User login
FDA approves first EpiPen generic
The , including anaphylaxis, for adults and children weighing more than 33 pounds, according to an announcement from the agency.

“Today’s approval of the first generic version of the most widely prescribed epinephrine autoinjector in the U.S. is part of our longstanding commitment to advance access to lower cost, safe, and effective generic alternatives once patents and other exclusivities no longer prevent approval,” FDA commissioner Scott Gottlieb, MD, said in the release.
Manufactured by Teva Pharmaceuticals USA, the two strengths of the generic versions are 0.3 mg and 0.15 mg.
The FDA has previously approved other epinephrine autoinjectors, which include brand-name products and so-called “authorized generic” versions of Epi-Pen and Adrenaclick. An authorized generic “is made under the brand name’s existing drug application using the same formulation, process, and manufacturing facilities that are used by the brand name manufacturer. The labeling or packaging is, however, changed to remove the brand name or other trade dress. In some cases, a company may choose to sell an authorized generic at a lower cost than the brand-name drug product,” according to the FDA statement.
“Complex” generics – those that, as with this generic, include both a drug and a delivery device – face a tougher path to approval because the FDA has to evaluate and approve both components. “We remain committed to doing our part to provide scientific and regulatory clarity for sponsors seeking to develop complex generics, as well as prioritize the approval of medicines with little or no generic competition, as part of our overarching effort to remove barriers to generic development and market entry of critically important medicines,” Dr. Gottlieb explained.
Side effects of epinephrine autoinjectors include anxiety, restlessness, palpitations, nausea, and weakness; rarely, serious skin and soft-tissue infections after use of epinephrine autoinjectors have been reported.
Find more information about this approval in the FDA press announcement.
The , including anaphylaxis, for adults and children weighing more than 33 pounds, according to an announcement from the agency.
“Today’s approval of the first generic version of the most widely prescribed epinephrine autoinjector in the U.S. is part of our longstanding commitment to advance access to lower cost, safe, and effective generic alternatives once patents and other exclusivities no longer prevent approval,” FDA commissioner Scott Gottlieb, MD, said in the release.
Manufactured by Teva Pharmaceuticals USA, the two strengths of the generic versions are 0.3 mg and 0.15 mg.
The FDA has previously approved other epinephrine autoinjectors, which include brand-name products and so-called “authorized generic” versions of Epi-Pen and Adrenaclick. An authorized generic “is made under the brand name’s existing drug application using the same formulation, process, and manufacturing facilities that are used by the brand name manufacturer. The labeling or packaging is, however, changed to remove the brand name or other trade dress. In some cases, a company may choose to sell an authorized generic at a lower cost than the brand-name drug product,” according to the FDA statement.
“Complex” generics – those that, as with this generic, include both a drug and a delivery device – face a tougher path to approval because the FDA has to evaluate and approve both components. “We remain committed to doing our part to provide scientific and regulatory clarity for sponsors seeking to develop complex generics, as well as prioritize the approval of medicines with little or no generic competition, as part of our overarching effort to remove barriers to generic development and market entry of critically important medicines,” Dr. Gottlieb explained.
Side effects of epinephrine autoinjectors include anxiety, restlessness, palpitations, nausea, and weakness; rarely, serious skin and soft-tissue infections after use of epinephrine autoinjectors have been reported.
Find more information about this approval in the FDA press announcement.
The , including anaphylaxis, for adults and children weighing more than 33 pounds, according to an announcement from the agency.
“Today’s approval of the first generic version of the most widely prescribed epinephrine autoinjector in the U.S. is part of our longstanding commitment to advance access to lower cost, safe, and effective generic alternatives once patents and other exclusivities no longer prevent approval,” FDA commissioner Scott Gottlieb, MD, said in the release.
Manufactured by Teva Pharmaceuticals USA, the two strengths of the generic versions are 0.3 mg and 0.15 mg.
The FDA has previously approved other epinephrine autoinjectors, which include brand-name products and so-called “authorized generic” versions of Epi-Pen and Adrenaclick. An authorized generic “is made under the brand name’s existing drug application using the same formulation, process, and manufacturing facilities that are used by the brand name manufacturer. The labeling or packaging is, however, changed to remove the brand name or other trade dress. In some cases, a company may choose to sell an authorized generic at a lower cost than the brand-name drug product,” according to the FDA statement.
“Complex” generics – those that, as with this generic, include both a drug and a delivery device – face a tougher path to approval because the FDA has to evaluate and approve both components. “We remain committed to doing our part to provide scientific and regulatory clarity for sponsors seeking to develop complex generics, as well as prioritize the approval of medicines with little or no generic competition, as part of our overarching effort to remove barriers to generic development and market entry of critically important medicines,” Dr. Gottlieb explained.
Side effects of epinephrine autoinjectors include anxiety, restlessness, palpitations, nausea, and weakness; rarely, serious skin and soft-tissue infections after use of epinephrine autoinjectors have been reported.
Find more information about this approval in the FDA press announcement.
Kalydeco approved for patients aged 1-2 years
The Food and Drug Administration has approved Kalydeco (ivacaftor) for the treatment of patients aged 12 to less than 24 months who have cystic fibrosis that is caused by any of 10 mutations in the CFTR gene and is responsive to the drug, the drug’s developer announced.
The drug was approved for patients aged 6 years and older in 2012 and in patients aged 2-5 years in 2015 and is the only approved drug that treats the underlying cause of cystic fibrosis rather than its symptoms.
The approval is based on the ongoing phase 3, open-label ARRIVAL trial (NCT02725567), which is assessing the drug’s safety in children aged 12 months to less than 24 months. The trial’s investigators have found that its safety profile in this age group is consistent with that seen in older children and adults. Most adverse events were mild to moderate; the most common (occurring in more than 30% of patients) were cough, pyrexia, elevated aspartate aminotransferase, elevated alanine aminotransferase, and runny nose. The trial found that, after 24 weeks of treatment, the mean sweat chloride levels decreased from 104.1 mmol/L (n = 14) to 33.8 mmol/L (n = 14).
Ivacaftor is contraindicated in patients taking certain antibiotics, seizure medications, or other medications; risk of drug interaction – affecting either the performance of ivacaftor or that of the other medication – is also a concern. Patients should inform their doctors if they are pregnant, planning to become pregnant, or breastfeeding; have liver or kidney problems; or drink grapefruit juice or eat grapefruit or Seville oranges. There also is a risk of high liver enzymes or cataracts. Ivacaftor is available in 150-mg tablets for adults and pediatric patients aged 6 years and older and in 50-mg and 75-mg granules for younger patients. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved Kalydeco (ivacaftor) for the treatment of patients aged 12 to less than 24 months who have cystic fibrosis that is caused by any of 10 mutations in the CFTR gene and is responsive to the drug, the drug’s developer announced.
The drug was approved for patients aged 6 years and older in 2012 and in patients aged 2-5 years in 2015 and is the only approved drug that treats the underlying cause of cystic fibrosis rather than its symptoms.
