User login
ID experts urge widespread flu vaccination for 2018-2019 season
WASHINGTON – The flu vaccine may not be perfect, but it can reduce the severity of illness and curb the risk of spreading the disease to others, William Schaffner, MD, emphasized at a press conference held by the National Foundation for Infectious Diseases.
“Give the vaccine credit for softening the blow,” said Dr. Schaffner, medical director of NFID and a professor at Vanderbilt University in Nashville.
Dr. Schaffner and a panel of experts including U.S. Surgeon General Jerome M. Adams, MD, encouraged the public and the health care community to follow recommendation from the Centers for Disease Control & Prevention that everyone aged 6 months and older receive an influenza vaccine.
Dr. Schaffner shared recent data showing that complications from the flu don’t stop when the acute illness resolves. Acute influenza causes a whole-body inflammatory reaction, and consequently “there is an increased risk of heart attack and stroke during the 2-4 weeks of recovery from acute influenza,” he said. In addition, older adults who experience acute flu and are already frail may never regain their pre-flu level of function, as the flu can start a “domino effect of decline and disability.”
Despite last year’s severe flu season that included 180 deaths in children, vaccination remains the most effective protection against the flu, Dr. Adams said.
This year, between 163 million and 168 million doses of vaccine will be available in the United States. The vaccine is available in a range of settings including doctors’ offices, pharmacies, grocery stores, and workplaces, said Dr. Adams.
Flu vaccine choices this year include a return of the live-attenuated influenza vaccine (LAIV) given via nasal spray, along with the standard influenza vaccine that includes either three influenza viruses (trivalent, with two influenza A and one influenza B) or four influenza viruses (quadrivalent, with two influenza A and two influenza B). Other options are adjuvanted vaccine and high-dose vaccine for adults aged 65 years and older, and a cell-based and recombinant vaccine as alternatives to egg-based vaccines.
Dr. Adams emphasized the importance of healthy people getting vaccinated to protect the community. “All the people who died from the flu caught it from someone else,” he said.

The message to health care providers remains the same: Recommend the flu vaccine to patients at every opportunity, and lead by example and get vaccinated yourself, Dr. Adams said. He noted this year’s strategies to promote flu vaccination on social media, and encouraged clinicians to recommend the flu shot to their patients and to showcase their own shots via the #FightFlu hashtag.
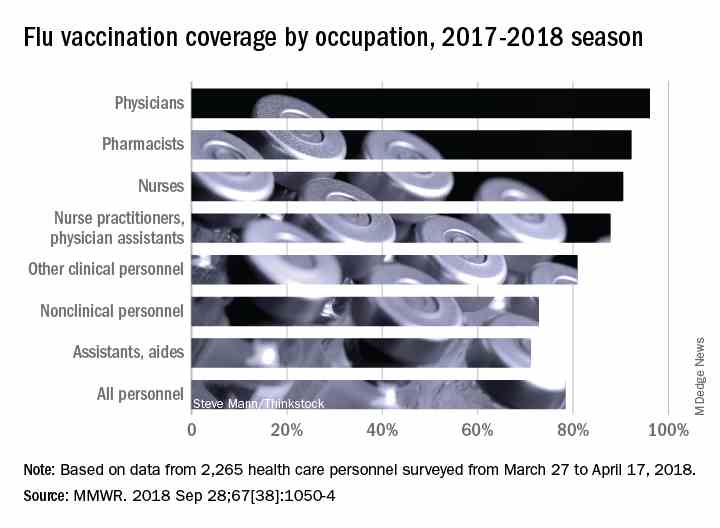
Vaccination among health care personnel last year was approximately 78%, which is a plateau over the past several years (MMWR 2018; 67:1050-54).
Be prepared to offer antivirals to patients as appropriate, and to promote the pneumococcal vaccine to eligible older adults as well, to protect not only themselves, but their contacts and the community, Dr. Adams emphasized. Currently approved antiviral drugs recommended for the 2018-2019 flu season: oseltamivir, zanamivir, and peramivir.
Wendy Sue Swanson, MD, of Seattle Children’s Hospital, stressed the importance of flu vaccination for all children, given their ability to spread viral infections. She noted a concerning 2% drop in vaccinations for children aged 6 months to 4 years, although vaccination coverage in this group was highest among children overall, at approximately 68% last season.
Last year, approximately 80% of the child deaths from flu occurred in unvaccinated children, but the vaccine has been shown to reduce the likelihood of hospitalization or death even if a child does become ill, Dr. Swanson said.
Laura E. Riley, MD, of Weill Cornell Medical Center, noted that vaccination of pregnant women has plateaued in recent years, and was 49% last year. “Our goal is 80% plus,” she said. Data show that pregnant women who received flu vaccination were 40% less likely to be hospitalized for the flu, she noted. The American College of Obstetricians and Gynecologists recommends flu vaccination as safe during any trimester, and valuable to both mothers and newborns because it provides protective antibodies during the first 6 months of life before babies can receive their own vaccinations, Dr. Riley said.
More information about this year’s flu season is available from the CDC and NFID.
WASHINGTON – The flu vaccine may not be perfect, but it can reduce the severity of illness and curb the risk of spreading the disease to others, William Schaffner, MD, emphasized at a press conference held by the National Foundation for Infectious Diseases.
“Give the vaccine credit for softening the blow,” said Dr. Schaffner, medical director of NFID and a professor at Vanderbilt University in Nashville.
Dr. Schaffner and a panel of experts including U.S. Surgeon General Jerome M. Adams, MD, encouraged the public and the health care community to follow recommendation from the Centers for Disease Control & Prevention that everyone aged 6 months and older receive an influenza vaccine.
Dr. Schaffner shared recent data showing that complications from the flu don’t stop when the acute illness resolves. Acute influenza causes a whole-body inflammatory reaction, and consequently “there is an increased risk of heart attack and stroke during the 2-4 weeks of recovery from acute influenza,” he said. In addition, older adults who experience acute flu and are already frail may never regain their pre-flu level of function, as the flu can start a “domino effect of decline and disability.”
Despite last year’s severe flu season that included 180 deaths in children, vaccination remains the most effective protection against the flu, Dr. Adams said.
This year, between 163 million and 168 million doses of vaccine will be available in the United States. The vaccine is available in a range of settings including doctors’ offices, pharmacies, grocery stores, and workplaces, said Dr. Adams.
