User login
Bipolar and seizure medication linked with serious immune system reaction
The Food and Drug Administration has issued a warning that the seizure and bipolar medication Lamictal (lamotrigine) can cause a rare but potentially life-threatening immune response.
This life-threatening immune response, known as hemophagocytic lymphohistiocytosis (HLH), causes an uncontrolled immune response and can present as a persistent fever greater than 101° F. HLH can also lead to severe issues with blood cells and organs like the liver, kidneys, and lungs.
Lamotrigine is commonly used as a maintenance treatment for patients with bipolar disorder to help manage depression and mood episodes of mania and hypomania. Patients who abruptly stop taking lamotrigine before talking to their physician can suffer seizures, as well as new or worsening mental health issues.
The FDA is recommending that health care providers be aware of the connection between lamotrigine and HLH and be able to recognize and treat the immune response promptly to improve outcomes and decrease mortality. This can be difficult because of the nonspecific nature of HLH symptoms like fever and rash. HLH is commonly confused with another immune-related reaction known as Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS). Patients should be evaluated if they develop fever or rash and immediately discontinue use of lamotrigine if HLH is suspected.
The basis for the new warning is eight cases worldwide of confirmed or suspected HLH involving “reasonable evidence that lamotrigine was the cause of HLH ... based on the timing of events and the order in which they occurred,” the agency said, noting that this number includes only reports submitted to the FDA and found in the medical literature during the 24-year approval history of the drug, so there are likely additional cases about which we are unaware. The eight patients were all hospitalized and received drug and other medical treatments, with one dying.
HLH can be diagnosed if a patient has at least five of the following eight signs or symptoms: fever and rash; enlarged spleen; cytopenias; elevated blood triglycerides and high levels of ferritin or low levels of fibrinogen; hemophagocytosis confirmed via bone marrow, spleen, or lymph node biopsy; decreased or absent natural killer (NK) cell activity; and elevated levels of CD25 in the blood.
Other signs and symptoms may include: enlarged liver, swollen lymph nodes, yellowing of the skin or eyes, unusual bleeding, disturbances in vision, and trouble walking.
The FDA encourages health care providers and patients to report adverse events to the FDA’s MedWatch Safety Information and Adverse Event Reporting Program.
The Food and Drug Administration has issued a warning that the seizure and bipolar medication Lamictal (lamotrigine) can cause a rare but potentially life-threatening immune response.
This life-threatening immune response, known as hemophagocytic lymphohistiocytosis (HLH), causes an uncontrolled immune response and can present as a persistent fever greater than 101° F. HLH can also lead to severe issues with blood cells and organs like the liver, kidneys, and lungs.
Lamotrigine is commonly used as a maintenance treatment for patients with bipolar disorder to help manage depression and mood episodes of mania and hypomania. Patients who abruptly stop taking lamotrigine before talking to their physician can suffer seizures, as well as new or worsening mental health issues.
The FDA is recommending that health care providers be aware of the connection between lamotrigine and HLH and be able to recognize and treat the immune response promptly to improve outcomes and decrease mortality. This can be difficult because of the nonspecific nature of HLH symptoms like fever and rash. HLH is commonly confused with another immune-related reaction known as Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS). Patients should be evaluated if they develop fever or rash and immediately discontinue use of lamotrigine if HLH is suspected.
The basis for the new warning is eight cases worldwide of confirmed or suspected HLH involving “reasonable evidence that lamotrigine was the cause of HLH ... based on the timing of events and the order in which they occurred,” the agency said, noting that this number includes only reports submitted to the FDA and found in the medical literature during the 24-year approval history of the drug, so there are likely additional cases about which we are unaware. The eight patients were all hospitalized and received drug and other medical treatments, with one dying.
HLH can be diagnosed if a patient has at least five of the following eight signs or symptoms: fever and rash; enlarged spleen; cytopenias; elevated blood triglycerides and high levels of ferritin or low levels of fibrinogen; hemophagocytosis confirmed via bone marrow, spleen, or lymph node biopsy; decreased or absent natural killer (NK) cell activity; and elevated levels of CD25 in the blood.
Other signs and symptoms may include: enlarged liver, swollen lymph nodes, yellowing of the skin or eyes, unusual bleeding, disturbances in vision, and trouble walking.
The FDA encourages health care providers and patients to report adverse events to the FDA’s MedWatch Safety Information and Adverse Event Reporting Program.
The Food and Drug Administration has issued a warning that the seizure and bipolar medication Lamictal (lamotrigine) can cause a rare but potentially life-threatening immune response.
This life-threatening immune response, known as hemophagocytic lymphohistiocytosis (HLH), causes an uncontrolled immune response and can present as a persistent fever greater than 101° F. HLH can also lead to severe issues with blood cells and organs like the liver, kidneys, and lungs.
Lamotrigine is commonly used as a maintenance treatment for patients with bipolar disorder to help manage depression and mood episodes of mania and hypomania. Patients who abruptly stop taking lamotrigine before talking to their physician can suffer seizures, as well as new or worsening mental health issues.
The FDA is recommending that health care providers be aware of the connection between lamotrigine and HLH and be able to recognize and treat the immune response promptly to improve outcomes and decrease mortality. This can be difficult because of the nonspecific nature of HLH symptoms like fever and rash. HLH is commonly confused with another immune-related reaction known as Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS). Patients should be evaluated if they develop fever or rash and immediately discontinue use of lamotrigine if HLH is suspected.
The basis for the new warning is eight cases worldwide of confirmed or suspected HLH involving “reasonable evidence that lamotrigine was the cause of HLH ... based on the timing of events and the order in which they occurred,” the agency said, noting that this number includes only reports submitted to the FDA and found in the medical literature during the 24-year approval history of the drug, so there are likely additional cases about which we are unaware. The eight patients were all hospitalized and received drug and other medical treatments, with one dying.
HLH can be diagnosed if a patient has at least five of the following eight signs or symptoms: fever and rash; enlarged spleen; cytopenias; elevated blood triglycerides and high levels of ferritin or low levels of fibrinogen; hemophagocytosis confirmed via bone marrow, spleen, or lymph node biopsy; decreased or absent natural killer (NK) cell activity; and elevated levels of CD25 in the blood.
Other signs and symptoms may include: enlarged liver, swollen lymph nodes, yellowing of the skin or eyes, unusual bleeding, disturbances in vision, and trouble walking.
The FDA encourages health care providers and patients to report adverse events to the FDA’s MedWatch Safety Information and Adverse Event Reporting Program.
Suicide on the minds of many Utah teens
according to the Centers for Disease Control and Prevention.
