User login
CDC opens Emergency Operations Center in advance of Hurricane Florence
The Centers for Disease Control and Prevention is activating its Emergency Operations Center (EOC) in advance of the landfall of Hurricane Florence, according to a CDC media statement.
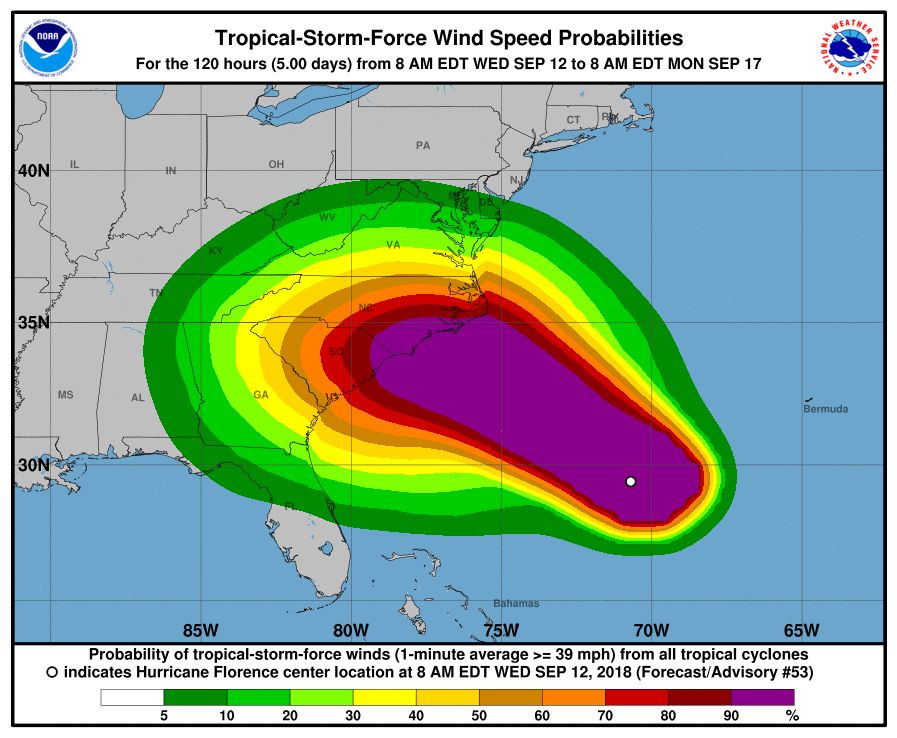
Activation of the center will allow for 24/7 management of all CDC activities before, during, and after the landfall of Hurricane Florence, including deployment of personnel and resources.
All CDC staff should be ready to provide infectious disease outbreak surveillance, public health messages, water/food safety evaluations, mold prevention and treatment, industrial contamination containment, and standing water control. The CDC is also sharing messages with the public on how to protect themselves from threats before, during, and after landfall, such as drowning and floodwater safety, carbon monoxide poisoning, downed power lines, unsafe food and water, and mold.
The EOC is the CDC’s command center for the coordination and monitoring of CDC activity with all other U.S. federal, state, and local agencies. The EOC also works with the Secretary of Health & Human Services Secretary’s Operations Center to ensure awareness and a coordinated public health and medical response, according to the statement.
Additional resources for interested parties can be found on the CDC website.
The Centers for Disease Control and Prevention is activating its Emergency Operations Center (EOC) in advance of the landfall of Hurricane Florence, according to a CDC media statement.
Activation of the center will allow for 24/7 management of all CDC activities before, during, and after the landfall of Hurricane Florence, including deployment of personnel and resources.
All CDC staff should be ready to provide infectious disease outbreak surveillance, public health messages, water/food safety evaluations, mold prevention and treatment, industrial contamination containment, and standing water control. The CDC is also sharing messages with the public on how to protect themselves from threats before, during, and after landfall, such as drowning and floodwater safety, carbon monoxide poisoning, downed power lines, unsafe food and water, and mold.
The EOC is the CDC’s command center for the coordination and monitoring of CDC activity with all other U.S. federal, state, and local agencies. The EOC also works with the Secretary of Health & Human Services Secretary’s Operations Center to ensure awareness and a coordinated public health and medical response, according to the statement.
Additional resources for interested parties can be found on the CDC website.
The Centers for Disease Control and Prevention is activating its Emergency Operations Center (EOC) in advance of the landfall of Hurricane Florence, according to a CDC media statement.
Activation of the center will allow for 24/7 management of all CDC activities before, during, and after the landfall of Hurricane Florence, including deployment of personnel and resources.
All CDC staff should be ready to provide infectious disease outbreak surveillance, public health messages, water/food safety evaluations, mold prevention and treatment, industrial contamination containment, and standing water control. The CDC is also sharing messages with the public on how to protect themselves from threats before, during, and after landfall, such as drowning and floodwater safety, carbon monoxide poisoning, downed power lines, unsafe food and water, and mold.
The EOC is the CDC’s command center for the coordination and monitoring of CDC activity with all other U.S. federal, state, and local agencies. The EOC also works with the Secretary of Health & Human Services Secretary’s Operations Center to ensure awareness and a coordinated public health and medical response, according to the statement.
Additional resources for interested parties can be found on the CDC website.
Venetoclax label now includes MRD data
The Food and Drug Administration has expanded the label for venetoclax tablets (Venclexta) to include data on minimal residual disease.
The drug’s prescribing information will now include details on minimal residual disease (MRD) negativity in previously treated patients with chronic lymphocytic leukemia (CLL) who received venetoclax in combination with rituximab in the phase 3 MURANO trial.