Flu vaccine choices this year include a return of the live-attenuated influenza vaccine (LAIV) given via nasal spray, along with the standard influenza vaccine that includes either three influenza viruses (trivalent, with two influenza A and one influenza B) or four influenza viruses (quadrivalent, with two influenza A and two influenza B). Other options are adjuvanted vaccine and high-dose vaccine for adults aged 65 years and older, and a cell-based and recombinant vaccine as alternatives to egg-based vaccines.
Dr. Adams emphasized the importance of healthy people getting vaccinated to protect the community. “All the people who died from the flu caught it from someone else,” he said.
The message to health care providers remains the same: Recommend the flu vaccine to patients at every opportunity, and lead by example and get vaccinated yourself, Dr. Adams said. He noted this year’s strategies to promote flu vaccination on social media, and encouraged clinicians to recommend the flu shot to their patients and to showcase their own shots via the #FightFlu hashtag.
Vaccination among health care personnel last year was approximately 78%, which is a plateau over the past several years (MMWR 2018; 67:1050-54).
Be prepared to offer antivirals to patients as appropriate, and to promote the pneumococcal vaccine to eligible older adults as well, to protect not only themselves, but their contacts and the community, Dr. Adams emphasized. Currently approved antiviral drugs recommended for the 2018-2019 flu season: oseltamivir, zanamivir, and peramivir.
Wendy Sue Swanson, MD, of Seattle Children’s Hospital, stressed the importance of flu vaccination for all children, given their ability to spread viral infections. She noted a concerning 2% drop in vaccinations for children aged 6 months to 4 years, although vaccination coverage in this group was highest among children overall, at approximately 68% last season.
Last year, approximately 80% of the child deaths from flu occurred in unvaccinated children, but the vaccine has been shown to reduce the likelihood of hospitalization or death even if a child does become ill, Dr. Swanson said.
Laura E. Riley, MD, of Weill Cornell Medical Center, noted that vaccination of pregnant women has plateaued in recent years, and was 49% last year. “Our goal is 80% plus,” she said. Data show that pregnant women who received flu vaccination were 40% less likely to be hospitalized for the flu, she noted. The American College of Obstetricians and Gynecologists recommends flu vaccination as safe during any trimester, and valuable to both mothers and newborns because it provides protective antibodies during the first 6 months of life before babies can receive their own vaccinations, Dr. Riley said.
More information about this year’s flu season is available from the CDC and NFID.
WASHINGTON – The flu vaccine may not be perfect, but it can reduce the severity of illness and curb the risk of spreading the disease to others, William Schaffner, MD, emphasized at a press conference held by the National Foundation for Infectious Diseases.
“Give the vaccine credit for softening the blow,” said Dr. Schaffner, medical director of NFID and a professor at Vanderbilt University in Nashville.
Dr. Schaffner and a panel of experts including U.S. Surgeon General Jerome M. Adams, MD, encouraged the public and the health care community to follow recommendation from the Centers for Disease Control & Prevention that everyone aged 6 months and older receive an influenza vaccine.
Dr. Schaffner shared recent data showing that complications from the flu don’t stop when the acute illness resolves. Acute influenza causes a whole-body inflammatory reaction, and consequently “there is an increased risk of heart attack and stroke during the 2-4 weeks of recovery from acute influenza,” he said. In addition, older adults who experience acute flu and are already frail may never regain their pre-flu level of function, as the flu can start a “domino effect of decline and disability.”
Despite last year’s severe flu season that included 180 deaths in children, vaccination remains the most effective protection against the flu, Dr. Adams said.
This year, between 163 million and 168 million doses of vaccine will be available in the United States. The vaccine is available in a range of settings including doctors’ offices, pharmacies, grocery stores, and workplaces, said Dr. Adams.
Flu vaccine choices this year include a return of the live-attenuated influenza vaccine (LAIV) given via nasal spray, along with the standard influenza vaccine that includes either three influenza viruses (trivalent, with two influenza A and one influenza B) or four influenza viruses (quadrivalent, with two influenza A and two influenza B). Other options are adjuvanted vaccine and high-dose vaccine for adults aged 65 years and older, and a cell-based and recombinant vaccine as alternatives to egg-based vaccines.
Dr. Adams emphasized the importance of healthy people getting vaccinated to protect the community. “All the people who died from the flu caught it from someone else,” he said.
The message to health care providers remains the same: Recommend the flu vaccine to patients at every opportunity, and lead by example and get vaccinated yourself, Dr. Adams said. He noted this year’s strategies to promote flu vaccination on social media, and encouraged clinicians to recommend the flu shot to their patients and to showcase their own shots via the #FightFlu hashtag.
Vaccination among health care personnel last year was approximately 78%, which is a plateau over the past several years (MMWR 2018; 67:1050-54).
Be prepared to offer antivirals to patients as appropriate, and to promote the pneumococcal vaccine to eligible older adults as well, to protect not only themselves, but their contacts and the community, Dr. Adams emphasized. Currently approved antiviral drugs recommended for the 2018-2019 flu season: oseltamivir, zanamivir, and peramivir.
Wendy Sue Swanson, MD, of Seattle Children’s Hospital, stressed the importance of flu vaccination for all children, given their ability to spread viral infections. She noted a concerning 2% drop in vaccinations for children aged 6 months to 4 years, although vaccination coverage in this group was highest among children overall, at approximately 68% last season.
Last year, approximately 80% of the child deaths from flu occurred in unvaccinated children, but the vaccine has been shown to reduce the likelihood of hospitalization or death even if a child does become ill, Dr. Swanson said.
Laura E. Riley, MD, of Weill Cornell Medical Center, noted that vaccination of pregnant women has plateaued in recent years, and was 49% last year. “Our goal is 80% plus,” she said. Data show that pregnant women who received flu vaccination were 40% less likely to be hospitalized for the flu, she noted. The American College of Obstetricians and Gynecologists recommends flu vaccination as safe during any trimester, and valuable to both mothers and newborns because it provides protective antibodies during the first 6 months of life before babies can receive their own vaccinations, Dr. Riley said.
More information about this year’s flu season is available from the CDC and NFID.
FROM AN NFID PRESS CONFERENCE
FDA approves new drug for CLL/SLL and follicular lymphoma
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.

who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
FDA grants OBI-3424 orphan designation for ALL
The Food and Drug Administration has granted orphan drug designation to OBI-3424 for the treatment of acute lymphoblastic leukemia (ALL).
OBI-3424 is a small-molecule prodrug that targets cancers overexpressing aldo-keto reductase 1C3 (AKR1C3) and selectively releases a DNA alkylating agent in the presence of the AKR1C3 enzyme.