Prevalence of suicidal ideation in 2015 was significantly higher among female students (25.5% vs. 13.7% for males), nonwhite students (23.4% vs. 18.7% for whites), those who were less religious (27.4% vs. 16.1% for religious students), and nonmembers of the Church of Latter Day Saints (27.1% vs. 15.3% for Mormons), Marissa L. Zwald, PhD, of the CDC’s Epidemic Intelligence Service and her associates reported in the Morbidity and Mortality Weekly Report.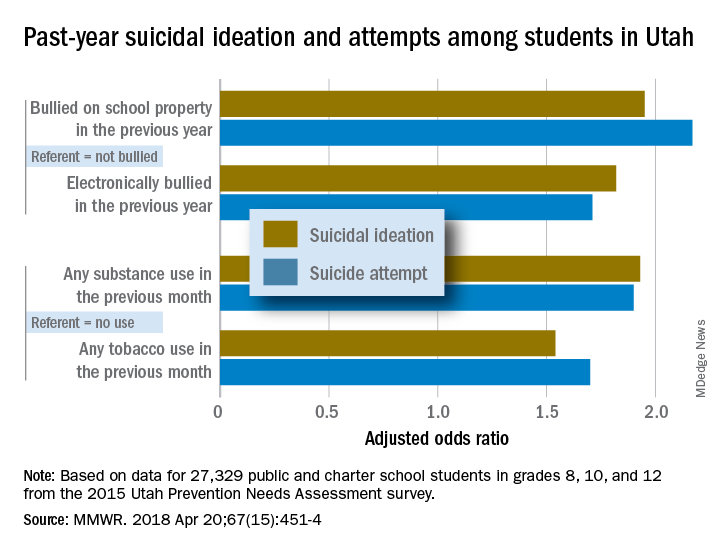
Risk factors included being bullied on school property in the previous year (adjusted odds ratios 1.95 for suicidal ideation and 2.17 for a suicide attempt, compared with those who were not bullied) and being electronically bullied (AOR of 1.82 for suicidal ideation and 1.71 for suicide attempt). Students who reported illicit substance use in the past month were more likely than nonusers to have suicidal ideation (AOR, 1.93) and to make a suicide attempt (AOR, 1.9); past-month use of tobacco, including e-cigarettes, increased risk, compared with no use, with an adjusted odds ratios of 1.54 for suicidal ideation and 1.7 for an attempt, the investigators reported.
They also looked at a number of possible protective factors, with a supportive family environment showing significance for both suicidal ideation and suicide attempts; the same was not true for prosocial behaviors or supportive community, school, or peer environments. “Possible prevention strategies to consider could include integrating family members and the home setting into existing or new interventions and identifying and addressing the needs of youths exhibiting risk factors identified in this investigation,” Dr. Zwald and her associates wrote.
SOURCE: Zwald ML et al. MMWR. 2018 Apr 20;67(15):451-4.
according to the Centers for Disease Control and Prevention.
Prevalence of suicidal ideation in 2015 was significantly higher among female students (25.5% vs. 13.7% for males), nonwhite students (23.4% vs. 18.7% for whites), those who were less religious (27.4% vs. 16.1% for religious students), and nonmembers of the Church of Latter Day Saints (27.1% vs. 15.3% for Mormons), Marissa L. Zwald, PhD, of the CDC’s Epidemic Intelligence Service and her associates reported in the Morbidity and Mortality Weekly Report.
Risk factors included being bullied on school property in the previous year (adjusted odds ratios 1.95 for suicidal ideation and 2.17 for a suicide attempt, compared with those who were not bullied) and being electronically bullied (AOR of 1.82 for suicidal ideation and 1.71 for suicide attempt). Students who reported illicit substance use in the past month were more likely than nonusers to have suicidal ideation (AOR, 1.93) and to make a suicide attempt (AOR, 1.9); past-month use of tobacco, including e-cigarettes, increased risk, compared with no use, with an adjusted odds ratios of 1.54 for suicidal ideation and 1.7 for an attempt, the investigators reported.
They also looked at a number of possible protective factors, with a supportive family environment showing significance for both suicidal ideation and suicide attempts; the same was not true for prosocial behaviors or supportive community, school, or peer environments. “Possible prevention strategies to consider could include integrating family members and the home setting into existing or new interventions and identifying and addressing the needs of youths exhibiting risk factors identified in this investigation,” Dr. Zwald and her associates wrote.
SOURCE: Zwald ML et al. MMWR. 2018 Apr 20;67(15):451-4.
according to the Centers for Disease Control and Prevention.
Prevalence of suicidal ideation in 2015 was significantly higher among female students (25.5% vs. 13.7% for males), nonwhite students (23.4% vs. 18.7% for whites), those who were less religious (27.4% vs. 16.1% for religious students), and nonmembers of the Church of Latter Day Saints (27.1% vs. 15.3% for Mormons), Marissa L. Zwald, PhD, of the CDC’s Epidemic Intelligence Service and her associates reported in the Morbidity and Mortality Weekly Report.
Risk factors included being bullied on school property in the previous year (adjusted odds ratios 1.95 for suicidal ideation and 2.17 for a suicide attempt, compared with those who were not bullied) and being electronically bullied (AOR of 1.82 for suicidal ideation and 1.71 for suicide attempt). Students who reported illicit substance use in the past month were more likely than nonusers to have suicidal ideation (AOR, 1.93) and to make a suicide attempt (AOR, 1.9); past-month use of tobacco, including e-cigarettes, increased risk, compared with no use, with an adjusted odds ratios of 1.54 for suicidal ideation and 1.7 for an attempt, the investigators reported.
They also looked at a number of possible protective factors, with a supportive family environment showing significance for both suicidal ideation and suicide attempts; the same was not true for prosocial behaviors or supportive community, school, or peer environments. “Possible prevention strategies to consider could include integrating family members and the home setting into existing or new interventions and identifying and addressing the needs of youths exhibiting risk factors identified in this investigation,” Dr. Zwald and her associates wrote.
SOURCE: Zwald ML et al. MMWR. 2018 Apr 20;67(15):451-4.
FROM MMWR
CDC: Marijuana use may spur industries to rethink current policies
according to a report from the Centers for Disease Control and Prevention.
As legal recreational marijuana use continues to expand across the United States, marijuana has been shown to inhibit certain motor skills, which has made it crucial for employers to have a better understanding how best to approach safety training, according to the study published in the Morbidity and Mortality Weekly Report.
“We have been looking at some of the behavioral risk factors associated with marijuana legalization and were interested in the data broken down by industry and occupation, which could help employers make decisions on any kind of safety and drug use policies in the workplace,” lead author Roberta Smith, RN, occupational health program manager at the Colorado Department of Public Health and Environment, said in a interview. “This doesn’t necessarily imply any impairment on the job, but these data will reinforce current policies and encourage employers to go back and see how their work places operate and make sure their employees are good to staff.”
To examine current marijuana use by working adults and the industries and occupations in which they are employed, the Colorado Department of Public Health and Environment analyzed data from the state’s Behavioral Risk Factor Surveillance System regarding current marijuana use (at least 1 day during the preceding 30 days) among 10,169 persons who had responded to the current marijuana use question.
Participants were over the age of 21 years old and were either employed at the time of the survey or had been unemployed for less than a year.
In the overall population, 14.6% reported using marijuana, with higher prevalence in men (17.2%) and those 18-25 years old (29.6%).
By industry, accommodation and food service workers reported the highest rate of use at 30.1%, followed by those in the arts, entertainment, and recreation industry with 28.3%.
While the highest percentage of reported users came from food services and entertainment, safety-sensitive jobs like construction saw rates as high as 20% when not adjusted for age, according to investigators,
Ms. Smith and her colleagues found use varied across safety-sensitive industries, with high rates in construction (19.7%), waste management (18.8%), and manufacturing (16.3%) that were above the total population prevalence. Meanwhile, mining, health care, and transportation were all 10% or lower, which may be because of more regular drug testing.
“It might be reassuring that our health care professionals are on the lower end of use,” said Ms. Smith. “Having worked in a medical facility, I know drug policies for workers are clear and employees are aware of drug testing and when it will occur.”
While the health care industry reported low usage, 15.8% of health care support management workers, such as x-ray technicians, reported marijuana use.
When adjusted for age, the prevalence among workers in certain industries – such as food services, arts, and construction industry – saw significant decreases, which lead investigators to conclude younger employees would be a key target for more marijuana-related drug-use policies.
