User login
Research and Reviews for the Practicing Oncologist
Symptom burdens related to chemotherapy-induced anemia in stage IV cancer
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).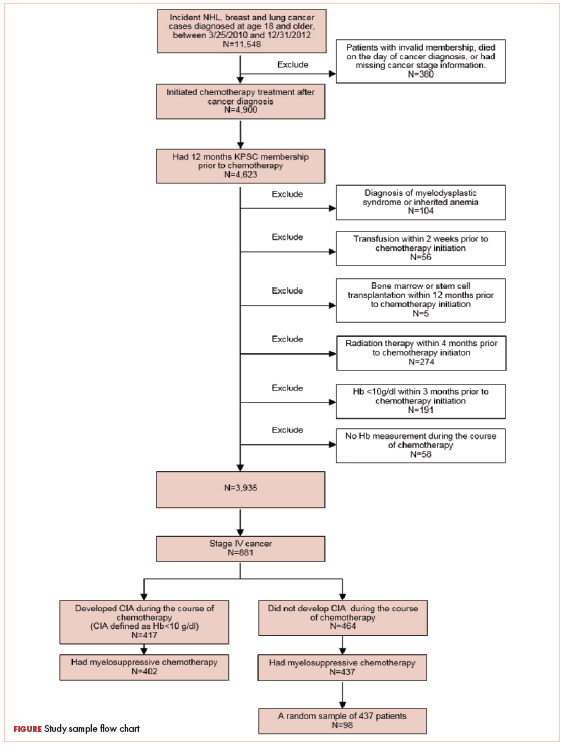
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).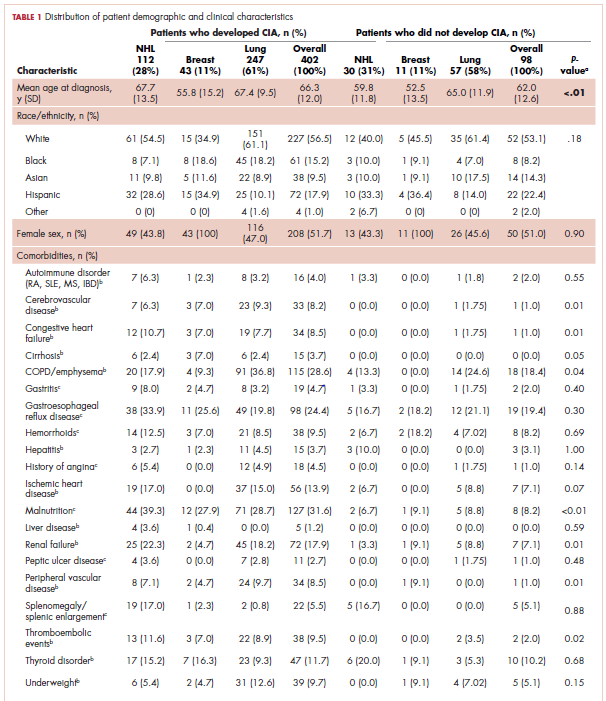
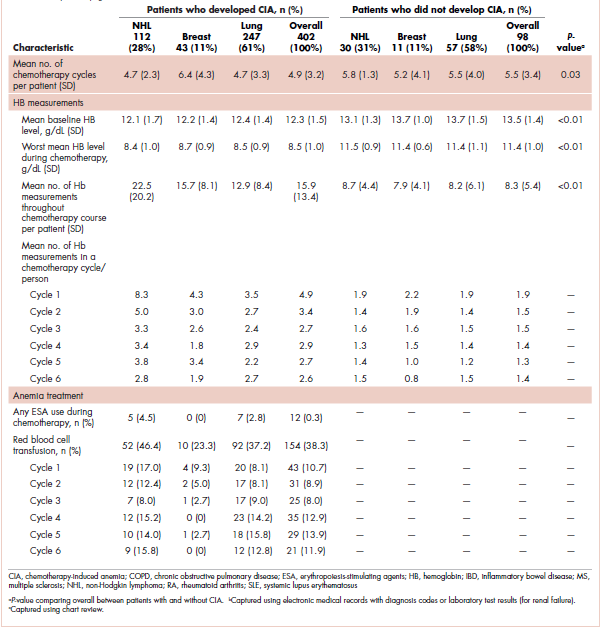
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2. 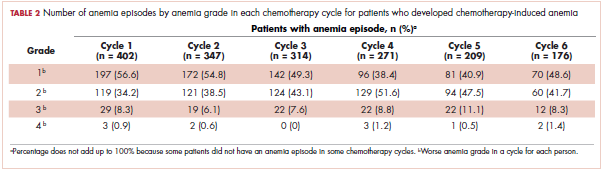
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA. 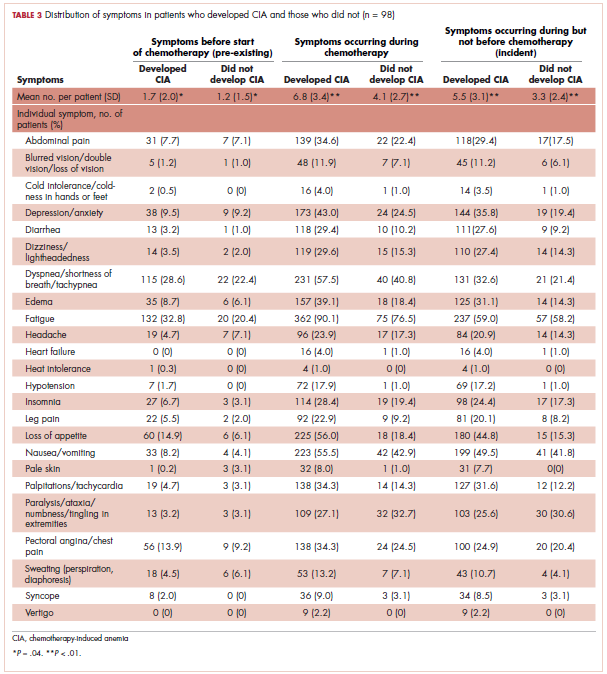
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).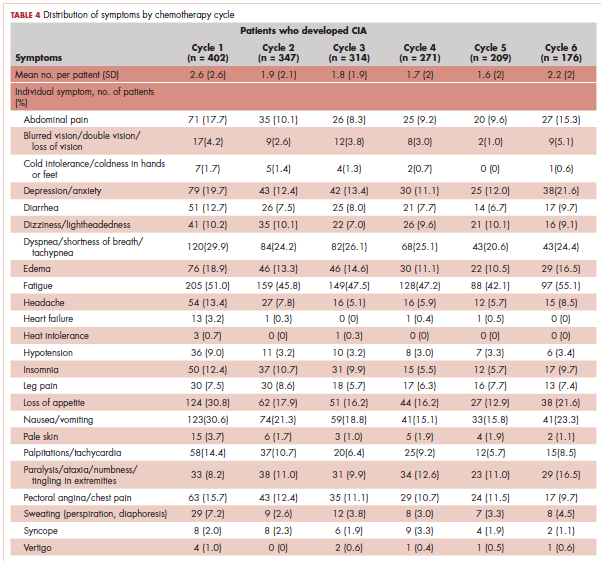
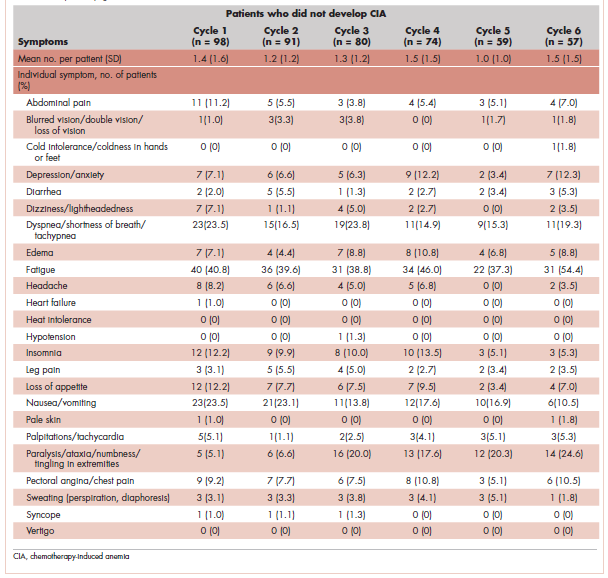
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).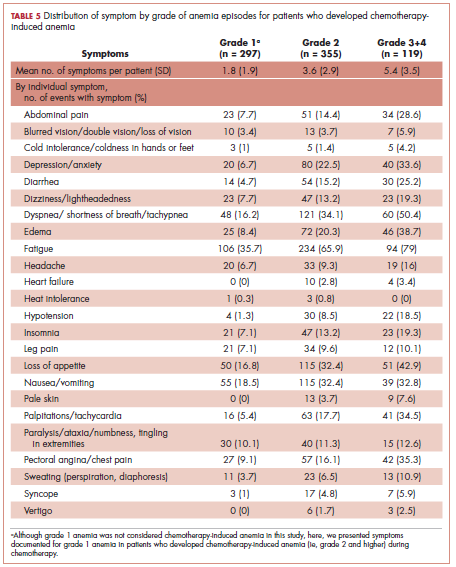
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2.
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA.
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2.
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA.
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018

CLL resistance mechanism to venetoclax identified
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The mutation was identified in samples from seven patients after venetoclax therapy, but not in any of the pretherapy samples.
Study details: Genetic analysis of CLL mutations in 15 patients enrolled in clinical trials of venetoclax.
Disclosures: The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
Source: Blombery P et al. ASH 2018, Abstract LBA-7.

In IDH-mutated AML, first-line IDH inhibitors plus chemo may boost remission
SAN DIEGO – In patients with newly diagnosed acute myeloid leukemia (AML) bearing IDH1 or IDH2 mutations, combinations of either ivosidenib (Tibsovo, for IDH1) or enasidenib (Idhifa, for IDH2) with standard induction and consolidation regimens are safe and well tolerated and are associated with encouraging remission rates, results of a phase 1 trial indicate.

In the open-label, phase 1 trial, ivosidenib plus chemotherapy was associated with elimination of minimal residual disease (MRD) by flow cytometry in 88% of treated patients and with IDH1-mutation clearance in 41% of patients.
Enasidenib plus chemotherapy was associated with elimination of MRD in 45% of patients and with IDH2-mutation clearance in 25% of patients, said Eytan M. Stein, MD, from Memorial Sloan Kettering Cancer Center in New York.
“The overall survival rates are robust, with greater than 75% 1-year survival in both ivosidenib- and enasidenib-treated patients,” he said at the annual meeting of the American Society of Hematology.
Both ivosidenib and enasidenib are approved in the United States for treatment of patients with relapsed or refractory AML bearing either IDH1 or IDH2 mutations, respectively. In this trial, the investigators explored the therapeutic potential of the IDH inhibitors in patients with previously untreated disease.
Dr. Stein and his colleagues investigated combining each of the agents with standard induction therapy with either daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day for 3 days, plus cytarabine 200 mg/m2 per day for 7 days. Patients with IDH1 mutations received ivosidenib 500 mg once daily, and those with IDH2 mutations received enasidenib 100 mg once daily.
After induction, patients with a complete remission (CR), CR with incomplete recovery of hematologic counts (CRi), or CR with incomplete recovery of platelets (CRp) could receive up to four cycles of consolidation therapy while continuing the IDH inhibitor. Patients who completed consolidation or were ineligible for consolidation could continue on maintenance therapy with their assigned drug until the end of the study.
The drugs were discontinued in patients who went on to hematopoietic stem cell transplant.
The most frequent co-occurring baseline mutations were DNMT3A, NPM1, and NRAS for patients with IDH1 mutations, and DNMT3A, SRSF2, and ASXL1 for patients with IDH2 mutations.
A total of 60 patients were assigned to ivosidenib and chemotherapy and 93 were assigned to enasidenib/chemotherapy. The median patient age was about 63 years in each arm.
All patients in each arm received a least one induction dose and about 48% in each arm received at least some consolidation dosing. In all, 18% of patients in the ivosidenib arm and 19% in the enasidenib arm went on to maintenance.
Treatment discontinuations occurred in 55% of patients in the ivosidenib group and 84% in the enasidenib group. The primary reason for discontinuation included HSCT, adverse events, progressive disease, and death (one and four patients in the respective arms).
Adverse events of interest, regardless of attribution, included the IDH differentiation syndrome in two patients on ivosidenib and one on enasidenib, leukocytosis, QT interval prolongation, and increased blood bilirubin.
The 30-day and 60-day mortality rates were 5% and 8% in the ivosidenib arm and 5% and 9% in the enasidenib arm, respectively.
Best overall response rates (CR+CRi+CRp) among all patients were 80% in the ivosidenib arm and 72% in the enasidenib arm. In each trial arm, the response rates were higher among patients with de novo AML, compared with secondary AML.
Of 12 ivosidenib-treated patients who had IDH1-mutation clearance, 10 had clearance at the end of induction therapy, and 2 achieved clearance during or after consolidation. Of 15 ivosidenib-treated patients who became MRD negative, 12 had achieved it by the end of induction, and 3 became MRD negative during consolidation.
In the enasidenib arm, 15 patients had IDH2 mutation clearance (11 after induction, 4 during consolidation) and 9 became MRD negative (7 after induction and 2 during or after consolidation).
The probability of surviving to 1 year after the start of induction among ivosidenib-treated patients was 79%; the median overall survival had not been reached and was not estimable at the time of data cutoff. The probability of surviving to 1 year among patients in the enasidenib arm was 75%. In this group, too, median overall survival had not been reached.
The clinical benefit of adding either IDH inhibitor to induction, consolidation, and maintenance therapy for patients with newly diagnosed AML with IDH mutations will be further evaluated in a randomized, phase 3 trial, Dr. Stein said.
The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
SOURCE: Stein EM et al. ASH 2018, Abstract 560.
SAN DIEGO – In patients with newly diagnosed acute myeloid leukemia (AML) bearing IDH1 or IDH2 mutations, combinations of either ivosidenib (Tibsovo, for IDH1) or enasidenib (Idhifa, for IDH2) with standard induction and consolidation regimens are safe and well tolerated and are associated with encouraging remission rates, results of a phase 1 trial indicate.
In the open-label, phase 1 trial, ivosidenib plus chemotherapy was associated with elimination of minimal residual disease (MRD) by flow cytometry in 88% of treated patients and with IDH1-mutation clearance in 41% of patients.
Enasidenib plus chemotherapy was associated with elimination of MRD in 45% of patients and with IDH2-mutation clearance in 25% of patients, said Eytan M. Stein, MD, from Memorial Sloan Kettering Cancer Center in New York.
“The overall survival rates are robust, with greater than 75% 1-year survival in both ivosidenib- and enasidenib-treated patients,” he said at the annual meeting of the American Society of Hematology.
Both ivosidenib and enasidenib are approved in the United States for treatment of patients with relapsed or refractory AML bearing either IDH1 or IDH2 mutations, respectively. In this trial, the investigators explored the therapeutic potential of the IDH inhibitors in patients with previously untreated disease.
Dr. Stein and his colleagues investigated combining each of the agents with standard induction therapy with either daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day for 3 days, plus cytarabine 200 mg/m2 per day for 7 days. Patients with IDH1 mutations received ivosidenib 500 mg once daily, and those with IDH2 mutations received enasidenib 100 mg once daily.
After induction, patients with a complete remission (CR), CR with incomplete recovery of hematologic counts (CRi), or CR with incomplete recovery of platelets (CRp) could receive up to four cycles of consolidation therapy while continuing the IDH inhibitor. Patients who completed consolidation or were ineligible for consolidation could continue on maintenance therapy with their assigned drug until the end of the study.
The drugs were discontinued in patients who went on to hematopoietic stem cell transplant.
The most frequent co-occurring baseline mutations were DNMT3A, NPM1, and NRAS for patients with IDH1 mutations, and DNMT3A, SRSF2, and ASXL1 for patients with IDH2 mutations.
A total of 60 patients were assigned to ivosidenib and chemotherapy and 93 were assigned to enasidenib/chemotherapy. The median patient age was about 63 years in each arm.
All patients in each arm received a least one induction dose and about 48% in each arm received at least some consolidation dosing. In all, 18% of patients in the ivosidenib arm and 19% in the enasidenib arm went on to maintenance.
Treatment discontinuations occurred in 55% of patients in the ivosidenib group and 84% in the enasidenib group. The primary reason for discontinuation included HSCT, adverse events, progressive disease, and death (one and four patients in the respective arms).
Adverse events of interest, regardless of attribution, included the IDH differentiation syndrome in two patients on ivosidenib and one on enasidenib, leukocytosis, QT interval prolongation, and increased blood bilirubin.
The 30-day and 60-day mortality rates were 5% and 8% in the ivosidenib arm and 5% and 9% in the enasidenib arm, respectively.
Best overall response rates (CR+CRi+CRp) among all patients were 80% in the ivosidenib arm and 72% in the enasidenib arm. In each trial arm, the response rates were higher among patients with de novo AML, compared with secondary AML.
Of 12 ivosidenib-treated patients who had IDH1-mutation clearance, 10 had clearance at the end of induction therapy, and 2 achieved clearance during or after consolidation. Of 15 ivosidenib-treated patients who became MRD negative, 12 had achieved it by the end of induction, and 3 became MRD negative during consolidation.
In the enasidenib arm, 15 patients had IDH2 mutation clearance (11 after induction, 4 during consolidation) and 9 became MRD negative (7 after induction and 2 during or after consolidation).
The probability of surviving to 1 year after the start of induction among ivosidenib-treated patients was 79%; the median overall survival had not been reached and was not estimable at the time of data cutoff. The probability of surviving to 1 year among patients in the enasidenib arm was 75%. In this group, too, median overall survival had not been reached.
The clinical benefit of adding either IDH inhibitor to induction, consolidation, and maintenance therapy for patients with newly diagnosed AML with IDH mutations will be further evaluated in a randomized, phase 3 trial, Dr. Stein said.
The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
SOURCE: Stein EM et al. ASH 2018, Abstract 560.
SAN DIEGO – In patients with newly diagnosed acute myeloid leukemia (AML) bearing IDH1 or IDH2 mutations, combinations of either ivosidenib (Tibsovo, for IDH1) or enasidenib (Idhifa, for IDH2) with standard induction and consolidation regimens are safe and well tolerated and are associated with encouraging remission rates, results of a phase 1 trial indicate.
In the open-label, phase 1 trial, ivosidenib plus chemotherapy was associated with elimination of minimal residual disease (MRD) by flow cytometry in 88% of treated patients and with IDH1-mutation clearance in 41% of patients.
Enasidenib plus chemotherapy was associated with elimination of MRD in 45% of patients and with IDH2-mutation clearance in 25% of patients, said Eytan M. Stein, MD, from Memorial Sloan Kettering Cancer Center in New York.
“The overall survival rates are robust, with greater than 75% 1-year survival in both ivosidenib- and enasidenib-treated patients,” he said at the annual meeting of the American Society of Hematology.
Both ivosidenib and enasidenib are approved in the United States for treatment of patients with relapsed or refractory AML bearing either IDH1 or IDH2 mutations, respectively. In this trial, the investigators explored the therapeutic potential of the IDH inhibitors in patients with previously untreated disease.
Dr. Stein and his colleagues investigated combining each of the agents with standard induction therapy with either daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day for 3 days, plus cytarabine 200 mg/m2 per day for 7 days. Patients with IDH1 mutations received ivosidenib 500 mg once daily, and those with IDH2 mutations received enasidenib 100 mg once daily.
After induction, patients with a complete remission (CR), CR with incomplete recovery of hematologic counts (CRi), or CR with incomplete recovery of platelets (CRp) could receive up to four cycles of consolidation therapy while continuing the IDH inhibitor. Patients who completed consolidation or were ineligible for consolidation could continue on maintenance therapy with their assigned drug until the end of the study.
The drugs were discontinued in patients who went on to hematopoietic stem cell transplant.
The most frequent co-occurring baseline mutations were DNMT3A, NPM1, and NRAS for patients with IDH1 mutations, and DNMT3A, SRSF2, and ASXL1 for patients with IDH2 mutations.
A total of 60 patients were assigned to ivosidenib and chemotherapy and 93 were assigned to enasidenib/chemotherapy. The median patient age was about 63 years in each arm.
All patients in each arm received a least one induction dose and about 48% in each arm received at least some consolidation dosing. In all, 18% of patients in the ivosidenib arm and 19% in the enasidenib arm went on to maintenance.
Treatment discontinuations occurred in 55% of patients in the ivosidenib group and 84% in the enasidenib group. The primary reason for discontinuation included HSCT, adverse events, progressive disease, and death (one and four patients in the respective arms).
Adverse events of interest, regardless of attribution, included the IDH differentiation syndrome in two patients on ivosidenib and one on enasidenib, leukocytosis, QT interval prolongation, and increased blood bilirubin.
The 30-day and 60-day mortality rates were 5% and 8% in the ivosidenib arm and 5% and 9% in the enasidenib arm, respectively.
Best overall response rates (CR+CRi+CRp) among all patients were 80% in the ivosidenib arm and 72% in the enasidenib arm. In each trial arm, the response rates were higher among patients with de novo AML, compared with secondary AML.
Of 12 ivosidenib-treated patients who had IDH1-mutation clearance, 10 had clearance at the end of induction therapy, and 2 achieved clearance during or after consolidation. Of 15 ivosidenib-treated patients who became MRD negative, 12 had achieved it by the end of induction, and 3 became MRD negative during consolidation.
In the enasidenib arm, 15 patients had IDH2 mutation clearance (11 after induction, 4 during consolidation) and 9 became MRD negative (7 after induction and 2 during or after consolidation).
The probability of surviving to 1 year after the start of induction among ivosidenib-treated patients was 79%; the median overall survival had not been reached and was not estimable at the time of data cutoff. The probability of surviving to 1 year among patients in the enasidenib arm was 75%. In this group, too, median overall survival had not been reached.
The clinical benefit of adding either IDH inhibitor to induction, consolidation, and maintenance therapy for patients with newly diagnosed AML with IDH mutations will be further evaluated in a randomized, phase 3 trial, Dr. Stein said.
The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
SOURCE: Stein EM et al. ASH 2018, Abstract 560.
REPORTING FROM ASH 2018
Key clinical point: The IDH1 inhibitor ivosidenib and IDH2 inhibitor enasidenib combined with induction and consolidation therapy showed promising efficacy against newly diagnosed acute myeloid leukemia.
Major finding: The 1-year survival rates were greater than 75% for patients with previously untreated acute myeloid leukemia who received either of the IDH-mutated inhibitors.
Study details: An open-label, prospective trial in 153 patients with newly diagnosed acute myeloid leukemia.
Disclosures: The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
Source: Stein EM et al. ASH 2018, Abstract 560.
Patterns of malignancies in patients with HIV-AIDS: a single institution observational study
India has the third largest HIV epidemic in the world because of its large population size, with 0.3% of the adult population infected with HIV. That translates to 2.1 million infected people, posing a significant challenge in the management of these individuals.1 In all, 43% of the infected are currently on highly active antiretroviral therapy (HAART).1 There has been a significant decrease in the number of HIV-AIDS–related deaths in recent years because of the remarkable increase in the use of antiretroviral therapy.2 However, the prolonged life expectancy in these patients has resulted in an increase in the risk of various new diseases such as cancers. With the complex interactions between altered immunity and infections, the risk of cancers is markedly increased in patients with HIV-AIDS.3 The spectrum of malignancies in this group of patients differs from that in the general population. In addition, the pattern and the magnitude of malignancies differ in different parts of the world.4 In this study, we have analyzed the pattern of malignancies in patients with HIV-AIDS in a regional cancer center in India. The aim of the study was to analyze the pattern of malignancies in patients with HIV-AIDS based on their age and sex and to document the CD4 counts at the time the malignancy was diagnosed.
Methods
We retrieved data from our institution’s medical records department on all patients who had HIV-AIDS and had been diagnosed with a malignancy. Data of all patients presenting with a malignancy and coexisting HIV-AIDS from January 2013 through December 2016 were analyzed initially. Only patients for whom there was a documented CD4 count were included in the final retrospective analysis. We analyzed the correlation between the patients’ CD4 counts and malignancies subclassified as AIDS-defining malignancies (ADMs; aggressive B-cell non-Hodgkin lymphoma [NHL] and cervical cancer) or non–AIDS-defining malignancies (NADMs; all other malignancies other than aggressive NHL and carcinoma cervix were defined as NADM). We also analyzed the correlation between the CD4 count and NHL and other malignancies. A statistical analysis was performed using SPSS Statistics for Windows, version 23 (IBM Corp, Armonk, NY). The independent sample Mann-Whitney U or Kruskal-Wallis tests were used for comparing the CD4 counts between the various subgroups of malignancies. The study was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
A total of 370 patients who were diagnosed with malignancy and have coexisting HIV-AIDS were identified. In all, 85 patients were excluded because there were no CD4 counts available for them, and the remaining 285 patients were included in the final analysis. Of that total, 136 patients (48%) were men, and 149 (52%) were women.
The median age of the population was 44.8 years (5-80 years) at the time of diagnosis with malignancy. The mean CD4 count of the entire population was 235.4 cells/mm3 (50-734 cells/mm3). There were 104 patients with CD4 counts of ≤200 cells/mm3, and 181 patients had CD4 counts of >200 cells/mm3 (Table 1). All patients received the HAART regimen, efavirenz-lamuvidine-tenofovir (600 mg/300 mg/300 mg Telura).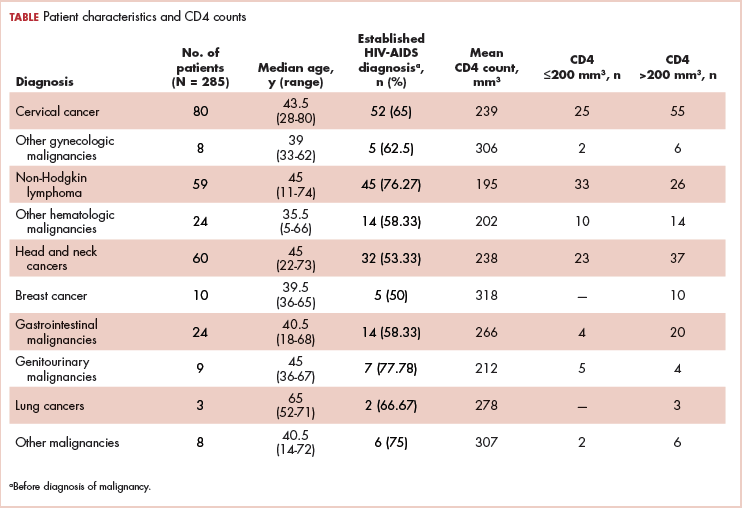
The most common malignancies in this population were gynecologic malignancies, followed by hematologic malignancies. Cervical cancer was the most common malignancy among women as well as in the overall study population. Among men, the most common malignancy was NHL. The second and third most common malignancies in men were carcinoma oral cavity and carcinoma oropharynx, respectively, whereas in women, they were NHL and breast cancer. The distribution of various hematologic, head and neck, and gastrointestinal malignancies in this group of patients is shown in Figures 1, 2, and 3.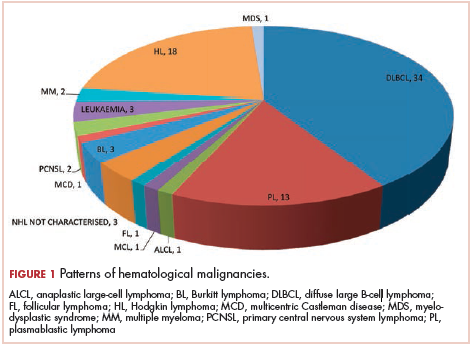
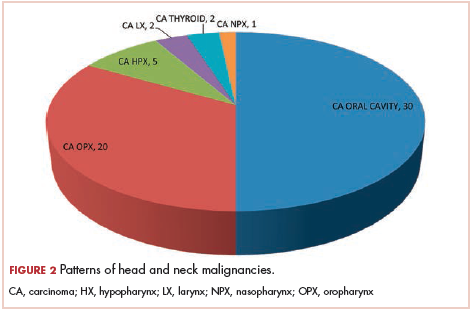
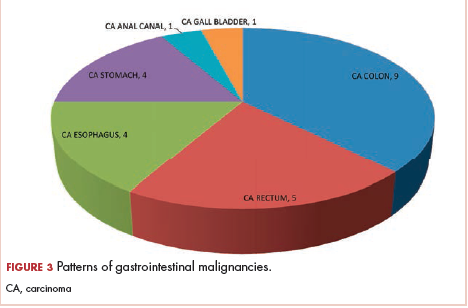
The ADMs in the study were NHL, including 2 patients diagnosed with primary central nervous system (CNS) lymphomas, and cervical cancer. No case of Kaposi sarcoma, also considered an ADM, was identified in this study. The common NADMs include head and neck malignancies (Figure 2), gastrointestinal malignancies (Figure 3), gynecological and genitourinary malignancies, and breast cancer. The mean CD4 count in the ADM subgroup was 221 cells/mm3, and in the NADM subgroup, it was 250 cells/mm3. There was a significant difference in the distribution of CD4 counts between the ADM and NADM subgroups (P = .03; Mann-Whitney U test). A statistical difference was also noted when the CD4 counts of the patients with NHL were compared with other malignancies (P = .0001; Mann-Whitney U test) There was no statistically significant difference noted when CD4 counts of patients with cervical cancer were compared with NADMs (P = .914).
Discussion
In 2015, a report from the Indian government estimated the prevalence of HIV in the country as 0.26% (0.22%-0.32%).5 The report also noted a decreasing trend in the number of new cases of HIV diagnosed and a decrease in the number of AIDS-related deaths.5 The decrease in deaths from AIDS is primarily attributed to the widespread use of HAART. With the introduction of HAART therapy, the survival of patients diagnosed with HIV-AIDS has increased markedly.6 However, newer challenges have emerged with improved survival, such as an increasing number of patients being diagnosed with malignancies. In the current HAART era, the pattern of malignancies in people living with HIV-AIDS has changed compared with the pre-HAART era.7 The literature suggests that worldwide, malignancies are encountered in about 30% patients with HIV-AIDS, but that percentage differs sharply from that encountered in India, where it is less than 5%.8 This may partly be explained by opportunistic infections such as tuberculosis in Indian patients, which remains the leading cause of death in the HIV-AIDS population. In our study, we retrospectively analyzed the pattern of malignancies in patients with HIV-AIDS.
Although few studies have quoted NHL as the predominant malignancy in their patients with HIV-AIDS, the predominant malignancy was cervical cancer in our patient population, as seen in few other studies.8-10 Head and neck malignancies also continue to be common malignancies in men with HIV-AIDS.10 Thus, an increase in malignancies induced by the human papillomavirus (HPV) can be seen in this group of patients. Only a few pediatric malignancies were noted in our study, and all of those patients had a vertical transmission of HIV.
Kaposi sarcoma is quite rare in the Indian population, and no case of Kaposi sarcoma was diagnosed in our study population. A similar finding was seen in several earlier publications from India. In the largest published series from India by Dhir and colleagues, evaluating 251 patients with HIV-AIDS and malignancy, no case of Kaposi sarcoma was reported.10 The authors mentioned that this finding might be because of the low seroprevalence of Kaposi sarcoma-associated herpesvirus in the Asian population.10 Three different studies from southern India have also not reported the incidence of Kaposi Sarcoma in their series of HIV-AIDS patients with malignancies,11-13 and similar findings were also reported in a study from northern India.9 The incidence of other immunodeficiency-related malignancies was identical to those reported in other studies in the literature.10,14
As seen in other studies, the CD4 counts in patients with ADM were significantly lower compared with those of patients with NADM, and that difference was not seen when CD4 counts of patients with cervical cancer were compared with patients in the NADM subgroup. The risk of NHL increases proportionally to the degree of immune suppression. The increased susceptibility to various infections in patients with low CD4 counts may also contribute to the occurrence of NHL in patients with low CD4 counts. The occurrence of various other rare cancers in patients with HIV-AIDS may be because of confounding rather than a direct HIV or immunosuppression effect.
An increasing incidence of NADMs has been noted in the Western literature.7,14 ADMs remain the most common malignancies in the HIV-AIDS population, accounting for about 48% of all malignancies.8 This is in concordance with previous publications from India.8,10 With the widespread availability of generic HAART, the incidence of ADMs may decrease even more in the future. In developing countries where the screening procedures for malignancies in both the general population and patients with HIV-AIDS have not yet been implemented at a national level, premalignant lesions of the cervix are not detected.10 Cervical cancer is the most common malignancy in our study population, which underscores the importance of cervical cancer screening in patients with HIV-AIDS.
In the developed countries, following the introduction of HAART in HIV-AIDS management, the incidence of Kaposi sarcoma decreased by 60% to 70%, and the incidence of NHL decreased by 30% to 50%, whereas the rates of cervical cancer remained either stable or declined.15,16 Despite the declining trend, the incidence of these malignancies continues to be high among patients with HIV-AIDS compared with the general population.17 A study from the United States showed increasing trends in various NADMs (such as anal, lung, and liver cancers and Hodgkin lymphoma) from 2006 to 2010.17 In 2003, the number of patients with NADM were higher than the number of patients with ADM in the United States.14 In a population-based study from Brazil, ADMs were the most common malignancies diagnosed in patients with HIV-AIDS. A declining trend was noted in the incidence of ADMs in the population and an increasing trend in the incidence of NADMs. This increase in NADM incidence was contributed by anal and lung cancers.18 Studies from developing countries such as Uganda and Botswana have also shown a decrease in the incidence of Kaposi sarcoma after the introduction of HAART.19-21
Kaposi sarcoma, cervical cancer, NHL (including Burkitt lymphoma, immunoblastic lymphoma, and primary CNS lymphoma [PCNSL]) comprise ADMs. All 3 ADMs have an underlying viral infection as the causative agent.22 Kaposi sarcoma is caused by the Kaposi sarcoma herpes virus, for which seroprevalence varies worldwide.23 As already noted in this article, the incidence of Kaposi sarcoma among the HIV-AIDS population has decreased worldwide since the introduction of HAART. The preinvasive uterine cervix lesions and carcinoma cervix are caused by HPV. NHL in patients with HIV-AIDS is a predominantly aggressive B-cell neoplasm. Epstein-Barr virus is implicated for most of the ADM NHLs.24 PCNSL occurs in patients with low CD4 counts and poses a diagnostic challenge. The treatment outcomes for patients with PCNSL before the HAART era were dismal. With the widespread use of HAART, the treatment outcomes of patients with HIV-AIDS and NHL improved, and, currently, these patients are managed the same way as other patients with NHL.22
The increasing incidence of the NADM is partly attributed to the increasing incidence of these malignancies in the general population. An elevated risk of certain NADMs is also attributable to viral infections. The common NADMs in the United States are lung, anal, oropharyngeal, and hepatocellular cancers and Hodgkin lymphoma.14 The common NADMs in our study population were oral, oropharyngeal, colon, and breast cancers and Hodgkin lymphoma. One-third of head and neck cancers, including most oropharyngeal cancers, and cervical and anal cancers in patients with HIV-AIDS are related to HPV.25 Patients with HIV-AIDS are at increased risk for chronic HPV infection from immunosuppression. Chronic HPV infections and prolonged immunosuppression cause premalignant high-grade squamous intraepithelial lesions and invasive cancers.22 The initiation of and adherence to HAART leads to immune recovery and reduces high-risk HPV-associated morbidity.26 Findings from previous studies have demonstrated the benefits of screening for cervical cancer in patients with HIV-AIDS.27 The HPV vaccine is immunogenic in patients with HIV-AIDS and might help prevent HPV-associated malignancies.28
Conclusions
With the wide use of HAART by patients with HIV-AIDS, we can expect an increase in the survival of that population. The incidence of malignancies may also increase significantly in these patients, and further longitudinal studies are needed, as malignancies may emerge as the most common cause of death in patients with HIV-AIDS. In addition, the extensive use of HAART therapy and implementation of screening programs for cervical cancer in patients with HIV-AIDS could result in a decrease in the incidence of ADMs.
1. UNAIDS. Prevention gap report. http://www.unaids.org/sites/default/files/media_asset/2016-prevention-gap-report_en.pdf. Released 2016. Accessed December 27, 2017.
3. Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol. 2012;24(5):506-516.
4. Biggar RJ, Chaturvedi AK, Bhatia K, Mbulaiteye SM. Cancer risk in persons with HIV-AIDS in India: a review and future directions for research. Infect Agent Cancer. 2009;4:4.
5. National AIDS Control Organisation & National Institute of Medical Statistics, ICMR, Ministry of Health & Family Welfare, Government of India. India HIV estimations 2015, technical report. http://www.naco.gov.in/sites/default/files/India%20HIV%20Estimations%202015.pdf. Published 2015. Accessed December 27, 2017.
6. Bonnet F, Lewden C, May T, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101(2):317-324.
7. Crum-Cianflone N, Hullsiek KH, Marconi V, et al. Trends in the incidence of cancers among HIV-infected persons and the impact of antiretroviral therapy: a 20-year cohort study. AIDS. 2009;23(1):41-50.
8. Sharma S, Soneja M, Ranjan S. Malignancies in human immunodeficiency virus infected patients in India: initial experience in the HAART era. Indian J Med Res. 2015;142(5):563-567.
9. Sachdeva RK, Sharma A, Singh S, Varma S. Spectrum of AIDS defining & non-AIDS defining malignancies in north India. In
10. Dhir AA, Sawant S, Dikshit RP, et al. Spectrum of HIV-AIDS related cancers in India. Cancer Causes Control. 2007;19(2):147-153.
11. Venkatesh KK, Saghayam S, Devaleenal B, et al. Spectrum of malignancies among HIV-infected patients in South India. Indian J Cancer. 2012;49(1):176-180.
12. Shruti P, Narayanan G, Puthuveettil J, Jayasree K, Vijayalakshmi K. Spectrum of HIV/AIDS-associated cancers in south India. J Clin Oncol. 2014;32(suppl):e12534.
13. Paul TR, Uppin MS, Uppin SG, et al. Spectrum of malignancies in human immunodeficiency virus–positive patients at a Tertiary Care Centre in South India. Indian J Cancer. 2014;51(4):459-463.
14. Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103(9):753-762.
15. Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148(10):728-736.
16. Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
17. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the United States. AIDS. 2014;28(6):881-890.
18. Tanaka LF, Latorre MDRD, Gutierrez EB, Heumann C, Herbinger KH, Froeschl G. Trends in the incidence of AIDS-defining and non-AIDS-defining cancers in people living with AIDS: a population-based study from São Paulo, Brazil. Int J STD AIDS. 2017;28(12):1190-1198.
19. Mutyaba I, Phipps W, Krantz EM, et al. A population-level evaluation of the effect of antiretroviral therapy on cancer incidence in Kyadondo County, Uganda, 1999–2008. J Acquir Immune Defic Syndr. 2015;69(4):481-486.
20. Dryden-Peterson S, Medhin H, Kebabonye-Pusoentsi M, et al. Cancer incidence following expansion of HIV treatment in Botswana. PLoS ONE. 2015;10(8):e0135602.
21. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12(1):6-11.
22. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041.
23. Gao SJ, Kingsley L, Li M, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2(8):925-928.
24. Epstein-Barr virus and AIDS-associated lymphomas. Lancet. 1991;338(8773):979-981.
25. Picard A, Badoual C, Hourseau M, et al. Human papilloma virus prevalence in HIV patients with head and neck squamous cell carcinoma. AIDS. 2016;30(8):1257-1266.
26. Minkoff H, Zhong Y, Burk RD, et al. Influence of adherent and effective antiretroviral therapy use on human papillomavirus infection and squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Infect Dis. 2010;201(5):681-690.
27. Ghebre RG, Grover S, Xu MJ, Chuang LT, Simonds H. Cervical cancer control in HIV-infected women: past, present and future. Gynecol Oncol Rep. 2017;21:101-108.
28. Kojic EM, Rana AI, Cu-Uvin S. Human papillomavirus vaccination in HIV-infected women: need for increased coverage. Expert Rev Vaccines. 2016;15(1):105-117.
India has the third largest HIV epidemic in the world because of its large population size, with 0.3% of the adult population infected with HIV. That translates to 2.1 million infected people, posing a significant challenge in the management of these individuals.1 In all, 43% of the infected are currently on highly active antiretroviral therapy (HAART).1 There has been a significant decrease in the number of HIV-AIDS–related deaths in recent years because of the remarkable increase in the use of antiretroviral therapy.2 However, the prolonged life expectancy in these patients has resulted in an increase in the risk of various new diseases such as cancers. With the complex interactions between altered immunity and infections, the risk of cancers is markedly increased in patients with HIV-AIDS.3 The spectrum of malignancies in this group of patients differs from that in the general population. In addition, the pattern and the magnitude of malignancies differ in different parts of the world.4 In this study, we have analyzed the pattern of malignancies in patients with HIV-AIDS in a regional cancer center in India. The aim of the study was to analyze the pattern of malignancies in patients with HIV-AIDS based on their age and sex and to document the CD4 counts at the time the malignancy was diagnosed.
Methods
We retrieved data from our institution’s medical records department on all patients who had HIV-AIDS and had been diagnosed with a malignancy. Data of all patients presenting with a malignancy and coexisting HIV-AIDS from January 2013 through December 2016 were analyzed initially. Only patients for whom there was a documented CD4 count were included in the final retrospective analysis. We analyzed the correlation between the patients’ CD4 counts and malignancies subclassified as AIDS-defining malignancies (ADMs; aggressive B-cell non-Hodgkin lymphoma [NHL] and cervical cancer) or non–AIDS-defining malignancies (NADMs; all other malignancies other than aggressive NHL and carcinoma cervix were defined as NADM). We also analyzed the correlation between the CD4 count and NHL and other malignancies. A statistical analysis was performed using SPSS Statistics for Windows, version 23 (IBM Corp, Armonk, NY). The independent sample Mann-Whitney U or Kruskal-Wallis tests were used for comparing the CD4 counts between the various subgroups of malignancies. The study was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
A total of 370 patients who were diagnosed with malignancy and have coexisting HIV-AIDS were identified. In all, 85 patients were excluded because there were no CD4 counts available for them, and the remaining 285 patients were included in the final analysis. Of that total, 136 patients (48%) were men, and 149 (52%) were women.
The median age of the population was 44.8 years (5-80 years) at the time of diagnosis with malignancy. The mean CD4 count of the entire population was 235.4 cells/mm3 (50-734 cells/mm3). There were 104 patients with CD4 counts of ≤200 cells/mm3, and 181 patients had CD4 counts of >200 cells/mm3 (Table 1). All patients received the HAART regimen, efavirenz-lamuvidine-tenofovir (600 mg/300 mg/300 mg Telura).
The most common malignancies in this population were gynecologic malignancies, followed by hematologic malignancies. Cervical cancer was the most common malignancy among women as well as in the overall study population. Among men, the most common malignancy was NHL. The second and third most common malignancies in men were carcinoma oral cavity and carcinoma oropharynx, respectively, whereas in women, they were NHL and breast cancer. The distribution of various hematologic, head and neck, and gastrointestinal malignancies in this group of patients is shown in Figures 1, 2, and 3.
The ADMs in the study were NHL, including 2 patients diagnosed with primary central nervous system (CNS) lymphomas, and cervical cancer. No case of Kaposi sarcoma, also considered an ADM, was identified in this study. The common NADMs include head and neck malignancies (Figure 2), gastrointestinal malignancies (Figure 3), gynecological and genitourinary malignancies, and breast cancer. The mean CD4 count in the ADM subgroup was 221 cells/mm3, and in the NADM subgroup, it was 250 cells/mm3. There was a significant difference in the distribution of CD4 counts between the ADM and NADM subgroups (P = .03; Mann-Whitney U test). A statistical difference was also noted when the CD4 counts of the patients with NHL were compared with other malignancies (P = .0001; Mann-Whitney U test) There was no statistically significant difference noted when CD4 counts of patients with cervical cancer were compared with NADMs (P = .914).
Discussion
In 2015, a report from the Indian government estimated the prevalence of HIV in the country as 0.26% (0.22%-0.32%).5 The report also noted a decreasing trend in the number of new cases of HIV diagnosed and a decrease in the number of AIDS-related deaths.5 The decrease in deaths from AIDS is primarily attributed to the widespread use of HAART. With the introduction of HAART therapy, the survival of patients diagnosed with HIV-AIDS has increased markedly.6 However, newer challenges have emerged with improved survival, such as an increasing number of patients being diagnosed with malignancies. In the current HAART era, the pattern of malignancies in people living with HIV-AIDS has changed compared with the pre-HAART era.7 The literature suggests that worldwide, malignancies are encountered in about 30% patients with HIV-AIDS, but that percentage differs sharply from that encountered in India, where it is less than 5%.8 This may partly be explained by opportunistic infections such as tuberculosis in Indian patients, which remains the leading cause of death in the HIV-AIDS population. In our study, we retrospectively analyzed the pattern of malignancies in patients with HIV-AIDS.
Although few studies have quoted NHL as the predominant malignancy in their patients with HIV-AIDS, the predominant malignancy was cervical cancer in our patient population, as seen in few other studies.8-10 Head and neck malignancies also continue to be common malignancies in men with HIV-AIDS.10 Thus, an increase in malignancies induced by the human papillomavirus (HPV) can be seen in this group of patients. Only a few pediatric malignancies were noted in our study, and all of those patients had a vertical transmission of HIV.
Kaposi sarcoma is quite rare in the Indian population, and no case of Kaposi sarcoma was diagnosed in our study population. A similar finding was seen in several earlier publications from India. In the largest published series from India by Dhir and colleagues, evaluating 251 patients with HIV-AIDS and malignancy, no case of Kaposi sarcoma was reported.10 The authors mentioned that this finding might be because of the low seroprevalence of Kaposi sarcoma-associated herpesvirus in the Asian population.10 Three different studies from southern India have also not reported the incidence of Kaposi Sarcoma in their series of HIV-AIDS patients with malignancies,11-13 and similar findings were also reported in a study from northern India.9 The incidence of other immunodeficiency-related malignancies was identical to those reported in other studies in the literature.10,14
As seen in other studies, the CD4 counts in patients with ADM were significantly lower compared with those of patients with NADM, and that difference was not seen when CD4 counts of patients with cervical cancer were compared with patients in the NADM subgroup. The risk of NHL increases proportionally to the degree of immune suppression. The increased susceptibility to various infections in patients with low CD4 counts may also contribute to the occurrence of NHL in patients with low CD4 counts. The occurrence of various other rare cancers in patients with HIV-AIDS may be because of confounding rather than a direct HIV or immunosuppression effect.
An increasing incidence of NADMs has been noted in the Western literature.7,14 ADMs remain the most common malignancies in the HIV-AIDS population, accounting for about 48% of all malignancies.8 This is in concordance with previous publications from India.8,10 With the widespread availability of generic HAART, the incidence of ADMs may decrease even more in the future. In developing countries where the screening procedures for malignancies in both the general population and patients with HIV-AIDS have not yet been implemented at a national level, premalignant lesions of the cervix are not detected.10 Cervical cancer is the most common malignancy in our study population, which underscores the importance of cervical cancer screening in patients with HIV-AIDS.
In the developed countries, following the introduction of HAART in HIV-AIDS management, the incidence of Kaposi sarcoma decreased by 60% to 70%, and the incidence of NHL decreased by 30% to 50%, whereas the rates of cervical cancer remained either stable or declined.15,16 Despite the declining trend, the incidence of these malignancies continues to be high among patients with HIV-AIDS compared with the general population.17 A study from the United States showed increasing trends in various NADMs (such as anal, lung, and liver cancers and Hodgkin lymphoma) from 2006 to 2010.17 In 2003, the number of patients with NADM were higher than the number of patients with ADM in the United States.14 In a population-based study from Brazil, ADMs were the most common malignancies diagnosed in patients with HIV-AIDS. A declining trend was noted in the incidence of ADMs in the population and an increasing trend in the incidence of NADMs. This increase in NADM incidence was contributed by anal and lung cancers.18 Studies from developing countries such as Uganda and Botswana have also shown a decrease in the incidence of Kaposi sarcoma after the introduction of HAART.19-21
Kaposi sarcoma, cervical cancer, NHL (including Burkitt lymphoma, immunoblastic lymphoma, and primary CNS lymphoma [PCNSL]) comprise ADMs. All 3 ADMs have an underlying viral infection as the causative agent.22 Kaposi sarcoma is caused by the Kaposi sarcoma herpes virus, for which seroprevalence varies worldwide.23 As already noted in this article, the incidence of Kaposi sarcoma among the HIV-AIDS population has decreased worldwide since the introduction of HAART. The preinvasive uterine cervix lesions and carcinoma cervix are caused by HPV. NHL in patients with HIV-AIDS is a predominantly aggressive B-cell neoplasm. Epstein-Barr virus is implicated for most of the ADM NHLs.24 PCNSL occurs in patients with low CD4 counts and poses a diagnostic challenge. The treatment outcomes for patients with PCNSL before the HAART era were dismal. With the widespread use of HAART, the treatment outcomes of patients with HIV-AIDS and NHL improved, and, currently, these patients are managed the same way as other patients with NHL.22
The increasing incidence of the NADM is partly attributed to the increasing incidence of these malignancies in the general population. An elevated risk of certain NADMs is also attributable to viral infections. The common NADMs in the United States are lung, anal, oropharyngeal, and hepatocellular cancers and Hodgkin lymphoma.14 The common NADMs in our study population were oral, oropharyngeal, colon, and breast cancers and Hodgkin lymphoma. One-third of head and neck cancers, including most oropharyngeal cancers, and cervical and anal cancers in patients with HIV-AIDS are related to HPV.25 Patients with HIV-AIDS are at increased risk for chronic HPV infection from immunosuppression. Chronic HPV infections and prolonged immunosuppression cause premalignant high-grade squamous intraepithelial lesions and invasive cancers.22 The initiation of and adherence to HAART leads to immune recovery and reduces high-risk HPV-associated morbidity.26 Findings from previous studies have demonstrated the benefits of screening for cervical cancer in patients with HIV-AIDS.27 The HPV vaccine is immunogenic in patients with HIV-AIDS and might help prevent HPV-associated malignancies.28
Conclusions
With the wide use of HAART by patients with HIV-AIDS, we can expect an increase in the survival of that population. The incidence of malignancies may also increase significantly in these patients, and further longitudinal studies are needed, as malignancies may emerge as the most common cause of death in patients with HIV-AIDS. In addition, the extensive use of HAART therapy and implementation of screening programs for cervical cancer in patients with HIV-AIDS could result in a decrease in the incidence of ADMs.
India has the third largest HIV epidemic in the world because of its large population size, with 0.3% of the adult population infected with HIV. That translates to 2.1 million infected people, posing a significant challenge in the management of these individuals.1 In all, 43% of the infected are currently on highly active antiretroviral therapy (HAART).1 There has been a significant decrease in the number of HIV-AIDS–related deaths in recent years because of the remarkable increase in the use of antiretroviral therapy.2 However, the prolonged life expectancy in these patients has resulted in an increase in the risk of various new diseases such as cancers. With the complex interactions between altered immunity and infections, the risk of cancers is markedly increased in patients with HIV-AIDS.3 The spectrum of malignancies in this group of patients differs from that in the general population. In addition, the pattern and the magnitude of malignancies differ in different parts of the world.4 In this study, we have analyzed the pattern of malignancies in patients with HIV-AIDS in a regional cancer center in India. The aim of the study was to analyze the pattern of malignancies in patients with HIV-AIDS based on their age and sex and to document the CD4 counts at the time the malignancy was diagnosed.
Methods
We retrieved data from our institution’s medical records department on all patients who had HIV-AIDS and had been diagnosed with a malignancy. Data of all patients presenting with a malignancy and coexisting HIV-AIDS from January 2013 through December 2016 were analyzed initially. Only patients for whom there was a documented CD4 count were included in the final retrospective analysis. We analyzed the correlation between the patients’ CD4 counts and malignancies subclassified as AIDS-defining malignancies (ADMs; aggressive B-cell non-Hodgkin lymphoma [NHL] and cervical cancer) or non–AIDS-defining malignancies (NADMs; all other malignancies other than aggressive NHL and carcinoma cervix were defined as NADM). We also analyzed the correlation between the CD4 count and NHL and other malignancies. A statistical analysis was performed using SPSS Statistics for Windows, version 23 (IBM Corp, Armonk, NY). The independent sample Mann-Whitney U or Kruskal-Wallis tests were used for comparing the CD4 counts between the various subgroups of malignancies. The study was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
A total of 370 patients who were diagnosed with malignancy and have coexisting HIV-AIDS were identified. In all, 85 patients were excluded because there were no CD4 counts available for them, and the remaining 285 patients were included in the final analysis. Of that total, 136 patients (48%) were men, and 149 (52%) were women.
The median age of the population was 44.8 years (5-80 years) at the time of diagnosis with malignancy. The mean CD4 count of the entire population was 235.4 cells/mm3 (50-734 cells/mm3). There were 104 patients with CD4 counts of ≤200 cells/mm3, and 181 patients had CD4 counts of >200 cells/mm3 (Table 1). All patients received the HAART regimen, efavirenz-lamuvidine-tenofovir (600 mg/300 mg/300 mg Telura).
The most common malignancies in this population were gynecologic malignancies, followed by hematologic malignancies. Cervical cancer was the most common malignancy among women as well as in the overall study population. Among men, the most common malignancy was NHL. The second and third most common malignancies in men were carcinoma oral cavity and carcinoma oropharynx, respectively, whereas in women, they were NHL and breast cancer. The distribution of various hematologic, head and neck, and gastrointestinal malignancies in this group of patients is shown in Figures 1, 2, and 3.
The ADMs in the study were NHL, including 2 patients diagnosed with primary central nervous system (CNS) lymphomas, and cervical cancer. No case of Kaposi sarcoma, also considered an ADM, was identified in this study. The common NADMs include head and neck malignancies (Figure 2), gastrointestinal malignancies (Figure 3), gynecological and genitourinary malignancies, and breast cancer. The mean CD4 count in the ADM subgroup was 221 cells/mm3, and in the NADM subgroup, it was 250 cells/mm3. There was a significant difference in the distribution of CD4 counts between the ADM and NADM subgroups (P = .03; Mann-Whitney U test). A statistical difference was also noted when the CD4 counts of the patients with NHL were compared with other malignancies (P = .0001; Mann-Whitney U test) There was no statistically significant difference noted when CD4 counts of patients with cervical cancer were compared with NADMs (P = .914).
Discussion
In 2015, a report from the Indian government estimated the prevalence of HIV in the country as 0.26% (0.22%-0.32%).5 The report also noted a decreasing trend in the number of new cases of HIV diagnosed and a decrease in the number of AIDS-related deaths.5 The decrease in deaths from AIDS is primarily attributed to the widespread use of HAART. With the introduction of HAART therapy, the survival of patients diagnosed with HIV-AIDS has increased markedly.6 However, newer challenges have emerged with improved survival, such as an increasing number of patients being diagnosed with malignancies. In the current HAART era, the pattern of malignancies in people living with HIV-AIDS has changed compared with the pre-HAART era.7 The literature suggests that worldwide, malignancies are encountered in about 30% patients with HIV-AIDS, but that percentage differs sharply from that encountered in India, where it is less than 5%.8 This may partly be explained by opportunistic infections such as tuberculosis in Indian patients, which remains the leading cause of death in the HIV-AIDS population. In our study, we retrospectively analyzed the pattern of malignancies in patients with HIV-AIDS.
Although few studies have quoted NHL as the predominant malignancy in their patients with HIV-AIDS, the predominant malignancy was cervical cancer in our patient population, as seen in few other studies.8-10 Head and neck malignancies also continue to be common malignancies in men with HIV-AIDS.10 Thus, an increase in malignancies induced by the human papillomavirus (HPV) can be seen in this group of patients. Only a few pediatric malignancies were noted in our study, and all of those patients had a vertical transmission of HIV.
Kaposi sarcoma is quite rare in the Indian population, and no case of Kaposi sarcoma was diagnosed in our study population. A similar finding was seen in several earlier publications from India. In the largest published series from India by Dhir and colleagues, evaluating 251 patients with HIV-AIDS and malignancy, no case of Kaposi sarcoma was reported.10 The authors mentioned that this finding might be because of the low seroprevalence of Kaposi sarcoma-associated herpesvirus in the Asian population.10 Three different studies from southern India have also not reported the incidence of Kaposi Sarcoma in their series of HIV-AIDS patients with malignancies,11-13 and similar findings were also reported in a study from northern India.9 The incidence of other immunodeficiency-related malignancies was identical to those reported in other studies in the literature.10,14
As seen in other studies, the CD4 counts in patients with ADM were significantly lower compared with those of patients with NADM, and that difference was not seen when CD4 counts of patients with cervical cancer were compared with patients in the NADM subgroup. The risk of NHL increases proportionally to the degree of immune suppression. The increased susceptibility to various infections in patients with low CD4 counts may also contribute to the occurrence of NHL in patients with low CD4 counts. The occurrence of various other rare cancers in patients with HIV-AIDS may be because of confounding rather than a direct HIV or immunosuppression effect.
An increasing incidence of NADMs has been noted in the Western literature.7,14 ADMs remain the most common malignancies in the HIV-AIDS population, accounting for about 48% of all malignancies.8 This is in concordance with previous publications from India.8,10 With the widespread availability of generic HAART, the incidence of ADMs may decrease even more in the future. In developing countries where the screening procedures for malignancies in both the general population and patients with HIV-AIDS have not yet been implemented at a national level, premalignant lesions of the cervix are not detected.10 Cervical cancer is the most common malignancy in our study population, which underscores the importance of cervical cancer screening in patients with HIV-AIDS.
In the developed countries, following the introduction of HAART in HIV-AIDS management, the incidence of Kaposi sarcoma decreased by 60% to 70%, and the incidence of NHL decreased by 30% to 50%, whereas the rates of cervical cancer remained either stable or declined.15,16 Despite the declining trend, the incidence of these malignancies continues to be high among patients with HIV-AIDS compared with the general population.17 A study from the United States showed increasing trends in various NADMs (such as anal, lung, and liver cancers and Hodgkin lymphoma) from 2006 to 2010.17 In 2003, the number of patients with NADM were higher than the number of patients with ADM in the United States.14 In a population-based study from Brazil, ADMs were the most common malignancies diagnosed in patients with HIV-AIDS. A declining trend was noted in the incidence of ADMs in the population and an increasing trend in the incidence of NADMs. This increase in NADM incidence was contributed by anal and lung cancers.18 Studies from developing countries such as Uganda and Botswana have also shown a decrease in the incidence of Kaposi sarcoma after the introduction of HAART.19-21
Kaposi sarcoma, cervical cancer, NHL (including Burkitt lymphoma, immunoblastic lymphoma, and primary CNS lymphoma [PCNSL]) comprise ADMs. All 3 ADMs have an underlying viral infection as the causative agent.22 Kaposi sarcoma is caused by the Kaposi sarcoma herpes virus, for which seroprevalence varies worldwide.23 As already noted in this article, the incidence of Kaposi sarcoma among the HIV-AIDS population has decreased worldwide since the introduction of HAART. The preinvasive uterine cervix lesions and carcinoma cervix are caused by HPV. NHL in patients with HIV-AIDS is a predominantly aggressive B-cell neoplasm. Epstein-Barr virus is implicated for most of the ADM NHLs.24 PCNSL occurs in patients with low CD4 counts and poses a diagnostic challenge. The treatment outcomes for patients with PCNSL before the HAART era were dismal. With the widespread use of HAART, the treatment outcomes of patients with HIV-AIDS and NHL improved, and, currently, these patients are managed the same way as other patients with NHL.22
The increasing incidence of the NADM is partly attributed to the increasing incidence of these malignancies in the general population. An elevated risk of certain NADMs is also attributable to viral infections. The common NADMs in the United States are lung, anal, oropharyngeal, and hepatocellular cancers and Hodgkin lymphoma.14 The common NADMs in our study population were oral, oropharyngeal, colon, and breast cancers and Hodgkin lymphoma. One-third of head and neck cancers, including most oropharyngeal cancers, and cervical and anal cancers in patients with HIV-AIDS are related to HPV.25 Patients with HIV-AIDS are at increased risk for chronic HPV infection from immunosuppression. Chronic HPV infections and prolonged immunosuppression cause premalignant high-grade squamous intraepithelial lesions and invasive cancers.22 The initiation of and adherence to HAART leads to immune recovery and reduces high-risk HPV-associated morbidity.26 Findings from previous studies have demonstrated the benefits of screening for cervical cancer in patients with HIV-AIDS.27 The HPV vaccine is immunogenic in patients with HIV-AIDS and might help prevent HPV-associated malignancies.28
Conclusions
With the wide use of HAART by patients with HIV-AIDS, we can expect an increase in the survival of that population. The incidence of malignancies may also increase significantly in these patients, and further longitudinal studies are needed, as malignancies may emerge as the most common cause of death in patients with HIV-AIDS. In addition, the extensive use of HAART therapy and implementation of screening programs for cervical cancer in patients with HIV-AIDS could result in a decrease in the incidence of ADMs.
1. UNAIDS. Prevention gap report. http://www.unaids.org/sites/default/files/media_asset/2016-prevention-gap-report_en.pdf. Released 2016. Accessed December 27, 2017.
3. Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol. 2012;24(5):506-516.
4. Biggar RJ, Chaturvedi AK, Bhatia K, Mbulaiteye SM. Cancer risk in persons with HIV-AIDS in India: a review and future directions for research. Infect Agent Cancer. 2009;4:4.
5. National AIDS Control Organisation & National Institute of Medical Statistics, ICMR, Ministry of Health & Family Welfare, Government of India. India HIV estimations 2015, technical report. http://www.naco.gov.in/sites/default/files/India%20HIV%20Estimations%202015.pdf. Published 2015. Accessed December 27, 2017.
6. Bonnet F, Lewden C, May T, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101(2):317-324.
7. Crum-Cianflone N, Hullsiek KH, Marconi V, et al. Trends in the incidence of cancers among HIV-infected persons and the impact of antiretroviral therapy: a 20-year cohort study. AIDS. 2009;23(1):41-50.
8. Sharma S, Soneja M, Ranjan S. Malignancies in human immunodeficiency virus infected patients in India: initial experience in the HAART era. Indian J Med Res. 2015;142(5):563-567.
9. Sachdeva RK, Sharma A, Singh S, Varma S. Spectrum of AIDS defining & non-AIDS defining malignancies in north India. In
10. Dhir AA, Sawant S, Dikshit RP, et al. Spectrum of HIV-AIDS related cancers in India. Cancer Causes Control. 2007;19(2):147-153.
11. Venkatesh KK, Saghayam S, Devaleenal B, et al. Spectrum of malignancies among HIV-infected patients in South India. Indian J Cancer. 2012;49(1):176-180.
12. Shruti P, Narayanan G, Puthuveettil J, Jayasree K, Vijayalakshmi K. Spectrum of HIV/AIDS-associated cancers in south India. J Clin Oncol. 2014;32(suppl):e12534.
13. Paul TR, Uppin MS, Uppin SG, et al. Spectrum of malignancies in human immunodeficiency virus–positive patients at a Tertiary Care Centre in South India. Indian J Cancer. 2014;51(4):459-463.
14. Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103(9):753-762.
15. Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148(10):728-736.
16. Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
17. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the United States. AIDS. 2014;28(6):881-890.
18. Tanaka LF, Latorre MDRD, Gutierrez EB, Heumann C, Herbinger KH, Froeschl G. Trends in the incidence of AIDS-defining and non-AIDS-defining cancers in people living with AIDS: a population-based study from São Paulo, Brazil. Int J STD AIDS. 2017;28(12):1190-1198.
19. Mutyaba I, Phipps W, Krantz EM, et al. A population-level evaluation of the effect of antiretroviral therapy on cancer incidence in Kyadondo County, Uganda, 1999–2008. J Acquir Immune Defic Syndr. 2015;69(4):481-486.
20. Dryden-Peterson S, Medhin H, Kebabonye-Pusoentsi M, et al. Cancer incidence following expansion of HIV treatment in Botswana. PLoS ONE. 2015;10(8):e0135602.
21. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12(1):6-11.
22. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041.
23. Gao SJ, Kingsley L, Li M, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2(8):925-928.
24. Epstein-Barr virus and AIDS-associated lymphomas. Lancet. 1991;338(8773):979-981.
25. Picard A, Badoual C, Hourseau M, et al. Human papilloma virus prevalence in HIV patients with head and neck squamous cell carcinoma. AIDS. 2016;30(8):1257-1266.
26. Minkoff H, Zhong Y, Burk RD, et al. Influence of adherent and effective antiretroviral therapy use on human papillomavirus infection and squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Infect Dis. 2010;201(5):681-690.
27. Ghebre RG, Grover S, Xu MJ, Chuang LT, Simonds H. Cervical cancer control in HIV-infected women: past, present and future. Gynecol Oncol Rep. 2017;21:101-108.
28. Kojic EM, Rana AI, Cu-Uvin S. Human papillomavirus vaccination in HIV-infected women: need for increased coverage. Expert Rev Vaccines. 2016;15(1):105-117.
1. UNAIDS. Prevention gap report. http://www.unaids.org/sites/default/files/media_asset/2016-prevention-gap-report_en.pdf. Released 2016. Accessed December 27, 2017.
3. Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol. 2012;24(5):506-516.
4. Biggar RJ, Chaturvedi AK, Bhatia K, Mbulaiteye SM. Cancer risk in persons with HIV-AIDS in India: a review and future directions for research. Infect Agent Cancer. 2009;4:4.
5. National AIDS Control Organisation & National Institute of Medical Statistics, ICMR, Ministry of Health & Family Welfare, Government of India. India HIV estimations 2015, technical report. http://www.naco.gov.in/sites/default/files/India%20HIV%20Estimations%202015.pdf. Published 2015. Accessed December 27, 2017.
6. Bonnet F, Lewden C, May T, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101(2):317-324.
7. Crum-Cianflone N, Hullsiek KH, Marconi V, et al. Trends in the incidence of cancers among HIV-infected persons and the impact of antiretroviral therapy: a 20-year cohort study. AIDS. 2009;23(1):41-50.
8. Sharma S, Soneja M, Ranjan S. Malignancies in human immunodeficiency virus infected patients in India: initial experience in the HAART era. Indian J Med Res. 2015;142(5):563-567.
9. Sachdeva RK, Sharma A, Singh S, Varma S. Spectrum of AIDS defining & non-AIDS defining malignancies in north India. In
10. Dhir AA, Sawant S, Dikshit RP, et al. Spectrum of HIV-AIDS related cancers in India. Cancer Causes Control. 2007;19(2):147-153.
11. Venkatesh KK, Saghayam S, Devaleenal B, et al. Spectrum of malignancies among HIV-infected patients in South India. Indian J Cancer. 2012;49(1):176-180.
12. Shruti P, Narayanan G, Puthuveettil J, Jayasree K, Vijayalakshmi K. Spectrum of HIV/AIDS-associated cancers in south India. J Clin Oncol. 2014;32(suppl):e12534.
13. Paul TR, Uppin MS, Uppin SG, et al. Spectrum of malignancies in human immunodeficiency virus–positive patients at a Tertiary Care Centre in South India. Indian J Cancer. 2014;51(4):459-463.
14. Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103(9):753-762.
15. Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148(10):728-736.
16. Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
17. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the United States. AIDS. 2014;28(6):881-890.
18. Tanaka LF, Latorre MDRD, Gutierrez EB, Heumann C, Herbinger KH, Froeschl G. Trends in the incidence of AIDS-defining and non-AIDS-defining cancers in people living with AIDS: a population-based study from São Paulo, Brazil. Int J STD AIDS. 2017;28(12):1190-1198.
19. Mutyaba I, Phipps W, Krantz EM, et al. A population-level evaluation of the effect of antiretroviral therapy on cancer incidence in Kyadondo County, Uganda, 1999–2008. J Acquir Immune Defic Syndr. 2015;69(4):481-486.
20. Dryden-Peterson S, Medhin H, Kebabonye-Pusoentsi M, et al. Cancer incidence following expansion of HIV treatment in Botswana. PLoS ONE. 2015;10(8):e0135602.
21. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12(1):6-11.
22. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041.
23. Gao SJ, Kingsley L, Li M, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2(8):925-928.
24. Epstein-Barr virus and AIDS-associated lymphomas. Lancet. 1991;338(8773):979-981.
25. Picard A, Badoual C, Hourseau M, et al. Human papilloma virus prevalence in HIV patients with head and neck squamous cell carcinoma. AIDS. 2016;30(8):1257-1266.
26. Minkoff H, Zhong Y, Burk RD, et al. Influence of adherent and effective antiretroviral therapy use on human papillomavirus infection and squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Infect Dis. 2010;201(5):681-690.
27. Ghebre RG, Grover S, Xu MJ, Chuang LT, Simonds H. Cervical cancer control in HIV-infected women: past, present and future. Gynecol Oncol Rep. 2017;21:101-108.
28. Kojic EM, Rana AI, Cu-Uvin S. Human papillomavirus vaccination in HIV-infected women: need for increased coverage. Expert Rev Vaccines. 2016;15(1):105-117.
Malignant olecranon bursitis in the setting of multiple myeloma relapse
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.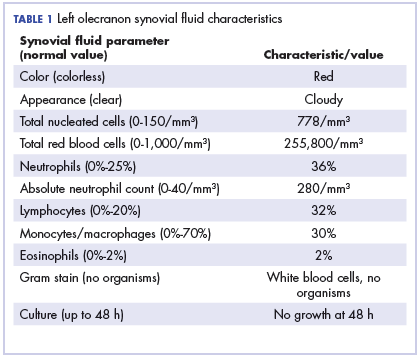
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).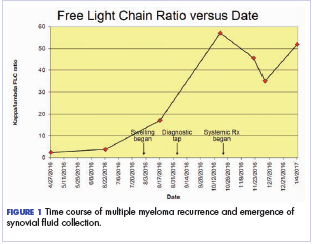
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.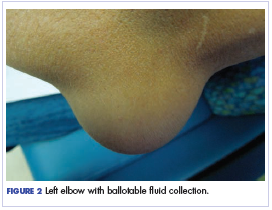
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).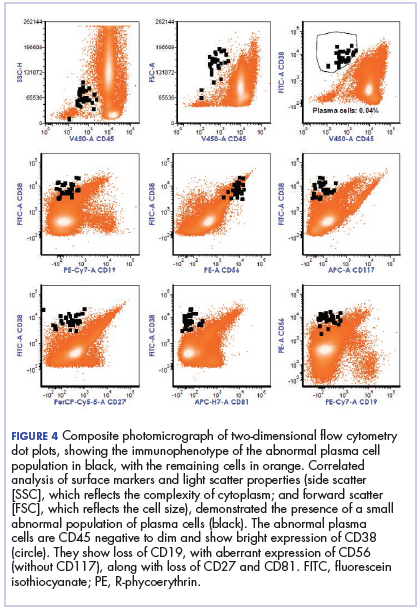
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
Immunotherapy may hold the key to defeating virally associated cancers
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.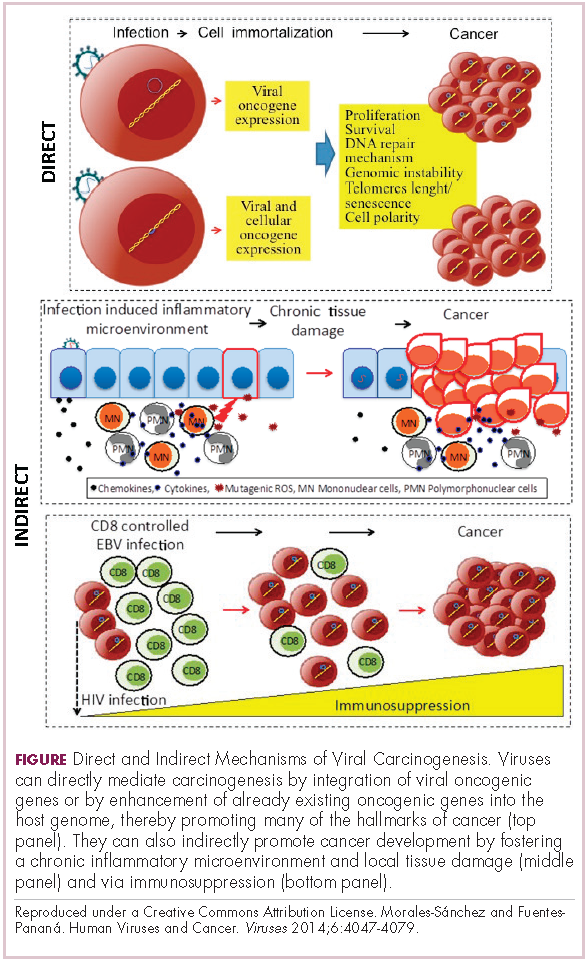
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).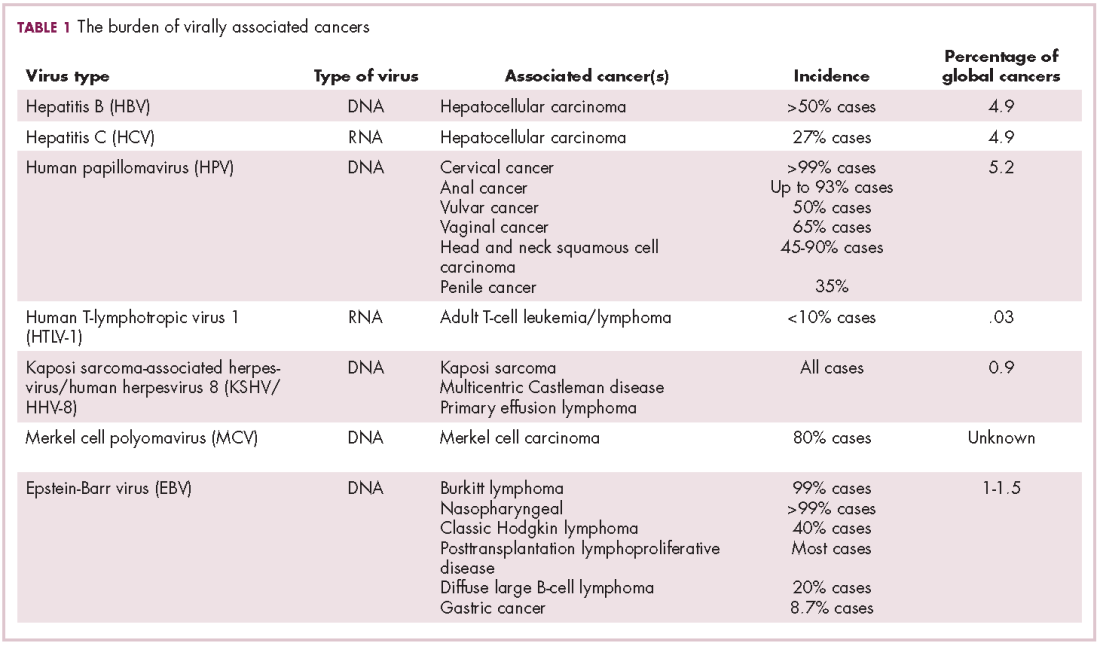
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9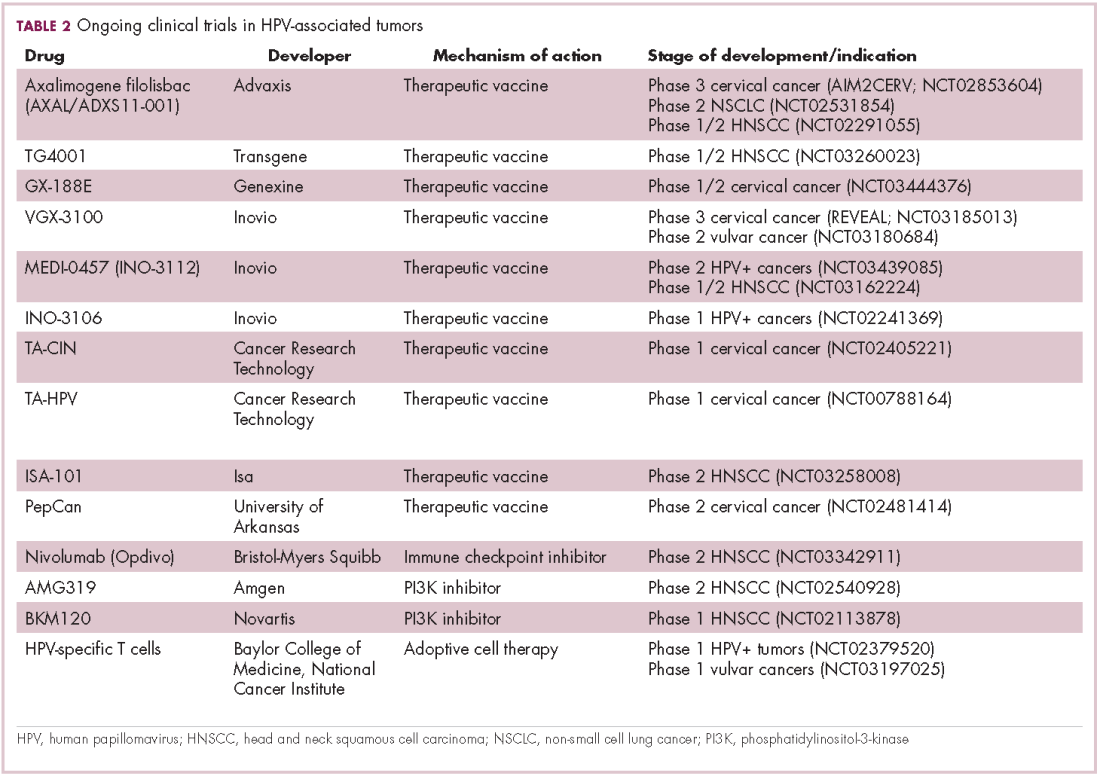
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).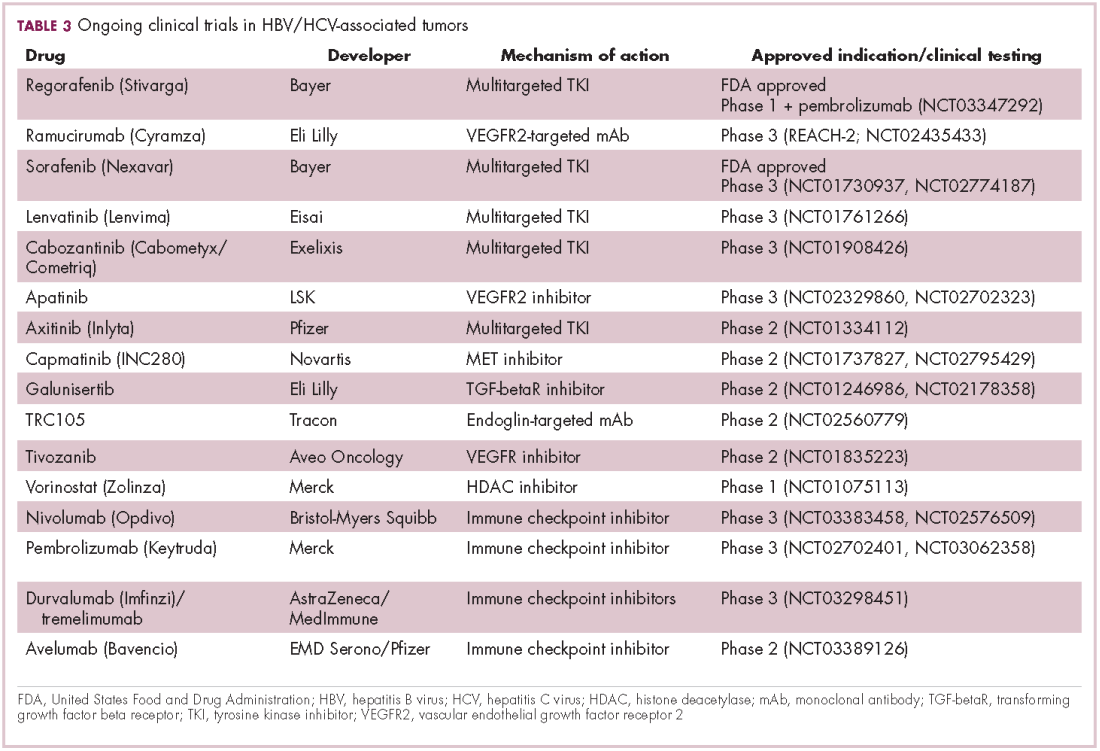
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).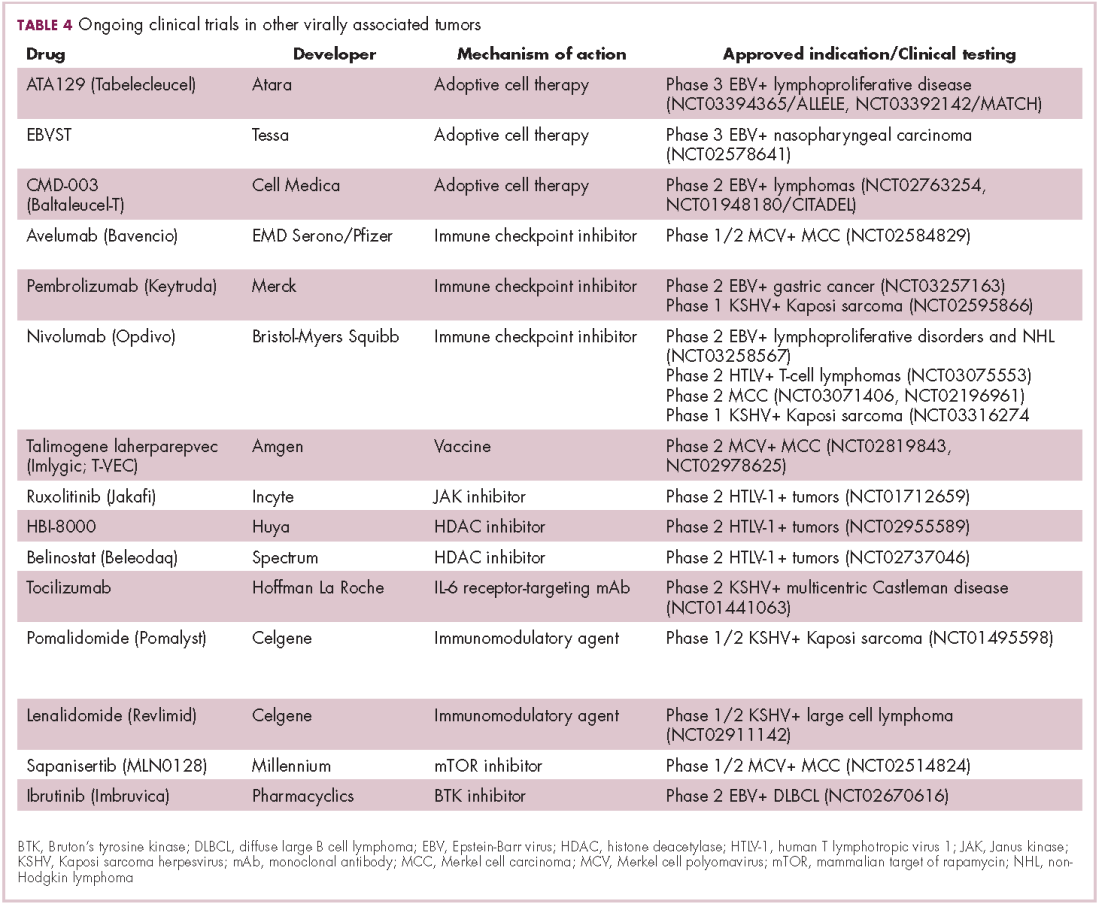
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
A ‘double-hit’ bone marrow rare co-occurrence of 2 different pathologies
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Collaboration is key to bridging the AYA cancer care divide
Survival gains among adolescents and young adults (AYAs) with cancer continue to lag behind outcomes for children and older adult patients. It’s a trend that spans decades, but clinicians and researchers are finally getting serious about trying to understand the underlying causes and are re-examining prevailing practices in an effort to address the discrepancies.
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist at Cleveland Clinic Children’s Hospital, Ohio, said in an interview. He’s specifically referring to the cancers that affect AYAs, who are broadly defined as patients aged 15 through 39 years. “A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults. The biology may be different in the adolescent and young adult patients, which may lead to different outcomes.”
In addition, the psychosocial needs in this age group differ vastly from those in other groups. “Many of these patients are in college or have just started their families, so we have to pay more attention to [issues related to] financial toxicity and fertility, for example,” said Dr Hanna, who is the director of pediatric bone marrow transplantation at the clinic. (The term “financial toxicity” describes the cumulative negative impact of the high cost of care, lost work time, and delays in reaching educational and career goals on patients with cancer and their families.)
Another factor that likely contributes to the outcome disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs.
A recent series of articles published in the journal Blood addressed these and other issues, among them, whether AYAs with acute lymphoblastic leukemia (ALL)1 or aggressive B-cell non-Hodgkin lymphomas (NHLs) 2 should be treated as children or adults; treatment strategies for those with acute myeloid leukemias (AMLs); 3 management of Hodgkin lymphoma;4 and psychosocial challenges and health-related quality of life (QoL) in AYAs with hematologic malignancies.5
In the introduction to the series, Jorge Cortes, MD, an assistant editor on the journal, wrote that hematologic malignancies in AYAs “represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population.”6
He noted, however, that “not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs often centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults,” noted Dr Cortes, professor and chair of the chronic myeloid leukemia section in the department of leukemia at the University of Texas MD Anderson Cancer Center, Houston.
Therapeutic options: pediatric or adult protocols?
In their article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15 through 20 years is supported by findings from numerous studies. In young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” because they have been shown to significantly improve outcomes, with long-term survival rates nearing 70%.1 Patients in these age groups require specific programs that factor in access to care and to trials, an increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
However, Kristen O’Dwyer, MD, and colleagues, argue in an article on AML treatment in AYAs that neither the pediatric nor adult approaches are ideally suited for AYAs because of the “distinguishing characteristics of AYAs with AML.” Rather, they conclude that AYA-specific approaches merit consideration.3
Similarly, Kieron Dunleavy, MD, and Thomas G Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that there is a “remarkable divide” in the treatment of patients younger than 18 years with lymphoma compared with their young adult counterparts, and that it underscores the need for collaboration in developing consensus regarding treatment of AYAs.2
Clinical setting: pediatric or adult?
Consideration is also being given to the clinical setting in which AYA patients receive their treatment. Lori Muffly, MD, MS, and colleagues have reported that survival was superior for AYA patients with ALL who were treated in pediatric cancer settings,7 and other researchers have reported similar findings.
However, those improved outcomes in the pediatric setting might be offset by a higher use of resources and therefore higher costs, based on recent findings in a Canadian study by Paul C Nathan, MD, and colleagues.8 Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the authors found that the cost of care was higher when treatment took place in a pediatric setting compared with in an adult institution, and that it was driven in part by higher hospitalization rates and longer hospital stays. These findings were true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
In an accompanying editorial, Helen M Parsons, PhD, and her co-authors wrote that adolescents who receive treatment in the pediatric setting “tended to seek more [emergency department (ED)] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship.9 They pointed out that the findings of higher inpatient days in the pediatric setting was not surprising given that induction therapies for pediatric ALL tend to be more complex and intensive than therapies commonly used in adults with ALL, and that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase,” they wrote, adding that further investigation was needed on this topic to better understand those trends. “The finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.” 9
The authors of the editorial emphasized that because of the differences in health care delivery and payment structures between the United States and Canada, where the Nathan study was done, it was important that similar studies are done in the United States to confirm these findings.
Disease and developmental biology
As Dr Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and that they may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr O'Dwyer and her colleagues noted,3 citing a recent European study of 5,564 patients with de novo AML that showed that the frequency of favorable cytogenetics was low in infants (13.7%), increased in children (25%) and young adults (44%), and decreased again in middle age and older patients.10
“Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr O'Dwyer and her colleagues wrote.3 It was also becoming more apparent that age influences the presence of AML-related molecular abnormalities, and recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr Boissel and Dr Baruchel also noted in their report that light was finally being shed on the “black hole” of understanding ALL biology in AYAs, and research has shown that there is a continuum between childhood and adult ALL.1 They concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, NC, said in an interview.
These patients have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, and in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added. “Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... these are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so.”
In a 2014 study, Dr Salsman and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life.11 The latter finding was surprising and highlights the importance of the social dimension in the lives of AYAs. “Patient after patient will say ‘I found out who my real friends are,’ ” he said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr Salsman and his colleagues are using those findings to develop interventions that can maximize self-care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves. For example, a randomized controlled pilot study that incorporates social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions is underway.
Another intervention targets emotional well-being through the use of web-based tools to increase positive affect. A proof-of-concept study showed that the approach was feasible and well received, and a larger-scale randomized controlled trial is being planned, he said.
Dr Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital. It was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr Cortes noted a paucity of clinical trials specifically designed for this population. “At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, describing AYA enrollment in clinical trials in cancer as “suboptimal at best.”6
Dr Salsman said these dismal enrolment numbers could in part be related to treatment setting. Data suggest that most AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers where the bulk of research is being done, he explained.
Dr Hanna agreed that more research involving AYAs was needed as is a better understanding of why enrollment is so much lower in this population. He pointed out that in 2017 the American Society of Clinical Oncology and Friends of Cancer Research released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.12 The organizations said that individuals aged 12 years and older should routinely be included in such trials because their drug metabolism is similar to adults, and inclusion of younger patients may also be appropriate if they are part of the population affected by the disease, depending on specific disease biology, action of the drug, and available safety information.
Officials at the Food and Drug Administration are considering that possibility, Dr Hanna said.
Dr Salsman added there has been an increase in recent years in the attention paid to disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups. For example, about 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population;13 the Institute of Medicine held a forum on the care of AYAs with cancer;14 and the National Cancer Institute held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology,15 he noted.
Dr Hanna added that “scientific groups such as Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now. One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically co-ordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.16
Next steps
Among the recommendations from authors in the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams and the involvement of multidisciplinary teams in care for this population.
Many centers are already working on models for collaborative care, Dr Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in helping clinical and supportive caregivers and their AYA patients “have a shared vision” as they work to maximize improvements in outcomes.
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr Hanna said. In the case of the community-powered advocacy organization Critical Mass, members have succeeded in getting lawmakers to introduce a bill in the US House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
1. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood. 2018;132:351-361.
2. Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375.
3. O’Dwyer K, Freyer DR, Horan JT. Treatment strategies for adolescent and young adult patients with acute myeloid leukemia. Blood. 2018;132:362-368.
4. Flerlage JE, Metzger ML, Bhakta N. The management of Hodgkin lymphoma in adolescents and young adults: burden of disease or burden of choice? Blood. 2018;132:376-384.
5. Husson O, Huijgens PC, van der Graaf WTA. Psychosocial challenges and health-related quality of life of adolescents and young adults with hematologic malignancies. Blood. 2018;132:385-392.
6. Cortes J. Introduction to a review series on adolescent and young adult malignant hematology. Blood. 2018;132:345-346.
7. Muffly L, Alvarez E, Lichtensztajn D, Abrahão R, Gomez SL, Keegan T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv. 2018;2(8):895-903.
8. Nathan PC, Bremner KE, Liu N, et al. Resource utilization and costs in adolescents treated for cancer in pediatric vs adult institutions. J Natl Cancer Inst. July 19, 2018. [Epub ahead of print.]
9. Parsons HM, Muffly L, Alvarez EM, Keegan THM. Does treatment setting matter? Evaluating resource utilization for adolescents treated in pediatric vs adult cancer institutions. https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djy123/5056313?searchresult=1. Published July 19, 2018. Last accessed October 12, 2018.
10. Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821-3830.
11. Salsman JM, Garcia SF, Yanez B, et al. Physical, emotional, and social health differences between posttreatment young adults with cancer and matched healthy controls. Cancer. 2014;120(15):2247-2254.
12. Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and Friends of Cancer Research joint research statement. J Clin Oncol. 2017;35(33):3737-3744.
13. Freyer DR, Seibel NL. The clinical trials gap for adolescents and young adults with cancer: recent progress and conceptual framework for continued research. Curr Pediatr Rep. Published online February 18, 2015. DOI 10.1007/s40124-015-0075-y.
14. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
15. Wilder Smith A, Seibel NL, Lewis DR, et al. Next steps for adolescent and young adult oncology workshop: An update on progress and recommendations for the future. Cancer. 2016;122(7):988-999.
16. Keegan THM, Ries LAG, Barr RD, et al. Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer. 2016;122(7):1009-1016.
Survival gains among adolescents and young adults (AYAs) with cancer continue to lag behind outcomes for children and older adult patients. It’s a trend that spans decades, but clinicians and researchers are finally getting serious about trying to understand the underlying causes and are re-examining prevailing practices in an effort to address the discrepancies.
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist at Cleveland Clinic Children’s Hospital, Ohio, said in an interview. He’s specifically referring to the cancers that affect AYAs, who are broadly defined as patients aged 15 through 39 years. “A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults. The biology may be different in the adolescent and young adult patients, which may lead to different outcomes.”
In addition, the psychosocial needs in this age group differ vastly from those in other groups. “Many of these patients are in college or have just started their families, so we have to pay more attention to [issues related to] financial toxicity and fertility, for example,” said Dr Hanna, who is the director of pediatric bone marrow transplantation at the clinic. (The term “financial toxicity” describes the cumulative negative impact of the high cost of care, lost work time, and delays in reaching educational and career goals on patients with cancer and their families.)
Another factor that likely contributes to the outcome disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs.
A recent series of articles published in the journal Blood addressed these and other issues, among them, whether AYAs with acute lymphoblastic leukemia (ALL)1 or aggressive B-cell non-Hodgkin lymphomas (NHLs) 2 should be treated as children or adults; treatment strategies for those with acute myeloid leukemias (AMLs); 3 management of Hodgkin lymphoma;4 and psychosocial challenges and health-related quality of life (QoL) in AYAs with hematologic malignancies.5
In the introduction to the series, Jorge Cortes, MD, an assistant editor on the journal, wrote that hematologic malignancies in AYAs “represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population.”6
He noted, however, that “not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs often centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults,” noted Dr Cortes, professor and chair of the chronic myeloid leukemia section in the department of leukemia at the University of Texas MD Anderson Cancer Center, Houston.
Therapeutic options: pediatric or adult protocols?
In their article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15 through 20 years is supported by findings from numerous studies. In young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” because they have been shown to significantly improve outcomes, with long-term survival rates nearing 70%.1 Patients in these age groups require specific programs that factor in access to care and to trials, an increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
However, Kristen O’Dwyer, MD, and colleagues, argue in an article on AML treatment in AYAs that neither the pediatric nor adult approaches are ideally suited for AYAs because of the “distinguishing characteristics of AYAs with AML.” Rather, they conclude that AYA-specific approaches merit consideration.3
Similarly, Kieron Dunleavy, MD, and Thomas G Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that there is a “remarkable divide” in the treatment of patients younger than 18 years with lymphoma compared with their young adult counterparts, and that it underscores the need for collaboration in developing consensus regarding treatment of AYAs.2
Clinical setting: pediatric or adult?
Consideration is also being given to the clinical setting in which AYA patients receive their treatment. Lori Muffly, MD, MS, and colleagues have reported that survival was superior for AYA patients with ALL who were treated in pediatric cancer settings,7 and other researchers have reported similar findings.
However, those improved outcomes in the pediatric setting might be offset by a higher use of resources and therefore higher costs, based on recent findings in a Canadian study by Paul C Nathan, MD, and colleagues.8 Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the authors found that the cost of care was higher when treatment took place in a pediatric setting compared with in an adult institution, and that it was driven in part by higher hospitalization rates and longer hospital stays. These findings were true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
In an accompanying editorial, Helen M Parsons, PhD, and her co-authors wrote that adolescents who receive treatment in the pediatric setting “tended to seek more [emergency department (ED)] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship.9 They pointed out that the findings of higher inpatient days in the pediatric setting was not surprising given that induction therapies for pediatric ALL tend to be more complex and intensive than therapies commonly used in adults with ALL, and that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase,” they wrote, adding that further investigation was needed on this topic to better understand those trends. “The finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.” 9
The authors of the editorial emphasized that because of the differences in health care delivery and payment structures between the United States and Canada, where the Nathan study was done, it was important that similar studies are done in the United States to confirm these findings.
Disease and developmental biology
As Dr Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and that they may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr O'Dwyer and her colleagues noted,3 citing a recent European study of 5,564 patients with de novo AML that showed that the frequency of favorable cytogenetics was low in infants (13.7%), increased in children (25%) and young adults (44%), and decreased again in middle age and older patients.10
“Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr O'Dwyer and her colleagues wrote.3 It was also becoming more apparent that age influences the presence of AML-related molecular abnormalities, and recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr Boissel and Dr Baruchel also noted in their report that light was finally being shed on the “black hole” of understanding ALL biology in AYAs, and research has shown that there is a continuum between childhood and adult ALL.1 They concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, NC, said in an interview.
These patients have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, and in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added. “Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... these are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so.”
In a 2014 study, Dr Salsman and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life.11 The latter finding was surprising and highlights the importance of the social dimension in the lives of AYAs. “Patient after patient will say ‘I found out who my real friends are,’ ” he said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr Salsman and his colleagues are using those findings to develop interventions that can maximize self-care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves. For example, a randomized controlled pilot study that incorporates social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions is underway.
Another intervention targets emotional well-being through the use of web-based tools to increase positive affect. A proof-of-concept study showed that the approach was feasible and well received, and a larger-scale randomized controlled trial is being planned, he said.
Dr Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital. It was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr Cortes noted a paucity of clinical trials specifically designed for this population. “At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, describing AYA enrollment in clinical trials in cancer as “suboptimal at best.”6
Dr Salsman said these dismal enrolment numbers could in part be related to treatment setting. Data suggest that most AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers where the bulk of research is being done, he explained.
Dr Hanna agreed that more research involving AYAs was needed as is a better understanding of why enrollment is so much lower in this population. He pointed out that in 2017 the American Society of Clinical Oncology and Friends of Cancer Research released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.12 The organizations said that individuals aged 12 years and older should routinely be included in such trials because their drug metabolism is similar to adults, and inclusion of younger patients may also be appropriate if they are part of the population affected by the disease, depending on specific disease biology, action of the drug, and available safety information.
Officials at the Food and Drug Administration are considering that possibility, Dr Hanna said.
Dr Salsman added there has been an increase in recent years in the attention paid to disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups. For example, about 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population;13 the Institute of Medicine held a forum on the care of AYAs with cancer;14 and the National Cancer Institute held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology,15 he noted.
Dr Hanna added that “scientific groups such as Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now. One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically co-ordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.16
Next steps
Among the recommendations from authors in the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams and the involvement of multidisciplinary teams in care for this population.
Many centers are already working on models for collaborative care, Dr Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in helping clinical and supportive caregivers and their AYA patients “have a shared vision” as they work to maximize improvements in outcomes.
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr Hanna said. In the case of the community-powered advocacy organization Critical Mass, members have succeeded in getting lawmakers to introduce a bill in the US House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
Survival gains among adolescents and young adults (AYAs) with cancer continue to lag behind outcomes for children and older adult patients. It’s a trend that spans decades, but clinicians and researchers are finally getting serious about trying to understand the underlying causes and are re-examining prevailing practices in an effort to address the discrepancies.
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist at Cleveland Clinic Children’s Hospital, Ohio, said in an interview. He’s specifically referring to the cancers that affect AYAs, who are broadly defined as patients aged 15 through 39 years. “A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults. The biology may be different in the adolescent and young adult patients, which may lead to different outcomes.”
In addition, the psychosocial needs in this age group differ vastly from those in other groups. “Many of these patients are in college or have just started their families, so we have to pay more attention to [issues related to] financial toxicity and fertility, for example,” said Dr Hanna, who is the director of pediatric bone marrow transplantation at the clinic. (The term “financial toxicity” describes the cumulative negative impact of the high cost of care, lost work time, and delays in reaching educational and career goals on patients with cancer and their families.)
Another factor that likely contributes to the outcome disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs.
A recent series of articles published in the journal Blood addressed these and other issues, among them, whether AYAs with acute lymphoblastic leukemia (ALL)1 or aggressive B-cell non-Hodgkin lymphomas (NHLs) 2 should be treated as children or adults; treatment strategies for those with acute myeloid leukemias (AMLs); 3 management of Hodgkin lymphoma;4 and psychosocial challenges and health-related quality of life (QoL) in AYAs with hematologic malignancies.5
In the introduction to the series, Jorge Cortes, MD, an assistant editor on the journal, wrote that hematologic malignancies in AYAs “represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population.”6
He noted, however, that “not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs often centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults,” noted Dr Cortes, professor and chair of the chronic myeloid leukemia section in the department of leukemia at the University of Texas MD Anderson Cancer Center, Houston.
Therapeutic options: pediatric or adult protocols?
In their article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15 through 20 years is supported by findings from numerous studies. In young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” because they have been shown to significantly improve outcomes, with long-term survival rates nearing 70%.1 Patients in these age groups require specific programs that factor in access to care and to trials, an increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
However, Kristen O’Dwyer, MD, and colleagues, argue in an article on AML treatment in AYAs that neither the pediatric nor adult approaches are ideally suited for AYAs because of the “distinguishing characteristics of AYAs with AML.” Rather, they conclude that AYA-specific approaches merit consideration.3
Similarly, Kieron Dunleavy, MD, and Thomas G Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that there is a “remarkable divide” in the treatment of patients younger than 18 years with lymphoma compared with their young adult counterparts, and that it underscores the need for collaboration in developing consensus regarding treatment of AYAs.2
Clinical setting: pediatric or adult?
Consideration is also being given to the clinical setting in which AYA patients receive their treatment. Lori Muffly, MD, MS, and colleagues have reported that survival was superior for AYA patients with ALL who were treated in pediatric cancer settings,7 and other researchers have reported similar findings.
However, those improved outcomes in the pediatric setting might be offset by a higher use of resources and therefore higher costs, based on recent findings in a Canadian study by Paul C Nathan, MD, and colleagues.8 Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the authors found that the cost of care was higher when treatment took place in a pediatric setting compared with in an adult institution, and that it was driven in part by higher hospitalization rates and longer hospital stays. These findings were true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
In an accompanying editorial, Helen M Parsons, PhD, and her co-authors wrote that adolescents who receive treatment in the pediatric setting “tended to seek more [emergency department (ED)] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship.9 They pointed out that the findings of higher inpatient days in the pediatric setting was not surprising given that induction therapies for pediatric ALL tend to be more complex and intensive than therapies commonly used in adults with ALL, and that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase,” they wrote, adding that further investigation was needed on this topic to better understand those trends. “The finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.” 9
The authors of the editorial emphasized that because of the differences in health care delivery and payment structures between the United States and Canada, where the Nathan study was done, it was important that similar studies are done in the United States to confirm these findings.
Disease and developmental biology
As Dr Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and that they may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr O'Dwyer and her colleagues noted,3 citing a recent European study of 5,564 patients with de novo AML that showed that the frequency of favorable cytogenetics was low in infants (13.7%), increased in children (25%) and young adults (44%), and decreased again in middle age and older patients.10
“Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr O'Dwyer and her colleagues wrote.3 It was also becoming more apparent that age influences the presence of AML-related molecular abnormalities, and recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr Boissel and Dr Baruchel also noted in their report that light was finally being shed on the “black hole” of understanding ALL biology in AYAs, and research has shown that there is a continuum between childhood and adult ALL.1 They concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, NC, said in an interview.
These patients have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, and in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added. “Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... these are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so.”
In a 2014 study, Dr Salsman and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life.11 The latter finding was surprising and highlights the importance of the social dimension in the lives of AYAs. “Patient after patient will say ‘I found out who my real friends are,’ ” he said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr Salsman and his colleagues are using those findings to develop interventions that can maximize self-care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves. For example, a randomized controlled pilot study that incorporates social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions is underway.
Another intervention targets emotional well-being through the use of web-based tools to increase positive affect. A proof-of-concept study showed that the approach was feasible and well received, and a larger-scale randomized controlled trial is being planned, he said.
Dr Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital. It was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr Cortes noted a paucity of clinical trials specifically designed for this population. “At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, describing AYA enrollment in clinical trials in cancer as “suboptimal at best.”6
Dr Salsman said these dismal enrolment numbers could in part be related to treatment setting. Data suggest that most AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers where the bulk of research is being done, he explained.
Dr Hanna agreed that more research involving AYAs was needed as is a better understanding of why enrollment is so much lower in this population. He pointed out that in 2017 the American Society of Clinical Oncology and Friends of Cancer Research released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.12 The organizations said that individuals aged 12 years and older should routinely be included in such trials because their drug metabolism is similar to adults, and inclusion of younger patients may also be appropriate if they are part of the population affected by the disease, depending on specific disease biology, action of the drug, and available safety information.
Officials at the Food and Drug Administration are considering that possibility, Dr Hanna said.
Dr Salsman added there has been an increase in recent years in the attention paid to disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups. For example, about 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population;13 the Institute of Medicine held a forum on the care of AYAs with cancer;14 and the National Cancer Institute held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology,15 he noted.
Dr Hanna added that “scientific groups such as Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now. One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically co-ordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.16
Next steps
Among the recommendations from authors in the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams and the involvement of multidisciplinary teams in care for this population.
Many centers are already working on models for collaborative care, Dr Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in helping clinical and supportive caregivers and their AYA patients “have a shared vision” as they work to maximize improvements in outcomes.
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr Hanna said. In the case of the community-powered advocacy organization Critical Mass, members have succeeded in getting lawmakers to introduce a bill in the US House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
1. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood. 2018;132:351-361.
2. Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375.
3. O’Dwyer K, Freyer DR, Horan JT. Treatment strategies for adolescent and young adult patients with acute myeloid leukemia. Blood. 2018;132:362-368.
4. Flerlage JE, Metzger ML, Bhakta N. The management of Hodgkin lymphoma in adolescents and young adults: burden of disease or burden of choice? Blood. 2018;132:376-384.
5. Husson O, Huijgens PC, van der Graaf WTA. Psychosocial challenges and health-related quality of life of adolescents and young adults with hematologic malignancies. Blood. 2018;132:385-392.
6. Cortes J. Introduction to a review series on adolescent and young adult malignant hematology. Blood. 2018;132:345-346.
7. Muffly L, Alvarez E, Lichtensztajn D, Abrahão R, Gomez SL, Keegan T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv. 2018;2(8):895-903.
8. Nathan PC, Bremner KE, Liu N, et al. Resource utilization and costs in adolescents treated for cancer in pediatric vs adult institutions. J Natl Cancer Inst. July 19, 2018. [Epub ahead of print.]
9. Parsons HM, Muffly L, Alvarez EM, Keegan THM. Does treatment setting matter? Evaluating resource utilization for adolescents treated in pediatric vs adult cancer institutions. https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djy123/5056313?searchresult=1. Published July 19, 2018. Last accessed October 12, 2018.
10. Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821-3830.
11. Salsman JM, Garcia SF, Yanez B, et al. Physical, emotional, and social health differences between posttreatment young adults with cancer and matched healthy controls. Cancer. 2014;120(15):2247-2254.
12. Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and Friends of Cancer Research joint research statement. J Clin Oncol. 2017;35(33):3737-3744.
13. Freyer DR, Seibel NL. The clinical trials gap for adolescents and young adults with cancer: recent progress and conceptual framework for continued research. Curr Pediatr Rep. Published online February 18, 2015. DOI 10.1007/s40124-015-0075-y.
14. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
15. Wilder Smith A, Seibel NL, Lewis DR, et al. Next steps for adolescent and young adult oncology workshop: An update on progress and recommendations for the future. Cancer. 2016;122(7):988-999.
16. Keegan THM, Ries LAG, Barr RD, et al. Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer. 2016;122(7):1009-1016.
1. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood. 2018;132:351-361.
2. Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375.
3. O’Dwyer K, Freyer DR, Horan JT. Treatment strategies for adolescent and young adult patients with acute myeloid leukemia. Blood. 2018;132:362-368.
4. Flerlage JE, Metzger ML, Bhakta N. The management of Hodgkin lymphoma in adolescents and young adults: burden of disease or burden of choice? Blood. 2018;132:376-384.
5. Husson O, Huijgens PC, van der Graaf WTA. Psychosocial challenges and health-related quality of life of adolescents and young adults with hematologic malignancies. Blood. 2018;132:385-392.
6. Cortes J. Introduction to a review series on adolescent and young adult malignant hematology. Blood. 2018;132:345-346.
7. Muffly L, Alvarez E, Lichtensztajn D, Abrahão R, Gomez SL, Keegan T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv. 2018;2(8):895-903.
8. Nathan PC, Bremner KE, Liu N, et al. Resource utilization and costs in adolescents treated for cancer in pediatric vs adult institutions. J Natl Cancer Inst. July 19, 2018. [Epub ahead of print.]
9. Parsons HM, Muffly L, Alvarez EM, Keegan THM. Does treatment setting matter? Evaluating resource utilization for adolescents treated in pediatric vs adult cancer institutions. https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djy123/5056313?searchresult=1. Published July 19, 2018. Last accessed October 12, 2018.
10. Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821-3830.
11. Salsman JM, Garcia SF, Yanez B, et al. Physical, emotional, and social health differences between posttreatment young adults with cancer and matched healthy controls. Cancer. 2014;120(15):2247-2254.
12. Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and Friends of Cancer Research joint research statement. J Clin Oncol. 2017;35(33):3737-3744.
13. Freyer DR, Seibel NL. The clinical trials gap for adolescents and young adults with cancer: recent progress and conceptual framework for continued research. Curr Pediatr Rep. Published online February 18, 2015. DOI 10.1007/s40124-015-0075-y.
14. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
15. Wilder Smith A, Seibel NL, Lewis DR, et al. Next steps for adolescent and young adult oncology workshop: An update on progress and recommendations for the future. Cancer. 2016;122(7):988-999.
16. Keegan THM, Ries LAG, Barr RD, et al. Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer. 2016;122(7):1009-1016.