AKR1C3 overexpression has been observed in ALL, particularly T-cell ALL.
OBI-3424 demonstrated activity against T-ALL in preclinical research presented as a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in October 2017.
Researchers reported that OBI-3424 “exerted profound in vivo efficacy” against T-ALL xenografts derived mainly from patients with aggressive and fatal T-ALL. In addition, OBI-3424 significantly reduced leukemia bone marrow infiltration in four of six evaluable T-ALL xenografts, and OBI-3424 was considered well tolerated.
The poster presentation describing this research is available for download from the website of OBI Pharma, the company developing OBI-3424 in cooperation with Ascenta Pharma.
OBI-3424 also has orphan drug designation from the FDA as a treatment for hepatocellular carcinoma.
The Food and Drug Administration has granted orphan drug designation to OBI-3424 for the treatment of acute lymphoblastic leukemia (ALL).
OBI-3424 is a small-molecule prodrug that targets cancers overexpressing aldo-keto reductase 1C3 (AKR1C3) and selectively releases a DNA alkylating agent in the presence of the AKR1C3 enzyme.
AKR1C3 overexpression has been observed in ALL, particularly T-cell ALL.
OBI-3424 demonstrated activity against T-ALL in preclinical research presented as a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in October 2017.
Researchers reported that OBI-3424 “exerted profound in vivo efficacy” against T-ALL xenografts derived mainly from patients with aggressive and fatal T-ALL. In addition, OBI-3424 significantly reduced leukemia bone marrow infiltration in four of six evaluable T-ALL xenografts, and OBI-3424 was considered well tolerated.
The poster presentation describing this research is available for download from the website of OBI Pharma, the company developing OBI-3424 in cooperation with Ascenta Pharma.
OBI-3424 also has orphan drug designation from the FDA as a treatment for hepatocellular carcinoma.
The Food and Drug Administration has granted orphan drug designation to OBI-3424 for the treatment of acute lymphoblastic leukemia (ALL).
OBI-3424 is a small-molecule prodrug that targets cancers overexpressing aldo-keto reductase 1C3 (AKR1C3) and selectively releases a DNA alkylating agent in the presence of the AKR1C3 enzyme.
AKR1C3 overexpression has been observed in ALL, particularly T-cell ALL.
OBI-3424 demonstrated activity against T-ALL in preclinical research presented as a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in October 2017.
Researchers reported that OBI-3424 “exerted profound in vivo efficacy” against T-ALL xenografts derived mainly from patients with aggressive and fatal T-ALL. In addition, OBI-3424 significantly reduced leukemia bone marrow infiltration in four of six evaluable T-ALL xenografts, and OBI-3424 was considered well tolerated.
The poster presentation describing this research is available for download from the website of OBI Pharma, the company developing OBI-3424 in cooperation with Ascenta Pharma.
OBI-3424 also has orphan drug designation from the FDA as a treatment for hepatocellular carcinoma.
HIV patients getting younger ... and older
Men who have sex with men (MSM) were younger at HIV diagnosis in 2016 than in 2008, but those living with the disease were older, according to the Centers for Disease Control and Prevention.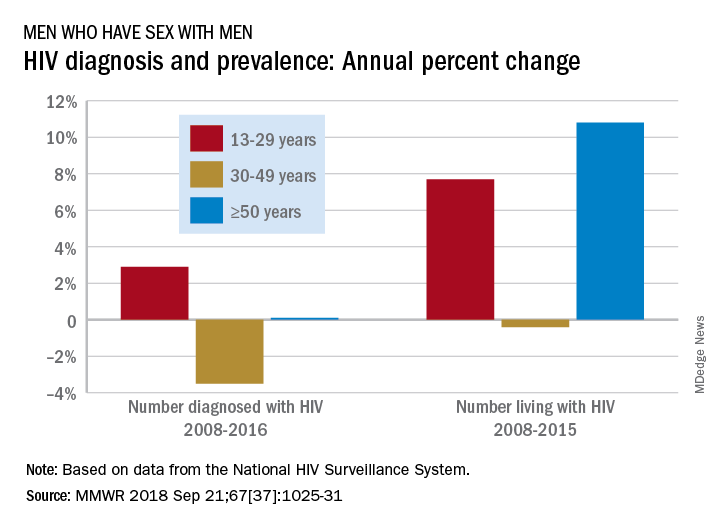
Among MSM aged 13-29 years, the number diagnosed with HIV increased by 2.9% per year from 2008 to 2016 but dropped 3.5% per year for those aged 30-49 and rose just 0.1% annually among those aged 50 and older. Over the period from 2008 to 2015, the number of MSM aged 50 and older who were living with HIV increased by 10.8% per year, compared with an annual percent change of 7.7% for those aged 13-29 and –0.4% for those aged 30-49, Andrew Mitsch, MPH, and his associates at the CDC’s National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention reported in Morbidity and Mortality Weekly Report.
The size of the population of MSM living with HIV went from 384,000 in 2008 to 523,000 in 2016, with 13- to 29-year-olds going from 10.7% of the population to 13.3%, 30- to 49-year-olds dropping from 61% to 44%, and the 50-and-older group increasing from 28.3% to 42.7%, they said.
“The increase in annual diagnosis of HIV infections among younger MSM might reflect increased HIV testing, in addition to ongoing transmission,” they suggested, and increased prevalence among older men is probably the “result of increased survival associated with widespread use of antiretroviral therapy, surviving middle age, and advancing to the older group.”
The investigators also noted the persistence of racial/ethnic disparities over the course of the study. Among the three largest groups, whites had the smallest increase in new diagnoses for 13- to 29-year-olds and the largest decrease for 30- to 49-year-olds, and they were second to blacks in the less-than-or-equal-to-50-years-of-age group, according to data from the National HIV Surveillance System.
“Promotion of care and treatment by public health agencies and private sector partners to achieve viral suppression among MSM with diagnosed HIV infection will improve health outcomes and reduce transmission to others, particularly if prevention efforts are tailored to specific age groups,” the researchers wrote.
SOURCE: Mitsch A et al. MMWR 2018 Sep 21;67(37):1025-31.
Men who have sex with men (MSM) were younger at HIV diagnosis in 2016 than in 2008, but those living with the disease were older, according to the Centers for Disease Control and Prevention.