Ms. Smith and her colleagues recognized the population used may not be a full representation of all Colorado employees and that missing data regarding how often individuals used marijuana within 30 days could offer different considerations for workplace impairment.
Investigators also noted the data may have been influenced by self-reported bias or recording errors by survey takers.
Moving forward, Ms. Smith and her colleagues are interested in how this data might shift as more states conduct their own research and as marijuana policy changes.
This report was funded by the CDC, and investigators report no relevant financial disclosures.
SOURCE: Smith R et al. MMWR. 2018 Apr 13;67(14):409-13.
according to a report from the Centers for Disease Control and Prevention.
As legal recreational marijuana use continues to expand across the United States, marijuana has been shown to inhibit certain motor skills, which has made it crucial for employers to have a better understanding how best to approach safety training, according to the study published in the Morbidity and Mortality Weekly Report.
“We have been looking at some of the behavioral risk factors associated with marijuana legalization and were interested in the data broken down by industry and occupation, which could help employers make decisions on any kind of safety and drug use policies in the workplace,” lead author Roberta Smith, RN, occupational health program manager at the Colorado Department of Public Health and Environment, said in a interview. “This doesn’t necessarily imply any impairment on the job, but these data will reinforce current policies and encourage employers to go back and see how their work places operate and make sure their employees are good to staff.”
To examine current marijuana use by working adults and the industries and occupations in which they are employed, the Colorado Department of Public Health and Environment analyzed data from the state’s Behavioral Risk Factor Surveillance System regarding current marijuana use (at least 1 day during the preceding 30 days) among 10,169 persons who had responded to the current marijuana use question.
Participants were over the age of 21 years old and were either employed at the time of the survey or had been unemployed for less than a year.
In the overall population, 14.6% reported using marijuana, with higher prevalence in men (17.2%) and those 18-25 years old (29.6%).
By industry, accommodation and food service workers reported the highest rate of use at 30.1%, followed by those in the arts, entertainment, and recreation industry with 28.3%.
While the highest percentage of reported users came from food services and entertainment, safety-sensitive jobs like construction saw rates as high as 20% when not adjusted for age, according to investigators,
Ms. Smith and her colleagues found use varied across safety-sensitive industries, with high rates in construction (19.7%), waste management (18.8%), and manufacturing (16.3%) that were above the total population prevalence. Meanwhile, mining, health care, and transportation were all 10% or lower, which may be because of more regular drug testing.
“It might be reassuring that our health care professionals are on the lower end of use,” said Ms. Smith. “Having worked in a medical facility, I know drug policies for workers are clear and employees are aware of drug testing and when it will occur.”
While the health care industry reported low usage, 15.8% of health care support management workers, such as x-ray technicians, reported marijuana use.
When adjusted for age, the prevalence among workers in certain industries – such as food services, arts, and construction industry – saw significant decreases, which lead investigators to conclude younger employees would be a key target for more marijuana-related drug-use policies.
Ms. Smith and her colleagues recognized the population used may not be a full representation of all Colorado employees and that missing data regarding how often individuals used marijuana within 30 days could offer different considerations for workplace impairment.
Investigators also noted the data may have been influenced by self-reported bias or recording errors by survey takers.
Moving forward, Ms. Smith and her colleagues are interested in how this data might shift as more states conduct their own research and as marijuana policy changes.
This report was funded by the CDC, and investigators report no relevant financial disclosures.
SOURCE: Smith R et al. MMWR. 2018 Apr 13;67(14):409-13.
according to a report from the Centers for Disease Control and Prevention.
As legal recreational marijuana use continues to expand across the United States, marijuana has been shown to inhibit certain motor skills, which has made it crucial for employers to have a better understanding how best to approach safety training, according to the study published in the Morbidity and Mortality Weekly Report.
“We have been looking at some of the behavioral risk factors associated with marijuana legalization and were interested in the data broken down by industry and occupation, which could help employers make decisions on any kind of safety and drug use policies in the workplace,” lead author Roberta Smith, RN, occupational health program manager at the Colorado Department of Public Health and Environment, said in a interview. “This doesn’t necessarily imply any impairment on the job, but these data will reinforce current policies and encourage employers to go back and see how their work places operate and make sure their employees are good to staff.”
To examine current marijuana use by working adults and the industries and occupations in which they are employed, the Colorado Department of Public Health and Environment analyzed data from the state’s Behavioral Risk Factor Surveillance System regarding current marijuana use (at least 1 day during the preceding 30 days) among 10,169 persons who had responded to the current marijuana use question.
Participants were over the age of 21 years old and were either employed at the time of the survey or had been unemployed for less than a year.
In the overall population, 14.6% reported using marijuana, with higher prevalence in men (17.2%) and those 18-25 years old (29.6%).
By industry, accommodation and food service workers reported the highest rate of use at 30.1%, followed by those in the arts, entertainment, and recreation industry with 28.3%.
While the highest percentage of reported users came from food services and entertainment, safety-sensitive jobs like construction saw rates as high as 20% when not adjusted for age, according to investigators,
Ms. Smith and her colleagues found use varied across safety-sensitive industries, with high rates in construction (19.7%), waste management (18.8%), and manufacturing (16.3%) that were above the total population prevalence. Meanwhile, mining, health care, and transportation were all 10% or lower, which may be because of more regular drug testing.
“It might be reassuring that our health care professionals are on the lower end of use,” said Ms. Smith. “Having worked in a medical facility, I know drug policies for workers are clear and employees are aware of drug testing and when it will occur.”
While the health care industry reported low usage, 15.8% of health care support management workers, such as x-ray technicians, reported marijuana use.
When adjusted for age, the prevalence among workers in certain industries – such as food services, arts, and construction industry – saw significant decreases, which lead investigators to conclude younger employees would be a key target for more marijuana-related drug-use policies.
Ms. Smith and her colleagues recognized the population used may not be a full representation of all Colorado employees and that missing data regarding how often individuals used marijuana within 30 days could offer different considerations for workplace impairment.
Investigators also noted the data may have been influenced by self-reported bias or recording errors by survey takers.
Moving forward, Ms. Smith and her colleagues are interested in how this data might shift as more states conduct their own research and as marijuana policy changes.
This report was funded by the CDC, and investigators report no relevant financial disclosures.
SOURCE: Smith R et al. MMWR. 2018 Apr 13;67(14):409-13.
FROM MMWR
FDA grants priority review of follicular lymphoma drug
by the Food and Drug Administration.
The biopharmaceutical company Verastem is seeking full approval for duvelisib for the treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and accelerated approval for the treatment of relapsed/refractory follicular lymphoma. The FDA has set Oct. 5, 2018, as the target action date, according to Verastem.
by the Food and Drug Administration.
The biopharmaceutical company Verastem is seeking full approval for duvelisib for the treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and accelerated approval for the treatment of relapsed/refractory follicular lymphoma. The FDA has set Oct. 5, 2018, as the target action date, according to Verastem.
by the Food and Drug Administration.
The biopharmaceutical company Verastem is seeking full approval for duvelisib for the treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and accelerated approval for the treatment of relapsed/refractory follicular lymphoma. The FDA has set Oct. 5, 2018, as the target action date, according to Verastem.
Asthma flourishing in its medical home
according to the Centers for Disease Control and Prevention.