The approval is based on the ongoing phase 3, open-label ARRIVAL trial (NCT02725567), which is assessing the drug’s safety in children aged 12 months to less than 24 months. The trial’s investigators have found that its safety profile in this age group is consistent with that seen in older children and adults. Most adverse events were mild to moderate; the most common (occurring in more than 30% of patients) were cough, pyrexia, elevated aspartate aminotransferase, elevated alanine aminotransferase, and runny nose. The trial found that, after 24 weeks of treatment, the mean sweat chloride levels decreased from 104.1 mmol/L (n = 14) to 33.8 mmol/L (n = 14).
Ivacaftor is contraindicated in patients taking certain antibiotics, seizure medications, or other medications; risk of drug interaction – affecting either the performance of ivacaftor or that of the other medication – is also a concern. Patients should inform their doctors if they are pregnant, planning to become pregnant, or breastfeeding; have liver or kidney problems; or drink grapefruit juice or eat grapefruit or Seville oranges. There also is a risk of high liver enzymes or cataracts. Ivacaftor is available in 150-mg tablets for adults and pediatric patients aged 6 years and older and in 50-mg and 75-mg granules for younger patients. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved Kalydeco (ivacaftor) for the treatment of patients aged 12 to less than 24 months who have cystic fibrosis that is caused by any of 10 mutations in the CFTR gene and is responsive to the drug, the drug’s developer announced.
The drug was approved for patients aged 6 years and older in 2012 and in patients aged 2-5 years in 2015 and is the only approved drug that treats the underlying cause of cystic fibrosis rather than its symptoms.
The approval is based on the ongoing phase 3, open-label ARRIVAL trial (NCT02725567), which is assessing the drug’s safety in children aged 12 months to less than 24 months. The trial’s investigators have found that its safety profile in this age group is consistent with that seen in older children and adults. Most adverse events were mild to moderate; the most common (occurring in more than 30% of patients) were cough, pyrexia, elevated aspartate aminotransferase, elevated alanine aminotransferase, and runny nose. The trial found that, after 24 weeks of treatment, the mean sweat chloride levels decreased from 104.1 mmol/L (n = 14) to 33.8 mmol/L (n = 14).
Ivacaftor is contraindicated in patients taking certain antibiotics, seizure medications, or other medications; risk of drug interaction – affecting either the performance of ivacaftor or that of the other medication – is also a concern. Patients should inform their doctors if they are pregnant, planning to become pregnant, or breastfeeding; have liver or kidney problems; or drink grapefruit juice or eat grapefruit or Seville oranges. There also is a risk of high liver enzymes or cataracts. Ivacaftor is available in 150-mg tablets for adults and pediatric patients aged 6 years and older and in 50-mg and 75-mg granules for younger patients. Full prescribing information can be found on the FDA website.
More deliveries now include opioid use disorder
according to the Centers for Disease Control and Prevention.

The national prevalence of opioid use disorder increased by 333% as it went from 1.5 cases per 1,000 delivery hospitalizations in 1999 to 6.5 cases per 1,000 in 2014. At the state level, there were significant increases in all 28 states with data available for at least 3 consecutive years during the study period, Sarah C. Haight, MPH, and her associates at the CDC in Atlanta said in the Morbidity and Mortality Weekly Report.
Average annual rate changes for those states ranged from a low of 0.01 per 1,000 delivery hospitalizations per year in California to 5.37 per year in Vermont, with the national rate change coming in at 0.39 per year. Of the 14 states with data available in 1999, Iowa had the lowest rate at 0.1 per 1,000 deliveries and Maryland had the highest at 8.2. In 2014, when data were available for 26 states and the District of Columbia, the highest rate was Vermont’s 48.6 per 1,000 deliveries and the lowest was 0.7 in Washington, D.C., the investigators reported.
Although “increasing trends might represent actual increases in prevalence or improved screening and diagnosis,” Ms. Haight and her associates added that “these estimates also correlate with state opioid prescribing rates in the general population. West Virginia, for example, had a prescribing rate estimated at 138 opioid prescriptions per 100 persons in 2012.”
“These findings illustrate the devastating impact of the opioid epidemic on families across the U.S., including on the very youngest,” said CDC Director Robert R. Redfield, MD. “Untreated opioid use disorder during pregnancy can lead to heartbreaking results. Each case represents a mother, a child, and a family in need of continued treatment and support.”
Data for the analysis came from the Agency for Healthcare Research and Quality’s National Inpatient Sample and State Inpatient Databases.
SOURCE: Haight SC et al. MMWR. 2018 Aug 10;67[31]:845-9.
according to the Centers for Disease Control and Prevention.
The national prevalence of opioid use disorder increased by 333% as it went from 1.5 cases per 1,000 delivery hospitalizations in 1999 to 6.5 cases per 1,000 in 2014. At the state level, there were significant increases in all 28 states with data available for at least 3 consecutive years during the study period, Sarah C. Haight, MPH, and her associates at the CDC in Atlanta said in the Morbidity and Mortality Weekly Report.
Average annual rate changes for those states ranged from a low of 0.01 per 1,000 delivery hospitalizations per year in California to 5.37 per year in Vermont, with the national rate change coming in at 0.39 per year. Of the 14 states with data available in 1999, Iowa had the lowest rate at 0.1 per 1,000 deliveries and Maryland had the highest at 8.2. In 2014, when data were available for 26 states and the District of Columbia, the highest rate was Vermont’s 48.6 per 1,000 deliveries and the lowest was 0.7 in Washington, D.C., the investigators reported.
Although “increasing trends might represent actual increases in prevalence or improved screening and diagnosis,” Ms. Haight and her associates added that “these estimates also correlate with state opioid prescribing rates in the general population. West Virginia, for example, had a prescribing rate estimated at 138 opioid prescriptions per 100 persons in 2012.”
“These findings illustrate the devastating impact of the opioid epidemic on families across the U.S., including on the very youngest,” said CDC Director Robert R. Redfield, MD. “Untreated opioid use disorder during pregnancy can lead to heartbreaking results. Each case represents a mother, a child, and a family in need of continued treatment and support.”
Data for the analysis came from the Agency for Healthcare Research and Quality’s National Inpatient Sample and State Inpatient Databases.
SOURCE: Haight SC et al. MMWR. 2018 Aug 10;67[31]:845-9.
according to the Centers for Disease Control and Prevention.
The national prevalence of opioid use disorder increased by 333% as it went from 1.5 cases per 1,000 delivery hospitalizations in 1999 to 6.5 cases per 1,000 in 2014. At the state level, there were significant increases in all 28 states with data available for at least 3 consecutive years during the study period, Sarah C. Haight, MPH, and her associates at the CDC in Atlanta said in the Morbidity and Mortality Weekly Report.
