User login
Pancreatic Adenocarcinoma: Update on Neoadjuvant and Adjuvant Treatment
Introduction
Exocrine pancreatic cancer refers to pancreatic adenocarcinomas that arise from ductal epithelial cells. Pancreatic ductal adenocarcinoma is a highly lethal malignancy, ranking as the fourth most common cause of cancer-related death in the United States1 and the eighth most common worldwide.2 In the United States, the pancreas is the second most common site of gastrointestinal malignancy after the colon.1 The only potentially curative modality for pancreatic adenocarcinomas is complete resection, followed by adjuvant therapy; unfortunately, only around 20% of patients are surgical candidates at the time of presentation due to delayed development of symptoms and consequently diagnosis.3 Most symptomatic patients with pancreatic cancer have locally advanced disease at diagnosis, and only a select group of patients with good performance status and borderline resectable disease can be offered neoadjuvant therapy. Adjuvant chemotherapy is typically recommended for patients who undergo potentially curative resection for pancreatic cancer.
Epidemiology
In the United States, pancreatic cancer has an annual estimated incidence of 55,440 new cases.1 It causes an estimated 44,330 deaths per year, with a 5-year overall survival (OS) rate of 8.2%.1 Worldwide an estimated 138,100 men and 127,900 women die of pancreatic cancer each year.2 In general, pancreatic cancers occur more commonly in persons living in Western/industrialized countries, older persons (age > 60 years), males (ratio 1.3:1 female), and African-Americans and native Hawaiians.4
Etiology
The major preventable environmental risk factor for pancreatic cancer is cigarette smoking, which accounts for 25% of all cases.5 A prospective study that estimated the excess incidence of pancreatic cancer among cigarette smokers and assessed the influence of smoking cessation on the risk for pancreatic cancer showed that persons who quit smoking reduced their risk of pancreatic cancer by 48% after 2 years of cessation, compared with smokers who did not quit, and reduced their risk to near the level of a never smoker after 10 years of cessation.5 Risk is higher for heavy smokers and those with homozygous deletions of the glutathione S-transferase theta 1 gene (GSTT1), which results in the absence of the carcinogen-metabolizing function of the glutathione S-transferase enzyme. High body mass index and sedentary lifestyle have been linked to pancreatic cancer.6 Data regarding aspirin, diet, coffee, and excess alcohol consumption are insufficient, inconclusive, and even conflicting, and thus the effect of these factors on risk for pancreatic cancer remains unclear. Infectious risk factors such as Helicobacter pylori and hepatitis B and C virus have weak associations with pancreatic cancer. Chronic pancreatitis and pancreatic cysts (eg, intraductal papillary mucinous neoplasm [IPMN] of the pancreas) carry a risk for malignant transformation, and hence may require surveillance. Multiple epidemiologic studies have shown a strong association between pancreatic cancer and recently diagnosed diabetes mellitus (relative risk [RR] 1.97 [95% confidence interval {CI} 1.78 to 2.18]); the presence of diabetes also may be a long-term predisposing factor for pancreatic cancer, and cancer screening needs to be considered for selected patients.7
A predisposing genetic anomaly accounts for 15% of all cases of pancreatic cancer.8 Hereditary risk factors are divided into 2 broad categories: defined genetic syndromes and familial pancreatic cancer. Familial predispositions that do not meet genetic syndrome criteria account for approximately 5% to 10% of all cases associated with hereditary factors; in one study, 29% of tested kindreds with an incident pancreatic cancer had a germline BRCA2 mutation.9 Other predisposing genetic syndromes that have been linked to pancreatic cancer include:
- Peutz-Jeghers syndrome with germline STK11 mutations (RR 132);
- Hereditary pancreatitis with germline PRSS1, SPINK1, and CFTR mutations (RR 26–87);
- Familial atypical multiple mole melanoma syndrome with CDKN2A mutations (RR 20–40);
- Familial breast and ovarian cancer with BRCA2 (RR 10) and BRCA1 (RR 2.8) mutations;
- Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch II syndrome) with MLH1, MSH2, MSH6, and PMS2 mutations (RR 9–11); and
- Familial adenomatous polyposis with APC mutations (RR 5).10
Other gene mutations with unknown relative risk for pancreatic cancer include mutations affecting PALB2, ATM, and TP53.
The International Cancer of the Pancreas Screening consortium consensus on screening for pancreatic cancer in patients with increased risk for familial pancreatic cancer recommends screening those at high risk: first-degree relatives (FDRs) of patients with pancreatic cancer from a familial pancreatic kindred with at least 2 affected FDRs; patients with Peutz-Jeghers syndrome; and p16, BRCA2, and HNPCC mutation carriers with 1 or more affected FDRs and hereditary pancreatitis. The guidelines emphasize that screening should be done only in those who are surgical candidates and are evaluated at an experienced multidisciplinary center.8
Deleterious germline mutations in pancreatic cancer can account for 33% of patients with apparent sporadic cancers and no hereditary risk. These include germline mutations affecting BRCA1/2, PALB2, ATM, MLH1, CHK-2, CDKN2A, and TP53.11
Pathogenesis
Pancreatic neoplasms can be benign or malignant and thus a tissue histologic diagnosis is paramount. Pancreatic adenocarcinomas with exocrine features represent more than 95% of all pancreatic neoplasms, with only 5% arising from the endocrine pancreas (ie, neuroendocrine tumors). Pancreatic neuroendocrine tumors and pancreatic adenocarcinoma must be distinguished histologically because treatment of the 2 neoplasms is completely different. Other malignant pancreatic tumors are signet ring cell carcinoma, adenosquamous carcinoma, undifferentiated (anaplastic) carcinoma, and mucinous noncystic (colloid) carcinoma; the latter tumor has a better prognosis.12 It is essential to characterize and distinguish among benign cystic neoplasms, as some require surgical resection due to the risk of malignant transformation. IPMN, pancreatic intraepithelial neoplasia, and mucinous cystic neoplasms are thought to be premalignant lesions of invasive ductal adenocarcinomas, and the pathological report should highlight the degree of dysplasia for adequate risk stratification.13 This information could be the deciding factor in whether a pancreatectomy is recommended by a multidisciplinary team.
Most pancreatic cancers harbor activating or silencing genetic mutations, and multiple combinations of altered genes can be detected by next-generation sequencing (average of 63 genetic alterations per cancer).14 Mutational activated KRAS is the most frequent (> 90%) genetic alteration in pancreatic cancer, even in early neoplastic precursors (IPMN > 75%). KRAS is a highly complex, dynamic proto-oncogene involved in signaling of various receptor kinases such as the epidermal growth factor receptor and the insulin-like growth factor receptor-I. It also engages in canonical downstream effector pathways, mainly Raf/MEK/ERK, PI3K/PDK1/Akt, and the Ral guanine nucleotide exchange factor pathway, which drive much of the pathogenesis of malignancy. These pathways lead to sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration, and metastasis. An activating point mutation in codon G12 is the most common (98%) locus of KRAS mutation in pancreatic adenocarcinoma, but all drugs targeting this mutation have failed in clinical practice.15 Additionally, inactivation of tumor suppressor genes such as p53, DPC4 (SMAD4/MADH4), CDKN2A (p16/MTS1), and BRCA2 can be found in 75%, 30%, 35%, and 4% of pancreatic adenocarcinoma cases, respectively.14 Another pancreatic cancer hallmark is inactivation of DNA damage repair genes, which include MLH1 and MSH2.16
Diagnosis and Staging
Case Presentation
A 71-year-old male veteran with no significant past medical history other than hypertension and hyperlipidemia and an excellent performance status presents to the emergency department after noticing a yellowish skin and sclera color. He denies weight loss, abdominal pain, or any other pertinent symptom or sign. Physical examination reveals a healthy developed man with yellowish discoloration of the skin and sclera and a soft, nontender benign abdomen; physical examination is otherwise unremarkable. Laboratory evaluation reveals a direct bilirubin level of 4.5 mg/dL and normal values for complete blood count and renal, liver, and coagulation panels. Abdominal and pelvis computed tomography (CT) with intravenous contrast shows a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein, which remains patent. Follow-up endoscopic ultrasound (EUS) confirms an irregular mass at the head of the pancreas measuring 3.2 × 2.6 cm with sonographic evidence suggesting invasion into the portal vein. During the procedure, the bile duct is successfully stented, the mass is biopsied, and bile duct brushing is performed. Pathology report is consistent with pancreatic adenocarcinoma.
- What is the typical presentation of pancreatic cancer?
The most common symptoms of pancreatic cancer at the time of presentation include weight loss (85%), asthenia/anorexia (86%), and/or abdominal pain (79%).17 The most frequent signs are jaundice (55%), hepatomegaly (39%), and cachexia (13%). Courvoisier sign, a nontender but palpable distended gallbladder at the right costal margin, is neither sensitive nor specific for pancreatic cancer (13% of cases). Trousseau syndrome, a superficial thrombophlebitis, is another classic sign that reflects the hypercoagulable nature of pancreatic cancer (3% of cases).17 The pathophysiology of this syndrome is not completely understood, but it may occur secondary to the release of cancer microparticles in the blood stream which in turn stimulate the coagulation cascade. Other nonspecific symptoms are dark urine, nausea, vomiting, diarrhea, steatorrhea, and epigastric and back pain. Because symptoms early in the course of the disease are nonspecific, pancreatic cancer is typically diagnosed late, after the cancer has invaded local structures or metastasized. The initial presentation varies depending on tumor location, with 70% of pancreatic head malignancies presenting with jaundice and pain correlating to an advanced stage.18 Although data supporting an association between new-onset diabetes mellitus and pancreatic cancer are inconclusive, pancreatic cancer should still be a consideration in patients with new-onset diabetes mellitus and other symptoms such as pain and weight loss. Early signs of incurable disease include a palpable mass, ascites, lymphadenopathy (classic Virchow node), and an umbilical mass (Sister Mary Joseph node). Incidentally discovered pancreatic masses on imaging are rare, but the incidence is increasing due to frequent imaging for other reasons and improved diagnostic techniques.
- What is the approach to diagnosis and staging?
History and physical examination findings are not sufficiently sensitive or specific to diagnose pancreatic cancer. High clinical suspicion in a patient with risk factors can lead to a comprehensive evaluation and potential early diagnosis. In general, an initial diagnostic work-up for suspected pancreatic cancer will include serologic evaluation (liver function test, lipase, tumor markers) and abdominal imaging (ultrasound, CT scans, or magnetic resonance imaging [MRI]). Ultrasound is a first-line diagnostic tool with a sensitivity of 90% and specificity of 98.8% for pancreatic cancer, but it is investigator-dependent and is less accurate in detecting tumors smaller than 3 cm in diameter.19 Multiphasic helical CT of the abdomen has better sensitivity (100%) and specificity (100%) for detecting tumors larger than 2 cm, but this modality is less accurate in detecting pancreatic masses smaller than 2 cm (77%).20 Percutaneous fine-needle aspiration (FNA) performed by ultrasound or CT guidance is avoided due to theoretical concerns about intraperitoneal seeding and bleeding.
If a pancreatic mass is detected by ultrasound or CT, additional interventions may be indicated depending on the clinical scenario. EUS-guided biopsy can provide histological confirmation and is currently utilized frequently for diagnosis and early resectability staging. Endoscopic retrograde cholangiopancreatography (ERCP) is indicated for patients with biliary obstruction requiring stent placement, and this procedure may provide tissue confirmation by forceps biopsy or brush cytology (lower accuracy than EUS). In a meta-analysis that evaluated the diagnostic value of tests for pancreatic cancer, ERCP had the highest sensitivity (92%) and specificity (96%) compared to ultrasound and CT,21 but this modality carries a risk for pancreatitis, bleeding, and cholangitis. Magnetic resonance cholangiopancreatography has not replaced ERCP, but it but may be an alternative for patients who cannot undergo ERCP (eg, gastric outlet obstruction, duodenal stenosis, anatomical surgical disruption, unsuccessful ERCP). ERCP is used frequently because many patients present with obstructive jaundice due to pancreatic mass compression, specifically if the mass is located in the head, and must undergo ERCP and stenting of the common bile duct.
The carbohydrate antigen (CA) 19-9 level has variable sensitivity and specificity in pancreatic cancer, as levels can be elevated in many benign pancreaticobiliary disorders. Elevated CA 19-9, in the appropriate clinical scenario (ie, a suspicious pancreatic mass and a value greater than 37 U/mL) demonstrated a sensitivity of 77% and specificity of 87% when differentiating pancreaticobiliary cancer from benign clinical conditions such as acute cholangitis or cholestasis.22 CA 19-9 level has prognostic value, as it may predict occult disease and correlates with survival rates, but no specific cutoff value has been established to guide perioperative therapy for high-risk resectable tumors.23
The American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) system is the preferred method for staging pancreatic cancer (Table 1). 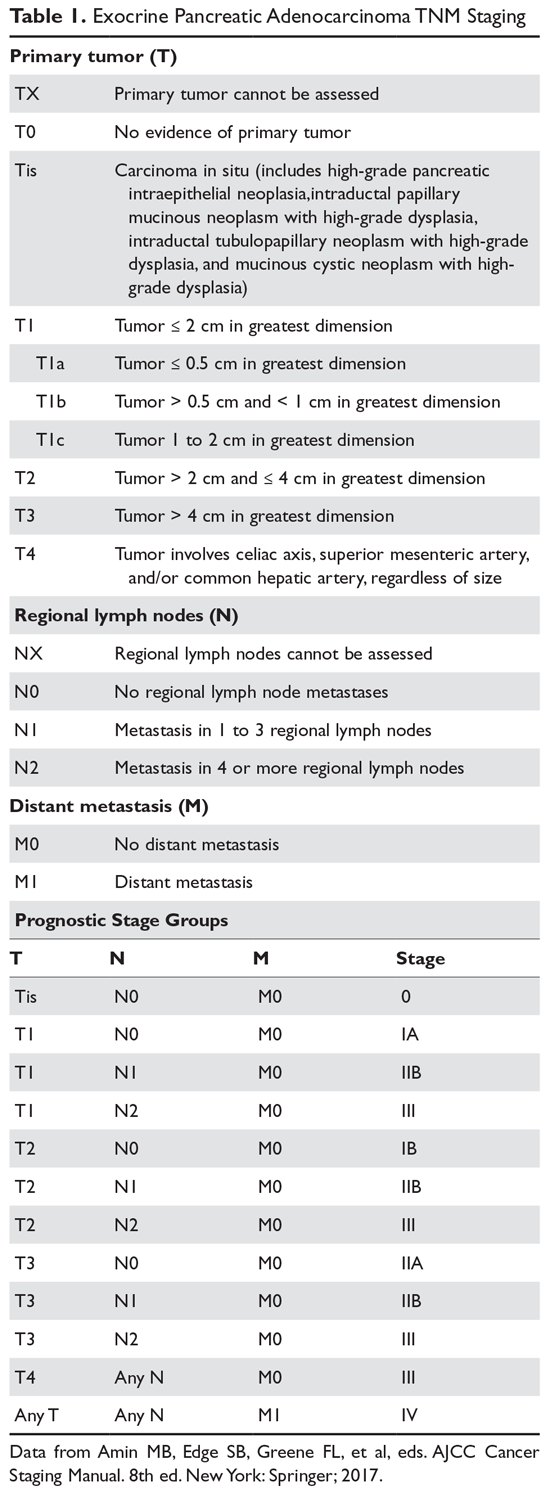

Positron emission tomography with CT scan is occasionally utilized in practice to assess tumor burden by evaluating anatomical structures and assessing physiologic uptake, which aids in establishing the extent of disease in equivocal cases. Staging laparoscopy with or without peritoneal biopsy is sometimes used to establish appropriate staging in cases that are questionable for occult metastatic disease. This procedure helps avoid unnecessary morbid surgeries.
Neoadjuvant Therapy
Case Continued
The patient is referred to oncology. Blood work reveals a CA 19-9 level of 100 U/mL (reference range < 35 U/mL) and a staging CT scan of the chest reveals a benign-appearing 3-mm nodule (no prior imaging for comparison). CT scan of the abdomen and pelvis does not define venous vasculature involvement appropriately and hence MRI of the abdomen and pelvis is performed. MRI reveals a pancreatic head mass measuring 3.0 × 2.7 cm, without arterial or venous vasculature invasion. However, the mass is abutting the portal vein and superior mesenteric vein and there is a new nonspecific 8-mm aortocaval lymph node.
- What are the current approaches to treating patients with resectable, unresectable, and metastatic disease?
Accurate staging and assessment of surgical resectability in pancreatic cancer are paramount as these steps prevent a futile morbid Whipple procedure in patients with advanced disease and a high risk of recurrence. Conversely, it allows patients with low-volume disease to undergo a potentially curative surgery. Approximately 20% of patients present with resectable disease, 40% present with locally advanced unresectable tumors (eg, involvement of critical vascular structures), and 40% present with metastatic disease.3 Treatment for resectable pancreatic cancer continues to be upfront surgery, although neoadjuvant therapy with either chemoradiation, radiation alone, or chemotherapy is an option per guidelines from the American Society of Clinical Oncology (ASCO),28 the NCCN,26 and the European Society for Medical Oncology (ESMO),29 particularly for patients with borderline resectable tumors (Table 3). 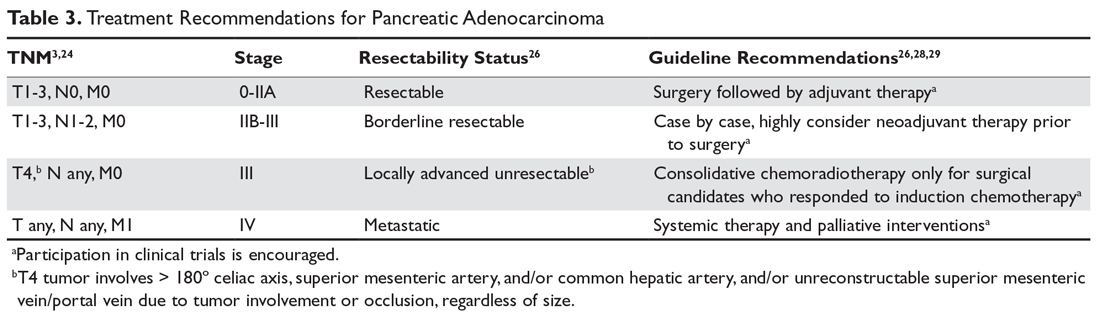
Systemic chemotherapy is recommended for fit candidates with locally advanced unresectable or metastatic disease, with an emphasis on supportive palliative measures. Palliative interventions include biliary stenting, duodenal stent for relieving gastric-outlet obstruction, and celiac axis nerve blocks, when indicated. Routine preoperative biliary stent placement/drainage in patients undergoing subsequent surgery for pancreatic cancer located in the head is associated with an increased risk of surgical complications when compared with up-front surgery without prior biliary drainage, and thus stent placement/drainage is not recommended.26 Aggressive supportive management of symptoms, such as cancer-associated pain, anorexia-cachexia syndromes, and anxiety-depression disorders, should remain a primary palliative focus.
Case Continued
A multidisciplinary tumor board discusses the patient’s case and deems the cancer borderline resectable; neoadjuvant therapy is recommended. The patient is started on treatment with gemcitabine and nab-paclitaxel as first-line neoadjuvant therapy. After 4 cycles, the CA 19-9 level drops to 14 U/mL, and MRI reveals a smaller head mass of 1.3 × 1.4 cm with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remains stable. At tumor board, it is evident that the patient has responded to therapy and the recommendation is to treat with gemcitabine chemoradiotherapy before surgery.
- What neoadjuvant therapy strategies are used in the treatment of pancreatic adenocarcinoma?
There are no established evidence-based recommendations for neoadjuvant therapy in patients with borderline resectable pancreatic cancer or patients with unresectable locally advanced pancreatic cancer. However, there are ongoing trials to investigate this treatment approach, and it is offered off-label in specific clinical scenarios, such as in the case patient described here. In patients with borderline resectable disease, preoperative chemotherapy followed by chemoradiation is a routine practice in most cancer centers,32 and ongoing clinical trials are an option for this cohort of patients (eg, Southwest Oncology Group Trial 1505, NCT02562716). The definitions of borderline resectable and unresectable pancreatic cancer have been described by the NCCN,26 although most surgeons consider involvement of the major upper abdominal blood vessels the main unresectability criterion; oncologists also consider other parameters such as suspicious lesions on scans, worsening performance status, and a significantly elevated CA 19-9 level suggestive of disseminated disease.28 The goal of a conversion approach by chemotherapy with or without radiation for borderline and unresectable cancers is to deliver a tolerable regimen leading to tumor downstaging, allowing for surgical resection. No randomized clinical trial has shown a survival advantage of this approach. Enrollment in clinical trials is preferred for patients with borderline and unresectable cancer, and there are trials that are currently enrolling patients.
The main treatment strategies for patients with locally advanced borderline and unresectable pancreatic cancer outside of a clinical trial are primary radiotherapy, systemic chemotherapy, and chemoradiation therapy. Guidelines from ASCO, NCCN, and ESMO recommend induction chemotherapy followed by restaging and consolidation chemoradiotherapy in the absence of progression.26,28,29 There is no standard chemoradiation regimen and the role of chemotherapy sensitizers, including fluorouracil, gemcitabine, and capecitabine (an oral fluoropyrimidine substitute), and targeted agents in combination with different radiation modalities is now being investigated.
Fluorouracil is a radio-sensitizer that has been used in locally advanced pancreatic cancer based on experience in other gastrointestinal malignancies; data shows conflicting results with this drug. Capecitabine and tegafur/gimeracil/oteracil (S-1) are oral prodrugs that can safely replace infusional fluorouracil. Gemcitabine, a more potent radiation sensitizer, is very toxic, even at low-doses twice weekly, and does not provide a survival benefit, as demonstrated in the Cancer and Leukemia Group B (CALGB) 89805 trial, a phase 2 study of patients with surgically staged locally advanced pancreatic cancer.33 Gemcitabine-based chemoradiotherapy was also evaluated in the Eastern Cooperative Group (ECOG) E4201 trial, which randomly assigned patients to receive gemcitabine alone (at 1000 mg/m2/wk for weeks 1 through 6, followed by 1 week rest, then weekly for 3 out of 4 weeks) or gemcitabine (600 mg/m2/wk for weeks 1 to 5, then 4 weeks later 1000 mg/m2 for 3 out of 4 weeks) plus radiotherapy (starting on day 1, 1.8 Gy/fraction for total of 50.4 Gy).34 Patients with locally advanced unresectable pancreatic cancer had a better OS outcome with gemcitabine in combination with radiation therapy (11.1 months) as compared with patients who received gemcitabine alone (9.2 months). Although there was a greater incidence of grade 4 and 5 treatment-related toxicities in the combination arm, no statistical differences in quality-of-life measurements were reported. Gemcitabine-based and capecitabine-based chemoradiotherapy were compared in the open-label phase 2 multicenter randomized SCALOP trial.35 Patients with locally advanced pancreatic cancer were assigned to receive 3 cycles of induction with gemcitabine 1000 mg/m2 days 1, 8, and 15 and capecitabine 830 mg/m2 days 1 to 21 every 28 days; patients who had stable or responding disease were randomly assigned to receive a fourth cycle followed by capecitabine (830 mg/m2 twice daily on weekdays only) or gemcitabine (300 mg/m2 weekly) with radiation (50.4 Gy over 28 fractions). Patients treated with capecitabine-based chemoradiotherapy had higher nonsignificant median OS (17.6 months) and median progression-free survival (12 months) compared to those treated with gemcitabine (14.6 months and 10.4 months, respectively).
The benefit of radiation therapy in the treatment of locally advanced pancreatic cancer was further explored by the Fédération Francophone de Cancérologie Digestive 2000-01 phase 3 trial. This study compared induction chemoradiotherapy (60 Gy, 2 Gy/fraction; concomitant fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) to gemcitabine alone (1000 mg/m2 weekly for 7 weeks) followed by maintenance gemcitabine in both arms.36 Unexpectedly, the median OS was significantly shorter in the chemoradiotherapy arm than in the chemotherapy alone arm (8.6 months versus 13 months, respectively, P = 0.03) and the combination arm had more toxicities. The phase 3 open-label LAP07 study explored the role of radiation therapy in patients with locally advanced pancreatic cancer who had controlled disease after 4 months of induction therapy.37 LAP07 had 2 randomizations: first, patients with locally advanced pancreatic cancer were assigned to receive weekly gemcitabine alone (1000 mg/m2) or this same dose of gemcitabine plus erlotinib 100 mg/day; second, patients with progression-free disease (61% of initial cohort) after 4 months of therapy were assigned to receive 2 months of the same chemotherapy or chemoradiotherapy (54 Gy plus capecitabine). This study showed that the addition of erlotinib to gemcitabine did not improve survival and in fact affected survival adversely. Of note, no survival benefit was observed after the first randomization from chemotherapy to consolidating chemoradiotherapy. Chemoradiotherapy achieved better locoregional tumor control with significantly less local tumor progression (32% versus 46%, P < 0.03) and no increase in toxicity. Based on prior moderate-quality evidence, guidelines recommend consolidative chemoradiotherapy only for surgical resection candidates following induction chemotherapy; for those who are not surgical candidates, guidelines recommend continuing systemic therapy.26,28,29
Gemcitabine and fluorouracil-based chemotherapies were the standard induction regimens until evidence from studies of metastatic systemic treatment protocols with FOLFIRINOX (ACCORD trial38) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) plus gemcitabine (MPACT trial39) was extrapolated to clinical practice. These regimens were shown to achieve higher objective response rates when compared to single-agent gemcitabine in patients with metastatic pancreatic cancer. Due to the broad heterogeneity of results in small retrospective series with neoadjuvant trials in borderline resectable pancreatic cancer, the quality of the evidence is low and any recommendation is limited. Many individual series have demonstrated improved complete resection rates and promising survival rates. In the largest single-institution retrospective review of patients with borderline resectable pancreatic adenocarcinoma who completed neoadjuvant gemcitabine-based chemoradiotherapy (50 Gy in 28 fractions or 30 Gy in 10 fractions), 94% achieved a margin-negative pancreatectomy; the median OS in those who completed preoperative therapy and had surgery was 40 months, with a 5-year OS of 36%.40 A meta-analysis by Andriulli and colleagues included 20 prospective studies of patients with initially resectable (366 lesions) or unresectable (341 lesions) disease who were treated with neoadjuvant/preoperative gemcitabine with or without radiotherapy.41 In the group with initially unresectable disease, 39% underwent surgery after restaging and 68% of explored patients were resected; the R0 resection rate was 60%. After restaging, 91% of patients with resectable disease underwent surgery, with 82% of explored patients undergoing surgical resection and 89% of these achieving R0 resection. The estimated 1- and 2-year survival probabilities after resection among patients with initially unresectable disease were 86.3% and 54.2%.41
The largest single-institution retrospective review of FOLFIRINOX (fluorouracil, oxaliplatin, irinotecan, and leucovorin), an alternative to gemcitabine, for neoadjuvant induction therapy for patients with locally advanced unresectable disease was conducted at Memorial Sloan Kettering Cancer Center. In this study (n = 101), 31% of patients initially deemed unresectable who completed FOLFIRINOX induction therapy with or without chemoradiation underwent resection. The R0 resection rate in these patients was 55%, and patients who did not progress during induction FOLFIRINOX therapy had a median OS of 26 months.42 A systematic review and meta-analysis of FOLFIRINOX chemotherapy with or without radiotherapy in patients with locally advanced unresectable pancreatic cancer reported that 25.9% of patients underwent resection after FOLFIRINOX therapy, and the R0 resection rate in these patients was 78.4%.43 The median OS in this study was 24.2 months, which was longer than the previously reported median OS rates for gemcitabine.
There is no strong evidence published for the use of combination nab-paclitaxel plus gemcitabine in the neoadjuvant setting, but it is used in clinical practice based on evidence from the MPACT trial, which showed the combination improved OS and progression-free survival in patients with metastatic pancreatic cancer.39 An early-phase 1-arm clinical trial of neoadjuvant gemcitabine, docetaxel, and capecitabine (GTX) followed by radiotherapy showed an increased response rate and survival for locally advanced disease; however, the NCCN expert panel has reached a consensus but not a uniform recommendation regarding this regimen due to significant toxicities and low patient accrual.26 Selected patients with pancreatic cancer with BRCA1/2 mutations are more sensitive to platinum-based chemotherapy. Although studies of neoadjuvant platinum-based chemotherapy in this population have not been reported, the NCCN guidelines list it as an alternative option based on extrapolated data.26 A clinical trial of gemcitabine, nab-paclitaxel, and cisplatin in the neoadjuvant setting in patients with resectable pancreatic cancer is currently enrolling patients (NGC triple regimen NCT0339257).
Summary
Chemotherapy alone or followed by chemoradiotherapy may be used as initial treatment for patients with borderline and unresectable pancreatic adenocarcinoma without distant metastases who are potential surgical candidates. Chemoradiotherapy remains a preferred treatment option for patients with poorly controlled pain from local tumor invasion, in view of the well-documented analgesic palliative effect of radiation therapy. FOLFIRINOX with or without radiation therapy may offer the highest documented response rates, but it also results in higher rates of treatment-related toxicities. FOLFIRINOX can be offered to selected fit patients (< 65 years old, no comorbidity contraindication, good functional status [ECOG 0–1]) who can tolerate triple therapy with a more toxic adverse-effect profile. A clinical trial evaluating neoadjuvant FOLFIRINOX with or without preoperative chemoradiotherapy in patients with borderline resectable pancreatic cancer is ongoing (PANDAS-PRODIGE 44, NCT02676349). Gemcitabine with or without radiation therapy is a tolerable combination, although it is potentially more toxic when combined with radiation. The addition of nab-paclitaxel to gemcitabine without radiation may emerge as a preferred neoadjuvant treatment for selected patients; a clinical trial investigating this modality in patients with resectable and borderline resectable disease is ongoing (NCT02723331).
Adjuvant Therapy
Case Continued
Prior to the planned surgical resection and after undergoing chemoradiation therapy, the patient has an excellent performance status and repeat MRI shows a 1.3 × 1.4 cm head mass with no further vasculature involvement, no evidence of lymphadenopathy, and no distant metastasis. The CA 19-9 level is stable at 18 U/mL. The patient undergoes an uncomplicated partial pancreaticoduodenectomy, and analysis of a surgical pathology specimen reveals T3N0 disease with closest margin of 0.1 cm.
- Would the patient benefit from adjuvant therapy?
Adjuvant chemotherapy for 6 months after pancreatic cancer resection should be offered to all patients based on mature data. Gemcitabine and capecitabine are the current standard of care in adjuvant therapy; alternatively, single-agent gemcitabine can be offered to patients with poor performance status or patients who cannot tolerate the toxicities associated with this combination.28 Adjuvant treatment should be initiated within approximately 8 weeks of surgical resection. The value of radiation therapy remains controversial, but it can be offered within the context of a clinical trial or to patients with positive margins after surgical resection and/or lymph node–positive disease. Based on low-quality supportive evidence, it is strongly recommended that patients who receive neoadjuvant therapy complete a total of 6 months of chemotherapy, factoring in the duration of the preoperative regimen.28 Different adjuvant strategies have been investigated, including chemotherapy alone with a fluoropyrimidine and/or gemcitabine with or without combined chemoradiation therapy.
The European Study Group for Pancreatic Cancer 1 (ESPAC)-1 trial was a randomized clinical trial that evaluated several adjuvant strategies in pancreatic cancer treatment. This trial assigned patients who underwent pancreatic adenocarcinoma resection to adjuvant chemotherapy alone (intravenous fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 daily for 5 days, monthly for 6 months), chemoradiotherapy (20 Gy in 10 daily fractions over 2 weeks with 500 mg/m2 intravenous fluorouracil on days 1–3, repeated after 2 weeks), both chemotherapy and chemoradiation, and observation.44 The results showed no added benefit for adjuvant chemoradiotherapy, with a median OS of 15.5 months in the chemoradiotherapy cohort, as compared to a median OS of 16.1 months in the chemotherapy-alone cohort (hazard ratio [HR] 1.18 [95% CI 0.90 to 1.55], P = 0.24). In addition, there was evidence of a survival benefit for the chemotherapy-alone arm when compared to the combined modality arm, with a median OS of 19.7 versus 14.0 months, respectively (HR 0.66 [95% CI 0.52 to 0.83], P = 0.0005). Although ESPAC-1 has been criticized for being underpowered to perform statistical comparison, it is still considered a landmark trial demonstrating benefit with single-agent chemotherapy alone. A follow-up analysis of ESPAC-1 showed that adjuvant chemotherapy alone conferred a significant 5-year survival benefit while the combined modality had a deleterious effect on survival. 45 Hence, adjuvant chemotherapy alone became the standard of care in the United States following resection.
The results of the multicenter randomized controlled phase 3 CONKO-001 (CharitéOnkologie 001) trial, which were reported in 2007, supported the use of adjuvant gemcitabine for 6 months in patients with resected pancreatic adenocarcinoma. In this study, patients treated with adjuvant gemcitabine (1000 mg/m2 days 1, 8, and 15 every 4 weeks for 6 months) had superior disease-free survival compared with those who received surgery alone.30 A long-term outcome update of this study demonstrated a significant improvement in 5-year OS for patients treated with adjuvant gemcitabine (20.7% [95% CI 14.7% to 26.6%]) compared to those who received surgical resection alone (10.4% [95% CI 5.9% to 15.0%]). This benefit persisted at 10-year follow-up, with an OS of 12.2% (95% CI 7.3% to 17.2%) in the adjuvant gemcitabine group, as compared to 7.7% (95% CI 3.6% to 11.8%) in the resection alone group.31
Fluorouracil and gemcitabine remained equivalent adjuvant treatment options until the results of the ESPAC-3 trial were reported in 2010.32 This large phase 3 trial, conducted mainly in the United Kingdom, compared weekly gemcitabine (1000 mg/m2 weekly for 3 of every 4 weeks) to leucovorin-modulated fluorouracil (Mayo Clinic regimen: leucovorin 20 mg/m2 followed by fluorouracil 425 mg/m2 intravenous bolus days 1 through 5 every 28 days) as adjuvant therapy in resected pancreatic adenocarcinoma. After a median follow-up of 34.2 months, the median OS was similar in the 2 groups (fluorouracil/leucovorin 23.0 months versus gemcitabine 23.6 months; P = 0.39). However, the fluorouracil/leucovorin group experienced more grade 3/4 treatment-related toxicities (mucositis, stomatitis, diarrhea, and hosptializations; 14% versus 7.5%; P < 0.001).46 Following this trial, gemcitabine became the standard of care for adjuvant chemotherapy for resected pancreatic cancer.
The U.S. Radiation Therapy Oncology Group (RTOG) 9704 trial was conducted to investigate the potential benefit of adding radiation therapy to gemcitabine. This trial demonstrated an improved trend among patients with pancreatic head tumors (but not with cancers of the pancreatic body or tail) who received adjuvant gemcitabine followed by chemoradiotherapy (50.4 Gy in 1.8 Gy daily fractions for 5.5 weeks with concurrent infusional fluorouracil 250 mg/m2 daily) and subsequent gemcitabine monotherapy compared to postoperative fluorouracil-based chemoradiotherapy. Results showed a 5-year OS of 22% versus 18%, respectively, although this improvement was not statistically significant (P = 0.08). This trial failed to show a benefit of adding radiotherapy to gemcitabine.47
The ESPAC-4 trial, reported in 2017, evaluated the combination of gemcitabine and capecitabine compared to gemcitabine alone as adjuvant therapy for resected pancreatic adenocarcinoma.48 Patients were randomly assigned after surgical resection, regardless of margin or node status, to 6 months of gemcitabine alone (1000 mg/m2/day on days 1, 8, and 15 of each 28-day cycle) or gemcitabine plus capecitabine (1660 mg/m2/day on days 1 through 21 of each 28-day cycle). Combination therapy had a significant survival benefit compared to single therapy, with median OS durations of 28 months and 25.5 months, respectively (HR for death 0.82 [95% CI 0.68 to 0.98]). The 5-year OS for patients who received combination treatment was 29 months (95% CI 22.9 to 35.2) versus 16 months (95% CI 10.2 to 23.7) for those in the monotherapy group. As expected, grade 3 or 4 treatment-related toxicities (diarrhea, hand-foot syndrome, and neutropenia) were significantly more common with combined therapy, although there were no significant differences in the rates of serious adverse events. The adjuvant combination of gemcitabine and capecitabine became the current and preferred new standard of care following resection of pancreatic ductal adenocarcinoma,28 but single-agent gemcitabine and fluorouracil/leucovorin continue to be viable options,26,28,29 particularly for elderly patients, patients with borderline performance status, or patients with multiple comorbidities.
Evidence showing that a more intensive regimen can improve outcome in the adjuvant setting remains elusive. The phase 3 APACT study (Adjuvant Therapy for Patients with Resected Pancreatic Cancer, NCT01964430) comparing gemcitabine alone to gemcitabine plus nab-paclitaxel in patients with surgically resected pancreatic adenocarcinoma has concluded, with the results projected to be released in 2018. Another phase 3 trial investigating the efficacy of FOLFIRINOX versus gemcitabine alone as adjuvant therapy is underway in France and Canada (PRODIGE24/ACCORD24, NCT01526135). Other strategies with newer targeted therapies and immunotherapy are in the development phase.
Follow-Up and Surveillance
Case Conclusion
After recovery from surgery, the patient is offered and completes 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine. He is started on surveillance at 3 and 6 months, and he maintains an excellent performance status. He develops clinical evidence of pancreatic enzyme insufficiency and is placed on oral replacement therapy. He has no other complaints, and there is no evidence of recurrence on MRI and CA 19-9 levels.
- What is the recommended duration of surveillance following curative resection?
Surveillance after curative resection of pancreatic adenocarcinoma is recommended by NCCN guidelines.26 However, pancreatic adenocarcinoma has a poor prognosis, and surveillance after curative surgical resection with or without perioperative therapy has not been shown to impact survival. Most recurrences will occur within 2 years after treatment. Surveillance recommendations differ among expert groups.26,28,29 NCCN guidelines recommend evaluating patients by history and physical examination every 3 to 6 months for the first 2 years, then every 6 to 12 months for 3 years. CA 19-9 level and CT scan should be obtained every 3 to 6 months for 2 years and then every 6 to 12 months for 3 years. Follow-up with CA 19-9 levels and CT scans after 5 years is not routinely performed unless guided by signs, symptoms, or laboratory findings that raise suspicion for recurrence. Follow-up visits should also include evaluation of treatment-related toxicities, symptom management, nutrition support of pancreatic insufficiency, and psychosocial support.
Conclusion
Pancreatic cancer is a leading cause of cancer-related death that frequently presents with locally advanced or metastatic disease due to nonspecific symptoms and lack of a screening modality. Histological tissue biopsy confirmation and accurate resectability staging guide treatment planning and prognosis. The only potentially curative therapy is surgical resection plus adjuvant therapy for those with resectable disease. Surgical candidates with borderline resectable and unresectable disease can be offered induction preoperative chemotherapy followed by consolidation chemoradiation, based on clinical consensus practice. Enrollment in clinical trials should be encouraged for all patients, as evidence from clinical trials is essential to making progress in pancreatic cancer treatment.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69.
3. Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th edition staging system for patients with pancreatic adenocarcinoma: a Surveillance, Epidemiology and End Results (SEER) analysis. Ann Surg Oncol 2017;24:2023–30.
4. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer.gov/statfacts/html/pancreas.html. Accessed 17 February 2018.
5. Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
6. Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
7. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62. Epub 2014 Mar 9.
8. Canto MI, Harinck F, Hruban RH, et al, on behalf of the International Cancer of the Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. Epub 2012 Nov 7.
9. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64:2634–8.
10. McKay SH,Humphris JL, Johns AL, et al. Inherited pancreatic cancer. Cancer Forum 2016;40(1).
11. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90.
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. AFIP Atlas of Tumor Pathology. 4th series, fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007.
13. Vege SS, Ziring B, Jain R, Moayyedi P, Clinical Guidelines Committee, American Gastroenterology Association. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22.
14. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501.
15. Choi M, Bien H, Mofunanya A, Powers S. Challenges in Ras therapeutics in pancreatic cancer. Semin Cancer Biol 2017 Nov 21. pii: S1044-579X(17)30235-3.
16. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68. Epub 2016 Nov 15.
17. Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
18. Modolell I, Guarner L, Malagelada JR. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann Oncol 1999;10 Suppl 4:82–4.
19. Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
20. Bronstein YL, Loyer EM, Kaur H, et al. Detection of small pancreatic tumors with multiphasic helical CT. AJR Am J Roentgenol 2004;182:619–23.
21. Niederau C, Grendell JH. Diagnosis of pancreatic carcinoma. Imaging techniques and tumor markers. Pancreas 1992;7:66–86.
22. Kim HJ, Kim MH, Myung SJ, et al. A new strategy for the application of CA19-9 in the differentiation of pancreaticobiliary cancer: analysis using a receiver operating characteristic curve. Am J Gastroenterol 1999;94:1941–6.
23. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2541–56.
24. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
25. Soriano A, Castells A, Ayuso C, et al. Preoperative staging and tumor resectability assessment of pancreatic cancer: prospective study comparing endoscopic ultrasonography, helical computed tomography, magnetic resonance imaging, and angiography. Am J Gastroenterol 2004;99:492–501.
26. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
27. Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014;270:248–60.
28. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol 2017;35:2324–8.
28. Ducreux M, Cuhna AS, Caramella C, et al; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v56–68.
30. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
31. Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 2013;310:1473–81.
32. Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
33. Blackstock AW, Tepper JE, Niedwiecki D, et al. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer 2003;34(2-3):107–16.
34. Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105–12.
35. Hurt CN, Falk S, Crosby T, et al. Long-term results and recurrence patterns from SCALOP: a phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br J Cancer 2017;116:1264–70.
36. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol 2008;19:1592–9.
37. Hammel P, Huguet F, van Laethem JL, et al, LAP07 Trial Group. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
38. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
39. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703.
40. Katz MH, Pisters PW, Evans DB, et al. Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 2008;206:833–46.
41. Andriulli A, Festa V, Botteri E, et al. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: a meta-analysis of prospective studies. Ann Surg Oncol 2012;19:1644–62.
42. Sadot E, Doussot A, O’Reilly EM, et al. FOLFIRINOX induction therapy for stage 3 pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:3512–21.
43. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
44. Neoptolemos JP, Dunn JA, Stocken DD, et al, European Study Group for Pancreatic Cancer. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet 2001;358:1576–85.
45. Neoptolemos JP, Stocken DD, Friess H, et al, European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
46. Neoptolemos JP, Stocken DD, Bassi C, et al, European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 2010;304:1073–81.
47. Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA 2008;299:1019–26.
48. Neoptolemos JP, Palmer DH, Ghaneh P, et al, European Study Group for Pancreatic Cancer. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. Epub 2017 Jan 25.
Introduction
Exocrine pancreatic cancer refers to pancreatic adenocarcinomas that arise from ductal epithelial cells. Pancreatic ductal adenocarcinoma is a highly lethal malignancy, ranking as the fourth most common cause of cancer-related death in the United States1 and the eighth most common worldwide.2 In the United States, the pancreas is the second most common site of gastrointestinal malignancy after the colon.1 The only potentially curative modality for pancreatic adenocarcinomas is complete resection, followed by adjuvant therapy; unfortunately, only around 20% of patients are surgical candidates at the time of presentation due to delayed development of symptoms and consequently diagnosis.3 Most symptomatic patients with pancreatic cancer have locally advanced disease at diagnosis, and only a select group of patients with good performance status and borderline resectable disease can be offered neoadjuvant therapy. Adjuvant chemotherapy is typically recommended for patients who undergo potentially curative resection for pancreatic cancer.
Epidemiology
In the United States, pancreatic cancer has an annual estimated incidence of 55,440 new cases.1 It causes an estimated 44,330 deaths per year, with a 5-year overall survival (OS) rate of 8.2%.1 Worldwide an estimated 138,100 men and 127,900 women die of pancreatic cancer each year.2 In general, pancreatic cancers occur more commonly in persons living in Western/industrialized countries, older persons (age > 60 years), males (ratio 1.3:1 female), and African-Americans and native Hawaiians.4
Etiology
The major preventable environmental risk factor for pancreatic cancer is cigarette smoking, which accounts for 25% of all cases.5 A prospective study that estimated the excess incidence of pancreatic cancer among cigarette smokers and assessed the influence of smoking cessation on the risk for pancreatic cancer showed that persons who quit smoking reduced their risk of pancreatic cancer by 48% after 2 years of cessation, compared with smokers who did not quit, and reduced their risk to near the level of a never smoker after 10 years of cessation.5 Risk is higher for heavy smokers and those with homozygous deletions of the glutathione S-transferase theta 1 gene (GSTT1), which results in the absence of the carcinogen-metabolizing function of the glutathione S-transferase enzyme. High body mass index and sedentary lifestyle have been linked to pancreatic cancer.6 Data regarding aspirin, diet, coffee, and excess alcohol consumption are insufficient, inconclusive, and even conflicting, and thus the effect of these factors on risk for pancreatic cancer remains unclear. Infectious risk factors such as Helicobacter pylori and hepatitis B and C virus have weak associations with pancreatic cancer. Chronic pancreatitis and pancreatic cysts (eg, intraductal papillary mucinous neoplasm [IPMN] of the pancreas) carry a risk for malignant transformation, and hence may require surveillance. Multiple epidemiologic studies have shown a strong association between pancreatic cancer and recently diagnosed diabetes mellitus (relative risk [RR] 1.97 [95% confidence interval {CI} 1.78 to 2.18]); the presence of diabetes also may be a long-term predisposing factor for pancreatic cancer, and cancer screening needs to be considered for selected patients.7
A predisposing genetic anomaly accounts for 15% of all cases of pancreatic cancer.8 Hereditary risk factors are divided into 2 broad categories: defined genetic syndromes and familial pancreatic cancer. Familial predispositions that do not meet genetic syndrome criteria account for approximately 5% to 10% of all cases associated with hereditary factors; in one study, 29% of tested kindreds with an incident pancreatic cancer had a germline BRCA2 mutation.9 Other predisposing genetic syndromes that have been linked to pancreatic cancer include:
- Peutz-Jeghers syndrome with germline STK11 mutations (RR 132);
- Hereditary pancreatitis with germline PRSS1, SPINK1, and CFTR mutations (RR 26–87);
- Familial atypical multiple mole melanoma syndrome with CDKN2A mutations (RR 20–40);
- Familial breast and ovarian cancer with BRCA2 (RR 10) and BRCA1 (RR 2.8) mutations;
- Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch II syndrome) with MLH1, MSH2, MSH6, and PMS2 mutations (RR 9–11); and
- Familial adenomatous polyposis with APC mutations (RR 5).10
Other gene mutations with unknown relative risk for pancreatic cancer include mutations affecting PALB2, ATM, and TP53.
The International Cancer of the Pancreas Screening consortium consensus on screening for pancreatic cancer in patients with increased risk for familial pancreatic cancer recommends screening those at high risk: first-degree relatives (FDRs) of patients with pancreatic cancer from a familial pancreatic kindred with at least 2 affected FDRs; patients with Peutz-Jeghers syndrome; and p16, BRCA2, and HNPCC mutation carriers with 1 or more affected FDRs and hereditary pancreatitis. The guidelines emphasize that screening should be done only in those who are surgical candidates and are evaluated at an experienced multidisciplinary center.8
Deleterious germline mutations in pancreatic cancer can account for 33% of patients with apparent sporadic cancers and no hereditary risk. These include germline mutations affecting BRCA1/2, PALB2, ATM, MLH1, CHK-2, CDKN2A, and TP53.11
Pathogenesis
Pancreatic neoplasms can be benign or malignant and thus a tissue histologic diagnosis is paramount. Pancreatic adenocarcinomas with exocrine features represent more than 95% of all pancreatic neoplasms, with only 5% arising from the endocrine pancreas (ie, neuroendocrine tumors). Pancreatic neuroendocrine tumors and pancreatic adenocarcinoma must be distinguished histologically because treatment of the 2 neoplasms is completely different. Other malignant pancreatic tumors are signet ring cell carcinoma, adenosquamous carcinoma, undifferentiated (anaplastic) carcinoma, and mucinous noncystic (colloid) carcinoma; the latter tumor has a better prognosis.12 It is essential to characterize and distinguish among benign cystic neoplasms, as some require surgical resection due to the risk of malignant transformation. IPMN, pancreatic intraepithelial neoplasia, and mucinous cystic neoplasms are thought to be premalignant lesions of invasive ductal adenocarcinomas, and the pathological report should highlight the degree of dysplasia for adequate risk stratification.13 This information could be the deciding factor in whether a pancreatectomy is recommended by a multidisciplinary team.
Most pancreatic cancers harbor activating or silencing genetic mutations, and multiple combinations of altered genes can be detected by next-generation sequencing (average of 63 genetic alterations per cancer).14 Mutational activated KRAS is the most frequent (> 90%) genetic alteration in pancreatic cancer, even in early neoplastic precursors (IPMN > 75%). KRAS is a highly complex, dynamic proto-oncogene involved in signaling of various receptor kinases such as the epidermal growth factor receptor and the insulin-like growth factor receptor-I. It also engages in canonical downstream effector pathways, mainly Raf/MEK/ERK, PI3K/PDK1/Akt, and the Ral guanine nucleotide exchange factor pathway, which drive much of the pathogenesis of malignancy. These pathways lead to sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration, and metastasis. An activating point mutation in codon G12 is the most common (98%) locus of KRAS mutation in pancreatic adenocarcinoma, but all drugs targeting this mutation have failed in clinical practice.15 Additionally, inactivation of tumor suppressor genes such as p53, DPC4 (SMAD4/MADH4), CDKN2A (p16/MTS1), and BRCA2 can be found in 75%, 30%, 35%, and 4% of pancreatic adenocarcinoma cases, respectively.14 Another pancreatic cancer hallmark is inactivation of DNA damage repair genes, which include MLH1 and MSH2.16
Diagnosis and Staging
Case Presentation
A 71-year-old male veteran with no significant past medical history other than hypertension and hyperlipidemia and an excellent performance status presents to the emergency department after noticing a yellowish skin and sclera color. He denies weight loss, abdominal pain, or any other pertinent symptom or sign. Physical examination reveals a healthy developed man with yellowish discoloration of the skin and sclera and a soft, nontender benign abdomen; physical examination is otherwise unremarkable. Laboratory evaluation reveals a direct bilirubin level of 4.5 mg/dL and normal values for complete blood count and renal, liver, and coagulation panels. Abdominal and pelvis computed tomography (CT) with intravenous contrast shows a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein, which remains patent. Follow-up endoscopic ultrasound (EUS) confirms an irregular mass at the head of the pancreas measuring 3.2 × 2.6 cm with sonographic evidence suggesting invasion into the portal vein. During the procedure, the bile duct is successfully stented, the mass is biopsied, and bile duct brushing is performed. Pathology report is consistent with pancreatic adenocarcinoma.
- What is the typical presentation of pancreatic cancer?
The most common symptoms of pancreatic cancer at the time of presentation include weight loss (85%), asthenia/anorexia (86%), and/or abdominal pain (79%).17 The most frequent signs are jaundice (55%), hepatomegaly (39%), and cachexia (13%). Courvoisier sign, a nontender but palpable distended gallbladder at the right costal margin, is neither sensitive nor specific for pancreatic cancer (13% of cases). Trousseau syndrome, a superficial thrombophlebitis, is another classic sign that reflects the hypercoagulable nature of pancreatic cancer (3% of cases).17 The pathophysiology of this syndrome is not completely understood, but it may occur secondary to the release of cancer microparticles in the blood stream which in turn stimulate the coagulation cascade. Other nonspecific symptoms are dark urine, nausea, vomiting, diarrhea, steatorrhea, and epigastric and back pain. Because symptoms early in the course of the disease are nonspecific, pancreatic cancer is typically diagnosed late, after the cancer has invaded local structures or metastasized. The initial presentation varies depending on tumor location, with 70% of pancreatic head malignancies presenting with jaundice and pain correlating to an advanced stage.18 Although data supporting an association between new-onset diabetes mellitus and pancreatic cancer are inconclusive, pancreatic cancer should still be a consideration in patients with new-onset diabetes mellitus and other symptoms such as pain and weight loss. Early signs of incurable disease include a palpable mass, ascites, lymphadenopathy (classic Virchow node), and an umbilical mass (Sister Mary Joseph node). Incidentally discovered pancreatic masses on imaging are rare, but the incidence is increasing due to frequent imaging for other reasons and improved diagnostic techniques.
- What is the approach to diagnosis and staging?
History and physical examination findings are not sufficiently sensitive or specific to diagnose pancreatic cancer. High clinical suspicion in a patient with risk factors can lead to a comprehensive evaluation and potential early diagnosis. In general, an initial diagnostic work-up for suspected pancreatic cancer will include serologic evaluation (liver function test, lipase, tumor markers) and abdominal imaging (ultrasound, CT scans, or magnetic resonance imaging [MRI]). Ultrasound is a first-line diagnostic tool with a sensitivity of 90% and specificity of 98.8% for pancreatic cancer, but it is investigator-dependent and is less accurate in detecting tumors smaller than 3 cm in diameter.19 Multiphasic helical CT of the abdomen has better sensitivity (100%) and specificity (100%) for detecting tumors larger than 2 cm, but this modality is less accurate in detecting pancreatic masses smaller than 2 cm (77%).20 Percutaneous fine-needle aspiration (FNA) performed by ultrasound or CT guidance is avoided due to theoretical concerns about intraperitoneal seeding and bleeding.
If a pancreatic mass is detected by ultrasound or CT, additional interventions may be indicated depending on the clinical scenario. EUS-guided biopsy can provide histological confirmation and is currently utilized frequently for diagnosis and early resectability staging. Endoscopic retrograde cholangiopancreatography (ERCP) is indicated for patients with biliary obstruction requiring stent placement, and this procedure may provide tissue confirmation by forceps biopsy or brush cytology (lower accuracy than EUS). In a meta-analysis that evaluated the diagnostic value of tests for pancreatic cancer, ERCP had the highest sensitivity (92%) and specificity (96%) compared to ultrasound and CT,21 but this modality carries a risk for pancreatitis, bleeding, and cholangitis. Magnetic resonance cholangiopancreatography has not replaced ERCP, but it but may be an alternative for patients who cannot undergo ERCP (eg, gastric outlet obstruction, duodenal stenosis, anatomical surgical disruption, unsuccessful ERCP). ERCP is used frequently because many patients present with obstructive jaundice due to pancreatic mass compression, specifically if the mass is located in the head, and must undergo ERCP and stenting of the common bile duct.
The carbohydrate antigen (CA) 19-9 level has variable sensitivity and specificity in pancreatic cancer, as levels can be elevated in many benign pancreaticobiliary disorders. Elevated CA 19-9, in the appropriate clinical scenario (ie, a suspicious pancreatic mass and a value greater than 37 U/mL) demonstrated a sensitivity of 77% and specificity of 87% when differentiating pancreaticobiliary cancer from benign clinical conditions such as acute cholangitis or cholestasis.22 CA 19-9 level has prognostic value, as it may predict occult disease and correlates with survival rates, but no specific cutoff value has been established to guide perioperative therapy for high-risk resectable tumors.23
The American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) system is the preferred method for staging pancreatic cancer (Table 1).
Positron emission tomography with CT scan is occasionally utilized in practice to assess tumor burden by evaluating anatomical structures and assessing physiologic uptake, which aids in establishing the extent of disease in equivocal cases. Staging laparoscopy with or without peritoneal biopsy is sometimes used to establish appropriate staging in cases that are questionable for occult metastatic disease. This procedure helps avoid unnecessary morbid surgeries.
Neoadjuvant Therapy
Case Continued
The patient is referred to oncology. Blood work reveals a CA 19-9 level of 100 U/mL (reference range < 35 U/mL) and a staging CT scan of the chest reveals a benign-appearing 3-mm nodule (no prior imaging for comparison). CT scan of the abdomen and pelvis does not define venous vasculature involvement appropriately and hence MRI of the abdomen and pelvis is performed. MRI reveals a pancreatic head mass measuring 3.0 × 2.7 cm, without arterial or venous vasculature invasion. However, the mass is abutting the portal vein and superior mesenteric vein and there is a new nonspecific 8-mm aortocaval lymph node.
- What are the current approaches to treating patients with resectable, unresectable, and metastatic disease?
Accurate staging and assessment of surgical resectability in pancreatic cancer are paramount as these steps prevent a futile morbid Whipple procedure in patients with advanced disease and a high risk of recurrence. Conversely, it allows patients with low-volume disease to undergo a potentially curative surgery. Approximately 20% of patients present with resectable disease, 40% present with locally advanced unresectable tumors (eg, involvement of critical vascular structures), and 40% present with metastatic disease.3 Treatment for resectable pancreatic cancer continues to be upfront surgery, although neoadjuvant therapy with either chemoradiation, radiation alone, or chemotherapy is an option per guidelines from the American Society of Clinical Oncology (ASCO),28 the NCCN,26 and the European Society for Medical Oncology (ESMO),29 particularly for patients with borderline resectable tumors (Table 3).
Systemic chemotherapy is recommended for fit candidates with locally advanced unresectable or metastatic disease, with an emphasis on supportive palliative measures. Palliative interventions include biliary stenting, duodenal stent for relieving gastric-outlet obstruction, and celiac axis nerve blocks, when indicated. Routine preoperative biliary stent placement/drainage in patients undergoing subsequent surgery for pancreatic cancer located in the head is associated with an increased risk of surgical complications when compared with up-front surgery without prior biliary drainage, and thus stent placement/drainage is not recommended.26 Aggressive supportive management of symptoms, such as cancer-associated pain, anorexia-cachexia syndromes, and anxiety-depression disorders, should remain a primary palliative focus.
Case Continued
A multidisciplinary tumor board discusses the patient’s case and deems the cancer borderline resectable; neoadjuvant therapy is recommended. The patient is started on treatment with gemcitabine and nab-paclitaxel as first-line neoadjuvant therapy. After 4 cycles, the CA 19-9 level drops to 14 U/mL, and MRI reveals a smaller head mass of 1.3 × 1.4 cm with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remains stable. At tumor board, it is evident that the patient has responded to therapy and the recommendation is to treat with gemcitabine chemoradiotherapy before surgery.
- What neoadjuvant therapy strategies are used in the treatment of pancreatic adenocarcinoma?
There are no established evidence-based recommendations for neoadjuvant therapy in patients with borderline resectable pancreatic cancer or patients with unresectable locally advanced pancreatic cancer. However, there are ongoing trials to investigate this treatment approach, and it is offered off-label in specific clinical scenarios, such as in the case patient described here. In patients with borderline resectable disease, preoperative chemotherapy followed by chemoradiation is a routine practice in most cancer centers,32 and ongoing clinical trials are an option for this cohort of patients (eg, Southwest Oncology Group Trial 1505, NCT02562716). The definitions of borderline resectable and unresectable pancreatic cancer have been described by the NCCN,26 although most surgeons consider involvement of the major upper abdominal blood vessels the main unresectability criterion; oncologists also consider other parameters such as suspicious lesions on scans, worsening performance status, and a significantly elevated CA 19-9 level suggestive of disseminated disease.28 The goal of a conversion approach by chemotherapy with or without radiation for borderline and unresectable cancers is to deliver a tolerable regimen leading to tumor downstaging, allowing for surgical resection. No randomized clinical trial has shown a survival advantage of this approach. Enrollment in clinical trials is preferred for patients with borderline and unresectable cancer, and there are trials that are currently enrolling patients.
The main treatment strategies for patients with locally advanced borderline and unresectable pancreatic cancer outside of a clinical trial are primary radiotherapy, systemic chemotherapy, and chemoradiation therapy. Guidelines from ASCO, NCCN, and ESMO recommend induction chemotherapy followed by restaging and consolidation chemoradiotherapy in the absence of progression.26,28,29 There is no standard chemoradiation regimen and the role of chemotherapy sensitizers, including fluorouracil, gemcitabine, and capecitabine (an oral fluoropyrimidine substitute), and targeted agents in combination with different radiation modalities is now being investigated.
Fluorouracil is a radio-sensitizer that has been used in locally advanced pancreatic cancer based on experience in other gastrointestinal malignancies; data shows conflicting results with this drug. Capecitabine and tegafur/gimeracil/oteracil (S-1) are oral prodrugs that can safely replace infusional fluorouracil. Gemcitabine, a more potent radiation sensitizer, is very toxic, even at low-doses twice weekly, and does not provide a survival benefit, as demonstrated in the Cancer and Leukemia Group B (CALGB) 89805 trial, a phase 2 study of patients with surgically staged locally advanced pancreatic cancer.33 Gemcitabine-based chemoradiotherapy was also evaluated in the Eastern Cooperative Group (ECOG) E4201 trial, which randomly assigned patients to receive gemcitabine alone (at 1000 mg/m2/wk for weeks 1 through 6, followed by 1 week rest, then weekly for 3 out of 4 weeks) or gemcitabine (600 mg/m2/wk for weeks 1 to 5, then 4 weeks later 1000 mg/m2 for 3 out of 4 weeks) plus radiotherapy (starting on day 1, 1.8 Gy/fraction for total of 50.4 Gy).34 Patients with locally advanced unresectable pancreatic cancer had a better OS outcome with gemcitabine in combination with radiation therapy (11.1 months) as compared with patients who received gemcitabine alone (9.2 months). Although there was a greater incidence of grade 4 and 5 treatment-related toxicities in the combination arm, no statistical differences in quality-of-life measurements were reported. Gemcitabine-based and capecitabine-based chemoradiotherapy were compared in the open-label phase 2 multicenter randomized SCALOP trial.35 Patients with locally advanced pancreatic cancer were assigned to receive 3 cycles of induction with gemcitabine 1000 mg/m2 days 1, 8, and 15 and capecitabine 830 mg/m2 days 1 to 21 every 28 days; patients who had stable or responding disease were randomly assigned to receive a fourth cycle followed by capecitabine (830 mg/m2 twice daily on weekdays only) or gemcitabine (300 mg/m2 weekly) with radiation (50.4 Gy over 28 fractions). Patients treated with capecitabine-based chemoradiotherapy had higher nonsignificant median OS (17.6 months) and median progression-free survival (12 months) compared to those treated with gemcitabine (14.6 months and 10.4 months, respectively).
The benefit of radiation therapy in the treatment of locally advanced pancreatic cancer was further explored by the Fédération Francophone de Cancérologie Digestive 2000-01 phase 3 trial. This study compared induction chemoradiotherapy (60 Gy, 2 Gy/fraction; concomitant fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) to gemcitabine alone (1000 mg/m2 weekly for 7 weeks) followed by maintenance gemcitabine in both arms.36 Unexpectedly, the median OS was significantly shorter in the chemoradiotherapy arm than in the chemotherapy alone arm (8.6 months versus 13 months, respectively, P = 0.03) and the combination arm had more toxicities. The phase 3 open-label LAP07 study explored the role of radiation therapy in patients with locally advanced pancreatic cancer who had controlled disease after 4 months of induction therapy.37 LAP07 had 2 randomizations: first, patients with locally advanced pancreatic cancer were assigned to receive weekly gemcitabine alone (1000 mg/m2) or this same dose of gemcitabine plus erlotinib 100 mg/day; second, patients with progression-free disease (61% of initial cohort) after 4 months of therapy were assigned to receive 2 months of the same chemotherapy or chemoradiotherapy (54 Gy plus capecitabine). This study showed that the addition of erlotinib to gemcitabine did not improve survival and in fact affected survival adversely. Of note, no survival benefit was observed after the first randomization from chemotherapy to consolidating chemoradiotherapy. Chemoradiotherapy achieved better locoregional tumor control with significantly less local tumor progression (32% versus 46%, P < 0.03) and no increase in toxicity. Based on prior moderate-quality evidence, guidelines recommend consolidative chemoradiotherapy only for surgical resection candidates following induction chemotherapy; for those who are not surgical candidates, guidelines recommend continuing systemic therapy.26,28,29
Gemcitabine and fluorouracil-based chemotherapies were the standard induction regimens until evidence from studies of metastatic systemic treatment protocols with FOLFIRINOX (ACCORD trial38) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) plus gemcitabine (MPACT trial39) was extrapolated to clinical practice. These regimens were shown to achieve higher objective response rates when compared to single-agent gemcitabine in patients with metastatic pancreatic cancer. Due to the broad heterogeneity of results in small retrospective series with neoadjuvant trials in borderline resectable pancreatic cancer, the quality of the evidence is low and any recommendation is limited. Many individual series have demonstrated improved complete resection rates and promising survival rates. In the largest single-institution retrospective review of patients with borderline resectable pancreatic adenocarcinoma who completed neoadjuvant gemcitabine-based chemoradiotherapy (50 Gy in 28 fractions or 30 Gy in 10 fractions), 94% achieved a margin-negative pancreatectomy; the median OS in those who completed preoperative therapy and had surgery was 40 months, with a 5-year OS of 36%.40 A meta-analysis by Andriulli and colleagues included 20 prospective studies of patients with initially resectable (366 lesions) or unresectable (341 lesions) disease who were treated with neoadjuvant/preoperative gemcitabine with or without radiotherapy.41 In the group with initially unresectable disease, 39% underwent surgery after restaging and 68% of explored patients were resected; the R0 resection rate was 60%. After restaging, 91% of patients with resectable disease underwent surgery, with 82% of explored patients undergoing surgical resection and 89% of these achieving R0 resection. The estimated 1- and 2-year survival probabilities after resection among patients with initially unresectable disease were 86.3% and 54.2%.41
The largest single-institution retrospective review of FOLFIRINOX (fluorouracil, oxaliplatin, irinotecan, and leucovorin), an alternative to gemcitabine, for neoadjuvant induction therapy for patients with locally advanced unresectable disease was conducted at Memorial Sloan Kettering Cancer Center. In this study (n = 101), 31% of patients initially deemed unresectable who completed FOLFIRINOX induction therapy with or without chemoradiation underwent resection. The R0 resection rate in these patients was 55%, and patients who did not progress during induction FOLFIRINOX therapy had a median OS of 26 months.42 A systematic review and meta-analysis of FOLFIRINOX chemotherapy with or without radiotherapy in patients with locally advanced unresectable pancreatic cancer reported that 25.9% of patients underwent resection after FOLFIRINOX therapy, and the R0 resection rate in these patients was 78.4%.43 The median OS in this study was 24.2 months, which was longer than the previously reported median OS rates for gemcitabine.
There is no strong evidence published for the use of combination nab-paclitaxel plus gemcitabine in the neoadjuvant setting, but it is used in clinical practice based on evidence from the MPACT trial, which showed the combination improved OS and progression-free survival in patients with metastatic pancreatic cancer.39 An early-phase 1-arm clinical trial of neoadjuvant gemcitabine, docetaxel, and capecitabine (GTX) followed by radiotherapy showed an increased response rate and survival for locally advanced disease; however, the NCCN expert panel has reached a consensus but not a uniform recommendation regarding this regimen due to significant toxicities and low patient accrual.26 Selected patients with pancreatic cancer with BRCA1/2 mutations are more sensitive to platinum-based chemotherapy. Although studies of neoadjuvant platinum-based chemotherapy in this population have not been reported, the NCCN guidelines list it as an alternative option based on extrapolated data.26 A clinical trial of gemcitabine, nab-paclitaxel, and cisplatin in the neoadjuvant setting in patients with resectable pancreatic cancer is currently enrolling patients (NGC triple regimen NCT0339257).
Summary
Chemotherapy alone or followed by chemoradiotherapy may be used as initial treatment for patients with borderline and unresectable pancreatic adenocarcinoma without distant metastases who are potential surgical candidates. Chemoradiotherapy remains a preferred treatment option for patients with poorly controlled pain from local tumor invasion, in view of the well-documented analgesic palliative effect of radiation therapy. FOLFIRINOX with or without radiation therapy may offer the highest documented response rates, but it also results in higher rates of treatment-related toxicities. FOLFIRINOX can be offered to selected fit patients (< 65 years old, no comorbidity contraindication, good functional status [ECOG 0–1]) who can tolerate triple therapy with a more toxic adverse-effect profile. A clinical trial evaluating neoadjuvant FOLFIRINOX with or without preoperative chemoradiotherapy in patients with borderline resectable pancreatic cancer is ongoing (PANDAS-PRODIGE 44, NCT02676349). Gemcitabine with or without radiation therapy is a tolerable combination, although it is potentially more toxic when combined with radiation. The addition of nab-paclitaxel to gemcitabine without radiation may emerge as a preferred neoadjuvant treatment for selected patients; a clinical trial investigating this modality in patients with resectable and borderline resectable disease is ongoing (NCT02723331).