Current asthma prevalence was 8.8% for adults aged 18 years and older who worked in health care and social assistance in 2011-2016, which put them above those in education services (8.2%); arts, entertainment, and recreation (8.1%); accommodation and food services (7.7%); and finance and insurance (7.5%). The overall rate for all working adults was 6.8%, Jacek M. Mazurek, MD, PhD, and Girija Syamlal, MBBS, reported in the Morbidity and Mortality Weekly Report.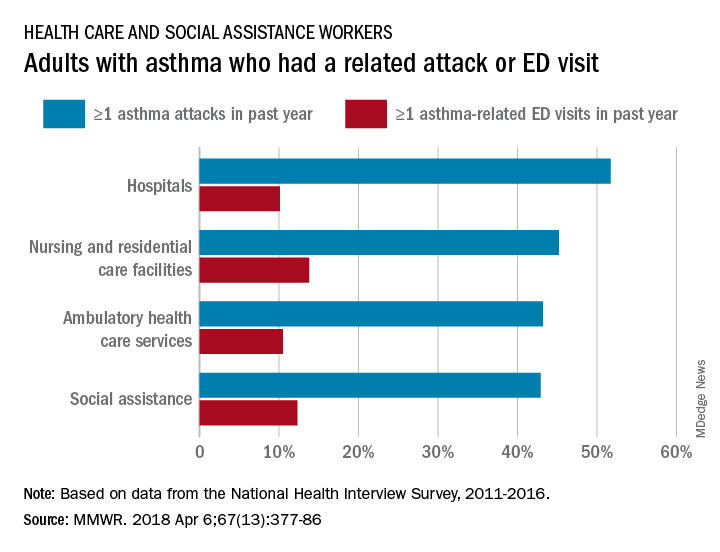
Among persons with asthma who were employed in health care and social assistance, 45.8% reported having at least one asthma attack in the previous year. Among the subgroups of the industry, those working in hospitals were highest with a 51.7% rate of past-year asthma attacks, followed by those working in nursing and residential care facilities at 45.2%, those working in ambulatory health care services at 43.2%, and those working in social assistance at 42.9%. The highest asthma attack rates among all industries were 57.3% for wood product manufacturing and 56.7% for plastics and rubber products manufacturing, the investigators said, based on data from the National Health Interview Survey.
Asthma-related visits to the emergency department in the past year were much less common for those in health care – 11.3% overall – and followed a pattern different from asthma attacks. Those working in nursing and residential care facilities were highest at 13.8%, with those in social assistance at 12.3%, those in ambulatory care at 10.5%, and those in hospitals the lowest at 10.1%. The highest ED-visit rate for any industry, 22.9%, was for workers in private households, said Dr. Mazurek and Dr. Syamlal, both of the respiratory health division at the CDC’s National Institute for Occupational Safety and Health in Morgantown, W.Va.
SOURCE: Mazurek JM, Syamlal G. MMWR. 2018 Apr 6;67(13):377-86.
according to the Centers for Disease Control and Prevention.
Current asthma prevalence was 8.8% for adults aged 18 years and older who worked in health care and social assistance in 2011-2016, which put them above those in education services (8.2%); arts, entertainment, and recreation (8.1%); accommodation and food services (7.7%); and finance and insurance (7.5%). The overall rate for all working adults was 6.8%, Jacek M. Mazurek, MD, PhD, and Girija Syamlal, MBBS, reported in the Morbidity and Mortality Weekly Report.
Among persons with asthma who were employed in health care and social assistance, 45.8% reported having at least one asthma attack in the previous year. Among the subgroups of the industry, those working in hospitals were highest with a 51.7% rate of past-year asthma attacks, followed by those working in nursing and residential care facilities at 45.2%, those working in ambulatory health care services at 43.2%, and those working in social assistance at 42.9%. The highest asthma attack rates among all industries were 57.3% for wood product manufacturing and 56.7% for plastics and rubber products manufacturing, the investigators said, based on data from the National Health Interview Survey.
Asthma-related visits to the emergency department in the past year were much less common for those in health care – 11.3% overall – and followed a pattern different from asthma attacks. Those working in nursing and residential care facilities were highest at 13.8%, with those in social assistance at 12.3%, those in ambulatory care at 10.5%, and those in hospitals the lowest at 10.1%. The highest ED-visit rate for any industry, 22.9%, was for workers in private households, said Dr. Mazurek and Dr. Syamlal, both of the respiratory health division at the CDC’s National Institute for Occupational Safety and Health in Morgantown, W.Va.
SOURCE: Mazurek JM, Syamlal G. MMWR. 2018 Apr 6;67(13):377-86.
according to the Centers for Disease Control and Prevention.
Current asthma prevalence was 8.8% for adults aged 18 years and older who worked in health care and social assistance in 2011-2016, which put them above those in education services (8.2%); arts, entertainment, and recreation (8.1%); accommodation and food services (7.7%); and finance and insurance (7.5%). The overall rate for all working adults was 6.8%, Jacek M. Mazurek, MD, PhD, and Girija Syamlal, MBBS, reported in the Morbidity and Mortality Weekly Report.
Among persons with asthma who were employed in health care and social assistance, 45.8% reported having at least one asthma attack in the previous year. Among the subgroups of the industry, those working in hospitals were highest with a 51.7% rate of past-year asthma attacks, followed by those working in nursing and residential care facilities at 45.2%, those working in ambulatory health care services at 43.2%, and those working in social assistance at 42.9%. The highest asthma attack rates among all industries were 57.3% for wood product manufacturing and 56.7% for plastics and rubber products manufacturing, the investigators said, based on data from the National Health Interview Survey.
Asthma-related visits to the emergency department in the past year were much less common for those in health care – 11.3% overall – and followed a pattern different from asthma attacks. Those working in nursing and residential care facilities were highest at 13.8%, with those in social assistance at 12.3%, those in ambulatory care at 10.5%, and those in hospitals the lowest at 10.1%. The highest ED-visit rate for any industry, 22.9%, was for workers in private households, said Dr. Mazurek and Dr. Syamlal, both of the respiratory health division at the CDC’s National Institute for Occupational Safety and Health in Morgantown, W.Va.
SOURCE: Mazurek JM, Syamlal G. MMWR. 2018 Apr 6;67(13):377-86.
FROM MMWR
Pregnant women in clinical trials: FDA questions how to include them
Pregnant women are rarely included in clinical drug trials, creating a significant and potentially dangerous gap in knowledge. Now, a new draft guidance from the Food and Drug Administration broadens the discussion about these trials, suggesting issues to consider – including ethics and risks – when testing medications in pregnant women.
“The guidance opens the possibility of ethical conduct of trials in pregnant women but carefully lays out the caveats to be considered,” Christina Chambers, PhD, a perinatal epidemiologist at the University of California, San Diego, said in an interview. “With proper planning and thoughtful consultation with the relevant experts, this change in regulatory limitations will benefit pregnant women and their children.”

Attitudes have evolved toward more acceptance of including pregnant women in drug trials, according to a 2015 committee opinion from the American College of Obstetricians and Gynecologists. Still, “concerns about the potential for pregnancy in research trial participants have led to practices involving overly burdensome contraception requirements,” the opinion states. “Although changes have been made to encourage and recruit more women into research studies, a gap still exists in the available data on health and disease in women, including those who are pregnant” (Obstet Gynecol 2015;126:e100-7).
[polldaddy:9979976]
The draft guidance, released April 6 by the FDA, is “intended to advance scientific research in pregnant women, and discusses issues that should be considered within the framework of human subject protection regulations,” according to posting comments in the Federal Register.