The combination of venetoclax and rituximab was approved by the FDA in June 2018 for the treatment of patients with CLL or small lymphocytic lymphoma, with or without 17p deletion, who received at least one prior therapy.
The MURANO trial (NCT02005471), which supported the FDA approval, included 389 patients with relapsed or refractory CLL. They were randomized to receive venetoclax plus rituximab or bendamustine plus rituximab (N Engl J Med. 2018; 378:1107-20).
Researchers evaluated MRD in patients who achieved a partial response or better. MRD was assessed using allele-specific oligonucleotide polymerase chain reaction; the definition of MRD negativity was less than one CLL cell per 10,000 lymphocytes.
The researchers assessed MRD in the peripheral blood after about 9 months on therapy (3 months after the last dose of rituximab). At that time, 53% (103/194) of patients in the venetoclax-rituximab arm were MRD negative, as were 12% (23/195) of patients in the bendamustine-rituximab arm.
The researchers also assessed MRD in the peripheral blood of patients with a complete response or complete response with incomplete marrow recovery. MRD negativity was achieved by 3% (6/194) of these patients in the venetoclax-rituximab arm and 2% (3/195) in the bendamustine-rituximab arm.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie outside of the United States.
The Food and Drug Administration has expanded the label for venetoclax tablets (Venclexta) to include data on minimal residual disease.
The drug’s prescribing information will now include details on minimal residual disease (MRD) negativity in previously treated patients with chronic lymphocytic leukemia (CLL) who received venetoclax in combination with rituximab in the phase 3 MURANO trial.
The combination of venetoclax and rituximab was approved by the FDA in June 2018 for the treatment of patients with CLL or small lymphocytic lymphoma, with or without 17p deletion, who received at least one prior therapy.
The MURANO trial (NCT02005471), which supported the FDA approval, included 389 patients with relapsed or refractory CLL. They were randomized to receive venetoclax plus rituximab or bendamustine plus rituximab (N Engl J Med. 2018; 378:1107-20).
Researchers evaluated MRD in patients who achieved a partial response or better. MRD was assessed using allele-specific oligonucleotide polymerase chain reaction; the definition of MRD negativity was less than one CLL cell per 10,000 lymphocytes.
The researchers assessed MRD in the peripheral blood after about 9 months on therapy (3 months after the last dose of rituximab). At that time, 53% (103/194) of patients in the venetoclax-rituximab arm were MRD negative, as were 12% (23/195) of patients in the bendamustine-rituximab arm.
The researchers also assessed MRD in the peripheral blood of patients with a complete response or complete response with incomplete marrow recovery. MRD negativity was achieved by 3% (6/194) of these patients in the venetoclax-rituximab arm and 2% (3/195) in the bendamustine-rituximab arm.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie outside of the United States.
The Food and Drug Administration has expanded the label for venetoclax tablets (Venclexta) to include data on minimal residual disease.
The drug’s prescribing information will now include details on minimal residual disease (MRD) negativity in previously treated patients with chronic lymphocytic leukemia (CLL) who received venetoclax in combination with rituximab in the phase 3 MURANO trial.
The combination of venetoclax and rituximab was approved by the FDA in June 2018 for the treatment of patients with CLL or small lymphocytic lymphoma, with or without 17p deletion, who received at least one prior therapy.
The MURANO trial (NCT02005471), which supported the FDA approval, included 389 patients with relapsed or refractory CLL. They were randomized to receive venetoclax plus rituximab or bendamustine plus rituximab (N Engl J Med. 2018; 378:1107-20).
Researchers evaluated MRD in patients who achieved a partial response or better. MRD was assessed using allele-specific oligonucleotide polymerase chain reaction; the definition of MRD negativity was less than one CLL cell per 10,000 lymphocytes.
The researchers assessed MRD in the peripheral blood after about 9 months on therapy (3 months after the last dose of rituximab). At that time, 53% (103/194) of patients in the venetoclax-rituximab arm were MRD negative, as were 12% (23/195) of patients in the bendamustine-rituximab arm.
The researchers also assessed MRD in the peripheral blood of patients with a complete response or complete response with incomplete marrow recovery. MRD negativity was achieved by 3% (6/194) of these patients in the venetoclax-rituximab arm and 2% (3/195) in the bendamustine-rituximab arm.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie outside of the United States.
FDA warns kratom vendors about unproven claims
The Food and Drug Administration has issued letters of warning to – and therefore breaking federal law – according to a statement from FDA commissioner Scott Gottlieb, MD. These vendors both claimed that their kratom products could, among other things, “relieve” or “treat” opium/opioid withdrawal.

“To date, there have been no adequate and well-controlled studies involving the use of kratom as a treatment for opioid use withdrawal or other diseases in humans,” noted Dr. Gottlieb.
As Dr. Gottlieb pointed out in his statement, not only can fraudulent health claims pose direct health risks, they can also, in the case of kratom, deter or delay people who’re suffering from opioid use disorder from seeking FDA-approved treatments that have been demonstrated to be safe and effective.

Kratom, also known more formally as Mitragyna speciosa, is a plant native to Thailand, Malaysia, Indonesia, and Papua New Guinea. Some compounds in the plant are believed to be opioids, some of which may have the potential for abuse. As Dr. Gottlieb pointed out in his statement, the substance is illegal or controlled in several countries and banned in some states and municipalities in the United States.