Among MSM aged 13-29 years, the number diagnosed with HIV increased by 2.9% per year from 2008 to 2016 but dropped 3.5% per year for those aged 30-49 and rose just 0.1% annually among those aged 50 and older. Over the period from 2008 to 2015, the number of MSM aged 50 and older who were living with HIV increased by 10.8% per year, compared with an annual percent change of 7.7% for those aged 13-29 and –0.4% for those aged 30-49, Andrew Mitsch, MPH, and his associates at the CDC’s National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention reported in Morbidity and Mortality Weekly Report.
The size of the population of MSM living with HIV went from 384,000 in 2008 to 523,000 in 2016, with 13- to 29-year-olds going from 10.7% of the population to 13.3%, 30- to 49-year-olds dropping from 61% to 44%, and the 50-and-older group increasing from 28.3% to 42.7%, they said.
“The increase in annual diagnosis of HIV infections among younger MSM might reflect increased HIV testing, in addition to ongoing transmission,” they suggested, and increased prevalence among older men is probably the “result of increased survival associated with widespread use of antiretroviral therapy, surviving middle age, and advancing to the older group.”
The investigators also noted the persistence of racial/ethnic disparities over the course of the study. Among the three largest groups, whites had the smallest increase in new diagnoses for 13- to 29-year-olds and the largest decrease for 30- to 49-year-olds, and they were second to blacks in the less-than-or-equal-to-50-years-of-age group, according to data from the National HIV Surveillance System.
“Promotion of care and treatment by public health agencies and private sector partners to achieve viral suppression among MSM with diagnosed HIV infection will improve health outcomes and reduce transmission to others, particularly if prevention efforts are tailored to specific age groups,” the researchers wrote.
SOURCE: Mitsch A et al. MMWR 2018 Sep 21;67(37):1025-31.
Men who have sex with men (MSM) were younger at HIV diagnosis in 2016 than in 2008, but those living with the disease were older, according to the Centers for Disease Control and Prevention.
Among MSM aged 13-29 years, the number diagnosed with HIV increased by 2.9% per year from 2008 to 2016 but dropped 3.5% per year for those aged 30-49 and rose just 0.1% annually among those aged 50 and older. Over the period from 2008 to 2015, the number of MSM aged 50 and older who were living with HIV increased by 10.8% per year, compared with an annual percent change of 7.7% for those aged 13-29 and –0.4% for those aged 30-49, Andrew Mitsch, MPH, and his associates at the CDC’s National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention reported in Morbidity and Mortality Weekly Report.
The size of the population of MSM living with HIV went from 384,000 in 2008 to 523,000 in 2016, with 13- to 29-year-olds going from 10.7% of the population to 13.3%, 30- to 49-year-olds dropping from 61% to 44%, and the 50-and-older group increasing from 28.3% to 42.7%, they said.
“The increase in annual diagnosis of HIV infections among younger MSM might reflect increased HIV testing, in addition to ongoing transmission,” they suggested, and increased prevalence among older men is probably the “result of increased survival associated with widespread use of antiretroviral therapy, surviving middle age, and advancing to the older group.”
The investigators also noted the persistence of racial/ethnic disparities over the course of the study. Among the three largest groups, whites had the smallest increase in new diagnoses for 13- to 29-year-olds and the largest decrease for 30- to 49-year-olds, and they were second to blacks in the less-than-or-equal-to-50-years-of-age group, according to data from the National HIV Surveillance System.
“Promotion of care and treatment by public health agencies and private sector partners to achieve viral suppression among MSM with diagnosed HIV infection will improve health outcomes and reduce transmission to others, particularly if prevention efforts are tailored to specific age groups,” the researchers wrote.
SOURCE: Mitsch A et al. MMWR 2018 Sep 21;67(37):1025-31.
FROM MMWR
FDA issues new REMS for immediate-release opioids
Opioid prescribers will need to be mindful of a new, expanded Risk Evaluation and Mitigation Strategy issued Sept. 18 by the Food and Drug Administration, covering immediate-release opioid analgesics used in the outpatient setting. The strategy also applies to extended-release and long-acting opioids, which have been subject to REMS since 2012.
![]()
The new REMS program requires for the first time that training be made available to health care providers who are involved in pain management. For the purposes of this REMS program, the training is not limited to just the prescriber, but includes nurses and pharmacists.
, including alternatives to opioids for pain management.
The FDA said it is in the process of approving a new label for opioids that will contain information about health care provider education that is now a part of the REMS.
“Our new effort is aimed at arming providers with the most current and comprehensive information on the appropriate management of pain,” FDA Commissioner Scott Gottlieb, MD, said in a statement. “Appropriate prescribing practices and education are important steps that we are prioritizing to help address the human and financial toll of this crisis.”

Dr. Gottlieb added that the goal of the new REMS is to help prescribers with the latest evidence on the appropriate amount of doses that should be prescribed for a given condition and that the “aim is to reduce overall dispensing as a way to further reduce exposure to these drugs. Our goal is to help prevent patients from becoming addicted by decreasing unnecessary or inappropriate exposure to opioids and fostering rational prescribing to enable appropriate access to those patients who have legitimate medical need for these medicines.”
The FDA also approved a new guidance document that includes updated educational content.
Opioid prescribers will need to be mindful of a new, expanded Risk Evaluation and Mitigation Strategy issued Sept. 18 by the Food and Drug Administration, covering immediate-release opioid analgesics used in the outpatient setting. The strategy also applies to extended-release and long-acting opioids, which have been subject to REMS since 2012.
![]()
The new REMS program requires for the first time that training be made available to health care providers who are involved in pain management. For the purposes of this REMS program, the training is not limited to just the prescriber, but includes nurses and pharmacists.
, including alternatives to opioids for pain management.
The FDA said it is in the process of approving a new label for opioids that will contain information about health care provider education that is now a part of the REMS.
“Our new effort is aimed at arming providers with the most current and comprehensive information on the appropriate management of pain,” FDA Commissioner Scott Gottlieb, MD, said in a statement. “Appropriate prescribing practices and education are important steps that we are prioritizing to help address the human and financial toll of this crisis.”
Dr. Gottlieb added that the goal of the new REMS is to help prescribers with the latest evidence on the appropriate amount of doses that should be prescribed for a given condition and that the “aim is to reduce overall dispensing as a way to further reduce exposure to these drugs. Our goal is to help prevent patients from becoming addicted by decreasing unnecessary or inappropriate exposure to opioids and fostering rational prescribing to enable appropriate access to those patients who have legitimate medical need for these medicines.”
The FDA also approved a new guidance document that includes updated educational content.
Opioid prescribers will need to be mindful of a new, expanded Risk Evaluation and Mitigation Strategy issued Sept. 18 by the Food and Drug Administration, covering immediate-release opioid analgesics used in the outpatient setting. The strategy also applies to extended-release and long-acting opioids, which have been subject to REMS since 2012.