The draft notes that in some cases, the lack of data about drugs may harm pregnant women and their fetuses by leading physicians to be fearful about prescribing medication. Conversely, physicians and pregnant women are often in the dark about the risks and benefits of medications that are prescribed and used, according to the draft.
In terms of research going forward, the guidance says “development of accessible treatment options for the pregnant population is a significant public health issue.”
The guidance, which recommends that clinical trial sponsors consider enlisting ethicists to take part in drug development program, offers these guidelines, among others, to drugmakers:
- It is “ethically justifiable” to include pregnant women in clinical trials under specific circumstances. “Sponsors should consider meeting with the appropriate FDA review division early in the development phase to discuss when and how to include pregnant women in the drug development plan. These discussions should involve FDA experts in bioethics and maternal health.”
- “Pregnant women can be enrolled in clinical trials that involve greater than minimal risk to the fetuses if the trials offer the potential for direct clinical benefit to the enrolled pregnant women and/or their fetuses.”
- A new pregnancy during a randomized, blinded clinical trial should prompt unblinding “so that counseling may be offered based on whether the fetus has been exposed to the investigational drug, placebo, or control.”
- The pregnant woman may continue the trial if potential benefits outweigh the risks.
- In general, pregnant women should not be enrolled in phase 1 and phase 2 clinical trials. Instead, those trials should be completed first “in a nonpregnant population that include females of reproductive potential.”
- Several types of events may call for the cessation of a clinical trial that includes pregnant women, such as serious maternal or fetal adverse events.

The draft guidance should take note of the fact that birth defects often don’t appear for months or even longer, according to Gerald Briggs, BPharm, FCCP, clinical professor of pharmacy at the University of California, San Francisco. “Until first year of life or later, the babies need to be monitored,” he said in an interview.
Mr. Briggs, who led a 2015 report examining the role of pregnant women in phase 4 clinical drug trials, added that the document should take note of recommendations from clinical teratologists regarding the design of animal studies that should be performed prior to human trials (Am J Obstet Gynecol. 2015;213(6):810-5).
Comments on the draft guidance can be made at www.federalregister.gov and are due by June 8, 2018.
Dr. Chambers and Mr. Briggs reported no relevant disclosures.
Pregnant women are rarely included in clinical drug trials, creating a significant and potentially dangerous gap in knowledge. Now, a new draft guidance from the Food and Drug Administration broadens the discussion about these trials, suggesting issues to consider – including ethics and risks – when testing medications in pregnant women.
“The guidance opens the possibility of ethical conduct of trials in pregnant women but carefully lays out the caveats to be considered,” Christina Chambers, PhD, a perinatal epidemiologist at the University of California, San Diego, said in an interview. “With proper planning and thoughtful consultation with the relevant experts, this change in regulatory limitations will benefit pregnant women and their children.”
Attitudes have evolved toward more acceptance of including pregnant women in drug trials, according to a 2015 committee opinion from the American College of Obstetricians and Gynecologists. Still, “concerns about the potential for pregnancy in research trial participants have led to practices involving overly burdensome contraception requirements,” the opinion states. “Although changes have been made to encourage and recruit more women into research studies, a gap still exists in the available data on health and disease in women, including those who are pregnant” (Obstet Gynecol 2015;126:e100-7).
[polldaddy:9979976]
The draft guidance, released April 6 by the FDA, is “intended to advance scientific research in pregnant women, and discusses issues that should be considered within the framework of human subject protection regulations,” according to posting comments in the Federal Register.
The draft notes that in some cases, the lack of data about drugs may harm pregnant women and their fetuses by leading physicians to be fearful about prescribing medication. Conversely, physicians and pregnant women are often in the dark about the risks and benefits of medications that are prescribed and used, according to the draft.
In terms of research going forward, the guidance says “development of accessible treatment options for the pregnant population is a significant public health issue.”
The guidance, which recommends that clinical trial sponsors consider enlisting ethicists to take part in drug development program, offers these guidelines, among others, to drugmakers:
- It is “ethically justifiable” to include pregnant women in clinical trials under specific circumstances. “Sponsors should consider meeting with the appropriate FDA review division early in the development phase to discuss when and how to include pregnant women in the drug development plan. These discussions should involve FDA experts in bioethics and maternal health.”
- “Pregnant women can be enrolled in clinical trials that involve greater than minimal risk to the fetuses if the trials offer the potential for direct clinical benefit to the enrolled pregnant women and/or their fetuses.”
- A new pregnancy during a randomized, blinded clinical trial should prompt unblinding “so that counseling may be offered based on whether the fetus has been exposed to the investigational drug, placebo, or control.”
- The pregnant woman may continue the trial if potential benefits outweigh the risks.
- In general, pregnant women should not be enrolled in phase 1 and phase 2 clinical trials. Instead, those trials should be completed first “in a nonpregnant population that include females of reproductive potential.”
- Several types of events may call for the cessation of a clinical trial that includes pregnant women, such as serious maternal or fetal adverse events.
The draft guidance should take note of the fact that birth defects often don’t appear for months or even longer, according to Gerald Briggs, BPharm, FCCP, clinical professor of pharmacy at the University of California, San Francisco. “Until first year of life or later, the babies need to be monitored,” he said in an interview.
Mr. Briggs, who led a 2015 report examining the role of pregnant women in phase 4 clinical drug trials, added that the document should take note of recommendations from clinical teratologists regarding the design of animal studies that should be performed prior to human trials (Am J Obstet Gynecol. 2015;213(6):810-5).
Comments on the draft guidance can be made at www.federalregister.gov and are due by June 8, 2018.
Dr. Chambers and Mr. Briggs reported no relevant disclosures.
Pregnant women are rarely included in clinical drug trials, creating a significant and potentially dangerous gap in knowledge. Now, a new draft guidance from the Food and Drug Administration broadens the discussion about these trials, suggesting issues to consider – including ethics and risks – when testing medications in pregnant women.
“The guidance opens the possibility of ethical conduct of trials in pregnant women but carefully lays out the caveats to be considered,” Christina Chambers, PhD, a perinatal epidemiologist at the University of California, San Diego, said in an interview. “With proper planning and thoughtful consultation with the relevant experts, this change in regulatory limitations will benefit pregnant women and their children.”
Attitudes have evolved toward more acceptance of including pregnant women in drug trials, according to a 2015 committee opinion from the American College of Obstetricians and Gynecologists. Still, “concerns about the potential for pregnancy in research trial participants have led to practices involving overly burdensome contraception requirements,” the opinion states. “Although changes have been made to encourage and recruit more women into research studies, a gap still exists in the available data on health and disease in women, including those who are pregnant” (Obstet Gynecol 2015;126:e100-7).
[polldaddy:9979976]
The draft guidance, released April 6 by the FDA, is “intended to advance scientific research in pregnant women, and discusses issues that should be considered within the framework of human subject protection regulations,” according to posting comments in the Federal Register.
The draft notes that in some cases, the lack of data about drugs may harm pregnant women and their fetuses by leading physicians to be fearful about prescribing medication. Conversely, physicians and pregnant women are often in the dark about the risks and benefits of medications that are prescribed and used, according to the draft.
In terms of research going forward, the guidance says “development of accessible treatment options for the pregnant population is a significant public health issue.”
The guidance, which recommends that clinical trial sponsors consider enlisting ethicists to take part in drug development program, offers these guidelines, among others, to drugmakers:
- It is “ethically justifiable” to include pregnant women in clinical trials under specific circumstances. “Sponsors should consider meeting with the appropriate FDA review division early in the development phase to discuss when and how to include pregnant women in the drug development plan. These discussions should involve FDA experts in bioethics and maternal health.”