Find out more in Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration has issued letters of warning to – and therefore breaking federal law – according to a statement from FDA commissioner Scott Gottlieb, MD. These vendors both claimed that their kratom products could, among other things, “relieve” or “treat” opium/opioid withdrawal.
“To date, there have been no adequate and well-controlled studies involving the use of kratom as a treatment for opioid use withdrawal or other diseases in humans,” noted Dr. Gottlieb.
As Dr. Gottlieb pointed out in his statement, not only can fraudulent health claims pose direct health risks, they can also, in the case of kratom, deter or delay people who’re suffering from opioid use disorder from seeking FDA-approved treatments that have been demonstrated to be safe and effective.
Kratom, also known more formally as Mitragyna speciosa, is a plant native to Thailand, Malaysia, Indonesia, and Papua New Guinea. Some compounds in the plant are believed to be opioids, some of which may have the potential for abuse. As Dr. Gottlieb pointed out in his statement, the substance is illegal or controlled in several countries and banned in some states and municipalities in the United States.
Find out more in Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration has issued letters of warning to – and therefore breaking federal law – according to a statement from FDA commissioner Scott Gottlieb, MD. These vendors both claimed that their kratom products could, among other things, “relieve” or “treat” opium/opioid withdrawal.
“To date, there have been no adequate and well-controlled studies involving the use of kratom as a treatment for opioid use withdrawal or other diseases in humans,” noted Dr. Gottlieb.
As Dr. Gottlieb pointed out in his statement, not only can fraudulent health claims pose direct health risks, they can also, in the case of kratom, deter or delay people who’re suffering from opioid use disorder from seeking FDA-approved treatments that have been demonstrated to be safe and effective.
Kratom, also known more formally as Mitragyna speciosa, is a plant native to Thailand, Malaysia, Indonesia, and Papua New Guinea. Some compounds in the plant are believed to be opioids, some of which may have the potential for abuse. As Dr. Gottlieb pointed out in his statement, the substance is illegal or controlled in several countries and banned in some states and municipalities in the United States.
Find out more in Dr. Gottlieb’s full statement on the FDA website.
Pediatric ED diagnoses vary with the season
The two most common reasons for pediatric emergency department visits exhibit considerable and opposing seasonal variations, according to the Agency for Healthcare Research and Quality.
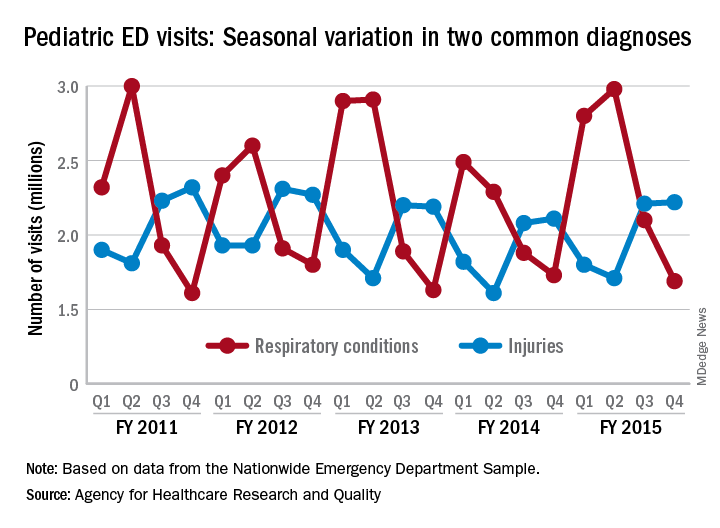
Of the 30 million ED visits by children aged 18 years and younger during fiscal year 2015, about 9.6 million, or just under 24%, were for respiratory disorders, making it the most common diagnosis by body system. The second-most common reason, injuries, was associated with approximately 7.9 million visits in 2015, the AHRQ reported recently in a statistical brief.
Over the 4-year period from 2011 to 2015, pediatric ED visits for respiratory disorders peaked during the months from October to March and dropped during April-September. In 2015, for example, there was a 43% difference between October-December, which was the highest-volume quarter, and July-September, which had the lowest volume of visits, the report showed.
The opposite pattern of seasonality was seen with visits for injury-related visits: The high point each year occurs in April-September, with the low in October-March. In 2015, there was a 30% difference between the lowest-volume quarter of January-March and the highest-volume quarter of July-September, based on the analysis of data from the Nationwide Emergency Department Sample.
The two most common reasons for pediatric emergency department visits exhibit considerable and opposing seasonal variations, according to the Agency for Healthcare Research and Quality.
Of the 30 million ED visits by children aged 18 years and younger during fiscal year 2015, about 9.6 million, or just under 24%, were for respiratory disorders, making it the most common diagnosis by body system. The second-most common reason, injuries, was associated with approximately 7.9 million visits in 2015, the AHRQ reported recently in a statistical brief.
Over the 4-year period from 2011 to 2015, pediatric ED visits for respiratory disorders peaked during the months from October to March and dropped during April-September. In 2015, for example, there was a 43% difference between October-December, which was the highest-volume quarter, and July-September, which had the lowest volume of visits, the report showed.
The opposite pattern of seasonality was seen with visits for injury-related visits: The high point each year occurs in April-September, with the low in October-March. In 2015, there was a 30% difference between the lowest-volume quarter of January-March and the highest-volume quarter of July-September, based on the analysis of data from the Nationwide Emergency Department Sample.
The two most common reasons for pediatric emergency department visits exhibit considerable and opposing seasonal variations, according to the Agency for Healthcare Research and Quality.
