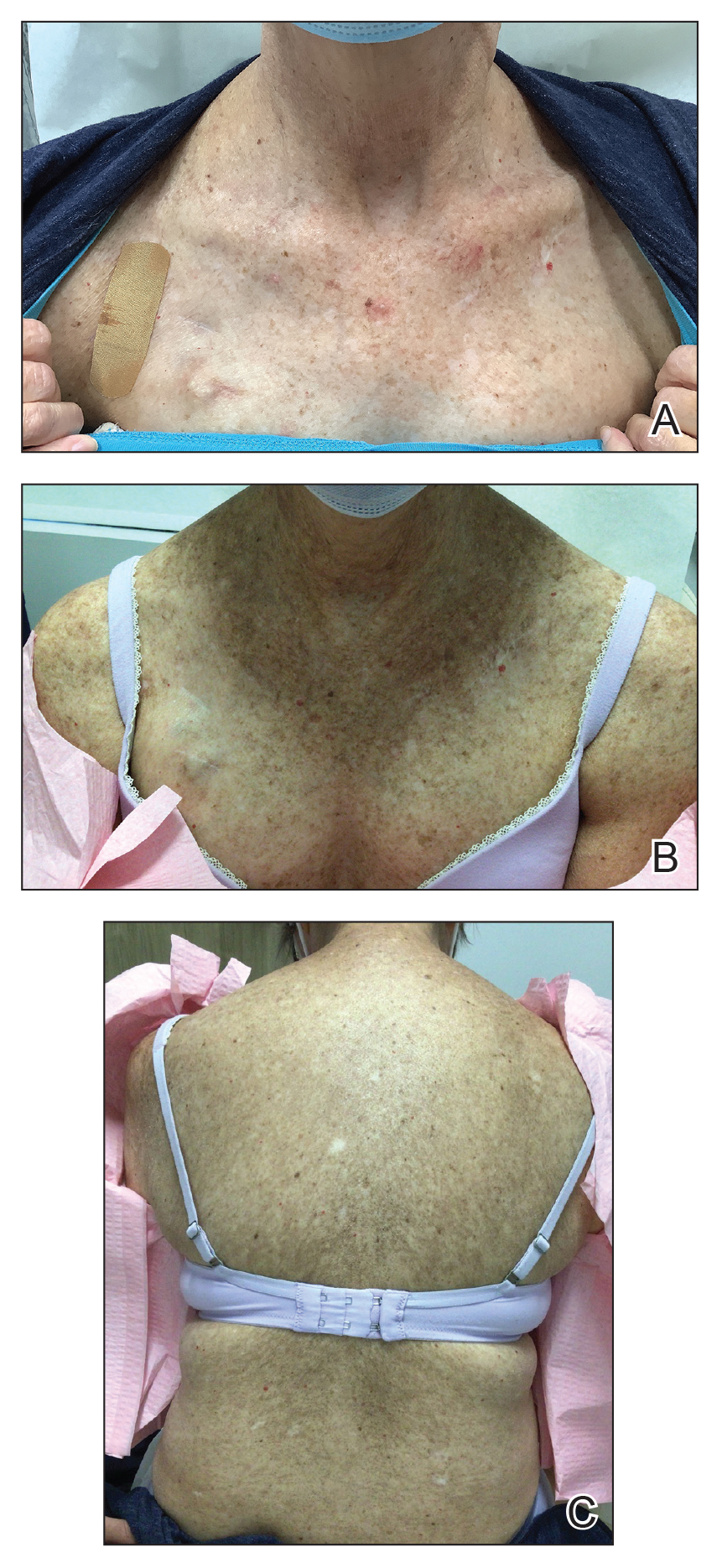
developed on the patient’s chest and back within 3 weeks of initiating treatment with belumosudil.
Again, the patient was offered biopsy for the new-onset pigmentation but declined. During this time, she had no notable sun exposure and primarily stayed indoors despite living in a region with a sunny semi-arid climate. Her medication and supplement list were reviewed and included acalabrutinib, a multivitamin, lutein, biotin, and a fish oil supplement. A compete blood cell count as well as ferritin, transferrin, cortisol, and adrenocorticotropic hormone levels were unremarkable.
The patient continued to take belumosudil for treatment of GVHD. The hyperpigmentation faded slightly by a 2-month follow-up visit but persisted and was stable. She has not tried other treatments for GVHD to manage the hyperpigmentation.
Conditions known to cause diffuse bronzing of the skin include Addison disease, hemochromatosis, Cushing disease, and medication adverse events. Our patient presented with an absence of systemic symptoms, normal laboratory results, and no clinical indicators suggesting alternate causes. Given that the onset of the hyperpigmentation was 3 weeks after she started a new medication, we hypothesized that the bronzing was an adverse effect of the belumosudil—though this correlation cannot be definitively proven by this case.
The most common offending agents for drug-induced skin hyperpigmentation are nonsteroidal anti- inflammatory drugs, antimalarials, amiodarone, cytotoxic drugs, and tetracyclines.1,2 Our patient’s medication list included the cytotoxic agent acalabrutinib, a Bruton tyrosine kinase inhibitor used for the treatment of non-Hodgkin lymphoma. It has been associated with dermatologic findings of ecchymosis, bruising, panniculitis, and cellulitis, but there are no known reports of hyperpigmentation.3 Our patient had been taking acalabrutinib for 6 months when the GVHD rash developed. At the time, she also was taking a multivitamin and lutein, biotin, and fish oil supplements, none of which have been associated with hyperpigmentation.
Polypharmacy adds a layer of difficulty in identifying the inciting cause of pigmentary change. In our case, symptoms began 3 weeks after the initiation of belumosudil. There were no cutaneous reactions observed in the ROCKstar study of belumosudil; the most common adverse events were upper respiratory tract infection, diarrhea, fatigue, nausea, increased liver enzymes, and dyspnea.4,5 Patients on belumosudil have developed aggressive cutaneous squamous cell carcinoma.6 However, a search of PubMed articles indexed for MEDLINE using the search terms acalabrutinib or belumosudil with hyperpigmentation or cutaneous reaction returned no reports of these medications causing hyperpigmentation or cutaneous deposits.
Treatment of drug-induced hyperpigmentation is difficult because discontinuation of the offending agent typically confirms diagnosis, but interruption of treatment is not always possible, as in our patient. The skin changes can fade over time, but effects typically are long lasting.
Dermatologists play a key role in the identification of drug-induced skin hyperpigmentation. After endocrine or metabolic causes of skin hyperpigmentation have been ruled out, a thorough review of the patient’s medication list should be done to assess for a drug-induced cause. Treatment is limited to sun avoidance, as interruption of treatment may not be possible, and lesions typically do fade over time. These chronic skin changes can have a psychosocial effect on patients and regular follow-up is recommended.
- Giménez García RM, Carrasco Molina S. Drug-induced hyperpigmentation: review and case series. J Am Board Fam Med. 2019;32:628-638. doi:10.3122/jabfm.2019.04.180212
- Dereure O. Drug-induced skin pigmentation. epidemiology, diagnosis and treatment. Am J Clin Dermatol. 2001;2:253-62. doi:10.2165/00128071-200102040-00006
- Sibaud V, Beylot-Barry M, Protin C, et al. Dermatological toxicities of Bruton’s tyrosine kinase inhibitors. Am J Clin Dermatol. 2020; 21:799-812. doi:10.1007/s40257-020-00535-x
- Cutler C, Lee SJ, Arai S, et al. Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study. Blood. 2021;138:2278-2289. doi:10.1182/blood.2021012021
- Jagasia M, Lazaryan A, Bachier CR, et al. ROCK2 inhibition with belumosudil (KD025) for the treatment of chronic graftversus- host disease. J Clin Oncol. 2021;39:1888-1898. doi:10.1200 /JCO.20.02754
- Lee GH, Guzman AK, Divito SJ, et al. Cutaneous squamous-cell carcinoma after treatment with ruxolitinib or belumosudil. N Engl J Med. 2023;389:188-190. doi:10.1056/NEJMc2304157
To the Editor:
Drug-induced hyperpigmentation is a common cause of an acquired increase in pigmentation. Belumosudil is an oral selective inhibitor of Rho-associated coiled-coil containing protein kinase (ROCK2) that is approved for the treatment of chronic graft-vs-host disease (GVHD). We describe a patient who developed diffuse skin bronzing 3 weeks after initiation of belumosudil treatment.
A 64-year-old fair-skinned woman presented to the dermatology clinic with bronzing of the skin and dystrophic nails 3 weeks after starting belumosudil for treatment of chronic GVHD. Six months prior to presentation, the patient had received a bone marrow transplant for chronic lymphoid leukemia. She presented to dermatology 6 months after the transplant with a new-onset rash that was suspicious for GVHD. Physical examination revealed pruritic pink papules diffusely scattered on the legs and forearms (Figure 1). The patient declined biopsy at that time and later followed up with oncology. The patient’s oncologist supported a diagnosis of GVHD, and the patient began treatment with belumosudil 200 mg/d which was intended to be taken until treatment failure due to progression of chronic GVHD.
Three weeks after starting belumosudil, the patient developed diffuse bronzing of the skin and brown, evenly colored patches scattered on the trunk, back, and upper and lower extremities on a background of the presumed GVHD rash (Figure 2). The hyperpigmentation was abrupt, starting on the chest and spreading to the abdomen, extremities, and back (Figure 3).
developed on the patient’s chest and back within 3 weeks of initiating treatment with belumosudil.
Again, the patient was offered biopsy for the new-onset pigmentation but declined. During this time, she had no notable sun exposure and primarily stayed indoors despite living in a region with a sunny semi-arid climate. Her medication and supplement list were reviewed and included acalabrutinib, a multivitamin, lutein, biotin, and a fish oil supplement. A compete blood cell count as well as ferritin, transferrin, cortisol, and adrenocorticotropic hormone levels were unremarkable.
The patient continued to take belumosudil for treatment of GVHD. The hyperpigmentation faded slightly by a 2-month follow-up visit but persisted and was stable. She has not tried other treatments for GVHD to manage the hyperpigmentation.
Conditions known to cause diffuse bronzing of the skin include Addison disease, hemochromatosis, Cushing disease, and medication adverse events. Our patient presented with an absence of systemic symptoms, normal laboratory results, and no clinical indicators suggesting alternate causes. Given that the onset of the hyperpigmentation was 3 weeks after she started a new medication, we hypothesized that the bronzing was an adverse effect of the belumosudil—though this correlation cannot be definitively proven by this case.
The most common offending agents for drug-induced skin hyperpigmentation are nonsteroidal anti- inflammatory drugs, antimalarials, amiodarone, cytotoxic drugs, and tetracyclines.1,2 Our patient’s medication list included the cytotoxic agent acalabrutinib, a Bruton tyrosine kinase inhibitor used for the treatment of non-Hodgkin lymphoma. It has been associated with dermatologic findings of ecchymosis, bruising, panniculitis, and cellulitis, but there are no known reports of hyperpigmentation.3 Our patient had been taking acalabrutinib for 6 months when the GVHD rash developed. At the time, she also was taking a multivitamin and lutein, biotin, and fish oil supplements, none of which have been associated with hyperpigmentation.
Polypharmacy adds a layer of difficulty in identifying the inciting cause of pigmentary change. In our case, symptoms began 3 weeks after the initiation of belumosudil. There were no cutaneous reactions observed in the ROCKstar study of belumosudil; the most common adverse events were upper respiratory tract infection, diarrhea, fatigue, nausea, increased liver enzymes, and dyspnea.4,5 Patients on belumosudil have developed aggressive cutaneous squamous cell carcinoma.6 However, a search of PubMed articles indexed for MEDLINE using the search terms acalabrutinib or belumosudil with hyperpigmentation or cutaneous reaction returned no reports of these medications causing hyperpigmentation or cutaneous deposits.
Treatment of drug-induced hyperpigmentation is difficult because discontinuation of the offending agent typically confirms diagnosis, but interruption of treatment is not always possible, as in our patient. The skin changes can fade over time, but effects typically are long lasting.
Dermatologists play a key role in the identification of drug-induced skin hyperpigmentation. After endocrine or metabolic causes of skin hyperpigmentation have been ruled out, a thorough review of the patient’s medication list should be done to assess for a drug-induced cause. Treatment is limited to sun avoidance, as interruption of treatment may not be possible, and lesions typically do fade over time. These chronic skin changes can have a psychosocial effect on patients and regular follow-up is recommended.
To the Editor:
Drug-induced hyperpigmentation is a common cause of an acquired increase in pigmentation. Belumosudil is an oral selective inhibitor of Rho-associated coiled-coil containing protein kinase (ROCK2) that is approved for the treatment of chronic graft-vs-host disease (GVHD). We describe a patient who developed diffuse skin bronzing 3 weeks after initiation of belumosudil treatment.
A 64-year-old fair-skinned woman presented to the dermatology clinic with bronzing of the skin and dystrophic nails 3 weeks after starting belumosudil for treatment of chronic GVHD. Six months prior to presentation, the patient had received a bone marrow transplant for chronic lymphoid leukemia. She presented to dermatology 6 months after the transplant with a new-onset rash that was suspicious for GVHD. Physical examination revealed pruritic pink papules diffusely scattered on the legs and forearms (Figure 1). The patient declined biopsy at that time and later followed up with oncology. The patient’s oncologist supported a diagnosis of GVHD, and the patient began treatment with belumosudil 200 mg/d which was intended to be taken until treatment failure due to progression of chronic GVHD.
Three weeks after starting belumosudil, the patient developed diffuse bronzing of the skin and brown, evenly colored patches scattered on the trunk, back, and upper and lower extremities on a background of the presumed GVHD rash (Figure 2). The hyperpigmentation was abrupt, starting on the chest and spreading to the abdomen, extremities, and back (Figure 3).
developed on the patient’s chest and back within 3 weeks of initiating treatment with belumosudil.
Again, the patient was offered biopsy for the new-onset pigmentation but declined. During this time, she had no notable sun exposure and primarily stayed indoors despite living in a region with a sunny semi-arid climate. Her medication and supplement list were reviewed and included acalabrutinib, a multivitamin, lutein, biotin, and a fish oil supplement. A compete blood cell count as well as ferritin, transferrin, cortisol, and adrenocorticotropic hormone levels were unremarkable.
The patient continued to take belumosudil for treatment of GVHD. The hyperpigmentation faded slightly by a 2-month follow-up visit but persisted and was stable. She has not tried other treatments for GVHD to manage the hyperpigmentation.
Conditions known to cause diffuse bronzing of the skin include Addison disease, hemochromatosis, Cushing disease, and medication adverse events. Our patient presented with an absence of systemic symptoms, normal laboratory results, and no clinical indicators suggesting alternate causes. Given that the onset of the hyperpigmentation was 3 weeks after she started a new medication, we hypothesized that the bronzing was an adverse effect of the belumosudil—though this correlation cannot be definitively proven by this case.
The most common offending agents for drug-induced skin hyperpigmentation are nonsteroidal anti- inflammatory drugs, antimalarials, amiodarone, cytotoxic drugs, and tetracyclines.1,2 Our patient’s medication list included the cytotoxic agent acalabrutinib, a Bruton tyrosine kinase inhibitor used for the treatment of non-Hodgkin lymphoma. It has been associated with dermatologic findings of ecchymosis, bruising, panniculitis, and cellulitis, but there are no known reports of hyperpigmentation.3 Our patient had been taking acalabrutinib for 6 months when the GVHD rash developed. At the time, she also was taking a multivitamin and lutein, biotin, and fish oil supplements, none of which have been associated with hyperpigmentation.
Polypharmacy adds a layer of difficulty in identifying the inciting cause of pigmentary change. In our case, symptoms began 3 weeks after the initiation of belumosudil. There were no cutaneous reactions observed in the ROCKstar study of belumosudil; the most common adverse events were upper respiratory tract infection, diarrhea, fatigue, nausea, increased liver enzymes, and dyspnea.4,5 Patients on belumosudil have developed aggressive cutaneous squamous cell carcinoma.6 However, a search of PubMed articles indexed for MEDLINE using the search terms acalabrutinib or belumosudil with hyperpigmentation or cutaneous reaction returned no reports of these medications causing hyperpigmentation or cutaneous deposits.
Treatment of drug-induced hyperpigmentation is difficult because discontinuation of the offending agent typically confirms diagnosis, but interruption of treatment is not always possible, as in our patient. The skin changes can fade over time, but effects typically are long lasting.
Dermatologists play a key role in the identification of drug-induced skin hyperpigmentation. After endocrine or metabolic causes of skin hyperpigmentation have been ruled out, a thorough review of the patient’s medication list should be done to assess for a drug-induced cause. Treatment is limited to sun avoidance, as interruption of treatment may not be possible, and lesions typically do fade over time. These chronic skin changes can have a psychosocial effect on patients and regular follow-up is recommended.
- Giménez García RM, Carrasco Molina S. Drug-induced hyperpigmentation: review and case series. J Am Board Fam Med. 2019;32:628-638. doi:10.3122/jabfm.2019.04.180212
- Dereure O. Drug-induced skin pigmentation. epidemiology, diagnosis and treatment. Am J Clin Dermatol. 2001;2:253-62. doi:10.2165/00128071-200102040-00006
- Sibaud V, Beylot-Barry M, Protin C, et al. Dermatological toxicities of Bruton’s tyrosine kinase inhibitors. Am J Clin Dermatol. 2020; 21:799-812. doi:10.1007/s40257-020-00535-x
- Cutler C, Lee SJ, Arai S, et al. Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study. Blood. 2021;138:2278-2289. doi:10.1182/blood.2021012021
- Jagasia M, Lazaryan A, Bachier CR, et al. ROCK2 inhibition with belumosudil (KD025) for the treatment of chronic graftversus- host disease. J Clin Oncol. 2021;39:1888-1898. doi:10.1200 /JCO.20.02754
- Lee GH, Guzman AK, Divito SJ, et al. Cutaneous squamous-cell carcinoma after treatment with ruxolitinib or belumosudil. N Engl J Med. 2023;389:188-190. doi:10.1056/NEJMc2304157
- Giménez García RM, Carrasco Molina S. Drug-induced hyperpigmentation: review and case series. J Am Board Fam Med. 2019;32:628-638. doi:10.3122/jabfm.2019.04.180212
- Dereure O. Drug-induced skin pigmentation. epidemiology, diagnosis and treatment. Am J Clin Dermatol. 2001;2:253-62. doi:10.2165/00128071-200102040-00006
- Sibaud V, Beylot-Barry M, Protin C, et al. Dermatological toxicities of Bruton’s tyrosine kinase inhibitors. Am J Clin Dermatol. 2020; 21:799-812. doi:10.1007/s40257-020-00535-x
- Cutler C, Lee SJ, Arai S, et al. Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study. Blood. 2021;138:2278-2289. doi:10.1182/blood.2021012021
- Jagasia M, Lazaryan A, Bachier CR, et al. ROCK2 inhibition with belumosudil (KD025) for the treatment of chronic graftversus- host disease. J Clin Oncol. 2021;39:1888-1898. doi:10.1200 /JCO.20.02754
- Lee GH, Guzman AK, Divito SJ, et al. Cutaneous squamous-cell carcinoma after treatment with ruxolitinib or belumosudil. N Engl J Med. 2023;389:188-190. doi:10.1056/NEJMc2304157
Atypical Skin Bronzing in Response to Belumosudil for Graft-vs-Host Disease
Atypical Skin Bronzing in Response to Belumosudil for Graft-vs-Host Disease
PRACTICE POINTS
- Drug-induced hyperpigmentation is a common cause of acquired hyperpigmentation and should be evaluated after metabolic or endocrine causes are ruled out.
- Belumosudil for chronic graft-vs-host disease can induce rapid-onset diffuse bronzing hyperpigmentation, even in the absence of other systemic or laboratory abnormalities.
- Treatment entails discontinuation of the offending agent and limitation of exacerbating factors such as sun exposure.

Actinic Keratosis Treatment With Diclofenac Gel 1%
Actinic Keratosis Treatment With Diclofenac Gel 1%
To the Editor:
Actinic keratoses (AKs) are keratinocyte neoplasms that manifest as rough, scaly, erythematous papules with ill-defined borders (commonly known as precancers) and develop due to long-term UV light exposure.1 They must be treated promptly due to the risk for progression to squamous cell carcinoma (SCC). One US Department of Veterans Affairs study reported that 0.6% of AKs progress to SCC in 1 year and 2.6% progressed to SCC in 4 years.2 In 10% of AKs that will progress to SCC, one study reported progression in approximately 2 years.3
The risk for progression also increases in patients with multiple AKs; the risk is 4-fold higher in patients with 6 to 20 AKs and 11-fold higher in patients with more than 20 AKs.4 Common treatment options include lesion-directed therapies such as cryotherapy, laser therapy, surgery, and curettage, as well as field-directed therapies such as topical 5-fluorouracil (5-FU), diclofenac gel 3%, chemical peeling, topical imiquimod, and photodynamic therapy (PDT).4 When diclofenac gel is chosen as a treatment modality, it is commonly prescribed in the 3% formulation. Diclofenac gel 3% has been shown to be effective in the treatment of AKs,5,6 but diclofenac gel 1% has not been well described in the literature. We report the case of a patient with AKs on the lower legs who was treated with diclofenac gel after other therapies failed.
A 55-year-old woman presented for a routine skin check due to a history of nonmelanoma skin cancer. Her medical history also included palmar hyperhidrosis, disseminated superficial actinic porokeratosis, and extensive actinic damage, as well as numerous biopsy-proven AKs. She had been evaluated every 3 months up to presentation due to the frequency of AK development over the past 5 years. The lesions were mainly localized to both lower legs, where the patient had acquired considerable lifetime sun exposure from tanning beds and sunbathing while boating. She also noted exposure to well water as a child, but none of her family members had a similar issue with AKs.
Prior to this visit, the patient had undergone 5 years of therapy for AKs. She initially was treated with multiple courses of topical 5-FU, but she consequently developed severe allergic contact dermatitis. Subsequent treatments included cryotherapy as well as application of tretinoin cream nightly for 2 weeks followed by PDT. She was unable to tolerate the tretinoin, which she reported led to dryness and irritation. She reported mild improvement after her first session of PDT but only minimal improvement after the next session. Ingenol mebutate was then prescribed for topical use on the legs for 2 days, which did not result in improvement. The patient continued to follow up for unresolved AKs on the legs and was prescribed acitretin to help reduce the risk for progression to SCC. At follow-up 3 months later, she reported decreased soreness from AKs after starting the acitretin and, aside from mild dryness, she tolerated the medication well; however, with continued use of acitretin, she began to experience adverse effects 6 months later, including thyroid suppression and hair loss, leading to discontinuation. Instead, 3 months later, she was recommended to start nicotinamide supplementation for prevention of SCC.
Due to continued AK development (Figure, A), we eventually prescribed diclofenac gel 3% twice daily for both legs 9 months after prescribing nicotinamide. This regimen was cost prohibitive, as the medication was not covered by her insurance and the cost was $300 for one tube. We recommended the patient instead apply the 3% gel to the right leg only due to greater severity of AKs on this leg and over-the-counter diclofenac gel 1% twice daily to the left leg. Approximately 5 months later, she reported a reduction in the discomfort from AKs as well as a reduction in the total number of AKs. She applied the 2 different products as instructed for the first month but did not notice a difference between them. She then continued to apply only the 1% gel on both legs for a total of 8 months with excellent response (Figure, B). At subsequent follow-up visits over a 2-year period, she has only required cryotherapy as spot treatment for AKs.
For 1 to a few discrete AKs, liquid nitrogen cryotherapy is considered first-line therapy.7 However, if multiple AKs are present, surrounding photodamaged skin also should be treated with field-directed therapy due to surrounding keratinocytes bearing a high mutational burden and risk of cancerization.8 Common field-directed therapies include topical 5-FU, topical imiquimod, topical tirbanibulin, PDT, retinoids, and topical diclofenac 3%.
One challenge in field-directed treatment of AKs is the side-effect profile seen in some patients, causing them to prematurely discontinue treatment. In our patient, 5-FU cream, tretinoin cream, and oral acitretin were not well tolerated. Topical diclofenac generally is well tolerated, with mostly mild local skin reactions and low risk for systemic adverse events. Adverse effects mainly consist of mild local skin reactions including pruritus (reported in 31%-52% of patients who used topical diclofenac), dryness (25%-27%), and irritation (less than 1%).9,10 Although diclofenac carries a black-box warning for serious cardiovascular thrombotic events and serious gastrointestinal tract bleeding, systemic absorption of topical diclofenac has been proven to be substantially lower (5- to 17-fold) compared to the oral formulation, and resulting serious adverse effects have been found to be largely reduced compared to the oral formulation.11,12 If allergic contact dermatitis develops, diclofenac should be discontinued.9,13
Diclofenac’s antineoplastic mechanism of action of cyclooxygenase-2 inhibition involves induction of apoptosis as well as reduction in tumor cell proliferation and tumor angiogenesis.14,15 Topical diclofenac may result in decreased levels of lactate and amino acid in AK lesions, particularly in lesions responding to treatment.16 Topical diclofenac may alter immune infiltration by inducing infiltration of dermal CD8+ T cells along with high IFN-γ messenger RNA expression, suggesting improvement of T-cell function after topical diclofenac treatment.16
Although diclofenac gel 3% has been shown to be effective in treatment of AKs,5,6 diclofenac gel 1% has not yet been well studied. Use of the 1% gel is indicated for osteoarthritis and musculoskeletal pain by the US Food and Drug Administration.10,17 Efficacy of the 1% gel has been documented for these and other conditions including seborrheic keratoses.18-20
Because the 1% diclofenac formulation is available over-the-counter, it is more accessible to patients compared to the 3% formulation and often substantially decreases the cost of the medication for the patient. The cost of diclofenac gel 1% in the United States ranges from $0.04 to $0.31 per gram compared to $1.07 to $11.79 per gram for the 3% gel prescription formulation.17 Efficacy of the 1% formulation compared to the 3% formulation could represent an avenue to increase accessibility to field-directed therapy in the population for the treatment of AKs with a potentially well-tolerated, effective, and low-cost medication formulation.
This case represents the effectiveness of diclofenac gel 1% in treating AKs. Several treatment modalities failed in our case, but she experienced improvement with use of over-the-counter diclofenac gel 1%. She also noted no difference in response between the prescription 3% diclofenac formulation and the over-the-counter 1% formulation. Diclofenac gel 1% may represent an excellent therapeutic option in treatment-refractory cases of AKs. Larger randomized trials should be considered to assess safety and efficacy.
- FEisen DB, Asgari MM, Bennett DD, et al. Guidelines of care for the management of actinic keratosis. J Am Acad Dermatol. 2021;85:e209-e233.
- Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
- Fuchs A, Marmur E. The kinetics of skin cancer: progression of actinic keratosis to squamous cell carcinoma. Dermatol Surg. 2007;33: 1099-1101.
- Dianzani C, Conforti C, Giuffrida R, et al. Current therapies for actinic keratosis. Int J Dermatol. 2020;59:677-684.
- Javor S, Cozzani E, Parodi A. Topical treatment of actinic keratosis with 3.0% diclofenac in 2.5% hyaluronan gel: review of the literature about the cumulative evidence of its efficacy and safety. G Ital Dermatol Venereol. 2016;151:275-280.
- Martin GM, Stockfleth E. Diclofenac sodium 3% gel for the management of actinic keratosis: 10+ years of cumulative evidence of efficacy and safety. J Drugs Dermatol. 2012;11:600-608.
- Arisi M, Guasco Pisani E, et al. Cryotherapy for actinic keratosis: basic principles and literature review. Clin Cosmet Investig Dermatol. 2022;15:357-365.
- Calzavara-Pinton P, Calzavara-Pinton I, Rovati C, et al. Topical pharmacotherapy for actinic keratoses in older adults. Drugs Aging. 2022;39:143-152.
- Beutner C, Forkel S, Kreipe K, et al. Contact allergy to topical diclofenac with systemic tolerance. Contact Dermatitis. 2022;86:41-43.
- Voltaren gel (diclofenac sodium topical gel). Prescribing information. Novartis Consumer Health, Inc; 2009. Accessed May 21, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2009/022122s006lbl.pdf
- Moreira SA, Liu DJ. Diclofenac systemic bioavailability of a topical 1% diclofenac + 3% menthol combination gel vs. an oral diclofenac tablet in healthy volunteers: a randomized, open-label, crossover study. Int J Clin Pharmacol Ther. 2017;55:368-372.
- Kienzler JL, Gold M, Nollevaux F. Systemic bioavailability of topical diclofenac sodium gel 1% versus oral diclofenac sodium in healthy volunteers. J Clin Pharmacol. 2010;50:50-61.
- Gulin SJ, Chiriac A. Diclofenac-induced allergic contact dermatitis: a series of four patients. Drug Saf Case Rep. 2016;3:15.
- Fecker LF, Stockfleth E, Nindl I, et al. The role of apoptosis in therapy and prophylaxis of epithelial tumours by nonsteroidal antiinflammatory drugs (NSAIDs). Br J Dermatol. 2007;156(Suppl 3):25-33.
- Thomas GJ, Herranz P, Cruz SB, et al. Treatment of actinic keratosis through inhibition of cyclooxygenase-2: potential mechanism of action of diclofenac sodium 3% in hyaluronic acid 2.5. Dermatol Ther. 2019;32:e12800.
- Singer K, Dettmer K, Unger P, et al. Topical diclofenac reprograms metabolism and immune cell infiltration in actinic keratosis. Front Oncol. 2019;9:605.
- Diclofenac (topical). Drug information. UpToDate. https://www-uptodate-com.libraryaccess.elpaso.ttuhsc.edu/contents/diclofenac-topical-drug-information?source=auto_suggest&selectedTitle=1~3---3~4---diclofenac&search=diclofenac%20topical#F8017265
- Afify AA, Hana MR. Comparative evaluation of topical diclofenac sodium versus topical ibuprofen in the treatment of seborrheic keratosis. Dermatol Ther. 2020;33:e14370.
- Yin F, Ma J, Xiao H, et al. Randomized, double-blind, noninferiority study of diclofenac diethylamine 2.32% gel applied twice daily versus diclofenac diethylamine 1.16% gel applied four times daily in patients with acute ankle sprain. BMC Musculoskelet Disord. 2022;23:1125.
- van Herwaarden N, van den Elsen GAH, de Jong ICA, et al. Topical NSAIDs: ineffective or undervalued? [in Dutch]. Ned Tijdschr Geneeskd. 2021;165:D5317.