- “Pregnant women can be enrolled in clinical trials that involve greater than minimal risk to the fetuses if the trials offer the potential for direct clinical benefit to the enrolled pregnant women and/or their fetuses.”
- A new pregnancy during a randomized, blinded clinical trial should prompt unblinding “so that counseling may be offered based on whether the fetus has been exposed to the investigational drug, placebo, or control.”
- The pregnant woman may continue the trial if potential benefits outweigh the risks.
- In general, pregnant women should not be enrolled in phase 1 and phase 2 clinical trials. Instead, those trials should be completed first “in a nonpregnant population that include females of reproductive potential.”
- Several types of events may call for the cessation of a clinical trial that includes pregnant women, such as serious maternal or fetal adverse events.
The draft guidance should take note of the fact that birth defects often don’t appear for months or even longer, according to Gerald Briggs, BPharm, FCCP, clinical professor of pharmacy at the University of California, San Francisco. “Until first year of life or later, the babies need to be monitored,” he said in an interview.
Mr. Briggs, who led a 2015 report examining the role of pregnant women in phase 4 clinical drug trials, added that the document should take note of recommendations from clinical teratologists regarding the design of animal studies that should be performed prior to human trials (Am J Obstet Gynecol. 2015;213(6):810-5).
Comments on the draft guidance can be made at www.federalregister.gov and are due by June 8, 2018.
Dr. Chambers and Mr. Briggs reported no relevant disclosures.
Lower heart disease mortality brings increased disparity
The overall death rate from heart disease is down 68% since 1968 in the United States, but the disparity between blacks and whites has increased over that time, according to the Centers for Disease Control and Prevention.
Overall, heart disease mortality for adults aged 35 years and older went from 1,035 per 100,000 population in 1968 to 327 per 100,000 in 2015, a drop of 68%. For whites, the story was very similar: The death rate dropped from 1,032 to 326, or 68%. For blacks, who had a higher death rate to begin with, at 1,072 per 100,000, the drop was 63% to 396 per 100,000, Miriam Van Dyke, MPH, of Emory University, Atlanta, and her associates reported in MMWR Surveillance Summaries.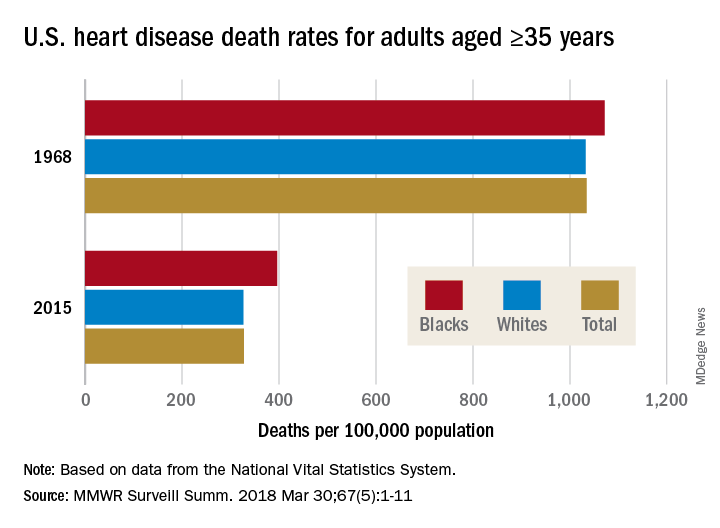
Disparities can be seen at the state level as well. For blacks aged 35 years and older, heart disease death rates ranged from 200 per 100,000 in Oregon to 516 per 100,000 in Arkansas in 2015. For whites in that age group, the death rate was lowest in the District of Columbia (198 per 100,000) and highest in Oklahoma (446 per 100,000), Ms. Van Dyke and her associates wrote.
To help them pinpoint differences by race within geographic areas, the investigators calculated ratios of black to white heart disease death rates. In 1968, the state with the highest ratio, or the largest excess of black mortality, was Rhode Island at 1.38, and the state with the lowest ratio, meaning the largest excess of white mortality, was Minnesota at 0.64. There were 27 states that year with a ratio over 1.0, 12 states with a ratio below 1.0, 1 state with a ratio of 1.0 (Wisconsin), and 11 states that did not have a black population large enough to make a reliable estimate, they said.
In 2015, the jurisdiction with the largest excess of black mortality was D.C., with a ratio of 2.42; the state with the lowest ratio was Rhode Island, at 0.69. That year, there were 34 states with a ratio over 1.0, 6 states with a ratio below 1.0, and 11 states – the same 11 as in 1968 – with black populations too small to reliably estimate death rates, the investigators noted.
“The elimination of racial disparities in heart disease death rates, along with continued decreases in heart disease death rates for all persons in the United States, is important for the overall state of health. The trends in black-white disparities in heart disease death rates … highlight the importance of continued surveillance of these trends at the national and state level,” the investigators wrote.
SOURCE: Van Dyke M et al. MMWR Surveill Summ. 2018 Mar 30;67(5):1-11.
The overall death rate from heart disease is down 68% since 1968 in the United States, but the disparity between blacks and whites has increased over that time, according to the Centers for Disease Control and Prevention.
Overall, heart disease mortality for adults aged 35 years and older went from 1,035 per 100,000 population in 1968 to 327 per 100,000 in 2015, a drop of 68%. For whites, the story was very similar: The death rate dropped from 1,032 to 326, or 68%. For blacks, who had a higher death rate to begin with, at 1,072 per 100,000, the drop was 63% to 396 per 100,000, Miriam Van Dyke, MPH, of Emory University, Atlanta, and her associates reported in MMWR Surveillance Summaries.
Disparities can be seen at the state level as well. For blacks aged 35 years and older, heart disease death rates ranged from 200 per 100,000 in Oregon to 516 per 100,000 in Arkansas in 2015. For whites in that age group, the death rate was lowest in the District of Columbia (198 per 100,000) and highest in Oklahoma (446 per 100,000), Ms. Van Dyke and her associates wrote.
To help them pinpoint differences by race within geographic areas, the investigators calculated ratios of black to white heart disease death rates. In 1968, the state with the highest ratio, or the largest excess of black mortality, was Rhode Island at 1.38, and the state with the lowest ratio, meaning the largest excess of white mortality, was Minnesota at 0.64. There were 27 states that year with a ratio over 1.0, 12 states with a ratio below 1.0, 1 state with a ratio of 1.0 (Wisconsin), and 11 states that did not have a black population large enough to make a reliable estimate, they said.
In 2015, the jurisdiction with the largest excess of black mortality was D.C., with a ratio of 2.42; the state with the lowest ratio was Rhode Island, at 0.69. That year, there were 34 states with a ratio over 1.0, 6 states with a ratio below 1.0, and 11 states – the same 11 as in 1968 – with black populations too small to reliably estimate death rates, the investigators noted.
“The elimination of racial disparities in heart disease death rates, along with continued decreases in heart disease death rates for all persons in the United States, is important for the overall state of health. The trends in black-white disparities in heart disease death rates … highlight the importance of continued surveillance of these trends at the national and state level,” the investigators wrote.
SOURCE: Van Dyke M et al. MMWR Surveill Summ. 2018 Mar 30;67(5):1-11.
The overall death rate from heart disease is down 68% since 1968 in the United States, but the disparity between blacks and whites has increased over that time, according to the Centers for Disease Control and Prevention.