Average annual rate changes for those states ranged from a low of 0.01 per 1,000 delivery hospitalizations per year in California to 5.37 per year in Vermont, with the national rate change coming in at 0.39 per year. Of the 14 states with data available in 1999, Iowa had the lowest rate at 0.1 per 1,000 deliveries and Maryland had the highest at 8.2. In 2014, when data were available for 26 states and the District of Columbia, the highest rate was Vermont’s 48.6 per 1,000 deliveries and the lowest was 0.7 in Washington, D.C., the investigators reported.
Although “increasing trends might represent actual increases in prevalence or improved screening and diagnosis,” Ms. Haight and her associates added that “these estimates also correlate with state opioid prescribing rates in the general population. West Virginia, for example, had a prescribing rate estimated at 138 opioid prescriptions per 100 persons in 2012.”
“These findings illustrate the devastating impact of the opioid epidemic on families across the U.S., including on the very youngest,” said CDC Director Robert R. Redfield, MD. “Untreated opioid use disorder during pregnancy can lead to heartbreaking results. Each case represents a mother, a child, and a family in need of continued treatment and support.”
Data for the analysis came from the Agency for Healthcare Research and Quality’s National Inpatient Sample and State Inpatient Databases.
SOURCE: Haight SC et al. MMWR. 2018 Aug 10;67[31]:845-9.
FROM MMWR
FDA gives Orkambi indication for younger patients
(CF), according to its manufacturer, Vertex Pharmaceuticals. Specifically, the drug is meant to treat the most common underlying cause of CF – having two copies of the F508del-CFTR mutation – and is the first drug to treat it.

The approval is based on a phase 3, two-part, open-label, multicenter study that assessed various doses in patents aged 2-5 years. The study demonstrated safety and tolerability in that age group equivalent to that seen in older patients. The drug is expected to be available for this age group within 2-4 weeks of this approval.
Available as oral granules in two doses for weight-based dosing (either lumacaftor 100 mg/ivacaftor 125 mg or lumacaftor 150 mg/ivacaftor 188 mg), the compound targets the defective chloride channels responsible for CF; the two halves work together to increase the number of chloride channels on cell surfaces and also improve their function.
Orkambi should be prescribed only for patients with CF who have the dual F508del-CFTR mutation; it is not indicated for other types of CF. Patients should not take this drug if they are taking drugs such as rifampin, phenytoin, triazolam, or cyclosporine because of possible drug interactions. It can also lead to worsening liver function and elevated blood liver enzymes, increased blood pressure, or cataracts. The most common side effects include breathing problems, nausea, fatigue, and rash. Full prescribing information is available on the FDA website.
(CF), according to its manufacturer, Vertex Pharmaceuticals. Specifically, the drug is meant to treat the most common underlying cause of CF – having two copies of the F508del-CFTR mutation – and is the first drug to treat it.
The approval is based on a phase 3, two-part, open-label, multicenter study that assessed various doses in patents aged 2-5 years. The study demonstrated safety and tolerability in that age group equivalent to that seen in older patients. The drug is expected to be available for this age group within 2-4 weeks of this approval.
Available as oral granules in two doses for weight-based dosing (either lumacaftor 100 mg/ivacaftor 125 mg or lumacaftor 150 mg/ivacaftor 188 mg), the compound targets the defective chloride channels responsible for CF; the two halves work together to increase the number of chloride channels on cell surfaces and also improve their function.
Orkambi should be prescribed only for patients with CF who have the dual F508del-CFTR mutation; it is not indicated for other types of CF. Patients should not take this drug if they are taking drugs such as rifampin, phenytoin, triazolam, or cyclosporine because of possible drug interactions. It can also lead to worsening liver function and elevated blood liver enzymes, increased blood pressure, or cataracts. The most common side effects include breathing problems, nausea, fatigue, and rash. Full prescribing information is available on the FDA website.
(CF), according to its manufacturer, Vertex Pharmaceuticals. Specifically, the drug is meant to treat the most common underlying cause of CF – having two copies of the F508del-CFTR mutation – and is the first drug to treat it.
The approval is based on a phase 3, two-part, open-label, multicenter study that assessed various doses in patents aged 2-5 years. The study demonstrated safety and tolerability in that age group equivalent to that seen in older patients. The drug is expected to be available for this age group within 2-4 weeks of this approval.
Available as oral granules in two doses for weight-based dosing (either lumacaftor 100 mg/ivacaftor 125 mg or lumacaftor 150 mg/ivacaftor 188 mg), the compound targets the defective chloride channels responsible for CF; the two halves work together to increase the number of chloride channels on cell surfaces and also improve their function.
Orkambi should be prescribed only for patients with CF who have the dual F508del-CFTR mutation; it is not indicated for other types of CF. Patients should not take this drug if they are taking drugs such as rifampin, phenytoin, triazolam, or cyclosporine because of possible drug interactions. It can also lead to worsening liver function and elevated blood liver enzymes, increased blood pressure, or cataracts. The most common side effects include breathing problems, nausea, fatigue, and rash. Full prescribing information is available on the FDA website.
FDA warns against azithromycin in blood or lymph node cancers
The Food and Drug Administration has issued a in patients with blood or lymph node cancers who have received donor stem cell transplants.
This use of azithromycin can lead to increased risk of cancer relapse and death in this population. The FDA is continuing to review data and is expected to issue further recommendations.
Patients with blood or lymph node cancers are at an increased risk of bronchiolitis obliterans syndrome after donor stem cell transplant; although azithromycin is not approved for prevention of this condition, the antibiotic is sometimes prescribed for that purpose.
A French study of 480 patients was undertaken to assess the effectiveness of this prophylaxis but revealed the increased risk of relapse and death and was halted 13 months after completing enrollment. The rate of cancer relapse was 32.9% in the azithromycin group and just 20.8% in the placebo group; the 2-year survival rate was 56.6% in the azithromycin group and 70.1% in the placebo group (JAMA 2017;318[6]:557-66).
Bronchiolitis obliterans syndrome is marked by inflammation and scarring of the airways that leads to severe shortness of breath and dry cough. There are no known effective antibiotic treatments for prophylaxis of the condition, according to the FDA.
FDA officials are advising physicians not to prescribe long-term azithromycin in this population. Patients who have had a stem cell transplant and are already taking the antibiotic, should consult a doctor before discontinuing.
The manufacturer of brand name azithromycin (Zithromax) has issued a Dear Healthcare Provider letter about the safety issue, and more information can be found in the FDA’s safety announcement.
The Food and Drug Administration has issued a in patients with blood or lymph node cancers who have received donor stem cell transplants.
This use of azithromycin can lead to increased risk of cancer relapse and death in this population. The FDA is continuing to review data and is expected to issue further recommendations.
Patients with blood or lymph node cancers are at an increased risk of bronchiolitis obliterans syndrome after donor stem cell transplant; although azithromycin is not approved for prevention of this condition, the antibiotic is sometimes prescribed for that purpose.
