User login
FDA alerts clinicians to gastric balloon deaths
according to an alert from the Food and Drug Administration issued on June 4.
Seven of these deaths occurred in patients in the United States; four involved the ORBERA Intragastric Balloon System, and three involved the ReShape Integrated Dual Balloon System.
The FDA has approved updated labeling for the ORBERA and ReShape balloon systems in the United States. The labels contain more information about possible death associated with the use of these devices in the United States. The manufacturers’ sites, Apollo Endosurgery and ReShape Lifesciences, provide more details about the new labeling.
In a letter to health care providers, the FDA advised clinicians to educate bariatric surgery patients about the symptoms of complications from balloon procedures, including not only gastric perforation but also esophageal perforation, balloon deflation, gastrointestinal obstruction, and ulceration. In addition, the FDA reminded clinicians to monitor patients during the entire course of treatment for additional complications, including acute pancreatitis and spontaneous hyperinflation.
Any adverse events involving intragastric balloon systems should be reported to the FDA through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
according to an alert from the Food and Drug Administration issued on June 4.
Seven of these deaths occurred in patients in the United States; four involved the ORBERA Intragastric Balloon System, and three involved the ReShape Integrated Dual Balloon System.
The FDA has approved updated labeling for the ORBERA and ReShape balloon systems in the United States. The labels contain more information about possible death associated with the use of these devices in the United States. The manufacturers’ sites, Apollo Endosurgery and ReShape Lifesciences, provide more details about the new labeling.
In a letter to health care providers, the FDA advised clinicians to educate bariatric surgery patients about the symptoms of complications from balloon procedures, including not only gastric perforation but also esophageal perforation, balloon deflation, gastrointestinal obstruction, and ulceration. In addition, the FDA reminded clinicians to monitor patients during the entire course of treatment for additional complications, including acute pancreatitis and spontaneous hyperinflation.
Any adverse events involving intragastric balloon systems should be reported to the FDA through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
according to an alert from the Food and Drug Administration issued on June 4.
Seven of these deaths occurred in patients in the United States; four involved the ORBERA Intragastric Balloon System, and three involved the ReShape Integrated Dual Balloon System.
The FDA has approved updated labeling for the ORBERA and ReShape balloon systems in the United States. The labels contain more information about possible death associated with the use of these devices in the United States. The manufacturers’ sites, Apollo Endosurgery and ReShape Lifesciences, provide more details about the new labeling.
In a letter to health care providers, the FDA advised clinicians to educate bariatric surgery patients about the symptoms of complications from balloon procedures, including not only gastric perforation but also esophageal perforation, balloon deflation, gastrointestinal obstruction, and ulceration. In addition, the FDA reminded clinicians to monitor patients during the entire course of treatment for additional complications, including acute pancreatitis and spontaneous hyperinflation.
Any adverse events involving intragastric balloon systems should be reported to the FDA through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
FDA approves Olumiant for treatment of rheumatoid arthritis
, an orally administered Janus kinase (JAK) inhibitor, to treat adults with moderate to severe rheumatoid arthritis (RA) who have responded inadequately or poorly to methotrexate, its manufacturer, Eli Lilly, announced June 1. The regulators voted against approval of the 4-mg dose because of concerns about the safety profile.
Olumiant is accompanied by a boxed warning about the risk of serious infections, malignancies, and thrombosis. Patients taking Olumiant also have experienced tuberculosis and opportunistic viral, fungal, and bacterial infections. These infections have led to hospitalization or death.
As part of the approval, Lilly and the original developer of baricitinib, Incyte, have agreed to conduct further randomized and controlled clinical trials to evaluate the long-term safety of Olumiant.
Lilly said in its announcement that it expects to launch Olumiant in the United States by the end of the second quarter of 2018 at a targeted price that is 60% less than “the leading TNF inhibitor.” Additionally, Lilly will offer patient support in the form of a patient support program called Olumiant Together. More information for the program can be obtained by calling 844-OLUMIANT.
, an orally administered Janus kinase (JAK) inhibitor, to treat adults with moderate to severe rheumatoid arthritis (RA) who have responded inadequately or poorly to methotrexate, its manufacturer, Eli Lilly, announced June 1. The regulators voted against approval of the 4-mg dose because of concerns about the safety profile.
Olumiant is accompanied by a boxed warning about the risk of serious infections, malignancies, and thrombosis. Patients taking Olumiant also have experienced tuberculosis and opportunistic viral, fungal, and bacterial infections. These infections have led to hospitalization or death.
As part of the approval, Lilly and the original developer of baricitinib, Incyte, have agreed to conduct further randomized and controlled clinical trials to evaluate the long-term safety of Olumiant.
Lilly said in its announcement that it expects to launch Olumiant in the United States by the end of the second quarter of 2018 at a targeted price that is 60% less than “the leading TNF inhibitor.” Additionally, Lilly will offer patient support in the form of a patient support program called Olumiant Together. More information for the program can be obtained by calling 844-OLUMIANT.
, an orally administered Janus kinase (JAK) inhibitor, to treat adults with moderate to severe rheumatoid arthritis (RA) who have responded inadequately or poorly to methotrexate, its manufacturer, Eli Lilly, announced June 1. The regulators voted against approval of the 4-mg dose because of concerns about the safety profile.
Olumiant is accompanied by a boxed warning about the risk of serious infections, malignancies, and thrombosis. Patients taking Olumiant also have experienced tuberculosis and opportunistic viral, fungal, and bacterial infections. These infections have led to hospitalization or death.
As part of the approval, Lilly and the original developer of baricitinib, Incyte, have agreed to conduct further randomized and controlled clinical trials to evaluate the long-term safety of Olumiant.
Lilly said in its announcement that it expects to launch Olumiant in the United States by the end of the second quarter of 2018 at a targeted price that is 60% less than “the leading TNF inhibitor.” Additionally, Lilly will offer patient support in the form of a patient support program called Olumiant Together. More information for the program can be obtained by calling 844-OLUMIANT.
FDA recalls HeartMate 3 LV assist device
The HeartMate 3 left ventricular assist device received a class 1 recall from the Food and Drug Administration on May 17, an action the agency publicly announced on May 22.
The FDA’s step formalized a “corrective action” that the HeartMate 3’s manufacturer, Abbott, first issued in early April and then announced in early May, which said the device used to treat patients with severe advanced heart failure was subject to “outflow graft twist occlusions” that trigger a persistent low-flow alarm and “can result in serious adverse events such as hemodynamic compromise, thrombus, and death,” according to the FDA’s May 17 statement.
