User login
Alan M. Rapoport, MD, on Medication Overuse Headache
Neurology Reviews recently shared two poll questions with our Facebook followers about treatment medication overuse headache (MOH). I was very interested to see the results of our poll. While the number of responses was somewhat low, we do get some sense of what respondents are saying. In this commentary, I will first tell you my perspective on the answers and then we will see what some other headache specialists say about the answers to these questions.
Poll Results:
Can MOH be treated with preventive medications without detoxification?
33 votes
YES, 39%
NO, 61%
Can MOH be treated with the new preventive medications (the monoclonal antibodies to CGRP ligand or receptor) without detoxification?
26 votes
YES, 38%
NO, 62%
My Commentary:
Let me explain in more detail my thoughts on the first question, “Can MOH be treated with preventive medications without detoxification?”
If a patient had the diagnosis of MOH – meaning 15 or more headache days per month for at least 3 months, with use of stronger medications (triptans, ergots, opiates, butalbital-containing medications) for 10 days per month or milder treatment (aspirin, acetaminophen, nonsteroidal anti-inflammatory drugs [NSAIDs]) for 15 days per month – can they improve by being put on a traditional preventive medication without intentionally reducing their overused acute medications by a detoxification protocol ordered by a doctor or nurse?
Only 39% of our audience said yes. Yet some studies have shown that patients placed on onabotulinumtoxinA or topiramate might improve without them going through a detoxification of the overused medications. As a physician, I would suggest simultaneously decreasing in their acute medications. I think in some cases this approach creates additional improvement and makes the patient feel better. It would be better for their quality of life, as well as for their kidneys and possibly even their brains.
Here are my thoughts on the second question, “Can MOH be treated with the new preventive medications (the monoclonal antibodies to CGRP ligand or receptor), without detoxification?”
If a patient has MOH, can you expect them to improve after being placed on 1 of the 4 monoclonal antibodies to CGRP ligand or its receptor – all of which are either recently approved or currently in development – without suggesting that they decrease their overused acute care medications? Note that erenumab (Aimovig-aaoe) has been approved by the FDA and marketed as of the time of this writing; we expect 2 more products to be approved very soon.
Almost an identical percentage of our audience (38%) said yes. There is evidence in published clinical trials that those patients given these new medications did about as well with or without the presence of MOH, and both groups did better than the placebo patients. Note that most trials prohibited overuse of opiates and butalbital.
I am a firm believer of detoxifying patients from their overused over-the-counter (OTC) or prescription medications. I believe that opiates and butalbital-containing medications, when overused, are worse for patients than OTCs, NSAIDs, ergot and triptans, but all of these can cause MOH. There are many studies showing that both inpatient and outpatient detoxification alone can really help. However, it is difficult to detoxify patients and some refuse to try this approach.
So, what should we do as physicians? If a patient has MOH, I educate them, try to detoxify them slowly on an outpatient basis, and if I feel it will help, start them on a preventive medication, even before the detoxification begins so they can reach therapeutic levels. In the future, will I use one of the standard preventives, approved or off-label, for migraine prevention (beta blockers, topiramate and other anticonvulsants, antidepressants, angiotensin receptor blockers, onabotulinumtoxinA and others)? It remains to be seen. I am leaning towards the anti CGRP ligand and receptor monoclonal antibodies and preventive small molecule oral CGRP receptor blockers. While that might be enough to start with, I will continue explaining to my patients why they should actively begin a slow detoxification.
Let us see what some headache specialists said about both questions.
Robert Cowan, MD, FAAN:
There have been studies that show migraine can improve without the discontinuation of medication overuse. But that is not what the question asks. The question as posed is whether MOH can be treated with a preventive medication without detoxification. Since the diagnosis of MOH has, in the past, required the cessation of overuse leading to an improvement in the underlying headache, then technically, the answer would be “no.” But that being said, there is ample evidence that the number of headache days/months and other measures of headache can, in fact, improve with the introduction of a preventive, along with other measures such as lifestyle modification. The other ambiguity in the question has to do with what is meant by “detoxification.” Is this a hospital-based detox, or is a gradual decrease in the offending medicine in combination with the addition of a preventive, still considered “detoxification?” Also, does the response imply a sequential relationship between the detoxification and initiation of the preventive? Without further clarification, this response ratio to the question is very difficult to interpret.
There is animal data that suggests acute migraine medications may promote MOH in susceptible individuals through CGRP-dependent mechanisms and anti-CGRP antibodies may be useful for the MOH (Cephalalgia. 2017;37(6):560-570. doi: 10.1177/0333102416650702). While there are no published CGRP antibody studies that did not exclude MOH patients to my knowledge, an abstract by Silberstein et al at the recent AHS Scientific Meeting reported decreased use of overused medication with fremanazumab (Headache. 2018;58(S2):76-78).
Ira Turner, MD
There is clear data to suggest that it is not necessary to detoxify these patients before starting preventive therapy. This is true for the older and newer medications. In fact, not only do these preventive therapies still work in the presence of medication overuse, but they also help to reduce medication overuse. The one caveat that must be mentioned is that this may not apply to opiate overuse. Opiate over-users were excluded from these studies.
While it is of course our goal to reduce and stop acute medication overuse, it should not be done at the expense of delaying preventive therapy. In fact, it is desirable to do both simultaneously. This applies to oral preventive medications, botulinum toxin and CGRP monoclonal antibodies.
In view of this well-established data, it was quite surprising to me to see the results of the 2 polls cited. It seems as if we still have a lot of educating to do regarding migraine prevention in general and with medication overuse in migraine in particular.
Stewart Tepper, MD
Dr. Tepper did not have time to comment, but suggested we show you an abstract presented at the recent AHS meeting. It shows that erenumab-aaoe helps patients with MOH who have not been detoxified (Headache. 2018;58(S2):160-162).
###
Please write to us at Neurology Reviews Migraine Resource Center (mrc@mdedge.com) with your opinions.
Alan M. Rapoport, MD
Editor-in-Chief
Migraine Resource Center
Clinical Professor of Neurology
The David Geffen School of Medicine at UCLA
Los Angeles, California
Neurology Reviews recently shared two poll questions with our Facebook followers about treatment medication overuse headache (MOH). I was very interested to see the results of our poll. While the number of responses was somewhat low, we do get some sense of what respondents are saying. In this commentary, I will first tell you my perspective on the answers and then we will see what some other headache specialists say about the answers to these questions.
Poll Results:
Can MOH be treated with preventive medications without detoxification?
33 votes
YES, 39%
NO, 61%
Can MOH be treated with the new preventive medications (the monoclonal antibodies to CGRP ligand or receptor) without detoxification?
26 votes
YES, 38%
NO, 62%
My Commentary:
Let me explain in more detail my thoughts on the first question, “Can MOH be treated with preventive medications without detoxification?”
If a patient had the diagnosis of MOH – meaning 15 or more headache days per month for at least 3 months, with use of stronger medications (triptans, ergots, opiates, butalbital-containing medications) for 10 days per month or milder treatment (aspirin, acetaminophen, nonsteroidal anti-inflammatory drugs [NSAIDs]) for 15 days per month – can they improve by being put on a traditional preventive medication without intentionally reducing their overused acute medications by a detoxification protocol ordered by a doctor or nurse?
Only 39% of our audience said yes. Yet some studies have shown that patients placed on onabotulinumtoxinA or topiramate might improve without them going through a detoxification of the overused medications. As a physician, I would suggest simultaneously decreasing in their acute medications. I think in some cases this approach creates additional improvement and makes the patient feel better. It would be better for their quality of life, as well as for their kidneys and possibly even their brains.
Here are my thoughts on the second question, “Can MOH be treated with the new preventive medications (the monoclonal antibodies to CGRP ligand or receptor), without detoxification?”
If a patient has MOH, can you expect them to improve after being placed on 1 of the 4 monoclonal antibodies to CGRP ligand or its receptor – all of which are either recently approved or currently in development – without suggesting that they decrease their overused acute care medications? Note that erenumab (Aimovig-aaoe) has been approved by the FDA and marketed as of the time of this writing; we expect 2 more products to be approved very soon.
Almost an identical percentage of our audience (38%) said yes. There is evidence in published clinical trials that those patients given these new medications did about as well with or without the presence of MOH, and both groups did better than the placebo patients. Note that most trials prohibited overuse of opiates and butalbital.
I am a firm believer of detoxifying patients from their overused over-the-counter (OTC) or prescription medications. I believe that opiates and butalbital-containing medications, when overused, are worse for patients than OTCs, NSAIDs, ergot and triptans, but all of these can cause MOH. There are many studies showing that both inpatient and outpatient detoxification alone can really help. However, it is difficult to detoxify patients and some refuse to try this approach.
So, what should we do as physicians? If a patient has MOH, I educate them, try to detoxify them slowly on an outpatient basis, and if I feel it will help, start them on a preventive medication, even before the detoxification begins so they can reach therapeutic levels. In the future, will I use one of the standard preventives, approved or off-label, for migraine prevention (beta blockers, topiramate and other anticonvulsants, antidepressants, angiotensin receptor blockers, onabotulinumtoxinA and others)? It remains to be seen. I am leaning towards the anti CGRP ligand and receptor monoclonal antibodies and preventive small molecule oral CGRP receptor blockers. While that might be enough to start with, I will continue explaining to my patients why they should actively begin a slow detoxification.
Let us see what some headache specialists said about both questions.
Robert Cowan, MD, FAAN:
There have been studies that show migraine can improve without the discontinuation of medication overuse. But that is not what the question asks. The question as posed is whether MOH can be treated with a preventive medication without detoxification. Since the diagnosis of MOH has, in the past, required the cessation of overuse leading to an improvement in the underlying headache, then technically, the answer would be “no.” But that being said, there is ample evidence that the number of headache days/months and other measures of headache can, in fact, improve with the introduction of a preventive, along with other measures such as lifestyle modification. The other ambiguity in the question has to do with what is meant by “detoxification.” Is this a hospital-based detox, or is a gradual decrease in the offending medicine in combination with the addition of a preventive, still considered “detoxification?” Also, does the response imply a sequential relationship between the detoxification and initiation of the preventive? Without further clarification, this response ratio to the question is very difficult to interpret.
There is animal data that suggests acute migraine medications may promote MOH in susceptible individuals through CGRP-dependent mechanisms and anti-CGRP antibodies may be useful for the MOH (Cephalalgia. 2017;37(6):560-570. doi: 10.1177/0333102416650702). While there are no published CGRP antibody studies that did not exclude MOH patients to my knowledge, an abstract by Silberstein et al at the recent AHS Scientific Meeting reported decreased use of overused medication with fremanazumab (Headache. 2018;58(S2):76-78).
Ira Turner, MD
There is clear data to suggest that it is not necessary to detoxify these patients before starting preventive therapy. This is true for the older and newer medications. In fact, not only do these preventive therapies still work in the presence of medication overuse, but they also help to reduce medication overuse. The one caveat that must be mentioned is that this may not apply to opiate overuse. Opiate over-users were excluded from these studies.
While it is of course our goal to reduce and stop acute medication overuse, it should not be done at the expense of delaying preventive therapy. In fact, it is desirable to do both simultaneously. This applies to oral preventive medications, botulinum toxin and CGRP monoclonal antibodies.
In view of this well-established data, it was quite surprising to me to see the results of the 2 polls cited. It seems as if we still have a lot of educating to do regarding migraine prevention in general and with medication overuse in migraine in particular.
Stewart Tepper, MD
Dr. Tepper did not have time to comment, but suggested we show you an abstract presented at the recent AHS meeting. It shows that erenumab-aaoe helps patients with MOH who have not been detoxified (Headache. 2018;58(S2):160-162).
###
Please write to us at Neurology Reviews Migraine Resource Center (mrc@mdedge.com) with your opinions.
Alan M. Rapoport, MD
Editor-in-Chief
Migraine Resource Center
Clinical Professor of Neurology
The David Geffen School of Medicine at UCLA
Los Angeles, California
Neurology Reviews recently shared two poll questions with our Facebook followers about treatment medication overuse headache (MOH). I was very interested to see the results of our poll. While the number of responses was somewhat low, we do get some sense of what respondents are saying. In this commentary, I will first tell you my perspective on the answers and then we will see what some other headache specialists say about the answers to these questions.
Poll Results:
Can MOH be treated with preventive medications without detoxification?
33 votes
YES, 39%
NO, 61%
Can MOH be treated with the new preventive medications (the monoclonal antibodies to CGRP ligand or receptor) without detoxification?
26 votes
YES, 38%
NO, 62%
My Commentary:
Let me explain in more detail my thoughts on the first question, “Can MOH be treated with preventive medications without detoxification?”
If a patient had the diagnosis of MOH – meaning 15 or more headache days per month for at least 3 months, with use of stronger medications (triptans, ergots, opiates, butalbital-containing medications) for 10 days per month or milder treatment (aspirin, acetaminophen, nonsteroidal anti-inflammatory drugs [NSAIDs]) for 15 days per month – can they improve by being put on a traditional preventive medication without intentionally reducing their overused acute medications by a detoxification protocol ordered by a doctor or nurse?
Only 39% of our audience said yes. Yet some studies have shown that patients placed on onabotulinumtoxinA or topiramate might improve without them going through a detoxification of the overused medications. As a physician, I would suggest simultaneously decreasing in their acute medications. I think in some cases this approach creates additional improvement and makes the patient feel better. It would be better for their quality of life, as well as for their kidneys and possibly even their brains.