A French study of 480 patients was undertaken to assess the effectiveness of this prophylaxis but revealed the increased risk of relapse and death and was halted 13 months after completing enrollment. The rate of cancer relapse was 32.9% in the azithromycin group and just 20.8% in the placebo group; the 2-year survival rate was 56.6% in the azithromycin group and 70.1% in the placebo group (JAMA 2017;318[6]:557-66).
Bronchiolitis obliterans syndrome is marked by inflammation and scarring of the airways that leads to severe shortness of breath and dry cough. There are no known effective antibiotic treatments for prophylaxis of the condition, according to the FDA.
FDA officials are advising physicians not to prescribe long-term azithromycin in this population. Patients who have had a stem cell transplant and are already taking the antibiotic, should consult a doctor before discontinuing.
The manufacturer of brand name azithromycin (Zithromax) has issued a Dear Healthcare Provider letter about the safety issue, and more information can be found in the FDA’s safety announcement.
The Food and Drug Administration has issued a in patients with blood or lymph node cancers who have received donor stem cell transplants.
This use of azithromycin can lead to increased risk of cancer relapse and death in this population. The FDA is continuing to review data and is expected to issue further recommendations.
Patients with blood or lymph node cancers are at an increased risk of bronchiolitis obliterans syndrome after donor stem cell transplant; although azithromycin is not approved for prevention of this condition, the antibiotic is sometimes prescribed for that purpose.
A French study of 480 patients was undertaken to assess the effectiveness of this prophylaxis but revealed the increased risk of relapse and death and was halted 13 months after completing enrollment. The rate of cancer relapse was 32.9% in the azithromycin group and just 20.8% in the placebo group; the 2-year survival rate was 56.6% in the azithromycin group and 70.1% in the placebo group (JAMA 2017;318[6]:557-66).
Bronchiolitis obliterans syndrome is marked by inflammation and scarring of the airways that leads to severe shortness of breath and dry cough. There are no known effective antibiotic treatments for prophylaxis of the condition, according to the FDA.
FDA officials are advising physicians not to prescribe long-term azithromycin in this population. Patients who have had a stem cell transplant and are already taking the antibiotic, should consult a doctor before discontinuing.
The manufacturer of brand name azithromycin (Zithromax) has issued a Dear Healthcare Provider letter about the safety issue, and more information can be found in the FDA’s safety announcement.
FDA rejects mepolizumab on efficacy, but supports safety for COPD
Asthma drug mepolizumab could be added safely to inhaled corticosteroids for maintenance therapy to help reduce exacerbations in chronic obstructive pulmonary disease (COPD) patients who meet criteria for eosinophil counts, but the current data do not support its efficacy strongly enough for approval, according to a majority of members of the Food and Drug Administration’s Pulmonary-Allergy Drugs Advisory Committee.
The committee voted 16-3 that there was insufficient evidence of efficacy to support guided by eosinophil levels; they also voted 16-3 that the risk-benefit profile was not adequate to support approval.
However, on a voting question of safety, the committee voted 17-2 that the safety data on mepolizumab were sufficient to support approval.
Mepolizumab, a humanized monoclonal antibody, is currently approved for the treatment of asthma with eosinophilic phenotype for patients aged 12 years and older and for adults with eosinophilic granulomatosis with polyangiitis. Manufacturer GlaxoSmithKline is seeking approval for its use as an add-on therapy in COPD patients at a subcutaneous dose of 100 mg every 4 weeks. Mepolizumab works by binding to interleukin-5 (IL-5) and reducing eosinophil maturation and survival, which prompted GlaxoSmithKline to pursue an indication for COPD patients in a high-eosinophil stratum.
The application was supported in part by two concurrent randomized trials of 52 weeks’ duration.
Banu A. Karimi-Shah, MD, clinical team leader of the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products, presented data from the two studies, referred to as Study 106 and Study 113.
In Study 106, researchers found statistically significant reductions in exacerbations for patients in the highest eosinophil group. However, challenges of the studies included a lack of consensus over the definition and possible relevance of an eosinophilic COPD phenotype, Dr. Karimi-Shah said in a presentation at the meeting.
In Study 113, mepolizumab had no significant impact on reducing moderate to severe exacerbations at either a 100-mg or 300-mg dose, Dr. Karimi-Shah said. In addition, most secondary endpoints, with the exception of reducing time to the first exacerbation among patients in the highest eosinophil group, did not consistently support the primary endpoint of exacerbation reduction in either study, she said.
Robert Busch, MD, also of the FDA’s Division of Pulmonary, Allergy, and Rheumatology products, served as a clinical reviewer and presented data on safety, efficacy, and risk-benefit profile of mepolizumab.
Dr. Busch noted that the variability in blood eosinophils make it challenging to use as a potential marker to identify patients who would benefit from mepolizumab as an add-on therapy.
Overall, most of the committee agreed on the existence of an eosinophilic COPD phenotype, but expressed concern about the threshold being used.
“The studies were not particularly well controlled regarding the characterization of patients,” said William J. Calhoun, MD, of the University of Texas Medical Branch, Galveston, who cast one of the ‘no’ votes on the question of efficacy.
By contrast, Jeffrey S. Wagener, MD, of the University of Colorado at Denver, Aurora, referenced his background in cystic fibrosis, and voted “yes” on the question of efficacy. “For patients that have no other option, this is a step forward,” he said.
Committee members on both sides of the vote emphasized the need for more research with larger numbers, better patient characterization, and more female patients. The committee members reported no relevant conflicts of interest.
Asthma drug mepolizumab could be added safely to inhaled corticosteroids for maintenance therapy to help reduce exacerbations in chronic obstructive pulmonary disease (COPD) patients who meet criteria for eosinophil counts, but the current data do not support its efficacy strongly enough for approval, according to a majority of members of the Food and Drug Administration’s Pulmonary-Allergy Drugs Advisory Committee.
The committee voted 16-3 that there was insufficient evidence of efficacy to support guided by eosinophil levels; they also voted 16-3 that the risk-benefit profile was not adequate to support approval.
However, on a voting question of safety, the committee voted 17-2 that the safety data on mepolizumab were sufficient to support approval.
Mepolizumab, a humanized monoclonal antibody, is currently approved for the treatment of asthma with eosinophilic phenotype for patients aged 12 years and older and for adults with eosinophilic granulomatosis with polyangiitis. Manufacturer GlaxoSmithKline is seeking approval for its use as an add-on therapy in COPD patients at a subcutaneous dose of 100 mg every 4 weeks. Mepolizumab works by binding to interleukin-5 (IL-5) and reducing eosinophil maturation and survival, which prompted GlaxoSmithKline to pursue an indication for COPD patients in a high-eosinophil stratum.
The application was supported in part by two concurrent randomized trials of 52 weeks’ duration.
Banu A. Karimi-Shah, MD, clinical team leader of the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products, presented data from the two studies, referred to as Study 106 and Study 113.
In Study 106, researchers found statistically significant reductions in exacerbations for patients in the highest eosinophil group. However, challenges of the studies included a lack of consensus over the definition and possible relevance of an eosinophilic COPD phenotype, Dr. Karimi-Shah said in a presentation at the meeting.
