User login
HF unlikely in pregnant cancer survivors without history of cardiotoxicity
The risk of adverse cardiac events in female cancer survivors during pregnancy is low unless there is a history of cardiotoxicity, according to Shiying Liu, MD, of the University of Toronto, and her associates.
In a research letter published in the Journal of the American College of Cardiology, Dr. Liu and her associates reported on a retrospective chart review of 78 women with 94 pregnancies who had previously received cancer therapy who were seen at Mount Sinai Hospital between 2005 and 2015. Of these, 15 pregnancies occurred in 13 women with a prior history of cardiotoxicity. The primary outcome was a composite of cardiac events including cardiac death, heart failure (HF), acute coronary syndrome, and sustained arrhythmia.
HF occurred during five pregnancies in four women; no other adverse cardiac events occurred during the study period. All four of the women who experienced HF had a history of cardiotoxicity. There was no difference in age at cancer diagnosis, age at pregnancy, cancer type, or exposure to anthracyclines between those who did and did not experience HF, but women who developed HF were more likely to have left ventricular systolic dysfunction at the first antenatal visit (75% vs. 8%; P = .004) and to be on cardiac medications (50% vs. 8%; P = .026).
“The risk of developing [HF] during pregnancy is rare in female cancer survivors without a history of cardiotoxicity. These women can be reassured that they are at a very low risk of developing [HF] during pregnancy. Women who have a history of cardiotoxicity have an approximately one in three chance of developing [HF] during pregnancy and should receive close cardiac surveillance during pregnancy at a center with expertise in cardiac disease in pregnancy,” the authors concluded.
Coauthor Paaladinesh Thavendiranathan, MD, reported support from the Canadian Institutes of Health Research New Investigator Award. None of the other authors had any relevant financial disclosures.
SOURCE: Liu S et al. J Am Coll Cardiol. 2018 Oct 15. doi: 10.1016/j.jacc.2018.07.085.
The risk of adverse cardiac events in female cancer survivors during pregnancy is low unless there is a history of cardiotoxicity, according to Shiying Liu, MD, of the University of Toronto, and her associates.
In a research letter published in the Journal of the American College of Cardiology, Dr. Liu and her associates reported on a retrospective chart review of 78 women with 94 pregnancies who had previously received cancer therapy who were seen at Mount Sinai Hospital between 2005 and 2015. Of these, 15 pregnancies occurred in 13 women with a prior history of cardiotoxicity. The primary outcome was a composite of cardiac events including cardiac death, heart failure (HF), acute coronary syndrome, and sustained arrhythmia.
HF occurred during five pregnancies in four women; no other adverse cardiac events occurred during the study period. All four of the women who experienced HF had a history of cardiotoxicity. There was no difference in age at cancer diagnosis, age at pregnancy, cancer type, or exposure to anthracyclines between those who did and did not experience HF, but women who developed HF were more likely to have left ventricular systolic dysfunction at the first antenatal visit (75% vs. 8%; P = .004) and to be on cardiac medications (50% vs. 8%; P = .026).
“The risk of developing [HF] during pregnancy is rare in female cancer survivors without a history of cardiotoxicity. These women can be reassured that they are at a very low risk of developing [HF] during pregnancy. Women who have a history of cardiotoxicity have an approximately one in three chance of developing [HF] during pregnancy and should receive close cardiac surveillance during pregnancy at a center with expertise in cardiac disease in pregnancy,” the authors concluded.
Coauthor Paaladinesh Thavendiranathan, MD, reported support from the Canadian Institutes of Health Research New Investigator Award. None of the other authors had any relevant financial disclosures.
SOURCE: Liu S et al. J Am Coll Cardiol. 2018 Oct 15. doi: 10.1016/j.jacc.2018.07.085.
The risk of adverse cardiac events in female cancer survivors during pregnancy is low unless there is a history of cardiotoxicity, according to Shiying Liu, MD, of the University of Toronto, and her associates.
In a research letter published in the Journal of the American College of Cardiology, Dr. Liu and her associates reported on a retrospective chart review of 78 women with 94 pregnancies who had previously received cancer therapy who were seen at Mount Sinai Hospital between 2005 and 2015. Of these, 15 pregnancies occurred in 13 women with a prior history of cardiotoxicity. The primary outcome was a composite of cardiac events including cardiac death, heart failure (HF), acute coronary syndrome, and sustained arrhythmia.
HF occurred during five pregnancies in four women; no other adverse cardiac events occurred during the study period. All four of the women who experienced HF had a history of cardiotoxicity. There was no difference in age at cancer diagnosis, age at pregnancy, cancer type, or exposure to anthracyclines between those who did and did not experience HF, but women who developed HF were more likely to have left ventricular systolic dysfunction at the first antenatal visit (75% vs. 8%; P = .004) and to be on cardiac medications (50% vs. 8%; P = .026).
“The risk of developing [HF] during pregnancy is rare in female cancer survivors without a history of cardiotoxicity. These women can be reassured that they are at a very low risk of developing [HF] during pregnancy. Women who have a history of cardiotoxicity have an approximately one in three chance of developing [HF] during pregnancy and should receive close cardiac surveillance during pregnancy at a center with expertise in cardiac disease in pregnancy,” the authors concluded.
Coauthor Paaladinesh Thavendiranathan, MD, reported support from the Canadian Institutes of Health Research New Investigator Award. None of the other authors had any relevant financial disclosures.
SOURCE: Liu S et al. J Am Coll Cardiol. 2018 Oct 15. doi: 10.1016/j.jacc.2018.07.085.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
States act to safeguard young cancer patients’ chances to have children
When Katherine Frega was diagnosed with Hodgkin lymphoma 8 years ago at the age 17 years, she was so sick that all she could focus on was starting chemotherapy to treat her aggressive blood cancer. It was her dad who thought to ask the oncologist, “How is this treatment going to affect her ability to have children?”
The oncologist discussed the risks but stressed that Ms. Frega needed to start treatment right away.
The question of fertility is often overlooked when young cancer patients are battling a life-threatening illness. And since health insurance doesn’t typically cover fertility preservation care, patients and their families may be deterred by the cost.
But a growing number of states now require plans to cover such services when medically necessary treatment jeopardizes fertility.
Treatment for cancer and other serious conditions involves toxic chemotherapy drugs, radiation, and surgery that can cause infertility in women and men.
The cost to freeze patients’ healthy eggs, sperm, or embryos for future use can be a major barrier, said Eden Cardozo, MD, a reproductive endocrinologist and director of the fertility preservation program at the Women & Infants Fertility Center in Providence, R.I. Dr. Cardozo was instrumental in getting Rhode Island’s law passed last year.
“[Patients] have to move quickly,” she said. “They don’t have time to raise funds from family and friends. They don’t have time to petition their insurance company.”
Reproductive health advocates argue that fertility preservation should be viewed as a core component of cancer care in younger people, not as an optional infertility offering. Some compare this type of coverage with the federal Women’s Health and Cancer Rights Act, which requires plans that cover a patient’s mastectomy to also provide for breast reconstruction.
New laws in Delaware, Illinois, and Maryland require plans to include this benefit. The Delaware law applies to plans issued or renewed after June of this year; the requirement in the other two states starts in 2019. Connecticut and Rhode Island passed similar laws last year. New Jersey lawmakers are considering a bill and advocates in New York plan to make another attempt after both legislative chambers passed fertility preservation bills in the last session but failed to reconcile them.
The state measures don’t apply to companies that are self-funded, meaning they pay their employee claims directly rather than buying state-regulated insurance policies for that purpose. They also don’t apply to government-funded programs such as Medicaid or Tricare.
Although freezing sperm and embryos has been common medical practice for decades, egg freezing was considered experimental by professional groups until 2012. As the technology has improved, the need for insurance coverage has grown, said Joyce Reinecke, executive director of the Alliance for Fertility Preservation, an advocacy group for cancer patients.
When Ms. Frega’s cancer didn’t respond to chemotherapy, her doctors recommended a bone marrow transplant in January 2012. Even if her eggs hadn’t been damaged by the chemotherapy, the transplant would likely cause permanent infertility, she was told. So she took hormones to stimulate her ovaries to produce more eggs, among other things, and seven were retrieved during an outpatient procedure days before her transplant.
Ms. Frega’s parents paid $10,000 for the medications and egg retrieval, a significant amount but less than what many pay. They were aided by Livestrong Fertility, a nonprofit group that provides access to discounted fertility preservation services for cancer patients who meet income guidelines.
Ms. Frega has good insurance through her mother’s employer plan. “They covered everything else, except for this,” she said. “They considered it not medically necessary.”
Cancer-free following two bone marrow transplants, Ms. Frega, now 25 years old, is a third-year medical student at the Upstate Medical University in Syracuse, N.Y. She plans to specialize in oncology.
Between 20% and 70% of cancer patients experience some degree of fertility impairment, according to Dr. Cardozo in Rhode Island. Though they make up the largest at-risk group, the complication isn’t unique to cancer patients. People with other conditions such as lupus and rheumatoid arthritis who are treated with chemotherapy drugs may be affected, as may patients with conditions such as endometriosis who require surgery.
Despite the much-ballyhooed examples of tech companies like Facebook, Apple, and Google that offer egg freezing as an employee perk, cryopreservation, as it’s called, isn’t a typical employee benefit.
Only 6% of large companies with 500 or more workers offer egg freezing for employees or their spouses, according to the 2017 annual employer survey by benefits consultant Mercer. About a quarter cover in vitro fertilization (IVF). About 44% of large employers don’t offer any infertility services, the survey found.
Men face the same infertility risk when they need cancer treatment.
When Blake Hornbrook, an Army medic at Fort Campbell, Ky., had surgery to remove a cancerous testicle in the fall of 2015, he and his wife, Kelsey, were stationed in Germany. Mr. Hornbrook, then 26 years old, looked into fertility preservation while overseas, but the annual storage fee of 1,000 euros (about $1,150) deterred the couple.
He required a second surgery several months later to see if the cancer had spread to his lymph nodes. The couple returned to the United States and drove directly from the airport to a sperm bank in Fairfax, Va. It cost roughly $400 for the initial appointment to provide a sperm specimen and store it, Mr. Hornbrook said.
Tricare covered Mr. Hornbrook’s cancer treatment, but it didn’t pay for fertility preservation or for IVF, which he estimated cost the couple $6,500 in clinic fees. Tricare provided discounts on some of the fertility drugs.
Their daughter, Harper, was born 7 months ago, and Mr. Hornbrook’s cancer remains in remission.
For young cancer patients, the cost of storing the eggs or sperm that have been preserved can add up. Even if a state has a fertility preservation law, it typically doesn’t cover those costs, Ms. Reinecke said.
The Hornbrooks pay $480 annually to store his sperm and $375 to store their remaining embryos. Ms. Frega pays $1,000 annually to store her eggs.
Ms. Frega hopes to be able to conceive naturally and knowing she has frozen eggs available is “relieving, but also anxiety producing,” she said. If she can’t get pregnant later on, she may have to pay $10,000 or more for IVF as well. “That’s what lies ahead,” she said.
A total of 16 states require insurers to offer or cover infertility services to some extent, according to infertility advocacy organization Resolve. Requirements vary: Insurers may have to cover diagnosis or testing for infertility, for example, but not treatments like IVF or fertility medications, said Barbara Collura, president and CEO of Resolve.
Typically, state infertility coverage laws require couples to try to get pregnant for a year or two before they’re eligible for insurance coverage of IVF or other treatments.
That requirement makes little sense for patients trying to preserve their fertility before undergoing medically necessary cancer or other treatment.
“These people aren’t infertile,” said Ms. Collura. “They need to undergo some sort of intervention that is going to impair their future fertility, and what we say is that if it’s medically necessary, they should have a right to have it covered.”
KHN’s coverage of women’s health care issues is supported in part by The David and Lucile Packard Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
When Katherine Frega was diagnosed with Hodgkin lymphoma 8 years ago at the age 17 years, she was so sick that all she could focus on was starting chemotherapy to treat her aggressive blood cancer. It was her dad who thought to ask the oncologist, “How is this treatment going to affect her ability to have children?”
The oncologist discussed the risks but stressed that Ms. Frega needed to start treatment right away.
The question of fertility is often overlooked when young cancer patients are battling a life-threatening illness. And since health insurance doesn’t typically cover fertility preservation care, patients and their families may be deterred by the cost.
But a growing number of states now require plans to cover such services when medically necessary treatment jeopardizes fertility.
Treatment for cancer and other serious conditions involves toxic chemotherapy drugs, radiation, and surgery that can cause infertility in women and men.
The cost to freeze patients’ healthy eggs, sperm, or embryos for future use can be a major barrier, said Eden Cardozo, MD, a reproductive endocrinologist and director of the fertility preservation program at the Women & Infants Fertility Center in Providence, R.I. Dr. Cardozo was instrumental in getting Rhode Island’s law passed last year.
“[Patients] have to move quickly,” she said. “They don’t have time to raise funds from family and friends. They don’t have time to petition their insurance company.”
Reproductive health advocates argue that fertility preservation should be viewed as a core component of cancer care in younger people, not as an optional infertility offering. Some compare this type of coverage with the federal Women’s Health and Cancer Rights Act, which requires plans that cover a patient’s mastectomy to also provide for breast reconstruction.
New laws in Delaware, Illinois, and Maryland require plans to include this benefit. The Delaware law applies to plans issued or renewed after June of this year; the requirement in the other two states starts in 2019. Connecticut and Rhode Island passed similar laws last year. New Jersey lawmakers are considering a bill and advocates in New York plan to make another attempt after both legislative chambers passed fertility preservation bills in the last session but failed to reconcile them.
The state measures don’t apply to companies that are self-funded, meaning they pay their employee claims directly rather than buying state-regulated insurance policies for that purpose. They also don’t apply to government-funded programs such as Medicaid or Tricare.
Although freezing sperm and embryos has been common medical practice for decades, egg freezing was considered experimental by professional groups until 2012. As the technology has improved, the need for insurance coverage has grown, said Joyce Reinecke, executive director of the Alliance for Fertility Preservation, an advocacy group for cancer patients.
When Ms. Frega’s cancer didn’t respond to chemotherapy, her doctors recommended a bone marrow transplant in January 2012. Even if her eggs hadn’t been damaged by the chemotherapy, the transplant would likely cause permanent infertility, she was told. So she took hormones to stimulate her ovaries to produce more eggs, among other things, and seven were retrieved during an outpatient procedure days before her transplant.