Here are my thoughts on the second question, “Can MOH be treated with the new preventive medications (the monoclonal antibodies to CGRP ligand or receptor), without detoxification?”
If a patient has MOH, can you expect them to improve after being placed on 1 of the 4 monoclonal antibodies to CGRP ligand or its receptor – all of which are either recently approved or currently in development – without suggesting that they decrease their overused acute care medications? Note that erenumab (Aimovig-aaoe) has been approved by the FDA and marketed as of the time of this writing; we expect 2 more products to be approved very soon.
Almost an identical percentage of our audience (38%) said yes. There is evidence in published clinical trials that those patients given these new medications did about as well with or without the presence of MOH, and both groups did better than the placebo patients. Note that most trials prohibited overuse of opiates and butalbital.
I am a firm believer of detoxifying patients from their overused over-the-counter (OTC) or prescription medications. I believe that opiates and butalbital-containing medications, when overused, are worse for patients than OTCs, NSAIDs, ergot and triptans, but all of these can cause MOH. There are many studies showing that both inpatient and outpatient detoxification alone can really help. However, it is difficult to detoxify patients and some refuse to try this approach.
So, what should we do as physicians? If a patient has MOH, I educate them, try to detoxify them slowly on an outpatient basis, and if I feel it will help, start them on a preventive medication, even before the detoxification begins so they can reach therapeutic levels. In the future, will I use one of the standard preventives, approved or off-label, for migraine prevention (beta blockers, topiramate and other anticonvulsants, antidepressants, angiotensin receptor blockers, onabotulinumtoxinA and others)? It remains to be seen. I am leaning towards the anti CGRP ligand and receptor monoclonal antibodies and preventive small molecule oral CGRP receptor blockers. While that might be enough to start with, I will continue explaining to my patients why they should actively begin a slow detoxification.
Let us see what some headache specialists said about both questions.
Robert Cowan, MD, FAAN:
There have been studies that show migraine can improve without the discontinuation of medication overuse. But that is not what the question asks. The question as posed is whether MOH can be treated with a preventive medication without detoxification. Since the diagnosis of MOH has, in the past, required the cessation of overuse leading to an improvement in the underlying headache, then technically, the answer would be “no.” But that being said, there is ample evidence that the number of headache days/months and other measures of headache can, in fact, improve with the introduction of a preventive, along with other measures such as lifestyle modification. The other ambiguity in the question has to do with what is meant by “detoxification.” Is this a hospital-based detox, or is a gradual decrease in the offending medicine in combination with the addition of a preventive, still considered “detoxification?” Also, does the response imply a sequential relationship between the detoxification and initiation of the preventive? Without further clarification, this response ratio to the question is very difficult to interpret.
There is animal data that suggests acute migraine medications may promote MOH in susceptible individuals through CGRP-dependent mechanisms and anti-CGRP antibodies may be useful for the MOH (Cephalalgia. 2017;37(6):560-570. doi: 10.1177/0333102416650702). While there are no published CGRP antibody studies that did not exclude MOH patients to my knowledge, an abstract by Silberstein et al at the recent AHS Scientific Meeting reported decreased use of overused medication with fremanazumab (Headache. 2018;58(S2):76-78).
Ira Turner, MD
There is clear data to suggest that it is not necessary to detoxify these patients before starting preventive therapy. This is true for the older and newer medications. In fact, not only do these preventive therapies still work in the presence of medication overuse, but they also help to reduce medication overuse. The one caveat that must be mentioned is that this may not apply to opiate overuse. Opiate over-users were excluded from these studies.
While it is of course our goal to reduce and stop acute medication overuse, it should not be done at the expense of delaying preventive therapy. In fact, it is desirable to do both simultaneously. This applies to oral preventive medications, botulinum toxin and CGRP monoclonal antibodies.
In view of this well-established data, it was quite surprising to me to see the results of the 2 polls cited. It seems as if we still have a lot of educating to do regarding migraine prevention in general and with medication overuse in migraine in particular.
Stewart Tepper, MD
Dr. Tepper did not have time to comment, but suggested we show you an abstract presented at the recent AHS meeting. It shows that erenumab-aaoe helps patients with MOH who have not been detoxified (Headache. 2018;58(S2):160-162).
###
Please write to us at Neurology Reviews Migraine Resource Center (mrc@mdedge.com) with your opinions.
Alan M. Rapoport, MD
Editor-in-Chief
Migraine Resource Center
Clinical Professor of Neurology
The David Geffen School of Medicine at UCLA
Los Angeles, California
Breast cancer patients getting unnecessary scans against recommendations
PHOENIX – Despite clear guidance on lack of benefit and potential harms, many patients with early-stage breast cancers at low metastasis risk have undergone imaging tests for staging, recent retrospective studies have shown.
Nearly one-third of early-stage breast cancer patients received the unnecessary and potentially harmful interventions in one of the two studies presented at a symposium on quality care sponsored by the American Society of Clinical Oncology (ASCO).
Low-risk patients in that study were more likely to undergo imaging if they were younger or had triple-negative disease, among other factors, said researcher Brett Barlow, of the University of Alabama at Birmingham.
Physicians should be further educated about the low-risk nature of early-stage breast cancer, even in subgroups that are perceived to be higher risk, Mr. Barlow said in an interview.
“I think we can reassure physicians that these patients will do well and that these guidelines are based on good data,” he said “There could potentially be an element of distrust in these guidelines in these higher-risk patients, and that may be what’s driving some of these extra tests.”
According to Mr. Barlow, imaging low-risk patients is inconsistent with guidance from Choosing Wisely, an initiative designed to promote discussions between clinicians and patients about medical tests or procedures that are unnecessary.
As part of that initiative, ASCO recommended that PET, CT, and radionuclide bone scans should not be performed for staging of early-stage breast cancer at low risk for metastasis.
There is a lack of evidence that demonstrates a benefit for those imaging modalities in patients with newly identified ductal carcinoma in situ (DCIS) or clinical stage I or II disease, the society said at the time.
Unnecessary imaging can result in unnecessary invasive procedures, overtreatment, radiation exposure, and misdiagnosis, the society said in the guidance, which was published in 2012.
Despite the guidance, 262 out of 872 patients with stage 0-II breast cancer (30%) seen during 2013-2015 underwent imaging, according to results of the single-center retrospective cohort study Mr. Barlow and his coauthors described in a poster presentation.
The median age of the patients who underwent unnecessary imaging was 55 years versus 60 years for patients who did not, according to the researchers.
Risk of inappropriate imaging was increased in younger patients, those with triple-negative disease versus those with any hormone receptor–positive disease, those with higher-stage breast cancer, and those without Medicare insurance, investigators found.
Although it’s unclear whether there were any formal, institution-level efforts to promote the ASCO recommendations during the 2013-2015 period, it was “definitely a topic of debate at the time,” Mr. Barlow said.
“Something we hope to evaluate further is whether we have improved,” he said. “It’s important to set a baseline and see how we did in this area. We look forward to reevaluating that in a few years to see.”
In a separate study, investigators reviewed records from Mount Sinai Health System in New York and found that unnecessary scans were performed in 19% of patients diagnosed with stage I-II breast cancer during 2014-2015.
No cases of metastatic disease were found in 733 patients included in the study, and 43% had false-positive findings, according to Ana I. Velazquez Manana, MD, MS, of Mount Sinai Beth Israel Foundation, New York, and her coinvestigators.
Imaging increased costs by $4,480 per patient, according to the investigators, who found in multivariate analysis that the unnecessary scans were associated with young age, presence of T2 tumor, positive lymph nodes, and triple-negative disease.
“Further educational efforts are needed to avoid unnecessary scans in patients with early-stage breast cancer,” the researchers wrote in an abstract describing the results.
Mr. Barlow reported no disclosures, while one coauthor reported disclosures related to Carevive Systems, Genentech/Roche, Medscape, Pack Health, and Pfizer. Dr. Velazquez Manana and her coauthors had no relationships to disclose, and their study was funded by the Medical Student Rotation for Underrepresented Populations.
SOURCE: Barlow B et al. Quality Care Symposium, Abstract 51. Velazquez Manana AI et al. Quality Care Symposium, Abstract 52.
PHOENIX – Despite clear guidance on lack of benefit and potential harms, many patients with early-stage breast cancers at low metastasis risk have undergone imaging tests for staging, recent retrospective studies have shown.
Nearly one-third of early-stage breast cancer patients received the unnecessary and potentially harmful interventions in one of the two studies presented at a symposium on quality care sponsored by the American Society of Clinical Oncology (ASCO).
Low-risk patients in that study were more likely to undergo imaging if they were younger or had triple-negative disease, among other factors, said researcher Brett Barlow, of the University of Alabama at Birmingham.
Physicians should be further educated about the low-risk nature of early-stage breast cancer, even in subgroups that are perceived to be higher risk, Mr. Barlow said in an interview.
“I think we can reassure physicians that these patients will do well and that these guidelines are based on good data,” he said “There could potentially be an element of distrust in these guidelines in these higher-risk patients, and that may be what’s driving some of these extra tests.”
According to Mr. Barlow, imaging low-risk patients is inconsistent with guidance from Choosing Wisely, an initiative designed to promote discussions between clinicians and patients about medical tests or procedures that are unnecessary.
As part of that initiative, ASCO recommended that PET, CT, and radionuclide bone scans should not be performed for staging of early-stage breast cancer at low risk for metastasis.
There is a lack of evidence that demonstrates a benefit for those imaging modalities in patients with newly identified ductal carcinoma in situ (DCIS) or clinical stage I or II disease, the society said at the time.
Unnecessary imaging can result in unnecessary invasive procedures, overtreatment, radiation exposure, and misdiagnosis, the society said in the guidance, which was published in 2012.
Despite the guidance, 262 out of 872 patients with stage 0-II breast cancer (30%) seen during 2013-2015 underwent imaging, according to results of the single-center retrospective cohort study Mr. Barlow and his coauthors described in a poster presentation.
The median age of the patients who underwent unnecessary imaging was 55 years versus 60 years for patients who did not, according to the researchers.
Risk of inappropriate imaging was increased in younger patients, those with triple-negative disease versus those with any hormone receptor–positive disease, those with higher-stage breast cancer, and those without Medicare insurance, investigators found.
Although it’s unclear whether there were any formal, institution-level efforts to promote the ASCO recommendations during the 2013-2015 period, it was “definitely a topic of debate at the time,” Mr. Barlow said.
“Something we hope to evaluate further is whether we have improved,” he said. “It’s important to set a baseline and see how we did in this area. We look forward to reevaluating that in a few years to see.”
In a separate study, investigators reviewed records from Mount Sinai Health System in New York and found that unnecessary scans were performed in 19% of patients diagnosed with stage I-II breast cancer during 2014-2015.
No cases of metastatic disease were found in 733 patients included in the study, and 43% had false-positive findings, according to Ana I. Velazquez Manana, MD, MS, of Mount Sinai Beth Israel Foundation, New York, and her coinvestigators.
Imaging increased costs by $4,480 per patient, according to the investigators, who found in multivariate analysis that the unnecessary scans were associated with young age, presence of T2 tumor, positive lymph nodes, and triple-negative disease.
“Further educational efforts are needed to avoid unnecessary scans in patients with early-stage breast cancer,” the researchers wrote in an abstract describing the results.
Mr. Barlow reported no disclosures, while one coauthor reported disclosures related to Carevive Systems, Genentech/Roche, Medscape, Pack Health, and Pfizer. Dr. Velazquez Manana and her coauthors had no relationships to disclose, and their study was funded by the Medical Student Rotation for Underrepresented Populations.
SOURCE: Barlow B et al. Quality Care Symposium, Abstract 51. Velazquez Manana AI et al. Quality Care Symposium, Abstract 52.
PHOENIX – Despite clear guidance on lack of benefit and potential harms, many patients with early-stage breast cancers at low metastasis risk have undergone imaging tests for staging, recent retrospective studies have shown.
Nearly one-third of early-stage breast cancer patients received the unnecessary and potentially harmful interventions in one of the two studies presented at a symposium on quality care sponsored by the American Society of Clinical Oncology (ASCO).
Low-risk patients in that study were more likely to undergo imaging if they were younger or had triple-negative disease, among other factors, said researcher Brett Barlow, of the University of Alabama at Birmingham.
Physicians should be further educated about the low-risk nature of early-stage breast cancer, even in subgroups that are perceived to be higher risk, Mr. Barlow said in an interview.
“I think we can reassure physicians that these patients will do well and that these guidelines are based on good data,” he said “There could potentially be an element of distrust in these guidelines in these higher-risk patients, and that may be what’s driving some of these extra tests.”
According to Mr. Barlow, imaging low-risk patients is inconsistent with guidance from Choosing Wisely, an initiative designed to promote discussions between clinicians and patients about medical tests or procedures that are unnecessary.
As part of that initiative, ASCO recommended that PET, CT, and radionuclide bone scans should not be performed for staging of early-stage breast cancer at low risk for metastasis.
There is a lack of evidence that demonstrates a benefit for those imaging modalities in patients with newly identified ductal carcinoma in situ (DCIS) or clinical stage I or II disease, the society said at the time.
Unnecessary imaging can result in unnecessary invasive procedures, overtreatment, radiation exposure, and misdiagnosis, the society said in the guidance, which was published in 2012.
Despite the guidance, 262 out of 872 patients with stage 0-II breast cancer (30%) seen during 2013-2015 underwent imaging, according to results of the single-center retrospective cohort study Mr. Barlow and his coauthors described in a poster presentation.