Adjuvant Therapy
Case Continued
Prior to the planned surgical resection and after undergoing chemoradiation therapy, the patient has an excellent performance status and repeat MRI shows a 1.3 × 1.4 cm head mass with no further vasculature involvement, no evidence of lymphadenopathy, and no distant metastasis. The CA 19-9 level is stable at 18 U/mL. The patient undergoes an uncomplicated partial pancreaticoduodenectomy, and analysis of a surgical pathology specimen reveals T3N0 disease with closest margin of 0.1 cm.
- Would the patient benefit from adjuvant therapy?
Adjuvant chemotherapy for 6 months after pancreatic cancer resection should be offered to all patients based on mature data. Gemcitabine and capecitabine are the current standard of care in adjuvant therapy; alternatively, single-agent gemcitabine can be offered to patients with poor performance status or patients who cannot tolerate the toxicities associated with this combination.28 Adjuvant treatment should be initiated within approximately 8 weeks of surgical resection. The value of radiation therapy remains controversial, but it can be offered within the context of a clinical trial or to patients with positive margins after surgical resection and/or lymph node–positive disease. Based on low-quality supportive evidence, it is strongly recommended that patients who receive neoadjuvant therapy complete a total of 6 months of chemotherapy, factoring in the duration of the preoperative regimen.28 Different adjuvant strategies have been investigated, including chemotherapy alone with a fluoropyrimidine and/or gemcitabine with or without combined chemoradiation therapy.
The European Study Group for Pancreatic Cancer 1 (ESPAC)-1 trial was a randomized clinical trial that evaluated several adjuvant strategies in pancreatic cancer treatment. This trial assigned patients who underwent pancreatic adenocarcinoma resection to adjuvant chemotherapy alone (intravenous fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 daily for 5 days, monthly for 6 months), chemoradiotherapy (20 Gy in 10 daily fractions over 2 weeks with 500 mg/m2 intravenous fluorouracil on days 1–3, repeated after 2 weeks), both chemotherapy and chemoradiation, and observation.44 The results showed no added benefit for adjuvant chemoradiotherapy, with a median OS of 15.5 months in the chemoradiotherapy cohort, as compared to a median OS of 16.1 months in the chemotherapy-alone cohort (hazard ratio [HR] 1.18 [95% CI 0.90 to 1.55], P = 0.24). In addition, there was evidence of a survival benefit for the chemotherapy-alone arm when compared to the combined modality arm, with a median OS of 19.7 versus 14.0 months, respectively (HR 0.66 [95% CI 0.52 to 0.83], P = 0.0005). Although ESPAC-1 has been criticized for being underpowered to perform statistical comparison, it is still considered a landmark trial demonstrating benefit with single-agent chemotherapy alone. A follow-up analysis of ESPAC-1 showed that adjuvant chemotherapy alone conferred a significant 5-year survival benefit while the combined modality had a deleterious effect on survival. 45 Hence, adjuvant chemotherapy alone became the standard of care in the United States following resection.
The results of the multicenter randomized controlled phase 3 CONKO-001 (CharitéOnkologie 001) trial, which were reported in 2007, supported the use of adjuvant gemcitabine for 6 months in patients with resected pancreatic adenocarcinoma. In this study, patients treated with adjuvant gemcitabine (1000 mg/m2 days 1, 8, and 15 every 4 weeks for 6 months) had superior disease-free survival compared with those who received surgery alone.30 A long-term outcome update of this study demonstrated a significant improvement in 5-year OS for patients treated with adjuvant gemcitabine (20.7% [95% CI 14.7% to 26.6%]) compared to those who received surgical resection alone (10.4% [95% CI 5.9% to 15.0%]). This benefit persisted at 10-year follow-up, with an OS of 12.2% (95% CI 7.3% to 17.2%) in the adjuvant gemcitabine group, as compared to 7.7% (95% CI 3.6% to 11.8%) in the resection alone group.31
Fluorouracil and gemcitabine remained equivalent adjuvant treatment options until the results of the ESPAC-3 trial were reported in 2010.32 This large phase 3 trial, conducted mainly in the United Kingdom, compared weekly gemcitabine (1000 mg/m2 weekly for 3 of every 4 weeks) to leucovorin-modulated fluorouracil (Mayo Clinic regimen: leucovorin 20 mg/m2 followed by fluorouracil 425 mg/m2 intravenous bolus days 1 through 5 every 28 days) as adjuvant therapy in resected pancreatic adenocarcinoma. After a median follow-up of 34.2 months, the median OS was similar in the 2 groups (fluorouracil/leucovorin 23.0 months versus gemcitabine 23.6 months; P = 0.39). However, the fluorouracil/leucovorin group experienced more grade 3/4 treatment-related toxicities (mucositis, stomatitis, diarrhea, and hosptializations; 14% versus 7.5%; P < 0.001).46 Following this trial, gemcitabine became the standard of care for adjuvant chemotherapy for resected pancreatic cancer.
The U.S. Radiation Therapy Oncology Group (RTOG) 9704 trial was conducted to investigate the potential benefit of adding radiation therapy to gemcitabine. This trial demonstrated an improved trend among patients with pancreatic head tumors (but not with cancers of the pancreatic body or tail) who received adjuvant gemcitabine followed by chemoradiotherapy (50.4 Gy in 1.8 Gy daily fractions for 5.5 weeks with concurrent infusional fluorouracil 250 mg/m2 daily) and subsequent gemcitabine monotherapy compared to postoperative fluorouracil-based chemoradiotherapy. Results showed a 5-year OS of 22% versus 18%, respectively, although this improvement was not statistically significant (P = 0.08). This trial failed to show a benefit of adding radiotherapy to gemcitabine.47
The ESPAC-4 trial, reported in 2017, evaluated the combination of gemcitabine and capecitabine compared to gemcitabine alone as adjuvant therapy for resected pancreatic adenocarcinoma.48 Patients were randomly assigned after surgical resection, regardless of margin or node status, to 6 months of gemcitabine alone (1000 mg/m2/day on days 1, 8, and 15 of each 28-day cycle) or gemcitabine plus capecitabine (1660 mg/m2/day on days 1 through 21 of each 28-day cycle). Combination therapy had a significant survival benefit compared to single therapy, with median OS durations of 28 months and 25.5 months, respectively (HR for death 0.82 [95% CI 0.68 to 0.98]). The 5-year OS for patients who received combination treatment was 29 months (95% CI 22.9 to 35.2) versus 16 months (95% CI 10.2 to 23.7) for those in the monotherapy group. As expected, grade 3 or 4 treatment-related toxicities (diarrhea, hand-foot syndrome, and neutropenia) were significantly more common with combined therapy, although there were no significant differences in the rates of serious adverse events. The adjuvant combination of gemcitabine and capecitabine became the current and preferred new standard of care following resection of pancreatic ductal adenocarcinoma,28 but single-agent gemcitabine and fluorouracil/leucovorin continue to be viable options,26,28,29 particularly for elderly patients, patients with borderline performance status, or patients with multiple comorbidities.
Evidence showing that a more intensive regimen can improve outcome in the adjuvant setting remains elusive. The phase 3 APACT study (Adjuvant Therapy for Patients with Resected Pancreatic Cancer, NCT01964430) comparing gemcitabine alone to gemcitabine plus nab-paclitaxel in patients with surgically resected pancreatic adenocarcinoma has concluded, with the results projected to be released in 2018. Another phase 3 trial investigating the efficacy of FOLFIRINOX versus gemcitabine alone as adjuvant therapy is underway in France and Canada (PRODIGE24/ACCORD24, NCT01526135). Other strategies with newer targeted therapies and immunotherapy are in the development phase.
Follow-Up and Surveillance
Case Conclusion
After recovery from surgery, the patient is offered and completes 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine. He is started on surveillance at 3 and 6 months, and he maintains an excellent performance status. He develops clinical evidence of pancreatic enzyme insufficiency and is placed on oral replacement therapy. He has no other complaints, and there is no evidence of recurrence on MRI and CA 19-9 levels.
- What is the recommended duration of surveillance following curative resection?
Surveillance after curative resection of pancreatic adenocarcinoma is recommended by NCCN guidelines.26 However, pancreatic adenocarcinoma has a poor prognosis, and surveillance after curative surgical resection with or without perioperative therapy has not been shown to impact survival. Most recurrences will occur within 2 years after treatment. Surveillance recommendations differ among expert groups.26,28,29 NCCN guidelines recommend evaluating patients by history and physical examination every 3 to 6 months for the first 2 years, then every 6 to 12 months for 3 years. CA 19-9 level and CT scan should be obtained every 3 to 6 months for 2 years and then every 6 to 12 months for 3 years. Follow-up with CA 19-9 levels and CT scans after 5 years is not routinely performed unless guided by signs, symptoms, or laboratory findings that raise suspicion for recurrence. Follow-up visits should also include evaluation of treatment-related toxicities, symptom management, nutrition support of pancreatic insufficiency, and psychosocial support.
Conclusion
Pancreatic cancer is a leading cause of cancer-related death that frequently presents with locally advanced or metastatic disease due to nonspecific symptoms and lack of a screening modality. Histological tissue biopsy confirmation and accurate resectability staging guide treatment planning and prognosis. The only potentially curative therapy is surgical resection plus adjuvant therapy for those with resectable disease. Surgical candidates with borderline resectable and unresectable disease can be offered induction preoperative chemotherapy followed by consolidation chemoradiation, based on clinical consensus practice. Enrollment in clinical trials should be encouraged for all patients, as evidence from clinical trials is essential to making progress in pancreatic cancer treatment.
Introduction
Exocrine pancreatic cancer refers to pancreatic adenocarcinomas that arise from ductal epithelial cells. Pancreatic ductal adenocarcinoma is a highly lethal malignancy, ranking as the fourth most common cause of cancer-related death in the United States1 and the eighth most common worldwide.2 In the United States, the pancreas is the second most common site of gastrointestinal malignancy after the colon.1 The only potentially curative modality for pancreatic adenocarcinomas is complete resection, followed by adjuvant therapy; unfortunately, only around 20% of patients are surgical candidates at the time of presentation due to delayed development of symptoms and consequently diagnosis.3 Most symptomatic patients with pancreatic cancer have locally advanced disease at diagnosis, and only a select group of patients with good performance status and borderline resectable disease can be offered neoadjuvant therapy. Adjuvant chemotherapy is typically recommended for patients who undergo potentially curative resection for pancreatic cancer.
Epidemiology
In the United States, pancreatic cancer has an annual estimated incidence of 55,440 new cases.1 It causes an estimated 44,330 deaths per year, with a 5-year overall survival (OS) rate of 8.2%.1 Worldwide an estimated 138,100 men and 127,900 women die of pancreatic cancer each year.2 In general, pancreatic cancers occur more commonly in persons living in Western/industrialized countries, older persons (age > 60 years), males (ratio 1.3:1 female), and African-Americans and native Hawaiians.4
Etiology
The major preventable environmental risk factor for pancreatic cancer is cigarette smoking, which accounts for 25% of all cases.5 A prospective study that estimated the excess incidence of pancreatic cancer among cigarette smokers and assessed the influence of smoking cessation on the risk for pancreatic cancer showed that persons who quit smoking reduced their risk of pancreatic cancer by 48% after 2 years of cessation, compared with smokers who did not quit, and reduced their risk to near the level of a never smoker after 10 years of cessation.5 Risk is higher for heavy smokers and those with homozygous deletions of the glutathione S-transferase theta 1 gene (GSTT1), which results in the absence of the carcinogen-metabolizing function of the glutathione S-transferase enzyme. High body mass index and sedentary lifestyle have been linked to pancreatic cancer.6 Data regarding aspirin, diet, coffee, and excess alcohol consumption are insufficient, inconclusive, and even conflicting, and thus the effect of these factors on risk for pancreatic cancer remains unclear. Infectious risk factors such as Helicobacter pylori and hepatitis B and C virus have weak associations with pancreatic cancer. Chronic pancreatitis and pancreatic cysts (eg, intraductal papillary mucinous neoplasm [IPMN] of the pancreas) carry a risk for malignant transformation, and hence may require surveillance. Multiple epidemiologic studies have shown a strong association between pancreatic cancer and recently diagnosed diabetes mellitus (relative risk [RR] 1.97 [95% confidence interval {CI} 1.78 to 2.18]); the presence of diabetes also may be a long-term predisposing factor for pancreatic cancer, and cancer screening needs to be considered for selected patients.7
A predisposing genetic anomaly accounts for 15% of all cases of pancreatic cancer.8 Hereditary risk factors are divided into 2 broad categories: defined genetic syndromes and familial pancreatic cancer. Familial predispositions that do not meet genetic syndrome criteria account for approximately 5% to 10% of all cases associated with hereditary factors; in one study, 29% of tested kindreds with an incident pancreatic cancer had a germline BRCA2 mutation.9 Other predisposing genetic syndromes that have been linked to pancreatic cancer include:
- Peutz-Jeghers syndrome with germline STK11 mutations (RR 132);
- Hereditary pancreatitis with germline PRSS1, SPINK1, and CFTR mutations (RR 26–87);
- Familial atypical multiple mole melanoma syndrome with CDKN2A mutations (RR 20–40);
- Familial breast and ovarian cancer with BRCA2 (RR 10) and BRCA1 (RR 2.8) mutations;
- Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch II syndrome) with MLH1, MSH2, MSH6, and PMS2 mutations (RR 9–11); and
- Familial adenomatous polyposis with APC mutations (RR 5).10
Other gene mutations with unknown relative risk for pancreatic cancer include mutations affecting PALB2, ATM, and TP53.
The International Cancer of the Pancreas Screening consortium consensus on screening for pancreatic cancer in patients with increased risk for familial pancreatic cancer recommends screening those at high risk: first-degree relatives (FDRs) of patients with pancreatic cancer from a familial pancreatic kindred with at least 2 affected FDRs; patients with Peutz-Jeghers syndrome; and p16, BRCA2, and HNPCC mutation carriers with 1 or more affected FDRs and hereditary pancreatitis. The guidelines emphasize that screening should be done only in those who are surgical candidates and are evaluated at an experienced multidisciplinary center.8
Deleterious germline mutations in pancreatic cancer can account for 33% of patients with apparent sporadic cancers and no hereditary risk. These include germline mutations affecting BRCA1/2, PALB2, ATM, MLH1, CHK-2, CDKN2A, and TP53.11
Pathogenesis
Pancreatic neoplasms can be benign or malignant and thus a tissue histologic diagnosis is paramount. Pancreatic adenocarcinomas with exocrine features represent more than 95% of all pancreatic neoplasms, with only 5% arising from the endocrine pancreas (ie, neuroendocrine tumors). Pancreatic neuroendocrine tumors and pancreatic adenocarcinoma must be distinguished histologically because treatment of the 2 neoplasms is completely different. Other malignant pancreatic tumors are signet ring cell carcinoma, adenosquamous carcinoma, undifferentiated (anaplastic) carcinoma, and mucinous noncystic (colloid) carcinoma; the latter tumor has a better prognosis.12 It is essential to characterize and distinguish among benign cystic neoplasms, as some require surgical resection due to the risk of malignant transformation. IPMN, pancreatic intraepithelial neoplasia, and mucinous cystic neoplasms are thought to be premalignant lesions of invasive ductal adenocarcinomas, and the pathological report should highlight the degree of dysplasia for adequate risk stratification.13 This information could be the deciding factor in whether a pancreatectomy is recommended by a multidisciplinary team.
Most pancreatic cancers harbor activating or silencing genetic mutations, and multiple combinations of altered genes can be detected by next-generation sequencing (average of 63 genetic alterations per cancer).14 Mutational activated KRAS is the most frequent (> 90%) genetic alteration in pancreatic cancer, even in early neoplastic precursors (IPMN > 75%). KRAS is a highly complex, dynamic proto-oncogene involved in signaling of various receptor kinases such as the epidermal growth factor receptor and the insulin-like growth factor receptor-I. It also engages in canonical downstream effector pathways, mainly Raf/MEK/ERK, PI3K/PDK1/Akt, and the Ral guanine nucleotide exchange factor pathway, which drive much of the pathogenesis of malignancy. These pathways lead to sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration, and metastasis. An activating point mutation in codon G12 is the most common (98%) locus of KRAS mutation in pancreatic adenocarcinoma, but all drugs targeting this mutation have failed in clinical practice.15 Additionally, inactivation of tumor suppressor genes such as p53, DPC4 (SMAD4/MADH4), CDKN2A (p16/MTS1), and BRCA2 can be found in 75%, 30%, 35%, and 4% of pancreatic adenocarcinoma cases, respectively.14 Another pancreatic cancer hallmark is inactivation of DNA damage repair genes, which include MLH1 and MSH2.16
Diagnosis and Staging
Case Presentation
A 71-year-old male veteran with no significant past medical history other than hypertension and hyperlipidemia and an excellent performance status presents to the emergency department after noticing a yellowish skin and sclera color. He denies weight loss, abdominal pain, or any other pertinent symptom or sign. Physical examination reveals a healthy developed man with yellowish discoloration of the skin and sclera and a soft, nontender benign abdomen; physical examination is otherwise unremarkable. Laboratory evaluation reveals a direct bilirubin level of 4.5 mg/dL and normal values for complete blood count and renal, liver, and coagulation panels. Abdominal and pelvis computed tomography (CT) with intravenous contrast shows a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein, which remains patent. Follow-up endoscopic ultrasound (EUS) confirms an irregular mass at the head of the pancreas measuring 3.2 × 2.6 cm with sonographic evidence suggesting invasion into the portal vein. During the procedure, the bile duct is successfully stented, the mass is biopsied, and bile duct brushing is performed. Pathology report is consistent with pancreatic adenocarcinoma.
- What is the typical presentation of pancreatic cancer?
The most common symptoms of pancreatic cancer at the time of presentation include weight loss (85%), asthenia/anorexia (86%), and/or abdominal pain (79%).17 The most frequent signs are jaundice (55%), hepatomegaly (39%), and cachexia (13%). Courvoisier sign, a nontender but palpable distended gallbladder at the right costal margin, is neither sensitive nor specific for pancreatic cancer (13% of cases). Trousseau syndrome, a superficial thrombophlebitis, is another classic sign that reflects the hypercoagulable nature of pancreatic cancer (3% of cases).17 The pathophysiology of this syndrome is not completely understood, but it may occur secondary to the release of cancer microparticles in the blood stream which in turn stimulate the coagulation cascade. Other nonspecific symptoms are dark urine, nausea, vomiting, diarrhea, steatorrhea, and epigastric and back pain. Because symptoms early in the course of the disease are nonspecific, pancreatic cancer is typically diagnosed late, after the cancer has invaded local structures or metastasized. The initial presentation varies depending on tumor location, with 70% of pancreatic head malignancies presenting with jaundice and pain correlating to an advanced stage.18 Although data supporting an association between new-onset diabetes mellitus and pancreatic cancer are inconclusive, pancreatic cancer should still be a consideration in patients with new-onset diabetes mellitus and other symptoms such as pain and weight loss. Early signs of incurable disease include a palpable mass, ascites, lymphadenopathy (classic Virchow node), and an umbilical mass (Sister Mary Joseph node). Incidentally discovered pancreatic masses on imaging are rare, but the incidence is increasing due to frequent imaging for other reasons and improved diagnostic techniques.
- What is the approach to diagnosis and staging?
History and physical examination findings are not sufficiently sensitive or specific to diagnose pancreatic cancer. High clinical suspicion in a patient with risk factors can lead to a comprehensive evaluation and potential early diagnosis. In general, an initial diagnostic work-up for suspected pancreatic cancer will include serologic evaluation (liver function test, lipase, tumor markers) and abdominal imaging (ultrasound, CT scans, or magnetic resonance imaging [MRI]). Ultrasound is a first-line diagnostic tool with a sensitivity of 90% and specificity of 98.8% for pancreatic cancer, but it is investigator-dependent and is less accurate in detecting tumors smaller than 3 cm in diameter.19 Multiphasic helical CT of the abdomen has better sensitivity (100%) and specificity (100%) for detecting tumors larger than 2 cm, but this modality is less accurate in detecting pancreatic masses smaller than 2 cm (77%).20 Percutaneous fine-needle aspiration (FNA) performed by ultrasound or CT guidance is avoided due to theoretical concerns about intraperitoneal seeding and bleeding.
If a pancreatic mass is detected by ultrasound or CT, additional interventions may be indicated depending on the clinical scenario. EUS-guided biopsy can provide histological confirmation and is currently utilized frequently for diagnosis and early resectability staging. Endoscopic retrograde cholangiopancreatography (ERCP) is indicated for patients with biliary obstruction requiring stent placement, and this procedure may provide tissue confirmation by forceps biopsy or brush cytology (lower accuracy than EUS). In a meta-analysis that evaluated the diagnostic value of tests for pancreatic cancer, ERCP had the highest sensitivity (92%) and specificity (96%) compared to ultrasound and CT,21 but this modality carries a risk for pancreatitis, bleeding, and cholangitis. Magnetic resonance cholangiopancreatography has not replaced ERCP, but it but may be an alternative for patients who cannot undergo ERCP (eg, gastric outlet obstruction, duodenal stenosis, anatomical surgical disruption, unsuccessful ERCP). ERCP is used frequently because many patients present with obstructive jaundice due to pancreatic mass compression, specifically if the mass is located in the head, and must undergo ERCP and stenting of the common bile duct.
The carbohydrate antigen (CA) 19-9 level has variable sensitivity and specificity in pancreatic cancer, as levels can be elevated in many benign pancreaticobiliary disorders. Elevated CA 19-9, in the appropriate clinical scenario (ie, a suspicious pancreatic mass and a value greater than 37 U/mL) demonstrated a sensitivity of 77% and specificity of 87% when differentiating pancreaticobiliary cancer from benign clinical conditions such as acute cholangitis or cholestasis.22 CA 19-9 level has prognostic value, as it may predict occult disease and correlates with survival rates, but no specific cutoff value has been established to guide perioperative therapy for high-risk resectable tumors.23
The American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) system is the preferred method for staging pancreatic cancer (Table 1).
Positron emission tomography with CT scan is occasionally utilized in practice to assess tumor burden by evaluating anatomical structures and assessing physiologic uptake, which aids in establishing the extent of disease in equivocal cases. Staging laparoscopy with or without peritoneal biopsy is sometimes used to establish appropriate staging in cases that are questionable for occult metastatic disease. This procedure helps avoid unnecessary morbid surgeries.
Neoadjuvant Therapy
Case Continued
The patient is referred to oncology. Blood work reveals a CA 19-9 level of 100 U/mL (reference range < 35 U/mL) and a staging CT scan of the chest reveals a benign-appearing 3-mm nodule (no prior imaging for comparison). CT scan of the abdomen and pelvis does not define venous vasculature involvement appropriately and hence MRI of the abdomen and pelvis is performed. MRI reveals a pancreatic head mass measuring 3.0 × 2.7 cm, without arterial or venous vasculature invasion. However, the mass is abutting the portal vein and superior mesenteric vein and there is a new nonspecific 8-mm aortocaval lymph node.
- What are the current approaches to treating patients with resectable, unresectable, and metastatic disease?
Accurate staging and assessment of surgical resectability in pancreatic cancer are paramount as these steps prevent a futile morbid Whipple procedure in patients with advanced disease and a high risk of recurrence. Conversely, it allows patients with low-volume disease to undergo a potentially curative surgery. Approximately 20% of patients present with resectable disease, 40% present with locally advanced unresectable tumors (eg, involvement of critical vascular structures), and 40% present with metastatic disease.3 Treatment for resectable pancreatic cancer continues to be upfront surgery, although neoadjuvant therapy with either chemoradiation, radiation alone, or chemotherapy is an option per guidelines from the American Society of Clinical Oncology (ASCO),28 the NCCN,26 and the European Society for Medical Oncology (ESMO),29 particularly for patients with borderline resectable tumors (Table 3).
Systemic chemotherapy is recommended for fit candidates with locally advanced unresectable or metastatic disease, with an emphasis on supportive palliative measures. Palliative interventions include biliary stenting, duodenal stent for relieving gastric-outlet obstruction, and celiac axis nerve blocks, when indicated. Routine preoperative biliary stent placement/drainage in patients undergoing subsequent surgery for pancreatic cancer located in the head is associated with an increased risk of surgical complications when compared with up-front surgery without prior biliary drainage, and thus stent placement/drainage is not recommended.26 Aggressive supportive management of symptoms, such as cancer-associated pain, anorexia-cachexia syndromes, and anxiety-depression disorders, should remain a primary palliative focus.
Case Continued
A multidisciplinary tumor board discusses the patient’s case and deems the cancer borderline resectable; neoadjuvant therapy is recommended. The patient is started on treatment with gemcitabine and nab-paclitaxel as first-line neoadjuvant therapy. After 4 cycles, the CA 19-9 level drops to 14 U/mL, and MRI reveals a smaller head mass of 1.3 × 1.4 cm with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remains stable. At tumor board, it is evident that the patient has responded to therapy and the recommendation is to treat with gemcitabine chemoradiotherapy before surgery.
- What neoadjuvant therapy strategies are used in the treatment of pancreatic adenocarcinoma?
There are no established evidence-based recommendations for neoadjuvant therapy in patients with borderline resectable pancreatic cancer or patients with unresectable locally advanced pancreatic cancer. However, there are ongoing trials to investigate this treatment approach, and it is offered off-label in specific clinical scenarios, such as in the case patient described here. In patients with borderline resectable disease, preoperative chemotherapy followed by chemoradiation is a routine practice in most cancer centers,32 and ongoing clinical trials are an option for this cohort of patients (eg, Southwest Oncology Group Trial 1505, NCT02562716). The definitions of borderline resectable and unresectable pancreatic cancer have been described by the NCCN,26 although most surgeons consider involvement of the major upper abdominal blood vessels the main unresectability criterion; oncologists also consider other parameters such as suspicious lesions on scans, worsening performance status, and a significantly elevated CA 19-9 level suggestive of disseminated disease.28 The goal of a conversion approach by chemotherapy with or without radiation for borderline and unresectable cancers is to deliver a tolerable regimen leading to tumor downstaging, allowing for surgical resection. No randomized clinical trial has shown a survival advantage of this approach. Enrollment in clinical trials is preferred for patients with borderline and unresectable cancer, and there are trials that are currently enrolling patients.
The main treatment strategies for patients with locally advanced borderline and unresectable pancreatic cancer outside of a clinical trial are primary radiotherapy, systemic chemotherapy, and chemoradiation therapy. Guidelines from ASCO, NCCN, and ESMO recommend induction chemotherapy followed by restaging and consolidation chemoradiotherapy in the absence of progression.26,28,29 There is no standard chemoradiation regimen and the role of chemotherapy sensitizers, including fluorouracil, gemcitabine, and capecitabine (an oral fluoropyrimidine substitute), and targeted agents in combination with different radiation modalities is now being investigated.
Fluorouracil is a radio-sensitizer that has been used in locally advanced pancreatic cancer based on experience in other gastrointestinal malignancies; data shows conflicting results with this drug. Capecitabine and tegafur/gimeracil/oteracil (S-1) are oral prodrugs that can safely replace infusional fluorouracil. Gemcitabine, a more potent radiation sensitizer, is very toxic, even at low-doses twice weekly, and does not provide a survival benefit, as demonstrated in the Cancer and Leukemia Group B (CALGB) 89805 trial, a phase 2 study of patients with surgically staged locally advanced pancreatic cancer.33 Gemcitabine-based chemoradiotherapy was also evaluated in the Eastern Cooperative Group (ECOG) E4201 trial, which randomly assigned patients to receive gemcitabine alone (at 1000 mg/m2/wk for weeks 1 through 6, followed by 1 week rest, then weekly for 3 out of 4 weeks) or gemcitabine (600 mg/m2/wk for weeks 1 to 5, then 4 weeks later 1000 mg/m2 for 3 out of 4 weeks) plus radiotherapy (starting on day 1, 1.8 Gy/fraction for total of 50.4 Gy).34 Patients with locally advanced unresectable pancreatic cancer had a better OS outcome with gemcitabine in combination with radiation therapy (11.1 months) as compared with patients who received gemcitabine alone (9.2 months). Although there was a greater incidence of grade 4 and 5 treatment-related toxicities in the combination arm, no statistical differences in quality-of-life measurements were reported. Gemcitabine-based and capecitabine-based chemoradiotherapy were compared in the open-label phase 2 multicenter randomized SCALOP trial.35 Patients with locally advanced pancreatic cancer were assigned to receive 3 cycles of induction with gemcitabine 1000 mg/m2 days 1, 8, and 15 and capecitabine 830 mg/m2 days 1 to 21 every 28 days; patients who had stable or responding disease were randomly assigned to receive a fourth cycle followed by capecitabine (830 mg/m2 twice daily on weekdays only) or gemcitabine (300 mg/m2 weekly) with radiation (50.4 Gy over 28 fractions). Patients treated with capecitabine-based chemoradiotherapy had higher nonsignificant median OS (17.6 months) and median progression-free survival (12 months) compared to those treated with gemcitabine (14.6 months and 10.4 months, respectively).
The benefit of radiation therapy in the treatment of locally advanced pancreatic cancer was further explored by the Fédération Francophone de Cancérologie Digestive 2000-01 phase 3 trial. This study compared induction chemoradiotherapy (60 Gy, 2 Gy/fraction; concomitant fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) to gemcitabine alone (1000 mg/m2 weekly for 7 weeks) followed by maintenance gemcitabine in both arms.36 Unexpectedly, the median OS was significantly shorter in the chemoradiotherapy arm than in the chemotherapy alone arm (8.6 months versus 13 months, respectively, P = 0.03) and the combination arm had more toxicities. The phase 3 open-label LAP07 study explored the role of radiation therapy in patients with locally advanced pancreatic cancer who had controlled disease after 4 months of induction therapy.37 LAP07 had 2 randomizations: first, patients with locally advanced pancreatic cancer were assigned to receive weekly gemcitabine alone (1000 mg/m2) or this same dose of gemcitabine plus erlotinib 100 mg/day; second, patients with progression-free disease (61% of initial cohort) after 4 months of therapy were assigned to receive 2 months of the same chemotherapy or chemoradiotherapy (54 Gy plus capecitabine). This study showed that the addition of erlotinib to gemcitabine did not improve survival and in fact affected survival adversely. Of note, no survival benefit was observed after the first randomization from chemotherapy to consolidating chemoradiotherapy. Chemoradiotherapy achieved better locoregional tumor control with significantly less local tumor progression (32% versus 46%, P < 0.03) and no increase in toxicity. Based on prior moderate-quality evidence, guidelines recommend consolidative chemoradiotherapy only for surgical resection candidates following induction chemotherapy; for those who are not surgical candidates, guidelines recommend continuing systemic therapy.26,28,29
Gemcitabine and fluorouracil-based chemotherapies were the standard induction regimens until evidence from studies of metastatic systemic treatment protocols with FOLFIRINOX (ACCORD trial38) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) plus gemcitabine (MPACT trial39) was extrapolated to clinical practice. These regimens were shown to achieve higher objective response rates when compared to single-agent gemcitabine in patients with metastatic pancreatic cancer. Due to the broad heterogeneity of results in small retrospective series with neoadjuvant trials in borderline resectable pancreatic cancer, the quality of the evidence is low and any recommendation is limited. Many individual series have demonstrated improved complete resection rates and promising survival rates. In the largest single-institution retrospective review of patients with borderline resectable pancreatic adenocarcinoma who completed neoadjuvant gemcitabine-based chemoradiotherapy (50 Gy in 28 fractions or 30 Gy in 10 fractions), 94% achieved a margin-negative pancreatectomy; the median OS in those who completed preoperative therapy and had surgery was 40 months, with a 5-year OS of 36%.40 A meta-analysis by Andriulli and colleagues included 20 prospective studies of patients with initially resectable (366 lesions) or unresectable (341 lesions) disease who were treated with neoadjuvant/preoperative gemcitabine with or without radiotherapy.41 In the group with initially unresectable disease, 39% underwent surgery after restaging and 68% of explored patients were resected; the R0 resection rate was 60%. After restaging, 91% of patients with resectable disease underwent surgery, with 82% of explored patients undergoing surgical resection and 89% of these achieving R0 resection. The estimated 1- and 2-year survival probabilities after resection among patients with initially unresectable disease were 86.3% and 54.2%.41
The largest single-institution retrospective review of FOLFIRINOX (fluorouracil, oxaliplatin, irinotecan, and leucovorin), an alternative to gemcitabine, for neoadjuvant induction therapy for patients with locally advanced unresectable disease was conducted at Memorial Sloan Kettering Cancer Center. In this study (n = 101), 31% of patients initially deemed unresectable who completed FOLFIRINOX induction therapy with or without chemoradiation underwent resection. The R0 resection rate in these patients was 55%, and patients who did not progress during induction FOLFIRINOX therapy had a median OS of 26 months.42 A systematic review and meta-analysis of FOLFIRINOX chemotherapy with or without radiotherapy in patients with locally advanced unresectable pancreatic cancer reported that 25.9% of patients underwent resection after FOLFIRINOX therapy, and the R0 resection rate in these patients was 78.4%.43 The median OS in this study was 24.2 months, which was longer than the previously reported median OS rates for gemcitabine.
There is no strong evidence published for the use of combination nab-paclitaxel plus gemcitabine in the neoadjuvant setting, but it is used in clinical practice based on evidence from the MPACT trial, which showed the combination improved OS and progression-free survival in patients with metastatic pancreatic cancer.39 An early-phase 1-arm clinical trial of neoadjuvant gemcitabine, docetaxel, and capecitabine (GTX) followed by radiotherapy showed an increased response rate and survival for locally advanced disease; however, the NCCN expert panel has reached a consensus but not a uniform recommendation regarding this regimen due to significant toxicities and low patient accrual.26 Selected patients with pancreatic cancer with BRCA1/2 mutations are more sensitive to platinum-based chemotherapy. Although studies of neoadjuvant platinum-based chemotherapy in this population have not been reported, the NCCN guidelines list it as an alternative option based on extrapolated data.26 A clinical trial of gemcitabine, nab-paclitaxel, and cisplatin in the neoadjuvant setting in patients with resectable pancreatic cancer is currently enrolling patients (NGC triple regimen NCT0339257).
Summary
Chemotherapy alone or followed by chemoradiotherapy may be used as initial treatment for patients with borderline and unresectable pancreatic adenocarcinoma without distant metastases who are potential surgical candidates. Chemoradiotherapy remains a preferred treatment option for patients with poorly controlled pain from local tumor invasion, in view of the well-documented analgesic palliative effect of radiation therapy. FOLFIRINOX with or without radiation therapy may offer the highest documented response rates, but it also results in higher rates of treatment-related toxicities. FOLFIRINOX can be offered to selected fit patients (< 65 years old, no comorbidity contraindication, good functional status [ECOG 0–1]) who can tolerate triple therapy with a more toxic adverse-effect profile. A clinical trial evaluating neoadjuvant FOLFIRINOX with or without preoperative chemoradiotherapy in patients with borderline resectable pancreatic cancer is ongoing (PANDAS-PRODIGE 44, NCT02676349). Gemcitabine with or without radiation therapy is a tolerable combination, although it is potentially more toxic when combined with radiation. The addition of nab-paclitaxel to gemcitabine without radiation may emerge as a preferred neoadjuvant treatment for selected patients; a clinical trial investigating this modality in patients with resectable and borderline resectable disease is ongoing (NCT02723331).
Adjuvant Therapy
Case Continued
Prior to the planned surgical resection and after undergoing chemoradiation therapy, the patient has an excellent performance status and repeat MRI shows a 1.3 × 1.4 cm head mass with no further vasculature involvement, no evidence of lymphadenopathy, and no distant metastasis. The CA 19-9 level is stable at 18 U/mL. The patient undergoes an uncomplicated partial pancreaticoduodenectomy, and analysis of a surgical pathology specimen reveals T3N0 disease with closest margin of 0.1 cm.
- Would the patient benefit from adjuvant therapy?
Adjuvant chemotherapy for 6 months after pancreatic cancer resection should be offered to all patients based on mature data. Gemcitabine and capecitabine are the current standard of care in adjuvant therapy; alternatively, single-agent gemcitabine can be offered to patients with poor performance status or patients who cannot tolerate the toxicities associated with this combination.28 Adjuvant treatment should be initiated within approximately 8 weeks of surgical resection. The value of radiation therapy remains controversial, but it can be offered within the context of a clinical trial or to patients with positive margins after surgical resection and/or lymph node–positive disease. Based on low-quality supportive evidence, it is strongly recommended that patients who receive neoadjuvant therapy complete a total of 6 months of chemotherapy, factoring in the duration of the preoperative regimen.28 Different adjuvant strategies have been investigated, including chemotherapy alone with a fluoropyrimidine and/or gemcitabine with or without combined chemoradiation therapy.
The European Study Group for Pancreatic Cancer 1 (ESPAC)-1 trial was a randomized clinical trial that evaluated several adjuvant strategies in pancreatic cancer treatment. This trial assigned patients who underwent pancreatic adenocarcinoma resection to adjuvant chemotherapy alone (intravenous fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 daily for 5 days, monthly for 6 months), chemoradiotherapy (20 Gy in 10 daily fractions over 2 weeks with 500 mg/m2 intravenous fluorouracil on days 1–3, repeated after 2 weeks), both chemotherapy and chemoradiation, and observation.44 The results showed no added benefit for adjuvant chemoradiotherapy, with a median OS of 15.5 months in the chemoradiotherapy cohort, as compared to a median OS of 16.1 months in the chemotherapy-alone cohort (hazard ratio [HR] 1.18 [95% CI 0.90 to 1.55], P = 0.24). In addition, there was evidence of a survival benefit for the chemotherapy-alone arm when compared to the combined modality arm, with a median OS of 19.7 versus 14.0 months, respectively (HR 0.66 [95% CI 0.52 to 0.83], P = 0.0005). Although ESPAC-1 has been criticized for being underpowered to perform statistical comparison, it is still considered a landmark trial demonstrating benefit with single-agent chemotherapy alone. A follow-up analysis of ESPAC-1 showed that adjuvant chemotherapy alone conferred a significant 5-year survival benefit while the combined modality had a deleterious effect on survival. 45 Hence, adjuvant chemotherapy alone became the standard of care in the United States following resection.
The results of the multicenter randomized controlled phase 3 CONKO-001 (CharitéOnkologie 001) trial, which were reported in 2007, supported the use of adjuvant gemcitabine for 6 months in patients with resected pancreatic adenocarcinoma. In this study, patients treated with adjuvant gemcitabine (1000 mg/m2 days 1, 8, and 15 every 4 weeks for 6 months) had superior disease-free survival compared with those who received surgery alone.30 A long-term outcome update of this study demonstrated a significant improvement in 5-year OS for patients treated with adjuvant gemcitabine (20.7% [95% CI 14.7% to 26.6%]) compared to those who received surgical resection alone (10.4% [95% CI 5.9% to 15.0%]). This benefit persisted at 10-year follow-up, with an OS of 12.2% (95% CI 7.3% to 17.2%) in the adjuvant gemcitabine group, as compared to 7.7% (95% CI 3.6% to 11.8%) in the resection alone group.31
Fluorouracil and gemcitabine remained equivalent adjuvant treatment options until the results of the ESPAC-3 trial were reported in 2010.32 This large phase 3 trial, conducted mainly in the United Kingdom, compared weekly gemcitabine (1000 mg/m2 weekly for 3 of every 4 weeks) to leucovorin-modulated fluorouracil (Mayo Clinic regimen: leucovorin 20 mg/m2 followed by fluorouracil 425 mg/m2 intravenous bolus days 1 through 5 every 28 days) as adjuvant therapy in resected pancreatic adenocarcinoma. After a median follow-up of 34.2 months, the median OS was similar in the 2 groups (fluorouracil/leucovorin 23.0 months versus gemcitabine 23.6 months; P = 0.39). However, the fluorouracil/leucovorin group experienced more grade 3/4 treatment-related toxicities (mucositis, stomatitis, diarrhea, and hosptializations; 14% versus 7.5%; P < 0.001).46 Following this trial, gemcitabine became the standard of care for adjuvant chemotherapy for resected pancreatic cancer.
The U.S. Radiation Therapy Oncology Group (RTOG) 9704 trial was conducted to investigate the potential benefit of adding radiation therapy to gemcitabine. This trial demonstrated an improved trend among patients with pancreatic head tumors (but not with cancers of the pancreatic body or tail) who received adjuvant gemcitabine followed by chemoradiotherapy (50.4 Gy in 1.8 Gy daily fractions for 5.5 weeks with concurrent infusional fluorouracil 250 mg/m2 daily) and subsequent gemcitabine monotherapy compared to postoperative fluorouracil-based chemoradiotherapy. Results showed a 5-year OS of 22% versus 18%, respectively, although this improvement was not statistically significant (P = 0.08). This trial failed to show a benefit of adding radiotherapy to gemcitabine.47
The ESPAC-4 trial, reported in 2017, evaluated the combination of gemcitabine and capecitabine compared to gemcitabine alone as adjuvant therapy for resected pancreatic adenocarcinoma.48 Patients were randomly assigned after surgical resection, regardless of margin or node status, to 6 months of gemcitabine alone (1000 mg/m2/day on days 1, 8, and 15 of each 28-day cycle) or gemcitabine plus capecitabine (1660 mg/m2/day on days 1 through 21 of each 28-day cycle). Combination therapy had a significant survival benefit compared to single therapy, with median OS durations of 28 months and 25.5 months, respectively (HR for death 0.82 [95% CI 0.68 to 0.98]). The 5-year OS for patients who received combination treatment was 29 months (95% CI 22.9 to 35.2) versus 16 months (95% CI 10.2 to 23.7) for those in the monotherapy group. As expected, grade 3 or 4 treatment-related toxicities (diarrhea, hand-foot syndrome, and neutropenia) were significantly more common with combined therapy, although there were no significant differences in the rates of serious adverse events. The adjuvant combination of gemcitabine and capecitabine became the current and preferred new standard of care following resection of pancreatic ductal adenocarcinoma,28 but single-agent gemcitabine and fluorouracil/leucovorin continue to be viable options,26,28,29 particularly for elderly patients, patients with borderline performance status, or patients with multiple comorbidities.
Evidence showing that a more intensive regimen can improve outcome in the adjuvant setting remains elusive. The phase 3 APACT study (Adjuvant Therapy for Patients with Resected Pancreatic Cancer, NCT01964430) comparing gemcitabine alone to gemcitabine plus nab-paclitaxel in patients with surgically resected pancreatic adenocarcinoma has concluded, with the results projected to be released in 2018. Another phase 3 trial investigating the efficacy of FOLFIRINOX versus gemcitabine alone as adjuvant therapy is underway in France and Canada (PRODIGE24/ACCORD24, NCT01526135). Other strategies with newer targeted therapies and immunotherapy are in the development phase.
Follow-Up and Surveillance
Case Conclusion
After recovery from surgery, the patient is offered and completes 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine. He is started on surveillance at 3 and 6 months, and he maintains an excellent performance status. He develops clinical evidence of pancreatic enzyme insufficiency and is placed on oral replacement therapy. He has no other complaints, and there is no evidence of recurrence on MRI and CA 19-9 levels.
- What is the recommended duration of surveillance following curative resection?
Surveillance after curative resection of pancreatic adenocarcinoma is recommended by NCCN guidelines.26 However, pancreatic adenocarcinoma has a poor prognosis, and surveillance after curative surgical resection with or without perioperative therapy has not been shown to impact survival. Most recurrences will occur within 2 years after treatment. Surveillance recommendations differ among expert groups.26,28,29 NCCN guidelines recommend evaluating patients by history and physical examination every 3 to 6 months for the first 2 years, then every 6 to 12 months for 3 years. CA 19-9 level and CT scan should be obtained every 3 to 6 months for 2 years and then every 6 to 12 months for 3 years. Follow-up with CA 19-9 levels and CT scans after 5 years is not routinely performed unless guided by signs, symptoms, or laboratory findings that raise suspicion for recurrence. Follow-up visits should also include evaluation of treatment-related toxicities, symptom management, nutrition support of pancreatic insufficiency, and psychosocial support.
Conclusion
Pancreatic cancer is a leading cause of cancer-related death that frequently presents with locally advanced or metastatic disease due to nonspecific symptoms and lack of a screening modality. Histological tissue biopsy confirmation and accurate resectability staging guide treatment planning and prognosis. The only potentially curative therapy is surgical resection plus adjuvant therapy for those with resectable disease. Surgical candidates with borderline resectable and unresectable disease can be offered induction preoperative chemotherapy followed by consolidation chemoradiation, based on clinical consensus practice. Enrollment in clinical trials should be encouraged for all patients, as evidence from clinical trials is essential to making progress in pancreatic cancer treatment.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69.
3. Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th edition staging system for patients with pancreatic adenocarcinoma: a Surveillance, Epidemiology and End Results (SEER) analysis. Ann Surg Oncol 2017;24:2023–30.
4. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer.gov/statfacts/html/pancreas.html. Accessed 17 February 2018.
5. Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
6. Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
7. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62. Epub 2014 Mar 9.
8. Canto MI, Harinck F, Hruban RH, et al, on behalf of the International Cancer of the Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. Epub 2012 Nov 7.
9. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64:2634–8.
10. McKay SH,Humphris JL, Johns AL, et al. Inherited pancreatic cancer. Cancer Forum 2016;40(1).
11. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90.
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. AFIP Atlas of Tumor Pathology. 4th series, fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007.
13. Vege SS, Ziring B, Jain R, Moayyedi P, Clinical Guidelines Committee, American Gastroenterology Association. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22.
14. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501.
15. Choi M, Bien H, Mofunanya A, Powers S. Challenges in Ras therapeutics in pancreatic cancer. Semin Cancer Biol 2017 Nov 21. pii: S1044-579X(17)30235-3.
16. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68. Epub 2016 Nov 15.
17. Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
18. Modolell I, Guarner L, Malagelada JR. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann Oncol 1999;10 Suppl 4:82–4.
19. Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
20. Bronstein YL, Loyer EM, Kaur H, et al. Detection of small pancreatic tumors with multiphasic helical CT. AJR Am J Roentgenol 2004;182:619–23.
21. Niederau C, Grendell JH. Diagnosis of pancreatic carcinoma. Imaging techniques and tumor markers. Pancreas 1992;7:66–86.
22. Kim HJ, Kim MH, Myung SJ, et al. A new strategy for the application of CA19-9 in the differentiation of pancreaticobiliary cancer: analysis using a receiver operating characteristic curve. Am J Gastroenterol 1999;94:1941–6.
23. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2541–56.
24. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
25. Soriano A, Castells A, Ayuso C, et al. Preoperative staging and tumor resectability assessment of pancreatic cancer: prospective study comparing endoscopic ultrasonography, helical computed tomography, magnetic resonance imaging, and angiography. Am J Gastroenterol 2004;99:492–501.
26. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
27. Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014;270:248–60.
28. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol 2017;35:2324–8.
28. Ducreux M, Cuhna AS, Caramella C, et al; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v56–68.
30. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
31. Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 2013;310:1473–81.
32. Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
33. Blackstock AW, Tepper JE, Niedwiecki D, et al. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer 2003;34(2-3):107–16.
34. Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105–12.
35. Hurt CN, Falk S, Crosby T, et al. Long-term results and recurrence patterns from SCALOP: a phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br J Cancer 2017;116:1264–70.
36. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol 2008;19:1592–9.
37. Hammel P, Huguet F, van Laethem JL, et al, LAP07 Trial Group. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
38. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
39. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703.
40. Katz MH, Pisters PW, Evans DB, et al. Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 2008;206:833–46.
41. Andriulli A, Festa V, Botteri E, et al. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: a meta-analysis of prospective studies. Ann Surg Oncol 2012;19:1644–62.
42. Sadot E, Doussot A, O’Reilly EM, et al. FOLFIRINOX induction therapy for stage 3 pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:3512–21.
43. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
44. Neoptolemos JP, Dunn JA, Stocken DD, et al, European Study Group for Pancreatic Cancer. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet 2001;358:1576–85.
45. Neoptolemos JP, Stocken DD, Friess H, et al, European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
46. Neoptolemos JP, Stocken DD, Bassi C, et al, European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 2010;304:1073–81.
47. Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA 2008;299:1019–26.
48. Neoptolemos JP, Palmer DH, Ghaneh P, et al, European Study Group for Pancreatic Cancer. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. Epub 2017 Jan 25.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69.
3. Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th edition staging system for patients with pancreatic adenocarcinoma: a Surveillance, Epidemiology and End Results (SEER) analysis. Ann Surg Oncol 2017;24:2023–30.
4. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer.gov/statfacts/html/pancreas.html. Accessed 17 February 2018.
5. Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
6. Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
7. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62. Epub 2014 Mar 9.
8. Canto MI, Harinck F, Hruban RH, et al, on behalf of the International Cancer of the Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. Epub 2012 Nov 7.
9. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64:2634–8.
10. McKay SH,Humphris JL, Johns AL, et al. Inherited pancreatic cancer. Cancer Forum 2016;40(1).
11. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90.
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. AFIP Atlas of Tumor Pathology. 4th series, fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007.
13. Vege SS, Ziring B, Jain R, Moayyedi P, Clinical Guidelines Committee, American Gastroenterology Association. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22.
14. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501.
15. Choi M, Bien H, Mofunanya A, Powers S. Challenges in Ras therapeutics in pancreatic cancer. Semin Cancer Biol 2017 Nov 21. pii: S1044-579X(17)30235-3.
16. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68. Epub 2016 Nov 15.
17. Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
18. Modolell I, Guarner L, Malagelada JR. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann Oncol 1999;10 Suppl 4:82–4.
19. Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
20. Bronstein YL, Loyer EM, Kaur H, et al. Detection of small pancreatic tumors with multiphasic helical CT. AJR Am J Roentgenol 2004;182:619–23.
21. Niederau C, Grendell JH. Diagnosis of pancreatic carcinoma. Imaging techniques and tumor markers. Pancreas 1992;7:66–86.
22. Kim HJ, Kim MH, Myung SJ, et al. A new strategy for the application of CA19-9 in the differentiation of pancreaticobiliary cancer: analysis using a receiver operating characteristic curve. Am J Gastroenterol 1999;94:1941–6.
23. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2541–56.
24. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
25. Soriano A, Castells A, Ayuso C, et al. Preoperative staging and tumor resectability assessment of pancreatic cancer: prospective study comparing endoscopic ultrasonography, helical computed tomography, magnetic resonance imaging, and angiography. Am J Gastroenterol 2004;99:492–501.
26. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
27. Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014;270:248–60.
28. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol 2017;35:2324–8.
28. Ducreux M, Cuhna AS, Caramella C, et al; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v56–68.
30. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
31. Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 2013;310:1473–81.
32. Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
33. Blackstock AW, Tepper JE, Niedwiecki D, et al. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer 2003;34(2-3):107–16.
34. Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105–12.
35. Hurt CN, Falk S, Crosby T, et al. Long-term results and recurrence patterns from SCALOP: a phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br J Cancer 2017;116:1264–70.
36. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol 2008;19:1592–9.
37. Hammel P, Huguet F, van Laethem JL, et al, LAP07 Trial Group. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
38. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
39. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703.
40. Katz MH, Pisters PW, Evans DB, et al. Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 2008;206:833–46.
41. Andriulli A, Festa V, Botteri E, et al. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: a meta-analysis of prospective studies. Ann Surg Oncol 2012;19:1644–62.
42. Sadot E, Doussot A, O’Reilly EM, et al. FOLFIRINOX induction therapy for stage 3 pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:3512–21.
43. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
44. Neoptolemos JP, Dunn JA, Stocken DD, et al, European Study Group for Pancreatic Cancer. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet 2001;358:1576–85.
45. Neoptolemos JP, Stocken DD, Friess H, et al, European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
46. Neoptolemos JP, Stocken DD, Bassi C, et al, European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 2010;304:1073–81.
47. Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA 2008;299:1019–26.
48. Neoptolemos JP, Palmer DH, Ghaneh P, et al, European Study Group for Pancreatic Cancer. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. Epub 2017 Jan 25.
Strategies for managing medication-induced hyperprolactinemia
Ms. E, age 23, presents to your office for a routine visit for management of bipolar I disorder and posttraumatic stress disorder with comorbid type 2 diabetes mellitus. She currently is taking
Ms. E has a history of self-discontinuing medication when adverse events occur. She has been hospitalized twice for psychosis and suicide attempts. Past psychotropic medications that have been discontinued due to adverse effects include ziprasidone (mild abnormal lip movement), olanzapine (ineffective and drowsy), valproic acid (tremor and abdominal discomfort), lithium (rash), and aripiprazole (increased fasting blood sugar and labile mood).
At her appointment today, Ms. E says she is concerned because she has been experiencing galactorrhea for the past 4 weeks. Her prolactin level is 14.4 ng/mL; a normal level for a woman who is not pregnant is <25 ng/mL. However, a repeat prolactin level is obtained, and is found to be elevated at 38 ng/mL.
Prolactin, a polypeptide hormone that is secreted from the pituitary gland, has many functions, including involvement in the synthesis and maintenance of breast milk production, in reproductive behavior, and in luteal function.1,2 Hyperprolactinemia—an elevated prolactin level—is a common endocrinologic disorder of the hypothalamic–pituitary–axis.3 Children, adolescents, premenopausal women, and women in the perinatal period are more vulnerable to medication-induced hyperprolactinemia.4 If not asymptomatic, patients with hyperprolactinemia may experience amenorrhea, galactorrhea, hypogonadism, sexual dysfunction, or infertility.1,4 Chronic hyperprolactinemia may increase the risk for long-term complications, such as decreased bone mineral density and osteoporosis, although available evidence has conflicting findings.1

Hyperprolactinemia is diagnosed by a prolactin concentration above the upper reference range.3 Various hormones and neurotransmitters can impact inhibition or stimulation of prolactin release.5 For example, dopamine tonically inhibits prolactin release and synthesis, whereas estrogen stimulates prolactin secretion.1,5 Prolactin also can be elevated under several physiologic and pathologic conditions, such as during stressful situations, meals, or sexual activity.1,5 A prolactin level >250 ng/mL is usually indicative of a prolactinoma; however, some medications, such as strong D2 receptor antagonists (eg, risperidone, haloperidol), can cause significant elevation without evidence of prolactinoma.3 In the absence of a tumor, medications are often identified as the cause of hyperprolactinemia.3 According to the Endocrinology Society clinical practice guideline, medication-induced elevated prolactin levels are typically between 25 to 100 ng/mL.3
Medication-induced hyperprolactinemia
Antipsychotics, antidepressants, hormonal preparations, antihypertensives, and gastrointestinal agents have been associated with hyperprolactinemia (Table 11,3,5-11). These medication classes increase prolactin by decreasing dopamine, which facilitates disinhibition of prolactin synthesis and release, or increasing prolactin stimulating hormones, such as serotonin or estrogen.5
Antipsychotics are the most common medication-related cause of hyperprolactinemia.3 Typical antipsychotics are more likely to cause hyperprolactinemia than atypical antipsychotics; the incidence among patients taking typical antipsychotics is 40% to 90%.3 Atypical antipsychotics, except risperidone and paliperidone, are considered to cause less endocrinologic effects than typical antipsychotics through various mechanisms: serotonergic receptor antagonism, fast dissociation from D2 receptors, D2 receptor partial agonism, and preferential binding of D3 vs D2 receptors.1,5 By having transient D2 receptor association, clozapine and quetiapine are considered to have less risk of hyperprolactinemia compared with other atypical antipsychotics.1,5 Aripiprazole, brexpiprazole, and cariprazine are partial D2 receptor agonists, and cariprazine is the only agent that exhibits preferential binding to D3 receptors.12,13 Based on limited data, brexpiprazole and cariprazine may have prolactin-sparing properties given their partial D2 receptor agonism.12,13 However, one study found increased prolactin levels in some patients after treatment with brexpiprazole, 4 mg/d.14 Similarly, another study found that cariprazine could increase prolactin levels as much as 4.1 ng/mL, depending on the dose.15 Except for aripiprazole, brexpiprazole, cariprazine, and clozapine, all other atypical antipsychotics marketed in the United States have a standard warning in the package insert regarding prolactin elevations.1,16,17
Because antidepressants are less well-studied as a cause of medication-induced hyperprolactinemia, drawing definitive conclusions regarding incidence rates is limited, but the incidence seems to be fairly low.6,18 A French pharmacovigilance study found that of 182,836 spontaneous adverse drug events reported between 1985 and 2009, there were 159 reports of selective serotonin reuptake inhibitors (SSRIs) inducing hyperprolactinemia.6 F
Mirtazapine and bupropion have been found to be prolactin-neutral.5 Bupropion also has been reported to decrease prolactin levels, potentially via its ability to block dopamine reuptake.19
Managing medication-induced hyperprolactinemia
Screening for and identifying clinically significant hyperprolactinemia is critical, because adverse effects of medications can lead to nonadherence and clinical decompensation.20 Patients must be informed of potential symptoms of hyperprolactinemia, and clinicians should inquire about such symptoms at each visit. Routine monitoring of prolactin levels in asymptomatic patients is not necessary, because the Endocrine Society Clinical Practice Guideline does not recommend treating patients with asymptomatic medication-induced hyperprolactinemia.3
In patients who report hyperprolactinemia symptoms, clinicians should review the patient’s prescribed medications and past medical history (eg, chronic renal failure, hypothyroidism) for potential causes or exacerbations, and address these factors accordingly.3 Order a measurement of prolactin level. A patient with a prolactin level >100 ng/mL should be referred to Endocrinology to rule out prolactinoma.1
If a patient’s prolactin level is between 25 and 100 ng/mL, review the patient’s medications (Table 11,3,5-11), because prolactin levels within this range usually signal a medication-induced cause.3 For patients with antipsychotic-induced hyperprolactinemia, there are several management strategies (Table 21,3,4,9,16,17,21-27):
- Watch and wait may be warranted when the patient is experiencing mild hyperprolactinemia symptoms.
- Discontinue. If the patient can be maintained without an antipsychotic, discontinuing the antipsychotic would be a first-line option.3
- Reduce the dose. Reducing the antipsychotic dose may be the preferred strategy for patients with moderate to severe hyperprolactinemia symptoms who responded to the antipsychotic and do not wish to start adjunctive therapy.4
- Switching to a prolactin-sparing antipsychotic may help normalize prolactin levels and may be preferred when the risk of relapse is low.3 Dopamine agonists can treat medication-induced hyperprolactinemia, but may worsen psychiatric symptoms.28,29 Therefore, this may be the preferred strategy if the offending medication cannot be discontinued or switched, or if the patient has a comorbid prolactinoma.

Less data exist on managing hyperprolactinemia that is induced by a medication other than an antipsychotic; however, it seems reasonable that the same strategies could be implemented. Specifically, for SSRI–induced hyperprolactinemia, if clinically appropriate, switching to or adding an alternative antidepressant that may be prolactin-sparing, such as mirtazapine or bupropion, could be attempted.8 One study found that fluoxetine-induced galactorrhea ceased within 10 days of discontinuing the medication.30
CASE CONTINUED
Because Ms. E has been on the same medication regimen for 3 years and recently developed galactorrhea, it seems unlikely that her hyperprolactinemia is medication-induced. However, a tumor-related cause is less likely because the prolactin level is <100 ng/mL. Based on the literature, the only possible medication-induced cause of her galactorrhea is risperidone. Ms. E agrees to a trial of adjunctive oral aripiprazole, 5 mg/d, with close monitoring of her type 2 diabetes mellitus. Because of the long elimination half-life of aripiprazole, 1 month is required to monitor for improvement in galactorrhea. Ms. E is advised to use breast pads as a nonpharmacologic strategy in the interim. After 1 month of treatment, Ms. E denies galactorrhea symptoms and no longer requires the use of breast pads.
1. Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs.2014;28(5):421-453.
2. Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523-1631.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
4. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64-73.
5. La Torre D, Falorni A. Pharmacological causes of hyperprolactinemia. Ther Clin Risk Manag. 2007;3(5):929-951.
6. Petit A, Piednoir D, Germain ML, et al. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database [in French]. Therapie. 2003;58(2):159-163.
7. Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29(5):833-846.
8. Coker F, Taylor D. Antidepressant-induced hyperprolactinaemia: incidence, mechanisms and management. CNS Drugs. 2010;24(7):563-574.
9. Molitch ME. Medication induced hyperprolactinemia. Mayo Clin Proc. 2005;80(8):1050-1057.
10. Xenazine (tetrabenazine) [package insert]. Washington, DC: Prestwick Pharmaceuticals, Inc.; 2008.
11. Peña KS, Rosenfeld JA. Evaluation and treatment of galactorrhea. Am Fam Physician 2001;63(9):1763-1770.
12. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
13. Das S, Barnwal P, Winston AB, et al. Brexpiprazole: so far so good. Ther Adv Psychopharmacol. 2016;6(1):39-54.
14. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
15. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Pscyhiatry. 2016;77(3):371-378.
16. Rexulti (brexpiprazole) [package insert]. Tokyo, Japan: Otsuka Pharmaceuticals Inc.; 2015.
17. Cariprazine (Vraylar) [package insert]. Parsippany, New Jersey: Actavis Pharmacueitcals Inc.; 2015.
18. Marken PA, Haykal RF, Fisher JN. Management of psychotropic-induced hyperprolactinemia. Clin Pharm. 1992;11(10):851-856.
19. Meltzer HY, Fang VS, Tricou BJ, et al. Effect of antidepressants on neuroendocrine axis in humans. Adv Biochem Psychopharmacol. 1982;32:303-316.
20. Tsuboi T, Bies RR, Suzuki T, et al. Hyperprolactinemia and estimated dopamine D2 receptor occupancy in patients with schizophrenia: analysis of the CATIE data. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:178-182.
21. Lee BH, Kim YK, Park SH. Using aripiprazole to resolve antipsychotic-induced symptomatic hyperprolactinemia: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):714-717.
22. Lu ML, Shen WW, Chen CH. Time course of the changes in antipsychotic-induced hyperprolactinemia following the switch to aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1978-1981.
23. Mendhekar DN, Andrade C. Galactorrhea with aripiprazole. Can J Psychiatry. 2005;50(4):243.
24. Joseph SP. Aripiprazole induced hyperprolactinemia in a young female with delusional disorder. Indian J Psychol Med. 2016;38(3):260-262.
25. Meng M, Li W, Zhang S, et al. Using aripiprazole to reduce antipsychotic-induced hyperprolactinemia: meta-analysis of currently available randomized controlled trials. Shaghai Arch Psychiatry. 2015;27(1):4-17.
26. Tollin SR. Use of the dopamine agonists bromocriptine and cabergoline in the management of risperidone induced hyperprolactinemia in patients with psychotic disorders. J Endocrinol Invest. 2000;23(11):765-70.
27. Yuan HN, Wang CY, Sze CW, et al. A randomized, crossover comparison of herbal medicine and bromocriptine against risperidone-induced hyperprolactinemia in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(3):264-370.
28. Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. General Hospital Psychiatry. 2008;30(4):378-380.
29. Ishitobi M, Kosaka H, Shukunami K, et al. Adjunctive treatment with low-dosage pramipexole for risperidone-associated hyperprolactinemia and sexual dysfunction in a male patient with schizophrenia. J Clin Psychopharmacol 2011;31(2):243-245.
30. Peterson MC. Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc. 2001;76(2):215-216.
Ms. E, age 23, presents to your office for a routine visit for management of bipolar I disorder and posttraumatic stress disorder with comorbid type 2 diabetes mellitus. She currently is taking
Ms. E has a history of self-discontinuing medication when adverse events occur. She has been hospitalized twice for psychosis and suicide attempts. Past psychotropic medications that have been discontinued due to adverse effects include ziprasidone (mild abnormal lip movement), olanzapine (ineffective and drowsy), valproic acid (tremor and abdominal discomfort), lithium (rash), and aripiprazole (increased fasting blood sugar and labile mood).
At her appointment today, Ms. E says she is concerned because she has been experiencing galactorrhea for the past 4 weeks. Her prolactin level is 14.4 ng/mL; a normal level for a woman who is not pregnant is <25 ng/mL. However, a repeat prolactin level is obtained, and is found to be elevated at 38 ng/mL.
Prolactin, a polypeptide hormone that is secreted from the pituitary gland, has many functions, including involvement in the synthesis and maintenance of breast milk production, in reproductive behavior, and in luteal function.1,2 Hyperprolactinemia—an elevated prolactin level—is a common endocrinologic disorder of the hypothalamic–pituitary–axis.3 Children, adolescents, premenopausal women, and women in the perinatal period are more vulnerable to medication-induced hyperprolactinemia.4 If not asymptomatic, patients with hyperprolactinemia may experience amenorrhea, galactorrhea, hypogonadism, sexual dysfunction, or infertility.1,4 Chronic hyperprolactinemia may increase the risk for long-term complications, such as decreased bone mineral density and osteoporosis, although available evidence has conflicting findings.1
Hyperprolactinemia is diagnosed by a prolactin concentration above the upper reference range.3 Various hormones and neurotransmitters can impact inhibition or stimulation of prolactin release.5 For example, dopamine tonically inhibits prolactin release and synthesis, whereas estrogen stimulates prolactin secretion.1,5 Prolactin also can be elevated under several physiologic and pathologic conditions, such as during stressful situations, meals, or sexual activity.1,5 A prolactin level >250 ng/mL is usually indicative of a prolactinoma; however, some medications, such as strong D2 receptor antagonists (eg, risperidone, haloperidol), can cause significant elevation without evidence of prolactinoma.3 In the absence of a tumor, medications are often identified as the cause of hyperprolactinemia.3 According to the Endocrinology Society clinical practice guideline, medication-induced elevated prolactin levels are typically between 25 to 100 ng/mL.3
Medication-induced hyperprolactinemia
Antipsychotics, antidepressants, hormonal preparations, antihypertensives, and gastrointestinal agents have been associated with hyperprolactinemia (Table 11,3,5-11). These medication classes increase prolactin by decreasing dopamine, which facilitates disinhibition of prolactin synthesis and release, or increasing prolactin stimulating hormones, such as serotonin or estrogen.5
Antipsychotics are the most common medication-related cause of hyperprolactinemia.3 Typical antipsychotics are more likely to cause hyperprolactinemia than atypical antipsychotics; the incidence among patients taking typical antipsychotics is 40% to 90%.3 Atypical antipsychotics, except risperidone and paliperidone, are considered to cause less endocrinologic effects than typical antipsychotics through various mechanisms: serotonergic receptor antagonism, fast dissociation from D2 receptors, D2 receptor partial agonism, and preferential binding of D3 vs D2 receptors.1,5 By having transient D2 receptor association, clozapine and quetiapine are considered to have less risk of hyperprolactinemia compared with other atypical antipsychotics.1,5 Aripiprazole, brexpiprazole, and cariprazine are partial D2 receptor agonists, and cariprazine is the only agent that exhibits preferential binding to D3 receptors.12,13 Based on limited data, brexpiprazole and cariprazine may have prolactin-sparing properties given their partial D2 receptor agonism.12,13 However, one study found increased prolactin levels in some patients after treatment with brexpiprazole, 4 mg/d.14 Similarly, another study found that cariprazine could increase prolactin levels as much as 4.1 ng/mL, depending on the dose.15 Except for aripiprazole, brexpiprazole, cariprazine, and clozapine, all other atypical antipsychotics marketed in the United States have a standard warning in the package insert regarding prolactin elevations.1,16,17
Because antidepressants are less well-studied as a cause of medication-induced hyperprolactinemia, drawing definitive conclusions regarding incidence rates is limited, but the incidence seems to be fairly low.6,18 A French pharmacovigilance study found that of 182,836 spontaneous adverse drug events reported between 1985 and 2009, there were 159 reports of selective serotonin reuptake inhibitors (SSRIs) inducing hyperprolactinemia.6 F
Mirtazapine and bupropion have been found to be prolactin-neutral.5 Bupropion also has been reported to decrease prolactin levels, potentially via its ability to block dopamine reuptake.19
Managing medication-induced hyperprolactinemia
Screening for and identifying clinically significant hyperprolactinemia is critical, because adverse effects of medications can lead to nonadherence and clinical decompensation.20 Patients must be informed of potential symptoms of hyperprolactinemia, and clinicians should inquire about such symptoms at each visit. Routine monitoring of prolactin levels in asymptomatic patients is not necessary, because the Endocrine Society Clinical Practice Guideline does not recommend treating patients with asymptomatic medication-induced hyperprolactinemia.3
In patients who report hyperprolactinemia symptoms, clinicians should review the patient’s prescribed medications and past medical history (eg, chronic renal failure, hypothyroidism) for potential causes or exacerbations, and address these factors accordingly.3 Order a measurement of prolactin level. A patient with a prolactin level >100 ng/mL should be referred to Endocrinology to rule out prolactinoma.1
If a patient’s prolactin level is between 25 and 100 ng/mL, review the patient’s medications (Table 11,3,5-11), because prolactin levels within this range usually signal a medication-induced cause.3 For patients with antipsychotic-induced hyperprolactinemia, there are several management strategies (Table 21,3,4,9,16,17,21-27):
- Watch and wait may be warranted when the patient is experiencing mild hyperprolactinemia symptoms.