To the Editor:
Actinic keratoses (AKs) are keratinocyte neoplasms that manifest as rough, scaly, erythematous papules with ill-defined borders (commonly known as precancers) and develop due to long-term UV light exposure.1 They must be treated promptly due to the risk for progression to squamous cell carcinoma (SCC). One US Department of Veterans Affairs study reported that 0.6% of AKs progress to SCC in 1 year and 2.6% progressed to SCC in 4 years.2 In 10% of AKs that will progress to SCC, one study reported progression in approximately 2 years.3
The risk for progression also increases in patients with multiple AKs; the risk is 4-fold higher in patients with 6 to 20 AKs and 11-fold higher in patients with more than 20 AKs.4 Common treatment options include lesion-directed therapies such as cryotherapy, laser therapy, surgery, and curettage, as well as field-directed therapies such as topical 5-fluorouracil (5-FU), diclofenac gel 3%, chemical peeling, topical imiquimod, and photodynamic therapy (PDT).4 When diclofenac gel is chosen as a treatment modality, it is commonly prescribed in the 3% formulation. Diclofenac gel 3% has been shown to be effective in the treatment of AKs,5,6 but diclofenac gel 1% has not been well described in the literature. We report the case of a patient with AKs on the lower legs who was treated with diclofenac gel after other therapies failed.
A 55-year-old woman presented for a routine skin check due to a history of nonmelanoma skin cancer. Her medical history also included palmar hyperhidrosis, disseminated superficial actinic porokeratosis, and extensive actinic damage, as well as numerous biopsy-proven AKs. She had been evaluated every 3 months up to presentation due to the frequency of AK development over the past 5 years. The lesions were mainly localized to both lower legs, where the patient had acquired considerable lifetime sun exposure from tanning beds and sunbathing while boating. She also noted exposure to well water as a child, but none of her family members had a similar issue with AKs.
Prior to this visit, the patient had undergone 5 years of therapy for AKs. She initially was treated with multiple courses of topical 5-FU, but she consequently developed severe allergic contact dermatitis. Subsequent treatments included cryotherapy as well as application of tretinoin cream nightly for 2 weeks followed by PDT. She was unable to tolerate the tretinoin, which she reported led to dryness and irritation. She reported mild improvement after her first session of PDT but only minimal improvement after the next session. Ingenol mebutate was then prescribed for topical use on the legs for 2 days, which did not result in improvement. The patient continued to follow up for unresolved AKs on the legs and was prescribed acitretin to help reduce the risk for progression to SCC. At follow-up 3 months later, she reported decreased soreness from AKs after starting the acitretin and, aside from mild dryness, she tolerated the medication well; however, with continued use of acitretin, she began to experience adverse effects 6 months later, including thyroid suppression and hair loss, leading to discontinuation. Instead, 3 months later, she was recommended to start nicotinamide supplementation for prevention of SCC.
Due to continued AK development (Figure, A), we eventually prescribed diclofenac gel 3% twice daily for both legs 9 months after prescribing nicotinamide. This regimen was cost prohibitive, as the medication was not covered by her insurance and the cost was $300 for one tube. We recommended the patient instead apply the 3% gel to the right leg only due to greater severity of AKs on this leg and over-the-counter diclofenac gel 1% twice daily to the left leg. Approximately 5 months later, she reported a reduction in the discomfort from AKs as well as a reduction in the total number of AKs. She applied the 2 different products as instructed for the first month but did not notice a difference between them. She then continued to apply only the 1% gel on both legs for a total of 8 months with excellent response (Figure, B). At subsequent follow-up visits over a 2-year period, she has only required cryotherapy as spot treatment for AKs.
For 1 to a few discrete AKs, liquid nitrogen cryotherapy is considered first-line therapy.7 However, if multiple AKs are present, surrounding photodamaged skin also should be treated with field-directed therapy due to surrounding keratinocytes bearing a high mutational burden and risk of cancerization.8 Common field-directed therapies include topical 5-FU, topical imiquimod, topical tirbanibulin, PDT, retinoids, and topical diclofenac 3%.
One challenge in field-directed treatment of AKs is the side-effect profile seen in some patients, causing them to prematurely discontinue treatment. In our patient, 5-FU cream, tretinoin cream, and oral acitretin were not well tolerated. Topical diclofenac generally is well tolerated, with mostly mild local skin reactions and low risk for systemic adverse events. Adverse effects mainly consist of mild local skin reactions including pruritus (reported in 31%-52% of patients who used topical diclofenac), dryness (25%-27%), and irritation (less than 1%).9,10 Although diclofenac carries a black-box warning for serious cardiovascular thrombotic events and serious gastrointestinal tract bleeding, systemic absorption of topical diclofenac has been proven to be substantially lower (5- to 17-fold) compared to the oral formulation, and resulting serious adverse effects have been found to be largely reduced compared to the oral formulation.11,12 If allergic contact dermatitis develops, diclofenac should be discontinued.9,13
Diclofenac’s antineoplastic mechanism of action of cyclooxygenase-2 inhibition involves induction of apoptosis as well as reduction in tumor cell proliferation and tumor angiogenesis.14,15 Topical diclofenac may result in decreased levels of lactate and amino acid in AK lesions, particularly in lesions responding to treatment.16 Topical diclofenac may alter immune infiltration by inducing infiltration of dermal CD8+ T cells along with high IFN-γ messenger RNA expression, suggesting improvement of T-cell function after topical diclofenac treatment.16
Although diclofenac gel 3% has been shown to be effective in treatment of AKs,5,6 diclofenac gel 1% has not yet been well studied. Use of the 1% gel is indicated for osteoarthritis and musculoskeletal pain by the US Food and Drug Administration.10,17 Efficacy of the 1% gel has been documented for these and other conditions including seborrheic keratoses.18-20
Because the 1% diclofenac formulation is available over-the-counter, it is more accessible to patients compared to the 3% formulation and often substantially decreases the cost of the medication for the patient. The cost of diclofenac gel 1% in the United States ranges from $0.04 to $0.31 per gram compared to $1.07 to $11.79 per gram for the 3% gel prescription formulation.17 Efficacy of the 1% formulation compared to the 3% formulation could represent an avenue to increase accessibility to field-directed therapy in the population for the treatment of AKs with a potentially well-tolerated, effective, and low-cost medication formulation.
This case represents the effectiveness of diclofenac gel 1% in treating AKs. Several treatment modalities failed in our case, but she experienced improvement with use of over-the-counter diclofenac gel 1%. She also noted no difference in response between the prescription 3% diclofenac formulation and the over-the-counter 1% formulation. Diclofenac gel 1% may represent an excellent therapeutic option in treatment-refractory cases of AKs. Larger randomized trials should be considered to assess safety and efficacy.
To the Editor:
Actinic keratoses (AKs) are keratinocyte neoplasms that manifest as rough, scaly, erythematous papules with ill-defined borders (commonly known as precancers) and develop due to long-term UV light exposure.1 They must be treated promptly due to the risk for progression to squamous cell carcinoma (SCC). One US Department of Veterans Affairs study reported that 0.6% of AKs progress to SCC in 1 year and 2.6% progressed to SCC in 4 years.2 In 10% of AKs that will progress to SCC, one study reported progression in approximately 2 years.3
The risk for progression also increases in patients with multiple AKs; the risk is 4-fold higher in patients with 6 to 20 AKs and 11-fold higher in patients with more than 20 AKs.4 Common treatment options include lesion-directed therapies such as cryotherapy, laser therapy, surgery, and curettage, as well as field-directed therapies such as topical 5-fluorouracil (5-FU), diclofenac gel 3%, chemical peeling, topical imiquimod, and photodynamic therapy (PDT).4 When diclofenac gel is chosen as a treatment modality, it is commonly prescribed in the 3% formulation. Diclofenac gel 3% has been shown to be effective in the treatment of AKs,5,6 but diclofenac gel 1% has not been well described in the literature. We report the case of a patient with AKs on the lower legs who was treated with diclofenac gel after other therapies failed.
A 55-year-old woman presented for a routine skin check due to a history of nonmelanoma skin cancer. Her medical history also included palmar hyperhidrosis, disseminated superficial actinic porokeratosis, and extensive actinic damage, as well as numerous biopsy-proven AKs. She had been evaluated every 3 months up to presentation due to the frequency of AK development over the past 5 years. The lesions were mainly localized to both lower legs, where the patient had acquired considerable lifetime sun exposure from tanning beds and sunbathing while boating. She also noted exposure to well water as a child, but none of her family members had a similar issue with AKs.
Prior to this visit, the patient had undergone 5 years of therapy for AKs. She initially was treated with multiple courses of topical 5-FU, but she consequently developed severe allergic contact dermatitis. Subsequent treatments included cryotherapy as well as application of tretinoin cream nightly for 2 weeks followed by PDT. She was unable to tolerate the tretinoin, which she reported led to dryness and irritation. She reported mild improvement after her first session of PDT but only minimal improvement after the next session. Ingenol mebutate was then prescribed for topical use on the legs for 2 days, which did not result in improvement. The patient continued to follow up for unresolved AKs on the legs and was prescribed acitretin to help reduce the risk for progression to SCC. At follow-up 3 months later, she reported decreased soreness from AKs after starting the acitretin and, aside from mild dryness, she tolerated the medication well; however, with continued use of acitretin, she began to experience adverse effects 6 months later, including thyroid suppression and hair loss, leading to discontinuation. Instead, 3 months later, she was recommended to start nicotinamide supplementation for prevention of SCC.
Due to continued AK development (Figure, A), we eventually prescribed diclofenac gel 3% twice daily for both legs 9 months after prescribing nicotinamide. This regimen was cost prohibitive, as the medication was not covered by her insurance and the cost was $300 for one tube. We recommended the patient instead apply the 3% gel to the right leg only due to greater severity of AKs on this leg and over-the-counter diclofenac gel 1% twice daily to the left leg. Approximately 5 months later, she reported a reduction in the discomfort from AKs as well as a reduction in the total number of AKs. She applied the 2 different products as instructed for the first month but did not notice a difference between them. She then continued to apply only the 1% gel on both legs for a total of 8 months with excellent response (Figure, B). At subsequent follow-up visits over a 2-year period, she has only required cryotherapy as spot treatment for AKs.
For 1 to a few discrete AKs, liquid nitrogen cryotherapy is considered first-line therapy.7 However, if multiple AKs are present, surrounding photodamaged skin also should be treated with field-directed therapy due to surrounding keratinocytes bearing a high mutational burden and risk of cancerization.8 Common field-directed therapies include topical 5-FU, topical imiquimod, topical tirbanibulin, PDT, retinoids, and topical diclofenac 3%.
One challenge in field-directed treatment of AKs is the side-effect profile seen in some patients, causing them to prematurely discontinue treatment. In our patient, 5-FU cream, tretinoin cream, and oral acitretin were not well tolerated. Topical diclofenac generally is well tolerated, with mostly mild local skin reactions and low risk for systemic adverse events. Adverse effects mainly consist of mild local skin reactions including pruritus (reported in 31%-52% of patients who used topical diclofenac), dryness (25%-27%), and irritation (less than 1%).9,10 Although diclofenac carries a black-box warning for serious cardiovascular thrombotic events and serious gastrointestinal tract bleeding, systemic absorption of topical diclofenac has been proven to be substantially lower (5- to 17-fold) compared to the oral formulation, and resulting serious adverse effects have been found to be largely reduced compared to the oral formulation.11,12 If allergic contact dermatitis develops, diclofenac should be discontinued.9,13
Diclofenac’s antineoplastic mechanism of action of cyclooxygenase-2 inhibition involves induction of apoptosis as well as reduction in tumor cell proliferation and tumor angiogenesis.14,15 Topical diclofenac may result in decreased levels of lactate and amino acid in AK lesions, particularly in lesions responding to treatment.16 Topical diclofenac may alter immune infiltration by inducing infiltration of dermal CD8+ T cells along with high IFN-γ messenger RNA expression, suggesting improvement of T-cell function after topical diclofenac treatment.16
Although diclofenac gel 3% has been shown to be effective in treatment of AKs,5,6 diclofenac gel 1% has not yet been well studied. Use of the 1% gel is indicated for osteoarthritis and musculoskeletal pain by the US Food and Drug Administration.10,17 Efficacy of the 1% gel has been documented for these and other conditions including seborrheic keratoses.18-20
Because the 1% diclofenac formulation is available over-the-counter, it is more accessible to patients compared to the 3% formulation and often substantially decreases the cost of the medication for the patient. The cost of diclofenac gel 1% in the United States ranges from $0.04 to $0.31 per gram compared to $1.07 to $11.79 per gram for the 3% gel prescription formulation.17 Efficacy of the 1% formulation compared to the 3% formulation could represent an avenue to increase accessibility to field-directed therapy in the population for the treatment of AKs with a potentially well-tolerated, effective, and low-cost medication formulation.
This case represents the effectiveness of diclofenac gel 1% in treating AKs. Several treatment modalities failed in our case, but she experienced improvement with use of over-the-counter diclofenac gel 1%. She also noted no difference in response between the prescription 3% diclofenac formulation and the over-the-counter 1% formulation. Diclofenac gel 1% may represent an excellent therapeutic option in treatment-refractory cases of AKs. Larger randomized trials should be considered to assess safety and efficacy.
- FEisen DB, Asgari MM, Bennett DD, et al. Guidelines of care for the management of actinic keratosis. J Am Acad Dermatol. 2021;85:e209-e233.
- Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
- Fuchs A, Marmur E. The kinetics of skin cancer: progression of actinic keratosis to squamous cell carcinoma. Dermatol Surg. 2007;33: 1099-1101.
- Dianzani C, Conforti C, Giuffrida R, et al. Current therapies for actinic keratosis. Int J Dermatol. 2020;59:677-684.
- Javor S, Cozzani E, Parodi A. Topical treatment of actinic keratosis with 3.0% diclofenac in 2.5% hyaluronan gel: review of the literature about the cumulative evidence of its efficacy and safety. G Ital Dermatol Venereol. 2016;151:275-280.
- Martin GM, Stockfleth E. Diclofenac sodium 3% gel for the management of actinic keratosis: 10+ years of cumulative evidence of efficacy and safety. J Drugs Dermatol. 2012;11:600-608.
- Arisi M, Guasco Pisani E, et al. Cryotherapy for actinic keratosis: basic principles and literature review. Clin Cosmet Investig Dermatol. 2022;15:357-365.
- Calzavara-Pinton P, Calzavara-Pinton I, Rovati C, et al. Topical pharmacotherapy for actinic keratoses in older adults. Drugs Aging. 2022;39:143-152.
- Beutner C, Forkel S, Kreipe K, et al. Contact allergy to topical diclofenac with systemic tolerance. Contact Dermatitis. 2022;86:41-43.
- Voltaren gel (diclofenac sodium topical gel). Prescribing information. Novartis Consumer Health, Inc; 2009. Accessed May 21, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2009/022122s006lbl.pdf
- Moreira SA, Liu DJ. Diclofenac systemic bioavailability of a topical 1% diclofenac + 3% menthol combination gel vs. an oral diclofenac tablet in healthy volunteers: a randomized, open-label, crossover study. Int J Clin Pharmacol Ther. 2017;55:368-372.
- Kienzler JL, Gold M, Nollevaux F. Systemic bioavailability of topical diclofenac sodium gel 1% versus oral diclofenac sodium in healthy volunteers. J Clin Pharmacol. 2010;50:50-61.
- Gulin SJ, Chiriac A. Diclofenac-induced allergic contact dermatitis: a series of four patients. Drug Saf Case Rep. 2016;3:15.
- Fecker LF, Stockfleth E, Nindl I, et al. The role of apoptosis in therapy and prophylaxis of epithelial tumours by nonsteroidal antiinflammatory drugs (NSAIDs). Br J Dermatol. 2007;156(Suppl 3):25-33.
- Thomas GJ, Herranz P, Cruz SB, et al. Treatment of actinic keratosis through inhibition of cyclooxygenase-2: potential mechanism of action of diclofenac sodium 3% in hyaluronic acid 2.5. Dermatol Ther. 2019;32:e12800.
- Singer K, Dettmer K, Unger P, et al. Topical diclofenac reprograms metabolism and immune cell infiltration in actinic keratosis. Front Oncol. 2019;9:605.
- Diclofenac (topical). Drug information. UpToDate. https://www-uptodate-com.libraryaccess.elpaso.ttuhsc.edu/contents/diclofenac-topical-drug-information?source=auto_suggest&selectedTitle=1~3---3~4---diclofenac&search=diclofenac%20topical#F8017265
- Afify AA, Hana MR. Comparative evaluation of topical diclofenac sodium versus topical ibuprofen in the treatment of seborrheic keratosis. Dermatol Ther. 2020;33:e14370.
- Yin F, Ma J, Xiao H, et al. Randomized, double-blind, noninferiority study of diclofenac diethylamine 2.32% gel applied twice daily versus diclofenac diethylamine 1.16% gel applied four times daily in patients with acute ankle sprain. BMC Musculoskelet Disord. 2022;23:1125.
- van Herwaarden N, van den Elsen GAH, de Jong ICA, et al. Topical NSAIDs: ineffective or undervalued? [in Dutch]. Ned Tijdschr Geneeskd. 2021;165:D5317.
- FEisen DB, Asgari MM, Bennett DD, et al. Guidelines of care for the management of actinic keratosis. J Am Acad Dermatol. 2021;85:e209-e233.
- Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
- Fuchs A, Marmur E. The kinetics of skin cancer: progression of actinic keratosis to squamous cell carcinoma. Dermatol Surg. 2007;33: 1099-1101.
- Dianzani C, Conforti C, Giuffrida R, et al. Current therapies for actinic keratosis. Int J Dermatol. 2020;59:677-684.
- Javor S, Cozzani E, Parodi A. Topical treatment of actinic keratosis with 3.0% diclofenac in 2.5% hyaluronan gel: review of the literature about the cumulative evidence of its efficacy and safety. G Ital Dermatol Venereol. 2016;151:275-280.
- Martin GM, Stockfleth E. Diclofenac sodium 3% gel for the management of actinic keratosis: 10+ years of cumulative evidence of efficacy and safety. J Drugs Dermatol. 2012;11:600-608.
- Arisi M, Guasco Pisani E, et al. Cryotherapy for actinic keratosis: basic principles and literature review. Clin Cosmet Investig Dermatol. 2022;15:357-365.
- Calzavara-Pinton P, Calzavara-Pinton I, Rovati C, et al. Topical pharmacotherapy for actinic keratoses in older adults. Drugs Aging. 2022;39:143-152.
- Beutner C, Forkel S, Kreipe K, et al. Contact allergy to topical diclofenac with systemic tolerance. Contact Dermatitis. 2022;86:41-43.
- Voltaren gel (diclofenac sodium topical gel). Prescribing information. Novartis Consumer Health, Inc; 2009. Accessed May 21, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2009/022122s006lbl.pdf
- Moreira SA, Liu DJ. Diclofenac systemic bioavailability of a topical 1% diclofenac + 3% menthol combination gel vs. an oral diclofenac tablet in healthy volunteers: a randomized, open-label, crossover study. Int J Clin Pharmacol Ther. 2017;55:368-372.
- Kienzler JL, Gold M, Nollevaux F. Systemic bioavailability of topical diclofenac sodium gel 1% versus oral diclofenac sodium in healthy volunteers. J Clin Pharmacol. 2010;50:50-61.
- Gulin SJ, Chiriac A. Diclofenac-induced allergic contact dermatitis: a series of four patients. Drug Saf Case Rep. 2016;3:15.
- Fecker LF, Stockfleth E, Nindl I, et al. The role of apoptosis in therapy and prophylaxis of epithelial tumours by nonsteroidal antiinflammatory drugs (NSAIDs). Br J Dermatol. 2007;156(Suppl 3):25-33.
- Thomas GJ, Herranz P, Cruz SB, et al. Treatment of actinic keratosis through inhibition of cyclooxygenase-2: potential mechanism of action of diclofenac sodium 3% in hyaluronic acid 2.5. Dermatol Ther. 2019;32:e12800.
- Singer K, Dettmer K, Unger P, et al. Topical diclofenac reprograms metabolism and immune cell infiltration in actinic keratosis. Front Oncol. 2019;9:605.
- Diclofenac (topical). Drug information. UpToDate. https://www-uptodate-com.libraryaccess.elpaso.ttuhsc.edu/contents/diclofenac-topical-drug-information?source=auto_suggest&selectedTitle=1~3---3~4---diclofenac&search=diclofenac%20topical#F8017265
- Afify AA, Hana MR. Comparative evaluation of topical diclofenac sodium versus topical ibuprofen in the treatment of seborrheic keratosis. Dermatol Ther. 2020;33:e14370.
- Yin F, Ma J, Xiao H, et al. Randomized, double-blind, noninferiority study of diclofenac diethylamine 2.32% gel applied twice daily versus diclofenac diethylamine 1.16% gel applied four times daily in patients with acute ankle sprain. BMC Musculoskelet Disord. 2022;23:1125.
- van Herwaarden N, van den Elsen GAH, de Jong ICA, et al. Topical NSAIDs: ineffective or undervalued? [in Dutch]. Ned Tijdschr Geneeskd. 2021;165:D5317.
Actinic Keratosis Treatment With Diclofenac Gel 1%
Actinic Keratosis Treatment With Diclofenac Gel 1%
PRACTICE POINTS
- There are numerous field-directed therapies for actinic keratoses (AKs); however, efficacy and tolerability vary among the available treatments.
- Diclofenac gel 1% is an affordable option that could potentially increase accessibility and decrease cost of field therapy for the treatment of AKs, while maintaining therapeutic efficacy.

Low-Dose Oral Naltrexone for Darier Disease
To the Editor:
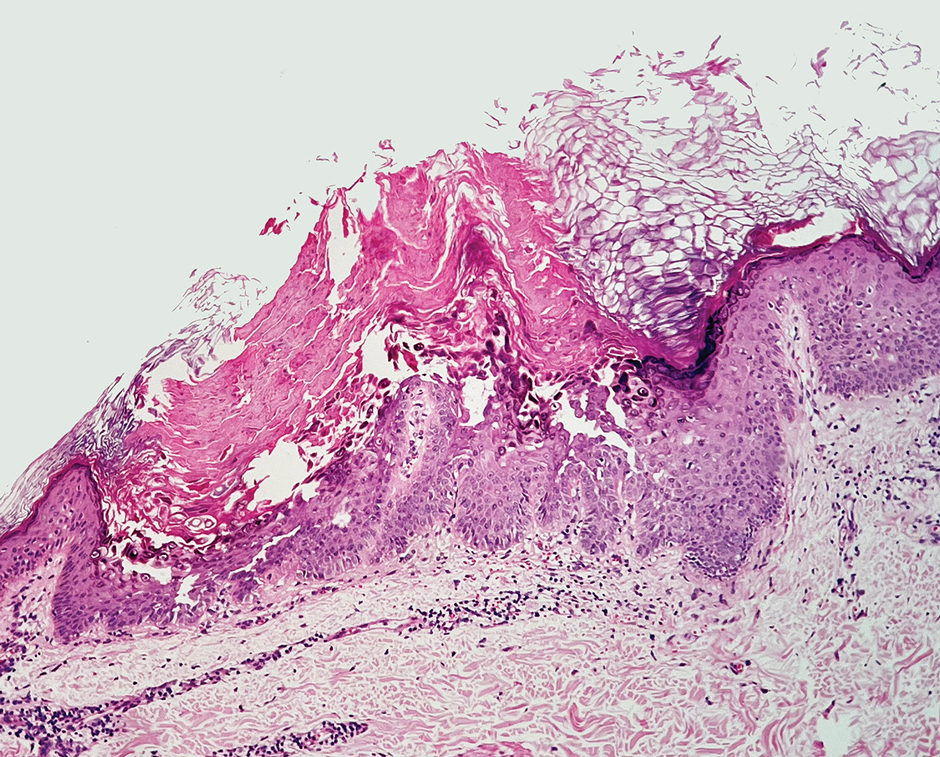
A 34-year-old Brazilian woman presented to the dermatology department with pruritic lesions on the neck and chest that had been present since adolescence. She reported a family history of Darier disease in her father. Physical examination revealed erythematous follicular papules on the neck, inframammary region, and abdomen (Figure 1A), as well as longitudinal bandlike leukonychia and distal nail splits on the fingernails (Figure 1B). Histopathology of a lesion on the back revealed compact hyperkeratosis and parakeratosis above an acantholytic cleft accompanied by dyskeratotic keratinocytes, including some corps ronds and grains, which supported the clinical impression of Darier disease (Figure 2). The typical clinical presentation along with the family history and histopathology confirmed the diagnosis. After therapeutic failure with topical corticosteroids and oral antibiotics for 3 months, low-dose oral naltrexone (4.5 mg/d) as monotherapy noticeably improved the lesions and pruritus within 2 months, with near-complete regression at 6 months, achieving disease stability (Figures 1C and 1D). The patient remained stable with no recurrence after 1 year of follow-up.
Darier disease is an autosomal-dominant genodermatosis caused by a mutation in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum calcium ATPase, leading to defective intracellular calcium signaling and alterations in epidermal adhesion and keratinization.1 Darier disease typically begins in adolescence and is aggravated by exposure to heat and friction. It is characterized by seborrheic distribution of painful and pruritic red-brown keratotic papules. Nail manifestations include longitudinal ridges—erythronychia and/or leukonychia—and grooves that end in a V-shaped notch. The differential diagnosis includes Hailey-Hailey disease, psoriasis, and pityriasis rubra pilaris.1,2 The diagnosis is clinical and is confirmed by histopathology, which reveals suprabasal cleavage, acantholytic dyskeratosis, corps ronds, and grains. Treatment options are limited and include corticosteroids, oral and/or topical antibiotics, and systemic retinoids.2
Oral naltrexone has been used in Darier disease based on its observed effectiveness in Hailey-Hailey disease, considering the histopathologic similarities and alterations in calcium homeostasis in both conditions. Low-dose oral naltrexone (1-5 mg/d) increases the expression of opioid receptors (δ, μ, κ), enhancing its immunomodulatory and antinociceptive effects. The δ opioid receptor regulates the expression of desmoglein, improving epidermal differentiation and wound healing.3 Activation of the δ and μ receptors increases intracellular calcium through the inositol phosphate pathway, which contributes to calcium homeostasis.4 Naltrexone blocks the nonopioid toll-like receptor 4 found in keratinocytes and macrophages, exerting an anti-inflammatory effect by reducing proinflammatory cytokines.3 Adverse events associated with low-dose naltrexone are minimal, mostly mild, and often related to sleep disorders3,5; however, patients should undergo screening for prior opioid dependence, recent opioid usage, and signs of opioid withdrawal before initiating naltrexone treatment.5
Boehmer et al6 used naltrexone (4.5 mg/d) and oral magnesium (200 mg/d) in 6 patients with inconsistent results, except for 1 case that concurrently used acitretin (25 mg/d) with satisfactory improvement. Pessoa et al7 added naltrexone (4.5 mg/d) to oral isotretinoin (0.5 mg/kg/d) in 1 patient, resulting in notable improvement of lesions within 3 months.
In our patient with Darier disease, low-dose naltrexone demonstrated a substantial response as monotherapy after 2 months of treatment and nearly complete regression of lesions within 6 months, with no reported side effects after 1 year of follow-up. The use of low-dose naltrexone could be a promising and safe treatment option as monotherapy or in combination with conventional therapy for Darier disease; however, further studies are needed.
Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271-277. doi:10.1038/6784
Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50. doi:10.1016/0190-9622(92)70154-8
Lee B, Elston DM. The uses of naltrexone in dermatologic conditions. Am Acad Dermatol. 2019;80:1746-1752. doi:10.1016/j.jaad.2018.12.031
Samways DSK, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151-161. doi:10.1016/j.cellsig.2005.08.005
Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236. doi:10.1001/jamadermatol.2018.4093
Boehmer D, Eyerich K, Darsow U, et al. Variable response to low‐dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol. 2019;33:950-953. doi:10.1111/jdv.15457
Pessoa T, Rebelo C, Gabriela Marques Pinto, et al. Combination of naltrexone and isotretinoin for the treatment of Darier disease. Cureus. 2023;15:E33321. doi:10.7759/cureus.33321
To the Editor:
A 34-year-old Brazilian woman presented to the dermatology department with pruritic lesions on the neck and chest that had been present since adolescence. She reported a family history of Darier disease in her father. Physical examination revealed erythematous follicular papules on the neck, inframammary region, and abdomen (Figure 1A), as well as longitudinal bandlike leukonychia and distal nail splits on the fingernails (Figure 1B). Histopathology of a lesion on the back revealed compact hyperkeratosis and parakeratosis above an acantholytic cleft accompanied by dyskeratotic keratinocytes, including some corps ronds and grains, which supported the clinical impression of Darier disease (Figure 2). The typical clinical presentation along with the family history and histopathology confirmed the diagnosis. After therapeutic failure with topical corticosteroids and oral antibiotics for 3 months, low-dose oral naltrexone (4.5 mg/d) as monotherapy noticeably improved the lesions and pruritus within 2 months, with near-complete regression at 6 months, achieving disease stability (Figures 1C and 1D). The patient remained stable with no recurrence after 1 year of follow-up.
Darier disease is an autosomal-dominant genodermatosis caused by a mutation in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum calcium ATPase, leading to defective intracellular calcium signaling and alterations in epidermal adhesion and keratinization.1 Darier disease typically begins in adolescence and is aggravated by exposure to heat and friction. It is characterized by seborrheic distribution of painful and pruritic red-brown keratotic papules. Nail manifestations include longitudinal ridges—erythronychia and/or leukonychia—and grooves that end in a V-shaped notch. The differential diagnosis includes Hailey-Hailey disease, psoriasis, and pityriasis rubra pilaris.1,2 The diagnosis is clinical and is confirmed by histopathology, which reveals suprabasal cleavage, acantholytic dyskeratosis, corps ronds, and grains. Treatment options are limited and include corticosteroids, oral and/or topical antibiotics, and systemic retinoids.2
Oral naltrexone has been used in Darier disease based on its observed effectiveness in Hailey-Hailey disease, considering the histopathologic similarities and alterations in calcium homeostasis in both conditions. Low-dose oral naltrexone (1-5 mg/d) increases the expression of opioid receptors (δ, μ, κ), enhancing its immunomodulatory and antinociceptive effects. The δ opioid receptor regulates the expression of desmoglein, improving epidermal differentiation and wound healing.3 Activation of the δ and μ receptors increases intracellular calcium through the inositol phosphate pathway, which contributes to calcium homeostasis.4 Naltrexone blocks the nonopioid toll-like receptor 4 found in keratinocytes and macrophages, exerting an anti-inflammatory effect by reducing proinflammatory cytokines.3 Adverse events associated with low-dose naltrexone are minimal, mostly mild, and often related to sleep disorders3,5; however, patients should undergo screening for prior opioid dependence, recent opioid usage, and signs of opioid withdrawal before initiating naltrexone treatment.5
Boehmer et al6 used naltrexone (4.5 mg/d) and oral magnesium (200 mg/d) in 6 patients with inconsistent results, except for 1 case that concurrently used acitretin (25 mg/d) with satisfactory improvement. Pessoa et al7 added naltrexone (4.5 mg/d) to oral isotretinoin (0.5 mg/kg/d) in 1 patient, resulting in notable improvement of lesions within 3 months.