Ms. Frega’s parents paid $10,000 for the medications and egg retrieval, a significant amount but less than what many pay. They were aided by Livestrong Fertility, a nonprofit group that provides access to discounted fertility preservation services for cancer patients who meet income guidelines.
Ms. Frega has good insurance through her mother’s employer plan. “They covered everything else, except for this,” she said. “They considered it not medically necessary.”
Cancer-free following two bone marrow transplants, Ms. Frega, now 25 years old, is a third-year medical student at the Upstate Medical University in Syracuse, N.Y. She plans to specialize in oncology.
Between 20% and 70% of cancer patients experience some degree of fertility impairment, according to Dr. Cardozo in Rhode Island. Though they make up the largest at-risk group, the complication isn’t unique to cancer patients. People with other conditions such as lupus and rheumatoid arthritis who are treated with chemotherapy drugs may be affected, as may patients with conditions such as endometriosis who require surgery.
Despite the much-ballyhooed examples of tech companies like Facebook, Apple, and Google that offer egg freezing as an employee perk, cryopreservation, as it’s called, isn’t a typical employee benefit.
Only 6% of large companies with 500 or more workers offer egg freezing for employees or their spouses, according to the 2017 annual employer survey by benefits consultant Mercer. About a quarter cover in vitro fertilization (IVF). About 44% of large employers don’t offer any infertility services, the survey found.
Men face the same infertility risk when they need cancer treatment.
When Blake Hornbrook, an Army medic at Fort Campbell, Ky., had surgery to remove a cancerous testicle in the fall of 2015, he and his wife, Kelsey, were stationed in Germany. Mr. Hornbrook, then 26 years old, looked into fertility preservation while overseas, but the annual storage fee of 1,000 euros (about $1,150) deterred the couple.
He required a second surgery several months later to see if the cancer had spread to his lymph nodes. The couple returned to the United States and drove directly from the airport to a sperm bank in Fairfax, Va. It cost roughly $400 for the initial appointment to provide a sperm specimen and store it, Mr. Hornbrook said.
Tricare covered Mr. Hornbrook’s cancer treatment, but it didn’t pay for fertility preservation or for IVF, which he estimated cost the couple $6,500 in clinic fees. Tricare provided discounts on some of the fertility drugs.
Their daughter, Harper, was born 7 months ago, and Mr. Hornbrook’s cancer remains in remission.
For young cancer patients, the cost of storing the eggs or sperm that have been preserved can add up. Even if a state has a fertility preservation law, it typically doesn’t cover those costs, Ms. Reinecke said.
The Hornbrooks pay $480 annually to store his sperm and $375 to store their remaining embryos. Ms. Frega pays $1,000 annually to store her eggs.
Ms. Frega hopes to be able to conceive naturally and knowing she has frozen eggs available is “relieving, but also anxiety producing,” she said. If she can’t get pregnant later on, she may have to pay $10,000 or more for IVF as well. “That’s what lies ahead,” she said.
A total of 16 states require insurers to offer or cover infertility services to some extent, according to infertility advocacy organization Resolve. Requirements vary: Insurers may have to cover diagnosis or testing for infertility, for example, but not treatments like IVF or fertility medications, said Barbara Collura, president and CEO of Resolve.
Typically, state infertility coverage laws require couples to try to get pregnant for a year or two before they’re eligible for insurance coverage of IVF or other treatments.
That requirement makes little sense for patients trying to preserve their fertility before undergoing medically necessary cancer or other treatment.
“These people aren’t infertile,” said Ms. Collura. “They need to undergo some sort of intervention that is going to impair their future fertility, and what we say is that if it’s medically necessary, they should have a right to have it covered.”
KHN’s coverage of women’s health care issues is supported in part by The David and Lucile Packard Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
When Katherine Frega was diagnosed with Hodgkin lymphoma 8 years ago at the age 17 years, she was so sick that all she could focus on was starting chemotherapy to treat her aggressive blood cancer. It was her dad who thought to ask the oncologist, “How is this treatment going to affect her ability to have children?”
The oncologist discussed the risks but stressed that Ms. Frega needed to start treatment right away.
The question of fertility is often overlooked when young cancer patients are battling a life-threatening illness. And since health insurance doesn’t typically cover fertility preservation care, patients and their families may be deterred by the cost.
But a growing number of states now require plans to cover such services when medically necessary treatment jeopardizes fertility.
Treatment for cancer and other serious conditions involves toxic chemotherapy drugs, radiation, and surgery that can cause infertility in women and men.
The cost to freeze patients’ healthy eggs, sperm, or embryos for future use can be a major barrier, said Eden Cardozo, MD, a reproductive endocrinologist and director of the fertility preservation program at the Women & Infants Fertility Center in Providence, R.I. Dr. Cardozo was instrumental in getting Rhode Island’s law passed last year.
“[Patients] have to move quickly,” she said. “They don’t have time to raise funds from family and friends. They don’t have time to petition their insurance company.”
Reproductive health advocates argue that fertility preservation should be viewed as a core component of cancer care in younger people, not as an optional infertility offering. Some compare this type of coverage with the federal Women’s Health and Cancer Rights Act, which requires plans that cover a patient’s mastectomy to also provide for breast reconstruction.
New laws in Delaware, Illinois, and Maryland require plans to include this benefit. The Delaware law applies to plans issued or renewed after June of this year; the requirement in the other two states starts in 2019. Connecticut and Rhode Island passed similar laws last year. New Jersey lawmakers are considering a bill and advocates in New York plan to make another attempt after both legislative chambers passed fertility preservation bills in the last session but failed to reconcile them.
The state measures don’t apply to companies that are self-funded, meaning they pay their employee claims directly rather than buying state-regulated insurance policies for that purpose. They also don’t apply to government-funded programs such as Medicaid or Tricare.
Although freezing sperm and embryos has been common medical practice for decades, egg freezing was considered experimental by professional groups until 2012. As the technology has improved, the need for insurance coverage has grown, said Joyce Reinecke, executive director of the Alliance for Fertility Preservation, an advocacy group for cancer patients.
When Ms. Frega’s cancer didn’t respond to chemotherapy, her doctors recommended a bone marrow transplant in January 2012. Even if her eggs hadn’t been damaged by the chemotherapy, the transplant would likely cause permanent infertility, she was told. So she took hormones to stimulate her ovaries to produce more eggs, among other things, and seven were retrieved during an outpatient procedure days before her transplant.
Ms. Frega’s parents paid $10,000 for the medications and egg retrieval, a significant amount but less than what many pay. They were aided by Livestrong Fertility, a nonprofit group that provides access to discounted fertility preservation services for cancer patients who meet income guidelines.
Ms. Frega has good insurance through her mother’s employer plan. “They covered everything else, except for this,” she said. “They considered it not medically necessary.”
Cancer-free following two bone marrow transplants, Ms. Frega, now 25 years old, is a third-year medical student at the Upstate Medical University in Syracuse, N.Y. She plans to specialize in oncology.
Between 20% and 70% of cancer patients experience some degree of fertility impairment, according to Dr. Cardozo in Rhode Island. Though they make up the largest at-risk group, the complication isn’t unique to cancer patients. People with other conditions such as lupus and rheumatoid arthritis who are treated with chemotherapy drugs may be affected, as may patients with conditions such as endometriosis who require surgery.
Despite the much-ballyhooed examples of tech companies like Facebook, Apple, and Google that offer egg freezing as an employee perk, cryopreservation, as it’s called, isn’t a typical employee benefit.
Only 6% of large companies with 500 or more workers offer egg freezing for employees or their spouses, according to the 2017 annual employer survey by benefits consultant Mercer. About a quarter cover in vitro fertilization (IVF). About 44% of large employers don’t offer any infertility services, the survey found.
Men face the same infertility risk when they need cancer treatment.
When Blake Hornbrook, an Army medic at Fort Campbell, Ky., had surgery to remove a cancerous testicle in the fall of 2015, he and his wife, Kelsey, were stationed in Germany. Mr. Hornbrook, then 26 years old, looked into fertility preservation while overseas, but the annual storage fee of 1,000 euros (about $1,150) deterred the couple.
He required a second surgery several months later to see if the cancer had spread to his lymph nodes. The couple returned to the United States and drove directly from the airport to a sperm bank in Fairfax, Va. It cost roughly $400 for the initial appointment to provide a sperm specimen and store it, Mr. Hornbrook said.
Tricare covered Mr. Hornbrook’s cancer treatment, but it didn’t pay for fertility preservation or for IVF, which he estimated cost the couple $6,500 in clinic fees. Tricare provided discounts on some of the fertility drugs.
Their daughter, Harper, was born 7 months ago, and Mr. Hornbrook’s cancer remains in remission.
For young cancer patients, the cost of storing the eggs or sperm that have been preserved can add up. Even if a state has a fertility preservation law, it typically doesn’t cover those costs, Ms. Reinecke said.
The Hornbrooks pay $480 annually to store his sperm and $375 to store their remaining embryos. Ms. Frega pays $1,000 annually to store her eggs.
Ms. Frega hopes to be able to conceive naturally and knowing she has frozen eggs available is “relieving, but also anxiety producing,” she said. If she can’t get pregnant later on, she may have to pay $10,000 or more for IVF as well. “That’s what lies ahead,” she said.
A total of 16 states require insurers to offer or cover infertility services to some extent, according to infertility advocacy organization Resolve. Requirements vary: Insurers may have to cover diagnosis or testing for infertility, for example, but not treatments like IVF or fertility medications, said Barbara Collura, president and CEO of Resolve.
Typically, state infertility coverage laws require couples to try to get pregnant for a year or two before they’re eligible for insurance coverage of IVF or other treatments.
That requirement makes little sense for patients trying to preserve their fertility before undergoing medically necessary cancer or other treatment.
“These people aren’t infertile,” said Ms. Collura. “They need to undergo some sort of intervention that is going to impair their future fertility, and what we say is that if it’s medically necessary, they should have a right to have it covered.”
KHN’s coverage of women’s health care issues is supported in part by The David and Lucile Packard Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Researchers consider R/R ALL drugs in the first-line setting
CHICAGO – Novel antibodies are improving outcomes in relapsed and refractory acute lymphoblastic leukemia (ALL), and the hope is that they will also show benefit in the up-front treatment setting and thereby improve overall outcomes, according to Anjali Advani, MD.
“It has been a really exciting time in ALL because several drugs have now been FDA approved: blinatumomab, inotuzumab, and now – for patients who are less than 26 years of age – we actually have CAR [chimeric antigen receptor] T cells that have been approved,” Dr. Advani, a hematologist and director of the inpatient leukemia program at the Cleveland Clinic said at the American Society of Hematology Meeting on Hematologic Malignancies.
At the time of relapse, however, the only known cure is allogeneic bone marrow transplant. That may change as more data regarding CAR T cells become available, but the typical goal at this time is to get patients into remission and then to transplant, she said.
Blinatumomab
“Blinatumomab is a very interesting antibody,” Dr. Advani said, explaining that it is a bispecific, T cell–engaging antibody with an anti-CD3 arm that engages the T cell and an anti-CD19 antibody that engages the B lymphoblast.
“Basically this drug then acts as a bridge between the lymphoblast and the T cell to lead to proliferation of the cytotoxic T cell and apoptosis of the lymphoblast,” she said. “It’s interesting because it’s an antibody but it actually works through the immune system through the T cells.”
The largest study to date of blinatumomab in the relapsed/refractory ALL setting showed a 43% complete remission (CR) or CR with partial hematological recovery of peripheral blood counts (CRi) in 189 treated patients with Philadelphia chromosome–negative ALL. It also demonstrated and a 39% rate of salvage status 2 or higher, she said, noting that the response was impressive given that about 30% of participants had a prior transplant (Lancet. 2015 Jan 1;16[1]:57-66).
Of the responders, 40% went on to allogeneic transplant. This was a “fairly impressive” rate given the 30% prior-transplant rate, Dr. Advani said.
“There also was a high minimal residual disease response in those patients achieving CR,” she said, adding that the only significant predictor of response was bone marrow blast count; patients with 50% or more blasts in the bone marrow had a reduced likelihood of responding to blinatumomab.
The agent was approved by the Food and Drug Administration in December 2014 based on these phase 2 findings.
Adverse events mainly included toxicities that are expected in leukemia patients; the most frequent were febrile neutropenia, neutropenia, and anemia. Two patients developed cytokine release syndrome, and about half of the blinatumomab-treated patients experienced neurological events, although the majority of those were grade 1 or 2 and were easily manageable, she noted.
Blinatumomab was further evaluated in the phase 3 TOWER study (NCT02013167), which compared it with standard-of-care chemotherapy regimens. This study showed much higher response rates with blinatumomab than with the chemotherapy regimens (CR with full, partial, or incomplete hematologic recovery, 44% vs. 25%, respectively), Dr. Advani said (N Engl J Med. 2017 Mar 2;376[9]:836-47).
“The main things to remember [are that blinatumomab is] generally very well tolerated and it has been shown to be superior over standard chemotherapy,” she said. “I think it’s a very good drug to use as a bridge to transplant.”
One setting where blinatumomab perhaps should not be used is in patients with central nervous system disease, she noted.
“There is some concern, at least theoretically, that if you have to use concurrent intrathecal chemo along with blinatumomab, there could be some neurotoxicity,” Dr. Advani said, adding that there are no clear data in that setting because patients with CNS disease were not included in the trials.
Patients with high tumor burden may also be poor candidates for blinatumomab because they tend to have lower response rates.
“That doesn’t mean you can’t use it, but you have to kind of think about what the best option would be,” she said.
Additionally, patients treated with CAR T-cell therapy may develop CD19 loss or CD19-negative disease, and blinatumomab should be avoided in these patients.
“The nice thing ... is you don’t have to worry about veno-occlusive disease [VOD] in patients who are proceeding to transplant,” she said, explaining that no increased risk of VOD was seen in these trials.
Inotuzumab
Inotuzumab, which was approved in 2017, differs from blinatumomab in that it is an anti-CD22-calicheamicin conjugate; however, it also showed high response rates in the initial phase 2 trial in relapsed/refractory ALL. The overall response rate was 57%, with 18% achieving a complete response and 63% achieving complete molecular remission.