- Discontinue. If the patient can be maintained without an antipsychotic, discontinuing the antipsychotic would be a first-line option.3
- Reduce the dose. Reducing the antipsychotic dose may be the preferred strategy for patients with moderate to severe hyperprolactinemia symptoms who responded to the antipsychotic and do not wish to start adjunctive therapy.4
- Switching to a prolactin-sparing antipsychotic may help normalize prolactin levels and may be preferred when the risk of relapse is low.3 Dopamine agonists can treat medication-induced hyperprolactinemia, but may worsen psychiatric symptoms.28,29 Therefore, this may be the preferred strategy if the offending medication cannot be discontinued or switched, or if the patient has a comorbid prolactinoma.
Less data exist on managing hyperprolactinemia that is induced by a medication other than an antipsychotic; however, it seems reasonable that the same strategies could be implemented. Specifically, for SSRI–induced hyperprolactinemia, if clinically appropriate, switching to or adding an alternative antidepressant that may be prolactin-sparing, such as mirtazapine or bupropion, could be attempted.8 One study found that fluoxetine-induced galactorrhea ceased within 10 days of discontinuing the medication.30
CASE CONTINUED
Because Ms. E has been on the same medication regimen for 3 years and recently developed galactorrhea, it seems unlikely that her hyperprolactinemia is medication-induced. However, a tumor-related cause is less likely because the prolactin level is <100 ng/mL. Based on the literature, the only possible medication-induced cause of her galactorrhea is risperidone. Ms. E agrees to a trial of adjunctive oral aripiprazole, 5 mg/d, with close monitoring of her type 2 diabetes mellitus. Because of the long elimination half-life of aripiprazole, 1 month is required to monitor for improvement in galactorrhea. Ms. E is advised to use breast pads as a nonpharmacologic strategy in the interim. After 1 month of treatment, Ms. E denies galactorrhea symptoms and no longer requires the use of breast pads.
Ms. E, age 23, presents to your office for a routine visit for management of bipolar I disorder and posttraumatic stress disorder with comorbid type 2 diabetes mellitus. She currently is taking
Ms. E has a history of self-discontinuing medication when adverse events occur. She has been hospitalized twice for psychosis and suicide attempts. Past psychotropic medications that have been discontinued due to adverse effects include ziprasidone (mild abnormal lip movement), olanzapine (ineffective and drowsy), valproic acid (tremor and abdominal discomfort), lithium (rash), and aripiprazole (increased fasting blood sugar and labile mood).
At her appointment today, Ms. E says she is concerned because she has been experiencing galactorrhea for the past 4 weeks. Her prolactin level is 14.4 ng/mL; a normal level for a woman who is not pregnant is <25 ng/mL. However, a repeat prolactin level is obtained, and is found to be elevated at 38 ng/mL.
Prolactin, a polypeptide hormone that is secreted from the pituitary gland, has many functions, including involvement in the synthesis and maintenance of breast milk production, in reproductive behavior, and in luteal function.1,2 Hyperprolactinemia—an elevated prolactin level—is a common endocrinologic disorder of the hypothalamic–pituitary–axis.3 Children, adolescents, premenopausal women, and women in the perinatal period are more vulnerable to medication-induced hyperprolactinemia.4 If not asymptomatic, patients with hyperprolactinemia may experience amenorrhea, galactorrhea, hypogonadism, sexual dysfunction, or infertility.1,4 Chronic hyperprolactinemia may increase the risk for long-term complications, such as decreased bone mineral density and osteoporosis, although available evidence has conflicting findings.1
Hyperprolactinemia is diagnosed by a prolactin concentration above the upper reference range.3 Various hormones and neurotransmitters can impact inhibition or stimulation of prolactin release.5 For example, dopamine tonically inhibits prolactin release and synthesis, whereas estrogen stimulates prolactin secretion.1,5 Prolactin also can be elevated under several physiologic and pathologic conditions, such as during stressful situations, meals, or sexual activity.1,5 A prolactin level >250 ng/mL is usually indicative of a prolactinoma; however, some medications, such as strong D2 receptor antagonists (eg, risperidone, haloperidol), can cause significant elevation without evidence of prolactinoma.3 In the absence of a tumor, medications are often identified as the cause of hyperprolactinemia.3 According to the Endocrinology Society clinical practice guideline, medication-induced elevated prolactin levels are typically between 25 to 100 ng/mL.3
Medication-induced hyperprolactinemia
Antipsychotics, antidepressants, hormonal preparations, antihypertensives, and gastrointestinal agents have been associated with hyperprolactinemia (Table 11,3,5-11). These medication classes increase prolactin by decreasing dopamine, which facilitates disinhibition of prolactin synthesis and release, or increasing prolactin stimulating hormones, such as serotonin or estrogen.5
Antipsychotics are the most common medication-related cause of hyperprolactinemia.3 Typical antipsychotics are more likely to cause hyperprolactinemia than atypical antipsychotics; the incidence among patients taking typical antipsychotics is 40% to 90%.3 Atypical antipsychotics, except risperidone and paliperidone, are considered to cause less endocrinologic effects than typical antipsychotics through various mechanisms: serotonergic receptor antagonism, fast dissociation from D2 receptors, D2 receptor partial agonism, and preferential binding of D3 vs D2 receptors.1,5 By having transient D2 receptor association, clozapine and quetiapine are considered to have less risk of hyperprolactinemia compared with other atypical antipsychotics.1,5 Aripiprazole, brexpiprazole, and cariprazine are partial D2 receptor agonists, and cariprazine is the only agent that exhibits preferential binding to D3 receptors.12,13 Based on limited data, brexpiprazole and cariprazine may have prolactin-sparing properties given their partial D2 receptor agonism.12,13 However, one study found increased prolactin levels in some patients after treatment with brexpiprazole, 4 mg/d.14 Similarly, another study found that cariprazine could increase prolactin levels as much as 4.1 ng/mL, depending on the dose.15 Except for aripiprazole, brexpiprazole, cariprazine, and clozapine, all other atypical antipsychotics marketed in the United States have a standard warning in the package insert regarding prolactin elevations.1,16,17
Because antidepressants are less well-studied as a cause of medication-induced hyperprolactinemia, drawing definitive conclusions regarding incidence rates is limited, but the incidence seems to be fairly low.6,18 A French pharmacovigilance study found that of 182,836 spontaneous adverse drug events reported between 1985 and 2009, there were 159 reports of selective serotonin reuptake inhibitors (SSRIs) inducing hyperprolactinemia.6 F
Mirtazapine and bupropion have been found to be prolactin-neutral.5 Bupropion also has been reported to decrease prolactin levels, potentially via its ability to block dopamine reuptake.19
Managing medication-induced hyperprolactinemia
Screening for and identifying clinically significant hyperprolactinemia is critical, because adverse effects of medications can lead to nonadherence and clinical decompensation.20 Patients must be informed of potential symptoms of hyperprolactinemia, and clinicians should inquire about such symptoms at each visit. Routine monitoring of prolactin levels in asymptomatic patients is not necessary, because the Endocrine Society Clinical Practice Guideline does not recommend treating patients with asymptomatic medication-induced hyperprolactinemia.3
In patients who report hyperprolactinemia symptoms, clinicians should review the patient’s prescribed medications and past medical history (eg, chronic renal failure, hypothyroidism) for potential causes or exacerbations, and address these factors accordingly.3 Order a measurement of prolactin level. A patient with a prolactin level >100 ng/mL should be referred to Endocrinology to rule out prolactinoma.1
If a patient’s prolactin level is between 25 and 100 ng/mL, review the patient’s medications (Table 11,3,5-11), because prolactin levels within this range usually signal a medication-induced cause.3 For patients with antipsychotic-induced hyperprolactinemia, there are several management strategies (Table 21,3,4,9,16,17,21-27):
- Watch and wait may be warranted when the patient is experiencing mild hyperprolactinemia symptoms.
- Discontinue. If the patient can be maintained without an antipsychotic, discontinuing the antipsychotic would be a first-line option.3
- Reduce the dose. Reducing the antipsychotic dose may be the preferred strategy for patients with moderate to severe hyperprolactinemia symptoms who responded to the antipsychotic and do not wish to start adjunctive therapy.4
- Switching to a prolactin-sparing antipsychotic may help normalize prolactin levels and may be preferred when the risk of relapse is low.3 Dopamine agonists can treat medication-induced hyperprolactinemia, but may worsen psychiatric symptoms.28,29 Therefore, this may be the preferred strategy if the offending medication cannot be discontinued or switched, or if the patient has a comorbid prolactinoma.
Less data exist on managing hyperprolactinemia that is induced by a medication other than an antipsychotic; however, it seems reasonable that the same strategies could be implemented. Specifically, for SSRI–induced hyperprolactinemia, if clinically appropriate, switching to or adding an alternative antidepressant that may be prolactin-sparing, such as mirtazapine or bupropion, could be attempted.8 One study found that fluoxetine-induced galactorrhea ceased within 10 days of discontinuing the medication.30
CASE CONTINUED
Because Ms. E has been on the same medication regimen for 3 years and recently developed galactorrhea, it seems unlikely that her hyperprolactinemia is medication-induced. However, a tumor-related cause is less likely because the prolactin level is <100 ng/mL. Based on the literature, the only possible medication-induced cause of her galactorrhea is risperidone. Ms. E agrees to a trial of adjunctive oral aripiprazole, 5 mg/d, with close monitoring of her type 2 diabetes mellitus. Because of the long elimination half-life of aripiprazole, 1 month is required to monitor for improvement in galactorrhea. Ms. E is advised to use breast pads as a nonpharmacologic strategy in the interim. After 1 month of treatment, Ms. E denies galactorrhea symptoms and no longer requires the use of breast pads.
1. Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs.2014;28(5):421-453.
2. Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523-1631.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
4. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64-73.
5. La Torre D, Falorni A. Pharmacological causes of hyperprolactinemia. Ther Clin Risk Manag. 2007;3(5):929-951.
6. Petit A, Piednoir D, Germain ML, et al. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database [in French]. Therapie. 2003;58(2):159-163.
7. Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29(5):833-846.
8. Coker F, Taylor D. Antidepressant-induced hyperprolactinaemia: incidence, mechanisms and management. CNS Drugs. 2010;24(7):563-574.
9. Molitch ME. Medication induced hyperprolactinemia. Mayo Clin Proc. 2005;80(8):1050-1057.
10. Xenazine (tetrabenazine) [package insert]. Washington, DC: Prestwick Pharmaceuticals, Inc.; 2008.
11. Peña KS, Rosenfeld JA. Evaluation and treatment of galactorrhea. Am Fam Physician 2001;63(9):1763-1770.
12. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
13. Das S, Barnwal P, Winston AB, et al. Brexpiprazole: so far so good. Ther Adv Psychopharmacol. 2016;6(1):39-54.
14. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
15. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Pscyhiatry. 2016;77(3):371-378.
16. Rexulti (brexpiprazole) [package insert]. Tokyo, Japan: Otsuka Pharmaceuticals Inc.; 2015.
17. Cariprazine (Vraylar) [package insert]. Parsippany, New Jersey: Actavis Pharmacueitcals Inc.; 2015.
18. Marken PA, Haykal RF, Fisher JN. Management of psychotropic-induced hyperprolactinemia. Clin Pharm. 1992;11(10):851-856.
19. Meltzer HY, Fang VS, Tricou BJ, et al. Effect of antidepressants on neuroendocrine axis in humans. Adv Biochem Psychopharmacol. 1982;32:303-316.
20. Tsuboi T, Bies RR, Suzuki T, et al. Hyperprolactinemia and estimated dopamine D2 receptor occupancy in patients with schizophrenia: analysis of the CATIE data. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:178-182.
21. Lee BH, Kim YK, Park SH. Using aripiprazole to resolve antipsychotic-induced symptomatic hyperprolactinemia: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):714-717.
22. Lu ML, Shen WW, Chen CH. Time course of the changes in antipsychotic-induced hyperprolactinemia following the switch to aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1978-1981.
23. Mendhekar DN, Andrade C. Galactorrhea with aripiprazole. Can J Psychiatry. 2005;50(4):243.
24. Joseph SP. Aripiprazole induced hyperprolactinemia in a young female with delusional disorder. Indian J Psychol Med. 2016;38(3):260-262.
25. Meng M, Li W, Zhang S, et al. Using aripiprazole to reduce antipsychotic-induced hyperprolactinemia: meta-analysis of currently available randomized controlled trials. Shaghai Arch Psychiatry. 2015;27(1):4-17.
26. Tollin SR. Use of the dopamine agonists bromocriptine and cabergoline in the management of risperidone induced hyperprolactinemia in patients with psychotic disorders. J Endocrinol Invest. 2000;23(11):765-70.
27. Yuan HN, Wang CY, Sze CW, et al. A randomized, crossover comparison of herbal medicine and bromocriptine against risperidone-induced hyperprolactinemia in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(3):264-370.
28. Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. General Hospital Psychiatry. 2008;30(4):378-380.
29. Ishitobi M, Kosaka H, Shukunami K, et al. Adjunctive treatment with low-dosage pramipexole for risperidone-associated hyperprolactinemia and sexual dysfunction in a male patient with schizophrenia. J Clin Psychopharmacol 2011;31(2):243-245.
30. Peterson MC. Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc. 2001;76(2):215-216.
1. Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs.2014;28(5):421-453.
2. Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523-1631.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
4. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64-73.
5. La Torre D, Falorni A. Pharmacological causes of hyperprolactinemia. Ther Clin Risk Manag. 2007;3(5):929-951.
6. Petit A, Piednoir D, Germain ML, et al. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database [in French]. Therapie. 2003;58(2):159-163.
7. Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29(5):833-846.
8. Coker F, Taylor D. Antidepressant-induced hyperprolactinaemia: incidence, mechanisms and management. CNS Drugs. 2010;24(7):563-574.
9. Molitch ME. Medication induced hyperprolactinemia. Mayo Clin Proc. 2005;80(8):1050-1057.
10. Xenazine (tetrabenazine) [package insert]. Washington, DC: Prestwick Pharmaceuticals, Inc.; 2008.
11. Peña KS, Rosenfeld JA. Evaluation and treatment of galactorrhea. Am Fam Physician 2001;63(9):1763-1770.
12. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
13. Das S, Barnwal P, Winston AB, et al. Brexpiprazole: so far so good. Ther Adv Psychopharmacol. 2016;6(1):39-54.
14. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
15. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Pscyhiatry. 2016;77(3):371-378.
16. Rexulti (brexpiprazole) [package insert]. Tokyo, Japan: Otsuka Pharmaceuticals Inc.; 2015.
17. Cariprazine (Vraylar) [package insert]. Parsippany, New Jersey: Actavis Pharmacueitcals Inc.; 2015.
18. Marken PA, Haykal RF, Fisher JN. Management of psychotropic-induced hyperprolactinemia. Clin Pharm. 1992;11(10):851-856.
19. Meltzer HY, Fang VS, Tricou BJ, et al. Effect of antidepressants on neuroendocrine axis in humans. Adv Biochem Psychopharmacol. 1982;32:303-316.
20. Tsuboi T, Bies RR, Suzuki T, et al. Hyperprolactinemia and estimated dopamine D2 receptor occupancy in patients with schizophrenia: analysis of the CATIE data. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:178-182.
21. Lee BH, Kim YK, Park SH. Using aripiprazole to resolve antipsychotic-induced symptomatic hyperprolactinemia: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):714-717.
22. Lu ML, Shen WW, Chen CH. Time course of the changes in antipsychotic-induced hyperprolactinemia following the switch to aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1978-1981.
23. Mendhekar DN, Andrade C. Galactorrhea with aripiprazole. Can J Psychiatry. 2005;50(4):243.
24. Joseph SP. Aripiprazole induced hyperprolactinemia in a young female with delusional disorder. Indian J Psychol Med. 2016;38(3):260-262.
25. Meng M, Li W, Zhang S, et al. Using aripiprazole to reduce antipsychotic-induced hyperprolactinemia: meta-analysis of currently available randomized controlled trials. Shaghai Arch Psychiatry. 2015;27(1):4-17.
26. Tollin SR. Use of the dopamine agonists bromocriptine and cabergoline in the management of risperidone induced hyperprolactinemia in patients with psychotic disorders. J Endocrinol Invest. 2000;23(11):765-70.
27. Yuan HN, Wang CY, Sze CW, et al. A randomized, crossover comparison of herbal medicine and bromocriptine against risperidone-induced hyperprolactinemia in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(3):264-370.
28. Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. General Hospital Psychiatry. 2008;30(4):378-380.
29. Ishitobi M, Kosaka H, Shukunami K, et al. Adjunctive treatment with low-dosage pramipexole for risperidone-associated hyperprolactinemia and sexual dysfunction in a male patient with schizophrenia. J Clin Psychopharmacol 2011;31(2):243-245.
30. Peterson MC. Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc. 2001;76(2):215-216.
Decompensation in a 51-year-old woman with schizophrenia
CASE Psychotic and reclusive
Ms. A, age 51, has schizophrenia and has been doing well living at a supervised residential facility. She was stable on haloperidol, 10 mg twice a day, for years but recently became agitated, threatening her roommate and yelling during the night. Ms. A begins to refuse to take her haloperidol. She also refuses to attend several outpatient appointments. As a result, Ms. A is admitted to the psychiatric unit on an involuntary basis.
In the hospital, Ms. A rarely comes out of her room. When she does come out, she usually sits in a chair, talking to herself and occasionally yelling or crying in apparent distress. Ms. A refuses to engage with her treatment team and lies mute in her bed when they attempt to interview her. Her records indicate that previous medication trials have included
Over the next week, Ms. A begins to interact more appropriately with nursing sta
[polldaddy:9945425]
The authors’ observations
As a class, antipsychotics lead to symptom reduction in approximately 70% of patients.1 However, the degree of response can vary markedly between individuals; although some patients may experience almost complete resolution of symptoms, others are still markedly impaired, as in Ms. A’s case.
A substantial amount of literature suggests that although the practice is common, use of >1 antipsychotic does not significantly increase efficacy but increases risk of adverse effects, such as type 2 diabetes mellitus, metabolic syndrome, cognitive impairment, and extrapyramidal symptoms.2-4 One exception is augmentation of clozapine with a second antipsychotic, which in certain cases appears to offer greater efficacy than clozapine alone.1 Practice guidelines and evidence generally do not support the use of multiple antipsychotics, but 20% of patients take >1 antipsychotic.5,6 Although antipsychotic polypharmacy may be appropriate for some patients, current literature suggests it is being done more often than recommended.
Clozapine is considered the most efficacious option for treatment-resistant schizophrenia.7 Because of Ms. A’s history of recurrent hospitalizations, her extensive list of trialed medications, and her ongoing symptoms despite a sufficient trial of haloperidol, the treatment team gives serious consideration to switching Ms. A to clozapine. However, Ms. A is not able to tolerate blood draws without significant support from nursing staff, and it is likely she would be unable to tolerate the frequent blood monitoring required of patients receiving clozapine.
Because many of Ms. A’s symptoms were negative or depressive, including hypersomnia, psychomotor retardation, sadness with frequent crying spells, and reduced interest in activities, adding an antidepressant to Ms. A’s medication regimen was considered. A recent systematic review and meta-analysis showed that adding an antidepressant to an antipsychotic in patients with schizophrenia had small but beneficial effects on depressive and negative symptoms and a low risk of adverse effects.8 However, Ms. A declined this option.
TREATMENT Adding long-acting haloperidol
Ms. A had previously achieved therapeutic blood levels9 with oral haloperidol. Data suggest that compared with the oral form, long-acting injectable antipsychotics can both improve compliance and decrease rehospitalization rates.10-12 Because Ms. A previously had done well with haloperidol decanoate, 200 mg every 2 weeks, achieving a blood level of 16.2 ng/mL, and because she had a partial response to oral haloperidol, we add haloperidol decanoate, 100 mg every 2 weeks, to her regimen, with the intention of transitioning her to all-depot dosing. In addition, the treatment team tries to engage Ms. A in a discussion of potential psychological contributions to her current presentation. They note that Ms. A has her basic needs met on the unit and reports feeling safe there; thus, a fear of discharge may be contributing to her lack of engagement with the team. However, because of her limited communication, it is challenging to investigate this hypothesis or explore other possible psychological issues.
Despite increasing the dosing of haloperidol, Ms. A shows minimal improvement. She continues to stonewall her treatment team, and is unwilling or unable to engage in meaningful conversation. A review of her chart suggests that this hospital course is different from previous ones in which her average stay was a few weeks, and she generally was able to converse with the treatment team, participate in discussions about her care, and make decisions about her desire for discharge.
The team considers if additional factors could be impacting Ms. A’s current presentation. They raise the possibility that she could be going through menopause, and hormonal fluctuations may be contributing to her symptoms. Despite being on the unit for nearly 2 months, Ms. A has not required the use of sanitary products. She also reports to nursing staff that at times she feels flushed and sweaty, but she is afebrile and does not have other signs or symptoms of infection.
[polldaddy:9945428]
The authors’ observations

Evidence suggests that estrogen levels can influence the development and severity of symptoms of schizophrenia (Table 113,14). Rates of schizophrenia are lower in women, and women typically have a later onset of illness with less severe symptoms.13 Women also have a second peak incidence of schizophrenia between ages 45 and 50, corresponding with the hormonal changes associated with menopause and the associated drop in estrogen.14 Symptoms also fluctuate with hormonal cycles—women experience worsening symptoms during the premenstrual phase of the menstrual cycle, when estrogen levels are low, and an improvement of symptoms during high-estrogen phases of the cycle.14 Overall, low levels of estrogen also have been observed in women with schizophrenia relative to controls, although this may be partially attributable to treatment with antipsychotics.14
Estrogen affects various regions of the brain implicated in schizophrenia and likely imparts its behavioral effects through several different mechanisms. Estrogen can act on cells to directly impact intracellular signaling and to alter gene expression.15 Although most often thought of as being related to reproductive functions, estrogen receptors can be found in many cortical and subcortical regions of the brain, such as the hippocampus, substantia nigra, and prefrontal cortex. Estrogen receptor expression levels in certain brain regions have been found to be altered in individuals with schizophrenia.15 Estrogen also enhances neurogenesis and neuroplasticity, playing a role in learning and memory.16 Particularly relevant, estrogen has been shown to directly impact both the dopaminergic and serotonergic systems.15,17 In animal models, estrogen has been shown to decrease the behavioral effects induced by dopamine agonists and decrease symptoms of schizophrenia.18 The underlying molecular mechanisms by which estrogen has these effects are uncertain.
Given estrogen’s potentially protective effects, clinical trials have explored the role of estrogen as an adjuvant to antipsychotics for treating schizophrenia. Studies have shown that estrogen can improve psychotic symptoms in patients with schizophrenia.19,20 However, because estrogen administration can increase the risk of breast and uterine cancer, researchers are instead investigating selective estrogen receptor modulators (SERMs).14 These medications have mixed agonist and antagonist effects, with different effects on different tissues. Raloxifene is a SERM that acts as an estrogen agonist in some tissues, but an antagonist in uterine and breast tissue, which may minimize potential deleterious adverse effects (Table 221-24). Repeated randomized controlled trials have found promising results for use of raloxifene as an adjunctive treatment in peri- and postmenopausal women with schizophrenia, including those refractory to antipsychotic treatment.13,25-27
TREATMENT Address symptoms

The treatment team takes steps to address Ms. A’s perimenopausal symptoms. For mild to moderate hot flashes, primary interventions are nonpharmacologic.28 Because Ms. A primarily reports her hot flashes at night, she is given lightweight pajamas and moved to the coolest room on the unit. Both bring some relief, and her hot flashes appear to be less distressing. The treatment team decides to consult Endocrinology to further investigate the feasibility of starting raloxifene (Table 3) because of their experience using this medication to manage osteoporosis.
[polldaddy:9945429]
The authors’ observations
Raloxifene is FDA-approved for treating osteoporosis and preventing invasive breast cancer.29 Because it is an estrogen antagonist in both breast and uterine tissues, raloxifene does not increase the risk of uterine or breast cancer. Large studies have shown rates of cardiovascular events are similar for raloxifene and placebo, and some studies have found that raloxifene treatment is associated with improvement in cardiovascular risk factors, including lower blood pressure, lower low-density lipoprotein cholesterol, and increased high-density lipoprotein cholesterol.29 Raloxifene does, however, increase risk of venous thromboembolism, including deep vein thrombosis and pulmonary embolism, and fatal stroke.29,30 Overall, the risk remains relatively low, with an absolute risk increase of fatal stroke of 0.7 per 1,000 woman-years (number needed to harm [NNH]: 250) and an absolute risk increase of venous thromboembolic events of 1.88 per 1,000 women-years (NNH: 158).31 However, raloxifene may not be appropriate for patients with independent risk factors for these events. Despite this, a large meta-analysis found a 10% decrease in mortality for patients taking raloxifene compared with those receiving placebo.32 Raloxifene also can cause hot flashes, muscle cramps, and flu-like symptoms.29
Diagnosis of menopause and perimenopause is largely clinical, with hormone testing generally recommended for women age <45 in whom the diagnosis may be unclear.28 Thus, Ms. A’s vasomotor symptoms and absence of a menstrual cycle for at least 2 months were diagnostic of perimenopause; a 12-month cessation in menstrual cycles is required for a diagnosis of menopause.28
OUTCOME Improvement with raloxifene
Because Ms. A is at relatively low risk for a thromboembolism or stroke, the benefit of raloxifene is thought to outweigh the risk, and she is started on raloxifene, 60 mg/d. Over the next 2 weeks, Ms. A becomes increasingly interactive, and is seen sitting at a table talking with other patients on multiple occasions. She spends time looking at fashion magazines, and engages in conversation about fashion with staff and other patients. She participates in group therapy for the first time during this hospital stay and begins to talk about discharge. She occasionally smiles and waves at her treatment team and participates more in the daily interview, although these interactions remain limited and on her terms. She maintains this improvement and is transferred to a psychiatric facility in her home county for ongoing care and discharge planning.
2. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
3. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
4. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
5. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
6. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
7. McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
8. Helfer B, Samara MT, Huhn M, et al. Efficacy and safety of antidepressants added to antipsychotics for schizophrenia: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(9);876-886.
9. Ulrich S, Neuhof S, Braun V, et al. Therapeutic window of serum haloperidol concentration in acute schizophrenia and schizoaffective disorder. Pharmacopsychiatry. 1998;31(5):163-169.
10. Lafeuille MH, Dean J, Carter V, et al. Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia. Curr Med Res Opin. 2014;30(8):1643-1655.
11. MacEwan JP, Kamat SA, Duffy RA, et al. Hospital readmission rates among patients with schizophrenia treated with long-acting injectables or oral antipsychotics. Psychiatr Serv. 2016;67(11):1183-1188.
12. Marcus SC, Zummo J, Pettit AR, et al. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768.
13. Usall J, Huerta-Ramos E, Iniesta R, et al; RALOPSYCAT Group. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
14. Seeman MV. Treating schizophrenia at the time of menopause. Maturitas. 2012;72(2):117-120.
15. Gogos A, Sbisa AM, Sun J, et al. A role for estrogen in schizophrenia: clinical and preclinical findings. Int J Endocrinol. 2015;2015:615356. doi: 10.1155/2015/615356.
16. Khan MM. Neurocognitive, neuroprotective, and cardiometabolic effects of raloxifene: potential for improving therapeutic outcomes in schizophrenia. CNS Drugs. 2016;30(7):589-601.
17. Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci. 2015;9:37.
18. Häfner H, Behrens S, De Vry J, et al. An animal model for the effects of estradiol on dopamine-mediated behavior: implications for sex differences in schizophrenia. Psychiatry Res. 1991;38(2):125-134.
19. Akhondzadeh S, Nejatisafa AA, Amini H, et al. Adjunctive estrogen treatment in women with chronic schizophrenia: a double-blind, randomized, and placebo-controlled trial. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(6):1007-1012.
20. Kulkarni J, de Castella A, Fitzgerald PB, et al. Estrogen in severe mental illness: a potential new treatment approach. Arch Gen Psychiatry. 2008;65(8):955-960.
21. Ellis AJ, Hendrick VM, Williams R, Komm BS. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf. 2015;14(6):921-934.
22. Morello KC, Wurz GT, DeGregorio MW. Pharmacokinetics of selective estrogen receptor modulators. Clin pharmacokinet. 2003;42(4):361-372.
23. Lewiecki EM, Miller PD, Harris ST, et al. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014;17(4):490-495.
24. Raloxifene Hydrochloride. Micromedex 2.0. Truven Health Analytics. www.micromedexsolutions.com. Accessed July 24, 2016.
25. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
26. Huerta-Ramos E, Iniesta R, Ochoa S, et al. Effects of raloxifene on cognition in postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. Eur Neuropsychopharmacol. 2014;24(2):223-231.
27. Kianimehr G, Fatehi F, Hashempoor S, et al. Raloxifene adjunctive therapy for postmenopausal women suffering from chronic schizophrenia: a randomized double-blind and placebo controlled trial. Daru. 2014;22:55.
28. Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(11):3975-4011.
29. Ellis AJ, Hendrick VM, Williams R, et al. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf. 2015;14(6):921-934.
30. Adomaityte J, Farooq M, Qayyum R. Effect of raloxifene therapy on venous thromboembolism in postmenopausal women. A meta-analysis. Thromb Haemost. 2008;99(2):338-342.
31. Lewiecki EM, Miller PD, Harris ST, et al. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014;17(4):490-495.
32. Grady D, Cauley JA, Stock JL, et al. Effect of raloxifene on all-cause mortality. Am J Med. 2010;123(5):469.e1-461.e7.
CASE Psychotic and reclusive
Ms. A, age 51, has schizophrenia and has been doing well living at a supervised residential facility. She was stable on haloperidol, 10 mg twice a day, for years but recently became agitated, threatening her roommate and yelling during the night. Ms. A begins to refuse to take her haloperidol. She also refuses to attend several outpatient appointments. As a result, Ms. A is admitted to the psychiatric unit on an involuntary basis.
In the hospital, Ms. A rarely comes out of her room. When she does come out, she usually sits in a chair, talking to herself and occasionally yelling or crying in apparent distress. Ms. A refuses to engage with her treatment team and lies mute in her bed when they attempt to interview her. Her records indicate that previous medication trials have included
Over the next week, Ms. A begins to interact more appropriately with nursing sta
[polldaddy:9945425]
The authors’ observations
As a class, antipsychotics lead to symptom reduction in approximately 70% of patients.1 However, the degree of response can vary markedly between individuals; although some patients may experience almost complete resolution of symptoms, others are still markedly impaired, as in Ms. A’s case.
A substantial amount of literature suggests that although the practice is common, use of >1 antipsychotic does not significantly increase efficacy but increases risk of adverse effects, such as type 2 diabetes mellitus, metabolic syndrome, cognitive impairment, and extrapyramidal symptoms.2-4 One exception is augmentation of clozapine with a second antipsychotic, which in certain cases appears to offer greater efficacy than clozapine alone.1 Practice guidelines and evidence generally do not support the use of multiple antipsychotics, but 20% of patients take >1 antipsychotic.5,6 Although antipsychotic polypharmacy may be appropriate for some patients, current literature suggests it is being done more often than recommended.
Clozapine is considered the most efficacious option for treatment-resistant schizophrenia.7 Because of Ms. A’s history of recurrent hospitalizations, her extensive list of trialed medications, and her ongoing symptoms despite a sufficient trial of haloperidol, the treatment team gives serious consideration to switching Ms. A to clozapine. However, Ms. A is not able to tolerate blood draws without significant support from nursing staff, and it is likely she would be unable to tolerate the frequent blood monitoring required of patients receiving clozapine.
Because many of Ms. A’s symptoms were negative or depressive, including hypersomnia, psychomotor retardation, sadness with frequent crying spells, and reduced interest in activities, adding an antidepressant to Ms. A’s medication regimen was considered. A recent systematic review and meta-analysis showed that adding an antidepressant to an antipsychotic in patients with schizophrenia had small but beneficial effects on depressive and negative symptoms and a low risk of adverse effects.8 However, Ms. A declined this option.
TREATMENT Adding long-acting haloperidol
Ms. A had previously achieved therapeutic blood levels9 with oral haloperidol. Data suggest that compared with the oral form, long-acting injectable antipsychotics can both improve compliance and decrease rehospitalization rates.10-12 Because Ms. A previously had done well with haloperidol decanoate, 200 mg every 2 weeks, achieving a blood level of 16.2 ng/mL, and because she had a partial response to oral haloperidol, we add haloperidol decanoate, 100 mg every 2 weeks, to her regimen, with the intention of transitioning her to all-depot dosing. In addition, the treatment team tries to engage Ms. A in a discussion of potential psychological contributions to her current presentation. They note that Ms. A has her basic needs met on the unit and reports feeling safe there; thus, a fear of discharge may be contributing to her lack of engagement with the team. However, because of her limited communication, it is challenging to investigate this hypothesis or explore other possible psychological issues.
Despite increasing the dosing of haloperidol, Ms. A shows minimal improvement. She continues to stonewall her treatment team, and is unwilling or unable to engage in meaningful conversation. A review of her chart suggests that this hospital course is different from previous ones in which her average stay was a few weeks, and she generally was able to converse with the treatment team, participate in discussions about her care, and make decisions about her desire for discharge.
The team considers if additional factors could be impacting Ms. A’s current presentation. They raise the possibility that she could be going through menopause, and hormonal fluctuations may be contributing to her symptoms. Despite being on the unit for nearly 2 months, Ms. A has not required the use of sanitary products. She also reports to nursing staff that at times she feels flushed and sweaty, but she is afebrile and does not have other signs or symptoms of infection.
[polldaddy:9945428]
The authors’ observations
Evidence suggests that estrogen levels can influence the development and severity of symptoms of schizophrenia (Table 113,14). Rates of schizophrenia are lower in women, and women typically have a later onset of illness with less severe symptoms.13 Women also have a second peak incidence of schizophrenia between ages 45 and 50, corresponding with the hormonal changes associated with menopause and the associated drop in estrogen.14 Symptoms also fluctuate with hormonal cycles—women experience worsening symptoms during the premenstrual phase of the menstrual cycle, when estrogen levels are low, and an improvement of symptoms during high-estrogen phases of the cycle.14 Overall, low levels of estrogen also have been observed in women with schizophrenia relative to controls, although this may be partially attributable to treatment with antipsychotics.14
Estrogen affects various regions of the brain implicated in schizophrenia and likely imparts its behavioral effects through several different mechanisms. Estrogen can act on cells to directly impact intracellular signaling and to alter gene expression.15 Although most often thought of as being related to reproductive functions, estrogen receptors can be found in many cortical and subcortical regions of the brain, such as the hippocampus, substantia nigra, and prefrontal cortex. Estrogen receptor expression levels in certain brain regions have been found to be altered in individuals with schizophrenia.15 Estrogen also enhances neurogenesis and neuroplasticity, playing a role in learning and memory.16 Particularly relevant, estrogen has been shown to directly impact both the dopaminergic and serotonergic systems.15,17 In animal models, estrogen has been shown to decrease the behavioral effects induced by dopamine agonists and decrease symptoms of schizophrenia.18 The underlying molecular mechanisms by which estrogen has these effects are uncertain.
Given estrogen’s potentially protective effects, clinical trials have explored the role of estrogen as an adjuvant to antipsychotics for treating schizophrenia. Studies have shown that estrogen can improve psychotic symptoms in patients with schizophrenia.19,20 However, because estrogen administration can increase the risk of breast and uterine cancer, researchers are instead investigating selective estrogen receptor modulators (SERMs).14 These medications have mixed agonist and antagonist effects, with different effects on different tissues. Raloxifene is a SERM that acts as an estrogen agonist in some tissues, but an antagonist in uterine and breast tissue, which may minimize potential deleterious adverse effects (Table 221-24). Repeated randomized controlled trials have found promising results for use of raloxifene as an adjunctive treatment in peri- and postmenopausal women with schizophrenia, including those refractory to antipsychotic treatment.13,25-27
TREATMENT Address symptoms
The treatment team takes steps to address Ms. A’s perimenopausal symptoms. For mild to moderate hot flashes, primary interventions are nonpharmacologic.28 Because Ms. A primarily reports her hot flashes at night, she is given lightweight pajamas and moved to the coolest room on the unit. Both bring some relief, and her hot flashes appear to be less distressing. The treatment team decides to consult Endocrinology to further investigate the feasibility of starting raloxifene (Table 3) because of their experience using this medication to manage osteoporosis.
[polldaddy:9945429]
The authors’ observations
Raloxifene is FDA-approved for treating osteoporosis and preventing invasive breast cancer.29 Because it is an estrogen antagonist in both breast and uterine tissues, raloxifene does not increase the risk of uterine or breast cancer. Large studies have shown rates of cardiovascular events are similar for raloxifene and placebo, and some studies have found that raloxifene treatment is associated with improvement in cardiovascular risk factors, including lower blood pressure, lower low-density lipoprotein cholesterol, and increased high-density lipoprotein cholesterol.29 Raloxifene does, however, increase risk of venous thromboembolism, including deep vein thrombosis and pulmonary embolism, and fatal stroke.29,30 Overall, the risk remains relatively low, with an absolute risk increase of fatal stroke of 0.7 per 1,000 woman-years (number needed to harm [NNH]: 250) and an absolute risk increase of venous thromboembolic events of 1.88 per 1,000 women-years (NNH: 158).31 However, raloxifene may not be appropriate for patients with independent risk factors for these events. Despite this, a large meta-analysis found a 10% decrease in mortality for patients taking raloxifene compared with those receiving placebo.32 Raloxifene also can cause hot flashes, muscle cramps, and flu-like symptoms.29
Diagnosis of menopause and perimenopause is largely clinical, with hormone testing generally recommended for women age <45 in whom the diagnosis may be unclear.28 Thus, Ms. A’s vasomotor symptoms and absence of a menstrual cycle for at least 2 months were diagnostic of perimenopause; a 12-month cessation in menstrual cycles is required for a diagnosis of menopause.28
OUTCOME Improvement with raloxifene
Because Ms. A is at relatively low risk for a thromboembolism or stroke, the benefit of raloxifene is thought to outweigh the risk, and she is started on raloxifene, 60 mg/d. Over the next 2 weeks, Ms. A becomes increasingly interactive, and is seen sitting at a table talking with other patients on multiple occasions. She spends time looking at fashion magazines, and engages in conversation about fashion with staff and other patients. She participates in group therapy for the first time during this hospital stay and begins to talk about discharge. She occasionally smiles and waves at her treatment team and participates more in the daily interview, although these interactions remain limited and on her terms. She maintains this improvement and is transferred to a psychiatric facility in her home county for ongoing care and discharge planning.
CASE Psychotic and reclusive
Ms. A, age 51, has schizophrenia and has been doing well living at a supervised residential facility. She was stable on haloperidol, 10 mg twice a day, for years but recently became agitated, threatening her roommate and yelling during the night. Ms. A begins to refuse to take her haloperidol. She also refuses to attend several outpatient appointments. As a result, Ms. A is admitted to the psychiatric unit on an involuntary basis.
In the hospital, Ms. A rarely comes out of her room. When she does come out, she usually sits in a chair, talking to herself and occasionally yelling or crying in apparent distress. Ms. A refuses to engage with her treatment team and lies mute in her bed when they attempt to interview her. Her records indicate that previous medication trials have included
Over the next week, Ms. A begins to interact more appropriately with nursing sta
[polldaddy:9945425]
The authors’ observations
As a class, antipsychotics lead to symptom reduction in approximately 70% of patients.1 However, the degree of response can vary markedly between individuals; although some patients may experience almost complete resolution of symptoms, others are still markedly impaired, as in Ms. A’s case.
A substantial amount of literature suggests that although the practice is common, use of >1 antipsychotic does not significantly increase efficacy but increases risk of adverse effects, such as type 2 diabetes mellitus, metabolic syndrome, cognitive impairment, and extrapyramidal symptoms.2-4 One exception is augmentation of clozapine with a second antipsychotic, which in certain cases appears to offer greater efficacy than clozapine alone.1 Practice guidelines and evidence generally do not support the use of multiple antipsychotics, but 20% of patients take >1 antipsychotic.5,6 Although antipsychotic polypharmacy may be appropriate for some patients, current literature suggests it is being done more often than recommended.
Clozapine is considered the most efficacious option for treatment-resistant schizophrenia.7 Because of Ms. A’s history of recurrent hospitalizations, her extensive list of trialed medications, and her ongoing symptoms despite a sufficient trial of haloperidol, the treatment team gives serious consideration to switching Ms. A to clozapine. However, Ms. A is not able to tolerate blood draws without significant support from nursing staff, and it is likely she would be unable to tolerate the frequent blood monitoring required of patients receiving clozapine.
Because many of Ms. A’s symptoms were negative or depressive, including hypersomnia, psychomotor retardation, sadness with frequent crying spells, and reduced interest in activities, adding an antidepressant to Ms. A’s medication regimen was considered. A recent systematic review and meta-analysis showed that adding an antidepressant to an antipsychotic in patients with schizophrenia had small but beneficial effects on depressive and negative symptoms and a low risk of adverse effects.8 However, Ms. A declined this option.
TREATMENT Adding long-acting haloperidol
Ms. A had previously achieved therapeutic blood levels9 with oral haloperidol. Data suggest that compared with the oral form, long-acting injectable antipsychotics can both improve compliance and decrease rehospitalization rates.10-12 Because Ms. A previously had done well with haloperidol decanoate, 200 mg every 2 weeks, achieving a blood level of 16.2 ng/mL, and because she had a partial response to oral haloperidol, we add haloperidol decanoate, 100 mg every 2 weeks, to her regimen, with the intention of transitioning her to all-depot dosing. In addition, the treatment team tries to engage Ms. A in a discussion of potential psychological contributions to her current presentation. They note that Ms. A has her basic needs met on the unit and reports feeling safe there; thus, a fear of discharge may be contributing to her lack of engagement with the team. However, because of her limited communication, it is challenging to investigate this hypothesis or explore other possible psychological issues.
Despite increasing the dosing of haloperidol, Ms. A shows minimal improvement. She continues to stonewall her treatment team, and is unwilling or unable to engage in meaningful conversation. A review of her chart suggests that this hospital course is different from previous ones in which her average stay was a few weeks, and she generally was able to converse with the treatment team, participate in discussions about her care, and make decisions about her desire for discharge.
The team considers if additional factors could be impacting Ms. A’s current presentation. They raise the possibility that she could be going through menopause, and hormonal fluctuations may be contributing to her symptoms. Despite being on the unit for nearly 2 months, Ms. A has not required the use of sanitary products. She also reports to nursing staff that at times she feels flushed and sweaty, but she is afebrile and does not have other signs or symptoms of infection.
[polldaddy:9945428]
The authors’ observations
Evidence suggests that estrogen levels can influence the development and severity of symptoms of schizophrenia (Table 113,14). Rates of schizophrenia are lower in women, and women typically have a later onset of illness with less severe symptoms.13 Women also have a second peak incidence of schizophrenia between ages 45 and 50, corresponding with the hormonal changes associated with menopause and the associated drop in estrogen.14 Symptoms also fluctuate with hormonal cycles—women experience worsening symptoms during the premenstrual phase of the menstrual cycle, when estrogen levels are low, and an improvement of symptoms during high-estrogen phases of the cycle.14 Overall, low levels of estrogen also have been observed in women with schizophrenia relative to controls, although this may be partially attributable to treatment with antipsychotics.14
Estrogen affects various regions of the brain implicated in schizophrenia and likely imparts its behavioral effects through several different mechanisms. Estrogen can act on cells to directly impact intracellular signaling and to alter gene expression.15 Although most often thought of as being related to reproductive functions, estrogen receptors can be found in many cortical and subcortical regions of the brain, such as the hippocampus, substantia nigra, and prefrontal cortex. Estrogen receptor expression levels in certain brain regions have been found to be altered in individuals with schizophrenia.15 Estrogen also enhances neurogenesis and neuroplasticity, playing a role in learning and memory.16 Particularly relevant, estrogen has been shown to directly impact both the dopaminergic and serotonergic systems.15,17 In animal models, estrogen has been shown to decrease the behavioral effects induced by dopamine agonists and decrease symptoms of schizophrenia.18 The underlying molecular mechanisms by which estrogen has these effects are uncertain.
Given estrogen’s potentially protective effects, clinical trials have explored the role of estrogen as an adjuvant to antipsychotics for treating schizophrenia. Studies have shown that estrogen can improve psychotic symptoms in patients with schizophrenia.19,20 However, because estrogen administration can increase the risk of breast and uterine cancer, researchers are instead investigating selective estrogen receptor modulators (SERMs).14 These medications have mixed agonist and antagonist effects, with different effects on different tissues. Raloxifene is a SERM that acts as an estrogen agonist in some tissues, but an antagonist in uterine and breast tissue, which may minimize potential deleterious adverse effects (Table 221-24). Repeated randomized controlled trials have found promising results for use of raloxifene as an adjunctive treatment in peri- and postmenopausal women with schizophrenia, including those refractory to antipsychotic treatment.13,25-27
TREATMENT Address symptoms
The treatment team takes steps to address Ms. A’s perimenopausal symptoms. For mild to moderate hot flashes, primary interventions are nonpharmacologic.28 Because Ms. A primarily reports her hot flashes at night, she is given lightweight pajamas and moved to the coolest room on the unit. Both bring some relief, and her hot flashes appear to be less distressing. The treatment team decides to consult Endocrinology to further investigate the feasibility of starting raloxifene (Table 3) because of their experience using this medication to manage osteoporosis.
[polldaddy:9945429]
The authors’ observations
Raloxifene is FDA-approved for treating osteoporosis and preventing invasive breast cancer.29 Because it is an estrogen antagonist in both breast and uterine tissues, raloxifene does not increase the risk of uterine or breast cancer. Large studies have shown rates of cardiovascular events are similar for raloxifene and placebo, and some studies have found that raloxifene treatment is associated with improvement in cardiovascular risk factors, including lower blood pressure, lower low-density lipoprotein cholesterol, and increased high-density lipoprotein cholesterol.29 Raloxifene does, however, increase risk of venous thromboembolism, including deep vein thrombosis and pulmonary embolism, and fatal stroke.29,30 Overall, the risk remains relatively low, with an absolute risk increase of fatal stroke of 0.7 per 1,000 woman-years (number needed to harm [NNH]: 250) and an absolute risk increase of venous thromboembolic events of 1.88 per 1,000 women-years (NNH: 158).31 However, raloxifene may not be appropriate for patients with independent risk factors for these events. Despite this, a large meta-analysis found a 10% decrease in mortality for patients taking raloxifene compared with those receiving placebo.32 Raloxifene also can cause hot flashes, muscle cramps, and flu-like symptoms.29
Diagnosis of menopause and perimenopause is largely clinical, with hormone testing generally recommended for women age <45 in whom the diagnosis may be unclear.28 Thus, Ms. A’s vasomotor symptoms and absence of a menstrual cycle for at least 2 months were diagnostic of perimenopause; a 12-month cessation in menstrual cycles is required for a diagnosis of menopause.28
OUTCOME Improvement with raloxifene
Because Ms. A is at relatively low risk for a thromboembolism or stroke, the benefit of raloxifene is thought to outweigh the risk, and she is started on raloxifene, 60 mg/d. Over the next 2 weeks, Ms. A becomes increasingly interactive, and is seen sitting at a table talking with other patients on multiple occasions. She spends time looking at fashion magazines, and engages in conversation about fashion with staff and other patients. She participates in group therapy for the first time during this hospital stay and begins to talk about discharge. She occasionally smiles and waves at her treatment team and participates more in the daily interview, although these interactions remain limited and on her terms. She maintains this improvement and is transferred to a psychiatric facility in her home county for ongoing care and discharge planning.
2. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
3. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
4. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
5. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
6. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
7. McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
8. Helfer B, Samara MT, Huhn M, et al. Efficacy and safety of antidepressants added to antipsychotics for schizophrenia: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(9);876-886.
9. Ulrich S, Neuhof S, Braun V, et al. Therapeutic window of serum haloperidol concentration in acute schizophrenia and schizoaffective disorder. Pharmacopsychiatry. 1998;31(5):163-169.
10. Lafeuille MH, Dean J, Carter V, et al. Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia. Curr Med Res Opin. 2014;30(8):1643-1655.
11. MacEwan JP, Kamat SA, Duffy RA, et al. Hospital readmission rates among patients with schizophrenia treated with long-acting injectables or oral antipsychotics. Psychiatr Serv. 2016;67(11):1183-1188.
12. Marcus SC, Zummo J, Pettit AR, et al. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768.
13. Usall J, Huerta-Ramos E, Iniesta R, et al; RALOPSYCAT Group. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
14. Seeman MV. Treating schizophrenia at the time of menopause. Maturitas. 2012;72(2):117-120.
15. Gogos A, Sbisa AM, Sun J, et al. A role for estrogen in schizophrenia: clinical and preclinical findings. Int J Endocrinol. 2015;2015:615356. doi: 10.1155/2015/615356.
16. Khan MM. Neurocognitive, neuroprotective, and cardiometabolic effects of raloxifene: potential for improving therapeutic outcomes in schizophrenia. CNS Drugs. 2016;30(7):589-601.
17. Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci. 2015;9:37.
18. Häfner H, Behrens S, De Vry J, et al. An animal model for the effects of estradiol on dopamine-mediated behavior: implications for sex differences in schizophrenia. Psychiatry Res. 1991;38(2):125-134.
19. Akhondzadeh S, Nejatisafa AA, Amini H, et al. Adjunctive estrogen treatment in women with chronic schizophrenia: a double-blind, randomized, and placebo-controlled trial. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(6):1007-1012.
20. Kulkarni J, de Castella A, Fitzgerald PB, et al. Estrogen in severe mental illness: a potential new treatment approach. Arch Gen Psychiatry. 2008;65(8):955-960.
21. Ellis AJ, Hendrick VM, Williams R, Komm BS. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf. 2015;14(6):921-934.
22. Morello KC, Wurz GT, DeGregorio MW. Pharmacokinetics of selective estrogen receptor modulators. Clin pharmacokinet. 2003;42(4):361-372.
23. Lewiecki EM, Miller PD, Harris ST, et al. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014;17(4):490-495.
24. Raloxifene Hydrochloride. Micromedex 2.0. Truven Health Analytics. www.micromedexsolutions.com. Accessed July 24, 2016.
25. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
26. Huerta-Ramos E, Iniesta R, Ochoa S, et al. Effects of raloxifene on cognition in postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. Eur Neuropsychopharmacol. 2014;24(2):223-231.
27. Kianimehr G, Fatehi F, Hashempoor S, et al. Raloxifene adjunctive therapy for postmenopausal women suffering from chronic schizophrenia: a randomized double-blind and placebo controlled trial. Daru. 2014;22:55.
28. Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(11):3975-4011.
29. Ellis AJ, Hendrick VM, Williams R, et al. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf. 2015;14(6):921-934.
30. Adomaityte J, Farooq M, Qayyum R. Effect of raloxifene therapy on venous thromboembolism in postmenopausal women. A meta-analysis. Thromb Haemost. 2008;99(2):338-342.
31. Lewiecki EM, Miller PD, Harris ST, et al. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014;17(4):490-495.
32. Grady D, Cauley JA, Stock JL, et al. Effect of raloxifene on all-cause mortality. Am J Med. 2010;123(5):469.e1-461.e7.
2. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
3. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
4. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
5. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
6. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
7. McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
8. Helfer B, Samara MT, Huhn M, et al. Efficacy and safety of antidepressants added to antipsychotics for schizophrenia: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(9);876-886.
9. Ulrich S, Neuhof S, Braun V, et al. Therapeutic window of serum haloperidol concentration in acute schizophrenia and schizoaffective disorder. Pharmacopsychiatry. 1998;31(5):163-169.
10. Lafeuille MH, Dean J, Carter V, et al. Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia. Curr Med Res Opin. 2014;30(8):1643-1655.
11. MacEwan JP, Kamat SA, Duffy RA, et al. Hospital readmission rates among patients with schizophrenia treated with long-acting injectables or oral antipsychotics. Psychiatr Serv. 2016;67(11):1183-1188.
12. Marcus SC, Zummo J, Pettit AR, et al. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768.
13. Usall J, Huerta-Ramos E, Iniesta R, et al; RALOPSYCAT Group. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
14. Seeman MV. Treating schizophrenia at the time of menopause. Maturitas. 2012;72(2):117-120.
15. Gogos A, Sbisa AM, Sun J, et al. A role for estrogen in schizophrenia: clinical and preclinical findings. Int J Endocrinol. 2015;2015:615356. doi: 10.1155/2015/615356.
16. Khan MM. Neurocognitive, neuroprotective, and cardiometabolic effects of raloxifene: potential for improving therapeutic outcomes in schizophrenia. CNS Drugs. 2016;30(7):589-601.
17. Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci. 2015;9:37.
18. Häfner H, Behrens S, De Vry J, et al. An animal model for the effects of estradiol on dopamine-mediated behavior: implications for sex differences in schizophrenia. Psychiatry Res. 1991;38(2):125-134.
19. Akhondzadeh S, Nejatisafa AA, Amini H, et al. Adjunctive estrogen treatment in women with chronic schizophrenia: a double-blind, randomized, and placebo-controlled trial. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(6):1007-1012.
20. Kulkarni J, de Castella A, Fitzgerald PB, et al. Estrogen in severe mental illness: a potential new treatment approach. Arch Gen Psychiatry. 2008;65(8):955-960.
21. Ellis AJ, Hendrick VM, Williams R, Komm BS. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf. 2015;14(6):921-934.
22. Morello KC, Wurz GT, DeGregorio MW. Pharmacokinetics of selective estrogen receptor modulators. Clin pharmacokinet. 2003;42(4):361-372.
23. Lewiecki EM, Miller PD, Harris ST, et al. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014;17(4):490-495.
24. Raloxifene Hydrochloride. Micromedex 2.0. Truven Health Analytics. www.micromedexsolutions.com. Accessed July 24, 2016.
25. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
26. Huerta-Ramos E, Iniesta R, Ochoa S, et al. Effects of raloxifene on cognition in postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. Eur Neuropsychopharmacol. 2014;24(2):223-231.
27. Kianimehr G, Fatehi F, Hashempoor S, et al. Raloxifene adjunctive therapy for postmenopausal women suffering from chronic schizophrenia: a randomized double-blind and placebo controlled trial. Daru. 2014;22:55.
28. Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(11):3975-4011.
29. Ellis AJ, Hendrick VM, Williams R, et al. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf. 2015;14(6):921-934.
30. Adomaityte J, Farooq M, Qayyum R. Effect of raloxifene therapy on venous thromboembolism in postmenopausal women. A meta-analysis. Thromb Haemost. 2008;99(2):338-342.
31. Lewiecki EM, Miller PD, Harris ST, et al. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014;17(4):490-495.
32. Grady D, Cauley JA, Stock JL, et al. Effect of raloxifene on all-cause mortality. Am J Med. 2010;123(5):469.e1-461.e7.
Treatment of Biliary Tract Cancers
Introduction
Biliary tract carcinoma (BTC) is th
Epidemiology
In the United States, BTC is rare and accounts for approximately 4% of all gastrointestinal malignancies, with an estimated 6000 to 7000 cases of carcinoma of the gallbladder and 3000 to 4000 cases of carcinoma of the bile duct diagnosed annually.4 Among women, there is a 26-fold variation in BTC mortality worldwide, ranging from 0.8 deaths per 100,000 in South Africa to 21.2 per 100,000 in Chile.1,5 Interestingly, for American Indians in New Mexico, gallbladder cancer mortality rates (8.9 per 100,000) surpass those for breast and pancreatic cancers.6 The incidence of anatomical cholangiocarcinoma subtypes also varies regionally, reflecting disparities in genetic and environmental predisposing factors.2,7 In a large, single-center study in the United States, intrahepatic cholangiocarcinoma accounted for less than 10% of cases, perihilar accounted for 50%, and distal accounted for the remaining 40%.8 Importantly, intrahepatic cholangiocarcinoma is the second most common primary malignancy of the liver, and its incidence seems to be rising in many western countries. In the United States, there has been an estimated 128% rise over the past 40 years.4,9
BTC is associated with high mortality rates.10 Median overall survival (OS) for cholangiocarcinoma is 20 to 28 months and 5-year survival is around 25%.10 Most cholangiocarcinomas are diagnosed at advanced stages with unresectable tumors.10 Furthermore, outcomes following resection with curative intent are poor—median disease-free survival (DFS) of 12 to 36 months has been reported.11,12 Patients with intrahepatic disease have a better prognosis when compared with patients who have extrahepatic tumors.12 Gallbladder cancer, likewise, carries a poor overall prognosis; median OS is 32 months and 5-year survival is as low as 13%.6
Risk factors for BTC include intrinsic and extrinsic elements.6 Incidence of BTC increases with age, and diagnosis typically occurs in the sixth to eighth decade of life.5,6,13 In contrast to gallbladder cancer, the incidence of cholangiocarcinoma is slightly higher in men.9 Obesity, diabetes, and consumption of sweetened drinks also increase the risk for BTC.14–16 Cholelithiasis is the most prevalent risk factor for gallbladder cancer, and the risk is greater for larger stones.5 Around 1 in 5 patients with porcelain gallbladder will develop gallbladder carcinoma.17 Primary sclerosing cholangitis (PSC), chronic calculi of the bile duct, choledochal cysts, cirrhosis, hepatitis C, and liver fluke infections are well established risk factors for cholangiocarcinoma.7,12,18 PSC is one of the best described entities among these predisposing conditions. Lifetime prevalence of cholangiocarcinoma among patients with PSC ranges from 5% to 10%.18,19 These patients also present at a younger age; in one series, the median age at diagnosis for BTC arising from PSC was 39 years.18 It is important to recognize, however, that in most patients diagnosed with cholangiocarcinoma, no predisposing factors are identified.8
Diagnosis
Clinical Presentation
Clinical presentation of BTC depends upon anatomic location.20 Patients with early invasive gallbladder cancer are most often asymptomatic.21 When symptoms occur, they may be nonspecific and mimic cholelithiasis.21 The most common clinical presentations include jaundice, weight loss, and abdominal pain.21 Prior to widespread availability of imaging studies, the preoperative diagnosis rate for gallbladder cancer was as low as 10%.22 However, the accuracy of computed tomography (CT) has changed this scenario, with sensitivity ranging from 73% to 87% and specificity from 88% to 100%.21 As a result of its silent clinical character, cholangiocarcinoma is frequently difficult to diagnose.23 Perihilar and distal cholangiocarcinoma characteristically present with signs of biliary obstruction, and imaging and laboratory data can corroborate the presence of cholestasis.24 On examination, patients with extrahepatic cholangiocarcinoma may present with jaundice, hepatomegaly, and a palpable right upper quadrant mass.25 A palpable gallbladder (Courvoisier sign) can also be present.25 Intrahepatic cholangiocarcinoma presents differently, and patients are less likely to be jaundiced.23 Typical clinical features are nonspecific and include dull right upper quadrant pain, weight loss, and an elevated alkaline phosphatase level.23 Alternatively, asymptomatic patients can present with incidentally detected lesions, when imaging is obtained as part of the workup for other causes or during screening for hepatocellular carcinoma in patients with viral hepatitis or cirrhosis.23,26 Uncommonly, BTC patients present because of signs or symptoms related to metastatic disease or evidence of metastatic disease on imaging.
Pathology and Grading
The majority of BTCs are adenocarcinomas, corresponding to 90% of cholangiocarcinomas and 99% of gallbladder cancers.27,28 They are graded as well, moderately, or poorly differentiated.2 Adenosquamous and squamous cell carcinoma are responsible for most of the remaining cases.2,29 Cholangiocarcinomas are divided into 3 types, defined by the Liver Cancer Study Group of Japan: (1) mass-forming, (2) periductal-infiltrating, and (3) intraductal-growing.30,31 Mass-forming intrahepatic cholangiocarcinomas are characterized morphologically by a homogeneous gray-yellow mass with frequent satellite nodules and irregular but well-defined margins.17,30 Central necrosis and fibrosis are also common.30 In the periductal-infiltrating type, tumor typically grows along the bile duct wall without mass formation, resulting in concentric mural thickening and proximal biliary dilation.30 Intraductal-growing papillary cholangiocarcinoma is characterized by the presence of intraluminal papillary or tubular polypoid tumors of the intra- or extrahepatic bile ducts, with partial obstruction and proximal biliary dilation.30
Cholangiocarcinoma
Case Presentation
A previously healthy 59-year-old man presents to his primary care physician with a 3-month history of dull right upper quadrant pain associated with weight loss. The patient is markedly cachectic and abdominal examination reveals upper quadrant tenderness. Laboratory exams are significant for elevated alkaline phosphatase (500 U/L; reference range 45–115 U/L), cancer antigen 19-9 (CA 19-9, 73 U/mL; reference range ≤ 37 U/mL), and carcinoembryonic antigen (CEA , 20 ng/mL; reference range for nonsmokers ≤ 3.0 ng/mL). Aspartate aminotransferase, alanine aminotransferase, total bilirubin, and coagulation studies are within normal range. Ultrasound demonstrates a homogeneous mass with irregular borders in the right lobe of the liver. Triphasic contrast-enhanced CT scan demonstrates a tumor with ragged rim enhancement at the periphery, and portal venous phase shows gradual centripetal enhancement of the tumor with capsular retraction. No abdominal lymph nodes or extrahepatic tumors are noted (Figure 1, Image A).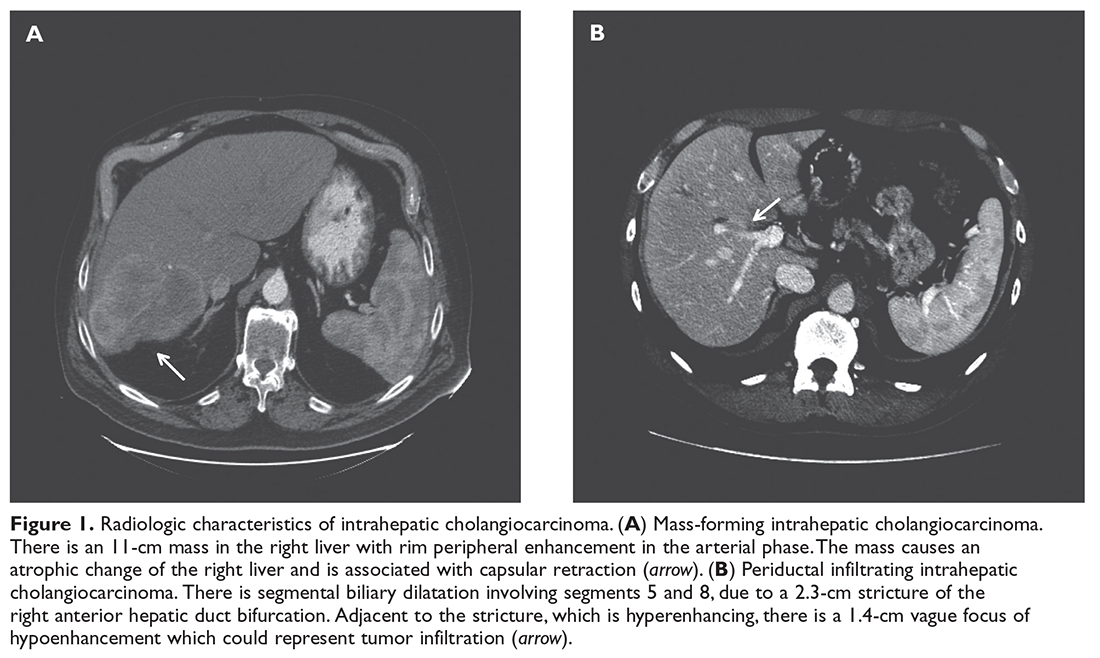
- What are the next diagnostic steps?
The most critical differential diagnosis of solid liver mass in patients without cirrhosis is cholangiocarcinoma and metastases from another primary site.32 Alternatively, when an intrahepatic lesion is noted on an imaging study in the setting of cirrhosis, the next diagnostic step is differentiation between cholangiocarcinoma and hepatocellular carcinoma (HCC).32 Triphasic contrast-enhanced CT and dynamic magnetic resonance imaging (MRI) are key diagnostic procedures.32,33 In the appropriate setting, classical imaging features in the arterial phase with washout in portal venous or delayed phase can be diagnostic of HCC and may obviate the need for a biopsy (Figure 2). 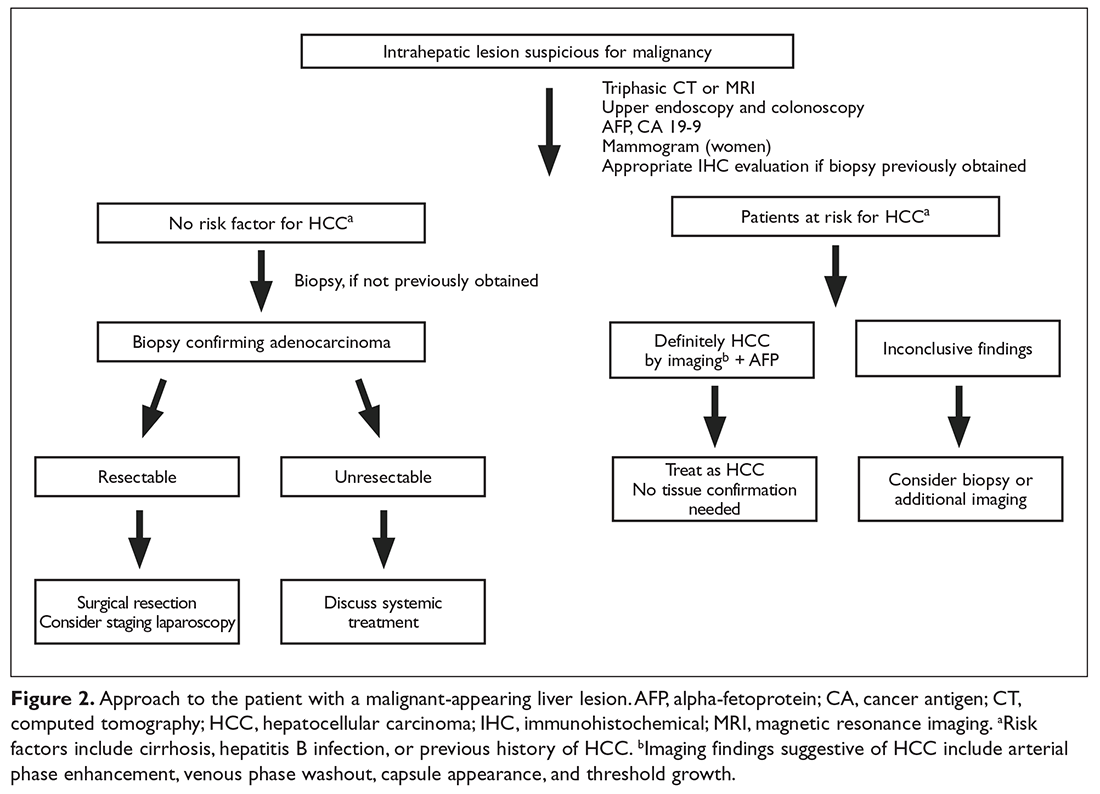
Typical radiographic features of cholangiocarcinoma include a hypodense hepatic lesion that can be either well-defined or infiltrative and is frequently associated with biliary dilatation (Figure 1, Image A).33 The dense fibrotic nature of the tumor may cause capsular retraction, which is seen in up to 20% of cases.17 This finding is highly suggestive of cholangiocarcinoma and is rarely present in HCC.33 Following contrast administration, there is peripheral (rim) enhancement throughout both arterial and venous phases.32–34 However, these classic features were present in only 70% of cases in one study.35 Although intrahepatic cholangiocarcinomas are most commonly hypovascular, small mass-forming intrahepatic cholangiocarcinomas can often be arterially hyperenhancing and mimic HCC.33 Tumor enhancement on delayed CT imaging has been correlated with survival. Asayama et al demonstrated that tumors that exhibited delayed enhancement on CT in more than two-thirds of their volume were associated with a worse prognosis.36
Patients without cirrhosis who present with a localized lesion of the liver should undergo extensive evaluation for a primary cancer site.37 CT of the chest, abdomen, and pelvis with contrast should be obtained.37 Additionally, mammogram and endoscopic evaluation with esophagogastroduodenoscopy (EGD) and colonoscopy should be included in the work-up.37
Preoperative tumor markers are also included in the work-up. All patients with a solid liver lesion should have serum alpha-fetoprotein (AFP) levels checked. AFP is a serum glycoprotein recognized as a marker for HCC and is reported to detect preclinical HCC.38 However, serum concentrations are normal in up to 40% of small HCCs.38 Although no specific marker for cholangiocarcinoma has yet been identified, the presence of certain tumor markers in the serum of patients may be of diagnostic value, especially in patients with PSC. CA 19-9 and CEA are the best studied. Elevated levels of CA 19-9 prior to treatment are associated with a poorer prognosis, and CA 19-9 concentrations greater than 1000 U/mL are consistent with advanced disease.39,40 One large series evaluated the diagnostic value of serum CEA levels in 333 patients with PSC, 13% of whom were diagnosed with cholangiocarcinoma.34 A serum CEA level greater than 5.2 ng/mL had a sensitivity of 68.0% and specificity of 81.5%.38
If a biopsy is obtained, appropriate immunohistochemistry (IHC) can facilitate the diagnosis. BTC is strongly positive for CK-7 and CK-19.41 CK-7 positivity is not specific and is also common among metastatic cancers of the lung and breast; therefore, in some cases cholangiocarcinoma may be a diagnosis of exclusion. Immunostaining for monoclonal CEA is diffusely positive in up to 75% of cases.41 An IHC panel consisting of Hep Par-1, arginase-1, monoclonal CEA, CK-7, CK-20, TTF-1, MOC-31, and CDX-2 has been proposed to optimize the differential diagnosis of HCC, metastatic adenocarcinoma, and cholangiocarcinoma.41
Case Continued
CT of the chest, abdomen, and pelvis reveals no concerns for metastasis and no evidence of primary cancer elsewhere. EGD and colonoscopy are clear. AFP levels are within normal limits (2 ng/mL). Biopsy is performed and demonstrates adenocarcinoma. IHC studies demonstrate cells positive for monoclonal CEA, CK-7, CK-19, and MOC-31, and negative for Napsin A, TTF-1, and CK-20.