![]()
The new REMS program requires for the first time that training be made available to health care providers who are involved in pain management. For the purposes of this REMS program, the training is not limited to just the prescriber, but includes nurses and pharmacists.
, including alternatives to opioids for pain management.
The FDA said it is in the process of approving a new label for opioids that will contain information about health care provider education that is now a part of the REMS.
“Our new effort is aimed at arming providers with the most current and comprehensive information on the appropriate management of pain,” FDA Commissioner Scott Gottlieb, MD, said in a statement. “Appropriate prescribing practices and education are important steps that we are prioritizing to help address the human and financial toll of this crisis.”
Dr. Gottlieb added that the goal of the new REMS is to help prescribers with the latest evidence on the appropriate amount of doses that should be prescribed for a given condition and that the “aim is to reduce overall dispensing as a way to further reduce exposure to these drugs. Our goal is to help prevent patients from becoming addicted by decreasing unnecessary or inappropriate exposure to opioids and fostering rational prescribing to enable appropriate access to those patients who have legitimate medical need for these medicines.”
The FDA also approved a new guidance document that includes updated educational content.
FDA approves device for coronary artery perforations
according to an announcement from the agency.
These small tears in the arterial wall are rare but life-threatening complications of certain procedures, such as percutaneous coronary interventions. The device uses a balloon catheter like that used with PCI and, when successful, it can spare patients more invasive procedures, such as open heart surgery.
The approval is based on survey data from 80 patients treated with this device. In 76 of the patients (95%), the device was successfully delivered to the site of the acute coronary arterial perforation, and it successfully healed the tear in 73 patients (91%). Two patients died during the procedure, and five whose perforations were sealed successfully died in the hospital after the procedure, as did one whose perforation was not sealed.
The agency noted that this is the first device approved for the indication in 17 years. More information can be found in the full FDA announcement.
according to an announcement from the agency.
These small tears in the arterial wall are rare but life-threatening complications of certain procedures, such as percutaneous coronary interventions. The device uses a balloon catheter like that used with PCI and, when successful, it can spare patients more invasive procedures, such as open heart surgery.
The approval is based on survey data from 80 patients treated with this device. In 76 of the patients (95%), the device was successfully delivered to the site of the acute coronary arterial perforation, and it successfully healed the tear in 73 patients (91%). Two patients died during the procedure, and five whose perforations were sealed successfully died in the hospital after the procedure, as did one whose perforation was not sealed.
The agency noted that this is the first device approved for the indication in 17 years. More information can be found in the full FDA announcement.
according to an announcement from the agency.
These small tears in the arterial wall are rare but life-threatening complications of certain procedures, such as percutaneous coronary interventions. The device uses a balloon catheter like that used with PCI and, when successful, it can spare patients more invasive procedures, such as open heart surgery.
The approval is based on survey data from 80 patients treated with this device. In 76 of the patients (95%), the device was successfully delivered to the site of the acute coronary arterial perforation, and it successfully healed the tear in 73 patients (91%). Two patients died during the procedure, and five whose perforations were sealed successfully died in the hospital after the procedure, as did one whose perforation was not sealed.
The agency noted that this is the first device approved for the indication in 17 years. More information can be found in the full FDA announcement.
FDA approves Ajovy for migraine prevention
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
FDA attacks antibiotic resistance with new strategy
WASHINGTON – A strategy combining stewardship and science is needed to help combat antimicrobial resistance, and updated plans from the U.S. Food and Drug Administration include four key components to address all aspects of product development and use, FDA commissioner Scott Gottlieb, MD, said in a press briefing in Washington on Sept. 14.
“The FDA plays a unique role in advancing human and animal health” that provides a unique vantage point for coordinating all aspects of product development and application, he said.
The FDA’s comprehensive approach to the challenge of antimicrobial resistance (AMR) includes:
- Facilitating product development.
- Promoting antimicrobial stewardship.
- Supporting the development of new tools for surveillance.
- Advancing scientific initiatives, including research for the development of alternative treatments.
The FDA’s product development plan to combat AMR includes the creation of incentives for companies to develop new antibiotic products and create a robust pipeline, which is a challenge because of the lack of immediate economic gain, Dr. Gottlieb said.
“It necessary to change the perception that the costs and risks of antibiotic innovation are too high relative to their expected gains,” he emphasized.
Strategies to incentivize companies include fast track designation, priority review, and breakthrough therapy designation. In addition, the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) is designed to promote development of antimicrobial drugs for limited and underserved populations, Dr. Gottlieb said. The FDA plan also calls for pursuing reimbursement options with the Centers for Medicare & Medicaid Services.
Promoting antimicrobial stewardship remains an ongoing element of the FDA’s plan to reduce AMR. In conjunction with the release of the FDA’s updated approach to AMR, the FDA’s Center for Veterinary Medicine CVM released a 5-year action plan to promote and support antimicrobial stewardship in not only the agricultural arena, but in companion animals as well.
The FDA plans to bring all antimicrobials of medical importance that are approved for use in animals under the oversight of CVM, which will pursue the improve labeling on antimicrobial drugs used in the feed and water of food-producing animals, including defining durations of use, Dr. Gottlieb noted.
Supporting the development and improvement of surveillance tools is “essential to understanding the drivers of resistance in human and veterinary settings and formulating appropriate responses” to outbreaks, Dr. Gottlieb said.
To help meet this goal, the FDA will expand sampling via the National Antimicrobial Resistance Monitoring System (NARMS) database, he said. Other surveillance goals include supporting genomics research and expanding AMR monitoring to include pathogens associated with animal feed and companion animals, he added.
As part of the final component of the FDA’s AMR strategy to advance scientific initiatives, the FDA has released a new Request for Information “to obtain additional, external input on how best to develop an annual list of regulatory science initiatives specific for antimicrobial products,” Dr. Gottlieb announced. The FDA intends to use the information gained from clinicians and others in its creation of guidance documents and recommendations to streamline the antibiotic development process. He also cited the FDA’s ongoing support of partnerships with public and private organizations such as the Clinical Trials Transformation Initiative, which focuses on drug development for severe bacterial infections with current unmet medical need.
“We need to harness science and policy to help our public health systems and researchers become nimbler in the battle against drug-resistant pathogens,” Dr. Gottlieb concluded.
In a panel discussion following the briefing, several experts offered perspective on the FDA’s goals and on the challenges of AMR.
