User login
Due Diligence: Denials
Before submitting a claim, hospitalists should ensure that the service is rendered, that it is completely and accurately documented in the medical record, that the correct information is entered on the claim form, and that it is a covered benefit and eligible for payment.
Although the latter two elements typically are delegated to the billing team, hospitalists should encourage or request feedback regarding payment and denials. The ensuing open dialogue between physicians and billers might prove helpful in understanding and resolving future billing issues. Less-experienced billers first respond to claim denials by submitting documentation (i.e. “appeal with paper”) despite the inappropriateness of this action. If the denial is upheld, this attempt is viewed as unsuccessful and, without further consideration, “written off.” However, careful examination of the payor’s initial claim determination could elicit a more suitable response.
Service Provider
Provider enrollment issues can sidetrack claim submissions. Physicians must register their NPI (national provider identifier) with the correct practice location and group assignment, particularly when previously practicing physicians join a new group practice. Failure to do so is an infrequent, yet valid, cause for denial.
Alternatively, enrollment issues play a greater role when services involve nurse practitioners (NPs) and physician assistants (PAs) who are enrolled with Medicare but might be prohibited from enrolling with other payors. For example, an NP independently provides subsequent hospital care (e.g. 99232) to a Medicare beneficiary. The claim is submitted in the NP’s name and reimbursed at the correct amount by Medicare as the primary insurer. The remaining balance is submitted to the secondary insurer, who does not enroll NPPs. The claim is rejected. If the physician group has a contractual agreement to recognize NPP services by reporting them under the collaborating physician’s name, the claim can be resubmitted in the physician’s name. In absence of such an agreement, the claim should be written off.
Location
The place of service (POS) must match the reported service/procedure code. For example, a hospitalist is asked to see a patient in the ED. The patient requires further testing but does not meet the criterion for an inpatient stay. The hospitalist admits the patient to observation, treats him, and discharges him to home.
Hospitalists need to avoid the common mistake of mismatching the service code with the location/POS. Observation services performed by the “physician of record” should be reported with the corresponding codes: initial observation care (99218-99220), subsequent observation care (99224-99226), or observation discharge (99217), as appropriate.1 The correct POS should be reported as outpatient hospital (POS 22), not inpatient hospital (POS 21). Trying to report outpatient codes with an inpatient POS will result in claim denial.
A similar denial occurs when trying to report inpatient codes (99231-99233) in an outpatient location (e.g. 23-ED). These denials require claim resubmission with the correct POS and/or service/procedure code. A complete list of POS codes and corresponding definitions can be obtained from Chapter 26, Section 10.5 of the Medicare Claims Processing Manual, available at www.cms.hhs.gov/manuals/downloads/clm104c26.pdf.
Diagnosis
Denials involving diagnoses produce issues of “medical necessity.”1 Examine these denials carefully. Consider the service/procedure code when trying to formulate a response to the denial. The diagnosis code represents the reason for the service or procedure and might be a sign, symptom, or condition with which the patient presents. Medicare reimburses for procedures and services that are deemed “reasonable and necessary.”
In an effort to unify standards, Medicare has developed national coverage determinations (NCDs) to identify coverage requirements for frequent or problematic procedures or services. These coverage requirements can identify specific conditions (i.e. ICD-9-CM codes) for which the services or procedures are considered medically necessary. In the absence of a national coverage policy, an item or service could be covered at the discretion of Medicare contractors based on a local coverage determination (LCD), which varies by contractor.
Medical necessity denials often involve a mismatched or missing diagnosis. For example, a payor might deny a claim for cardiopulmonary resuscitation (92950) that is associated with a diagnosis code of congestive heart failure (428.0), despite this being the underlying condition that prompted the decline in the patient’s condition. The payor might only accept “cardiac arrest” (427.5) as the “medically necessary” diagnosis for cardiopulmonary resuscitation, as this is the direct reason necessitating the procedure. After reviewing the documentation to ensure that the documentation supports the diagnosis, the claim can be resubmitted with a confirmed and corrected diagnosis code.
Initial-Request Response
While diagnoses can lead to medical necessity issues, not all medical necessity denials are due to incorrect diagnoses. Some “medical necessity” denials result from a failure to respond to a payor request. More specifically, if the “medical necessity” denial involves a covered evaluation and management visit, the denial is more likely the result of a failure to respond to a prepayment request for documentation.
Medicare typically issues prepayment requests for documentation for the following inpatient CPT codes: 99223, 99233, 99232, 99239, and 99292.1 If the documentation is not provided to the Medicare review department within a designated time frame (e.g. 30-45 days), the claim is automatically denied. The reason for denial is cited as being “not deemed a medical necessity.” These claims do not require electronic resubmission, and instead require submission of documentation to the Medicare appeals department. Once the supporting documentation is reviewed, reimbursement is issued.
Supportive Documentation
There are times when payor requests for additional information or documentation is handled in a timely fashion. However, the paper submission might have been incomplete, as the encounter note itself might not contain the cumulative information representing the reported service.
For example, other pieces of pertinent information may be obtained from the data or order section of the chart. If the individual responsible for gathering the requested documentation does not review it before submission, important or referenced entries may be missed, and the complexity of the billed service might be sacrificed. The provider should submit any entry with the same date as the requested documentation in support: labs, diagnostic testing, physician orders, patient instructions, nursing notes, resident notes, notes by other physicians in the same group, discharge summaries, etc.
Legibility of the encounter note is crucial when the documentation is sent for review. Most reviewers will seek another reviewer’s assistance in translating, but they are not obligated to do this. If the note is deemed incomprehensible, the service is denied, resulting in a nonpayment or a refund. Electronic medical records (EMRs) are assisting physicians and other providers with legibility issues and improving review findings. If a physician is still writing notes by hand, a transcription might be sent along with the documentation to prevent unnecessary denials. Only consider this for requests involving providers with problematic handwriting. A legible signature is required. If a denial ensues in absence of a signature, the provider can submit an appeal with an acceptable attestation.
Modifier Considerations
Some services are denied for being “incidental/integral” to another reimbursed service (i.e. bundled). Payors implement electronic payment edits that disallow separate payment for “related” services. The industry standard, known as the National Correct Coding Initiative (NCCI), identifies code pairs that should not be reported together on the same date by either a single physician or physicians of the same specialty within a provider group.
When a claim is denied for this reason, billers tend to automatically and erroneously resubmit the claim with a modifier appended to the disallowed or “bundled” procedure code. Documentation should be reviewed to determine if the denied service is separately reportable from the paid service. The biller might append the appropriate modifier and resubmit the claim only when well supported by documentation.
For example, the hospitalist evaluated a patient with congestive heart failure and pleural effusions. The hospitalist determined that the patient requires placement of a central venous catheter (36556). Because the patient’s underlying condition was evaluated, and resulted in the decision to place a catheter, both the visit (99233) and the procedure (36556) can be reported. If submitted without modifiers, some payors will deny payment for the visit for being integral to the catheter placement. In this case, the claim should be resubmitted with modifier 25 appended to the visit. Payors might still require documentation review to ensure legitimacy of this modifier before the claim is paid. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
Reference
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: AMA Press; 2011.
Before submitting a claim, hospitalists should ensure that the service is rendered, that it is completely and accurately documented in the medical record, that the correct information is entered on the claim form, and that it is a covered benefit and eligible for payment.
Although the latter two elements typically are delegated to the billing team, hospitalists should encourage or request feedback regarding payment and denials. The ensuing open dialogue between physicians and billers might prove helpful in understanding and resolving future billing issues. Less-experienced billers first respond to claim denials by submitting documentation (i.e. “appeal with paper”) despite the inappropriateness of this action. If the denial is upheld, this attempt is viewed as unsuccessful and, without further consideration, “written off.” However, careful examination of the payor’s initial claim determination could elicit a more suitable response.
Service Provider
Provider enrollment issues can sidetrack claim submissions. Physicians must register their NPI (national provider identifier) with the correct practice location and group assignment, particularly when previously practicing physicians join a new group practice. Failure to do so is an infrequent, yet valid, cause for denial.
Alternatively, enrollment issues play a greater role when services involve nurse practitioners (NPs) and physician assistants (PAs) who are enrolled with Medicare but might be prohibited from enrolling with other payors. For example, an NP independently provides subsequent hospital care (e.g. 99232) to a Medicare beneficiary. The claim is submitted in the NP’s name and reimbursed at the correct amount by Medicare as the primary insurer. The remaining balance is submitted to the secondary insurer, who does not enroll NPPs. The claim is rejected. If the physician group has a contractual agreement to recognize NPP services by reporting them under the collaborating physician’s name, the claim can be resubmitted in the physician’s name. In absence of such an agreement, the claim should be written off.
Location
The place of service (POS) must match the reported service/procedure code. For example, a hospitalist is asked to see a patient in the ED. The patient requires further testing but does not meet the criterion for an inpatient stay. The hospitalist admits the patient to observation, treats him, and discharges him to home.
Hospitalists need to avoid the common mistake of mismatching the service code with the location/POS. Observation services performed by the “physician of record” should be reported with the corresponding codes: initial observation care (99218-99220), subsequent observation care (99224-99226), or observation discharge (99217), as appropriate.1 The correct POS should be reported as outpatient hospital (POS 22), not inpatient hospital (POS 21). Trying to report outpatient codes with an inpatient POS will result in claim denial.
A similar denial occurs when trying to report inpatient codes (99231-99233) in an outpatient location (e.g. 23-ED). These denials require claim resubmission with the correct POS and/or service/procedure code. A complete list of POS codes and corresponding definitions can be obtained from Chapter 26, Section 10.5 of the Medicare Claims Processing Manual, available at www.cms.hhs.gov/manuals/downloads/clm104c26.pdf.
Diagnosis
Denials involving diagnoses produce issues of “medical necessity.”1 Examine these denials carefully. Consider the service/procedure code when trying to formulate a response to the denial. The diagnosis code represents the reason for the service or procedure and might be a sign, symptom, or condition with which the patient presents. Medicare reimburses for procedures and services that are deemed “reasonable and necessary.”
In an effort to unify standards, Medicare has developed national coverage determinations (NCDs) to identify coverage requirements for frequent or problematic procedures or services. These coverage requirements can identify specific conditions (i.e. ICD-9-CM codes) for which the services or procedures are considered medically necessary. In the absence of a national coverage policy, an item or service could be covered at the discretion of Medicare contractors based on a local coverage determination (LCD), which varies by contractor.
Medical necessity denials often involve a mismatched or missing diagnosis. For example, a payor might deny a claim for cardiopulmonary resuscitation (92950) that is associated with a diagnosis code of congestive heart failure (428.0), despite this being the underlying condition that prompted the decline in the patient’s condition. The payor might only accept “cardiac arrest” (427.5) as the “medically necessary” diagnosis for cardiopulmonary resuscitation, as this is the direct reason necessitating the procedure. After reviewing the documentation to ensure that the documentation supports the diagnosis, the claim can be resubmitted with a confirmed and corrected diagnosis code.
Initial-Request Response
While diagnoses can lead to medical necessity issues, not all medical necessity denials are due to incorrect diagnoses. Some “medical necessity” denials result from a failure to respond to a payor request. More specifically, if the “medical necessity” denial involves a covered evaluation and management visit, the denial is more likely the result of a failure to respond to a prepayment request for documentation.
Medicare typically issues prepayment requests for documentation for the following inpatient CPT codes: 99223, 99233, 99232, 99239, and 99292.1 If the documentation is not provided to the Medicare review department within a designated time frame (e.g. 30-45 days), the claim is automatically denied. The reason for denial is cited as being “not deemed a medical necessity.” These claims do not require electronic resubmission, and instead require submission of documentation to the Medicare appeals department. Once the supporting documentation is reviewed, reimbursement is issued.
Supportive Documentation
There are times when payor requests for additional information or documentation is handled in a timely fashion. However, the paper submission might have been incomplete, as the encounter note itself might not contain the cumulative information representing the reported service.
For example, other pieces of pertinent information may be obtained from the data or order section of the chart. If the individual responsible for gathering the requested documentation does not review it before submission, important or referenced entries may be missed, and the complexity of the billed service might be sacrificed. The provider should submit any entry with the same date as the requested documentation in support: labs, diagnostic testing, physician orders, patient instructions, nursing notes, resident notes, notes by other physicians in the same group, discharge summaries, etc.
Legibility of the encounter note is crucial when the documentation is sent for review. Most reviewers will seek another reviewer’s assistance in translating, but they are not obligated to do this. If the note is deemed incomprehensible, the service is denied, resulting in a nonpayment or a refund. Electronic medical records (EMRs) are assisting physicians and other providers with legibility issues and improving review findings. If a physician is still writing notes by hand, a transcription might be sent along with the documentation to prevent unnecessary denials. Only consider this for requests involving providers with problematic handwriting. A legible signature is required. If a denial ensues in absence of a signature, the provider can submit an appeal with an acceptable attestation.
Modifier Considerations
Some services are denied for being “incidental/integral” to another reimbursed service (i.e. bundled). Payors implement electronic payment edits that disallow separate payment for “related” services. The industry standard, known as the National Correct Coding Initiative (NCCI), identifies code pairs that should not be reported together on the same date by either a single physician or physicians of the same specialty within a provider group.
When a claim is denied for this reason, billers tend to automatically and erroneously resubmit the claim with a modifier appended to the disallowed or “bundled” procedure code. Documentation should be reviewed to determine if the denied service is separately reportable from the paid service. The biller might append the appropriate modifier and resubmit the claim only when well supported by documentation.
For example, the hospitalist evaluated a patient with congestive heart failure and pleural effusions. The hospitalist determined that the patient requires placement of a central venous catheter (36556). Because the patient’s underlying condition was evaluated, and resulted in the decision to place a catheter, both the visit (99233) and the procedure (36556) can be reported. If submitted without modifiers, some payors will deny payment for the visit for being integral to the catheter placement. In this case, the claim should be resubmitted with modifier 25 appended to the visit. Payors might still require documentation review to ensure legitimacy of this modifier before the claim is paid. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
Reference
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: AMA Press; 2011.
Before submitting a claim, hospitalists should ensure that the service is rendered, that it is completely and accurately documented in the medical record, that the correct information is entered on the claim form, and that it is a covered benefit and eligible for payment.
Although the latter two elements typically are delegated to the billing team, hospitalists should encourage or request feedback regarding payment and denials. The ensuing open dialogue between physicians and billers might prove helpful in understanding and resolving future billing issues. Less-experienced billers first respond to claim denials by submitting documentation (i.e. “appeal with paper”) despite the inappropriateness of this action. If the denial is upheld, this attempt is viewed as unsuccessful and, without further consideration, “written off.” However, careful examination of the payor’s initial claim determination could elicit a more suitable response.
Service Provider
Provider enrollment issues can sidetrack claim submissions. Physicians must register their NPI (national provider identifier) with the correct practice location and group assignment, particularly when previously practicing physicians join a new group practice. Failure to do so is an infrequent, yet valid, cause for denial.
Alternatively, enrollment issues play a greater role when services involve nurse practitioners (NPs) and physician assistants (PAs) who are enrolled with Medicare but might be prohibited from enrolling with other payors. For example, an NP independently provides subsequent hospital care (e.g. 99232) to a Medicare beneficiary. The claim is submitted in the NP’s name and reimbursed at the correct amount by Medicare as the primary insurer. The remaining balance is submitted to the secondary insurer, who does not enroll NPPs. The claim is rejected. If the physician group has a contractual agreement to recognize NPP services by reporting them under the collaborating physician’s name, the claim can be resubmitted in the physician’s name. In absence of such an agreement, the claim should be written off.