- How is cholangiocarcinoma staged and classified?
The purpose of the staging system is to provide information on prognosis and guidance for therapy. Prognostic factors and the therapeutic approaches for BTC differ depending upon their location in the biliary tree. Accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated (Table 1 and Table 2).23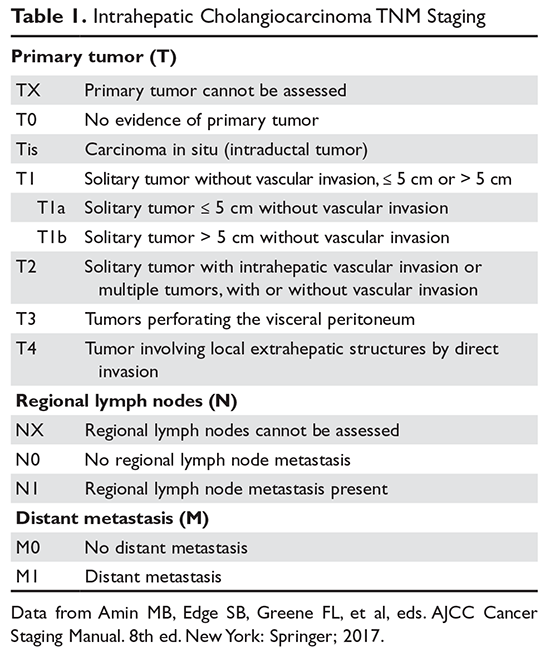
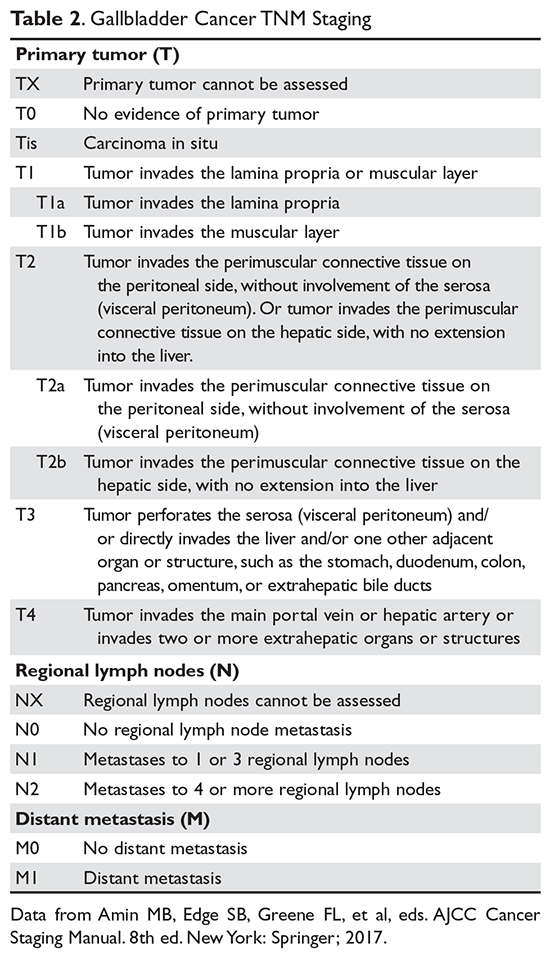
The Bismuth-Corlette classification is used to further classify perihilar cholangiocarcinoma according to patterns of hepatic duct involvement. Type I tumors are located below the confluence of the left and right hepatic ducts.42 Type II reach the confluence of the hepatic ducts.42 Type III occlude the common hepatic duct and either the right or left hepatic duct (IIIa and IIIb, respectively).42 Finally, type IV are multicentric, or involve the confluence and both the right and left hepatic ducts.42 Tumors that involve the common hepatic duct bifurcation are named Klatskin tumors.42
- What is the first-line treatment for localized cholangiocarcinomas?
Surgical resection is the only potentially curative treatment for localized cholangiocarcinoma, although fewer than 20% of patients are suitable for curative treatment, due to the presence of advanced disease at diagnosis.43,44 Available evidence supports the recommendation that resection with negative margins, regardless of extent, should be the goal of therapy for patients with potentially resectable disease.44 Extensive hepatic resections are often necessary to achieve clear margins since the majority of patients present with large masses. Substantial evidence corroborates that R0 resection is associated with better survival, whereas the benefit of wide compared to narrow (< 5–10 mm) margins is unclear.45 A recent analysis of 96 patients suggests that the proximal resection margin has more prognostic implications than distal margins.45
Surgical options and resectability criteria depend upon tumor location. Extent of tumor in the bile duct is one of the most important factors that determine resectability.17 Although multifocal liver tumors (including satellite lesions), lymph node metastases to the porta hepatis, and distant metastases are considered relative contraindications to surgery, surgical approaches can be considered in selected patients.43 Patient selection for surgery is facilitated by careful preoperative staging, which may include laparoscopy. Laparoscopic staging prior to resection may prevent unnecessary laparotomy in 30% to 45% of patients.42,46
- Is there a role for adjuvant treatment?
Recurrence following complete resection is a primary limitation for cure in BTC, which provides a rationale for the use of adjuvant therapy.47,48 In a sample of 79 patients with extrahepatic cholangiocarcinoma who underwent curative resection, the cumulative recurrence rate after 4 years was 56%.47 Initial recurrence at a distant site occurs in 40% to 50% of patients.48
Lymphovascular and perineural invasion, lymph node metastasis, and tumor size ≥ 5 cm have been reported as independent predictors of recurrence and mortality following resection.49 A 2017 meta-analysis which included 30 studies involving more than 22,499 patients reported a 41% reduction in the risk of death with adjuvant chemotherapy, which translated to a mean OS benefit of 4 months in an unselected population.49 Moreover, this study revealed inferior OS in patients given adjuvant radiation therapy (RT) in combination with chemotherapy.49 These results are in line with the previous meta-analysis by Horgan et al, which demonstrated that adjuvant RT seems to benefit only patients with R1 resections, with a possible detrimental effect in R0 disease.50 Therefore, adjuvant chemoradiation cannot be viewed as a standard practice following R0 resection, and should be reserved for those patients with positive margins (R1/ 2) to reduce local progression.
In the phase 3 BILCAP trial presented at ASCO 2017, 447 patients with completely resected cholangiocarcinoma or gallbladder cancer with adequate biliary drainage and Eastern Cooperative Oncology Group (ECOG) performance score ≤ 2 were randomly assigned to observation or capecitabine (1250 mg/m2 twice daily for days 1–14 every 21 days for 8 cycles).51 Surgical treatment achieved R0 resection in 62% of patients and 46% were node-negative. Median OS was 51 months for the capecitabine group and 36 months for the control arm (hazard ratio [HR] 0.80, 95% CI 0.63 to 1.04, P = 0.097). Analyses with adjustment for nodal status, grade of disease, and gender indicated a HR of 0.71 (P < 0.01). Median DFS was 25 months versus 18 months favoring the capecitabine group, and rates of grade 3 or 4 toxicity were less than anticipated. Following the results of this trial, adjuvant capecitabine should become the new standard of care.
- What is the treatment for locally advanced cholangiocarcinoma?
The optimal approach to patients with locally advanced unresectable cholangiocarcinoma has not been established. The prognosis for patients with either locally unresectable or locally recurrent disease is typically measured in months. Goals of palliative therapy are relief of symptoms and improvement in quality of life, and there is no role for surgical debulking.
Liver transplantation is a potentially curative option for selected patients with hilar or intrahepatic cholangiocarcinoma. Patients with lymph node-negative, non-disseminated, locally advanced hilar cholangiocarcinomas have 5-year survival rates ranging from 25% to 42% following transplantation.52 Retrospective data suggests that neoadjuvant chemoradiation followed by liver transplantation is highly effective for selected patients with hilar cholangiocarcinoma.52 However, these results require confirmation from prospective clinical evidence. It is important to recognize that liver transplantation plays no role in the management of distal cholangiocarcinoma or gallbladder cancer.
Rarely, patients with borderline resectable intrahepatic cholangiocarcinoma will have a sufficient response to chemotherapy to permit later resection, and, in such cases, starting with chemotherapy and then restaging to evaluate resectability is appropriate.54 A single-center, retrospective analysis including 186 patients by Le Roy et al evaluated survival in patients with locally advanced, unresectable intrahepatic cholangiocarcinoma who received primary chemotherapy, followed by surgery in those with secondary resectability.54 After a median of 6 cycles of chemotherapy, 53% of patients achieved resectability and underwent surgery with curative intent. These patients had similar short- and long-term results compared to patients with initially resectable intrahepatic cholangiocarcinoma who had surgery alone, with median OS reaching 24 months.54
Ablative radiotherapy is an additional option for localized inoperable intrahepatic cholangiocarcinoma. Tao and colleagues evaluated 79 consecutive patients with inoperable intrahepatic cholangiocarcinoma treated with definitive RT.55 Median tumor size was 7.9 cm and 89% of patients received chemotherapy before RT. Median OS was 30 months and 3-year OS was 44%. Radiation dose was the single most important prognostic factor, and higher doses correlated with improved local control and OS. A biologic equivalent dose (BED) greater than 80.5 Gy was identified as an ablative dose of RT for large intrahepatic cholangiocarcinomas. The 3-year OS for patients receiving BED greater than 80.5 Gy was 73% versus 38% for those receiving lower doses.
Case Continued
The patient is deemed to have resectable disease and undergoes surgical resection followed by adjuvant capecitabine for 8 cycles. Unfortunately, after 1 year, follow-up imaging identifies bilateral enlarging lung nodules. Biopsy is performed and confirms metastatic cholangiocarcinoma.
- What is the treatment for metastatic BTC?
The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. A benefit of chemotherapy over best supportive care for cholangiocarcinoma was demonstrated in an early phase 3 trial that randomly assigned 90 patients with advanced pancreatic or biliary cancer (37 with bile duct cancer) to receive either fluorouracil (FU) -based systemic chemotherapy or best supportive care. Results showed that chemotherapy significantly improved OS (6 months versus 2.5 months).56 Chemotherapy is also beneficial for patients with unresectable gallbladder cancer. In a single-center randomized study including 81 patients with unresectable gallbladder cancer, gemcitabine and oxaliplatin (GEMOX) improved progression-free survival (PFS) and OS compared to best supportive care.57 Treatment for metastatic cholangiocarcinoma and gallbladder cancer follows the same algorithm.
In 2010, cisplatin plus gemcitabine was established as a reference regimen for first-line therapy by the ABC-02 study, in which 410 patients with locally advanced or metastatic bile duct, gallbladder, or ampullary cancer were randomly assigned to 6 courses of cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2 on days 1 and 8, every 21 days) or gemcitabine alone (1000 mg/m2 days 1, 8, 15, every 28 days).58 OS was significantly greater with combination therapy (11.7 versus 8.1 months), and PFS also favored the combination arm (8 versus 5 months). Toxicity was comparable in both groups, with the exception of significantly higher rates of grade 3 or 4 neutropenia with gemcitabine plus cisplatin (25% versus 17%), and higher rates of grade 3 or 4 abnormal liver function with gemcitabine alone (27% versus 17%). Most quality-of-life scales showed a trend favoring combined therapy.58 A smaller, identically designed Japanese phase 3 randomized trial achieved similar results, demonstrating greater OS with cisplatin plus gemcitabine compared to gemcitabine alone (11.2 versus 7.7 months).59
The gemcitabine plus cisplatin combination has not been directly compared with other gemcitabine combinations in phase 3 trials. A pooled analysis of 104 trials of a variety of chemotherapy regimens in advanced biliary cancer concluded that the gemcitabine plus cisplatin regimen offered the highest rates of objective response and tumor control compared with either gemcitabine-free or cisplatin-free regimens.60 However, this did not translate into significant benefit in terms of either time to tumor progression or median OS. It is important to note that this analysis did not include results of the subsequent ABC-02 trial.
There is no standard treatment for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. There are no completed prospective phase 3 trials supporting the use of second-line chemotherapy after failure of first-line chemotherapy in BTC, and the selection of candidates for second-line therapy as well as the optimal regimen are not established.61 The ongoing phase 2 multicenter ABC-06 trial is evaluating oxaliplatin plus short-term infusional FU and leucovorin (FOLFOX) versus best supportive care for second-line therapy. In a systematic review including 23 studies (14 phase 2 clinical trials and 9 retrospective studies) with 761 patients with BTC, the median OS was 7.2 months.
The optimal selection of candidates for second-line chemotherapy is not established. Two independent studies suggest that patients who have a good performance status (0 or 1), disease control with the first-line chemotherapy, low CA 19-9 level, and possibly previous surgery on their primary tumor, have the longest survival with second-line chemotherapy. However, whether these characteristics predict for chemotherapy responsiveness or more favorable biologic behavior is not clear.62,63 No particular regimen has proved superior to any other, and the choice of second-line regimen remains empiric.
For patients with adequate performance status, examples of other conventional chemotherapy regimens with demonstrated activity that could be considered for second-line therapy include: FOLFOX or capecitabine, gemcitabine plus capecitabine, capecitabine plus cisplatin, or irinotecan plus short-term infusional FU and leucovorin (FOLFIRI) with or without bevacizumab.64 For selected patients, second-line molecularly targeted therapy using erlotinib plus bevacizumab may be considered. However, this regimen is very costly.64 Examples of other regimens with demonstrated activity in phase 2 trials include GEMOX, gemcitabine plus fluoropyrimidine, and fluoropyrimidine plus oxaliplatin or cisplatin.64
There is promising data from studies of targeted therapy for specific molecular subgroups. A recent phase 2 trial evaluated the activity of BGJ398, an orally bioavailable, selective, ATP-competitive pan inhibitor of human fibroblast growth factor receptor (FGFR) kinase, in patients with FGFR-altered advanced cholangiocarcinoma.65 The overall response rate was 14.8% (18.8% FGFR2 fusions only) and disease control rate was 75.4% (83.3% FGFR2 fusions only). All responsive tumors contained FGFR2 fusions. Adverse events were manageable, and grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%). Those included hyperphosphatemia, stomatitis, and palmar-plantar erythrodysesthesia. Javle and colleagues also identified HER2/neu blockade as a promising treatment strategy for gallbladder cancer patients with this gene amplification.66 This retrospective analysis included 9 patients with gallbladder cancer and 5 patients with cholangiocarcinoma who received HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab). In the gallbladder cancer group, HER2/neu gene amplification or overexpression was detected in 8 cases. These patients experienced disease stability (n = 3), partial response (n = 4), or complete response (n = 1) with HER2/neu–directed therapy. Median duration of response was 40 weeks. The cholangiocarcinoma cases treated in this series had no radiological responses despite HER2/neu mutations or amplification.
Gallbladder Cancer
Case Presentation
A 57-year-old woman from Chile presents with a 3-week history of progressive right upper quadrant abdominal pain. She denies nausea, vomiting, dysphagia, odynophagia, alterations in bowel habits, fever, or jaundice. Her past medical history is significant for obesity and hypertension. She has no history of smoking, alcohol, or illicit drug use. Laboratory studies show marked leukocytosis (23,800/µL) with neutrophilia (91%). Liver function test results are within normal limits. Ultrasound of the abdomen reveals gallbladder wall thickening and cholelithiasis.
The patient undergoes an uneventful laparoscopic cholecystectomy and is discharged from the hospital after 48 hours. Pathology report reveals a moderately differentiated adenocarcinoma of the gallbladder invading the perimuscular connective tissue (T2). No lymph nodes are identified in the specimen.
- What is the appropriate surgical management of gallbladder cancer?
Gallbladder cancer can be diagnosed preoperatively or can be found incidentally by intraoperative or pathological findings. In one large series, gallbladder cancer was incidentally found during 0.25% of laparoscopic cholecystectomies.67
For patients who are diagnosed with previously unsuspected gallbladder cancer by pathology findings, the extent of tumor invasion (T stage) indicates the need for re-resection (Figure 3).64
Alternatively, when gallbladder cancer is documented or suspected preoperatively, adequate imaging is important to identify patients with absolute contraindications to resection. Contraindications to surgery include metastasis, extensive involvement of the hepatoduodenal ligament, encasement of major vessels, and involvement of celiac, peripancreatic, periduodenal, or superior mesenteric nodes.72 Notwithstanding, retrospective series suggest individual patients may benefit, and surgical indications in advanced disease should be determined on an individual basis.73 Staging imaging should be obtained using multiphasic contrast-enhanced CT or MRI of the chest, abdomen, and pelvis. PET-scan can be used in selected cases where metastatic disease is suspected.64 Laparoscopic diagnostic staging should be considered prior to resection.64 This procedure can identify previously unknown contraindications to tumor resection in as much as 23% of patients, and the yield is significantly higher in locally advanced tumors.73
Patients with a diagnosis of potentially resectable, localized gallbladder cancer should be offered definitive surgery. Extended cholecystectomy is recommended for patients stage T2 or above. This procedure involves wedge resection of the gallbladder bed or a segmentectomy IVb/V and lymph node dissection, which should include the cystic duct, common bile duct, posterior superior pancreaticoduodenal lymph nodes, and those around the hepatoduodenal ligament.72 Bile duct excision should be performed if there is malignant involvement.64
Conclusion
BTCs are anatomically and clinically heterogeneous tumors. Prognostic factors and therapeutic approaches for BTCs differ depending upon their location in the biliary tree and, accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated. Surgical resection is the only potentially curative treatment for localized BTC. However, recurrence following complete resection is a primary limitation for cure, which provides a rationale for the use of adjuvant therapy. The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. Multiple randomized clinical trials have demonstrated a benefit of chemotherapy for metastatic disease. For patients with adequate performance status, second-line therapy can be considered, and data from studies that evaluated targeted therapy for specific molecular subgroups is promising.
1. Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2(Suppl 1):e000152.
2. Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2017 Oct 10. doi: 10.1038/nrclinonc.2017.157.
3. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist 2008;13:415–23.
4. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2014 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2017.
5. Torre LA, Siegel RL, Islami F, et al. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol 2017 Aug 18. doi: 10.1016/j.cgh.2017.08.017.
6. Lau CSM, Zywot A, Mahendraraj K, Chamberlain CS. Gallbladder carcinoma in the United States: a population based clinical outcomes study involving 22,343 patients from the Surveillance, Epidemiology, and End Result Database (1973–2013). HPB Surg 2017;2017:1532835. doi:10.1155/2017/1532835.
7. Hughes T, O’Connor T, Techasen A, et al. Opisthorchiasis and cholangiocarcinoma in Southeast Asia: an unresolved problem. Int J Gen Med 2017;10:227–37.
8. DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–62.
9. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9.
10. Yao KJ, Jabbour S, Parekh N, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117.
11. Choi SB, Kim KS, Choi JY, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol 2009;16:3048–56.
12. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96.
13. Duffy A, Capanu M, Abou-Alfa GK, et al. Gallbladder cancer (GBC): 10-year experience at Memorial Sloan-Kettering Cancer Centre (MSKCC). J Surg Oncol 2008;98:485–9.
14. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer — viewpoint of the IARC Working Group. N Engl J Med 2016;375:794–8.
15. Chen J, Han Y, Xu C, et al. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases. Eur J Cancer Prev 2015;24:89–99.
16. Larsson SC, Giovannucci EL, Wolk A. Sweetened beverage consumption and risk of biliary tract and gallbladder cancer in a prospective study. J Natl Cancer Inst 2016;108: doi: 10.1093/jnci/djw125.
17. Gore RM. Biliary tract neoplasms: diagnosis and staging. Cancer Imaging 2007;7(Special Issue A):S15–23.
18. Broome U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610–5.
19. Burak K, Angulo P, Pasha T, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523–6.
20. Rodrigues J, Diehl DL. Cholangiocarcinoma: clinical manifestations and diagnosis. Tech Gastrointest Endosc 2016;18:75–82.
21. Mitchell CH, Johnson PT, Fishman EK, et al. Features suggestive of gallbladder malignancy. J Comput Assist Tomogr 2014;38:235–41.
22. Beltz WR, Condon RE. Primary carcinoma of the gallbladder. Ann Surg 1974;180:180–4.
23. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512–22.
24. Patel T. Cholangiocarcinoma—controversies and challenges. Nat Rev Gastroenterol Hepatol 2011;8:189–200.
25. Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 1996;224:463–73.
26. Bartella I, Dufour JF. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J Gastrointestin Liver Dis 2015;24:481-9.
27. Yamaguchi K, Enjoji M. Carcinoma of the gallbladder: a clinicopathology of 103 patients and a newly proposed staging. Cancer 1988;62:1425–32.
28. Esposito I, Schirmacher P. Pathological aspects of cholangiocarcinoma. HPB. 2008;10:83–6.
29. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol 2016;5:62.
30. Chung YE, Kim MJ, Park YN, et al. Varying appearances of cholangiocarcinoma: radiologic-pathologic correlation. Radiographics 2009;29:683–700.
31. Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg 2003;10:288–91.
32. Rao PN. Nodule in liver: investigations, differential diagnosis and follow-up. J Clin Exp Hepatol 2014;4(Suppl 3):S57–62.
33. Kim TK, Lee E, Jang HJ. Imaging findings of mimickers of hepatocellular carcinoma. Clin Mol Hepatol 2015;21:326–43.
34. Hennedige TP, Neo WT, Venkatesh SK. Imaging of malignancies of the biliary tract- an update. Cancer Imaging 2014;14:14.
35. Kim SH, Lee CH, Kim BH, et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J Comput Assist Tomogr 2012;36:704–9.
36. Asayama Y, Yoshimitsu K, Irie H, et al. Delayed-phase dynamic CT enhancement as a prognostic factor for mass-forming intrahepatic cholangiocarcinoma. Radiology 2006;238:150–5.
37. National Comprehensive Cancer Network. Cancer of unknown primary. www.nccn.org/professionals/physician_gls/pdf/bone.pdf. Accessed 1 Dec 2017.
38. Kefeli A, Basyigit S, Yeniova AO. Diagnosis of hepatocellular carcinoma. In: Abdeldayem HM, ed. Updates in liver cancer. London: InTech; 2017.
39. Bergquist JR, Ivanics T, Storlie CB, et al. Implications of CA19-9 elevation for survival, staging, and treatment sequencing in intrahepatic cholangiocarcinoma: A national cohort analysis. J Surg Oncol 2016;114:475–82.
40. Chung YJ, Choi DW, Choi SH, et al. Prognostic factors following surgical resection of distal bile duct cancer. J Korean Surg Soc 2013;85:212–8.
41. Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol 2002;33:1175–81.
42. Paul A, Kaiser GM, Molmenti EP, et al. Klatskin tumors and the accuracy of the Bismuth-Corlette classification. Am Surg 2011;77:1695–9.
43. Cannavale A, Santoni M, Gazzetti M, et al. Updated management of malignant biliary tract tumors: an illustrative review. J Vasc Interv Radiol 2016;27:1056–69.
44. Matsuo K, Rocha FG, Ito K, et al. The Blumgart preoperative staging system for hilar cholangiocarcinoma: analysis of resectability and outcomes in 380 patients. J Am Coll Surg 2012;215:343–55.
45. Yoo T, Park SJ, Han SS, et al. Proximal resection margins: more prognostic than distal resection margins in patients undergoing hilar cholangiocarcinoma resection. Cancer Res Treat 2017 Nov 16; doi.org/10.4143/crt.2017.320.
46. Joseph S, Connor S, Garden OJ. Staging laparoscopy for cholangiocarcinoma. HPB 2008;10:116–9.
47. Jarnagin WR, Ruo L, Little SA, et al. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: implications for adjuvant therapeutic strategies. Cancer 2003;98:1689–700.
48. Kobayashi A, Miwa S, Nakata T, Miyagawa S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br J Surg 2010;97:56–64.
49. Ghidini M, Tomasello G, Botticelli A, et al. Adjuvant chemotherapy for resected biliary tract cancers: a systematic review and meta-analysis. HPB 2017;19:741–8.
50. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934–40.
51. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study [abstract]. J Clin Oncol 2017 35:15_suppl:4006-4006.
52. Darwish Murad S, Kim WR, Darnois DM, et al. Efficacy of neoadjuvant chemoradiation followed by liver transplantation for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88–98.
53. Sapisochin G, Facciuto M, Rubbia-Brandt L, et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016;64:1178–88.
54. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg 2017 Aug 31. doi: 10.1002/bjs.10641.
55. Tao R, Krishnan S, Bhosale PR, et al. Ablative radiotherapy doses lead to a substantial prolongation of survival in patients with inoperable intrahepatic cholangiocarcinoma: a retrospective dose response analysis. J Clin Oncol 2016;34:219–26.
56. Glimelius B, Hoffman K, SjÓdén PO, et al. 555 Palliative chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Eur J Cancer 1995;31:S118.
57. Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol 2010;28:4581–6.
58. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
59. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469–74.
60. Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
61. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38.
62. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290–7.
63. Fornaro L, Cereda S, Aprile G, et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br J Cancer 2014;110:2165–9.
64. National Comprehensive Cancer Network. Hepatobiliary cancer. www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. Accessed 12 Nov 2017.
65. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2017 Nov 28;JCO2017755009.
66. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58.
67. Konstantinidis IT, Deshpande V, Genevay M, et al. Trends in presentation and survival for gallbladder cancer during a period of more than 4 decades: a single-institution experience. Arch Surg 2009;144:441–47.
68. Singh S, Agarwal AK. Gallbladder cancer: the role of laparoscopy and radical resection. Ann Surg 2009;250:494–5.
69. Kapoor VK, Haribhakti SP. Extended cholecystectomy for carcinoma of the gall bladder. Trop Gastroenterol 1995;16:74–5.
70. Ethun CG, Postlewait LM, Le N, et al. Association of optimal time Interval to re-resection for incidental gallbladder cancer with overall survival: a multi-Institution analysis from the US extrahepatic biliary malignancy consortium. JAMA Surg 2017;152:143–9.
71. Goetze TO, Paolucci V. Benefits of reoperation of T2 and more advanced incidental gallbladder carcinoma: analysis of the German registry. Ann Surg 2008;247:104–8.
72. Nishio H, Nagino M, Ebata T, et al. Aggressive surgery for stage IV gallbladder carcinoma; what are the contraindications? J Hepatobiliary Pancreat Surg 2007;14:351–7.
73. Agarwal AK, Kalayarasan R, Javed A, et al. The role of staging laparoscopy in primary gallbladder cancer--an analysis of 409 patients: a prospective study to evaluate the role of staging laparoscopy in the management of gallbladder cancer. Ann Surg 2013;258:318–23.
Introduction
Biliary tract carcinoma (BTC) is th
Epidemiology
In the United States, BTC is rare and accounts for approximately 4% of all gastrointestinal malignancies, with an estimated 6000 to 7000 cases of carcinoma of the gallbladder and 3000 to 4000 cases of carcinoma of the bile duct diagnosed annually.4 Among women, there is a 26-fold variation in BTC mortality worldwide, ranging from 0.8 deaths per 100,000 in South Africa to 21.2 per 100,000 in Chile.1,5 Interestingly, for American Indians in New Mexico, gallbladder cancer mortality rates (8.9 per 100,000) surpass those for breast and pancreatic cancers.6 The incidence of anatomical cholangiocarcinoma subtypes also varies regionally, reflecting disparities in genetic and environmental predisposing factors.2,7 In a large, single-center study in the United States, intrahepatic cholangiocarcinoma accounted for less than 10% of cases, perihilar accounted for 50%, and distal accounted for the remaining 40%.8 Importantly, intrahepatic cholangiocarcinoma is the second most common primary malignancy of the liver, and its incidence seems to be rising in many western countries. In the United States, there has been an estimated 128% rise over the past 40 years.4,9
BTC is associated with high mortality rates.10 Median overall survival (OS) for cholangiocarcinoma is 20 to 28 months and 5-year survival is around 25%.10 Most cholangiocarcinomas are diagnosed at advanced stages with unresectable tumors.10 Furthermore, outcomes following resection with curative intent are poor—median disease-free survival (DFS) of 12 to 36 months has been reported.11,12 Patients with intrahepatic disease have a better prognosis when compared with patients who have extrahepatic tumors.12 Gallbladder cancer, likewise, carries a poor overall prognosis; median OS is 32 months and 5-year survival is as low as 13%.6
Risk factors for BTC include intrinsic and extrinsic elements.6 Incidence of BTC increases with age, and diagnosis typically occurs in the sixth to eighth decade of life.5,6,13 In contrast to gallbladder cancer, the incidence of cholangiocarcinoma is slightly higher in men.9 Obesity, diabetes, and consumption of sweetened drinks also increase the risk for BTC.14–16 Cholelithiasis is the most prevalent risk factor for gallbladder cancer, and the risk is greater for larger stones.5 Around 1 in 5 patients with porcelain gallbladder will develop gallbladder carcinoma.17 Primary sclerosing cholangitis (PSC), chronic calculi of the bile duct, choledochal cysts, cirrhosis, hepatitis C, and liver fluke infections are well established risk factors for cholangiocarcinoma.7,12,18 PSC is one of the best described entities among these predisposing conditions. Lifetime prevalence of cholangiocarcinoma among patients with PSC ranges from 5% to 10%.18,19 These patients also present at a younger age; in one series, the median age at diagnosis for BTC arising from PSC was 39 years.18 It is important to recognize, however, that in most patients diagnosed with cholangiocarcinoma, no predisposing factors are identified.8
Diagnosis
Clinical Presentation
Clinical presentation of BTC depends upon anatomic location.20 Patients with early invasive gallbladder cancer are most often asymptomatic.21 When symptoms occur, they may be nonspecific and mimic cholelithiasis.21 The most common clinical presentations include jaundice, weight loss, and abdominal pain.21 Prior to widespread availability of imaging studies, the preoperative diagnosis rate for gallbladder cancer was as low as 10%.22 However, the accuracy of computed tomography (CT) has changed this scenario, with sensitivity ranging from 73% to 87% and specificity from 88% to 100%.21 As a result of its silent clinical character, cholangiocarcinoma is frequently difficult to diagnose.23 Perihilar and distal cholangiocarcinoma characteristically present with signs of biliary obstruction, and imaging and laboratory data can corroborate the presence of cholestasis.24 On examination, patients with extrahepatic cholangiocarcinoma may present with jaundice, hepatomegaly, and a palpable right upper quadrant mass.25 A palpable gallbladder (Courvoisier sign) can also be present.25 Intrahepatic cholangiocarcinoma presents differently, and patients are less likely to be jaundiced.23 Typical clinical features are nonspecific and include dull right upper quadrant pain, weight loss, and an elevated alkaline phosphatase level.23 Alternatively, asymptomatic patients can present with incidentally detected lesions, when imaging is obtained as part of the workup for other causes or during screening for hepatocellular carcinoma in patients with viral hepatitis or cirrhosis.23,26 Uncommonly, BTC patients present because of signs or symptoms related to metastatic disease or evidence of metastatic disease on imaging.
Pathology and Grading
The majority of BTCs are adenocarcinomas, corresponding to 90% of cholangiocarcinomas and 99% of gallbladder cancers.27,28 They are graded as well, moderately, or poorly differentiated.2 Adenosquamous and squamous cell carcinoma are responsible for most of the remaining cases.2,29 Cholangiocarcinomas are divided into 3 types, defined by the Liver Cancer Study Group of Japan: (1) mass-forming, (2) periductal-infiltrating, and (3) intraductal-growing.30,31 Mass-forming intrahepatic cholangiocarcinomas are characterized morphologically by a homogeneous gray-yellow mass with frequent satellite nodules and irregular but well-defined margins.17,30 Central necrosis and fibrosis are also common.30 In the periductal-infiltrating type, tumor typically grows along the bile duct wall without mass formation, resulting in concentric mural thickening and proximal biliary dilation.30 Intraductal-growing papillary cholangiocarcinoma is characterized by the presence of intraluminal papillary or tubular polypoid tumors of the intra- or extrahepatic bile ducts, with partial obstruction and proximal biliary dilation.30
Cholangiocarcinoma
Case Presentation
A previously healthy 59-year-old man presents to his primary care physician with a 3-month history of dull right upper quadrant pain associated with weight loss. The patient is markedly cachectic and abdominal examination reveals upper quadrant tenderness. Laboratory exams are significant for elevated alkaline phosphatase (500 U/L; reference range 45–115 U/L), cancer antigen 19-9 (CA 19-9, 73 U/mL; reference range ≤ 37 U/mL), and carcinoembryonic antigen (CEA , 20 ng/mL; reference range for nonsmokers ≤ 3.0 ng/mL). Aspartate aminotransferase, alanine aminotransferase, total bilirubin, and coagulation studies are within normal range. Ultrasound demonstrates a homogeneous mass with irregular borders in the right lobe of the liver. Triphasic contrast-enhanced CT scan demonstrates a tumor with ragged rim enhancement at the periphery, and portal venous phase shows gradual centripetal enhancement of the tumor with capsular retraction. No abdominal lymph nodes or extrahepatic tumors are noted (Figure 1, Image A).
- What are the next diagnostic steps?
The most critical differential diagnosis of solid liver mass in patients without cirrhosis is cholangiocarcinoma and metastases from another primary site.32 Alternatively, when an intrahepatic lesion is noted on an imaging study in the setting of cirrhosis, the next diagnostic step is differentiation between cholangiocarcinoma and hepatocellular carcinoma (HCC).32 Triphasic contrast-enhanced CT and dynamic magnetic resonance imaging (MRI) are key diagnostic procedures.32,33 In the appropriate setting, classical imaging features in the arterial phase with washout in portal venous or delayed phase can be diagnostic of HCC and may obviate the need for a biopsy (Figure 2).
Typical radiographic features of cholangiocarcinoma include a hypodense hepatic lesion that can be either well-defined or infiltrative and is frequently associated with biliary dilatation (Figure 1, Image A).33 The dense fibrotic nature of the tumor may cause capsular retraction, which is seen in up to 20% of cases.17 This finding is highly suggestive of cholangiocarcinoma and is rarely present in HCC.33 Following contrast administration, there is peripheral (rim) enhancement throughout both arterial and venous phases.32–34 However, these classic features were present in only 70% of cases in one study.35 Although intrahepatic cholangiocarcinomas are most commonly hypovascular, small mass-forming intrahepatic cholangiocarcinomas can often be arterially hyperenhancing and mimic HCC.33 Tumor enhancement on delayed CT imaging has been correlated with survival. Asayama et al demonstrated that tumors that exhibited delayed enhancement on CT in more than two-thirds of their volume were associated with a worse prognosis.36
Patients without cirrhosis who present with a localized lesion of the liver should undergo extensive evaluation for a primary cancer site.37 CT of the chest, abdomen, and pelvis with contrast should be obtained.37 Additionally, mammogram and endoscopic evaluation with esophagogastroduodenoscopy (EGD) and colonoscopy should be included in the work-up.37
Preoperative tumor markers are also included in the work-up. All patients with a solid liver lesion should have serum alpha-fetoprotein (AFP) levels checked. AFP is a serum glycoprotein recognized as a marker for HCC and is reported to detect preclinical HCC.38 However, serum concentrations are normal in up to 40% of small HCCs.38 Although no specific marker for cholangiocarcinoma has yet been identified, the presence of certain tumor markers in the serum of patients may be of diagnostic value, especially in patients with PSC. CA 19-9 and CEA are the best studied. Elevated levels of CA 19-9 prior to treatment are associated with a poorer prognosis, and CA 19-9 concentrations greater than 1000 U/mL are consistent with advanced disease.39,40 One large series evaluated the diagnostic value of serum CEA levels in 333 patients with PSC, 13% of whom were diagnosed with cholangiocarcinoma.34 A serum CEA level greater than 5.2 ng/mL had a sensitivity of 68.0% and specificity of 81.5%.38
If a biopsy is obtained, appropriate immunohistochemistry (IHC) can facilitate the diagnosis. BTC is strongly positive for CK-7 and CK-19.41 CK-7 positivity is not specific and is also common among metastatic cancers of the lung and breast; therefore, in some cases cholangiocarcinoma may be a diagnosis of exclusion. Immunostaining for monoclonal CEA is diffusely positive in up to 75% of cases.41 An IHC panel consisting of Hep Par-1, arginase-1, monoclonal CEA, CK-7, CK-20, TTF-1, MOC-31, and CDX-2 has been proposed to optimize the differential diagnosis of HCC, metastatic adenocarcinoma, and cholangiocarcinoma.41
Case Continued
CT of the chest, abdomen, and pelvis reveals no concerns for metastasis and no evidence of primary cancer elsewhere. EGD and colonoscopy are clear. AFP levels are within normal limits (2 ng/mL). Biopsy is performed and demonstrates adenocarcinoma. IHC studies demonstrate cells positive for monoclonal CEA, CK-7, CK-19, and MOC-31, and negative for Napsin A, TTF-1, and CK-20.
- How is cholangiocarcinoma staged and classified?
The purpose of the staging system is to provide information on prognosis and guidance for therapy. Prognostic factors and the therapeutic approaches for BTC differ depending upon their location in the biliary tree. Accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated (Table 1 and Table 2).23
The Bismuth-Corlette classification is used to further classify perihilar cholangiocarcinoma according to patterns of hepatic duct involvement. Type I tumors are located below the confluence of the left and right hepatic ducts.42 Type II reach the confluence of the hepatic ducts.42 Type III occlude the common hepatic duct and either the right or left hepatic duct (IIIa and IIIb, respectively).42 Finally, type IV are multicentric, or involve the confluence and both the right and left hepatic ducts.42 Tumors that involve the common hepatic duct bifurcation are named Klatskin tumors.42
- What is the first-line treatment for localized cholangiocarcinomas?
Surgical resection is the only potentially curative treatment for localized cholangiocarcinoma, although fewer than 20% of patients are suitable for curative treatment, due to the presence of advanced disease at diagnosis.43,44 Available evidence supports the recommendation that resection with negative margins, regardless of extent, should be the goal of therapy for patients with potentially resectable disease.44 Extensive hepatic resections are often necessary to achieve clear margins since the majority of patients present with large masses. Substantial evidence corroborates that R0 resection is associated with better survival, whereas the benefit of wide compared to narrow (< 5–10 mm) margins is unclear.45 A recent analysis of 96 patients suggests that the proximal resection margin has more prognostic implications than distal margins.45
Surgical options and resectability criteria depend upon tumor location. Extent of tumor in the bile duct is one of the most important factors that determine resectability.17 Although multifocal liver tumors (including satellite lesions), lymph node metastases to the porta hepatis, and distant metastases are considered relative contraindications to surgery, surgical approaches can be considered in selected patients.43 Patient selection for surgery is facilitated by careful preoperative staging, which may include laparoscopy. Laparoscopic staging prior to resection may prevent unnecessary laparotomy in 30% to 45% of patients.42,46
- Is there a role for adjuvant treatment?
Recurrence following complete resection is a primary limitation for cure in BTC, which provides a rationale for the use of adjuvant therapy.47,48 In a sample of 79 patients with extrahepatic cholangiocarcinoma who underwent curative resection, the cumulative recurrence rate after 4 years was 56%.47 Initial recurrence at a distant site occurs in 40% to 50% of patients.48
Lymphovascular and perineural invasion, lymph node metastasis, and tumor size ≥ 5 cm have been reported as independent predictors of recurrence and mortality following resection.49 A 2017 meta-analysis which included 30 studies involving more than 22,499 patients reported a 41% reduction in the risk of death with adjuvant chemotherapy, which translated to a mean OS benefit of 4 months in an unselected population.49 Moreover, this study revealed inferior OS in patients given adjuvant radiation therapy (RT) in combination with chemotherapy.49 These results are in line with the previous meta-analysis by Horgan et al, which demonstrated that adjuvant RT seems to benefit only patients with R1 resections, with a possible detrimental effect in R0 disease.50 Therefore, adjuvant chemoradiation cannot be viewed as a standard practice following R0 resection, and should be reserved for those patients with positive margins (R1/ 2) to reduce local progression.
In the phase 3 BILCAP trial presented at ASCO 2017, 447 patients with completely resected cholangiocarcinoma or gallbladder cancer with adequate biliary drainage and Eastern Cooperative Oncology Group (ECOG) performance score ≤ 2 were randomly assigned to observation or capecitabine (1250 mg/m2 twice daily for days 1–14 every 21 days for 8 cycles).51 Surgical treatment achieved R0 resection in 62% of patients and 46% were node-negative. Median OS was 51 months for the capecitabine group and 36 months for the control arm (hazard ratio [HR] 0.80, 95% CI 0.63 to 1.04, P = 0.097). Analyses with adjustment for nodal status, grade of disease, and gender indicated a HR of 0.71 (P < 0.01). Median DFS was 25 months versus 18 months favoring the capecitabine group, and rates of grade 3 or 4 toxicity were less than anticipated. Following the results of this trial, adjuvant capecitabine should become the new standard of care.
- What is the treatment for locally advanced cholangiocarcinoma?
The optimal approach to patients with locally advanced unresectable cholangiocarcinoma has not been established. The prognosis for patients with either locally unresectable or locally recurrent disease is typically measured in months. Goals of palliative therapy are relief of symptoms and improvement in quality of life, and there is no role for surgical debulking.
Liver transplantation is a potentially curative option for selected patients with hilar or intrahepatic cholangiocarcinoma. Patients with lymph node-negative, non-disseminated, locally advanced hilar cholangiocarcinomas have 5-year survival rates ranging from 25% to 42% following transplantation.52 Retrospective data suggests that neoadjuvant chemoradiation followed by liver transplantation is highly effective for selected patients with hilar cholangiocarcinoma.52 However, these results require confirmation from prospective clinical evidence. It is important to recognize that liver transplantation plays no role in the management of distal cholangiocarcinoma or gallbladder cancer.
Rarely, patients with borderline resectable intrahepatic cholangiocarcinoma will have a sufficient response to chemotherapy to permit later resection, and, in such cases, starting with chemotherapy and then restaging to evaluate resectability is appropriate.54 A single-center, retrospective analysis including 186 patients by Le Roy et al evaluated survival in patients with locally advanced, unresectable intrahepatic cholangiocarcinoma who received primary chemotherapy, followed by surgery in those with secondary resectability.54 After a median of 6 cycles of chemotherapy, 53% of patients achieved resectability and underwent surgery with curative intent. These patients had similar short- and long-term results compared to patients with initially resectable intrahepatic cholangiocarcinoma who had surgery alone, with median OS reaching 24 months.54
Ablative radiotherapy is an additional option for localized inoperable intrahepatic cholangiocarcinoma. Tao and colleagues evaluated 79 consecutive patients with inoperable intrahepatic cholangiocarcinoma treated with definitive RT.55 Median tumor size was 7.9 cm and 89% of patients received chemotherapy before RT. Median OS was 30 months and 3-year OS was 44%. Radiation dose was the single most important prognostic factor, and higher doses correlated with improved local control and OS. A biologic equivalent dose (BED) greater than 80.5 Gy was identified as an ablative dose of RT for large intrahepatic cholangiocarcinomas. The 3-year OS for patients receiving BED greater than 80.5 Gy was 73% versus 38% for those receiving lower doses.
Case Continued
The patient is deemed to have resectable disease and undergoes surgical resection followed by adjuvant capecitabine for 8 cycles. Unfortunately, after 1 year, follow-up imaging identifies bilateral enlarging lung nodules. Biopsy is performed and confirms metastatic cholangiocarcinoma.
- What is the treatment for metastatic BTC?
The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. A benefit of chemotherapy over best supportive care for cholangiocarcinoma was demonstrated in an early phase 3 trial that randomly assigned 90 patients with advanced pancreatic or biliary cancer (37 with bile duct cancer) to receive either fluorouracil (FU) -based systemic chemotherapy or best supportive care. Results showed that chemotherapy significantly improved OS (6 months versus 2.5 months).56 Chemotherapy is also beneficial for patients with unresectable gallbladder cancer. In a single-center randomized study including 81 patients with unresectable gallbladder cancer, gemcitabine and oxaliplatin (GEMOX) improved progression-free survival (PFS) and OS compared to best supportive care.57 Treatment for metastatic cholangiocarcinoma and gallbladder cancer follows the same algorithm.
In 2010, cisplatin plus gemcitabine was established as a reference regimen for first-line therapy by the ABC-02 study, in which 410 patients with locally advanced or metastatic bile duct, gallbladder, or ampullary cancer were randomly assigned to 6 courses of cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2 on days 1 and 8, every 21 days) or gemcitabine alone (1000 mg/m2 days 1, 8, 15, every 28 days).58 OS was significantly greater with combination therapy (11.7 versus 8.1 months), and PFS also favored the combination arm (8 versus 5 months). Toxicity was comparable in both groups, with the exception of significantly higher rates of grade 3 or 4 neutropenia with gemcitabine plus cisplatin (25% versus 17%), and higher rates of grade 3 or 4 abnormal liver function with gemcitabine alone (27% versus 17%). Most quality-of-life scales showed a trend favoring combined therapy.58 A smaller, identically designed Japanese phase 3 randomized trial achieved similar results, demonstrating greater OS with cisplatin plus gemcitabine compared to gemcitabine alone (11.2 versus 7.7 months).59
The gemcitabine plus cisplatin combination has not been directly compared with other gemcitabine combinations in phase 3 trials. A pooled analysis of 104 trials of a variety of chemotherapy regimens in advanced biliary cancer concluded that the gemcitabine plus cisplatin regimen offered the highest rates of objective response and tumor control compared with either gemcitabine-free or cisplatin-free regimens.60 However, this did not translate into significant benefit in terms of either time to tumor progression or median OS. It is important to note that this analysis did not include results of the subsequent ABC-02 trial.
There is no standard treatment for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. There are no completed prospective phase 3 trials supporting the use of second-line chemotherapy after failure of first-line chemotherapy in BTC, and the selection of candidates for second-line therapy as well as the optimal regimen are not established.61 The ongoing phase 2 multicenter ABC-06 trial is evaluating oxaliplatin plus short-term infusional FU and leucovorin (FOLFOX) versus best supportive care for second-line therapy. In a systematic review including 23 studies (14 phase 2 clinical trials and 9 retrospective studies) with 761 patients with BTC, the median OS was 7.2 months.
The optimal selection of candidates for second-line chemotherapy is not established. Two independent studies suggest that patients who have a good performance status (0 or 1), disease control with the first-line chemotherapy, low CA 19-9 level, and possibly previous surgery on their primary tumor, have the longest survival with second-line chemotherapy. However, whether these characteristics predict for chemotherapy responsiveness or more favorable biologic behavior is not clear.62,63 No particular regimen has proved superior to any other, and the choice of second-line regimen remains empiric.
For patients with adequate performance status, examples of other conventional chemotherapy regimens with demonstrated activity that could be considered for second-line therapy include: FOLFOX or capecitabine, gemcitabine plus capecitabine, capecitabine plus cisplatin, or irinotecan plus short-term infusional FU and leucovorin (FOLFIRI) with or without bevacizumab.64 For selected patients, second-line molecularly targeted therapy using erlotinib plus bevacizumab may be considered. However, this regimen is very costly.64 Examples of other regimens with demonstrated activity in phase 2 trials include GEMOX, gemcitabine plus fluoropyrimidine, and fluoropyrimidine plus oxaliplatin or cisplatin.64
There is promising data from studies of targeted therapy for specific molecular subgroups. A recent phase 2 trial evaluated the activity of BGJ398, an orally bioavailable, selective, ATP-competitive pan inhibitor of human fibroblast growth factor receptor (FGFR) kinase, in patients with FGFR-altered advanced cholangiocarcinoma.65 The overall response rate was 14.8% (18.8% FGFR2 fusions only) and disease control rate was 75.4% (83.3% FGFR2 fusions only). All responsive tumors contained FGFR2 fusions. Adverse events were manageable, and grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%). Those included hyperphosphatemia, stomatitis, and palmar-plantar erythrodysesthesia. Javle and colleagues also identified HER2/neu blockade as a promising treatment strategy for gallbladder cancer patients with this gene amplification.66 This retrospective analysis included 9 patients with gallbladder cancer and 5 patients with cholangiocarcinoma who received HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab). In the gallbladder cancer group, HER2/neu gene amplification or overexpression was detected in 8 cases. These patients experienced disease stability (n = 3), partial response (n = 4), or complete response (n = 1) with HER2/neu–directed therapy. Median duration of response was 40 weeks. The cholangiocarcinoma cases treated in this series had no radiological responses despite HER2/neu mutations or amplification.
Gallbladder Cancer
Case Presentation
A 57-year-old woman from Chile presents with a 3-week history of progressive right upper quadrant abdominal pain. She denies nausea, vomiting, dysphagia, odynophagia, alterations in bowel habits, fever, or jaundice. Her past medical history is significant for obesity and hypertension. She has no history of smoking, alcohol, or illicit drug use. Laboratory studies show marked leukocytosis (23,800/µL) with neutrophilia (91%). Liver function test results are within normal limits. Ultrasound of the abdomen reveals gallbladder wall thickening and cholelithiasis.
The patient undergoes an uneventful laparoscopic cholecystectomy and is discharged from the hospital after 48 hours. Pathology report reveals a moderately differentiated adenocarcinoma of the gallbladder invading the perimuscular connective tissue (T2). No lymph nodes are identified in the specimen.
- What is the appropriate surgical management of gallbladder cancer?
Gallbladder cancer can be diagnosed preoperatively or can be found incidentally by intraoperative or pathological findings. In one large series, gallbladder cancer was incidentally found during 0.25% of laparoscopic cholecystectomies.67
For patients who are diagnosed with previously unsuspected gallbladder cancer by pathology findings, the extent of tumor invasion (T stage) indicates the need for re-resection (Figure 3).64
Alternatively, when gallbladder cancer is documented or suspected preoperatively, adequate imaging is important to identify patients with absolute contraindications to resection. Contraindications to surgery include metastasis, extensive involvement of the hepatoduodenal ligament, encasement of major vessels, and involvement of celiac, peripancreatic, periduodenal, or superior mesenteric nodes.72 Notwithstanding, retrospective series suggest individual patients may benefit, and surgical indications in advanced disease should be determined on an individual basis.73 Staging imaging should be obtained using multiphasic contrast-enhanced CT or MRI of the chest, abdomen, and pelvis. PET-scan can be used in selected cases where metastatic disease is suspected.64 Laparoscopic diagnostic staging should be considered prior to resection.64 This procedure can identify previously unknown contraindications to tumor resection in as much as 23% of patients, and the yield is significantly higher in locally advanced tumors.73
Patients with a diagnosis of potentially resectable, localized gallbladder cancer should be offered definitive surgery. Extended cholecystectomy is recommended for patients stage T2 or above. This procedure involves wedge resection of the gallbladder bed or a segmentectomy IVb/V and lymph node dissection, which should include the cystic duct, common bile duct, posterior superior pancreaticoduodenal lymph nodes, and those around the hepatoduodenal ligament.72 Bile duct excision should be performed if there is malignant involvement.64
Conclusion
BTCs are anatomically and clinically heterogeneous tumors. Prognostic factors and therapeutic approaches for BTCs differ depending upon their location in the biliary tree and, accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated. Surgical resection is the only potentially curative treatment for localized BTC. However, recurrence following complete resection is a primary limitation for cure, which provides a rationale for the use of adjuvant therapy. The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. Multiple randomized clinical trials have demonstrated a benefit of chemotherapy for metastatic disease. For patients with adequate performance status, second-line therapy can be considered, and data from studies that evaluated targeted therapy for specific molecular subgroups is promising.
Introduction
Biliary tract carcinoma (BTC) is th
Epidemiology
In the United States, BTC is rare and accounts for approximately 4% of all gastrointestinal malignancies, with an estimated 6000 to 7000 cases of carcinoma of the gallbladder and 3000 to 4000 cases of carcinoma of the bile duct diagnosed annually.4 Among women, there is a 26-fold variation in BTC mortality worldwide, ranging from 0.8 deaths per 100,000 in South Africa to 21.2 per 100,000 in Chile.1,5 Interestingly, for American Indians in New Mexico, gallbladder cancer mortality rates (8.9 per 100,000) surpass those for breast and pancreatic cancers.6 The incidence of anatomical cholangiocarcinoma subtypes also varies regionally, reflecting disparities in genetic and environmental predisposing factors.2,7 In a large, single-center study in the United States, intrahepatic cholangiocarcinoma accounted for less than 10% of cases, perihilar accounted for 50%, and distal accounted for the remaining 40%.8 Importantly, intrahepatic cholangiocarcinoma is the second most common primary malignancy of the liver, and its incidence seems to be rising in many western countries. In the United States, there has been an estimated 128% rise over the past 40 years.4,9
BTC is associated with high mortality rates.10 Median overall survival (OS) for cholangiocarcinoma is 20 to 28 months and 5-year survival is around 25%.10 Most cholangiocarcinomas are diagnosed at advanced stages with unresectable tumors.10 Furthermore, outcomes following resection with curative intent are poor—median disease-free survival (DFS) of 12 to 36 months has been reported.11,12 Patients with intrahepatic disease have a better prognosis when compared with patients who have extrahepatic tumors.12 Gallbladder cancer, likewise, carries a poor overall prognosis; median OS is 32 months and 5-year survival is as low as 13%.6
Risk factors for BTC include intrinsic and extrinsic elements.6 Incidence of BTC increases with age, and diagnosis typically occurs in the sixth to eighth decade of life.5,6,13 In contrast to gallbladder cancer, the incidence of cholangiocarcinoma is slightly higher in men.9 Obesity, diabetes, and consumption of sweetened drinks also increase the risk for BTC.14–16 Cholelithiasis is the most prevalent risk factor for gallbladder cancer, and the risk is greater for larger stones.5 Around 1 in 5 patients with porcelain gallbladder will develop gallbladder carcinoma.17 Primary sclerosing cholangitis (PSC), chronic calculi of the bile duct, choledochal cysts, cirrhosis, hepatitis C, and liver fluke infections are well established risk factors for cholangiocarcinoma.7,12,18 PSC is one of the best described entities among these predisposing conditions. Lifetime prevalence of cholangiocarcinoma among patients with PSC ranges from 5% to 10%.18,19 These patients also present at a younger age; in one series, the median age at diagnosis for BTC arising from PSC was 39 years.18 It is important to recognize, however, that in most patients diagnosed with cholangiocarcinoma, no predisposing factors are identified.8
Diagnosis
Clinical Presentation
Clinical presentation of BTC depends upon anatomic location.20 Patients with early invasive gallbladder cancer are most often asymptomatic.21 When symptoms occur, they may be nonspecific and mimic cholelithiasis.21 The most common clinical presentations include jaundice, weight loss, and abdominal pain.21 Prior to widespread availability of imaging studies, the preoperative diagnosis rate for gallbladder cancer was as low as 10%.22 However, the accuracy of computed tomography (CT) has changed this scenario, with sensitivity ranging from 73% to 87% and specificity from 88% to 100%.21 As a result of its silent clinical character, cholangiocarcinoma is frequently difficult to diagnose.23 Perihilar and distal cholangiocarcinoma characteristically present with signs of biliary obstruction, and imaging and laboratory data can corroborate the presence of cholestasis.24 On examination, patients with extrahepatic cholangiocarcinoma may present with jaundice, hepatomegaly, and a palpable right upper quadrant mass.25 A palpable gallbladder (Courvoisier sign) can also be present.25 Intrahepatic cholangiocarcinoma presents differently, and patients are less likely to be jaundiced.23 Typical clinical features are nonspecific and include dull right upper quadrant pain, weight loss, and an elevated alkaline phosphatase level.23 Alternatively, asymptomatic patients can present with incidentally detected lesions, when imaging is obtained as part of the workup for other causes or during screening for hepatocellular carcinoma in patients with viral hepatitis or cirrhosis.23,26 Uncommonly, BTC patients present because of signs or symptoms related to metastatic disease or evidence of metastatic disease on imaging.
Pathology and Grading
The majority of BTCs are adenocarcinomas, corresponding to 90% of cholangiocarcinomas and 99% of gallbladder cancers.27,28 They are graded as well, moderately, or poorly differentiated.2 Adenosquamous and squamous cell carcinoma are responsible for most of the remaining cases.2,29 Cholangiocarcinomas are divided into 3 types, defined by the Liver Cancer Study Group of Japan: (1) mass-forming, (2) periductal-infiltrating, and (3) intraductal-growing.30,31 Mass-forming intrahepatic cholangiocarcinomas are characterized morphologically by a homogeneous gray-yellow mass with frequent satellite nodules and irregular but well-defined margins.17,30 Central necrosis and fibrosis are also common.30 In the periductal-infiltrating type, tumor typically grows along the bile duct wall without mass formation, resulting in concentric mural thickening and proximal biliary dilation.30 Intraductal-growing papillary cholangiocarcinoma is characterized by the presence of intraluminal papillary or tubular polypoid tumors of the intra- or extrahepatic bile ducts, with partial obstruction and proximal biliary dilation.30
Cholangiocarcinoma
Case Presentation
A previously healthy 59-year-old man presents to his primary care physician with a 3-month history of dull right upper quadrant pain associated with weight loss. The patient is markedly cachectic and abdominal examination reveals upper quadrant tenderness. Laboratory exams are significant for elevated alkaline phosphatase (500 U/L; reference range 45–115 U/L), cancer antigen 19-9 (CA 19-9, 73 U/mL; reference range ≤ 37 U/mL), and carcinoembryonic antigen (CEA , 20 ng/mL; reference range for nonsmokers ≤ 3.0 ng/mL). Aspartate aminotransferase, alanine aminotransferase, total bilirubin, and coagulation studies are within normal range. Ultrasound demonstrates a homogeneous mass with irregular borders in the right lobe of the liver. Triphasic contrast-enhanced CT scan demonstrates a tumor with ragged rim enhancement at the periphery, and portal venous phase shows gradual centripetal enhancement of the tumor with capsular retraction. No abdominal lymph nodes or extrahepatic tumors are noted (Figure 1, Image A).
- What are the next diagnostic steps?
The most critical differential diagnosis of solid liver mass in patients without cirrhosis is cholangiocarcinoma and metastases from another primary site.32 Alternatively, when an intrahepatic lesion is noted on an imaging study in the setting of cirrhosis, the next diagnostic step is differentiation between cholangiocarcinoma and hepatocellular carcinoma (HCC).32 Triphasic contrast-enhanced CT and dynamic magnetic resonance imaging (MRI) are key diagnostic procedures.32,33 In the appropriate setting, classical imaging features in the arterial phase with washout in portal venous or delayed phase can be diagnostic of HCC and may obviate the need for a biopsy (Figure 2).
Typical radiographic features of cholangiocarcinoma include a hypodense hepatic lesion that can be either well-defined or infiltrative and is frequently associated with biliary dilatation (Figure 1, Image A).33 The dense fibrotic nature of the tumor may cause capsular retraction, which is seen in up to 20% of cases.17 This finding is highly suggestive of cholangiocarcinoma and is rarely present in HCC.33 Following contrast administration, there is peripheral (rim) enhancement throughout both arterial and venous phases.32–34 However, these classic features were present in only 70% of cases in one study.35 Although intrahepatic cholangiocarcinomas are most commonly hypovascular, small mass-forming intrahepatic cholangiocarcinomas can often be arterially hyperenhancing and mimic HCC.33 Tumor enhancement on delayed CT imaging has been correlated with survival. Asayama et al demonstrated that tumors that exhibited delayed enhancement on CT in more than two-thirds of their volume were associated with a worse prognosis.36
Patients without cirrhosis who present with a localized lesion of the liver should undergo extensive evaluation for a primary cancer site.37 CT of the chest, abdomen, and pelvis with contrast should be obtained.37 Additionally, mammogram and endoscopic evaluation with esophagogastroduodenoscopy (EGD) and colonoscopy should be included in the work-up.37
Preoperative tumor markers are also included in the work-up. All patients with a solid liver lesion should have serum alpha-fetoprotein (AFP) levels checked. AFP is a serum glycoprotein recognized as a marker for HCC and is reported to detect preclinical HCC.38 However, serum concentrations are normal in up to 40% of small HCCs.38 Although no specific marker for cholangiocarcinoma has yet been identified, the presence of certain tumor markers in the serum of patients may be of diagnostic value, especially in patients with PSC. CA 19-9 and CEA are the best studied. Elevated levels of CA 19-9 prior to treatment are associated with a poorer prognosis, and CA 19-9 concentrations greater than 1000 U/mL are consistent with advanced disease.39,40 One large series evaluated the diagnostic value of serum CEA levels in 333 patients with PSC, 13% of whom were diagnosed with cholangiocarcinoma.34 A serum CEA level greater than 5.2 ng/mL had a sensitivity of 68.0% and specificity of 81.5%.38
If a biopsy is obtained, appropriate immunohistochemistry (IHC) can facilitate the diagnosis. BTC is strongly positive for CK-7 and CK-19.41 CK-7 positivity is not specific and is also common among metastatic cancers of the lung and breast; therefore, in some cases cholangiocarcinoma may be a diagnosis of exclusion. Immunostaining for monoclonal CEA is diffusely positive in up to 75% of cases.41 An IHC panel consisting of Hep Par-1, arginase-1, monoclonal CEA, CK-7, CK-20, TTF-1, MOC-31, and CDX-2 has been proposed to optimize the differential diagnosis of HCC, metastatic adenocarcinoma, and cholangiocarcinoma.41
Case Continued
CT of the chest, abdomen, and pelvis reveals no concerns for metastasis and no evidence of primary cancer elsewhere. EGD and colonoscopy are clear. AFP levels are within normal limits (2 ng/mL). Biopsy is performed and demonstrates adenocarcinoma. IHC studies demonstrate cells positive for monoclonal CEA, CK-7, CK-19, and MOC-31, and negative for Napsin A, TTF-1, and CK-20.
- How is cholangiocarcinoma staged and classified?
The purpose of the staging system is to provide information on prognosis and guidance for therapy. Prognostic factors and the therapeutic approaches for BTC differ depending upon their location in the biliary tree. Accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated (Table 1 and Table 2).23
The Bismuth-Corlette classification is used to further classify perihilar cholangiocarcinoma according to patterns of hepatic duct involvement. Type I tumors are located below the confluence of the left and right hepatic ducts.42 Type II reach the confluence of the hepatic ducts.42 Type III occlude the common hepatic duct and either the right or left hepatic duct (IIIa and IIIb, respectively).42 Finally, type IV are multicentric, or involve the confluence and both the right and left hepatic ducts.42 Tumors that involve the common hepatic duct bifurcation are named Klatskin tumors.42
- What is the first-line treatment for localized cholangiocarcinomas?
Surgical resection is the only potentially curative treatment for localized cholangiocarcinoma, although fewer than 20% of patients are suitable for curative treatment, due to the presence of advanced disease at diagnosis.43,44 Available evidence supports the recommendation that resection with negative margins, regardless of extent, should be the goal of therapy for patients with potentially resectable disease.44 Extensive hepatic resections are often necessary to achieve clear margins since the majority of patients present with large masses. Substantial evidence corroborates that R0 resection is associated with better survival, whereas the benefit of wide compared to narrow (< 5–10 mm) margins is unclear.45 A recent analysis of 96 patients suggests that the proximal resection margin has more prognostic implications than distal margins.45
Surgical options and resectability criteria depend upon tumor location. Extent of tumor in the bile duct is one of the most important factors that determine resectability.17 Although multifocal liver tumors (including satellite lesions), lymph node metastases to the porta hepatis, and distant metastases are considered relative contraindications to surgery, surgical approaches can be considered in selected patients.43 Patient selection for surgery is facilitated by careful preoperative staging, which may include laparoscopy. Laparoscopic staging prior to resection may prevent unnecessary laparotomy in 30% to 45% of patients.42,46
- Is there a role for adjuvant treatment?
Recurrence following complete resection is a primary limitation for cure in BTC, which provides a rationale for the use of adjuvant therapy.47,48 In a sample of 79 patients with extrahepatic cholangiocarcinoma who underwent curative resection, the cumulative recurrence rate after 4 years was 56%.47 Initial recurrence at a distant site occurs in 40% to 50% of patients.48
Lymphovascular and perineural invasion, lymph node metastasis, and tumor size ≥ 5 cm have been reported as independent predictors of recurrence and mortality following resection.49 A 2017 meta-analysis which included 30 studies involving more than 22,499 patients reported a 41% reduction in the risk of death with adjuvant chemotherapy, which translated to a mean OS benefit of 4 months in an unselected population.49 Moreover, this study revealed inferior OS in patients given adjuvant radiation therapy (RT) in combination with chemotherapy.49 These results are in line with the previous meta-analysis by Horgan et al, which demonstrated that adjuvant RT seems to benefit only patients with R1 resections, with a possible detrimental effect in R0 disease.50 Therefore, adjuvant chemoradiation cannot be viewed as a standard practice following R0 resection, and should be reserved for those patients with positive margins (R1/ 2) to reduce local progression.
In the phase 3 BILCAP trial presented at ASCO 2017, 447 patients with completely resected cholangiocarcinoma or gallbladder cancer with adequate biliary drainage and Eastern Cooperative Oncology Group (ECOG) performance score ≤ 2 were randomly assigned to observation or capecitabine (1250 mg/m2 twice daily for days 1–14 every 21 days for 8 cycles).51 Surgical treatment achieved R0 resection in 62% of patients and 46% were node-negative. Median OS was 51 months for the capecitabine group and 36 months for the control arm (hazard ratio [HR] 0.80, 95% CI 0.63 to 1.04, P = 0.097). Analyses with adjustment for nodal status, grade of disease, and gender indicated a HR of 0.71 (P < 0.01). Median DFS was 25 months versus 18 months favoring the capecitabine group, and rates of grade 3 or 4 toxicity were less than anticipated. Following the results of this trial, adjuvant capecitabine should become the new standard of care.