William Flynn, DVM, deputy director of science policy for the Center of Veterinary Medicine, noted some goals for reducing the use of antibiotics in the veterinary arena.
“We are trying to focus on the driver: What are the disease conditions that drive use of the product,” he said. Ideally, better management of disease conditions can reduce reliance on antibiotics, he added.
Also in the panel discussion, Steven Gitterman, MD, deputy director of the division of microbiology devices at the Center for Devices and Radiological Health, emphasized the value of sustainable trial databases so AMR research can continue on an ongoing basis. Finally, Carolyn Wilson, PhD, associate director of research at the Center for Biologics Evaluation and Research, noted that the FDA’s research and development efforts include antibiotic alternatives, including live biotherapeutic products, fecal microbiota transplantation, and bacteriophage therapy.
Visit www.fda.gov for a transcript of Dr. Gottlieb’s talk, and for the updated FDA website page with more details on the agency’s plans to combat antimicrobial resistance.
Dr. Gottlieb and the panelists had no financial conflicts to disclose.
WASHINGTON – A strategy combining stewardship and science is needed to help combat antimicrobial resistance, and updated plans from the U.S. Food and Drug Administration include four key components to address all aspects of product development and use, FDA commissioner Scott Gottlieb, MD, said in a press briefing in Washington on Sept. 14.
“The FDA plays a unique role in advancing human and animal health” that provides a unique vantage point for coordinating all aspects of product development and application, he said.
The FDA’s comprehensive approach to the challenge of antimicrobial resistance (AMR) includes:
- Facilitating product development.
- Promoting antimicrobial stewardship.
- Supporting the development of new tools for surveillance.
- Advancing scientific initiatives, including research for the development of alternative treatments.
The FDA’s product development plan to combat AMR includes the creation of incentives for companies to develop new antibiotic products and create a robust pipeline, which is a challenge because of the lack of immediate economic gain, Dr. Gottlieb said.
“It necessary to change the perception that the costs and risks of antibiotic innovation are too high relative to their expected gains,” he emphasized.
Strategies to incentivize companies include fast track designation, priority review, and breakthrough therapy designation. In addition, the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) is designed to promote development of antimicrobial drugs for limited and underserved populations, Dr. Gottlieb said. The FDA plan also calls for pursuing reimbursement options with the Centers for Medicare & Medicaid Services.
Promoting antimicrobial stewardship remains an ongoing element of the FDA’s plan to reduce AMR. In conjunction with the release of the FDA’s updated approach to AMR, the FDA’s Center for Veterinary Medicine CVM released a 5-year action plan to promote and support antimicrobial stewardship in not only the agricultural arena, but in companion animals as well.
The FDA plans to bring all antimicrobials of medical importance that are approved for use in animals under the oversight of CVM, which will pursue the improve labeling on antimicrobial drugs used in the feed and water of food-producing animals, including defining durations of use, Dr. Gottlieb noted.
Supporting the development and improvement of surveillance tools is “essential to understanding the drivers of resistance in human and veterinary settings and formulating appropriate responses” to outbreaks, Dr. Gottlieb said.
To help meet this goal, the FDA will expand sampling via the National Antimicrobial Resistance Monitoring System (NARMS) database, he said. Other surveillance goals include supporting genomics research and expanding AMR monitoring to include pathogens associated with animal feed and companion animals, he added.
As part of the final component of the FDA’s AMR strategy to advance scientific initiatives, the FDA has released a new Request for Information “to obtain additional, external input on how best to develop an annual list of regulatory science initiatives specific for antimicrobial products,” Dr. Gottlieb announced. The FDA intends to use the information gained from clinicians and others in its creation of guidance documents and recommendations to streamline the antibiotic development process. He also cited the FDA’s ongoing support of partnerships with public and private organizations such as the Clinical Trials Transformation Initiative, which focuses on drug development for severe bacterial infections with current unmet medical need.
“We need to harness science and policy to help our public health systems and researchers become nimbler in the battle against drug-resistant pathogens,” Dr. Gottlieb concluded.
In a panel discussion following the briefing, several experts offered perspective on the FDA’s goals and on the challenges of AMR.
William Flynn, DVM, deputy director of science policy for the Center of Veterinary Medicine, noted some goals for reducing the use of antibiotics in the veterinary arena.
“We are trying to focus on the driver: What are the disease conditions that drive use of the product,” he said. Ideally, better management of disease conditions can reduce reliance on antibiotics, he added.
Also in the panel discussion, Steven Gitterman, MD, deputy director of the division of microbiology devices at the Center for Devices and Radiological Health, emphasized the value of sustainable trial databases so AMR research can continue on an ongoing basis. Finally, Carolyn Wilson, PhD, associate director of research at the Center for Biologics Evaluation and Research, noted that the FDA’s research and development efforts include antibiotic alternatives, including live biotherapeutic products, fecal microbiota transplantation, and bacteriophage therapy.
Visit www.fda.gov for a transcript of Dr. Gottlieb’s talk, and for the updated FDA website page with more details on the agency’s plans to combat antimicrobial resistance.
Dr. Gottlieb and the panelists had no financial conflicts to disclose.
WASHINGTON – A strategy combining stewardship and science is needed to help combat antimicrobial resistance, and updated plans from the U.S. Food and Drug Administration include four key components to address all aspects of product development and use, FDA commissioner Scott Gottlieb, MD, said in a press briefing in Washington on Sept. 14.
“The FDA plays a unique role in advancing human and animal health” that provides a unique vantage point for coordinating all aspects of product development and application, he said.
The FDA’s comprehensive approach to the challenge of antimicrobial resistance (AMR) includes:
- Facilitating product development.
- Promoting antimicrobial stewardship.
- Supporting the development of new tools for surveillance.
- Advancing scientific initiatives, including research for the development of alternative treatments.
The FDA’s product development plan to combat AMR includes the creation of incentives for companies to develop new antibiotic products and create a robust pipeline, which is a challenge because of the lack of immediate economic gain, Dr. Gottlieb said.
“It necessary to change the perception that the costs and risks of antibiotic innovation are too high relative to their expected gains,” he emphasized.
Strategies to incentivize companies include fast track designation, priority review, and breakthrough therapy designation. In addition, the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) is designed to promote development of antimicrobial drugs for limited and underserved populations, Dr. Gottlieb said. The FDA plan also calls for pursuing reimbursement options with the Centers for Medicare & Medicaid Services.
