User login
Genentech submits sNDA for venetoclax in untreated AML
A supplemental new drug application (sNDA) for venetoclax (Venclexta) used in combination with either a hypomethylating agent or low-dose cytarabine (LDAC) for previously untreated acute myeloid leukemia has been submitted to the Food and Drug Administration by Genentech, which developed it.
Specifically, the sNDA is for these drug combinations in the treatment of AML patients ineligible for intensive chemotherapy, according to the announcement from Genentech.
The sNDA is based on results of two trials that included patients in this population. In the phase 1b M14-358 (NCT02203773), venetoclax was combined with either azacitidine or decitabine; patients treated with 400 mg of venetoclax had a complete remission rate of 73%, and the median overall survival across all doses of venetoclax was 17.5 months. Low white blood cell count with fever, low white blood cell count, anemia, low platelet count, and decreased potassium levels were the most common grade 3/4 adverse events (occurring in 10% or more of patients). In the phase 1b/2 study M14-387 (NCT02287233), venetoclax was used in combination with LDAC; patients treated with a 600-mg dose of venetoclax showed a complete response rate of 62%, and a median overall survival of 11.4 months. Low white blood cell count with fever, decreased potassium levels, pneumonia, disease progression, decreased phosphate levels, high blood pressure, and sepsis were the most common grade 3/4 adverse events seen in this study.
This sNDA follows FDA breakthrough therapy designations, based on these same trials, for these uses of venetoclax with either hypomethylating agents or LDAC. The FDA also recently approved venetoclax in combination with rituximab (Rituxan) for treatment of patients who have chronic lymphocytic leukemia or small lymphocytic lymphoma, with or without 17p depletion, and have been treated with at least one prior therapy.
“AML is an aggressive disease with the lowest survival rate of all leukemias, and we look forward to working closely with the FDA to bring this potential option to patients with this very difficult-to-treat blood cancer as soon as possible,” said Sandra Horning, MD, chief medical officer at Genentech.
More information is included in the full release.
A supplemental new drug application (sNDA) for venetoclax (Venclexta) used in combination with either a hypomethylating agent or low-dose cytarabine (LDAC) for previously untreated acute myeloid leukemia has been submitted to the Food and Drug Administration by Genentech, which developed it.
Specifically, the sNDA is for these drug combinations in the treatment of AML patients ineligible for intensive chemotherapy, according to the announcement from Genentech.
The sNDA is based on results of two trials that included patients in this population. In the phase 1b M14-358 (NCT02203773), venetoclax was combined with either azacitidine or decitabine; patients treated with 400 mg of venetoclax had a complete remission rate of 73%, and the median overall survival across all doses of venetoclax was 17.5 months. Low white blood cell count with fever, low white blood cell count, anemia, low platelet count, and decreased potassium levels were the most common grade 3/4 adverse events (occurring in 10% or more of patients). In the phase 1b/2 study M14-387 (NCT02287233), venetoclax was used in combination with LDAC; patients treated with a 600-mg dose of venetoclax showed a complete response rate of 62%, and a median overall survival of 11.4 months. Low white blood cell count with fever, decreased potassium levels, pneumonia, disease progression, decreased phosphate levels, high blood pressure, and sepsis were the most common grade 3/4 adverse events seen in this study.
This sNDA follows FDA breakthrough therapy designations, based on these same trials, for these uses of venetoclax with either hypomethylating agents or LDAC. The FDA also recently approved venetoclax in combination with rituximab (Rituxan) for treatment of patients who have chronic lymphocytic leukemia or small lymphocytic lymphoma, with or without 17p depletion, and have been treated with at least one prior therapy.
“AML is an aggressive disease with the lowest survival rate of all leukemias, and we look forward to working closely with the FDA to bring this potential option to patients with this very difficult-to-treat blood cancer as soon as possible,” said Sandra Horning, MD, chief medical officer at Genentech.
More information is included in the full release.
A supplemental new drug application (sNDA) for venetoclax (Venclexta) used in combination with either a hypomethylating agent or low-dose cytarabine (LDAC) for previously untreated acute myeloid leukemia has been submitted to the Food and Drug Administration by Genentech, which developed it.
Specifically, the sNDA is for these drug combinations in the treatment of AML patients ineligible for intensive chemotherapy, according to the announcement from Genentech.
The sNDA is based on results of two trials that included patients in this population. In the phase 1b M14-358 (NCT02203773), venetoclax was combined with either azacitidine or decitabine; patients treated with 400 mg of venetoclax had a complete remission rate of 73%, and the median overall survival across all doses of venetoclax was 17.5 months. Low white blood cell count with fever, low white blood cell count, anemia, low platelet count, and decreased potassium levels were the most common grade 3/4 adverse events (occurring in 10% or more of patients). In the phase 1b/2 study M14-387 (NCT02287233), venetoclax was used in combination with LDAC; patients treated with a 600-mg dose of venetoclax showed a complete response rate of 62%, and a median overall survival of 11.4 months. Low white blood cell count with fever, decreased potassium levels, pneumonia, disease progression, decreased phosphate levels, high blood pressure, and sepsis were the most common grade 3/4 adverse events seen in this study.
This sNDA follows FDA breakthrough therapy designations, based on these same trials, for these uses of venetoclax with either hypomethylating agents or LDAC. The FDA also recently approved venetoclax in combination with rituximab (Rituxan) for treatment of patients who have chronic lymphocytic leukemia or small lymphocytic lymphoma, with or without 17p depletion, and have been treated with at least one prior therapy.
“AML is an aggressive disease with the lowest survival rate of all leukemias, and we look forward to working closely with the FDA to bring this potential option to patients with this very difficult-to-treat blood cancer as soon as possible,” said Sandra Horning, MD, chief medical officer at Genentech.
More information is included in the full release.
Fluoroquinolones can cause fatal hypoglycemia, FDA warns
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
FDA gives green light to freeze-dried plasma in combat
The Department of Defense has received emergency use authorization from the Food and Drug Administration to use pathogen-reduced, leukocyte-depleted, freeze-dried plasma for the emergency treatment of hemorrhage and coagulopathy in combat situations.
Hemorrhage and coagulopathy are the leading causes of preventable deaths among combat trauma casualties. While plasma contains proteins that help clot blood and thus can treat these conditions, it isn’t feasible to keep it on hand for combat emergencies in the field because of logistical and operational requirements, such as refrigeration or thawing periods. This freeze-dried plasma product, on the other hand, can be easily reconstituted in situations in which refrigeration isn’t possible.
The FDA authorization allows for the use of a French-made, powdered, freeze-dried product. This emergency use authorization came about in part because of a joint program established between the FDA and the Department of Defense in January 2018.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country, by expediting the development and availability of safe and effective, priority medical products that are essential to the health of our military service members,” said FDA commissioner Scott Gottlieb, MD. “This is especially true when it comes to products used to treat injuries in a potential battlefield setting.”
More information about this emergency use authorization can be found in the FDA’s full press announcement.
The Department of Defense has received emergency use authorization from the Food and Drug Administration to use pathogen-reduced, leukocyte-depleted, freeze-dried plasma for the emergency treatment of hemorrhage and coagulopathy in combat situations.
Hemorrhage and coagulopathy are the leading causes of preventable deaths among combat trauma casualties. While plasma contains proteins that help clot blood and thus can treat these conditions, it isn’t feasible to keep it on hand for combat emergencies in the field because of logistical and operational requirements, such as refrigeration or thawing periods. This freeze-dried plasma product, on the other hand, can be easily reconstituted in situations in which refrigeration isn’t possible.
The FDA authorization allows for the use of a French-made, powdered, freeze-dried product. This emergency use authorization came about in part because of a joint program established between the FDA and the Department of Defense in January 2018.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country, by expediting the development and availability of safe and effective, priority medical products that are essential to the health of our military service members,” said FDA commissioner Scott Gottlieb, MD. “This is especially true when it comes to products used to treat injuries in a potential battlefield setting.”
More information about this emergency use authorization can be found in the FDA’s full press announcement.
The Department of Defense has received emergency use authorization from the Food and Drug Administration to use pathogen-reduced, leukocyte-depleted, freeze-dried plasma for the emergency treatment of hemorrhage and coagulopathy in combat situations.
Hemorrhage and coagulopathy are the leading causes of preventable deaths among combat trauma casualties. While plasma contains proteins that help clot blood and thus can treat these conditions, it isn’t feasible to keep it on hand for combat emergencies in the field because of logistical and operational requirements, such as refrigeration or thawing periods. This freeze-dried plasma product, on the other hand, can be easily reconstituted in situations in which refrigeration isn’t possible.
The FDA authorization allows for the use of a French-made, powdered, freeze-dried product. This emergency use authorization came about in part because of a joint program established between the FDA and the Department of Defense in January 2018.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country, by expediting the development and availability of safe and effective, priority medical products that are essential to the health of our military service members,” said FDA commissioner Scott Gottlieb, MD. “This is especially true when it comes to products used to treat injuries in a potential battlefield setting.”
More information about this emergency use authorization can be found in the FDA’s full press announcement.
FDA approves Aristada Initio for schizophrenia
The Food and Drug Administration has approved aripiprazole lauroxil (Aristada Initio) for the initiation of aripiprazole lauroxil (Aristada) for treating schizophrenia in adults, the drug’s developer, Alkermes, announced July 2 in a press release.
“,” Craig Hopkinson, MD, chief medical officer at Alkermes, said in the press release.
The standard initiation regimen for aripiprazole lauroxil previously was 21 consecutive days of oral aripiprazole starting with the first dose of Aristada. The alternative initiation regimen allows patients to achieve “relevant levels of aripiprazole within 4 days of initiation,” according to Alkermes. The result gives health care providers “an additional tool to support patients.”
One important advantage of Aristada Initio is that, in addition to monthly and 6-week dosing options, it offers a 2-month option.
Aristada and Aristada Initio both contain aripiprazole lauroxil, but the drugs are not interchangeable because they have different pharmacokinetic profiles, the company said. In addition, Aristada Initio is to be administered a single time only.
Aristada Initio has exhibited some of the same adverse events as other atypical antipsychotics, including neuroleptic malignant syndrome, tardive dyskinesia, and metabolic changes. Injection-site reactions also have been observed.
Aristada Initio is expected to become available by mid-July.
Full prescribing information and boxed warnings can be found on the Alkermes website.
The Food and Drug Administration has approved aripiprazole lauroxil (Aristada Initio) for the initiation of aripiprazole lauroxil (Aristada) for treating schizophrenia in adults, the drug’s developer, Alkermes, announced July 2 in a press release.
“,” Craig Hopkinson, MD, chief medical officer at Alkermes, said in the press release.
The standard initiation regimen for aripiprazole lauroxil previously was 21 consecutive days of oral aripiprazole starting with the first dose of Aristada. The alternative initiation regimen allows patients to achieve “relevant levels of aripiprazole within 4 days of initiation,” according to Alkermes. The result gives health care providers “an additional tool to support patients.”
One important advantage of Aristada Initio is that, in addition to monthly and 6-week dosing options, it offers a 2-month option.
Aristada and Aristada Initio both contain aripiprazole lauroxil, but the drugs are not interchangeable because they have different pharmacokinetic profiles, the company said. In addition, Aristada Initio is to be administered a single time only.
Aristada Initio has exhibited some of the same adverse events as other atypical antipsychotics, including neuroleptic malignant syndrome, tardive dyskinesia, and metabolic changes. Injection-site reactions also have been observed.
Aristada Initio is expected to become available by mid-July.
Full prescribing information and boxed warnings can be found on the Alkermes website.
The Food and Drug Administration has approved aripiprazole lauroxil (Aristada Initio) for the initiation of aripiprazole lauroxil (Aristada) for treating schizophrenia in adults, the drug’s developer, Alkermes, announced July 2 in a press release.
“,” Craig Hopkinson, MD, chief medical officer at Alkermes, said in the press release.
The standard initiation regimen for aripiprazole lauroxil previously was 21 consecutive days of oral aripiprazole starting with the first dose of Aristada. The alternative initiation regimen allows patients to achieve “relevant levels of aripiprazole within 4 days of initiation,” according to Alkermes. The result gives health care providers “an additional tool to support patients.”
One important advantage of Aristada Initio is that, in addition to monthly and 6-week dosing options, it offers a 2-month option.
Aristada and Aristada Initio both contain aripiprazole lauroxil, but the drugs are not interchangeable because they have different pharmacokinetic profiles, the company said. In addition, Aristada Initio is to be administered a single time only.
Aristada Initio has exhibited some of the same adverse events as other atypical antipsychotics, including neuroleptic malignant syndrome, tardive dyskinesia, and metabolic changes. Injection-site reactions also have been observed.
Aristada Initio is expected to become available by mid-July.
Full prescribing information and boxed warnings can be found on the Alkermes website.
Death rates rising for 10- to 19-year-olds
Mortality in this age group, which had dropped nearly 33% from 1999 to 2013, climbed from 29.6 per 100,000 population aged 10-19 years in 2013 to 33.1 per 100,000 in 2016, the last year for which data are available. Meanwhile, deaths from injuries – unintentional injuries, suicides, homicides, and legal intervention – went from 19.8 per 100,000 to 23.3, an increase of almost 18%, from 2013 to 2016, and the noninjury death rate “was relatively stable,” Sally C. Curtin and her associates at the NCHS Division of Vital Statistics said in a National Vital Statistics Report.
The recent surge in injury deaths was more substantial in the older half of the age group. The mortality rate for children aged 10-14 years went from a low of 6.4 per 100,000 in 2012 to 7.1 in 2016, an increase of 11%, while the rate for those aged 15-19 rose 19% as it jumped from 32.8 per 100,000 in 2013 to 39.0 in 2016, the investigators wrote in the report.
The rate of unintentional injury deaths in 10- to 19-year-olds shows the same pattern as all deaths and injury deaths: Decline from 1999 to 2013 and then a rise for the last 3 years. That recent rise also can be seen in the most common form of unintentional injury deaths, motor vehicle traffic accidents, and in poisoning deaths, although that uptick began a year later. Homicide deaths declined by one-third from 2007 to 2014 and then increased, while suicide rates have been rising since 2007, the investigators said. Legal intervention deaths, defined as those caused by law enforcement actions, were not included because of relatively small annual numbers.
“Although progress was made in reducing injury deaths among children and adolescents aged 10-19 years during 1999-2013, the recent upturn shows that persistent as well as emerging challenges remain. … Further reductions will require renewed focus and effort,” Ms. Curtin and her associates wrote.
SOURCE: Curtin SC et al. Natl Vital Stat Rep. 2018 Jun;67(4):1-16.
Mortality in this age group, which had dropped nearly 33% from 1999 to 2013, climbed from 29.6 per 100,000 population aged 10-19 years in 2013 to 33.1 per 100,000 in 2016, the last year for which data are available. Meanwhile, deaths from injuries – unintentional injuries, suicides, homicides, and legal intervention – went from 19.8 per 100,000 to 23.3, an increase of almost 18%, from 2013 to 2016, and the noninjury death rate “was relatively stable,” Sally C. Curtin and her associates at the NCHS Division of Vital Statistics said in a National Vital Statistics Report.
The recent surge in injury deaths was more substantial in the older half of the age group. The mortality rate for children aged 10-14 years went from a low of 6.4 per 100,000 in 2012 to 7.1 in 2016, an increase of 11%, while the rate for those aged 15-19 rose 19% as it jumped from 32.8 per 100,000 in 2013 to 39.0 in 2016, the investigators wrote in the report.
The rate of unintentional injury deaths in 10- to 19-year-olds shows the same pattern as all deaths and injury deaths: Decline from 1999 to 2013 and then a rise for the last 3 years. That recent rise also can be seen in the most common form of unintentional injury deaths, motor vehicle traffic accidents, and in poisoning deaths, although that uptick began a year later. Homicide deaths declined by one-third from 2007 to 2014 and then increased, while suicide rates have been rising since 2007, the investigators said. Legal intervention deaths, defined as those caused by law enforcement actions, were not included because of relatively small annual numbers.
“Although progress was made in reducing injury deaths among children and adolescents aged 10-19 years during 1999-2013, the recent upturn shows that persistent as well as emerging challenges remain. … Further reductions will require renewed focus and effort,” Ms. Curtin and her associates wrote.
SOURCE: Curtin SC et al. Natl Vital Stat Rep. 2018 Jun;67(4):1-16.
Mortality in this age group, which had dropped nearly 33% from 1999 to 2013, climbed from 29.6 per 100,000 population aged 10-19 years in 2013 to 33.1 per 100,000 in 2016, the last year for which data are available. Meanwhile, deaths from injuries – unintentional injuries, suicides, homicides, and legal intervention – went from 19.8 per 100,000 to 23.3, an increase of almost 18%, from 2013 to 2016, and the noninjury death rate “was relatively stable,” Sally C. Curtin and her associates at the NCHS Division of Vital Statistics said in a National Vital Statistics Report.
The recent surge in injury deaths was more substantial in the older half of the age group. The mortality rate for children aged 10-14 years went from a low of 6.4 per 100,000 in 2012 to 7.1 in 2016, an increase of 11%, while the rate for those aged 15-19 rose 19% as it jumped from 32.8 per 100,000 in 2013 to 39.0 in 2016, the investigators wrote in the report.
The rate of unintentional injury deaths in 10- to 19-year-olds shows the same pattern as all deaths and injury deaths: Decline from 1999 to 2013 and then a rise for the last 3 years. That recent rise also can be seen in the most common form of unintentional injury deaths, motor vehicle traffic accidents, and in poisoning deaths, although that uptick began a year later. Homicide deaths declined by one-third from 2007 to 2014 and then increased, while suicide rates have been rising since 2007, the investigators said. Legal intervention deaths, defined as those caused by law enforcement actions, were not included because of relatively small annual numbers.
“Although progress was made in reducing injury deaths among children and adolescents aged 10-19 years during 1999-2013, the recent upturn shows that persistent as well as emerging challenges remain. … Further reductions will require renewed focus and effort,” Ms. Curtin and her associates wrote.
SOURCE: Curtin SC et al. Natl Vital Stat Rep. 2018 Jun;67(4):1-16.
FROM NATIONAL VITAL STATISTICS REPORTS
FDA approves Zephyr endobronchial valve to treat severe emphysema
The valve is the first minimally invasive device approved in the United States for treating such patients, according to Pulmonx, the device manufacturer.
The FDA previously granted the novel device expedited review, as patients who did not respond to drug treatment had only limited alternative options, including lung volume reduction and lung transplant, Tina Kiang, PhD, of the FDA’s Center for Devices and Radiological Health, said in a press release. “This novel device is a less invasive treatment that expands the options available to patients,” said Dr. Kiang, acting director of the center’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices.![]()
The approval is based on a multicenter study of 190 patients with severe emphysema. A total of 128 received Zephyr valves and medical management, while 62 received medical management only. The primary measure was the number of patients who achieved at least a 15% improvement in their pulmonary function score: At 1 year, 47.7% of the Zephyr valve patients had achieved such improvement versus 16.8% of the control group, according to the FDA.
Adverse events included death, pneumothorax, pneumonia, worsening of emphysema, coughing up blood, shortness of breath, and chest pain. The valve is contraindicated in patients with active lung infections; those allergic to nitinol, nickel, titanium, or silicone; and active smokers.
Read more about this approval in the full FDA press announcement.
The valve is the first minimally invasive device approved in the United States for treating such patients, according to Pulmonx, the device manufacturer.
The FDA previously granted the novel device expedited review, as patients who did not respond to drug treatment had only limited alternative options, including lung volume reduction and lung transplant, Tina Kiang, PhD, of the FDA’s Center for Devices and Radiological Health, said in a press release. “This novel device is a less invasive treatment that expands the options available to patients,” said Dr. Kiang, acting director of the center’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices.![]()
The approval is based on a multicenter study of 190 patients with severe emphysema. A total of 128 received Zephyr valves and medical management, while 62 received medical management only. The primary measure was the number of patients who achieved at least a 15% improvement in their pulmonary function score: At 1 year, 47.7% of the Zephyr valve patients had achieved such improvement versus 16.8% of the control group, according to the FDA.
Adverse events included death, pneumothorax, pneumonia, worsening of emphysema, coughing up blood, shortness of breath, and chest pain. The valve is contraindicated in patients with active lung infections; those allergic to nitinol, nickel, titanium, or silicone; and active smokers.
Read more about this approval in the full FDA press announcement.
The valve is the first minimally invasive device approved in the United States for treating such patients, according to Pulmonx, the device manufacturer.
The FDA previously granted the novel device expedited review, as patients who did not respond to drug treatment had only limited alternative options, including lung volume reduction and lung transplant, Tina Kiang, PhD, of the FDA’s Center for Devices and Radiological Health, said in a press release. “This novel device is a less invasive treatment that expands the options available to patients,” said Dr. Kiang, acting director of the center’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices.![]()
The approval is based on a multicenter study of 190 patients with severe emphysema. A total of 128 received Zephyr valves and medical management, while 62 received medical management only. The primary measure was the number of patients who achieved at least a 15% improvement in their pulmonary function score: At 1 year, 47.7% of the Zephyr valve patients had achieved such improvement versus 16.8% of the control group, according to the FDA.
Adverse events included death, pneumothorax, pneumonia, worsening of emphysema, coughing up blood, shortness of breath, and chest pain. The valve is contraindicated in patients with active lung infections; those allergic to nitinol, nickel, titanium, or silicone; and active smokers.
Read more about this approval in the full FDA press announcement.
Cost led to missed care for 4.5% of Americans in 2017
The percentage of Americans who went without medical care due to cost rose to 4.5% in 2017, reversing a 6-year trend, the National Center for Health Statistics reported.
The rate was 4.4% in 2016, which represented a slowdown in what had been steady decline over the previous 5 years, according to data from the National Health Interview Survey. Declining rates corresponded with the implementation of early provisions of the Affordable Care Act in 2010.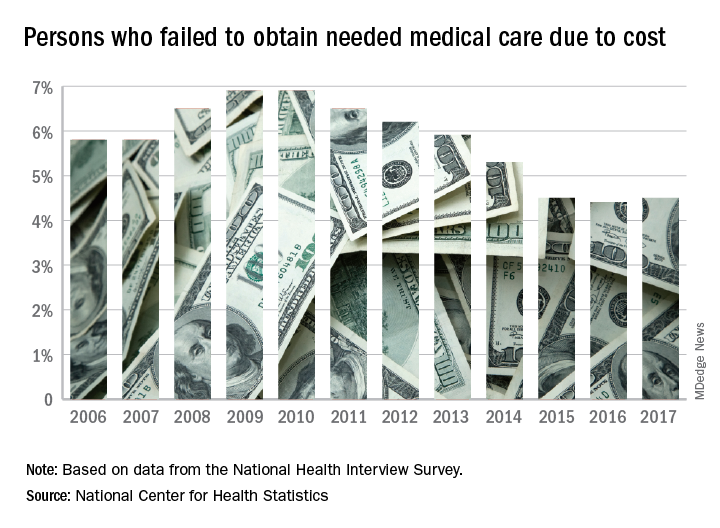
The 2017 rate varied considerably by age group. Not surprisingly, more working-age people – those aged 18-64 years – reported that they did not seek medical care at some point in the previous 12 months due to cost (6.1%). The rate was 1.2% for those under 18 years and 2.7% for the Medicare eligible – those aged 65 years and older.
For 2016, the rates were 6.2% for those aged 18-64 years, 1.2% for the under-18 group, and 2.1% for the 65+ group, the data show.
Among females of all ages in 2017, 4.8% failed to get needed care at some point in the previous year, compared with 4.1% of men. Those numbers were unchanged from 2016 but down from 4.9% for females in 2015 and up from 4.0% for males that year, the NCHS said.
In 2017, the rate also varied by race/ethnicity – 4.1% for whites, 5.3% for Hispanics, 6.1% for blacks – and by location – 4.1% for large metropolitan areas, 4.9% for small metro areas, and 5.5% for rural locales, according to the early release of survey data.
The percentage of Americans who went without medical care due to cost rose to 4.5% in 2017, reversing a 6-year trend, the National Center for Health Statistics reported.
The rate was 4.4% in 2016, which represented a slowdown in what had been steady decline over the previous 5 years, according to data from the National Health Interview Survey. Declining rates corresponded with the implementation of early provisions of the Affordable Care Act in 2010.
The 2017 rate varied considerably by age group. Not surprisingly, more working-age people – those aged 18-64 years – reported that they did not seek medical care at some point in the previous 12 months due to cost (6.1%). The rate was 1.2% for those under 18 years and 2.7% for the Medicare eligible – those aged 65 years and older.
For 2016, the rates were 6.2% for those aged 18-64 years, 1.2% for the under-18 group, and 2.1% for the 65+ group, the data show.
Among females of all ages in 2017, 4.8% failed to get needed care at some point in the previous year, compared with 4.1% of men. Those numbers were unchanged from 2016 but down from 4.9% for females in 2015 and up from 4.0% for males that year, the NCHS said.
In 2017, the rate also varied by race/ethnicity – 4.1% for whites, 5.3% for Hispanics, 6.1% for blacks – and by location – 4.1% for large metropolitan areas, 4.9% for small metro areas, and 5.5% for rural locales, according to the early release of survey data.
The percentage of Americans who went without medical care due to cost rose to 4.5% in 2017, reversing a 6-year trend, the National Center for Health Statistics reported.
The rate was 4.4% in 2016, which represented a slowdown in what had been steady decline over the previous 5 years, according to data from the National Health Interview Survey. Declining rates corresponded with the implementation of early provisions of the Affordable Care Act in 2010.
The 2017 rate varied considerably by age group. Not surprisingly, more working-age people – those aged 18-64 years – reported that they did not seek medical care at some point in the previous 12 months due to cost (6.1%). The rate was 1.2% for those under 18 years and 2.7% for the Medicare eligible – those aged 65 years and older.
For 2016, the rates were 6.2% for those aged 18-64 years, 1.2% for the under-18 group, and 2.1% for the 65+ group, the data show.
Among females of all ages in 2017, 4.8% failed to get needed care at some point in the previous year, compared with 4.1% of men. Those numbers were unchanged from 2016 but down from 4.9% for females in 2015 and up from 4.0% for males that year, the NCHS said.
In 2017, the rate also varied by race/ethnicity – 4.1% for whites, 5.3% for Hispanics, 6.1% for blacks – and by location – 4.1% for large metropolitan areas, 4.9% for small metro areas, and 5.5% for rural locales, according to the early release of survey data.
FDA issues recommendations to avoid surgical fires
The Food and Drug Administration on May 29 issued a set of recommendations to medical professionals and health care facility staff to reduce the occurrence of surgical fires on or near a patient.
Surgical fires most often occur when there is an oxygen-enriched environment (a concentration of greater than 30%). In addition to an oxygen source, the other two necessary elements of the “fire triangle” are an ignition source and a fuel source.
The recommendations discuss the safe use of devices or items that may serve as a source of any one of those three elements.
Oxygen: Evaluate if supplemental oxygen is needed. If it is, titrate to the minimum concentration needed for adequate saturation. Closed oxygen delivery systems (such as a laryngeal mask or endotracheal tube) are safer than open oxygen delivery systems (such as a nasal cannula or mask). If you must use an open system, take additional precautions to exclude oxygen and flammable/combustible gases from the operative field, such as draping techniques that avoid accumulation of oxygen.
Ignition sources: Consider alternatives to using an ignition source for surgery of the head, neck, and upper chest if high concentrations of supplemental oxygen are being delivered. Check for insulation failure before use, and keep devices clean of char and tissue. When not in use, place the devices safely away from the patient and drapes. Devices are safer to use if you can allow time for the oxygen concentration in the room to decrease.
Fuel sources: Ensure dry conditions prior to draping, avoiding pooling of alcohol-based antiseptics during skin preparation. Use the appropriate-sized applicator for the surgical site. Be aware of products that may serve as a fuel source, such as oxygen-trapping gauze, plastic laryngeal masks, and aware of potential patient sources such as hair or gastrointestinal gases.
Training should include how to manage fires that do occur – stop the ignition source, then extinguish the fire – and evacuation procedures.
Read the full recommendations here.
The Food and Drug Administration on May 29 issued a set of recommendations to medical professionals and health care facility staff to reduce the occurrence of surgical fires on or near a patient.
Surgical fires most often occur when there is an oxygen-enriched environment (a concentration of greater than 30%). In addition to an oxygen source, the other two necessary elements of the “fire triangle” are an ignition source and a fuel source.
The recommendations discuss the safe use of devices or items that may serve as a source of any one of those three elements.
Oxygen: Evaluate if supplemental oxygen is needed. If it is, titrate to the minimum concentration needed for adequate saturation. Closed oxygen delivery systems (such as a laryngeal mask or endotracheal tube) are safer than open oxygen delivery systems (such as a nasal cannula or mask). If you must use an open system, take additional precautions to exclude oxygen and flammable/combustible gases from the operative field, such as draping techniques that avoid accumulation of oxygen.
Ignition sources: Consider alternatives to using an ignition source for surgery of the head, neck, and upper chest if high concentrations of supplemental oxygen are being delivered. Check for insulation failure before use, and keep devices clean of char and tissue. When not in use, place the devices safely away from the patient and drapes. Devices are safer to use if you can allow time for the oxygen concentration in the room to decrease.
Fuel sources: Ensure dry conditions prior to draping, avoiding pooling of alcohol-based antiseptics during skin preparation. Use the appropriate-sized applicator for the surgical site. Be aware of products that may serve as a fuel source, such as oxygen-trapping gauze, plastic laryngeal masks, and aware of potential patient sources such as hair or gastrointestinal gases.
Training should include how to manage fires that do occur – stop the ignition source, then extinguish the fire – and evacuation procedures.
Read the full recommendations here.
The Food and Drug Administration on May 29 issued a set of recommendations to medical professionals and health care facility staff to reduce the occurrence of surgical fires on or near a patient.
Surgical fires most often occur when there is an oxygen-enriched environment (a concentration of greater than 30%). In addition to an oxygen source, the other two necessary elements of the “fire triangle” are an ignition source and a fuel source.
The recommendations discuss the safe use of devices or items that may serve as a source of any one of those three elements.
Oxygen: Evaluate if supplemental oxygen is needed. If it is, titrate to the minimum concentration needed for adequate saturation. Closed oxygen delivery systems (such as a laryngeal mask or endotracheal tube) are safer than open oxygen delivery systems (such as a nasal cannula or mask). If you must use an open system, take additional precautions to exclude oxygen and flammable/combustible gases from the operative field, such as draping techniques that avoid accumulation of oxygen.
Ignition sources: Consider alternatives to using an ignition source for surgery of the head, neck, and upper chest if high concentrations of supplemental oxygen are being delivered. Check for insulation failure before use, and keep devices clean of char and tissue. When not in use, place the devices safely away from the patient and drapes. Devices are safer to use if you can allow time for the oxygen concentration in the room to decrease.
Fuel sources: Ensure dry conditions prior to draping, avoiding pooling of alcohol-based antiseptics during skin preparation. Use the appropriate-sized applicator for the surgical site. Be aware of products that may serve as a fuel source, such as oxygen-trapping gauze, plastic laryngeal masks, and aware of potential patient sources such as hair or gastrointestinal gases.
Training should include how to manage fires that do occur – stop the ignition source, then extinguish the fire – and evacuation procedures.
Read the full recommendations here.
FDA approves long-acting ESA for dialysis-related anemia in children, adolescents
The patients aged 5-17 years with chronic kidney disease whose hemoglobin had been stabilized with an erythropoiesis-stimulating agent (ESA).
Efficacy was based partly on how well target hemoglobin levels were maintained in this trial, but also on extrapolation from results of trials in adults. The safety profile in these pediatric patients was consistent with those previously observed in adults. Mircera is manufactured by Vifor Pharma.
More information on the approval of Mircera in this population can be found in the FDA release. The prescribing information for Mircera, initially approved in 2007, has also been updated.
The patients aged 5-17 years with chronic kidney disease whose hemoglobin had been stabilized with an erythropoiesis-stimulating agent (ESA).
Efficacy was based partly on how well target hemoglobin levels were maintained in this trial, but also on extrapolation from results of trials in adults. The safety profile in these pediatric patients was consistent with those previously observed in adults. Mircera is manufactured by Vifor Pharma.
More information on the approval of Mircera in this population can be found in the FDA release. The prescribing information for Mircera, initially approved in 2007, has also been updated.
The patients aged 5-17 years with chronic kidney disease whose hemoglobin had been stabilized with an erythropoiesis-stimulating agent (ESA).
Efficacy was based partly on how well target hemoglobin levels were maintained in this trial, but also on extrapolation from results of trials in adults. The safety profile in these pediatric patients was consistent with those previously observed in adults. Mircera is manufactured by Vifor Pharma.
More information on the approval of Mircera in this population can be found in the FDA release. The prescribing information for Mircera, initially approved in 2007, has also been updated.
Youth tobacco use shows ‘promising declines’
according to the Centers for Disease Control and Prevention.
The prevalence of current tobacco use – defined as use on 1 or more days in the past 30 days – among high schoolers fell from 24.2% in 2011 to 19.6% in 2017, and middle school use decreased from 7.5% to 5.6% over that same time. That means the number of youth tobacco users went from almost 4.6 million in 2011 to slightly more than 3.6 million in 2017, Teresa W. Wang, PhD, and her associates said in the Morbidity and Mortality Weekly Report.
Almost half (47%) of the high school students who used tobacco in 2017 used two or more products, as did two out of five (42%) middle schoolers. That year, black high school students were less likely to use any tobacco product (14.2%) than were whites (22.7%) and Hispanics (16.7%). E-cigarettes were the most popular form of tobacco among white and Hispanic high schoolers, while cigars were the most commonly used form among blacks, they reported based on data from the National Youth Tobacco Surveys, which had sample sizes of 18,766 in 2011 and 17,872 in 2017.
“Despite promising declines in tobacco use, far too many young people continue to use tobacco products, including e-cigarettes,” CDC Director Robert R. Redfield, MD, said in a written statement accompanying the report. “Comprehensive, sustained strategies can help prevent and reduce tobacco use and protect our nation’s youth from this preventable health risk.”
In a separate statement, FDA Commissioner Scott Gottlieb, MD, said, “We are working hard to develop a pathway to put products like e-cigarettes through an appropriate series of regulatory gates to properly evaluate them as an alternative for adults who still want to get access to satisfying levels of nicotine, without all the risks associated with lighting tobacco on fire. And we will continue to encourage the development of potentially less harmful forms of nicotine delivery for currently addicted adult smokers. … But these public health opportunities are put at risk if all we do is hook another generation of kids on nicotine and tobacco products through alternatives like e-cigarettes.”
SOURCE: Wang TW et al. MMWR. 2018;67(22):629-33.
according to the Centers for Disease Control and Prevention.
The prevalence of current tobacco use – defined as use on 1 or more days in the past 30 days – among high schoolers fell from 24.2% in 2011 to 19.6% in 2017, and middle school use decreased from 7.5% to 5.6% over that same time. That means the number of youth tobacco users went from almost 4.6 million in 2011 to slightly more than 3.6 million in 2017, Teresa W. Wang, PhD, and her associates said in the Morbidity and Mortality Weekly Report.
Almost half (47%) of the high school students who used tobacco in 2017 used two or more products, as did two out of five (42%) middle schoolers. That year, black high school students were less likely to use any tobacco product (14.2%) than were whites (22.7%) and Hispanics (16.7%). E-cigarettes were the most popular form of tobacco among white and Hispanic high schoolers, while cigars were the most commonly used form among blacks, they reported based on data from the National Youth Tobacco Surveys, which had sample sizes of 18,766 in 2011 and 17,872 in 2017.
“Despite promising declines in tobacco use, far too many young people continue to use tobacco products, including e-cigarettes,” CDC Director Robert R. Redfield, MD, said in a written statement accompanying the report. “Comprehensive, sustained strategies can help prevent and reduce tobacco use and protect our nation’s youth from this preventable health risk.”
In a separate statement, FDA Commissioner Scott Gottlieb, MD, said, “We are working hard to develop a pathway to put products like e-cigarettes through an appropriate series of regulatory gates to properly evaluate them as an alternative for adults who still want to get access to satisfying levels of nicotine, without all the risks associated with lighting tobacco on fire. And we will continue to encourage the development of potentially less harmful forms of nicotine delivery for currently addicted adult smokers. … But these public health opportunities are put at risk if all we do is hook another generation of kids on nicotine and tobacco products through alternatives like e-cigarettes.”
SOURCE: Wang TW et al. MMWR. 2018;67(22):629-33.
according to the Centers for Disease Control and Prevention.
The prevalence of current tobacco use – defined as use on 1 or more days in the past 30 days – among high schoolers fell from 24.2% in 2011 to 19.6% in 2017, and middle school use decreased from 7.5% to 5.6% over that same time. That means the number of youth tobacco users went from almost 4.6 million in 2011 to slightly more than 3.6 million in 2017, Teresa W. Wang, PhD, and her associates said in the Morbidity and Mortality Weekly Report.
Almost half (47%) of the high school students who used tobacco in 2017 used two or more products, as did two out of five (42%) middle schoolers. That year, black high school students were less likely to use any tobacco product (14.2%) than were whites (22.7%) and Hispanics (16.7%). E-cigarettes were the most popular form of tobacco among white and Hispanic high schoolers, while cigars were the most commonly used form among blacks, they reported based on data from the National Youth Tobacco Surveys, which had sample sizes of 18,766 in 2011 and 17,872 in 2017.
“Despite promising declines in tobacco use, far too many young people continue to use tobacco products, including e-cigarettes,” CDC Director Robert R. Redfield, MD, said in a written statement accompanying the report. “Comprehensive, sustained strategies can help prevent and reduce tobacco use and protect our nation’s youth from this preventable health risk.”
In a separate statement, FDA Commissioner Scott Gottlieb, MD, said, “We are working hard to develop a pathway to put products like e-cigarettes through an appropriate series of regulatory gates to properly evaluate them as an alternative for adults who still want to get access to satisfying levels of nicotine, without all the risks associated with lighting tobacco on fire. And we will continue to encourage the development of potentially less harmful forms of nicotine delivery for currently addicted adult smokers. … But these public health opportunities are put at risk if all we do is hook another generation of kids on nicotine and tobacco products through alternatives like e-cigarettes.”
SOURCE: Wang TW et al. MMWR. 2018;67(22):629-33.
FROM MMWR