- What is the treatment for locally advanced cholangiocarcinoma?
The optimal approach to patients with locally advanced unresectable cholangiocarcinoma has not been established. The prognosis for patients with either locally unresectable or locally recurrent disease is typically measured in months. Goals of palliative therapy are relief of symptoms and improvement in quality of life, and there is no role for surgical debulking.
Liver transplantation is a potentially curative option for selected patients with hilar or intrahepatic cholangiocarcinoma. Patients with lymph node-negative, non-disseminated, locally advanced hilar cholangiocarcinomas have 5-year survival rates ranging from 25% to 42% following transplantation.52 Retrospective data suggests that neoadjuvant chemoradiation followed by liver transplantation is highly effective for selected patients with hilar cholangiocarcinoma.52 However, these results require confirmation from prospective clinical evidence. It is important to recognize that liver transplantation plays no role in the management of distal cholangiocarcinoma or gallbladder cancer.
Rarely, patients with borderline resectable intrahepatic cholangiocarcinoma will have a sufficient response to chemotherapy to permit later resection, and, in such cases, starting with chemotherapy and then restaging to evaluate resectability is appropriate.54 A single-center, retrospective analysis including 186 patients by Le Roy et al evaluated survival in patients with locally advanced, unresectable intrahepatic cholangiocarcinoma who received primary chemotherapy, followed by surgery in those with secondary resectability.54 After a median of 6 cycles of chemotherapy, 53% of patients achieved resectability and underwent surgery with curative intent. These patients had similar short- and long-term results compared to patients with initially resectable intrahepatic cholangiocarcinoma who had surgery alone, with median OS reaching 24 months.54
Ablative radiotherapy is an additional option for localized inoperable intrahepatic cholangiocarcinoma. Tao and colleagues evaluated 79 consecutive patients with inoperable intrahepatic cholangiocarcinoma treated with definitive RT.55 Median tumor size was 7.9 cm and 89% of patients received chemotherapy before RT. Median OS was 30 months and 3-year OS was 44%. Radiation dose was the single most important prognostic factor, and higher doses correlated with improved local control and OS. A biologic equivalent dose (BED) greater than 80.5 Gy was identified as an ablative dose of RT for large intrahepatic cholangiocarcinomas. The 3-year OS for patients receiving BED greater than 80.5 Gy was 73% versus 38% for those receiving lower doses.
Case Continued
The patient is deemed to have resectable disease and undergoes surgical resection followed by adjuvant capecitabine for 8 cycles. Unfortunately, after 1 year, follow-up imaging identifies bilateral enlarging lung nodules. Biopsy is performed and confirms metastatic cholangiocarcinoma.
- What is the treatment for metastatic BTC?
The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. A benefit of chemotherapy over best supportive care for cholangiocarcinoma was demonstrated in an early phase 3 trial that randomly assigned 90 patients with advanced pancreatic or biliary cancer (37 with bile duct cancer) to receive either fluorouracil (FU) -based systemic chemotherapy or best supportive care. Results showed that chemotherapy significantly improved OS (6 months versus 2.5 months).56 Chemotherapy is also beneficial for patients with unresectable gallbladder cancer. In a single-center randomized study including 81 patients with unresectable gallbladder cancer, gemcitabine and oxaliplatin (GEMOX) improved progression-free survival (PFS) and OS compared to best supportive care.57 Treatment for metastatic cholangiocarcinoma and gallbladder cancer follows the same algorithm.
In 2010, cisplatin plus gemcitabine was established as a reference regimen for first-line therapy by the ABC-02 study, in which 410 patients with locally advanced or metastatic bile duct, gallbladder, or ampullary cancer were randomly assigned to 6 courses of cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2 on days 1 and 8, every 21 days) or gemcitabine alone (1000 mg/m2 days 1, 8, 15, every 28 days).58 OS was significantly greater with combination therapy (11.7 versus 8.1 months), and PFS also favored the combination arm (8 versus 5 months). Toxicity was comparable in both groups, with the exception of significantly higher rates of grade 3 or 4 neutropenia with gemcitabine plus cisplatin (25% versus 17%), and higher rates of grade 3 or 4 abnormal liver function with gemcitabine alone (27% versus 17%). Most quality-of-life scales showed a trend favoring combined therapy.58 A smaller, identically designed Japanese phase 3 randomized trial achieved similar results, demonstrating greater OS with cisplatin plus gemcitabine compared to gemcitabine alone (11.2 versus 7.7 months).59
The gemcitabine plus cisplatin combination has not been directly compared with other gemcitabine combinations in phase 3 trials. A pooled analysis of 104 trials of a variety of chemotherapy regimens in advanced biliary cancer concluded that the gemcitabine plus cisplatin regimen offered the highest rates of objective response and tumor control compared with either gemcitabine-free or cisplatin-free regimens.60 However, this did not translate into significant benefit in terms of either time to tumor progression or median OS. It is important to note that this analysis did not include results of the subsequent ABC-02 trial.
There is no standard treatment for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. There are no completed prospective phase 3 trials supporting the use of second-line chemotherapy after failure of first-line chemotherapy in BTC, and the selection of candidates for second-line therapy as well as the optimal regimen are not established.61 The ongoing phase 2 multicenter ABC-06 trial is evaluating oxaliplatin plus short-term infusional FU and leucovorin (FOLFOX) versus best supportive care for second-line therapy. In a systematic review including 23 studies (14 phase 2 clinical trials and 9 retrospective studies) with 761 patients with BTC, the median OS was 7.2 months.
The optimal selection of candidates for second-line chemotherapy is not established. Two independent studies suggest that patients who have a good performance status (0 or 1), disease control with the first-line chemotherapy, low CA 19-9 level, and possibly previous surgery on their primary tumor, have the longest survival with second-line chemotherapy. However, whether these characteristics predict for chemotherapy responsiveness or more favorable biologic behavior is not clear.62,63 No particular regimen has proved superior to any other, and the choice of second-line regimen remains empiric.
For patients with adequate performance status, examples of other conventional chemotherapy regimens with demonstrated activity that could be considered for second-line therapy include: FOLFOX or capecitabine, gemcitabine plus capecitabine, capecitabine plus cisplatin, or irinotecan plus short-term infusional FU and leucovorin (FOLFIRI) with or without bevacizumab.64 For selected patients, second-line molecularly targeted therapy using erlotinib plus bevacizumab may be considered. However, this regimen is very costly.64 Examples of other regimens with demonstrated activity in phase 2 trials include GEMOX, gemcitabine plus fluoropyrimidine, and fluoropyrimidine plus oxaliplatin or cisplatin.64
There is promising data from studies of targeted therapy for specific molecular subgroups. A recent phase 2 trial evaluated the activity of BGJ398, an orally bioavailable, selective, ATP-competitive pan inhibitor of human fibroblast growth factor receptor (FGFR) kinase, in patients with FGFR-altered advanced cholangiocarcinoma.65 The overall response rate was 14.8% (18.8% FGFR2 fusions only) and disease control rate was 75.4% (83.3% FGFR2 fusions only). All responsive tumors contained FGFR2 fusions. Adverse events were manageable, and grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%). Those included hyperphosphatemia, stomatitis, and palmar-plantar erythrodysesthesia. Javle and colleagues also identified HER2/neu blockade as a promising treatment strategy for gallbladder cancer patients with this gene amplification.66 This retrospective analysis included 9 patients with gallbladder cancer and 5 patients with cholangiocarcinoma who received HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab). In the gallbladder cancer group, HER2/neu gene amplification or overexpression was detected in 8 cases. These patients experienced disease stability (n = 3), partial response (n = 4), or complete response (n = 1) with HER2/neu–directed therapy. Median duration of response was 40 weeks. The cholangiocarcinoma cases treated in this series had no radiological responses despite HER2/neu mutations or amplification.
Gallbladder Cancer
Case Presentation
A 57-year-old woman from Chile presents with a 3-week history of progressive right upper quadrant abdominal pain. She denies nausea, vomiting, dysphagia, odynophagia, alterations in bowel habits, fever, or jaundice. Her past medical history is significant for obesity and hypertension. She has no history of smoking, alcohol, or illicit drug use. Laboratory studies show marked leukocytosis (23,800/µL) with neutrophilia (91%). Liver function test results are within normal limits. Ultrasound of the abdomen reveals gallbladder wall thickening and cholelithiasis.
The patient undergoes an uneventful laparoscopic cholecystectomy and is discharged from the hospital after 48 hours. Pathology report reveals a moderately differentiated adenocarcinoma of the gallbladder invading the perimuscular connective tissue (T2). No lymph nodes are identified in the specimen.
- What is the appropriate surgical management of gallbladder cancer?
Gallbladder cancer can be diagnosed preoperatively or can be found incidentally by intraoperative or pathological findings. In one large series, gallbladder cancer was incidentally found during 0.25% of laparoscopic cholecystectomies.67
For patients who are diagnosed with previously unsuspected gallbladder cancer by pathology findings, the extent of tumor invasion (T stage) indicates the need for re-resection (Figure 3).64
Alternatively, when gallbladder cancer is documented or suspected preoperatively, adequate imaging is important to identify patients with absolute contraindications to resection. Contraindications to surgery include metastasis, extensive involvement of the hepatoduodenal ligament, encasement of major vessels, and involvement of celiac, peripancreatic, periduodenal, or superior mesenteric nodes.72 Notwithstanding, retrospective series suggest individual patients may benefit, and surgical indications in advanced disease should be determined on an individual basis.73 Staging imaging should be obtained using multiphasic contrast-enhanced CT or MRI of the chest, abdomen, and pelvis. PET-scan can be used in selected cases where metastatic disease is suspected.64 Laparoscopic diagnostic staging should be considered prior to resection.64 This procedure can identify previously unknown contraindications to tumor resection in as much as 23% of patients, and the yield is significantly higher in locally advanced tumors.73
Patients with a diagnosis of potentially resectable, localized gallbladder cancer should be offered definitive surgery. Extended cholecystectomy is recommended for patients stage T2 or above. This procedure involves wedge resection of the gallbladder bed or a segmentectomy IVb/V and lymph node dissection, which should include the cystic duct, common bile duct, posterior superior pancreaticoduodenal lymph nodes, and those around the hepatoduodenal ligament.72 Bile duct excision should be performed if there is malignant involvement.64
Conclusion
BTCs are anatomically and clinically heterogeneous tumors. Prognostic factors and therapeutic approaches for BTCs differ depending upon their location in the biliary tree and, accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated. Surgical resection is the only potentially curative treatment for localized BTC. However, recurrence following complete resection is a primary limitation for cure, which provides a rationale for the use of adjuvant therapy. The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. Multiple randomized clinical trials have demonstrated a benefit of chemotherapy for metastatic disease. For patients with adequate performance status, second-line therapy can be considered, and data from studies that evaluated targeted therapy for specific molecular subgroups is promising.
1. Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2(Suppl 1):e000152.
2. Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2017 Oct 10. doi: 10.1038/nrclinonc.2017.157.
3. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist 2008;13:415–23.
4. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2014 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2017.
5. Torre LA, Siegel RL, Islami F, et al. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol 2017 Aug 18. doi: 10.1016/j.cgh.2017.08.017.
6. Lau CSM, Zywot A, Mahendraraj K, Chamberlain CS. Gallbladder carcinoma in the United States: a population based clinical outcomes study involving 22,343 patients from the Surveillance, Epidemiology, and End Result Database (1973–2013). HPB Surg 2017;2017:1532835. doi:10.1155/2017/1532835.
7. Hughes T, O’Connor T, Techasen A, et al. Opisthorchiasis and cholangiocarcinoma in Southeast Asia: an unresolved problem. Int J Gen Med 2017;10:227–37.
8. DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–62.
9. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9.
10. Yao KJ, Jabbour S, Parekh N, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117.
11. Choi SB, Kim KS, Choi JY, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol 2009;16:3048–56.
12. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96.
13. Duffy A, Capanu M, Abou-Alfa GK, et al. Gallbladder cancer (GBC): 10-year experience at Memorial Sloan-Kettering Cancer Centre (MSKCC). J Surg Oncol 2008;98:485–9.
14. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer — viewpoint of the IARC Working Group. N Engl J Med 2016;375:794–8.
15. Chen J, Han Y, Xu C, et al. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases. Eur J Cancer Prev 2015;24:89–99.
16. Larsson SC, Giovannucci EL, Wolk A. Sweetened beverage consumption and risk of biliary tract and gallbladder cancer in a prospective study. J Natl Cancer Inst 2016;108: doi: 10.1093/jnci/djw125.
17. Gore RM. Biliary tract neoplasms: diagnosis and staging. Cancer Imaging 2007;7(Special Issue A):S15–23.
18. Broome U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610–5.
19. Burak K, Angulo P, Pasha T, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523–6.
20. Rodrigues J, Diehl DL. Cholangiocarcinoma: clinical manifestations and diagnosis. Tech Gastrointest Endosc 2016;18:75–82.
21. Mitchell CH, Johnson PT, Fishman EK, et al. Features suggestive of gallbladder malignancy. J Comput Assist Tomogr 2014;38:235–41.
22. Beltz WR, Condon RE. Primary carcinoma of the gallbladder. Ann Surg 1974;180:180–4.
23. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512–22.
24. Patel T. Cholangiocarcinoma—controversies and challenges. Nat Rev Gastroenterol Hepatol 2011;8:189–200.
25. Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 1996;224:463–73.
26. Bartella I, Dufour JF. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J Gastrointestin Liver Dis 2015;24:481-9.
27. Yamaguchi K, Enjoji M. Carcinoma of the gallbladder: a clinicopathology of 103 patients and a newly proposed staging. Cancer 1988;62:1425–32.
28. Esposito I, Schirmacher P. Pathological aspects of cholangiocarcinoma. HPB. 2008;10:83–6.
29. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol 2016;5:62.
30. Chung YE, Kim MJ, Park YN, et al. Varying appearances of cholangiocarcinoma: radiologic-pathologic correlation. Radiographics 2009;29:683–700.
31. Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg 2003;10:288–91.
32. Rao PN. Nodule in liver: investigations, differential diagnosis and follow-up. J Clin Exp Hepatol 2014;4(Suppl 3):S57–62.
33. Kim TK, Lee E, Jang HJ. Imaging findings of mimickers of hepatocellular carcinoma. Clin Mol Hepatol 2015;21:326–43.
34. Hennedige TP, Neo WT, Venkatesh SK. Imaging of malignancies of the biliary tract- an update. Cancer Imaging 2014;14:14.
35. Kim SH, Lee CH, Kim BH, et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J Comput Assist Tomogr 2012;36:704–9.
36. Asayama Y, Yoshimitsu K, Irie H, et al. Delayed-phase dynamic CT enhancement as a prognostic factor for mass-forming intrahepatic cholangiocarcinoma. Radiology 2006;238:150–5.
37. National Comprehensive Cancer Network. Cancer of unknown primary. www.nccn.org/professionals/physician_gls/pdf/bone.pdf. Accessed 1 Dec 2017.
38. Kefeli A, Basyigit S, Yeniova AO. Diagnosis of hepatocellular carcinoma. In: Abdeldayem HM, ed. Updates in liver cancer. London: InTech; 2017.
39. Bergquist JR, Ivanics T, Storlie CB, et al. Implications of CA19-9 elevation for survival, staging, and treatment sequencing in intrahepatic cholangiocarcinoma: A national cohort analysis. J Surg Oncol 2016;114:475–82.
40. Chung YJ, Choi DW, Choi SH, et al. Prognostic factors following surgical resection of distal bile duct cancer. J Korean Surg Soc 2013;85:212–8.
41. Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol 2002;33:1175–81.
42. Paul A, Kaiser GM, Molmenti EP, et al. Klatskin tumors and the accuracy of the Bismuth-Corlette classification. Am Surg 2011;77:1695–9.
43. Cannavale A, Santoni M, Gazzetti M, et al. Updated management of malignant biliary tract tumors: an illustrative review. J Vasc Interv Radiol 2016;27:1056–69.
44. Matsuo K, Rocha FG, Ito K, et al. The Blumgart preoperative staging system for hilar cholangiocarcinoma: analysis of resectability and outcomes in 380 patients. J Am Coll Surg 2012;215:343–55.
45. Yoo T, Park SJ, Han SS, et al. Proximal resection margins: more prognostic than distal resection margins in patients undergoing hilar cholangiocarcinoma resection. Cancer Res Treat 2017 Nov 16; doi.org/10.4143/crt.2017.320.
46. Joseph S, Connor S, Garden OJ. Staging laparoscopy for cholangiocarcinoma. HPB 2008;10:116–9.
47. Jarnagin WR, Ruo L, Little SA, et al. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: implications for adjuvant therapeutic strategies. Cancer 2003;98:1689–700.
48. Kobayashi A, Miwa S, Nakata T, Miyagawa S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br J Surg 2010;97:56–64.
49. Ghidini M, Tomasello G, Botticelli A, et al. Adjuvant chemotherapy for resected biliary tract cancers: a systematic review and meta-analysis. HPB 2017;19:741–8.
50. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934–40.
51. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study [abstract]. J Clin Oncol 2017 35:15_suppl:4006-4006.
52. Darwish Murad S, Kim WR, Darnois DM, et al. Efficacy of neoadjuvant chemoradiation followed by liver transplantation for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88–98.
53. Sapisochin G, Facciuto M, Rubbia-Brandt L, et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016;64:1178–88.
54. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg 2017 Aug 31. doi: 10.1002/bjs.10641.
55. Tao R, Krishnan S, Bhosale PR, et al. Ablative radiotherapy doses lead to a substantial prolongation of survival in patients with inoperable intrahepatic cholangiocarcinoma: a retrospective dose response analysis. J Clin Oncol 2016;34:219–26.
56. Glimelius B, Hoffman K, SjÓdén PO, et al. 555 Palliative chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Eur J Cancer 1995;31:S118.
57. Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol 2010;28:4581–6.
58. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
59. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469–74.
60. Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
61. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38.
62. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290–7.
63. Fornaro L, Cereda S, Aprile G, et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br J Cancer 2014;110:2165–9.
64. National Comprehensive Cancer Network. Hepatobiliary cancer. www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. Accessed 12 Nov 2017.
65. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2017 Nov 28;JCO2017755009.
66. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58.
67. Konstantinidis IT, Deshpande V, Genevay M, et al. Trends in presentation and survival for gallbladder cancer during a period of more than 4 decades: a single-institution experience. Arch Surg 2009;144:441–47.
68. Singh S, Agarwal AK. Gallbladder cancer: the role of laparoscopy and radical resection. Ann Surg 2009;250:494–5.
69. Kapoor VK, Haribhakti SP. Extended cholecystectomy for carcinoma of the gall bladder. Trop Gastroenterol 1995;16:74–5.
70. Ethun CG, Postlewait LM, Le N, et al. Association of optimal time Interval to re-resection for incidental gallbladder cancer with overall survival: a multi-Institution analysis from the US extrahepatic biliary malignancy consortium. JAMA Surg 2017;152:143–9.
71. Goetze TO, Paolucci V. Benefits of reoperation of T2 and more advanced incidental gallbladder carcinoma: analysis of the German registry. Ann Surg 2008;247:104–8.
72. Nishio H, Nagino M, Ebata T, et al. Aggressive surgery for stage IV gallbladder carcinoma; what are the contraindications? J Hepatobiliary Pancreat Surg 2007;14:351–7.
73. Agarwal AK, Kalayarasan R, Javed A, et al. The role of staging laparoscopy in primary gallbladder cancer--an analysis of 409 patients: a prospective study to evaluate the role of staging laparoscopy in the management of gallbladder cancer. Ann Surg 2013;258:318–23.
1. Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2(Suppl 1):e000152.
2. Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2017 Oct 10. doi: 10.1038/nrclinonc.2017.157.
3. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist 2008;13:415–23.
4. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2014 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2017.
5. Torre LA, Siegel RL, Islami F, et al. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol 2017 Aug 18. doi: 10.1016/j.cgh.2017.08.017.
6. Lau CSM, Zywot A, Mahendraraj K, Chamberlain CS. Gallbladder carcinoma in the United States: a population based clinical outcomes study involving 22,343 patients from the Surveillance, Epidemiology, and End Result Database (1973–2013). HPB Surg 2017;2017:1532835. doi:10.1155/2017/1532835.
7. Hughes T, O’Connor T, Techasen A, et al. Opisthorchiasis and cholangiocarcinoma in Southeast Asia: an unresolved problem. Int J Gen Med 2017;10:227–37.
8. DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–62.
9. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9.
10. Yao KJ, Jabbour S, Parekh N, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117.
11. Choi SB, Kim KS, Choi JY, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol 2009;16:3048–56.
12. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96.
13. Duffy A, Capanu M, Abou-Alfa GK, et al. Gallbladder cancer (GBC): 10-year experience at Memorial Sloan-Kettering Cancer Centre (MSKCC). J Surg Oncol 2008;98:485–9.
14. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer — viewpoint of the IARC Working Group. N Engl J Med 2016;375:794–8.
15. Chen J, Han Y, Xu C, et al. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases. Eur J Cancer Prev 2015;24:89–99.
16. Larsson SC, Giovannucci EL, Wolk A. Sweetened beverage consumption and risk of biliary tract and gallbladder cancer in a prospective study. J Natl Cancer Inst 2016;108: doi: 10.1093/jnci/djw125.
17. Gore RM. Biliary tract neoplasms: diagnosis and staging. Cancer Imaging 2007;7(Special Issue A):S15–23.
18. Broome U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610–5.
19. Burak K, Angulo P, Pasha T, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523–6.
20. Rodrigues J, Diehl DL. Cholangiocarcinoma: clinical manifestations and diagnosis. Tech Gastrointest Endosc 2016;18:75–82.
21. Mitchell CH, Johnson PT, Fishman EK, et al. Features suggestive of gallbladder malignancy. J Comput Assist Tomogr 2014;38:235–41.
22. Beltz WR, Condon RE. Primary carcinoma of the gallbladder. Ann Surg 1974;180:180–4.
23. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512–22.
24. Patel T. Cholangiocarcinoma—controversies and challenges. Nat Rev Gastroenterol Hepatol 2011;8:189–200.
25. Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 1996;224:463–73.
26. Bartella I, Dufour JF. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J Gastrointestin Liver Dis 2015;24:481-9.
27. Yamaguchi K, Enjoji M. Carcinoma of the gallbladder: a clinicopathology of 103 patients and a newly proposed staging. Cancer 1988;62:1425–32.
28. Esposito I, Schirmacher P. Pathological aspects of cholangiocarcinoma. HPB. 2008;10:83–6.
29. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol 2016;5:62.
30. Chung YE, Kim MJ, Park YN, et al. Varying appearances of cholangiocarcinoma: radiologic-pathologic correlation. Radiographics 2009;29:683–700.
31. Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg 2003;10:288–91.
32. Rao PN. Nodule in liver: investigations, differential diagnosis and follow-up. J Clin Exp Hepatol 2014;4(Suppl 3):S57–62.
33. Kim TK, Lee E, Jang HJ. Imaging findings of mimickers of hepatocellular carcinoma. Clin Mol Hepatol 2015;21:326–43.
34. Hennedige TP, Neo WT, Venkatesh SK. Imaging of malignancies of the biliary tract- an update. Cancer Imaging 2014;14:14.
35. Kim SH, Lee CH, Kim BH, et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J Comput Assist Tomogr 2012;36:704–9.
36. Asayama Y, Yoshimitsu K, Irie H, et al. Delayed-phase dynamic CT enhancement as a prognostic factor for mass-forming intrahepatic cholangiocarcinoma. Radiology 2006;238:150–5.
37. National Comprehensive Cancer Network. Cancer of unknown primary. www.nccn.org/professionals/physician_gls/pdf/bone.pdf. Accessed 1 Dec 2017.
38. Kefeli A, Basyigit S, Yeniova AO. Diagnosis of hepatocellular carcinoma. In: Abdeldayem HM, ed. Updates in liver cancer. London: InTech; 2017.
39. Bergquist JR, Ivanics T, Storlie CB, et al. Implications of CA19-9 elevation for survival, staging, and treatment sequencing in intrahepatic cholangiocarcinoma: A national cohort analysis. J Surg Oncol 2016;114:475–82.
40. Chung YJ, Choi DW, Choi SH, et al. Prognostic factors following surgical resection of distal bile duct cancer. J Korean Surg Soc 2013;85:212–8.
41. Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol 2002;33:1175–81.
42. Paul A, Kaiser GM, Molmenti EP, et al. Klatskin tumors and the accuracy of the Bismuth-Corlette classification. Am Surg 2011;77:1695–9.
43. Cannavale A, Santoni M, Gazzetti M, et al. Updated management of malignant biliary tract tumors: an illustrative review. J Vasc Interv Radiol 2016;27:1056–69.
44. Matsuo K, Rocha FG, Ito K, et al. The Blumgart preoperative staging system for hilar cholangiocarcinoma: analysis of resectability and outcomes in 380 patients. J Am Coll Surg 2012;215:343–55.
45. Yoo T, Park SJ, Han SS, et al. Proximal resection margins: more prognostic than distal resection margins in patients undergoing hilar cholangiocarcinoma resection. Cancer Res Treat 2017 Nov 16; doi.org/10.4143/crt.2017.320.
46. Joseph S, Connor S, Garden OJ. Staging laparoscopy for cholangiocarcinoma. HPB 2008;10:116–9.
47. Jarnagin WR, Ruo L, Little SA, et al. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: implications for adjuvant therapeutic strategies. Cancer 2003;98:1689–700.
48. Kobayashi A, Miwa S, Nakata T, Miyagawa S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br J Surg 2010;97:56–64.
49. Ghidini M, Tomasello G, Botticelli A, et al. Adjuvant chemotherapy for resected biliary tract cancers: a systematic review and meta-analysis. HPB 2017;19:741–8.
50. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934–40.
51. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study [abstract]. J Clin Oncol 2017 35:15_suppl:4006-4006.
52. Darwish Murad S, Kim WR, Darnois DM, et al. Efficacy of neoadjuvant chemoradiation followed by liver transplantation for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88–98.
53. Sapisochin G, Facciuto M, Rubbia-Brandt L, et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016;64:1178–88.
54. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg 2017 Aug 31. doi: 10.1002/bjs.10641.
55. Tao R, Krishnan S, Bhosale PR, et al. Ablative radiotherapy doses lead to a substantial prolongation of survival in patients with inoperable intrahepatic cholangiocarcinoma: a retrospective dose response analysis. J Clin Oncol 2016;34:219–26.
56. Glimelius B, Hoffman K, SjÓdén PO, et al. 555 Palliative chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Eur J Cancer 1995;31:S118.
57. Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol 2010;28:4581–6.
58. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
59. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469–74.
60. Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
61. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38.
62. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290–7.
63. Fornaro L, Cereda S, Aprile G, et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br J Cancer 2014;110:2165–9.
64. National Comprehensive Cancer Network. Hepatobiliary cancer. www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. Accessed 12 Nov 2017.
65. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2017 Nov 28;JCO2017755009.
66. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58.
67. Konstantinidis IT, Deshpande V, Genevay M, et al. Trends in presentation and survival for gallbladder cancer during a period of more than 4 decades: a single-institution experience. Arch Surg 2009;144:441–47.
68. Singh S, Agarwal AK. Gallbladder cancer: the role of laparoscopy and radical resection. Ann Surg 2009;250:494–5.
69. Kapoor VK, Haribhakti SP. Extended cholecystectomy for carcinoma of the gall bladder. Trop Gastroenterol 1995;16:74–5.
70. Ethun CG, Postlewait LM, Le N, et al. Association of optimal time Interval to re-resection for incidental gallbladder cancer with overall survival: a multi-Institution analysis from the US extrahepatic biliary malignancy consortium. JAMA Surg 2017;152:143–9.
71. Goetze TO, Paolucci V. Benefits of reoperation of T2 and more advanced incidental gallbladder carcinoma: analysis of the German registry. Ann Surg 2008;247:104–8.
72. Nishio H, Nagino M, Ebata T, et al. Aggressive surgery for stage IV gallbladder carcinoma; what are the contraindications? J Hepatobiliary Pancreat Surg 2007;14:351–7.
73. Agarwal AK, Kalayarasan R, Javed A, et al. The role of staging laparoscopy in primary gallbladder cancer--an analysis of 409 patients: a prospective study to evaluate the role of staging laparoscopy in the management of gallbladder cancer. Ann Surg 2013;258:318–23.
Polycythemia Vera and Essential Thrombocythemia: Current Management
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1. 
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4. 
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1.
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4.
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1.
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4.
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
Rapid weight loss, irritability, and nausea after restarting ADHD treatment
CASE Medication management
Mr. L, age 58, presents to the outpatient psychiatric clinic seeking treatment for attention-deficit/hyperactivity disorder (ADHD), which was first diagnosed 11 years ago. Since discontinuing his ADHD medication, lisdexamfetamine 60 mg/d, 8 months ago, he has not been completing tasks and has been distracted in his job as a limousine driver. Mr. L says that when he was taking the medication, “I could focus and prioritize.” He reports that he has trouble retaining information and is easily distracted. He says he generally is organized with appointments and keeping track of things but is messy, forgetful, tardy, and impatient. Procrastination is an ongoing problem. He denies misplacing things or being impulsive. Mr. L reports that as a child he was frequently reprimanded for talking in class. He states, “I get in trouble even now for talking too much.”
Mr. L is cooperative and polite, maintains good eye contact, and is alert. No psychomotor abnormalities are noted. His speech is spontaneous and coherent, with normal rate, rhythm, and volume. He reports that his mood is “all right,” and denies suicidal or homicidal ideation. His insight is full, judgment is intact, and thought is linear and logical. Mr. L sleeps 5 hours at night and takes a nap during the day, but his energy varies.
His psychiatric history is negative for suicide attempts or hospitalizations. Mr. L denies a history of major depressive episodes, manic symptoms, hallucinations, or delusions. Anxiety history is negative for excessive worrying, obsessions and compulsions, and panic attacks. Mr. L has no family history of mental illness or substance abuse, and he denies any personal history of drug use. He stopped using tobacco 14 years ago. Mr. L says he drinks 3 caffeinated drinks a day and 2 glasses of wine once a week. Previous medications included
A review of systems is negative. Vital signs are unremarkable. A recent electrocardiogram (EKG) showed normal sinus rhythm. Thyroid-stimulating hormone, comprehensive metabolic panel (CMP), lipids, iron, vitamin B12, folate, complete blood count (CBC), hemoglobin A1c, and urine analysis are normal, except for mildly elevated low-density lipoprotein. Testing for hepatitis C is negative.
The previous diagnosis of ADHD is confirmed, and Mr. L is started on
[polldaddy:9928295]
The author’s observations
Anxiety, irritability, agitation, and palpitations can all be symptoms of stimulant medications.1,2 There are numerous other iatrogenic causes, including steroid-based asthma treatments, thyroid medications, antidepressants in bipolar patients, and caffeine-based migraine treatments. Mr. L’s theory that his 15-lb weight loss was the result of his methylphenidate ER dose being too high was a reasonable one. Often, medication doses need to be adjusted with weight changes. His decrease in energy during the day could be explained by the methylphenidate ER controlling his hyperactive symptoms, which include high energy. At night, when the medication wears off, his hyperactivity symptoms could be returning, which would account for the increase in energy when he gets home from work. Although longer-acting stimulants tend to have a more benign adverse effects profile, they can cause insomnia if they are still in the patient’s system at bedtime. Shorter-acting stimulants wear off quickly but can be advantageous for patients who want to target concentration during certain times of day, such as for school and homework.
TREATMENT A surprising cause
The next month, Mr. L presents to the emergency room complaining of jitteriness, headache, and tingling in his fingers, and is evaluated for suspected carbon monoxide (CO) poisoning. Three months earlier, he had noted the odor of exhaust fumes in the limousine he drives 7 days a week. He took it to the mechanic twice for evaluation, but no cause was found. Despite his concerns, he continued to drive the car until an older client, in frail health, suddenly became short of breath and developed chest pain shortly after entering his vehicle, on a day when the odor was particularly bad. Before that, a family of passengers had complained of headaches upon entering his vehicle. The third time he brought his car to be checked, the mechanic identified an exhaust system leak.

[polldaddy:9928298]
The author’s observations

Work-up for suspected CO poisoning includes ABG, COHb level, CBC, basic metabolic panel, EKG, cardiac enzymes, and chest radiography, as well as other laboratory tests as deemed appropriate. Treatment includes oxygen by mask for low-level poisoning.
High levels of poisoning may require hyperbaric oxygen, which should be considered for patients who are unconscious or have an abnormal score on the Carbon Monoxide Neuropsychological Screening Battery, COHb of >40%, signs of cardiac ischemia or arrhythmia, history of ischemic heart disease with COHb level >20%, recurrent symptoms for up to 3 weeks, or symptoms that have not resolved with normobaric oxygen after 4 to 6 hours.9 Any pregnant woman with CO poisoning should receive hyperbaric therapy.10
OUTCOME Lasting improvement
Mr. L presents for follow-up in the psychiatric clinic 3 weeks after his emergency room visit. After his limousine was repaired, his symptoms resolved. He no longer experiences fatigue during the day with higher energy at night, palpitations, jitteriness, headache, or tingling. His concentration has improved, so he opts to stick with the 18-mg dose of methylphenidate ER rather than increase it to the initial dose. He places a CO detector in his vehicle, which proves to be a good decision when it gives him a warning that the exhaust leak had not been properly repaired.
[polldaddy:9928299]
The author’s observations
Although the correct cause of Mr. L’s symptoms was found incidentally, this case is an important reminder to always consider medical causes in the differential diagnosis. We are taught in medical school to look first for horses (more likely causes), not zebras (less likely causes), but sometimes zebras do occur. Be mindful that medical causes should be considered not only for symptoms of primary illnesses, but also for symptoms thought to be caused by adverse effects of medications. The differential diagnosis for Mr. L’s symptoms (palpitations, agitation, anxiety, irritability, weight loss, fatigue, nausea, and headache) included metabolic and endocrine abnormalities (thyroid disease, pheochromocytoma, hypoglycemia); psychiatric conditions (panic, bipolar disorder, depression); substance abuse (caffeine, cocaine, amphetamines); immune disorders; cardiac disorders; malignancy; toxic exposure; infectious sources; and nutritional deficiencies. CO poisoning can cause many of these symptoms (Table 2).1,2,8
Intentional CO poisoning should be considered in an obtunded or unconscious patient with depression. Patients may consider CO poisoning a more peaceful way to complete suicide than shooting, cutting, or hanging. As for unintentional poisoning, clinical suspicion can be increased by time of year, occupation, locale, and smoking status. Winter months increase risk because of the high use of heating devices, cars warming up in the garage, closed fireplace flues, and vehicle tailpipes blocked by snow. As in Mr. L’s case, occupation also may increase suspicion; drivers, mechanics, tollbooth operators, parking attendants, miners, and firefighters are all at increased risk for CO poisoning. Regarding locale, polluted urban environments as well as cold climates requiring heating sources cause higher risks for CO exposure. Rarely, excessive smoking can result in CO poisoning. The author once had a patient with schizophrenia who was admitted to the hospital with delirium. It was determined that he had CO poisoning from his 5-pack-a-day smoking habit.
Psychiatric patients often have the frustrating experience of their physical symptoms being attributed to psychiatric causes, which results in major medical issues being overlooked. We psychiatrists can fall into the same trap of overlooking medical illnesses, as indicated in this case, where Mr. L’s CO poisoning initially was attributed to adverse effects of his psychiatric medication.
1. Drugs.com. Amphetamine side effects. https://www.drugs.com/sfx/amphetamine-side-effects.html. Accessed December 7, 2017.
2. Golmirzaei J, Mahboobi H, Yazdanparast M, et al. Psychopharmacology of attention-deficit hyperactivity disorder: effects and side effects. Curr Pharm Des. 2016;22(5):590-594.
3. Bleecker ML. Carbon monoxide intoxication. Handb Clin Neurol. 2015;131(3):191-203.
4. Carter D. Carbon monoxide: the forgotten killer. http://scot.nhs.uk/sehd/cmo/CMO(1998)19.pdf. Published September 7, 1998. Accessed January 10, 2018.
5. Stewart RD, Baretta ED, Platte LR, et al. Carboxyhemoglobin levels in American blood donors. JAMA. 1974;229(9):1187-1195.
6. AA1Car. Troubleshoot odors & smells inside your car. http://www.aa1car.com/library/troubleshoot_odors.htm. Accessed December 7, 2017.
7. Rodkey FL, O’Neal JD, Collison HA, et al. Relative affinity of hemoglobin S and hemoglobin A for carbon monoxide and oxygen. Clin Chem. 1974;20(1):83-84.
8. Kirkpatrick JN. Occult carbon monoxide poisoning. West J Med. 1987;146(1):52-56.
9. Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med. 1998;339(22):1603-1608.
10. Guzman JA. Carbon monoxide poisoning. Critical Care Clin. 2012;28(4):537-548.
CASE Medication management
Mr. L, age 58, presents to the outpatient psychiatric clinic seeking treatment for attention-deficit/hyperactivity disorder (ADHD), which was first diagnosed 11 years ago. Since discontinuing his ADHD medication, lisdexamfetamine 60 mg/d, 8 months ago, he has not been completing tasks and has been distracted in his job as a limousine driver. Mr. L says that when he was taking the medication, “I could focus and prioritize.” He reports that he has trouble retaining information and is easily distracted. He says he generally is organized with appointments and keeping track of things but is messy, forgetful, tardy, and impatient. Procrastination is an ongoing problem. He denies misplacing things or being impulsive. Mr. L reports that as a child he was frequently reprimanded for talking in class. He states, “I get in trouble even now for talking too much.”
Mr. L is cooperative and polite, maintains good eye contact, and is alert. No psychomotor abnormalities are noted. His speech is spontaneous and coherent, with normal rate, rhythm, and volume. He reports that his mood is “all right,” and denies suicidal or homicidal ideation. His insight is full, judgment is intact, and thought is linear and logical. Mr. L sleeps 5 hours at night and takes a nap during the day, but his energy varies.
His psychiatric history is negative for suicide attempts or hospitalizations. Mr. L denies a history of major depressive episodes, manic symptoms, hallucinations, or delusions. Anxiety history is negative for excessive worrying, obsessions and compulsions, and panic attacks. Mr. L has no family history of mental illness or substance abuse, and he denies any personal history of drug use. He stopped using tobacco 14 years ago. Mr. L says he drinks 3 caffeinated drinks a day and 2 glasses of wine once a week. Previous medications included
A review of systems is negative. Vital signs are unremarkable. A recent electrocardiogram (EKG) showed normal sinus rhythm. Thyroid-stimulating hormone, comprehensive metabolic panel (CMP), lipids, iron, vitamin B12, folate, complete blood count (CBC), hemoglobin A1c, and urine analysis are normal, except for mildly elevated low-density lipoprotein. Testing for hepatitis C is negative.
The previous diagnosis of ADHD is confirmed, and Mr. L is started on
[polldaddy:9928295]
The author’s observations
Anxiety, irritability, agitation, and palpitations can all be symptoms of stimulant medications.1,2 There are numerous other iatrogenic causes, including steroid-based asthma treatments, thyroid medications, antidepressants in bipolar patients, and caffeine-based migraine treatments. Mr. L’s theory that his 15-lb weight loss was the result of his methylphenidate ER dose being too high was a reasonable one. Often, medication doses need to be adjusted with weight changes. His decrease in energy during the day could be explained by the methylphenidate ER controlling his hyperactive symptoms, which include high energy. At night, when the medication wears off, his hyperactivity symptoms could be returning, which would account for the increase in energy when he gets home from work. Although longer-acting stimulants tend to have a more benign adverse effects profile, they can cause insomnia if they are still in the patient’s system at bedtime. Shorter-acting stimulants wear off quickly but can be advantageous for patients who want to target concentration during certain times of day, such as for school and homework.
TREATMENT A surprising cause
The next month, Mr. L presents to the emergency room complaining of jitteriness, headache, and tingling in his fingers, and is evaluated for suspected carbon monoxide (CO) poisoning. Three months earlier, he had noted the odor of exhaust fumes in the limousine he drives 7 days a week. He took it to the mechanic twice for evaluation, but no cause was found. Despite his concerns, he continued to drive the car until an older client, in frail health, suddenly became short of breath and developed chest pain shortly after entering his vehicle, on a day when the odor was particularly bad. Before that, a family of passengers had complained of headaches upon entering his vehicle. The third time he brought his car to be checked, the mechanic identified an exhaust system leak.
[polldaddy:9928298]
The author’s observations
Work-up for suspected CO poisoning includes ABG, COHb level, CBC, basic metabolic panel, EKG, cardiac enzymes, and chest radiography, as well as other laboratory tests as deemed appropriate. Treatment includes oxygen by mask for low-level poisoning.
High levels of poisoning may require hyperbaric oxygen, which should be considered for patients who are unconscious or have an abnormal score on the Carbon Monoxide Neuropsychological Screening Battery, COHb of >40%, signs of cardiac ischemia or arrhythmia, history of ischemic heart disease with COHb level >20%, recurrent symptoms for up to 3 weeks, or symptoms that have not resolved with normobaric oxygen after 4 to 6 hours.9 Any pregnant woman with CO poisoning should receive hyperbaric therapy.10
OUTCOME Lasting improvement
Mr. L presents for follow-up in the psychiatric clinic 3 weeks after his emergency room visit. After his limousine was repaired, his symptoms resolved. He no longer experiences fatigue during the day with higher energy at night, palpitations, jitteriness, headache, or tingling. His concentration has improved, so he opts to stick with the 18-mg dose of methylphenidate ER rather than increase it to the initial dose. He places a CO detector in his vehicle, which proves to be a good decision when it gives him a warning that the exhaust leak had not been properly repaired.
[polldaddy:9928299]
The author’s observations
Although the correct cause of Mr. L’s symptoms was found incidentally, this case is an important reminder to always consider medical causes in the differential diagnosis. We are taught in medical school to look first for horses (more likely causes), not zebras (less likely causes), but sometimes zebras do occur. Be mindful that medical causes should be considered not only for symptoms of primary illnesses, but also for symptoms thought to be caused by adverse effects of medications. The differential diagnosis for Mr. L’s symptoms (palpitations, agitation, anxiety, irritability, weight loss, fatigue, nausea, and headache) included metabolic and endocrine abnormalities (thyroid disease, pheochromocytoma, hypoglycemia); psychiatric conditions (panic, bipolar disorder, depression); substance abuse (caffeine, cocaine, amphetamines); immune disorders; cardiac disorders; malignancy; toxic exposure; infectious sources; and nutritional deficiencies. CO poisoning can cause many of these symptoms (Table 2).1,2,8
Intentional CO poisoning should be considered in an obtunded or unconscious patient with depression. Patients may consider CO poisoning a more peaceful way to complete suicide than shooting, cutting, or hanging. As for unintentional poisoning, clinical suspicion can be increased by time of year, occupation, locale, and smoking status. Winter months increase risk because of the high use of heating devices, cars warming up in the garage, closed fireplace flues, and vehicle tailpipes blocked by snow. As in Mr. L’s case, occupation also may increase suspicion; drivers, mechanics, tollbooth operators, parking attendants, miners, and firefighters are all at increased risk for CO poisoning. Regarding locale, polluted urban environments as well as cold climates requiring heating sources cause higher risks for CO exposure. Rarely, excessive smoking can result in CO poisoning. The author once had a patient with schizophrenia who was admitted to the hospital with delirium. It was determined that he had CO poisoning from his 5-pack-a-day smoking habit.
Psychiatric patients often have the frustrating experience of their physical symptoms being attributed to psychiatric causes, which results in major medical issues being overlooked. We psychiatrists can fall into the same trap of overlooking medical illnesses, as indicated in this case, where Mr. L’s CO poisoning initially was attributed to adverse effects of his psychiatric medication.
CASE Medication management
Mr. L, age 58, presents to the outpatient psychiatric clinic seeking treatment for attention-deficit/hyperactivity disorder (ADHD), which was first diagnosed 11 years ago. Since discontinuing his ADHD medication, lisdexamfetamine 60 mg/d, 8 months ago, he has not been completing tasks and has been distracted in his job as a limousine driver. Mr. L says that when he was taking the medication, “I could focus and prioritize.” He reports that he has trouble retaining information and is easily distracted. He says he generally is organized with appointments and keeping track of things but is messy, forgetful, tardy, and impatient. Procrastination is an ongoing problem. He denies misplacing things or being impulsive. Mr. L reports that as a child he was frequently reprimanded for talking in class. He states, “I get in trouble even now for talking too much.”
Mr. L is cooperative and polite, maintains good eye contact, and is alert. No psychomotor abnormalities are noted. His speech is spontaneous and coherent, with normal rate, rhythm, and volume. He reports that his mood is “all right,” and denies suicidal or homicidal ideation. His insight is full, judgment is intact, and thought is linear and logical. Mr. L sleeps 5 hours at night and takes a nap during the day, but his energy varies.
His psychiatric history is negative for suicide attempts or hospitalizations. Mr. L denies a history of major depressive episodes, manic symptoms, hallucinations, or delusions. Anxiety history is negative for excessive worrying, obsessions and compulsions, and panic attacks. Mr. L has no family history of mental illness or substance abuse, and he denies any personal history of drug use. He stopped using tobacco 14 years ago. Mr. L says he drinks 3 caffeinated drinks a day and 2 glasses of wine once a week. Previous medications included
A review of systems is negative. Vital signs are unremarkable. A recent electrocardiogram (EKG) showed normal sinus rhythm. Thyroid-stimulating hormone, comprehensive metabolic panel (CMP), lipids, iron, vitamin B12, folate, complete blood count (CBC), hemoglobin A1c, and urine analysis are normal, except for mildly elevated low-density lipoprotein. Testing for hepatitis C is negative.
The previous diagnosis of ADHD is confirmed, and Mr. L is started on
[polldaddy:9928295]
The author’s observations
Anxiety, irritability, agitation, and palpitations can all be symptoms of stimulant medications.1,2 There are numerous other iatrogenic causes, including steroid-based asthma treatments, thyroid medications, antidepressants in bipolar patients, and caffeine-based migraine treatments. Mr. L’s theory that his 15-lb weight loss was the result of his methylphenidate ER dose being too high was a reasonable one. Often, medication doses need to be adjusted with weight changes. His decrease in energy during the day could be explained by the methylphenidate ER controlling his hyperactive symptoms, which include high energy. At night, when the medication wears off, his hyperactivity symptoms could be returning, which would account for the increase in energy when he gets home from work. Although longer-acting stimulants tend to have a more benign adverse effects profile, they can cause insomnia if they are still in the patient’s system at bedtime. Shorter-acting stimulants wear off quickly but can be advantageous for patients who want to target concentration during certain times of day, such as for school and homework.
TREATMENT A surprising cause
The next month, Mr. L presents to the emergency room complaining of jitteriness, headache, and tingling in his fingers, and is evaluated for suspected carbon monoxide (CO) poisoning. Three months earlier, he had noted the odor of exhaust fumes in the limousine he drives 7 days a week. He took it to the mechanic twice for evaluation, but no cause was found. Despite his concerns, he continued to drive the car until an older client, in frail health, suddenly became short of breath and developed chest pain shortly after entering his vehicle, on a day when the odor was particularly bad. Before that, a family of passengers had complained of headaches upon entering his vehicle. The third time he brought his car to be checked, the mechanic identified an exhaust system leak.
[polldaddy:9928298]
The author’s observations
Work-up for suspected CO poisoning includes ABG, COHb level, CBC, basic metabolic panel, EKG, cardiac enzymes, and chest radiography, as well as other laboratory tests as deemed appropriate. Treatment includes oxygen by mask for low-level poisoning.
High levels of poisoning may require hyperbaric oxygen, which should be considered for patients who are unconscious or have an abnormal score on the Carbon Monoxide Neuropsychological Screening Battery, COHb of >40%, signs of cardiac ischemia or arrhythmia, history of ischemic heart disease with COHb level >20%, recurrent symptoms for up to 3 weeks, or symptoms that have not resolved with normobaric oxygen after 4 to 6 hours.9 Any pregnant woman with CO poisoning should receive hyperbaric therapy.10
OUTCOME Lasting improvement
Mr. L presents for follow-up in the psychiatric clinic 3 weeks after his emergency room visit. After his limousine was repaired, his symptoms resolved. He no longer experiences fatigue during the day with higher energy at night, palpitations, jitteriness, headache, or tingling. His concentration has improved, so he opts to stick with the 18-mg dose of methylphenidate ER rather than increase it to the initial dose. He places a CO detector in his vehicle, which proves to be a good decision when it gives him a warning that the exhaust leak had not been properly repaired.
[polldaddy:9928299]
The author’s observations
Although the correct cause of Mr. L’s symptoms was found incidentally, this case is an important reminder to always consider medical causes in the differential diagnosis. We are taught in medical school to look first for horses (more likely causes), not zebras (less likely causes), but sometimes zebras do occur. Be mindful that medical causes should be considered not only for symptoms of primary illnesses, but also for symptoms thought to be caused by adverse effects of medications. The differential diagnosis for Mr. L’s symptoms (palpitations, agitation, anxiety, irritability, weight loss, fatigue, nausea, and headache) included metabolic and endocrine abnormalities (thyroid disease, pheochromocytoma, hypoglycemia); psychiatric conditions (panic, bipolar disorder, depression); substance abuse (caffeine, cocaine, amphetamines); immune disorders; cardiac disorders; malignancy; toxic exposure; infectious sources; and nutritional deficiencies. CO poisoning can cause many of these symptoms (Table 2).1,2,8
Intentional CO poisoning should be considered in an obtunded or unconscious patient with depression. Patients may consider CO poisoning a more peaceful way to complete suicide than shooting, cutting, or hanging. As for unintentional poisoning, clinical suspicion can be increased by time of year, occupation, locale, and smoking status. Winter months increase risk because of the high use of heating devices, cars warming up in the garage, closed fireplace flues, and vehicle tailpipes blocked by snow. As in Mr. L’s case, occupation also may increase suspicion; drivers, mechanics, tollbooth operators, parking attendants, miners, and firefighters are all at increased risk for CO poisoning. Regarding locale, polluted urban environments as well as cold climates requiring heating sources cause higher risks for CO exposure. Rarely, excessive smoking can result in CO poisoning. The author once had a patient with schizophrenia who was admitted to the hospital with delirium. It was determined that he had CO poisoning from his 5-pack-a-day smoking habit.
Psychiatric patients often have the frustrating experience of their physical symptoms being attributed to psychiatric causes, which results in major medical issues being overlooked. We psychiatrists can fall into the same trap of overlooking medical illnesses, as indicated in this case, where Mr. L’s CO poisoning initially was attributed to adverse effects of his psychiatric medication.
1. Drugs.com. Amphetamine side effects. https://www.drugs.com/sfx/amphetamine-side-effects.html. Accessed December 7, 2017.
2. Golmirzaei J, Mahboobi H, Yazdanparast M, et al. Psychopharmacology of attention-deficit hyperactivity disorder: effects and side effects. Curr Pharm Des. 2016;22(5):590-594.
3. Bleecker ML. Carbon monoxide intoxication. Handb Clin Neurol. 2015;131(3):191-203.
4. Carter D. Carbon monoxide: the forgotten killer. http://scot.nhs.uk/sehd/cmo/CMO(1998)19.pdf. Published September 7, 1998. Accessed January 10, 2018.
5. Stewart RD, Baretta ED, Platte LR, et al. Carboxyhemoglobin levels in American blood donors. JAMA. 1974;229(9):1187-1195.
6. AA1Car. Troubleshoot odors & smells inside your car. http://www.aa1car.com/library/troubleshoot_odors.htm. Accessed December 7, 2017.
7. Rodkey FL, O’Neal JD, Collison HA, et al. Relative affinity of hemoglobin S and hemoglobin A for carbon monoxide and oxygen. Clin Chem. 1974;20(1):83-84.
8. Kirkpatrick JN. Occult carbon monoxide poisoning. West J Med. 1987;146(1):52-56.
9. Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med. 1998;339(22):1603-1608.
10. Guzman JA. Carbon monoxide poisoning. Critical Care Clin. 2012;28(4):537-548.
1. Drugs.com. Amphetamine side effects. https://www.drugs.com/sfx/amphetamine-side-effects.html. Accessed December 7, 2017.
2. Golmirzaei J, Mahboobi H, Yazdanparast M, et al. Psychopharmacology of attention-deficit hyperactivity disorder: effects and side effects. Curr Pharm Des. 2016;22(5):590-594.
3. Bleecker ML. Carbon monoxide intoxication. Handb Clin Neurol. 2015;131(3):191-203.
4. Carter D. Carbon monoxide: the forgotten killer. http://scot.nhs.uk/sehd/cmo/CMO(1998)19.pdf. Published September 7, 1998. Accessed January 10, 2018.
5. Stewart RD, Baretta ED, Platte LR, et al. Carboxyhemoglobin levels in American blood donors. JAMA. 1974;229(9):1187-1195.
6. AA1Car. Troubleshoot odors & smells inside your car. http://www.aa1car.com/library/troubleshoot_odors.htm. Accessed December 7, 2017.
7. Rodkey FL, O’Neal JD, Collison HA, et al. Relative affinity of hemoglobin S and hemoglobin A for carbon monoxide and oxygen. Clin Chem. 1974;20(1):83-84.
8. Kirkpatrick JN. Occult carbon monoxide poisoning. West J Med. 1987;146(1):52-56.
9. Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med. 1998;339(22):1603-1608.
10. Guzman JA. Carbon monoxide poisoning. Critical Care Clin. 2012;28(4):537-548.
Antipsychotics for obsessive-compulsive disorder: Weighing risks vs benefits
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of

Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of
Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of
Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
Using pharmacogenetics guidelines when prescribing: What’s available
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3

CPIC provides guidance for implementing pharmacogenomics

In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6

Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3
CPIC provides guidance for implementing pharmacogenomics
In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6
Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3
CPIC provides guidance for implementing pharmacogenomics
In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6
Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9

A 95-year-old man with treatment-resistant depression
CASE Depressed, avoidant
Mr. R, age 95, has a history of recurrent major depressive disorder. He presents to the emergency department with depressive symptoms that began 6 weeks ago. His symptoms include depressed mood, hopelessness, anhedonia, anxiety, and insomnia. Co-occurring anorexia nervosa has resulted in a 20-lb weight loss. He denies suicidal ideation.
A mental status examination reveals profound psychomotor agitation, dysphoric mood, tearfulness, and mood-congruent delusions. Mr. R’s Mini-Mental State Examination (MMSE) score is 14/30; his Hamilton Depression Rating Scale (HAM-D) score is 21, indicating severe depression (19 to 22). However, the examiner feels that these scores may not reflect an accurate assessment because Mr. R gave flippant responses and did not cooperate during the interview. Physical examination is unremarkable. Previous medication trials included buspirone, escitalopram, and risperidone; none of these medications successfully alleviated his depressive symptoms.
On admission, Mr. R is given oral mirtazapine, 15 mg/d, and quetiapine, 25 mg/d, to target depressive mood, insomnia, and weight loss. Urgent intervention is indicated because his depressive symptoms are profoundly causing failure to thrive and are compromising his physical health. Mr. R’s deterioration concerns the physician team. Because of a history of failed pharmacotherapy trials, the team reassesses Mr. R’s treatment options.
[polldaddy:9903171]
The authors’ observations
The physician team recommends that Mr. R undergo ECT to obtain rapid relief from his depressive symptoms. After discussion of the potential risks and benefits, Mr. R agrees to this treatment. Quetiapine is discontinued prior to initiating ECT to avoid unnecessary medications; mirtazapine is continued.
Mr. R’s lack of response to previous antidepressants and significant deterioration were concerning. The physicians wanted to avoid higher-dose medications because of the risk of falls or somnolence. Their clinical experience and the literature supporting ECT for patients of Mr. R’s age lead them to select ECT as the most appropriate therapeutic option.
ECT has no absolute contraindications.1 The rate of ECT use in the United States has fluctuated over time because of factors unrelated to the efficacy and availability of ECT or alternative treatments.2 This form of intervention is also somewhat stigmatized.
Some psychiatrists are reluctant to prescribe ECT for geriatric patients because of concerns of potential neurocognitive or medical complications and risks during anesthesia. However, in the United States, older patients with depression are more likely to be treated with ECT than their younger counterparts.3 ECT usually induces greater immediate efficacy than antidepressants.4
Evidence supports using ECT in older patients
Multiple studies have found that ECT is a rapid, safe, and efficacious intervention for treating older persons with depression. Patients age >60 who receive ECT plus pharmacotherapy have lower HAM-D scores than those receiving pharmacotherapy alone.5 Overall, the rates of remission for depression range from 50% to 70%; yet geriatric patients who receive only ECT have response rates around 90%.6 Older age, presence of psychotic symptoms, and shorter duration of illness can predict a rapidly positive ECT response.7
When treated with ECT, older patients, including those age >85, have fewer subsequent episodes of depression compared with those who receive pharmacotherapy alone.1 Older individuals with physical illness or cognitive impairment respond to and tolerate ECT much like younger patients.6 Older patients receiving ECT may experience less cognitive decline than younger ones.7 Those in their ninth decade of life with treatment-resistant depression, psychotic features, and post-stroke depression often respond robustly with improvement following ECT.8
Remission rates also depend on the technique of administration. Interactions between electrode placement and stimulus parameter dosage affect efficacy and adverse effects.9 Right-sided, unilateral ECT induces less cognitive dysfunction compared with bilateral electrode placement,9 but bilateral ECT is more clinically effective.10 However, the efficacy of right-sided ECT is more dose-sensitive, and some data suggest that suboptimal response is due to insufficient stimulus dosages.11 One double-blind randomized controlled trial documented that when using a high-dose stimulus parameter, unilateral ECT is as effective as bilateral ECT.12 When there is a suboptimal response to unilateral ECT, bilateral ECT might be beneficial.12,13 For preventing relapse in older patients, increasing the interval between ECT treatments is more effective than stopping ECT abruptly.13
[polldaddy:9903172]

Indications of ECT

ECT is indicated for patients with severe depression, mania, and other conditions (Table).14 The most common indication for ECT in older persons is a history of treatment-resistant depression, with melancholia, psychosis, or suicidal ideations.1-6,12 There are also age-related and clinical factors to consider with ECT. This treatment provides a safe, rapid remission for patients age >65, even after adjusting for somatic conditions, duration of illness, medication resistance, or case severity.15 Compared with younger patients, older adults may not tolerate antidepressants as well because of age-related pharmacokinetic alterations, including increased sensitivity to anticholinergic and/or hypotensive effects.1
Factors that favor ECT include a previous good response to it; patient preference; and an indication for rapid intervention, such as suicidality, catatonia, dehydration, malnutrition, or a suboptimal result from pharmacotherapy.3 Mortality among individuals age >85 who receive ECT reportedly is lower than that among their counterparts who receive alternative treatments.16 ECT has been administered safely and effectively in patients with comorbid medical illnesses such as stroke, cerebral aneurysm, cardiovascular disease with ischemia or arrhythmia, dementia, and osteoporosis.17
Neurocognitive effects
Reports on the effects of ECT on neurocognitive functioning have varied. In some studies, performance improved or did not change in severely depressed older patients who received ECT.18,19 In older people who receive ECT, MMSE scores often return to baseline by the end of treatment.20 There often is only mild transient cognitive impairment in patients with late-life depression who receive ECT. Areas of concern include attention span, orientation, and speed of mental processing.20 Physicians should conduct cognitive tests before, during, and after ECT sessions to monitor their patient’s mental status.20
Cognitive stability can be maintained by administering ECT twice a week; applying right-sided, unilateral electrode placement; and using short, ultra-brief stimulus pulse width parameters.21 Cognitive impairment induced by ECT is not associated with age in geriatric patients with depression.22 Older adults who experienced longer postictal reorientation time periods have better outcomes than others who reach orientation faster; their intellectual impairment returned to baseline.20 Falling is another complication associated with ECT. A longitudinal cohort study found the incident of falls among patients receiving ECT was 13%.22 Risk factors for falls during a course of ECT include the number of treatments and the presence of coexisting Parkinson’s disease.23
OUTCOME Improvement
Mr. R receives 8 sessions of right-sided, unilateral ECT with an individualized dosage titration method. Treatments are completed with a stimulus intensity at 6 times seizure threshold, with an ultra-brief pulse width at 0.3 milliseconds. Mr. R’s mood and affect begin to improve after 3 ECT sessions. His MMSE score increases to 28/30 (Figure). His clinical improvement is progressively sustained; he develops an increasingly jovial attitude and experiences less anxiety. Mr. R’s confidence, appetite, and sleep also improve. There are no complications with treatment, and Mr. R has no complaints. After 8 ECT sessions, Mr. R has no affective symptoms and does not experience any cognitive impairment.
The authors’ observations
Depression among older people is a growing public health concern. It is a leading cause of disability, and often leads to nursing home placement.24 ECT is a safe, effective treatment for late-life depression, but is underutilized in patients age >75 because of concerns for cognitive impairment.6 However, there is evidence that response rates to ECT are higher in patients ages 45 to 85, compared with young individuals ages 18 to 45.25 ECT is a viable intervention for older depressed patients, particularly for those who do not tolerate or fail to respond to pharmacotherapy. Many of these patients are at risk for drug-induced toxicities or interactions or suicide.1
1. Kerner N, Prudic J. Current electroconvulsive therapy practice and research in the geriatric population. Neuropsychiatry (London). 2014;4(1):33-54.
2. Dombrovski AY, Mulsant BH. The evidence for electroconvulsive therapy (ECT) in the treatment of severe late-life depression. ECT: the preferred treatment for severe depression in late life. Int Psychogeriatr. 2007;19(1):10-14,27-35; discussion 24-26.
3. Olfson M, Marcus S, Sackeim HA, et al. Use of ECT for the inpatient treatment of recurrent major depression. Am J Psychiatry. 1998;155(1):22-29.
4. Salzman C, Wong E, Wright BC. Drug and ECT treatment of depression in the elderly, 1996-2001: a literature review. Biol Psychiatry. 2002;52(3):265-284.
5. Kellner CH, Husain MM, Knapp RG, et al; CORE/PRIDE Work Group. A novel strategy for continuation ect in geriatric depression: phase 2 of the PRIDE study. A
6. Tew JD Jr, Mulsant BH, Haskett RF, et al. Acute efficacy of ECT in the treatment of major depression in the old-old. Am J Psychiatry. 1999;156(12):1865-1870.
7. Dombrovski AY, Mulsant BH, Haskett RF, et al. Predictors of remission after electroconvulsive therapy in unipolar major depression. J Clin Psychiatry. 2005;66(8):1043-1049.
8. Charles K. UpToDate. Unipolar major depression in adults: indications for efficacy of electroconvulsive therapy (ECT). https://www.uptodate.com/contents/unipolar-major-depression-in-adults-indications-for-and-efficacy-of-electroconvulsive-therapy-ect. Updated May 16, 2017. Accessed November 26, 2017.
9. Sackeim HA, Prudic J, Devanand DP, et al. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425-434.
10. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
11. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357(19):1939-1945.
12. Stoppe A, Louzã M, Rosa M, et al. Fixed high dose electroconvulsive therapy in elderly with depression: a double-blind, randomized comparison of efficacy and tolerability between unilateral and bilateral electrode placement. J ECT. 2006;22(2):92-99.
13. Geduldig ET, Kellner CH. Electroconvulsive therapy in the elderly: new findings in geriatric depression. Curr Psychiatry Rep. 2016;18(4):40.
14. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(suppl 4):1-45.
15. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23(3):274-282.
16. Philibert RA, Richards L, Lynch CF, et al. Effect of ECT on mortality and clinical outcome in geriatric unipolar depression. J Clin Psychiatry. 1995;56(9):390-394.
17. Tomac TA, Rummans TA, Pileggi TS, et al. Safety and efficacy of electroconvulsive therapy in patients over age 85. Am J Geriatr Psychiatry. 1997;5(2):126-130.
18. Verwijk E, Comijs HC, Kok RM, et al. Short and long-term neurocognitive functioning after electroconvulsive therapy in depressed elderly: a prospective naturalistic study. Int Psychogeriatr. 2014;26(2):315-324.
19. Flint AJ, Gagnon N. Effective use of electroconvulsive therapy in late life depression. Can J Psychiatry. 2002;47(8):734-741.
20. Bjolseth TM, Engedal K, Benth JS, et al. Speed of recovery from disorientation may predict the treatment outcome of electroconvulsive therapy (ECT) in elderly patients with major depression. J Affect Disord. 2016;190:178-186.
21. Sackeim HA, Prudic J, Nobler MS, et al. Ultra-brief pulse ECT and the affective and cognitive consequences of ECT. J ECT. 2001;17(1):77.
22. Bjolseth TM, Engedal K, Benth JS, et al. Baseline cognitive function does not predict the treatment outcome of electroconvulsive therapy (ECT) in late-life depression. J Affect Disord. 2015;185:67-75.
23. de Carle AJ, Kohn R. Electroconvulsive therapy and falls in the elderly. J ECT. 2000;16(3):252-257.
24. Hoover DR, Siegel M, Lucas J, et al. Depression in the first year of stay for elderly long-term nursing home residents in the USA. Int Psychogeriatr. 2010;22:1161.
25. O’Connor MK, Knapp R, Husain M, et al. The influence of age on the response of major depression to electroconvulsive therapy: a C.O.R.E. Report. Am J Geriatr Psychiatry. 2001; 9:382.
CASE Depressed, avoidant
Mr. R, age 95, has a history of recurrent major depressive disorder. He presents to the emergency department with depressive symptoms that began 6 weeks ago. His symptoms include depressed mood, hopelessness, anhedonia, anxiety, and insomnia. Co-occurring anorexia nervosa has resulted in a 20-lb weight loss. He denies suicidal ideation.
A mental status examination reveals profound psychomotor agitation, dysphoric mood, tearfulness, and mood-congruent delusions. Mr. R’s Mini-Mental State Examination (MMSE) score is 14/30; his Hamilton Depression Rating Scale (HAM-D) score is 21, indicating severe depression (19 to 22). However, the examiner feels that these scores may not reflect an accurate assessment because Mr. R gave flippant responses and did not cooperate during the interview. Physical examination is unremarkable. Previous medication trials included buspirone, escitalopram, and risperidone; none of these medications successfully alleviated his depressive symptoms.
On admission, Mr. R is given oral mirtazapine, 15 mg/d, and quetiapine, 25 mg/d, to target depressive mood, insomnia, and weight loss. Urgent intervention is indicated because his depressive symptoms are profoundly causing failure to thrive and are compromising his physical health. Mr. R’s deterioration concerns the physician team. Because of a history of failed pharmacotherapy trials, the team reassesses Mr. R’s treatment options.
[polldaddy:9903171]
The authors’ observations
The physician team recommends that Mr. R undergo ECT to obtain rapid relief from his depressive symptoms. After discussion of the potential risks and benefits, Mr. R agrees to this treatment. Quetiapine is discontinued prior to initiating ECT to avoid unnecessary medications; mirtazapine is continued.
Mr. R’s lack of response to previous antidepressants and significant deterioration were concerning. The physicians wanted to avoid higher-dose medications because of the risk of falls or somnolence. Their clinical experience and the literature supporting ECT for patients of Mr. R’s age lead them to select ECT as the most appropriate therapeutic option.
ECT has no absolute contraindications.1 The rate of ECT use in the United States has fluctuated over time because of factors unrelated to the efficacy and availability of ECT or alternative treatments.2 This form of intervention is also somewhat stigmatized.