Promoting antimicrobial stewardship remains an ongoing element of the FDA’s plan to reduce AMR. In conjunction with the release of the FDA’s updated approach to AMR, the FDA’s Center for Veterinary Medicine CVM released a 5-year action plan to promote and support antimicrobial stewardship in not only the agricultural arena, but in companion animals as well.
The FDA plans to bring all antimicrobials of medical importance that are approved for use in animals under the oversight of CVM, which will pursue the improve labeling on antimicrobial drugs used in the feed and water of food-producing animals, including defining durations of use, Dr. Gottlieb noted.
Supporting the development and improvement of surveillance tools is “essential to understanding the drivers of resistance in human and veterinary settings and formulating appropriate responses” to outbreaks, Dr. Gottlieb said.
To help meet this goal, the FDA will expand sampling via the National Antimicrobial Resistance Monitoring System (NARMS) database, he said. Other surveillance goals include supporting genomics research and expanding AMR monitoring to include pathogens associated with animal feed and companion animals, he added.
As part of the final component of the FDA’s AMR strategy to advance scientific initiatives, the FDA has released a new Request for Information “to obtain additional, external input on how best to develop an annual list of regulatory science initiatives specific for antimicrobial products,” Dr. Gottlieb announced. The FDA intends to use the information gained from clinicians and others in its creation of guidance documents and recommendations to streamline the antibiotic development process. He also cited the FDA’s ongoing support of partnerships with public and private organizations such as the Clinical Trials Transformation Initiative, which focuses on drug development for severe bacterial infections with current unmet medical need.
“We need to harness science and policy to help our public health systems and researchers become nimbler in the battle against drug-resistant pathogens,” Dr. Gottlieb concluded.
In a panel discussion following the briefing, several experts offered perspective on the FDA’s goals and on the challenges of AMR.
William Flynn, DVM, deputy director of science policy for the Center of Veterinary Medicine, noted some goals for reducing the use of antibiotics in the veterinary arena.
“We are trying to focus on the driver: What are the disease conditions that drive use of the product,” he said. Ideally, better management of disease conditions can reduce reliance on antibiotics, he added.
Also in the panel discussion, Steven Gitterman, MD, deputy director of the division of microbiology devices at the Center for Devices and Radiological Health, emphasized the value of sustainable trial databases so AMR research can continue on an ongoing basis. Finally, Carolyn Wilson, PhD, associate director of research at the Center for Biologics Evaluation and Research, noted that the FDA’s research and development efforts include antibiotic alternatives, including live biotherapeutic products, fecal microbiota transplantation, and bacteriophage therapy.
Visit www.fda.gov for a transcript of Dr. Gottlieb’s talk, and for the updated FDA website page with more details on the agency’s plans to combat antimicrobial resistance.
Dr. Gottlieb and the panelists had no financial conflicts to disclose.
FDA approves new hairy cell leukemia drug
The Food and Drug Administration (FDA) has approved moxetumomab pasudotox-tdfk (Lumoxiti), a CD22-directed cytotoxin, to treat hairy cell leukemia (HCL).
Moxetumomab pasudotox is approved to treat adults with relapsed or refractory HCL who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog.
The prescribing information for moxetumomab pasudotox includes a boxed warning noting that the drug poses risks of capillary leak syndrome and hemolytic uremic syndrome. Other serious warnings include the risk of decreased renal function, infusion-related reactions, and electrolyte abnormalities.
The FDA granted the application for moxetumomab pasudotox fast track, priority review, and an orphan drug designation.
The agency approved AstraZeneca’s moxetumomab pasudotox based on results from a phase 3 trial (NCT01829711). Data from this study were presented at the 2018 annual meeting of the American Society of Clinical Oncology (abstract 7004).
The trial included 80 patients with relapsed or refractory HCL who had received at least two prior lines of therapy.
At a median of 16.7 months of follow-up, the objective response rate was 75%, the complete response (CR) rate was 41%, and the durable CR rate was 30%. Durable CR was defined as CR with hematologic remission for more than 180 days.
Most patients with a CR achieved minimal residual disease negativity (82%; 27/33).
The median duration of response was not reached, nor was the median progression-free survival.
The most common treatment-related adverse events (AEs) were nausea, peripheral edema, headache, and pyrexia. Other treatment-related AEs included infections and neutropenia.
Treatment-related AEs that led to discontinuation included capillary leak syndrome, hemolytic uremic syndrome, and increased blood creatinine.
There were three deaths in this trial, but none of them were considered treatment related.
The Food and Drug Administration (FDA) has approved moxetumomab pasudotox-tdfk (Lumoxiti), a CD22-directed cytotoxin, to treat hairy cell leukemia (HCL).
Moxetumomab pasudotox is approved to treat adults with relapsed or refractory HCL who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog.
The prescribing information for moxetumomab pasudotox includes a boxed warning noting that the drug poses risks of capillary leak syndrome and hemolytic uremic syndrome. Other serious warnings include the risk of decreased renal function, infusion-related reactions, and electrolyte abnormalities.
The FDA granted the application for moxetumomab pasudotox fast track, priority review, and an orphan drug designation.
The agency approved AstraZeneca’s moxetumomab pasudotox based on results from a phase 3 trial (NCT01829711). Data from this study were presented at the 2018 annual meeting of the American Society of Clinical Oncology (abstract 7004).
The trial included 80 patients with relapsed or refractory HCL who had received at least two prior lines of therapy.
At a median of 16.7 months of follow-up, the objective response rate was 75%, the complete response (CR) rate was 41%, and the durable CR rate was 30%. Durable CR was defined as CR with hematologic remission for more than 180 days.
Most patients with a CR achieved minimal residual disease negativity (82%; 27/33).
The median duration of response was not reached, nor was the median progression-free survival.
The most common treatment-related adverse events (AEs) were nausea, peripheral edema, headache, and pyrexia. Other treatment-related AEs included infections and neutropenia.
Treatment-related AEs that led to discontinuation included capillary leak syndrome, hemolytic uremic syndrome, and increased blood creatinine.
There were three deaths in this trial, but none of them were considered treatment related.
The Food and Drug Administration (FDA) has approved moxetumomab pasudotox-tdfk (Lumoxiti), a CD22-directed cytotoxin, to treat hairy cell leukemia (HCL).
Moxetumomab pasudotox is approved to treat adults with relapsed or refractory HCL who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog.
The prescribing information for moxetumomab pasudotox includes a boxed warning noting that the drug poses risks of capillary leak syndrome and hemolytic uremic syndrome. Other serious warnings include the risk of decreased renal function, infusion-related reactions, and electrolyte abnormalities.