In our patient with Darier disease, low-dose naltrexone demonstrated a substantial response as monotherapy after 2 months of treatment and nearly complete regression of lesions within 6 months, with no reported side effects after 1 year of follow-up. The use of low-dose naltrexone could be a promising and safe treatment option as monotherapy or in combination with conventional therapy for Darier disease; however, further studies are needed.
To the Editor:
A 34-year-old Brazilian woman presented to the dermatology department with pruritic lesions on the neck and chest that had been present since adolescence. She reported a family history of Darier disease in her father. Physical examination revealed erythematous follicular papules on the neck, inframammary region, and abdomen (Figure 1A), as well as longitudinal bandlike leukonychia and distal nail splits on the fingernails (Figure 1B). Histopathology of a lesion on the back revealed compact hyperkeratosis and parakeratosis above an acantholytic cleft accompanied by dyskeratotic keratinocytes, including some corps ronds and grains, which supported the clinical impression of Darier disease (Figure 2). The typical clinical presentation along with the family history and histopathology confirmed the diagnosis. After therapeutic failure with topical corticosteroids and oral antibiotics for 3 months, low-dose oral naltrexone (4.5 mg/d) as monotherapy noticeably improved the lesions and pruritus within 2 months, with near-complete regression at 6 months, achieving disease stability (Figures 1C and 1D). The patient remained stable with no recurrence after 1 year of follow-up.
Darier disease is an autosomal-dominant genodermatosis caused by a mutation in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum calcium ATPase, leading to defective intracellular calcium signaling and alterations in epidermal adhesion and keratinization.1 Darier disease typically begins in adolescence and is aggravated by exposure to heat and friction. It is characterized by seborrheic distribution of painful and pruritic red-brown keratotic papules. Nail manifestations include longitudinal ridges—erythronychia and/or leukonychia—and grooves that end in a V-shaped notch. The differential diagnosis includes Hailey-Hailey disease, psoriasis, and pityriasis rubra pilaris.1,2 The diagnosis is clinical and is confirmed by histopathology, which reveals suprabasal cleavage, acantholytic dyskeratosis, corps ronds, and grains. Treatment options are limited and include corticosteroids, oral and/or topical antibiotics, and systemic retinoids.2
Oral naltrexone has been used in Darier disease based on its observed effectiveness in Hailey-Hailey disease, considering the histopathologic similarities and alterations in calcium homeostasis in both conditions. Low-dose oral naltrexone (1-5 mg/d) increases the expression of opioid receptors (δ, μ, κ), enhancing its immunomodulatory and antinociceptive effects. The δ opioid receptor regulates the expression of desmoglein, improving epidermal differentiation and wound healing.3 Activation of the δ and μ receptors increases intracellular calcium through the inositol phosphate pathway, which contributes to calcium homeostasis.4 Naltrexone blocks the nonopioid toll-like receptor 4 found in keratinocytes and macrophages, exerting an anti-inflammatory effect by reducing proinflammatory cytokines.3 Adverse events associated with low-dose naltrexone are minimal, mostly mild, and often related to sleep disorders3,5; however, patients should undergo screening for prior opioid dependence, recent opioid usage, and signs of opioid withdrawal before initiating naltrexone treatment.5
Boehmer et al6 used naltrexone (4.5 mg/d) and oral magnesium (200 mg/d) in 6 patients with inconsistent results, except for 1 case that concurrently used acitretin (25 mg/d) with satisfactory improvement. Pessoa et al7 added naltrexone (4.5 mg/d) to oral isotretinoin (0.5 mg/kg/d) in 1 patient, resulting in notable improvement of lesions within 3 months.
In our patient with Darier disease, low-dose naltrexone demonstrated a substantial response as monotherapy after 2 months of treatment and nearly complete regression of lesions within 6 months, with no reported side effects after 1 year of follow-up. The use of low-dose naltrexone could be a promising and safe treatment option as monotherapy or in combination with conventional therapy for Darier disease; however, further studies are needed.
Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271-277. doi:10.1038/6784
Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50. doi:10.1016/0190-9622(92)70154-8
Lee B, Elston DM. The uses of naltrexone in dermatologic conditions. Am Acad Dermatol. 2019;80:1746-1752. doi:10.1016/j.jaad.2018.12.031
Samways DSK, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151-161. doi:10.1016/j.cellsig.2005.08.005
Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236. doi:10.1001/jamadermatol.2018.4093
Boehmer D, Eyerich K, Darsow U, et al. Variable response to low‐dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol. 2019;33:950-953. doi:10.1111/jdv.15457
Pessoa T, Rebelo C, Gabriela Marques Pinto, et al. Combination of naltrexone and isotretinoin for the treatment of Darier disease. Cureus. 2023;15:E33321. doi:10.7759/cureus.33321
Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271-277. doi:10.1038/6784
Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50. doi:10.1016/0190-9622(92)70154-8
Lee B, Elston DM. The uses of naltrexone in dermatologic conditions. Am Acad Dermatol. 2019;80:1746-1752. doi:10.1016/j.jaad.2018.12.031
Samways DSK, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151-161. doi:10.1016/j.cellsig.2005.08.005
Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236. doi:10.1001/jamadermatol.2018.4093
Boehmer D, Eyerich K, Darsow U, et al. Variable response to low‐dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol. 2019;33:950-953. doi:10.1111/jdv.15457
Pessoa T, Rebelo C, Gabriela Marques Pinto, et al. Combination of naltrexone and isotretinoin for the treatment of Darier disease. Cureus. 2023;15:E33321. doi:10.7759/cureus.33321
Practice Points
- Consider low-dose naltrexone as a potential treatment option for patients with Darier disease, as it regulates opioid receptors and has shown benefits in enhancing epidermal differentiation, wound healing, and anti-inflammatory effects.
- Further research is needed to validate the efficacy and safety of low-dose naltrexone in treating Darier disease considering its observed clinical improvement in this single patient case.
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
To the Editor:
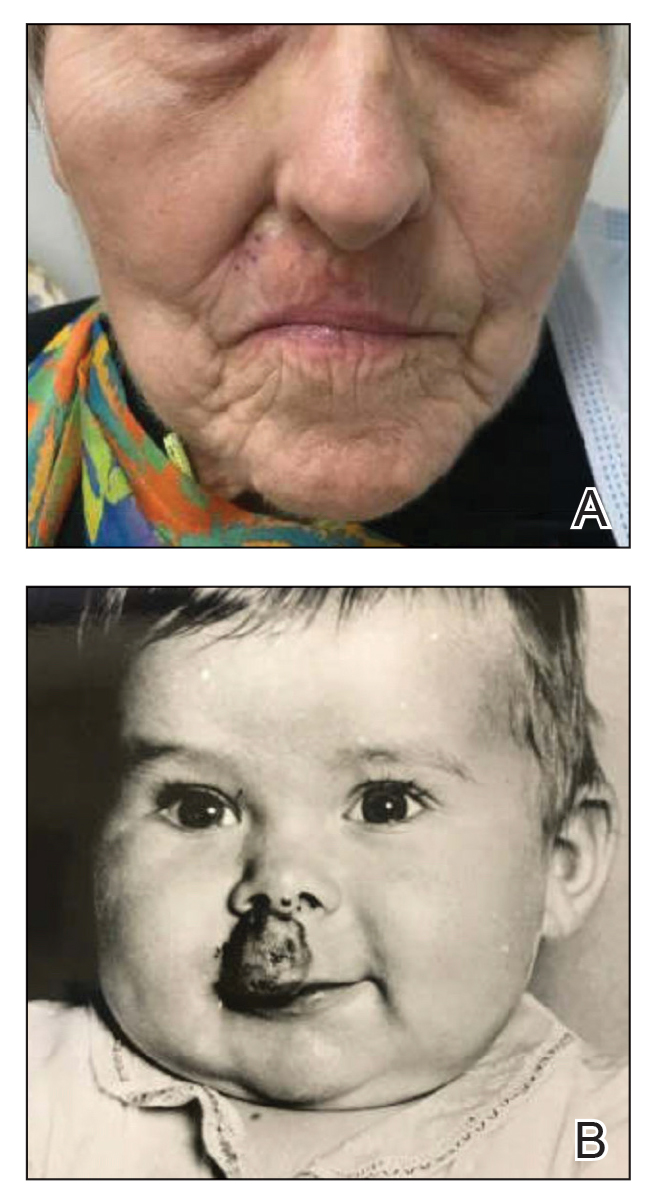
Basal cell carcinoma (BCC), which is the most common type of skin cancer, typically arises on sun-damaged skin as a result of long-term exposure to UV radiation. Another known risk factor for BCC is exposure to ionizing radiation, though this is less commonly encountered.1 We present a unique case of a BCC arising at the site of an involuted infantile hemangioma that had been treated with implanted and retained gold radon seeds more than 7 decades prior. This case highlights the importance of obtaining a detailed history of radiation exposures to better counsel patients about skin cancer risk and manage disease in complex skin locations.
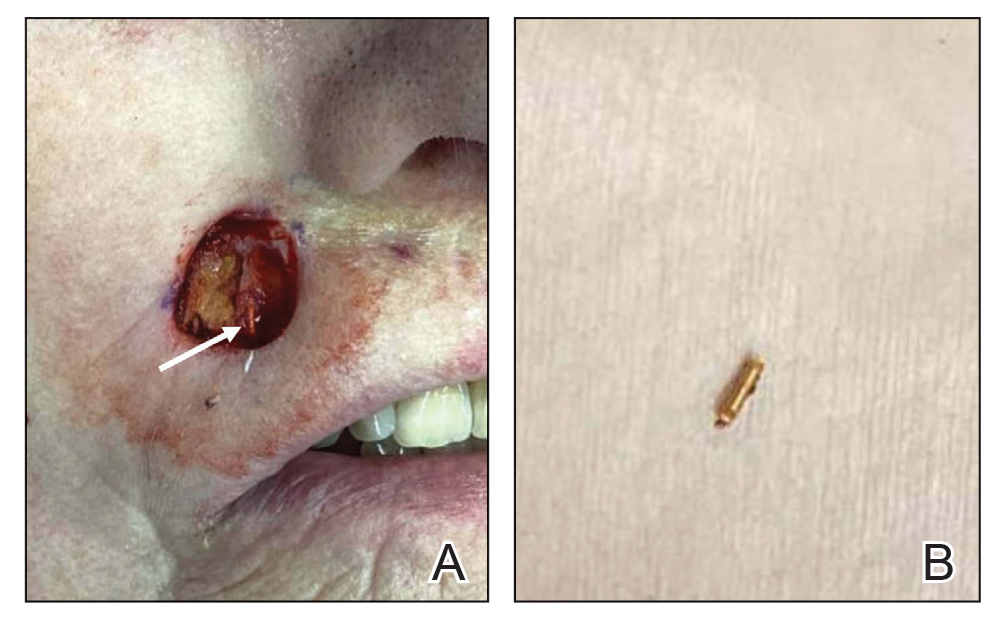
A 75-year-old woman presented to an outside dermatologist for evaluation of a pink papule on the right upper cutaneous lip that had enlarged over several months (Figure 1). The patient’s medical history was remarkable for an infantile hemangioma present since shortly after birth in the same location that had been treated with 10 implanted gold radon seeds when she was 6 years old. Over her lifetime, several seeds had self-extruded from the area, but some remained within the subcutaneous tissue as confirmed by dental radiographs. A shave biopsy of the papule demonstrated a superficial BCC, and the patient was referred to our institution for Mohs micrographic surgery.
Intraoperative frozen sections revealed both superficial and nodular BCC, and the tumor was cleared in 3 stages. During surgery, a gold radon seed was visualized at the base of the excised BCC and was removed from the subcutaneous tissue (Figure 2). The primary defect on the upper lip was closed with a rotation flap. The patient returned for follow-up 2 months later and showed good healing and cosmetic outcome.
Although not commonly encountered, ionizing radiation is a known risk factor for BCC.1 Basal cell carcinoma arising from implanted gold radon seeds represents a minority of reported cases.2,3 Radium was first used to treat skin disease in the early 1900s.1 The radioactive decay of radium produced tissue destruction via alpha, beta, and gamma particles, which slowly released over weeks when radium was packaged into a capsule.4 Following implantation of the capsule, DNA damage occurred due to double-stranded breaks, chromosomal aberrations, and generation of reactive oxygen species. The downstream effect of these cellular insults resulted in cell-cycle shortening, apoptosis, and carcinogenesis.5
Gold radon seeds were used to treat infantile hemangiomas in the United States and Europe from the early 1940s to the 1960s; their use declined dramatically in the 1950s due to adverse effects and discovery of the potential for future malignancies as well as the development of safer and more effective treatments.1,3 Our patient received a substantial dose of ionizing radiation from the implantation of gold radon seeds at the site of the infantile hemangioma, which dramatically increased her risk for BCC in this location.
Infantile hemangiomas are the most common vascular tumors in children. Most infantile hemangiomas regress spontaneously and are stably involuted by about 5 or 6 years of age.6 Treatment is indicated for rapidly growing hemangiomas that are at risk for ulceration or are located by critical structures (eg, the eyes or airway). Hemangiomas located on or near the lips should be treated to avoid disfigurement and loss of function as a consequence of rapid growth and involution.7 The treatment of choice for large or high-risk infantile hemangiomas over the past 10 to 15 years has been beta blockers.6-8 Propranolol hydrochloride, a systemic beta blocker, was approved by the US Food and Drug Administration in 2014 for the treatment of infantile hemangiomas and has demonstrated safety and effectiveness in promoting involution in these lesions.8 Unlike radiation therapy from implanted gold radon seeds, propranolol does not increase the risk for BCC. Although other risk factors such as skin type and cumulative UV exposure contribute to the development of BCC, the exact location of the BCC overlying the residual gold radon seeds was highly suggestive of ionizing radiation playing a major role in the carcinogenesis of the tumor in our patient.
Our case highlights the importance of screening elderly patients for exposures that may increase the risk for skin carcinogenesis. Dermatologists are accustomed to asking about history of UV exposure, sunburns, and use of sun-protective measures; however, direct questioning about less common sources of radiation exposure also may help stratify a patient’s risk for developing BCC. Although the US Preventive Services Task Force 2023 guidelines determined there is insufficient evidence to recommend visual skin cancer screening examinations in asymptomatic adults,9 we advocate for verbal screening of radiation exposure in both primary care and dermatology office settings. At a time when access to care, particularly dermatology services, is challenging, determining the appropriate interval for follow-up based on the patient’s skin cancer risk is imperative.
- Fürst CJ, Lundell M, Holm LE. Radiation therapy of hemangiomas, 1909- 1959. a cohort based on 50 years of clinical practice at Radiumhemmet, Stockholm. Acta Oncol. 1987;26:33-36. doi:10.3109/02841868709092974
- Bräuner EV, Loft S, Sørensen M, et al. Residential radon exposure and skin cancer incidence in a prospective Danish cohort. PLoS ONE. 2015;10:E0135642. doi:10.1371/journal.pone.0135642
- Weiss E, Sukal SA, Zimbler MS, et al. Basal cell carcinoma arising 57 years after interstitial radiotherapy of a nasal hemangioma. Dermatol Surg. 2008;34:1137-1140. doi:10.1111/j.1524-4725.2008.34229.x
- Lavery MJ, Lorenzelli D, Crema J. A radon seed identified during skin surgery: an unusual finding. Clin Exp Dermatol. 2021;46:604-606. doi:10.1111/ced.14454
- Robertson A, Allen J, Laney R, et al. The cellular and molecular carcinogenic effects of radon exposure: a review. Int J Mol Sci. 2013;14:14024-14063. doi:10.3390/ijms140714024
- Rodríguez Bandera AI, Sebaratnam DF, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392. doi:10.1016 /j.jaad.2021.08.019
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475. doi:10.1542/peds.2018-3475
- Sebaratnam DF, Rodríguez Bandera AL, Wong LF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85: 1395-1404. doi:10.1016/j.jaad.2021.08.020
- US Preventive Services Task Force, Mangione CM, Barry MJ, Nicholson WK, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2023;329:1290-1295. doi:10.1001/jama.2023.4342
To the Editor:
Basal cell carcinoma (BCC), which is the most common type of skin cancer, typically arises on sun-damaged skin as a result of long-term exposure to UV radiation. Another known risk factor for BCC is exposure to ionizing radiation, though this is less commonly encountered.1 We present a unique case of a BCC arising at the site of an involuted infantile hemangioma that had been treated with implanted and retained gold radon seeds more than 7 decades prior. This case highlights the importance of obtaining a detailed history of radiation exposures to better counsel patients about skin cancer risk and manage disease in complex skin locations.
A 75-year-old woman presented to an outside dermatologist for evaluation of a pink papule on the right upper cutaneous lip that had enlarged over several months (Figure 1). The patient’s medical history was remarkable for an infantile hemangioma present since shortly after birth in the same location that had been treated with 10 implanted gold radon seeds when she was 6 years old. Over her lifetime, several seeds had self-extruded from the area, but some remained within the subcutaneous tissue as confirmed by dental radiographs. A shave biopsy of the papule demonstrated a superficial BCC, and the patient was referred to our institution for Mohs micrographic surgery.
Intraoperative frozen sections revealed both superficial and nodular BCC, and the tumor was cleared in 3 stages. During surgery, a gold radon seed was visualized at the base of the excised BCC and was removed from the subcutaneous tissue (Figure 2). The primary defect on the upper lip was closed with a rotation flap. The patient returned for follow-up 2 months later and showed good healing and cosmetic outcome.
Although not commonly encountered, ionizing radiation is a known risk factor for BCC.1 Basal cell carcinoma arising from implanted gold radon seeds represents a minority of reported cases.2,3 Radium was first used to treat skin disease in the early 1900s.1 The radioactive decay of radium produced tissue destruction via alpha, beta, and gamma particles, which slowly released over weeks when radium was packaged into a capsule.4 Following implantation of the capsule, DNA damage occurred due to double-stranded breaks, chromosomal aberrations, and generation of reactive oxygen species. The downstream effect of these cellular insults resulted in cell-cycle shortening, apoptosis, and carcinogenesis.5
Gold radon seeds were used to treat infantile hemangiomas in the United States and Europe from the early 1940s to the 1960s; their use declined dramatically in the 1950s due to adverse effects and discovery of the potential for future malignancies as well as the development of safer and more effective treatments.1,3 Our patient received a substantial dose of ionizing radiation from the implantation of gold radon seeds at the site of the infantile hemangioma, which dramatically increased her risk for BCC in this location.
Infantile hemangiomas are the most common vascular tumors in children. Most infantile hemangiomas regress spontaneously and are stably involuted by about 5 or 6 years of age.6 Treatment is indicated for rapidly growing hemangiomas that are at risk for ulceration or are located by critical structures (eg, the eyes or airway). Hemangiomas located on or near the lips should be treated to avoid disfigurement and loss of function as a consequence of rapid growth and involution.7 The treatment of choice for large or high-risk infantile hemangiomas over the past 10 to 15 years has been beta blockers.6-8 Propranolol hydrochloride, a systemic beta blocker, was approved by the US Food and Drug Administration in 2014 for the treatment of infantile hemangiomas and has demonstrated safety and effectiveness in promoting involution in these lesions.8 Unlike radiation therapy from implanted gold radon seeds, propranolol does not increase the risk for BCC. Although other risk factors such as skin type and cumulative UV exposure contribute to the development of BCC, the exact location of the BCC overlying the residual gold radon seeds was highly suggestive of ionizing radiation playing a major role in the carcinogenesis of the tumor in our patient.
Our case highlights the importance of screening elderly patients for exposures that may increase the risk for skin carcinogenesis. Dermatologists are accustomed to asking about history of UV exposure, sunburns, and use of sun-protective measures; however, direct questioning about less common sources of radiation exposure also may help stratify a patient’s risk for developing BCC. Although the US Preventive Services Task Force 2023 guidelines determined there is insufficient evidence to recommend visual skin cancer screening examinations in asymptomatic adults,9 we advocate for verbal screening of radiation exposure in both primary care and dermatology office settings. At a time when access to care, particularly dermatology services, is challenging, determining the appropriate interval for follow-up based on the patient’s skin cancer risk is imperative.
To the Editor:
Basal cell carcinoma (BCC), which is the most common type of skin cancer, typically arises on sun-damaged skin as a result of long-term exposure to UV radiation. Another known risk factor for BCC is exposure to ionizing radiation, though this is less commonly encountered.1 We present a unique case of a BCC arising at the site of an involuted infantile hemangioma that had been treated with implanted and retained gold radon seeds more than 7 decades prior. This case highlights the importance of obtaining a detailed history of radiation exposures to better counsel patients about skin cancer risk and manage disease in complex skin locations.
A 75-year-old woman presented to an outside dermatologist for evaluation of a pink papule on the right upper cutaneous lip that had enlarged over several months (Figure 1). The patient’s medical history was remarkable for an infantile hemangioma present since shortly after birth in the same location that had been treated with 10 implanted gold radon seeds when she was 6 years old. Over her lifetime, several seeds had self-extruded from the area, but some remained within the subcutaneous tissue as confirmed by dental radiographs. A shave biopsy of the papule demonstrated a superficial BCC, and the patient was referred to our institution for Mohs micrographic surgery.
Intraoperative frozen sections revealed both superficial and nodular BCC, and the tumor was cleared in 3 stages. During surgery, a gold radon seed was visualized at the base of the excised BCC and was removed from the subcutaneous tissue (Figure 2). The primary defect on the upper lip was closed with a rotation flap. The patient returned for follow-up 2 months later and showed good healing and cosmetic outcome.
Although not commonly encountered, ionizing radiation is a known risk factor for BCC.1 Basal cell carcinoma arising from implanted gold radon seeds represents a minority of reported cases.2,3 Radium was first used to treat skin disease in the early 1900s.1 The radioactive decay of radium produced tissue destruction via alpha, beta, and gamma particles, which slowly released over weeks when radium was packaged into a capsule.4 Following implantation of the capsule, DNA damage occurred due to double-stranded breaks, chromosomal aberrations, and generation of reactive oxygen species. The downstream effect of these cellular insults resulted in cell-cycle shortening, apoptosis, and carcinogenesis.5
Gold radon seeds were used to treat infantile hemangiomas in the United States and Europe from the early 1940s to the 1960s; their use declined dramatically in the 1950s due to adverse effects and discovery of the potential for future malignancies as well as the development of safer and more effective treatments.1,3 Our patient received a substantial dose of ionizing radiation from the implantation of gold radon seeds at the site of the infantile hemangioma, which dramatically increased her risk for BCC in this location.
Infantile hemangiomas are the most common vascular tumors in children. Most infantile hemangiomas regress spontaneously and are stably involuted by about 5 or 6 years of age.6 Treatment is indicated for rapidly growing hemangiomas that are at risk for ulceration or are located by critical structures (eg, the eyes or airway). Hemangiomas located on or near the lips should be treated to avoid disfigurement and loss of function as a consequence of rapid growth and involution.7 The treatment of choice for large or high-risk infantile hemangiomas over the past 10 to 15 years has been beta blockers.6-8 Propranolol hydrochloride, a systemic beta blocker, was approved by the US Food and Drug Administration in 2014 for the treatment of infantile hemangiomas and has demonstrated safety and effectiveness in promoting involution in these lesions.8 Unlike radiation therapy from implanted gold radon seeds, propranolol does not increase the risk for BCC. Although other risk factors such as skin type and cumulative UV exposure contribute to the development of BCC, the exact location of the BCC overlying the residual gold radon seeds was highly suggestive of ionizing radiation playing a major role in the carcinogenesis of the tumor in our patient.
Our case highlights the importance of screening elderly patients for exposures that may increase the risk for skin carcinogenesis. Dermatologists are accustomed to asking about history of UV exposure, sunburns, and use of sun-protective measures; however, direct questioning about less common sources of radiation exposure also may help stratify a patient’s risk for developing BCC. Although the US Preventive Services Task Force 2023 guidelines determined there is insufficient evidence to recommend visual skin cancer screening examinations in asymptomatic adults,9 we advocate for verbal screening of radiation exposure in both primary care and dermatology office settings. At a time when access to care, particularly dermatology services, is challenging, determining the appropriate interval for follow-up based on the patient’s skin cancer risk is imperative.
- Fürst CJ, Lundell M, Holm LE. Radiation therapy of hemangiomas, 1909- 1959. a cohort based on 50 years of clinical practice at Radiumhemmet, Stockholm. Acta Oncol. 1987;26:33-36. doi:10.3109/02841868709092974
- Bräuner EV, Loft S, Sørensen M, et al. Residential radon exposure and skin cancer incidence in a prospective Danish cohort. PLoS ONE. 2015;10:E0135642. doi:10.1371/journal.pone.0135642
- Weiss E, Sukal SA, Zimbler MS, et al. Basal cell carcinoma arising 57 years after interstitial radiotherapy of a nasal hemangioma. Dermatol Surg. 2008;34:1137-1140. doi:10.1111/j.1524-4725.2008.34229.x
- Lavery MJ, Lorenzelli D, Crema J. A radon seed identified during skin surgery: an unusual finding. Clin Exp Dermatol. 2021;46:604-606. doi:10.1111/ced.14454
- Robertson A, Allen J, Laney R, et al. The cellular and molecular carcinogenic effects of radon exposure: a review. Int J Mol Sci. 2013;14:14024-14063. doi:10.3390/ijms140714024
- Rodríguez Bandera AI, Sebaratnam DF, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392. doi:10.1016 /j.jaad.2021.08.019
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475. doi:10.1542/peds.2018-3475
- Sebaratnam DF, Rodríguez Bandera AL, Wong LF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85: 1395-1404. doi:10.1016/j.jaad.2021.08.020
- US Preventive Services Task Force, Mangione CM, Barry MJ, Nicholson WK, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2023;329:1290-1295. doi:10.1001/jama.2023.4342
- Fürst CJ, Lundell M, Holm LE. Radiation therapy of hemangiomas, 1909- 1959. a cohort based on 50 years of clinical practice at Radiumhemmet, Stockholm. Acta Oncol. 1987;26:33-36. doi:10.3109/02841868709092974
- Bräuner EV, Loft S, Sørensen M, et al. Residential radon exposure and skin cancer incidence in a prospective Danish cohort. PLoS ONE. 2015;10:E0135642. doi:10.1371/journal.pone.0135642
- Weiss E, Sukal SA, Zimbler MS, et al. Basal cell carcinoma arising 57 years after interstitial radiotherapy of a nasal hemangioma. Dermatol Surg. 2008;34:1137-1140. doi:10.1111/j.1524-4725.2008.34229.x
- Lavery MJ, Lorenzelli D, Crema J. A radon seed identified during skin surgery: an unusual finding. Clin Exp Dermatol. 2021;46:604-606. doi:10.1111/ced.14454
- Robertson A, Allen J, Laney R, et al. The cellular and molecular carcinogenic effects of radon exposure: a review. Int J Mol Sci. 2013;14:14024-14063. doi:10.3390/ijms140714024
- Rodríguez Bandera AI, Sebaratnam DF, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392. doi:10.1016 /j.jaad.2021.08.019
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475. doi:10.1542/peds.2018-3475
- Sebaratnam DF, Rodríguez Bandera AL, Wong LF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85: 1395-1404. doi:10.1016/j.jaad.2021.08.020
- US Preventive Services Task Force, Mangione CM, Barry MJ, Nicholson WK, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2023;329:1290-1295. doi:10.1001/jama.2023.4342
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
PRACTICE POINTS
- Historical use of ionizing radiation to treat skin disease is a risk factor for basal cell carcinoma (BCC).
- Mohs micrographic surgery is the treatment of choice for BCC in high-risk areas such as the nose, eyelids, and lips, where tissue conservation and complete margin control are essential.
- Elderly patients should be screened for less common sources of radiation exposure for better risk stratification and to determine appropriate intervals for follow-up with a dermatologist.

Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
To the Editor:
Uveitis associated with tattoos is common, yet the etiology and optimal treatment options for this phenomenon remain unclear. Possible causes include a delayed hypersensitivity reaction to tattoo ink antigen or systemic sarcoidosis localized to the skin.1 Long-term treatment options include topical, intralesional, and systemic corticosteroids or immunosuppressants.2 Short-term options often include direct surgical excision and laser treatment. However, laser removal of tattoo pigment typically involves multiple sessions over the course of years, and there is a risk for antigen dispersal that may lead to anaphylaxis. Determining the most effective and safe treatment for a patient with progressive and severe ocular symptoms can be challenging. We describe a patient with cutaneous blue ink tattoos who developed chronic bilateral glaucoma, iritis, uveitis, and ocular hypertension that was refractory to multiple systemic medications and ophthalmologic procedures but responded to CO2 laser ablation.
A 27-year-old man with an active smoking history presented to our laser surgery center with a rash of approximately 4 years’ duration in areas with blue tattoo ink on both forearms. He was referred by his ophthalmologist due to bilateral uveitis and iritis and subsequent ocular hypertension and glaucoma that developed approximately 5 years after tattoo placement on the bilateral forearms. When the rash first appeared, the skin in the areas of the blue tattoo ink had hyperpigmented pruritic plaques. The patient was treated by a dermatologist with topical steroids to help reduce the itching and inflammation. Around the same time, he also started having ocular symptoms—vitreous floaters, erythema, eye pain, and blurriness—and was diagnosed with iritis of unclear etiology by ophthalmology. Figure 1 documents the patient’s clinical course. Due to escalating intraocular pressure and symptoms, he was referred to a glaucoma specialist and a rheumatologist. Systemic and rheumatologic medical conditions were ruled out with negative results on a series of blood tests (eg, rheumatoid factor, HLA-B27, antinuclear antibody, lysozyme, interferon gamma release assay, erythrocyte sedimentation rate, C-reactive protein, hepatitis B/C virus, Treponema pallidum, HIV), and magnetic resonance imaging of the brain was negative, ruling out demyelinating disease. Laboratory workup for sarcoidosis also was performed. The angiotensin-converting enzyme level was 30 U/L (reference range, 9-67 U/L), and a chest radiograph and computed tomography with contrast indicated no evidence of cardiopulmonary involvement. Although sarcoidosis could not be definitively ruled out, no other cause could be determined, and the patient’s glaucoma specialist diagnosed him with tattoo-associated uveitis. The patient was started on brimonidine, latanoprost, prednisolone, and dorzolamidetimolol eye drops, as well as acetazolamide (500 mg twice daily) and oral prednisone (various doses). Over the next 3 years, the patient continued to have symptoms, and immunosuppressant medications—methotrexate 20-25 mg weekly and adalimumab 40 mg every 2 weeks—were added to his treatment regimen. The patient also underwent bilateral ophthalmologic procedures, including a Baerveldt glaucoma implant procedure in the left eye and circumferential trabeculectomy in the right eye.