Some psychiatrists are reluctant to prescribe ECT for geriatric patients because of concerns of potential neurocognitive or medical complications and risks during anesthesia. However, in the United States, older patients with depression are more likely to be treated with ECT than their younger counterparts.3 ECT usually induces greater immediate efficacy than antidepressants.4
Evidence supports using ECT in older patients
Multiple studies have found that ECT is a rapid, safe, and efficacious intervention for treating older persons with depression. Patients age >60 who receive ECT plus pharmacotherapy have lower HAM-D scores than those receiving pharmacotherapy alone.5 Overall, the rates of remission for depression range from 50% to 70%; yet geriatric patients who receive only ECT have response rates around 90%.6 Older age, presence of psychotic symptoms, and shorter duration of illness can predict a rapidly positive ECT response.7
When treated with ECT, older patients, including those age >85, have fewer subsequent episodes of depression compared with those who receive pharmacotherapy alone.1 Older individuals with physical illness or cognitive impairment respond to and tolerate ECT much like younger patients.6 Older patients receiving ECT may experience less cognitive decline than younger ones.7 Those in their ninth decade of life with treatment-resistant depression, psychotic features, and post-stroke depression often respond robustly with improvement following ECT.8
Remission rates also depend on the technique of administration. Interactions between electrode placement and stimulus parameter dosage affect efficacy and adverse effects.9 Right-sided, unilateral ECT induces less cognitive dysfunction compared with bilateral electrode placement,9 but bilateral ECT is more clinically effective.10 However, the efficacy of right-sided ECT is more dose-sensitive, and some data suggest that suboptimal response is due to insufficient stimulus dosages.11 One double-blind randomized controlled trial documented that when using a high-dose stimulus parameter, unilateral ECT is as effective as bilateral ECT.12 When there is a suboptimal response to unilateral ECT, bilateral ECT might be beneficial.12,13 For preventing relapse in older patients, increasing the interval between ECT treatments is more effective than stopping ECT abruptly.13
[polldaddy:9903172]
Indications of ECT
ECT is indicated for patients with severe depression, mania, and other conditions (Table).14 The most common indication for ECT in older persons is a history of treatment-resistant depression, with melancholia, psychosis, or suicidal ideations.1-6,12 There are also age-related and clinical factors to consider with ECT. This treatment provides a safe, rapid remission for patients age >65, even after adjusting for somatic conditions, duration of illness, medication resistance, or case severity.15 Compared with younger patients, older adults may not tolerate antidepressants as well because of age-related pharmacokinetic alterations, including increased sensitivity to anticholinergic and/or hypotensive effects.1
Factors that favor ECT include a previous good response to it; patient preference; and an indication for rapid intervention, such as suicidality, catatonia, dehydration, malnutrition, or a suboptimal result from pharmacotherapy.3 Mortality among individuals age >85 who receive ECT reportedly is lower than that among their counterparts who receive alternative treatments.16 ECT has been administered safely and effectively in patients with comorbid medical illnesses such as stroke, cerebral aneurysm, cardiovascular disease with ischemia or arrhythmia, dementia, and osteoporosis.17
Neurocognitive effects
Reports on the effects of ECT on neurocognitive functioning have varied. In some studies, performance improved or did not change in severely depressed older patients who received ECT.18,19 In older people who receive ECT, MMSE scores often return to baseline by the end of treatment.20 There often is only mild transient cognitive impairment in patients with late-life depression who receive ECT. Areas of concern include attention span, orientation, and speed of mental processing.20 Physicians should conduct cognitive tests before, during, and after ECT sessions to monitor their patient’s mental status.20
Cognitive stability can be maintained by administering ECT twice a week; applying right-sided, unilateral electrode placement; and using short, ultra-brief stimulus pulse width parameters.21 Cognitive impairment induced by ECT is not associated with age in geriatric patients with depression.22 Older adults who experienced longer postictal reorientation time periods have better outcomes than others who reach orientation faster; their intellectual impairment returned to baseline.20 Falling is another complication associated with ECT. A longitudinal cohort study found the incident of falls among patients receiving ECT was 13%.22 Risk factors for falls during a course of ECT include the number of treatments and the presence of coexisting Parkinson’s disease.23
OUTCOME Improvement
Mr. R receives 8 sessions of right-sided, unilateral ECT with an individualized dosage titration method. Treatments are completed with a stimulus intensity at 6 times seizure threshold, with an ultra-brief pulse width at 0.3 milliseconds. Mr. R’s mood and affect begin to improve after 3 ECT sessions. His MMSE score increases to 28/30 (Figure). His clinical improvement is progressively sustained; he develops an increasingly jovial attitude and experiences less anxiety. Mr. R’s confidence, appetite, and sleep also improve. There are no complications with treatment, and Mr. R has no complaints. After 8 ECT sessions, Mr. R has no affective symptoms and does not experience any cognitive impairment.
The authors’ observations
Depression among older people is a growing public health concern. It is a leading cause of disability, and often leads to nursing home placement.24 ECT is a safe, effective treatment for late-life depression, but is underutilized in patients age >75 because of concerns for cognitive impairment.6 However, there is evidence that response rates to ECT are higher in patients ages 45 to 85, compared with young individuals ages 18 to 45.25 ECT is a viable intervention for older depressed patients, particularly for those who do not tolerate or fail to respond to pharmacotherapy. Many of these patients are at risk for drug-induced toxicities or interactions or suicide.1
CASE Depressed, avoidant
Mr. R, age 95, has a history of recurrent major depressive disorder. He presents to the emergency department with depressive symptoms that began 6 weeks ago. His symptoms include depressed mood, hopelessness, anhedonia, anxiety, and insomnia. Co-occurring anorexia nervosa has resulted in a 20-lb weight loss. He denies suicidal ideation.
A mental status examination reveals profound psychomotor agitation, dysphoric mood, tearfulness, and mood-congruent delusions. Mr. R’s Mini-Mental State Examination (MMSE) score is 14/30; his Hamilton Depression Rating Scale (HAM-D) score is 21, indicating severe depression (19 to 22). However, the examiner feels that these scores may not reflect an accurate assessment because Mr. R gave flippant responses and did not cooperate during the interview. Physical examination is unremarkable. Previous medication trials included buspirone, escitalopram, and risperidone; none of these medications successfully alleviated his depressive symptoms.
On admission, Mr. R is given oral mirtazapine, 15 mg/d, and quetiapine, 25 mg/d, to target depressive mood, insomnia, and weight loss. Urgent intervention is indicated because his depressive symptoms are profoundly causing failure to thrive and are compromising his physical health. Mr. R’s deterioration concerns the physician team. Because of a history of failed pharmacotherapy trials, the team reassesses Mr. R’s treatment options.
[polldaddy:9903171]
The authors’ observations
The physician team recommends that Mr. R undergo ECT to obtain rapid relief from his depressive symptoms. After discussion of the potential risks and benefits, Mr. R agrees to this treatment. Quetiapine is discontinued prior to initiating ECT to avoid unnecessary medications; mirtazapine is continued.
Mr. R’s lack of response to previous antidepressants and significant deterioration were concerning. The physicians wanted to avoid higher-dose medications because of the risk of falls or somnolence. Their clinical experience and the literature supporting ECT for patients of Mr. R’s age lead them to select ECT as the most appropriate therapeutic option.
ECT has no absolute contraindications.1 The rate of ECT use in the United States has fluctuated over time because of factors unrelated to the efficacy and availability of ECT or alternative treatments.2 This form of intervention is also somewhat stigmatized.
Some psychiatrists are reluctant to prescribe ECT for geriatric patients because of concerns of potential neurocognitive or medical complications and risks during anesthesia. However, in the United States, older patients with depression are more likely to be treated with ECT than their younger counterparts.3 ECT usually induces greater immediate efficacy than antidepressants.4
Evidence supports using ECT in older patients
Multiple studies have found that ECT is a rapid, safe, and efficacious intervention for treating older persons with depression. Patients age >60 who receive ECT plus pharmacotherapy have lower HAM-D scores than those receiving pharmacotherapy alone.5 Overall, the rates of remission for depression range from 50% to 70%; yet geriatric patients who receive only ECT have response rates around 90%.6 Older age, presence of psychotic symptoms, and shorter duration of illness can predict a rapidly positive ECT response.7
When treated with ECT, older patients, including those age >85, have fewer subsequent episodes of depression compared with those who receive pharmacotherapy alone.1 Older individuals with physical illness or cognitive impairment respond to and tolerate ECT much like younger patients.6 Older patients receiving ECT may experience less cognitive decline than younger ones.7 Those in their ninth decade of life with treatment-resistant depression, psychotic features, and post-stroke depression often respond robustly with improvement following ECT.8
Remission rates also depend on the technique of administration. Interactions between electrode placement and stimulus parameter dosage affect efficacy and adverse effects.9 Right-sided, unilateral ECT induces less cognitive dysfunction compared with bilateral electrode placement,9 but bilateral ECT is more clinically effective.10 However, the efficacy of right-sided ECT is more dose-sensitive, and some data suggest that suboptimal response is due to insufficient stimulus dosages.11 One double-blind randomized controlled trial documented that when using a high-dose stimulus parameter, unilateral ECT is as effective as bilateral ECT.12 When there is a suboptimal response to unilateral ECT, bilateral ECT might be beneficial.12,13 For preventing relapse in older patients, increasing the interval between ECT treatments is more effective than stopping ECT abruptly.13
[polldaddy:9903172]
Indications of ECT
ECT is indicated for patients with severe depression, mania, and other conditions (Table).14 The most common indication for ECT in older persons is a history of treatment-resistant depression, with melancholia, psychosis, or suicidal ideations.1-6,12 There are also age-related and clinical factors to consider with ECT. This treatment provides a safe, rapid remission for patients age >65, even after adjusting for somatic conditions, duration of illness, medication resistance, or case severity.15 Compared with younger patients, older adults may not tolerate antidepressants as well because of age-related pharmacokinetic alterations, including increased sensitivity to anticholinergic and/or hypotensive effects.1
Factors that favor ECT include a previous good response to it; patient preference; and an indication for rapid intervention, such as suicidality, catatonia, dehydration, malnutrition, or a suboptimal result from pharmacotherapy.3 Mortality among individuals age >85 who receive ECT reportedly is lower than that among their counterparts who receive alternative treatments.16 ECT has been administered safely and effectively in patients with comorbid medical illnesses such as stroke, cerebral aneurysm, cardiovascular disease with ischemia or arrhythmia, dementia, and osteoporosis.17
Neurocognitive effects
Reports on the effects of ECT on neurocognitive functioning have varied. In some studies, performance improved or did not change in severely depressed older patients who received ECT.18,19 In older people who receive ECT, MMSE scores often return to baseline by the end of treatment.20 There often is only mild transient cognitive impairment in patients with late-life depression who receive ECT. Areas of concern include attention span, orientation, and speed of mental processing.20 Physicians should conduct cognitive tests before, during, and after ECT sessions to monitor their patient’s mental status.20
Cognitive stability can be maintained by administering ECT twice a week; applying right-sided, unilateral electrode placement; and using short, ultra-brief stimulus pulse width parameters.21 Cognitive impairment induced by ECT is not associated with age in geriatric patients with depression.22 Older adults who experienced longer postictal reorientation time periods have better outcomes than others who reach orientation faster; their intellectual impairment returned to baseline.20 Falling is another complication associated with ECT. A longitudinal cohort study found the incident of falls among patients receiving ECT was 13%.22 Risk factors for falls during a course of ECT include the number of treatments and the presence of coexisting Parkinson’s disease.23
OUTCOME Improvement
Mr. R receives 8 sessions of right-sided, unilateral ECT with an individualized dosage titration method. Treatments are completed with a stimulus intensity at 6 times seizure threshold, with an ultra-brief pulse width at 0.3 milliseconds. Mr. R’s mood and affect begin to improve after 3 ECT sessions. His MMSE score increases to 28/30 (Figure). His clinical improvement is progressively sustained; he develops an increasingly jovial attitude and experiences less anxiety. Mr. R’s confidence, appetite, and sleep also improve. There are no complications with treatment, and Mr. R has no complaints. After 8 ECT sessions, Mr. R has no affective symptoms and does not experience any cognitive impairment.
The authors’ observations
Depression among older people is a growing public health concern. It is a leading cause of disability, and often leads to nursing home placement.24 ECT is a safe, effective treatment for late-life depression, but is underutilized in patients age >75 because of concerns for cognitive impairment.6 However, there is evidence that response rates to ECT are higher in patients ages 45 to 85, compared with young individuals ages 18 to 45.25 ECT is a viable intervention for older depressed patients, particularly for those who do not tolerate or fail to respond to pharmacotherapy. Many of these patients are at risk for drug-induced toxicities or interactions or suicide.1
1. Kerner N, Prudic J. Current electroconvulsive therapy practice and research in the geriatric population. Neuropsychiatry (London). 2014;4(1):33-54.
2. Dombrovski AY, Mulsant BH. The evidence for electroconvulsive therapy (ECT) in the treatment of severe late-life depression. ECT: the preferred treatment for severe depression in late life. Int Psychogeriatr. 2007;19(1):10-14,27-35; discussion 24-26.
3. Olfson M, Marcus S, Sackeim HA, et al. Use of ECT for the inpatient treatment of recurrent major depression. Am J Psychiatry. 1998;155(1):22-29.
4. Salzman C, Wong E, Wright BC. Drug and ECT treatment of depression in the elderly, 1996-2001: a literature review. Biol Psychiatry. 2002;52(3):265-284.
5. Kellner CH, Husain MM, Knapp RG, et al; CORE/PRIDE Work Group. A novel strategy for continuation ect in geriatric depression: phase 2 of the PRIDE study. A
6. Tew JD Jr, Mulsant BH, Haskett RF, et al. Acute efficacy of ECT in the treatment of major depression in the old-old. Am J Psychiatry. 1999;156(12):1865-1870.
7. Dombrovski AY, Mulsant BH, Haskett RF, et al. Predictors of remission after electroconvulsive therapy in unipolar major depression. J Clin Psychiatry. 2005;66(8):1043-1049.
8. Charles K. UpToDate. Unipolar major depression in adults: indications for efficacy of electroconvulsive therapy (ECT). https://www.uptodate.com/contents/unipolar-major-depression-in-adults-indications-for-and-efficacy-of-electroconvulsive-therapy-ect. Updated May 16, 2017. Accessed November 26, 2017.
9. Sackeim HA, Prudic J, Devanand DP, et al. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425-434.
10. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
11. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357(19):1939-1945.
12. Stoppe A, Louzã M, Rosa M, et al. Fixed high dose electroconvulsive therapy in elderly with depression: a double-blind, randomized comparison of efficacy and tolerability between unilateral and bilateral electrode placement. J ECT. 2006;22(2):92-99.
13. Geduldig ET, Kellner CH. Electroconvulsive therapy in the elderly: new findings in geriatric depression. Curr Psychiatry Rep. 2016;18(4):40.
14. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(suppl 4):1-45.
15. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23(3):274-282.
16. Philibert RA, Richards L, Lynch CF, et al. Effect of ECT on mortality and clinical outcome in geriatric unipolar depression. J Clin Psychiatry. 1995;56(9):390-394.
17. Tomac TA, Rummans TA, Pileggi TS, et al. Safety and efficacy of electroconvulsive therapy in patients over age 85. Am J Geriatr Psychiatry. 1997;5(2):126-130.
18. Verwijk E, Comijs HC, Kok RM, et al. Short and long-term neurocognitive functioning after electroconvulsive therapy in depressed elderly: a prospective naturalistic study. Int Psychogeriatr. 2014;26(2):315-324.
19. Flint AJ, Gagnon N. Effective use of electroconvulsive therapy in late life depression. Can J Psychiatry. 2002;47(8):734-741.
20. Bjolseth TM, Engedal K, Benth JS, et al. Speed of recovery from disorientation may predict the treatment outcome of electroconvulsive therapy (ECT) in elderly patients with major depression. J Affect Disord. 2016;190:178-186.
21. Sackeim HA, Prudic J, Nobler MS, et al. Ultra-brief pulse ECT and the affective and cognitive consequences of ECT. J ECT. 2001;17(1):77.
22. Bjolseth TM, Engedal K, Benth JS, et al. Baseline cognitive function does not predict the treatment outcome of electroconvulsive therapy (ECT) in late-life depression. J Affect Disord. 2015;185:67-75.
23. de Carle AJ, Kohn R. Electroconvulsive therapy and falls in the elderly. J ECT. 2000;16(3):252-257.
24. Hoover DR, Siegel M, Lucas J, et al. Depression in the first year of stay for elderly long-term nursing home residents in the USA. Int Psychogeriatr. 2010;22:1161.
25. O’Connor MK, Knapp R, Husain M, et al. The influence of age on the response of major depression to electroconvulsive therapy: a C.O.R.E. Report. Am J Geriatr Psychiatry. 2001; 9:382.
1. Kerner N, Prudic J. Current electroconvulsive therapy practice and research in the geriatric population. Neuropsychiatry (London). 2014;4(1):33-54.
2. Dombrovski AY, Mulsant BH. The evidence for electroconvulsive therapy (ECT) in the treatment of severe late-life depression. ECT: the preferred treatment for severe depression in late life. Int Psychogeriatr. 2007;19(1):10-14,27-35; discussion 24-26.
3. Olfson M, Marcus S, Sackeim HA, et al. Use of ECT for the inpatient treatment of recurrent major depression. Am J Psychiatry. 1998;155(1):22-29.
4. Salzman C, Wong E, Wright BC. Drug and ECT treatment of depression in the elderly, 1996-2001: a literature review. Biol Psychiatry. 2002;52(3):265-284.
5. Kellner CH, Husain MM, Knapp RG, et al; CORE/PRIDE Work Group. A novel strategy for continuation ect in geriatric depression: phase 2 of the PRIDE study. A
6. Tew JD Jr, Mulsant BH, Haskett RF, et al. Acute efficacy of ECT in the treatment of major depression in the old-old. Am J Psychiatry. 1999;156(12):1865-1870.
7. Dombrovski AY, Mulsant BH, Haskett RF, et al. Predictors of remission after electroconvulsive therapy in unipolar major depression. J Clin Psychiatry. 2005;66(8):1043-1049.
8. Charles K. UpToDate. Unipolar major depression in adults: indications for efficacy of electroconvulsive therapy (ECT). https://www.uptodate.com/contents/unipolar-major-depression-in-adults-indications-for-and-efficacy-of-electroconvulsive-therapy-ect. Updated May 16, 2017. Accessed November 26, 2017.
9. Sackeim HA, Prudic J, Devanand DP, et al. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425-434.
10. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
11. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357(19):1939-1945.
12. Stoppe A, Louzã M, Rosa M, et al. Fixed high dose electroconvulsive therapy in elderly with depression: a double-blind, randomized comparison of efficacy and tolerability between unilateral and bilateral electrode placement. J ECT. 2006;22(2):92-99.
13. Geduldig ET, Kellner CH. Electroconvulsive therapy in the elderly: new findings in geriatric depression. Curr Psychiatry Rep. 2016;18(4):40.
14. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(suppl 4):1-45.
15. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23(3):274-282.
16. Philibert RA, Richards L, Lynch CF, et al. Effect of ECT on mortality and clinical outcome in geriatric unipolar depression. J Clin Psychiatry. 1995;56(9):390-394.
17. Tomac TA, Rummans TA, Pileggi TS, et al. Safety and efficacy of electroconvulsive therapy in patients over age 85. Am J Geriatr Psychiatry. 1997;5(2):126-130.
18. Verwijk E, Comijs HC, Kok RM, et al. Short and long-term neurocognitive functioning after electroconvulsive therapy in depressed elderly: a prospective naturalistic study. Int Psychogeriatr. 2014;26(2):315-324.
19. Flint AJ, Gagnon N. Effective use of electroconvulsive therapy in late life depression. Can J Psychiatry. 2002;47(8):734-741.
20. Bjolseth TM, Engedal K, Benth JS, et al. Speed of recovery from disorientation may predict the treatment outcome of electroconvulsive therapy (ECT) in elderly patients with major depression. J Affect Disord. 2016;190:178-186.
21. Sackeim HA, Prudic J, Nobler MS, et al. Ultra-brief pulse ECT and the affective and cognitive consequences of ECT. J ECT. 2001;17(1):77.
22. Bjolseth TM, Engedal K, Benth JS, et al. Baseline cognitive function does not predict the treatment outcome of electroconvulsive therapy (ECT) in late-life depression. J Affect Disord. 2015;185:67-75.
23. de Carle AJ, Kohn R. Electroconvulsive therapy and falls in the elderly. J ECT. 2000;16(3):252-257.
24. Hoover DR, Siegel M, Lucas J, et al. Depression in the first year of stay for elderly long-term nursing home residents in the USA. Int Psychogeriatr. 2010;22:1161.
25. O’Connor MK, Knapp R, Husain M, et al. The influence of age on the response of major depression to electroconvulsive therapy: a C.O.R.E. Report. Am J Geriatr Psychiatry. 2001; 9:382.
Metabolic Complications of HIV Infection
From the University of Connecticut School of Medicine, Farmington, CT.
Abstract
- Objective: To review the metabolic complications of HIV infection.
- Methods: Review of the literature in the context of 3 clinical cases.
- Results: People with HIV infection are living longer thanks to the advent of potent antiretroviral therapy. This has led to increased incidence of age-related metabolic complications, including a higher risk of cardiovascular disease, hyperlipidemia, metabolic syndrome, and osteoporosis. Appropriate management of these complications requires an understanding of disease-related and drug-related side effects as well as the potential for drug-drug interactions. A multidisciplinary approach to patient management is most effective.
- Conclusion: Awareness of the metabolic complications frequently encountered in HIV infection, drug interactions, and possible toxicities is critical to the successful management of HIV-infected individuals.
Key words: HIV; antiretroviral therapy; hyperlipidemia; metabolic syndrome; diabetes; hypogonadism.
According to the most recent data from the Joint United Nations Programme on HIV/AIDS (UNAIDS), 36 million people worldwide are living with HIV/AIDS, with 18 million accessing effective antiretroviral therapy (ART) [1]. The past 2 decades have witnessed enormous advances in the field from prevention to diagnosis and therapeutics, and modern ART largely allows HIV-infected persons to live near-normal life spans [2,3]. However, from the beginning of the epidemic, HIV-infected persons on effective therapy have suffered from myriad metabolic consequences, many of which affect quality of life and result in excess mortality [4]. It is also true that untreated HIV infection portends an increased risk of metabolic complications, likely related to abnormal immune activation, as demonstrated in structured interruption trials [5,6]. It is worth noting, however, that while many of these metabolic dyscrasias and associated risks have historically been attributed primarily to the treatment of HIV infection with ART, data from cohort studies and randomized clinical trials have repeatedly demonstrated significant reductions in morbidity and mortality when ART is initiated early [7]. In this paper, we review HIV-related metabolic complications frequently encountered in clinical practice (hyperlipidemia, diabetes, and bone disease) and best practice considerations in the context of 3 clinical cases.
Case Patient 1
Initial Presentation and History
A 58-year-old male with a history of hypertension and mixed hyperlipidemia is referred for evaluation of newly diagnosed HIV infection. He has no history of intravenous drug use but has had multiple male and female sex partners in the past few years, and requested testing after a partner tested positive. His last negative test was 2 years ago. The patient does not smoke cigarettes. Overall he feels well and tolerates his regimen of lisinopril 10 mg and simvastatin 20 mg daily. On initial evaluation, his exam is unremarkable other than subtle white plaques on the dorsal surface of the tongue and buccal mucosa, and moderate central obesity. Vital signs including blood pressure are normal. Initial laboratory evaluation reveals a CD4 cell count of 150 cells/mm3 and an HIV RNA level of 200,000 copies/mL. Fasting serum total cholesterol is 220 mg/dL, triglycerides 250 mg/dL, LDL 170 mg/dL, and HDL 35 mg/dL. Serum BUN, creatinine, and liver function testing results are normal.
What initial regimen might be recommended based on the status of his HIV infection and comorbidities?
The most recent iteration of the US Department of Health and Human Services (DHHS) guidelines on use of antiretroviral agents (ARVs) in HIV recommends an initial ART regimen that includes a backbone of 2 nucleoside reverse transcriptase inhibitors (NRTIs), generally tenofovir disoproxil fumarate or tenofovir alafenamide, abacavir (ABC), emtricitabine (FTC), or lamivudine (3TC) [2]. To this backbone should be added a third agent; the majority of those currently recommended are integrase strand transfer inhibitors (INSTIs) (dolutegravir, elvitegravir, raltegravir); one recommended protease inhibitor (PI) (ritonavir-boosted darunavir) is also an option. Some of these initial recommended regimens are available as fixed-dosed combinations in 1 pill, making them attractive options.
The latest guidelines also clearly recommend starting ART in all HIV-infected individuals, irrespective of CD4 count. The patient described above has a very low CD4 count, so there is no question he needs to begin therapy promptly. Given his low CD4 count and relatively high viral load, one may consider a ritonavir-boosted PI as perhaps the most robust option and with a relatively high barrier to resistance development, in contrast to other options. Assuming the patient’s baseline resistance testing reveals a fully sensitive wild-type virus without meaningful resistance mutations, he will begin a regimen of TDF/FTC plus ritonavir-boosted darunavir, 3 pills once daily. Given his low CD4 count (below 200 cells/mm3), he will also need prophylaxis for Pneumocystis jirovecii pneumonia, in the form of trimethoprim/sulfamethoxazole (TMP/SMX) daily. Given the potential for interaction between the boosted PI and simvastatin, his lipid-lowering agent is switched to atorvastatin 10 mg daily.
What is the association between hyperlipidemia and HIV infection and treatment?
Hyperlipidemia represents a key modifiable risk factor for the development of cardiovascular disease (CVD) in HIV-infected individuals [8]. Indeed, a multicenter cross-sectional study of older HIV-infected individuals performed in Spain revealed a 54% prevalence of dyslipidemia and 23% CVD [9]. Most experts believe that metabolic abnormalities observed in HIV-infected individuals are the result of a combination of factors: those resulting from abnormal immune activation and inflammation related to viral replication, and those related to certain ARVs [10].
Early after HIV seroconversion, decline in HDL is one of the first proatherogenic changes observed. This, along with increased triglyceride and LDL levels, likely contribute to increased risk of CVD in this population. Increased microbial translocation, evidenced by increased levels of lipopolysaccharide (LPS), may drive immune activation, leading to dyslipidemia via a multitude of hypothesized mechanisms [4]. It has been theorized that HDL lipoproteins are less stable on ART, leading to potentially impaired plasma lipolytic activities or hepatic cholesteryl ester uptake [6,11]. Increased VLDL from release of free fatty acids may lead to higher triglyceride levels and triglyceride-rich LDL and HDL, all associated with increased risk of CVD [11].
In terms of effects of specific ARV classes, although newer agents have less of a propensity to cause dyslipidemia, the PI class arguably remains most problematic. In comparison to other classes, the PIs tend to result in greater increases in triglycerides, total cholesterol, and LDL, and have frequent drug-drug interactions with lipid-lowering agents [10,12]. Estimated prevalence of dyslipidemia in patients receiving PI therapy varies from 28% to 80% [13]. The prospective multinational cohort Data collection on Adverse events of Anti-HIV Drugs (DAD) study found significantly higher rates of hypertriglyceridemia, hypercholesterolemia, and low HDL in patients on PIs in comparison to non–nucleoside reverse transcriptase inhibitors (NNRTIs) [14]. Various mechanisms have been proposed to explain the PIs adverse effects on lipids, including inhibition of lipogenesis and adipocyte differentiation, decreased hepatocyte clearance of chylomicrons and VLDL, and increased hepatic synthesis of triglycerides [15]. Of the available PIs, atazanavir and darunavir have less potential to lead to dyslipidemia [10], while lopinavir/ritonavir, fosamprenavir, and tipranavir may have the highest [13]. Of the NNRTIs, efavirenz is most frequently associated with dyslipidemia, specifically increased triglycerides and total cholesterol [13]. However, these increased values seen on efavirenz therapy may be offset by relative increases in HDL, with little resultant effect on the total cholesterol:HDL ratio. Rilpivirine, etravirine, and nevirapine are relatively less likely to drive lipid changes, although certain drug interactions are important to recognize in clinical practice, such as the interaction between rilpivirine and proton pump inhibitors [2,13,16]. It is also worth noting that no NNRTIs are included in current guidelines as preferred therapy [2].
Historically, the thymidine analogue NRTIs (stavudine, didanosine, zidovudine) have been associated with lipid dyscrasias and lipoatrophy, but fortunately these are no longer used frequenty except in cases requiring deep salvage therapy for highly treatment-experienced patients. Two newer NRTIs, tenofovir and abacavir, have relatively neutral to favorable effects on lipids. The combination of tenofovir disoproxil (TDF) and emtricitabine (trade name Truvada) was associated with significantly lower triglycerides, total cholesterol and LDL than other NRTI pairs [6]. TDF has been postulated to have lipid-lowering effects. Switch studies in which patients were taken off thymidine analogues and placed on TDF, demonstrated recovery of limb fat in patients with lipoatrophy, and those switched off abacavir-based ART to TDF showed statistically significant lower fasting total cholesterol at week 12, without differences of viral suppression [8]. Tenofovir alafenamide (TAF) is a next-generation prodrug of tenofovir that results in improved stability in plasma and higher intracellular levels in comparison to TDF [17]. Although randomized controlled trials of TAF vs TDF-based ARV regimens have suggested statistically higher total cholesterol, serum HDL is also increased resulting in unchanged total:HDL ratios and no differences in risk classifications [18]. Integrase inhibitors (INSTI) now represent first-line therapy in combination with an NRTI backbone, and since their availability in 2007 have been evaluated in comparison to various PIs and NNRTIs. Both raltegravir and dolutegravir have consistently shown broad neutral effects on lipids and are among the most metabolically friendly agents available [19–21]. Because it is given in fixed-dose combination with non-ritonavir pharmacologic booster cobicistat, elvitegravir has effects similar to ritonavir-boosted PIs on lipids [6].
What are management considerations in the treatment of hyperlipidemia in HIV-infected patients?
Patients with HIV and hyperlipidemia may benefit from lipid-lowering therapy in addition to ART, although in certain cases appropriate switches may make a difference. Careful consideration of drug interactions between ARVs and lipid-lowering agents, in addition to ARV history and known drug resistance, is warranted prior to selecting a regimen in these patients. In addition, the latest American College of Cardiology/American Heart Association guidelines suggest evaluating 10-year risk of atherosclerotic cardiovascular disease (ASCVD) using the pooled cohort equation to determine the type and dose of statin required (moderate vs high intensity) [22]. It is noteworthy that HIV infection and its therapies are not taken into account as potential risk factors in this model. Primary prevention in non-diabetic patients with a statin is recommended for patients with a 10-year absolute risk of ≥ 7.5% [22]. This patient’s risk is estimated at between 12% and 13% based on this equation, so primary prevention with a moderate-or high-intensity statin is recommended (Table 1) [23]. Data from more than 80,000 patients in the Veterans Aging Cohort Study (VACS) showed that HIV-infected patients with no baseline ASCVD had 50% increased risk of acute myocardial infarction when compared to HIV-uninfected patients over 6 years of follow-up [24]. Thus, consideration of the virus itself or its therapy as an additional risk factor may be valid. 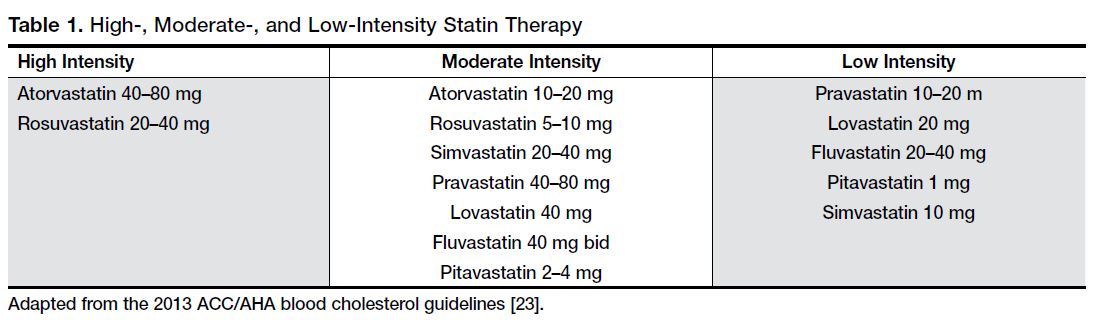
Screening and Monitoring of Hyperlipidemia
The most recent iteration of the DHHS primary care guidelines for the management of HIV-infected individuals recommends obtaining fasting (ideally 12 hours) lipid profiles upon initiation of care, and within 1 to 3 months of beginning therapy [12,13]. These initial levels, along with other elements of the patient’s history and calculation of risk may help determine whether lipid-lowering therapy is indicated, and if so, which therapy would be best. In general, after regimen switches or additions of either ARV or statin therapy, repeating fasting lipid levels 6 weeks later is recommended to gauge the effects of the switch. This is especially critical when interactions between ARVs and lipid-lowering therapies are possible. Some experts recommend performing annual screening of patients with normal baseline lipids or with well-controlled hyperlipidemia on therapy. Assessment of 10-year ASCVD risk is also recommended annually, in addition to baseline risk assessment, to determine the need and appropriateness of statin therapy [25]. The question of primary prevention in HIV has yet to be definitively answered. Small studies in this population have demonstrated that statins have the potential to slow progression of carotid intima media thickness and reduce noncalcified plaque volume [24]. An NIH/AIDS Clinical Trial Group–sponsored randomized clinical trial (“REPRIEVE”) is currently underway to address this question. More than 6000 HIV-infected men and women with no history of ASCVD at 100 sites in several countries are enrolled to assess the benefit of pitavastatin as primary prevention in this risk group [24]. Metabolized via glucuronidation primarily, as opposed to cytochrome p450 (CYP 3A4 isoenzyme), pitavastatin is thought to have fewer drug interactions with ARVs in general [6] (Table 2). 
Relevant Drug-Drug Interactions
Deciding which statin to begin in HIV-infected patients depends on whether moderate- or high-intensity therapy is warranted and whether the potential for drug interaction with ARVs exists. Table 2 [6,12] depicts available statins and the potential for pharmacokinetic interaction with the primary ARV classes. Simvastatin and lovastatin are heavily metabolized via the CYP 3A4 pathway, resulting in the highest potential risk of interaction with CYP 3A4 inhibitors, such as the PIs, or inducers (eg, NNRTIs, in particular efavirenz) [6]. The former may inhibit metabolism of these statins, resulting in increased risk of toxicity, while co-administration with efavirenz, for example, may result in inadequate serum concentration and therefore inadequate lipid-lowering effects. Although less lipophilic, atorvastatin results in similar interactions with PIs and NNRTIs, and therefore low starting doses with close monitoring is recommended [6]. Fewer interactions have been noted with rosuvastatin, pravastatin, and pitavastatin, as these do not require CYP 3A4 for their metabolism and are thus less likely to be affected by ARVs. These therefore represent potentially safer first choices for certain patients on ARVs, although of these, only rosuvastatin is classified as a high-intensity statin [22,23] (Table 1). When compared directly to pravastatin 40 mg daily in patients receiving ritonavir-boosted PIs, rosuvastatin performed superiorly at 10 mg per day, resulting in more significant reductions in LDL and triglyceride levels [15]. Although it is eliminated largely unchanged through the kidney and liver, pravastatin has been reported to idiosyncratically interact with darunavir, resulting in potentially increased pravastatin levels and associated toxicity [25]. Treatment of pure hypertriglyceridemia in HIV-infected patients should begin with fibrates, which have little to no risk of interaction with most clinically relevant ARVs [6,10]. Alternatives to lower triglycerides include niacin and N-3 polyunsaturated fatty acids [25].
Case 1 Continued
The patient has an impressive response to his initial regimen of TDF/FTC plus boosted darunavir, with repeat CD4 count after 12 weeks of 275 (18%) cells/mm3 and an undetectable viral load (< 20 copies/mL). Other lab parameters are favorable and he is tolerating the regimen well without notable side effects. However, at his next visit, although his viral load remains undetectable, his triglyceride level has increased to 350 mg/dL, although other lipid parameters are comparable to the prior result. He complains of diffuse body aches, concentrated in large muscle groups of the extremities, and dark-colored urine. A creatine phosphokinase (CPK) level is elevated at 300 IU/L (normal, 22–269, negative MB fraction). Serum creatinine is 1.4 mg/dL (had been 1.1 mg/dL at baseline). Given he has done so well otherwise on these ARVs, he is reluctant to make any changes.
What drug-drug interaction is most likely causing this patient's problem, and how should it be managed?
This scenario is not uncommon in clinical practice, and changes to regimens are sometimes necessary in order to avoid drug interactions. Care must be taken to thoroughly review antiretroviral history and available resistance testing (in this case a relatively short history) in order to ensure a fully active and suppressive regimen is chosen. This description could be the result of an interaction between lipid-lowering therapy and ARVs resulting in increased relative concentrations of one drug or the other and therefore leading to toxicity. Given this possibility, and suboptimal control of hyperlipidemia, consideration should be given to switching both his ART and his statin therapy.
Safety and Potential Toxicities of Lipid-Lowering Therapy
Increased serum concentration of certain statins when co-administered with CYP 3A4 inhibitors like the PIs leads to heightened risk of statin-associated toxicities. In general, this includes muscle inflammation, leading to increases in serum CPK level and associated symptoms, including myalgias, myositis, or in extreme cases, rhabdomyolysis [6]. Although rare, this toxicity can be serious and may lead to acute renal injury if not recognized and managed appropriately. In theory, the potential for statin-associated hepatotoxicity may also be increased in patients receiving PIs, although this has not been borne out in clinical trials [26]. In fact, quite the opposite may be true, in that statins have been shown to improve liver function in patients with hepatitis C virus (HCV) coinfection and with nonalcoholic fatty liver disease [6,15].
Case 1 Conclusion
The patient does well on his new ARV regimen of TAF/FTC and dolutegravir, 2 pills once daily. He no longer requires TMP/SMX, as his CD4 count has been reliably above 200 cells/mm3 on several occasions. Serum creatinine is back down to baseline and CPK has normalized. Fasting lipids have improved since the switch, and he no longer has symptoms of myositis on rosuvastatin 10 mg daily.
Summary
Consideration of statin therapy is complicated by potential drug interactions with ARVs and associated toxicity. However, given known effects of ARVs on lipids, and of immune activation and inflammation related to the virus itself, these patients should be carefully evaluated for statin therapy for their anti-inflammatory and immune modulatory effects as much as for their lipid-lowering ability. Utilization of HIV infection and its therapies as additional cardiovascular risk factors when calculating 10-year risk deserves further consideration; forthcoming results of the REPRIEVE trial are certain to contribute valuable information to this field of study.
Case Patient 2
Initial Presentation and History
A 45-year-old female with history of HIV infection since 2008 presents to the office for new-onset diabetes, diagnosed 2 weeks ago. She has had symptoms of polyuria and polydipsia for the last 1 month. She denies diarrhea, nausea, vomiting or weight loss. She is currently on a regimen consisting of zidovudine/lamivudine plus lopinavir/ritonavir. There is no family history of diabetes. Her examination is unremarkable, including normal vital signs (weight 150 lb, blood pressure 114/70, heart rate 76) and no evidence of insulin resistance, including acanthosis nigricans or striae. Glycosylated hemoglobin level (HbA1c) is 8%. Creatinine and liver function tests are within reference ranges.
Do HIV-infected patients have a higher incidence of type 2 diabetes mellitus (DM)?
Prevalence of type 2 DM in HIV-infected patients varies between 2% to 14% [27]. This variation is due to the different cutoffs used for diagnosis, differences in cohorts studied, and how risk factors are analyzed [28–31]. In a recent nationally representative estimate of DM prevalence among HIV-infected adults receiving medical care in the United States in 2009–2010, the prevalence of DM was noted to be 10.3%. In comparison to the general adult US population, HIV-infected individuals have a 3.8% higher prevalence of DM after adjusting for age, sex, race/ethnicity, education, poverty-level, obesity, and HCV infection [27].
There is controversy over whether HIV infection itself increases the risk of type 2 DM, with some studies showing increased risk [28,32,33] and others showing no independent effect or an inverse effect [30,34,35]. Studies on the impact of ethnicity and race on prevalence of DM are limited [36].
Certain traditional risk factors (age, ethnicity, obesity) are still responsible for most of the increased risk of diabetes in the HIV-infected population [35,37]. HIV infection itself is associated with metabolic dysfunction, independent of ARV. In HIV-infected patients, impaired glucose metabolism is associated with altered levels of adipokines, increased adiponectin and soluble-tumor necrosis factor receptor 1 (sTNFR1) and decreased leptin [38,39]. HIV-associated alterations in CD4+ and CD8+ T cell function also impair glycolysis, which may adversely impact glucose metabolism [40].
Other contributing factors in HIV-infected patients are HCV co-infection [41], medications (atypical antipsychotics, corticosteroids), opiates, and low testosterone [42]. HCV co-infection may lead to hepatic steatosis and liver fibrosis, and increasing insulin resistance.
Recent genomic studies show several common single-nucleotide polymorphisms (SNPs) associated with diabetes in the general population. In the Swiss HIV Cohort Study, SNPs accounted for 14% of type 2 DM risk variability, whereas ARV exposure accounted for 3% and age for 19% of the variability in DM [43].
ARVs also increase the risk of type 2 DM by both direct and indirect effects. Certain ARVs causes lipoatrophy [30] and visceral fat accumulation/lipohypertrophy [29,44]. PIs increase insulin resistance via effects on GLUT-4 transporter and decrease insulin secretion through effects on B cell function [45]. NRTIs (eg, stavudine, zidovudine and didanosine) can cause direct mitochondrial toxicity [46–48]. Utilization of newer ARV agents has decreased the prevalence of severe lipoatrophy, but lipohypertrophy and the underlying metabolic abnormalities persist. The DHHS “preferred” nucleoside analogues, tenofovir and abacavir, do not induce mitochondrial toxicity and have more favorable metabolic profiles [49,50]. In ACTG Study 5142, thymidine-sparing regimens were found to cause less lipoatrophy [51]. In addition, darunavir and atazanavir, the preferred and alternative PIs and the integrase strand transfer inhibitor have limited or modest impact on insulin sensitivity [20,52,53]. This has led to a recent decline in the incidence of type 2 DM in HIV-infected patients.
Statins can also increase insulin resistance and DM [54], although studies have shown mixed results [55–57]. The benefits of statin therapy likely outweigh the risk of DM since there is a significant cardiovascular event reduction with their use [58,59].
How is diabetes diagnosed in HIV-infected patients?
Optimal diabetes screening guidelines have not been established specifically for HIV-infected patients. The American Diabetes Association (ADA) guidelines recommend that diabetes in the general population be diagnosed by 2 elevated fasting blood glucose levels, HbA1c, oral glucose tolerance test (OGTT), or high random glucose with classic symptoms of hyperglycemia [60]. Repeat testing is recommended every 3 years. The OGTT is recommended for diagnosis in pregnant women.
HbA1c may underestimate glycemic burden in HIV-infected individual due to higher mean corpuscular volume, NRTI use (specifically abacavir), or lower CD4 count [61–65]. The Infectious Diseases Society of America (IDSA) 2013 primary care guidelines for HIV-infected patients recommends obtaining a fasting glucose and/or HbA1c prior to and within 1–3 months after starting ARV [12]. Use of HbA1c threshold cutoff of 5.8% for the diagnosis of DM and testing every 6–12 months are recommended.
How should this patient’s diabetes be managed?
The ADA guidelines suggest a patient-centered approach to management of diabetes [66]. All patients should be educated about lifestyle modifications with medical nutrition therapy and moderate-intensity aerobic activity and weight loss [67]. If a patient is on lopinavir/ritonavir or a thymidine analogue (zidovudine, stavudine), one should consider switching the ARV regimen [2].
There are currently no randomized controlled trials of diabetes treatment specific to patients with HIV infection. Metformin is the first-line agent. It improves insulin sensitivity by reducing hepatic glucose production and improving peripheral glucose uptake and lipid parameters [68,69]. Other oral hypoglycemic agents used in the treatment of type 2 diabetes are shown in Table 3. 
Case 2 Continued
The patient is switched to TAF/FTC plus dolutegravir with improvement in blood sugars. She is also started on metformin. Co-administration of metformin and dolutegravir will be carefully monitored since dolutegravir increases metformin concentration [70]. When dolutegravir is used with metformin, the total daily dose of metformin should be limited to 1000 mg.
• How should this patient be followed?
If the patient is still not at goal HbAb1c at follow-up, there are multiple other treatment options, including use of insulin. Goal HbA1c for most patients with type 2 DM is < 7%; however, this goal should be individualized for each patient in accordance with the ADA guidelines [12]. A longitudinal cohort study of 11,346 veterans with type 2 diabetes compared the glycemic effectiveness of oral diabetic medications ( metformin, sulfonylurea and a thiazolidinedione) among veterans with and without HIV infection. This study did not find any significant difference in HbA1c based on different diabetes medications. However the HBA1c reduction was less in black and Hispanic patients. The mechanism for the poorer response among these patients need to be evaluated further [71]. In addition to management of blood sugar, other CVD risk factors, hyperlipidemia, hypertension, smoking, etc, should be assessed and managed aggressively.
Case Patient 3
Initial Presentation and History
A 45-year-old male with a history of HIV infection diagnosed 10 years ago, on TDF/FTC/efavirenz (trade name Atripla) for the last 7 years, presents with a left femoral neck fracture after he missed the pavement and fell on his left hip. His history is significant for IV drug abuse for 10 years prior to diagnosis of HIV, and he has been on methadone for the last 6 years.
Is HIV infection associated with increased prevalence of osteopenia and osteoporosis and increased risk of fractures?
With recent advancements in antiretroviral therapy and improved survival of the HIV-infected population, osteoporosis and increased fracture risk have become important causes of morbidity and mortality. Osteoporosis is a skeletal disorder characterized by compromised bone strength, which predisposes to an increased risk of fracture. The World Health Organization defines osteoporosis as a bone mineral density (BMD) measurement by dual X-ray absorptiometry (DXA) at the spine, hip, or forearm that is more than 2.5 standard deviations below that of a "young normal" adult (T-score < –2.5) or a history of one or more fragility fractures. Fragility fractures result from mechanical forces that would not ordinarily result in fracture, such as fall from standing height [40]. Osteopenia is characterized by low BMD (T-score between –1.0 and –2.5) and can be a precursor to osteoporosis.
Several observational, retrospective, and prospective studies have shown lower bone density and an increased risk of fractures in the HIV-infected population compared to age-, race- and sex-matched HIV-negative adults. In a large meta-analysis of pooled prevalence data on 884 HIV-infected patients compared with 654 HIV-uninfected age- and sex-matched controls [72], overall, HIV-infected patients had a significant 6.4-fold increased odds of reduced BMD and a 3.7-fold increased odds of osteoporosis compared to the control population. This meta-analysis also compared ARV-treated subjects to ARV-naive subjects and showed that ARV-treated subjects (n = 824) had a higher prevalence of reduced BMD compared with ARV-naive subjects (n= 202; odds ratio 2.5, 95% CI 1.8–3.7). The odds of osteoporosis was increased 2.4 times (95% CI 1.2 – 4.8) in ARV-treated subjects compared with ARV-naive subjects. None of the studies adjusted for potentially important confounding factors, such as age or duration of HIV infection. PI-treated patients (n = 791) were also found to have a higher prevalence of reduced BMD compared with PI-untreated patients (n = 410; OR 1.5, 95% CI 1.1–2.0). The odds of osteoporosis in PI-treated patients (n = 666) was also 1.6-fold greater (95% CI 1.1–2.3) than those not treated with PI (n = 367).
Low bone density has also been reported in HIV- positive premenopausal women irrespective of ARV status. In a recent study of 89 premenopausal women (mean age, 37 years) predominantly of African origin with HIV infection, osteopenia and osteoporosis were prevalent in one-third of these women, irrespective of ARV use and were associated with low BMI [73]. In a sub-study of the INSIGHT trial evaluating prevalence of and risk factors for low BMD in untreated HIV infection, performed at several sites across 6 continents involving 424 subjects, osteopenia was present in a third of this relatively young predominantly non-white ART-naive population (mean age 34 + 10 years) with normal CD4 cell counts, while only 2% had osteoporosis. Factors independently associated with lower BMD at the hip and spine were female sex, Latino/Hispanic ethnicity, lower BMI, and higher estimated glomerular filtration rate. Longer duration of HIV infection was also associated with lower hip BMD. Current or nadir CD4 cell count and HIV viral load were not associated with low BMD [74].
Many studies have reported increased fracture prevalence in the HIV population. In a retrospective study of fracture prevalence in a large US health care system, a significantly higher rate of fractures was reported in HIV-infected men and women compared to non-HIV-infected controls (2.87 vs. 1.77 fractures per 100 persons, P < 0.001). The difference in the increased fracture prevalence was greater in HIV positive men compared to women (3.08 vs. 1.83; P < 0.001). Vertebral, wrist and hip fractures were more prevalent in men compared to vertebral and wrist fractures only in women. Fracture prevalence was higher in both Caucasian females and males and only in African-American women [75].
In the HIV Outpatient Study (HOPS) [76], age-adjusted fracture rates in the HIV population were noted to be 1.98 to 3.69 times higher than rates in the general population. The HOPS was an open prospective cohort study of HIV-infected adults who were followed at 10 US HIV clinics. Rates of first fractures at any anatomic site from 2000–2008 were assessed among 5826 active HOPS patients (median age 40 years, 79% male, 52% Caucasian, and 73% exposed to ART). Among persons aged 25–54 years, both fracture rates and relative proportion of fragility fractures were higher among HOPS patients than among outpatient controls. Older age, substance abuse, nadir CD4+ cell count <200 cells/mm, HCV infection and DM were associated with incident fractures [76].
What factors contribute to poor bone health in the HIV population?
Several factors that contribute to low bone density are present at a higher rate in the HIV population (Table 4). These include poor nutritional status in terms of suboptimal calcium and vitamin D intake, hypogonadism, low body weight, and alcohol, tobacco and substance abuse. 
Vitamin D deficiency is very common in HIV-infected patients, with a prevalence of up to 60% to 75% [77]. Hypogonadism is also relatively common among HIV population [78], contributing to lower bone density. Co-infection with HCV is also associated with increased risk of fractures. In a large cohort of Medicaid beneficiaries, a significant increase in the risk of hip fracture was demonstrated in HCV/HIV co-infected subjects compared either with HCV mono-infected, HIV mono-infected or non-infected individuals [79]. In another large database study, a significantly higher risk of osteoporotic fracture (closed wrist, vertebral or hip fracture) was reported in HCV/HIV co-infected versus HIV mono-infected individuals [80] with fracture rates of 2.57 and 2.07/1000 patient-years (P < 0.001). Dual treatment for HIV/hepatitis B co-infection has also been shown to be associated with a higher risk of hip fracture compared to treatment of HIV mono-infected individuals [81].
HIV infection itself can increase bone loss and reduce bone formation through direct effects related to the HIV antigen load or indirect effects related to activation of the pro-inflammatory cytokines resulting in bone resorption and loss [82]. Co-infection with HCV and/or hepatitis B also contributes to lower bone density in this population. Certain ARVs may also contribute to low bone density in the HIV population. Lipoatrophy related to HIV may also mediate bone loss through complex relationship between central signaling of adipocyte hormones [82,83].
Direct Viral Effects
Several HIV viral proteins have been shown to promote osteoclast activity (vpr and gp120), suppress osteoblast activity (p55-gag) and increase osteoblast apoptosis [84], resulting in increased bone resorption and reduced bone formation, leading to low bone mass. High HIV RNA viral load and T-cell activation are also associated with elevated levels of receptor activator of nuclear factor kappa-B ligand (RANKL), which results in osteoclast formation and increased bone resorption [85]. Other endogenous physiological inhibitors of osteoclastogenesis such as osteoprotegrin and interferon-γ levels are also remarkably downregulated in advanced HIV infection, resulting in increased bone resorption [86]. At a cellular level, HIV proteins including Tat and Nef reduce the number of available mesenchymal stem cell (MSC) precursors that proliferate into osteoblasts by inducing MSC senescence, due to increased oxidative stress and mitochondrial dysfunction resulting in reduced proliferation of osteoblasts and lower rates of bone formation [87]. Collectively, these mechanisms result in significant uncoupling of bone formation and resorption, resulting in less bone formation and greater rate of bone loss and lower bone density.
Pro-inflammatory Pathways
Cytokines and other soluble immune factors play a major role in the physiology of osteoblast maturation and osteoclastic bone resorption [88,89]. Immune dysfunction and persistent inflammation in HIV result in increased levels of several inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin-6 (IL-6), and RANKL, resulting in stimulation of osteoclastogenesis and bone resorption [90]. Due to a disruption between T and B cells in HIV and decreased osteoprotegrin (OPG) production and increased RANKL level, RANKL/OPG ratio is elevated, favoring osteoclastogenesis [91].
Is antiretroviral therapy associated with bone loss?
The initiation of ART has been reported to cause 2% to 6% bone loss irrespective of the regimen used, similar to that sustained in the first 2 years after menopause [92]. Certain NRTIs and PIs are associated with higher rates of bone loss than others. TDF has been associated most commonly with decreased bone mineral density, which usually stabilizes with continued use [93]. In a randomized trial comparing 4 treatment arms of ABC/3TC or TDF/FTC with EFV or ATV/ritonavir, TDF was associated with a greater reduction in BMD compared to abacavir-based regimens [94]. The likely cause of this may be TDF-mediated renal toxicity, including proximal tubular dysfunction and hypophosphatemia, resulting in increased PTH and bone resorption, and nephrogenic diabetes insipidus [95]. TAF is another prodrug of tenofovir diphosphate associated with less renal and bone toxicity compared with TDF. TAF has been associated with significantly less decrease in bone mineral density and renal dysfunction in randomized studies compared to regimen using TDF [17]. Vitamin D deficiency and hypophosphatemia associated with TDF therapy may present with osteomalacia, which predisposes to bone pain and fractures. Treatment with TDF may rarely be associated with the development of Fanconi syndrome and osteomalacia [96]. BMD is often severely reduced and bone pain and pathological fractures are characteristic features. Certain PI regimens containing ritonavir-boosted atazanavir have also been associated with greater bone loss in the spine than the hip, compared to efavirenz-containing regimens [97].
The universal bone loss associated with ART is thought to be a result of the "immune reconstitution inflammatory syndrome" (IRIS). This occurs as a result of rapid improvement in immune function after the commencement of ARV as a result of systemic or local inflammation, resulting in increased levels of cytokines that may contribute to bone loss. This has been shown in animal studies where T cell transplantation into immunocompromised mice to mimic ARV-induced T-cell expansion resulted in increased RANKL and TNF-α production by B cells and/or T cells, accompanied by enhanced bone resorption and BMD loss. When TNF-α or RANKL-null T-cells or TNF-α antagonists were used instead, the loss of cortical bone was prevented [98]. In a prospective study evaluating changes in bone turnover markers and inflammatory cytokines with ARV therapy in HIV infected subjects, a significant increase in bone resorption markers, RANKL and TNF-α were seen after initiation of ARV. The magnitude of CD4-cell recovery correlated with the increase in markers of bone resorption [99], suggesting that recovery of the immune system contributes to the increase in cytokine-mediated bone resorption.
How is bone health and fracture risk assessed in the HIV-positive population?
The predictive value of low BMD for fracture risk assessment in the HIV-positive population has not been established. In the absence of definitive data, the fracture risk assessment and standard methods of measuring bone density using DXA are utilized. In a large study of 1000 men and women, osteoporosis defined as a BMD T-score –2.5 as measured by DXA, was associated with a significantly increased risk of incident fractures but was not a good predictor of morphometric vertebral fractures [100]. In the absence of prospective longitudinal studies evaluating the bone density parameters at which fracture risk is significantly increased in the HIV population, it is reasonable to follow the guidelines used in the non-HIV population.
The approach to treatment of osteopenia and osteoporosis is similar to that in non HIV-infected population and is directed at lifestyle changes and treatment of secondary causes of osteoporosis [101], followed by initiation of antiresorptive therapy.
Management of Bone Disease
There are several guidelines available for the management of bone disease in the HIV population. The most recent guidelines from the IDSA [12] recommend assessing the risk of fragility fracture using the Fracture Risk Assessment Tool (FRAX), without DXA, in all HIV-infected men aged 40–49 years and HIV-infected premenopausal women aged ≥ 40 years. DXA should be performed in men aged ≥ 50 years, postmenopausal women, patients with a history of fragility fracture, patients receiving chronic glucocorticoid treatment, and patients at high risk of falls. In resource-limited settings, FRAX without bone mineral density can be substituted for DXA. ART guidelines should be followed. TDF and boosted PIs should be avoided if possible in at-risk patients. Dietary and lifestyle management strategies for high-risk patients should be employed and anti-osteoporosis treatment initiated if indicated [102].
The FRAX tool is available at www.shef.ac.uk/FRAX/ and is used to calculate 10-year fracture risk using patient clinical data, including presence of risk factors for osteoporosis. The tool is population-specific by race and region. It has not been validated for the HIV-positive population and may underestimate fracture risk [103]. HIV status is considered a secondary cause of osteoporosis in FRAX calculation.
The National Osteoporosis Foundation recommends screening with DXA for all women > 65 years of age, all men > 70 years of age, and adults > 50 years of age with additional risk factors for osteoporosis. Evaluation for secondary causes for low BMD should always be considered in the HIV-positive population including evaluation of calcium and vitamin D intake. Laboratory testing may include complete blood count, calcium, phosphate, albumin, creatinine, PTH, 25 hydroxy vitamin D (25,OHD) and 24 hour urine for evaluation of calcium, creatinine and phosphate (especially if on TDF) excretion. Testosterone level can be checked in men and estradiol, prolactin, FSH and LH in women for evaluation of hypogonadism. Bone turnover markers (bone specific alkaline phosphatase and serum C-terminal telopeptide) can also be assessed at baseline.
Studies using high-resolution peripheral quantitative computed tomography (HSPQCT) have shown significant reductions in tibial trabecular bone density and trabecular number in pre-menopausal and postmenopausal HIV-infected women [104], with reduced bone stiffness measured using finite element analysis [105]. Co-infection with HCV is also associated with significantly lower trabecular volumetric BMD and smaller cortical dimensions in the tibia, compared to healthy subjects [106]. HSPQCT is not widely available for clinical use at this time. Lateral imaging of the spine or vertebral morphometric analysis may be done in cases of height loss to assess for occult vertebral compression fractures.
There is a high prevalence of vitamin D deficiency in the HIV-infected population [107]. Treatment goal is to have a vitamin D level of at least 30 ng/mL, based on Endocrine Society practice guidelines [108], and may require supplementation with 1000–2000 units of vitamin D daily. Calcium intake should be optimized, averaging 1000 mg per day including diet and supplements, to be taken in divided amounts through the day for optimal absorption. Secondary causes of low bone density as mentioned in Table 4 should also be addressed. Patients should be counseled on tobacco and alcohol abuse. Corticosteroids should be dosed at the lowest dose needed. Medications such as proton pump inhibitors can impair the absorption of calcium carbonate, in which case calcium citrate supplements should be used if there is suboptimal calcium intake in the diet.
Which medications have been shown to be effective in treatment of osteoporosis in the HIV population?
Bisphosphonates are the mainstay of therapy for osteoporosis in the HIV-infected population. Only alendronate and zoledronate have substantial evidence of safety and effectiveness in the HIV-infected population, but these studies have been small and of limited duration.
Bisphosphonates are pyrophosphate analogues that inhibit bone resorption by binding to the hydroxyapatite crystals in the bone. Several prospective studies have shown alendronate to increase bone density compared to calcium and vitamin D alone in the HIV infected patients with reduced bone density [109,110], with significant reduction in markers of bone resorption [111].
Zoledronic acid (ZA), an amino-bisphosphonate which is infused intravenously, has also been used in smaller studies in HIV-infected persons. In a prospective study evaluating yearly ZA infusion to biennial ZA infusion in subjects with HIV and low bone density [112], biennial ZA infusions were found to be effective in improving and maintaining bone density in the HIV population. In another prospective study evaluating the effects of ZA in HIV-positive men, ZA infusion was given at baseline and at 12 months. Compared to placebo, treatment group had significantly higher bone density and lower bone turnover markers till 5 years after the last infusion [113].
In a meta-analysis evaluating the effect of bisphosphonates on bone mineral density in 328 adults with HIV infection from 8 randomized controlled trials (5 with alendronate and 3 with ZA as the intervention), a significant increase in BMD at the lumbar spine and hip was observed in the treatment groups at 48 and 96 weeks. However, these studies were not long enough to detect the impact of bisphosphonates on fracture risk [114]. ZA has also been shown to be effective in preventing ARV induced bone loss after a single infusion [115].
These studies confirm that both alendronate and ZA are effective in improving BMD in the HIV-infected population, with early studies showing a beneficial effect of ZA in mitigating ARV-induced bone loss as well. DXA may be repeated 1 to 2 years after initiation of osteoporosis therapy and less often subsequently if BMD is stable to improved [116].
Although these studies show significant improvement in bone density with treatment, longitudinal data on fracture reduction with these medications in the HIV-infected population are not available. Additionally, these patients have onset of osteoporosis at a younger age and the need for osteoporosis treatment needs to be assessed carefully before initiating treatment. There are other medications available for the treatment of osteoporosis in the non-HIV population such as raloxifene, teriparatide and denosumab, but no randomized controlled studies of these agents are available in the HIV-infected population.
Summary
The advent of highly potent antiretroviral therapy capable of early and prolonged viral suppression in HIV-infected patients has resulted in significant increases in life span. As we have already seen, this will likely lead to a rising incidence of various metabolic complications of HIV and ARV, including hyperlipidemia and diabetes with associated cardiovascular disease risk. A keen awareness of these potential complications, drug interactions, and possible toxicities will be paramount to their successful management. Appropriate care of HIV-infected individuals going forward will likely require multidisciplinary collaboration as the epidemic evolves to allow our patients to live not only longer, but healthier lives.
Corresponding author: Lisa M. Chirch, MD, UCONN Health, Farmington, CT 06030, chirch@uchc.edu.
Financial disclosures: None
Author contributions: All authors contributed equally to this article
1. UNAIDS. Fact sheet - Latest statistics on the status of the AIDS epidemic. Accessed 10 Nov 2017 at http://www.unaids.org/en/resources/fact-sheet.
2. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Accessed 10 Nov 2017 at https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf.
3. Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PloS One 2013;8:e81355.
4. Pedersen KK, Pedersen M, Troseid M, et al. Microbial translocation in HIV infection is associated with dyslipidemia, insulin resistance, and risk of myocardial infarction. J Acquir Immune Defic Syndr 2013;64:425–33.
5. Strategies for Management of Antiretroviral Therapy Study Group, El-Sadr WM, Lundgren J, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006;355:2283–96.
6. Chastain DB, Henderson H, Stover KR. Epidemiology and management of antiretroviral-associated cardiovascular disease. Open AIDS J 2015;9:23–37.
7. Group ISS, Lundgren JD, Babiker AG, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med 2015;373:795–807.
8. Moyle GJ, Orkin C, Fisher M, et al. A randomized comparative trial of continued abacavir/lamivudine plus efavirenz or replacement with efavirenz/emtricitabine/tenofovir DF in hypercholesterolemic HIV-1 infected individuals. PloS One 2015;10:e0116297.
9. Mothe B, Perez I, Domingo P, et al. HIV-1 infection in subjects older than 70: a multicenter cross-sectional assessment in Catalonia, Spain. Curr HIV Res 2009;7:597–600.
10. Calvo M, Martinez E. Update on metabolic issues in HIV patients. Curr Opin HIV AIDS 2014;9:332–9.
11. Gillard BK, Raya JL, Ruiz-Esponda R, et al. Impaired lipoprotein processing in HIV patients on antiretroviral therapy: aberrant high-density lipoprotein lipids, stability, and function. Arterioscler Thromb Vasc Biol 2013;33:1714-21.
12. Aberg JA, Gallant JE, Ghanem KG, et al. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 2014;58:1–10.
13. Calza L, Colangeli V, Manfredi R, et al. Clinical management of dyslipidaemia associated with combination antiretroviral therapy in HIV-infected patients. J Antimicrob Chemother 2016;71:1451–65.
14. Friis-Moller N, Weber R, Reiss P, et al. Cardiovascular disease risk factors in HIV patients--association with antiretroviral therapy. Results from the DAD study. AIDS 2003;17:1179–93.
15. Husain NE, Ahmed MH. Managing dyslipidemia in HIV/AIDS patients: challenges and solutions. HIV/AIDS (Auckl) 2015;7:1–10.
16. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. Department of Health and Human Services. Accessed at www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
17. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 2015;385:2606–15.
18. Sax PE, Zolopa A, Brar I, et al. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr 2014;67:52–8.
19. Eron JJ, Young B, Cooper DA, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet 2010;375:396–407.
20. Quercia R, Roberts J, Martin-Carpenter L, Zala C. Comparative changes of lipid levels in treatment-naive, HIV-1-infected adults treated with dolutegravir vs. efavirenz, raltegravir, and ritonavir-boosted darunavir-based regimens over 48 weeks. Clin Drug Investig 2015;35:211–9.
21. Lennox JL, DeJesus E, Berger DS, et al. Raltegravir versus efavirenz regimens in treatment-naive hiv-1–infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune Defic Syndr 2010;55:39–48.
22. Nayor M, Vasan RS. Recent update to the US cholesterol treatment guidelines. a comparison with international guidelines. Circulation 2016;133:1795–806.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:2889–934.
24. Mitka M. Exploring statins to decrease HIV-related heart disease risk. JAMA 2015;314:657–9.
25. Dube MP, Stein JH, Aberg JA, et al. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Diseases Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003;37:613–27.
26. Milazzo L, Menzaghi B, Corvasce S, et al. Safety of statin therapy in HIV/hepatitis C virus-coinfected patients. J Acquir Immune Defic Syndr 2007;46:258–60.
27. Hernandez-Romieu AC, Garg S, Rosenberg ES, et al. Is diabetes prevalence higher among HIV-infected individuals compared with the general population? Evidence from MMP and NHANES 2009-2010. BMJ Open Diab Res Care 2017;5:e000304.
28. Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005;165:1179–84.
29. De Wit S, Sabin CA, Weber R, et al. Incidence and risk factors for new-onset diabetes in HIV-infected patients: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. Diabetes Care 2008;31:1224–9.
30. Rasmussen LD, Mathiesen ER, Kronborg G, et al. Risk of diabetes mellitus in persons with and without HIV: a Danish nationwide population-based cohort study. PloS One 2012;7:e44575.
31. Polsky S, Floris-Moore M, Schoenbaum EE, et al. Incident hyperglycaemia among older adults with or at-risk for HIV infection. Antivir Ther 2011;16:181–8.
32. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007;92:2506–12.
33. Galli L, Salpietro S, Pellicciotta G, et al. Risk of type 2 diabetes among HIV-infected and healthy subjects in Italy. Eur J Epidemiol 2012;27:657–65.
34. Howard AA, Hoover DR, Anastos K, et al. The effects of opiate use and hepatitis C virus infection on risk of diabetes mellitus in the Women’s Interagency HIV Study. J Acquir Immune Defic Syndr 2010;54:152–9.
35. Butt AA, McGinnis K, Rodriguez-Barradas MC, et al. HIV infection and the risk of diabetes mellitus. AIDS 2009;23:1227–34.
36. Hadigan C, Kattakuzhy S. Diabetes mellitus type 2 and abnormal glucose metabolism in the setting of human immunodeficiency virus. Endocrin Metab Clin North Am 2014;43:685–96.
37. Butt AA, Fultz SL, Kwoh CK, Kelley D, et al. Risk of diabetes in HIV infected veterans pre- and post-HAART and the role of HCV coinfection. Hepatology 2004;40:115–9.
38. Veloso S, Escote X, Ceperuelo-Mallafre V, et al. Leptin and adiponectin, but not IL18, are related with insulin resistance in treated HIV-1-infected patients with lipodystrophy. Cytokine 2012;58:253–60.
39. Vigouroux C, Maachi M, Nguyen TH, et al. Serum adipocytokines are related to lipodystrophy and metabolic disorders in HIV-infected men under antiretroviral therapy. AIDS 2003;17:1503–11.
40. Palmer CS, Hussain T, Duette G, et al. Regulators of glucose metabolism in CD4+ and CD8+ T cells. Int Rev Immunol 2016;35:477–88.
41. Mehta SH, Moore RD, Thomas DL, et al. The effect of HAART and HCV infection on the development of hyperglycemia among HIV-infected persons. J Acquir Immune Defic Syndr 2003;33:577–84.