Of 49 treated patients, 22 patients proceeded to allogeneic transplant, and 5 of those developed VOD.
“Interestingly, four out of five of these patients had received a clofarabine-based preparative regimen, and this likely explains why there was a higher risk of VOD in this study,” she said, noting that the VOD risk has been lower in subsequent studies of inotuzumab.
The international INO-VATE ALL study (NCT01564784) that led to FDA approval was similar in design to the TOWER study in that it compared inotuzumab with standard chemotherapy regimens, and response rates were clearly higher (81% vs. 33%) with inotuzumab (N Engl J Med. 2016 Aug 25;375[8]:740-53).
The VOD risk in the INO-VATE trial was 11%, and it seemed to be higher in those who received dual alkylator–conditioning regimens, which are commonly used in Europe.
Longer-term outcomes after transplant in INO-VATE participants show that median survival has not been reached.
“It’s encouraging that with longer follow-up these patients actually look like they’re doing well,” Dr. Advani said, adding that inotuzumab is a good treatment option for relapsed patients with high disease burden or with CNS disease.
The continuous hookup required for this treatment may be problematic for some younger and older patients, but it is generally not an issue, she noted.
It is important, though, to give as few cycles prior to transplant as possible and to “really think about the preparative regimen to decrease the risk of VOD.”
CAR T-cell therapy
As for CAR T-cell therapy in the relapsed/refractory ALL setting, tisagenlecleucel was approved in 2017 for those up to age 25 years with B-cell precursor ALL that is refractory or in second or later relapse.
Approval was based on a single-arm trial of 63 patients with relapsed or refractory pediatric precursor B-cell ALL, including 35 patients who had prior transplant. The confirmed overall remission rate was 82%, with a 63% CR rate and 19% CRi rate.
“This is a very exciting area,” Dr. Advani said. “There are multiple trials being done in adults with ALL to really look at the older subgroup of patients.”
Overall outcomes
“These treatments we have now really seem to be effective in the relapse setting, but the problem is that once patients relapse and then go to transplant, their overall survival is still poor,” Dr. Advani said. “So the question is how can we improve the up-front treatment of patients so that hopefully they don’t relapse, and hopefully we also can send a smaller number of patients to transplant.”
Two trials seek to address this, she said.
The A041501 study (NCT03150693) is comparing C10403 chemotherapy with C10403 induction followed by two cycles of inotuzumab before continuing with chemotherapy in adults under age 40 years with previously untreated B ALL.
The primary objective is improved 3-year event-free survival, she said, adding that minimal residual disease (MRD) testing will be used and that CD20-positive patients will receive rituximab, as is now standard.
The phase 3 E1910 study (NCT02003222) is evaluating up-front blinatumomab in patients aged 30-70 years with newly diagnosed BCR-ABL–negative B-lineage ALL. This trial was complicated by the recent approval of blinatumomab for MRD-positive disease, which rendered randomization of MRD-positive patients unethical. MRD-negative patients will be randomized, however.
“The hope is that, by incorporating blinatumomab up front, this will again improve outcomes for patients,” she said.
Dr. Advani reported consultancy for Pfizer; research funding from Genzyme, Novartis, Pfizer, and Sigma Tau; and honoraria from Genzyme, Pfizer, and Sigma Tau. She is also on the speakers bureau for Sigma Tau.
CHICAGO – Novel antibodies are improving outcomes in relapsed and refractory acute lymphoblastic leukemia (ALL), and the hope is that they will also show benefit in the up-front treatment setting and thereby improve overall outcomes, according to Anjali Advani, MD.
“It has been a really exciting time in ALL because several drugs have now been FDA approved: blinatumomab, inotuzumab, and now – for patients who are less than 26 years of age – we actually have CAR [chimeric antigen receptor] T cells that have been approved,” Dr. Advani, a hematologist and director of the inpatient leukemia program at the Cleveland Clinic said at the American Society of Hematology Meeting on Hematologic Malignancies.
At the time of relapse, however, the only known cure is allogeneic bone marrow transplant. That may change as more data regarding CAR T cells become available, but the typical goal at this time is to get patients into remission and then to transplant, she said.
Blinatumomab
“Blinatumomab is a very interesting antibody,” Dr. Advani said, explaining that it is a bispecific, T cell–engaging antibody with an anti-CD3 arm that engages the T cell and an anti-CD19 antibody that engages the B lymphoblast.
“Basically this drug then acts as a bridge between the lymphoblast and the T cell to lead to proliferation of the cytotoxic T cell and apoptosis of the lymphoblast,” she said. “It’s interesting because it’s an antibody but it actually works through the immune system through the T cells.”
The largest study to date of blinatumomab in the relapsed/refractory ALL setting showed a 43% complete remission (CR) or CR with partial hematological recovery of peripheral blood counts (CRi) in 189 treated patients with Philadelphia chromosome–negative ALL. It also demonstrated and a 39% rate of salvage status 2 or higher, she said, noting that the response was impressive given that about 30% of participants had a prior transplant (Lancet. 2015 Jan 1;16[1]:57-66).
Of the responders, 40% went on to allogeneic transplant. This was a “fairly impressive” rate given the 30% prior-transplant rate, Dr. Advani said.
“There also was a high minimal residual disease response in those patients achieving CR,” she said, adding that the only significant predictor of response was bone marrow blast count; patients with 50% or more blasts in the bone marrow had a reduced likelihood of responding to blinatumomab.
The agent was approved by the Food and Drug Administration in December 2014 based on these phase 2 findings.
Adverse events mainly included toxicities that are expected in leukemia patients; the most frequent were febrile neutropenia, neutropenia, and anemia. Two patients developed cytokine release syndrome, and about half of the blinatumomab-treated patients experienced neurological events, although the majority of those were grade 1 or 2 and were easily manageable, she noted.
Blinatumomab was further evaluated in the phase 3 TOWER study (NCT02013167), which compared it with standard-of-care chemotherapy regimens. This study showed much higher response rates with blinatumomab than with the chemotherapy regimens (CR with full, partial, or incomplete hematologic recovery, 44% vs. 25%, respectively), Dr. Advani said (N Engl J Med. 2017 Mar 2;376[9]:836-47).
“The main things to remember [are that blinatumomab is] generally very well tolerated and it has been shown to be superior over standard chemotherapy,” she said. “I think it’s a very good drug to use as a bridge to transplant.”
One setting where blinatumomab perhaps should not be used is in patients with central nervous system disease, she noted.
“There is some concern, at least theoretically, that if you have to use concurrent intrathecal chemo along with blinatumomab, there could be some neurotoxicity,” Dr. Advani said, adding that there are no clear data in that setting because patients with CNS disease were not included in the trials.
Patients with high tumor burden may also be poor candidates for blinatumomab because they tend to have lower response rates.
“That doesn’t mean you can’t use it, but you have to kind of think about what the best option would be,” she said.
Additionally, patients treated with CAR T-cell therapy may develop CD19 loss or CD19-negative disease, and blinatumomab should be avoided in these patients.
“The nice thing ... is you don’t have to worry about veno-occlusive disease [VOD] in patients who are proceeding to transplant,” she said, explaining that no increased risk of VOD was seen in these trials.
Inotuzumab
Inotuzumab, which was approved in 2017, differs from blinatumomab in that it is an anti-CD22-calicheamicin conjugate; however, it also showed high response rates in the initial phase 2 trial in relapsed/refractory ALL. The overall response rate was 57%, with 18% achieving a complete response and 63% achieving complete molecular remission.
Of 49 treated patients, 22 patients proceeded to allogeneic transplant, and 5 of those developed VOD.
“Interestingly, four out of five of these patients had received a clofarabine-based preparative regimen, and this likely explains why there was a higher risk of VOD in this study,” she said, noting that the VOD risk has been lower in subsequent studies of inotuzumab.
The international INO-VATE ALL study (NCT01564784) that led to FDA approval was similar in design to the TOWER study in that it compared inotuzumab with standard chemotherapy regimens, and response rates were clearly higher (81% vs. 33%) with inotuzumab (N Engl J Med. 2016 Aug 25;375[8]:740-53).
The VOD risk in the INO-VATE trial was 11%, and it seemed to be higher in those who received dual alkylator–conditioning regimens, which are commonly used in Europe.
Longer-term outcomes after transplant in INO-VATE participants show that median survival has not been reached.
“It’s encouraging that with longer follow-up these patients actually look like they’re doing well,” Dr. Advani said, adding that inotuzumab is a good treatment option for relapsed patients with high disease burden or with CNS disease.
The continuous hookup required for this treatment may be problematic for some younger and older patients, but it is generally not an issue, she noted.
It is important, though, to give as few cycles prior to transplant as possible and to “really think about the preparative regimen to decrease the risk of VOD.”
CAR T-cell therapy
As for CAR T-cell therapy in the relapsed/refractory ALL setting, tisagenlecleucel was approved in 2017 for those up to age 25 years with B-cell precursor ALL that is refractory or in second or later relapse.
Approval was based on a single-arm trial of 63 patients with relapsed or refractory pediatric precursor B-cell ALL, including 35 patients who had prior transplant. The confirmed overall remission rate was 82%, with a 63% CR rate and 19% CRi rate.
“This is a very exciting area,” Dr. Advani said. “There are multiple trials being done in adults with ALL to really look at the older subgroup of patients.”
Overall outcomes
“These treatments we have now really seem to be effective in the relapse setting, but the problem is that once patients relapse and then go to transplant, their overall survival is still poor,” Dr. Advani said. “So the question is how can we improve the up-front treatment of patients so that hopefully they don’t relapse, and hopefully we also can send a smaller number of patients to transplant.”
Two trials seek to address this, she said.
The A041501 study (NCT03150693) is comparing C10403 chemotherapy with C10403 induction followed by two cycles of inotuzumab before continuing with chemotherapy in adults under age 40 years with previously untreated B ALL.
The primary objective is improved 3-year event-free survival, she said, adding that minimal residual disease (MRD) testing will be used and that CD20-positive patients will receive rituximab, as is now standard.
The phase 3 E1910 study (NCT02003222) is evaluating up-front blinatumomab in patients aged 30-70 years with newly diagnosed BCR-ABL–negative B-lineage ALL. This trial was complicated by the recent approval of blinatumomab for MRD-positive disease, which rendered randomization of MRD-positive patients unethical. MRD-negative patients will be randomized, however.
“The hope is that, by incorporating blinatumomab up front, this will again improve outcomes for patients,” she said.
Dr. Advani reported consultancy for Pfizer; research funding from Genzyme, Novartis, Pfizer, and Sigma Tau; and honoraria from Genzyme, Pfizer, and Sigma Tau. She is also on the speakers bureau for Sigma Tau.
CHICAGO – Novel antibodies are improving outcomes in relapsed and refractory acute lymphoblastic leukemia (ALL), and the hope is that they will also show benefit in the up-front treatment setting and thereby improve overall outcomes, according to Anjali Advani, MD.
“It has been a really exciting time in ALL because several drugs have now been FDA approved: blinatumomab, inotuzumab, and now – for patients who are less than 26 years of age – we actually have CAR [chimeric antigen receptor] T cells that have been approved,” Dr. Advani, a hematologist and director of the inpatient leukemia program at the Cleveland Clinic said at the American Society of Hematology Meeting on Hematologic Malignancies.
At the time of relapse, however, the only known cure is allogeneic bone marrow transplant. That may change as more data regarding CAR T cells become available, but the typical goal at this time is to get patients into remission and then to transplant, she said.
Blinatumomab
“Blinatumomab is a very interesting antibody,” Dr. Advani said, explaining that it is a bispecific, T cell–engaging antibody with an anti-CD3 arm that engages the T cell and an anti-CD19 antibody that engages the B lymphoblast.
“Basically this drug then acts as a bridge between the lymphoblast and the T cell to lead to proliferation of the cytotoxic T cell and apoptosis of the lymphoblast,” she said. “It’s interesting because it’s an antibody but it actually works through the immune system through the T cells.”
The largest study to date of blinatumomab in the relapsed/refractory ALL setting showed a 43% complete remission (CR) or CR with partial hematological recovery of peripheral blood counts (CRi) in 189 treated patients with Philadelphia chromosome–negative ALL. It also demonstrated and a 39% rate of salvage status 2 or higher, she said, noting that the response was impressive given that about 30% of participants had a prior transplant (Lancet. 2015 Jan 1;16[1]:57-66).
Of the responders, 40% went on to allogeneic transplant. This was a “fairly impressive” rate given the 30% prior-transplant rate, Dr. Advani said.
“There also was a high minimal residual disease response in those patients achieving CR,” she said, adding that the only significant predictor of response was bone marrow blast count; patients with 50% or more blasts in the bone marrow had a reduced likelihood of responding to blinatumomab.
The agent was approved by the Food and Drug Administration in December 2014 based on these phase 2 findings.
Adverse events mainly included toxicities that are expected in leukemia patients; the most frequent were febrile neutropenia, neutropenia, and anemia. Two patients developed cytokine release syndrome, and about half of the blinatumomab-treated patients experienced neurological events, although the majority of those were grade 1 or 2 and were easily manageable, she noted.
Blinatumomab was further evaluated in the phase 3 TOWER study (NCT02013167), which compared it with standard-of-care chemotherapy regimens. This study showed much higher response rates with blinatumomab than with the chemotherapy regimens (CR with full, partial, or incomplete hematologic recovery, 44% vs. 25%, respectively), Dr. Advani said (N Engl J Med. 2017 Mar 2;376[9]:836-47).
“The main things to remember [are that blinatumomab is] generally very well tolerated and it has been shown to be superior over standard chemotherapy,” she said. “I think it’s a very good drug to use as a bridge to transplant.”
One setting where blinatumomab perhaps should not be used is in patients with central nervous system disease, she noted.
“There is some concern, at least theoretically, that if you have to use concurrent intrathecal chemo along with blinatumomab, there could be some neurotoxicity,” Dr. Advani said, adding that there are no clear data in that setting because patients with CNS disease were not included in the trials.
Patients with high tumor burden may also be poor candidates for blinatumomab because they tend to have lower response rates.
“That doesn’t mean you can’t use it, but you have to kind of think about what the best option would be,” she said.
Additionally, patients treated with CAR T-cell therapy may develop CD19 loss or CD19-negative disease, and blinatumomab should be avoided in these patients.