In Study 113, mepolizumab had no significant impact on reducing moderate to severe exacerbations at either a 100-mg or 300-mg dose, Dr. Karimi-Shah said. In addition, most secondary endpoints, with the exception of reducing time to the first exacerbation among patients in the highest eosinophil group, did not consistently support the primary endpoint of exacerbation reduction in either study, she said.
Robert Busch, MD, also of the FDA’s Division of Pulmonary, Allergy, and Rheumatology products, served as a clinical reviewer and presented data on safety, efficacy, and risk-benefit profile of mepolizumab.
Dr. Busch noted that the variability in blood eosinophils make it challenging to use as a potential marker to identify patients who would benefit from mepolizumab as an add-on therapy.
Overall, most of the committee agreed on the existence of an eosinophilic COPD phenotype, but expressed concern about the threshold being used.
“The studies were not particularly well controlled regarding the characterization of patients,” said William J. Calhoun, MD, of the University of Texas Medical Branch, Galveston, who cast one of the ‘no’ votes on the question of efficacy.
By contrast, Jeffrey S. Wagener, MD, of the University of Colorado at Denver, Aurora, referenced his background in cystic fibrosis, and voted “yes” on the question of efficacy. “For patients that have no other option, this is a step forward,” he said.
Committee members on both sides of the vote emphasized the need for more research with larger numbers, better patient characterization, and more female patients. The committee members reported no relevant conflicts of interest.
Asthma drug mepolizumab could be added safely to inhaled corticosteroids for maintenance therapy to help reduce exacerbations in chronic obstructive pulmonary disease (COPD) patients who meet criteria for eosinophil counts, but the current data do not support its efficacy strongly enough for approval, according to a majority of members of the Food and Drug Administration’s Pulmonary-Allergy Drugs Advisory Committee.
The committee voted 16-3 that there was insufficient evidence of efficacy to support guided by eosinophil levels; they also voted 16-3 that the risk-benefit profile was not adequate to support approval.
However, on a voting question of safety, the committee voted 17-2 that the safety data on mepolizumab were sufficient to support approval.
Mepolizumab, a humanized monoclonal antibody, is currently approved for the treatment of asthma with eosinophilic phenotype for patients aged 12 years and older and for adults with eosinophilic granulomatosis with polyangiitis. Manufacturer GlaxoSmithKline is seeking approval for its use as an add-on therapy in COPD patients at a subcutaneous dose of 100 mg every 4 weeks. Mepolizumab works by binding to interleukin-5 (IL-5) and reducing eosinophil maturation and survival, which prompted GlaxoSmithKline to pursue an indication for COPD patients in a high-eosinophil stratum.
The application was supported in part by two concurrent randomized trials of 52 weeks’ duration.
Banu A. Karimi-Shah, MD, clinical team leader of the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products, presented data from the two studies, referred to as Study 106 and Study 113.
In Study 106, researchers found statistically significant reductions in exacerbations for patients in the highest eosinophil group. However, challenges of the studies included a lack of consensus over the definition and possible relevance of an eosinophilic COPD phenotype, Dr. Karimi-Shah said in a presentation at the meeting.
In Study 113, mepolizumab had no significant impact on reducing moderate to severe exacerbations at either a 100-mg or 300-mg dose, Dr. Karimi-Shah said. In addition, most secondary endpoints, with the exception of reducing time to the first exacerbation among patients in the highest eosinophil group, did not consistently support the primary endpoint of exacerbation reduction in either study, she said.
Robert Busch, MD, also of the FDA’s Division of Pulmonary, Allergy, and Rheumatology products, served as a clinical reviewer and presented data on safety, efficacy, and risk-benefit profile of mepolizumab.
Dr. Busch noted that the variability in blood eosinophils make it challenging to use as a potential marker to identify patients who would benefit from mepolizumab as an add-on therapy.
Overall, most of the committee agreed on the existence of an eosinophilic COPD phenotype, but expressed concern about the threshold being used.
“The studies were not particularly well controlled regarding the characterization of patients,” said William J. Calhoun, MD, of the University of Texas Medical Branch, Galveston, who cast one of the ‘no’ votes on the question of efficacy.
By contrast, Jeffrey S. Wagener, MD, of the University of Colorado at Denver, Aurora, referenced his background in cystic fibrosis, and voted “yes” on the question of efficacy. “For patients that have no other option, this is a step forward,” he said.
Committee members on both sides of the vote emphasized the need for more research with larger numbers, better patient characterization, and more female patients. The committee members reported no relevant conflicts of interest.
FROM AN FDA ADVISORY COMMITTEE MEETING
Preterm birth rate ‘is on the rise again’
After several years of decline, the incidence of preterm births in the United States “is on the rise again,” according to the National Center for Health Statistics.
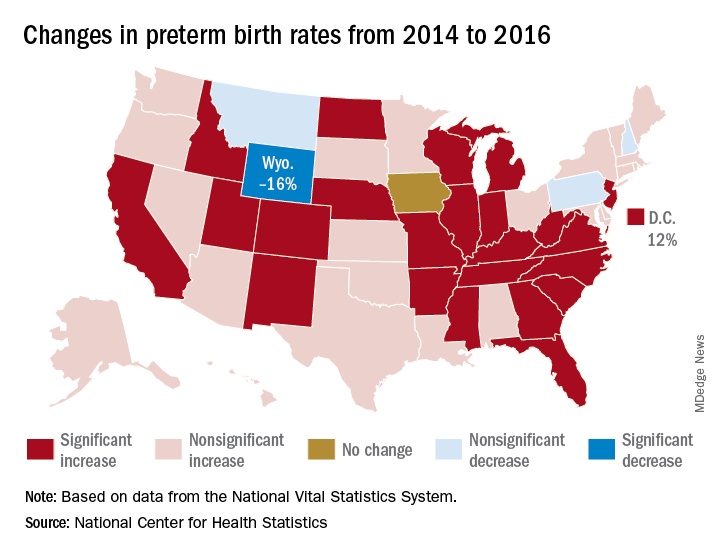
That 3% increase was spread pretty evenly: 23 states and the District of Columbia experienced statistically significant increases from 2014 to 2016, and 22 other states also had increases, although these were not statistically significant. One state, Iowa, had no change; three states – Montana, New Hampshire, and Pennsylvania – had nonsignificant declines, and Wyoming was the only state with a statistically significant drop (16%) in preterm birth incidence, they said based on data from the National Vital Statistics System.
The largest increase, 12%, was seen in the District of Columbia, followed by Idaho and North Dakota at 10% and Arkansas, New Mexico, and West Virginia at 9%, the researchers reported.
After several years of decline, the incidence of preterm births in the United States “is on the rise again,” according to the National Center for Health Statistics.