Location
The place of service (POS) must match the reported service/procedure code. For example, a hospitalist is asked to see a patient in the ED. The patient requires further testing but does not meet the criterion for an inpatient stay. The hospitalist admits the patient to observation, treats him, and discharges him to home.
Hospitalists need to avoid the common mistake of mismatching the service code with the location/POS. Observation services performed by the “physician of record” should be reported with the corresponding codes: initial observation care (99218-99220), subsequent observation care (99224-99226), or observation discharge (99217), as appropriate.1 The correct POS should be reported as outpatient hospital (POS 22), not inpatient hospital (POS 21). Trying to report outpatient codes with an inpatient POS will result in claim denial.
A similar denial occurs when trying to report inpatient codes (99231-99233) in an outpatient location (e.g. 23-ED). These denials require claim resubmission with the correct POS and/or service/procedure code. A complete list of POS codes and corresponding definitions can be obtained from Chapter 26, Section 10.5 of the Medicare Claims Processing Manual, available at www.cms.hhs.gov/manuals/downloads/clm104c26.pdf.
Diagnosis
Denials involving diagnoses produce issues of “medical necessity.”1 Examine these denials carefully. Consider the service/procedure code when trying to formulate a response to the denial. The diagnosis code represents the reason for the service or procedure and might be a sign, symptom, or condition with which the patient presents. Medicare reimburses for procedures and services that are deemed “reasonable and necessary.”
In an effort to unify standards, Medicare has developed national coverage determinations (NCDs) to identify coverage requirements for frequent or problematic procedures or services. These coverage requirements can identify specific conditions (i.e. ICD-9-CM codes) for which the services or procedures are considered medically necessary. In the absence of a national coverage policy, an item or service could be covered at the discretion of Medicare contractors based on a local coverage determination (LCD), which varies by contractor.
Medical necessity denials often involve a mismatched or missing diagnosis. For example, a payor might deny a claim for cardiopulmonary resuscitation (92950) that is associated with a diagnosis code of congestive heart failure (428.0), despite this being the underlying condition that prompted the decline in the patient’s condition. The payor might only accept “cardiac arrest” (427.5) as the “medically necessary” diagnosis for cardiopulmonary resuscitation, as this is the direct reason necessitating the procedure. After reviewing the documentation to ensure that the documentation supports the diagnosis, the claim can be resubmitted with a confirmed and corrected diagnosis code.
Initial-Request Response
While diagnoses can lead to medical necessity issues, not all medical necessity denials are due to incorrect diagnoses. Some “medical necessity” denials result from a failure to respond to a payor request. More specifically, if the “medical necessity” denial involves a covered evaluation and management visit, the denial is more likely the result of a failure to respond to a prepayment request for documentation.
Medicare typically issues prepayment requests for documentation for the following inpatient CPT codes: 99223, 99233, 99232, 99239, and 99292.1 If the documentation is not provided to the Medicare review department within a designated time frame (e.g. 30-45 days), the claim is automatically denied. The reason for denial is cited as being “not deemed a medical necessity.” These claims do not require electronic resubmission, and instead require submission of documentation to the Medicare appeals department. Once the supporting documentation is reviewed, reimbursement is issued.
Supportive Documentation
There are times when payor requests for additional information or documentation is handled in a timely fashion. However, the paper submission might have been incomplete, as the encounter note itself might not contain the cumulative information representing the reported service.
For example, other pieces of pertinent information may be obtained from the data or order section of the chart. If the individual responsible for gathering the requested documentation does not review it before submission, important or referenced entries may be missed, and the complexity of the billed service might be sacrificed. The provider should submit any entry with the same date as the requested documentation in support: labs, diagnostic testing, physician orders, patient instructions, nursing notes, resident notes, notes by other physicians in the same group, discharge summaries, etc.
Legibility of the encounter note is crucial when the documentation is sent for review. Most reviewers will seek another reviewer’s assistance in translating, but they are not obligated to do this. If the note is deemed incomprehensible, the service is denied, resulting in a nonpayment or a refund. Electronic medical records (EMRs) are assisting physicians and other providers with legibility issues and improving review findings. If a physician is still writing notes by hand, a transcription might be sent along with the documentation to prevent unnecessary denials. Only consider this for requests involving providers with problematic handwriting. A legible signature is required. If a denial ensues in absence of a signature, the provider can submit an appeal with an acceptable attestation.
Modifier Considerations
Some services are denied for being “incidental/integral” to another reimbursed service (i.e. bundled). Payors implement electronic payment edits that disallow separate payment for “related” services. The industry standard, known as the National Correct Coding Initiative (NCCI), identifies code pairs that should not be reported together on the same date by either a single physician or physicians of the same specialty within a provider group.
When a claim is denied for this reason, billers tend to automatically and erroneously resubmit the claim with a modifier appended to the disallowed or “bundled” procedure code. Documentation should be reviewed to determine if the denied service is separately reportable from the paid service. The biller might append the appropriate modifier and resubmit the claim only when well supported by documentation.
For example, the hospitalist evaluated a patient with congestive heart failure and pleural effusions. The hospitalist determined that the patient requires placement of a central venous catheter (36556). Because the patient’s underlying condition was evaluated, and resulted in the decision to place a catheter, both the visit (99233) and the procedure (36556) can be reported. If submitted without modifiers, some payors will deny payment for the visit for being integral to the catheter placement. In this case, the claim should be resubmitted with modifier 25 appended to the visit. Payors might still require documentation review to ensure legitimacy of this modifier before the claim is paid. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
Reference
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: AMA Press; 2011.
Accurate Measures?
Everyone’s talking about quality. Encouraging high-value care is one of the stated objectives of the value-based purchasing program being rolled out by the Centers for Medicare & Medicaid Services (CMS). It’s also the subject of a new report to Congress from the Department of Health and Human Services (HHS), “National Strategy for Quality Improvement in Health Care” (www.healthcare.gov/center/reports/quality03212011a.html). For its part, SHM is placing added emphasis on a range of mentored quality-improvement (QI) initiatives for hospitalists.
Amid the flurry of activity, researchers are still attempting to address a central question that could determine the success or failure of many such efforts: How do you accurately measure what constitutes high-quality care?
Chris Murray, MD, DPhil, director of the Seattle-based Institute for Health Metrics and Evaluation, says the healthcare field traditionally has tried to assess quality in three main ways. One is to ask patients about their own experience: Were they satisfied with the level of care they received? Another is to assess what are known as process of care measures: Did the providers follow guidelines in providing patients with appropriate care? The third is to look at risk-adjusted outcomes: How did the patients ultimately fare?
Focused on Facts
CMS’s value-based purchasing program, at least initially, is focusing on the first two types of metrics. Process measures, Dr. Murray says, are popular in part because they’re relatively easy to gauge. For many of them, however, “the connection to improved health is a bit weak,” he says. Whether heart patients get a prescription for a beta-blocker drug, for example, doesn’t address the outcome. “The problem there is that we don’t know if they ever filled the prescription or if the patient takes the beta-blocker,” Dr. Murray says.
That uncertainty feeds into the larger question of how broadly to consider the accountability of providers when measuring quality. “Should we be thinking that quality means putting in place the supports required for a patient to actually achieve a good outcome, or just offering them?” Dr. Murray asks. The debate might be far from settled, but a growing number of tools and studies are at least helping researchers to connect the dots on how care is delivered, on what kind of practices might affect outcomes the most, and how a community’s underlying risks could influence both considerations.
A recent Annals of Internal Medicine study that scrutinized 30-day mortality rates for heart-attack patients found few quantitative differences between the top 5% and bottom 5% of hospitals, based on rates published on the CMS Hospital Compare website.1 Site visits and in-depth interviews with nearly 160 medical staff members, however, uncovered some telling distinctions.
The study found that following evidence-based protocols and processes, while important, likely is not sufficient to attain a high performance level in caring for heart-attack patients. Instead, “high-performing hospitals were characterized by an organizational culture that supported efforts to improve AMI [acute myocardial infarction] care across the hospital.” In other words, everyone from management to the medical staff was fully invested in QI efforts. Notably, the staff “reported the presence of physician champions and empowered nursing staff, pharmacist involvement in patient care, and high qualification standards for all staff.”
For its 13th annual HealthGrades Quality in America study, the Denver-based ratings organization HealthGrades tried to look more quantitatively at the link between top hospitals and patient outcomes. Its study coauthors culled data from roughly 40 million Medicare discharges from 2007 through 2009 for most of the nation’s 5,000 hospitals, and assigned ratings based on 26 measures related to mortality and complication rates (www.healthgrades.com/business/news/press-releases/hospital-quality-2010.aspx).
If all hospitals were performing on par with what HealthGrades terms a five-star hospital, the study suggests the U.S. healthcare system could have saved the lives of more than 230,000 Medicare beneficiaries over the three-year period. More than half of the preventable deaths were associated with sepsis, pneumonia, respiratory failure, and heart failure.
Although the high number raises the question of whether some preventable deaths might exist only on paper, the study does raise other eye-popping calculations. Typical patients who went to a five-star hospital instead of a one-star hospital had a 72% lower risk of dying and reduced their risk by 53% compared with U.S. hospitals overall. The survival advantage persisted after hospitalization, too: Patients discharged from five-star-rated hospitals were 57% less likely to die within 30 days than all patients.
Ali Mokdad, PhD, professor of global health at the Institute for Health Metrics and Evaluation, says one big caveat to such rankings is the matter of adjusted risk. What kind of patient populations are these hospitals treating? Are people in the area inherently less healthy? Are significant barriers to healthcare blocking access to preventive medicine?
Dr. Murray says measuring quality with risk-adjusted outcomes has periodically fallen in and out of favor, due in part to concerns over how the risk is calculated and whether the assessments could be biased against providers that see more difficult patients. Nonetheless, he believes the metric is underused in the U.S. “I think the pendulum went way away from risk-adjusted outcomes to process measures too much, and we need to have a mixed combination,” he says.
With improvements to the methodology, he sees a wealth of potential in picking out risk predictors from large data sets. “The world is getting better at predicting rehospitalization, predicting death from attributes of the patient,” he says. “If you can do a better job at risk adjustment, you can do a better job on identifying quality.”
Risk Adjustment
One area in which the U.S. has lagged is in integrating the risk of death due to chronic conditions into broader measures of healthcare. At the recent Global Health Metrics & Evaluation Conference in Seattle, Dr. Mokdad pointed out the stringent oversight applied to commercial airliners. An avoidable crash and loss of life would quickly lead to a full-scale investigation. Why, he wondered, can’t the same scrutiny be brought to bear on preventable deaths due to chronic conditions such as diabetes and heart disease?
An ambitious new surveillance project, in fact, is trying to do exactly that. Known as the Monitoring Disparities in Chronic Conditions (MDCC) Study, the effort will use Washington state’s King County as a test case to hone the necessary data collection techniques. If it pans out, the study could become a national model for how to assess a population’s health status. “You know how a physician takes your pulse?” Dr. Mokdad says. “We’re doing that for the community.”
The research team, which includes Dr. Mokdad, Dr. Murray, and collaborators from Dartmouth and Harvard universities, will administer in-depth, culturally sensitive surveys to more than 3,000 county residents. A subset of 750 participants also will receive physical exams that measure markers of health and activity.
One goal is to work out how to efficiently integrate data from multiple sources so researchers can apply their risk adjustment strategies. For example, can they get enough information to ask how many heart attack patients are on beta-blockers one year after a hospital discharge? “There is also this big question of community background health risk,” Dr. Murray says. “Is this a community where people are just sicker, and how do you factor that in addition to taking into account the comorbidities that individuals have when they show up in the hospital?”
Researchers are close to obtaining enough information on such key factors as blood pressure, cholesterol, tobacco use, and obesity to actually rate communities according to risk, he says.2 “That’s never been done at the local level, and I think it’s where we need to go to truly put things on a level playing field when you’re assessing quality.” TH
Bryn Nelson is a freelance medical writer based in Seattle.
References
- Curry LA, Spatz E, Cherlin E, et al. What distinguishes top-performing hospitals in acute myocardial infarction mortality rates? Ann Intern Med. 2001;154(6):384-390.
- Murray CJ, Kulkarni SC, Michaud C, et al. Eight Americas: investigating mortality disparities across races, counties, and race-counties in the United States. PLoS Med. 2006;3(9):e260.
Everyone’s talking about quality. Encouraging high-value care is one of the stated objectives of the value-based purchasing program being rolled out by the Centers for Medicare & Medicaid Services (CMS). It’s also the subject of a new report to Congress from the Department of Health and Human Services (HHS), “National Strategy for Quality Improvement in Health Care” (www.healthcare.gov/center/reports/quality03212011a.html). For its part, SHM is placing added emphasis on a range of mentored quality-improvement (QI) initiatives for hospitalists.
Amid the flurry of activity, researchers are still attempting to address a central question that could determine the success or failure of many such efforts: How do you accurately measure what constitutes high-quality care?
Chris Murray, MD, DPhil, director of the Seattle-based Institute for Health Metrics and Evaluation, says the healthcare field traditionally has tried to assess quality in three main ways. One is to ask patients about their own experience: Were they satisfied with the level of care they received? Another is to assess what are known as process of care measures: Did the providers follow guidelines in providing patients with appropriate care? The third is to look at risk-adjusted outcomes: How did the patients ultimately fare?
Focused on Facts
CMS’s value-based purchasing program, at least initially, is focusing on the first two types of metrics. Process measures, Dr. Murray says, are popular in part because they’re relatively easy to gauge. For many of them, however, “the connection to improved health is a bit weak,” he says. Whether heart patients get a prescription for a beta-blocker drug, for example, doesn’t address the outcome. “The problem there is that we don’t know if they ever filled the prescription or if the patient takes the beta-blocker,” Dr. Murray says.
That uncertainty feeds into the larger question of how broadly to consider the accountability of providers when measuring quality. “Should we be thinking that quality means putting in place the supports required for a patient to actually achieve a good outcome, or just offering them?” Dr. Murray asks. The debate might be far from settled, but a growing number of tools and studies are at least helping researchers to connect the dots on how care is delivered, on what kind of practices might affect outcomes the most, and how a community’s underlying risks could influence both considerations.
A recent Annals of Internal Medicine study that scrutinized 30-day mortality rates for heart-attack patients found few quantitative differences between the top 5% and bottom 5% of hospitals, based on rates published on the CMS Hospital Compare website.1 Site visits and in-depth interviews with nearly 160 medical staff members, however, uncovered some telling distinctions.
The study found that following evidence-based protocols and processes, while important, likely is not sufficient to attain a high performance level in caring for heart-attack patients. Instead, “high-performing hospitals were characterized by an organizational culture that supported efforts to improve AMI [acute myocardial infarction] care across the hospital.” In other words, everyone from management to the medical staff was fully invested in QI efforts. Notably, the staff “reported the presence of physician champions and empowered nursing staff, pharmacist involvement in patient care, and high qualification standards for all staff.”
For its 13th annual HealthGrades Quality in America study, the Denver-based ratings organization HealthGrades tried to look more quantitatively at the link between top hospitals and patient outcomes. Its study coauthors culled data from roughly 40 million Medicare discharges from 2007 through 2009 for most of the nation’s 5,000 hospitals, and assigned ratings based on 26 measures related to mortality and complication rates (www.healthgrades.com/business/news/press-releases/hospital-quality-2010.aspx).
If all hospitals were performing on par with what HealthGrades terms a five-star hospital, the study suggests the U.S. healthcare system could have saved the lives of more than 230,000 Medicare beneficiaries over the three-year period. More than half of the preventable deaths were associated with sepsis, pneumonia, respiratory failure, and heart failure.