“The nice thing ... is you don’t have to worry about veno-occlusive disease [VOD] in patients who are proceeding to transplant,” she said, explaining that no increased risk of VOD was seen in these trials.
Inotuzumab
Inotuzumab, which was approved in 2017, differs from blinatumomab in that it is an anti-CD22-calicheamicin conjugate; however, it also showed high response rates in the initial phase 2 trial in relapsed/refractory ALL. The overall response rate was 57%, with 18% achieving a complete response and 63% achieving complete molecular remission.
Of 49 treated patients, 22 patients proceeded to allogeneic transplant, and 5 of those developed VOD.
“Interestingly, four out of five of these patients had received a clofarabine-based preparative regimen, and this likely explains why there was a higher risk of VOD in this study,” she said, noting that the VOD risk has been lower in subsequent studies of inotuzumab.
The international INO-VATE ALL study (NCT01564784) that led to FDA approval was similar in design to the TOWER study in that it compared inotuzumab with standard chemotherapy regimens, and response rates were clearly higher (81% vs. 33%) with inotuzumab (N Engl J Med. 2016 Aug 25;375[8]:740-53).
The VOD risk in the INO-VATE trial was 11%, and it seemed to be higher in those who received dual alkylator–conditioning regimens, which are commonly used in Europe.
Longer-term outcomes after transplant in INO-VATE participants show that median survival has not been reached.
“It’s encouraging that with longer follow-up these patients actually look like they’re doing well,” Dr. Advani said, adding that inotuzumab is a good treatment option for relapsed patients with high disease burden or with CNS disease.
The continuous hookup required for this treatment may be problematic for some younger and older patients, but it is generally not an issue, she noted.
It is important, though, to give as few cycles prior to transplant as possible and to “really think about the preparative regimen to decrease the risk of VOD.”
CAR T-cell therapy
As for CAR T-cell therapy in the relapsed/refractory ALL setting, tisagenlecleucel was approved in 2017 for those up to age 25 years with B-cell precursor ALL that is refractory or in second or later relapse.
Approval was based on a single-arm trial of 63 patients with relapsed or refractory pediatric precursor B-cell ALL, including 35 patients who had prior transplant. The confirmed overall remission rate was 82%, with a 63% CR rate and 19% CRi rate.
“This is a very exciting area,” Dr. Advani said. “There are multiple trials being done in adults with ALL to really look at the older subgroup of patients.”
Overall outcomes
“These treatments we have now really seem to be effective in the relapse setting, but the problem is that once patients relapse and then go to transplant, their overall survival is still poor,” Dr. Advani said. “So the question is how can we improve the up-front treatment of patients so that hopefully they don’t relapse, and hopefully we also can send a smaller number of patients to transplant.”
Two trials seek to address this, she said.
The A041501 study (NCT03150693) is comparing C10403 chemotherapy with C10403 induction followed by two cycles of inotuzumab before continuing with chemotherapy in adults under age 40 years with previously untreated B ALL.
The primary objective is improved 3-year event-free survival, she said, adding that minimal residual disease (MRD) testing will be used and that CD20-positive patients will receive rituximab, as is now standard.
The phase 3 E1910 study (NCT02003222) is evaluating up-front blinatumomab in patients aged 30-70 years with newly diagnosed BCR-ABL–negative B-lineage ALL. This trial was complicated by the recent approval of blinatumomab for MRD-positive disease, which rendered randomization of MRD-positive patients unethical. MRD-negative patients will be randomized, however.
“The hope is that, by incorporating blinatumomab up front, this will again improve outcomes for patients,” she said.
Dr. Advani reported consultancy for Pfizer; research funding from Genzyme, Novartis, Pfizer, and Sigma Tau; and honoraria from Genzyme, Pfizer, and Sigma Tau. She is also on the speakers bureau for Sigma Tau.
EXPERT ANALYSIS FROM MHM 2018
Ibrutinib plus obinutuzumab gets priority review in CLL/SLL
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).

The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).
The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).
The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
Mysterious polio-like illness baffles medical experts while frightening parents
A spike in the number of children with a rare neurological disease that causes polio-like symptoms has health officials across the country scrambling to understand the illness. Yet, more than 4 years after health officials first recorded the most recent uptick in cases, much about the national outbreak remains a mystery.
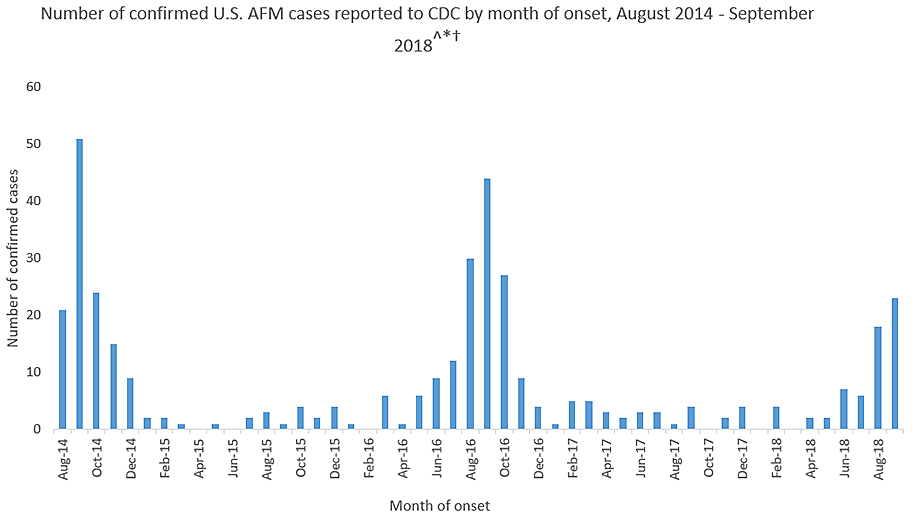
Acute flaccid myelitis (AFM) affects the gray matter in the spinal cord, causing sudden muscle weakness and a loss of reflexes. The illness can lead to serious complications – including paralysis or respiratory failure – and requires immediate medical attention.
The Centers for Disease Control and Prevention is investigating 127 cases of possible AFM, including 62 that have been confirmed in 22 states this year. At least 90% of the cases are among patients 18 years old and younger. The average age of a patient is 4 years old.
AFM remains extremely rare, even with the recent increase. The CDC estimates fewer than 1 in a million Americans will get the disease. Officials advised parents not to panic but remain vigilant for any sudden onset of symptoms. They also suggested that children stay up to date with their vaccines and practice good hand washing habits.
This year’s outbreak marks the third spike of AFM in 4 years. From August 2014 to September 2018, 386 cases have been confirmed. Yet, experts still do not understand crucial aspects of the disease, including its origins and who is most at risk.
“There is a lot we don’t know about AFM,” said Nancy Messonnier, MD, director of the National Center for Immunization and Respiratory Diseases. Here’s what puzzles health officials about AFM:
The cause is still unknown
Acute flaccid myelitis can be caused by viruses, such as polio or West Nile. But federal officials said that those viruses have not been linked to the U.S. outbreak over the past 4 years. They have not isolated the cause of these cases.
Despite symptoms reminiscent of polio, no AFM cases have tested positive for that virus, according to the CDC. Investigators have also ruled out a variety of germs. Environmental agents, viruses, and other pathogens are still being considered.
The 2014 outbreak of AFM coincided with a surge of another virus that caused severe respiratory problems, called EV-D68. However, the CDC could not establish a causal link between AFM and the virus. Since then, no large outbreaks of the virus have occurred, according to the CDC.
Carlos Pardo-Villamizar, MD, a neurologist and director of the Johns Hopkins Transverse Myelitis Center, said that the mystery lies in whether the damage seen in AFM is caused by an external agent or the body’s own defenses.
“At this moment, we don’t know if it’s a virus that is coming and producing direct damage of the gray matter in the spinal cord,” he said, “or if a virus is triggering immunological responses that produce a secondary damage in the spinal cord.”
It’s not clear who is at risk
Although the disease appears to target a certain age group, federal disease experts do not know who is likely to get acute flaccid myelitis.
Dr. Pardo-Villamizar said identifying vulnerable populations is “a work in progress.”
Mary Anne Jackson, MD, a pediatric infectious disease specialist and interim dean of the school of medicine at the University of Missouri–Kansas City, said many of the patients she saw were healthy children before falling ill with the disease. She suspects that a host of factors play a role in the likelihood of getting AFM, but more cases must be reviewed in order to find an answer.
The long-term effects are unknown
The CDC said it doesn’t know how long symptoms of the disease will last for patients. However, experts say that initial indications from a small number of cases suggest a grim outlook.
A study published last year found six of eight children in Colorado with acute flaccid myelitis still struggled with motor skills 1 year after their diagnosis. Nonetheless, the researchers found that the patients and families “demonstrated a high degree of resilience and recovery.”
“The majority of these patients are left with extensive problems,” said Dr. Pardo-Villamizar, who was not involved in the study.
Dr. Jackson, who also saw persistent muscle weakness in her patients, said she believes the CDC may be hesitant to specify the long-term effects of the disease because existing studies have included only small numbers of patients. More studies that include a larger proportion of confirmed cases are needed to better understand long-term outcomes, she said.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
A spike in the number of children with a rare neurological disease that causes polio-like symptoms has health officials across the country scrambling to understand the illness. Yet, more than 4 years after health officials first recorded the most recent uptick in cases, much about the national outbreak remains a mystery.
Acute flaccid myelitis (AFM) affects the gray matter in the spinal cord, causing sudden muscle weakness and a loss of reflexes. The illness can lead to serious complications – including paralysis or respiratory failure – and requires immediate medical attention.
The Centers for Disease Control and Prevention is investigating 127 cases of possible AFM, including 62 that have been confirmed in 22 states this year. At least 90% of the cases are among patients 18 years old and younger. The average age of a patient is 4 years old.
AFM remains extremely rare, even with the recent increase. The CDC estimates fewer than 1 in a million Americans will get the disease. Officials advised parents not to panic but remain vigilant for any sudden onset of symptoms. They also suggested that children stay up to date with their vaccines and practice good hand washing habits.
This year’s outbreak marks the third spike of AFM in 4 years. From August 2014 to September 2018, 386 cases have been confirmed. Yet, experts still do not understand crucial aspects of the disease, including its origins and who is most at risk.
“There is a lot we don’t know about AFM,” said Nancy Messonnier, MD, director of the National Center for Immunization and Respiratory Diseases. Here’s what puzzles health officials about AFM:
The cause is still unknown
Acute flaccid myelitis can be caused by viruses, such as polio or West Nile. But federal officials said that those viruses have not been linked to the U.S. outbreak over the past 4 years. They have not isolated the cause of these cases.
Despite symptoms reminiscent of polio, no AFM cases have tested positive for that virus, according to the CDC. Investigators have also ruled out a variety of germs. Environmental agents, viruses, and other pathogens are still being considered.
The 2014 outbreak of AFM coincided with a surge of another virus that caused severe respiratory problems, called EV-D68. However, the CDC could not establish a causal link between AFM and the virus. Since then, no large outbreaks of the virus have occurred, according to the CDC.
Carlos Pardo-Villamizar, MD, a neurologist and director of the Johns Hopkins Transverse Myelitis Center, said that the mystery lies in whether the damage seen in AFM is caused by an external agent or the body’s own defenses.
“At this moment, we don’t know if it’s a virus that is coming and producing direct damage of the gray matter in the spinal cord,” he said, “or if a virus is triggering immunological responses that produce a secondary damage in the spinal cord.”
It’s not clear who is at risk
Although the disease appears to target a certain age group, federal disease experts do not know who is likely to get acute flaccid myelitis.
Dr. Pardo-Villamizar said identifying vulnerable populations is “a work in progress.”
Mary Anne Jackson, MD, a pediatric infectious disease specialist and interim dean of the school of medicine at the University of Missouri–Kansas City, said many of the patients she saw were healthy children before falling ill with the disease. She suspects that a host of factors play a role in the likelihood of getting AFM, but more cases must be reviewed in order to find an answer.
The long-term effects are unknown
The CDC said it doesn’t know how long symptoms of the disease will last for patients. However, experts say that initial indications from a small number of cases suggest a grim outlook.
A study published last year found six of eight children in Colorado with acute flaccid myelitis still struggled with motor skills 1 year after their diagnosis. Nonetheless, the researchers found that the patients and families “demonstrated a high degree of resilience and recovery.”
“The majority of these patients are left with extensive problems,” said Dr. Pardo-Villamizar, who was not involved in the study.
Dr. Jackson, who also saw persistent muscle weakness in her patients, said she believes the CDC may be hesitant to specify the long-term effects of the disease because existing studies have included only small numbers of patients. More studies that include a larger proportion of confirmed cases are needed to better understand long-term outcomes, she said.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
A spike in the number of children with a rare neurological disease that causes polio-like symptoms has health officials across the country scrambling to understand the illness. Yet, more than 4 years after health officials first recorded the most recent uptick in cases, much about the national outbreak remains a mystery.
Acute flaccid myelitis (AFM) affects the gray matter in the spinal cord, causing sudden muscle weakness and a loss of reflexes. The illness can lead to serious complications – including paralysis or respiratory failure – and requires immediate medical attention.
The Centers for Disease Control and Prevention is investigating 127 cases of possible AFM, including 62 that have been confirmed in 22 states this year. At least 90% of the cases are among patients 18 years old and younger. The average age of a patient is 4 years old.
AFM remains extremely rare, even with the recent increase. The CDC estimates fewer than 1 in a million Americans will get the disease. Officials advised parents not to panic but remain vigilant for any sudden onset of symptoms. They also suggested that children stay up to date with their vaccines and practice good hand washing habits.
This year’s outbreak marks the third spike of AFM in 4 years. From August 2014 to September 2018, 386 cases have been confirmed. Yet, experts still do not understand crucial aspects of the disease, including its origins and who is most at risk.
“There is a lot we don’t know about AFM,” said Nancy Messonnier, MD, director of the National Center for Immunization and Respiratory Diseases. Here’s what puzzles health officials about AFM:
The cause is still unknown
Acute flaccid myelitis can be caused by viruses, such as polio or West Nile. But federal officials said that those viruses have not been linked to the U.S. outbreak over the past 4 years. They have not isolated the cause of these cases.