That 3% increase was spread pretty evenly: 23 states and the District of Columbia experienced statistically significant increases from 2014 to 2016, and 22 other states also had increases, although these were not statistically significant. One state, Iowa, had no change; three states – Montana, New Hampshire, and Pennsylvania – had nonsignificant declines, and Wyoming was the only state with a statistically significant drop (16%) in preterm birth incidence, they said based on data from the National Vital Statistics System.
The largest increase, 12%, was seen in the District of Columbia, followed by Idaho and North Dakota at 10% and Arkansas, New Mexico, and West Virginia at 9%, the researchers reported.
After several years of decline, the incidence of preterm births in the United States “is on the rise again,” according to the National Center for Health Statistics.
That 3% increase was spread pretty evenly: 23 states and the District of Columbia experienced statistically significant increases from 2014 to 2016, and 22 other states also had increases, although these were not statistically significant. One state, Iowa, had no change; three states – Montana, New Hampshire, and Pennsylvania – had nonsignificant declines, and Wyoming was the only state with a statistically significant drop (16%) in preterm birth incidence, they said based on data from the National Vital Statistics System.
The largest increase, 12%, was seen in the District of Columbia, followed by Idaho and North Dakota at 10% and Arkansas, New Mexico, and West Virginia at 9%, the researchers reported.
Liver cancer death rates down for Asians and Pacific Islanders
Liver cancer death rates are dropping for Asians/Pacific Islanders, but that is the exception to a larger trend, according to the National Center for Health Statistics.
The age-adjusted death rate for liver cancer is down 22% among Asian or Pacific Islander adults aged 25 years and older since the turn of the century, falling from 17.5 per 100,000 population in 2000 – when it was the highest of the four racial/ethnic groups included in the report – to 13.6 per 100,000 in 2016, by which time it was just middle of the pack, the NCHS reported.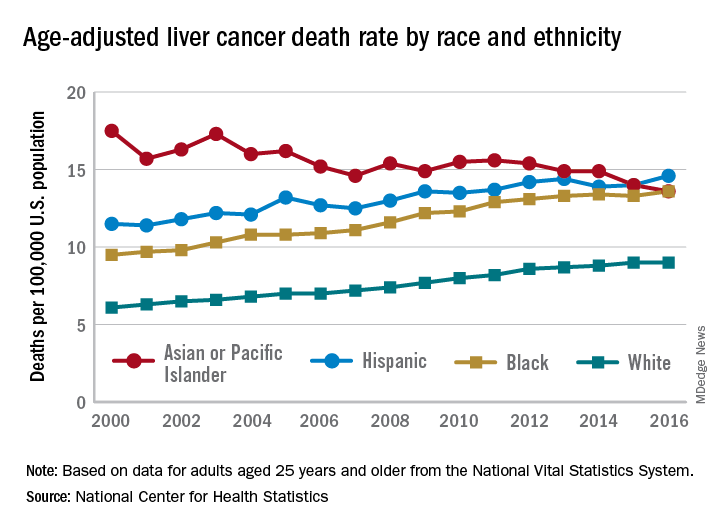
That shift resulted as much from increases for the other groups as from the decreased rate for Asians/Pacific Islanders. White adults were dying of liver cancer at a 48% higher rate in 2016 (9.0 per 100,000) than they were in 2000 (6.1), blacks saw their death rate go from 9.5 to 13.6 – a 43% increase – and the rate for Hispanics rose by 27% from 2000 (11.5) to 2016 (14.6), said Jiaquan Xu, MD, of the NCHS mortality statistics branch.
The adjusted death rate from liver cancer for all adults went from 7.2 per 100,000 in 2000 to 10.3 in 2016 for an increase of 43%. Over that period, the rate for men was always at least twice as high as it was for women: It rose from 10.5 per 100,000 for men and 4.9 for women in 2000 to 15.0 for men and 6.3 for women in 2016, Dr. Xu said based on data from the mortality files of the National Vital Statistics System.
Geographically, the District of Columbia had the highest rate at 16.8 per 100,000 in 2016, followed by Louisiana (13.8), Hawaii (12.7), and Mississippi and New Mexico (12.4 each). Vermont’s rate of 6.0 was the lowest in the country, with Maine second at 7.4, Montana third at 7.7, and Utah and Nebraska tied for fourth at 7.8, according to Dr. Xu.
Liver cancer death rates are dropping for Asians/Pacific Islanders, but that is the exception to a larger trend, according to the National Center for Health Statistics.
The age-adjusted death rate for liver cancer is down 22% among Asian or Pacific Islander adults aged 25 years and older since the turn of the century, falling from 17.5 per 100,000 population in 2000 – when it was the highest of the four racial/ethnic groups included in the report – to 13.6 per 100,000 in 2016, by which time it was just middle of the pack, the NCHS reported.
That shift resulted as much from increases for the other groups as from the decreased rate for Asians/Pacific Islanders. White adults were dying of liver cancer at a 48% higher rate in 2016 (9.0 per 100,000) than they were in 2000 (6.1), blacks saw their death rate go from 9.5 to 13.6 – a 43% increase – and the rate for Hispanics rose by 27% from 2000 (11.5) to 2016 (14.6), said Jiaquan Xu, MD, of the NCHS mortality statistics branch.
The adjusted death rate from liver cancer for all adults went from 7.2 per 100,000 in 2000 to 10.3 in 2016 for an increase of 43%. Over that period, the rate for men was always at least twice as high as it was for women: It rose from 10.5 per 100,000 for men and 4.9 for women in 2000 to 15.0 for men and 6.3 for women in 2016, Dr. Xu said based on data from the mortality files of the National Vital Statistics System.
Geographically, the District of Columbia had the highest rate at 16.8 per 100,000 in 2016, followed by Louisiana (13.8), Hawaii (12.7), and Mississippi and New Mexico (12.4 each). Vermont’s rate of 6.0 was the lowest in the country, with Maine second at 7.4, Montana third at 7.7, and Utah and Nebraska tied for fourth at 7.8, according to Dr. Xu.
Liver cancer death rates are dropping for Asians/Pacific Islanders, but that is the exception to a larger trend, according to the National Center for Health Statistics.
The age-adjusted death rate for liver cancer is down 22% among Asian or Pacific Islander adults aged 25 years and older since the turn of the century, falling from 17.5 per 100,000 population in 2000 – when it was the highest of the four racial/ethnic groups included in the report – to 13.6 per 100,000 in 2016, by which time it was just middle of the pack, the NCHS reported.
That shift resulted as much from increases for the other groups as from the decreased rate for Asians/Pacific Islanders. White adults were dying of liver cancer at a 48% higher rate in 2016 (9.0 per 100,000) than they were in 2000 (6.1), blacks saw their death rate go from 9.5 to 13.6 – a 43% increase – and the rate for Hispanics rose by 27% from 2000 (11.5) to 2016 (14.6), said Jiaquan Xu, MD, of the NCHS mortality statistics branch.