The median age of the patients who underwent unnecessary imaging was 55 years versus 60 years for patients who did not, according to the researchers.
Risk of inappropriate imaging was increased in younger patients, those with triple-negative disease versus those with any hormone receptor–positive disease, those with higher-stage breast cancer, and those without Medicare insurance, investigators found.
Although it’s unclear whether there were any formal, institution-level efforts to promote the ASCO recommendations during the 2013-2015 period, it was “definitely a topic of debate at the time,” Mr. Barlow said.
“Something we hope to evaluate further is whether we have improved,” he said. “It’s important to set a baseline and see how we did in this area. We look forward to reevaluating that in a few years to see.”
In a separate study, investigators reviewed records from Mount Sinai Health System in New York and found that unnecessary scans were performed in 19% of patients diagnosed with stage I-II breast cancer during 2014-2015.
No cases of metastatic disease were found in 733 patients included in the study, and 43% had false-positive findings, according to Ana I. Velazquez Manana, MD, MS, of Mount Sinai Beth Israel Foundation, New York, and her coinvestigators.
Imaging increased costs by $4,480 per patient, according to the investigators, who found in multivariate analysis that the unnecessary scans were associated with young age, presence of T2 tumor, positive lymph nodes, and triple-negative disease.
“Further educational efforts are needed to avoid unnecessary scans in patients with early-stage breast cancer,” the researchers wrote in an abstract describing the results.
Mr. Barlow reported no disclosures, while one coauthor reported disclosures related to Carevive Systems, Genentech/Roche, Medscape, Pack Health, and Pfizer. Dr. Velazquez Manana and her coauthors had no relationships to disclose, and their study was funded by the Medical Student Rotation for Underrepresented Populations.
SOURCE: Barlow B et al. Quality Care Symposium, Abstract 51. Velazquez Manana AI et al. Quality Care Symposium, Abstract 52.
REPORTING FROM THE QUALITY CARE SYMPOSIUM
Key clinical point: Many patients with early breast cancers at low metastasis risk received imaging tests for staging despite ASCO recommendations against such testing.
Major finding: In two studies, 30% and 19% of low-risk breast cancer patients underwent imaging for staging.
Study details: Two single-center, retrospective cohort studies that included 872 and 733 patients, respectively.
Disclosures: In one study, researchers reported disclosures related to Carevive Systems, Genentech/Roche, Medscape, Pack Health, and Pfizer. The second study was funded by the Medical Student Rotation for Underrepresented Populations.
Source: Barlow B et al. Quality Care Symposium, Abstract 51; Velazquez Manana AI et al. Quality Care Symposium, Abstract 52.
No evidence of subclinical axial involvement seen in skin psoriasis
A study of individuals with longstanding skin psoriasis but no clinical arthritis or spondylitis has found no evidence of subclinical involvement of the sacroiliac joint or spine.
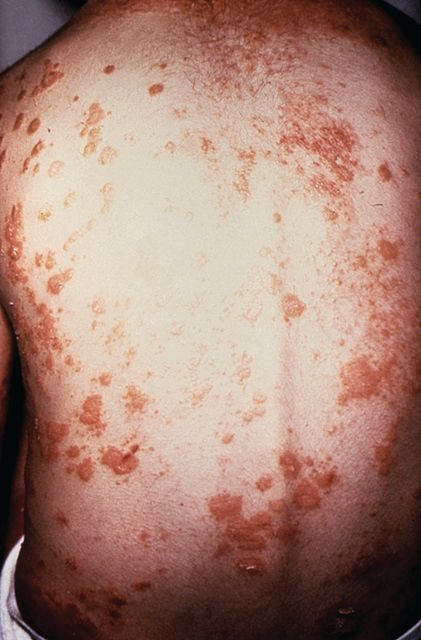
The prevalence of sacroiliac lesions on blinded MRI assessment was similar in 20 patients who had skin psoriasis for a median of 23 years and in 22 healthy controls, and no sacroiliac ankylosis was seen in either group. Similarly, there was no significant difference between the two groups in spinal lesions on MRI, nor in any of the five levels of lesion frequency, Vlad A. Bratu, MD, of the department of radiology at University Hospital Basel (Switzerland) and his coauthors reported in Arthritis Care & Research.
On blinded MRI assessment, five (25%) patients with skin psoriasis and two (9.1%) controls were classified as having inflammation of the sacroiliac joint. Three of these patients in the psoriasis group and one in the control group were older than 50, and the three with psoriasis had had the condition for 26-35 years.
Dr. Bratu and his colleagues said that subclinical peripheral joint inflammation on MRI had previously been a common finding in patients who had skin psoriasis but no clinical signs of psoriatic arthritis. But given the limited evidence of concomitant subclinical axial or spinal inflammation in their study, the authors argued there was no support for routine screening for potential subclinical axial inflammation in patients with longstanding skin psoriasis.
They noted that bone marrow edema lesions in at least two sacroiliac joint quadrants were seen in 35% of patients with psoriasis and 23% of healthy controls, a finding that reflected those seen in other studies in healthy individuals.
“If a specificity threshold for a given MRI lesion of at least 0.9 is applied for axial MRI to discriminate between axial SpA [spondyloarthritis] and background variation in healthy controls or in differential diagnostic conditions, no more than 10% of healthy controls in our study should meet this criterion by an individual level data analysis,” they wrote.
The authors also pointed out the impact of age on lesion frequency, which was more evident in spinal lesions.
“This observation supports the hypothesis that some spinal alterations in higher age may reflect degenerative rather than inflammatory changes,” they wrote. “However, there is a gap in knowledge with virtually no evidence about presence and pattern of degenerative versus inflammatory spinal lesions in subjects beyond 50 years of age.”
The study was supported by the Gottfried and Julia Bangerter-Rhyner Foundation. No conflicts of interest were declared.
SOURCE: Bratu V et al. Arthritis Care Res. 2018 Sep 22. doi: 10.1002/acr.23767.
A study of individuals with longstanding skin psoriasis but no clinical arthritis or spondylitis has found no evidence of subclinical involvement of the sacroiliac joint or spine.
The prevalence of sacroiliac lesions on blinded MRI assessment was similar in 20 patients who had skin psoriasis for a median of 23 years and in 22 healthy controls, and no sacroiliac ankylosis was seen in either group. Similarly, there was no significant difference between the two groups in spinal lesions on MRI, nor in any of the five levels of lesion frequency, Vlad A. Bratu, MD, of the department of radiology at University Hospital Basel (Switzerland) and his coauthors reported in Arthritis Care & Research.
On blinded MRI assessment, five (25%) patients with skin psoriasis and two (9.1%) controls were classified as having inflammation of the sacroiliac joint. Three of these patients in the psoriasis group and one in the control group were older than 50, and the three with psoriasis had had the condition for 26-35 years.
Dr. Bratu and his colleagues said that subclinical peripheral joint inflammation on MRI had previously been a common finding in patients who had skin psoriasis but no clinical signs of psoriatic arthritis. But given the limited evidence of concomitant subclinical axial or spinal inflammation in their study, the authors argued there was no support for routine screening for potential subclinical axial inflammation in patients with longstanding skin psoriasis.
They noted that bone marrow edema lesions in at least two sacroiliac joint quadrants were seen in 35% of patients with psoriasis and 23% of healthy controls, a finding that reflected those seen in other studies in healthy individuals.
“If a specificity threshold for a given MRI lesion of at least 0.9 is applied for axial MRI to discriminate between axial SpA [spondyloarthritis] and background variation in healthy controls or in differential diagnostic conditions, no more than 10% of healthy controls in our study should meet this criterion by an individual level data analysis,” they wrote.
The authors also pointed out the impact of age on lesion frequency, which was more evident in spinal lesions.
“This observation supports the hypothesis that some spinal alterations in higher age may reflect degenerative rather than inflammatory changes,” they wrote. “However, there is a gap in knowledge with virtually no evidence about presence and pattern of degenerative versus inflammatory spinal lesions in subjects beyond 50 years of age.”
The study was supported by the Gottfried and Julia Bangerter-Rhyner Foundation. No conflicts of interest were declared.
SOURCE: Bratu V et al. Arthritis Care Res. 2018 Sep 22. doi: 10.1002/acr.23767.
A study of individuals with longstanding skin psoriasis but no clinical arthritis or spondylitis has found no evidence of subclinical involvement of the sacroiliac joint or spine.
The prevalence of sacroiliac lesions on blinded MRI assessment was similar in 20 patients who had skin psoriasis for a median of 23 years and in 22 healthy controls, and no sacroiliac ankylosis was seen in either group. Similarly, there was no significant difference between the two groups in spinal lesions on MRI, nor in any of the five levels of lesion frequency, Vlad A. Bratu, MD, of the department of radiology at University Hospital Basel (Switzerland) and his coauthors reported in Arthritis Care & Research.
On blinded MRI assessment, five (25%) patients with skin psoriasis and two (9.1%) controls were classified as having inflammation of the sacroiliac joint. Three of these patients in the psoriasis group and one in the control group were older than 50, and the three with psoriasis had had the condition for 26-35 years.
Dr. Bratu and his colleagues said that subclinical peripheral joint inflammation on MRI had previously been a common finding in patients who had skin psoriasis but no clinical signs of psoriatic arthritis. But given the limited evidence of concomitant subclinical axial or spinal inflammation in their study, the authors argued there was no support for routine screening for potential subclinical axial inflammation in patients with longstanding skin psoriasis.
They noted that bone marrow edema lesions in at least two sacroiliac joint quadrants were seen in 35% of patients with psoriasis and 23% of healthy controls, a finding that reflected those seen in other studies in healthy individuals.
“If a specificity threshold for a given MRI lesion of at least 0.9 is applied for axial MRI to discriminate between axial SpA [spondyloarthritis] and background variation in healthy controls or in differential diagnostic conditions, no more than 10% of healthy controls in our study should meet this criterion by an individual level data analysis,” they wrote.
The authors also pointed out the impact of age on lesion frequency, which was more evident in spinal lesions.
“This observation supports the hypothesis that some spinal alterations in higher age may reflect degenerative rather than inflammatory changes,” they wrote. “However, there is a gap in knowledge with virtually no evidence about presence and pattern of degenerative versus inflammatory spinal lesions in subjects beyond 50 years of age.”
The study was supported by the Gottfried and Julia Bangerter-Rhyner Foundation. No conflicts of interest were declared.
SOURCE: Bratu V et al. Arthritis Care Res. 2018 Sep 22. doi: 10.1002/acr.23767.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: The prevalence of sacroiliac bone marrow lesions was similar between patients with skin psoriasis and healthy controls.
Study details: Case-control study in 20 patients with skin psoriasis and 22 healthy controls.
Disclosures: The study was supported by the Gottfried and Julia Bangerter-Rhyner Foundation. No conflicts of interest were declared.
Source: Bratu V et al. Arthritis Care Res. 2018 Sep 22. doi: 10.1002/acr.23767.
Conference News Roundup—European Society of Cardiology
Patients With High Blood Pressure Should Undergo Clock Drawing Test
A clock drawing test for detecting cognitive dysfunction should be conducted routinely in patients with high blood pressure, according to researchers.
Patients with high blood pressure who have impaired cognitive function are at increased risk of developing dementia within five years. Despite this known link, cognitive function is not routinely measured in patients with high blood pressure.
“The ability to draw the numbers of a clock and a particular time is an easy way to find out if a patient with high blood pressure has cognitive impairment,” said Augusto Vicario, MD, a cardiologist at the Cardiovascular Institute of Buenos Aires. “Identifying these patients provides the opportunity to intervene before dementia develops.”
The Heart-Brain Study in Argentina evaluated the usefulness of the clock drawing test, compared with the Mini-Mental State Examination (MMSE), to detect cognitive impairment in 1,414 adults with high blood pressure recruited from 18 cardiology centers in Argentina. The population’s average blood pressure was 144/84 mm Hg, average age was 60, and 62% were women.
For the clock drawing test, patients were given a piece of paper with a circle on it (diameter, 10 cm). They were asked to write the numbers of the clock in the correct positions inside the circle and draw hands on the clock indicating the time “20 to four.” Patients were scored as having normal cognition or moderate or severe cognitive impairment.
The researchers found a higher prevalence of cognitive impairment with the clock drawing test (36%), compared with the MMSE (21%). Three of 10 patients who had a normal MMSE score had an abnormal clock drawing result. The disparity in results between the two tests was greatest in middle-aged patients.
“Untreated high blood pressure silently and progressively damages the arteries in the subcortex of the brain and stops communication between the subcortex and frontal lobe,” said Dr. Vicario. “This disconnect leads to impaired executive functions such as planning, visuospatial abilities, remembering details, and decision-making. The clock drawing test is known to evaluate executive functions. The MMSE evaluates several other cognitive abilities but is weakly correlated with executive functions.
“Our study suggests that the clock drawing test should be preferred over the MMSE for early detection of executive dysfunction in patients with high blood pressure, particularly in middle age. We think the score on the clock drawing test can be considered a surrogate measure of silent vascular damage in the brain and identifies patients at greater risk of developing dementia. In our study, more than one-third of patients were at risk.
“The clock drawing test should be adopted as a routine screening tool for cognitive decline in patients with high blood pressure. Further studies are needed to determine whether lowering blood pressure can prevent progression to dementia,” Dr. Vicario concluded.