Despite symptoms reminiscent of polio, no AFM cases have tested positive for that virus, according to the CDC. Investigators have also ruled out a variety of germs. Environmental agents, viruses, and other pathogens are still being considered.
The 2014 outbreak of AFM coincided with a surge of another virus that caused severe respiratory problems, called EV-D68. However, the CDC could not establish a causal link between AFM and the virus. Since then, no large outbreaks of the virus have occurred, according to the CDC.
Carlos Pardo-Villamizar, MD, a neurologist and director of the Johns Hopkins Transverse Myelitis Center, said that the mystery lies in whether the damage seen in AFM is caused by an external agent or the body’s own defenses.
“At this moment, we don’t know if it’s a virus that is coming and producing direct damage of the gray matter in the spinal cord,” he said, “or if a virus is triggering immunological responses that produce a secondary damage in the spinal cord.”
It’s not clear who is at risk
Although the disease appears to target a certain age group, federal disease experts do not know who is likely to get acute flaccid myelitis.
Dr. Pardo-Villamizar said identifying vulnerable populations is “a work in progress.”
Mary Anne Jackson, MD, a pediatric infectious disease specialist and interim dean of the school of medicine at the University of Missouri–Kansas City, said many of the patients she saw were healthy children before falling ill with the disease. She suspects that a host of factors play a role in the likelihood of getting AFM, but more cases must be reviewed in order to find an answer.
The long-term effects are unknown
The CDC said it doesn’t know how long symptoms of the disease will last for patients. However, experts say that initial indications from a small number of cases suggest a grim outlook.
A study published last year found six of eight children in Colorado with acute flaccid myelitis still struggled with motor skills 1 year after their diagnosis. Nonetheless, the researchers found that the patients and families “demonstrated a high degree of resilience and recovery.”
“The majority of these patients are left with extensive problems,” said Dr. Pardo-Villamizar, who was not involved in the study.
Dr. Jackson, who also saw persistent muscle weakness in her patients, said she believes the CDC may be hesitant to specify the long-term effects of the disease because existing studies have included only small numbers of patients. More studies that include a larger proportion of confirmed cases are needed to better understand long-term outcomes, she said.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Third ACR Plenary presentations set to make an impact in rheumatology
A new study that validates the use of the Lupus Low Disease Activity State as a treatment outcome in systemic lupus erythematosus clinical trials is one of the highly-rated abstracts that will be presented in the third Plenary Session at the annual meeting of the American College of Rheumatology on Tuesday, Oct. 23.
The prospective, multicenter validation study, to be presented by Vera Golder, MBBS, of Monash University in Melbourne, builds on the results of previously reported studies using the Lupus Low Disease Activity State as a treatment target at EULAR this year and at last year’s International Congress on Systemic Lupus Erythematosus.
Among other presentations in the session will be the results of the PEXIVAS trial. Peter A. Merkel, MD, of the University of Pennsylvania, Philadelphia, will present findings from the randomized trial assessing oral glucocorticoid use and plasma exchange in patients with ANCA-associated vasculitis. The results have been highly anticipated as being among several research efforts to support reduction of corticosteroids in these patients.
In addition, attendees will hear results of a phase 3 study of apremilast for the treatment of oral ulcers in patients with Behçet’s syndrome. In the study, presented by Gulen Hatemi, MD, of Cerrahpasa Medical School, Istanbul, benefits of the drug were sustained for 28 weeks. Findings from a phase 2 study were reported in 2015.
A new study that validates the use of the Lupus Low Disease Activity State as a treatment outcome in systemic lupus erythematosus clinical trials is one of the highly-rated abstracts that will be presented in the third Plenary Session at the annual meeting of the American College of Rheumatology on Tuesday, Oct. 23.
The prospective, multicenter validation study, to be presented by Vera Golder, MBBS, of Monash University in Melbourne, builds on the results of previously reported studies using the Lupus Low Disease Activity State as a treatment target at EULAR this year and at last year’s International Congress on Systemic Lupus Erythematosus.
Among other presentations in the session will be the results of the PEXIVAS trial. Peter A. Merkel, MD, of the University of Pennsylvania, Philadelphia, will present findings from the randomized trial assessing oral glucocorticoid use and plasma exchange in patients with ANCA-associated vasculitis. The results have been highly anticipated as being among several research efforts to support reduction of corticosteroids in these patients.
In addition, attendees will hear results of a phase 3 study of apremilast for the treatment of oral ulcers in patients with Behçet’s syndrome. In the study, presented by Gulen Hatemi, MD, of Cerrahpasa Medical School, Istanbul, benefits of the drug were sustained for 28 weeks. Findings from a phase 2 study were reported in 2015.
A new study that validates the use of the Lupus Low Disease Activity State as a treatment outcome in systemic lupus erythematosus clinical trials is one of the highly-rated abstracts that will be presented in the third Plenary Session at the annual meeting of the American College of Rheumatology on Tuesday, Oct. 23.
The prospective, multicenter validation study, to be presented by Vera Golder, MBBS, of Monash University in Melbourne, builds on the results of previously reported studies using the Lupus Low Disease Activity State as a treatment target at EULAR this year and at last year’s International Congress on Systemic Lupus Erythematosus.
Among other presentations in the session will be the results of the PEXIVAS trial. Peter A. Merkel, MD, of the University of Pennsylvania, Philadelphia, will present findings from the randomized trial assessing oral glucocorticoid use and plasma exchange in patients with ANCA-associated vasculitis. The results have been highly anticipated as being among several research efforts to support reduction of corticosteroids in these patients.
In addition, attendees will hear results of a phase 3 study of apremilast for the treatment of oral ulcers in patients with Behçet’s syndrome. In the study, presented by Gulen Hatemi, MD, of Cerrahpasa Medical School, Istanbul, benefits of the drug were sustained for 28 weeks. Findings from a phase 2 study were reported in 2015.
REPORTING FROM THE ACR ANNUAL MEETING
Pediatric Skin Care: Survey of the Cutis Editorial Board
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on pediatric skin care. Here’s what we found.
Do you recommend sunscreen in babies younger than 6 months?
More than half (64%) of dermatologists we surveyed do not recommend using sunscreens in babies younger than 6 months; they should stay out of the sun. They recommended sun-protective clothing, hats, and sunglasses in this age group.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Babies younger than 6 months are still developing barrier functionality and have a higher surface area to body weight ratio compared to older children and adults. There is decreased UV light barrier protection and increased risk for systemic drug absorption. Therefore, it is best to avoid sunscreen in babies younger than 6 months. Instead, I recommend avoiding sunlight during peak hours, keeping babies in the shade, and dressing them in sun-protective clothing and hats.
Next page: Bathing and eczema
What advice do you give parents/guardians on bathing for babies with eczema?
Two-thirds of dermatologists (68%) indicated that moisturizers should be applied after bathing babies with eczema. More than half (64%) suggested using fragrance-free cleansers. Results varied on the frequency of bathing; 32% said bathe once daily for short periods with warm water, and 41% suggested to reduce bathing to a few nights a week.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Dry skin is a characteristic feature of atopic dermatitis. We now know that skin barrier dysfunction, such as filaggrin deficiency, contributes to the pathophysiology in some patients. In addition, a paucity of stratum corneum and intercellular lipids increases transepidermal water loss. Therefore, emollients containing humectants to augment stratum corneum hydration and occludents to reduce evaporation are extremely helpful in maintenance treatment of atopic dermatitis.
Next page: Nonsteroidal agents
Do you prescribe nonsteroidal agents for children younger than 2 years?
Almost two-thirds (64%) of dermatologists do prescribe nonsteroidal agents for children younger than 2 years.
If yes, which nonsteroidal agents do you prescribe for children?
Of those dermatologists who indicated that they do prescribe nonsteroidal agents for children younger than 2 years, almost half (45%) prescribe pimecrolimus, while 36% prescribe crisaborole or tacrolimus.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
We have limited options for US Food and Drug Administration (FDA)–approved therapies for atopic dermatitis in children younger than 2 years. The risks of adverse effects also are higher in younger children compared to older children and adults, particularly hypothalamic-pituitary-adrenal suppression with corticosteroids. Pimecrolimus was the most common nonsteroidal prescribed in this group. It should be noted that pimecrolimus is not FDA approved for use in children younger than 2 years; however, 2 phase 3 studies were conducted with 436 infants aged 3 months to 23 months and they demonstrated safety and efficacy.
Next page: Procedures in adolescents
Which procedures do you perform on adolescents?
Forty-one percent of dermatologists surveyed indicated that they do not perform procedures on adolescents. Of those that do, 32% use laser hair removal or chemical peels in adolescents, while only 23% each use light therapy for acne or onabotulinumtoxinA for hyperhidrosis. Pulsed dye laser use was reported in only 18% of dermatologists.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The results of this survey are a reflection of the relatively recent trends in dermatology training. Now that there is board certification in pediatric dermatology, many dermatologists now refer to pediatric dermatologists for children who need or want procedures.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Keep the regimen as simple as possible and make sure your skin care advice is culturally relevant.—Craig Burkhart, MD (Chapel Hill, North Carolina)
Please, avoid direct sun exposure as much as possible by wearing protective garments and finding shades in the outside.—Jisun Cha, MD (New Brunswick, New Jersey)
Good habits start early. Teach children how to care for their skin and they will carry that practice with them over the course of their lifetime, and hopefully pass it on.—James Q. Del Rosso, DO (Las Vegas, Nevada)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from September 13, 2018, to October 4, 2018. A total of 22 usable responses were received.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on pediatric skin care. Here’s what we found.
Do you recommend sunscreen in babies younger than 6 months?
More than half (64%) of dermatologists we surveyed do not recommend using sunscreens in babies younger than 6 months; they should stay out of the sun. They recommended sun-protective clothing, hats, and sunglasses in this age group.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Babies younger than 6 months are still developing barrier functionality and have a higher surface area to body weight ratio compared to older children and adults. There is decreased UV light barrier protection and increased risk for systemic drug absorption. Therefore, it is best to avoid sunscreen in babies younger than 6 months. Instead, I recommend avoiding sunlight during peak hours, keeping babies in the shade, and dressing them in sun-protective clothing and hats.
Next page: Bathing and eczema
What advice do you give parents/guardians on bathing for babies with eczema?
Two-thirds of dermatologists (68%) indicated that moisturizers should be applied after bathing babies with eczema. More than half (64%) suggested using fragrance-free cleansers. Results varied on the frequency of bathing; 32% said bathe once daily for short periods with warm water, and 41% suggested to reduce bathing to a few nights a week.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Dry skin is a characteristic feature of atopic dermatitis. We now know that skin barrier dysfunction, such as filaggrin deficiency, contributes to the pathophysiology in some patients. In addition, a paucity of stratum corneum and intercellular lipids increases transepidermal water loss. Therefore, emollients containing humectants to augment stratum corneum hydration and occludents to reduce evaporation are extremely helpful in maintenance treatment of atopic dermatitis.
Next page: Nonsteroidal agents
Do you prescribe nonsteroidal agents for children younger than 2 years?
Almost two-thirds (64%) of dermatologists do prescribe nonsteroidal agents for children younger than 2 years.
If yes, which nonsteroidal agents do you prescribe for children?
Of those dermatologists who indicated that they do prescribe nonsteroidal agents for children younger than 2 years, almost half (45%) prescribe pimecrolimus, while 36% prescribe crisaborole or tacrolimus.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
We have limited options for US Food and Drug Administration (FDA)–approved therapies for atopic dermatitis in children younger than 2 years. The risks of adverse effects also are higher in younger children compared to older children and adults, particularly hypothalamic-pituitary-adrenal suppression with corticosteroids. Pimecrolimus was the most common nonsteroidal prescribed in this group. It should be noted that pimecrolimus is not FDA approved for use in children younger than 2 years; however, 2 phase 3 studies were conducted with 436 infants aged 3 months to 23 months and they demonstrated safety and efficacy.
Next page: Procedures in adolescents
Which procedures do you perform on adolescents?
Forty-one percent of dermatologists surveyed indicated that they do not perform procedures on adolescents. Of those that do, 32% use laser hair removal or chemical peels in adolescents, while only 23% each use light therapy for acne or onabotulinumtoxinA for hyperhidrosis. Pulsed dye laser use was reported in only 18% of dermatologists.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The results of this survey are a reflection of the relatively recent trends in dermatology training. Now that there is board certification in pediatric dermatology, many dermatologists now refer to pediatric dermatologists for children who need or want procedures.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Keep the regimen as simple as possible and make sure your skin care advice is culturally relevant.—Craig Burkhart, MD (Chapel Hill, North Carolina)
Please, avoid direct sun exposure as much as possible by wearing protective garments and finding shades in the outside.—Jisun Cha, MD (New Brunswick, New Jersey)
Good habits start early. Teach children how to care for their skin and they will carry that practice with them over the course of their lifetime, and hopefully pass it on.—James Q. Del Rosso, DO (Las Vegas, Nevada)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from September 13, 2018, to October 4, 2018. A total of 22 usable responses were received.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on pediatric skin care. Here’s what we found.
Do you recommend sunscreen in babies younger than 6 months?
More than half (64%) of dermatologists we surveyed do not recommend using sunscreens in babies younger than 6 months; they should stay out of the sun. They recommended sun-protective clothing, hats, and sunglasses in this age group.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Babies younger than 6 months are still developing barrier functionality and have a higher surface area to body weight ratio compared to older children and adults. There is decreased UV light barrier protection and increased risk for systemic drug absorption. Therefore, it is best to avoid sunscreen in babies younger than 6 months. Instead, I recommend avoiding sunlight during peak hours, keeping babies in the shade, and dressing them in sun-protective clothing and hats.
Next page: Bathing and eczema
What advice do you give parents/guardians on bathing for babies with eczema?
Two-thirds of dermatologists (68%) indicated that moisturizers should be applied after bathing babies with eczema. More than half (64%) suggested using fragrance-free cleansers. Results varied on the frequency of bathing; 32% said bathe once daily for short periods with warm water, and 41% suggested to reduce bathing to a few nights a week.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Dry skin is a characteristic feature of atopic dermatitis. We now know that skin barrier dysfunction, such as filaggrin deficiency, contributes to the pathophysiology in some patients. In addition, a paucity of stratum corneum and intercellular lipids increases transepidermal water loss. Therefore, emollients containing humectants to augment stratum corneum hydration and occludents to reduce evaporation are extremely helpful in maintenance treatment of atopic dermatitis.
Next page: Nonsteroidal agents
Do you prescribe nonsteroidal agents for children younger than 2 years?
Almost two-thirds (64%) of dermatologists do prescribe nonsteroidal agents for children younger than 2 years.
If yes, which nonsteroidal agents do you prescribe for children?
Of those dermatologists who indicated that they do prescribe nonsteroidal agents for children younger than 2 years, almost half (45%) prescribe pimecrolimus, while 36% prescribe crisaborole or tacrolimus.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
We have limited options for US Food and Drug Administration (FDA)–approved therapies for atopic dermatitis in children younger than 2 years. The risks of adverse effects also are higher in younger children compared to older children and adults, particularly hypothalamic-pituitary-adrenal suppression with corticosteroids. Pimecrolimus was the most common nonsteroidal prescribed in this group. It should be noted that pimecrolimus is not FDA approved for use in children younger than 2 years; however, 2 phase 3 studies were conducted with 436 infants aged 3 months to 23 months and they demonstrated safety and efficacy.
Next page: Procedures in adolescents
Which procedures do you perform on adolescents?
Forty-one percent of dermatologists surveyed indicated that they do not perform procedures on adolescents. Of those that do, 32% use laser hair removal or chemical peels in adolescents, while only 23% each use light therapy for acne or onabotulinumtoxinA for hyperhidrosis. Pulsed dye laser use was reported in only 18% of dermatologists.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The results of this survey are a reflection of the relatively recent trends in dermatology training. Now that there is board certification in pediatric dermatology, many dermatologists now refer to pediatric dermatologists for children who need or want procedures.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Keep the regimen as simple as possible and make sure your skin care advice is culturally relevant.—Craig Burkhart, MD (Chapel Hill, North Carolina)
Please, avoid direct sun exposure as much as possible by wearing protective garments and finding shades in the outside.—Jisun Cha, MD (New Brunswick, New Jersey)
Good habits start early. Teach children how to care for their skin and they will carry that practice with them over the course of their lifetime, and hopefully pass it on.—James Q. Del Rosso, DO (Las Vegas, Nevada)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from September 13, 2018, to October 4, 2018. A total of 22 usable responses were received.
Book Review: DuPont’s approach to addiction is tough, yet compassionate
What do Queen Silvia of Sweden, Pope Francis, and the first director of the National Institute on Drug Abuse (NIDA) have in common? They all share a deep and sobering commitment to fighting the global disease of addiction.

Robert L. DuPont, MD, has written a beautiful and surprisingly spiritual guide into the American addiction epidemic. He is the author of “The Selfish Brain: Learning From Addiction” (Center City, Minn.: Hazelden, 2000) and with his newest publication, “Chemical Slavery: Understanding Addiction and Stopping the Drug Epidemic” (Institute for Behavior and Health, 2018), he writes a clear-eyed tome detailing the history of drug and alcohol use within the United States and the current state of America’s drug epidemic.
Dr. DuPont is well known within the American addiction community as NIDA’s first director and as the second drug czar, under two presidents, Richard M. Nixon and Gerald R. Ford. His breadth of experience, spanning 50-plus years, dates from his early career with the District of Columbia Department of Corrections, into his work in public policy on drugs and alcohol. This experience infuses his book with the hard science of addiction and the common-sense compassion required to shepherd people into recovery. One of us has worked with and been influenced by him since the 1970s. The other, also an addiction psychiatrist, finished this book both invigorated and compelled to say thank you to Dr. DuPont and his life’s work! He remains relevant, insightful, and always optimistic about the future of addiction treatment.
Harm reduction explored
The book begins with a cogent history of drug and alcohol use in America. Dr. DuPont details this as well as the public policies that have evolved to address them. He weaves into our national history reasoning behind why, as a “mass consumer” culture, we are more prone to addiction than ever before. The loss of cultural and societal pressures has a role to play in the rise along with genetics and environmental stress. The adolescent brain is prominently discussed throughout this first section as a highly vulnerable organ that can lead to lifelong addiction if primed early by addictive chemicals.
Dr. DuPont addresses the biology of addiction, delving into both the biological mechanisms within the brain, making those details accessible and understandable – to a practicing physician as well as a family member or patient struggling with addiction.
He also addresses harm reduction, a fairly new concept within the field. Harm reduction has taken on more prominence with localities across the country providing people with addictions with safe places and clean needles to continue their substance use without risks of serious or life-threatening diseases and crime. He challenges the idea that harm reduction is active recovery from substance addiction. Instead, he opines that harm reduction must be tethered to and must lead to real recovery work or it risks becoming an organizational enabling of the addict’s behavior.
Dr. DuPont pulls no punches with his language. He uses words such as “fatal,” “addict,” and “alcoholic.” He addresses the concerns by some in the field that those kinds of words are harsh, derogatory, and prejudicial by calling out addiction as a disease hallmarked primarily by loss of control and by dishonesty. To shun those words is to perpetuate the disease, delaying life-saving treatment.
Compassion is a theme throughout. He says we must stigmatize the addiction but not the addict. He advocates for real consequences to addictive behaviors as a key to getting addicted physicians and others with this disease into recovery. Treatment works, but we also have studied specific approaches that work and why.1 His writing conveys a genuine empathy for his patients and argues that treatment is delayed when serious and negative consequences for patients are removed.
Focus on prevention
A clear passion for prevention is evident within his chapters on youth addiction. For adolescents, Dr. DuPont presents a “One Choice approach,” which requires complete abstinence from alcohol, tobacco, and marijuana. He also presents science showing that patients younger than age 21 will have a greater risk of developing lifelong and debilitating addictions if they use these chemicals prior to this age. His emphasis for the One Choice approach carries ramifications throughout the primary care, pediatric, and family practice communities.
Dr. DuPont is nothing if not an optimist. Though he clearly defines addiction as an often-fatal disease, he remains positive about the future of addiction treatment, both with changes in public policy and with advancements in the medicine of addiction. He makes a compelling argument regarding Sweden’s approach to the drug problem, citing Queen Silvia’s lifelong commitment to prevent, control, and treat drug and alcohol addiction. Sweden’s model is, indeed, intriguing, and that country’s outcomes present a strong argument for the marriage of the criminal justice system with medical intervention – an approach that the United States has adopted only in a patchwork fashion.

The book describes controversies throughout, such as Dr. DuPont’s furtherance of our work on how best to treat dual disorders.2 He is not impressed with the self-medication hypothesis as well as the dive into the U.S. national medical marijuana experiment. We had looked at college students having new-onset memory or attention-deficit/hyperactivity problems, only to find that it was likely psychostimulant seeking to reverse marijuana effects.3 Physicians, families, and patients would be well served to review his arguments on the clear definition of “medicine” with regard to marijuana and the risks taken when we medicalize a known addictive chemical. He tackles the push for legalization as well, and the risks of increased societal acceptance and commercialization power that comes with this. He has led the field in thinking about the role of early drug exposure, brain training, and hijacking in the addictive process. This gateway hypothesis can occur whether the first teen drugs are cannabis or tobacco4 or alcohol.

Dr. DuPont finishes his work with a detailed biography, in which he addresses his family history of addiction, and his own overuse of alcohol in his late teens and early 20s as well as a confession of onetime use of marijuana during medical school. He always has led the field in trying to explain the disease of addiction, intervention, treatment, and recovery. But this self-reflection and brutal honesty is refreshing and in step with themes in his book, which promote the idea of recovery as an embrace of honesty, in mind, body, and spirit.
Dr. Jorandby trained in addiction psychiatry at Yale University, New Haven, Conn., and works as an addiction psychiatrist with Amen Clinics in Washington. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis.
References
1. J Subst Abuse Treat. 2009 Mar;36(2):159-71.
2. J Addict Dis. 2007;26 Suppl 1:13-23.
3. Am J Psychiatry. 2007 Jun;164(6):973.
4. J Addict Dis. 2003;22(3):51-62.
What do Queen Silvia of Sweden, Pope Francis, and the first director of the National Institute on Drug Abuse (NIDA) have in common? They all share a deep and sobering commitment to fighting the global disease of addiction.
Robert L. DuPont, MD, has written a beautiful and surprisingly spiritual guide into the American addiction epidemic. He is the author of “The Selfish Brain: Learning From Addiction” (Center City, Minn.: Hazelden, 2000) and with his newest publication, “Chemical Slavery: Understanding Addiction and Stopping the Drug Epidemic” (Institute for Behavior and Health, 2018), he writes a clear-eyed tome detailing the history of drug and alcohol use within the United States and the current state of America’s drug epidemic.
Dr. DuPont is well known within the American addiction community as NIDA’s first director and as the second drug czar, under two presidents, Richard M. Nixon and Gerald R. Ford. His breadth of experience, spanning 50-plus years, dates from his early career with the District of Columbia Department of Corrections, into his work in public policy on drugs and alcohol. This experience infuses his book with the hard science of addiction and the common-sense compassion required to shepherd people into recovery. One of us has worked with and been influenced by him since the 1970s. The other, also an addiction psychiatrist, finished this book both invigorated and compelled to say thank you to Dr. DuPont and his life’s work! He remains relevant, insightful, and always optimistic about the future of addiction treatment.
Harm reduction explored
The book begins with a cogent history of drug and alcohol use in America. Dr. DuPont details this as well as the public policies that have evolved to address them. He weaves into our national history reasoning behind why, as a “mass consumer” culture, we are more prone to addiction than ever before. The loss of cultural and societal pressures has a role to play in the rise along with genetics and environmental stress. The adolescent brain is prominently discussed throughout this first section as a highly vulnerable organ that can lead to lifelong addiction if primed early by addictive chemicals.
Dr. DuPont addresses the biology of addiction, delving into both the biological mechanisms within the brain, making those details accessible and understandable – to a practicing physician as well as a family member or patient struggling with addiction.
He also addresses harm reduction, a fairly new concept within the field. Harm reduction has taken on more prominence with localities across the country providing people with addictions with safe places and clean needles to continue their substance use without risks of serious or life-threatening diseases and crime. He challenges the idea that harm reduction is active recovery from substance addiction. Instead, he opines that harm reduction must be tethered to and must lead to real recovery work or it risks becoming an organizational enabling of the addict’s behavior.
Dr. DuPont pulls no punches with his language. He uses words such as “fatal,” “addict,” and “alcoholic.” He addresses the concerns by some in the field that those kinds of words are harsh, derogatory, and prejudicial by calling out addiction as a disease hallmarked primarily by loss of control and by dishonesty. To shun those words is to perpetuate the disease, delaying life-saving treatment.
Compassion is a theme throughout. He says we must stigmatize the addiction but not the addict. He advocates for real consequences to addictive behaviors as a key to getting addicted physicians and others with this disease into recovery. Treatment works, but we also have studied specific approaches that work and why.1 His writing conveys a genuine empathy for his patients and argues that treatment is delayed when serious and negative consequences for patients are removed.
Focus on prevention
A clear passion for prevention is evident within his chapters on youth addiction. For adolescents, Dr. DuPont presents a “One Choice approach,” which requires complete abstinence from alcohol, tobacco, and marijuana. He also presents science showing that patients younger than age 21 will have a greater risk of developing lifelong and debilitating addictions if they use these chemicals prior to this age. His emphasis for the One Choice approach carries ramifications throughout the primary care, pediatric, and family practice communities.
Dr. DuPont is nothing if not an optimist. Though he clearly defines addiction as an often-fatal disease, he remains positive about the future of addiction treatment, both with changes in public policy and with advancements in the medicine of addiction. He makes a compelling argument regarding Sweden’s approach to the drug problem, citing Queen Silvia’s lifelong commitment to prevent, control, and treat drug and alcohol addiction. Sweden’s model is, indeed, intriguing, and that country’s outcomes present a strong argument for the marriage of the criminal justice system with medical intervention – an approach that the United States has adopted only in a patchwork fashion.
The book describes controversies throughout, such as Dr. DuPont’s furtherance of our work on how best to treat dual disorders.2 He is not impressed with the self-medication hypothesis as well as the dive into the U.S. national medical marijuana experiment. We had looked at college students having new-onset memory or attention-deficit/hyperactivity problems, only to find that it was likely psychostimulant seeking to reverse marijuana effects.3 Physicians, families, and patients would be well served to review his arguments on the clear definition of “medicine” with regard to marijuana and the risks taken when we medicalize a known addictive chemical. He tackles the push for legalization as well, and the risks of increased societal acceptance and commercialization power that comes with this. He has led the field in thinking about the role of early drug exposure, brain training, and hijacking in the addictive process. This gateway hypothesis can occur whether the first teen drugs are cannabis or tobacco4 or alcohol.
Dr. DuPont finishes his work with a detailed biography, in which he addresses his family history of addiction, and his own overuse of alcohol in his late teens and early 20s as well as a confession of onetime use of marijuana during medical school. He always has led the field in trying to explain the disease of addiction, intervention, treatment, and recovery. But this self-reflection and brutal honesty is refreshing and in step with themes in his book, which promote the idea of recovery as an embrace of honesty, in mind, body, and spirit.
Dr. Jorandby trained in addiction psychiatry at Yale University, New Haven, Conn., and works as an addiction psychiatrist with Amen Clinics in Washington. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis.
References
1. J Subst Abuse Treat. 2009 Mar;36(2):159-71.
2. J Addict Dis. 2007;26 Suppl 1:13-23.
3. Am J Psychiatry. 2007 Jun;164(6):973.
4. J Addict Dis. 2003;22(3):51-62.
What do Queen Silvia of Sweden, Pope Francis, and the first director of the National Institute on Drug Abuse (NIDA) have in common? They all share a deep and sobering commitment to fighting the global disease of addiction.
Robert L. DuPont, MD, has written a beautiful and surprisingly spiritual guide into the American addiction epidemic. He is the author of “The Selfish Brain: Learning From Addiction” (Center City, Minn.: Hazelden, 2000) and with his newest publication, “Chemical Slavery: Understanding Addiction and Stopping the Drug Epidemic” (Institute for Behavior and Health, 2018), he writes a clear-eyed tome detailing the history of drug and alcohol use within the United States and the current state of America’s drug epidemic.
Dr. DuPont is well known within the American addiction community as NIDA’s first director and as the second drug czar, under two presidents, Richard M. Nixon and Gerald R. Ford. His breadth of experience, spanning 50-plus years, dates from his early career with the District of Columbia Department of Corrections, into his work in public policy on drugs and alcohol. This experience infuses his book with the hard science of addiction and the common-sense compassion required to shepherd people into recovery. One of us has worked with and been influenced by him since the 1970s. The other, also an addiction psychiatrist, finished this book both invigorated and compelled to say thank you to Dr. DuPont and his life’s work! He remains relevant, insightful, and always optimistic about the future of addiction treatment.
Harm reduction explored
The book begins with a cogent history of drug and alcohol use in America. Dr. DuPont details this as well as the public policies that have evolved to address them. He weaves into our national history reasoning behind why, as a “mass consumer” culture, we are more prone to addiction than ever before. The loss of cultural and societal pressures has a role to play in the rise along with genetics and environmental stress. The adolescent brain is prominently discussed throughout this first section as a highly vulnerable organ that can lead to lifelong addiction if primed early by addictive chemicals.
Dr. DuPont addresses the biology of addiction, delving into both the biological mechanisms within the brain, making those details accessible and understandable – to a practicing physician as well as a family member or patient struggling with addiction.
He also addresses harm reduction, a fairly new concept within the field. Harm reduction has taken on more prominence with localities across the country providing people with addictions with safe places and clean needles to continue their substance use without risks of serious or life-threatening diseases and crime. He challenges the idea that harm reduction is active recovery from substance addiction. Instead, he opines that harm reduction must be tethered to and must lead to real recovery work or it risks becoming an organizational enabling of the addict’s behavior.
Dr. DuPont pulls no punches with his language. He uses words such as “fatal,” “addict,” and “alcoholic.” He addresses the concerns by some in the field that those kinds of words are harsh, derogatory, and prejudicial by calling out addiction as a disease hallmarked primarily by loss of control and by dishonesty. To shun those words is to perpetuate the disease, delaying life-saving treatment.
Compassion is a theme throughout. He says we must stigmatize the addiction but not the addict. He advocates for real consequences to addictive behaviors as a key to getting addicted physicians and others with this disease into recovery. Treatment works, but we also have studied specific approaches that work and why.1 His writing conveys a genuine empathy for his patients and argues that treatment is delayed when serious and negative consequences for patients are removed.
Focus on prevention
A clear passion for prevention is evident within his chapters on youth addiction. For adolescents, Dr. DuPont presents a “One Choice approach,” which requires complete abstinence from alcohol, tobacco, and marijuana. He also presents science showing that patients younger than age 21 will have a greater risk of developing lifelong and debilitating addictions if they use these chemicals prior to this age. His emphasis for the One Choice approach carries ramifications throughout the primary care, pediatric, and family practice communities.
Dr. DuPont is nothing if not an optimist. Though he clearly defines addiction as an often-fatal disease, he remains positive about the future of addiction treatment, both with changes in public policy and with advancements in the medicine of addiction. He makes a compelling argument regarding Sweden’s approach to the drug problem, citing Queen Silvia’s lifelong commitment to prevent, control, and treat drug and alcohol addiction. Sweden’s model is, indeed, intriguing, and that country’s outcomes present a strong argument for the marriage of the criminal justice system with medical intervention – an approach that the United States has adopted only in a patchwork fashion.
The book describes controversies throughout, such as Dr. DuPont’s furtherance of our work on how best to treat dual disorders.2 He is not impressed with the self-medication hypothesis as well as the dive into the U.S. national medical marijuana experiment. We had looked at college students having new-onset memory or attention-deficit/hyperactivity problems, only to find that it was likely psychostimulant seeking to reverse marijuana effects.3 Physicians, families, and patients would be well served to review his arguments on the clear definition of “medicine” with regard to marijuana and the risks taken when we medicalize a known addictive chemical. He tackles the push for legalization as well, and the risks of increased societal acceptance and commercialization power that comes with this. He has led the field in thinking about the role of early drug exposure, brain training, and hijacking in the addictive process. This gateway hypothesis can occur whether the first teen drugs are cannabis or tobacco4 or alcohol.
Dr. DuPont finishes his work with a detailed biography, in which he addresses his family history of addiction, and his own overuse of alcohol in his late teens and early 20s as well as a confession of onetime use of marijuana during medical school. He always has led the field in trying to explain the disease of addiction, intervention, treatment, and recovery. But this self-reflection and brutal honesty is refreshing and in step with themes in his book, which promote the idea of recovery as an embrace of honesty, in mind, body, and spirit.
Dr. Jorandby trained in addiction psychiatry at Yale University, New Haven, Conn., and works as an addiction psychiatrist with Amen Clinics in Washington. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis.
References
1. J Subst Abuse Treat. 2009 Mar;36(2):159-71.
2. J Addict Dis. 2007;26 Suppl 1:13-23.
3. Am J Psychiatry. 2007 Jun;164(6):973.
4. J Addict Dis. 2003;22(3):51-62.
Gene-Replacement Therapy for SMA1 May Necessitate New Rating Measures to Capture Patients’ Motor Function Gains
CHOP-INTEND scores plateaued in a phase I trial of AVXS-101 even as patients reached new motor milestones.
CHICAGO—The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) was designed to track motor function in patients with spinal muscular atrophy type 1 (SMA1), but the test may not be sensitive to advances in motor function made by patients who receive an investigational gene-replacement therapy, researchers said at the 47th Annual Meeting of the Child Neurology Society.
AVXS-101 is an adeno-associated virus serotype 9 (AAV9)-based gene-replacement therapy that contains a copy of the SMN1 gene. AVXS-101 crosses the blood–brain barrier and is meant to treat the deletion or loss of function of the SMN1 gene in patients with SMA. The treatment may result in sustained SMN protein expression with a one-time dose.
Researchers conducted a phase I, open-label, dose-escalation study to study the treatment. Fifteen patients received AVXS-101 intravenously, and 12 participants received the proposed therapeutic dose.
“Patients treated with AVXS-101 achieved and maintained motor milestones unprecedented in the natural history of SMA1,” said Linda P. Lowes, PT, PhD, Principal Investigator for the Center for Gene Therapy at Nationwide Children’s Hospital in Columbus, Ohio, and Associate Professor of Pediatrics at Ohio State University, and colleagues. “CHOP-INTEND is a reliable tool for detecting early treatment impact, but appears insensitive to further motor function gains among AVXS-101–treated children as they age and develop.”

For example, the scale did not capture skills such as independent rolling and sitting that had not been seen in patients with SMA1, the researchers said.
CHOP-INTEND uses a 0–64-point scale where higher scores indicate better motor function. By six months, essentially no untreated patients with SMA1 achieve a score of 40 or greater points on the CHOP-INTEND, and scores may decrease between age 6 and 12 months. In the phase I trial of AVXS-101, CHOP-INTEND score increased from baseline by a mean of 9.8 points at one month postdose, 15.4 points at three months postdose, and 25.4 points at 24 months (n = 12 at all time points). Eleven of the 12 patients maintained a CHOP-INTEND score greater than 40 points for a mean of 19.5 months. Two children were able to crawl, pull to a stand, and stand and walk independently.
In the study, patients who achieved additional sitting milestones did not have accompanying increases on their CHOP-INTEND scores. For example, the 11 patients who sat for at least 5 seconds had a mean CHOP-INTEND plateau score of 56.9, while the 10 patients who sat for at least 10 seconds had a mean CHOP-INTEND plateau score of 57.0 and the nine patients who sat for at least 30 seconds had a mean CHOP-INTEND plateau score of 57.7.
The investigators suggested that the addition of scoring items such as supported sitting, head lifting, and walking could “mitigate the artificial ceiling effect” with the current scoring system. Other modifications to CHOP-INTEND, such as assessing one side of a patient instead of both sides, could reduce test administration time without affecting scores.
Furthermore, several CHOP-INTEND items may not perform well in assessing patients treated with AVXS-101 because they test primitive reflexes that fade as children live longer (eg, spinal incurvation) or children may become too heavy for the tests (eg, ventral suspension).
The study was sponsored by AveXis.
CHOP-INTEND scores plateaued in a phase I trial of AVXS-101 even as patients reached new motor milestones.
CHOP-INTEND scores plateaued in a phase I trial of AVXS-101 even as patients reached new motor milestones.
CHICAGO—The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) was designed to track motor function in patients with spinal muscular atrophy type 1 (SMA1), but the test may not be sensitive to advances in motor function made by patients who receive an investigational gene-replacement therapy, researchers said at the 47th Annual Meeting of the Child Neurology Society.
AVXS-101 is an adeno-associated virus serotype 9 (AAV9)-based gene-replacement therapy that contains a copy of the SMN1 gene. AVXS-101 crosses the blood–brain barrier and is meant to treat the deletion or loss of function of the SMN1 gene in patients with SMA. The treatment may result in sustained SMN protein expression with a one-time dose.
Researchers conducted a phase I, open-label, dose-escalation study to study the treatment. Fifteen patients received AVXS-101 intravenously, and 12 participants received the proposed therapeutic dose.
“Patients treated with AVXS-101 achieved and maintained motor milestones unprecedented in the natural history of SMA1,” said Linda P. Lowes, PT, PhD, Principal Investigator for the Center for Gene Therapy at Nationwide Children’s Hospital in Columbus, Ohio, and Associate Professor of Pediatrics at Ohio State University, and colleagues. “CHOP-INTEND is a reliable tool for detecting early treatment impact, but appears insensitive to further motor function gains among AVXS-101–treated children as they age and develop.”
For example, the scale did not capture skills such as independent rolling and sitting that had not been seen in patients with SMA1, the researchers said.
CHOP-INTEND uses a 0–64-point scale where higher scores indicate better motor function. By six months, essentially no untreated patients with SMA1 achieve a score of 40 or greater points on the CHOP-INTEND, and scores may decrease between age 6 and 12 months. In the phase I trial of AVXS-101, CHOP-INTEND score increased from baseline by a mean of 9.8 points at one month postdose, 15.4 points at three months postdose, and 25.4 points at 24 months (n = 12 at all time points). Eleven of the 12 patients maintained a CHOP-INTEND score greater than 40 points for a mean of 19.5 months. Two children were able to crawl, pull to a stand, and stand and walk independently.
In the study, patients who achieved additional sitting milestones did not have accompanying increases on their CHOP-INTEND scores. For example, the 11 patients who sat for at least 5 seconds had a mean CHOP-INTEND plateau score of 56.9, while the 10 patients who sat for at least 10 seconds had a mean CHOP-INTEND plateau score of 57.0 and the nine patients who sat for at least 30 seconds had a mean CHOP-INTEND plateau score of 57.7.
The investigators suggested that the addition of scoring items such as supported sitting, head lifting, and walking could “mitigate the artificial ceiling effect” with the current scoring system. Other modifications to CHOP-INTEND, such as assessing one side of a patient instead of both sides, could reduce test administration time without affecting scores.
Furthermore, several CHOP-INTEND items may not perform well in assessing patients treated with AVXS-101 because they test primitive reflexes that fade as children live longer (eg, spinal incurvation) or children may become too heavy for the tests (eg, ventral suspension).
The study was sponsored by AveXis.
CHICAGO—The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) was designed to track motor function in patients with spinal muscular atrophy type 1 (SMA1), but the test may not be sensitive to advances in motor function made by patients who receive an investigational gene-replacement therapy, researchers said at the 47th Annual Meeting of the Child Neurology Society.
AVXS-101 is an adeno-associated virus serotype 9 (AAV9)-based gene-replacement therapy that contains a copy of the SMN1 gene. AVXS-101 crosses the blood–brain barrier and is meant to treat the deletion or loss of function of the SMN1 gene in patients with SMA. The treatment may result in sustained SMN protein expression with a one-time dose.
Researchers conducted a phase I, open-label, dose-escalation study to study the treatment. Fifteen patients received AVXS-101 intravenously, and 12 participants received the proposed therapeutic dose.
“Patients treated with AVXS-101 achieved and maintained motor milestones unprecedented in the natural history of SMA1,” said Linda P. Lowes, PT, PhD, Principal Investigator for the Center for Gene Therapy at Nationwide Children’s Hospital in Columbus, Ohio, and Associate Professor of Pediatrics at Ohio State University, and colleagues. “CHOP-INTEND is a reliable tool for detecting early treatment impact, but appears insensitive to further motor function gains among AVXS-101–treated children as they age and develop.”
For example, the scale did not capture skills such as independent rolling and sitting that had not been seen in patients with SMA1, the researchers said.
CHOP-INTEND uses a 0–64-point scale where higher scores indicate better motor function. By six months, essentially no untreated patients with SMA1 achieve a score of 40 or greater points on the CHOP-INTEND, and scores may decrease between age 6 and 12 months. In the phase I trial of AVXS-101, CHOP-INTEND score increased from baseline by a mean of 9.8 points at one month postdose, 15.4 points at three months postdose, and 25.4 points at 24 months (n = 12 at all time points). Eleven of the 12 patients maintained a CHOP-INTEND score greater than 40 points for a mean of 19.5 months. Two children were able to crawl, pull to a stand, and stand and walk independently.
In the study, patients who achieved additional sitting milestones did not have accompanying increases on their CHOP-INTEND scores. For example, the 11 patients who sat for at least 5 seconds had a mean CHOP-INTEND plateau score of 56.9, while the 10 patients who sat for at least 10 seconds had a mean CHOP-INTEND plateau score of 57.0 and the nine patients who sat for at least 30 seconds had a mean CHOP-INTEND plateau score of 57.7.
The investigators suggested that the addition of scoring items such as supported sitting, head lifting, and walking could “mitigate the artificial ceiling effect” with the current scoring system. Other modifications to CHOP-INTEND, such as assessing one side of a patient instead of both sides, could reduce test administration time without affecting scores.
Furthermore, several CHOP-INTEND items may not perform well in assessing patients treated with AVXS-101 because they test primitive reflexes that fade as children live longer (eg, spinal incurvation) or children may become too heavy for the tests (eg, ventral suspension).
The study was sponsored by AveXis.
Can ultrasound screening improve survival in ovarian cancer?
Annual ultrasound screening of asymptomatic women at risk of epithelial ovarian cancer can lead to lower staging of cancer at detection and improved survival, compared with no screening, according to a prospective clinical trial that followed more than 46,000 women over 2 decades.