42. Monroe AK, Dobs AS, Xu X, et al. Sex hormones, insulin resistance, and diabetes mellitus among men with or at risk for HIV infection. J Acquir Immune Defic Syndr 2011;58:173–80.
43. Rotger M, Gsponer T, Martinez R, et al. Impact of single nucleotide polymorphisms and of clinical risk factors on new-onset diabetes mellitus in HIV-infected individuals. Clin Infect Dis 2010;51:1090–8.
44. Ledergerber B, Furrer H, Rickenbach M, et al. Factors associated with the incidence of type 2 diabetes mellitus in HIV-infected participants in the Swiss HIV Cohort Study. Clin Infect Dis 2007;45:111–9.
45. Hruz PW. Molecular mechanisms for insulin resistance in treated HIV-infection. Best practice & research. Clin Endocrin Metab 2011;25:459–68.
46. Brown TT, Li X, Cole SR, et al. Cumulative exposure to nucleoside analogue reverse transcriptase inhibitors is associated with insulin resistance markers in the Multicenter AIDS Cohort Study. AIDS 2005;19:1375–83.
47. Cossarizza A, Moyle G. Antiretroviral nucleoside and nucleotide analogues and mitochondria. AIDS 2004;18:137–51.
48. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384-7.
49. McComsey GA, Paulsen DM, Lonergan JT, et al. Improvements in lipoatrophy, mitochondrial DNA levels and fat apoptosis after replacing stavudine with abacavir or zidovudine. AIDS 2005;19:15–23.
50. Venhoff N, Setzer B, Melkaoui K, Walker UA. Mitochondrial toxicity of tenofovir, emtricitabine and abacavir alone and in combination with additional nucleoside reverse transcriptase inhibitors. Antivir Ther 2007;12:1075–85.
51. Haubrich RH, Riddler SA, DiRienzo AG, et al. Metabolic outcomes in a randomized trial of nucleoside, nonnucleoside and protease inhibitor-sparing regimens for initial HIV treatment. AIDS 2009;23:1109–18.
52. Aberg JA, Tebas P, Overton ET, et al. Metabolic effects of darunavir/ritonavir versus atazanavir/ritonavir in treatment-naive, HIV type 1-infected subjects over 48 weeks. AIDS Res Hum Retrovir 2012;28:1184–95.
53. Overton ET, Tebas P, Coate B, et al. Effects of once-daily darunavir/ritonavir versus atazanavir/ritonavir on insulin sensitivity in HIV-infected persons over 48 weeks: results of an exploratory substudy of METABOLIK, a phase 4, randomized trial. HIV Clin Trials 2016;17:72–7.
54. Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010;375:735–42.
55. McComsey G, Jiang Y, Erlandson KM, et al. Rosuvastatin improves hip bone mineral density but worsens insulin resistance. Boston, MA: Conference on Retroviruses and Opportunistic Infections; 2014.
56. Lichtenstein K DR, Wood K, et al. Statin use is associated with incident diabetes mellitus among patients in the HIV Outpatient Study. Atlanta, GA: Conference on Retroviruses and Opportunistic Infections; 2013.
57. Spagnuolo V GL, Poli A, et al. Association between statin use and type 2 diabetes mellitus occurrence among HIV-1+ patients receiving ART. Atlanta, GA: Conference on Retroviruses and Opportunistic Infections; 2013.
58. Wang KL, Liu CJ, Chao TF, et al. Statins, risk of diabetes, and implications on outcomes in the general population. J Am Coll Cardiol 2012;60:1231–8.
59. Ridker PM, Pradhan A, MacFadyen JG, et al. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012;380:565–71.
60. Bloomgarden ZT, Handelsman Y. Approaches to treatment 2: Comparison of American Association of Clinical Endocrinologists (AACE) and American Diabetes Association (ADA) type 2 diabetes treatment guidelines. J Diabetes 2016;8:4–6.
61. Kim PS, Woods C, Georgoff P, et al. A1C underestimates glycemia in HIV infection. Diabetes Care 2009;32:1591–3.
62. Diop ME, Bastard JP, Meunier N, et al. Inappropriately low glycated hemoglobin values and hemolysis in HIV-infected patients. AIDS Res Hum Retrovir 2006;22:1242–7.
63. Polgreen PM, Putz D, Stapleton JT. Inaccurate glycosylated hemoglobin A1C measurements in human immunodeficiency virus-positive patients with diabetes mellitus. Clin Infect Dis 2003;37:e53–56.
64. Glesby MJ, Hoover DR, Shi Q, et al. Glycated haemoglobin in diabetic women with and without HIV infection: data from the Women’s Interagency HIV Study. Antivir Ther 2010;15:571–7.
65. Slama L, Palella FJ Jr, Abraham AG, et al. Inaccuracy of haemoglobin A1c among HIV-infected men: effects of CD4 cell count, antiretroviral therapies and haematological parameters. J Antimicrob Chemother 2014;69:3360–7.
66. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012;55:1577–96.
67. Look ARG, Pi-Sunyer X, Blackburn G, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care 2007;30:1374–83.
68. Hajjar J, Habra MA, Naing A. Metformin: an old drug with new potential. Expert Opin Investig Drugs 2013;22:1511–7.
69. Kohli R, Shevitz A, Gorbach S, Wanke C. A randomized placebo-controlled trial of metformin for the treatment of HIV lipodystrophy. HIV Med 2007;8:420–6.
70. Tivicay prescribing information. Accessed at www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Tivicay/pdf/TIVICAY-PI-PIL.PDF.
71. Han JH, Gordon K, Womack JA, et al. Comparative effectiveness of diabetic oral medications among HIV-infected and HIV-uninfected veterans. Diabetes Care 2017;40:218–25.
72. Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006;20:2165–74.
73. Libois A, Clumeck N, Kabeya K, et al. Risk factors of osteopenia in HIV-infected women: no role of antiretroviral therapy. Maturitas 2010;65:51–4.
74. Carr A, Grund B, Neuhaus J, et al. Prevalence of and risk factors for low bone mineral density in untreated HIV infection: a substudy of the INSIGHT Strategic Timing of AntiRetroviral Treatment (START) trial. HIV Med 2015;16 Suppl 1:137–46.
75. Triant VA, Brown TT, Lee H, Grinspoon SK. Fracture prevalence among human immunodeficiency virus (HIV)-infected versus non-HIV-infected patients in a large U.S. healthcare system. J Clin Endocrinol Metab 2008;93:3499–504.
76. Young B, Dao CN, Buchacz K, Baker R, Brooks JT, Investigators HIVOS. Increased rates of bone fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared with the US general population, 2000-2006. Clin Infect Dis 2011;52:1061–8.
77. Rodriguez M, Daniels B, Gunawardene S, Robbins GK. High frequency of vitamin D deficiency in ambulatory HIV-Positive patients. AIDS Res Hum Retrovir 2009;25:9–14.
78. Teichmann J, Lange U, Discher T, et al. Bone mineral density in human immunodeficiency virus-1 infected men with hypogonadism prior to highly-active-antiretroviral-therapy (HAART). Eur J Med Res 2009;14:59–64.
79. Lo Re V 3rd, Volk J, Newcomb CW, et al. Risk of hip fracture associated with hepatitis C virus infection and hepatitis C/human immunodeficiency virus coinfection. Hepatology 2012;56:1688–98.
80. Maalouf NM, Zhang S, Drechsler H, et al. Hepatitis C co-infection and severity of liver disease as risk factors for osteoporotic fractures among HIV-infected patients. J Bone Miner Res 2013;28:2577–83.
81. Byrne DD, Newcomb CW, Carbonari DM, et al. Increased risk of hip fracture associated with dually treated HIV/hepatitis B virus coinfection. J Viral Hepat 2015;22:936–47.
82. Gilsanz V, Chalfant J, Mo AO, et al. Reciprocal relations of subcutaneous and visceral fat to bone structure and strength. J Clin Endocrinol Metab 2009;94:3387–93.
83. Rosen CJ, Klibanski A. Bone, fat, and body composition: evolving concepts in the pathogenesis of osteoporosis. Am J Med 2009;122:409–14.
84. Fakruddin JM, Laurence J. HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk. J Biol Chem 2003;278:48251–8.
85. Gazzola L, Bellistri GM, Tincati C, et al. Association between peripheral T-Lymphocyte activation and impaired bone mineral density in HIV-infected patients. J Transl Med 2013;11:51.
86. Clerici M, Shearer GM. A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol Today 1993;14:107–11.
87. Chew N, Tan E, Li L, Lim R. HIV-1 tat and rev upregulates osteoclast bone resorption. J Int AIDS Soc 2014;17(4 Suppl 3):19724.
88. McComsey GA, Tebas P, Shane E, et al. Bone disease in HIV infection: a practical review and recommendations for HIV care providers. Clin Infect Dis 2010;51:937–46.
89. Panayiotopoulos A, Bhat N, Bhangoo A. Bone and vitamin D metabolism in HIV. Rev Endocr Metab Disord 2013;14:119–25.
90. Fakruddin JM, Laurence J. Interactions among human immunodeficiency virus (HIV)-1, interferon-gamma and receptor of activated NF-kappa B ligand (RANKL): implications for HIV pathogenesis. Clin Exp Immunol 2004;137:538–45.
91. Li Y, Toraldo G, Li A, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007;109:3839–48.
92. Finkelstein JS, Brockwell SE, Mehta V, et al. Bone mineral density changes during the menopause transition in a multiethnic cohort of women. J Clin Endocrinol Metab 2008;93:861–8.
93. Huang JS, Hughes MD, Riddler SA, Haubrich RH, Aids Clinical Trials Group AST. Bone mineral density effects of randomized regimen and nucleoside reverse transcriptase inhibitor selection from ACTG A5142. HIV Clin Trials 2013;145:224–34.
94. McComsey GA, Kitch D, Daar ES, et al. Bone mineral density and fractures in antiretroviral-naive persons randomized to receive abacavir-lamivudine or tenofovir disoproxil fumarate-emtricitabine along with efavirenz or atazanavir-ritonavir: Aids Clinical Trials Group A5224s, a substudy of ACTG A5202. J Infect Dis 2011;203:1791–801.
95. Schafer JJ, Manlangit K, Squires KE. Bone health and human immunodeficiency virus infection. Pharmacotherapy 2013;33:665–82.
96. Mateo L, Holgado S, Marinoso ML, et al. Hypophosphatemic osteomalacia induced by tenofovir in HIV-infected patients. Clin Rheumatol 2016;35:1271–9.
97. Brown TT, Moser C, Currier JS, et al. Changes in bone mineral density after initiation of antiretroviral treatment with tenofovir disoproxil fumarate/emtricitabine plus atazanavir/ritonavir, darunavir/ritonavir, or raltegravir. J Infect Dis 2015;212:1241–9.
98. Ofotokun I, Titanji K, Vikulina T, et al. Role of T-cell reconstitution in HIV-1 antiretroviral therapy-induced bone loss. Nat Commun 2015;6:8282.
99. Ofotokun I, Titanji K, Vunnava A, et al. Antiretroviral therapy induces a rapid increase in bone resorption that is positively associated with the magnitude of immune reconstitution in HIV infection. AIDS 2016;30:405–14.
100. Stephens KI, Rubinsztain L, Payan J, et al. Dual-energy x-ray absorptiometry and calculated FRAX risk scores may underestimate osteoporotic fracture risk in vitamin d-deficient veterans with HIV infection. Endocr Pract 2016;22:440–6.
101. Mirza F, Canalis E. Management of endocrine disease: Secondary osteoporosis: pathophysiology and management. Eur J Endocrinol 2015;173:R131–151.
102. Brown TT, Hoy J, Borderi M, et al. Recommendations for evaluation and management of bone disease in HIV. Clin Infect Dis 2015;60:1242–51.
103. Mazzotta E, Ursini T, Agostinone A, et al. Prevalence and predictors of low bone mineral density and fragility fractures among HIV-infected patients at one Italian center after universal DXA screening: sensitivity and specificity of current guidelines on bone mineral density management. AIDS Patient Care STDS 2015;29:169–80.
104. Calmy A, Chevalley T, Delhumeau C, et al. Long-term HIV infection and antiretroviral therapy are associated with bone microstructure alterations in premenopausal women. Osteoporos Int 2013;24:1843–52.
105. Yin MT, Lund E, Shah J, et al. Lower peak bone mass and abnormal trabecular and cortical microarchitecture in young men infected with HIV early in life. AIDS 2014;28:345–53.
106. Lo Re V, 3rd, Lynn K, Stumm ER, et al. Structural bone deficits in HIV/HCV-coinfected, HCV-monoinfected, and HIV-monoinfected women. J Infect Dis 2015;212:924–33.
107. Allavena C, Delpierre C, Cuzin L, et al. High frequency of vitamin D deficiency in HIV-infected patients: effects of HIV-related factors and antiretroviral drugs. J Antimicrob Chemother 2012;67:2222–30.
108. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011;96:1911–30.
109. Mondy K, Powderly WG, Claxton SA, et al. Alendronate, vitamin D, and calcium for the treatment of osteopenia/osteoporosis associated with HIV infection. J Acquir Immune Defic Syndr 2005;38:426–31.
110. McComsey GA, Kendall MA, Tebas P, et al. Alendronate with calcium and vitamin D supplementation is safe and effective for the treatment of decreased bone mineral density in HIV. AIDS 2007;21:2473–82.
111. Guaraldi G, Orlando G, Madeddu G, et al. Alendronate reduces bone resorption in HIV-associated osteopenia/osteoporosis. HIV Clin Trials 2004;5:269–77.
112. Negredo E, Bonjoch A, Perez-Alvarez N, et al. Comparison of two different strategies of treatment with zoledronate in HIV-infected patients with low bone mineral density: single dose versus two doses in 2 years. HIV Med 2015;16:441–8.
113. Bolland MJ, Grey A, Horne AM, et al. Effects of intravenous zoledronate on bone turnover and bone density persist for at least five years in HIV-infected men. J Clin Endocrinol Metab 2012;97:1922–8.
114. Pinzone MR, Moreno S, Cacopardo B, Nunnari G. Is there enough evidence to use bisphosphonates in HIV-infected patients? A systematic review and meta-analysis. AIDS Rev 2014;16:213–22.
115. Ofotokun I, Titanji K, Lahiri CD, et al. A Single-dose zoledronic acid infusion prevents antiretroviral therapy-induced bone loss in treatment-naive HIV-infected patients: a phase IIb trial. Clin Infect Dis 2016;63:663–71.
116. Lewiecki EM, Gordon CM, Baim S, et al. International Society for Clinical Densitometry 2007 adult and pediatric official positions. Bone 2008;43:1115–21.
From the University of Connecticut School of Medicine, Farmington, CT.
Abstract
- Objective: To review the metabolic complications of HIV infection.
- Methods: Review of the literature in the context of 3 clinical cases.
- Results: People with HIV infection are living longer thanks to the advent of potent antiretroviral therapy. This has led to increased incidence of age-related metabolic complications, including a higher risk of cardiovascular disease, hyperlipidemia, metabolic syndrome, and osteoporosis. Appropriate management of these complications requires an understanding of disease-related and drug-related side effects as well as the potential for drug-drug interactions. A multidisciplinary approach to patient management is most effective.
- Conclusion: Awareness of the metabolic complications frequently encountered in HIV infection, drug interactions, and possible toxicities is critical to the successful management of HIV-infected individuals.
Key words: HIV; antiretroviral therapy; hyperlipidemia; metabolic syndrome; diabetes; hypogonadism.
According to the most recent data from the Joint United Nations Programme on HIV/AIDS (UNAIDS), 36 million people worldwide are living with HIV/AIDS, with 18 million accessing effective antiretroviral therapy (ART) [1]. The past 2 decades have witnessed enormous advances in the field from prevention to diagnosis and therapeutics, and modern ART largely allows HIV-infected persons to live near-normal life spans [2,3]. However, from the beginning of the epidemic, HIV-infected persons on effective therapy have suffered from myriad metabolic consequences, many of which affect quality of life and result in excess mortality [4]. It is also true that untreated HIV infection portends an increased risk of metabolic complications, likely related to abnormal immune activation, as demonstrated in structured interruption trials [5,6]. It is worth noting, however, that while many of these metabolic dyscrasias and associated risks have historically been attributed primarily to the treatment of HIV infection with ART, data from cohort studies and randomized clinical trials have repeatedly demonstrated significant reductions in morbidity and mortality when ART is initiated early [7]. In this paper, we review HIV-related metabolic complications frequently encountered in clinical practice (hyperlipidemia, diabetes, and bone disease) and best practice considerations in the context of 3 clinical cases.
Case Patient 1
Initial Presentation and History
A 58-year-old male with a history of hypertension and mixed hyperlipidemia is referred for evaluation of newly diagnosed HIV infection. He has no history of intravenous drug use but has had multiple male and female sex partners in the past few years, and requested testing after a partner tested positive. His last negative test was 2 years ago. The patient does not smoke cigarettes. Overall he feels well and tolerates his regimen of lisinopril 10 mg and simvastatin 20 mg daily. On initial evaluation, his exam is unremarkable other than subtle white plaques on the dorsal surface of the tongue and buccal mucosa, and moderate central obesity. Vital signs including blood pressure are normal. Initial laboratory evaluation reveals a CD4 cell count of 150 cells/mm3 and an HIV RNA level of 200,000 copies/mL. Fasting serum total cholesterol is 220 mg/dL, triglycerides 250 mg/dL, LDL 170 mg/dL, and HDL 35 mg/dL. Serum BUN, creatinine, and liver function testing results are normal.
What initial regimen might be recommended based on the status of his HIV infection and comorbidities?
The most recent iteration of the US Department of Health and Human Services (DHHS) guidelines on use of antiretroviral agents (ARVs) in HIV recommends an initial ART regimen that includes a backbone of 2 nucleoside reverse transcriptase inhibitors (NRTIs), generally tenofovir disoproxil fumarate or tenofovir alafenamide, abacavir (ABC), emtricitabine (FTC), or lamivudine (3TC) [2]. To this backbone should be added a third agent; the majority of those currently recommended are integrase strand transfer inhibitors (INSTIs) (dolutegravir, elvitegravir, raltegravir); one recommended protease inhibitor (PI) (ritonavir-boosted darunavir) is also an option. Some of these initial recommended regimens are available as fixed-dosed combinations in 1 pill, making them attractive options.
The latest guidelines also clearly recommend starting ART in all HIV-infected individuals, irrespective of CD4 count. The patient described above has a very low CD4 count, so there is no question he needs to begin therapy promptly. Given his low CD4 count and relatively high viral load, one may consider a ritonavir-boosted PI as perhaps the most robust option and with a relatively high barrier to resistance development, in contrast to other options. Assuming the patient’s baseline resistance testing reveals a fully sensitive wild-type virus without meaningful resistance mutations, he will begin a regimen of TDF/FTC plus ritonavir-boosted darunavir, 3 pills once daily. Given his low CD4 count (below 200 cells/mm3), he will also need prophylaxis for Pneumocystis jirovecii pneumonia, in the form of trimethoprim/sulfamethoxazole (TMP/SMX) daily. Given the potential for interaction between the boosted PI and simvastatin, his lipid-lowering agent is switched to atorvastatin 10 mg daily.
What is the association between hyperlipidemia and HIV infection and treatment?
Hyperlipidemia represents a key modifiable risk factor for the development of cardiovascular disease (CVD) in HIV-infected individuals [8]. Indeed, a multicenter cross-sectional study of older HIV-infected individuals performed in Spain revealed a 54% prevalence of dyslipidemia and 23% CVD [9]. Most experts believe that metabolic abnormalities observed in HIV-infected individuals are the result of a combination of factors: those resulting from abnormal immune activation and inflammation related to viral replication, and those related to certain ARVs [10].
Early after HIV seroconversion, decline in HDL is one of the first proatherogenic changes observed. This, along with increased triglyceride and LDL levels, likely contribute to increased risk of CVD in this population. Increased microbial translocation, evidenced by increased levels of lipopolysaccharide (LPS), may drive immune activation, leading to dyslipidemia via a multitude of hypothesized mechanisms [4]. It has been theorized that HDL lipoproteins are less stable on ART, leading to potentially impaired plasma lipolytic activities or hepatic cholesteryl ester uptake [6,11]. Increased VLDL from release of free fatty acids may lead to higher triglyceride levels and triglyceride-rich LDL and HDL, all associated with increased risk of CVD [11].
In terms of effects of specific ARV classes, although newer agents have less of a propensity to cause dyslipidemia, the PI class arguably remains most problematic. In comparison to other classes, the PIs tend to result in greater increases in triglycerides, total cholesterol, and LDL, and have frequent drug-drug interactions with lipid-lowering agents [10,12]. Estimated prevalence of dyslipidemia in patients receiving PI therapy varies from 28% to 80% [13]. The prospective multinational cohort Data collection on Adverse events of Anti-HIV Drugs (DAD) study found significantly higher rates of hypertriglyceridemia, hypercholesterolemia, and low HDL in patients on PIs in comparison to non–nucleoside reverse transcriptase inhibitors (NNRTIs) [14]. Various mechanisms have been proposed to explain the PIs adverse effects on lipids, including inhibition of lipogenesis and adipocyte differentiation, decreased hepatocyte clearance of chylomicrons and VLDL, and increased hepatic synthesis of triglycerides [15]. Of the available PIs, atazanavir and darunavir have less potential to lead to dyslipidemia [10], while lopinavir/ritonavir, fosamprenavir, and tipranavir may have the highest [13]. Of the NNRTIs, efavirenz is most frequently associated with dyslipidemia, specifically increased triglycerides and total cholesterol [13]. However, these increased values seen on efavirenz therapy may be offset by relative increases in HDL, with little resultant effect on the total cholesterol:HDL ratio. Rilpivirine, etravirine, and nevirapine are relatively less likely to drive lipid changes, although certain drug interactions are important to recognize in clinical practice, such as the interaction between rilpivirine and proton pump inhibitors [2,13,16]. It is also worth noting that no NNRTIs are included in current guidelines as preferred therapy [2].
Historically, the thymidine analogue NRTIs (stavudine, didanosine, zidovudine) have been associated with lipid dyscrasias and lipoatrophy, but fortunately these are no longer used frequenty except in cases requiring deep salvage therapy for highly treatment-experienced patients. Two newer NRTIs, tenofovir and abacavir, have relatively neutral to favorable effects on lipids. The combination of tenofovir disoproxil (TDF) and emtricitabine (trade name Truvada) was associated with significantly lower triglycerides, total cholesterol and LDL than other NRTI pairs [6]. TDF has been postulated to have lipid-lowering effects. Switch studies in which patients were taken off thymidine analogues and placed on TDF, demonstrated recovery of limb fat in patients with lipoatrophy, and those switched off abacavir-based ART to TDF showed statistically significant lower fasting total cholesterol at week 12, without differences of viral suppression [8]. Tenofovir alafenamide (TAF) is a next-generation prodrug of tenofovir that results in improved stability in plasma and higher intracellular levels in comparison to TDF [17]. Although randomized controlled trials of TAF vs TDF-based ARV regimens have suggested statistically higher total cholesterol, serum HDL is also increased resulting in unchanged total:HDL ratios and no differences in risk classifications [18]. Integrase inhibitors (INSTI) now represent first-line therapy in combination with an NRTI backbone, and since their availability in 2007 have been evaluated in comparison to various PIs and NNRTIs. Both raltegravir and dolutegravir have consistently shown broad neutral effects on lipids and are among the most metabolically friendly agents available [19–21]. Because it is given in fixed-dose combination with non-ritonavir pharmacologic booster cobicistat, elvitegravir has effects similar to ritonavir-boosted PIs on lipids [6].
What are management considerations in the treatment of hyperlipidemia in HIV-infected patients?
Patients with HIV and hyperlipidemia may benefit from lipid-lowering therapy in addition to ART, although in certain cases appropriate switches may make a difference. Careful consideration of drug interactions between ARVs and lipid-lowering agents, in addition to ARV history and known drug resistance, is warranted prior to selecting a regimen in these patients. In addition, the latest American College of Cardiology/American Heart Association guidelines suggest evaluating 10-year risk of atherosclerotic cardiovascular disease (ASCVD) using the pooled cohort equation to determine the type and dose of statin required (moderate vs high intensity) [22]. It is noteworthy that HIV infection and its therapies are not taken into account as potential risk factors in this model. Primary prevention in non-diabetic patients with a statin is recommended for patients with a 10-year absolute risk of ≥ 7.5% [22]. This patient’s risk is estimated at between 12% and 13% based on this equation, so primary prevention with a moderate-or high-intensity statin is recommended (Table 1) [23]. Data from more than 80,000 patients in the Veterans Aging Cohort Study (VACS) showed that HIV-infected patients with no baseline ASCVD had 50% increased risk of acute myocardial infarction when compared to HIV-uninfected patients over 6 years of follow-up [24]. Thus, consideration of the virus itself or its therapy as an additional risk factor may be valid.
Screening and Monitoring of Hyperlipidemia
The most recent iteration of the DHHS primary care guidelines for the management of HIV-infected individuals recommends obtaining fasting (ideally 12 hours) lipid profiles upon initiation of care, and within 1 to 3 months of beginning therapy [12,13]. These initial levels, along with other elements of the patient’s history and calculation of risk may help determine whether lipid-lowering therapy is indicated, and if so, which therapy would be best. In general, after regimen switches or additions of either ARV or statin therapy, repeating fasting lipid levels 6 weeks later is recommended to gauge the effects of the switch. This is especially critical when interactions between ARVs and lipid-lowering therapies are possible. Some experts recommend performing annual screening of patients with normal baseline lipids or with well-controlled hyperlipidemia on therapy. Assessment of 10-year ASCVD risk is also recommended annually, in addition to baseline risk assessment, to determine the need and appropriateness of statin therapy [25]. The question of primary prevention in HIV has yet to be definitively answered. Small studies in this population have demonstrated that statins have the potential to slow progression of carotid intima media thickness and reduce noncalcified plaque volume [24]. An NIH/AIDS Clinical Trial Group–sponsored randomized clinical trial (“REPRIEVE”) is currently underway to address this question. More than 6000 HIV-infected men and women with no history of ASCVD at 100 sites in several countries are enrolled to assess the benefit of pitavastatin as primary prevention in this risk group [24]. Metabolized via glucuronidation primarily, as opposed to cytochrome p450 (CYP 3A4 isoenzyme), pitavastatin is thought to have fewer drug interactions with ARVs in general [6] (Table 2).
Relevant Drug-Drug Interactions
Deciding which statin to begin in HIV-infected patients depends on whether moderate- or high-intensity therapy is warranted and whether the potential for drug interaction with ARVs exists. Table 2 [6,12] depicts available statins and the potential for pharmacokinetic interaction with the primary ARV classes. Simvastatin and lovastatin are heavily metabolized via the CYP 3A4 pathway, resulting in the highest potential risk of interaction with CYP 3A4 inhibitors, such as the PIs, or inducers (eg, NNRTIs, in particular efavirenz) [6]. The former may inhibit metabolism of these statins, resulting in increased risk of toxicity, while co-administration with efavirenz, for example, may result in inadequate serum concentration and therefore inadequate lipid-lowering effects. Although less lipophilic, atorvastatin results in similar interactions with PIs and NNRTIs, and therefore low starting doses with close monitoring is recommended [6]. Fewer interactions have been noted with rosuvastatin, pravastatin, and pitavastatin, as these do not require CYP 3A4 for their metabolism and are thus less likely to be affected by ARVs. These therefore represent potentially safer first choices for certain patients on ARVs, although of these, only rosuvastatin is classified as a high-intensity statin [22,23] (Table 1). When compared directly to pravastatin 40 mg daily in patients receiving ritonavir-boosted PIs, rosuvastatin performed superiorly at 10 mg per day, resulting in more significant reductions in LDL and triglyceride levels [15]. Although it is eliminated largely unchanged through the kidney and liver, pravastatin has been reported to idiosyncratically interact with darunavir, resulting in potentially increased pravastatin levels and associated toxicity [25]. Treatment of pure hypertriglyceridemia in HIV-infected patients should begin with fibrates, which have little to no risk of interaction with most clinically relevant ARVs [6,10]. Alternatives to lower triglycerides include niacin and N-3 polyunsaturated fatty acids [25].
Case 1 Continued
The patient has an impressive response to his initial regimen of TDF/FTC plus boosted darunavir, with repeat CD4 count after 12 weeks of 275 (18%) cells/mm3 and an undetectable viral load (< 20 copies/mL). Other lab parameters are favorable and he is tolerating the regimen well without notable side effects. However, at his next visit, although his viral load remains undetectable, his triglyceride level has increased to 350 mg/dL, although other lipid parameters are comparable to the prior result. He complains of diffuse body aches, concentrated in large muscle groups of the extremities, and dark-colored urine. A creatine phosphokinase (CPK) level is elevated at 300 IU/L (normal, 22–269, negative MB fraction). Serum creatinine is 1.4 mg/dL (had been 1.1 mg/dL at baseline). Given he has done so well otherwise on these ARVs, he is reluctant to make any changes.
What drug-drug interaction is most likely causing this patient's problem, and how should it be managed?
This scenario is not uncommon in clinical practice, and changes to regimens are sometimes necessary in order to avoid drug interactions. Care must be taken to thoroughly review antiretroviral history and available resistance testing (in this case a relatively short history) in order to ensure a fully active and suppressive regimen is chosen. This description could be the result of an interaction between lipid-lowering therapy and ARVs resulting in increased relative concentrations of one drug or the other and therefore leading to toxicity. Given this possibility, and suboptimal control of hyperlipidemia, consideration should be given to switching both his ART and his statin therapy.
Safety and Potential Toxicities of Lipid-Lowering Therapy
Increased serum concentration of certain statins when co-administered with CYP 3A4 inhibitors like the PIs leads to heightened risk of statin-associated toxicities. In general, this includes muscle inflammation, leading to increases in serum CPK level and associated symptoms, including myalgias, myositis, or in extreme cases, rhabdomyolysis [6]. Although rare, this toxicity can be serious and may lead to acute renal injury if not recognized and managed appropriately. In theory, the potential for statin-associated hepatotoxicity may also be increased in patients receiving PIs, although this has not been borne out in clinical trials [26]. In fact, quite the opposite may be true, in that statins have been shown to improve liver function in patients with hepatitis C virus (HCV) coinfection and with nonalcoholic fatty liver disease [6,15].
Case 1 Conclusion
The patient does well on his new ARV regimen of TAF/FTC and dolutegravir, 2 pills once daily. He no longer requires TMP/SMX, as his CD4 count has been reliably above 200 cells/mm3 on several occasions. Serum creatinine is back down to baseline and CPK has normalized. Fasting lipids have improved since the switch, and he no longer has symptoms of myositis on rosuvastatin 10 mg daily.
Summary
Consideration of statin therapy is complicated by potential drug interactions with ARVs and associated toxicity. However, given known effects of ARVs on lipids, and of immune activation and inflammation related to the virus itself, these patients should be carefully evaluated for statin therapy for their anti-inflammatory and immune modulatory effects as much as for their lipid-lowering ability. Utilization of HIV infection and its therapies as additional cardiovascular risk factors when calculating 10-year risk deserves further consideration; forthcoming results of the REPRIEVE trial are certain to contribute valuable information to this field of study.
Case Patient 2
Initial Presentation and History
A 45-year-old female with history of HIV infection since 2008 presents to the office for new-onset diabetes, diagnosed 2 weeks ago. She has had symptoms of polyuria and polydipsia for the last 1 month. She denies diarrhea, nausea, vomiting or weight loss. She is currently on a regimen consisting of zidovudine/lamivudine plus lopinavir/ritonavir. There is no family history of diabetes. Her examination is unremarkable, including normal vital signs (weight 150 lb, blood pressure 114/70, heart rate 76) and no evidence of insulin resistance, including acanthosis nigricans or striae. Glycosylated hemoglobin level (HbA1c) is 8%. Creatinine and liver function tests are within reference ranges.
Do HIV-infected patients have a higher incidence of type 2 diabetes mellitus (DM)?
Prevalence of type 2 DM in HIV-infected patients varies between 2% to 14% [27]. This variation is due to the different cutoffs used for diagnosis, differences in cohorts studied, and how risk factors are analyzed [28–31]. In a recent nationally representative estimate of DM prevalence among HIV-infected adults receiving medical care in the United States in 2009–2010, the prevalence of DM was noted to be 10.3%. In comparison to the general adult US population, HIV-infected individuals have a 3.8% higher prevalence of DM after adjusting for age, sex, race/ethnicity, education, poverty-level, obesity, and HCV infection [27].
There is controversy over whether HIV infection itself increases the risk of type 2 DM, with some studies showing increased risk [28,32,33] and others showing no independent effect or an inverse effect [30,34,35]. Studies on the impact of ethnicity and race on prevalence of DM are limited [36].
Certain traditional risk factors (age, ethnicity, obesity) are still responsible for most of the increased risk of diabetes in the HIV-infected population [35,37]. HIV infection itself is associated with metabolic dysfunction, independent of ARV. In HIV-infected patients, impaired glucose metabolism is associated with altered levels of adipokines, increased adiponectin and soluble-tumor necrosis factor receptor 1 (sTNFR1) and decreased leptin [38,39]. HIV-associated alterations in CD4+ and CD8+ T cell function also impair glycolysis, which may adversely impact glucose metabolism [40].
Other contributing factors in HIV-infected patients are HCV co-infection [41], medications (atypical antipsychotics, corticosteroids), opiates, and low testosterone [42]. HCV co-infection may lead to hepatic steatosis and liver fibrosis, and increasing insulin resistance.
Recent genomic studies show several common single-nucleotide polymorphisms (SNPs) associated with diabetes in the general population. In the Swiss HIV Cohort Study, SNPs accounted for 14% of type 2 DM risk variability, whereas ARV exposure accounted for 3% and age for 19% of the variability in DM [43].
ARVs also increase the risk of type 2 DM by both direct and indirect effects. Certain ARVs causes lipoatrophy [30] and visceral fat accumulation/lipohypertrophy [29,44]. PIs increase insulin resistance via effects on GLUT-4 transporter and decrease insulin secretion through effects on B cell function [45]. NRTIs (eg, stavudine, zidovudine and didanosine) can cause direct mitochondrial toxicity [46–48]. Utilization of newer ARV agents has decreased the prevalence of severe lipoatrophy, but lipohypertrophy and the underlying metabolic abnormalities persist. The DHHS “preferred” nucleoside analogues, tenofovir and abacavir, do not induce mitochondrial toxicity and have more favorable metabolic profiles [49,50]. In ACTG Study 5142, thymidine-sparing regimens were found to cause less lipoatrophy [51]. In addition, darunavir and atazanavir, the preferred and alternative PIs and the integrase strand transfer inhibitor have limited or modest impact on insulin sensitivity [20,52,53]. This has led to a recent decline in the incidence of type 2 DM in HIV-infected patients.
Statins can also increase insulin resistance and DM [54], although studies have shown mixed results [55–57]. The benefits of statin therapy likely outweigh the risk of DM since there is a significant cardiovascular event reduction with their use [58,59].
How is diabetes diagnosed in HIV-infected patients?
Optimal diabetes screening guidelines have not been established specifically for HIV-infected patients. The American Diabetes Association (ADA) guidelines recommend that diabetes in the general population be diagnosed by 2 elevated fasting blood glucose levels, HbA1c, oral glucose tolerance test (OGTT), or high random glucose with classic symptoms of hyperglycemia [60]. Repeat testing is recommended every 3 years. The OGTT is recommended for diagnosis in pregnant women.
HbA1c may underestimate glycemic burden in HIV-infected individual due to higher mean corpuscular volume, NRTI use (specifically abacavir), or lower CD4 count [61–65]. The Infectious Diseases Society of America (IDSA) 2013 primary care guidelines for HIV-infected patients recommends obtaining a fasting glucose and/or HbA1c prior to and within 1–3 months after starting ARV [12]. Use of HbA1c threshold cutoff of 5.8% for the diagnosis of DM and testing every 6–12 months are recommended.
How should this patient’s diabetes be managed?
The ADA guidelines suggest a patient-centered approach to management of diabetes [66]. All patients should be educated about lifestyle modifications with medical nutrition therapy and moderate-intensity aerobic activity and weight loss [67]. If a patient is on lopinavir/ritonavir or a thymidine analogue (zidovudine, stavudine), one should consider switching the ARV regimen [2].
There are currently no randomized controlled trials of diabetes treatment specific to patients with HIV infection. Metformin is the first-line agent. It improves insulin sensitivity by reducing hepatic glucose production and improving peripheral glucose uptake and lipid parameters [68,69]. Other oral hypoglycemic agents used in the treatment of type 2 diabetes are shown in Table 3.
Case 2 Continued
The patient is switched to TAF/FTC plus dolutegravir with improvement in blood sugars. She is also started on metformin. Co-administration of metformin and dolutegravir will be carefully monitored since dolutegravir increases metformin concentration [70]. When dolutegravir is used with metformin, the total daily dose of metformin should be limited to 1000 mg.
• How should this patient be followed?
If the patient is still not at goal HbAb1c at follow-up, there are multiple other treatment options, including use of insulin. Goal HbA1c for most patients with type 2 DM is < 7%; however, this goal should be individualized for each patient in accordance with the ADA guidelines [12]. A longitudinal cohort study of 11,346 veterans with type 2 diabetes compared the glycemic effectiveness of oral diabetic medications ( metformin, sulfonylurea and a thiazolidinedione) among veterans with and without HIV infection. This study did not find any significant difference in HbA1c based on different diabetes medications. However the HBA1c reduction was less in black and Hispanic patients. The mechanism for the poorer response among these patients need to be evaluated further [71]. In addition to management of blood sugar, other CVD risk factors, hyperlipidemia, hypertension, smoking, etc, should be assessed and managed aggressively.
Case Patient 3
Initial Presentation and History
A 45-year-old male with a history of HIV infection diagnosed 10 years ago, on TDF/FTC/efavirenz (trade name Atripla) for the last 7 years, presents with a left femoral neck fracture after he missed the pavement and fell on his left hip. His history is significant for IV drug abuse for 10 years prior to diagnosis of HIV, and he has been on methadone for the last 6 years.
Is HIV infection associated with increased prevalence of osteopenia and osteoporosis and increased risk of fractures?
With recent advancements in antiretroviral therapy and improved survival of the HIV-infected population, osteoporosis and increased fracture risk have become important causes of morbidity and mortality. Osteoporosis is a skeletal disorder characterized by compromised bone strength, which predisposes to an increased risk of fracture. The World Health Organization defines osteoporosis as a bone mineral density (BMD) measurement by dual X-ray absorptiometry (DXA) at the spine, hip, or forearm that is more than 2.5 standard deviations below that of a "young normal" adult (T-score < –2.5) or a history of one or more fragility fractures. Fragility fractures result from mechanical forces that would not ordinarily result in fracture, such as fall from standing height [40]. Osteopenia is characterized by low BMD (T-score between –1.0 and –2.5) and can be a precursor to osteoporosis.
Several observational, retrospective, and prospective studies have shown lower bone density and an increased risk of fractures in the HIV-infected population compared to age-, race- and sex-matched HIV-negative adults. In a large meta-analysis of pooled prevalence data on 884 HIV-infected patients compared with 654 HIV-uninfected age- and sex-matched controls [72], overall, HIV-infected patients had a significant 6.4-fold increased odds of reduced BMD and a 3.7-fold increased odds of osteoporosis compared to the control population. This meta-analysis also compared ARV-treated subjects to ARV-naive subjects and showed that ARV-treated subjects (n = 824) had a higher prevalence of reduced BMD compared with ARV-naive subjects (n= 202; odds ratio 2.5, 95% CI 1.8–3.7). The odds of osteoporosis was increased 2.4 times (95% CI 1.2 – 4.8) in ARV-treated subjects compared with ARV-naive subjects. None of the studies adjusted for potentially important confounding factors, such as age or duration of HIV infection. PI-treated patients (n = 791) were also found to have a higher prevalence of reduced BMD compared with PI-untreated patients (n = 410; OR 1.5, 95% CI 1.1–2.0). The odds of osteoporosis in PI-treated patients (n = 666) was also 1.6-fold greater (95% CI 1.1–2.3) than those not treated with PI (n = 367).
Low bone density has also been reported in HIV- positive premenopausal women irrespective of ARV status. In a recent study of 89 premenopausal women (mean age, 37 years) predominantly of African origin with HIV infection, osteopenia and osteoporosis were prevalent in one-third of these women, irrespective of ARV use and were associated with low BMI [73]. In a sub-study of the INSIGHT trial evaluating prevalence of and risk factors for low BMD in untreated HIV infection, performed at several sites across 6 continents involving 424 subjects, osteopenia was present in a third of this relatively young predominantly non-white ART-naive population (mean age 34 + 10 years) with normal CD4 cell counts, while only 2% had osteoporosis. Factors independently associated with lower BMD at the hip and spine were female sex, Latino/Hispanic ethnicity, lower BMI, and higher estimated glomerular filtration rate. Longer duration of HIV infection was also associated with lower hip BMD. Current or nadir CD4 cell count and HIV viral load were not associated with low BMD [74].
Many studies have reported increased fracture prevalence in the HIV population. In a retrospective study of fracture prevalence in a large US health care system, a significantly higher rate of fractures was reported in HIV-infected men and women compared to non-HIV-infected controls (2.87 vs. 1.77 fractures per 100 persons, P < 0.001). The difference in the increased fracture prevalence was greater in HIV positive men compared to women (3.08 vs. 1.83; P < 0.001). Vertebral, wrist and hip fractures were more prevalent in men compared to vertebral and wrist fractures only in women. Fracture prevalence was higher in both Caucasian females and males and only in African-American women [75].
In the HIV Outpatient Study (HOPS) [76], age-adjusted fracture rates in the HIV population were noted to be 1.98 to 3.69 times higher than rates in the general population. The HOPS was an open prospective cohort study of HIV-infected adults who were followed at 10 US HIV clinics. Rates of first fractures at any anatomic site from 2000–2008 were assessed among 5826 active HOPS patients (median age 40 years, 79% male, 52% Caucasian, and 73% exposed to ART). Among persons aged 25–54 years, both fracture rates and relative proportion of fragility fractures were higher among HOPS patients than among outpatient controls. Older age, substance abuse, nadir CD4+ cell count <200 cells/mm, HCV infection and DM were associated with incident fractures [76].
What factors contribute to poor bone health in the HIV population?
Several factors that contribute to low bone density are present at a higher rate in the HIV population (Table 4). These include poor nutritional status in terms of suboptimal calcium and vitamin D intake, hypogonadism, low body weight, and alcohol, tobacco and substance abuse.
Vitamin D deficiency is very common in HIV-infected patients, with a prevalence of up to 60% to 75% [77]. Hypogonadism is also relatively common among HIV population [78], contributing to lower bone density. Co-infection with HCV is also associated with increased risk of fractures. In a large cohort of Medicaid beneficiaries, a significant increase in the risk of hip fracture was demonstrated in HCV/HIV co-infected subjects compared either with HCV mono-infected, HIV mono-infected or non-infected individuals [79]. In another large database study, a significantly higher risk of osteoporotic fracture (closed wrist, vertebral or hip fracture) was reported in HCV/HIV co-infected versus HIV mono-infected individuals [80] with fracture rates of 2.57 and 2.07/1000 patient-years (P < 0.001). Dual treatment for HIV/hepatitis B co-infection has also been shown to be associated with a higher risk of hip fracture compared to treatment of HIV mono-infected individuals [81].
HIV infection itself can increase bone loss and reduce bone formation through direct effects related to the HIV antigen load or indirect effects related to activation of the pro-inflammatory cytokines resulting in bone resorption and loss [82]. Co-infection with HCV and/or hepatitis B also contributes to lower bone density in this population. Certain ARVs may also contribute to low bone density in the HIV population. Lipoatrophy related to HIV may also mediate bone loss through complex relationship between central signaling of adipocyte hormones [82,83].
Direct Viral Effects
Several HIV viral proteins have been shown to promote osteoclast activity (vpr and gp120), suppress osteoblast activity (p55-gag) and increase osteoblast apoptosis [84], resulting in increased bone resorption and reduced bone formation, leading to low bone mass. High HIV RNA viral load and T-cell activation are also associated with elevated levels of receptor activator of nuclear factor kappa-B ligand (RANKL), which results in osteoclast formation and increased bone resorption [85]. Other endogenous physiological inhibitors of osteoclastogenesis such as osteoprotegrin and interferon-γ levels are also remarkably downregulated in advanced HIV infection, resulting in increased bone resorption [86]. At a cellular level, HIV proteins including Tat and Nef reduce the number of available mesenchymal stem cell (MSC) precursors that proliferate into osteoblasts by inducing MSC senescence, due to increased oxidative stress and mitochondrial dysfunction resulting in reduced proliferation of osteoblasts and lower rates of bone formation [87]. Collectively, these mechanisms result in significant uncoupling of bone formation and resorption, resulting in less bone formation and greater rate of bone loss and lower bone density.
Pro-inflammatory Pathways
Cytokines and other soluble immune factors play a major role in the physiology of osteoblast maturation and osteoclastic bone resorption [88,89]. Immune dysfunction and persistent inflammation in HIV result in increased levels of several inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin-6 (IL-6), and RANKL, resulting in stimulation of osteoclastogenesis and bone resorption [90]. Due to a disruption between T and B cells in HIV and decreased osteoprotegrin (OPG) production and increased RANKL level, RANKL/OPG ratio is elevated, favoring osteoclastogenesis [91].
Is antiretroviral therapy associated with bone loss?
The initiation of ART has been reported to cause 2% to 6% bone loss irrespective of the regimen used, similar to that sustained in the first 2 years after menopause [92]. Certain NRTIs and PIs are associated with higher rates of bone loss than others. TDF has been associated most commonly with decreased bone mineral density, which usually stabilizes with continued use [93]. In a randomized trial comparing 4 treatment arms of ABC/3TC or TDF/FTC with EFV or ATV/ritonavir, TDF was associated with a greater reduction in BMD compared to abacavir-based regimens [94]. The likely cause of this may be TDF-mediated renal toxicity, including proximal tubular dysfunction and hypophosphatemia, resulting in increased PTH and bone resorption, and nephrogenic diabetes insipidus [95]. TAF is another prodrug of tenofovir diphosphate associated with less renal and bone toxicity compared with TDF. TAF has been associated with significantly less decrease in bone mineral density and renal dysfunction in randomized studies compared to regimen using TDF [17]. Vitamin D deficiency and hypophosphatemia associated with TDF therapy may present with osteomalacia, which predisposes to bone pain and fractures. Treatment with TDF may rarely be associated with the development of Fanconi syndrome and osteomalacia [96]. BMD is often severely reduced and bone pain and pathological fractures are characteristic features. Certain PI regimens containing ritonavir-boosted atazanavir have also been associated with greater bone loss in the spine than the hip, compared to efavirenz-containing regimens [97].
The universal bone loss associated with ART is thought to be a result of the "immune reconstitution inflammatory syndrome" (IRIS). This occurs as a result of rapid improvement in immune function after the commencement of ARV as a result of systemic or local inflammation, resulting in increased levels of cytokines that may contribute to bone loss. This has been shown in animal studies where T cell transplantation into immunocompromised mice to mimic ARV-induced T-cell expansion resulted in increased RANKL and TNF-α production by B cells and/or T cells, accompanied by enhanced bone resorption and BMD loss. When TNF-α or RANKL-null T-cells or TNF-α antagonists were used instead, the loss of cortical bone was prevented [98]. In a prospective study evaluating changes in bone turnover markers and inflammatory cytokines with ARV therapy in HIV infected subjects, a significant increase in bone resorption markers, RANKL and TNF-α were seen after initiation of ARV. The magnitude of CD4-cell recovery correlated with the increase in markers of bone resorption [99], suggesting that recovery of the immune system contributes to the increase in cytokine-mediated bone resorption.
How is bone health and fracture risk assessed in the HIV-positive population?
The predictive value of low BMD for fracture risk assessment in the HIV-positive population has not been established. In the absence of definitive data, the fracture risk assessment and standard methods of measuring bone density using DXA are utilized. In a large study of 1000 men and women, osteoporosis defined as a BMD T-score –2.5 as measured by DXA, was associated with a significantly increased risk of incident fractures but was not a good predictor of morphometric vertebral fractures [100]. In the absence of prospective longitudinal studies evaluating the bone density parameters at which fracture risk is significantly increased in the HIV population, it is reasonable to follow the guidelines used in the non-HIV population.
The approach to treatment of osteopenia and osteoporosis is similar to that in non HIV-infected population and is directed at lifestyle changes and treatment of secondary causes of osteoporosis [101], followed by initiation of antiresorptive therapy.
Management of Bone Disease
There are several guidelines available for the management of bone disease in the HIV population. The most recent guidelines from the IDSA [12] recommend assessing the risk of fragility fracture using the Fracture Risk Assessment Tool (FRAX), without DXA, in all HIV-infected men aged 40–49 years and HIV-infected premenopausal women aged ≥ 40 years. DXA should be performed in men aged ≥ 50 years, postmenopausal women, patients with a history of fragility fracture, patients receiving chronic glucocorticoid treatment, and patients at high risk of falls. In resource-limited settings, FRAX without bone mineral density can be substituted for DXA. ART guidelines should be followed. TDF and boosted PIs should be avoided if possible in at-risk patients. Dietary and lifestyle management strategies for high-risk patients should be employed and anti-osteoporosis treatment initiated if indicated [102].
The FRAX tool is available at www.shef.ac.uk/FRAX/ and is used to calculate 10-year fracture risk using patient clinical data, including presence of risk factors for osteoporosis. The tool is population-specific by race and region. It has not been validated for the HIV-positive population and may underestimate fracture risk [103]. HIV status is considered a secondary cause of osteoporosis in FRAX calculation.
The National Osteoporosis Foundation recommends screening with DXA for all women > 65 years of age, all men > 70 years of age, and adults > 50 years of age with additional risk factors for osteoporosis. Evaluation for secondary causes for low BMD should always be considered in the HIV-positive population including evaluation of calcium and vitamin D intake. Laboratory testing may include complete blood count, calcium, phosphate, albumin, creatinine, PTH, 25 hydroxy vitamin D (25,OHD) and 24 hour urine for evaluation of calcium, creatinine and phosphate (especially if on TDF) excretion. Testosterone level can be checked in men and estradiol, prolactin, FSH and LH in women for evaluation of hypogonadism. Bone turnover markers (bone specific alkaline phosphatase and serum C-terminal telopeptide) can also be assessed at baseline.
Studies using high-resolution peripheral quantitative computed tomography (HSPQCT) have shown significant reductions in tibial trabecular bone density and trabecular number in pre-menopausal and postmenopausal HIV-infected women [104], with reduced bone stiffness measured using finite element analysis [105]. Co-infection with HCV is also associated with significantly lower trabecular volumetric BMD and smaller cortical dimensions in the tibia, compared to healthy subjects [106]. HSPQCT is not widely available for clinical use at this time. Lateral imaging of the spine or vertebral morphometric analysis may be done in cases of height loss to assess for occult vertebral compression fractures.
There is a high prevalence of vitamin D deficiency in the HIV-infected population [107]. Treatment goal is to have a vitamin D level of at least 30 ng/mL, based on Endocrine Society practice guidelines [108], and may require supplementation with 1000–2000 units of vitamin D daily. Calcium intake should be optimized, averaging 1000 mg per day including diet and supplements, to be taken in divided amounts through the day for optimal absorption. Secondary causes of low bone density as mentioned in Table 4 should also be addressed. Patients should be counseled on tobacco and alcohol abuse. Corticosteroids should be dosed at the lowest dose needed. Medications such as proton pump inhibitors can impair the absorption of calcium carbonate, in which case calcium citrate supplements should be used if there is suboptimal calcium intake in the diet.
Which medications have been shown to be effective in treatment of osteoporosis in the HIV population?
Bisphosphonates are the mainstay of therapy for osteoporosis in the HIV-infected population. Only alendronate and zoledronate have substantial evidence of safety and effectiveness in the HIV-infected population, but these studies have been small and of limited duration.
Bisphosphonates are pyrophosphate analogues that inhibit bone resorption by binding to the hydroxyapatite crystals in the bone. Several prospective studies have shown alendronate to increase bone density compared to calcium and vitamin D alone in the HIV infected patients with reduced bone density [109,110], with significant reduction in markers of bone resorption [111].
Zoledronic acid (ZA), an amino-bisphosphonate which is infused intravenously, has also been used in smaller studies in HIV-infected persons. In a prospective study evaluating yearly ZA infusion to biennial ZA infusion in subjects with HIV and low bone density [112], biennial ZA infusions were found to be effective in improving and maintaining bone density in the HIV population. In another prospective study evaluating the effects of ZA in HIV-positive men, ZA infusion was given at baseline and at 12 months. Compared to placebo, treatment group had significantly higher bone density and lower bone turnover markers till 5 years after the last infusion [113].
In a meta-analysis evaluating the effect of bisphosphonates on bone mineral density in 328 adults with HIV infection from 8 randomized controlled trials (5 with alendronate and 3 with ZA as the intervention), a significant increase in BMD at the lumbar spine and hip was observed in the treatment groups at 48 and 96 weeks. However, these studies were not long enough to detect the impact of bisphosphonates on fracture risk [114]. ZA has also been shown to be effective in preventing ARV induced bone loss after a single infusion [115].
These studies confirm that both alendronate and ZA are effective in improving BMD in the HIV-infected population, with early studies showing a beneficial effect of ZA in mitigating ARV-induced bone loss as well. DXA may be repeated 1 to 2 years after initiation of osteoporosis therapy and less often subsequently if BMD is stable to improved [116].
Although these studies show significant improvement in bone density with treatment, longitudinal data on fracture reduction with these medications in the HIV-infected population are not available. Additionally, these patients have onset of osteoporosis at a younger age and the need for osteoporosis treatment needs to be assessed carefully before initiating treatment. There are other medications available for the treatment of osteoporosis in the non-HIV population such as raloxifene, teriparatide and denosumab, but no randomized controlled studies of these agents are available in the HIV-infected population.
Summary
The advent of highly potent antiretroviral therapy capable of early and prolonged viral suppression in HIV-infected patients has resulted in significant increases in life span. As we have already seen, this will likely lead to a rising incidence of various metabolic complications of HIV and ARV, including hyperlipidemia and diabetes with associated cardiovascular disease risk. A keen awareness of these potential complications, drug interactions, and possible toxicities will be paramount to their successful management. Appropriate care of HIV-infected individuals going forward will likely require multidisciplinary collaboration as the epidemic evolves to allow our patients to live not only longer, but healthier lives.
Corresponding author: Lisa M. Chirch, MD, UCONN Health, Farmington, CT 06030, chirch@uchc.edu.
Financial disclosures: None
Author contributions: All authors contributed equally to this article
From the University of Connecticut School of Medicine, Farmington, CT.
Abstract
- Objective: To review the metabolic complications of HIV infection.
- Methods: Review of the literature in the context of 3 clinical cases.
- Results: People with HIV infection are living longer thanks to the advent of potent antiretroviral therapy. This has led to increased incidence of age-related metabolic complications, including a higher risk of cardiovascular disease, hyperlipidemia, metabolic syndrome, and osteoporosis. Appropriate management of these complications requires an understanding of disease-related and drug-related side effects as well as the potential for drug-drug interactions. A multidisciplinary approach to patient management is most effective.
- Conclusion: Awareness of the metabolic complications frequently encountered in HIV infection, drug interactions, and possible toxicities is critical to the successful management of HIV-infected individuals.
Key words: HIV; antiretroviral therapy; hyperlipidemia; metabolic syndrome; diabetes; hypogonadism.
According to the most recent data from the Joint United Nations Programme on HIV/AIDS (UNAIDS), 36 million people worldwide are living with HIV/AIDS, with 18 million accessing effective antiretroviral therapy (ART) [1]. The past 2 decades have witnessed enormous advances in the field from prevention to diagnosis and therapeutics, and modern ART largely allows HIV-infected persons to live near-normal life spans [2,3]. However, from the beginning of the epidemic, HIV-infected persons on effective therapy have suffered from myriad metabolic consequences, many of which affect quality of life and result in excess mortality [4]. It is also true that untreated HIV infection portends an increased risk of metabolic complications, likely related to abnormal immune activation, as demonstrated in structured interruption trials [5,6]. It is worth noting, however, that while many of these metabolic dyscrasias and associated risks have historically been attributed primarily to the treatment of HIV infection with ART, data from cohort studies and randomized clinical trials have repeatedly demonstrated significant reductions in morbidity and mortality when ART is initiated early [7]. In this paper, we review HIV-related metabolic complications frequently encountered in clinical practice (hyperlipidemia, diabetes, and bone disease) and best practice considerations in the context of 3 clinical cases.
Case Patient 1
Initial Presentation and History
A 58-year-old male with a history of hypertension and mixed hyperlipidemia is referred for evaluation of newly diagnosed HIV infection. He has no history of intravenous drug use but has had multiple male and female sex partners in the past few years, and requested testing after a partner tested positive. His last negative test was 2 years ago. The patient does not smoke cigarettes. Overall he feels well and tolerates his regimen of lisinopril 10 mg and simvastatin 20 mg daily. On initial evaluation, his exam is unremarkable other than subtle white plaques on the dorsal surface of the tongue and buccal mucosa, and moderate central obesity. Vital signs including blood pressure are normal. Initial laboratory evaluation reveals a CD4 cell count of 150 cells/mm3 and an HIV RNA level of 200,000 copies/mL. Fasting serum total cholesterol is 220 mg/dL, triglycerides 250 mg/dL, LDL 170 mg/dL, and HDL 35 mg/dL. Serum BUN, creatinine, and liver function testing results are normal.
What initial regimen might be recommended based on the status of his HIV infection and comorbidities?
The most recent iteration of the US Department of Health and Human Services (DHHS) guidelines on use of antiretroviral agents (ARVs) in HIV recommends an initial ART regimen that includes a backbone of 2 nucleoside reverse transcriptase inhibitors (NRTIs), generally tenofovir disoproxil fumarate or tenofovir alafenamide, abacavir (ABC), emtricitabine (FTC), or lamivudine (3TC) [2]. To this backbone should be added a third agent; the majority of those currently recommended are integrase strand transfer inhibitors (INSTIs) (dolutegravir, elvitegravir, raltegravir); one recommended protease inhibitor (PI) (ritonavir-boosted darunavir) is also an option. Some of these initial recommended regimens are available as fixed-dosed combinations in 1 pill, making them attractive options.
The latest guidelines also clearly recommend starting ART in all HIV-infected individuals, irrespective of CD4 count. The patient described above has a very low CD4 count, so there is no question he needs to begin therapy promptly. Given his low CD4 count and relatively high viral load, one may consider a ritonavir-boosted PI as perhaps the most robust option and with a relatively high barrier to resistance development, in contrast to other options. Assuming the patient’s baseline resistance testing reveals a fully sensitive wild-type virus without meaningful resistance mutations, he will begin a regimen of TDF/FTC plus ritonavir-boosted darunavir, 3 pills once daily. Given his low CD4 count (below 200 cells/mm3), he will also need prophylaxis for Pneumocystis jirovecii pneumonia, in the form of trimethoprim/sulfamethoxazole (TMP/SMX) daily. Given the potential for interaction between the boosted PI and simvastatin, his lipid-lowering agent is switched to atorvastatin 10 mg daily.
What is the association between hyperlipidemia and HIV infection and treatment?
Hyperlipidemia represents a key modifiable risk factor for the development of cardiovascular disease (CVD) in HIV-infected individuals [8]. Indeed, a multicenter cross-sectional study of older HIV-infected individuals performed in Spain revealed a 54% prevalence of dyslipidemia and 23% CVD [9]. Most experts believe that metabolic abnormalities observed in HIV-infected individuals are the result of a combination of factors: those resulting from abnormal immune activation and inflammation related to viral replication, and those related to certain ARVs [10].
Early after HIV seroconversion, decline in HDL is one of the first proatherogenic changes observed. This, along with increased triglyceride and LDL levels, likely contribute to increased risk of CVD in this population. Increased microbial translocation, evidenced by increased levels of lipopolysaccharide (LPS), may drive immune activation, leading to dyslipidemia via a multitude of hypothesized mechanisms [4]. It has been theorized that HDL lipoproteins are less stable on ART, leading to potentially impaired plasma lipolytic activities or hepatic cholesteryl ester uptake [6,11]. Increased VLDL from release of free fatty acids may lead to higher triglyceride levels and triglyceride-rich LDL and HDL, all associated with increased risk of CVD [11].
In terms of effects of specific ARV classes, although newer agents have less of a propensity to cause dyslipidemia, the PI class arguably remains most problematic. In comparison to other classes, the PIs tend to result in greater increases in triglycerides, total cholesterol, and LDL, and have frequent drug-drug interactions with lipid-lowering agents [10,12]. Estimated prevalence of dyslipidemia in patients receiving PI therapy varies from 28% to 80% [13]. The prospective multinational cohort Data collection on Adverse events of Anti-HIV Drugs (DAD) study found significantly higher rates of hypertriglyceridemia, hypercholesterolemia, and low HDL in patients on PIs in comparison to non–nucleoside reverse transcriptase inhibitors (NNRTIs) [14]. Various mechanisms have been proposed to explain the PIs adverse effects on lipids, including inhibition of lipogenesis and adipocyte differentiation, decreased hepatocyte clearance of chylomicrons and VLDL, and increased hepatic synthesis of triglycerides [15]. Of the available PIs, atazanavir and darunavir have less potential to lead to dyslipidemia [10], while lopinavir/ritonavir, fosamprenavir, and tipranavir may have the highest [13]. Of the NNRTIs, efavirenz is most frequently associated with dyslipidemia, specifically increased triglycerides and total cholesterol [13]. However, these increased values seen on efavirenz therapy may be offset by relative increases in HDL, with little resultant effect on the total cholesterol:HDL ratio. Rilpivirine, etravirine, and nevirapine are relatively less likely to drive lipid changes, although certain drug interactions are important to recognize in clinical practice, such as the interaction between rilpivirine and proton pump inhibitors [2,13,16]. It is also worth noting that no NNRTIs are included in current guidelines as preferred therapy [2].
Historically, the thymidine analogue NRTIs (stavudine, didanosine, zidovudine) have been associated with lipid dyscrasias and lipoatrophy, but fortunately these are no longer used frequenty except in cases requiring deep salvage therapy for highly treatment-experienced patients. Two newer NRTIs, tenofovir and abacavir, have relatively neutral to favorable effects on lipids. The combination of tenofovir disoproxil (TDF) and emtricitabine (trade name Truvada) was associated with significantly lower triglycerides, total cholesterol and LDL than other NRTI pairs [6]. TDF has been postulated to have lipid-lowering effects. Switch studies in which patients were taken off thymidine analogues and placed on TDF, demonstrated recovery of limb fat in patients with lipoatrophy, and those switched off abacavir-based ART to TDF showed statistically significant lower fasting total cholesterol at week 12, without differences of viral suppression [8]. Tenofovir alafenamide (TAF) is a next-generation prodrug of tenofovir that results in improved stability in plasma and higher intracellular levels in comparison to TDF [17]. Although randomized controlled trials of TAF vs TDF-based ARV regimens have suggested statistically higher total cholesterol, serum HDL is also increased resulting in unchanged total:HDL ratios and no differences in risk classifications [18]. Integrase inhibitors (INSTI) now represent first-line therapy in combination with an NRTI backbone, and since their availability in 2007 have been evaluated in comparison to various PIs and NNRTIs. Both raltegravir and dolutegravir have consistently shown broad neutral effects on lipids and are among the most metabolically friendly agents available [19–21]. Because it is given in fixed-dose combination with non-ritonavir pharmacologic booster cobicistat, elvitegravir has effects similar to ritonavir-boosted PIs on lipids [6].
What are management considerations in the treatment of hyperlipidemia in HIV-infected patients?
Patients with HIV and hyperlipidemia may benefit from lipid-lowering therapy in addition to ART, although in certain cases appropriate switches may make a difference. Careful consideration of drug interactions between ARVs and lipid-lowering agents, in addition to ARV history and known drug resistance, is warranted prior to selecting a regimen in these patients. In addition, the latest American College of Cardiology/American Heart Association guidelines suggest evaluating 10-year risk of atherosclerotic cardiovascular disease (ASCVD) using the pooled cohort equation to determine the type and dose of statin required (moderate vs high intensity) [22]. It is noteworthy that HIV infection and its therapies are not taken into account as potential risk factors in this model. Primary prevention in non-diabetic patients with a statin is recommended for patients with a 10-year absolute risk of ≥ 7.5% [22]. This patient’s risk is estimated at between 12% and 13% based on this equation, so primary prevention with a moderate-or high-intensity statin is recommended (Table 1) [23]. Data from more than 80,000 patients in the Veterans Aging Cohort Study (VACS) showed that HIV-infected patients with no baseline ASCVD had 50% increased risk of acute myocardial infarction when compared to HIV-uninfected patients over 6 years of follow-up [24]. Thus, consideration of the virus itself or its therapy as an additional risk factor may be valid.
Screening and Monitoring of Hyperlipidemia
The most recent iteration of the DHHS primary care guidelines for the management of HIV-infected individuals recommends obtaining fasting (ideally 12 hours) lipid profiles upon initiation of care, and within 1 to 3 months of beginning therapy [12,13]. These initial levels, along with other elements of the patient’s history and calculation of risk may help determine whether lipid-lowering therapy is indicated, and if so, which therapy would be best. In general, after regimen switches or additions of either ARV or statin therapy, repeating fasting lipid levels 6 weeks later is recommended to gauge the effects of the switch. This is especially critical when interactions between ARVs and lipid-lowering therapies are possible. Some experts recommend performing annual screening of patients with normal baseline lipids or with well-controlled hyperlipidemia on therapy. Assessment of 10-year ASCVD risk is also recommended annually, in addition to baseline risk assessment, to determine the need and appropriateness of statin therapy [25]. The question of primary prevention in HIV has yet to be definitively answered. Small studies in this population have demonstrated that statins have the potential to slow progression of carotid intima media thickness and reduce noncalcified plaque volume [24]. An NIH/AIDS Clinical Trial Group–sponsored randomized clinical trial (“REPRIEVE”) is currently underway to address this question. More than 6000 HIV-infected men and women with no history of ASCVD at 100 sites in several countries are enrolled to assess the benefit of pitavastatin as primary prevention in this risk group [24]. Metabolized via glucuronidation primarily, as opposed to cytochrome p450 (CYP 3A4 isoenzyme), pitavastatin is thought to have fewer drug interactions with ARVs in general [6] (Table 2).
Relevant Drug-Drug Interactions
Deciding which statin to begin in HIV-infected patients depends on whether moderate- or high-intensity therapy is warranted and whether the potential for drug interaction with ARVs exists. Table 2 [6,12] depicts available statins and the potential for pharmacokinetic interaction with the primary ARV classes. Simvastatin and lovastatin are heavily metabolized via the CYP 3A4 pathway, resulting in the highest potential risk of interaction with CYP 3A4 inhibitors, such as the PIs, or inducers (eg, NNRTIs, in particular efavirenz) [6]. The former may inhibit metabolism of these statins, resulting in increased risk of toxicity, while co-administration with efavirenz, for example, may result in inadequate serum concentration and therefore inadequate lipid-lowering effects. Although less lipophilic, atorvastatin results in similar interactions with PIs and NNRTIs, and therefore low starting doses with close monitoring is recommended [6]. Fewer interactions have been noted with rosuvastatin, pravastatin, and pitavastatin, as these do not require CYP 3A4 for their metabolism and are thus less likely to be affected by ARVs. These therefore represent potentially safer first choices for certain patients on ARVs, although of these, only rosuvastatin is classified as a high-intensity statin [22,23] (Table 1). When compared directly to pravastatin 40 mg daily in patients receiving ritonavir-boosted PIs, rosuvastatin performed superiorly at 10 mg per day, resulting in more significant reductions in LDL and triglyceride levels [15]. Although it is eliminated largely unchanged through the kidney and liver, pravastatin has been reported to idiosyncratically interact with darunavir, resulting in potentially increased pravastatin levels and associated toxicity [25]. Treatment of pure hypertriglyceridemia in HIV-infected patients should begin with fibrates, which have little to no risk of interaction with most clinically relevant ARVs [6,10]. Alternatives to lower triglycerides include niacin and N-3 polyunsaturated fatty acids [25].