Of the 30 million ED visits by children aged 18 years and younger during fiscal year 2015, about 9.6 million, or just under 24%, were for respiratory disorders, making it the most common diagnosis by body system. The second-most common reason, injuries, was associated with approximately 7.9 million visits in 2015, the AHRQ reported recently in a statistical brief.
Over the 4-year period from 2011 to 2015, pediatric ED visits for respiratory disorders peaked during the months from October to March and dropped during April-September. In 2015, for example, there was a 43% difference between October-December, which was the highest-volume quarter, and July-September, which had the lowest volume of visits, the report showed.
The opposite pattern of seasonality was seen with visits for injury-related visits: The high point each year occurs in April-September, with the low in October-March. In 2015, there was a 30% difference between the lowest-volume quarter of January-March and the highest-volume quarter of July-September, based on the analysis of data from the Nationwide Emergency Department Sample.
Service, please: Hospital setting matters for pneumonia
the National Center for Health Statistics (NCHS) reported.
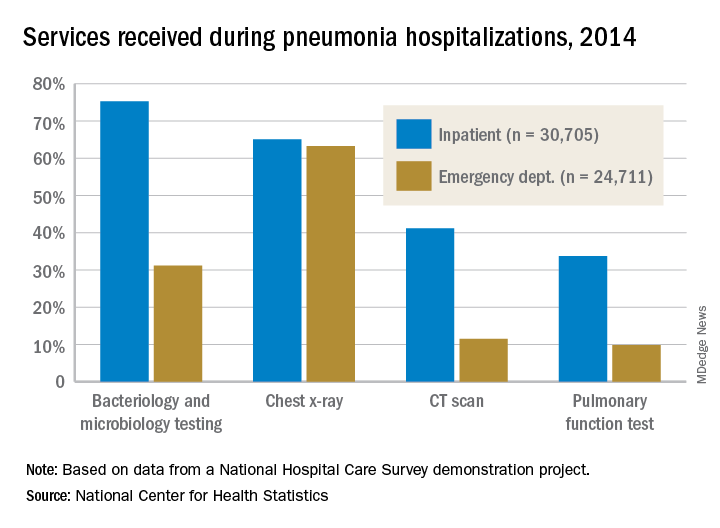
The percentages were not as close, however, for other diagnostic services. Inpatient stays were much more likely than ED encounters to involve bacteriology and microbiology testing (75.3% vs. 31.2%), CT scans (41.2% vs. 11.5%), and pulmonary function tests (33.7% vs. 9.8%), investigators from the NCHS said.
The age distribution of the two patient populations also were quite different, with those aged 65 years and older making up the largest share (46%) of pneumonia inpatients and the 15-and-under group representing the largest proportion (47%) of ED visits. For the inpatient setting, the smallest age group was those aged 15-44 years (10%), and for the ED it was those aged 65 years and older (14%), they reported.
The National Hospital Care Survey “is not yet nationally representative,” the NCHS investigators wrote – the overall sample for 2014 consisted of 581 hospitals – but “the number of encounters and the inclusion of [personally identifiable information] allow an example of analysis that was not previously possible.”
the National Center for Health Statistics (NCHS) reported.
The percentages were not as close, however, for other diagnostic services. Inpatient stays were much more likely than ED encounters to involve bacteriology and microbiology testing (75.3% vs. 31.2%), CT scans (41.2% vs. 11.5%), and pulmonary function tests (33.7% vs. 9.8%), investigators from the NCHS said.
The age distribution of the two patient populations also were quite different, with those aged 65 years and older making up the largest share (46%) of pneumonia inpatients and the 15-and-under group representing the largest proportion (47%) of ED visits. For the inpatient setting, the smallest age group was those aged 15-44 years (10%), and for the ED it was those aged 65 years and older (14%), they reported.
The National Hospital Care Survey “is not yet nationally representative,” the NCHS investigators wrote – the overall sample for 2014 consisted of 581 hospitals – but “the number of encounters and the inclusion of [personally identifiable information] allow an example of analysis that was not previously possible.”
the National Center for Health Statistics (NCHS) reported.
The percentages were not as close, however, for other diagnostic services. Inpatient stays were much more likely than ED encounters to involve bacteriology and microbiology testing (75.3% vs. 31.2%), CT scans (41.2% vs. 11.5%), and pulmonary function tests (33.7% vs. 9.8%), investigators from the NCHS said.
The age distribution of the two patient populations also were quite different, with those aged 65 years and older making up the largest share (46%) of pneumonia inpatients and the 15-and-under group representing the largest proportion (47%) of ED visits. For the inpatient setting, the smallest age group was those aged 15-44 years (10%), and for the ED it was those aged 65 years and older (14%), they reported.
The National Hospital Care Survey “is not yet nationally representative,” the NCHS investigators wrote – the overall sample for 2014 consisted of 581 hospitals – but “the number of encounters and the inclusion of [personally identifiable information] allow an example of analysis that was not previously possible.”
Valsartan recalls: FDA, manufacturers issue advisories
This story updated Aug. 23 to include additional recall and release of new test. The FDA added an updated statement on their ongoing investigation on Aug. 30 and again on Oct. 24.
for recognizing the recalled products and prescribing replacement products.

The affected products containing the active ingredient valsartan were voluntarily recalled because of the detection of N-nitrosodimethylamine (NDMA), an impurity that is classified as a probable carcinogen. The presence of NDMA was unexpected and is thought to be related to changes in the manufacturing process, the FDA announced in a press release.
The voluntary recall affects all lots of nonexpired products that contain the ingredient valsartan supplied to companies by Zhejiang Huahai Pharmaceuticals, Linhai, China. This company has stopped distributing valsartan. The FDA is working with the affected manufacturers – Major Pharmaceuticals, Solco Healthcare, and Teva Pharmaceuticals – to reduce or eliminate impure valsartan from future products. The voluntary recall also applies to Solco and Teva valsartan/hydrochlorothiazide (HCTZ) combination products.
On Aug. 23, the FDA announced that the recall was extended to all valsartan-containing products from Torrent Pharmaceuticals Limited.
The agency said its review is ongoing and includes investigating the levels of NDMA in the recalled products, assessing the possible effect on patients who have been taking them, and what measures can be taken to reduce or eliminate the impurity from future batches.
“Our drug shortages team is also working hard to ensure patients’ therapeutic needs are met in the United States with an adequate supply of unaffected medications,” FDA Commissioner Scott Gottlieb, MD, said.
Additionally, the FDA on Aug. 23 released a gas chromatography-mass spectrometry (GC/MS) headspace method that drug manufacturers and regulators can use to detect and quantify NDMA in valsartan and finished drug products.
In the interim, patients taking the recalled valsartan-containing medicines should continue taking their medicine until they have a replacement product, the statement said. To determine whether a specific product has been recalled, patients should be instructed to look at the drug name and company name on the label of their prescription bottle. If the information is not on the bottle, patients should contact the pharmacy that dispensed the medicine.
If a patient is taking one of the recalled medicines, they should follow the recall instructions provided by the specific company, according to the FDA.
Contact information for each manufacturer can be found at the following links:
Major Pharmaceuticals: www.fda.gov/Safety/Recalls/ucm613625.htm.
Solco Healthcare: www.fda.gov/Safety/Recalls/ucm613504.htm.
Teva Pharmaceuticals: www.fda.gov/Safety/Recalls/ucm613729.htm.
Torrent Pharmaceuticals Limited: https://www.fda.gov/Safety/Recalls/ucm617821.htm
This story updated Aug. 23 to include additional recall and release of new test. The FDA added an updated statement on their ongoing investigation on Aug. 30 and again on Oct. 24.
for recognizing the recalled products and prescribing replacement products.
The affected products containing the active ingredient valsartan were voluntarily recalled because of the detection of N-nitrosodimethylamine (NDMA), an impurity that is classified as a probable carcinogen. The presence of NDMA was unexpected and is thought to be related to changes in the manufacturing process, the FDA announced in a press release.
The voluntary recall affects all lots of nonexpired products that contain the ingredient valsartan supplied to companies by Zhejiang Huahai Pharmaceuticals, Linhai, China. This company has stopped distributing valsartan. The FDA is working with the affected manufacturers – Major Pharmaceuticals, Solco Healthcare, and Teva Pharmaceuticals – to reduce or eliminate impure valsartan from future products. The voluntary recall also applies to Solco and Teva valsartan/hydrochlorothiazide (HCTZ) combination products.
On Aug. 23, the FDA announced that the recall was extended to all valsartan-containing products from Torrent Pharmaceuticals Limited.
The agency said its review is ongoing and includes investigating the levels of NDMA in the recalled products, assessing the possible effect on patients who have been taking them, and what measures can be taken to reduce or eliminate the impurity from future batches.
“Our drug shortages team is also working hard to ensure patients’ therapeutic needs are met in the United States with an adequate supply of unaffected medications,” FDA Commissioner Scott Gottlieb, MD, said.
Additionally, the FDA on Aug. 23 released a gas chromatography-mass spectrometry (GC/MS) headspace method that drug manufacturers and regulators can use to detect and quantify NDMA in valsartan and finished drug products.
In the interim, patients taking the recalled valsartan-containing medicines should continue taking their medicine until they have a replacement product, the statement said. To determine whether a specific product has been recalled, patients should be instructed to look at the drug name and company name on the label of their prescription bottle. If the information is not on the bottle, patients should contact the pharmacy that dispensed the medicine.
If a patient is taking one of the recalled medicines, they should follow the recall instructions provided by the specific company, according to the FDA.
Contact information for each manufacturer can be found at the following links:
Major Pharmaceuticals: www.fda.gov/Safety/Recalls/ucm613625.htm.
Solco Healthcare: www.fda.gov/Safety/Recalls/ucm613504.htm.
Teva Pharmaceuticals: www.fda.gov/Safety/Recalls/ucm613729.htm.
Torrent Pharmaceuticals Limited: https://www.fda.gov/Safety/Recalls/ucm617821.htm
This story updated Aug. 23 to include additional recall and release of new test. The FDA added an updated statement on their ongoing investigation on Aug. 30 and again on Oct. 24.
for recognizing the recalled products and prescribing replacement products.
The affected products containing the active ingredient valsartan were voluntarily recalled because of the detection of N-nitrosodimethylamine (NDMA), an impurity that is classified as a probable carcinogen. The presence of NDMA was unexpected and is thought to be related to changes in the manufacturing process, the FDA announced in a press release.
The voluntary recall affects all lots of nonexpired products that contain the ingredient valsartan supplied to companies by Zhejiang Huahai Pharmaceuticals, Linhai, China. This company has stopped distributing valsartan. The FDA is working with the affected manufacturers – Major Pharmaceuticals, Solco Healthcare, and Teva Pharmaceuticals – to reduce or eliminate impure valsartan from future products. The voluntary recall also applies to Solco and Teva valsartan/hydrochlorothiazide (HCTZ) combination products.
On Aug. 23, the FDA announced that the recall was extended to all valsartan-containing products from Torrent Pharmaceuticals Limited.
The agency said its review is ongoing and includes investigating the levels of NDMA in the recalled products, assessing the possible effect on patients who have been taking them, and what measures can be taken to reduce or eliminate the impurity from future batches.
“Our drug shortages team is also working hard to ensure patients’ therapeutic needs are met in the United States with an adequate supply of unaffected medications,” FDA Commissioner Scott Gottlieb, MD, said.
Additionally, the FDA on Aug. 23 released a gas chromatography-mass spectrometry (GC/MS) headspace method that drug manufacturers and regulators can use to detect and quantify NDMA in valsartan and finished drug products.
In the interim, patients taking the recalled valsartan-containing medicines should continue taking their medicine until they have a replacement product, the statement said. To determine whether a specific product has been recalled, patients should be instructed to look at the drug name and company name on the label of their prescription bottle. If the information is not on the bottle, patients should contact the pharmacy that dispensed the medicine.
If a patient is taking one of the recalled medicines, they should follow the recall instructions provided by the specific company, according to the FDA.
Contact information for each manufacturer can be found at the following links:
Major Pharmaceuticals: www.fda.gov/Safety/Recalls/ucm613625.htm.
Solco Healthcare: www.fda.gov/Safety/Recalls/ucm613504.htm.
Teva Pharmaceuticals: www.fda.gov/Safety/Recalls/ucm613729.htm.
Torrent Pharmaceuticals Limited: https://www.fda.gov/Safety/Recalls/ucm617821.htm
Feds take baseline on EHR interoperability
Office-based physicians who have electronic health records are most likely to send patient health information (PHI) in the form of referrals and laboratory results and receive it as lab results and imaging reports, according to national estimates of electronic PHI using four aspects of interoperability.
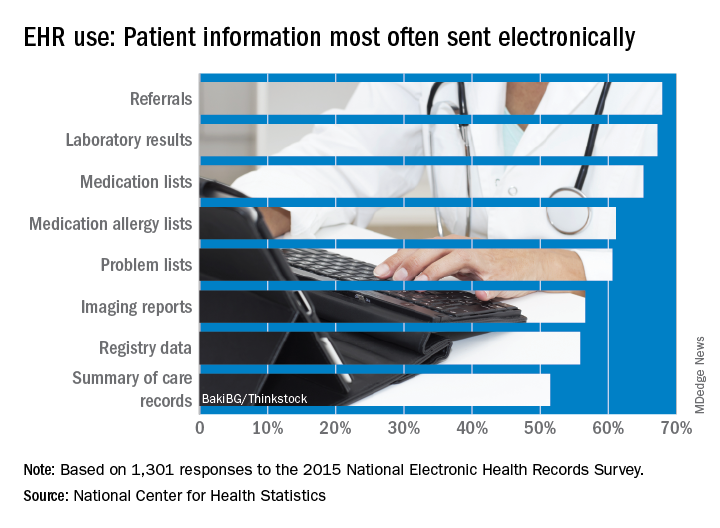
Those aspects of sharing PHI are sending, receiving, integrating, and searching. With federal estimates of EHR adoption at 78% for office-based physicians, data from the 2015 National Electronic Health Records Just over 65% are sending medication lists electronically, 61% are sending medication allergy lists, and almost 61% are sending problem lists, Ninee S. Yang, PhD, and her associates at the Centers for Disease Control and Prevention said in National Health Statistics Reports.
Communication in the other direction showed a somewhat different distribution. Of the 1,525 physicians who reported receiving PHI electronically, 79% were the recipients of laboratory results, with imaging reports well behind at 61%, followed by medication lists and referrals at 54% and summary of care records at 52%, the investigators reported.
The ability to integrate information into an EHR – reported by 959 survey respondents – is another key element of interoperability, and 73% of those physicians said that they could integrate lab results into their systems. Other types of PHI, however, did not fare as well: imaging reports came in at 50%, hospital discharge summaries at 49%, summary of care records at 41%, and emergency department notifications at 40%, Dr. Yang and her associates said.
The fourth aspect of interoperability, ability to search for PHI from sources outside the practice, was reported by 1,335 respondents in 2015, with 90% looking for medication lists, 88% for medication allergy lists, 80% for hospital discharge summaries, 59% for imaging reports, and 49% for lab results, they said.
These first national estimates of PHI type will be used as baseline data “in tracking progress outlined in the federal plan for achieving interoperability” among physicians with EHR systems, Dr. Yang and her associates wrote.
SOURCE: Yang NS et al. Natl Health Stat Report. 2018 Aug 15;(115):1-9.
Office-based physicians who have electronic health records are most likely to send patient health information (PHI) in the form of referrals and laboratory results and receive it as lab results and imaging reports, according to national estimates of electronic PHI using four aspects of interoperability.
Those aspects of sharing PHI are sending, receiving, integrating, and searching. With federal estimates of EHR adoption at 78% for office-based physicians, data from the 2015 National Electronic Health Records Just over 65% are sending medication lists electronically, 61% are sending medication allergy lists, and almost 61% are sending problem lists, Ninee S. Yang, PhD, and her associates at the Centers for Disease Control and Prevention said in National Health Statistics Reports.
Communication in the other direction showed a somewhat different distribution. Of the 1,525 physicians who reported receiving PHI electronically, 79% were the recipients of laboratory results, with imaging reports well behind at 61%, followed by medication lists and referrals at 54% and summary of care records at 52%, the investigators reported.
The ability to integrate information into an EHR – reported by 959 survey respondents – is another key element of interoperability, and 73% of those physicians said that they could integrate lab results into their systems. Other types of PHI, however, did not fare as well: imaging reports came in at 50%, hospital discharge summaries at 49%, summary of care records at 41%, and emergency department notifications at 40%, Dr. Yang and her associates said.
The fourth aspect of interoperability, ability to search for PHI from sources outside the practice, was reported by 1,335 respondents in 2015, with 90% looking for medication lists, 88% for medication allergy lists, 80% for hospital discharge summaries, 59% for imaging reports, and 49% for lab results, they said.
These first national estimates of PHI type will be used as baseline data “in tracking progress outlined in the federal plan for achieving interoperability” among physicians with EHR systems, Dr. Yang and her associates wrote.
SOURCE: Yang NS et al. Natl Health Stat Report. 2018 Aug 15;(115):1-9.
Office-based physicians who have electronic health records are most likely to send patient health information (PHI) in the form of referrals and laboratory results and receive it as lab results and imaging reports, according to national estimates of electronic PHI using four aspects of interoperability.
Those aspects of sharing PHI are sending, receiving, integrating, and searching. With federal estimates of EHR adoption at 78% for office-based physicians, data from the 2015 National Electronic Health Records Just over 65% are sending medication lists electronically, 61% are sending medication allergy lists, and almost 61% are sending problem lists, Ninee S. Yang, PhD, and her associates at the Centers for Disease Control and Prevention said in National Health Statistics Reports.
Communication in the other direction showed a somewhat different distribution. Of the 1,525 physicians who reported receiving PHI electronically, 79% were the recipients of laboratory results, with imaging reports well behind at 61%, followed by medication lists and referrals at 54% and summary of care records at 52%, the investigators reported.
The ability to integrate information into an EHR – reported by 959 survey respondents – is another key element of interoperability, and 73% of those physicians said that they could integrate lab results into their systems. Other types of PHI, however, did not fare as well: imaging reports came in at 50%, hospital discharge summaries at 49%, summary of care records at 41%, and emergency department notifications at 40%, Dr. Yang and her associates said.
The fourth aspect of interoperability, ability to search for PHI from sources outside the practice, was reported by 1,335 respondents in 2015, with 90% looking for medication lists, 88% for medication allergy lists, 80% for hospital discharge summaries, 59% for imaging reports, and 49% for lab results, they said.
These first national estimates of PHI type will be used as baseline data “in tracking progress outlined in the federal plan for achieving interoperability” among physicians with EHR systems, Dr. Yang and her associates wrote.
SOURCE: Yang NS et al. Natl Health Stat Report. 2018 Aug 15;(115):1-9.
FROM NATIONAL HEALTH STATISTICS REPORTS
FDA grants full approval to pembrolizumab for advanced NSCLC
(NSCLC), with no EGFR or ALK genomic tumor aberrations.

The checkpoint inhibitor was previously approved for patients with metastatic nonsquamous NSCLC in 2017, under the accelerated approval process, based on phase 2 results. Approval is now converted to a full approval, based on the results of the phase 3 Keynote-189 trial.
Patients in Keynote-189 who received pembrolizumab in combination with pemetrexed and platinum chemotherapy demonstrated a statistically significant and clinically meaningful improvement in overall survival (hazard ratio, 0.49 [95% confidence interval, 0.38-0.64]; P less than .00001), according to the company press statement.
There was also a significant improvement in progression-free survival (PFS) with the pembrolizumab plus chemotherapy combination, compared with chemotherapy alone (HR, 0.52 [95% CI, 0.43-0.64]; P less than .00001).
Patients with metastatic NSCLC, regardless of PD-L1 tumor expression status and with no EGFR or ALK genomic tumor aberrations were randomized to receive pembrolizumab 200 mg, cisplatin or carboplatin, and pemetrexed intravenously every 3 weeks for four cycles followed by pembrolizumab 200 mg for up to 24 months and pemetrexed every 3 weeks (n = 410); or cisplatin or carboplatin and pemetrexed intravenously every 3 weeks for four cycles followed by pemetrexed every 3 weeks (n = 206). Treatment continued until progression of disease or unacceptable toxicity.
The most common adverse reactions with pembrolizumab, resulting in discontinuation, were pneumonitis (3%) and acute kidney injury (2%). The most common adverse reactions or laboratory abnormalities resulting in interruption of treatment were neutropenia (13%), asthenia/fatigue (7%), anemia (7%), and thrombocytopenia (5%).
(NSCLC), with no EGFR or ALK genomic tumor aberrations.
The checkpoint inhibitor was previously approved for patients with metastatic nonsquamous NSCLC in 2017, under the accelerated approval process, based on phase 2 results. Approval is now converted to a full approval, based on the results of the phase 3 Keynote-189 trial.
Patients in Keynote-189 who received pembrolizumab in combination with pemetrexed and platinum chemotherapy demonstrated a statistically significant and clinically meaningful improvement in overall survival (hazard ratio, 0.49 [95% confidence interval, 0.38-0.64]; P less than .00001), according to the company press statement.
There was also a significant improvement in progression-free survival (PFS) with the pembrolizumab plus chemotherapy combination, compared with chemotherapy alone (HR, 0.52 [95% CI, 0.43-0.64]; P less than .00001).