The adjusted death rate from liver cancer for all adults went from 7.2 per 100,000 in 2000 to 10.3 in 2016 for an increase of 43%. Over that period, the rate for men was always at least twice as high as it was for women: It rose from 10.5 per 100,000 for men and 4.9 for women in 2000 to 15.0 for men and 6.3 for women in 2016, Dr. Xu said based on data from the mortality files of the National Vital Statistics System.
Geographically, the District of Columbia had the highest rate at 16.8 per 100,000 in 2016, followed by Louisiana (13.8), Hawaii (12.7), and Mississippi and New Mexico (12.4 each). Vermont’s rate of 6.0 was the lowest in the country, with Maine second at 7.4, Montana third at 7.7, and Utah and Nebraska tied for fourth at 7.8, according to Dr. Xu.
CDC reports Salmonella outbreak
A total of 90 people in 26 states have been infected with multidrug-resistant Salmonella in an outbreak linked to raw turkey products, according to the Centers for Disease Control and Prevention.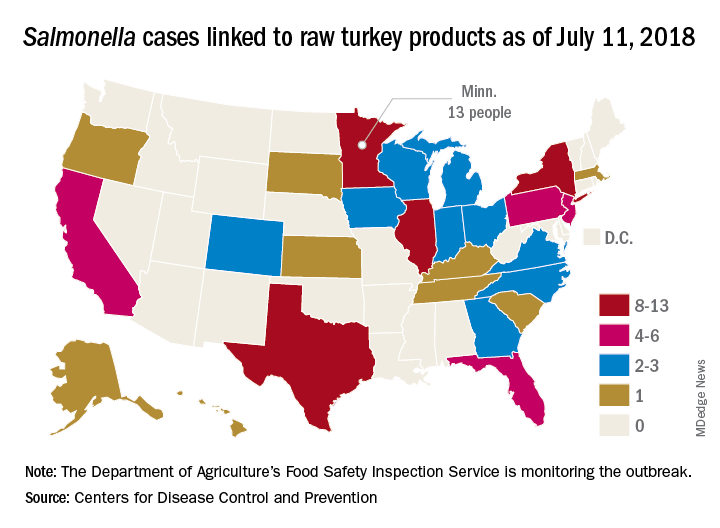
As of July 11, 2018, 40 of the 78 people with available information who were infected with the outbreak strain of Salmonella Reading have been hospitalized, but no deaths have been reported. Of the 61 ill people who have been interviewed, most have reported preparing or eating turkey products from a number of sources, although two lived in households where raw turkey was given to pets: No common supplier has been identified, the CDC reported in an investigation notice posted July 19.
The first illness in this outbreak started on Nov. 20, 2017, and the most recent one started on June 29, 2018. The U.S. Department of Agriculture’s Food Safety Inspection Service is monitoring the outbreak, and public health and regulatory agency efforts are being coordinated by the CDC through its PulseNet national subtyping network. DNA fingerprinting “performed on Salmonella from ill people in this outbreak showed that they are closely related genetically. This means that the ill people are more likely to share a common source of infection,” the CDC said.
Consumers should handle raw turkey carefully and cook it thoroughly to prevent Salmonella, the CDC advised. Raw food of any type should not be given to pets. At this time, the CDC said that it is “not advising that consumers avoid eating properly cooked turkey products, or that retailers stop selling raw turkey products.”
A total of 90 people in 26 states have been infected with multidrug-resistant Salmonella in an outbreak linked to raw turkey products, according to the Centers for Disease Control and Prevention.
As of July 11, 2018, 40 of the 78 people with available information who were infected with the outbreak strain of Salmonella Reading have been hospitalized, but no deaths have been reported. Of the 61 ill people who have been interviewed, most have reported preparing or eating turkey products from a number of sources, although two lived in households where raw turkey was given to pets: No common supplier has been identified, the CDC reported in an investigation notice posted July 19.
The first illness in this outbreak started on Nov. 20, 2017, and the most recent one started on June 29, 2018. The U.S. Department of Agriculture’s Food Safety Inspection Service is monitoring the outbreak, and public health and regulatory agency efforts are being coordinated by the CDC through its PulseNet national subtyping network. DNA fingerprinting “performed on Salmonella from ill people in this outbreak showed that they are closely related genetically. This means that the ill people are more likely to share a common source of infection,” the CDC said.
Consumers should handle raw turkey carefully and cook it thoroughly to prevent Salmonella, the CDC advised. Raw food of any type should not be given to pets. At this time, the CDC said that it is “not advising that consumers avoid eating properly cooked turkey products, or that retailers stop selling raw turkey products.”
A total of 90 people in 26 states have been infected with multidrug-resistant Salmonella in an outbreak linked to raw turkey products, according to the Centers for Disease Control and Prevention.
As of July 11, 2018, 40 of the 78 people with available information who were infected with the outbreak strain of Salmonella Reading have been hospitalized, but no deaths have been reported. Of the 61 ill people who have been interviewed, most have reported preparing or eating turkey products from a number of sources, although two lived in households where raw turkey was given to pets: No common supplier has been identified, the CDC reported in an investigation notice posted July 19.
The first illness in this outbreak started on Nov. 20, 2017, and the most recent one started on June 29, 2018. The U.S. Department of Agriculture’s Food Safety Inspection Service is monitoring the outbreak, and public health and regulatory agency efforts are being coordinated by the CDC through its PulseNet national subtyping network. DNA fingerprinting “performed on Salmonella from ill people in this outbreak showed that they are closely related genetically. This means that the ill people are more likely to share a common source of infection,” the CDC said.
Consumers should handle raw turkey carefully and cook it thoroughly to prevent Salmonella, the CDC advised. Raw food of any type should not be given to pets. At this time, the CDC said that it is “not advising that consumers avoid eating properly cooked turkey products, or that retailers stop selling raw turkey products.”
Fentanyl analogs nearly double their overdose death toll
, according to preliminary data from 10 states.
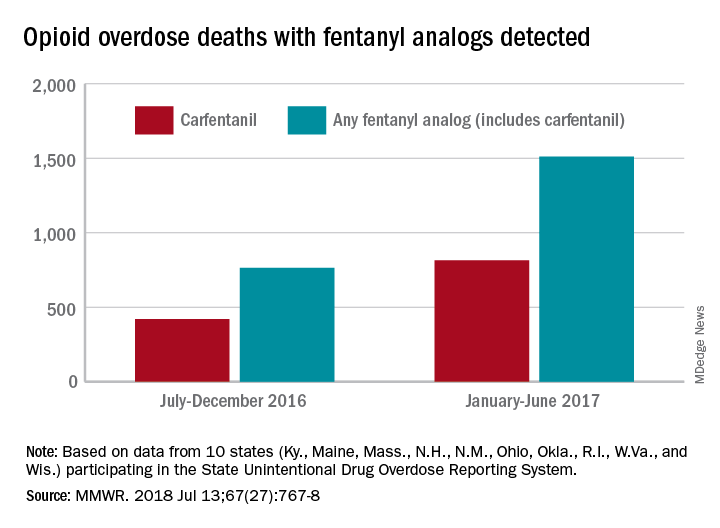
During July 2016 to December 2016, there were 764 opioid overdose deaths that tested positive for any fentanyl analog, with carfentanil being the most common (421 deaths). From January 2017 to June 2017, the respective numbers increased by 98% (1,511) and 94% (815), wrote Julie O’Donnell, PhD, and her associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control. The report was published in the Morbidity and Mortality Weekly Report.
“The increasing array of fentanyl analogs highlights the need to build forensic toxicological testing capabilities to identify and report emerging threats, and to enhance capacity to rapidly respond to evolving drug trends,” Dr. O’Donnell and her associates said.
Along with carfentanil, 13 other analogs were detected in decedents during the 12-month period: 3-methylfentanyl, 4-fluorobutyrfentanyl, 4-fluorofentanyl, 4-fluoroisobutyrfentanyl, acetylfentanyl, acrylfentanyl, butyrylfentanyl, cyclopropylfentanyl, cyclopentylfentanyl, furanylethylfentanyl, furanylfentanyl, isobutyrylfentanyl, and tetrahydrofuranylfentanyl. Deaths may have involved “more than one analog, as well as ... other opioid and nonopioid substances,” they noted.
The 10 states reporting data to the State Unintentional Drug Overdose Reporting System (SUDORS) were Kentucky, Maine, Massachusetts, New Hampshire, New Mexico, Ohio, Oklahoma, Rhode Island, West Virginia, and Wisconsin. Two other SUDORS-reporting states – Missouri and Pennsylvania – did not have their data ready in time to be included in this analysis.
The increasing availability of fentanyl analogs hit Ohio especially hard: More deaths occurred there than in the other 10 states combined. Of the 421 carfentanil-related deaths in July 2016 to December 2016, nearly 400 were in Ohio, and there were 218 Ohio deaths in April 2017 alone. A look at the bigger picture shows that 3 of the 10 states reported carfentanil-related overdose deaths in the second half of 2016, compared with 7 in the first half of 2017, the investigators said.
Carfentanil, which is the most potent of the 14 fentanyl analogs that have been detected so far, “is intended for sedation of large animals, and is estimated to have 10,000 times the potency of morphine,” Dr. O’Donnell and her associates wrote.
SOURCE: O’Donnell J et al. MMWR. 2018 Jul 13;67(27):767-8.
, according to preliminary data from 10 states.
During July 2016 to December 2016, there were 764 opioid overdose deaths that tested positive for any fentanyl analog, with carfentanil being the most common (421 deaths). From January 2017 to June 2017, the respective numbers increased by 98% (1,511) and 94% (815), wrote Julie O’Donnell, PhD, and her associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control. The report was published in the Morbidity and Mortality Weekly Report.
“The increasing array of fentanyl analogs highlights the need to build forensic toxicological testing capabilities to identify and report emerging threats, and to enhance capacity to rapidly respond to evolving drug trends,” Dr. O’Donnell and her associates said.
Along with carfentanil, 13 other analogs were detected in decedents during the 12-month period: 3-methylfentanyl, 4-fluorobutyrfentanyl, 4-fluorofentanyl, 4-fluoroisobutyrfentanyl, acetylfentanyl, acrylfentanyl, butyrylfentanyl, cyclopropylfentanyl, cyclopentylfentanyl, furanylethylfentanyl, furanylfentanyl, isobutyrylfentanyl, and tetrahydrofuranylfentanyl. Deaths may have involved “more than one analog, as well as ... other opioid and nonopioid substances,” they noted.
The 10 states reporting data to the State Unintentional Drug Overdose Reporting System (SUDORS) were Kentucky, Maine, Massachusetts, New Hampshire, New Mexico, Ohio, Oklahoma, Rhode Island, West Virginia, and Wisconsin. Two other SUDORS-reporting states – Missouri and Pennsylvania – did not have their data ready in time to be included in this analysis.
The increasing availability of fentanyl analogs hit Ohio especially hard: More deaths occurred there than in the other 10 states combined. Of the 421 carfentanil-related deaths in July 2016 to December 2016, nearly 400 were in Ohio, and there were 218 Ohio deaths in April 2017 alone. A look at the bigger picture shows that 3 of the 10 states reported carfentanil-related overdose deaths in the second half of 2016, compared with 7 in the first half of 2017, the investigators said.
Carfentanil, which is the most potent of the 14 fentanyl analogs that have been detected so far, “is intended for sedation of large animals, and is estimated to have 10,000 times the potency of morphine,” Dr. O’Donnell and her associates wrote.
SOURCE: O’Donnell J et al. MMWR. 2018 Jul 13;67(27):767-8.
, according to preliminary data from 10 states.
During July 2016 to December 2016, there were 764 opioid overdose deaths that tested positive for any fentanyl analog, with carfentanil being the most common (421 deaths). From January 2017 to June 2017, the respective numbers increased by 98% (1,511) and 94% (815), wrote Julie O’Donnell, PhD, and her associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control. The report was published in the Morbidity and Mortality Weekly Report.
“The increasing array of fentanyl analogs highlights the need to build forensic toxicological testing capabilities to identify and report emerging threats, and to enhance capacity to rapidly respond to evolving drug trends,” Dr. O’Donnell and her associates said.
Along with carfentanil, 13 other analogs were detected in decedents during the 12-month period: 3-methylfentanyl, 4-fluorobutyrfentanyl, 4-fluorofentanyl, 4-fluoroisobutyrfentanyl, acetylfentanyl, acrylfentanyl, butyrylfentanyl, cyclopropylfentanyl, cyclopentylfentanyl, furanylethylfentanyl, furanylfentanyl, isobutyrylfentanyl, and tetrahydrofuranylfentanyl. Deaths may have involved “more than one analog, as well as ... other opioid and nonopioid substances,” they noted.
The 10 states reporting data to the State Unintentional Drug Overdose Reporting System (SUDORS) were Kentucky, Maine, Massachusetts, New Hampshire, New Mexico, Ohio, Oklahoma, Rhode Island, West Virginia, and Wisconsin. Two other SUDORS-reporting states – Missouri and Pennsylvania – did not have their data ready in time to be included in this analysis.
The increasing availability of fentanyl analogs hit Ohio especially hard: More deaths occurred there than in the other 10 states combined. Of the 421 carfentanil-related deaths in July 2016 to December 2016, nearly 400 were in Ohio, and there were 218 Ohio deaths in April 2017 alone. A look at the bigger picture shows that 3 of the 10 states reported carfentanil-related overdose deaths in the second half of 2016, compared with 7 in the first half of 2017, the investigators said.
Carfentanil, which is the most potent of the 14 fentanyl analogs that have been detected so far, “is intended for sedation of large animals, and is estimated to have 10,000 times the potency of morphine,” Dr. O’Donnell and her associates wrote.
SOURCE: O’Donnell J et al. MMWR. 2018 Jul 13;67(27):767-8.
FROM MMWR