“The findings of this study support the concept that a major predictor of ovarian cancer survival is stage at detection,” said John R. van Nagell Jr., MD, of the University of Kentucky–Markey Cancer Center, Lexington, and his coauthors. “The 10-year survival of women whose ovarian cancer was detected at an early stage (I or II) was 35% higher than that of women diagnosed with stage III cancer.” Study results were published in the November issue of Obstetrics & Gynecology.
The study evaluated 46,101 women enrolled in the University of Kentucky Ovarian Cancer Screening Trial over 30.5 years. Trial participants, all of whom had annual ultrasound screening, were age 50 and older, or 25 and older with a family history of ovarian cancer. Overall, 23% and 44% of the women had a family history of either ovarian or breast cancer, respectively. Women in the study had an average of seven scans each. The unscreened comparator group was women with ovarian cancer referred to the UK Markey Cancer Center.
The study detected 71 cases of invasive epithelial ovarian cancers and 17 epithelial ovarian tumors of low malignant potential. None of the women with these tumors had a recurrence. Among the invasive cancers, the majority were either stage I (42%) or II (21%), and none were stage IV. The median age of these patients was 66 years. Of the low-malignancy tumors, 27% were stage I and 73% stage II, with none stage III or IV. A total of 699 women (1.5%) with persistent ovarian tumors had surgery.
Screened women also had improved survival compared to unscreened women: 86% vs. 45% at 5 years, 68% vs. 31% at 10 years (P less than .001).
However, the study also showed a high overall incidence rate for ovarian cancer, including false-positive and false-negative cases, compared with National Cancer Institute reports in the Kentucky state cancer profile: 271 per 100,000 vs. 10.4/100,000.
The study also looked at the economics of annual screening. “Ovarian screening reduced the 10-year mortality by 37% and produced 416 life years gained,” Dr. van Nagell and his coauthors said. Based on an estimated cost of $56 for each transvaginal ultrasound scan, that translates into a cost of $40,731 for each life year gained.
One concern of screening ultrasound is the high false-positive rate. “Although the sensitivity of transvaginal ultrasonography in detecting an ovarian abnormality is high, it has been unreliable in differentiating benign from malignant ovarian tumors,” they said. While they noted the accuracy of assessing malignancy has improved, the risk of complications in women who have surgery for benign tumors is an ongoing concern. “Additional research is necessary to identify high-risk populations who will benefit most from screening.”
Dr. van Nagell and his coauthors reported having no financial relationships. The study was supported by research grants from the Kentucky Department of Health and Human Services and the Telford Foundation.
SOURCE: Van Nagell JR Jr et al. Obstet Gynecol. 2018 Nov;132[5]:1091-100. doi: 10.1097/AOG.0000000000002921.
Use a measure of caution when interpreting the results of the study by van Nagell et al., Sharon E. Robertson, MD, MPH, and Jeffery E. Peipert, MD, PhD, said in an invited commentary. First, they noted the “surprisingly high” rate of ovarian cancer (271 per 100,000) – although it may improve the predictive value of ultrasound screening, when one applies the test sensitivity and specificity the trial reported to the general population, “the positive predictive value falls to an unacceptable 0.7%.”
They also questioned the rationale for comparing the study population to an unscreened cohort of women with ovarian cancer referred to the University of Kentucky. “However, are we really comparing apples to apples?” they asked, noting “important key baseline differences” between the two groups, including that the screened cohort “could have contained an unbalanced proportion of genetically related ovarian cancers.” They also noted the risk profile of the unscreened cohort is unknown.
Addressing the differences in survival rates between screened and unscreened patients, Dr. Robertson and Dr. Peipert noted the study population had a higher rate of type I tumors than that seen in National Cancer Institute data for the general population (27% vs. 11%), along with the absence of genetic analysis. “For these reasons, we cannot confidently agree with the authors’ conclusion that the ultrasound screening reduced ovarian cancer mortality,” they stated.
They commended the Kentucky group for a “landmark study,” but added, “the evidence that screening for ovarian cancer improves survival remains elusive.” They called for more evidence before widespread screening programs are implemented.
Dr. Robertson and Dr. Peipert of Indiana University, Indianapolis, commented on the article by van Nagell et al. in Obstetrics and Gynecology (2018 Nov;132[5]:1089-90. doi: 10.1097/AOG.0000000000002962). Dr. Peipert disclosed relationships with Cooper Surgical/Teva, Merck, and Bayer. Dr. Robertson had no relevant financial relationships.
Use a measure of caution when interpreting the results of the study by van Nagell et al., Sharon E. Robertson, MD, MPH, and Jeffery E. Peipert, MD, PhD, said in an invited commentary. First, they noted the “surprisingly high” rate of ovarian cancer (271 per 100,000) – although it may improve the predictive value of ultrasound screening, when one applies the test sensitivity and specificity the trial reported to the general population, “the positive predictive value falls to an unacceptable 0.7%.”
They also questioned the rationale for comparing the study population to an unscreened cohort of women with ovarian cancer referred to the University of Kentucky. “However, are we really comparing apples to apples?” they asked, noting “important key baseline differences” between the two groups, including that the screened cohort “could have contained an unbalanced proportion of genetically related ovarian cancers.” They also noted the risk profile of the unscreened cohort is unknown.
Addressing the differences in survival rates between screened and unscreened patients, Dr. Robertson and Dr. Peipert noted the study population had a higher rate of type I tumors than that seen in National Cancer Institute data for the general population (27% vs. 11%), along with the absence of genetic analysis. “For these reasons, we cannot confidently agree with the authors’ conclusion that the ultrasound screening reduced ovarian cancer mortality,” they stated.
They commended the Kentucky group for a “landmark study,” but added, “the evidence that screening for ovarian cancer improves survival remains elusive.” They called for more evidence before widespread screening programs are implemented.
Dr. Robertson and Dr. Peipert of Indiana University, Indianapolis, commented on the article by van Nagell et al. in Obstetrics and Gynecology (2018 Nov;132[5]:1089-90. doi: 10.1097/AOG.0000000000002962). Dr. Peipert disclosed relationships with Cooper Surgical/Teva, Merck, and Bayer. Dr. Robertson had no relevant financial relationships.
Use a measure of caution when interpreting the results of the study by van Nagell et al., Sharon E. Robertson, MD, MPH, and Jeffery E. Peipert, MD, PhD, said in an invited commentary. First, they noted the “surprisingly high” rate of ovarian cancer (271 per 100,000) – although it may improve the predictive value of ultrasound screening, when one applies the test sensitivity and specificity the trial reported to the general population, “the positive predictive value falls to an unacceptable 0.7%.”
They also questioned the rationale for comparing the study population to an unscreened cohort of women with ovarian cancer referred to the University of Kentucky. “However, are we really comparing apples to apples?” they asked, noting “important key baseline differences” between the two groups, including that the screened cohort “could have contained an unbalanced proportion of genetically related ovarian cancers.” They also noted the risk profile of the unscreened cohort is unknown.
Addressing the differences in survival rates between screened and unscreened patients, Dr. Robertson and Dr. Peipert noted the study population had a higher rate of type I tumors than that seen in National Cancer Institute data for the general population (27% vs. 11%), along with the absence of genetic analysis. “For these reasons, we cannot confidently agree with the authors’ conclusion that the ultrasound screening reduced ovarian cancer mortality,” they stated.
They commended the Kentucky group for a “landmark study,” but added, “the evidence that screening for ovarian cancer improves survival remains elusive.” They called for more evidence before widespread screening programs are implemented.
Dr. Robertson and Dr. Peipert of Indiana University, Indianapolis, commented on the article by van Nagell et al. in Obstetrics and Gynecology (2018 Nov;132[5]:1089-90. doi: 10.1097/AOG.0000000000002962). Dr. Peipert disclosed relationships with Cooper Surgical/Teva, Merck, and Bayer. Dr. Robertson had no relevant financial relationships.
Annual ultrasound screening of asymptomatic women at risk of epithelial ovarian cancer can lead to lower staging of cancer at detection and improved survival, compared with no screening, according to a prospective clinical trial that followed more than 46,000 women over 2 decades.
“The findings of this study support the concept that a major predictor of ovarian cancer survival is stage at detection,” said John R. van Nagell Jr., MD, of the University of Kentucky–Markey Cancer Center, Lexington, and his coauthors. “The 10-year survival of women whose ovarian cancer was detected at an early stage (I or II) was 35% higher than that of women diagnosed with stage III cancer.” Study results were published in the November issue of Obstetrics & Gynecology.
The study evaluated 46,101 women enrolled in the University of Kentucky Ovarian Cancer Screening Trial over 30.5 years. Trial participants, all of whom had annual ultrasound screening, were age 50 and older, or 25 and older with a family history of ovarian cancer. Overall, 23% and 44% of the women had a family history of either ovarian or breast cancer, respectively. Women in the study had an average of seven scans each. The unscreened comparator group was women with ovarian cancer referred to the UK Markey Cancer Center.
The study detected 71 cases of invasive epithelial ovarian cancers and 17 epithelial ovarian tumors of low malignant potential. None of the women with these tumors had a recurrence. Among the invasive cancers, the majority were either stage I (42%) or II (21%), and none were stage IV. The median age of these patients was 66 years. Of the low-malignancy tumors, 27% were stage I and 73% stage II, with none stage III or IV. A total of 699 women (1.5%) with persistent ovarian tumors had surgery.
Screened women also had improved survival compared to unscreened women: 86% vs. 45% at 5 years, 68% vs. 31% at 10 years (P less than .001).
However, the study also showed a high overall incidence rate for ovarian cancer, including false-positive and false-negative cases, compared with National Cancer Institute reports in the Kentucky state cancer profile: 271 per 100,000 vs. 10.4/100,000.
The study also looked at the economics of annual screening. “Ovarian screening reduced the 10-year mortality by 37% and produced 416 life years gained,” Dr. van Nagell and his coauthors said. Based on an estimated cost of $56 for each transvaginal ultrasound scan, that translates into a cost of $40,731 for each life year gained.
One concern of screening ultrasound is the high false-positive rate. “Although the sensitivity of transvaginal ultrasonography in detecting an ovarian abnormality is high, it has been unreliable in differentiating benign from malignant ovarian tumors,” they said. While they noted the accuracy of assessing malignancy has improved, the risk of complications in women who have surgery for benign tumors is an ongoing concern. “Additional research is necessary to identify high-risk populations who will benefit most from screening.”
Dr. van Nagell and his coauthors reported having no financial relationships. The study was supported by research grants from the Kentucky Department of Health and Human Services and the Telford Foundation.
SOURCE: Van Nagell JR Jr et al. Obstet Gynecol. 2018 Nov;132[5]:1091-100. doi: 10.1097/AOG.0000000000002921.
Annual ultrasound screening of asymptomatic women at risk of epithelial ovarian cancer can lead to lower staging of cancer at detection and improved survival, compared with no screening, according to a prospective clinical trial that followed more than 46,000 women over 2 decades.
“The findings of this study support the concept that a major predictor of ovarian cancer survival is stage at detection,” said John R. van Nagell Jr., MD, of the University of Kentucky–Markey Cancer Center, Lexington, and his coauthors. “The 10-year survival of women whose ovarian cancer was detected at an early stage (I or II) was 35% higher than that of women diagnosed with stage III cancer.” Study results were published in the November issue of Obstetrics & Gynecology.
The study evaluated 46,101 women enrolled in the University of Kentucky Ovarian Cancer Screening Trial over 30.5 years. Trial participants, all of whom had annual ultrasound screening, were age 50 and older, or 25 and older with a family history of ovarian cancer. Overall, 23% and 44% of the women had a family history of either ovarian or breast cancer, respectively. Women in the study had an average of seven scans each. The unscreened comparator group was women with ovarian cancer referred to the UK Markey Cancer Center.
The study detected 71 cases of invasive epithelial ovarian cancers and 17 epithelial ovarian tumors of low malignant potential. None of the women with these tumors had a recurrence. Among the invasive cancers, the majority were either stage I (42%) or II (21%), and none were stage IV. The median age of these patients was 66 years. Of the low-malignancy tumors, 27% were stage I and 73% stage II, with none stage III or IV. A total of 699 women (1.5%) with persistent ovarian tumors had surgery.
Screened women also had improved survival compared to unscreened women: 86% vs. 45% at 5 years, 68% vs. 31% at 10 years (P less than .001).
However, the study also showed a high overall incidence rate for ovarian cancer, including false-positive and false-negative cases, compared with National Cancer Institute reports in the Kentucky state cancer profile: 271 per 100,000 vs. 10.4/100,000.
The study also looked at the economics of annual screening. “Ovarian screening reduced the 10-year mortality by 37% and produced 416 life years gained,” Dr. van Nagell and his coauthors said. Based on an estimated cost of $56 for each transvaginal ultrasound scan, that translates into a cost of $40,731 for each life year gained.
One concern of screening ultrasound is the high false-positive rate. “Although the sensitivity of transvaginal ultrasonography in detecting an ovarian abnormality is high, it has been unreliable in differentiating benign from malignant ovarian tumors,” they said. While they noted the accuracy of assessing malignancy has improved, the risk of complications in women who have surgery for benign tumors is an ongoing concern. “Additional research is necessary to identify high-risk populations who will benefit most from screening.”
Dr. van Nagell and his coauthors reported having no financial relationships. The study was supported by research grants from the Kentucky Department of Health and Human Services and the Telford Foundation.
SOURCE: Van Nagell JR Jr et al. Obstet Gynecol. 2018 Nov;132[5]:1091-100. doi: 10.1097/AOG.0000000000002921.
FROM OBSTETRICS & GYNECOLOGY
Key clinical point: Annual ultrasound may improve outcomes for women at risk for epithelial ovarian cancer.
Major finding: Five-year survival was 86% for screened women vs. 45% for unscreened women (P less than .001).
Study details: Analysis of 46,101 at-risk women enrolled in the University of Kentucky Ovarian Cancer Screening Trial, a prospective cohort trial, from January 1987 to June 2017.
Disclosures: Dr. van Nagell and coauthors reported having no financial relationships. The study was supported by research grants from the Kentucky Department of Health and Human Services and the Telford Foundation.
Source: Van Nagell JR Jr et al. Obstet Gynecol. 2018 Nov;132[5]:1091-100. doi: 10.1097/AOG.0000000000002921.