Short Sleep Associated With Doubled Risk of Cardiovascular Disease
Middle-aged men who sleep for five hours or fewer per night have twice the risk of developing a major cardiovascular event during the following two decades, compared with men who sleep for seven to eight hours, according to a study.
“For people with busy lives, sleeping may feel like a waste of time, but our study suggests that short sleep could be linked with future cardiovascular disease,” said Moa Bengtsson of the University of Gothenburg, Sweden.
Previous studies have generated conflicting evidence on whether short sleep is associated with a greater chance of having a future cardiovascular event. This study investigated this relationship in 50-year-old men.
In 1993, 50% of all men born in 1943 and living in Gothenburg were randomly selected to participate in the study. Of the 1,463 invited, 798 (55%) men agreed to take part. Participants underwent a physical examination and completed a questionnaire on current health conditions, average sleep duration, physical activity, and smoking. The men were divided into four groups according to their self-estimated average sleep duration at the start of the study: five or fewer hours, six hours, seven to eight hours (considered normal sleep duration), and more than eight hours.
Participants were followed up for 21 years for the occurrence of major cardiovascular events, which included heart attack, stroke, hospitalization due to heart failure, coronary revascularization, or death from cardiovascular disease. Data on cardiovascular events were collected from medical records, the Swedish Hospital Discharge Registry, and the Swedish Cause of Death Register.
Men with incomplete data on sleep duration, incomplete follow-up information, or who had a major cardiovascular event before the start of the study were excluded, leaving a total of 759 men for the analyses.
High blood pressure, diabetes, obesity, current smoking, low physical activity, and poor sleep quality were more common in men who slept for five or fewer hours per night, compared with those who slept for seven to eight hours.
Compared with those with normal sleep duration, men who slept for five or fewer hours per night had a twofold higher risk of having a major cardiovascular event by age 71. The risk remained doubled after adjusting for cardiovascular risk factors at the start of the study, including obesity, diabetes, and smoking.
“Men with the shortest sleep duration at the age of 50 were twice as likely to have had a cardiovascular event by age 71 as those who slept a normal amount, even when other risk factors were taken into account,” said Ms. Bengtsson.
“In our study, the magnitude of increased cardiovascular risk associated with insufficient sleep is similar to that of smoking or having diabetes at age 50. This was an observational study, so, based on our findings, we cannot conclude that short sleep causes cardiovascular disease or say definitively that sleeping more will reduce risk. However, the findings do suggest that sleep is important, and that should be a wake-up call to all of us.”
How Long Is a Good Night’s Sleep?
Researchers have found that a sweet spot of six to eight hours’ sleep per night is most beneficial for heart health. More or less sleep is detrimental.
“We spend one-third of our lives sleeping, yet we know little about the impact of this biologic need on the cardiovascular system,” said Epameinondas Fountas, MD, of the Onassis Cardiac Surgery Centre in Athens.
The study investigated the relationship between sleep duration and cardiovascular disease using a meta-analysis that included 11 prospective studies published within the past five years of more than one million adults without cardiovascular disease.
Two groups, one with short (ie, fewer than six hours) and another with long (ie, more than eight hours) nightly sleep duration, were compared with the reference group (ie, six to eight hours’ sleep).
Short and long sleepers had a greater risk of developing or dying from coronary artery disease or stroke. Compared with adults who slept for six to eight hours per night, short and long sleepers had 11% and 33% greater risks, respectively, of developing or dying from coronary artery disease or stroke during an average follow-up of 9.3 years.
“Our findings suggest that too much or too little sleep may be bad for the heart,” said Dr. Fountas. “More research is needed to clarify exactly why, but we do know that sleep influences biologic processes like glucose metabolism, blood pressure, and inflammation, all of which have an impact on cardiovascular disease.”
A strength of the current analysis is that only prospective studies were included, noted Dr. Fountas. This technique avoids recall bias.
“Having the odd short night or lie-in is unlikely to be detrimental to health, but evidence is accumulating that prolonged nightly sleep deprivation or excessive sleeping should be avoided,” said Dr. Fountas. “The good news is that there are plenty of ways to get into the habit of getting six to eight hours a night: for example, by going to bed and getting up at the same time every day, avoiding alcohol and caffeine before bed, eating healthily, and being physically active. Getting the right amount of sleep is an important part of a healthy lifestyle.”
Four in 10 Patients With Atrial Fibrillation Have Unrecognized Brain Damage
Four out of 10 patients with atrial fibrillation but no history of stroke or transient ischemic attack have previously unrecognized brain damage, according to the first results of the Swiss Atrial Fibrillation Cohort Study (Swiss-AF).
“Our results suggest that clinically unrecognized brain damage may explain the association between dementia and atrial fibrillation in patients without prior stroke,” said David Conen, MD, MPH, Associate Professor of Cardiology at McMaster University in Hamilton, Canada.
Patients with atrial fibrillation have a significantly increased risk of stroke, which is why most are treated with oral anticoagulation. This increased stroke risk is probably the main reason why patients with atrial fibrillation also face an increased risk of cognitive dysfunction and dementia. The relationship between atrial fibrillation and dementia has also been shown among patients without prior strokes, however, meaning that additional mechanisms must be involved. Clarifying the mechanisms by which atrial fibrillation increases the risk of cognitive dysfunction and dementia is a first step towards developing preventive measures.
Swiss-AF is a prospective, observational study designed to pinpoint the mechanisms of cognitive decline in patients with atrial fibrillation. Dr. Conen’s analysis investigated the prevalence of silent brain damage in patients with atrial fibrillation.
The study enrolled 2,415 patients older than 65 with atrial fibrillation between 2014 and 2017 from 14 centers in Switzerland. All patients without contraindications underwent standardized brain MRI, and the images were analyzed in a central core laboratory. Scans were available in 1,736 patients. Of those patients, 347 (20%) had a history of stroke or transient ischemic attack and were excluded from the analysis.
The final analysis included 1,389 patients with atrial fibrillation but no history of stroke or transient ischemic attack. The average age of participants was 72, and 26% were women. The scans showed that 569 (41%) patients had at least one type of previously unrecognized brain damage: 207 (15%) had a cerebral infarct, 269 (19%) had microbleeds, and 222 (16%) had lacunes.
“Four in 10 patients with atrial fibrillation but no history of stroke or transient ischemic attack had clinically unrecognized silent brain lesions,” said Dr. Conen. “This brain damage could trigger cognitive decline.”
Most study participants (1,234; 89%) were treated with oral anticoagulants. Stefan Osswald, MD, Chief of Cardiology at University Hospital Basel in Switzerland, noted that the cross-sectional analysis looked at the data at a single point in time and cannot address the question of whether the cerebral infarcts and other brain lesions occurred before or after initiation of oral anticoagulation. “The findings nevertheless raise the issue that oral anticoagulation might not prevent all brain damage in patients with atrial fibrillation,” he said.
“All Swiss-AF participants underwent extensive cognitive testing. These data will be analyzed to see whether patients with silent brain lesions also have impaired cognitive function,” said Dr. Conen. Collaborations with other study groups will help determine whether these findings are specific to patients with atrial fibrillation.
Impaired Mental Status Doubles Elderly’s Risk of Death After Heart Attack
Impaired mental status is associated with a doubled risk of death one year after a heart attack in elderly patients, according to researchers.
“Cardiologists should consider conducting simple tests to assess mental status in elderly people after a heart attack,” said Farzin Beygui, MD, hospital practitioner at Caen University Hospital in France. “Patients with reduced mental status can then receive more intensive management such as regular follow-up appointments with their general practitioners or nurses, more specific assessment for an early diagnosis of dementia, and tailored therapy.”
The risks of dementia, Alzheimer’s disease, confusion, and delirium increase with age. Elderly people are also at higher risk of having a heart attack and dying afterwards. People aged 75 and older account for approximately one-third of heart attack admissions and more than half of those dying in the hospital after admission for a heart attack. Until now, it was not known whether impaired mental status affected the prognosis of elderly patients with heart attack.
This study assessed the impact of mental status on the risk of death in 600 patients age 75 and above consecutively admitted for heart attack and followed up for at least one year. Mental status was assessed using the Mini-Mental State Examination (MMSE) and the Confusion Assessment Method (CAM).Cognitive impairment was detected in 174 (29%) patients. Patients with impaired mental function were more than twice as likely to be dead one year after their heart attack than those with healthy mental function. The association was independent of other potential predictors of death such as age, sex, invasive treatment, type of myocardial infarction, heart failure, and severity of the heart attack.
Impaired mental status was also associated with a nearly fourfold higher rate of bleeding complications while in the hospital and a more than twofold higher risk of being readmitted to the hospital for cardiovascular causes within three months after discharge.
“Almost one-third of elderly heart attack patients in our study had reduced mental capacity,” said Dr. Beygui. “These patients had higher risks of bleeding, rehospitalization, and death. This may be because they forget to take their medicines or take them more than prescribed, rather than because of poor cognitive function itself.
“Assessing mental status is a simple way to identify elderly patients at particularly high risk of poor outcomes following a heart attack. Identifying these patients may help us target treatment to those who need it most.”
Patients With High Blood Pressure Should Undergo Clock Drawing Test
A clock drawing test for detecting cognitive dysfunction should be conducted routinely in patients with high blood pressure, according to researchers.
Patients with high blood pressure who have impaired cognitive function are at increased risk of developing dementia within five years. Despite this known link, cognitive function is not routinely measured in patients with high blood pressure.
“The ability to draw the numbers of a clock and a particular time is an easy way to find out if a patient with high blood pressure has cognitive impairment,” said Augusto Vicario, MD, a cardiologist at the Cardiovascular Institute of Buenos Aires. “Identifying these patients provides the opportunity to intervene before dementia develops.”
The Heart-Brain Study in Argentina evaluated the usefulness of the clock drawing test, compared with the Mini-Mental State Examination (MMSE), to detect cognitive impairment in 1,414 adults with high blood pressure recruited from 18 cardiology centers in Argentina. The population’s average blood pressure was 144/84 mm Hg, average age was 60, and 62% were women.
For the clock drawing test, patients were given a piece of paper with a circle on it (diameter, 10 cm). They were asked to write the numbers of the clock in the correct positions inside the circle and draw hands on the clock indicating the time “20 to four.” Patients were scored as having normal cognition or moderate or severe cognitive impairment.
The researchers found a higher prevalence of cognitive impairment with the clock drawing test (36%), compared with the MMSE (21%). Three of 10 patients who had a normal MMSE score had an abnormal clock drawing result. The disparity in results between the two tests was greatest in middle-aged patients.
“Untreated high blood pressure silently and progressively damages the arteries in the subcortex of the brain and stops communication between the subcortex and frontal lobe,” said Dr. Vicario. “This disconnect leads to impaired executive functions such as planning, visuospatial abilities, remembering details, and decision-making. The clock drawing test is known to evaluate executive functions. The MMSE evaluates several other cognitive abilities but is weakly correlated with executive functions.
“Our study suggests that the clock drawing test should be preferred over the MMSE for early detection of executive dysfunction in patients with high blood pressure, particularly in middle age. We think the score on the clock drawing test can be considered a surrogate measure of silent vascular damage in the brain and identifies patients at greater risk of developing dementia. In our study, more than one-third of patients were at risk.
“The clock drawing test should be adopted as a routine screening tool for cognitive decline in patients with high blood pressure. Further studies are needed to determine whether lowering blood pressure can prevent progression to dementia,” Dr. Vicario concluded.
Short Sleep Associated With Doubled Risk of Cardiovascular Disease
Middle-aged men who sleep for five hours or fewer per night have twice the risk of developing a major cardiovascular event during the following two decades, compared with men who sleep for seven to eight hours, according to a study.
“For people with busy lives, sleeping may feel like a waste of time, but our study suggests that short sleep could be linked with future cardiovascular disease,” said Moa Bengtsson of the University of Gothenburg, Sweden.
Previous studies have generated conflicting evidence on whether short sleep is associated with a greater chance of having a future cardiovascular event. This study investigated this relationship in 50-year-old men.
In 1993, 50% of all men born in 1943 and living in Gothenburg were randomly selected to participate in the study. Of the 1,463 invited, 798 (55%) men agreed to take part. Participants underwent a physical examination and completed a questionnaire on current health conditions, average sleep duration, physical activity, and smoking. The men were divided into four groups according to their self-estimated average sleep duration at the start of the study: five or fewer hours, six hours, seven to eight hours (considered normal sleep duration), and more than eight hours.
Participants were followed up for 21 years for the occurrence of major cardiovascular events, which included heart attack, stroke, hospitalization due to heart failure, coronary revascularization, or death from cardiovascular disease. Data on cardiovascular events were collected from medical records, the Swedish Hospital Discharge Registry, and the Swedish Cause of Death Register.
Men with incomplete data on sleep duration, incomplete follow-up information, or who had a major cardiovascular event before the start of the study were excluded, leaving a total of 759 men for the analyses.
High blood pressure, diabetes, obesity, current smoking, low physical activity, and poor sleep quality were more common in men who slept for five or fewer hours per night, compared with those who slept for seven to eight hours.
Compared with those with normal sleep duration, men who slept for five or fewer hours per night had a twofold higher risk of having a major cardiovascular event by age 71. The risk remained doubled after adjusting for cardiovascular risk factors at the start of the study, including obesity, diabetes, and smoking.
“Men with the shortest sleep duration at the age of 50 were twice as likely to have had a cardiovascular event by age 71 as those who slept a normal amount, even when other risk factors were taken into account,” said Ms. Bengtsson.
“In our study, the magnitude of increased cardiovascular risk associated with insufficient sleep is similar to that of smoking or having diabetes at age 50. This was an observational study, so, based on our findings, we cannot conclude that short sleep causes cardiovascular disease or say definitively that sleeping more will reduce risk. However, the findings do suggest that sleep is important, and that should be a wake-up call to all of us.”
How Long Is a Good Night’s Sleep?
Researchers have found that a sweet spot of six to eight hours’ sleep per night is most beneficial for heart health. More or less sleep is detrimental.
“We spend one-third of our lives sleeping, yet we know little about the impact of this biologic need on the cardiovascular system,” said Epameinondas Fountas, MD, of the Onassis Cardiac Surgery Centre in Athens.
The study investigated the relationship between sleep duration and cardiovascular disease using a meta-analysis that included 11 prospective studies published within the past five years of more than one million adults without cardiovascular disease.
Two groups, one with short (ie, fewer than six hours) and another with long (ie, more than eight hours) nightly sleep duration, were compared with the reference group (ie, six to eight hours’ sleep).
Short and long sleepers had a greater risk of developing or dying from coronary artery disease or stroke. Compared with adults who slept for six to eight hours per night, short and long sleepers had 11% and 33% greater risks, respectively, of developing or dying from coronary artery disease or stroke during an average follow-up of 9.3 years.
“Our findings suggest that too much or too little sleep may be bad for the heart,” said Dr. Fountas. “More research is needed to clarify exactly why, but we do know that sleep influences biologic processes like glucose metabolism, blood pressure, and inflammation, all of which have an impact on cardiovascular disease.”
A strength of the current analysis is that only prospective studies were included, noted Dr. Fountas. This technique avoids recall bias.
“Having the odd short night or lie-in is unlikely to be detrimental to health, but evidence is accumulating that prolonged nightly sleep deprivation or excessive sleeping should be avoided,” said Dr. Fountas. “The good news is that there are plenty of ways to get into the habit of getting six to eight hours a night: for example, by going to bed and getting up at the same time every day, avoiding alcohol and caffeine before bed, eating healthily, and being physically active. Getting the right amount of sleep is an important part of a healthy lifestyle.”
Four in 10 Patients With Atrial Fibrillation Have Unrecognized Brain Damage
Four out of 10 patients with atrial fibrillation but no history of stroke or transient ischemic attack have previously unrecognized brain damage, according to the first results of the Swiss Atrial Fibrillation Cohort Study (Swiss-AF).
“Our results suggest that clinically unrecognized brain damage may explain the association between dementia and atrial fibrillation in patients without prior stroke,” said David Conen, MD, MPH, Associate Professor of Cardiology at McMaster University in Hamilton, Canada.
Patients with atrial fibrillation have a significantly increased risk of stroke, which is why most are treated with oral anticoagulation. This increased stroke risk is probably the main reason why patients with atrial fibrillation also face an increased risk of cognitive dysfunction and dementia. The relationship between atrial fibrillation and dementia has also been shown among patients without prior strokes, however, meaning that additional mechanisms must be involved. Clarifying the mechanisms by which atrial fibrillation increases the risk of cognitive dysfunction and dementia is a first step towards developing preventive measures.
Swiss-AF is a prospective, observational study designed to pinpoint the mechanisms of cognitive decline in patients with atrial fibrillation. Dr. Conen’s analysis investigated the prevalence of silent brain damage in patients with atrial fibrillation.
The study enrolled 2,415 patients older than 65 with atrial fibrillation between 2014 and 2017 from 14 centers in Switzerland. All patients without contraindications underwent standardized brain MRI, and the images were analyzed in a central core laboratory. Scans were available in 1,736 patients. Of those patients, 347 (20%) had a history of stroke or transient ischemic attack and were excluded from the analysis.
The final analysis included 1,389 patients with atrial fibrillation but no history of stroke or transient ischemic attack. The average age of participants was 72, and 26% were women. The scans showed that 569 (41%) patients had at least one type of previously unrecognized brain damage: 207 (15%) had a cerebral infarct, 269 (19%) had microbleeds, and 222 (16%) had lacunes.
“Four in 10 patients with atrial fibrillation but no history of stroke or transient ischemic attack had clinically unrecognized silent brain lesions,” said Dr. Conen. “This brain damage could trigger cognitive decline.”
Most study participants (1,234; 89%) were treated with oral anticoagulants. Stefan Osswald, MD, Chief of Cardiology at University Hospital Basel in Switzerland, noted that the cross-sectional analysis looked at the data at a single point in time and cannot address the question of whether the cerebral infarcts and other brain lesions occurred before or after initiation of oral anticoagulation. “The findings nevertheless raise the issue that oral anticoagulation might not prevent all brain damage in patients with atrial fibrillation,” he said.
“All Swiss-AF participants underwent extensive cognitive testing. These data will be analyzed to see whether patients with silent brain lesions also have impaired cognitive function,” said Dr. Conen. Collaborations with other study groups will help determine whether these findings are specific to patients with atrial fibrillation.
Impaired Mental Status Doubles Elderly’s Risk of Death After Heart Attack
Impaired mental status is associated with a doubled risk of death one year after a heart attack in elderly patients, according to researchers.
“Cardiologists should consider conducting simple tests to assess mental status in elderly people after a heart attack,” said Farzin Beygui, MD, hospital practitioner at Caen University Hospital in France. “Patients with reduced mental status can then receive more intensive management such as regular follow-up appointments with their general practitioners or nurses, more specific assessment for an early diagnosis of dementia, and tailored therapy.”
The risks of dementia, Alzheimer’s disease, confusion, and delirium increase with age. Elderly people are also at higher risk of having a heart attack and dying afterwards. People aged 75 and older account for approximately one-third of heart attack admissions and more than half of those dying in the hospital after admission for a heart attack. Until now, it was not known whether impaired mental status affected the prognosis of elderly patients with heart attack.
This study assessed the impact of mental status on the risk of death in 600 patients age 75 and above consecutively admitted for heart attack and followed up for at least one year. Mental status was assessed using the Mini-Mental State Examination (MMSE) and the Confusion Assessment Method (CAM).Cognitive impairment was detected in 174 (29%) patients. Patients with impaired mental function were more than twice as likely to be dead one year after their heart attack than those with healthy mental function. The association was independent of other potential predictors of death such as age, sex, invasive treatment, type of myocardial infarction, heart failure, and severity of the heart attack.
Impaired mental status was also associated with a nearly fourfold higher rate of bleeding complications while in the hospital and a more than twofold higher risk of being readmitted to the hospital for cardiovascular causes within three months after discharge.
“Almost one-third of elderly heart attack patients in our study had reduced mental capacity,” said Dr. Beygui. “These patients had higher risks of bleeding, rehospitalization, and death. This may be because they forget to take their medicines or take them more than prescribed, rather than because of poor cognitive function itself.
“Assessing mental status is a simple way to identify elderly patients at particularly high risk of poor outcomes following a heart attack. Identifying these patients may help us target treatment to those who need it most.”
Patients With High Blood Pressure Should Undergo Clock Drawing Test
A clock drawing test for detecting cognitive dysfunction should be conducted routinely in patients with high blood pressure, according to researchers.
Patients with high blood pressure who have impaired cognitive function are at increased risk of developing dementia within five years. Despite this known link, cognitive function is not routinely measured in patients with high blood pressure.
“The ability to draw the numbers of a clock and a particular time is an easy way to find out if a patient with high blood pressure has cognitive impairment,” said Augusto Vicario, MD, a cardiologist at the Cardiovascular Institute of Buenos Aires. “Identifying these patients provides the opportunity to intervene before dementia develops.”
The Heart-Brain Study in Argentina evaluated the usefulness of the clock drawing test, compared with the Mini-Mental State Examination (MMSE), to detect cognitive impairment in 1,414 adults with high blood pressure recruited from 18 cardiology centers in Argentina. The population’s average blood pressure was 144/84 mm Hg, average age was 60, and 62% were women.
For the clock drawing test, patients were given a piece of paper with a circle on it (diameter, 10 cm). They were asked to write the numbers of the clock in the correct positions inside the circle and draw hands on the clock indicating the time “20 to four.” Patients were scored as having normal cognition or moderate or severe cognitive impairment.
The researchers found a higher prevalence of cognitive impairment with the clock drawing test (36%), compared with the MMSE (21%). Three of 10 patients who had a normal MMSE score had an abnormal clock drawing result. The disparity in results between the two tests was greatest in middle-aged patients.
“Untreated high blood pressure silently and progressively damages the arteries in the subcortex of the brain and stops communication between the subcortex and frontal lobe,” said Dr. Vicario. “This disconnect leads to impaired executive functions such as planning, visuospatial abilities, remembering details, and decision-making. The clock drawing test is known to evaluate executive functions. The MMSE evaluates several other cognitive abilities but is weakly correlated with executive functions.
“Our study suggests that the clock drawing test should be preferred over the MMSE for early detection of executive dysfunction in patients with high blood pressure, particularly in middle age. We think the score on the clock drawing test can be considered a surrogate measure of silent vascular damage in the brain and identifies patients at greater risk of developing dementia. In our study, more than one-third of patients were at risk.
“The clock drawing test should be adopted as a routine screening tool for cognitive decline in patients with high blood pressure. Further studies are needed to determine whether lowering blood pressure can prevent progression to dementia,” Dr. Vicario concluded.
Short Sleep Associated With Doubled Risk of Cardiovascular Disease
Middle-aged men who sleep for five hours or fewer per night have twice the risk of developing a major cardiovascular event during the following two decades, compared with men who sleep for seven to eight hours, according to a study.
“For people with busy lives, sleeping may feel like a waste of time, but our study suggests that short sleep could be linked with future cardiovascular disease,” said Moa Bengtsson of the University of Gothenburg, Sweden.
Previous studies have generated conflicting evidence on whether short sleep is associated with a greater chance of having a future cardiovascular event. This study investigated this relationship in 50-year-old men.
In 1993, 50% of all men born in 1943 and living in Gothenburg were randomly selected to participate in the study. Of the 1,463 invited, 798 (55%) men agreed to take part. Participants underwent a physical examination and completed a questionnaire on current health conditions, average sleep duration, physical activity, and smoking. The men were divided into four groups according to their self-estimated average sleep duration at the start of the study: five or fewer hours, six hours, seven to eight hours (considered normal sleep duration), and more than eight hours.
Participants were followed up for 21 years for the occurrence of major cardiovascular events, which included heart attack, stroke, hospitalization due to heart failure, coronary revascularization, or death from cardiovascular disease. Data on cardiovascular events were collected from medical records, the Swedish Hospital Discharge Registry, and the Swedish Cause of Death Register.
Men with incomplete data on sleep duration, incomplete follow-up information, or who had a major cardiovascular event before the start of the study were excluded, leaving a total of 759 men for the analyses.
High blood pressure, diabetes, obesity, current smoking, low physical activity, and poor sleep quality were more common in men who slept for five or fewer hours per night, compared with those who slept for seven to eight hours.
Compared with those with normal sleep duration, men who slept for five or fewer hours per night had a twofold higher risk of having a major cardiovascular event by age 71. The risk remained doubled after adjusting for cardiovascular risk factors at the start of the study, including obesity, diabetes, and smoking.
“Men with the shortest sleep duration at the age of 50 were twice as likely to have had a cardiovascular event by age 71 as those who slept a normal amount, even when other risk factors were taken into account,” said Ms. Bengtsson.
“In our study, the magnitude of increased cardiovascular risk associated with insufficient sleep is similar to that of smoking or having diabetes at age 50. This was an observational study, so, based on our findings, we cannot conclude that short sleep causes cardiovascular disease or say definitively that sleeping more will reduce risk. However, the findings do suggest that sleep is important, and that should be a wake-up call to all of us.”
How Long Is a Good Night’s Sleep?
Researchers have found that a sweet spot of six to eight hours’ sleep per night is most beneficial for heart health. More or less sleep is detrimental.
“We spend one-third of our lives sleeping, yet we know little about the impact of this biologic need on the cardiovascular system,” said Epameinondas Fountas, MD, of the Onassis Cardiac Surgery Centre in Athens.
The study investigated the relationship between sleep duration and cardiovascular disease using a meta-analysis that included 11 prospective studies published within the past five years of more than one million adults without cardiovascular disease.
Two groups, one with short (ie, fewer than six hours) and another with long (ie, more than eight hours) nightly sleep duration, were compared with the reference group (ie, six to eight hours’ sleep).
Short and long sleepers had a greater risk of developing or dying from coronary artery disease or stroke. Compared with adults who slept for six to eight hours per night, short and long sleepers had 11% and 33% greater risks, respectively, of developing or dying from coronary artery disease or stroke during an average follow-up of 9.3 years.
“Our findings suggest that too much or too little sleep may be bad for the heart,” said Dr. Fountas. “More research is needed to clarify exactly why, but we do know that sleep influences biologic processes like glucose metabolism, blood pressure, and inflammation, all of which have an impact on cardiovascular disease.”
A strength of the current analysis is that only prospective studies were included, noted Dr. Fountas. This technique avoids recall bias.
“Having the odd short night or lie-in is unlikely to be detrimental to health, but evidence is accumulating that prolonged nightly sleep deprivation or excessive sleeping should be avoided,” said Dr. Fountas. “The good news is that there are plenty of ways to get into the habit of getting six to eight hours a night: for example, by going to bed and getting up at the same time every day, avoiding alcohol and caffeine before bed, eating healthily, and being physically active. Getting the right amount of sleep is an important part of a healthy lifestyle.”
Four in 10 Patients With Atrial Fibrillation Have Unrecognized Brain Damage
Four out of 10 patients with atrial fibrillation but no history of stroke or transient ischemic attack have previously unrecognized brain damage, according to the first results of the Swiss Atrial Fibrillation Cohort Study (Swiss-AF).
“Our results suggest that clinically unrecognized brain damage may explain the association between dementia and atrial fibrillation in patients without prior stroke,” said David Conen, MD, MPH, Associate Professor of Cardiology at McMaster University in Hamilton, Canada.
Patients with atrial fibrillation have a significantly increased risk of stroke, which is why most are treated with oral anticoagulation. This increased stroke risk is probably the main reason why patients with atrial fibrillation also face an increased risk of cognitive dysfunction and dementia. The relationship between atrial fibrillation and dementia has also been shown among patients without prior strokes, however, meaning that additional mechanisms must be involved. Clarifying the mechanisms by which atrial fibrillation increases the risk of cognitive dysfunction and dementia is a first step towards developing preventive measures.
Swiss-AF is a prospective, observational study designed to pinpoint the mechanisms of cognitive decline in patients with atrial fibrillation. Dr. Conen’s analysis investigated the prevalence of silent brain damage in patients with atrial fibrillation.
The study enrolled 2,415 patients older than 65 with atrial fibrillation between 2014 and 2017 from 14 centers in Switzerland. All patients without contraindications underwent standardized brain MRI, and the images were analyzed in a central core laboratory. Scans were available in 1,736 patients. Of those patients, 347 (20%) had a history of stroke or transient ischemic attack and were excluded from the analysis.
The final analysis included 1,389 patients with atrial fibrillation but no history of stroke or transient ischemic attack. The average age of participants was 72, and 26% were women. The scans showed that 569 (41%) patients had at least one type of previously unrecognized brain damage: 207 (15%) had a cerebral infarct, 269 (19%) had microbleeds, and 222 (16%) had lacunes.
“Four in 10 patients with atrial fibrillation but no history of stroke or transient ischemic attack had clinically unrecognized silent brain lesions,” said Dr. Conen. “This brain damage could trigger cognitive decline.”
Most study participants (1,234; 89%) were treated with oral anticoagulants. Stefan Osswald, MD, Chief of Cardiology at University Hospital Basel in Switzerland, noted that the cross-sectional analysis looked at the data at a single point in time and cannot address the question of whether the cerebral infarcts and other brain lesions occurred before or after initiation of oral anticoagulation. “The findings nevertheless raise the issue that oral anticoagulation might not prevent all brain damage in patients with atrial fibrillation,” he said.
“All Swiss-AF participants underwent extensive cognitive testing. These data will be analyzed to see whether patients with silent brain lesions also have impaired cognitive function,” said Dr. Conen. Collaborations with other study groups will help determine whether these findings are specific to patients with atrial fibrillation.
Impaired Mental Status Doubles Elderly’s Risk of Death After Heart Attack
Impaired mental status is associated with a doubled risk of death one year after a heart attack in elderly patients, according to researchers.
“Cardiologists should consider conducting simple tests to assess mental status in elderly people after a heart attack,” said Farzin Beygui, MD, hospital practitioner at Caen University Hospital in France. “Patients with reduced mental status can then receive more intensive management such as regular follow-up appointments with their general practitioners or nurses, more specific assessment for an early diagnosis of dementia, and tailored therapy.”
The risks of dementia, Alzheimer’s disease, confusion, and delirium increase with age. Elderly people are also at higher risk of having a heart attack and dying afterwards. People aged 75 and older account for approximately one-third of heart attack admissions and more than half of those dying in the hospital after admission for a heart attack. Until now, it was not known whether impaired mental status affected the prognosis of elderly patients with heart attack.
This study assessed the impact of mental status on the risk of death in 600 patients age 75 and above consecutively admitted for heart attack and followed up for at least one year. Mental status was assessed using the Mini-Mental State Examination (MMSE) and the Confusion Assessment Method (CAM).Cognitive impairment was detected in 174 (29%) patients. Patients with impaired mental function were more than twice as likely to be dead one year after their heart attack than those with healthy mental function. The association was independent of other potential predictors of death such as age, sex, invasive treatment, type of myocardial infarction, heart failure, and severity of the heart attack.
Impaired mental status was also associated with a nearly fourfold higher rate of bleeding complications while in the hospital and a more than twofold higher risk of being readmitted to the hospital for cardiovascular causes within three months after discharge.
“Almost one-third of elderly heart attack patients in our study had reduced mental capacity,” said Dr. Beygui. “These patients had higher risks of bleeding, rehospitalization, and death. This may be because they forget to take their medicines or take them more than prescribed, rather than because of poor cognitive function itself.
“Assessing mental status is a simple way to identify elderly patients at particularly high risk of poor outcomes following a heart attack. Identifying these patients may help us target treatment to those who need it most.”

Lemborexant May Not Impair Postural Stability or Driving
The investigational drug is well tolerated and does not increase patients’ auditory awakening threshold.
BALTIMORE—Lemborexant, an investigational treatment for insomnia, does not impair postural stability or driving ability on the morning after dosing, according to the results of phase I studies presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies. In addition, lemborexant does not impair the ability to be awakened at night and helps patients return to sleep faster than placebo. Lemborexant is a dual orexin receptor antagonist in development for the treatment of insomnia and irregular sleep–wake rhythm disorder.
Comparing Lemborexant With Zolpidem
In one double-blind study, Patricia Murphy, PhD, Director of Clinical Research at Eisai, and colleagues randomized 63 healthy volunteers to one of four treatment sequences. The treatments were 5 mg of lemborexant, 10 mg of lemborexant, 6.25 mg of zolpidem, and placebo. Treatments were administered five minutes before bedtime.
At about four hours after bedtime, participants were exposed through earbuds to a 15-dB tone that became 5 dB louder every 15 seconds. Investigators recorded the volume required to awaken the participants. Within five minutes of awakening, participants had their postural stability measured by ataxiameter. After this assessment, the investigators used polysomnography to measure the time it took for participants to fall asleep again. Finally, participants underwent a second postural stability measurement after awakening in the morning. Participants entered a washout period of at least 14 days after each treatment and repeated the same tests for each treatment.
Approximately 78% of the population was female, and the average age was 63.6. Neither lemborexant nor zolpidem significantly increased participants’ auditory awakening threshold (AAT), compared with placebo. The mean AAT was 50 dB for placebo, 51.8 dB for 5 mg of lemborexant, 51.7 dB for 10 mg of lemborexant, and 58.4 dB for zolpidem.
The 5-mg dose of lemborexant did not significantly decrease postural stability, compared with placebo, when participants were awakened at night. The 10-mg dose of lemborexant caused a clinically meaningful decrease in postural stability, and zolpidem was associated with a decrease in stability that was twice that seen with 10 mg of lemborexant.
The mean latency to return to sleep was 40.9 minutes for placebo, 18.1 minutes for 5 mg of lemborexant, 12.1 minutes for 10 mg of lemborexant, and 19.6 minutes for zolpidem. Neither dose of lemborexant significantly affected postural stability after eight hours of sleep. Zolpidem was associated with a significant decrease in postural stability, compared with placebo, but the increase was not clinically meaningful.

Comparing Lemborexant With Zopiclone
In a separate study, Annemiek Vermeeren, PhD, Assistant Professor of Neuropsychology and Psychopharmacology at Maastricht University in the Netherlands, and colleagues evaluated whether lemborexant affected driving performance on the morning after dosing. They enrolled 48 healthy adults into the double-blind, incomplete crossover trial. Participants were randomized to one of 12 treatment sequences, and treatments included lemborexant (2.5 mg, 5 mg, or 10 mg) for eight consecutive days, zopiclone (7.5 mg) on Days 1 and 8 (with placebo on intervening days), and placebo for eight consecutive days. A washout period of at least 14 days followed each eight-day treatment period. Each participant received two of the three lemborexant doses.
Participants completed a practice driving test during screening, and driving performance was assessed at approximately nine hours after dosing on Days 2 and 9. Subjects were instructed to drive at 95 km/h within one lane for approximately one hour, while instruments on the vehicle measured their standard deviation of lateral position (SDLP).
The population’s mean age was 58.5, and 54.2% of participants were male. The mean increases from placebo in SDLP were less than 0.75 cm on both test days after all doses of lemborexant. These changes were neither statistically significant nor clinically meaningful. Zopiclone was associated with increases in SDLP of 2.04 cm on Day 2 and 1.88 cm on Day 9. These changes were statistically significant and clinically meaningful on both days.
The investigators categorized patients as between ages 21 and 65 or age 65 or older. The effects of lemborexant did not differ between adults older and younger than 65 or between males and females.
The driving instructor terminated three of 384 driving tests early. All participants whose tests were terminated had taken zopiclone.
The investigators did not observe any serious adverse events, severe treatment-emergent adverse events, or treatment-emergent adverse events leading to study discontinuation. The most common treatment-emergent adverse events reported after treatment with lemborexant were somnolence, headache, and dry mouth. The most common treatment-emergent adverse events reported after treatment with zopiclone were somnolence, dysgeusia, and dizziness.
Eisai and Purdue Pharma, the developers of lemborexant, funded the studies.
—Erik Greb
Suggested Reading
Murphy P, Moline M, Mayleben D, et al. Lemborexant, A dual orexin receptor antagonist (DORA) for the treatment of insomnia disorder: results from a Bayesian, adaptive, randomized, double-blind, placebo-controlled study. J Clin Sleep Med. 2017;13(11):1289-1299.
The investigational drug is well tolerated and does not increase patients’ auditory awakening threshold.
The investigational drug is well tolerated and does not increase patients’ auditory awakening threshold.
BALTIMORE—Lemborexant, an investigational treatment for insomnia, does not impair postural stability or driving ability on the morning after dosing, according to the results of phase I studies presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies. In addition, lemborexant does not impair the ability to be awakened at night and helps patients return to sleep faster than placebo. Lemborexant is a dual orexin receptor antagonist in development for the treatment of insomnia and irregular sleep–wake rhythm disorder.
Comparing Lemborexant With Zolpidem
In one double-blind study, Patricia Murphy, PhD, Director of Clinical Research at Eisai, and colleagues randomized 63 healthy volunteers to one of four treatment sequences. The treatments were 5 mg of lemborexant, 10 mg of lemborexant, 6.25 mg of zolpidem, and placebo. Treatments were administered five minutes before bedtime.
At about four hours after bedtime, participants were exposed through earbuds to a 15-dB tone that became 5 dB louder every 15 seconds. Investigators recorded the volume required to awaken the participants. Within five minutes of awakening, participants had their postural stability measured by ataxiameter. After this assessment, the investigators used polysomnography to measure the time it took for participants to fall asleep again. Finally, participants underwent a second postural stability measurement after awakening in the morning. Participants entered a washout period of at least 14 days after each treatment and repeated the same tests for each treatment.
Approximately 78% of the population was female, and the average age was 63.6. Neither lemborexant nor zolpidem significantly increased participants’ auditory awakening threshold (AAT), compared with placebo. The mean AAT was 50 dB for placebo, 51.8 dB for 5 mg of lemborexant, 51.7 dB for 10 mg of lemborexant, and 58.4 dB for zolpidem.
The 5-mg dose of lemborexant did not significantly decrease postural stability, compared with placebo, when participants were awakened at night. The 10-mg dose of lemborexant caused a clinically meaningful decrease in postural stability, and zolpidem was associated with a decrease in stability that was twice that seen with 10 mg of lemborexant.
The mean latency to return to sleep was 40.9 minutes for placebo, 18.1 minutes for 5 mg of lemborexant, 12.1 minutes for 10 mg of lemborexant, and 19.6 minutes for zolpidem. Neither dose of lemborexant significantly affected postural stability after eight hours of sleep. Zolpidem was associated with a significant decrease in postural stability, compared with placebo, but the increase was not clinically meaningful.
Comparing Lemborexant With Zopiclone
In a separate study, Annemiek Vermeeren, PhD, Assistant Professor of Neuropsychology and Psychopharmacology at Maastricht University in the Netherlands, and colleagues evaluated whether lemborexant affected driving performance on the morning after dosing. They enrolled 48 healthy adults into the double-blind, incomplete crossover trial. Participants were randomized to one of 12 treatment sequences, and treatments included lemborexant (2.5 mg, 5 mg, or 10 mg) for eight consecutive days, zopiclone (7.5 mg) on Days 1 and 8 (with placebo on intervening days), and placebo for eight consecutive days. A washout period of at least 14 days followed each eight-day treatment period. Each participant received two of the three lemborexant doses.
Participants completed a practice driving test during screening, and driving performance was assessed at approximately nine hours after dosing on Days 2 and 9. Subjects were instructed to drive at 95 km/h within one lane for approximately one hour, while instruments on the vehicle measured their standard deviation of lateral position (SDLP).
The population’s mean age was 58.5, and 54.2% of participants were male. The mean increases from placebo in SDLP were less than 0.75 cm on both test days after all doses of lemborexant. These changes were neither statistically significant nor clinically meaningful. Zopiclone was associated with increases in SDLP of 2.04 cm on Day 2 and 1.88 cm on Day 9. These changes were statistically significant and clinically meaningful on both days.
The investigators categorized patients as between ages 21 and 65 or age 65 or older. The effects of lemborexant did not differ between adults older and younger than 65 or between males and females.
The driving instructor terminated three of 384 driving tests early. All participants whose tests were terminated had taken zopiclone.
The investigators did not observe any serious adverse events, severe treatment-emergent adverse events, or treatment-emergent adverse events leading to study discontinuation. The most common treatment-emergent adverse events reported after treatment with lemborexant were somnolence, headache, and dry mouth. The most common treatment-emergent adverse events reported after treatment with zopiclone were somnolence, dysgeusia, and dizziness.
Eisai and Purdue Pharma, the developers of lemborexant, funded the studies.
—Erik Greb
Suggested Reading
Murphy P, Moline M, Mayleben D, et al. Lemborexant, A dual orexin receptor antagonist (DORA) for the treatment of insomnia disorder: results from a Bayesian, adaptive, randomized, double-blind, placebo-controlled study. J Clin Sleep Med. 2017;13(11):1289-1299.
BALTIMORE—Lemborexant, an investigational treatment for insomnia, does not impair postural stability or driving ability on the morning after dosing, according to the results of phase I studies presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies. In addition, lemborexant does not impair the ability to be awakened at night and helps patients return to sleep faster than placebo. Lemborexant is a dual orexin receptor antagonist in development for the treatment of insomnia and irregular sleep–wake rhythm disorder.
Comparing Lemborexant With Zolpidem
In one double-blind study, Patricia Murphy, PhD, Director of Clinical Research at Eisai, and colleagues randomized 63 healthy volunteers to one of four treatment sequences. The treatments were 5 mg of lemborexant, 10 mg of lemborexant, 6.25 mg of zolpidem, and placebo. Treatments were administered five minutes before bedtime.
At about four hours after bedtime, participants were exposed through earbuds to a 15-dB tone that became 5 dB louder every 15 seconds. Investigators recorded the volume required to awaken the participants. Within five minutes of awakening, participants had their postural stability measured by ataxiameter. After this assessment, the investigators used polysomnography to measure the time it took for participants to fall asleep again. Finally, participants underwent a second postural stability measurement after awakening in the morning. Participants entered a washout period of at least 14 days after each treatment and repeated the same tests for each treatment.
Approximately 78% of the population was female, and the average age was 63.6. Neither lemborexant nor zolpidem significantly increased participants’ auditory awakening threshold (AAT), compared with placebo. The mean AAT was 50 dB for placebo, 51.8 dB for 5 mg of lemborexant, 51.7 dB for 10 mg of lemborexant, and 58.4 dB for zolpidem.
The 5-mg dose of lemborexant did not significantly decrease postural stability, compared with placebo, when participants were awakened at night. The 10-mg dose of lemborexant caused a clinically meaningful decrease in postural stability, and zolpidem was associated with a decrease in stability that was twice that seen with 10 mg of lemborexant.
The mean latency to return to sleep was 40.9 minutes for placebo, 18.1 minutes for 5 mg of lemborexant, 12.1 minutes for 10 mg of lemborexant, and 19.6 minutes for zolpidem. Neither dose of lemborexant significantly affected postural stability after eight hours of sleep. Zolpidem was associated with a significant decrease in postural stability, compared with placebo, but the increase was not clinically meaningful.
Comparing Lemborexant With Zopiclone
In a separate study, Annemiek Vermeeren, PhD, Assistant Professor of Neuropsychology and Psychopharmacology at Maastricht University in the Netherlands, and colleagues evaluated whether lemborexant affected driving performance on the morning after dosing. They enrolled 48 healthy adults into the double-blind, incomplete crossover trial. Participants were randomized to one of 12 treatment sequences, and treatments included lemborexant (2.5 mg, 5 mg, or 10 mg) for eight consecutive days, zopiclone (7.5 mg) on Days 1 and 8 (with placebo on intervening days), and placebo for eight consecutive days. A washout period of at least 14 days followed each eight-day treatment period. Each participant received two of the three lemborexant doses.
Participants completed a practice driving test during screening, and driving performance was assessed at approximately nine hours after dosing on Days 2 and 9. Subjects were instructed to drive at 95 km/h within one lane for approximately one hour, while instruments on the vehicle measured their standard deviation of lateral position (SDLP).
The population’s mean age was 58.5, and 54.2% of participants were male. The mean increases from placebo in SDLP were less than 0.75 cm on both test days after all doses of lemborexant. These changes were neither statistically significant nor clinically meaningful. Zopiclone was associated with increases in SDLP of 2.04 cm on Day 2 and 1.88 cm on Day 9. These changes were statistically significant and clinically meaningful on both days.
The investigators categorized patients as between ages 21 and 65 or age 65 or older. The effects of lemborexant did not differ between adults older and younger than 65 or between males and females.
The driving instructor terminated three of 384 driving tests early. All participants whose tests were terminated had taken zopiclone.
The investigators did not observe any serious adverse events, severe treatment-emergent adverse events, or treatment-emergent adverse events leading to study discontinuation. The most common treatment-emergent adverse events reported after treatment with lemborexant were somnolence, headache, and dry mouth. The most common treatment-emergent adverse events reported after treatment with zopiclone were somnolence, dysgeusia, and dizziness.
Eisai and Purdue Pharma, the developers of lemborexant, funded the studies.
—Erik Greb
Suggested Reading
Murphy P, Moline M, Mayleben D, et al. Lemborexant, A dual orexin receptor antagonist (DORA) for the treatment of insomnia disorder: results from a Bayesian, adaptive, randomized, double-blind, placebo-controlled study. J Clin Sleep Med. 2017;13(11):1289-1299.
Emgality approved for migraine prevention in adults
The Food and Drug Administration has approved galcanezumab-gnlm (Emgality) for the preventive treatment of migraine in adults, according to an announcement from Eli Lilly, the drug’s manufacturer.

FDA approval is based on results of three different phase 3 clinical trials, EVOLVE-1, EVOLVE-2, and REGAIN. EVOLVE-1 and EVOLVE-2 were 6-month, double-blind, placebo-controlled studies that included adults with episodic migraine (4-14 headache days per month). REGAIN was a 3-month, double-blind, placebo-controlled study of adults with chronic migraine (at least 15 headache days per month). The primary endpoint of all trials was mean change from baseline in the number of monthly headache days.
In all trials, patients received either placebo, 120 mg galcanezumab-gnlm after an initial loading dose of 240 mg, or 240 mg galcanezumab-gnlm. In EVOLVE-1 and EVOLVE-2, people who received galcanezumab-gnlm had significantly fewer headache days per month than with placebo, and those who received galcanezumab-gnlm were also more likely to achieve a 50%, 75%, and 100% reduction in headache days.
In REGAIN, patients who received galcanezumab-gnlm experienced fewer headache days than those who received placebo, and were also more likely to achieve a 50% reduction in headache days. There was no difference between groups in likelihood of achieving a 75% or 100% reduction.
The recommended dosage, according to the label, is a monthly, 120-mg subcutaneous injection, with an initial loading dose of 240 mg. The most common adverse event associated with galcanezumab-gnlm was injection site reaction.
Galcanezumab-gnlm, a calcitonin gene–related peptide antagonist, is also under final review by the European Commission for approval in Europe.
The U.S. list price of galcanezumab-gnlm is $575 once-monthly, or $6,900 annually, and patients with commercial insurance are candidates to receive it free for up to 12 months as part of Lilly’s patient support program (subject to specific terms and conditions), according to the announcement.
The Food and Drug Administration has approved galcanezumab-gnlm (Emgality) for the preventive treatment of migraine in adults, according to an announcement from Eli Lilly, the drug’s manufacturer.
FDA approval is based on results of three different phase 3 clinical trials, EVOLVE-1, EVOLVE-2, and REGAIN. EVOLVE-1 and EVOLVE-2 were 6-month, double-blind, placebo-controlled studies that included adults with episodic migraine (4-14 headache days per month). REGAIN was a 3-month, double-blind, placebo-controlled study of adults with chronic migraine (at least 15 headache days per month). The primary endpoint of all trials was mean change from baseline in the number of monthly headache days.
In all trials, patients received either placebo, 120 mg galcanezumab-gnlm after an initial loading dose of 240 mg, or 240 mg galcanezumab-gnlm. In EVOLVE-1 and EVOLVE-2, people who received galcanezumab-gnlm had significantly fewer headache days per month than with placebo, and those who received galcanezumab-gnlm were also more likely to achieve a 50%, 75%, and 100% reduction in headache days.
In REGAIN, patients who received galcanezumab-gnlm experienced fewer headache days than those who received placebo, and were also more likely to achieve a 50% reduction in headache days. There was no difference between groups in likelihood of achieving a 75% or 100% reduction.
The recommended dosage, according to the label, is a monthly, 120-mg subcutaneous injection, with an initial loading dose of 240 mg. The most common adverse event associated with galcanezumab-gnlm was injection site reaction.
Galcanezumab-gnlm, a calcitonin gene–related peptide antagonist, is also under final review by the European Commission for approval in Europe.
The U.S. list price of galcanezumab-gnlm is $575 once-monthly, or $6,900 annually, and patients with commercial insurance are candidates to receive it free for up to 12 months as part of Lilly’s patient support program (subject to specific terms and conditions), according to the announcement.
The Food and Drug Administration has approved galcanezumab-gnlm (Emgality) for the preventive treatment of migraine in adults, according to an announcement from Eli Lilly, the drug’s manufacturer.
FDA approval is based on results of three different phase 3 clinical trials, EVOLVE-1, EVOLVE-2, and REGAIN. EVOLVE-1 and EVOLVE-2 were 6-month, double-blind, placebo-controlled studies that included adults with episodic migraine (4-14 headache days per month). REGAIN was a 3-month, double-blind, placebo-controlled study of adults with chronic migraine (at least 15 headache days per month). The primary endpoint of all trials was mean change from baseline in the number of monthly headache days.
In all trials, patients received either placebo, 120 mg galcanezumab-gnlm after an initial loading dose of 240 mg, or 240 mg galcanezumab-gnlm. In EVOLVE-1 and EVOLVE-2, people who received galcanezumab-gnlm had significantly fewer headache days per month than with placebo, and those who received galcanezumab-gnlm were also more likely to achieve a 50%, 75%, and 100% reduction in headache days.
In REGAIN, patients who received galcanezumab-gnlm experienced fewer headache days than those who received placebo, and were also more likely to achieve a 50% reduction in headache days. There was no difference between groups in likelihood of achieving a 75% or 100% reduction.
The recommended dosage, according to the label, is a monthly, 120-mg subcutaneous injection, with an initial loading dose of 240 mg. The most common adverse event associated with galcanezumab-gnlm was injection site reaction.
Galcanezumab-gnlm, a calcitonin gene–related peptide antagonist, is also under final review by the European Commission for approval in Europe.
The U.S. list price of galcanezumab-gnlm is $575 once-monthly, or $6,900 annually, and patients with commercial insurance are candidates to receive it free for up to 12 months as part of Lilly’s patient support program (subject to specific terms and conditions), according to the announcement.
Abnormal Sleep Staging Predicts Fatigue in Patients With MS
REM sleep onset latency, sleep-related movement, and subjective insomnia are associated with MS-related fatigue.
BALTIMORE—Objective and subjective sleep measures are associated with fatigue in people with multiple sclerosis (MS), according to a meta-analysis presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies.

Evidence suggests that individuals with MS have disruptions in sleep that are associated with and may contribute to fatigue, said Jagriti “Jackie” Bhattarai, PhD, a postdoctoral fellow in the MS Rehabilitation Research Laboratory at the Johns Hopkins School of Medicine’s Department of Physical Medicine and Rehabilitation in Baltimore. Among the sleep parameters investigated, sleep staging, sleep-related movement, and subjective insomnia had moderate associations with fatigue. Other sleep parameters examined in the study were not statistically significantly associated with fatigue, she said.
A Major Factor in Disability
Fatigue is a leading contributor to disability in patients with MS. Although fatigue is a complex symptom with multiple neurologic and behavioral causes, emerging evidence suggests that “sleep disturbance may have a significant role in the development or maintenance of fatigue in MS,” Dr. Bhattarai said. The relationship between sleep parameters and fatigue in people with MS is not well understood, however, she said.
To examine the relationship between commonly used sleep parameters and fatigue in MS, Dr. Bhattarai and colleagues identified studies that included at least one validated measure of fatigue and one validated measure of sleep disturbance. They included studies that reported the effect sizes of the associations between sleep and fatigue or provided enough data for the investigators to compute the effect sizes.
Their meta-analysis included 37 studies with 6,129 participants. Participants had an average age of 42 and MS duration of 9.5 years. About 80% of the sample had relapsing-remitting MS.
Sleep measures included polysomnography, actigraphy, and the multiple sleep latency test, as well as subjective measures such as the Insomnia Severity Index, the Pittsburgh Sleep Quality Index, and the Medical Outcomes Study Sleep Scale.
Fatigue was measured using the Fatigue Severity Scale, the modified Fatigue Impact Scale, or the Neurological Fatigue Index for MS.