Case 1 Continued
The patient has an impressive response to his initial regimen of TDF/FTC plus boosted darunavir, with repeat CD4 count after 12 weeks of 275 (18%) cells/mm3 and an undetectable viral load (< 20 copies/mL). Other lab parameters are favorable and he is tolerating the regimen well without notable side effects. However, at his next visit, although his viral load remains undetectable, his triglyceride level has increased to 350 mg/dL, although other lipid parameters are comparable to the prior result. He complains of diffuse body aches, concentrated in large muscle groups of the extremities, and dark-colored urine. A creatine phosphokinase (CPK) level is elevated at 300 IU/L (normal, 22–269, negative MB fraction). Serum creatinine is 1.4 mg/dL (had been 1.1 mg/dL at baseline). Given he has done so well otherwise on these ARVs, he is reluctant to make any changes.
What drug-drug interaction is most likely causing this patient's problem, and how should it be managed?
This scenario is not uncommon in clinical practice, and changes to regimens are sometimes necessary in order to avoid drug interactions. Care must be taken to thoroughly review antiretroviral history and available resistance testing (in this case a relatively short history) in order to ensure a fully active and suppressive regimen is chosen. This description could be the result of an interaction between lipid-lowering therapy and ARVs resulting in increased relative concentrations of one drug or the other and therefore leading to toxicity. Given this possibility, and suboptimal control of hyperlipidemia, consideration should be given to switching both his ART and his statin therapy.
Safety and Potential Toxicities of Lipid-Lowering Therapy
Increased serum concentration of certain statins when co-administered with CYP 3A4 inhibitors like the PIs leads to heightened risk of statin-associated toxicities. In general, this includes muscle inflammation, leading to increases in serum CPK level and associated symptoms, including myalgias, myositis, or in extreme cases, rhabdomyolysis [6]. Although rare, this toxicity can be serious and may lead to acute renal injury if not recognized and managed appropriately. In theory, the potential for statin-associated hepatotoxicity may also be increased in patients receiving PIs, although this has not been borne out in clinical trials [26]. In fact, quite the opposite may be true, in that statins have been shown to improve liver function in patients with hepatitis C virus (HCV) coinfection and with nonalcoholic fatty liver disease [6,15].
Case 1 Conclusion
The patient does well on his new ARV regimen of TAF/FTC and dolutegravir, 2 pills once daily. He no longer requires TMP/SMX, as his CD4 count has been reliably above 200 cells/mm3 on several occasions. Serum creatinine is back down to baseline and CPK has normalized. Fasting lipids have improved since the switch, and he no longer has symptoms of myositis on rosuvastatin 10 mg daily.
Summary
Consideration of statin therapy is complicated by potential drug interactions with ARVs and associated toxicity. However, given known effects of ARVs on lipids, and of immune activation and inflammation related to the virus itself, these patients should be carefully evaluated for statin therapy for their anti-inflammatory and immune modulatory effects as much as for their lipid-lowering ability. Utilization of HIV infection and its therapies as additional cardiovascular risk factors when calculating 10-year risk deserves further consideration; forthcoming results of the REPRIEVE trial are certain to contribute valuable information to this field of study.
Case Patient 2
Initial Presentation and History
A 45-year-old female with history of HIV infection since 2008 presents to the office for new-onset diabetes, diagnosed 2 weeks ago. She has had symptoms of polyuria and polydipsia for the last 1 month. She denies diarrhea, nausea, vomiting or weight loss. She is currently on a regimen consisting of zidovudine/lamivudine plus lopinavir/ritonavir. There is no family history of diabetes. Her examination is unremarkable, including normal vital signs (weight 150 lb, blood pressure 114/70, heart rate 76) and no evidence of insulin resistance, including acanthosis nigricans or striae. Glycosylated hemoglobin level (HbA1c) is 8%. Creatinine and liver function tests are within reference ranges.
Do HIV-infected patients have a higher incidence of type 2 diabetes mellitus (DM)?
Prevalence of type 2 DM in HIV-infected patients varies between 2% to 14% [27]. This variation is due to the different cutoffs used for diagnosis, differences in cohorts studied, and how risk factors are analyzed [28–31]. In a recent nationally representative estimate of DM prevalence among HIV-infected adults receiving medical care in the United States in 2009–2010, the prevalence of DM was noted to be 10.3%. In comparison to the general adult US population, HIV-infected individuals have a 3.8% higher prevalence of DM after adjusting for age, sex, race/ethnicity, education, poverty-level, obesity, and HCV infection [27].
There is controversy over whether HIV infection itself increases the risk of type 2 DM, with some studies showing increased risk [28,32,33] and others showing no independent effect or an inverse effect [30,34,35]. Studies on the impact of ethnicity and race on prevalence of DM are limited [36].
Certain traditional risk factors (age, ethnicity, obesity) are still responsible for most of the increased risk of diabetes in the HIV-infected population [35,37]. HIV infection itself is associated with metabolic dysfunction, independent of ARV. In HIV-infected patients, impaired glucose metabolism is associated with altered levels of adipokines, increased adiponectin and soluble-tumor necrosis factor receptor 1 (sTNFR1) and decreased leptin [38,39]. HIV-associated alterations in CD4+ and CD8+ T cell function also impair glycolysis, which may adversely impact glucose metabolism [40].
Other contributing factors in HIV-infected patients are HCV co-infection [41], medications (atypical antipsychotics, corticosteroids), opiates, and low testosterone [42]. HCV co-infection may lead to hepatic steatosis and liver fibrosis, and increasing insulin resistance.
Recent genomic studies show several common single-nucleotide polymorphisms (SNPs) associated with diabetes in the general population. In the Swiss HIV Cohort Study, SNPs accounted for 14% of type 2 DM risk variability, whereas ARV exposure accounted for 3% and age for 19% of the variability in DM [43].
ARVs also increase the risk of type 2 DM by both direct and indirect effects. Certain ARVs causes lipoatrophy [30] and visceral fat accumulation/lipohypertrophy [29,44]. PIs increase insulin resistance via effects on GLUT-4 transporter and decrease insulin secretion through effects on B cell function [45]. NRTIs (eg, stavudine, zidovudine and didanosine) can cause direct mitochondrial toxicity [46–48]. Utilization of newer ARV agents has decreased the prevalence of severe lipoatrophy, but lipohypertrophy and the underlying metabolic abnormalities persist. The DHHS “preferred” nucleoside analogues, tenofovir and abacavir, do not induce mitochondrial toxicity and have more favorable metabolic profiles [49,50]. In ACTG Study 5142, thymidine-sparing regimens were found to cause less lipoatrophy [51]. In addition, darunavir and atazanavir, the preferred and alternative PIs and the integrase strand transfer inhibitor have limited or modest impact on insulin sensitivity [20,52,53]. This has led to a recent decline in the incidence of type 2 DM in HIV-infected patients.
Statins can also increase insulin resistance and DM [54], although studies have shown mixed results [55–57]. The benefits of statin therapy likely outweigh the risk of DM since there is a significant cardiovascular event reduction with their use [58,59].
How is diabetes diagnosed in HIV-infected patients?
Optimal diabetes screening guidelines have not been established specifically for HIV-infected patients. The American Diabetes Association (ADA) guidelines recommend that diabetes in the general population be diagnosed by 2 elevated fasting blood glucose levels, HbA1c, oral glucose tolerance test (OGTT), or high random glucose with classic symptoms of hyperglycemia [60]. Repeat testing is recommended every 3 years. The OGTT is recommended for diagnosis in pregnant women.
HbA1c may underestimate glycemic burden in HIV-infected individual due to higher mean corpuscular volume, NRTI use (specifically abacavir), or lower CD4 count [61–65]. The Infectious Diseases Society of America (IDSA) 2013 primary care guidelines for HIV-infected patients recommends obtaining a fasting glucose and/or HbA1c prior to and within 1–3 months after starting ARV [12]. Use of HbA1c threshold cutoff of 5.8% for the diagnosis of DM and testing every 6–12 months are recommended.
How should this patient’s diabetes be managed?
The ADA guidelines suggest a patient-centered approach to management of diabetes [66]. All patients should be educated about lifestyle modifications with medical nutrition therapy and moderate-intensity aerobic activity and weight loss [67]. If a patient is on lopinavir/ritonavir or a thymidine analogue (zidovudine, stavudine), one should consider switching the ARV regimen [2].
There are currently no randomized controlled trials of diabetes treatment specific to patients with HIV infection. Metformin is the first-line agent. It improves insulin sensitivity by reducing hepatic glucose production and improving peripheral glucose uptake and lipid parameters [68,69]. Other oral hypoglycemic agents used in the treatment of type 2 diabetes are shown in Table 3.
Case 2 Continued
The patient is switched to TAF/FTC plus dolutegravir with improvement in blood sugars. She is also started on metformin. Co-administration of metformin and dolutegravir will be carefully monitored since dolutegravir increases metformin concentration [70]. When dolutegravir is used with metformin, the total daily dose of metformin should be limited to 1000 mg.
• How should this patient be followed?
If the patient is still not at goal HbAb1c at follow-up, there are multiple other treatment options, including use of insulin. Goal HbA1c for most patients with type 2 DM is < 7%; however, this goal should be individualized for each patient in accordance with the ADA guidelines [12]. A longitudinal cohort study of 11,346 veterans with type 2 diabetes compared the glycemic effectiveness of oral diabetic medications ( metformin, sulfonylurea and a thiazolidinedione) among veterans with and without HIV infection. This study did not find any significant difference in HbA1c based on different diabetes medications. However the HBA1c reduction was less in black and Hispanic patients. The mechanism for the poorer response among these patients need to be evaluated further [71]. In addition to management of blood sugar, other CVD risk factors, hyperlipidemia, hypertension, smoking, etc, should be assessed and managed aggressively.
Case Patient 3
Initial Presentation and History
A 45-year-old male with a history of HIV infection diagnosed 10 years ago, on TDF/FTC/efavirenz (trade name Atripla) for the last 7 years, presents with a left femoral neck fracture after he missed the pavement and fell on his left hip. His history is significant for IV drug abuse for 10 years prior to diagnosis of HIV, and he has been on methadone for the last 6 years.
Is HIV infection associated with increased prevalence of osteopenia and osteoporosis and increased risk of fractures?
With recent advancements in antiretroviral therapy and improved survival of the HIV-infected population, osteoporosis and increased fracture risk have become important causes of morbidity and mortality. Osteoporosis is a skeletal disorder characterized by compromised bone strength, which predisposes to an increased risk of fracture. The World Health Organization defines osteoporosis as a bone mineral density (BMD) measurement by dual X-ray absorptiometry (DXA) at the spine, hip, or forearm that is more than 2.5 standard deviations below that of a "young normal" adult (T-score < –2.5) or a history of one or more fragility fractures. Fragility fractures result from mechanical forces that would not ordinarily result in fracture, such as fall from standing height [40]. Osteopenia is characterized by low BMD (T-score between –1.0 and –2.5) and can be a precursor to osteoporosis.
Several observational, retrospective, and prospective studies have shown lower bone density and an increased risk of fractures in the HIV-infected population compared to age-, race- and sex-matched HIV-negative adults. In a large meta-analysis of pooled prevalence data on 884 HIV-infected patients compared with 654 HIV-uninfected age- and sex-matched controls [72], overall, HIV-infected patients had a significant 6.4-fold increased odds of reduced BMD and a 3.7-fold increased odds of osteoporosis compared to the control population. This meta-analysis also compared ARV-treated subjects to ARV-naive subjects and showed that ARV-treated subjects (n = 824) had a higher prevalence of reduced BMD compared with ARV-naive subjects (n= 202; odds ratio 2.5, 95% CI 1.8–3.7). The odds of osteoporosis was increased 2.4 times (95% CI 1.2 – 4.8) in ARV-treated subjects compared with ARV-naive subjects. None of the studies adjusted for potentially important confounding factors, such as age or duration of HIV infection. PI-treated patients (n = 791) were also found to have a higher prevalence of reduced BMD compared with PI-untreated patients (n = 410; OR 1.5, 95% CI 1.1–2.0). The odds of osteoporosis in PI-treated patients (n = 666) was also 1.6-fold greater (95% CI 1.1–2.3) than those not treated with PI (n = 367).
Low bone density has also been reported in HIV- positive premenopausal women irrespective of ARV status. In a recent study of 89 premenopausal women (mean age, 37 years) predominantly of African origin with HIV infection, osteopenia and osteoporosis were prevalent in one-third of these women, irrespective of ARV use and were associated with low BMI [73]. In a sub-study of the INSIGHT trial evaluating prevalence of and risk factors for low BMD in untreated HIV infection, performed at several sites across 6 continents involving 424 subjects, osteopenia was present in a third of this relatively young predominantly non-white ART-naive population (mean age 34 + 10 years) with normal CD4 cell counts, while only 2% had osteoporosis. Factors independently associated with lower BMD at the hip and spine were female sex, Latino/Hispanic ethnicity, lower BMI, and higher estimated glomerular filtration rate. Longer duration of HIV infection was also associated with lower hip BMD. Current or nadir CD4 cell count and HIV viral load were not associated with low BMD [74].
Many studies have reported increased fracture prevalence in the HIV population. In a retrospective study of fracture prevalence in a large US health care system, a significantly higher rate of fractures was reported in HIV-infected men and women compared to non-HIV-infected controls (2.87 vs. 1.77 fractures per 100 persons, P < 0.001). The difference in the increased fracture prevalence was greater in HIV positive men compared to women (3.08 vs. 1.83; P < 0.001). Vertebral, wrist and hip fractures were more prevalent in men compared to vertebral and wrist fractures only in women. Fracture prevalence was higher in both Caucasian females and males and only in African-American women [75].
In the HIV Outpatient Study (HOPS) [76], age-adjusted fracture rates in the HIV population were noted to be 1.98 to 3.69 times higher than rates in the general population. The HOPS was an open prospective cohort study of HIV-infected adults who were followed at 10 US HIV clinics. Rates of first fractures at any anatomic site from 2000–2008 were assessed among 5826 active HOPS patients (median age 40 years, 79% male, 52% Caucasian, and 73% exposed to ART). Among persons aged 25–54 years, both fracture rates and relative proportion of fragility fractures were higher among HOPS patients than among outpatient controls. Older age, substance abuse, nadir CD4+ cell count <200 cells/mm, HCV infection and DM were associated with incident fractures [76].
What factors contribute to poor bone health in the HIV population?
Several factors that contribute to low bone density are present at a higher rate in the HIV population (Table 4). These include poor nutritional status in terms of suboptimal calcium and vitamin D intake, hypogonadism, low body weight, and alcohol, tobacco and substance abuse.
Vitamin D deficiency is very common in HIV-infected patients, with a prevalence of up to 60% to 75% [77]. Hypogonadism is also relatively common among HIV population [78], contributing to lower bone density. Co-infection with HCV is also associated with increased risk of fractures. In a large cohort of Medicaid beneficiaries, a significant increase in the risk of hip fracture was demonstrated in HCV/HIV co-infected subjects compared either with HCV mono-infected, HIV mono-infected or non-infected individuals [79]. In another large database study, a significantly higher risk of osteoporotic fracture (closed wrist, vertebral or hip fracture) was reported in HCV/HIV co-infected versus HIV mono-infected individuals [80] with fracture rates of 2.57 and 2.07/1000 patient-years (P < 0.001). Dual treatment for HIV/hepatitis B co-infection has also been shown to be associated with a higher risk of hip fracture compared to treatment of HIV mono-infected individuals [81].
HIV infection itself can increase bone loss and reduce bone formation through direct effects related to the HIV antigen load or indirect effects related to activation of the pro-inflammatory cytokines resulting in bone resorption and loss [82]. Co-infection with HCV and/or hepatitis B also contributes to lower bone density in this population. Certain ARVs may also contribute to low bone density in the HIV population. Lipoatrophy related to HIV may also mediate bone loss through complex relationship between central signaling of adipocyte hormones [82,83].
Direct Viral Effects
Several HIV viral proteins have been shown to promote osteoclast activity (vpr and gp120), suppress osteoblast activity (p55-gag) and increase osteoblast apoptosis [84], resulting in increased bone resorption and reduced bone formation, leading to low bone mass. High HIV RNA viral load and T-cell activation are also associated with elevated levels of receptor activator of nuclear factor kappa-B ligand (RANKL), which results in osteoclast formation and increased bone resorption [85]. Other endogenous physiological inhibitors of osteoclastogenesis such as osteoprotegrin and interferon-γ levels are also remarkably downregulated in advanced HIV infection, resulting in increased bone resorption [86]. At a cellular level, HIV proteins including Tat and Nef reduce the number of available mesenchymal stem cell (MSC) precursors that proliferate into osteoblasts by inducing MSC senescence, due to increased oxidative stress and mitochondrial dysfunction resulting in reduced proliferation of osteoblasts and lower rates of bone formation [87]. Collectively, these mechanisms result in significant uncoupling of bone formation and resorption, resulting in less bone formation and greater rate of bone loss and lower bone density.
Pro-inflammatory Pathways
Cytokines and other soluble immune factors play a major role in the physiology of osteoblast maturation and osteoclastic bone resorption [88,89]. Immune dysfunction and persistent inflammation in HIV result in increased levels of several inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin-6 (IL-6), and RANKL, resulting in stimulation of osteoclastogenesis and bone resorption [90]. Due to a disruption between T and B cells in HIV and decreased osteoprotegrin (OPG) production and increased RANKL level, RANKL/OPG ratio is elevated, favoring osteoclastogenesis [91].
Is antiretroviral therapy associated with bone loss?
The initiation of ART has been reported to cause 2% to 6% bone loss irrespective of the regimen used, similar to that sustained in the first 2 years after menopause [92]. Certain NRTIs and PIs are associated with higher rates of bone loss than others. TDF has been associated most commonly with decreased bone mineral density, which usually stabilizes with continued use [93]. In a randomized trial comparing 4 treatment arms of ABC/3TC or TDF/FTC with EFV or ATV/ritonavir, TDF was associated with a greater reduction in BMD compared to abacavir-based regimens [94]. The likely cause of this may be TDF-mediated renal toxicity, including proximal tubular dysfunction and hypophosphatemia, resulting in increased PTH and bone resorption, and nephrogenic diabetes insipidus [95]. TAF is another prodrug of tenofovir diphosphate associated with less renal and bone toxicity compared with TDF. TAF has been associated with significantly less decrease in bone mineral density and renal dysfunction in randomized studies compared to regimen using TDF [17]. Vitamin D deficiency and hypophosphatemia associated with TDF therapy may present with osteomalacia, which predisposes to bone pain and fractures. Treatment with TDF may rarely be associated with the development of Fanconi syndrome and osteomalacia [96]. BMD is often severely reduced and bone pain and pathological fractures are characteristic features. Certain PI regimens containing ritonavir-boosted atazanavir have also been associated with greater bone loss in the spine than the hip, compared to efavirenz-containing regimens [97].
The universal bone loss associated with ART is thought to be a result of the "immune reconstitution inflammatory syndrome" (IRIS). This occurs as a result of rapid improvement in immune function after the commencement of ARV as a result of systemic or local inflammation, resulting in increased levels of cytokines that may contribute to bone loss. This has been shown in animal studies where T cell transplantation into immunocompromised mice to mimic ARV-induced T-cell expansion resulted in increased RANKL and TNF-α production by B cells and/or T cells, accompanied by enhanced bone resorption and BMD loss. When TNF-α or RANKL-null T-cells or TNF-α antagonists were used instead, the loss of cortical bone was prevented [98]. In a prospective study evaluating changes in bone turnover markers and inflammatory cytokines with ARV therapy in HIV infected subjects, a significant increase in bone resorption markers, RANKL and TNF-α were seen after initiation of ARV. The magnitude of CD4-cell recovery correlated with the increase in markers of bone resorption [99], suggesting that recovery of the immune system contributes to the increase in cytokine-mediated bone resorption.
How is bone health and fracture risk assessed in the HIV-positive population?
The predictive value of low BMD for fracture risk assessment in the HIV-positive population has not been established. In the absence of definitive data, the fracture risk assessment and standard methods of measuring bone density using DXA are utilized. In a large study of 1000 men and women, osteoporosis defined as a BMD T-score –2.5 as measured by DXA, was associated with a significantly increased risk of incident fractures but was not a good predictor of morphometric vertebral fractures [100]. In the absence of prospective longitudinal studies evaluating the bone density parameters at which fracture risk is significantly increased in the HIV population, it is reasonable to follow the guidelines used in the non-HIV population.
The approach to treatment of osteopenia and osteoporosis is similar to that in non HIV-infected population and is directed at lifestyle changes and treatment of secondary causes of osteoporosis [101], followed by initiation of antiresorptive therapy.
Management of Bone Disease
There are several guidelines available for the management of bone disease in the HIV population. The most recent guidelines from the IDSA [12] recommend assessing the risk of fragility fracture using the Fracture Risk Assessment Tool (FRAX), without DXA, in all HIV-infected men aged 40–49 years and HIV-infected premenopausal women aged ≥ 40 years. DXA should be performed in men aged ≥ 50 years, postmenopausal women, patients with a history of fragility fracture, patients receiving chronic glucocorticoid treatment, and patients at high risk of falls. In resource-limited settings, FRAX without bone mineral density can be substituted for DXA. ART guidelines should be followed. TDF and boosted PIs should be avoided if possible in at-risk patients. Dietary and lifestyle management strategies for high-risk patients should be employed and anti-osteoporosis treatment initiated if indicated [102].
The FRAX tool is available at www.shef.ac.uk/FRAX/ and is used to calculate 10-year fracture risk using patient clinical data, including presence of risk factors for osteoporosis. The tool is population-specific by race and region. It has not been validated for the HIV-positive population and may underestimate fracture risk [103]. HIV status is considered a secondary cause of osteoporosis in FRAX calculation.
The National Osteoporosis Foundation recommends screening with DXA for all women > 65 years of age, all men > 70 years of age, and adults > 50 years of age with additional risk factors for osteoporosis. Evaluation for secondary causes for low BMD should always be considered in the HIV-positive population including evaluation of calcium and vitamin D intake. Laboratory testing may include complete blood count, calcium, phosphate, albumin, creatinine, PTH, 25 hydroxy vitamin D (25,OHD) and 24 hour urine for evaluation of calcium, creatinine and phosphate (especially if on TDF) excretion. Testosterone level can be checked in men and estradiol, prolactin, FSH and LH in women for evaluation of hypogonadism. Bone turnover markers (bone specific alkaline phosphatase and serum C-terminal telopeptide) can also be assessed at baseline.
Studies using high-resolution peripheral quantitative computed tomography (HSPQCT) have shown significant reductions in tibial trabecular bone density and trabecular number in pre-menopausal and postmenopausal HIV-infected women [104], with reduced bone stiffness measured using finite element analysis [105]. Co-infection with HCV is also associated with significantly lower trabecular volumetric BMD and smaller cortical dimensions in the tibia, compared to healthy subjects [106]. HSPQCT is not widely available for clinical use at this time. Lateral imaging of the spine or vertebral morphometric analysis may be done in cases of height loss to assess for occult vertebral compression fractures.
There is a high prevalence of vitamin D deficiency in the HIV-infected population [107]. Treatment goal is to have a vitamin D level of at least 30 ng/mL, based on Endocrine Society practice guidelines [108], and may require supplementation with 1000–2000 units of vitamin D daily. Calcium intake should be optimized, averaging 1000 mg per day including diet and supplements, to be taken in divided amounts through the day for optimal absorption. Secondary causes of low bone density as mentioned in Table 4 should also be addressed. Patients should be counseled on tobacco and alcohol abuse. Corticosteroids should be dosed at the lowest dose needed. Medications such as proton pump inhibitors can impair the absorption of calcium carbonate, in which case calcium citrate supplements should be used if there is suboptimal calcium intake in the diet.
Which medications have been shown to be effective in treatment of osteoporosis in the HIV population?
Bisphosphonates are the mainstay of therapy for osteoporosis in the HIV-infected population. Only alendronate and zoledronate have substantial evidence of safety and effectiveness in the HIV-infected population, but these studies have been small and of limited duration.
Bisphosphonates are pyrophosphate analogues that inhibit bone resorption by binding to the hydroxyapatite crystals in the bone. Several prospective studies have shown alendronate to increase bone density compared to calcium and vitamin D alone in the HIV infected patients with reduced bone density [109,110], with significant reduction in markers of bone resorption [111].
Zoledronic acid (ZA), an amino-bisphosphonate which is infused intravenously, has also been used in smaller studies in HIV-infected persons. In a prospective study evaluating yearly ZA infusion to biennial ZA infusion in subjects with HIV and low bone density [112], biennial ZA infusions were found to be effective in improving and maintaining bone density in the HIV population. In another prospective study evaluating the effects of ZA in HIV-positive men, ZA infusion was given at baseline and at 12 months. Compared to placebo, treatment group had significantly higher bone density and lower bone turnover markers till 5 years after the last infusion [113].
In a meta-analysis evaluating the effect of bisphosphonates on bone mineral density in 328 adults with HIV infection from 8 randomized controlled trials (5 with alendronate and 3 with ZA as the intervention), a significant increase in BMD at the lumbar spine and hip was observed in the treatment groups at 48 and 96 weeks. However, these studies were not long enough to detect the impact of bisphosphonates on fracture risk [114]. ZA has also been shown to be effective in preventing ARV induced bone loss after a single infusion [115].
These studies confirm that both alendronate and ZA are effective in improving BMD in the HIV-infected population, with early studies showing a beneficial effect of ZA in mitigating ARV-induced bone loss as well. DXA may be repeated 1 to 2 years after initiation of osteoporosis therapy and less often subsequently if BMD is stable to improved [116].
Although these studies show significant improvement in bone density with treatment, longitudinal data on fracture reduction with these medications in the HIV-infected population are not available. Additionally, these patients have onset of osteoporosis at a younger age and the need for osteoporosis treatment needs to be assessed carefully before initiating treatment. There are other medications available for the treatment of osteoporosis in the non-HIV population such as raloxifene, teriparatide and denosumab, but no randomized controlled studies of these agents are available in the HIV-infected population.
Summary
The advent of highly potent antiretroviral therapy capable of early and prolonged viral suppression in HIV-infected patients has resulted in significant increases in life span. As we have already seen, this will likely lead to a rising incidence of various metabolic complications of HIV and ARV, including hyperlipidemia and diabetes with associated cardiovascular disease risk. A keen awareness of these potential complications, drug interactions, and possible toxicities will be paramount to their successful management. Appropriate care of HIV-infected individuals going forward will likely require multidisciplinary collaboration as the epidemic evolves to allow our patients to live not only longer, but healthier lives.
Corresponding author: Lisa M. Chirch, MD, UCONN Health, Farmington, CT 06030, chirch@uchc.edu.
Financial disclosures: None
Author contributions: All authors contributed equally to this article
1. UNAIDS. Fact sheet - Latest statistics on the status of the AIDS epidemic. Accessed 10 Nov 2017 at http://www.unaids.org/en/resources/fact-sheet.
2. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Accessed 10 Nov 2017 at https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf.
3. Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PloS One 2013;8:e81355.
4. Pedersen KK, Pedersen M, Troseid M, et al. Microbial translocation in HIV infection is associated with dyslipidemia, insulin resistance, and risk of myocardial infarction. J Acquir Immune Defic Syndr 2013;64:425–33.
5. Strategies for Management of Antiretroviral Therapy Study Group, El-Sadr WM, Lundgren J, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006;355:2283–96.
6. Chastain DB, Henderson H, Stover KR. Epidemiology and management of antiretroviral-associated cardiovascular disease. Open AIDS J 2015;9:23–37.
7. Group ISS, Lundgren JD, Babiker AG, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med 2015;373:795–807.
8. Moyle GJ, Orkin C, Fisher M, et al. A randomized comparative trial of continued abacavir/lamivudine plus efavirenz or replacement with efavirenz/emtricitabine/tenofovir DF in hypercholesterolemic HIV-1 infected individuals. PloS One 2015;10:e0116297.
9. Mothe B, Perez I, Domingo P, et al. HIV-1 infection in subjects older than 70: a multicenter cross-sectional assessment in Catalonia, Spain. Curr HIV Res 2009;7:597–600.
10. Calvo M, Martinez E. Update on metabolic issues in HIV patients. Curr Opin HIV AIDS 2014;9:332–9.
11. Gillard BK, Raya JL, Ruiz-Esponda R, et al. Impaired lipoprotein processing in HIV patients on antiretroviral therapy: aberrant high-density lipoprotein lipids, stability, and function. Arterioscler Thromb Vasc Biol 2013;33:1714-21.
12. Aberg JA, Gallant JE, Ghanem KG, et al. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 2014;58:1–10.
13. Calza L, Colangeli V, Manfredi R, et al. Clinical management of dyslipidaemia associated with combination antiretroviral therapy in HIV-infected patients. J Antimicrob Chemother 2016;71:1451–65.
14. Friis-Moller N, Weber R, Reiss P, et al. Cardiovascular disease risk factors in HIV patients--association with antiretroviral therapy. Results from the DAD study. AIDS 2003;17:1179–93.
15. Husain NE, Ahmed MH. Managing dyslipidemia in HIV/AIDS patients: challenges and solutions. HIV/AIDS (Auckl) 2015;7:1–10.
16. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. Department of Health and Human Services. Accessed at www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
17. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 2015;385:2606–15.
18. Sax PE, Zolopa A, Brar I, et al. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr 2014;67:52–8.
19. Eron JJ, Young B, Cooper DA, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet 2010;375:396–407.
20. Quercia R, Roberts J, Martin-Carpenter L, Zala C. Comparative changes of lipid levels in treatment-naive, HIV-1-infected adults treated with dolutegravir vs. efavirenz, raltegravir, and ritonavir-boosted darunavir-based regimens over 48 weeks. Clin Drug Investig 2015;35:211–9.
21. Lennox JL, DeJesus E, Berger DS, et al. Raltegravir versus efavirenz regimens in treatment-naive hiv-1–infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune Defic Syndr 2010;55:39–48.
22. Nayor M, Vasan RS. Recent update to the US cholesterol treatment guidelines. a comparison with international guidelines. Circulation 2016;133:1795–806.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:2889–934.
24. Mitka M. Exploring statins to decrease HIV-related heart disease risk. JAMA 2015;314:657–9.
25. Dube MP, Stein JH, Aberg JA, et al. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Diseases Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003;37:613–27.
26. Milazzo L, Menzaghi B, Corvasce S, et al. Safety of statin therapy in HIV/hepatitis C virus-coinfected patients. J Acquir Immune Defic Syndr 2007;46:258–60.
27. Hernandez-Romieu AC, Garg S, Rosenberg ES, et al. Is diabetes prevalence higher among HIV-infected individuals compared with the general population? Evidence from MMP and NHANES 2009-2010. BMJ Open Diab Res Care 2017;5:e000304.
28. Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005;165:1179–84.
29. De Wit S, Sabin CA, Weber R, et al. Incidence and risk factors for new-onset diabetes in HIV-infected patients: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. Diabetes Care 2008;31:1224–9.
30. Rasmussen LD, Mathiesen ER, Kronborg G, et al. Risk of diabetes mellitus in persons with and without HIV: a Danish nationwide population-based cohort study. PloS One 2012;7:e44575.
31. Polsky S, Floris-Moore M, Schoenbaum EE, et al. Incident hyperglycaemia among older adults with or at-risk for HIV infection. Antivir Ther 2011;16:181–8.
32. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007;92:2506–12.
33. Galli L, Salpietro S, Pellicciotta G, et al. Risk of type 2 diabetes among HIV-infected and healthy subjects in Italy. Eur J Epidemiol 2012;27:657–65.
34. Howard AA, Hoover DR, Anastos K, et al. The effects of opiate use and hepatitis C virus infection on risk of diabetes mellitus in the Women’s Interagency HIV Study. J Acquir Immune Defic Syndr 2010;54:152–9.
35. Butt AA, McGinnis K, Rodriguez-Barradas MC, et al. HIV infection and the risk of diabetes mellitus. AIDS 2009;23:1227–34.
36. Hadigan C, Kattakuzhy S. Diabetes mellitus type 2 and abnormal glucose metabolism in the setting of human immunodeficiency virus. Endocrin Metab Clin North Am 2014;43:685–96.
37. Butt AA, Fultz SL, Kwoh CK, Kelley D, et al. Risk of diabetes in HIV infected veterans pre- and post-HAART and the role of HCV coinfection. Hepatology 2004;40:115–9.
38. Veloso S, Escote X, Ceperuelo-Mallafre V, et al. Leptin and adiponectin, but not IL18, are related with insulin resistance in treated HIV-1-infected patients with lipodystrophy. Cytokine 2012;58:253–60.
39. Vigouroux C, Maachi M, Nguyen TH, et al. Serum adipocytokines are related to lipodystrophy and metabolic disorders in HIV-infected men under antiretroviral therapy. AIDS 2003;17:1503–11.
40. Palmer CS, Hussain T, Duette G, et al. Regulators of glucose metabolism in CD4+ and CD8+ T cells. Int Rev Immunol 2016;35:477–88.
41. Mehta SH, Moore RD, Thomas DL, et al. The effect of HAART and HCV infection on the development of hyperglycemia among HIV-infected persons. J Acquir Immune Defic Syndr 2003;33:577–84.
42. Monroe AK, Dobs AS, Xu X, et al. Sex hormones, insulin resistance, and diabetes mellitus among men with or at risk for HIV infection. J Acquir Immune Defic Syndr 2011;58:173–80.
43. Rotger M, Gsponer T, Martinez R, et al. Impact of single nucleotide polymorphisms and of clinical risk factors on new-onset diabetes mellitus in HIV-infected individuals. Clin Infect Dis 2010;51:1090–8.
44. Ledergerber B, Furrer H, Rickenbach M, et al. Factors associated with the incidence of type 2 diabetes mellitus in HIV-infected participants in the Swiss HIV Cohort Study. Clin Infect Dis 2007;45:111–9.
45. Hruz PW. Molecular mechanisms for insulin resistance in treated HIV-infection. Best practice & research. Clin Endocrin Metab 2011;25:459–68.
46. Brown TT, Li X, Cole SR, et al. Cumulative exposure to nucleoside analogue reverse transcriptase inhibitors is associated with insulin resistance markers in the Multicenter AIDS Cohort Study. AIDS 2005;19:1375–83.
47. Cossarizza A, Moyle G. Antiretroviral nucleoside and nucleotide analogues and mitochondria. AIDS 2004;18:137–51.
48. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384-7.
49. McComsey GA, Paulsen DM, Lonergan JT, et al. Improvements in lipoatrophy, mitochondrial DNA levels and fat apoptosis after replacing stavudine with abacavir or zidovudine. AIDS 2005;19:15–23.
50. Venhoff N, Setzer B, Melkaoui K, Walker UA. Mitochondrial toxicity of tenofovir, emtricitabine and abacavir alone and in combination with additional nucleoside reverse transcriptase inhibitors. Antivir Ther 2007;12:1075–85.
51. Haubrich RH, Riddler SA, DiRienzo AG, et al. Metabolic outcomes in a randomized trial of nucleoside, nonnucleoside and protease inhibitor-sparing regimens for initial HIV treatment. AIDS 2009;23:1109–18.
52. Aberg JA, Tebas P, Overton ET, et al. Metabolic effects of darunavir/ritonavir versus atazanavir/ritonavir in treatment-naive, HIV type 1-infected subjects over 48 weeks. AIDS Res Hum Retrovir 2012;28:1184–95.
53. Overton ET, Tebas P, Coate B, et al. Effects of once-daily darunavir/ritonavir versus atazanavir/ritonavir on insulin sensitivity in HIV-infected persons over 48 weeks: results of an exploratory substudy of METABOLIK, a phase 4, randomized trial. HIV Clin Trials 2016;17:72–7.
54. Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010;375:735–42.
55. McComsey G, Jiang Y, Erlandson KM, et al. Rosuvastatin improves hip bone mineral density but worsens insulin resistance. Boston, MA: Conference on Retroviruses and Opportunistic Infections; 2014.
56. Lichtenstein K DR, Wood K, et al. Statin use is associated with incident diabetes mellitus among patients in the HIV Outpatient Study. Atlanta, GA: Conference on Retroviruses and Opportunistic Infections; 2013.
57. Spagnuolo V GL, Poli A, et al. Association between statin use and type 2 diabetes mellitus occurrence among HIV-1+ patients receiving ART. Atlanta, GA: Conference on Retroviruses and Opportunistic Infections; 2013.
58. Wang KL, Liu CJ, Chao TF, et al. Statins, risk of diabetes, and implications on outcomes in the general population. J Am Coll Cardiol 2012;60:1231–8.
59. Ridker PM, Pradhan A, MacFadyen JG, et al. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012;380:565–71.
60. Bloomgarden ZT, Handelsman Y. Approaches to treatment 2: Comparison of American Association of Clinical Endocrinologists (AACE) and American Diabetes Association (ADA) type 2 diabetes treatment guidelines. J Diabetes 2016;8:4–6.
61. Kim PS, Woods C, Georgoff P, et al. A1C underestimates glycemia in HIV infection. Diabetes Care 2009;32:1591–3.
62. Diop ME, Bastard JP, Meunier N, et al. Inappropriately low glycated hemoglobin values and hemolysis in HIV-infected patients. AIDS Res Hum Retrovir 2006;22:1242–7.
63. Polgreen PM, Putz D, Stapleton JT. Inaccurate glycosylated hemoglobin A1C measurements in human immunodeficiency virus-positive patients with diabetes mellitus. Clin Infect Dis 2003;37:e53–56.
64. Glesby MJ, Hoover DR, Shi Q, et al. Glycated haemoglobin in diabetic women with and without HIV infection: data from the Women’s Interagency HIV Study. Antivir Ther 2010;15:571–7.
65. Slama L, Palella FJ Jr, Abraham AG, et al. Inaccuracy of haemoglobin A1c among HIV-infected men: effects of CD4 cell count, antiretroviral therapies and haematological parameters. J Antimicrob Chemother 2014;69:3360–7.
66. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012;55:1577–96.
67. Look ARG, Pi-Sunyer X, Blackburn G, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care 2007;30:1374–83.
68. Hajjar J, Habra MA, Naing A. Metformin: an old drug with new potential. Expert Opin Investig Drugs 2013;22:1511–7.
69. Kohli R, Shevitz A, Gorbach S, Wanke C. A randomized placebo-controlled trial of metformin for the treatment of HIV lipodystrophy. HIV Med 2007;8:420–6.
70. Tivicay prescribing information. Accessed at www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Tivicay/pdf/TIVICAY-PI-PIL.PDF.
71. Han JH, Gordon K, Womack JA, et al. Comparative effectiveness of diabetic oral medications among HIV-infected and HIV-uninfected veterans. Diabetes Care 2017;40:218–25.
72. Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006;20:2165–74.
73. Libois A, Clumeck N, Kabeya K, et al. Risk factors of osteopenia in HIV-infected women: no role of antiretroviral therapy. Maturitas 2010;65:51–4.
74. Carr A, Grund B, Neuhaus J, et al. Prevalence of and risk factors for low bone mineral density in untreated HIV infection: a substudy of the INSIGHT Strategic Timing of AntiRetroviral Treatment (START) trial. HIV Med 2015;16 Suppl 1:137–46.
75. Triant VA, Brown TT, Lee H, Grinspoon SK. Fracture prevalence among human immunodeficiency virus (HIV)-infected versus non-HIV-infected patients in a large U.S. healthcare system. J Clin Endocrinol Metab 2008;93:3499–504.
76. Young B, Dao CN, Buchacz K, Baker R, Brooks JT, Investigators HIVOS. Increased rates of bone fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared with the US general population, 2000-2006. Clin Infect Dis 2011;52:1061–8.
77. Rodriguez M, Daniels B, Gunawardene S, Robbins GK. High frequency of vitamin D deficiency in ambulatory HIV-Positive patients. AIDS Res Hum Retrovir 2009;25:9–14.
78. Teichmann J, Lange U, Discher T, et al. Bone mineral density in human immunodeficiency virus-1 infected men with hypogonadism prior to highly-active-antiretroviral-therapy (HAART). Eur J Med Res 2009;14:59–64.
79. Lo Re V 3rd, Volk J, Newcomb CW, et al. Risk of hip fracture associated with hepatitis C virus infection and hepatitis C/human immunodeficiency virus coinfection. Hepatology 2012;56:1688–98.
80. Maalouf NM, Zhang S, Drechsler H, et al. Hepatitis C co-infection and severity of liver disease as risk factors for osteoporotic fractures among HIV-infected patients. J Bone Miner Res 2013;28:2577–83.
81. Byrne DD, Newcomb CW, Carbonari DM, et al. Increased risk of hip fracture associated with dually treated HIV/hepatitis B virus coinfection. J Viral Hepat 2015;22:936–47.
82. Gilsanz V, Chalfant J, Mo AO, et al. Reciprocal relations of subcutaneous and visceral fat to bone structure and strength. J Clin Endocrinol Metab 2009;94:3387–93.
83. Rosen CJ, Klibanski A. Bone, fat, and body composition: evolving concepts in the pathogenesis of osteoporosis. Am J Med 2009;122:409–14.
84. Fakruddin JM, Laurence J. HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk. J Biol Chem 2003;278:48251–8.
85. Gazzola L, Bellistri GM, Tincati C, et al. Association between peripheral T-Lymphocyte activation and impaired bone mineral density in HIV-infected patients. J Transl Med 2013;11:51.
86. Clerici M, Shearer GM. A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol Today 1993;14:107–11.
87. Chew N, Tan E, Li L, Lim R. HIV-1 tat and rev upregulates osteoclast bone resorption. J Int AIDS Soc 2014;17(4 Suppl 3):19724.
88. McComsey GA, Tebas P, Shane E, et al. Bone disease in HIV infection: a practical review and recommendations for HIV care providers. Clin Infect Dis 2010;51:937–46.
89. Panayiotopoulos A, Bhat N, Bhangoo A. Bone and vitamin D metabolism in HIV. Rev Endocr Metab Disord 2013;14:119–25.
90. Fakruddin JM, Laurence J. Interactions among human immunodeficiency virus (HIV)-1, interferon-gamma and receptor of activated NF-kappa B ligand (RANKL): implications for HIV pathogenesis. Clin Exp Immunol 2004;137:538–45.
91. Li Y, Toraldo G, Li A, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007;109:3839–48.
92. Finkelstein JS, Brockwell SE, Mehta V, et al. Bone mineral density changes during the menopause transition in a multiethnic cohort of women. J Clin Endocrinol Metab 2008;93:861–8.
93. Huang JS, Hughes MD, Riddler SA, Haubrich RH, Aids Clinical Trials Group AST. Bone mineral density effects of randomized regimen and nucleoside reverse transcriptase inhibitor selection from ACTG A5142. HIV Clin Trials 2013;145:224–34.
94. McComsey GA, Kitch D, Daar ES, et al. Bone mineral density and fractures in antiretroviral-naive persons randomized to receive abacavir-lamivudine or tenofovir disoproxil fumarate-emtricitabine along with efavirenz or atazanavir-ritonavir: Aids Clinical Trials Group A5224s, a substudy of ACTG A5202. J Infect Dis 2011;203:1791–801.
95. Schafer JJ, Manlangit K, Squires KE. Bone health and human immunodeficiency virus infection. Pharmacotherapy 2013;33:665–82.
96. Mateo L, Holgado S, Marinoso ML, et al. Hypophosphatemic osteomalacia induced by tenofovir in HIV-infected patients. Clin Rheumatol 2016;35:1271–9.
97. Brown TT, Moser C, Currier JS, et al. Changes in bone mineral density after initiation of antiretroviral treatment with tenofovir disoproxil fumarate/emtricitabine plus atazanavir/ritonavir, darunavir/ritonavir, or raltegravir. J Infect Dis 2015;212:1241–9.
98. Ofotokun I, Titanji K, Vikulina T, et al. Role of T-cell reconstitution in HIV-1 antiretroviral therapy-induced bone loss. Nat Commun 2015;6:8282.
99. Ofotokun I, Titanji K, Vunnava A, et al. Antiretroviral therapy induces a rapid increase in bone resorption that is positively associated with the magnitude of immune reconstitution in HIV infection. AIDS 2016;30:405–14.
100. Stephens KI, Rubinsztain L, Payan J, et al. Dual-energy x-ray absorptiometry and calculated FRAX risk scores may underestimate osteoporotic fracture risk in vitamin d-deficient veterans with HIV infection. Endocr Pract 2016;22:440–6.
101. Mirza F, Canalis E. Management of endocrine disease: Secondary osteoporosis: pathophysiology and management. Eur J Endocrinol 2015;173:R131–151.
102. Brown TT, Hoy J, Borderi M, et al. Recommendations for evaluation and management of bone disease in HIV. Clin Infect Dis 2015;60:1242–51.
103. Mazzotta E, Ursini T, Agostinone A, et al. Prevalence and predictors of low bone mineral density and fragility fractures among HIV-infected patients at one Italian center after universal DXA screening: sensitivity and specificity of current guidelines on bone mineral density management. AIDS Patient Care STDS 2015;29:169–80.
104. Calmy A, Chevalley T, Delhumeau C, et al. Long-term HIV infection and antiretroviral therapy are associated with bone microstructure alterations in premenopausal women. Osteoporos Int 2013;24:1843–52.
105. Yin MT, Lund E, Shah J, et al. Lower peak bone mass and abnormal trabecular and cortical microarchitecture in young men infected with HIV early in life. AIDS 2014;28:345–53.
106. Lo Re V, 3rd, Lynn K, Stumm ER, et al. Structural bone deficits in HIV/HCV-coinfected, HCV-monoinfected, and HIV-monoinfected women. J Infect Dis 2015;212:924–33.
107. Allavena C, Delpierre C, Cuzin L, et al. High frequency of vitamin D deficiency in HIV-infected patients: effects of HIV-related factors and antiretroviral drugs. J Antimicrob Chemother 2012;67:2222–30.
108. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011;96:1911–30.
109. Mondy K, Powderly WG, Claxton SA, et al. Alendronate, vitamin D, and calcium for the treatment of osteopenia/osteoporosis associated with HIV infection. J Acquir Immune Defic Syndr 2005;38:426–31.
110. McComsey GA, Kendall MA, Tebas P, et al. Alendronate with calcium and vitamin D supplementation is safe and effective for the treatment of decreased bone mineral density in HIV. AIDS 2007;21:2473–82.
111. Guaraldi G, Orlando G, Madeddu G, et al. Alendronate reduces bone resorption in HIV-associated osteopenia/osteoporosis. HIV Clin Trials 2004;5:269–77.
112. Negredo E, Bonjoch A, Perez-Alvarez N, et al. Comparison of two different strategies of treatment with zoledronate in HIV-infected patients with low bone mineral density: single dose versus two doses in 2 years. HIV Med 2015;16:441–8.
113. Bolland MJ, Grey A, Horne AM, et al. Effects of intravenous zoledronate on bone turnover and bone density persist for at least five years in HIV-infected men. J Clin Endocrinol Metab 2012;97:1922–8.
114. Pinzone MR, Moreno S, Cacopardo B, Nunnari G. Is there enough evidence to use bisphosphonates in HIV-infected patients? A systematic review and meta-analysis. AIDS Rev 2014;16:213–22.
115. Ofotokun I, Titanji K, Lahiri CD, et al. A Single-dose zoledronic acid infusion prevents antiretroviral therapy-induced bone loss in treatment-naive HIV-infected patients: a phase IIb trial. Clin Infect Dis 2016;63:663–71.
116. Lewiecki EM, Gordon CM, Baim S, et al. International Society for Clinical Densitometry 2007 adult and pediatric official positions. Bone 2008;43:1115–21.
1. UNAIDS. Fact sheet - Latest statistics on the status of the AIDS epidemic. Accessed 10 Nov 2017 at http://www.unaids.org/en/resources/fact-sheet.
2. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Accessed 10 Nov 2017 at https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf.
3. Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PloS One 2013;8:e81355.
4. Pedersen KK, Pedersen M, Troseid M, et al. Microbial translocation in HIV infection is associated with dyslipidemia, insulin resistance, and risk of myocardial infarction. J Acquir Immune Defic Syndr 2013;64:425–33.
5. Strategies for Management of Antiretroviral Therapy Study Group, El-Sadr WM, Lundgren J, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006;355:2283–96.
6. Chastain DB, Henderson H, Stover KR. Epidemiology and management of antiretroviral-associated cardiovascular disease. Open AIDS J 2015;9:23–37.
7. Group ISS, Lundgren JD, Babiker AG, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med 2015;373:795–807.
8. Moyle GJ, Orkin C, Fisher M, et al. A randomized comparative trial of continued abacavir/lamivudine plus efavirenz or replacement with efavirenz/emtricitabine/tenofovir DF in hypercholesterolemic HIV-1 infected individuals. PloS One 2015;10:e0116297.
9. Mothe B, Perez I, Domingo P, et al. HIV-1 infection in subjects older than 70: a multicenter cross-sectional assessment in Catalonia, Spain. Curr HIV Res 2009;7:597–600.
10. Calvo M, Martinez E. Update on metabolic issues in HIV patients. Curr Opin HIV AIDS 2014;9:332–9.
11. Gillard BK, Raya JL, Ruiz-Esponda R, et al. Impaired lipoprotein processing in HIV patients on antiretroviral therapy: aberrant high-density lipoprotein lipids, stability, and function. Arterioscler Thromb Vasc Biol 2013;33:1714-21.
12. Aberg JA, Gallant JE, Ghanem KG, et al. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 2014;58:1–10.
13. Calza L, Colangeli V, Manfredi R, et al. Clinical management of dyslipidaemia associated with combination antiretroviral therapy in HIV-infected patients. J Antimicrob Chemother 2016;71:1451–65.
14. Friis-Moller N, Weber R, Reiss P, et al. Cardiovascular disease risk factors in HIV patients--association with antiretroviral therapy. Results from the DAD study. AIDS 2003;17:1179–93.
15. Husain NE, Ahmed MH. Managing dyslipidemia in HIV/AIDS patients: challenges and solutions. HIV/AIDS (Auckl) 2015;7:1–10.
16. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. Department of Health and Human Services. Accessed at www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
17. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 2015;385:2606–15.
18. Sax PE, Zolopa A, Brar I, et al. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr 2014;67:52–8.
19. Eron JJ, Young B, Cooper DA, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet 2010;375:396–407.
20. Quercia R, Roberts J, Martin-Carpenter L, Zala C. Comparative changes of lipid levels in treatment-naive, HIV-1-infected adults treated with dolutegravir vs. efavirenz, raltegravir, and ritonavir-boosted darunavir-based regimens over 48 weeks. Clin Drug Investig 2015;35:211–9.
21. Lennox JL, DeJesus E, Berger DS, et al. Raltegravir versus efavirenz regimens in treatment-naive hiv-1–infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune Defic Syndr 2010;55:39–48.
22. Nayor M, Vasan RS. Recent update to the US cholesterol treatment guidelines. a comparison with international guidelines. Circulation 2016;133:1795–806.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:2889–934.
24. Mitka M. Exploring statins to decrease HIV-related heart disease risk. JAMA 2015;314:657–9.
25. Dube MP, Stein JH, Aberg JA, et al. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Diseases Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003;37:613–27.
26. Milazzo L, Menzaghi B, Corvasce S, et al. Safety of statin therapy in HIV/hepatitis C virus-coinfected patients. J Acquir Immune Defic Syndr 2007;46:258–60.
27. Hernandez-Romieu AC, Garg S, Rosenberg ES, et al. Is diabetes prevalence higher among HIV-infected individuals compared with the general population? Evidence from MMP and NHANES 2009-2010. BMJ Open Diab Res Care 2017;5:e000304.
28. Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005;165:1179–84.
29. De Wit S, Sabin CA, Weber R, et al. Incidence and risk factors for new-onset diabetes in HIV-infected patients: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. Diabetes Care 2008;31:1224–9.
30. Rasmussen LD, Mathiesen ER, Kronborg G, et al. Risk of diabetes mellitus in persons with and without HIV: a Danish nationwide population-based cohort study. PloS One 2012;7:e44575.
31. Polsky S, Floris-Moore M, Schoenbaum EE, et al. Incident hyperglycaemia among older adults with or at-risk for HIV infection. Antivir Ther 2011;16:181–8.
32. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007;92:2506–12.
33. Galli L, Salpietro S, Pellicciotta G, et al. Risk of type 2 diabetes among HIV-infected and healthy subjects in Italy. Eur J Epidemiol 2012;27:657–65.
34. Howard AA, Hoover DR, Anastos K, et al. The effects of opiate use and hepatitis C virus infection on risk of diabetes mellitus in the Women’s Interagency HIV Study. J Acquir Immune Defic Syndr 2010;54:152–9.
35. Butt AA, McGinnis K, Rodriguez-Barradas MC, et al. HIV infection and the risk of diabetes mellitus. AIDS 2009;23:1227–34.
36. Hadigan C, Kattakuzhy S. Diabetes mellitus type 2 and abnormal glucose metabolism in the setting of human immunodeficiency virus. Endocrin Metab Clin North Am 2014;43:685–96.
37. Butt AA, Fultz SL, Kwoh CK, Kelley D, et al. Risk of diabetes in HIV infected veterans pre- and post-HAART and the role of HCV coinfection. Hepatology 2004;40:115–9.
38. Veloso S, Escote X, Ceperuelo-Mallafre V, et al. Leptin and adiponectin, but not IL18, are related with insulin resistance in treated HIV-1-infected patients with lipodystrophy. Cytokine 2012;58:253–60.
39. Vigouroux C, Maachi M, Nguyen TH, et al. Serum adipocytokines are related to lipodystrophy and metabolic disorders in HIV-infected men under antiretroviral therapy. AIDS 2003;17:1503–11.
40. Palmer CS, Hussain T, Duette G, et al. Regulators of glucose metabolism in CD4+ and CD8+ T cells. Int Rev Immunol 2016;35:477–88.
41. Mehta SH, Moore RD, Thomas DL, et al. The effect of HAART and HCV infection on the development of hyperglycemia among HIV-infected persons. J Acquir Immune Defic Syndr 2003;33:577–84.
42. Monroe AK, Dobs AS, Xu X, et al. Sex hormones, insulin resistance, and diabetes mellitus among men with or at risk for HIV infection. J Acquir Immune Defic Syndr 2011;58:173–80.
43. Rotger M, Gsponer T, Martinez R, et al. Impact of single nucleotide polymorphisms and of clinical risk factors on new-onset diabetes mellitus in HIV-infected individuals. Clin Infect Dis 2010;51:1090–8.
44. Ledergerber B, Furrer H, Rickenbach M, et al. Factors associated with the incidence of type 2 diabetes mellitus in HIV-infected participants in the Swiss HIV Cohort Study. Clin Infect Dis 2007;45:111–9.
45. Hruz PW. Molecular mechanisms for insulin resistance in treated HIV-infection. Best practice & research. Clin Endocrin Metab 2011;25:459–68.
46. Brown TT, Li X, Cole SR, et al. Cumulative exposure to nucleoside analogue reverse transcriptase inhibitors is associated with insulin resistance markers in the Multicenter AIDS Cohort Study. AIDS 2005;19:1375–83.
47. Cossarizza A, Moyle G. Antiretroviral nucleoside and nucleotide analogues and mitochondria. AIDS 2004;18:137–51.
48. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384-7.
49. McComsey GA, Paulsen DM, Lonergan JT, et al. Improvements in lipoatrophy, mitochondrial DNA levels and fat apoptosis after replacing stavudine with abacavir or zidovudine. AIDS 2005;19:15–23.
50. Venhoff N, Setzer B, Melkaoui K, Walker UA. Mitochondrial toxicity of tenofovir, emtricitabine and abacavir alone and in combination with additional nucleoside reverse transcriptase inhibitors. Antivir Ther 2007;12:1075–85.
51. Haubrich RH, Riddler SA, DiRienzo AG, et al. Metabolic outcomes in a randomized trial of nucleoside, nonnucleoside and protease inhibitor-sparing regimens for initial HIV treatment. AIDS 2009;23:1109–18.
52. Aberg JA, Tebas P, Overton ET, et al. Metabolic effects of darunavir/ritonavir versus atazanavir/ritonavir in treatment-naive, HIV type 1-infected subjects over 48 weeks. AIDS Res Hum Retrovir 2012;28:1184–95.
53. Overton ET, Tebas P, Coate B, et al. Effects of once-daily darunavir/ritonavir versus atazanavir/ritonavir on insulin sensitivity in HIV-infected persons over 48 weeks: results of an exploratory substudy of METABOLIK, a phase 4, randomized trial. HIV Clin Trials 2016;17:72–7.
54. Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010;375:735–42.
55. McComsey G, Jiang Y, Erlandson KM, et al. Rosuvastatin improves hip bone mineral density but worsens insulin resistance. Boston, MA: Conference on Retroviruses and Opportunistic Infections; 2014.
56. Lichtenstein K DR, Wood K, et al. Statin use is associated with incident diabetes mellitus among patients in the HIV Outpatient Study. Atlanta, GA: Conference on Retroviruses and Opportunistic Infections; 2013.
57. Spagnuolo V GL, Poli A, et al. Association between statin use and type 2 diabetes mellitus occurrence among HIV-1+ patients receiving ART. Atlanta, GA: Conference on Retroviruses and Opportunistic Infections; 2013.
58. Wang KL, Liu CJ, Chao TF, et al. Statins, risk of diabetes, and implications on outcomes in the general population. J Am Coll Cardiol 2012;60:1231–8.
59. Ridker PM, Pradhan A, MacFadyen JG, et al. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012;380:565–71.
60. Bloomgarden ZT, Handelsman Y. Approaches to treatment 2: Comparison of American Association of Clinical Endocrinologists (AACE) and American Diabetes Association (ADA) type 2 diabetes treatment guidelines. J Diabetes 2016;8:4–6.
61. Kim PS, Woods C, Georgoff P, et al. A1C underestimates glycemia in HIV infection. Diabetes Care 2009;32:1591–3.
62. Diop ME, Bastard JP, Meunier N, et al. Inappropriately low glycated hemoglobin values and hemolysis in HIV-infected patients. AIDS Res Hum Retrovir 2006;22:1242–7.
63. Polgreen PM, Putz D, Stapleton JT. Inaccurate glycosylated hemoglobin A1C measurements in human immunodeficiency virus-positive patients with diabetes mellitus. Clin Infect Dis 2003;37:e53–56.
64. Glesby MJ, Hoover DR, Shi Q, et al. Glycated haemoglobin in diabetic women with and without HIV infection: data from the Women’s Interagency HIV Study. Antivir Ther 2010;15:571–7.
65. Slama L, Palella FJ Jr, Abraham AG, et al. Inaccuracy of haemoglobin A1c among HIV-infected men: effects of CD4 cell count, antiretroviral therapies and haematological parameters. J Antimicrob Chemother 2014;69:3360–7.
66. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012;55:1577–96.
67. Look ARG, Pi-Sunyer X, Blackburn G, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care 2007;30:1374–83.
68. Hajjar J, Habra MA, Naing A. Metformin: an old drug with new potential. Expert Opin Investig Drugs 2013;22:1511–7.
69. Kohli R, Shevitz A, Gorbach S, Wanke C. A randomized placebo-controlled trial of metformin for the treatment of HIV lipodystrophy. HIV Med 2007;8:420–6.
70. Tivicay prescribing information. Accessed at www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Tivicay/pdf/TIVICAY-PI-PIL.PDF.
71. Han JH, Gordon K, Womack JA, et al. Comparative effectiveness of diabetic oral medications among HIV-infected and HIV-uninfected veterans. Diabetes Care 2017;40:218–25.
72. Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006;20:2165–74.
73. Libois A, Clumeck N, Kabeya K, et al. Risk factors of osteopenia in HIV-infected women: no role of antiretroviral therapy. Maturitas 2010;65:51–4.
74. Carr A, Grund B, Neuhaus J, et al. Prevalence of and risk factors for low bone mineral density in untreated HIV infection: a substudy of the INSIGHT Strategic Timing of AntiRetroviral Treatment (START) trial. HIV Med 2015;16 Suppl 1:137–46.
75. Triant VA, Brown TT, Lee H, Grinspoon SK. Fracture prevalence among human immunodeficiency virus (HIV)-infected versus non-HIV-infected patients in a large U.S. healthcare system. J Clin Endocrinol Metab 2008;93:3499–504.
76. Young B, Dao CN, Buchacz K, Baker R, Brooks JT, Investigators HIVOS. Increased rates of bone fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared with the US general population, 2000-2006. Clin Infect Dis 2011;52:1061–8.
77. Rodriguez M, Daniels B, Gunawardene S, Robbins GK. High frequency of vitamin D deficiency in ambulatory HIV-Positive patients. AIDS Res Hum Retrovir 2009;25:9–14.
78. Teichmann J, Lange U, Discher T, et al. Bone mineral density in human immunodeficiency virus-1 infected men with hypogonadism prior to highly-active-antiretroviral-therapy (HAART). Eur J Med Res 2009;14:59–64.
79. Lo Re V 3rd, Volk J, Newcomb CW, et al. Risk of hip fracture associated with hepatitis C virus infection and hepatitis C/human immunodeficiency virus coinfection. Hepatology 2012;56:1688–98.
80. Maalouf NM, Zhang S, Drechsler H, et al. Hepatitis C co-infection and severity of liver disease as risk factors for osteoporotic fractures among HIV-infected patients. J Bone Miner Res 2013;28:2577–83.
81. Byrne DD, Newcomb CW, Carbonari DM, et al. Increased risk of hip fracture associated with dually treated HIV/hepatitis B virus coinfection. J Viral Hepat 2015;22:936–47.
82. Gilsanz V, Chalfant J, Mo AO, et al. Reciprocal relations of subcutaneous and visceral fat to bone structure and strength. J Clin Endocrinol Metab 2009;94:3387–93.
83. Rosen CJ, Klibanski A. Bone, fat, and body composition: evolving concepts in the pathogenesis of osteoporosis. Am J Med 2009;122:409–14.
84. Fakruddin JM, Laurence J. HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk. J Biol Chem 2003;278:48251–8.
85. Gazzola L, Bellistri GM, Tincati C, et al. Association between peripheral T-Lymphocyte activation and impaired bone mineral density in HIV-infected patients. J Transl Med 2013;11:51.
86. Clerici M, Shearer GM. A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol Today 1993;14:107–11.
87. Chew N, Tan E, Li L, Lim R. HIV-1 tat and rev upregulates osteoclast bone resorption. J Int AIDS Soc 2014;17(4 Suppl 3):19724.
88. McComsey GA, Tebas P, Shane E, et al. Bone disease in HIV infection: a practical review and recommendations for HIV care providers. Clin Infect Dis 2010;51:937–46.
89. Panayiotopoulos A, Bhat N, Bhangoo A. Bone and vitamin D metabolism in HIV. Rev Endocr Metab Disord 2013;14:119–25.
90. Fakruddin JM, Laurence J. Interactions among human immunodeficiency virus (HIV)-1, interferon-gamma and receptor of activated NF-kappa B ligand (RANKL): implications for HIV pathogenesis. Clin Exp Immunol 2004;137:538–45.
91. Li Y, Toraldo G, Li A, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007;109:3839–48.
92. Finkelstein JS, Brockwell SE, Mehta V, et al. Bone mineral density changes during the menopause transition in a multiethnic cohort of women. J Clin Endocrinol Metab 2008;93:861–8.
93. Huang JS, Hughes MD, Riddler SA, Haubrich RH, Aids Clinical Trials Group AST. Bone mineral density effects of randomized regimen and nucleoside reverse transcriptase inhibitor selection from ACTG A5142. HIV Clin Trials 2013;145:224–34.
94. McComsey GA, Kitch D, Daar ES, et al. Bone mineral density and fractures in antiretroviral-naive persons randomized to receive abacavir-lamivudine or tenofovir disoproxil fumarate-emtricitabine along with efavirenz or atazanavir-ritonavir: Aids Clinical Trials Group A5224s, a substudy of ACTG A5202. J Infect Dis 2011;203:1791–801.
95. Schafer JJ, Manlangit K, Squires KE. Bone health and human immunodeficiency virus infection. Pharmacotherapy 2013;33:665–82.
96. Mateo L, Holgado S, Marinoso ML, et al. Hypophosphatemic osteomalacia induced by tenofovir in HIV-infected patients. Clin Rheumatol 2016;35:1271–9.
97. Brown TT, Moser C, Currier JS, et al. Changes in bone mineral density after initiation of antiretroviral treatment with tenofovir disoproxil fumarate/emtricitabine plus atazanavir/ritonavir, darunavir/ritonavir, or raltegravir. J Infect Dis 2015;212:1241–9.
98. Ofotokun I, Titanji K, Vikulina T, et al. Role of T-cell reconstitution in HIV-1 antiretroviral therapy-induced bone loss. Nat Commun 2015;6:8282.
99. Ofotokun I, Titanji K, Vunnava A, et al. Antiretroviral therapy induces a rapid increase in bone resorption that is positively associated with the magnitude of immune reconstitution in HIV infection. AIDS 2016;30:405–14.
100. Stephens KI, Rubinsztain L, Payan J, et al. Dual-energy x-ray absorptiometry and calculated FRAX risk scores may underestimate osteoporotic fracture risk in vitamin d-deficient veterans with HIV infection. Endocr Pract 2016;22:440–6.
101. Mirza F, Canalis E. Management of endocrine disease: Secondary osteoporosis: pathophysiology and management. Eur J Endocrinol 2015;173:R131–151.
102. Brown TT, Hoy J, Borderi M, et al. Recommendations for evaluation and management of bone disease in HIV. Clin Infect Dis 2015;60:1242–51.
103. Mazzotta E, Ursini T, Agostinone A, et al. Prevalence and predictors of low bone mineral density and fragility fractures among HIV-infected patients at one Italian center after universal DXA screening: sensitivity and specificity of current guidelines on bone mineral density management. AIDS Patient Care STDS 2015;29:169–80.
104. Calmy A, Chevalley T, Delhumeau C, et al. Long-term HIV infection and antiretroviral therapy are associated with bone microstructure alterations in premenopausal women. Osteoporos Int 2013;24:1843–52.
105. Yin MT, Lund E, Shah J, et al. Lower peak bone mass and abnormal trabecular and cortical microarchitecture in young men infected with HIV early in life. AIDS 2014;28:345–53.
106. Lo Re V, 3rd, Lynn K, Stumm ER, et al. Structural bone deficits in HIV/HCV-coinfected, HCV-monoinfected, and HIV-monoinfected women. J Infect Dis 2015;212:924–33.
107. Allavena C, Delpierre C, Cuzin L, et al. High frequency of vitamin D deficiency in HIV-infected patients: effects of HIV-related factors and antiretroviral drugs. J Antimicrob Chemother 2012;67:2222–30.
108. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011;96:1911–30.
109. Mondy K, Powderly WG, Claxton SA, et al. Alendronate, vitamin D, and calcium for the treatment of osteopenia/osteoporosis associated with HIV infection. J Acquir Immune Defic Syndr 2005;38:426–31.
110. McComsey GA, Kendall MA, Tebas P, et al. Alendronate with calcium and vitamin D supplementation is safe and effective for the treatment of decreased bone mineral density in HIV. AIDS 2007;21:2473–82.
111. Guaraldi G, Orlando G, Madeddu G, et al. Alendronate reduces bone resorption in HIV-associated osteopenia/osteoporosis. HIV Clin Trials 2004;5:269–77.
112. Negredo E, Bonjoch A, Perez-Alvarez N, et al. Comparison of two different strategies of treatment with zoledronate in HIV-infected patients with low bone mineral density: single dose versus two doses in 2 years. HIV Med 2015;16:441–8.
113. Bolland MJ, Grey A, Horne AM, et al. Effects of intravenous zoledronate on bone turnover and bone density persist for at least five years in HIV-infected men. J Clin Endocrinol Metab 2012;97:1922–8.
114. Pinzone MR, Moreno S, Cacopardo B, Nunnari G. Is there enough evidence to use bisphosphonates in HIV-infected patients? A systematic review and meta-analysis. AIDS Rev 2014;16:213–22.
115. Ofotokun I, Titanji K, Lahiri CD, et al. A Single-dose zoledronic acid infusion prevents antiretroviral therapy-induced bone loss in treatment-naive HIV-infected patients: a phase IIb trial. Clin Infect Dis 2016;63:663–71.
116. Lewiecki EM, Gordon CM, Baim S, et al. International Society for Clinical Densitometry 2007 adult and pediatric official positions. Bone 2008;43:1115–21.