Although the high number raises the question of whether some preventable deaths might exist only on paper, the study does raise other eye-popping calculations. Typical patients who went to a five-star hospital instead of a one-star hospital had a 72% lower risk of dying and reduced their risk by 53% compared with U.S. hospitals overall. The survival advantage persisted after hospitalization, too: Patients discharged from five-star-rated hospitals were 57% less likely to die within 30 days than all patients.
Ali Mokdad, PhD, professor of global health at the Institute for Health Metrics and Evaluation, says one big caveat to such rankings is the matter of adjusted risk. What kind of patient populations are these hospitals treating? Are people in the area inherently less healthy? Are significant barriers to healthcare blocking access to preventive medicine?
Dr. Murray says measuring quality with risk-adjusted outcomes has periodically fallen in and out of favor, due in part to concerns over how the risk is calculated and whether the assessments could be biased against providers that see more difficult patients. Nonetheless, he believes the metric is underused in the U.S. “I think the pendulum went way away from risk-adjusted outcomes to process measures too much, and we need to have a mixed combination,” he says.
With improvements to the methodology, he sees a wealth of potential in picking out risk predictors from large data sets. “The world is getting better at predicting rehospitalization, predicting death from attributes of the patient,” he says. “If you can do a better job at risk adjustment, you can do a better job on identifying quality.”
Risk Adjustment
One area in which the U.S. has lagged is in integrating the risk of death due to chronic conditions into broader measures of healthcare. At the recent Global Health Metrics & Evaluation Conference in Seattle, Dr. Mokdad pointed out the stringent oversight applied to commercial airliners. An avoidable crash and loss of life would quickly lead to a full-scale investigation. Why, he wondered, can’t the same scrutiny be brought to bear on preventable deaths due to chronic conditions such as diabetes and heart disease?
An ambitious new surveillance project, in fact, is trying to do exactly that. Known as the Monitoring Disparities in Chronic Conditions (MDCC) Study, the effort will use Washington state’s King County as a test case to hone the necessary data collection techniques. If it pans out, the study could become a national model for how to assess a population’s health status. “You know how a physician takes your pulse?” Dr. Mokdad says. “We’re doing that for the community.”
The research team, which includes Dr. Mokdad, Dr. Murray, and collaborators from Dartmouth and Harvard universities, will administer in-depth, culturally sensitive surveys to more than 3,000 county residents. A subset of 750 participants also will receive physical exams that measure markers of health and activity.
One goal is to work out how to efficiently integrate data from multiple sources so researchers can apply their risk adjustment strategies. For example, can they get enough information to ask how many heart attack patients are on beta-blockers one year after a hospital discharge? “There is also this big question of community background health risk,” Dr. Murray says. “Is this a community where people are just sicker, and how do you factor that in addition to taking into account the comorbidities that individuals have when they show up in the hospital?”
Researchers are close to obtaining enough information on such key factors as blood pressure, cholesterol, tobacco use, and obesity to actually rate communities according to risk, he says.2 “That’s never been done at the local level, and I think it’s where we need to go to truly put things on a level playing field when you’re assessing quality.” TH
Bryn Nelson is a freelance medical writer based in Seattle.
References
- Curry LA, Spatz E, Cherlin E, et al. What distinguishes top-performing hospitals in acute myocardial infarction mortality rates? Ann Intern Med. 2001;154(6):384-390.
- Murray CJ, Kulkarni SC, Michaud C, et al. Eight Americas: investigating mortality disparities across races, counties, and race-counties in the United States. PLoS Med. 2006;3(9):e260.
Everyone’s talking about quality. Encouraging high-value care is one of the stated objectives of the value-based purchasing program being rolled out by the Centers for Medicare & Medicaid Services (CMS). It’s also the subject of a new report to Congress from the Department of Health and Human Services (HHS), “National Strategy for Quality Improvement in Health Care” (www.healthcare.gov/center/reports/quality03212011a.html). For its part, SHM is placing added emphasis on a range of mentored quality-improvement (QI) initiatives for hospitalists.
Amid the flurry of activity, researchers are still attempting to address a central question that could determine the success or failure of many such efforts: How do you accurately measure what constitutes high-quality care?
Chris Murray, MD, DPhil, director of the Seattle-based Institute for Health Metrics and Evaluation, says the healthcare field traditionally has tried to assess quality in three main ways. One is to ask patients about their own experience: Were they satisfied with the level of care they received? Another is to assess what are known as process of care measures: Did the providers follow guidelines in providing patients with appropriate care? The third is to look at risk-adjusted outcomes: How did the patients ultimately fare?
Focused on Facts
CMS’s value-based purchasing program, at least initially, is focusing on the first two types of metrics. Process measures, Dr. Murray says, are popular in part because they’re relatively easy to gauge. For many of them, however, “the connection to improved health is a bit weak,” he says. Whether heart patients get a prescription for a beta-blocker drug, for example, doesn’t address the outcome. “The problem there is that we don’t know if they ever filled the prescription or if the patient takes the beta-blocker,” Dr. Murray says.
That uncertainty feeds into the larger question of how broadly to consider the accountability of providers when measuring quality. “Should we be thinking that quality means putting in place the supports required for a patient to actually achieve a good outcome, or just offering them?” Dr. Murray asks. The debate might be far from settled, but a growing number of tools and studies are at least helping researchers to connect the dots on how care is delivered, on what kind of practices might affect outcomes the most, and how a community’s underlying risks could influence both considerations.
A recent Annals of Internal Medicine study that scrutinized 30-day mortality rates for heart-attack patients found few quantitative differences between the top 5% and bottom 5% of hospitals, based on rates published on the CMS Hospital Compare website.1 Site visits and in-depth interviews with nearly 160 medical staff members, however, uncovered some telling distinctions.
The study found that following evidence-based protocols and processes, while important, likely is not sufficient to attain a high performance level in caring for heart-attack patients. Instead, “high-performing hospitals were characterized by an organizational culture that supported efforts to improve AMI [acute myocardial infarction] care across the hospital.” In other words, everyone from management to the medical staff was fully invested in QI efforts. Notably, the staff “reported the presence of physician champions and empowered nursing staff, pharmacist involvement in patient care, and high qualification standards for all staff.”
For its 13th annual HealthGrades Quality in America study, the Denver-based ratings organization HealthGrades tried to look more quantitatively at the link between top hospitals and patient outcomes. Its study coauthors culled data from roughly 40 million Medicare discharges from 2007 through 2009 for most of the nation’s 5,000 hospitals, and assigned ratings based on 26 measures related to mortality and complication rates (www.healthgrades.com/business/news/press-releases/hospital-quality-2010.aspx).
If all hospitals were performing on par with what HealthGrades terms a five-star hospital, the study suggests the U.S. healthcare system could have saved the lives of more than 230,000 Medicare beneficiaries over the three-year period. More than half of the preventable deaths were associated with sepsis, pneumonia, respiratory failure, and heart failure.
Although the high number raises the question of whether some preventable deaths might exist only on paper, the study does raise other eye-popping calculations. Typical patients who went to a five-star hospital instead of a one-star hospital had a 72% lower risk of dying and reduced their risk by 53% compared with U.S. hospitals overall. The survival advantage persisted after hospitalization, too: Patients discharged from five-star-rated hospitals were 57% less likely to die within 30 days than all patients.
Ali Mokdad, PhD, professor of global health at the Institute for Health Metrics and Evaluation, says one big caveat to such rankings is the matter of adjusted risk. What kind of patient populations are these hospitals treating? Are people in the area inherently less healthy? Are significant barriers to healthcare blocking access to preventive medicine?
Dr. Murray says measuring quality with risk-adjusted outcomes has periodically fallen in and out of favor, due in part to concerns over how the risk is calculated and whether the assessments could be biased against providers that see more difficult patients. Nonetheless, he believes the metric is underused in the U.S. “I think the pendulum went way away from risk-adjusted outcomes to process measures too much, and we need to have a mixed combination,” he says.
With improvements to the methodology, he sees a wealth of potential in picking out risk predictors from large data sets. “The world is getting better at predicting rehospitalization, predicting death from attributes of the patient,” he says. “If you can do a better job at risk adjustment, you can do a better job on identifying quality.”
Risk Adjustment
One area in which the U.S. has lagged is in integrating the risk of death due to chronic conditions into broader measures of healthcare. At the recent Global Health Metrics & Evaluation Conference in Seattle, Dr. Mokdad pointed out the stringent oversight applied to commercial airliners. An avoidable crash and loss of life would quickly lead to a full-scale investigation. Why, he wondered, can’t the same scrutiny be brought to bear on preventable deaths due to chronic conditions such as diabetes and heart disease?
An ambitious new surveillance project, in fact, is trying to do exactly that. Known as the Monitoring Disparities in Chronic Conditions (MDCC) Study, the effort will use Washington state’s King County as a test case to hone the necessary data collection techniques. If it pans out, the study could become a national model for how to assess a population’s health status. “You know how a physician takes your pulse?” Dr. Mokdad says. “We’re doing that for the community.”
The research team, which includes Dr. Mokdad, Dr. Murray, and collaborators from Dartmouth and Harvard universities, will administer in-depth, culturally sensitive surveys to more than 3,000 county residents. A subset of 750 participants also will receive physical exams that measure markers of health and activity.
One goal is to work out how to efficiently integrate data from multiple sources so researchers can apply their risk adjustment strategies. For example, can they get enough information to ask how many heart attack patients are on beta-blockers one year after a hospital discharge? “There is also this big question of community background health risk,” Dr. Murray says. “Is this a community where people are just sicker, and how do you factor that in addition to taking into account the comorbidities that individuals have when they show up in the hospital?”
Researchers are close to obtaining enough information on such key factors as blood pressure, cholesterol, tobacco use, and obesity to actually rate communities according to risk, he says.2 “That’s never been done at the local level, and I think it’s where we need to go to truly put things on a level playing field when you’re assessing quality.” TH
Bryn Nelson is a freelance medical writer based in Seattle.
References
- Curry LA, Spatz E, Cherlin E, et al. What distinguishes top-performing hospitals in acute myocardial infarction mortality rates? Ann Intern Med. 2001;154(6):384-390.
- Murray CJ, Kulkarni SC, Michaud C, et al. Eight Americas: investigating mortality disparities across races, counties, and race-counties in the United States. PLoS Med. 2006;3(9):e260.
Multiple Choices
Seven on, seven off. That’s what you can expect as a hospitalist, right? Maybe. As you consider an HM career, your first thought should be just that—is this a career or a job? Is this a year between residency and fellowship, clinical shifts that allow you to have 26 weeks off every year, or is it something else?
Either way, HM offers abundant opportunities conducive to work-life balance and career satisfaction. HM careers reside in clinical, academic, and administrative settings.
Clinical
As a physician entering the workplace, clinical practice often is the most familiar, but it does not have to be an extension of your residency floor rotation. Your individual schedule and employer will play a role in determining when you provide clinical service. Some employers, whether hospital-based, a private physician practice, or a national hospitalist company, utilize fixed schedules, which might include night shifts. The popular seven-on, seven-off model allows you to provide direct patient care every other week, creating personal or administrative time in between. You can opt to become a nocturnist and limit yourself to clinical service at night. Some hospital-based programs implement individualized schedules to meet the nonclinical demands of academic or administrative hospitalists. These schedules might combine Monday-through-Friday weeks, weekends, and nights to create flexibility within your group.
Clinical service can be carved out to fit both your interests and the setting in which you provide care. If placing central lines is your thing, a career as a proceduralist might be for you. Other hospitalists find themselves in clinical niches in specialty collaboration and specific care settings, including surgical comanagement, intensive care, emergency, clinical decision or observation units, or pre-admission and post-hospitalization clinics.
Hospitalists can improve quality, patient safety, and efficiency when working in specialized areas like a clinical decision or observation unit. In these settings, hospitalists often collaborate with midlevel providers, such as nurse practitioners or physician assistants, to provide observation or outpatient care to patients with medical conditions that require more than an ED visit. For example, many patients who present to the ED with chest pain are ideal patients for these settings to evaluate their symptoms and provide an optimal care transition out of the hospital or to an inpatient unit, if needed.
Perhaps you enjoy patient care, but just not all the time involved: You might find a fulfilling career that blends clinical service with research, teaching, or administrative work.
Academic
As an academic hospitalist, you have various options. Hospitalists provide education and oversight to trainees, both medical students and residents, in academic medical centers and community teaching hospitals. You might join an academic center and receive a faculty appointment, either as clinical instructor or assistant professor for first-time candidates.
Clinician educators generally serve as internal-medicine-ward attendings, teaching inpatient care to house staff and students in a traditional sense. Studies have demonstrated that students and house staff are more satisfied and feel they learn more when their ward attending is a hospitalist.1 Academic hospitalists foster career development by serving as mentors or residency program directors. Academic hospitalists also educate fellow physicians through faculty development series and programs.
Hospitalists can have roles in academics as clinician-researchers, usually following formal research training. Hospitalist researchers focus on numerous areas, including basic science, specific disease states, and hospital outcomes. A focus on hospital outcomes allows clinician-researchers to link “evidence-based medicine with quality improvement by systematically studying hospital care. The outcomes are used to optimize healthcare delivery at the level of both the individual patient and the hospital.”2
Administrative
Hospitalists can pursue leadership opportunities in academic, hospital-based, or community-based settings. Administrative hospitalists develop and guide programs: hospitalist, hospital-based, and multidisciplinary. Hospitalist-leaders serve as program managers, division heads, and medical directors in operational leadership. Aside from running the day-to-day operations of physician groups and hospital units, hospitalists lead in other arenas, such as utilization management, QI and patient safety, medical informatics, and hospital operations.
When serving as physician advisor or utilization management director, it is an opportunity for the hospitalist to lead care coordination within an organization and identify where opportunities related to hospital utilization exist. Many hospitalists lead multidisciplinary hospital committees in QI and patient safety. Hospitalists are perfectly positioned to identify areas within patient care where existing practices need improvement. As quality leaders, hospitalists facilitate the process changes necessary to implement evidence-based care. Some of the hospitalist-led QI areas include care transitions (patients moving from one setting to another; for example, inpatient to outpatient), VTE prophylaxis, inpatient glycemic control, and reduction of hospital-associated conditions.
Directing or guiding medical informatics as health systems across the country implement electronic health records (EHR) is an area where hospitalists can impact both quality and efficiency of care. Medical informaticists can guide clinical-decision support systems within an EHR, easing evidence-based, disease-specific care for other clinicians. As EHR becomes more available, this opportunity for hospitalists will grow.
In addition to these areas, hospitalists can manage or direct a hospital’s patient flow or throughput. Considering that more EDs and hospitals are overcrowded, improving patient flow is an area where hospitalists can join or lead a hospital’s throughput initiative. Evidence has shown that hospitalist-driven active bed management can improve ED crowding and overall hospital flow.3
So now that you know there is more to an HM career than the seven-on, seven-off job that you get between residency and fellowship, determining how and where to find that just-right combination is up to you—with a little help from your local hospitalist mentor. TH
Dr. McAllister is assistant professor in the division of hospital medicine at Cooper University Hospital/UMDNJ-Robert Wood Johnson Medical School in Camden, N.J. Dr. Kupersmith is assistant professor of medicine, UMDNJ-Robert Wood Johnson Medical School, division head, hospital medicine, medical director throughput, Cooper Health System.
References
- Hauer KE, Wachter RM, McCulloch CE, Woo GA, Auerbach AD. Effects of hospitalist attending physicians on trainee satisfaction with teaching and with internal medicine rotations. Arch Intern Med. 2004;164(17):1866-1871.