Patients with metastatic NSCLC, regardless of PD-L1 tumor expression status and with no EGFR or ALK genomic tumor aberrations were randomized to receive pembrolizumab 200 mg, cisplatin or carboplatin, and pemetrexed intravenously every 3 weeks for four cycles followed by pembrolizumab 200 mg for up to 24 months and pemetrexed every 3 weeks (n = 410); or cisplatin or carboplatin and pemetrexed intravenously every 3 weeks for four cycles followed by pemetrexed every 3 weeks (n = 206). Treatment continued until progression of disease or unacceptable toxicity.
The most common adverse reactions with pembrolizumab, resulting in discontinuation, were pneumonitis (3%) and acute kidney injury (2%). The most common adverse reactions or laboratory abnormalities resulting in interruption of treatment were neutropenia (13%), asthenia/fatigue (7%), anemia (7%), and thrombocytopenia (5%).
(NSCLC), with no EGFR or ALK genomic tumor aberrations.
The checkpoint inhibitor was previously approved for patients with metastatic nonsquamous NSCLC in 2017, under the accelerated approval process, based on phase 2 results. Approval is now converted to a full approval, based on the results of the phase 3 Keynote-189 trial.
Patients in Keynote-189 who received pembrolizumab in combination with pemetrexed and platinum chemotherapy demonstrated a statistically significant and clinically meaningful improvement in overall survival (hazard ratio, 0.49 [95% confidence interval, 0.38-0.64]; P less than .00001), according to the company press statement.
There was also a significant improvement in progression-free survival (PFS) with the pembrolizumab plus chemotherapy combination, compared with chemotherapy alone (HR, 0.52 [95% CI, 0.43-0.64]; P less than .00001).
Patients with metastatic NSCLC, regardless of PD-L1 tumor expression status and with no EGFR or ALK genomic tumor aberrations were randomized to receive pembrolizumab 200 mg, cisplatin or carboplatin, and pemetrexed intravenously every 3 weeks for four cycles followed by pembrolizumab 200 mg for up to 24 months and pemetrexed every 3 weeks (n = 410); or cisplatin or carboplatin and pemetrexed intravenously every 3 weeks for four cycles followed by pemetrexed every 3 weeks (n = 206). Treatment continued until progression of disease or unacceptable toxicity.
The most common adverse reactions with pembrolizumab, resulting in discontinuation, were pneumonitis (3%) and acute kidney injury (2%). The most common adverse reactions or laboratory abnormalities resulting in interruption of treatment were neutropenia (13%), asthenia/fatigue (7%), anemia (7%), and thrombocytopenia (5%).
FDA alert: Artificial heart driver linked to higher mortality
Postapproval results for SynCardia Systems’ Companion 2 (C2) driver system for temporary total artificial hearts (TAH-t) have shown higher mortality and stroke rates than were seen with the previous system, the circulatory support system. As a result, the Food and Drug Administration has issued a safety alert cautioning them to weigh the risks and benefits carefully. The alert, issued on August 17, is based on a postapproval study conducted by SynCardia Systems.
Furthermore, patients and health care professionals are encouraged to report any adverse events using the FDA’s Medwatch reporting form, as well as return any devices associated with adverse events to the SynCardia Systems to help them and the FDA better understand the issue.
The C2 driver system is an external pneumatic system that activates an implanted TAH-t in eligible heart failure patients who have severe biventricular failure and are waiting for transplant. It is smaller than its predecessor, but per the device’s approved use, patients must still remain in the hospital while on the device. Since its approval in 2012, the Freedom driver system was approved in 2014, which allows patients to return home.
The full safety alert can be found on the FDA website.
Postapproval results for SynCardia Systems’ Companion 2 (C2) driver system for temporary total artificial hearts (TAH-t) have shown higher mortality and stroke rates than were seen with the previous system, the circulatory support system. As a result, the Food and Drug Administration has issued a safety alert cautioning them to weigh the risks and benefits carefully. The alert, issued on August 17, is based on a postapproval study conducted by SynCardia Systems.
Furthermore, patients and health care professionals are encouraged to report any adverse events using the FDA’s Medwatch reporting form, as well as return any devices associated with adverse events to the SynCardia Systems to help them and the FDA better understand the issue.
The C2 driver system is an external pneumatic system that activates an implanted TAH-t in eligible heart failure patients who have severe biventricular failure and are waiting for transplant. It is smaller than its predecessor, but per the device’s approved use, patients must still remain in the hospital while on the device. Since its approval in 2012, the Freedom driver system was approved in 2014, which allows patients to return home.
The full safety alert can be found on the FDA website.
Postapproval results for SynCardia Systems’ Companion 2 (C2) driver system for temporary total artificial hearts (TAH-t) have shown higher mortality and stroke rates than were seen with the previous system, the circulatory support system. As a result, the Food and Drug Administration has issued a safety alert cautioning them to weigh the risks and benefits carefully. The alert, issued on August 17, is based on a postapproval study conducted by SynCardia Systems.
Furthermore, patients and health care professionals are encouraged to report any adverse events using the FDA’s Medwatch reporting form, as well as return any devices associated with adverse events to the SynCardia Systems to help them and the FDA better understand the issue.
The C2 driver system is an external pneumatic system that activates an implanted TAH-t in eligible heart failure patients who have severe biventricular failure and are waiting for transplant. It is smaller than its predecessor, but per the device’s approved use, patients must still remain in the hospital while on the device. Since its approval in 2012, the Freedom driver system was approved in 2014, which allows patients to return home.
The full safety alert can be found on the FDA website.
CDC: 2017 worst year yet for drug overdoses
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.