The class 1 recall category the FDA applied means the agency rates the danger posed as a “situation where there is a reasonable chance that a product will cause serious health problems or death.” However, in its statements, the agency has not suggested removing a well-functioning device from patients, nor has the agency called for discontinuing new placements of the HeartMate 3. The FDA gave a full endorsement to the approach Abbott suggested in its April 5 “Dear Physician” letter and then followed with a second letter on May 21.
The first letter attributed development of these twists in the tube that directs blood out from the device to accumulated mechanical forces from heartbeats, respiration, and activity, and also provided some management guidance that Abbott then expanded in its second letter.
The FDA cited some of the key steps Abbott recommended clinicians take with patients who receive a HeartMate 3. This included regular surveillance with transthoracic echocardiography, although echo is not considered definitive for identifying a graft twist obstruction and hence other investigations may also be needed. If the low-flow alarm sounds, a CT scan is “urgently” needed – as long as it’s not contraindicated – to identify a possible outflow twist. Abbott noted that surgical intervention may be needed to correct a twist.
Researchers recently reported 2-year follow-up data from 257 patients in MOMENTUM 3, the randomized pivotal trial for the HeartMate 3 that compared this fully magnetically levitated centrifugal-flow pump to the prior-generation, axial-flow pump. The composite endpoint of freedom from death, stroke, or need for repeat surgery after 2 years was 80% in the HeartMate 3 recipients and 60% among patients in the control arm who received the older-model device (N Engl J Med. 2018 April 12; 378[15]:1386-95).
The HeartMate 3 left ventricular assist device received a class 1 recall from the Food and Drug Administration on May 17, an action the agency publicly announced on May 22.
The FDA’s step formalized a “corrective action” that the HeartMate 3’s manufacturer, Abbott, first issued in early April and then announced in early May, which said the device used to treat patients with severe advanced heart failure was subject to “outflow graft twist occlusions” that trigger a persistent low-flow alarm and “can result in serious adverse events such as hemodynamic compromise, thrombus, and death,” according to the FDA’s May 17 statement.
The class 1 recall category the FDA applied means the agency rates the danger posed as a “situation where there is a reasonable chance that a product will cause serious health problems or death.” However, in its statements, the agency has not suggested removing a well-functioning device from patients, nor has the agency called for discontinuing new placements of the HeartMate 3. The FDA gave a full endorsement to the approach Abbott suggested in its April 5 “Dear Physician” letter and then followed with a second letter on May 21.
The first letter attributed development of these twists in the tube that directs blood out from the device to accumulated mechanical forces from heartbeats, respiration, and activity, and also provided some management guidance that Abbott then expanded in its second letter.
The FDA cited some of the key steps Abbott recommended clinicians take with patients who receive a HeartMate 3. This included regular surveillance with transthoracic echocardiography, although echo is not considered definitive for identifying a graft twist obstruction and hence other investigations may also be needed. If the low-flow alarm sounds, a CT scan is “urgently” needed – as long as it’s not contraindicated – to identify a possible outflow twist. Abbott noted that surgical intervention may be needed to correct a twist.
Researchers recently reported 2-year follow-up data from 257 patients in MOMENTUM 3, the randomized pivotal trial for the HeartMate 3 that compared this fully magnetically levitated centrifugal-flow pump to the prior-generation, axial-flow pump. The composite endpoint of freedom from death, stroke, or need for repeat surgery after 2 years was 80% in the HeartMate 3 recipients and 60% among patients in the control arm who received the older-model device (N Engl J Med. 2018 April 12; 378[15]:1386-95).
The HeartMate 3 left ventricular assist device received a class 1 recall from the Food and Drug Administration on May 17, an action the agency publicly announced on May 22.
The FDA’s step formalized a “corrective action” that the HeartMate 3’s manufacturer, Abbott, first issued in early April and then announced in early May, which said the device used to treat patients with severe advanced heart failure was subject to “outflow graft twist occlusions” that trigger a persistent low-flow alarm and “can result in serious adverse events such as hemodynamic compromise, thrombus, and death,” according to the FDA’s May 17 statement.
The class 1 recall category the FDA applied means the agency rates the danger posed as a “situation where there is a reasonable chance that a product will cause serious health problems or death.” However, in its statements, the agency has not suggested removing a well-functioning device from patients, nor has the agency called for discontinuing new placements of the HeartMate 3. The FDA gave a full endorsement to the approach Abbott suggested in its April 5 “Dear Physician” letter and then followed with a second letter on May 21.
The first letter attributed development of these twists in the tube that directs blood out from the device to accumulated mechanical forces from heartbeats, respiration, and activity, and also provided some management guidance that Abbott then expanded in its second letter.
The FDA cited some of the key steps Abbott recommended clinicians take with patients who receive a HeartMate 3. This included regular surveillance with transthoracic echocardiography, although echo is not considered definitive for identifying a graft twist obstruction and hence other investigations may also be needed. If the low-flow alarm sounds, a CT scan is “urgently” needed – as long as it’s not contraindicated – to identify a possible outflow twist. Abbott noted that surgical intervention may be needed to correct a twist.
Researchers recently reported 2-year follow-up data from 257 patients in MOMENTUM 3, the randomized pivotal trial for the HeartMate 3 that compared this fully magnetically levitated centrifugal-flow pump to the prior-generation, axial-flow pump. The composite endpoint of freedom from death, stroke, or need for repeat surgery after 2 years was 80% in the HeartMate 3 recipients and 60% among patients in the control arm who received the older-model device (N Engl J Med. 2018 April 12; 378[15]:1386-95).
FDA approves Prolia for glucocorticoid-induced osteoporosis
at high risk of fracture, the drug’s manufacturer Amgen announced May 21.
FDA approval was based on 12-month primary analysis results from a randomized, double-blind, phase 3 trial. Patients who received a 60-mg dose of Prolia subcutaneously every 6 months had greater lumbar spine bone mineral density at 1 year than did those who received a 5-mg dose of risedronate daily in all study subpopulations. These results were maintained after researchers controlled for gender, race, geographic region, and menopausal status, as well as baseline age, lumbar spine bone mineral density T score, and glucocorticoid dose within each subpopulation.
The most common adverse events associated with Prolia during the phase 3 study were back pain, hypertension, bronchitis, and headache, which are in line with previously reported safety data.
“Patients on long-term systemic glucocorticoid medications can experience a rapid reduction in bone mineral density within a few months of beginning treatment. With this approval, patients who receive treatment with glucocorticoids now have a new option to help improve their bone mineral density,” lead study author Kenneth F. Saag, MD, professor of medicine at the University of Alabama, Birmingham, said in Amgen’s news release.
at high risk of fracture, the drug’s manufacturer Amgen announced May 21.
FDA approval was based on 12-month primary analysis results from a randomized, double-blind, phase 3 trial. Patients who received a 60-mg dose of Prolia subcutaneously every 6 months had greater lumbar spine bone mineral density at 1 year than did those who received a 5-mg dose of risedronate daily in all study subpopulations. These results were maintained after researchers controlled for gender, race, geographic region, and menopausal status, as well as baseline age, lumbar spine bone mineral density T score, and glucocorticoid dose within each subpopulation.
The most common adverse events associated with Prolia during the phase 3 study were back pain, hypertension, bronchitis, and headache, which are in line with previously reported safety data.
“Patients on long-term systemic glucocorticoid medications can experience a rapid reduction in bone mineral density within a few months of beginning treatment. With this approval, patients who receive treatment with glucocorticoids now have a new option to help improve their bone mineral density,” lead study author Kenneth F. Saag, MD, professor of medicine at the University of Alabama, Birmingham, said in Amgen’s news release.
at high risk of fracture, the drug’s manufacturer Amgen announced May 21.
FDA approval was based on 12-month primary analysis results from a randomized, double-blind, phase 3 trial. Patients who received a 60-mg dose of Prolia subcutaneously every 6 months had greater lumbar spine bone mineral density at 1 year than did those who received a 5-mg dose of risedronate daily in all study subpopulations. These results were maintained after researchers controlled for gender, race, geographic region, and menopausal status, as well as baseline age, lumbar spine bone mineral density T score, and glucocorticoid dose within each subpopulation.
The most common adverse events associated with Prolia during the phase 3 study were back pain, hypertension, bronchitis, and headache, which are in line with previously reported safety data.
“Patients on long-term systemic glucocorticoid medications can experience a rapid reduction in bone mineral density within a few months of beginning treatment. With this approval, patients who receive treatment with glucocorticoids now have a new option to help improve their bone mineral density,” lead study author Kenneth F. Saag, MD, professor of medicine at the University of Alabama, Birmingham, said in Amgen’s news release.
FDA approves Doptelet for liver disease patients undergoing procedures
Doptelet (avatrombopag) is the first drug to be approved by the Food and Drug Administration for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure, the FDA announced in a statement.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
The safety and efficacy of two different doses of Doptelet administered orally over 5 days, as compared with placebo, was studied in the ADAPT trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. At both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to 7 days following the procedure as compared with those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and edema in the hands or feet. People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet, the FDA said in a press release announcing the approval.
The FDA granted the Doptelet approval to AkaRx.
Doptelet (avatrombopag) is the first drug to be approved by the Food and Drug Administration for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure, the FDA announced in a statement.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
The safety and efficacy of two different doses of Doptelet administered orally over 5 days, as compared with placebo, was studied in the ADAPT trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. At both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to 7 days following the procedure as compared with those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and edema in the hands or feet. People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet, the FDA said in a press release announcing the approval.
The FDA granted the Doptelet approval to AkaRx.
Doptelet (avatrombopag) is the first drug to be approved by the Food and Drug Administration for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure, the FDA announced in a statement.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
The safety and efficacy of two different doses of Doptelet administered orally over 5 days, as compared with placebo, was studied in the ADAPT trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. At both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to 7 days following the procedure as compared with those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and edema in the hands or feet. People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet, the FDA said in a press release announcing the approval.
The FDA granted the Doptelet approval to AkaRx.
FDA: PrEP indication updated to include adolescents at risk of HIV infection
The following is the text of an announcement by the U.S. Food and Drug Administration regarding a revision in the Truvada label to expand the PrEP indication to include at-risk adolescents. The new label change has not yet been posted.
The Food and Drug Administration approved revisions to the Truvada (emtricitabine and tenofovir disoproxil fumarate) labeling to expand the Pre-Exposure Prophylaxis (PrEP) indication to include adolescents weighing at least 35 kg who are at risk of HIV-1 acquisition. The major labeling changes with respect to this expanded indication are summarized below. In addition, Section 8 was reformatted per the Pregnancy and Lactation Labeling Rule (PLLR) and includes updated information specific to the use of Truvada for PrEP during pregnancy and breastfeeding. Other sections of labeling were reformatted for consistency with current and best labeling practices, as well as with labeling for other HIV fixed-dose combination products.
Indications and usage
1.2 HIV-1 pre-exposure prophylaxis (PrEP)
Truvada is indicated in combination with safer sex practices for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV-1 in at-risk adults and adolescents weighing at least 35 kg. Individuals must have a negative HIV-1 test immediately prior to initiating Truvada for HIV-1 PrEP.
If clinical symptoms consistent with acute viral infection are present and recent (less than 1 month) exposures are suspected, delay starting PrEP for at least one month and reconfirm HIV-1 status or use a test cleared by the FDA as an aid in the diagnosis of HIV-1 infection, including acute or primary HIV-1 infection
When considering Truvada for HIV-1 PrEP, factors that help to identify individuals at risk may include:
– has partner(s) known to be HIV-1 infected, or
– engages in sexual activity within a high prevalence area or social network and has additional risk factors for HIV-1 acquisition, such as:
- inconsistent or no condom use.
- diagnosis of sexually transmitted infections.
- exchange of sex for commodities (such as money, food, shelter, or drugs).
- use of illicit drugs or alcohol dependence.
- incarceration.
- partner(s) of unknown HIV-1 status with any of the factors listed above.
Dosage and administration
2.1 Testing prior to initiation of Truvada for treatment of HIV-1 infection or for HIV-1 PrEP
Prior to or when initiating Truvada, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to initiation and during use of Truvada, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus
2.2 HIV-1 screening for individuals receiving Truvada for HIV-1 PrEP
Screen all patients for HIV-1 infection before initiating Truvada for HIV-1 PrEP and at least once every 3 months while taking Truvada
2.5 Recommended dosage for HIV-1 PrEP
The dosage of Truvada in HIV-1 uninfected adults and adolescents weighing at least 35 kg is one tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food.
6.0 Adverse reactions
Clinical trials in adolescent subjects
In a single-arm, open-label clinical trial (ATN113), in which 67 HIV-1 uninfected adolescent (15 to 18 years of age) men who have sex with men received Truvada once daily for HIV-1 PrEP, the safety profile of Truvada was similar to that observed in adults. Median duration to exposure of Truvada was 47 weeks.
In the ATN113 trial, median BMD increased from baseline to Week 48, +2.58% for lumbar spine and +0.72% for total body. One subject had significant (greater than or equal to 4%) total body BMD loss at Week 24. Median changes from baseline BMD Z-scores were 0.0 for lumbar spine and −0.2 for total body at Week 48. Three subjects showed a worsening (change from greater than −2 to less than or equal to −2) from baseline in their lumbar spine or total body BMD Z-scores at Week 24 or 48. Interpretation of these data, however, may be limited by the low rate of adherence to Truvada by Week 48.
8.4 Pediatric use
HIV-1 PrEP
The safety and effectiveness of Truvada for HIV-1 PrEP in at-risk adolescents weighing at least 35 kg is supported by data from adequate and well-controlled studies of Truvada for HIV-1 PrEP in adults with additional data from safety and pharmacokinetic studies in previously conducted trials with the individual drug products, FTC and TDF, in HIV-1 infected adults and pediatric subjects.
Safety, adherence, and resistance were evaluated in a single-arm, open-label clinical trial (ATN113) in which 67 HIV-1 uninfected at-risk adolescent men who have sex with men received Truvada once daily for HIV-1 PrEP. The mean age of subjects was 17 years (range, 15-18 years); 46% were Hispanic, 52% black, and 37% white. The safety profile of Truvada in ATN113 was similar to that observed in the adult HIV-1 PrEP trials.
In the ATN113 trial, HIV-1 seroconversion occurred in three subjects. Tenofovir diphosphate levels in dried blood spot assays indicate that these subjects had poor adherence. No tenofovir- or FTC-associated HIV-1 resistance substitutions were detected in virus isolated from the three subjects who seroconverted.
Adherence to study drug, as demonstrated by tenofovir diphosphate levels in dried blood spot assays, declined markedly after Week 12 once subjects switched from monthly to quarterly visits, suggesting that adolescents may benefit from more frequent visits and counseling.
12.0 Clinical pharmacology
HIV-1 PrEP
The pharmacokinetic data for tenofovir and FTC following administration of Truvada in HIV-1 uninfected adolescents weighing 35 kg and above are not available. The dosage recommendations of Truvada for HIV-1 PrEP in this population are based on safety and adherence data from the ATN113 trial [see Use in Specific Populations (8.4)] and known pharmacokinetic information in HIV-infected adolescents taking TDF and FTC for treatment.
ResistanceATN113 Trial
In ATN113, a clinical trial of HIV-1 seronegative adolescent subjects [see Use in Specific Populations (8.4)], no amino acid substitutions associated with resistance to FTC or TDF were detected at the time of seroconversion from any of the 3 subjects who became infected with HIV-1 during the trial. All 3 subjects who seroconverted were nonadherent to the recommended Truvada dosage.
The updated label will soon be available at drugs@fda or DailyMed.
The following is the text of an announcement by the U.S. Food and Drug Administration regarding a revision in the Truvada label to expand the PrEP indication to include at-risk adolescents. The new label change has not yet been posted.
The Food and Drug Administration approved revisions to the Truvada (emtricitabine and tenofovir disoproxil fumarate) labeling to expand the Pre-Exposure Prophylaxis (PrEP) indication to include adolescents weighing at least 35 kg who are at risk of HIV-1 acquisition. The major labeling changes with respect to this expanded indication are summarized below. In addition, Section 8 was reformatted per the Pregnancy and Lactation Labeling Rule (PLLR) and includes updated information specific to the use of Truvada for PrEP during pregnancy and breastfeeding. Other sections of labeling were reformatted for consistency with current and best labeling practices, as well as with labeling for other HIV fixed-dose combination products.
Indications and usage
1.2 HIV-1 pre-exposure prophylaxis (PrEP)
Truvada is indicated in combination with safer sex practices for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV-1 in at-risk adults and adolescents weighing at least 35 kg. Individuals must have a negative HIV-1 test immediately prior to initiating Truvada for HIV-1 PrEP.
If clinical symptoms consistent with acute viral infection are present and recent (less than 1 month) exposures are suspected, delay starting PrEP for at least one month and reconfirm HIV-1 status or use a test cleared by the FDA as an aid in the diagnosis of HIV-1 infection, including acute or primary HIV-1 infection
When considering Truvada for HIV-1 PrEP, factors that help to identify individuals at risk may include:
– has partner(s) known to be HIV-1 infected, or
– engages in sexual activity within a high prevalence area or social network and has additional risk factors for HIV-1 acquisition, such as:
- inconsistent or no condom use.
- diagnosis of sexually transmitted infections.
- exchange of sex for commodities (such as money, food, shelter, or drugs).
- use of illicit drugs or alcohol dependence.
- incarceration.
- partner(s) of unknown HIV-1 status with any of the factors listed above.
Dosage and administration
2.1 Testing prior to initiation of Truvada for treatment of HIV-1 infection or for HIV-1 PrEP
Prior to or when initiating Truvada, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to initiation and during use of Truvada, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus
2.2 HIV-1 screening for individuals receiving Truvada for HIV-1 PrEP
Screen all patients for HIV-1 infection before initiating Truvada for HIV-1 PrEP and at least once every 3 months while taking Truvada
2.5 Recommended dosage for HIV-1 PrEP
The dosage of Truvada in HIV-1 uninfected adults and adolescents weighing at least 35 kg is one tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food.
6.0 Adverse reactions
Clinical trials in adolescent subjects
In a single-arm, open-label clinical trial (ATN113), in which 67 HIV-1 uninfected adolescent (15 to 18 years of age) men who have sex with men received Truvada once daily for HIV-1 PrEP, the safety profile of Truvada was similar to that observed in adults. Median duration to exposure of Truvada was 47 weeks.
In the ATN113 trial, median BMD increased from baseline to Week 48, +2.58% for lumbar spine and +0.72% for total body. One subject had significant (greater than or equal to 4%) total body BMD loss at Week 24. Median changes from baseline BMD Z-scores were 0.0 for lumbar spine and −0.2 for total body at Week 48. Three subjects showed a worsening (change from greater than −2 to less than or equal to −2) from baseline in their lumbar spine or total body BMD Z-scores at Week 24 or 48. Interpretation of these data, however, may be limited by the low rate of adherence to Truvada by Week 48.
8.4 Pediatric use
HIV-1 PrEP
The safety and effectiveness of Truvada for HIV-1 PrEP in at-risk adolescents weighing at least 35 kg is supported by data from adequate and well-controlled studies of Truvada for HIV-1 PrEP in adults with additional data from safety and pharmacokinetic studies in previously conducted trials with the individual drug products, FTC and TDF, in HIV-1 infected adults and pediatric subjects.
Safety, adherence, and resistance were evaluated in a single-arm, open-label clinical trial (ATN113) in which 67 HIV-1 uninfected at-risk adolescent men who have sex with men received Truvada once daily for HIV-1 PrEP. The mean age of subjects was 17 years (range, 15-18 years); 46% were Hispanic, 52% black, and 37% white. The safety profile of Truvada in ATN113 was similar to that observed in the adult HIV-1 PrEP trials.
In the ATN113 trial, HIV-1 seroconversion occurred in three subjects. Tenofovir diphosphate levels in dried blood spot assays indicate that these subjects had poor adherence. No tenofovir- or FTC-associated HIV-1 resistance substitutions were detected in virus isolated from the three subjects who seroconverted.
Adherence to study drug, as demonstrated by tenofovir diphosphate levels in dried blood spot assays, declined markedly after Week 12 once subjects switched from monthly to quarterly visits, suggesting that adolescents may benefit from more frequent visits and counseling.
12.0 Clinical pharmacology
HIV-1 PrEP
The pharmacokinetic data for tenofovir and FTC following administration of Truvada in HIV-1 uninfected adolescents weighing 35 kg and above are not available. The dosage recommendations of Truvada for HIV-1 PrEP in this population are based on safety and adherence data from the ATN113 trial [see Use in Specific Populations (8.4)] and known pharmacokinetic information in HIV-infected adolescents taking TDF and FTC for treatment.
ResistanceATN113 Trial
In ATN113, a clinical trial of HIV-1 seronegative adolescent subjects [see Use in Specific Populations (8.4)], no amino acid substitutions associated with resistance to FTC or TDF were detected at the time of seroconversion from any of the 3 subjects who became infected with HIV-1 during the trial. All 3 subjects who seroconverted were nonadherent to the recommended Truvada dosage.
The updated label will soon be available at drugs@fda or DailyMed.
The following is the text of an announcement by the U.S. Food and Drug Administration regarding a revision in the Truvada label to expand the PrEP indication to include at-risk adolescents. The new label change has not yet been posted.
The Food and Drug Administration approved revisions to the Truvada (emtricitabine and tenofovir disoproxil fumarate) labeling to expand the Pre-Exposure Prophylaxis (PrEP) indication to include adolescents weighing at least 35 kg who are at risk of HIV-1 acquisition. The major labeling changes with respect to this expanded indication are summarized below. In addition, Section 8 was reformatted per the Pregnancy and Lactation Labeling Rule (PLLR) and includes updated information specific to the use of Truvada for PrEP during pregnancy and breastfeeding. Other sections of labeling were reformatted for consistency with current and best labeling practices, as well as with labeling for other HIV fixed-dose combination products.
Indications and usage
1.2 HIV-1 pre-exposure prophylaxis (PrEP)
Truvada is indicated in combination with safer sex practices for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV-1 in at-risk adults and adolescents weighing at least 35 kg. Individuals must have a negative HIV-1 test immediately prior to initiating Truvada for HIV-1 PrEP.
If clinical symptoms consistent with acute viral infection are present and recent (less than 1 month) exposures are suspected, delay starting PrEP for at least one month and reconfirm HIV-1 status or use a test cleared by the FDA as an aid in the diagnosis of HIV-1 infection, including acute or primary HIV-1 infection
When considering Truvada for HIV-1 PrEP, factors that help to identify individuals at risk may include:
– has partner(s) known to be HIV-1 infected, or
– engages in sexual activity within a high prevalence area or social network and has additional risk factors for HIV-1 acquisition, such as:
- inconsistent or no condom use.
- diagnosis of sexually transmitted infections.
- exchange of sex for commodities (such as money, food, shelter, or drugs).
- use of illicit drugs or alcohol dependence.
- incarceration.
- partner(s) of unknown HIV-1 status with any of the factors listed above.
Dosage and administration
2.1 Testing prior to initiation of Truvada for treatment of HIV-1 infection or for HIV-1 PrEP
Prior to or when initiating Truvada, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to initiation and during use of Truvada, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus
2.2 HIV-1 screening for individuals receiving Truvada for HIV-1 PrEP
Screen all patients for HIV-1 infection before initiating Truvada for HIV-1 PrEP and at least once every 3 months while taking Truvada
2.5 Recommended dosage for HIV-1 PrEP
The dosage of Truvada in HIV-1 uninfected adults and adolescents weighing at least 35 kg is one tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food.
6.0 Adverse reactions
Clinical trials in adolescent subjects
In a single-arm, open-label clinical trial (ATN113), in which 67 HIV-1 uninfected adolescent (15 to 18 years of age) men who have sex with men received Truvada once daily for HIV-1 PrEP, the safety profile of Truvada was similar to that observed in adults. Median duration to exposure of Truvada was 47 weeks.
In the ATN113 trial, median BMD increased from baseline to Week 48, +2.58% for lumbar spine and +0.72% for total body. One subject had significant (greater than or equal to 4%) total body BMD loss at Week 24. Median changes from baseline BMD Z-scores were 0.0 for lumbar spine and −0.2 for total body at Week 48. Three subjects showed a worsening (change from greater than −2 to less than or equal to −2) from baseline in their lumbar spine or total body BMD Z-scores at Week 24 or 48. Interpretation of these data, however, may be limited by the low rate of adherence to Truvada by Week 48.
8.4 Pediatric use
HIV-1 PrEP
The safety and effectiveness of Truvada for HIV-1 PrEP in at-risk adolescents weighing at least 35 kg is supported by data from adequate and well-controlled studies of Truvada for HIV-1 PrEP in adults with additional data from safety and pharmacokinetic studies in previously conducted trials with the individual drug products, FTC and TDF, in HIV-1 infected adults and pediatric subjects.
Safety, adherence, and resistance were evaluated in a single-arm, open-label clinical trial (ATN113) in which 67 HIV-1 uninfected at-risk adolescent men who have sex with men received Truvada once daily for HIV-1 PrEP. The mean age of subjects was 17 years (range, 15-18 years); 46% were Hispanic, 52% black, and 37% white. The safety profile of Truvada in ATN113 was similar to that observed in the adult HIV-1 PrEP trials.
In the ATN113 trial, HIV-1 seroconversion occurred in three subjects. Tenofovir diphosphate levels in dried blood spot assays indicate that these subjects had poor adherence. No tenofovir- or FTC-associated HIV-1 resistance substitutions were detected in virus isolated from the three subjects who seroconverted.
Adherence to study drug, as demonstrated by tenofovir diphosphate levels in dried blood spot assays, declined markedly after Week 12 once subjects switched from monthly to quarterly visits, suggesting that adolescents may benefit from more frequent visits and counseling.
12.0 Clinical pharmacology
HIV-1 PrEP
The pharmacokinetic data for tenofovir and FTC following administration of Truvada in HIV-1 uninfected adolescents weighing 35 kg and above are not available. The dosage recommendations of Truvada for HIV-1 PrEP in this population are based on safety and adherence data from the ATN113 trial [see Use in Specific Populations (8.4)] and known pharmacokinetic information in HIV-infected adolescents taking TDF and FTC for treatment.
ResistanceATN113 Trial
In ATN113, a clinical trial of HIV-1 seronegative adolescent subjects [see Use in Specific Populations (8.4)], no amino acid substitutions associated with resistance to FTC or TDF were detected at the time of seroconversion from any of the 3 subjects who became infected with HIV-1 during the trial. All 3 subjects who seroconverted were nonadherent to the recommended Truvada dosage.
The updated label will soon be available at drugs@fda or DailyMed.
FDA approves marketing of device to control GI bleeding
The Food and Drug Administration announced May 7 that it has permitted marketing of .
Hemospray is an aerosolized spray device that delivers a mineral blend to the bleeding site in the GI tract and is applied during endoscopic procedures and can cover large ulcers or tumors.
The FDA evaluated data from clinical studies consisting of 228 patients with upper and lower GI bleeding, supplemented with evidence from medical literature, including an additional 522 patients. The studies found that Hemospray stopped GI bleeding in 95% of patients within 5 minutes of device usage. Results also found that bleeding recurred, usually within 72 hours, and up to 30 days following device usage, in 20% of patients. Bowel perforation was observed as a serious side effect in approximately 1% of patients.
“The device provides an additional, nonsurgical option for treating upper and lower GI bleeding in certain patients, and may help reduce the risk of death from a GI bleed for many patients,” said Binita Ashar, MD, director, division of surgical devices, in the FDA’s Center for Devices and Radiological Health in a press release.
Hemospray is not intended for patients who have a gastrointestinal fistula or are at high risk for GI perforation. The device is not intended for use in patients with variceal bleeding. The FDA permitted the marketing of the Hemospray device to Wilson-Cook Medical.*
Read the full press release here.
Correction, 5/10/18: An earlier version of this article incorrectly described the patient population that should not be treated with Hemospray.
The Food and Drug Administration announced May 7 that it has permitted marketing of .
Hemospray is an aerosolized spray device that delivers a mineral blend to the bleeding site in the GI tract and is applied during endoscopic procedures and can cover large ulcers or tumors.
The FDA evaluated data from clinical studies consisting of 228 patients with upper and lower GI bleeding, supplemented with evidence from medical literature, including an additional 522 patients. The studies found that Hemospray stopped GI bleeding in 95% of patients within 5 minutes of device usage. Results also found that bleeding recurred, usually within 72 hours, and up to 30 days following device usage, in 20% of patients. Bowel perforation was observed as a serious side effect in approximately 1% of patients.
“The device provides an additional, nonsurgical option for treating upper and lower GI bleeding in certain patients, and may help reduce the risk of death from a GI bleed for many patients,” said Binita Ashar, MD, director, division of surgical devices, in the FDA’s Center for Devices and Radiological Health in a press release.
Hemospray is not intended for patients who have a gastrointestinal fistula or are at high risk for GI perforation. The device is not intended for use in patients with variceal bleeding. The FDA permitted the marketing of the Hemospray device to Wilson-Cook Medical.*
Read the full press release here.
Correction, 5/10/18: An earlier version of this article incorrectly described the patient population that should not be treated with Hemospray.
The Food and Drug Administration announced May 7 that it has permitted marketing of .
Hemospray is an aerosolized spray device that delivers a mineral blend to the bleeding site in the GI tract and is applied during endoscopic procedures and can cover large ulcers or tumors.
The FDA evaluated data from clinical studies consisting of 228 patients with upper and lower GI bleeding, supplemented with evidence from medical literature, including an additional 522 patients. The studies found that Hemospray stopped GI bleeding in 95% of patients within 5 minutes of device usage. Results also found that bleeding recurred, usually within 72 hours, and up to 30 days following device usage, in 20% of patients. Bowel perforation was observed as a serious side effect in approximately 1% of patients.
“The device provides an additional, nonsurgical option for treating upper and lower GI bleeding in certain patients, and may help reduce the risk of death from a GI bleed for many patients,” said Binita Ashar, MD, director, division of surgical devices, in the FDA’s Center for Devices and Radiological Health in a press release.
Hemospray is not intended for patients who have a gastrointestinal fistula or are at high risk for GI perforation. The device is not intended for use in patients with variceal bleeding. The FDA permitted the marketing of the Hemospray device to Wilson-Cook Medical.*
Read the full press release here.
Correction, 5/10/18: An earlier version of this article incorrectly described the patient population that should not be treated with Hemospray.
Zika topped Lyme in 2016
Ticks are the arthropod ride of choice for vector-borne diseases in the United States, but the Zika virus and its mosquito minions gave the ticks and their bacterial passengers a run for their money in 2016, according to the Centers for Disease Control and Prevention.
There were 41,680 cases of Zika virus that year, more than any other vector-borne disease, including Lyme disease, which had been the most common transmissible pathogen going back to at least 2004, when arthropod-borne viral diseases became nationally notifiable, said Ronald Rosenberg, ScD, and his associates at the CDC’s National Center for Emerging and Zoonotic Infectious Diseases in Fort Collins, Colo.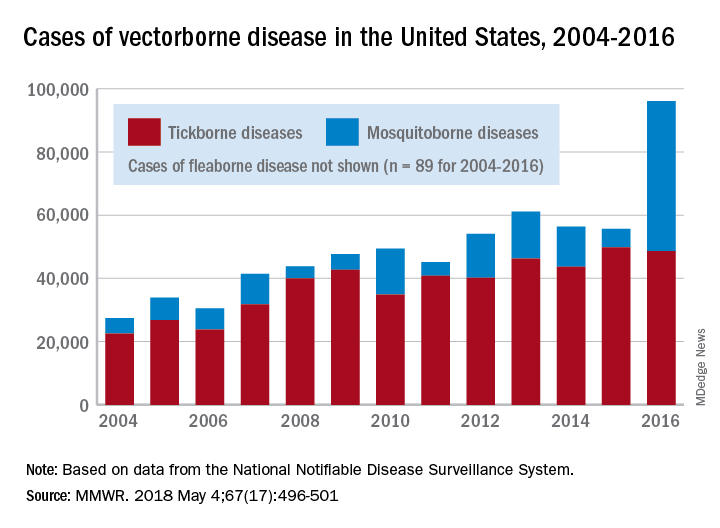
Since 2004, there have been 643,000 reported cases of vector-borne disease in the United States: 492,000 cases of tick-borne disease, of which over 402,000 were Lyme disease; 151,000 cases of mosquito-borne disease; and 89 cases of plague carried by the third type of vector, fleas, Dr. Rosenberg and his associates said based on data from the National Notifiable Disease Surveillance System.
In 2004, there were 22,527 cases of tick-borne disease and 4,858 cases of mosquito-borne disease, and the increases since then reflect the dynamics of the pathogens and vectors involved. Growth of tick-borne disease has been gradual: “Tick-borne pathogens rarely cause sudden epidemics because humans are typically incidental hosts who do not transmit further, and tick mobility is mostly limited to that of its animal hosts,” the researchers explained.
The number of mosquito-borne disease cases, on the other hand, varies considerably from year to year: There were 5,800 cases in 2015, almost 15,000 in 2013, and only 4,400 in 2011. Unlike ticks, which may feed on blood only once in a year, the more mobile mosquitoes feed every 48-72 hours and transmit their pathogens “directly between humans … resulting in explosive epidemics,” the investigators wrote.
SOURCE: Rosenberg R et al. MMWR 2018 May 4;67(17):496-501.
Ticks are the arthropod ride of choice for vector-borne diseases in the United States, but the Zika virus and its mosquito minions gave the ticks and their bacterial passengers a run for their money in 2016, according to the Centers for Disease Control and Prevention.
There were 41,680 cases of Zika virus that year, more than any other vector-borne disease, including Lyme disease, which had been the most common transmissible pathogen going back to at least 2004, when arthropod-borne viral diseases became nationally notifiable, said Ronald Rosenberg, ScD, and his associates at the CDC’s National Center for Emerging and Zoonotic Infectious Diseases in Fort Collins, Colo.
Since 2004, there have been 643,000 reported cases of vector-borne disease in the United States: 492,000 cases of tick-borne disease, of which over 402,000 were Lyme disease; 151,000 cases of mosquito-borne disease; and 89 cases of plague carried by the third type of vector, fleas, Dr. Rosenberg and his associates said based on data from the National Notifiable Disease Surveillance System.
In 2004, there were 22,527 cases of tick-borne disease and 4,858 cases of mosquito-borne disease, and the increases since then reflect the dynamics of the pathogens and vectors involved. Growth of tick-borne disease has been gradual: “Tick-borne pathogens rarely cause sudden epidemics because humans are typically incidental hosts who do not transmit further, and tick mobility is mostly limited to that of its animal hosts,” the researchers explained.
The number of mosquito-borne disease cases, on the other hand, varies considerably from year to year: There were 5,800 cases in 2015, almost 15,000 in 2013, and only 4,400 in 2011. Unlike ticks, which may feed on blood only once in a year, the more mobile mosquitoes feed every 48-72 hours and transmit their pathogens “directly between humans … resulting in explosive epidemics,” the investigators wrote.
SOURCE: Rosenberg R et al. MMWR 2018 May 4;67(17):496-501.
Ticks are the arthropod ride of choice for vector-borne diseases in the United States, but the Zika virus and its mosquito minions gave the ticks and their bacterial passengers a run for their money in 2016, according to the Centers for Disease Control and Prevention.
There were 41,680 cases of Zika virus that year, more than any other vector-borne disease, including Lyme disease, which had been the most common transmissible pathogen going back to at least 2004, when arthropod-borne viral diseases became nationally notifiable, said Ronald Rosenberg, ScD, and his associates at the CDC’s National Center for Emerging and Zoonotic Infectious Diseases in Fort Collins, Colo.
Since 2004, there have been 643,000 reported cases of vector-borne disease in the United States: 492,000 cases of tick-borne disease, of which over 402,000 were Lyme disease; 151,000 cases of mosquito-borne disease; and 89 cases of plague carried by the third type of vector, fleas, Dr. Rosenberg and his associates said based on data from the National Notifiable Disease Surveillance System.
In 2004, there were 22,527 cases of tick-borne disease and 4,858 cases of mosquito-borne disease, and the increases since then reflect the dynamics of the pathogens and vectors involved. Growth of tick-borne disease has been gradual: “Tick-borne pathogens rarely cause sudden epidemics because humans are typically incidental hosts who do not transmit further, and tick mobility is mostly limited to that of its animal hosts,” the researchers explained.
The number of mosquito-borne disease cases, on the other hand, varies considerably from year to year: There were 5,800 cases in 2015, almost 15,000 in 2013, and only 4,400 in 2011. Unlike ticks, which may feed on blood only once in a year, the more mobile mosquitoes feed every 48-72 hours and transmit their pathogens “directly between humans … resulting in explosive epidemics,” the investigators wrote.
SOURCE: Rosenberg R et al. MMWR 2018 May 4;67(17):496-501.
FROM MMWR
Inadequate antibiotic use persists in gonorrhea
Inappropriate treatment of gonorrhea persists despite growing antibiotic resistance, investigators reported in the Morbidity and Mortality Weekly Report.
In 2016, about 19% of gonorrhea cases diagnosed in the United States were not treated according to recommendations from the Centers for Disease Control and Prevention, wrote Emily J. Weston, MPH, and her associates. They recommended additional training and education on the need for providers to follow treatment recommendations.
![]()
To assess adherence to this recommendation, Ms. Weston and her associates analyzed data for 3,213 gonorrhea cases with complete treatment data reported in the United States in 2016. The cases spanned seven CDC sentinel surveillance jurisdictions, including California (excluding San Francisco), Florida, Massachusetts, Minnesota, Philadelphia (Pa.), Baltimore (Md.), and Multnomah County (Oregon).
In all, 18.7% of patients did not receive CDC-recommended treatment, the investigators reported. Inappropriate treatment most often consisted of ceftriaxone (250 mg) alone (5.9%), ceftriaxone with doxycycline (4.4%), azithromycin only (3.1%), ceftriaxone with azithromycin of other or unknown dosages (2.1%), or doxycycline only (1.2%).
Rates of appropriate treatment varied from 79% to 83% among individual jurisdictions and were unrelated to patient race, ethnicity, sex, or age, the researchers found. Men who had sex with men were more likely to receive recommended treatment (85%) than were heterosexual men and women (79%). Patients also were more likely to receive appropriate treatment at family planning, reproductive health, or sexually transmitted disease clinics than in other health care settings.
The results highlight the need for state and local departments to identify and educate providers who are inadequately treating gonorrhea, the researchers concluded. “State and local health departments should continue to work with providers and patients to assure timely detection and treatment of gonorrhea according to current CDC treatment recommendations.”
The study received no external funding. The investigators reported having no conflicts of interest.
SOURCE: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Inappropriate treatment of gonorrhea persists despite growing antibiotic resistance, investigators reported in the Morbidity and Mortality Weekly Report.
In 2016, about 19% of gonorrhea cases diagnosed in the United States were not treated according to recommendations from the Centers for Disease Control and Prevention, wrote Emily J. Weston, MPH, and her associates. They recommended additional training and education on the need for providers to follow treatment recommendations.
![]()
To assess adherence to this recommendation, Ms. Weston and her associates analyzed data for 3,213 gonorrhea cases with complete treatment data reported in the United States in 2016. The cases spanned seven CDC sentinel surveillance jurisdictions, including California (excluding San Francisco), Florida, Massachusetts, Minnesota, Philadelphia (Pa.), Baltimore (Md.), and Multnomah County (Oregon).
In all, 18.7% of patients did not receive CDC-recommended treatment, the investigators reported. Inappropriate treatment most often consisted of ceftriaxone (250 mg) alone (5.9%), ceftriaxone with doxycycline (4.4%), azithromycin only (3.1%), ceftriaxone with azithromycin of other or unknown dosages (2.1%), or doxycycline only (1.2%).
Rates of appropriate treatment varied from 79% to 83% among individual jurisdictions and were unrelated to patient race, ethnicity, sex, or age, the researchers found. Men who had sex with men were more likely to receive recommended treatment (85%) than were heterosexual men and women (79%). Patients also were more likely to receive appropriate treatment at family planning, reproductive health, or sexually transmitted disease clinics than in other health care settings.
The results highlight the need for state and local departments to identify and educate providers who are inadequately treating gonorrhea, the researchers concluded. “State and local health departments should continue to work with providers and patients to assure timely detection and treatment of gonorrhea according to current CDC treatment recommendations.”
The study received no external funding. The investigators reported having no conflicts of interest.
SOURCE: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Inappropriate treatment of gonorrhea persists despite growing antibiotic resistance, investigators reported in the Morbidity and Mortality Weekly Report.
In 2016, about 19% of gonorrhea cases diagnosed in the United States were not treated according to recommendations from the Centers for Disease Control and Prevention, wrote Emily J. Weston, MPH, and her associates. They recommended additional training and education on the need for providers to follow treatment recommendations.
![]()
To assess adherence to this recommendation, Ms. Weston and her associates analyzed data for 3,213 gonorrhea cases with complete treatment data reported in the United States in 2016. The cases spanned seven CDC sentinel surveillance jurisdictions, including California (excluding San Francisco), Florida, Massachusetts, Minnesota, Philadelphia (Pa.), Baltimore (Md.), and Multnomah County (Oregon).
In all, 18.7% of patients did not receive CDC-recommended treatment, the investigators reported. Inappropriate treatment most often consisted of ceftriaxone (250 mg) alone (5.9%), ceftriaxone with doxycycline (4.4%), azithromycin only (3.1%), ceftriaxone with azithromycin of other or unknown dosages (2.1%), or doxycycline only (1.2%).
Rates of appropriate treatment varied from 79% to 83% among individual jurisdictions and were unrelated to patient race, ethnicity, sex, or age, the researchers found. Men who had sex with men were more likely to receive recommended treatment (85%) than were heterosexual men and women (79%). Patients also were more likely to receive appropriate treatment at family planning, reproductive health, or sexually transmitted disease clinics than in other health care settings.
The results highlight the need for state and local departments to identify and educate providers who are inadequately treating gonorrhea, the researchers concluded. “State and local health departments should continue to work with providers and patients to assure timely detection and treatment of gonorrhea according to current CDC treatment recommendations.”
The study received no external funding. The investigators reported having no conflicts of interest.
SOURCE: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Key clinical point: Inadequate treatment of gonorrhea persists despite its growing antibiotic resistance.
Major finding: In all, 18.7% patients did not receive CDC-recommended treatment.
Study details: Analyses of 3,213 cases from seven U.S. jurisdictions.
Disclosures: The investigators reported no external funding sources. They reported having no conflicts of interest.
Source: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
FDA: More COPD patients can use triple therapy
The Food and Drug Administration has approved a new indication for the chronic obstructive pulmonary disease (COPD) therapy fluticasone furoate/umeclidinium/vilanterol (Trelegy Ellipta), which allows physicians to prescribe the drug to a broader class of COPD patients, according to a statement from two pharmaceutical companies.

“Following the initial approval of Trelegy Ellipta in September, we have analysed the data from the IMPACT study and identified additional benefits that this important medicine offers patients with [COPD],” said Hal Barron, MD, chief scientific officer and president of research and development at GlaxoSmithKline, in the statement. “We are pleased that the robust data from the IMPACT study has enabled the expanded indication announced today and the FDA action has been taken so swiftly.”
The results of the IMPACT trial, which was the first study to compare a single-inhaler triple therapy with two dual therapies, were published on April 18 (N Engl J Med 2018. doi: 10.1056/NEJMoa1713901).
This study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction, compared with vilanterol-umeclidinium (P less than .001 for both).
The Food and Drug Administration has approved a new indication for the chronic obstructive pulmonary disease (COPD) therapy fluticasone furoate/umeclidinium/vilanterol (Trelegy Ellipta), which allows physicians to prescribe the drug to a broader class of COPD patients, according to a statement from two pharmaceutical companies.
“Following the initial approval of Trelegy Ellipta in September, we have analysed the data from the IMPACT study and identified additional benefits that this important medicine offers patients with [COPD],” said Hal Barron, MD, chief scientific officer and president of research and development at GlaxoSmithKline, in the statement. “We are pleased that the robust data from the IMPACT study has enabled the expanded indication announced today and the FDA action has been taken so swiftly.”
The results of the IMPACT trial, which was the first study to compare a single-inhaler triple therapy with two dual therapies, were published on April 18 (N Engl J Med 2018. doi: 10.1056/NEJMoa1713901).
This study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction, compared with vilanterol-umeclidinium (P less than .001 for both).
The Food and Drug Administration has approved a new indication for the chronic obstructive pulmonary disease (COPD) therapy fluticasone furoate/umeclidinium/vilanterol (Trelegy Ellipta), which allows physicians to prescribe the drug to a broader class of COPD patients, according to a statement from two pharmaceutical companies.
“Following the initial approval of Trelegy Ellipta in September, we have analysed the data from the IMPACT study and identified additional benefits that this important medicine offers patients with [COPD],” said Hal Barron, MD, chief scientific officer and president of research and development at GlaxoSmithKline, in the statement. “We are pleased that the robust data from the IMPACT study has enabled the expanded indication announced today and the FDA action has been taken so swiftly.”
The results of the IMPACT trial, which was the first study to compare a single-inhaler triple therapy with two dual therapies, were published on April 18 (N Engl J Med 2018. doi: 10.1056/NEJMoa1713901).
This study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction, compared with vilanterol-umeclidinium (P less than .001 for both).