- Career options. SHM website. Available at: www.hospitalmedicine.org/AM/Template.cfm?Section=Young_Physicians&Template=/CM/HTMLDisplay.cfm&ContentID=22474. Accessed Dec. 28, 2010.
- Howell E, Bessman E, Kravet S, Kolodner K, Marshall R, Wright S. Active bed management by hospitalists and emergency department throughput. Ann Intern Med. 2008;149(11):804-811.
Seven on, seven off. That’s what you can expect as a hospitalist, right? Maybe. As you consider an HM career, your first thought should be just that—is this a career or a job? Is this a year between residency and fellowship, clinical shifts that allow you to have 26 weeks off every year, or is it something else?
Either way, HM offers abundant opportunities conducive to work-life balance and career satisfaction. HM careers reside in clinical, academic, and administrative settings.
Clinical
As a physician entering the workplace, clinical practice often is the most familiar, but it does not have to be an extension of your residency floor rotation. Your individual schedule and employer will play a role in determining when you provide clinical service. Some employers, whether hospital-based, a private physician practice, or a national hospitalist company, utilize fixed schedules, which might include night shifts. The popular seven-on, seven-off model allows you to provide direct patient care every other week, creating personal or administrative time in between. You can opt to become a nocturnist and limit yourself to clinical service at night. Some hospital-based programs implement individualized schedules to meet the nonclinical demands of academic or administrative hospitalists. These schedules might combine Monday-through-Friday weeks, weekends, and nights to create flexibility within your group.
Clinical service can be carved out to fit both your interests and the setting in which you provide care. If placing central lines is your thing, a career as a proceduralist might be for you. Other hospitalists find themselves in clinical niches in specialty collaboration and specific care settings, including surgical comanagement, intensive care, emergency, clinical decision or observation units, or pre-admission and post-hospitalization clinics.
Hospitalists can improve quality, patient safety, and efficiency when working in specialized areas like a clinical decision or observation unit. In these settings, hospitalists often collaborate with midlevel providers, such as nurse practitioners or physician assistants, to provide observation or outpatient care to patients with medical conditions that require more than an ED visit. For example, many patients who present to the ED with chest pain are ideal patients for these settings to evaluate their symptoms and provide an optimal care transition out of the hospital or to an inpatient unit, if needed.
Perhaps you enjoy patient care, but just not all the time involved: You might find a fulfilling career that blends clinical service with research, teaching, or administrative work.
Academic
As an academic hospitalist, you have various options. Hospitalists provide education and oversight to trainees, both medical students and residents, in academic medical centers and community teaching hospitals. You might join an academic center and receive a faculty appointment, either as clinical instructor or assistant professor for first-time candidates.
Clinician educators generally serve as internal-medicine-ward attendings, teaching inpatient care to house staff and students in a traditional sense. Studies have demonstrated that students and house staff are more satisfied and feel they learn more when their ward attending is a hospitalist.1 Academic hospitalists foster career development by serving as mentors or residency program directors. Academic hospitalists also educate fellow physicians through faculty development series and programs.
Hospitalists can have roles in academics as clinician-researchers, usually following formal research training. Hospitalist researchers focus on numerous areas, including basic science, specific disease states, and hospital outcomes. A focus on hospital outcomes allows clinician-researchers to link “evidence-based medicine with quality improvement by systematically studying hospital care. The outcomes are used to optimize healthcare delivery at the level of both the individual patient and the hospital.”2
Administrative
Hospitalists can pursue leadership opportunities in academic, hospital-based, or community-based settings. Administrative hospitalists develop and guide programs: hospitalist, hospital-based, and multidisciplinary. Hospitalist-leaders serve as program managers, division heads, and medical directors in operational leadership. Aside from running the day-to-day operations of physician groups and hospital units, hospitalists lead in other arenas, such as utilization management, QI and patient safety, medical informatics, and hospital operations.
When serving as physician advisor or utilization management director, it is an opportunity for the hospitalist to lead care coordination within an organization and identify where opportunities related to hospital utilization exist. Many hospitalists lead multidisciplinary hospital committees in QI and patient safety. Hospitalists are perfectly positioned to identify areas within patient care where existing practices need improvement. As quality leaders, hospitalists facilitate the process changes necessary to implement evidence-based care. Some of the hospitalist-led QI areas include care transitions (patients moving from one setting to another; for example, inpatient to outpatient), VTE prophylaxis, inpatient glycemic control, and reduction of hospital-associated conditions.
Directing or guiding medical informatics as health systems across the country implement electronic health records (EHR) is an area where hospitalists can impact both quality and efficiency of care. Medical informaticists can guide clinical-decision support systems within an EHR, easing evidence-based, disease-specific care for other clinicians. As EHR becomes more available, this opportunity for hospitalists will grow.
In addition to these areas, hospitalists can manage or direct a hospital’s patient flow or throughput. Considering that more EDs and hospitals are overcrowded, improving patient flow is an area where hospitalists can join or lead a hospital’s throughput initiative. Evidence has shown that hospitalist-driven active bed management can improve ED crowding and overall hospital flow.3
So now that you know there is more to an HM career than the seven-on, seven-off job that you get between residency and fellowship, determining how and where to find that just-right combination is up to you—with a little help from your local hospitalist mentor. TH
Dr. McAllister is assistant professor in the division of hospital medicine at Cooper University Hospital/UMDNJ-Robert Wood Johnson Medical School in Camden, N.J. Dr. Kupersmith is assistant professor of medicine, UMDNJ-Robert Wood Johnson Medical School, division head, hospital medicine, medical director throughput, Cooper Health System.
References
- Hauer KE, Wachter RM, McCulloch CE, Woo GA, Auerbach AD. Effects of hospitalist attending physicians on trainee satisfaction with teaching and with internal medicine rotations. Arch Intern Med. 2004;164(17):1866-1871.
- Career options. SHM website. Available at: www.hospitalmedicine.org/AM/Template.cfm?Section=Young_Physicians&Template=/CM/HTMLDisplay.cfm&ContentID=22474. Accessed Dec. 28, 2010.
- Howell E, Bessman E, Kravet S, Kolodner K, Marshall R, Wright S. Active bed management by hospitalists and emergency department throughput. Ann Intern Med. 2008;149(11):804-811.
Seven on, seven off. That’s what you can expect as a hospitalist, right? Maybe. As you consider an HM career, your first thought should be just that—is this a career or a job? Is this a year between residency and fellowship, clinical shifts that allow you to have 26 weeks off every year, or is it something else?
Either way, HM offers abundant opportunities conducive to work-life balance and career satisfaction. HM careers reside in clinical, academic, and administrative settings.
Clinical
As a physician entering the workplace, clinical practice often is the most familiar, but it does not have to be an extension of your residency floor rotation. Your individual schedule and employer will play a role in determining when you provide clinical service. Some employers, whether hospital-based, a private physician practice, or a national hospitalist company, utilize fixed schedules, which might include night shifts. The popular seven-on, seven-off model allows you to provide direct patient care every other week, creating personal or administrative time in between. You can opt to become a nocturnist and limit yourself to clinical service at night. Some hospital-based programs implement individualized schedules to meet the nonclinical demands of academic or administrative hospitalists. These schedules might combine Monday-through-Friday weeks, weekends, and nights to create flexibility within your group.
Clinical service can be carved out to fit both your interests and the setting in which you provide care. If placing central lines is your thing, a career as a proceduralist might be for you. Other hospitalists find themselves in clinical niches in specialty collaboration and specific care settings, including surgical comanagement, intensive care, emergency, clinical decision or observation units, or pre-admission and post-hospitalization clinics.
Hospitalists can improve quality, patient safety, and efficiency when working in specialized areas like a clinical decision or observation unit. In these settings, hospitalists often collaborate with midlevel providers, such as nurse practitioners or physician assistants, to provide observation or outpatient care to patients with medical conditions that require more than an ED visit. For example, many patients who present to the ED with chest pain are ideal patients for these settings to evaluate their symptoms and provide an optimal care transition out of the hospital or to an inpatient unit, if needed.
Perhaps you enjoy patient care, but just not all the time involved: You might find a fulfilling career that blends clinical service with research, teaching, or administrative work.
Academic
As an academic hospitalist, you have various options. Hospitalists provide education and oversight to trainees, both medical students and residents, in academic medical centers and community teaching hospitals. You might join an academic center and receive a faculty appointment, either as clinical instructor or assistant professor for first-time candidates.
Clinician educators generally serve as internal-medicine-ward attendings, teaching inpatient care to house staff and students in a traditional sense. Studies have demonstrated that students and house staff are more satisfied and feel they learn more when their ward attending is a hospitalist.1 Academic hospitalists foster career development by serving as mentors or residency program directors. Academic hospitalists also educate fellow physicians through faculty development series and programs.
Hospitalists can have roles in academics as clinician-researchers, usually following formal research training. Hospitalist researchers focus on numerous areas, including basic science, specific disease states, and hospital outcomes. A focus on hospital outcomes allows clinician-researchers to link “evidence-based medicine with quality improvement by systematically studying hospital care. The outcomes are used to optimize healthcare delivery at the level of both the individual patient and the hospital.”2
Administrative
Hospitalists can pursue leadership opportunities in academic, hospital-based, or community-based settings. Administrative hospitalists develop and guide programs: hospitalist, hospital-based, and multidisciplinary. Hospitalist-leaders serve as program managers, division heads, and medical directors in operational leadership. Aside from running the day-to-day operations of physician groups and hospital units, hospitalists lead in other arenas, such as utilization management, QI and patient safety, medical informatics, and hospital operations.
When serving as physician advisor or utilization management director, it is an opportunity for the hospitalist to lead care coordination within an organization and identify where opportunities related to hospital utilization exist. Many hospitalists lead multidisciplinary hospital committees in QI and patient safety. Hospitalists are perfectly positioned to identify areas within patient care where existing practices need improvement. As quality leaders, hospitalists facilitate the process changes necessary to implement evidence-based care. Some of the hospitalist-led QI areas include care transitions (patients moving from one setting to another; for example, inpatient to outpatient), VTE prophylaxis, inpatient glycemic control, and reduction of hospital-associated conditions.
Directing or guiding medical informatics as health systems across the country implement electronic health records (EHR) is an area where hospitalists can impact both quality and efficiency of care. Medical informaticists can guide clinical-decision support systems within an EHR, easing evidence-based, disease-specific care for other clinicians. As EHR becomes more available, this opportunity for hospitalists will grow.
In addition to these areas, hospitalists can manage or direct a hospital’s patient flow or throughput. Considering that more EDs and hospitals are overcrowded, improving patient flow is an area where hospitalists can join or lead a hospital’s throughput initiative. Evidence has shown that hospitalist-driven active bed management can improve ED crowding and overall hospital flow.3
So now that you know there is more to an HM career than the seven-on, seven-off job that you get between residency and fellowship, determining how and where to find that just-right combination is up to you—with a little help from your local hospitalist mentor. TH
Dr. McAllister is assistant professor in the division of hospital medicine at Cooper University Hospital/UMDNJ-Robert Wood Johnson Medical School in Camden, N.J. Dr. Kupersmith is assistant professor of medicine, UMDNJ-Robert Wood Johnson Medical School, division head, hospital medicine, medical director throughput, Cooper Health System.
References
- Hauer KE, Wachter RM, McCulloch CE, Woo GA, Auerbach AD. Effects of hospitalist attending physicians on trainee satisfaction with teaching and with internal medicine rotations. Arch Intern Med. 2004;164(17):1866-1871.
- Career options. SHM website. Available at: www.hospitalmedicine.org/AM/Template.cfm?Section=Young_Physicians&Template=/CM/HTMLDisplay.cfm&ContentID=22474. Accessed Dec. 28, 2010.
- Howell E, Bessman E, Kravet S, Kolodner K, Marshall R, Wright S. Active bed management by hospitalists and emergency department throughput. Ann Intern Med. 2008;149(11):804-811.
CON: Should Hospitals Get Reimbursements Based on Quality Performance?

If the road to hell is paved with good intentions, then hell is full of unintended consequences.
The debate about whether hospitals’ reimbursements should be based on quality performance is not a unique concept. Similar systems have been implemented in other fields (e.g. education), and we in the medical field can learn from their experiences. In education, testing students is the driving force in measuring a “quality outcome.”
A growing number of educators now believe that the focus on testing to measure quality has actually reduced the quality of education; they cite the bureaucratic, inflexible, and cumbersome requirements placed on the educators, and the diversion of precious resources to focus on standardized test scores. The actual education of the students becomes secondary, and there are allegations of school systems manipulating their data to ensure maximum funding.
With the drive to pay-for-performance in the medical field, will the actual medical care of the patient become secondary to hitting the “quality” metrics set by the government? Add in a volatile mix of money, and this becomes a recipe for disaster.
Questions are many:
- What standards of quality are we going to use?
- Do these metrics truly translate into “quality”?
- Will the goal of reaching these metrics become the main focus of the hospitals instead of actual patient care?
- Is the goal to really improve the quality of healthcare, or is it just another vehicle for government and private third parties to come up with another excuse to reduce reimbursement in the name of quality?
Even now, ED physicians are giving antibiotics liberally for fear that they will be admonished for “missing” the pneumonia core measures. Whether this is appropriate care for the patient is irrelevant to hitting the statistical goal. Where is the incentive to deliver appropriate care? Are we even asking the right questions? Do bureaucrats know that even if appropriate, timely, quality care is given that a positive outcome is not guaranteed?
The field of medicine has made incredible advances in patient care, but the fact remains that a certain percentage of the sick and elderly become sicker and eventually die, especially in hospitals.
Potential Problems are Many
Unintended consequences pose a real danger to a system that rewards and penalizes. One potential issue is the “rich getting richer and the poor getting poorer,” as more provider focus is placed on buffering statistics by keeping healthy people healthier to achieve better outcomes, meanwhile shunning or diverting the seriously sick patients in order to keep quality metrics within goal.
How will the government and insurance providers guarantee the accuracy of each hospital’s statistics?
Think of the money and resources that will be diverted away from the clinical arena and into the bureaucratic nightmare of record-keeping needed to implement this pay-for-performance system. How many billions of dollars will be needed to fund this new bureaucracy? Do we need another bureaucratic, punitive layer in our already cumbersome medical system?
My answer is clear: No! TH
Dr. Yu is medical director of adult hospitalist services at Presbyterian Medical Group in Albuquerque, N.M., and a Team Hospitalist member.
If the road to hell is paved with good intentions, then hell is full of unintended consequences.
The debate about whether hospitals’ reimbursements should be based on quality performance is not a unique concept. Similar systems have been implemented in other fields (e.g. education), and we in the medical field can learn from their experiences. In education, testing students is the driving force in measuring a “quality outcome.”
A growing number of educators now believe that the focus on testing to measure quality has actually reduced the quality of education; they cite the bureaucratic, inflexible, and cumbersome requirements placed on the educators, and the diversion of precious resources to focus on standardized test scores. The actual education of the students becomes secondary, and there are allegations of school systems manipulating their data to ensure maximum funding.
With the drive to pay-for-performance in the medical field, will the actual medical care of the patient become secondary to hitting the “quality” metrics set by the government? Add in a volatile mix of money, and this becomes a recipe for disaster.
Questions are many:
- What standards of quality are we going to use?
- Do these metrics truly translate into “quality”?
- Will the goal of reaching these metrics become the main focus of the hospitals instead of actual patient care?
- Is the goal to really improve the quality of healthcare, or is it just another vehicle for government and private third parties to come up with another excuse to reduce reimbursement in the name of quality?