Despite these medications and procedures, the patient’s symptoms and intraocular pressure had not improved. At the current visit, punch biopsy of the tattooed skin and histologic examination showed dermal lymphoplasmacytic inflammation with scattered foreign-body giant cells associated with blue tattoo ink and overlying hyperkeratosis and spongiosis, consistent with allergic contact dermatitis (Figure 2). Because both immunosuppressant medications and ophthalmologic procedures had failed to control the progression of the ocular symptoms and the patient was at risk for permanent blindness, surgical excision and laser tattoo removal were considered as potential treatment options. Due to the large surface area and circumferential nature of the tattoos, there was a notable risk for disfiguring scars at both recipient and donor sites with surgical excision followed by graft placement. Thus, CO2 laser ablation was the preferred treatment option. However, this procedure was not without risk for anaphylaxis if the tattoo pigment were to be released into systemic circulation. Thus, at the first visit, ablation was performed on 3 test spots and the patient was prescribed cetirizine, diphenhydramine, and prophylactic prednisone for a few days. The patient then received a total of 5 fully ablative CO2 laser sessions (pulse energy: 200 mJ [15 J/cm2]; computerized pattern generator: 2-8-9 [85.2 J/cm2]; rate: 200 Hz [20 W], 3 passes) over 13 months to remove all visible blue ink in stages (Figure 3). Even with a shortened time course (as more time between laser sessions typically is preferred), the treatments were well tolerated with only mild hypertrophic scarring that responded to intralesional steroids (triamcinolone 10 mg/mL). On repeat skin biopsy during the treatment course, the superficial dermis demonstrated mostly scar tissue and near-total pigment removal—a 90% to 95% reduction in blue ink from prior biopsy—and minimal inflammation (Figure 4). Scant fine to coarse pigment deposition was seen in the deep dermis next to subcutaneous fat, which was unchanged from the previous biopsy. The patient’s ophthalmologic symptoms were tracked via improvement in intraocular pressure and stabilization of his vision, indicating rapid and complete resolution of the glaucoma after the last laser treatment. With resolution of his ocular symptoms, the patient was tapered off all immunosuppressant medications. The patient was lost to follow-up approximately 2 years after the final laser treatment.

Tattoo-associated uveitis initially was described in 1969 in 3 patients with light blue tattoos who developed tattoo granulomas and simultaneous uveitis. These cases were successfully treated with excision.3 Multiple cases have been reported since, often with bilateral uveitis and tattoos demonstrating noncaseating granulomatous inflammation that were treated with steroids.4 In 2018, a diagnosis of exclusion was proposed for uveitis associated with granulomatous tattoo reaction without sarcoidosis: tattoo granulomas with uveitis (TAGU).1
In this case, sarcoidosis initially was high on the differential diagnosis. Sarcoidosis is an immune-mediated systemic disease of unknown etiology characterized by the presence of widespread noncaseating epithelioid cell granulomas, primarily seen in the pulmonary and lymphatic systems. However, it often initially manifests with cutaneous involvement with noncaseating “naked” granulomas in the dermis and subcutaneous tissue. Although TAGU cases have demonstrated noncaseating granulomas in association with dermal tattoo pigment on histopathology,1,4 dermal lymphoplasmacytic inflammation with scattered foreign body giant cells was noted in our patient, which was more consistent with allergic contact dermatitis. Thus, it is important to consider that TAGU can be seen with varying histologic patterns. In patients with tattoos, sarcoidosis can manifest grossly as a papulonodular cutaneous reaction.5 Active smoking is associated with a decreased risk for sarcoidosis, and those who smoke are statistically more likely to have tattoos than the general population,6,7 so our patient’s smoking history may be relevant. However, sarcoidosis was an unlikely diagnosis due to the serum angiotensin-converting enzyme level; results of a chest radiograph (bilateral adenopathy and coarse reticular opacities) and computed tomography (hilar and mediastinal adenopathy); and nonsarcoidal histopathology.
An allergic reaction to tattoo ink is caused by a delayed-type hypersensitivity reaction to a pigment hapten that can develop abruptly months to years after tattoo placement—1 year after tattoo placement in our patient. This reaction was seen in our patient’s blue pigment tattoos, although it is more commonly seen in red pigment tattoos.8 Although the etiology of TAGU is poorly understood, it also is hypothesized to be a delayed-type hypersensitivity response to tattoo ink particles, suggested by the pattern of lymphocytes infiltrating the tattoo and atypical T-cell infiltrate on vitreous biopsy.9,10 Further research is required to elucidate the relationship between tattoos and uveitis.
Q-switched lasers (eg, 532-nm or 1064-nm Nd:YAG, alexandrite, or ruby lasers) are the standard treatment options for uncomplicated tattoo removal and employ a high-intensity, ultrashort pulse duration.11 However Q-switched lasers require multiple sessions and target pigment-containing cells, releasing the tattoo particles into systemic circulation, which can potentially induce a severe allergic response.12 In contrast, CO2 lasers use a different mechanism, emitting energy at a wavelength of 10,600 nm, which is absorbed by intracellular water and allows for the ablation of the superficial epidermis along with the embedded ink with subsequent re-epithelialization, as well as heat-mediated thermal injury to allow for dermal collagen remodeling.13 In a 2021 retrospective study of ablative laser therapy for allergic tattoo reactions, patients were treated with the 10,600-nm ablative CO2 laser and noted improvements in itching and burning with minimal adverse events.12 Although using a CO2 laser may not be considered a firstline treatment option for TAGU, the refractory clinical course and notable morbidity of surgical excision necessitated the use of ablative laser in our case.
Tattoo granulomas with uveitis is a rare diagnosis with the potential for serious permanent sequelae including blindness. Existing treatments such as topical and oral corticosteroids, immunosuppressants, surgical excision, and Q-switched lasers all are possible options, but in a patient with progressive ocular symptoms with other potential rheumatologic conditions and sarcoidosis ruled out, fully ablative CO2 laser may be an effective treatment option. Our case demonstrated the successful treatment of TAGU with CO2 laser ablation. Given the unclear etiology of TAGU and the limited evidence on treatment options and efficacy, our case contributes to the body of literature that can inform clinical management of this unusual and serious reaction.
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32:1852-1861. doi:10.1111/jdv.15070
- Tiew S. Tattoo-associated panuveitis: a 10-year follow-up. Eur J Ophthalmol. 2019;29(1 suppl):18-21. doi:10.1177/1120672119846341
- Rorsman H, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2:27-28. doi:10.1016/s0140-6736(69)92600-2
- Ostheimer TA, Burkholder BM, Leung TG, et al. Tattoo-associated uveitis. Am J Ophthalmol. 2014;158:637-643.e1. doi:10.1016/j.ajo.2014.05.019
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-nodular reactions in black tattoos as markers of sarcoidosis: study of 92 tattoo reactions from a hospital material. Dermatology. 2016;232:679-686. doi:10.1159/000453315
- Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet. 2014;383: 1155-1167. doi:10.1016/S0140-6736(13)60680-7
- Kluger N. Epidemiology of tattoos in industrialized countries. Curr Probl Dermatol. 2015;48:6-20. doi:10.1159/000369175
- Serup J, Hutton Carlsen K, Dommershausen N, et al. Identification of pigments related to allergic tattoo reactions in 104 human skin biopsies. Contact Dermatitis. 2020;82:73-82. doi:10.1111/cod.13423
- Mansour AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. Am J Ophthalmol. 1991;111:515-516. doi:10.1016/s0002-9394(14)72395-5
- Reddy AK, Shildkrot Y, Newman SA, et al. T-lymphocyte predominance and cellular atypia in tattoo-associated uveitis. JAMA Ophthalmol. 2015;133:1356-1357. doi:10.1001/jamaophthalmol.2015.3354
- Wenzel SM. Current concepts in laser tattoo removal. Skin Therapy Lett. 2010;15:3-5.
- van der Bent SAS, Huisman S, Rustemeyer T, et al. Ablative laser surgery for allergic tattoo reactions: a retrospective study. mLasers Med Sci. 2021;36:1241-1248. doi:10.1007/s10103-020-03164-2
- Yumeen S, Khan T. Laser carbon dioxide resurfacing. In: StatPearls. StatPearls Publishing; April 23, 2023. Accessed March 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560544/
To the Editor:
Uveitis associated with tattoos is common, yet the etiology and optimal treatment options for this phenomenon remain unclear. Possible causes include a delayed hypersensitivity reaction to tattoo ink antigen or systemic sarcoidosis localized to the skin.1 Long-term treatment options include topical, intralesional, and systemic corticosteroids or immunosuppressants.2 Short-term options often include direct surgical excision and laser treatment. However, laser removal of tattoo pigment typically involves multiple sessions over the course of years, and there is a risk for antigen dispersal that may lead to anaphylaxis. Determining the most effective and safe treatment for a patient with progressive and severe ocular symptoms can be challenging. We describe a patient with cutaneous blue ink tattoos who developed chronic bilateral glaucoma, iritis, uveitis, and ocular hypertension that was refractory to multiple systemic medications and ophthalmologic procedures but responded to CO2 laser ablation.
A 27-year-old man with an active smoking history presented to our laser surgery center with a rash of approximately 4 years’ duration in areas with blue tattoo ink on both forearms. He was referred by his ophthalmologist due to bilateral uveitis and iritis and subsequent ocular hypertension and glaucoma that developed approximately 5 years after tattoo placement on the bilateral forearms. When the rash first appeared, the skin in the areas of the blue tattoo ink had hyperpigmented pruritic plaques. The patient was treated by a dermatologist with topical steroids to help reduce the itching and inflammation. Around the same time, he also started having ocular symptoms—vitreous floaters, erythema, eye pain, and blurriness—and was diagnosed with iritis of unclear etiology by ophthalmology. Figure 1 documents the patient’s clinical course. Due to escalating intraocular pressure and symptoms, he was referred to a glaucoma specialist and a rheumatologist. Systemic and rheumatologic medical conditions were ruled out with negative results on a series of blood tests (eg, rheumatoid factor, HLA-B27, antinuclear antibody, lysozyme, interferon gamma release assay, erythrocyte sedimentation rate, C-reactive protein, hepatitis B/C virus, Treponema pallidum, HIV), and magnetic resonance imaging of the brain was negative, ruling out demyelinating disease. Laboratory workup for sarcoidosis also was performed. The angiotensin-converting enzyme level was 30 U/L (reference range, 9-67 U/L), and a chest radiograph and computed tomography with contrast indicated no evidence of cardiopulmonary involvement. Although sarcoidosis could not be definitively ruled out, no other cause could be determined, and the patient’s glaucoma specialist diagnosed him with tattoo-associated uveitis. The patient was started on brimonidine, latanoprost, prednisolone, and dorzolamidetimolol eye drops, as well as acetazolamide (500 mg twice daily) and oral prednisone (various doses). Over the next 3 years, the patient continued to have symptoms, and immunosuppressant medications—methotrexate 20-25 mg weekly and adalimumab 40 mg every 2 weeks—were added to his treatment regimen. The patient also underwent bilateral ophthalmologic procedures, including a Baerveldt glaucoma implant procedure in the left eye and circumferential trabeculectomy in the right eye.
Despite these medications and procedures, the patient’s symptoms and intraocular pressure had not improved. At the current visit, punch biopsy of the tattooed skin and histologic examination showed dermal lymphoplasmacytic inflammation with scattered foreign-body giant cells associated with blue tattoo ink and overlying hyperkeratosis and spongiosis, consistent with allergic contact dermatitis (Figure 2). Because both immunosuppressant medications and ophthalmologic procedures had failed to control the progression of the ocular symptoms and the patient was at risk for permanent blindness, surgical excision and laser tattoo removal were considered as potential treatment options. Due to the large surface area and circumferential nature of the tattoos, there was a notable risk for disfiguring scars at both recipient and donor sites with surgical excision followed by graft placement. Thus, CO2 laser ablation was the preferred treatment option. However, this procedure was not without risk for anaphylaxis if the tattoo pigment were to be released into systemic circulation. Thus, at the first visit, ablation was performed on 3 test spots and the patient was prescribed cetirizine, diphenhydramine, and prophylactic prednisone for a few days. The patient then received a total of 5 fully ablative CO2 laser sessions (pulse energy: 200 mJ [15 J/cm2]; computerized pattern generator: 2-8-9 [85.2 J/cm2]; rate: 200 Hz [20 W], 3 passes) over 13 months to remove all visible blue ink in stages (Figure 3). Even with a shortened time course (as more time between laser sessions typically is preferred), the treatments were well tolerated with only mild hypertrophic scarring that responded to intralesional steroids (triamcinolone 10 mg/mL). On repeat skin biopsy during the treatment course, the superficial dermis demonstrated mostly scar tissue and near-total pigment removal—a 90% to 95% reduction in blue ink from prior biopsy—and minimal inflammation (Figure 4). Scant fine to coarse pigment deposition was seen in the deep dermis next to subcutaneous fat, which was unchanged from the previous biopsy. The patient’s ophthalmologic symptoms were tracked via improvement in intraocular pressure and stabilization of his vision, indicating rapid and complete resolution of the glaucoma after the last laser treatment. With resolution of his ocular symptoms, the patient was tapered off all immunosuppressant medications. The patient was lost to follow-up approximately 2 years after the final laser treatment.
Tattoo-associated uveitis initially was described in 1969 in 3 patients with light blue tattoos who developed tattoo granulomas and simultaneous uveitis. These cases were successfully treated with excision.3 Multiple cases have been reported since, often with bilateral uveitis and tattoos demonstrating noncaseating granulomatous inflammation that were treated with steroids.4 In 2018, a diagnosis of exclusion was proposed for uveitis associated with granulomatous tattoo reaction without sarcoidosis: tattoo granulomas with uveitis (TAGU).1
In this case, sarcoidosis initially was high on the differential diagnosis. Sarcoidosis is an immune-mediated systemic disease of unknown etiology characterized by the presence of widespread noncaseating epithelioid cell granulomas, primarily seen in the pulmonary and lymphatic systems. However, it often initially manifests with cutaneous involvement with noncaseating “naked” granulomas in the dermis and subcutaneous tissue. Although TAGU cases have demonstrated noncaseating granulomas in association with dermal tattoo pigment on histopathology,1,4 dermal lymphoplasmacytic inflammation with scattered foreign body giant cells was noted in our patient, which was more consistent with allergic contact dermatitis. Thus, it is important to consider that TAGU can be seen with varying histologic patterns. In patients with tattoos, sarcoidosis can manifest grossly as a papulonodular cutaneous reaction.5 Active smoking is associated with a decreased risk for sarcoidosis, and those who smoke are statistically more likely to have tattoos than the general population,6,7 so our patient’s smoking history may be relevant. However, sarcoidosis was an unlikely diagnosis due to the serum angiotensin-converting enzyme level; results of a chest radiograph (bilateral adenopathy and coarse reticular opacities) and computed tomography (hilar and mediastinal adenopathy); and nonsarcoidal histopathology.
An allergic reaction to tattoo ink is caused by a delayed-type hypersensitivity reaction to a pigment hapten that can develop abruptly months to years after tattoo placement—1 year after tattoo placement in our patient. This reaction was seen in our patient’s blue pigment tattoos, although it is more commonly seen in red pigment tattoos.8 Although the etiology of TAGU is poorly understood, it also is hypothesized to be a delayed-type hypersensitivity response to tattoo ink particles, suggested by the pattern of lymphocytes infiltrating the tattoo and atypical T-cell infiltrate on vitreous biopsy.9,10 Further research is required to elucidate the relationship between tattoos and uveitis.
Q-switched lasers (eg, 532-nm or 1064-nm Nd:YAG, alexandrite, or ruby lasers) are the standard treatment options for uncomplicated tattoo removal and employ a high-intensity, ultrashort pulse duration.11 However Q-switched lasers require multiple sessions and target pigment-containing cells, releasing the tattoo particles into systemic circulation, which can potentially induce a severe allergic response.12 In contrast, CO2 lasers use a different mechanism, emitting energy at a wavelength of 10,600 nm, which is absorbed by intracellular water and allows for the ablation of the superficial epidermis along with the embedded ink with subsequent re-epithelialization, as well as heat-mediated thermal injury to allow for dermal collagen remodeling.13 In a 2021 retrospective study of ablative laser therapy for allergic tattoo reactions, patients were treated with the 10,600-nm ablative CO2 laser and noted improvements in itching and burning with minimal adverse events.12 Although using a CO2 laser may not be considered a firstline treatment option for TAGU, the refractory clinical course and notable morbidity of surgical excision necessitated the use of ablative laser in our case.
Tattoo granulomas with uveitis is a rare diagnosis with the potential for serious permanent sequelae including blindness. Existing treatments such as topical and oral corticosteroids, immunosuppressants, surgical excision, and Q-switched lasers all are possible options, but in a patient with progressive ocular symptoms with other potential rheumatologic conditions and sarcoidosis ruled out, fully ablative CO2 laser may be an effective treatment option. Our case demonstrated the successful treatment of TAGU with CO2 laser ablation. Given the unclear etiology of TAGU and the limited evidence on treatment options and efficacy, our case contributes to the body of literature that can inform clinical management of this unusual and serious reaction.
To the Editor:
Uveitis associated with tattoos is common, yet the etiology and optimal treatment options for this phenomenon remain unclear. Possible causes include a delayed hypersensitivity reaction to tattoo ink antigen or systemic sarcoidosis localized to the skin.1 Long-term treatment options include topical, intralesional, and systemic corticosteroids or immunosuppressants.2 Short-term options often include direct surgical excision and laser treatment. However, laser removal of tattoo pigment typically involves multiple sessions over the course of years, and there is a risk for antigen dispersal that may lead to anaphylaxis. Determining the most effective and safe treatment for a patient with progressive and severe ocular symptoms can be challenging. We describe a patient with cutaneous blue ink tattoos who developed chronic bilateral glaucoma, iritis, uveitis, and ocular hypertension that was refractory to multiple systemic medications and ophthalmologic procedures but responded to CO2 laser ablation.
A 27-year-old man with an active smoking history presented to our laser surgery center with a rash of approximately 4 years’ duration in areas with blue tattoo ink on both forearms. He was referred by his ophthalmologist due to bilateral uveitis and iritis and subsequent ocular hypertension and glaucoma that developed approximately 5 years after tattoo placement on the bilateral forearms. When the rash first appeared, the skin in the areas of the blue tattoo ink had hyperpigmented pruritic plaques. The patient was treated by a dermatologist with topical steroids to help reduce the itching and inflammation. Around the same time, he also started having ocular symptoms—vitreous floaters, erythema, eye pain, and blurriness—and was diagnosed with iritis of unclear etiology by ophthalmology. Figure 1 documents the patient’s clinical course. Due to escalating intraocular pressure and symptoms, he was referred to a glaucoma specialist and a rheumatologist. Systemic and rheumatologic medical conditions were ruled out with negative results on a series of blood tests (eg, rheumatoid factor, HLA-B27, antinuclear antibody, lysozyme, interferon gamma release assay, erythrocyte sedimentation rate, C-reactive protein, hepatitis B/C virus, Treponema pallidum, HIV), and magnetic resonance imaging of the brain was negative, ruling out demyelinating disease. Laboratory workup for sarcoidosis also was performed. The angiotensin-converting enzyme level was 30 U/L (reference range, 9-67 U/L), and a chest radiograph and computed tomography with contrast indicated no evidence of cardiopulmonary involvement. Although sarcoidosis could not be definitively ruled out, no other cause could be determined, and the patient’s glaucoma specialist diagnosed him with tattoo-associated uveitis. The patient was started on brimonidine, latanoprost, prednisolone, and dorzolamidetimolol eye drops, as well as acetazolamide (500 mg twice daily) and oral prednisone (various doses). Over the next 3 years, the patient continued to have symptoms, and immunosuppressant medications—methotrexate 20-25 mg weekly and adalimumab 40 mg every 2 weeks—were added to his treatment regimen. The patient also underwent bilateral ophthalmologic procedures, including a Baerveldt glaucoma implant procedure in the left eye and circumferential trabeculectomy in the right eye.
Despite these medications and procedures, the patient’s symptoms and intraocular pressure had not improved. At the current visit, punch biopsy of the tattooed skin and histologic examination showed dermal lymphoplasmacytic inflammation with scattered foreign-body giant cells associated with blue tattoo ink and overlying hyperkeratosis and spongiosis, consistent with allergic contact dermatitis (Figure 2). Because both immunosuppressant medications and ophthalmologic procedures had failed to control the progression of the ocular symptoms and the patient was at risk for permanent blindness, surgical excision and laser tattoo removal were considered as potential treatment options. Due to the large surface area and circumferential nature of the tattoos, there was a notable risk for disfiguring scars at both recipient and donor sites with surgical excision followed by graft placement. Thus, CO2 laser ablation was the preferred treatment option. However, this procedure was not without risk for anaphylaxis if the tattoo pigment were to be released into systemic circulation. Thus, at the first visit, ablation was performed on 3 test spots and the patient was prescribed cetirizine, diphenhydramine, and prophylactic prednisone for a few days. The patient then received a total of 5 fully ablative CO2 laser sessions (pulse energy: 200 mJ [15 J/cm2]; computerized pattern generator: 2-8-9 [85.2 J/cm2]; rate: 200 Hz [20 W], 3 passes) over 13 months to remove all visible blue ink in stages (Figure 3). Even with a shortened time course (as more time between laser sessions typically is preferred), the treatments were well tolerated with only mild hypertrophic scarring that responded to intralesional steroids (triamcinolone 10 mg/mL). On repeat skin biopsy during the treatment course, the superficial dermis demonstrated mostly scar tissue and near-total pigment removal—a 90% to 95% reduction in blue ink from prior biopsy—and minimal inflammation (Figure 4). Scant fine to coarse pigment deposition was seen in the deep dermis next to subcutaneous fat, which was unchanged from the previous biopsy. The patient’s ophthalmologic symptoms were tracked via improvement in intraocular pressure and stabilization of his vision, indicating rapid and complete resolution of the glaucoma after the last laser treatment. With resolution of his ocular symptoms, the patient was tapered off all immunosuppressant medications. The patient was lost to follow-up approximately 2 years after the final laser treatment.
Tattoo-associated uveitis initially was described in 1969 in 3 patients with light blue tattoos who developed tattoo granulomas and simultaneous uveitis. These cases were successfully treated with excision.3 Multiple cases have been reported since, often with bilateral uveitis and tattoos demonstrating noncaseating granulomatous inflammation that were treated with steroids.4 In 2018, a diagnosis of exclusion was proposed for uveitis associated with granulomatous tattoo reaction without sarcoidosis: tattoo granulomas with uveitis (TAGU).1
In this case, sarcoidosis initially was high on the differential diagnosis. Sarcoidosis is an immune-mediated systemic disease of unknown etiology characterized by the presence of widespread noncaseating epithelioid cell granulomas, primarily seen in the pulmonary and lymphatic systems. However, it often initially manifests with cutaneous involvement with noncaseating “naked” granulomas in the dermis and subcutaneous tissue. Although TAGU cases have demonstrated noncaseating granulomas in association with dermal tattoo pigment on histopathology,1,4 dermal lymphoplasmacytic inflammation with scattered foreign body giant cells was noted in our patient, which was more consistent with allergic contact dermatitis. Thus, it is important to consider that TAGU can be seen with varying histologic patterns. In patients with tattoos, sarcoidosis can manifest grossly as a papulonodular cutaneous reaction.5 Active smoking is associated with a decreased risk for sarcoidosis, and those who smoke are statistically more likely to have tattoos than the general population,6,7 so our patient’s smoking history may be relevant. However, sarcoidosis was an unlikely diagnosis due to the serum angiotensin-converting enzyme level; results of a chest radiograph (bilateral adenopathy and coarse reticular opacities) and computed tomography (hilar and mediastinal adenopathy); and nonsarcoidal histopathology.
An allergic reaction to tattoo ink is caused by a delayed-type hypersensitivity reaction to a pigment hapten that can develop abruptly months to years after tattoo placement—1 year after tattoo placement in our patient. This reaction was seen in our patient’s blue pigment tattoos, although it is more commonly seen in red pigment tattoos.8 Although the etiology of TAGU is poorly understood, it also is hypothesized to be a delayed-type hypersensitivity response to tattoo ink particles, suggested by the pattern of lymphocytes infiltrating the tattoo and atypical T-cell infiltrate on vitreous biopsy.9,10 Further research is required to elucidate the relationship between tattoos and uveitis.
Q-switched lasers (eg, 532-nm or 1064-nm Nd:YAG, alexandrite, or ruby lasers) are the standard treatment options for uncomplicated tattoo removal and employ a high-intensity, ultrashort pulse duration.11 However Q-switched lasers require multiple sessions and target pigment-containing cells, releasing the tattoo particles into systemic circulation, which can potentially induce a severe allergic response.12 In contrast, CO2 lasers use a different mechanism, emitting energy at a wavelength of 10,600 nm, which is absorbed by intracellular water and allows for the ablation of the superficial epidermis along with the embedded ink with subsequent re-epithelialization, as well as heat-mediated thermal injury to allow for dermal collagen remodeling.13 In a 2021 retrospective study of ablative laser therapy for allergic tattoo reactions, patients were treated with the 10,600-nm ablative CO2 laser and noted improvements in itching and burning with minimal adverse events.12 Although using a CO2 laser may not be considered a firstline treatment option for TAGU, the refractory clinical course and notable morbidity of surgical excision necessitated the use of ablative laser in our case.
Tattoo granulomas with uveitis is a rare diagnosis with the potential for serious permanent sequelae including blindness. Existing treatments such as topical and oral corticosteroids, immunosuppressants, surgical excision, and Q-switched lasers all are possible options, but in a patient with progressive ocular symptoms with other potential rheumatologic conditions and sarcoidosis ruled out, fully ablative CO2 laser may be an effective treatment option. Our case demonstrated the successful treatment of TAGU with CO2 laser ablation. Given the unclear etiology of TAGU and the limited evidence on treatment options and efficacy, our case contributes to the body of literature that can inform clinical management of this unusual and serious reaction.
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32:1852-1861. doi:10.1111/jdv.15070
- Tiew S. Tattoo-associated panuveitis: a 10-year follow-up. Eur J Ophthalmol. 2019;29(1 suppl):18-21. doi:10.1177/1120672119846341
- Rorsman H, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2:27-28. doi:10.1016/s0140-6736(69)92600-2
- Ostheimer TA, Burkholder BM, Leung TG, et al. Tattoo-associated uveitis. Am J Ophthalmol. 2014;158:637-643.e1. doi:10.1016/j.ajo.2014.05.019
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-nodular reactions in black tattoos as markers of sarcoidosis: study of 92 tattoo reactions from a hospital material. Dermatology. 2016;232:679-686. doi:10.1159/000453315
- Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet. 2014;383: 1155-1167. doi:10.1016/S0140-6736(13)60680-7
- Kluger N. Epidemiology of tattoos in industrialized countries. Curr Probl Dermatol. 2015;48:6-20. doi:10.1159/000369175
- Serup J, Hutton Carlsen K, Dommershausen N, et al. Identification of pigments related to allergic tattoo reactions in 104 human skin biopsies. Contact Dermatitis. 2020;82:73-82. doi:10.1111/cod.13423
- Mansour AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. Am J Ophthalmol. 1991;111:515-516. doi:10.1016/s0002-9394(14)72395-5
- Reddy AK, Shildkrot Y, Newman SA, et al. T-lymphocyte predominance and cellular atypia in tattoo-associated uveitis. JAMA Ophthalmol. 2015;133:1356-1357. doi:10.1001/jamaophthalmol.2015.3354
- Wenzel SM. Current concepts in laser tattoo removal. Skin Therapy Lett. 2010;15:3-5.
- van der Bent SAS, Huisman S, Rustemeyer T, et al. Ablative laser surgery for allergic tattoo reactions: a retrospective study. mLasers Med Sci. 2021;36:1241-1248. doi:10.1007/s10103-020-03164-2
- Yumeen S, Khan T. Laser carbon dioxide resurfacing. In: StatPearls. StatPearls Publishing; April 23, 2023. Accessed March 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560544/
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32:1852-1861. doi:10.1111/jdv.15070
- Tiew S. Tattoo-associated panuveitis: a 10-year follow-up. Eur J Ophthalmol. 2019;29(1 suppl):18-21. doi:10.1177/1120672119846341
- Rorsman H, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2:27-28. doi:10.1016/s0140-6736(69)92600-2
- Ostheimer TA, Burkholder BM, Leung TG, et al. Tattoo-associated uveitis. Am J Ophthalmol. 2014;158:637-643.e1. doi:10.1016/j.ajo.2014.05.019
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-nodular reactions in black tattoos as markers of sarcoidosis: study of 92 tattoo reactions from a hospital material. Dermatology. 2016;232:679-686. doi:10.1159/000453315
- Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet. 2014;383: 1155-1167. doi:10.1016/S0140-6736(13)60680-7
- Kluger N. Epidemiology of tattoos in industrialized countries. Curr Probl Dermatol. 2015;48:6-20. doi:10.1159/000369175
- Serup J, Hutton Carlsen K, Dommershausen N, et al. Identification of pigments related to allergic tattoo reactions in 104 human skin biopsies. Contact Dermatitis. 2020;82:73-82. doi:10.1111/cod.13423
- Mansour AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. Am J Ophthalmol. 1991;111:515-516. doi:10.1016/s0002-9394(14)72395-5
- Reddy AK, Shildkrot Y, Newman SA, et al. T-lymphocyte predominance and cellular atypia in tattoo-associated uveitis. JAMA Ophthalmol. 2015;133:1356-1357. doi:10.1001/jamaophthalmol.2015.3354
- Wenzel SM. Current concepts in laser tattoo removal. Skin Therapy Lett. 2010;15:3-5.
- van der Bent SAS, Huisman S, Rustemeyer T, et al. Ablative laser surgery for allergic tattoo reactions: a retrospective study. mLasers Med Sci. 2021;36:1241-1248. doi:10.1007/s10103-020-03164-2
- Yumeen S, Khan T. Laser carbon dioxide resurfacing. In: StatPearls. StatPearls Publishing; April 23, 2023. Accessed March 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560544/
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
PRACTICE POINTS
- Dermatologists should be aware that uveitis can develop as a delayed hypersensitivity reaction to tattoo ink, particularly in patients with blue ink tattoos.
- It is important to rule out systemic conditions such as sarcoidosis in patients presenting with uveitis and a history of tattoos.
- In a patient with progressive ocular symptoms, carbon dioxide laser ablation may be an effective treatment option if other potential rheumatologic conditions and sarcoidosis have been ruled out and other therapies have not resulted in improvement of symptoms.