The FDA granted the application for moxetumomab pasudotox fast track, priority review, and an orphan drug designation.
The agency approved AstraZeneca’s moxetumomab pasudotox based on results from a phase 3 trial (NCT01829711). Data from this study were presented at the 2018 annual meeting of the American Society of Clinical Oncology (abstract 7004).
The trial included 80 patients with relapsed or refractory HCL who had received at least two prior lines of therapy.
At a median of 16.7 months of follow-up, the objective response rate was 75%, the complete response (CR) rate was 41%, and the durable CR rate was 30%. Durable CR was defined as CR with hematologic remission for more than 180 days.
Most patients with a CR achieved minimal residual disease negativity (82%; 27/33).
The median duration of response was not reached, nor was the median progression-free survival.
The most common treatment-related adverse events (AEs) were nausea, peripheral edema, headache, and pyrexia. Other treatment-related AEs included infections and neutropenia.
Treatment-related AEs that led to discontinuation included capillary leak syndrome, hemolytic uremic syndrome, and increased blood creatinine.
There were three deaths in this trial, but none of them were considered treatment related.
CDC: Obesity affects over 35% in 7 states
Iowa and Oklahoma, the two newest states with prevalences at or exceeding 35%, joined Alabama, Arkansas, Louisiana, Mississippi, and West Virginia, which has the country’s highest rate of adult obesity at 38.1%. Colorado’s 22.6% rate is the lowest prevalence among all states. The District of Columbia and Hawaii also have prevalences under 25%; previously, Massachusetts also was in this group, but its prevalence went up to 25.9% last year, the CDC reported.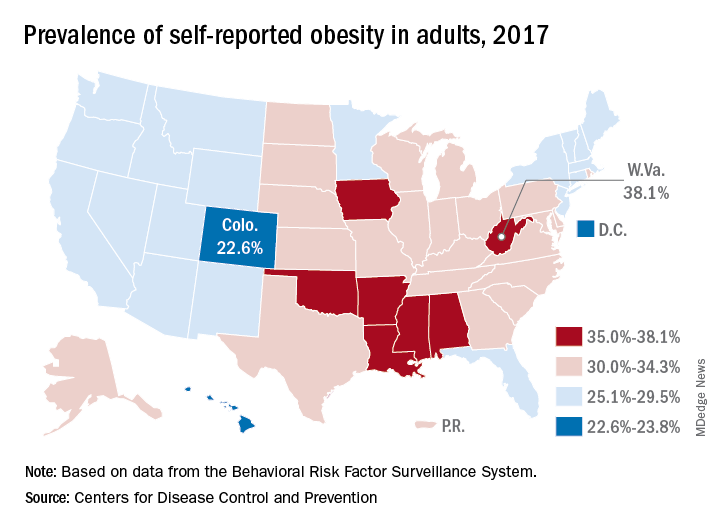
Regional disparities in self-reported adult obesity put the South (32.4%) and the Midwest (32.3%) well ahead of the Northeast (27.7%) and the West (26.1%) in 2017. Racial and ethnic disparities also were seen, with large gaps between blacks, who had a prevalence of 39%, and Hispanics (32.4%) and whites (29.3%). Obesity prevalence was 35% or higher among black adults in 31 states and D.C., while this was true among Hispanics in eight states and among whites in one (West Virginia), although the prevalence was at or above 35% for multiple racial groups in some of these states, the CDC reported based on data from the Behavioral Risk Factor Surveillance System.
“Obesity costs the United States health care system over $147 billion a year [and] research has shown that obesity affects work productivity and military readiness,” the CDC said in a written statement. “To protect the health of the next generation, support for healthy behaviors such as healthy eating, better sleep, stress management, and physical activity should start early and expand to reach Americans across the lifespan in the communities where they live, learn, work, and play.”
Iowa and Oklahoma, the two newest states with prevalences at or exceeding 35%, joined Alabama, Arkansas, Louisiana, Mississippi, and West Virginia, which has the country’s highest rate of adult obesity at 38.1%. Colorado’s 22.6% rate is the lowest prevalence among all states. The District of Columbia and Hawaii also have prevalences under 25%; previously, Massachusetts also was in this group, but its prevalence went up to 25.9% last year, the CDC reported.
Regional disparities in self-reported adult obesity put the South (32.4%) and the Midwest (32.3%) well ahead of the Northeast (27.7%) and the West (26.1%) in 2017. Racial and ethnic disparities also were seen, with large gaps between blacks, who had a prevalence of 39%, and Hispanics (32.4%) and whites (29.3%). Obesity prevalence was 35% or higher among black adults in 31 states and D.C., while this was true among Hispanics in eight states and among whites in one (West Virginia), although the prevalence was at or above 35% for multiple racial groups in some of these states, the CDC reported based on data from the Behavioral Risk Factor Surveillance System.
“Obesity costs the United States health care system over $147 billion a year [and] research has shown that obesity affects work productivity and military readiness,” the CDC said in a written statement. “To protect the health of the next generation, support for healthy behaviors such as healthy eating, better sleep, stress management, and physical activity should start early and expand to reach Americans across the lifespan in the communities where they live, learn, work, and play.”
Iowa and Oklahoma, the two newest states with prevalences at or exceeding 35%, joined Alabama, Arkansas, Louisiana, Mississippi, and West Virginia, which has the country’s highest rate of adult obesity at 38.1%. Colorado’s 22.6% rate is the lowest prevalence among all states. The District of Columbia and Hawaii also have prevalences under 25%; previously, Massachusetts also was in this group, but its prevalence went up to 25.9% last year, the CDC reported.
Regional disparities in self-reported adult obesity put the South (32.4%) and the Midwest (32.3%) well ahead of the Northeast (27.7%) and the West (26.1%) in 2017. Racial and ethnic disparities also were seen, with large gaps between blacks, who had a prevalence of 39%, and Hispanics (32.4%) and whites (29.3%). Obesity prevalence was 35% or higher among black adults in 31 states and D.C., while this was true among Hispanics in eight states and among whites in one (West Virginia), although the prevalence was at or above 35% for multiple racial groups in some of these states, the CDC reported based on data from the Behavioral Risk Factor Surveillance System.
“Obesity costs the United States health care system over $147 billion a year [and] research has shown that obesity affects work productivity and military readiness,” the CDC said in a written statement. “To protect the health of the next generation, support for healthy behaviors such as healthy eating, better sleep, stress management, and physical activity should start early and expand to reach Americans across the lifespan in the communities where they live, learn, work, and play.”