Even now, ED physicians are giving antibiotics liberally for fear that they will be admonished for “missing” the pneumonia core measures. Whether this is appropriate care for the patient is irrelevant to hitting the statistical goal. Where is the incentive to deliver appropriate care? Are we even asking the right questions? Do bureaucrats know that even if appropriate, timely, quality care is given that a positive outcome is not guaranteed?
The field of medicine has made incredible advances in patient care, but the fact remains that a certain percentage of the sick and elderly become sicker and eventually die, especially in hospitals.
Potential Problems are Many
Unintended consequences pose a real danger to a system that rewards and penalizes. One potential issue is the “rich getting richer and the poor getting poorer,” as more provider focus is placed on buffering statistics by keeping healthy people healthier to achieve better outcomes, meanwhile shunning or diverting the seriously sick patients in order to keep quality metrics within goal.
How will the government and insurance providers guarantee the accuracy of each hospital’s statistics?
Think of the money and resources that will be diverted away from the clinical arena and into the bureaucratic nightmare of record-keeping needed to implement this pay-for-performance system. How many billions of dollars will be needed to fund this new bureaucracy? Do we need another bureaucratic, punitive layer in our already cumbersome medical system?
My answer is clear: No! TH
Dr. Yu is medical director of adult hospitalist services at Presbyterian Medical Group in Albuquerque, N.M., and a Team Hospitalist member.
If the road to hell is paved with good intentions, then hell is full of unintended consequences.
The debate about whether hospitals’ reimbursements should be based on quality performance is not a unique concept. Similar systems have been implemented in other fields (e.g. education), and we in the medical field can learn from their experiences. In education, testing students is the driving force in measuring a “quality outcome.”
A growing number of educators now believe that the focus on testing to measure quality has actually reduced the quality of education; they cite the bureaucratic, inflexible, and cumbersome requirements placed on the educators, and the diversion of precious resources to focus on standardized test scores. The actual education of the students becomes secondary, and there are allegations of school systems manipulating their data to ensure maximum funding.
With the drive to pay-for-performance in the medical field, will the actual medical care of the patient become secondary to hitting the “quality” metrics set by the government? Add in a volatile mix of money, and this becomes a recipe for disaster.
Questions are many:
- What standards of quality are we going to use?
- Do these metrics truly translate into “quality”?
- Will the goal of reaching these metrics become the main focus of the hospitals instead of actual patient care?
- Is the goal to really improve the quality of healthcare, or is it just another vehicle for government and private third parties to come up with another excuse to reduce reimbursement in the name of quality?
Even now, ED physicians are giving antibiotics liberally for fear that they will be admonished for “missing” the pneumonia core measures. Whether this is appropriate care for the patient is irrelevant to hitting the statistical goal. Where is the incentive to deliver appropriate care? Are we even asking the right questions? Do bureaucrats know that even if appropriate, timely, quality care is given that a positive outcome is not guaranteed?
The field of medicine has made incredible advances in patient care, but the fact remains that a certain percentage of the sick and elderly become sicker and eventually die, especially in hospitals.
Potential Problems are Many
Unintended consequences pose a real danger to a system that rewards and penalizes. One potential issue is the “rich getting richer and the poor getting poorer,” as more provider focus is placed on buffering statistics by keeping healthy people healthier to achieve better outcomes, meanwhile shunning or diverting the seriously sick patients in order to keep quality metrics within goal.
How will the government and insurance providers guarantee the accuracy of each hospital’s statistics?
Think of the money and resources that will be diverted away from the clinical arena and into the bureaucratic nightmare of record-keeping needed to implement this pay-for-performance system. How many billions of dollars will be needed to fund this new bureaucracy? Do we need another bureaucratic, punitive layer in our already cumbersome medical system?
My answer is clear: No! TH
Dr. Yu is medical director of adult hospitalist services at Presbyterian Medical Group in Albuquerque, N.M., and a Team Hospitalist member.
PRO: Should Hospitals Get Reimbursements Based on Quality Performance?

Ask a hospitalist “Is quality important?” and most will answer “Yes.” Now ask the hospital’s CEO/CFO that same question, and you’ll get a resounding “Yes.”
Quality, as the primary determinant of value, has become priority No. 1 for hospitals.1 And with the Centers for Medicare & Medicaid Services’ (CMS) proposed rules for value-based purchasing (VBP), starting with a 1% withholding of Medicare reimbursement for demonstration of quality-measure performance, big dollars are at risk for hospitals.2
As the key providers of inpatient care, hospitalists will share in this financial accountability. The next-generation HM program must show value not only through efficiency and cost reduction, but also expanded services and quality.
Quality is a means of defining good care. Historically, the medical profession has escaped external accountability for quality as part of practitioner autonomy. Today, more than ever, consumer groups, payors, and regulatory bodies are demanding demonstration of quality outcomes, which impacts reimbursement and market share.
Is this demand for quality performance negative? Misused, it can be a mechanism for cost control through seemingly arbitrary indicators. Considered more broadly, it can be positive: We will be able to evaluate our practices to improve care.
Either way, the quality ship has sailed. Accepting this change, we see that the direction and execution are largely left open-ended, which brings another positive: HM has an opportunity to charter the course.
Hospitalists are inpatient care experts; we understand and improve health systems to provide excellent care. Above all else, quality is what we stand for. As a field, we are at the leading edge of change. Getting ahead of quality at each of our institutions is a great opportunity, and helping hospitals implement and deliver on quality initiatives is job security. Being held to what we value, hospitalists should be incentivized by quality performance.
Quality and Compensation
Why tie compensation to quality outcomes? First, hospitals are financially accountable for performance, and HM is financially accountable to hospitals. Second, we incent important objectives, in addition to other mechanisms (e.g. transparent reporting), to drive performance.
The majority of HM programs have an incentive component to their compensation structure, and quality is the leading performance incentive (hospitalists in these programs also have higher incomes).3 We can expect to see HM compensation structures evolve toward pay-for-performance or gainsharing models. HM groups should turn their focus to using incentives or bonuses. Here are some tips:
- Lead quality initiatives. Participate in hospital-based patient safety and satisfaction projects. Communicate the importance to your group to achieve buy-in.
- Define mutual goals. Choose two or three measurable areas that are the top priority items for the hospital and your group, and put them on your scorecard. Consider measuring team performance.
- Make it count. Make the amount of financial incentive a portion of compensation that is meaningful. Share data—and the effect on compensation—regularly to drive performance.
Quality is ours to lead. Define and deliver it, and you’ll find your group to be indispensable to the hospital, with dollars to gain—for all the right reasons. TH
Dr. Wright is senior medical officer at Hospitalists Management Company in Wisconsin and a Team Hospitalist member.
References
- Wachter RM, Goldman L. The hospitalist movement 5 years later. JAMA. 2002;287:487-494.
- Centers for Medicare & Medicaid Services. Medicare program: hospital patient value-based purchasing program. Federal Register. 2011;76(9).
- State of Hospital Medicine: 2010 Report Based on 2009 Data. SHM website. Available at: www.hospitalmedicine.org/survey. Accessed April 2, 2011.
Ask a hospitalist “Is quality important?” and most will answer “Yes.” Now ask the hospital’s CEO/CFO that same question, and you’ll get a resounding “Yes.”
Quality, as the primary determinant of value, has become priority No. 1 for hospitals.1 And with the Centers for Medicare & Medicaid Services’ (CMS) proposed rules for value-based purchasing (VBP), starting with a 1% withholding of Medicare reimbursement for demonstration of quality-measure performance, big dollars are at risk for hospitals.2
As the key providers of inpatient care, hospitalists will share in this financial accountability. The next-generation HM program must show value not only through efficiency and cost reduction, but also expanded services and quality.
Quality is a means of defining good care. Historically, the medical profession has escaped external accountability for quality as part of practitioner autonomy. Today, more than ever, consumer groups, payors, and regulatory bodies are demanding demonstration of quality outcomes, which impacts reimbursement and market share.
Is this demand for quality performance negative? Misused, it can be a mechanism for cost control through seemingly arbitrary indicators. Considered more broadly, it can be positive: We will be able to evaluate our practices to improve care.
Either way, the quality ship has sailed. Accepting this change, we see that the direction and execution are largely left open-ended, which brings another positive: HM has an opportunity to charter the course.
Hospitalists are inpatient care experts; we understand and improve health systems to provide excellent care. Above all else, quality is what we stand for. As a field, we are at the leading edge of change. Getting ahead of quality at each of our institutions is a great opportunity, and helping hospitals implement and deliver on quality initiatives is job security. Being held to what we value, hospitalists should be incentivized by quality performance.
Quality and Compensation
Why tie compensation to quality outcomes? First, hospitals are financially accountable for performance, and HM is financially accountable to hospitals. Second, we incent important objectives, in addition to other mechanisms (e.g. transparent reporting), to drive performance.
The majority of HM programs have an incentive component to their compensation structure, and quality is the leading performance incentive (hospitalists in these programs also have higher incomes).3 We can expect to see HM compensation structures evolve toward pay-for-performance or gainsharing models. HM groups should turn their focus to using incentives or bonuses. Here are some tips:
- Lead quality initiatives. Participate in hospital-based patient safety and satisfaction projects. Communicate the importance to your group to achieve buy-in.
- Define mutual goals. Choose two or three measurable areas that are the top priority items for the hospital and your group, and put them on your scorecard. Consider measuring team performance.
- Make it count. Make the amount of financial incentive a portion of compensation that is meaningful. Share data—and the effect on compensation—regularly to drive performance.
Quality is ours to lead. Define and deliver it, and you’ll find your group to be indispensable to the hospital, with dollars to gain—for all the right reasons. TH
Dr. Wright is senior medical officer at Hospitalists Management Company in Wisconsin and a Team Hospitalist member.
References
- Wachter RM, Goldman L. The hospitalist movement 5 years later. JAMA. 2002;287:487-494.
- Centers for Medicare & Medicaid Services. Medicare program: hospital patient value-based purchasing program. Federal Register. 2011;76(9).
- State of Hospital Medicine: 2010 Report Based on 2009 Data. SHM website. Available at: www.hospitalmedicine.org/survey. Accessed April 2, 2011.
Ask a hospitalist “Is quality important?” and most will answer “Yes.” Now ask the hospital’s CEO/CFO that same question, and you’ll get a resounding “Yes.”
Quality, as the primary determinant of value, has become priority No. 1 for hospitals.1 And with the Centers for Medicare & Medicaid Services’ (CMS) proposed rules for value-based purchasing (VBP), starting with a 1% withholding of Medicare reimbursement for demonstration of quality-measure performance, big dollars are at risk for hospitals.2
As the key providers of inpatient care, hospitalists will share in this financial accountability. The next-generation HM program must show value not only through efficiency and cost reduction, but also expanded services and quality.
Quality is a means of defining good care. Historically, the medical profession has escaped external accountability for quality as part of practitioner autonomy. Today, more than ever, consumer groups, payors, and regulatory bodies are demanding demonstration of quality outcomes, which impacts reimbursement and market share.
Is this demand for quality performance negative? Misused, it can be a mechanism for cost control through seemingly arbitrary indicators. Considered more broadly, it can be positive: We will be able to evaluate our practices to improve care.
Either way, the quality ship has sailed. Accepting this change, we see that the direction and execution are largely left open-ended, which brings another positive: HM has an opportunity to charter the course.
Hospitalists are inpatient care experts; we understand and improve health systems to provide excellent care. Above all else, quality is what we stand for. As a field, we are at the leading edge of change. Getting ahead of quality at each of our institutions is a great opportunity, and helping hospitals implement and deliver on quality initiatives is job security. Being held to what we value, hospitalists should be incentivized by quality performance.
Quality and Compensation
Why tie compensation to quality outcomes? First, hospitals are financially accountable for performance, and HM is financially accountable to hospitals. Second, we incent important objectives, in addition to other mechanisms (e.g. transparent reporting), to drive performance.
The majority of HM programs have an incentive component to their compensation structure, and quality is the leading performance incentive (hospitalists in these programs also have higher incomes).3 We can expect to see HM compensation structures evolve toward pay-for-performance or gainsharing models. HM groups should turn their focus to using incentives or bonuses. Here are some tips:
- Lead quality initiatives. Participate in hospital-based patient safety and satisfaction projects. Communicate the importance to your group to achieve buy-in.
- Define mutual goals. Choose two or three measurable areas that are the top priority items for the hospital and your group, and put them on your scorecard. Consider measuring team performance.
- Make it count. Make the amount of financial incentive a portion of compensation that is meaningful. Share data—and the effect on compensation—regularly to drive performance.
Quality is ours to lead. Define and deliver it, and you’ll find your group to be indispensable to the hospital, with dollars to gain—for all the right reasons. TH
Dr. Wright is senior medical officer at Hospitalists Management Company in Wisconsin and a Team Hospitalist member.
References
- Wachter RM, Goldman L. The hospitalist movement 5 years later. JAMA. 2002;287:487-494.
- Centers for Medicare & Medicaid Services. Medicare program: hospital patient value-based purchasing program. Federal Register. 2011;76(9).
- State of Hospital Medicine: 2010 Report Based on 2009 Data. SHM website. Available at: www.hospitalmedicine.org/survey. Accessed April 2, 2011.
ONLINE EXCLUSIVE: Hospitals Forced to Adapt Amid Shifting Slate of Quality Measures
With the arrival of value-based purchasing (VBP), many hospitals have faced hard decisions about where to allocate scarce resources to maximize their performance potential. In some cases, those decisions have been made even more difficult by a set of core measures still very much in flux. For hospitalists, that means the ability to quickly adapt and reprioritize will be in high demand as measures are added or subtracted from the value-based purchasing program.
In the proposed program rules released in mid-January, CMS initially selected 17 core clinical measures from a list of 28 eligible candidates included in the Hospital Inpatient Quality Reporting (IQR) Program (https://www.cms.gov/HospitalQualityInits/08_HospitalRHQDAPU.asp) and also listed on the public Hospital Compare website (www.hospitalcompare.hhs.gov/) for at least one year. CMS explained its decision to withhold some of the remaining core measures from the program by noting that they were “topped out” and thus provided a poor basis of comparison among hospitals. Others, according to the agency, were of little value or were encouraging bad behavior.
CMS used the latter reason in its explanation for why it was not including an eligible core measure on administering antibiotics to pneumonia patients within six hours of their arrival at a hospital. “We do not believe that this measure is appropriate for inclusion because it could lead to inappropriate antibiotic use,” the rules stated. For that measure and others not recommended for inclusion in the hospital VBP program, CMS added that it would propose retiring them “in the near future.”

—Laura M. Dietzel, program director for High-Tech Meaningful Use, PeaceHealth
Laura M. Dietzel, program director for High-Tech Meaningful Use at PeaceHealth, a faith-based healthcare system that operates eight hospitals in three Western states, says CMS’ short statement caught her health system off-guard and forced a rapid shift in its priorities. “We were really focusing on moving some numbers and making some technology changes for some core measures that now, based on what CMS has in the proposed value-based purchasing, will not be around even as of next year,” she says.
Dietzel says the move to high-tech tools like electronic medical records (EMRs) has further complicated how hospitals and healthcare systems like PeaceHealth are preparing for the VBP program. For two other measures that relate to pneumonia immunization, she says, some of PeaceHealth’s hospitals have “less than ideal” scores that could potentially create some financial risk.
An EMR system on tap for next year, she says, likely will help the hospitals significantly improve their scores. But what should they do between now and then: Reinvent the wheel to prop up their numbers, only to do it again in a year? Or should they take a temporary hit until the more comprehensive system is in place?
Similar questions are likely to confront more healthcare providers as the program expands and evolves. Even as it will likely retire some measures in fiscal year 2014, CMS will add more from a list of 20 related to hospital-acquired conditions and complications, patient safety indicators, inpatient quality indicators, and mortality rates.
With the arrival of value-based purchasing (VBP), many hospitals have faced hard decisions about where to allocate scarce resources to maximize their performance potential. In some cases, those decisions have been made even more difficult by a set of core measures still very much in flux. For hospitalists, that means the ability to quickly adapt and reprioritize will be in high demand as measures are added or subtracted from the value-based purchasing program.
In the proposed program rules released in mid-January, CMS initially selected 17 core clinical measures from a list of 28 eligible candidates included in the Hospital Inpatient Quality Reporting (IQR) Program (https://www.cms.gov/HospitalQualityInits/08_HospitalRHQDAPU.asp) and also listed on the public Hospital Compare website (www.hospitalcompare.hhs.gov/) for at least one year. CMS explained its decision to withhold some of the remaining core measures from the program by noting that they were “topped out” and thus provided a poor basis of comparison among hospitals. Others, according to the agency, were of little value or were encouraging bad behavior.
CMS used the latter reason in its explanation for why it was not including an eligible core measure on administering antibiotics to pneumonia patients within six hours of their arrival at a hospital. “We do not believe that this measure is appropriate for inclusion because it could lead to inappropriate antibiotic use,” the rules stated. For that measure and others not recommended for inclusion in the hospital VBP program, CMS added that it would propose retiring them “in the near future.”
—Laura M. Dietzel, program director for High-Tech Meaningful Use, PeaceHealth
Laura M. Dietzel, program director for High-Tech Meaningful Use at PeaceHealth, a faith-based healthcare system that operates eight hospitals in three Western states, says CMS’ short statement caught her health system off-guard and forced a rapid shift in its priorities. “We were really focusing on moving some numbers and making some technology changes for some core measures that now, based on what CMS has in the proposed value-based purchasing, will not be around even as of next year,” she says.
Dietzel says the move to high-tech tools like electronic medical records (EMRs) has further complicated how hospitals and healthcare systems like PeaceHealth are preparing for the VBP program. For two other measures that relate to pneumonia immunization, she says, some of PeaceHealth’s hospitals have “less than ideal” scores that could potentially create some financial risk.
An EMR system on tap for next year, she says, likely will help the hospitals significantly improve their scores. But what should they do between now and then: Reinvent the wheel to prop up their numbers, only to do it again in a year? Or should they take a temporary hit until the more comprehensive system is in place?
Similar questions are likely to confront more healthcare providers as the program expands and evolves. Even as it will likely retire some measures in fiscal year 2014, CMS will add more from a list of 20 related to hospital-acquired conditions and complications, patient safety indicators, inpatient quality indicators, and mortality rates.
With the arrival of value-based purchasing (VBP), many hospitals have faced hard decisions about where to allocate scarce resources to maximize their performance potential. In some cases, those decisions have been made even more difficult by a set of core measures still very much in flux. For hospitalists, that means the ability to quickly adapt and reprioritize will be in high demand as measures are added or subtracted from the value-based purchasing program.
In the proposed program rules released in mid-January, CMS initially selected 17 core clinical measures from a list of 28 eligible candidates included in the Hospital Inpatient Quality Reporting (IQR) Program (https://www.cms.gov/HospitalQualityInits/08_HospitalRHQDAPU.asp) and also listed on the public Hospital Compare website (www.hospitalcompare.hhs.gov/) for at least one year. CMS explained its decision to withhold some of the remaining core measures from the program by noting that they were “topped out” and thus provided a poor basis of comparison among hospitals. Others, according to the agency, were of little value or were encouraging bad behavior.
CMS used the latter reason in its explanation for why it was not including an eligible core measure on administering antibiotics to pneumonia patients within six hours of their arrival at a hospital. “We do not believe that this measure is appropriate for inclusion because it could lead to inappropriate antibiotic use,” the rules stated. For that measure and others not recommended for inclusion in the hospital VBP program, CMS added that it would propose retiring them “in the near future.”
—Laura M. Dietzel, program director for High-Tech Meaningful Use, PeaceHealth
Laura M. Dietzel, program director for High-Tech Meaningful Use at PeaceHealth, a faith-based healthcare system that operates eight hospitals in three Western states, says CMS’ short statement caught her health system off-guard and forced a rapid shift in its priorities. “We were really focusing on moving some numbers and making some technology changes for some core measures that now, based on what CMS has in the proposed value-based purchasing, will not be around even as of next year,” she says.
Dietzel says the move to high-tech tools like electronic medical records (EMRs) has further complicated how hospitals and healthcare systems like PeaceHealth are preparing for the VBP program. For two other measures that relate to pneumonia immunization, she says, some of PeaceHealth’s hospitals have “less than ideal” scores that could potentially create some financial risk.
An EMR system on tap for next year, she says, likely will help the hospitals significantly improve their scores. But what should they do between now and then: Reinvent the wheel to prop up their numbers, only to do it again in a year? Or should they take a temporary hit until the more comprehensive system is in place?
Similar questions are likely to confront more healthcare providers as the program expands and evolves. Even as it will likely retire some measures in fiscal year 2014, CMS will add more from a list of 20 related to hospital-acquired conditions and complications, patient safety indicators, inpatient quality indicators, and mortality rates.
ONLINE EXCLUSIVE: Experts explain how hospitalists can thrive in a new era of payment reform
ONLINE EXCLUSIVE: Listen to billing and coding consultants discuss the importance of provider buy-in
Click here to listen to Dr. Pinson
Click here to listen to Ms. Leong
Click here to listen to Dr. Pinson
Click here to listen to Ms. Leong
Click here to listen to Dr. Pinson
Click here to listen to Ms. Leong
ONLINE EXCLUSIVE: PEERist Program Provides Rural Nebraska Hospital 24/7 HM Coverage
Hospitalist programs take many shapes, and the organizational flowchart typically depends on the number and makeup of the physicians, the hospital, the patients, and the community. Adequately covering the needs of inpatient units can be especially frustrating in rural communities, some hospitalists say.
“There are only five physicians total in our town,” says Gary Ensz, MD, a partner in the Auburn Family Health Center in Auburn, Neb. “In addition to being family practitioners, we are our own hospitalists. Covering the emergency room, seeing patients in clinic, and following hospitalized patients is a big burden.”
To address these issues, Dr. Ensz and his partners developed the Physician Extender Emergency Room Hospitalist (PEERist) program. They utilize physician assistants (PAs) to serve many hospitalist functions under their supervision.
“We hired PAs to work in the hospital and the emergency room,” Dr. Ensz explains. “They do not work in the clinic and then take call. Their only responsibilities are to the hospital and ER.”

—Gary Ensz, MD, partner, Auburn (Neb.) Family Health Center
PEERists work under protocols addressing treatment concerns and give guidance on when physicians should be called. On-call doctors round in the morning with the PA. The physicians like this approach, Dr. Ensz says, because they see the PEERist when it is convenient for them; before, they would leave clinic patients to attend to concerns at the hospital.
Impact of ER Call
“For most of us practicing in rural situations, the demands of ER call force people to retire,” Dr. Ensz says. “Now we have someone at the hospital highly trained and, as they get more experience, actually do some work better simply because they are there all the time. At worst, you now have two sets of hands working on the patient.”
PAs work a rotating schedule of up to 72 hours with as many as nine days off in a row. PAs can be recruited from a much larger area; in fact, one commutes four hours each way. PEERists say the time off makes it easier to do things with their families or work another job.
Other Benefits
In addition to pluses associated with the practice, Dr. Ensz is seeing other benefits, such as closer working relationships with the nurses. He also stresses that his hospital now has 24/7 in-house coverage, something almost unheard of in small rural hospitals.
One of the more subtle improvements might have been in getting people to the hospital quicker. “It wasn’t unusual for someone to come to clinic with symptoms they had for a while,” Dr. Ensz says. “The PEERists being in-house all the time have done away with these concerns.”
Hospitalist programs take many shapes, and the organizational flowchart typically depends on the number and makeup of the physicians, the hospital, the patients, and the community. Adequately covering the needs of inpatient units can be especially frustrating in rural communities, some hospitalists say.
“There are only five physicians total in our town,” says Gary Ensz, MD, a partner in the Auburn Family Health Center in Auburn, Neb. “In addition to being family practitioners, we are our own hospitalists. Covering the emergency room, seeing patients in clinic, and following hospitalized patients is a big burden.”
To address these issues, Dr. Ensz and his partners developed the Physician Extender Emergency Room Hospitalist (PEERist) program. They utilize physician assistants (PAs) to serve many hospitalist functions under their supervision.
“We hired PAs to work in the hospital and the emergency room,” Dr. Ensz explains. “They do not work in the clinic and then take call. Their only responsibilities are to the hospital and ER.”
—Gary Ensz, MD, partner, Auburn (Neb.) Family Health Center
PEERists work under protocols addressing treatment concerns and give guidance on when physicians should be called. On-call doctors round in the morning with the PA. The physicians like this approach, Dr. Ensz says, because they see the PEERist when it is convenient for them; before, they would leave clinic patients to attend to concerns at the hospital.
Impact of ER Call
“For most of us practicing in rural situations, the demands of ER call force people to retire,” Dr. Ensz says. “Now we have someone at the hospital highly trained and, as they get more experience, actually do some work better simply because they are there all the time. At worst, you now have two sets of hands working on the patient.”
PAs work a rotating schedule of up to 72 hours with as many as nine days off in a row. PAs can be recruited from a much larger area; in fact, one commutes four hours each way. PEERists say the time off makes it easier to do things with their families or work another job.
Other Benefits
In addition to pluses associated with the practice, Dr. Ensz is seeing other benefits, such as closer working relationships with the nurses. He also stresses that his hospital now has 24/7 in-house coverage, something almost unheard of in small rural hospitals.
One of the more subtle improvements might have been in getting people to the hospital quicker. “It wasn’t unusual for someone to come to clinic with symptoms they had for a while,” Dr. Ensz says. “The PEERists being in-house all the time have done away with these concerns.”
Hospitalist programs take many shapes, and the organizational flowchart typically depends on the number and makeup of the physicians, the hospital, the patients, and the community. Adequately covering the needs of inpatient units can be especially frustrating in rural communities, some hospitalists say.
“There are only five physicians total in our town,” says Gary Ensz, MD, a partner in the Auburn Family Health Center in Auburn, Neb. “In addition to being family practitioners, we are our own hospitalists. Covering the emergency room, seeing patients in clinic, and following hospitalized patients is a big burden.”
To address these issues, Dr. Ensz and his partners developed the Physician Extender Emergency Room Hospitalist (PEERist) program. They utilize physician assistants (PAs) to serve many hospitalist functions under their supervision.
“We hired PAs to work in the hospital and the emergency room,” Dr. Ensz explains. “They do not work in the clinic and then take call. Their only responsibilities are to the hospital and ER.”
—Gary Ensz, MD, partner, Auburn (Neb.) Family Health Center
PEERists work under protocols addressing treatment concerns and give guidance on when physicians should be called. On-call doctors round in the morning with the PA. The physicians like this approach, Dr. Ensz says, because they see the PEERist when it is convenient for them; before, they would leave clinic patients to attend to concerns at the hospital.
Impact of ER Call
“For most of us practicing in rural situations, the demands of ER call force people to retire,” Dr. Ensz says. “Now we have someone at the hospital highly trained and, as they get more experience, actually do some work better simply because they are there all the time. At worst, you now have two sets of hands working on the patient.”
PAs work a rotating schedule of up to 72 hours with as many as nine days off in a row. PAs can be recruited from a much larger area; in fact, one commutes four hours each way. PEERists say the time off makes it easier to do things with their families or work another job.
Other Benefits
In addition to pluses associated with the practice, Dr. Ensz is seeing other benefits, such as closer working relationships with the nurses. He also stresses that his hospital now has 24/7 in-house coverage, something almost unheard of in small rural hospitals.
One of the more subtle improvements might have been in getting people to the hospital quicker. “It wasn’t unusual for someone to come to clinic with symptoms they had for a while,” Dr. Ensz says. “The PEERists being in-house all the time have done away with these concerns.”
Nurse Practitioners, Physician Assistants to the Rescue

In 2004, the hospitalist group at the University of Michigan Health System in Ann Arbor faced a manpower problem: In a refrain common to hospitalist groups around the country, changes in duty-hour regulations were making it harder for medical residents to continue to provide inpatient coverage at the same levels as before.
Addressing the issue was difficult for the HM group and hospital administrators; they were going to need a significant number of new providers, and qualified physicians were in short supply. To address these issues, the HM group chose to add nonphysician providers (NPP) to their service.
“NPPs had worked at UM for a long time in other areas,” says Vikas Parekh, MD, SFHM, associate director of hospitalist management. “We had just created a new service that was hiring new people and thought NPPs would help in providing services.”
Hiring NPPs helped solve the University of Michigan’s problem, and the tactic has helped solve manpower issues at numerous HM groups around the country. But deciding whether your HM group should hire physicians, NPPs—usually nurse practitioners (NPs) and physician assistants (PAs)—or some combination of the two will not be easy. It is a complex decision, one that requires following state-level licensing and practice laws as well as local hospital bylaws and federal and private insurance payment rules. Such decisions also mean HM group directors need to keep in mind case mixes and the personalities of the physicians in the practice.
“There is no one-size-fits-all solution,” says Mitchell Wilson, MD, SFHM, corporate medical director for Eagle Hospital Physicians in Atlanta. “Not all environments are well-suited to NPP practice. Even when it is, you can’t just throw an NPP into the mix on their own with the expectation they will be successful.”
Whether it’s covering admissions, streamlining discharges, or working as an integral part of a care team, NPPs can be the solution expanding HM groups are looking for.

“Our physicians depend on NPPs to help them complete patient care in a more efficient manner and work to enhance continuity of care,” says Mary Whitehead, RN, APRN-BC, FNP, of Hospital Medicine Associates in Fort Worth, Texas. “We lower physician rounding time so patients are seen sooner and tests are requested sooner. In addition, the patients really appreciate the extra time we can spend with them.”
Trained, Licensed, Available
NPs must be registered nurses with clinical experience before they can enroll in an advanced degree program, which usually results in a master’s degree or doctorate. Generally, a state board of nursing, or a state board jointly with the state medical board, regulates NPs.
PAs are trained in more of a traditional medical model. They have a variable education level all the way up to a PhD, although more states are requiring at least a master’s degree. Practice and other legal parameters most often come under the authority of state medical boards.
NPPs can provide additional medical expertise to patients, says Ryan Genzink, PA, a physician assistant with Hospitalists of Western Michigan in Grand Rapids and the American Academy of Physician Assistants’ medical liaison to SHM. “One of the challenges faced by physicians is that they often have to be in two places at once,” he says. “There is a recognition that teams provide better care for complicated patients.”
NPPs generally practice under the supervision of a physician hospitalist. Some states allow a greater degree of independence for NPs. However, most NPs and PAs are required to have a practice agreement outlining their responsibilities and the amount of oversight required (see Table 1, p. 39). There is no such thing as a “fire and forget” NPP.
“The practice needs to thoroughly understand the legal environment early in the process,” says John Nelson, MD, MHM, hospitalist director at Bellevue (Wash.) Medical Center, partner in Nelson Flores Hospital Medicine Consultants, and SHM cofounder. “NPPs are not a ‘hospitalist lite’ that can function entirely like a hospitalist.”
Hospital bylaws, which can vary greatly by city, county, or state, are another important consideration before you hire an NPP.
“In some areas, NPPs may not be able to practice in the ICU,” Dr. Wilson says. “In others, the physician may be required to see the patient instead of consulting with the NPP. The idiosyncrasies of the individual hospital’s bylaws may impact the efficiency of the NPP/MD team.”
Physician Characteristics
Environmental variables—namely, the personality of the physicians within the practice—should be considered before you head down the NPP path. It makes little practical or financial sense to spend the time and effort of hiring an NPP if the physicians still insist on doing all the work.
“[It’s] one of the most significant factors in successfully integrating an NPP program,” Dr. Wilson says. “Will [physicians] be able to tolerate some degree of uncertainty when letting others see their patients? Are they open to adapting to different practice styles? The thing we see most often in practices that use NPPs to their advantage is the recognition that there is an important role for the nonphysician at the bedside.”
Some physicians hesitate to work with NPPs, while others welcome the extra help and unique experience NPPs offer. Experts agree that forcing NPPs on a physician is not a good idea. They also agree that, especially when beginning a new program, group directors should let physicians who are interested in working with NPPs take the lead. As NPP use in the group matures, many of those who were at first unwilling can decide that there is a place for NPPs in their practice.
Case Mix Is Key

The types and kinds of patient seen might limit the use of NPPs in hospitalist practice. “Our experience is that acuity and complexity of the care, especially as it relates to diagnostic and therapeutic decision-making, makes it difficult for NPPs to function independently,” Dr. Parekh says.
Dr. Wilson agrees. “Depending on the specific attributes of the setting, a service with both high-complexity and high-acuity patients may be a more challenging environment to realize the efficiencies of NPPs,” he says. “There is a relationship between complexity, acuity, and physician involvement.”
Even so, a continuum of NPP use in HM practice is achievable. For example, as a patient improves, an NPP might be able to take on a larger role in treatment by participating in discharge planning. In more acute patients, the NPP can save valuable physician time by coordinating with consultants, staying on top of treatments, and consolidating clinically important data for the physician.
Many Models in Use
Historically, the widespread use of alternative providers began in 2004 as a result of the changes to resident duty-hours. The restrictions created a workforce gap, which led to a large number of new positions in hospitals nationwide. Many of HM’s early adopters essentially went with what they knew.
“We work in teams where the physician, NPP, and nurse see a group of patients similar in function to an attending, resident, and RN,” Genzink says. “We see ‘our’ patients in a collaborative fashion.”
There are other models that have proven successful in the correct setting. Some HM groups use specialist NPPs to cover specific clinical areas, such as orthopedics or oncology. This not only develops a cadre of providers with excellent understanding of their patients, but it also frees up physician time for more acute and complicated patients.
“Our physicians depend on us helping them get patient care completed more efficiently, so that length of stay is acceptable, and to enhance continuity of care,” says Whitehead, the American Academy of Nurse Practitioners’ liaison to SHM. “Having an NPP visit the patient daily, documenting progress, greatly enhances communications between physicians and consultants.”
Other groups have NPPs specialize by function—for example, they cover all admissions or work mainly with discharging a patient. Some groups have the physician see the patient on admission, work out a care plan, then turn over management to the NPP. Many agree that most NPPs are best utilized by having them cover specific shifts, such as overnight call or on a swing shift, to help during peak demand.
Monetary and Time Commitments
The financial impact of NPPs on a hospitalist practice depends on many factors. Groups will need to look not only at the salary and benefit costs associated with the position, but also how best to fit that person into the billing system.
Salary and benefit comparisons are fairly straightforward: The State of Hospital Medicine: 2010 Report Based on 2009 Data, produced by SHM and the Medical Group Management Association, shows median total compensation for adult hospitalists at $215,000 per year; NPP compensation is around $87,000.1
The general cost of benefits (health insurance, retirement, etc.) is fairly typical throughout a hospitalist practice, so there should be little difference between a new FTE hospitalist or NPP. Other considerations, including office space and support staff, would be roughly the same if the group hired a physician. The cost of continuing education and malpractice insurance likely will be less with an NPP, but it is best to check before making a new hire.
After the outgo has been established, the next step is to look at the differences in reimbursement for NPPs vs. physicians. Here, again, the math gets tricky. The Centers for Medicare & Medicaid Services (CMS) pay NPPs at 85% of the physician rate for a specific diagnosis. However, if there is direct physician involvement, the claim can be filed as “shared billing” and reimbursed at 100%.
For some hospitalist practices, adding NPPs is an easy decision to make. Dr. Parekh says his group already has policies in place that require a physician to see the patient every day. In that case, no extra physician time is necessary, so shared billing makes sense. Other hospitals’ bylaws might have similar requirements.
For practices in which the NPP is able to work with less oversight, it might be better to bill at 85% rather than use the physician time to meet shared-billing criteria. Even in practices with greater NPP autonomy, such variables as case mix and patient acuity might enter into the equation. If the patient is sick enough that the physician is involved for a significant amount of time, then shared billing probably is best.
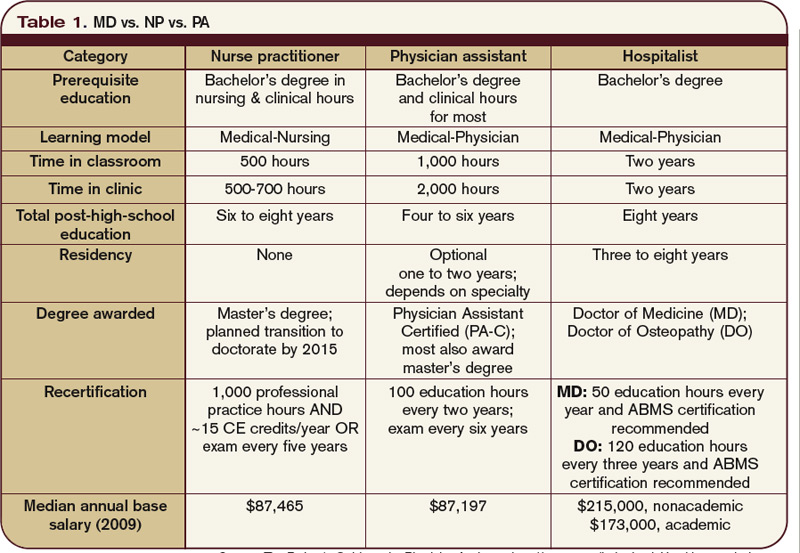
Experts say group directors and hiring managers should look carefully at contracts with private insurers, too. There most likely will be considerable variation in how each plan handles NPP claims.
Managing performance expectations can have an impact on the successful use of NPPs in a hospitalist practice. Setting realistic goals and groupwide understanding of what the NPPs’ roles will be is crucial. The practice should look at the work that needs to be done and decide if that work provides a genuinely valuable role for an NPP.
Hire for Need, Not Desperation

“The mistake I see most often is hiring an NPP because a practice is desperate for help,” says Martin Buser, MPH, FACHE, a partner in Hospitalist Management Resources, LLC, in San Diego. “Smart practices are looking at NPPs, evaluating where they do the most good, and then setting out their role and expectations based on these needs and the practice environment.”
Hiring mistakes can be compounded if the NPP is not a good match to the job description or group expectations. If the practice hires an NPP fresh out of school, the group will need to establish training and have the new hire work more closely with physicians. If, on the other hand, an NP has 10 years of experience in an ICU, or a PA has worked in the ED for the past five years, a higher level of autonomy can be granted sooner. However, NPPs with established backgrounds are almost as rare as experienced hospitalists (see “Integrating NPPs Into HM Practice,” p. 38).
Inevitably, there will be changes in the interactions between patients and the hospitalists, as both physicians and NPPs become more comfortable with the other’s practice style, as well as each other’s strengths and weaknesses.
MD-to-NPP Ratio Varies
The practice structure and optimal mix of NPPs to MDs is something that will be specific to the hospitalist group. “We don’t really have good studies on this subject,” Buser says. “I usually get worried when we exceed two NPPs to one MD.”
Others disagree. Dr. Parekh, who works in an academic center, says his group has been successful having one MD work with as many as three NPPs. At the other end, Dr. Wilson says his 10 years of experience suggest 1:1 is the most efficient ratio.
However, all of them agree that having one NPP work with more than one physician is not sustainable. The NPP will be less familiar with each doctor’s practice style, what kind of information they need, and how things should be presented. If two or more hospitalists share an NPP, there can be internal friction over division of the NPP’s time, as well as extending the time before the MDs have a good feel for the NPP’s strengths and weaknesses.
In the final analysis, the HM group has to look at the amount and type of work available. In some cases, it will make financial and clinical sense to bring on an NPP. Under other circumstances, an FTE hospitalist is the best fit.
“Sustainability, quality, and efficiency are the drivers for NPP/MD teams. Increasing pressure to offset program costs is not,” Dr. Wilson says. “You do it because it helps sustain the program, helps with recruiting, and effects your efficiency.” TH
Kurt Ullman is a freelance medical writer based in Indiana.
Reference
- Medical Group Management Association and the Society of Hospital Medicine. State of Hospital Medicine: 2010 Report Based on 2009 Data. 2010. Philadelphia and Englewood, Colo.
In 2004, the hospitalist group at the University of Michigan Health System in Ann Arbor faced a manpower problem: In a refrain common to hospitalist groups around the country, changes in duty-hour regulations were making it harder for medical residents to continue to provide inpatient coverage at the same levels as before.
Addressing the issue was difficult for the HM group and hospital administrators; they were going to need a significant number of new providers, and qualified physicians were in short supply. To address these issues, the HM group chose to add nonphysician providers (NPP) to their service.
“NPPs had worked at UM for a long time in other areas,” says Vikas Parekh, MD, SFHM, associate director of hospitalist management. “We had just created a new service that was hiring new people and thought NPPs would help in providing services.”
Hiring NPPs helped solve the University of Michigan’s problem, and the tactic has helped solve manpower issues at numerous HM groups around the country. But deciding whether your HM group should hire physicians, NPPs—usually nurse practitioners (NPs) and physician assistants (PAs)—or some combination of the two will not be easy. It is a complex decision, one that requires following state-level licensing and practice laws as well as local hospital bylaws and federal and private insurance payment rules. Such decisions also mean HM group directors need to keep in mind case mixes and the personalities of the physicians in the practice.
“There is no one-size-fits-all solution,” says Mitchell Wilson, MD, SFHM, corporate medical director for Eagle Hospital Physicians in Atlanta. “Not all environments are well-suited to NPP practice. Even when it is, you can’t just throw an NPP into the mix on their own with the expectation they will be successful.”
Whether it’s covering admissions, streamlining discharges, or working as an integral part of a care team, NPPs can be the solution expanding HM groups are looking for.
“Our physicians depend on NPPs to help them complete patient care in a more efficient manner and work to enhance continuity of care,” says Mary Whitehead, RN, APRN-BC, FNP, of Hospital Medicine Associates in Fort Worth, Texas. “We lower physician rounding time so patients are seen sooner and tests are requested sooner. In addition, the patients really appreciate the extra time we can spend with them.”
Trained, Licensed, Available
NPs must be registered nurses with clinical experience before they can enroll in an advanced degree program, which usually results in a master’s degree or doctorate. Generally, a state board of nursing, or a state board jointly with the state medical board, regulates NPs.
PAs are trained in more of a traditional medical model. They have a variable education level all the way up to a PhD, although more states are requiring at least a master’s degree. Practice and other legal parameters most often come under the authority of state medical boards.
NPPs can provide additional medical expertise to patients, says Ryan Genzink, PA, a physician assistant with Hospitalists of Western Michigan in Grand Rapids and the American Academy of Physician Assistants’ medical liaison to SHM. “One of the challenges faced by physicians is that they often have to be in two places at once,” he says. “There is a recognition that teams provide better care for complicated patients.”
NPPs generally practice under the supervision of a physician hospitalist. Some states allow a greater degree of independence for NPs. However, most NPs and PAs are required to have a practice agreement outlining their responsibilities and the amount of oversight required (see Table 1, p. 39). There is no such thing as a “fire and forget” NPP.
“The practice needs to thoroughly understand the legal environment early in the process,” says John Nelson, MD, MHM, hospitalist director at Bellevue (Wash.) Medical Center, partner in Nelson Flores Hospital Medicine Consultants, and SHM cofounder. “NPPs are not a ‘hospitalist lite’ that can function entirely like a hospitalist.”
Hospital bylaws, which can vary greatly by city, county, or state, are another important consideration before you hire an NPP.
“In some areas, NPPs may not be able to practice in the ICU,” Dr. Wilson says. “In others, the physician may be required to see the patient instead of consulting with the NPP. The idiosyncrasies of the individual hospital’s bylaws may impact the efficiency of the NPP/MD team.”
Physician Characteristics
Environmental variables—namely, the personality of the physicians within the practice—should be considered before you head down the NPP path. It makes little practical or financial sense to spend the time and effort of hiring an NPP if the physicians still insist on doing all the work.
“[It’s] one of the most significant factors in successfully integrating an NPP program,” Dr. Wilson says. “Will [physicians] be able to tolerate some degree of uncertainty when letting others see their patients? Are they open to adapting to different practice styles? The thing we see most often in practices that use NPPs to their advantage is the recognition that there is an important role for the nonphysician at the bedside.”
Some physicians hesitate to work with NPPs, while others welcome the extra help and unique experience NPPs offer. Experts agree that forcing NPPs on a physician is not a good idea. They also agree that, especially when beginning a new program, group directors should let physicians who are interested in working with NPPs take the lead. As NPP use in the group matures, many of those who were at first unwilling can decide that there is a place for NPPs in their practice.
Case Mix Is Key
The types and kinds of patient seen might limit the use of NPPs in hospitalist practice. “Our experience is that acuity and complexity of the care, especially as it relates to diagnostic and therapeutic decision-making, makes it difficult for NPPs to function independently,” Dr. Parekh says.
Dr. Wilson agrees. “Depending on the specific attributes of the setting, a service with both high-complexity and high-acuity patients may be a more challenging environment to realize the efficiencies of NPPs,” he says. “There is a relationship between complexity, acuity, and physician involvement.”
Even so, a continuum of NPP use in HM practice is achievable. For example, as a patient improves, an NPP might be able to take on a larger role in treatment by participating in discharge planning. In more acute patients, the NPP can save valuable physician time by coordinating with consultants, staying on top of treatments, and consolidating clinically important data for the physician.
Many Models in Use
Historically, the widespread use of alternative providers began in 2004 as a result of the changes to resident duty-hours. The restrictions created a workforce gap, which led to a large number of new positions in hospitals nationwide. Many of HM’s early adopters essentially went with what they knew.
“We work in teams where the physician, NPP, and nurse see a group of patients similar in function to an attending, resident, and RN,” Genzink says. “We see ‘our’ patients in a collaborative fashion.”
There are other models that have proven successful in the correct setting. Some HM groups use specialist NPPs to cover specific clinical areas, such as orthopedics or oncology. This not only develops a cadre of providers with excellent understanding of their patients, but it also frees up physician time for more acute and complicated patients.
“Our physicians depend on us helping them get patient care completed more efficiently, so that length of stay is acceptable, and to enhance continuity of care,” says Whitehead, the American Academy of Nurse Practitioners’ liaison to SHM. “Having an NPP visit the patient daily, documenting progress, greatly enhances communications between physicians and consultants.”
Other groups have NPPs specialize by function—for example, they cover all admissions or work mainly with discharging a patient. Some groups have the physician see the patient on admission, work out a care plan, then turn over management to the NPP. Many agree that most NPPs are best utilized by having them cover specific shifts, such as overnight call or on a swing shift, to help during peak demand.
Monetary and Time Commitments
The financial impact of NPPs on a hospitalist practice depends on many factors. Groups will need to look not only at the salary and benefit costs associated with the position, but also how best to fit that person into the billing system.
Salary and benefit comparisons are fairly straightforward: The State of Hospital Medicine: 2010 Report Based on 2009 Data, produced by SHM and the Medical Group Management Association, shows median total compensation for adult hospitalists at $215,000 per year; NPP compensation is around $87,000.1
The general cost of benefits (health insurance, retirement, etc.) is fairly typical throughout a hospitalist practice, so there should be little difference between a new FTE hospitalist or NPP. Other considerations, including office space and support staff, would be roughly the same if the group hired a physician. The cost of continuing education and malpractice insurance likely will be less with an NPP, but it is best to check before making a new hire.
After the outgo has been established, the next step is to look at the differences in reimbursement for NPPs vs. physicians. Here, again, the math gets tricky. The Centers for Medicare & Medicaid Services (CMS) pay NPPs at 85% of the physician rate for a specific diagnosis. However, if there is direct physician involvement, the claim can be filed as “shared billing” and reimbursed at 100%.
For some hospitalist practices, adding NPPs is an easy decision to make. Dr. Parekh says his group already has policies in place that require a physician to see the patient every day. In that case, no extra physician time is necessary, so shared billing makes sense. Other hospitals’ bylaws might have similar requirements.
For practices in which the NPP is able to work with less oversight, it might be better to bill at 85% rather than use the physician time to meet shared-billing criteria. Even in practices with greater NPP autonomy, such variables as case mix and patient acuity might enter into the equation. If the patient is sick enough that the physician is involved for a significant amount of time, then shared billing probably is best.
Experts say group directors and hiring managers should look carefully at contracts with private insurers, too. There most likely will be considerable variation in how each plan handles NPP claims.
Managing performance expectations can have an impact on the successful use of NPPs in a hospitalist practice. Setting realistic goals and groupwide understanding of what the NPPs’ roles will be is crucial. The practice should look at the work that needs to be done and decide if that work provides a genuinely valuable role for an NPP.
Hire for Need, Not Desperation
“The mistake I see most often is hiring an NPP because a practice is desperate for help,” says Martin Buser, MPH, FACHE, a partner in Hospitalist Management Resources, LLC, in San Diego. “Smart practices are looking at NPPs, evaluating where they do the most good, and then setting out their role and expectations based on these needs and the practice environment.”
Hiring mistakes can be compounded if the NPP is not a good match to the job description or group expectations. If the practice hires an NPP fresh out of school, the group will need to establish training and have the new hire work more closely with physicians. If, on the other hand, an NP has 10 years of experience in an ICU, or a PA has worked in the ED for the past five years, a higher level of autonomy can be granted sooner. However, NPPs with established backgrounds are almost as rare as experienced hospitalists (see “Integrating NPPs Into HM Practice,” p. 38).
Inevitably, there will be changes in the interactions between patients and the hospitalists, as both physicians and NPPs become more comfortable with the other’s practice style, as well as each other’s strengths and weaknesses.
MD-to-NPP Ratio Varies
The practice structure and optimal mix of NPPs to MDs is something that will be specific to the hospitalist group. “We don’t really have good studies on this subject,” Buser says. “I usually get worried when we exceed two NPPs to one MD.”
Others disagree. Dr. Parekh, who works in an academic center, says his group has been successful having one MD work with as many as three NPPs. At the other end, Dr. Wilson says his 10 years of experience suggest 1:1 is the most efficient ratio.
However, all of them agree that having one NPP work with more than one physician is not sustainable. The NPP will be less familiar with each doctor’s practice style, what kind of information they need, and how things should be presented. If two or more hospitalists share an NPP, there can be internal friction over division of the NPP’s time, as well as extending the time before the MDs have a good feel for the NPP’s strengths and weaknesses.
In the final analysis, the HM group has to look at the amount and type of work available. In some cases, it will make financial and clinical sense to bring on an NPP. Under other circumstances, an FTE hospitalist is the best fit.
“Sustainability, quality, and efficiency are the drivers for NPP/MD teams. Increasing pressure to offset program costs is not,” Dr. Wilson says. “You do it because it helps sustain the program, helps with recruiting, and effects your efficiency.” TH
Kurt Ullman is a freelance medical writer based in Indiana.
Reference
- Medical Group Management Association and the Society of Hospital Medicine. State of Hospital Medicine: 2010 Report Based on 2009 Data. 2010. Philadelphia and Englewood, Colo.
In 2004, the hospitalist group at the University of Michigan Health System in Ann Arbor faced a manpower problem: In a refrain common to hospitalist groups around the country, changes in duty-hour regulations were making it harder for medical residents to continue to provide inpatient coverage at the same levels as before.
Addressing the issue was difficult for the HM group and hospital administrators; they were going to need a significant number of new providers, and qualified physicians were in short supply. To address these issues, the HM group chose to add nonphysician providers (NPP) to their service.
“NPPs had worked at UM for a long time in other areas,” says Vikas Parekh, MD, SFHM, associate director of hospitalist management. “We had just created a new service that was hiring new people and thought NPPs would help in providing services.”
Hiring NPPs helped solve the University of Michigan’s problem, and the tactic has helped solve manpower issues at numerous HM groups around the country. But deciding whether your HM group should hire physicians, NPPs—usually nurse practitioners (NPs) and physician assistants (PAs)—or some combination of the two will not be easy. It is a complex decision, one that requires following state-level licensing and practice laws as well as local hospital bylaws and federal and private insurance payment rules. Such decisions also mean HM group directors need to keep in mind case mixes and the personalities of the physicians in the practice.
“There is no one-size-fits-all solution,” says Mitchell Wilson, MD, SFHM, corporate medical director for Eagle Hospital Physicians in Atlanta. “Not all environments are well-suited to NPP practice. Even when it is, you can’t just throw an NPP into the mix on their own with the expectation they will be successful.”
Whether it’s covering admissions, streamlining discharges, or working as an integral part of a care team, NPPs can be the solution expanding HM groups are looking for.
“Our physicians depend on NPPs to help them complete patient care in a more efficient manner and work to enhance continuity of care,” says Mary Whitehead, RN, APRN-BC, FNP, of Hospital Medicine Associates in Fort Worth, Texas. “We lower physician rounding time so patients are seen sooner and tests are requested sooner. In addition, the patients really appreciate the extra time we can spend with them.”
Trained, Licensed, Available
NPs must be registered nurses with clinical experience before they can enroll in an advanced degree program, which usually results in a master’s degree or doctorate. Generally, a state board of nursing, or a state board jointly with the state medical board, regulates NPs.
PAs are trained in more of a traditional medical model. They have a variable education level all the way up to a PhD, although more states are requiring at least a master’s degree. Practice and other legal parameters most often come under the authority of state medical boards.
NPPs can provide additional medical expertise to patients, says Ryan Genzink, PA, a physician assistant with Hospitalists of Western Michigan in Grand Rapids and the American Academy of Physician Assistants’ medical liaison to SHM. “One of the challenges faced by physicians is that they often have to be in two places at once,” he says. “There is a recognition that teams provide better care for complicated patients.”
NPPs generally practice under the supervision of a physician hospitalist. Some states allow a greater degree of independence for NPs. However, most NPs and PAs are required to have a practice agreement outlining their responsibilities and the amount of oversight required (see Table 1, p. 39). There is no such thing as a “fire and forget” NPP.
“The practice needs to thoroughly understand the legal environment early in the process,” says John Nelson, MD, MHM, hospitalist director at Bellevue (Wash.) Medical Center, partner in Nelson Flores Hospital Medicine Consultants, and SHM cofounder. “NPPs are not a ‘hospitalist lite’ that can function entirely like a hospitalist.”
Hospital bylaws, which can vary greatly by city, county, or state, are another important consideration before you hire an NPP.
“In some areas, NPPs may not be able to practice in the ICU,” Dr. Wilson says. “In others, the physician may be required to see the patient instead of consulting with the NPP. The idiosyncrasies of the individual hospital’s bylaws may impact the efficiency of the NPP/MD team.”
Physician Characteristics
Environmental variables—namely, the personality of the physicians within the practice—should be considered before you head down the NPP path. It makes little practical or financial sense to spend the time and effort of hiring an NPP if the physicians still insist on doing all the work.
“[It’s] one of the most significant factors in successfully integrating an NPP program,” Dr. Wilson says. “Will [physicians] be able to tolerate some degree of uncertainty when letting others see their patients? Are they open to adapting to different practice styles? The thing we see most often in practices that use NPPs to their advantage is the recognition that there is an important role for the nonphysician at the bedside.”
Some physicians hesitate to work with NPPs, while others welcome the extra help and unique experience NPPs offer. Experts agree that forcing NPPs on a physician is not a good idea. They also agree that, especially when beginning a new program, group directors should let physicians who are interested in working with NPPs take the lead. As NPP use in the group matures, many of those who were at first unwilling can decide that there is a place for NPPs in their practice.
Case Mix Is Key
The types and kinds of patient seen might limit the use of NPPs in hospitalist practice. “Our experience is that acuity and complexity of the care, especially as it relates to diagnostic and therapeutic decision-making, makes it difficult for NPPs to function independently,” Dr. Parekh says.
Dr. Wilson agrees. “Depending on the specific attributes of the setting, a service with both high-complexity and high-acuity patients may be a more challenging environment to realize the efficiencies of NPPs,” he says. “There is a relationship between complexity, acuity, and physician involvement.”
Even so, a continuum of NPP use in HM practice is achievable. For example, as a patient improves, an NPP might be able to take on a larger role in treatment by participating in discharge planning. In more acute patients, the NPP can save valuable physician time by coordinating with consultants, staying on top of treatments, and consolidating clinically important data for the physician.
Many Models in Use
Historically, the widespread use of alternative providers began in 2004 as a result of the changes to resident duty-hours. The restrictions created a workforce gap, which led to a large number of new positions in hospitals nationwide. Many of HM’s early adopters essentially went with what they knew.
“We work in teams where the physician, NPP, and nurse see a group of patients similar in function to an attending, resident, and RN,” Genzink says. “We see ‘our’ patients in a collaborative fashion.”
There are other models that have proven successful in the correct setting. Some HM groups use specialist NPPs to cover specific clinical areas, such as orthopedics or oncology. This not only develops a cadre of providers with excellent understanding of their patients, but it also frees up physician time for more acute and complicated patients.
“Our physicians depend on us helping them get patient care completed more efficiently, so that length of stay is acceptable, and to enhance continuity of care,” says Whitehead, the American Academy of Nurse Practitioners’ liaison to SHM. “Having an NPP visit the patient daily, documenting progress, greatly enhances communications between physicians and consultants.”
Other groups have NPPs specialize by function—for example, they cover all admissions or work mainly with discharging a patient. Some groups have the physician see the patient on admission, work out a care plan, then turn over management to the NPP. Many agree that most NPPs are best utilized by having them cover specific shifts, such as overnight call or on a swing shift, to help during peak demand.
Monetary and Time Commitments
The financial impact of NPPs on a hospitalist practice depends on many factors. Groups will need to look not only at the salary and benefit costs associated with the position, but also how best to fit that person into the billing system.
Salary and benefit comparisons are fairly straightforward: The State of Hospital Medicine: 2010 Report Based on 2009 Data, produced by SHM and the Medical Group Management Association, shows median total compensation for adult hospitalists at $215,000 per year; NPP compensation is around $87,000.1
The general cost of benefits (health insurance, retirement, etc.) is fairly typical throughout a hospitalist practice, so there should be little difference between a new FTE hospitalist or NPP. Other considerations, including office space and support staff, would be roughly the same if the group hired a physician. The cost of continuing education and malpractice insurance likely will be less with an NPP, but it is best to check before making a new hire.
After the outgo has been established, the next step is to look at the differences in reimbursement for NPPs vs. physicians. Here, again, the math gets tricky. The Centers for Medicare & Medicaid Services (CMS) pay NPPs at 85% of the physician rate for a specific diagnosis. However, if there is direct physician involvement, the claim can be filed as “shared billing” and reimbursed at 100%.
For some hospitalist practices, adding NPPs is an easy decision to make. Dr. Parekh says his group already has policies in place that require a physician to see the patient every day. In that case, no extra physician time is necessary, so shared billing makes sense. Other hospitals’ bylaws might have similar requirements.
For practices in which the NPP is able to work with less oversight, it might be better to bill at 85% rather than use the physician time to meet shared-billing criteria. Even in practices with greater NPP autonomy, such variables as case mix and patient acuity might enter into the equation. If the patient is sick enough that the physician is involved for a significant amount of time, then shared billing probably is best.
Experts say group directors and hiring managers should look carefully at contracts with private insurers, too. There most likely will be considerable variation in how each plan handles NPP claims.
Managing performance expectations can have an impact on the successful use of NPPs in a hospitalist practice. Setting realistic goals and groupwide understanding of what the NPPs’ roles will be is crucial. The practice should look at the work that needs to be done and decide if that work provides a genuinely valuable role for an NPP.
Hire for Need, Not Desperation
“The mistake I see most often is hiring an NPP because a practice is desperate for help,” says Martin Buser, MPH, FACHE, a partner in Hospitalist Management Resources, LLC, in San Diego. “Smart practices are looking at NPPs, evaluating where they do the most good, and then setting out their role and expectations based on these needs and the practice environment.”
Hiring mistakes can be compounded if the NPP is not a good match to the job description or group expectations. If the practice hires an NPP fresh out of school, the group will need to establish training and have the new hire work more closely with physicians. If, on the other hand, an NP has 10 years of experience in an ICU, or a PA has worked in the ED for the past five years, a higher level of autonomy can be granted sooner. However, NPPs with established backgrounds are almost as rare as experienced hospitalists (see “Integrating NPPs Into HM Practice,” p. 38).
Inevitably, there will be changes in the interactions between patients and the hospitalists, as both physicians and NPPs become more comfortable with the other’s practice style, as well as each other’s strengths and weaknesses.
MD-to-NPP Ratio Varies
The practice structure and optimal mix of NPPs to MDs is something that will be specific to the hospitalist group. “We don’t really have good studies on this subject,” Buser says. “I usually get worried when we exceed two NPPs to one MD.”
Others disagree. Dr. Parekh, who works in an academic center, says his group has been successful having one MD work with as many as three NPPs. At the other end, Dr. Wilson says his 10 years of experience suggest 1:1 is the most efficient ratio.
However, all of them agree that having one NPP work with more than one physician is not sustainable. The NPP will be less familiar with each doctor’s practice style, what kind of information they need, and how things should be presented. If two or more hospitalists share an NPP, there can be internal friction over division of the NPP’s time, as well as extending the time before the MDs have a good feel for the NPP’s strengths and weaknesses.
In the final analysis, the HM group has to look at the amount and type of work available. In some cases, it will make financial and clinical sense to bring on an NPP. Under other circumstances, an FTE hospitalist is the best fit.
“Sustainability, quality, and efficiency are the drivers for NPP/MD teams. Increasing pressure to offset program costs is not,” Dr. Wilson says. “You do it because it helps sustain the program, helps with recruiting, and effects your efficiency.” TH
Kurt Ullman is a freelance medical writer based in Indiana.
Reference
- Medical Group Management Association and the Society of Hospital Medicine. State of Hospital Medicine: 2010 Report Based on 2009 Data. 2010. Philadelphia and Englewood, Colo.