- Continuous monitoring of ocular symptoms and intraocular pressure is vital to prevent complications such as glaucoma and potential blindness.

Pseudoverrucous Papules and Nodules Around a Surgical Stoma
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
To the Editor:
A 22-year-old man was referred to our dermatology outpatient department for wartlike growths that gradually developed around a postoperative enteroatmospheric fistula and stoma over the past 4 months. The patient presented for an emergency exploratory laparotomy with a history of perforation peritonitis 1.5 years prior to the current presentation. He also had a small bowel obstruction 5 months prior to the current presentation that resulted in the resection of a large segment of the small bowel. He underwent a diverting loop ileostomy when the abdominal closure was not achieved because of bowel edema, following which he developed a postoperative enteroatmospheric fistula. In addition, the stoma retracted and was followed by dermal dehiscence, which led to notable leakage and resulted in heavy fecal contamination of the midline wound.
At the current presentation, physical examination revealed multiple grayish-white, dome-shaped, moist papules coalescing to form a peristomal pseudoverrucous mass on the lower side of the stoma (Figure 1). The patient experienced mild itching. The lesion showed no signs of erosion, bleeding, or purulent discharge, and there were no nearby lumps or enlarged lymph nodes. The differential diagnosis included peristomal pyoderma gangrenosum, human papillomavirus (HPV) infection, pseudoverrucous papules and nodules (PPNs), squamous cell carcinoma, and exuberant granulation tissue. A skin biopsy was performed, and histopathology revealed hyperkeratosis, moderate papillomatosis, and marked acanthotic hyperplasia seen as downgrowths into the dermis (Figure 2). No koilocytes, atypia, or mitotic figures were present. Abundant neutrophils and few eosinophils were seen in the dermal infiltrate. A final diagnosis of PPN was made based on clinicopathologic correlation. The patient was advised to use a smaller stoma bag and to change the collection pouch frequently to reduce skin contact with fecal matter.
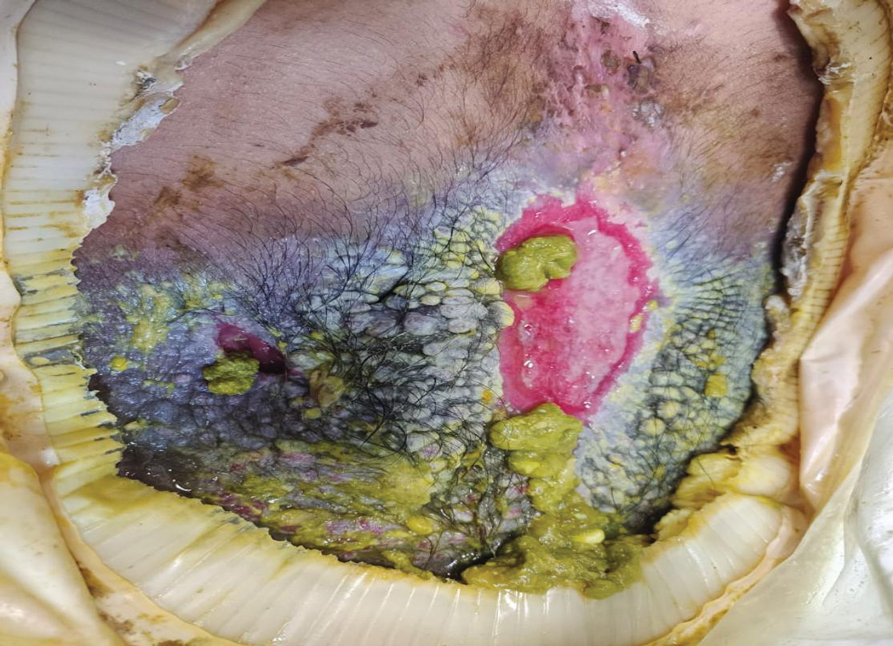
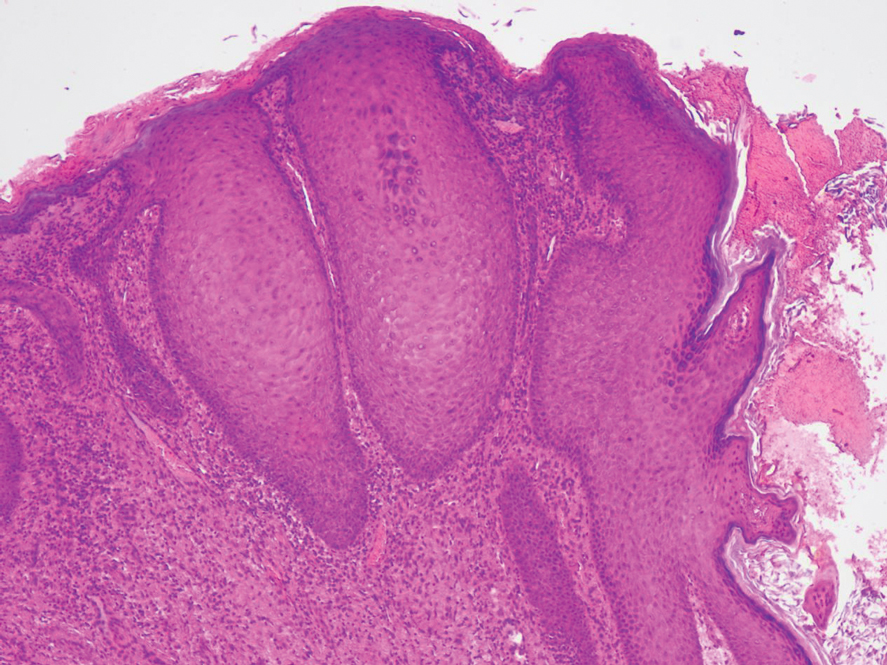
Peristomal skin conditions are reported in 18% to 55% of patients with stomas and include allergic contact dermatitis, mechanical dermatitis, infections, pyoderma gangrenosum, and irritant contact dermatitis.1,2 Pseudoverrucous papules (also called chronic papillomatous dermatitis or pseudoverrucous lesions) is a rare dermatologic complication found on the skin around stomas,3 most commonly around urostomy stomas. The presence of PPNs around colostomy stomas and the perianal region is extremely rare.2,4 This condition is the result of chronic irritant dermatitis from frequent exposure to urine or feces, leading to maceration and epidermal hyperplasia. It occurs because of improper sizing of the stoma bag or incorrect positioning or construction of the stoma.5
the overuse of topical benzocaine-resorcinol, leading to chronic irritation.6 It is clinically characterized by multiple grayish-white, wartlike, confluent papulonodules around areas chronically exposed to moisture. Differential diagnoses such as secondary neoplasms, HPV infection, exuberant granulation tissue, and candidal infections should be considered.3 Final diagnosis is based on clinicopathologic findings, similar to our case. Epidermal growth factor and transforming growth factor are thought to play a role in the pathophysiology of pseudoepitheliomatous hyperplasia. Increased expression of these mediators leads to proliferation of the epidermis into the dermis.7 The role of HPV in PPN remains unclear, as not all PPN lesions are positive for HPV and the cutaneous lesions resolve once the source of irritation is removed. Recommended treatment includes local skin care; stoma refitting; and, in severe cases, excision and revision of the stoma.2 Dermatologists must be aware of this often-underdiagnosed condition.
- Alslaim F, Al Farajat F, Alslaim HS, et al. Etiology and management of peristomal pseudoepitheliomatous hyperplasia. Cureus. 2021;13 :E20196. doi:10.7759/cureus.20196
- Rambhia PH, Conic RZ, Honda K, et al. Chronic papillomatous dermatitis in a patient with a urinary ileal diversion: a case report and review of the literature. Dermatol Arch. 2017;1:47-50. doi:10.36959/661/297
- Latour-Álvarez I, García-Peris E, Pestana-Eliche MM, et al. Nodular peristomal lesions. Actas Dermosifiliogr. 2016;108:363-364. doi:10.1016/j.ad.2016.02.018
- Dandale A, Dhurat R, Ghate S. Perianal pseudoverrucous papules and nodules. Indian J Sex Transm Dis AIDS. 2013;34:44-46. doi:10.4103/0253-7184.112939
- Brogna L. Prevention and management of pseudoverrucous lesions: a review and case scenarios. Adv Skin Wound Care. 2021;34:461-471. doi:10.1097/01.ASW.0000758620.93518.39
- Robson KJ, Maughan JA, Purcell SD, et al. Erosive papulonodular dermatosis associated with topical benzocaine: a report of two cases and evidence that granuloma gluteale, pseudoverrucous papules, and Jacquet’s erosive dermatitis are a disease spectrum. J Am Acad Dermatol. 2006;55(5 suppl):S74-S80. doi:10.1016/j .jaad.2005.12.025
- Oğuz ID, Vural S, Cinar E, et al. Peristomal pseudoverrucous lesions: a rare skin complication of colostomy. Cureus. 2023;15:E38068. doi:10.7759/cureus.38068
To the Editor:
A 22-year-old man was referred to our dermatology outpatient department for wartlike growths that gradually developed around a postoperative enteroatmospheric fistula and stoma over the past 4 months. The patient presented for an emergency exploratory laparotomy with a history of perforation peritonitis 1.5 years prior to the current presentation. He also had a small bowel obstruction 5 months prior to the current presentation that resulted in the resection of a large segment of the small bowel. He underwent a diverting loop ileostomy when the abdominal closure was not achieved because of bowel edema, following which he developed a postoperative enteroatmospheric fistula. In addition, the stoma retracted and was followed by dermal dehiscence, which led to notable leakage and resulted in heavy fecal contamination of the midline wound.
At the current presentation, physical examination revealed multiple grayish-white, dome-shaped, moist papules coalescing to form a peristomal pseudoverrucous mass on the lower side of the stoma (Figure 1). The patient experienced mild itching. The lesion showed no signs of erosion, bleeding, or purulent discharge, and there were no nearby lumps or enlarged lymph nodes. The differential diagnosis included peristomal pyoderma gangrenosum, human papillomavirus (HPV) infection, pseudoverrucous papules and nodules (PPNs), squamous cell carcinoma, and exuberant granulation tissue. A skin biopsy was performed, and histopathology revealed hyperkeratosis, moderate papillomatosis, and marked acanthotic hyperplasia seen as downgrowths into the dermis (Figure 2). No koilocytes, atypia, or mitotic figures were present. Abundant neutrophils and few eosinophils were seen in the dermal infiltrate. A final diagnosis of PPN was made based on clinicopathologic correlation. The patient was advised to use a smaller stoma bag and to change the collection pouch frequently to reduce skin contact with fecal matter.
Peristomal skin conditions are reported in 18% to 55% of patients with stomas and include allergic contact dermatitis, mechanical dermatitis, infections, pyoderma gangrenosum, and irritant contact dermatitis.1,2 Pseudoverrucous papules (also called chronic papillomatous dermatitis or pseudoverrucous lesions) is a rare dermatologic complication found on the skin around stomas,3 most commonly around urostomy stomas. The presence of PPNs around colostomy stomas and the perianal region is extremely rare.2,4 This condition is the result of chronic irritant dermatitis from frequent exposure to urine or feces, leading to maceration and epidermal hyperplasia. It occurs because of improper sizing of the stoma bag or incorrect positioning or construction of the stoma.5
the overuse of topical benzocaine-resorcinol, leading to chronic irritation.6 It is clinically characterized by multiple grayish-white, wartlike, confluent papulonodules around areas chronically exposed to moisture. Differential diagnoses such as secondary neoplasms, HPV infection, exuberant granulation tissue, and candidal infections should be considered.3 Final diagnosis is based on clinicopathologic findings, similar to our case. Epidermal growth factor and transforming growth factor are thought to play a role in the pathophysiology of pseudoepitheliomatous hyperplasia. Increased expression of these mediators leads to proliferation of the epidermis into the dermis.7 The role of HPV in PPN remains unclear, as not all PPN lesions are positive for HPV and the cutaneous lesions resolve once the source of irritation is removed. Recommended treatment includes local skin care; stoma refitting; and, in severe cases, excision and revision of the stoma.2 Dermatologists must be aware of this often-underdiagnosed condition.
To the Editor:
A 22-year-old man was referred to our dermatology outpatient department for wartlike growths that gradually developed around a postoperative enteroatmospheric fistula and stoma over the past 4 months. The patient presented for an emergency exploratory laparotomy with a history of perforation peritonitis 1.5 years prior to the current presentation. He also had a small bowel obstruction 5 months prior to the current presentation that resulted in the resection of a large segment of the small bowel. He underwent a diverting loop ileostomy when the abdominal closure was not achieved because of bowel edema, following which he developed a postoperative enteroatmospheric fistula. In addition, the stoma retracted and was followed by dermal dehiscence, which led to notable leakage and resulted in heavy fecal contamination of the midline wound.
At the current presentation, physical examination revealed multiple grayish-white, dome-shaped, moist papules coalescing to form a peristomal pseudoverrucous mass on the lower side of the stoma (Figure 1). The patient experienced mild itching. The lesion showed no signs of erosion, bleeding, or purulent discharge, and there were no nearby lumps or enlarged lymph nodes. The differential diagnosis included peristomal pyoderma gangrenosum, human papillomavirus (HPV) infection, pseudoverrucous papules and nodules (PPNs), squamous cell carcinoma, and exuberant granulation tissue. A skin biopsy was performed, and histopathology revealed hyperkeratosis, moderate papillomatosis, and marked acanthotic hyperplasia seen as downgrowths into the dermis (Figure 2). No koilocytes, atypia, or mitotic figures were present. Abundant neutrophils and few eosinophils were seen in the dermal infiltrate. A final diagnosis of PPN was made based on clinicopathologic correlation. The patient was advised to use a smaller stoma bag and to change the collection pouch frequently to reduce skin contact with fecal matter.
Peristomal skin conditions are reported in 18% to 55% of patients with stomas and include allergic contact dermatitis, mechanical dermatitis, infections, pyoderma gangrenosum, and irritant contact dermatitis.1,2 Pseudoverrucous papules (also called chronic papillomatous dermatitis or pseudoverrucous lesions) is a rare dermatologic complication found on the skin around stomas,3 most commonly around urostomy stomas. The presence of PPNs around colostomy stomas and the perianal region is extremely rare.2,4 This condition is the result of chronic irritant dermatitis from frequent exposure to urine or feces, leading to maceration and epidermal hyperplasia. It occurs because of improper sizing of the stoma bag or incorrect positioning or construction of the stoma.5
the overuse of topical benzocaine-resorcinol, leading to chronic irritation.6 It is clinically characterized by multiple grayish-white, wartlike, confluent papulonodules around areas chronically exposed to moisture. Differential diagnoses such as secondary neoplasms, HPV infection, exuberant granulation tissue, and candidal infections should be considered.3 Final diagnosis is based on clinicopathologic findings, similar to our case. Epidermal growth factor and transforming growth factor are thought to play a role in the pathophysiology of pseudoepitheliomatous hyperplasia. Increased expression of these mediators leads to proliferation of the epidermis into the dermis.7 The role of HPV in PPN remains unclear, as not all PPN lesions are positive for HPV and the cutaneous lesions resolve once the source of irritation is removed. Recommended treatment includes local skin care; stoma refitting; and, in severe cases, excision and revision of the stoma.2 Dermatologists must be aware of this often-underdiagnosed condition.
- Alslaim F, Al Farajat F, Alslaim HS, et al. Etiology and management of peristomal pseudoepitheliomatous hyperplasia. Cureus. 2021;13 :E20196. doi:10.7759/cureus.20196
- Rambhia PH, Conic RZ, Honda K, et al. Chronic papillomatous dermatitis in a patient with a urinary ileal diversion: a case report and review of the literature. Dermatol Arch. 2017;1:47-50. doi:10.36959/661/297
- Latour-Álvarez I, García-Peris E, Pestana-Eliche MM, et al. Nodular peristomal lesions. Actas Dermosifiliogr. 2016;108:363-364. doi:10.1016/j.ad.2016.02.018
- Dandale A, Dhurat R, Ghate S. Perianal pseudoverrucous papules and nodules. Indian J Sex Transm Dis AIDS. 2013;34:44-46. doi:10.4103/0253-7184.112939
- Brogna L. Prevention and management of pseudoverrucous lesions: a review and case scenarios. Adv Skin Wound Care. 2021;34:461-471. doi:10.1097/01.ASW.0000758620.93518.39
- Robson KJ, Maughan JA, Purcell SD, et al. Erosive papulonodular dermatosis associated with topical benzocaine: a report of two cases and evidence that granuloma gluteale, pseudoverrucous papules, and Jacquet’s erosive dermatitis are a disease spectrum. J Am Acad Dermatol. 2006;55(5 suppl):S74-S80. doi:10.1016/j .jaad.2005.12.025
- Oğuz ID, Vural S, Cinar E, et al. Peristomal pseudoverrucous lesions: a rare skin complication of colostomy. Cureus. 2023;15:E38068. doi:10.7759/cureus.38068
- Alslaim F, Al Farajat F, Alslaim HS, et al. Etiology and management of peristomal pseudoepitheliomatous hyperplasia. Cureus. 2021;13 :E20196. doi:10.7759/cureus.20196
- Rambhia PH, Conic RZ, Honda K, et al. Chronic papillomatous dermatitis in a patient with a urinary ileal diversion: a case report and review of the literature. Dermatol Arch. 2017;1:47-50. doi:10.36959/661/297
- Latour-Álvarez I, García-Peris E, Pestana-Eliche MM, et al. Nodular peristomal lesions. Actas Dermosifiliogr. 2016;108:363-364. doi:10.1016/j.ad.2016.02.018
- Dandale A, Dhurat R, Ghate S. Perianal pseudoverrucous papules and nodules. Indian J Sex Transm Dis AIDS. 2013;34:44-46. doi:10.4103/0253-7184.112939
- Brogna L. Prevention and management of pseudoverrucous lesions: a review and case scenarios. Adv Skin Wound Care. 2021;34:461-471. doi:10.1097/01.ASW.0000758620.93518.39
- Robson KJ, Maughan JA, Purcell SD, et al. Erosive papulonodular dermatosis associated with topical benzocaine: a report of two cases and evidence that granuloma gluteale, pseudoverrucous papules, and Jacquet’s erosive dermatitis are a disease spectrum. J Am Acad Dermatol. 2006;55(5 suppl):S74-S80. doi:10.1016/j .jaad.2005.12.025
- Oğuz ID, Vural S, Cinar E, et al. Peristomal pseudoverrucous lesions: a rare skin complication of colostomy. Cureus. 2023;15:E38068. doi:10.7759/cureus.38068
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
PRACTICE POINTS
- Pseudoverrucous papules and nodules (PPNs) can develop around stomas due to chronic irritant dermatitis from fecal or urinary exposure.
- Proper stoma management, including the use of appropriately sized stoma bags and frequent changes, is essential to prevent skin complications such as PPN.
- When evaluating peristomal lesions, consider a broad differential diagnosis, including infections, neoplasms, and dermatitis, and ensure thorough clinicopathologic correlation for accurate diagnosis and treatment.

Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
To the Editor:
Idiopathic gingival papillokeratosis with crypt formation (IGPC) is an uncommon benign condition that first was reported in 1967.1 The condition manifests as white plaques with a papillary appearance on the gingival tissue. While data on the prevalence of IGPC are limited, it is known to occur more frequently in younger patients (ie, 9-24 years1-3) and has been linked to use of orthodontic appliances.3,4 The lesions typically are asymptomatic with a bilateral appearance along the mucogingival junction. Research on IGPC has not identified the underlying mechanisms that trigger the hyperkeratinization and papillary alterations within the gingival tissue.
Management of IGPC can be challenging due to the rarity of the condition and its uncertain pathogenesis. Wiping or brushing the affected area offers only temporary improvement of symptoms and the appearance of the lesions. Surgical excision is another option; however, it can result in aesthetic and/or functional periodontal defects.2 Alternately, employing methods such as wiping or brushing the affected area offers only transient and temporary results in managing the condition. Additional investigative approaches and clinical studies are needed to identify more effective therapeutic modalities for the management of IGPC, particularly in pediatric patients, in whom aesthetic results may take on a heightened importance.1-3 We report a case of IGPC in which cryotherapy yielded satisfactory results with no recurrence of the lesions.
A 32-year-old woman presented to the dental clinic with white spots on the gingiva of 5 months’ duration. The patient reported a history of smoking cigarettes (3 packs per year) and drinking alcohol in social situations; her medical history was otherwise unremarkable. Clinical examination of the oral cavity revealed a bilateral, irregular, verrucouslike plaque throughout the vestibular upper attached gingiva. An incisional biopsy from the attached gingiva between teeth 13 and 23 was performed. Histopathologic analysis revealed parakeratosis and papillary acanthosis of the gingival mucosa associated with multifocal epithelial invaginations resembling crypts as well as long tapered epithelial ridges with no inflammation in the lamina propria. Based on the histopathologic findings, a diagnosis of IGPC was made (Figure 1).
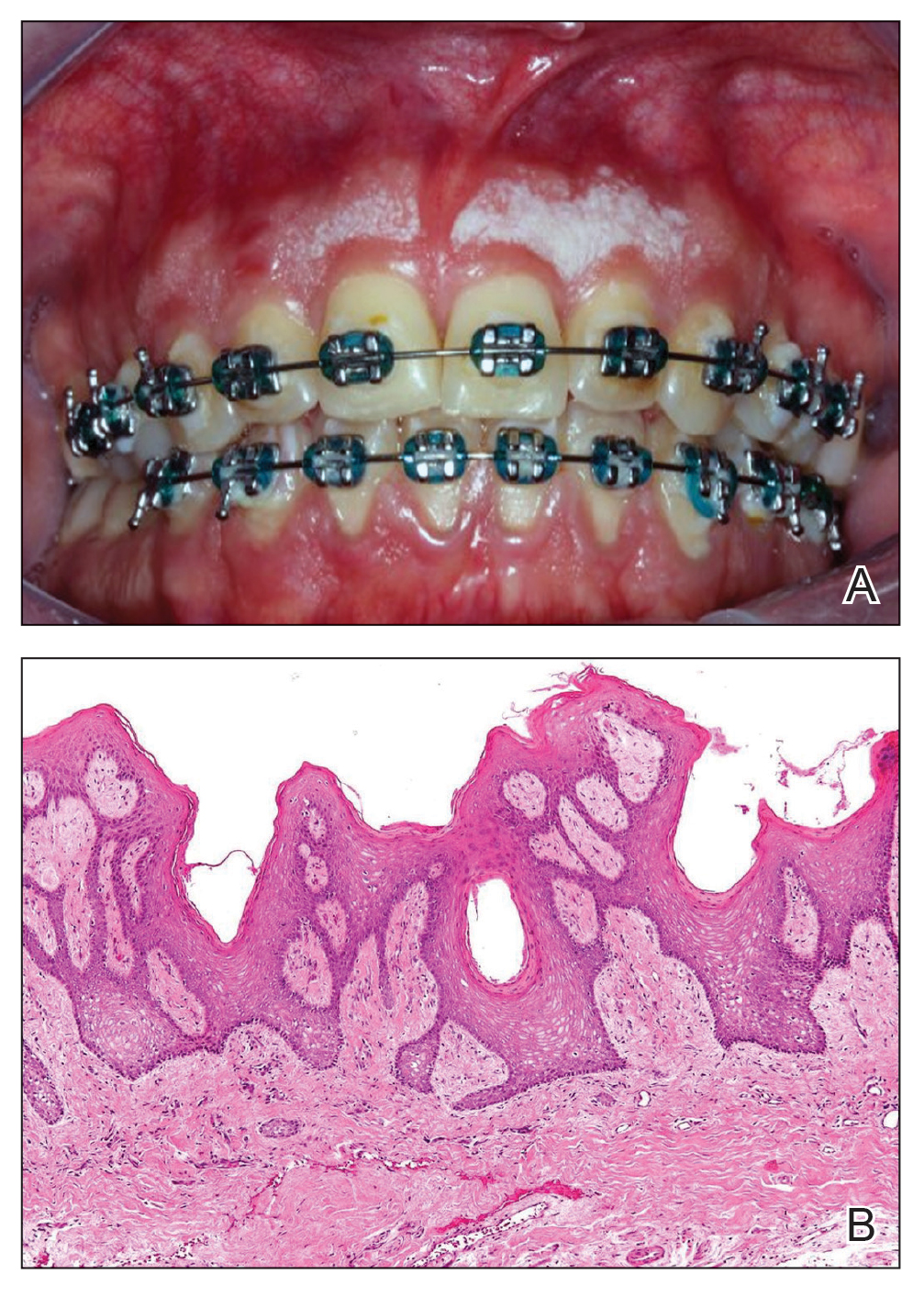
Given the patient’s clinical presentation, we suggested treatment with cryotherapy as a minimally invasive option that would preserve the gingival architecture and aesthetics while avoiding the potential complications of surgical excision. The patient consented to the procedure, and liquid nitrogen was administered through a handheld device using a 0.6-mm aperture spray tip. During application, the spray tip was positioned at a distance of 0.5 to 1.0 cm from the labial marginal gingiva at about a 45° angle. The freeze/thaw cycle involved a continuous one-way spray application of liquid nitrogen onto the lesion until solid ice formed over the entire area, followed by a waiting period until gradual thawing occurred.
A total of 5 cryotherapy sessions were conducted over an 8-week period; no recurrence of the lesions was observed during a 2-year follow-up period (Figure 2).
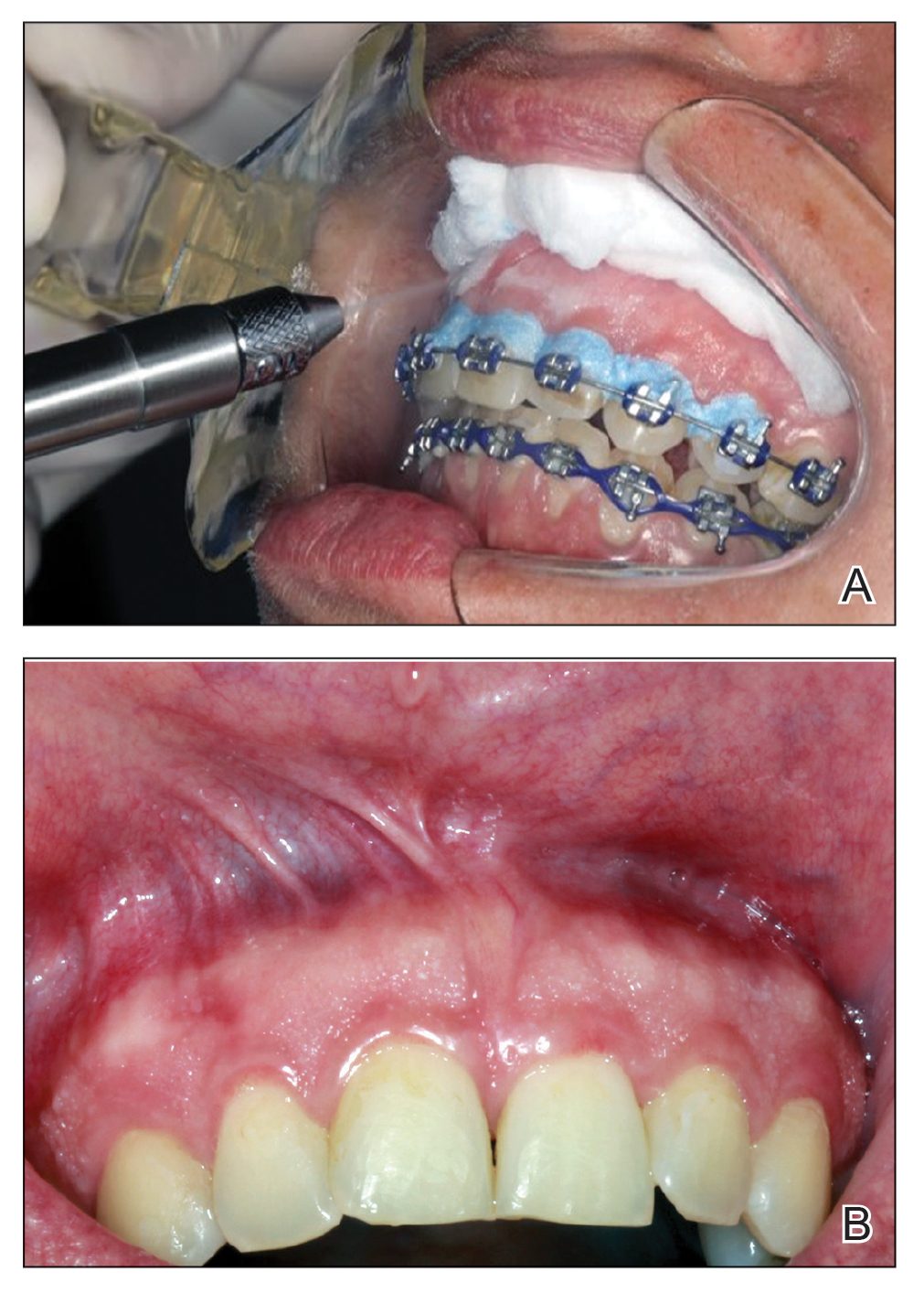
We present our case to add to the body of knowledge regarding management options for IGPC, specifically cryotherapy. Historically, brushing with a toothbrush and surgical excision have been the most commonly used interventions.2 Gently brushing the affected areas can help stimulate local blood circulation, which can improve the health of the gingival tissue, promote oxygenation and delivery of nutrients to the cells, and aid in the removal of metabolic waste. Surgical excision is the most commonly used treatment method for IGPC to ensure that the lesions are safely and completely removed; however, this option can result in aesthetic and/or functional periodontal defects. There also is a risk for recurrence, although Noonan et al2 reported no recurrence 4 years after performing a surgical excision for IGPC.
Cryotherapy reduces tissue sensitivity, provides local anesthesia, and reduces inflammation in the oral mucosa. Moreover, cryotherapy accelerates healing by stimulating vasoconstriction and reactive vasodilation, thus enhancing blood flow, oxygenation, and nutrient delivery for faster cell regeneration of the oral mucosa.4,5 Cryotherapy generally is regarded as a simple noninvasive procedure that is relatively safe when performed by qualified professionals.4,5 It can provide benefits such as minimal patient discomfort, rapid recovery, and potential reduction of complications associated with more invasive procedures.5
The efficacy of cryotherapy for IGPC may vary based on lesion severity, individual patient response, and the need for repeated treatment sessions. Robust scientific evidence concerning the long-term efficacy of cryotherapy as a treatment for IGPC is limited due to the rarity of this condition.
The etiopathogenesis of IGPC has been hypothesized to involve both genetic and environmental factors with equal significance. This suggestion is based on reports of IGPC occurring in multiple members of the same family and animal model studies indicating that gingival tissue is sensitive to environmental influences, such as nutritional factors.1,6 However, it is important to emphasize that these hypotheses remain speculative, and the true etiopathogenesis of IGPC remains uncertain.6 Microscopically, biopsy fragments from suspected cases of IGPC reveal gingival mucosa characterized by parakeratosis and papillary acanthosis accompanied by multifocal epithelial invaginations resembling crypts.2 Additionally, elongated and tapered epithelial ridges without inflammation in the lamina propria may be observed (as in our case), favoring the diagnosis of IGPC.3 The absence of inflammation is noteworthy because it suggests that the observed alterations are not attributed to typical inflammatory processes seen in some gingival conditions.
The limited number of studies reporting successful treatment outcomes with long-term follow-up for IGPC cases underscores the need for further exploration of effective treatment options. Cryotherapy emerges as a promising minimally invasive therapeutic approach, with our case offering support for its potential application. Additional research and clinical trials are essential to validate its efficacy and improve our understanding of cryotherapy as a treatment modality for IGPC lesions.
- Bennett JS, Grupe HE. Epithelial adnexal formations in human gingiva. Oral Surg Oral Med Oral Pathol. 1967;23:789-795. doi:10.1016/0030-4220(67)90371-4
- Noonan VL, Woo SB, Sundararajan D, et al. Idiopathic gingival papillokeratosis with crypt formation, a report of 7 cases of a previously undescribed entity: possible unusual oral epithelial nevus? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:358-364. doi:10.1016/j.oooo.2016.10.018
- Romo SA, de Arruda JAA, Nava FJT, et al. Idiopathic gingival papillokeratosis with crypt formation: a clinicopathological entity in the young population? Int J Dermatol. 2023;62:E291-E293. doi: 10.1111/ijd.16579
- Farah CS, Savage NW. Cryotherapy for treatment of oral lesions. Aust Dent J. 2006;51:2-5. doi:10.1111/j.1834-7819.2006.tb00392.x
- Nogueira VKC, Fernandes D, Navarro CM, et al. Cryotherapy for localized juvenile spongiotic gingival hyperplasia: preliminary findings on two cases. Int J Paediatr Dent. 2017;27:231-235. doi:10.1111/ipd.12278
- Bernick S, Bavetta LA. The development of gingival sebaceous-like glands and cysts in rats of the Holtzman strain. Oral Surg Oral Med Oral Pathol Oral Radiol. 1962;15:351-354. doi:10.1016/0030-4220(62)90116-0
To the Editor:
Idiopathic gingival papillokeratosis with crypt formation (IGPC) is an uncommon benign condition that first was reported in 1967.1 The condition manifests as white plaques with a papillary appearance on the gingival tissue. While data on the prevalence of IGPC are limited, it is known to occur more frequently in younger patients (ie, 9-24 years1-3) and has been linked to use of orthodontic appliances.3,4 The lesions typically are asymptomatic with a bilateral appearance along the mucogingival junction. Research on IGPC has not identified the underlying mechanisms that trigger the hyperkeratinization and papillary alterations within the gingival tissue.
Management of IGPC can be challenging due to the rarity of the condition and its uncertain pathogenesis. Wiping or brushing the affected area offers only temporary improvement of symptoms and the appearance of the lesions. Surgical excision is another option; however, it can result in aesthetic and/or functional periodontal defects.2 Alternately, employing methods such as wiping or brushing the affected area offers only transient and temporary results in managing the condition. Additional investigative approaches and clinical studies are needed to identify more effective therapeutic modalities for the management of IGPC, particularly in pediatric patients, in whom aesthetic results may take on a heightened importance.1-3 We report a case of IGPC in which cryotherapy yielded satisfactory results with no recurrence of the lesions.
A 32-year-old woman presented to the dental clinic with white spots on the gingiva of 5 months’ duration. The patient reported a history of smoking cigarettes (3 packs per year) and drinking alcohol in social situations; her medical history was otherwise unremarkable. Clinical examination of the oral cavity revealed a bilateral, irregular, verrucouslike plaque throughout the vestibular upper attached gingiva. An incisional biopsy from the attached gingiva between teeth 13 and 23 was performed. Histopathologic analysis revealed parakeratosis and papillary acanthosis of the gingival mucosa associated with multifocal epithelial invaginations resembling crypts as well as long tapered epithelial ridges with no inflammation in the lamina propria. Based on the histopathologic findings, a diagnosis of IGPC was made (Figure 1).
Given the patient’s clinical presentation, we suggested treatment with cryotherapy as a minimally invasive option that would preserve the gingival architecture and aesthetics while avoiding the potential complications of surgical excision. The patient consented to the procedure, and liquid nitrogen was administered through a handheld device using a 0.6-mm aperture spray tip. During application, the spray tip was positioned at a distance of 0.5 to 1.0 cm from the labial marginal gingiva at about a 45° angle. The freeze/thaw cycle involved a continuous one-way spray application of liquid nitrogen onto the lesion until solid ice formed over the entire area, followed by a waiting period until gradual thawing occurred.
A total of 5 cryotherapy sessions were conducted over an 8-week period; no recurrence of the lesions was observed during a 2-year follow-up period (Figure 2).
We present our case to add to the body of knowledge regarding management options for IGPC, specifically cryotherapy. Historically, brushing with a toothbrush and surgical excision have been the most commonly used interventions.2 Gently brushing the affected areas can help stimulate local blood circulation, which can improve the health of the gingival tissue, promote oxygenation and delivery of nutrients to the cells, and aid in the removal of metabolic waste. Surgical excision is the most commonly used treatment method for IGPC to ensure that the lesions are safely and completely removed; however, this option can result in aesthetic and/or functional periodontal defects. There also is a risk for recurrence, although Noonan et al2 reported no recurrence 4 years after performing a surgical excision for IGPC.
Cryotherapy reduces tissue sensitivity, provides local anesthesia, and reduces inflammation in the oral mucosa. Moreover, cryotherapy accelerates healing by stimulating vasoconstriction and reactive vasodilation, thus enhancing blood flow, oxygenation, and nutrient delivery for faster cell regeneration of the oral mucosa.4,5 Cryotherapy generally is regarded as a simple noninvasive procedure that is relatively safe when performed by qualified professionals.4,5 It can provide benefits such as minimal patient discomfort, rapid recovery, and potential reduction of complications associated with more invasive procedures.5
The efficacy of cryotherapy for IGPC may vary based on lesion severity, individual patient response, and the need for repeated treatment sessions. Robust scientific evidence concerning the long-term efficacy of cryotherapy as a treatment for IGPC is limited due to the rarity of this condition.
The etiopathogenesis of IGPC has been hypothesized to involve both genetic and environmental factors with equal significance. This suggestion is based on reports of IGPC occurring in multiple members of the same family and animal model studies indicating that gingival tissue is sensitive to environmental influences, such as nutritional factors.1,6 However, it is important to emphasize that these hypotheses remain speculative, and the true etiopathogenesis of IGPC remains uncertain.6 Microscopically, biopsy fragments from suspected cases of IGPC reveal gingival mucosa characterized by parakeratosis and papillary acanthosis accompanied by multifocal epithelial invaginations resembling crypts.2 Additionally, elongated and tapered epithelial ridges without inflammation in the lamina propria may be observed (as in our case), favoring the diagnosis of IGPC.3 The absence of inflammation is noteworthy because it suggests that the observed alterations are not attributed to typical inflammatory processes seen in some gingival conditions.
The limited number of studies reporting successful treatment outcomes with long-term follow-up for IGPC cases underscores the need for further exploration of effective treatment options. Cryotherapy emerges as a promising minimally invasive therapeutic approach, with our case offering support for its potential application. Additional research and clinical trials are essential to validate its efficacy and improve our understanding of cryotherapy as a treatment modality for IGPC lesions.
To the Editor:
Idiopathic gingival papillokeratosis with crypt formation (IGPC) is an uncommon benign condition that first was reported in 1967.1 The condition manifests as white plaques with a papillary appearance on the gingival tissue. While data on the prevalence of IGPC are limited, it is known to occur more frequently in younger patients (ie, 9-24 years1-3) and has been linked to use of orthodontic appliances.3,4 The lesions typically are asymptomatic with a bilateral appearance along the mucogingival junction. Research on IGPC has not identified the underlying mechanisms that trigger the hyperkeratinization and papillary alterations within the gingival tissue.
Management of IGPC can be challenging due to the rarity of the condition and its uncertain pathogenesis. Wiping or brushing the affected area offers only temporary improvement of symptoms and the appearance of the lesions. Surgical excision is another option; however, it can result in aesthetic and/or functional periodontal defects.2 Alternately, employing methods such as wiping or brushing the affected area offers only transient and temporary results in managing the condition. Additional investigative approaches and clinical studies are needed to identify more effective therapeutic modalities for the management of IGPC, particularly in pediatric patients, in whom aesthetic results may take on a heightened importance.1-3 We report a case of IGPC in which cryotherapy yielded satisfactory results with no recurrence of the lesions.
A 32-year-old woman presented to the dental clinic with white spots on the gingiva of 5 months’ duration. The patient reported a history of smoking cigarettes (3 packs per year) and drinking alcohol in social situations; her medical history was otherwise unremarkable. Clinical examination of the oral cavity revealed a bilateral, irregular, verrucouslike plaque throughout the vestibular upper attached gingiva. An incisional biopsy from the attached gingiva between teeth 13 and 23 was performed. Histopathologic analysis revealed parakeratosis and papillary acanthosis of the gingival mucosa associated with multifocal epithelial invaginations resembling crypts as well as long tapered epithelial ridges with no inflammation in the lamina propria. Based on the histopathologic findings, a diagnosis of IGPC was made (Figure 1).
Given the patient’s clinical presentation, we suggested treatment with cryotherapy as a minimally invasive option that would preserve the gingival architecture and aesthetics while avoiding the potential complications of surgical excision. The patient consented to the procedure, and liquid nitrogen was administered through a handheld device using a 0.6-mm aperture spray tip. During application, the spray tip was positioned at a distance of 0.5 to 1.0 cm from the labial marginal gingiva at about a 45° angle. The freeze/thaw cycle involved a continuous one-way spray application of liquid nitrogen onto the lesion until solid ice formed over the entire area, followed by a waiting period until gradual thawing occurred.
A total of 5 cryotherapy sessions were conducted over an 8-week period; no recurrence of the lesions was observed during a 2-year follow-up period (Figure 2).
We present our case to add to the body of knowledge regarding management options for IGPC, specifically cryotherapy. Historically, brushing with a toothbrush and surgical excision have been the most commonly used interventions.2 Gently brushing the affected areas can help stimulate local blood circulation, which can improve the health of the gingival tissue, promote oxygenation and delivery of nutrients to the cells, and aid in the removal of metabolic waste. Surgical excision is the most commonly used treatment method for IGPC to ensure that the lesions are safely and completely removed; however, this option can result in aesthetic and/or functional periodontal defects. There also is a risk for recurrence, although Noonan et al2 reported no recurrence 4 years after performing a surgical excision for IGPC.
Cryotherapy reduces tissue sensitivity, provides local anesthesia, and reduces inflammation in the oral mucosa. Moreover, cryotherapy accelerates healing by stimulating vasoconstriction and reactive vasodilation, thus enhancing blood flow, oxygenation, and nutrient delivery for faster cell regeneration of the oral mucosa.4,5 Cryotherapy generally is regarded as a simple noninvasive procedure that is relatively safe when performed by qualified professionals.4,5 It can provide benefits such as minimal patient discomfort, rapid recovery, and potential reduction of complications associated with more invasive procedures.5
The efficacy of cryotherapy for IGPC may vary based on lesion severity, individual patient response, and the need for repeated treatment sessions. Robust scientific evidence concerning the long-term efficacy of cryotherapy as a treatment for IGPC is limited due to the rarity of this condition.
The etiopathogenesis of IGPC has been hypothesized to involve both genetic and environmental factors with equal significance. This suggestion is based on reports of IGPC occurring in multiple members of the same family and animal model studies indicating that gingival tissue is sensitive to environmental influences, such as nutritional factors.1,6 However, it is important to emphasize that these hypotheses remain speculative, and the true etiopathogenesis of IGPC remains uncertain.6 Microscopically, biopsy fragments from suspected cases of IGPC reveal gingival mucosa characterized by parakeratosis and papillary acanthosis accompanied by multifocal epithelial invaginations resembling crypts.2 Additionally, elongated and tapered epithelial ridges without inflammation in the lamina propria may be observed (as in our case), favoring the diagnosis of IGPC.3 The absence of inflammation is noteworthy because it suggests that the observed alterations are not attributed to typical inflammatory processes seen in some gingival conditions.
The limited number of studies reporting successful treatment outcomes with long-term follow-up for IGPC cases underscores the need for further exploration of effective treatment options. Cryotherapy emerges as a promising minimally invasive therapeutic approach, with our case offering support for its potential application. Additional research and clinical trials are essential to validate its efficacy and improve our understanding of cryotherapy as a treatment modality for IGPC lesions.
- Bennett JS, Grupe HE. Epithelial adnexal formations in human gingiva. Oral Surg Oral Med Oral Pathol. 1967;23:789-795. doi:10.1016/0030-4220(67)90371-4
- Noonan VL, Woo SB, Sundararajan D, et al. Idiopathic gingival papillokeratosis with crypt formation, a report of 7 cases of a previously undescribed entity: possible unusual oral epithelial nevus? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:358-364. doi:10.1016/j.oooo.2016.10.018
- Romo SA, de Arruda JAA, Nava FJT, et al. Idiopathic gingival papillokeratosis with crypt formation: a clinicopathological entity in the young population? Int J Dermatol. 2023;62:E291-E293. doi: 10.1111/ijd.16579
- Farah CS, Savage NW. Cryotherapy for treatment of oral lesions. Aust Dent J. 2006;51:2-5. doi:10.1111/j.1834-7819.2006.tb00392.x
- Nogueira VKC, Fernandes D, Navarro CM, et al. Cryotherapy for localized juvenile spongiotic gingival hyperplasia: preliminary findings on two cases. Int J Paediatr Dent. 2017;27:231-235. doi:10.1111/ipd.12278
- Bernick S, Bavetta LA. The development of gingival sebaceous-like glands and cysts in rats of the Holtzman strain. Oral Surg Oral Med Oral Pathol Oral Radiol. 1962;15:351-354. doi:10.1016/0030-4220(62)90116-0
- Bennett JS, Grupe HE. Epithelial adnexal formations in human gingiva. Oral Surg Oral Med Oral Pathol. 1967;23:789-795. doi:10.1016/0030-4220(67)90371-4
- Noonan VL, Woo SB, Sundararajan D, et al. Idiopathic gingival papillokeratosis with crypt formation, a report of 7 cases of a previously undescribed entity: possible unusual oral epithelial nevus? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:358-364. doi:10.1016/j.oooo.2016.10.018
- Romo SA, de Arruda JAA, Nava FJT, et al. Idiopathic gingival papillokeratosis with crypt formation: a clinicopathological entity in the young population? Int J Dermatol. 2023;62:E291-E293. doi: 10.1111/ijd.16579
- Farah CS, Savage NW. Cryotherapy for treatment of oral lesions. Aust Dent J. 2006;51:2-5. doi:10.1111/j.1834-7819.2006.tb00392.x
- Nogueira VKC, Fernandes D, Navarro CM, et al. Cryotherapy for localized juvenile spongiotic gingival hyperplasia: preliminary findings on two cases. Int J Paediatr Dent. 2017;27:231-235. doi:10.1111/ipd.12278
- Bernick S, Bavetta LA. The development of gingival sebaceous-like glands and cysts in rats of the Holtzman strain. Oral Surg Oral Med Oral Pathol Oral Radiol. 1962;15:351-354. doi:10.1016/0030-4220(62)90116-0
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
PRACTICE POINTS
- Surgical excision is an effective treatment for idiopathic gingival papillokeratosis with crypt formation (IGPC) but may result in periodontal defects that impact the aesthetic outcome.
- Cryotherapy is a novel therapeutic intervention for IGPC.

Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
To the Editor:
Cutaneous metastasis of prostate cancer is rare and portends a bleak prognosis. Diagnosis of the primary cancer can be challenging, as skin metastasis can mimic a variety of conditions. We report a case of metastatic prostatic adenocarcinoma confirmed via biopsy of a new skin lesion.
A 97-year-old man presented to the dermatology clinic for routine follow-up of psoriasis. During the visit, a family member mentioned a new bleeding lesion on the left shoulder. It was not known how long the lesion had been present. Four months prior, the patient had a prostate-specific antigen (PSA) level of 582 ng/mL (reference range, 0-6.5 ng/mL), and computed tomography of the chest had shown innumerable pulmonary nodules in addition to lymphadenopathy of the left axilla, clavicle, and mediastinum. The imaging was ordered by the patient’s urologist as part of routine workup, as he had a history of obstructive renal failure and was being monitored for an indwelling catheter. Two months later, a bone scan ordered by the urologist due to high PSA levels showed extensive osteoblastic metastatic disease throughout the axial and proximal appendicular skeleton. The elevated PSA levels and findings of pulmonary and osteoblastic metastasis suggested a diagnosis of metastatic prostatic adenocarcinoma, but no confirmatory biopsy was performed following the imaging because the patient’s family declined additional workup or intervention.
Physical examination at the current presentation revealed an 8-mm brown papule with an overlying blue-white veil (Figure 1). There were no other skin findings. Primary differential diagnoses included metastatic prostate cancer, nodular melanoma, and traumatized seborrheic keratosis. A shave biopsy of the lesion showed multiple glandular structures infiltrating the dermis lined by monomorphic epithelial cells with prominent eosinophilic nucleoli (Figures 2 and 3). Focal cribriform architecture of the glands was present as well as dermal hemorrhage and a lymphohistiocytic infiltrate (Figure 2A). Interestingly, in-transit vascular metastases were confirmed with the support of ERG, CD34, and CD31 immunohistochemical staining of the vessels.
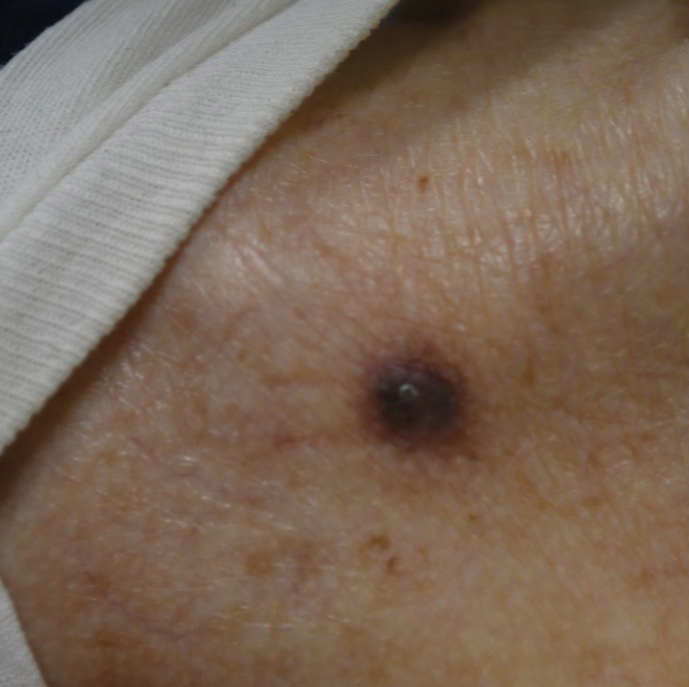
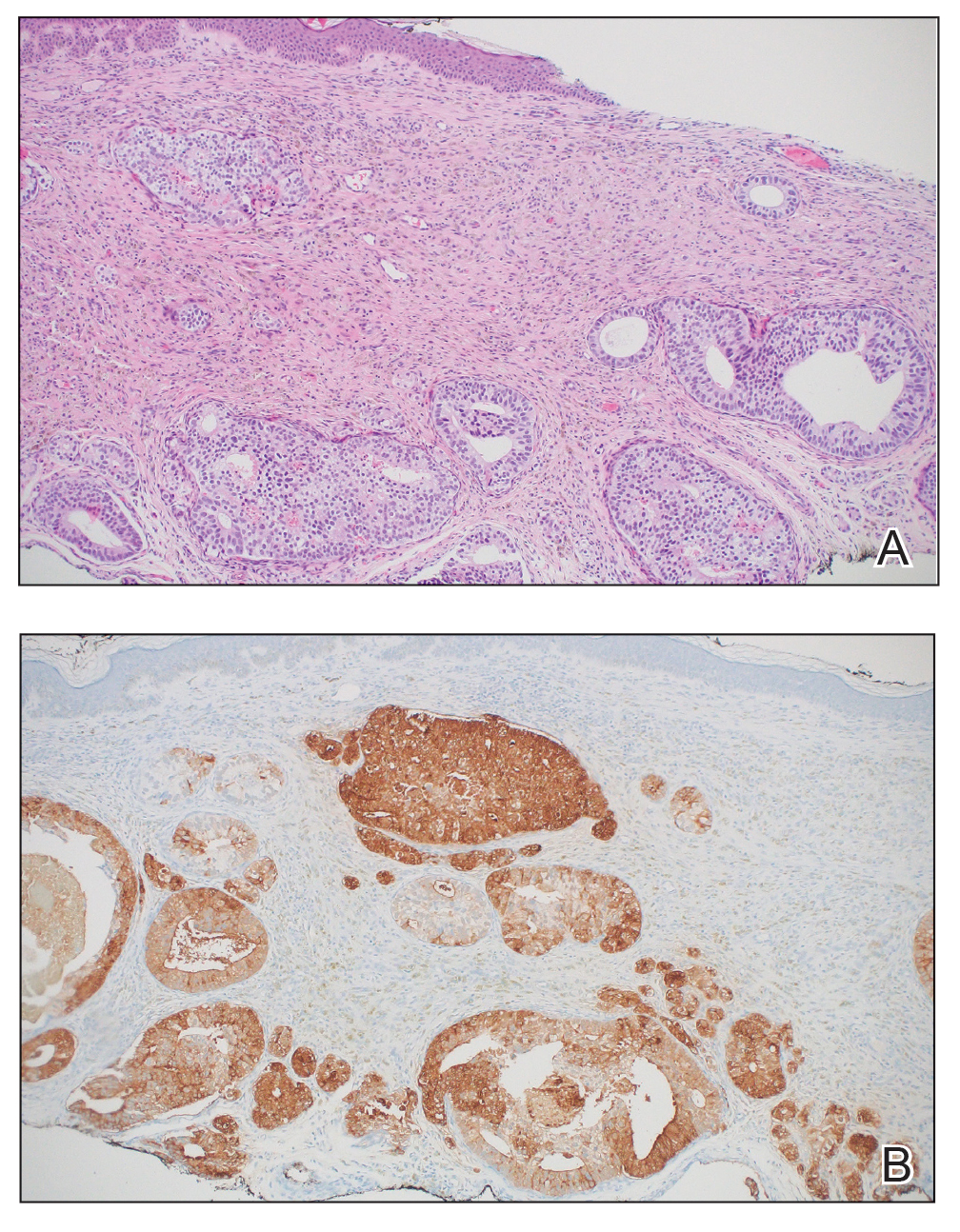
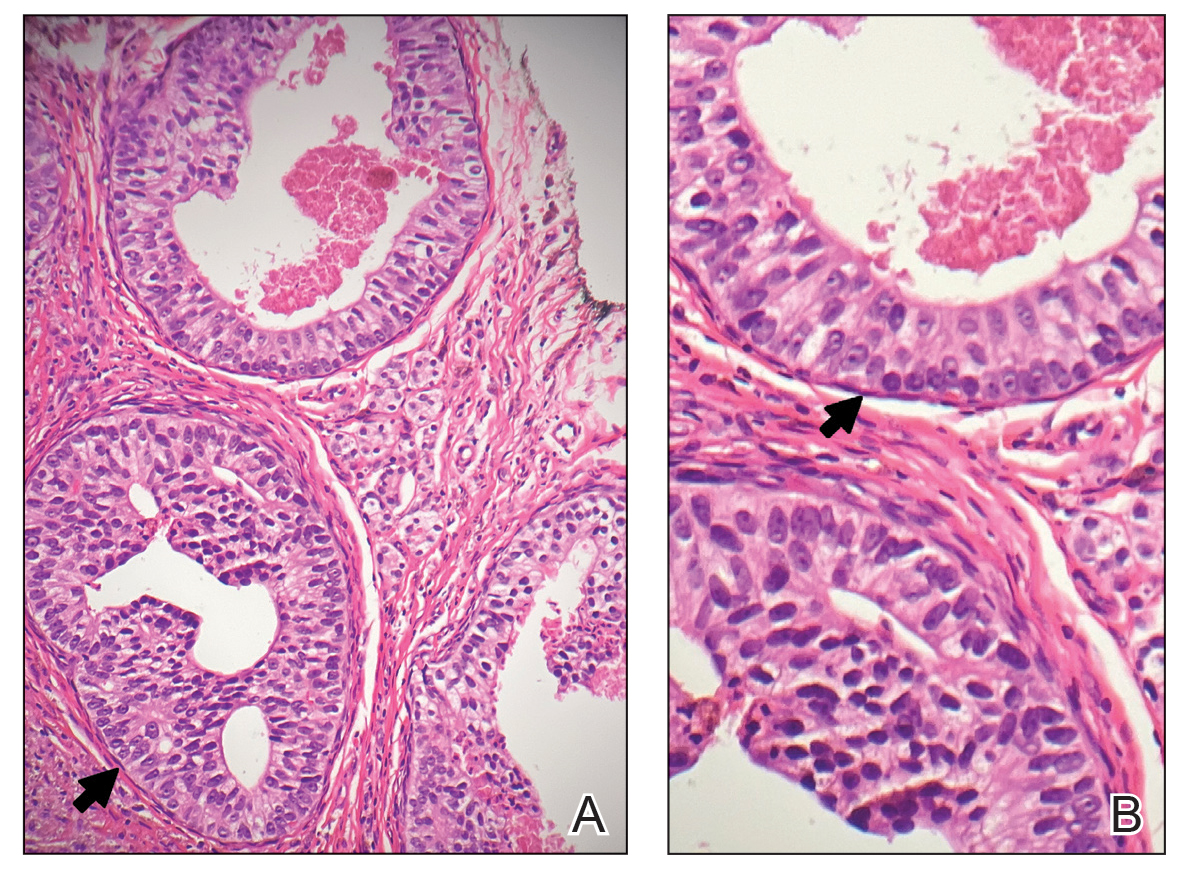
Immunohistochemical staining was positive for PSA (Figure 2B), NKX 3.1, and ERG in the invasive glandular structures, which also displayed patchy weak staining with AMACR. Staining was negative for prostein, cytokeratin (CK) 7, CK20, CK5/6, p63, p40, CDX2, and thyroid transcription factor 1. These findings were consistent with a diagnosis of cutaneous metastatic prostatic adenocarcinoma. Next-generation sequencing showed trans-membrane protease serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG) fusion compatible with the positive ERG immunohistochemical staining. The patient and family declined any treatment due to his age, comorbidities, and rapid decline. He died 2 months after diagnosis of the skin metastasis.
Aside from nonmelanoma skin cancer, prostate cancer is the most common cancer and the second leading cause of cancer-related deaths among men in the United States.1 It most commonly metastasizes to the bones, nonregional lymph nodes, liver, and thorax.2 Metastasis to the skin is very rare, with only a 0.36% incidence.3 When prostate cancer does metastasize to the skin, the prognosis is poor, with an estimated mean survival of 7 months after diagnosis of cutaneous metastasis.4 Our patient’s survival time was even shorter—only 2 months after diagnosis of cutaneous metastasis, likely the result of his late diagnosis.
Clinically, cutaneous metastasis of prostate cancer can manifest as a wide variety of lesions; in one report of 78 cases, 56 (72%) were hard nodules, 11 (14%) were single nodules, 5 (7%) were edema or lymphedema, and 5 (7%) were an unspecific rash.4 Diagnosis of cutaneous metastasis of prostate cancer can be challenging, as it often is mistaken for other skin conditions including herpes zoster, basal cell carcinoma, angiosarcoma, cellulitis, mammary Paget disease, telangiectasia, pyoderma, morphea, and trichoepithelioma.5 In our patient, the clinical appearance of the lesion resembled a nodular melanoma. Thus, in patients with a history of prostate cancer, it is important to keep cutaneous metastasis in the differential when examining the skin because of the prognostic implications. Cutaneous metastasis of prostate cancer often indicates a poor prognosis.
In a report of 78 patients, the most common sites of skin metastasis for prostate cancer were the inguinal area and penis (28% [22/78]), abdomen (23% [18/78]), head and neck (16% [12/78]), and chest (14% [11/78]); the extremities and back were less frequently involved (10% [8/78] and 9% [7/78], respectively).4 Generally, cutaneous metastasis of internal malignancies involves the deep dermis and the subcutaneous tissue. It is common for cutaneous metastases to show histologic features of the primary tumor, as we saw in our patient. In a case series with 45 histologic diagnoses of cutaneous metastases from internal malignancies, 75.5% (34/45) of cases showed morphologic features of the primary tumor.6 However, this is not always the case, and the histologic appearance may vary. Metastatic prostate cancer may manifest as sheets, nests, or cords and often may have nuclear pleomorphism with prominent nucleoli.7
Immunohistochemical staining can help make a definitive diagnosis and differentiate the source of the tumor. Prostate cancer metastases often will stain positive for NKX3.1, PSA, AMACR, ERG, PSMA, and prosaposin, with PSA being the most specific marker.7,8 In our patient, no prostate biopsy had been performed, thus the skin biopsy was the diagnostic tissue for the prostatic adenocarcinoma.
Next-generation sequencing showed a TMPRSS2- ERG fusion, which commonly is seen in prostate cancer.9 A search of Google Scholar using the terms next-generation sequencing, cutaneous metastasis, and prostate adenocarcinoma yielded 3 additional cases of cutaneous metastasis of prostate cancer in which next-generation sequencing was performed.10-12 One case showed mutations of the tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN) genes; one showed just a TP53 mutation; and one showed inactivation of the breast cancer predisposition gene 2 (BRCA2) and amplification of MYC proto-oncogene, BHLH transcription factor (MYC) and fibroblast growth factor receptor 1 (FGFR1).10,11,12 While limited by a small number of reported cases, there does not appear to be a repeating mutation to suggest a genetic mechanism of skin metastasis.
The route of cutaneous metastasis of prostate cancer still is unclear, but hypothesized mechanisms include hematogenous or lymphatic spread, direct infiltration, or implantation from a surgical scar.11 When cutaneous involvement occurs in an area far from the primary tumor, it is thought to be the result of hematogenous spread, which would be consistent with our patient’s findings.13 Given the role of Batson venous plexus as a conduit from the prostate to the vertebral column for metastatic spread and considering the location of the lesion on our patient’s back, we hypothesized that the mechanism of metastasis to the skin was from vascular extension of the metastatic foci involving the vertebrae.
Our case highlights the importance of considering cutaneous involvement of prostatic adenocarcinoma in patients with new skin lesions, particularly in the setting of a known or suspected prostate malignancy. Skin metastasis can have a range of manifestations and provides prognostic information that can help determine the course of treatment.
- US Cancer Statistics Working Group. US cancer statistics data visualizations tool, based on 2022 submission data (1999-2020). US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. November 2023. Accessed November 11, 2024. https://www.cdc.gov/cancer/dataviz
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74:210-216. doi:10.1002/pros.22742
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026. doi:10.1016/j.urology.2004.01.014
- Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684. doi:10.1111/j.1600-0560.2007.00873.x
- Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600. doi:10.1111/j.1365-2133.2006.07696.x
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614. doi:10.1684/ejd.2017.3142
- Onalaja-Underwood AA, Sokumbi O. Eruptive papules as a cutaneous manifestation of metastatic prostate adenocarcinoma. Am J Dermatopathol. 2023;45:828-830. doi:10.1097/DAD.0000000000002559
- Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991;145:907-923. doi:10.1016/s0022-5347(17)38491-4
- Wang Z, Wang Y, Zhang J, et al. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol Med Rep. 2017;16:5450-5458. doi:10.3892/mmr.2017.7281
- Sharma H, Franklin M, Braunberger R, et al. Cutaneous metastasis from prostate cancer: a case report with literature review. Curr Probl Cancer Case Rep. 2022;7:100175. doi:10.1016/j.cpccr.2022.100175
- Dills A, Obi O, Bustos K, et al. Cutaneous manifestation of prostate adenocarcinoma: a rare presentation of a common disease. J Investig Med High Impact Case Rep. 2021;9:2324709621990769. doi:10.1177/2324709621990769
- Fadel CA, Kallab AM. Cutaneous scrotal metastasis secondary to primary prostate adenocarcinoma responding to immunotherapy. Ann Intern Med: Clinical Cases. 2022;1. doi:10.7326/aimcc.2022.0682
- Powell FC, Venencie PY, Winkelmann RK. Metastatic prostate carcinoma manifesting as penile nodules. Arch Dermatol. 1984;120:1604- 1606. doi:10.1001/archderm.1984.01650480066022
To the Editor:
Cutaneous metastasis of prostate cancer is rare and portends a bleak prognosis. Diagnosis of the primary cancer can be challenging, as skin metastasis can mimic a variety of conditions. We report a case of metastatic prostatic adenocarcinoma confirmed via biopsy of a new skin lesion.
A 97-year-old man presented to the dermatology clinic for routine follow-up of psoriasis. During the visit, a family member mentioned a new bleeding lesion on the left shoulder. It was not known how long the lesion had been present. Four months prior, the patient had a prostate-specific antigen (PSA) level of 582 ng/mL (reference range, 0-6.5 ng/mL), and computed tomography of the chest had shown innumerable pulmonary nodules in addition to lymphadenopathy of the left axilla, clavicle, and mediastinum. The imaging was ordered by the patient’s urologist as part of routine workup, as he had a history of obstructive renal failure and was being monitored for an indwelling catheter. Two months later, a bone scan ordered by the urologist due to high PSA levels showed extensive osteoblastic metastatic disease throughout the axial and proximal appendicular skeleton. The elevated PSA levels and findings of pulmonary and osteoblastic metastasis suggested a diagnosis of metastatic prostatic adenocarcinoma, but no confirmatory biopsy was performed following the imaging because the patient’s family declined additional workup or intervention.
Physical examination at the current presentation revealed an 8-mm brown papule with an overlying blue-white veil (Figure 1). There were no other skin findings. Primary differential diagnoses included metastatic prostate cancer, nodular melanoma, and traumatized seborrheic keratosis. A shave biopsy of the lesion showed multiple glandular structures infiltrating the dermis lined by monomorphic epithelial cells with prominent eosinophilic nucleoli (Figures 2 and 3). Focal cribriform architecture of the glands was present as well as dermal hemorrhage and a lymphohistiocytic infiltrate (Figure 2A). Interestingly, in-transit vascular metastases were confirmed with the support of ERG, CD34, and CD31 immunohistochemical staining of the vessels.
Immunohistochemical staining was positive for PSA (Figure 2B), NKX 3.1, and ERG in the invasive glandular structures, which also displayed patchy weak staining with AMACR. Staining was negative for prostein, cytokeratin (CK) 7, CK20, CK5/6, p63, p40, CDX2, and thyroid transcription factor 1. These findings were consistent with a diagnosis of cutaneous metastatic prostatic adenocarcinoma. Next-generation sequencing showed trans-membrane protease serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG) fusion compatible with the positive ERG immunohistochemical staining. The patient and family declined any treatment due to his age, comorbidities, and rapid decline. He died 2 months after diagnosis of the skin metastasis.
Aside from nonmelanoma skin cancer, prostate cancer is the most common cancer and the second leading cause of cancer-related deaths among men in the United States.1 It most commonly metastasizes to the bones, nonregional lymph nodes, liver, and thorax.2 Metastasis to the skin is very rare, with only a 0.36% incidence.3 When prostate cancer does metastasize to the skin, the prognosis is poor, with an estimated mean survival of 7 months after diagnosis of cutaneous metastasis.4 Our patient’s survival time was even shorter—only 2 months after diagnosis of cutaneous metastasis, likely the result of his late diagnosis.
Clinically, cutaneous metastasis of prostate cancer can manifest as a wide variety of lesions; in one report of 78 cases, 56 (72%) were hard nodules, 11 (14%) were single nodules, 5 (7%) were edema or lymphedema, and 5 (7%) were an unspecific rash.4 Diagnosis of cutaneous metastasis of prostate cancer can be challenging, as it often is mistaken for other skin conditions including herpes zoster, basal cell carcinoma, angiosarcoma, cellulitis, mammary Paget disease, telangiectasia, pyoderma, morphea, and trichoepithelioma.5 In our patient, the clinical appearance of the lesion resembled a nodular melanoma. Thus, in patients with a history of prostate cancer, it is important to keep cutaneous metastasis in the differential when examining the skin because of the prognostic implications. Cutaneous metastasis of prostate cancer often indicates a poor prognosis.
In a report of 78 patients, the most common sites of skin metastasis for prostate cancer were the inguinal area and penis (28% [22/78]), abdomen (23% [18/78]), head and neck (16% [12/78]), and chest (14% [11/78]); the extremities and back were less frequently involved (10% [8/78] and 9% [7/78], respectively).4 Generally, cutaneous metastasis of internal malignancies involves the deep dermis and the subcutaneous tissue. It is common for cutaneous metastases to show histologic features of the primary tumor, as we saw in our patient. In a case series with 45 histologic diagnoses of cutaneous metastases from internal malignancies, 75.5% (34/45) of cases showed morphologic features of the primary tumor.6 However, this is not always the case, and the histologic appearance may vary. Metastatic prostate cancer may manifest as sheets, nests, or cords and often may have nuclear pleomorphism with prominent nucleoli.7
Immunohistochemical staining can help make a definitive diagnosis and differentiate the source of the tumor. Prostate cancer metastases often will stain positive for NKX3.1, PSA, AMACR, ERG, PSMA, and prosaposin, with PSA being the most specific marker.7,8 In our patient, no prostate biopsy had been performed, thus the skin biopsy was the diagnostic tissue for the prostatic adenocarcinoma.
Next-generation sequencing showed a TMPRSS2- ERG fusion, which commonly is seen in prostate cancer.9 A search of Google Scholar using the terms next-generation sequencing, cutaneous metastasis, and prostate adenocarcinoma yielded 3 additional cases of cutaneous metastasis of prostate cancer in which next-generation sequencing was performed.10-12 One case showed mutations of the tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN) genes; one showed just a TP53 mutation; and one showed inactivation of the breast cancer predisposition gene 2 (BRCA2) and amplification of MYC proto-oncogene, BHLH transcription factor (MYC) and fibroblast growth factor receptor 1 (FGFR1).10,11,12 While limited by a small number of reported cases, there does not appear to be a repeating mutation to suggest a genetic mechanism of skin metastasis.
The route of cutaneous metastasis of prostate cancer still is unclear, but hypothesized mechanisms include hematogenous or lymphatic spread, direct infiltration, or implantation from a surgical scar.11 When cutaneous involvement occurs in an area far from the primary tumor, it is thought to be the result of hematogenous spread, which would be consistent with our patient’s findings.13 Given the role of Batson venous plexus as a conduit from the prostate to the vertebral column for metastatic spread and considering the location of the lesion on our patient’s back, we hypothesized that the mechanism of metastasis to the skin was from vascular extension of the metastatic foci involving the vertebrae.
Our case highlights the importance of considering cutaneous involvement of prostatic adenocarcinoma in patients with new skin lesions, particularly in the setting of a known or suspected prostate malignancy. Skin metastasis can have a range of manifestations and provides prognostic information that can help determine the course of treatment.
To the Editor:
Cutaneous metastasis of prostate cancer is rare and portends a bleak prognosis. Diagnosis of the primary cancer can be challenging, as skin metastasis can mimic a variety of conditions. We report a case of metastatic prostatic adenocarcinoma confirmed via biopsy of a new skin lesion.
A 97-year-old man presented to the dermatology clinic for routine follow-up of psoriasis. During the visit, a family member mentioned a new bleeding lesion on the left shoulder. It was not known how long the lesion had been present. Four months prior, the patient had a prostate-specific antigen (PSA) level of 582 ng/mL (reference range, 0-6.5 ng/mL), and computed tomography of the chest had shown innumerable pulmonary nodules in addition to lymphadenopathy of the left axilla, clavicle, and mediastinum. The imaging was ordered by the patient’s urologist as part of routine workup, as he had a history of obstructive renal failure and was being monitored for an indwelling catheter. Two months later, a bone scan ordered by the urologist due to high PSA levels showed extensive osteoblastic metastatic disease throughout the axial and proximal appendicular skeleton. The elevated PSA levels and findings of pulmonary and osteoblastic metastasis suggested a diagnosis of metastatic prostatic adenocarcinoma, but no confirmatory biopsy was performed following the imaging because the patient’s family declined additional workup or intervention.
Physical examination at the current presentation revealed an 8-mm brown papule with an overlying blue-white veil (Figure 1). There were no other skin findings. Primary differential diagnoses included metastatic prostate cancer, nodular melanoma, and traumatized seborrheic keratosis. A shave biopsy of the lesion showed multiple glandular structures infiltrating the dermis lined by monomorphic epithelial cells with prominent eosinophilic nucleoli (Figures 2 and 3). Focal cribriform architecture of the glands was present as well as dermal hemorrhage and a lymphohistiocytic infiltrate (Figure 2A). Interestingly, in-transit vascular metastases were confirmed with the support of ERG, CD34, and CD31 immunohistochemical staining of the vessels.
Immunohistochemical staining was positive for PSA (Figure 2B), NKX 3.1, and ERG in the invasive glandular structures, which also displayed patchy weak staining with AMACR. Staining was negative for prostein, cytokeratin (CK) 7, CK20, CK5/6, p63, p40, CDX2, and thyroid transcription factor 1. These findings were consistent with a diagnosis of cutaneous metastatic prostatic adenocarcinoma. Next-generation sequencing showed trans-membrane protease serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG) fusion compatible with the positive ERG immunohistochemical staining. The patient and family declined any treatment due to his age, comorbidities, and rapid decline. He died 2 months after diagnosis of the skin metastasis.
Aside from nonmelanoma skin cancer, prostate cancer is the most common cancer and the second leading cause of cancer-related deaths among men in the United States.1 It most commonly metastasizes to the bones, nonregional lymph nodes, liver, and thorax.2 Metastasis to the skin is very rare, with only a 0.36% incidence.3 When prostate cancer does metastasize to the skin, the prognosis is poor, with an estimated mean survival of 7 months after diagnosis of cutaneous metastasis.4 Our patient’s survival time was even shorter—only 2 months after diagnosis of cutaneous metastasis, likely the result of his late diagnosis.
Clinically, cutaneous metastasis of prostate cancer can manifest as a wide variety of lesions; in one report of 78 cases, 56 (72%) were hard nodules, 11 (14%) were single nodules, 5 (7%) were edema or lymphedema, and 5 (7%) were an unspecific rash.4 Diagnosis of cutaneous metastasis of prostate cancer can be challenging, as it often is mistaken for other skin conditions including herpes zoster, basal cell carcinoma, angiosarcoma, cellulitis, mammary Paget disease, telangiectasia, pyoderma, morphea, and trichoepithelioma.5 In our patient, the clinical appearance of the lesion resembled a nodular melanoma. Thus, in patients with a history of prostate cancer, it is important to keep cutaneous metastasis in the differential when examining the skin because of the prognostic implications. Cutaneous metastasis of prostate cancer often indicates a poor prognosis.
In a report of 78 patients, the most common sites of skin metastasis for prostate cancer were the inguinal area and penis (28% [22/78]), abdomen (23% [18/78]), head and neck (16% [12/78]), and chest (14% [11/78]); the extremities and back were less frequently involved (10% [8/78] and 9% [7/78], respectively).4 Generally, cutaneous metastasis of internal malignancies involves the deep dermis and the subcutaneous tissue. It is common for cutaneous metastases to show histologic features of the primary tumor, as we saw in our patient. In a case series with 45 histologic diagnoses of cutaneous metastases from internal malignancies, 75.5% (34/45) of cases showed morphologic features of the primary tumor.6 However, this is not always the case, and the histologic appearance may vary. Metastatic prostate cancer may manifest as sheets, nests, or cords and often may have nuclear pleomorphism with prominent nucleoli.7
Immunohistochemical staining can help make a definitive diagnosis and differentiate the source of the tumor. Prostate cancer metastases often will stain positive for NKX3.1, PSA, AMACR, ERG, PSMA, and prosaposin, with PSA being the most specific marker.7,8 In our patient, no prostate biopsy had been performed, thus the skin biopsy was the diagnostic tissue for the prostatic adenocarcinoma.
Next-generation sequencing showed a TMPRSS2- ERG fusion, which commonly is seen in prostate cancer.9 A search of Google Scholar using the terms next-generation sequencing, cutaneous metastasis, and prostate adenocarcinoma yielded 3 additional cases of cutaneous metastasis of prostate cancer in which next-generation sequencing was performed.10-12 One case showed mutations of the tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN) genes; one showed just a TP53 mutation; and one showed inactivation of the breast cancer predisposition gene 2 (BRCA2) and amplification of MYC proto-oncogene, BHLH transcription factor (MYC) and fibroblast growth factor receptor 1 (FGFR1).10,11,12 While limited by a small number of reported cases, there does not appear to be a repeating mutation to suggest a genetic mechanism of skin metastasis.
The route of cutaneous metastasis of prostate cancer still is unclear, but hypothesized mechanisms include hematogenous or lymphatic spread, direct infiltration, or implantation from a surgical scar.11 When cutaneous involvement occurs in an area far from the primary tumor, it is thought to be the result of hematogenous spread, which would be consistent with our patient’s findings.13 Given the role of Batson venous plexus as a conduit from the prostate to the vertebral column for metastatic spread and considering the location of the lesion on our patient’s back, we hypothesized that the mechanism of metastasis to the skin was from vascular extension of the metastatic foci involving the vertebrae.
Our case highlights the importance of considering cutaneous involvement of prostatic adenocarcinoma in patients with new skin lesions, particularly in the setting of a known or suspected prostate malignancy. Skin metastasis can have a range of manifestations and provides prognostic information that can help determine the course of treatment.
- US Cancer Statistics Working Group. US cancer statistics data visualizations tool, based on 2022 submission data (1999-2020). US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. November 2023. Accessed November 11, 2024. https://www.cdc.gov/cancer/dataviz
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74:210-216. doi:10.1002/pros.22742
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026. doi:10.1016/j.urology.2004.01.014
- Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684. doi:10.1111/j.1600-0560.2007.00873.x
- Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600. doi:10.1111/j.1365-2133.2006.07696.x
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614. doi:10.1684/ejd.2017.3142
- Onalaja-Underwood AA, Sokumbi O. Eruptive papules as a cutaneous manifestation of metastatic prostate adenocarcinoma. Am J Dermatopathol. 2023;45:828-830. doi:10.1097/DAD.0000000000002559
- Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991;145:907-923. doi:10.1016/s0022-5347(17)38491-4
- Wang Z, Wang Y, Zhang J, et al. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol Med Rep. 2017;16:5450-5458. doi:10.3892/mmr.2017.7281
- Sharma H, Franklin M, Braunberger R, et al. Cutaneous metastasis from prostate cancer: a case report with literature review. Curr Probl Cancer Case Rep. 2022;7:100175. doi:10.1016/j.cpccr.2022.100175
- Dills A, Obi O, Bustos K, et al. Cutaneous manifestation of prostate adenocarcinoma: a rare presentation of a common disease. J Investig Med High Impact Case Rep. 2021;9:2324709621990769. doi:10.1177/2324709621990769
- Fadel CA, Kallab AM. Cutaneous scrotal metastasis secondary to primary prostate adenocarcinoma responding to immunotherapy. Ann Intern Med: Clinical Cases. 2022;1. doi:10.7326/aimcc.2022.0682
- Powell FC, Venencie PY, Winkelmann RK. Metastatic prostate carcinoma manifesting as penile nodules. Arch Dermatol. 1984;120:1604- 1606. doi:10.1001/archderm.1984.01650480066022
- US Cancer Statistics Working Group. US cancer statistics data visualizations tool, based on 2022 submission data (1999-2020). US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. November 2023. Accessed November 11, 2024. https://www.cdc.gov/cancer/dataviz
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74:210-216. doi:10.1002/pros.22742
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026. doi:10.1016/j.urology.2004.01.014
- Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684. doi:10.1111/j.1600-0560.2007.00873.x
- Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600. doi:10.1111/j.1365-2133.2006.07696.x
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614. doi:10.1684/ejd.2017.3142
- Onalaja-Underwood AA, Sokumbi O. Eruptive papules as a cutaneous manifestation of metastatic prostate adenocarcinoma. Am J Dermatopathol. 2023;45:828-830. doi:10.1097/DAD.0000000000002559
- Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991;145:907-923. doi:10.1016/s0022-5347(17)38491-4
- Wang Z, Wang Y, Zhang J, et al. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol Med Rep. 2017;16:5450-5458. doi:10.3892/mmr.2017.7281
- Sharma H, Franklin M, Braunberger R, et al. Cutaneous metastasis from prostate cancer: a case report with literature review. Curr Probl Cancer Case Rep. 2022;7:100175. doi:10.1016/j.cpccr.2022.100175
- Dills A, Obi O, Bustos K, et al. Cutaneous manifestation of prostate adenocarcinoma: a rare presentation of a common disease. J Investig Med High Impact Case Rep. 2021;9:2324709621990769. doi:10.1177/2324709621990769
- Fadel CA, Kallab AM. Cutaneous scrotal metastasis secondary to primary prostate adenocarcinoma responding to immunotherapy. Ann Intern Med: Clinical Cases. 2022;1. doi:10.7326/aimcc.2022.0682
- Powell FC, Venencie PY, Winkelmann RK. Metastatic prostate carcinoma manifesting as penile nodules. Arch Dermatol. 1984;120:1604- 1606. doi:10.1001/archderm.1984.01650480066022
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
PRACTICE POINTS
- Cutaneous metastasis of prostate cancer can have various manifestations and portends a poor prognosis.
- New skin lesions that develop in patients with a high clinical suspicion for prostate cancer warrant consideration of cutaneous metastasis.

Indeterminate Cell Histiocytosis and a Review of Current Treatment
Indeterminate Cell Histiocytosis and a Review of Current Treatment
To the Editor:
Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder with a poorly understood histogenesis and pathogenesis.1 The clinical manifestation of ICH is broad and can include isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs. Our case demonstrates a rare occurrence of ICH that initially was misdiagnosed and highlights the use of cobimetinib, a MEK inhibitor, as a potential new therapeutic option for ICH.
A 74-year-old man with a history of type 2 diabetes mellitus presented for evaluation of a progressive pruritic rash of approximately 5 years’ duration. The eruption previously had been diagnosed as Langerhans cell histiocytosis. It started on the chest and spread to the face, neck, trunk, and arms. The patient denied systemic symptoms and had no known history of malignancy.
Physical examination revealed pink to orange smooth papules, nodules, and small plaques on the ears, cheeks, trunk, neck, and arms (Figure 1). Baseline laboratory results showed a normal complete blood count and comprehensive metabolic panel, elevated lactate dehydrogenase and erythrocyte sedimentation rate, and hyperlipidemia. Serology for hepatitis B and C was negative. Bone marrow biopsy was normal, and positron emission tomography/ computed tomography demonstrated no evidence of extracutaneous disease. A punch biopsy of a lesion on the left forearm revealed epithelioid histiocytic proliferation in the dermis extending into the subcutis with a background infiltrate of small lymphocytes. Immunohistochemistry was positive for CD1a and CD56 and was variably positive for CD4 but negative for CD163, CD68, S100, Langerin, cyclin D1, myeloperoxidase, CD21, and CD23. No mutation was detected in BRAF codon 600. Given the negative Langerin stain, these findings were compatible with a diagnosis of ICH. After considering the lack of standard treatment options as well as the recent approval of cobimetinib for histiocytic disorders, we initiated treatment with cobimetinib at the standard dose of 60 mg daily for 21 days followed by a 7-day break.

One month into treatment, the patient’s lesions were less erythematous, and he reported improvement in pruritus. Two months into treatment, there was continued improvement in cutaneous symptoms with flattening of the lesions on the chest and back. At this time, the patient developed edema of the face and ears (Figure 2) and reported weakness, blurred vision, and decreased appetite. He was advised to take an additional 7-day treatment break before resuming cobimetinib at a decreased dose of 40 mg daily. The patient returned to the clinic 1 month later with improved systemic symptoms and continued flattening of the lesions. Five months into treatment, the lesions had continued to improve with complete resolution of the facial plaques (Figure 3).


Indeterminate cell histiocytosis is a rarely diagnosed condition characterized by the proliferation of indeterminate histiocytes that morphologically and immunophenotypically resemble Langerhans cells but lack their characteristic Birbeck granules.2 There is no standard treatment for ICH, but previous reports have described improvement with a variety of treatment options including methotrexate,3,4 UVB phototherapy,5 and topical delgocitinib 0.5%.6
Because histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway, it is possible that they would be responsive to MEK inhibition. Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.7 The approval followed the release of data from a phase 2 trial of cobimetinib in 18 adults with various histiocytic disorders, which demonstrated an 89% (16/18) overall response rate with 94% (17/18) of patients remaining progression free at 1 year.8 While cobimetinib has not specifically been studied in ICH, given the high response rate in histiocytic disorders and the lack of standard treatment options for ICH, the decision was made to initiate treatment with cobimetinib in our patient. Based on the observed improvement in our patient, we propose cobimetinib as a treatment option for patients with cutaneous ICH and recommend additional studies to confirm its safety and efficacy in patients with this disorder.
- Bakry OA, Samaka RM, Kandil MA, et al. Indeterminate cell histiocytosis with naïve cells. Rare Tumors. 2013;5:e13. doi:10.4081 /rt.2013.e13
- Manente L, Cotellessa C, Schmitt I, et al. Indeterminate cell histiocytosis: a rare histiocytic disorder. Am J Dermatopathol. 1997; 19:276-283. doi:10.1097/00000372-199706000-00014
- Lie E, Jedrych J, Sweren R, et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep. 2022;25:93-96. doi:10.1016/j.jdcr.2022.05.027
- Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38:937-939. doi:10.1111/j.1346-8138.2010.01148.x
- Logemann N, Thomas B, Yetto T. Indeterminate cell histiocytosis successfully treated with narrowband UVB. Dermatol Online J. 2013;19:20031. doi:10.5070/D31910020031
- Fujimoto RFT, Miura H, Takata M, et al. Indeterminate cell histiocytosis treated with 0.5% delgocitinib ointment. Br J Dermatol. 2023;188:E39. doi:10.1093/bjd/ljad029
- Diamond EL, Durham B, Dogan A, et al. Phase 2 trial of single-agent cobimetinib for adults with histiocytic neoplasms. Blood. 2023;142:1812. doi:10.1182/blood-2023-187508
- Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521-524. doi:10.1038/s41586-019-1012-y
To the Editor:
Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder with a poorly understood histogenesis and pathogenesis.1 The clinical manifestation of ICH is broad and can include isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs. Our case demonstrates a rare occurrence of ICH that initially was misdiagnosed and highlights the use of cobimetinib, a MEK inhibitor, as a potential new therapeutic option for ICH.
A 74-year-old man with a history of type 2 diabetes mellitus presented for evaluation of a progressive pruritic rash of approximately 5 years’ duration. The eruption previously had been diagnosed as Langerhans cell histiocytosis. It started on the chest and spread to the face, neck, trunk, and arms. The patient denied systemic symptoms and had no known history of malignancy.
Physical examination revealed pink to orange smooth papules, nodules, and small plaques on the ears, cheeks, trunk, neck, and arms (Figure 1). Baseline laboratory results showed a normal complete blood count and comprehensive metabolic panel, elevated lactate dehydrogenase and erythrocyte sedimentation rate, and hyperlipidemia. Serology for hepatitis B and C was negative. Bone marrow biopsy was normal, and positron emission tomography/ computed tomography demonstrated no evidence of extracutaneous disease. A punch biopsy of a lesion on the left forearm revealed epithelioid histiocytic proliferation in the dermis extending into the subcutis with a background infiltrate of small lymphocytes. Immunohistochemistry was positive for CD1a and CD56 and was variably positive for CD4 but negative for CD163, CD68, S100, Langerin, cyclin D1, myeloperoxidase, CD21, and CD23. No mutation was detected in BRAF codon 600. Given the negative Langerin stain, these findings were compatible with a diagnosis of ICH. After considering the lack of standard treatment options as well as the recent approval of cobimetinib for histiocytic disorders, we initiated treatment with cobimetinib at the standard dose of 60 mg daily for 21 days followed by a 7-day break.
One month into treatment, the patient’s lesions were less erythematous, and he reported improvement in pruritus. Two months into treatment, there was continued improvement in cutaneous symptoms with flattening of the lesions on the chest and back. At this time, the patient developed edema of the face and ears (Figure 2) and reported weakness, blurred vision, and decreased appetite. He was advised to take an additional 7-day treatment break before resuming cobimetinib at a decreased dose of 40 mg daily. The patient returned to the clinic 1 month later with improved systemic symptoms and continued flattening of the lesions. Five months into treatment, the lesions had continued to improve with complete resolution of the facial plaques (Figure 3).
Indeterminate cell histiocytosis is a rarely diagnosed condition characterized by the proliferation of indeterminate histiocytes that morphologically and immunophenotypically resemble Langerhans cells but lack their characteristic Birbeck granules.2 There is no standard treatment for ICH, but previous reports have described improvement with a variety of treatment options including methotrexate,3,4 UVB phototherapy,5 and topical delgocitinib 0.5%.6
Because histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway, it is possible that they would be responsive to MEK inhibition. Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.7 The approval followed the release of data from a phase 2 trial of cobimetinib in 18 adults with various histiocytic disorders, which demonstrated an 89% (16/18) overall response rate with 94% (17/18) of patients remaining progression free at 1 year.8 While cobimetinib has not specifically been studied in ICH, given the high response rate in histiocytic disorders and the lack of standard treatment options for ICH, the decision was made to initiate treatment with cobimetinib in our patient. Based on the observed improvement in our patient, we propose cobimetinib as a treatment option for patients with cutaneous ICH and recommend additional studies to confirm its safety and efficacy in patients with this disorder.
To the Editor:
Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder with a poorly understood histogenesis and pathogenesis.1 The clinical manifestation of ICH is broad and can include isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs. Our case demonstrates a rare occurrence of ICH that initially was misdiagnosed and highlights the use of cobimetinib, a MEK inhibitor, as a potential new therapeutic option for ICH.
A 74-year-old man with a history of type 2 diabetes mellitus presented for evaluation of a progressive pruritic rash of approximately 5 years’ duration. The eruption previously had been diagnosed as Langerhans cell histiocytosis. It started on the chest and spread to the face, neck, trunk, and arms. The patient denied systemic symptoms and had no known history of malignancy.
Physical examination revealed pink to orange smooth papules, nodules, and small plaques on the ears, cheeks, trunk, neck, and arms (Figure 1). Baseline laboratory results showed a normal complete blood count and comprehensive metabolic panel, elevated lactate dehydrogenase and erythrocyte sedimentation rate, and hyperlipidemia. Serology for hepatitis B and C was negative. Bone marrow biopsy was normal, and positron emission tomography/ computed tomography demonstrated no evidence of extracutaneous disease. A punch biopsy of a lesion on the left forearm revealed epithelioid histiocytic proliferation in the dermis extending into the subcutis with a background infiltrate of small lymphocytes. Immunohistochemistry was positive for CD1a and CD56 and was variably positive for CD4 but negative for CD163, CD68, S100, Langerin, cyclin D1, myeloperoxidase, CD21, and CD23. No mutation was detected in BRAF codon 600. Given the negative Langerin stain, these findings were compatible with a diagnosis of ICH. After considering the lack of standard treatment options as well as the recent approval of cobimetinib for histiocytic disorders, we initiated treatment with cobimetinib at the standard dose of 60 mg daily for 21 days followed by a 7-day break.
One month into treatment, the patient’s lesions were less erythematous, and he reported improvement in pruritus. Two months into treatment, there was continued improvement in cutaneous symptoms with flattening of the lesions on the chest and back. At this time, the patient developed edema of the face and ears (Figure 2) and reported weakness, blurred vision, and decreased appetite. He was advised to take an additional 7-day treatment break before resuming cobimetinib at a decreased dose of 40 mg daily. The patient returned to the clinic 1 month later with improved systemic symptoms and continued flattening of the lesions. Five months into treatment, the lesions had continued to improve with complete resolution of the facial plaques (Figure 3).
Indeterminate cell histiocytosis is a rarely diagnosed condition characterized by the proliferation of indeterminate histiocytes that morphologically and immunophenotypically resemble Langerhans cells but lack their characteristic Birbeck granules.2 There is no standard treatment for ICH, but previous reports have described improvement with a variety of treatment options including methotrexate,3,4 UVB phototherapy,5 and topical delgocitinib 0.5%.6
Because histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway, it is possible that they would be responsive to MEK inhibition. Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.7 The approval followed the release of data from a phase 2 trial of cobimetinib in 18 adults with various histiocytic disorders, which demonstrated an 89% (16/18) overall response rate with 94% (17/18) of patients remaining progression free at 1 year.8 While cobimetinib has not specifically been studied in ICH, given the high response rate in histiocytic disorders and the lack of standard treatment options for ICH, the decision was made to initiate treatment with cobimetinib in our patient. Based on the observed improvement in our patient, we propose cobimetinib as a treatment option for patients with cutaneous ICH and recommend additional studies to confirm its safety and efficacy in patients with this disorder.
- Bakry OA, Samaka RM, Kandil MA, et al. Indeterminate cell histiocytosis with naïve cells. Rare Tumors. 2013;5:e13. doi:10.4081 /rt.2013.e13
- Manente L, Cotellessa C, Schmitt I, et al. Indeterminate cell histiocytosis: a rare histiocytic disorder. Am J Dermatopathol. 1997; 19:276-283. doi:10.1097/00000372-199706000-00014
- Lie E, Jedrych J, Sweren R, et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep. 2022;25:93-96. doi:10.1016/j.jdcr.2022.05.027
- Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38:937-939. doi:10.1111/j.1346-8138.2010.01148.x
- Logemann N, Thomas B, Yetto T. Indeterminate cell histiocytosis successfully treated with narrowband UVB. Dermatol Online J. 2013;19:20031. doi:10.5070/D31910020031
- Fujimoto RFT, Miura H, Takata M, et al. Indeterminate cell histiocytosis treated with 0.5% delgocitinib ointment. Br J Dermatol. 2023;188:E39. doi:10.1093/bjd/ljad029
- Diamond EL, Durham B, Dogan A, et al. Phase 2 trial of single-agent cobimetinib for adults with histiocytic neoplasms. Blood. 2023;142:1812. doi:10.1182/blood-2023-187508
- Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521-524. doi:10.1038/s41586-019-1012-y
- Bakry OA, Samaka RM, Kandil MA, et al. Indeterminate cell histiocytosis with naïve cells. Rare Tumors. 2013;5:e13. doi:10.4081 /rt.2013.e13
- Manente L, Cotellessa C, Schmitt I, et al. Indeterminate cell histiocytosis: a rare histiocytic disorder. Am J Dermatopathol. 1997; 19:276-283. doi:10.1097/00000372-199706000-00014
- Lie E, Jedrych J, Sweren R, et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep. 2022;25:93-96. doi:10.1016/j.jdcr.2022.05.027
- Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38:937-939. doi:10.1111/j.1346-8138.2010.01148.x
- Logemann N, Thomas B, Yetto T. Indeterminate cell histiocytosis successfully treated with narrowband UVB. Dermatol Online J. 2013;19:20031. doi:10.5070/D31910020031
- Fujimoto RFT, Miura H, Takata M, et al. Indeterminate cell histiocytosis treated with 0.5% delgocitinib ointment. Br J Dermatol. 2023;188:E39. doi:10.1093/bjd/ljad029
- Diamond EL, Durham B, Dogan A, et al. Phase 2 trial of single-agent cobimetinib for adults with histiocytic neoplasms. Blood. 2023;142:1812. doi:10.1182/blood-2023-187508
- Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521-524. doi:10.1038/s41586-019-1012-y
Indeterminate Cell Histiocytosis and a Review of Current Treatment
Indeterminate Cell Histiocytosis and a Review of Current Treatment
PRACTICE POINTS
- Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder that can manifest as isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs.
- Although there is no standard treatment for ICH, histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway and may be responsive to MEK inhibition.
- Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.

Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
To the Editor:
Psoriasis is a chronic inflammatory skin condition that commonly affects the nail matrix and/or nail bed.1 Nail involvement is present in up to 50% of patients with cutaneous psoriasis and 80% of patients with psoriatic arthritis.1 Approximately 5% to 10% of patients with psoriasis demonstrate isolated nail involvement with no skin or joint manifestations.1 Nail psoriasis can cause severe pain and psychological distress, and extreme cases may cause considerable morbidity and functional impairment.2,3 Treatment often requires a long duration and may not result in complete recovery due to the slow rate of nail growth. Patients can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.1,2 Despite the availability of various treatment options, many cases remain refractory to standard interventions, which underscores the need for novel therapeutic approaches. Herein, we present a severe case of refractory isolated nail psoriasis that was successfully treated with deucravacitinib, an oral tyrosine kinase 2 (TYK2) inhibitor.
A 59-year-old woman presented with a progressive, yellow, hyperkeratotic lesion on the left thumbnail of 2 years’ duration. The patient noted initial discoloration and peeling at the distal end of the nail. Over time, the discoloration progressed to encompass the entire nail. Previous treatments performed by outside physicians including topical corticosteroids, calcineurin inhibitors, and 2 surgeries to remove the nail plate and nail bed all were unsuccessful. The patient also reported severe left thumbnail pain and pruritus that considerably impaired her ability to work. The rest of the nails were unaffected, and she had no personal or family history of psoriasis. Her medical history was notable for hypertension, gastroesophageal reflux disease, and osteomyelitis of the right thumb without nail involvement. Drug allergies included penicillin G benzathine, sulfonamides, amoxicillin, and ciprofloxacin.
Physical examination of the left thumbnail revealed severe yellow, hyperkeratotic, dystrophic changes with a large, yellow, crumbling hyperkeratotic plaque that extended from approximately 1 cm beyond the nail plate to the proximal end of the distal interphalangeal joint, to and along the lateral nail folds, with extensive distal onycholysis. The proximal and lateral nail folds demonstrated erythema as well as maceration that was extremely tender to minimal palpation (Figure 1). No cutaneous lesions were noted elsewhere on the body. The patient had no tenderness, swelling, or stiffness in any of the joints. The differential diagnosis at the time included squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.

Radiography of the left thumb revealed irregular swelling and nonspecific soft tissue enlargement at the tip of the digit. A nail clipping from the left thumbnail and 3-mm punch biopsies of the lateral and proximal nail folds as well as the horn of the proximal nail fold (Figure 2) were negative for fungus and confirmed psoriasiform dermatitis of the nail.

The patient was started on vinegar soaks (1:1 ratio of vinegar to water) every other day as well as urea cream 10%, ammonium lactate 15%, and petrolatum twice daily for 2 months without considerable improvement. Due to lack of improvement during this 2-month period, the patient subsequently was started on oral deucravacitinib 6 mg/d along with continued use of petrolatum twice daily and vinegar soaks every other day. We selected a trial of deucravacitinib for our patient because of its convenient daily oral dosing and promising clinical evidence.4,5 After 2 months of treatment with deucravacitinib, the patient reported substantial improvement and satisfaction with the treatment results. Physical examination of the left thumbnail after 2 months of deucravacitinib treatment revealed mildly hyperkeratotic, yellow, dystrophic changes of the nail with notable improvement of the yellow hyperkeratotic plaque on the distal thumbnail. Normal-appearing nail growth was noted at the proximal nail fold, demonstrating considerable improvement from the initial presentation (Figure 3). However, the patient had developed multiple oral ulcers, generalized pruritus, and an annular urticarial plaque on the left arm. As such, deucravacitinib was discontinued after 2 months of treatment. These symptoms resolved within a week of discontinuing deucravacitinib.

While the etiology of nail psoriasis remains unclear, it is believed to be due to a combination of immunologic, genetic, and environmental factors.3 Classical clinical features include nail pitting, leukonychia, onycholysis, nail bed hyperkeratosis, and splinter hemorrhages.1,3 Our patient exhibited a severe form of nail psoriasis, encompassing the entire nail matrix and bed and extending to the distal interphalangeal joint and lateral nail folds. Previous surgical interventions may have triggered the Koebner phenomenon—which commonly is associated with psoriasis—and resulted in new skin lesions as a secondary response to the surgical trauma.6 The severity of the condition profoundly impacted her quality of life and considerably hindered her ability to work.
Treatment for nail psoriasis includes topical or systemic therapies such as corticosteroids, vitamin D analogs, tacrolimus, and tumor necrosis factor α inhibitors.1,3 Topical treatment is challenging because it is difficult to deliver medication effectively to the nail bed and nail matrix, and patient adherence may be poor.2 Although it has been shown to be effective, intralesional triamcinolone can be associated with pain as the most common adverse effect.7 Systemic medications such as oral methotrexate also may be effective but are contraindicated in pregnant patients and are associated with potential adverse events (AEs), including hepatotoxicity and acute kidney injury.8 The use of biologics may be challenging due to potential AEs and patient reluctance toward injection-based treatments.9
Deucravacitinib is a TYK2 inhibitor approved for treatment of plaque psoriasis.10 Tyrosine kinase 2 is an intracellular kinase that mediates the signaling of IL-23 and other cytokines involved in psoriasis pathogenesis.10 Deucravacitinib selectively binds to the regulatory domain of TYK2, leading to targeted allosteric inhibition of TYK2-mediated IL-23 and type I interferon signaling.4,5,10 Compared with biologics, deucravacitinib is advantageous because it can be administered as a daily oral pill, encouraging high patient compliance.
In the POETYK PSO-1 and PSO-2 phase 3 randomized controlled trials, 20.9% (n=332) and 20.3% (n=510) of deucravacitinib-treated patients with moderate to severe nail involvement achieved a Physician’s Global Assessment of Fingernail score of 0/1 compared with 8.8% (n=165) and 7.9% (n=254) of patients in the placebo group, respectively. All patients in these trials had a diagnosis of plaque psoriasis with at least 10% body surface area involvement; none of the patients had isolated nail psoriasis.4,5
The phase 3 POETYK PSO-1 and PSO-2 trials demonstrated deucravacitinib to be safe and well tolerated with minimal AEs.4,5 However, the development of AEs in our patient, including oral ulcers and generalized pruritus, underscores the need for close monitoring and consideration of potential risks of treatment. Common AEs associated with deucravacitinib include upper respiratory infections (19.2% [n=840]), increased blood creatine phosphokinase levels (2.7% [n=840]), herpes simplex virus (2.0% [n=840]), and mouth ulcers (1.9% [n=840]).11
Patient education also is a crucial component in the treatment of nail psoriasis. Physicians should emphasize the slow growth of nails and need for prolonged treatment. Clear communication and realistic expectations are essential for ensuring patient adherence to treatment.
Our case highlights the potential efficacy and safety of deucravacitinib for treatment of nail psoriasis, potentially laying the groundwork for future clinical studies. Our patient had a severe case of nail psoriasis that involved the entire nail bed and nail plate, resulting in extreme pain, pruritus, and functional impairment. Her case was unique because involvement was isolated to the nail without any accompanying skin or joint manifestations. She showed a favorable response to deucravacitinib within only 2 months of treatment and exhibited considerable improvement of nail psoriasis, with a reported high level of satisfaction with the treatment. We plan to continue to monitor the patient for long-term results. Future randomized clinical trials with longer follow-up periods are crucial to further establish the efficacy and safety of deucravacitinib for treatment of nail psoriasis.
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Ji C, Wang H, Bao C, et al. Challenge of nail psoriasis: an update review. Clin Rev Allergy Immunol. 2021;61:377-402. doi:10.1007/s12016-021-08896-9
- Muneer H, Sathe NC, Masood S. Nail psoriasis. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated March 1, 2024. Accessed October 24, 2024. https://www.ncbi.nlm.nih.gov/books/NBK559260/
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88:29-39. doi:10.1016/j.jaad.2022.07.002
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J Am Acad Dermatol. 2023;88:40-51. doi:10.1016/j.jaad.2022.08.061
- Sanchez DP, Sonthalia S. Koebner phenomenon. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated November 14, 2022. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553108/
- Grover C, Kharghoria G, Bansal S. Triamcinolone acetonide injections in nail psoriasis: a pragmatic analysis. Skin Appendage Disord. 2024;10:50-59. doi:10.1159/000534699
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated August 16, 2023. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;2011:Cd008794. doi:10.1002/14651858.CD008794.pub2
- Thaçi D, Strober B, Gordon KB, et al. Deucravacitinib in moderate to severe psoriasis: clinical and quality-of-life outcomes in a phase 2 trial. Dermatol Ther (Heidelb). 2022;12:495-510. doi:10.1007/s13555-021-00649-y
- Week 0-16: demonstrated safety profile. Bristol-Myers Squibb. 2024. Accessed October 24, 2024. https://www.sotyktuhcp.com/safety-profile?cid=sem_2465603&gclid=CjwKCAiA9ourBhAVEiwA3L5RFnyYqmxbqkz1_zBNPz3dcyHKCSFf1XQ-7acznV0XbR5DDJHYkZcKJxoCWN0QAvD_BwE&gclsrc=aw.ds
To the Editor:
Psoriasis is a chronic inflammatory skin condition that commonly affects the nail matrix and/or nail bed.1 Nail involvement is present in up to 50% of patients with cutaneous psoriasis and 80% of patients with psoriatic arthritis.1 Approximately 5% to 10% of patients with psoriasis demonstrate isolated nail involvement with no skin or joint manifestations.1 Nail psoriasis can cause severe pain and psychological distress, and extreme cases may cause considerable morbidity and functional impairment.2,3 Treatment often requires a long duration and may not result in complete recovery due to the slow rate of nail growth. Patients can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.1,2 Despite the availability of various treatment options, many cases remain refractory to standard interventions, which underscores the need for novel therapeutic approaches. Herein, we present a severe case of refractory isolated nail psoriasis that was successfully treated with deucravacitinib, an oral tyrosine kinase 2 (TYK2) inhibitor.
A 59-year-old woman presented with a progressive, yellow, hyperkeratotic lesion on the left thumbnail of 2 years’ duration. The patient noted initial discoloration and peeling at the distal end of the nail. Over time, the discoloration progressed to encompass the entire nail. Previous treatments performed by outside physicians including topical corticosteroids, calcineurin inhibitors, and 2 surgeries to remove the nail plate and nail bed all were unsuccessful. The patient also reported severe left thumbnail pain and pruritus that considerably impaired her ability to work. The rest of the nails were unaffected, and she had no personal or family history of psoriasis. Her medical history was notable for hypertension, gastroesophageal reflux disease, and osteomyelitis of the right thumb without nail involvement. Drug allergies included penicillin G benzathine, sulfonamides, amoxicillin, and ciprofloxacin.
Physical examination of the left thumbnail revealed severe yellow, hyperkeratotic, dystrophic changes with a large, yellow, crumbling hyperkeratotic plaque that extended from approximately 1 cm beyond the nail plate to the proximal end of the distal interphalangeal joint, to and along the lateral nail folds, with extensive distal onycholysis. The proximal and lateral nail folds demonstrated erythema as well as maceration that was extremely tender to minimal palpation (Figure 1). No cutaneous lesions were noted elsewhere on the body. The patient had no tenderness, swelling, or stiffness in any of the joints. The differential diagnosis at the time included squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.
Radiography of the left thumb revealed irregular swelling and nonspecific soft tissue enlargement at the tip of the digit. A nail clipping from the left thumbnail and 3-mm punch biopsies of the lateral and proximal nail folds as well as the horn of the proximal nail fold (Figure 2) were negative for fungus and confirmed psoriasiform dermatitis of the nail.
The patient was started on vinegar soaks (1:1 ratio of vinegar to water) every other day as well as urea cream 10%, ammonium lactate 15%, and petrolatum twice daily for 2 months without considerable improvement. Due to lack of improvement during this 2-month period, the patient subsequently was started on oral deucravacitinib 6 mg/d along with continued use of petrolatum twice daily and vinegar soaks every other day. We selected a trial of deucravacitinib for our patient because of its convenient daily oral dosing and promising clinical evidence.4,5 After 2 months of treatment with deucravacitinib, the patient reported substantial improvement and satisfaction with the treatment results. Physical examination of the left thumbnail after 2 months of deucravacitinib treatment revealed mildly hyperkeratotic, yellow, dystrophic changes of the nail with notable improvement of the yellow hyperkeratotic plaque on the distal thumbnail. Normal-appearing nail growth was noted at the proximal nail fold, demonstrating considerable improvement from the initial presentation (Figure 3). However, the patient had developed multiple oral ulcers, generalized pruritus, and an annular urticarial plaque on the left arm. As such, deucravacitinib was discontinued after 2 months of treatment. These symptoms resolved within a week of discontinuing deucravacitinib.
While the etiology of nail psoriasis remains unclear, it is believed to be due to a combination of immunologic, genetic, and environmental factors.3 Classical clinical features include nail pitting, leukonychia, onycholysis, nail bed hyperkeratosis, and splinter hemorrhages.1,3 Our patient exhibited a severe form of nail psoriasis, encompassing the entire nail matrix and bed and extending to the distal interphalangeal joint and lateral nail folds. Previous surgical interventions may have triggered the Koebner phenomenon—which commonly is associated with psoriasis—and resulted in new skin lesions as a secondary response to the surgical trauma.6 The severity of the condition profoundly impacted her quality of life and considerably hindered her ability to work.
Treatment for nail psoriasis includes topical or systemic therapies such as corticosteroids, vitamin D analogs, tacrolimus, and tumor necrosis factor α inhibitors.1,3 Topical treatment is challenging because it is difficult to deliver medication effectively to the nail bed and nail matrix, and patient adherence may be poor.2 Although it has been shown to be effective, intralesional triamcinolone can be associated with pain as the most common adverse effect.7 Systemic medications such as oral methotrexate also may be effective but are contraindicated in pregnant patients and are associated with potential adverse events (AEs), including hepatotoxicity and acute kidney injury.8 The use of biologics may be challenging due to potential AEs and patient reluctance toward injection-based treatments.9
Deucravacitinib is a TYK2 inhibitor approved for treatment of plaque psoriasis.10 Tyrosine kinase 2 is an intracellular kinase that mediates the signaling of IL-23 and other cytokines involved in psoriasis pathogenesis.10 Deucravacitinib selectively binds to the regulatory domain of TYK2, leading to targeted allosteric inhibition of TYK2-mediated IL-23 and type I interferon signaling.4,5,10 Compared with biologics, deucravacitinib is advantageous because it can be administered as a daily oral pill, encouraging high patient compliance.
In the POETYK PSO-1 and PSO-2 phase 3 randomized controlled trials, 20.9% (n=332) and 20.3% (n=510) of deucravacitinib-treated patients with moderate to severe nail involvement achieved a Physician’s Global Assessment of Fingernail score of 0/1 compared with 8.8% (n=165) and 7.9% (n=254) of patients in the placebo group, respectively. All patients in these trials had a diagnosis of plaque psoriasis with at least 10% body surface area involvement; none of the patients had isolated nail psoriasis.4,5
The phase 3 POETYK PSO-1 and PSO-2 trials demonstrated deucravacitinib to be safe and well tolerated with minimal AEs.4,5 However, the development of AEs in our patient, including oral ulcers and generalized pruritus, underscores the need for close monitoring and consideration of potential risks of treatment. Common AEs associated with deucravacitinib include upper respiratory infections (19.2% [n=840]), increased blood creatine phosphokinase levels (2.7% [n=840]), herpes simplex virus (2.0% [n=840]), and mouth ulcers (1.9% [n=840]).11
Patient education also is a crucial component in the treatment of nail psoriasis. Physicians should emphasize the slow growth of nails and need for prolonged treatment. Clear communication and realistic expectations are essential for ensuring patient adherence to treatment.
Our case highlights the potential efficacy and safety of deucravacitinib for treatment of nail psoriasis, potentially laying the groundwork for future clinical studies. Our patient had a severe case of nail psoriasis that involved the entire nail bed and nail plate, resulting in extreme pain, pruritus, and functional impairment. Her case was unique because involvement was isolated to the nail without any accompanying skin or joint manifestations. She showed a favorable response to deucravacitinib within only 2 months of treatment and exhibited considerable improvement of nail psoriasis, with a reported high level of satisfaction with the treatment. We plan to continue to monitor the patient for long-term results. Future randomized clinical trials with longer follow-up periods are crucial to further establish the efficacy and safety of deucravacitinib for treatment of nail psoriasis.
To the Editor:
Psoriasis is a chronic inflammatory skin condition that commonly affects the nail matrix and/or nail bed.1 Nail involvement is present in up to 50% of patients with cutaneous psoriasis and 80% of patients with psoriatic arthritis.1 Approximately 5% to 10% of patients with psoriasis demonstrate isolated nail involvement with no skin or joint manifestations.1 Nail psoriasis can cause severe pain and psychological distress, and extreme cases may cause considerable morbidity and functional impairment.2,3 Treatment often requires a long duration and may not result in complete recovery due to the slow rate of nail growth. Patients can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.1,2 Despite the availability of various treatment options, many cases remain refractory to standard interventions, which underscores the need for novel therapeutic approaches. Herein, we present a severe case of refractory isolated nail psoriasis that was successfully treated with deucravacitinib, an oral tyrosine kinase 2 (TYK2) inhibitor.
A 59-year-old woman presented with a progressive, yellow, hyperkeratotic lesion on the left thumbnail of 2 years’ duration. The patient noted initial discoloration and peeling at the distal end of the nail. Over time, the discoloration progressed to encompass the entire nail. Previous treatments performed by outside physicians including topical corticosteroids, calcineurin inhibitors, and 2 surgeries to remove the nail plate and nail bed all were unsuccessful. The patient also reported severe left thumbnail pain and pruritus that considerably impaired her ability to work. The rest of the nails were unaffected, and she had no personal or family history of psoriasis. Her medical history was notable for hypertension, gastroesophageal reflux disease, and osteomyelitis of the right thumb without nail involvement. Drug allergies included penicillin G benzathine, sulfonamides, amoxicillin, and ciprofloxacin.
Physical examination of the left thumbnail revealed severe yellow, hyperkeratotic, dystrophic changes with a large, yellow, crumbling hyperkeratotic plaque that extended from approximately 1 cm beyond the nail plate to the proximal end of the distal interphalangeal joint, to and along the lateral nail folds, with extensive distal onycholysis. The proximal and lateral nail folds demonstrated erythema as well as maceration that was extremely tender to minimal palpation (Figure 1). No cutaneous lesions were noted elsewhere on the body. The patient had no tenderness, swelling, or stiffness in any of the joints. The differential diagnosis at the time included squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.
Radiography of the left thumb revealed irregular swelling and nonspecific soft tissue enlargement at the tip of the digit. A nail clipping from the left thumbnail and 3-mm punch biopsies of the lateral and proximal nail folds as well as the horn of the proximal nail fold (Figure 2) were negative for fungus and confirmed psoriasiform dermatitis of the nail.
The patient was started on vinegar soaks (1:1 ratio of vinegar to water) every other day as well as urea cream 10%, ammonium lactate 15%, and petrolatum twice daily for 2 months without considerable improvement. Due to lack of improvement during this 2-month period, the patient subsequently was started on oral deucravacitinib 6 mg/d along with continued use of petrolatum twice daily and vinegar soaks every other day. We selected a trial of deucravacitinib for our patient because of its convenient daily oral dosing and promising clinical evidence.4,5 After 2 months of treatment with deucravacitinib, the patient reported substantial improvement and satisfaction with the treatment results. Physical examination of the left thumbnail after 2 months of deucravacitinib treatment revealed mildly hyperkeratotic, yellow, dystrophic changes of the nail with notable improvement of the yellow hyperkeratotic plaque on the distal thumbnail. Normal-appearing nail growth was noted at the proximal nail fold, demonstrating considerable improvement from the initial presentation (Figure 3). However, the patient had developed multiple oral ulcers, generalized pruritus, and an annular urticarial plaque on the left arm. As such, deucravacitinib was discontinued after 2 months of treatment. These symptoms resolved within a week of discontinuing deucravacitinib.
While the etiology of nail psoriasis remains unclear, it is believed to be due to a combination of immunologic, genetic, and environmental factors.3 Classical clinical features include nail pitting, leukonychia, onycholysis, nail bed hyperkeratosis, and splinter hemorrhages.1,3 Our patient exhibited a severe form of nail psoriasis, encompassing the entire nail matrix and bed and extending to the distal interphalangeal joint and lateral nail folds. Previous surgical interventions may have triggered the Koebner phenomenon—which commonly is associated with psoriasis—and resulted in new skin lesions as a secondary response to the surgical trauma.6 The severity of the condition profoundly impacted her quality of life and considerably hindered her ability to work.
Treatment for nail psoriasis includes topical or systemic therapies such as corticosteroids, vitamin D analogs, tacrolimus, and tumor necrosis factor α inhibitors.1,3 Topical treatment is challenging because it is difficult to deliver medication effectively to the nail bed and nail matrix, and patient adherence may be poor.2 Although it has been shown to be effective, intralesional triamcinolone can be associated with pain as the most common adverse effect.7 Systemic medications such as oral methotrexate also may be effective but are contraindicated in pregnant patients and are associated with potential adverse events (AEs), including hepatotoxicity and acute kidney injury.8 The use of biologics may be challenging due to potential AEs and patient reluctance toward injection-based treatments.9
Deucravacitinib is a TYK2 inhibitor approved for treatment of plaque psoriasis.10 Tyrosine kinase 2 is an intracellular kinase that mediates the signaling of IL-23 and other cytokines involved in psoriasis pathogenesis.10 Deucravacitinib selectively binds to the regulatory domain of TYK2, leading to targeted allosteric inhibition of TYK2-mediated IL-23 and type I interferon signaling.4,5,10 Compared with biologics, deucravacitinib is advantageous because it can be administered as a daily oral pill, encouraging high patient compliance.
In the POETYK PSO-1 and PSO-2 phase 3 randomized controlled trials, 20.9% (n=332) and 20.3% (n=510) of deucravacitinib-treated patients with moderate to severe nail involvement achieved a Physician’s Global Assessment of Fingernail score of 0/1 compared with 8.8% (n=165) and 7.9% (n=254) of patients in the placebo group, respectively. All patients in these trials had a diagnosis of plaque psoriasis with at least 10% body surface area involvement; none of the patients had isolated nail psoriasis.4,5
The phase 3 POETYK PSO-1 and PSO-2 trials demonstrated deucravacitinib to be safe and well tolerated with minimal AEs.4,5 However, the development of AEs in our patient, including oral ulcers and generalized pruritus, underscores the need for close monitoring and consideration of potential risks of treatment. Common AEs associated with deucravacitinib include upper respiratory infections (19.2% [n=840]), increased blood creatine phosphokinase levels (2.7% [n=840]), herpes simplex virus (2.0% [n=840]), and mouth ulcers (1.9% [n=840]).11
Patient education also is a crucial component in the treatment of nail psoriasis. Physicians should emphasize the slow growth of nails and need for prolonged treatment. Clear communication and realistic expectations are essential for ensuring patient adherence to treatment.
Our case highlights the potential efficacy and safety of deucravacitinib for treatment of nail psoriasis, potentially laying the groundwork for future clinical studies. Our patient had a severe case of nail psoriasis that involved the entire nail bed and nail plate, resulting in extreme pain, pruritus, and functional impairment. Her case was unique because involvement was isolated to the nail without any accompanying skin or joint manifestations. She showed a favorable response to deucravacitinib within only 2 months of treatment and exhibited considerable improvement of nail psoriasis, with a reported high level of satisfaction with the treatment. We plan to continue to monitor the patient for long-term results. Future randomized clinical trials with longer follow-up periods are crucial to further establish the efficacy and safety of deucravacitinib for treatment of nail psoriasis.
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Ji C, Wang H, Bao C, et al. Challenge of nail psoriasis: an update review. Clin Rev Allergy Immunol. 2021;61:377-402. doi:10.1007/s12016-021-08896-9
- Muneer H, Sathe NC, Masood S. Nail psoriasis. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated March 1, 2024. Accessed October 24, 2024. https://www.ncbi.nlm.nih.gov/books/NBK559260/
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88:29-39. doi:10.1016/j.jaad.2022.07.002
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J Am Acad Dermatol. 2023;88:40-51. doi:10.1016/j.jaad.2022.08.061
- Sanchez DP, Sonthalia S. Koebner phenomenon. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated November 14, 2022. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553108/
- Grover C, Kharghoria G, Bansal S. Triamcinolone acetonide injections in nail psoriasis: a pragmatic analysis. Skin Appendage Disord. 2024;10:50-59. doi:10.1159/000534699
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated August 16, 2023. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;2011:Cd008794. doi:10.1002/14651858.CD008794.pub2
- Thaçi D, Strober B, Gordon KB, et al. Deucravacitinib in moderate to severe psoriasis: clinical and quality-of-life outcomes in a phase 2 trial. Dermatol Ther (Heidelb). 2022;12:495-510. doi:10.1007/s13555-021-00649-y
- Week 0-16: demonstrated safety profile. Bristol-Myers Squibb. 2024. Accessed October 24, 2024. https://www.sotyktuhcp.com/safety-profile?cid=sem_2465603&gclid=CjwKCAiA9ourBhAVEiwA3L5RFnyYqmxbqkz1_zBNPz3dcyHKCSFf1XQ-7acznV0XbR5DDJHYkZcKJxoCWN0QAvD_BwE&gclsrc=aw.ds
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Ji C, Wang H, Bao C, et al. Challenge of nail psoriasis: an update review. Clin Rev Allergy Immunol. 2021;61:377-402. doi:10.1007/s12016-021-08896-9
- Muneer H, Sathe NC, Masood S. Nail psoriasis. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated March 1, 2024. Accessed October 24, 2024. https://www.ncbi.nlm.nih.gov/books/NBK559260/
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88:29-39. doi:10.1016/j.jaad.2022.07.002
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J Am Acad Dermatol. 2023;88:40-51. doi:10.1016/j.jaad.2022.08.061
- Sanchez DP, Sonthalia S. Koebner phenomenon. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated November 14, 2022. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553108/
- Grover C, Kharghoria G, Bansal S. Triamcinolone acetonide injections in nail psoriasis: a pragmatic analysis. Skin Appendage Disord. 2024;10:50-59. doi:10.1159/000534699
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated August 16, 2023. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;2011:Cd008794. doi:10.1002/14651858.CD008794.pub2
- Thaçi D, Strober B, Gordon KB, et al. Deucravacitinib in moderate to severe psoriasis: clinical and quality-of-life outcomes in a phase 2 trial. Dermatol Ther (Heidelb). 2022;12:495-510. doi:10.1007/s13555-021-00649-y
- Week 0-16: demonstrated safety profile. Bristol-Myers Squibb. 2024. Accessed October 24, 2024. https://www.sotyktuhcp.com/safety-profile?cid=sem_2465603&gclid=CjwKCAiA9ourBhAVEiwA3L5RFnyYqmxbqkz1_zBNPz3dcyHKCSFf1XQ-7acznV0XbR5DDJHYkZcKJxoCWN0QAvD_BwE&gclsrc=aw.ds
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
PRACTICE POINTS
- Nail psoriasis can masquerade as other dermatologic conditions, including squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.
- Nail psoriasis can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.
- Deucravacitinib, an oral tyrosine kinase 2 inhibitor approved for the treatment of plaque psoriasis, has shown promise as an effective treatment for nail psoriasis in cases that are refractory to standard therapies.