Effect sizes varied across studies, as did the parameters examined within each study. The mean effect size was the largest for the association between REM sleep onset latency (ie, the time between sleep onset and initiation of REM sleep) and fatigue (r = 0.42), though this effect size was based on only two studies, and the association requires further investigation. “Shorter REM sleep onset latency has been associated with depression and narcolepsy,” both of which are more common in people with MS than in the general population, Dr. Bhattarai said. Subjective insomnia (r = 0.36) and objective sleep-related movement (r = 0.34) also yielded moderate effect sizes. “The shorter the REM sleep onset latency and the more subjective complaints that individuals have about their sleep, the greater the level of fatigue people with MS reported,” she said. “The more sleep-related movement that people have, the greater their level of MS-related fatigue.”
Potential to Inform Treatment Approach
The association between fatigue and objective sleep-disordered breathing was not statistically significant. Subjective sleep-disordered breathing, objective nocturnal arousals, sleep efficiency, sleep onset latency, and sleep duration had weak effect sizes. These problems are common and require treatment, however, Dr. Bhattarai said.
“When treating patients with MS who are experiencing MS-related fatigue, interventions geared toward improving patients’ perceived sleep quality and addressing MS symptoms that might disrupt sleep (eg, nocturia and periodic leg movements) may offer an avenue for improving fatigue,” said Dr. Bhattarai. “However, these effects remain to be shown in a randomized controlled trial.”
—Jake Remaly
REM sleep onset latency, sleep-related movement, and subjective insomnia are associated with MS-related fatigue.
REM sleep onset latency, sleep-related movement, and subjective insomnia are associated with MS-related fatigue.
BALTIMORE—Objective and subjective sleep measures are associated with fatigue in people with multiple sclerosis (MS), according to a meta-analysis presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies.
Evidence suggests that individuals with MS have disruptions in sleep that are associated with and may contribute to fatigue, said Jagriti “Jackie” Bhattarai, PhD, a postdoctoral fellow in the MS Rehabilitation Research Laboratory at the Johns Hopkins School of Medicine’s Department of Physical Medicine and Rehabilitation in Baltimore. Among the sleep parameters investigated, sleep staging, sleep-related movement, and subjective insomnia had moderate associations with fatigue. Other sleep parameters examined in the study were not statistically significantly associated with fatigue, she said.
A Major Factor in Disability
Fatigue is a leading contributor to disability in patients with MS. Although fatigue is a complex symptom with multiple neurologic and behavioral causes, emerging evidence suggests that “sleep disturbance may have a significant role in the development or maintenance of fatigue in MS,” Dr. Bhattarai said. The relationship between sleep parameters and fatigue in people with MS is not well understood, however, she said.
To examine the relationship between commonly used sleep parameters and fatigue in MS, Dr. Bhattarai and colleagues identified studies that included at least one validated measure of fatigue and one validated measure of sleep disturbance. They included studies that reported the effect sizes of the associations between sleep and fatigue or provided enough data for the investigators to compute the effect sizes.
Their meta-analysis included 37 studies with 6,129 participants. Participants had an average age of 42 and MS duration of 9.5 years. About 80% of the sample had relapsing-remitting MS.
Sleep measures included polysomnography, actigraphy, and the multiple sleep latency test, as well as subjective measures such as the Insomnia Severity Index, the Pittsburgh Sleep Quality Index, and the Medical Outcomes Study Sleep Scale.
Fatigue was measured using the Fatigue Severity Scale, the modified Fatigue Impact Scale, or the Neurological Fatigue Index for MS.
Effect sizes varied across studies, as did the parameters examined within each study. The mean effect size was the largest for the association between REM sleep onset latency (ie, the time between sleep onset and initiation of REM sleep) and fatigue (r = 0.42), though this effect size was based on only two studies, and the association requires further investigation. “Shorter REM sleep onset latency has been associated with depression and narcolepsy,” both of which are more common in people with MS than in the general population, Dr. Bhattarai said. Subjective insomnia (r = 0.36) and objective sleep-related movement (r = 0.34) also yielded moderate effect sizes. “The shorter the REM sleep onset latency and the more subjective complaints that individuals have about their sleep, the greater the level of fatigue people with MS reported,” she said. “The more sleep-related movement that people have, the greater their level of MS-related fatigue.”
Potential to Inform Treatment Approach
The association between fatigue and objective sleep-disordered breathing was not statistically significant. Subjective sleep-disordered breathing, objective nocturnal arousals, sleep efficiency, sleep onset latency, and sleep duration had weak effect sizes. These problems are common and require treatment, however, Dr. Bhattarai said.
“When treating patients with MS who are experiencing MS-related fatigue, interventions geared toward improving patients’ perceived sleep quality and addressing MS symptoms that might disrupt sleep (eg, nocturia and periodic leg movements) may offer an avenue for improving fatigue,” said Dr. Bhattarai. “However, these effects remain to be shown in a randomized controlled trial.”
—Jake Remaly
BALTIMORE—Objective and subjective sleep measures are associated with fatigue in people with multiple sclerosis (MS), according to a meta-analysis presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies.
Evidence suggests that individuals with MS have disruptions in sleep that are associated with and may contribute to fatigue, said Jagriti “Jackie” Bhattarai, PhD, a postdoctoral fellow in the MS Rehabilitation Research Laboratory at the Johns Hopkins School of Medicine’s Department of Physical Medicine and Rehabilitation in Baltimore. Among the sleep parameters investigated, sleep staging, sleep-related movement, and subjective insomnia had moderate associations with fatigue. Other sleep parameters examined in the study were not statistically significantly associated with fatigue, she said.
A Major Factor in Disability
Fatigue is a leading contributor to disability in patients with MS. Although fatigue is a complex symptom with multiple neurologic and behavioral causes, emerging evidence suggests that “sleep disturbance may have a significant role in the development or maintenance of fatigue in MS,” Dr. Bhattarai said. The relationship between sleep parameters and fatigue in people with MS is not well understood, however, she said.
To examine the relationship between commonly used sleep parameters and fatigue in MS, Dr. Bhattarai and colleagues identified studies that included at least one validated measure of fatigue and one validated measure of sleep disturbance. They included studies that reported the effect sizes of the associations between sleep and fatigue or provided enough data for the investigators to compute the effect sizes.
Their meta-analysis included 37 studies with 6,129 participants. Participants had an average age of 42 and MS duration of 9.5 years. About 80% of the sample had relapsing-remitting MS.
Sleep measures included polysomnography, actigraphy, and the multiple sleep latency test, as well as subjective measures such as the Insomnia Severity Index, the Pittsburgh Sleep Quality Index, and the Medical Outcomes Study Sleep Scale.
Fatigue was measured using the Fatigue Severity Scale, the modified Fatigue Impact Scale, or the Neurological Fatigue Index for MS.
Effect sizes varied across studies, as did the parameters examined within each study. The mean effect size was the largest for the association between REM sleep onset latency (ie, the time between sleep onset and initiation of REM sleep) and fatigue (r = 0.42), though this effect size was based on only two studies, and the association requires further investigation. “Shorter REM sleep onset latency has been associated with depression and narcolepsy,” both of which are more common in people with MS than in the general population, Dr. Bhattarai said. Subjective insomnia (r = 0.36) and objective sleep-related movement (r = 0.34) also yielded moderate effect sizes. “The shorter the REM sleep onset latency and the more subjective complaints that individuals have about their sleep, the greater the level of fatigue people with MS reported,” she said. “The more sleep-related movement that people have, the greater their level of MS-related fatigue.”
Potential to Inform Treatment Approach
The association between fatigue and objective sleep-disordered breathing was not statistically significant. Subjective sleep-disordered breathing, objective nocturnal arousals, sleep efficiency, sleep onset latency, and sleep duration had weak effect sizes. These problems are common and require treatment, however, Dr. Bhattarai said.
“When treating patients with MS who are experiencing MS-related fatigue, interventions geared toward improving patients’ perceived sleep quality and addressing MS symptoms that might disrupt sleep (eg, nocturia and periodic leg movements) may offer an avenue for improving fatigue,” said Dr. Bhattarai. “However, these effects remain to be shown in a randomized controlled trial.”
—Jake Remaly
Pediatric Vaccines & Infectious Diseases: Fall 2018

Disrupted Sleep, Cardiorespiratory Dysfunction Linked to SUDEP
Periictal cardiorespiratory dysfunction, sleep-disordered breathing, and endocrine dysfunction have all been linked to sudden unexpected death from epilepsy (SUDEP).
- Investigators conducted a prospective observational study on 30 patients in the Columbia University Medical Center’s adult epilepsy monitoring unit.
- Their analysis found that patients who were at high risk for SUDEP were more likely to experience cardiorespiratory dysfunction (60% vs 27%).
- Sleep-disordered breathing was found in 88% of patients with inpatient or outpatient polysomnography results that were fully scorable.
- The researchers also found endocrine dysfunction in 35% of patients, with men more likely to experience the problem.
- The analysis did not detect a significant relationship between cardiorespiratory dysfunction, sleep-disordered breathing, and neuroendocrine status.
Billakota S, Odom N, Westwood AJ, et al. Sleep-disordered breathing, neuroendocrine function, and clinical SUDEP risk in patients with epilepsy. Epilepsy Behav. 2018;87:78-82.
Periictal cardiorespiratory dysfunction, sleep-disordered breathing, and endocrine dysfunction have all been linked to sudden unexpected death from epilepsy (SUDEP).
- Investigators conducted a prospective observational study on 30 patients in the Columbia University Medical Center’s adult epilepsy monitoring unit.
- Their analysis found that patients who were at high risk for SUDEP were more likely to experience cardiorespiratory dysfunction (60% vs 27%).
- Sleep-disordered breathing was found in 88% of patients with inpatient or outpatient polysomnography results that were fully scorable.
- The researchers also found endocrine dysfunction in 35% of patients, with men more likely to experience the problem.
- The analysis did not detect a significant relationship between cardiorespiratory dysfunction, sleep-disordered breathing, and neuroendocrine status.
Billakota S, Odom N, Westwood AJ, et al. Sleep-disordered breathing, neuroendocrine function, and clinical SUDEP risk in patients with epilepsy. Epilepsy Behav. 2018;87:78-82.
Periictal cardiorespiratory dysfunction, sleep-disordered breathing, and endocrine dysfunction have all been linked to sudden unexpected death from epilepsy (SUDEP).
- Investigators conducted a prospective observational study on 30 patients in the Columbia University Medical Center’s adult epilepsy monitoring unit.
- Their analysis found that patients who were at high risk for SUDEP were more likely to experience cardiorespiratory dysfunction (60% vs 27%).
- Sleep-disordered breathing was found in 88% of patients with inpatient or outpatient polysomnography results that were fully scorable.
- The researchers also found endocrine dysfunction in 35% of patients, with men more likely to experience the problem.
- The analysis did not detect a significant relationship between cardiorespiratory dysfunction, sleep-disordered breathing, and neuroendocrine status.
Billakota S, Odom N, Westwood AJ, et al. Sleep-disordered breathing, neuroendocrine function, and clinical SUDEP risk in patients with epilepsy. Epilepsy Behav. 2018;87:78-82.
Psychogenic Testing Helps Separate PNES from Epilepsy
Neuropsychological testing may help differentiate epileptic seizures from psychogenic nonepileptic seizures (PNES) according to a study that evaluated data from 72 patients with epilepsy and 33 patients with PNES.
- In the past, psychometric testing has been shown to have limited utility in differentiating PNES from epileptic seizures.
- The new research suggests that multivariate assessment using several psychological tests is more effective in making the differential diagnosis.
- Using logistic regression, investigators found that a combination of 7 neuropsychological tests accurately classified about 85% of patients.
- The researchers acknowledged that video-EEG monitoring remains the gold standard for separating PNES from epileptic seizures but suggest that a standardized battery of neuropsychological tests may improve the clinical decision-making process.
Tyson BT, Baker S, Greenacre M, et al. Differentiating epilepsy from psychogenic nonepileptic seizures using neuropsychological test data. Epilepsy Behav. 2018;87:39-45.
Neuropsychological testing may help differentiate epileptic seizures from psychogenic nonepileptic seizures (PNES) according to a study that evaluated data from 72 patients with epilepsy and 33 patients with PNES.
- In the past, psychometric testing has been shown to have limited utility in differentiating PNES from epileptic seizures.
- The new research suggests that multivariate assessment using several psychological tests is more effective in making the differential diagnosis.
- Using logistic regression, investigators found that a combination of 7 neuropsychological tests accurately classified about 85% of patients.
- The researchers acknowledged that video-EEG monitoring remains the gold standard for separating PNES from epileptic seizures but suggest that a standardized battery of neuropsychological tests may improve the clinical decision-making process.
Tyson BT, Baker S, Greenacre M, et al. Differentiating epilepsy from psychogenic nonepileptic seizures using neuropsychological test data. Epilepsy Behav. 2018;87:39-45.
Neuropsychological testing may help differentiate epileptic seizures from psychogenic nonepileptic seizures (PNES) according to a study that evaluated data from 72 patients with epilepsy and 33 patients with PNES.
- In the past, psychometric testing has been shown to have limited utility in differentiating PNES from epileptic seizures.
- The new research suggests that multivariate assessment using several psychological tests is more effective in making the differential diagnosis.
- Using logistic regression, investigators found that a combination of 7 neuropsychological tests accurately classified about 85% of patients.
- The researchers acknowledged that video-EEG monitoring remains the gold standard for separating PNES from epileptic seizures but suggest that a standardized battery of neuropsychological tests may improve the clinical decision-making process.
Tyson BT, Baker S, Greenacre M, et al. Differentiating epilepsy from psychogenic nonepileptic seizures using neuropsychological test data. Epilepsy Behav. 2018;87:39-45.