Overall, heart disease mortality for adults aged 35 years and older went from 1,035 per 100,000 population in 1968 to 327 per 100,000 in 2015, a drop of 68%. For whites, the story was very similar: The death rate dropped from 1,032 to 326, or 68%. For blacks, who had a higher death rate to begin with, at 1,072 per 100,000, the drop was 63% to 396 per 100,000, Miriam Van Dyke, MPH, of Emory University, Atlanta, and her associates reported in MMWR Surveillance Summaries.
Disparities can be seen at the state level as well. For blacks aged 35 years and older, heart disease death rates ranged from 200 per 100,000 in Oregon to 516 per 100,000 in Arkansas in 2015. For whites in that age group, the death rate was lowest in the District of Columbia (198 per 100,000) and highest in Oklahoma (446 per 100,000), Ms. Van Dyke and her associates wrote.
To help them pinpoint differences by race within geographic areas, the investigators calculated ratios of black to white heart disease death rates. In 1968, the state with the highest ratio, or the largest excess of black mortality, was Rhode Island at 1.38, and the state with the lowest ratio, meaning the largest excess of white mortality, was Minnesota at 0.64. There were 27 states that year with a ratio over 1.0, 12 states with a ratio below 1.0, 1 state with a ratio of 1.0 (Wisconsin), and 11 states that did not have a black population large enough to make a reliable estimate, they said.
In 2015, the jurisdiction with the largest excess of black mortality was D.C., with a ratio of 2.42; the state with the lowest ratio was Rhode Island, at 0.69. That year, there were 34 states with a ratio over 1.0, 6 states with a ratio below 1.0, and 11 states – the same 11 as in 1968 – with black populations too small to reliably estimate death rates, the investigators noted.
“The elimination of racial disparities in heart disease death rates, along with continued decreases in heart disease death rates for all persons in the United States, is important for the overall state of health. The trends in black-white disparities in heart disease death rates … highlight the importance of continued surveillance of these trends at the national and state level,” the investigators wrote.
SOURCE: Van Dyke M et al. MMWR Surveill Summ. 2018 Mar 30;67(5):1-11.
FROM MMWR SURVEILLANCE SUMMARIES
FDA recalls kratom products for salmonella contamination
The Food and Drug Administration on April 3 recalled all products containing kratom manufactured by Triangle Pharmanaturals LLC, after a number of supplements tested positive for salmonella.
The FDA advises consumers to get rid of products including Raw Form Organics Maeng Da Kratom Emerald Green, Raw Form Organics Maeng Da Kratom Ivory White, and Raw Form Organics Maeng Da Kratom Ruby Red.
Evidence of the contamination was found after two samples were collected from a retail store in Oregon by the Oregon Public Health Division and tested positive for salmonella.

The recall was ordered after Triangle Pharmanaturals did not comply with a March 30 formal request from the FDA to voluntarily recall their products.
“This action is based on the imminent health risk posed by the contamination of this product with salmonella, and the refusal of this company to voluntarily act to protect its customers and issue a recall, despite our repeated requests and actions,” said FDA Commissioner Scott Gottlieb, M.D., said in a statement. “The action today is based on the risks posed by the contamination of this particular product with a potentially dangerous pathogen.”
At press time, Triangle Pharmanaturals did not respond to a request for comment.
This is the most recent in a list recalls of kratom products as part of an ongoing investigation of a salmonella outbreak by the FDA; however Triangle Pharmanaturals’ noncompliance is unique to the agency, according to an FDA representative.
“This is the first time the agency has issued a mandatory recall order to protect Americans from contaminated food products,” Michael Felberbaum, an FDA press officer, said in an interview. “This is the third time the FDA has invoked its mandatory recall authority, but the first time the agency ordered a mandatory recall because a company has opted not to voluntarily recall after the FDA’s notification of an opportunity to initiate a voluntary recall.”
Earlier in March, the CDC reported 87 people in 35 states infected with either Salmonella Javiana, Salmonela Okatie, or Salmonella Thompson, which have been associated with the outbreak.
While salmonella was identified in Triangle Pharmanaturals’ products, the strains identified are not currently linked to the outbreak.
Kratom, a plant that commonly grows in South East Asian countries like Thailand, Malaysia, Indonesia, and Papua New Guinea, has recently been used to produce food supplements and marketed as an alternative to addictive pain medication like opioids, as well as used to help treat opioid withdrawal symptoms.
Use of the food supplement has fired debate among physicians, patients, and public officials as all sides continue to determine its efficacy and how, or whether, it should be given a drug classification.
The Food and Drug Administration on April 3 recalled all products containing kratom manufactured by Triangle Pharmanaturals LLC, after a number of supplements tested positive for salmonella.
The FDA advises consumers to get rid of products including Raw Form Organics Maeng Da Kratom Emerald Green, Raw Form Organics Maeng Da Kratom Ivory White, and Raw Form Organics Maeng Da Kratom Ruby Red.
Evidence of the contamination was found after two samples were collected from a retail store in Oregon by the Oregon Public Health Division and tested positive for salmonella.
The recall was ordered after Triangle Pharmanaturals did not comply with a March 30 formal request from the FDA to voluntarily recall their products.
“This action is based on the imminent health risk posed by the contamination of this product with salmonella, and the refusal of this company to voluntarily act to protect its customers and issue a recall, despite our repeated requests and actions,” said FDA Commissioner Scott Gottlieb, M.D., said in a statement. “The action today is based on the risks posed by the contamination of this particular product with a potentially dangerous pathogen.”
At press time, Triangle Pharmanaturals did not respond to a request for comment.
This is the most recent in a list recalls of kratom products as part of an ongoing investigation of a salmonella outbreak by the FDA; however Triangle Pharmanaturals’ noncompliance is unique to the agency, according to an FDA representative.
“This is the first time the agency has issued a mandatory recall order to protect Americans from contaminated food products,” Michael Felberbaum, an FDA press officer, said in an interview. “This is the third time the FDA has invoked its mandatory recall authority, but the first time the agency ordered a mandatory recall because a company has opted not to voluntarily recall after the FDA’s notification of an opportunity to initiate a voluntary recall.”
Earlier in March, the CDC reported 87 people in 35 states infected with either Salmonella Javiana, Salmonela Okatie, or Salmonella Thompson, which have been associated with the outbreak.
While salmonella was identified in Triangle Pharmanaturals’ products, the strains identified are not currently linked to the outbreak.
Kratom, a plant that commonly grows in South East Asian countries like Thailand, Malaysia, Indonesia, and Papua New Guinea, has recently been used to produce food supplements and marketed as an alternative to addictive pain medication like opioids, as well as used to help treat opioid withdrawal symptoms.
Use of the food supplement has fired debate among physicians, patients, and public officials as all sides continue to determine its efficacy and how, or whether, it should be given a drug classification.
The Food and Drug Administration on April 3 recalled all products containing kratom manufactured by Triangle Pharmanaturals LLC, after a number of supplements tested positive for salmonella.
The FDA advises consumers to get rid of products including Raw Form Organics Maeng Da Kratom Emerald Green, Raw Form Organics Maeng Da Kratom Ivory White, and Raw Form Organics Maeng Da Kratom Ruby Red.
Evidence of the contamination was found after two samples were collected from a retail store in Oregon by the Oregon Public Health Division and tested positive for salmonella.
The recall was ordered after Triangle Pharmanaturals did not comply with a March 30 formal request from the FDA to voluntarily recall their products.
“This action is based on the imminent health risk posed by the contamination of this product with salmonella, and the refusal of this company to voluntarily act to protect its customers and issue a recall, despite our repeated requests and actions,” said FDA Commissioner Scott Gottlieb, M.D., said in a statement. “The action today is based on the risks posed by the contamination of this particular product with a potentially dangerous pathogen.”
At press time, Triangle Pharmanaturals did not respond to a request for comment.
This is the most recent in a list recalls of kratom products as part of an ongoing investigation of a salmonella outbreak by the FDA; however Triangle Pharmanaturals’ noncompliance is unique to the agency, according to an FDA representative.
“This is the first time the agency has issued a mandatory recall order to protect Americans from contaminated food products,” Michael Felberbaum, an FDA press officer, said in an interview. “This is the third time the FDA has invoked its mandatory recall authority, but the first time the agency ordered a mandatory recall because a company has opted not to voluntarily recall after the FDA’s notification of an opportunity to initiate a voluntary recall.”
Earlier in March, the CDC reported 87 people in 35 states infected with either Salmonella Javiana, Salmonela Okatie, or Salmonella Thompson, which have been associated with the outbreak.
While salmonella was identified in Triangle Pharmanaturals’ products, the strains identified are not currently linked to the outbreak.
Kratom, a plant that commonly grows in South East Asian countries like Thailand, Malaysia, Indonesia, and Papua New Guinea, has recently been used to produce food supplements and marketed as an alternative to addictive pain medication like opioids, as well as used to help treat opioid withdrawal symptoms.
Use of the food supplement has fired debate among physicians, patients, and public officials as all sides continue to determine its efficacy and how, or whether, it should be given a drug classification.
Synthetic opioids drive increase in overdose deaths
Opioid-related drug overdose deaths jumped 28% from 2015 to 2016, with the largest increase coming from synthetic opioids, such as illicitly manufactured fentanyl, according to the Centers for Disease Control and Prevention.
The age-adjusted death rate for opioid overdoses increased from 10.4 per 100,000 population in 2015 to 13.3 per 100,000 in 2016, and the 42,249 opioid deaths in 2016 represented more than 66% of all overdose deaths that year, Puja Seth, PhD, and her associates at the CDC reported in the Morbidity and Mortality Weekly Report.
Illegally manufactured fentanyl “is now being mixed into counterfeit opioid and benzodiazepine pills, heroin, and cocaine, likely contributing to increases in overdose death rates involving other substances,” they wrote. To illustrate that point, they reported that cocaine overdose deaths increased 52.4% from 2.1 per 100,000 in 2015 to 3.2 in 2016. The death rate for the other drug category covered in the report – psychostimulants with abuse potential – climbed from 1.8 per 100,000 in 2015 to 2.4 in 2016, for an increase of 33.3%, Dr. Seth and her associates noted.
Data presented from 31 states and the District of Columbia show that
CDC Principal Deputy Director Anne Schuchat, MD, said in a written statement.
Death rates from overdoses involving synthetic opioids increased in 21 states, with 10 states doubling their rates from 2015 to 2016, and 14 states had significant increases in death rates involving heroin. In D.C., for example, the death rate increased 392% (3.9 per 100,000 to 19.2) from synthetic opioid overdoses and 75% (9.9 per 100,000 to 17.3) for deaths related to heroin, the report showed.
“Effective, synchronized programs to prevent drug overdoses will require coordination of law enforcement, first responders, mental health/substance-abuse providers, public health agencies, and community partners,” Dr. Seth and her associates said.
SOURCE: Seth P et al. MMWR. 2018 Mar 30;67(12):349-58.
Opioid-related drug overdose deaths jumped 28% from 2015 to 2016, with the largest increase coming from synthetic opioids, such as illicitly manufactured fentanyl, according to the Centers for Disease Control and Prevention.
The age-adjusted death rate for opioid overdoses increased from 10.4 per 100,000 population in 2015 to 13.3 per 100,000 in 2016, and the 42,249 opioid deaths in 2016 represented more than 66% of all overdose deaths that year, Puja Seth, PhD, and her associates at the CDC reported in the Morbidity and Mortality Weekly Report.
Illegally manufactured fentanyl “is now being mixed into counterfeit opioid and benzodiazepine pills, heroin, and cocaine, likely contributing to increases in overdose death rates involving other substances,” they wrote. To illustrate that point, they reported that cocaine overdose deaths increased 52.4% from 2.1 per 100,000 in 2015 to 3.2 in 2016. The death rate for the other drug category covered in the report – psychostimulants with abuse potential – climbed from 1.8 per 100,000 in 2015 to 2.4 in 2016, for an increase of 33.3%, Dr. Seth and her associates noted.
Data presented from 31 states and the District of Columbia show that
CDC Principal Deputy Director Anne Schuchat, MD, said in a written statement.
Death rates from overdoses involving synthetic opioids increased in 21 states, with 10 states doubling their rates from 2015 to 2016, and 14 states had significant increases in death rates involving heroin. In D.C., for example, the death rate increased 392% (3.9 per 100,000 to 19.2) from synthetic opioid overdoses and 75% (9.9 per 100,000 to 17.3) for deaths related to heroin, the report showed.
“Effective, synchronized programs to prevent drug overdoses will require coordination of law enforcement, first responders, mental health/substance-abuse providers, public health agencies, and community partners,” Dr. Seth and her associates said.
SOURCE: Seth P et al. MMWR. 2018 Mar 30;67(12):349-58.
Opioid-related drug overdose deaths jumped 28% from 2015 to 2016, with the largest increase coming from synthetic opioids, such as illicitly manufactured fentanyl, according to the Centers for Disease Control and Prevention.
The age-adjusted death rate for opioid overdoses increased from 10.4 per 100,000 population in 2015 to 13.3 per 100,000 in 2016, and the 42,249 opioid deaths in 2016 represented more than 66% of all overdose deaths that year, Puja Seth, PhD, and her associates at the CDC reported in the Morbidity and Mortality Weekly Report.
Illegally manufactured fentanyl “is now being mixed into counterfeit opioid and benzodiazepine pills, heroin, and cocaine, likely contributing to increases in overdose death rates involving other substances,” they wrote. To illustrate that point, they reported that cocaine overdose deaths increased 52.4% from 2.1 per 100,000 in 2015 to 3.2 in 2016. The death rate for the other drug category covered in the report – psychostimulants with abuse potential – climbed from 1.8 per 100,000 in 2015 to 2.4 in 2016, for an increase of 33.3%, Dr. Seth and her associates noted.
Data presented from 31 states and the District of Columbia show that
CDC Principal Deputy Director Anne Schuchat, MD, said in a written statement.
Death rates from overdoses involving synthetic opioids increased in 21 states, with 10 states doubling their rates from 2015 to 2016, and 14 states had significant increases in death rates involving heroin. In D.C., for example, the death rate increased 392% (3.9 per 100,000 to 19.2) from synthetic opioid overdoses and 75% (9.9 per 100,000 to 17.3) for deaths related to heroin, the report showed.
“Effective, synchronized programs to prevent drug overdoses will require coordination of law enforcement, first responders, mental health/substance-abuse providers, public health agencies, and community partners,” Dr. Seth and her associates said.
SOURCE: Seth P et al. MMWR. 2018 Mar 30;67(12):349-58.
FROM MMWR
FDA advisors recommend lofexidine for opioid withdrawal
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.

Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.
Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.
Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING