User login
Ibrutinib plus obinutuzumab gets priority review in CLL/SLL
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).

The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).
The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).
The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
Talazoparib approved for HER2-negative advanced breast cancer
The for patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm), HER2-negative locally advanced or metastatic breast cancer.
The FDA also approved BRACAnalysis CDx from Myriad Genetic Laboratories as the companion diagnostic to identify patients with deleterious or suspected deleterious gBRCAm who are eligible for talazoparib, the agency announced.
Approval was based on improved progression-free survival in EMBRACA, a phase 3, open-label trial of 431 patients with gBRCAm HER2-negative locally advanced or metastatic breast cancer. Patients in the trial were randomized 2:1 to receive either talazoparib (1 mg) or a physician’s choice of chemotherapy (capecitabine, eribulin, gemcitabine, or vinorelbine). All patients had received treatment with an anthracycline and/or a taxane previously.
Median progression-free survival was 8.6 months in the talazoparib arm, compared with 5.6 months in the chemotherapy arm (HR, 0.54; 95% CI, 0.41-0.71; P less than .0001).
Anemia was the most common hematologic adverse event, with grade 3 or greater occurring in 39.2% of patients on the PARP inhibitor, compared with 4.8% of patients treated with other agents, according to results of the trial presented at the 2017 San Antonio Breast Cancer Symposium.
Warnings and precautions for myelodysplastic syndrome/acute myeloid leukemia, myelosuppression, and embryo-fetal toxicity are included in the drug’s prescribing information. The PARP inhibitor’s most common adverse reactions of any grade are fatigue, anemia, nausea, neutropenia, headache, thrombocytopenia, vomiting, alopecia, diarrhea, and decreased appetite, the FDA said.
The recommended dose for talazoparib, marketed as Talzenna by Pfizer, is 1 mg taken as a single oral daily dose, with or without food.
The for patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm), HER2-negative locally advanced or metastatic breast cancer.
The FDA also approved BRACAnalysis CDx from Myriad Genetic Laboratories as the companion diagnostic to identify patients with deleterious or suspected deleterious gBRCAm who are eligible for talazoparib, the agency announced.
Approval was based on improved progression-free survival in EMBRACA, a phase 3, open-label trial of 431 patients with gBRCAm HER2-negative locally advanced or metastatic breast cancer. Patients in the trial were randomized 2:1 to receive either talazoparib (1 mg) or a physician’s choice of chemotherapy (capecitabine, eribulin, gemcitabine, or vinorelbine). All patients had received treatment with an anthracycline and/or a taxane previously.
Median progression-free survival was 8.6 months in the talazoparib arm, compared with 5.6 months in the chemotherapy arm (HR, 0.54; 95% CI, 0.41-0.71; P less than .0001).
Anemia was the most common hematologic adverse event, with grade 3 or greater occurring in 39.2% of patients on the PARP inhibitor, compared with 4.8% of patients treated with other agents, according to results of the trial presented at the 2017 San Antonio Breast Cancer Symposium.
Warnings and precautions for myelodysplastic syndrome/acute myeloid leukemia, myelosuppression, and embryo-fetal toxicity are included in the drug’s prescribing information. The PARP inhibitor’s most common adverse reactions of any grade are fatigue, anemia, nausea, neutropenia, headache, thrombocytopenia, vomiting, alopecia, diarrhea, and decreased appetite, the FDA said.
The recommended dose for talazoparib, marketed as Talzenna by Pfizer, is 1 mg taken as a single oral daily dose, with or without food.
The for patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm), HER2-negative locally advanced or metastatic breast cancer.
The FDA also approved BRACAnalysis CDx from Myriad Genetic Laboratories as the companion diagnostic to identify patients with deleterious or suspected deleterious gBRCAm who are eligible for talazoparib, the agency announced.
Approval was based on improved progression-free survival in EMBRACA, a phase 3, open-label trial of 431 patients with gBRCAm HER2-negative locally advanced or metastatic breast cancer. Patients in the trial were randomized 2:1 to receive either talazoparib (1 mg) or a physician’s choice of chemotherapy (capecitabine, eribulin, gemcitabine, or vinorelbine). All patients had received treatment with an anthracycline and/or a taxane previously.
Median progression-free survival was 8.6 months in the talazoparib arm, compared with 5.6 months in the chemotherapy arm (HR, 0.54; 95% CI, 0.41-0.71; P less than .0001).
Anemia was the most common hematologic adverse event, with grade 3 or greater occurring in 39.2% of patients on the PARP inhibitor, compared with 4.8% of patients treated with other agents, according to results of the trial presented at the 2017 San Antonio Breast Cancer Symposium.
Warnings and precautions for myelodysplastic syndrome/acute myeloid leukemia, myelosuppression, and embryo-fetal toxicity are included in the drug’s prescribing information. The PARP inhibitor’s most common adverse reactions of any grade are fatigue, anemia, nausea, neutropenia, headache, thrombocytopenia, vomiting, alopecia, diarrhea, and decreased appetite, the FDA said.
The recommended dose for talazoparib, marketed as Talzenna by Pfizer, is 1 mg taken as a single oral daily dose, with or without food.
FDA offers guidance on MRD assessment in blood cancer trials
The of patients with hematologic malignancies.

The FDA said it developed the document to assist drug sponsors who are planning to use minimal residual disease (MRD) as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
MRD could potentially be used as a biomarker in clinical trials – specifically as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker, according to the draft guidance. Additionally, MRD could be used as a surrogate endpoint or “to select patients at high risk or to enrich the trial population.”
The draft guidance also provides specific considerations for MRD assessment in individual hematologic malignancies, including acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, and multiple myeloma.
The full document is available on the FDA website.
The of patients with hematologic malignancies.
The FDA said it developed the document to assist drug sponsors who are planning to use minimal residual disease (MRD) as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
MRD could potentially be used as a biomarker in clinical trials – specifically as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker, according to the draft guidance. Additionally, MRD could be used as a surrogate endpoint or “to select patients at high risk or to enrich the trial population.”
The draft guidance also provides specific considerations for MRD assessment in individual hematologic malignancies, including acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, and multiple myeloma.
The full document is available on the FDA website.
The of patients with hematologic malignancies.
The FDA said it developed the document to assist drug sponsors who are planning to use minimal residual disease (MRD) as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
MRD could potentially be used as a biomarker in clinical trials – specifically as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker, according to the draft guidance. Additionally, MRD could be used as a surrogate endpoint or “to select patients at high risk or to enrich the trial population.”
The draft guidance also provides specific considerations for MRD assessment in individual hematologic malignancies, including acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, and multiple myeloma.
The full document is available on the FDA website.
FDA approves DNA-based blood type test
The for use in transfusion.
It’s the second molecular test for blood compatibility but the first to report genotype in its results, according to an announcement from the agency.
The test is important because it evaluates patients – especially those who receive repeated blood transfusions for conditions such as sickle cell anemia – for non-ABO antigens, but it does so without using antisera, which is sometimes unavailable.
A study found comparable performance between the ID CORE XT, licensed serologic reagents, and DNA sequencing tests, according to the FDA.
The ID CORE XT test is marketed by Progenika Biopharma, a Grifols company.
More information can be found in the full FDA press announcement.
The for use in transfusion.
It’s the second molecular test for blood compatibility but the first to report genotype in its results, according to an announcement from the agency.
The test is important because it evaluates patients – especially those who receive repeated blood transfusions for conditions such as sickle cell anemia – for non-ABO antigens, but it does so without using antisera, which is sometimes unavailable.
A study found comparable performance between the ID CORE XT, licensed serologic reagents, and DNA sequencing tests, according to the FDA.
The ID CORE XT test is marketed by Progenika Biopharma, a Grifols company.
More information can be found in the full FDA press announcement.
The for use in transfusion.
It’s the second molecular test for blood compatibility but the first to report genotype in its results, according to an announcement from the agency.
The test is important because it evaluates patients – especially those who receive repeated blood transfusions for conditions such as sickle cell anemia – for non-ABO antigens, but it does so without using antisera, which is sometimes unavailable.
A study found comparable performance between the ID CORE XT, licensed serologic reagents, and DNA sequencing tests, according to the FDA.
The ID CORE XT test is marketed by Progenika Biopharma, a Grifols company.
More information can be found in the full FDA press announcement.
Rivaroxaban gains indication for prevention of major cardiovascular events in CAD/PAD
when taken with aspirin, Janssen Pharmaceuticals announced on October 11.
The Food and Drug Administration’s approval was based on a review of the 27,000-patient COMPASS trial, which showed last year that a low dosage of rivaroxaban (Xarelto) plus aspirin reduced the combined rate of cardiovascular disease events by 24% in patients with coronary artery disease and by 28% in participants with peripheral artery disease, compared with aspirin alone. (N Engl J Med. 2017 Oct 5;377[14]:1319-30)
The flip side to the reduction in COMPASS’s combined primary endpoint was a 51% increase in major bleeding. However, that bump did not translate to increases in fatal bleeds, intracerebral bleeds, or bleeding in other critical organs.
COMPASS (Cardiovascular Outcomes for People Using Anticoagulation Strategies) studied two dosages of rivaroxaban, 2.5 mg and 5 mg twice daily, and it was the lower dosage that did the trick. Until this approval, that formulation wasn’t available; Janssen announced the coming of the 2.5-mg pill in its release.
The new prescribing information states specifically that Xarelto 2.5 mg is indicated, in combination with aspirin, to reduce the risk of major cardiovascular events, cardiovascular death, MI, and stroke in patients with chronic coronary artery disease or peripheral artery disease.
This is the sixth indication for rivaroxaban, a factor Xa inhibitor that was first approved in 2011. It is also the first indication for cardiovascular prevention for any factor Xa inhibitor. Others on the U.S. market are apixaban (Eliquis), edoxaban (Savaysa), and betrixaban (Bevyxxa).
COMPASS was presented at the 2017 annual congress of the European Society of Cardiology. At that time, Eugene Braunwald, MD, of Harvard Medical School and Brigham and Women’s Hospital in Boston, commented that the trial produced “unambiguous results that should change guidelines and the management of stable coronary artery disease.” He added that the results are “an important step for thrombocardiology.”
when taken with aspirin, Janssen Pharmaceuticals announced on October 11.
The Food and Drug Administration’s approval was based on a review of the 27,000-patient COMPASS trial, which showed last year that a low dosage of rivaroxaban (Xarelto) plus aspirin reduced the combined rate of cardiovascular disease events by 24% in patients with coronary artery disease and by 28% in participants with peripheral artery disease, compared with aspirin alone. (N Engl J Med. 2017 Oct 5;377[14]:1319-30)
The flip side to the reduction in COMPASS’s combined primary endpoint was a 51% increase in major bleeding. However, that bump did not translate to increases in fatal bleeds, intracerebral bleeds, or bleeding in other critical organs.
COMPASS (Cardiovascular Outcomes for People Using Anticoagulation Strategies) studied two dosages of rivaroxaban, 2.5 mg and 5 mg twice daily, and it was the lower dosage that did the trick. Until this approval, that formulation wasn’t available; Janssen announced the coming of the 2.5-mg pill in its release.
The new prescribing information states specifically that Xarelto 2.5 mg is indicated, in combination with aspirin, to reduce the risk of major cardiovascular events, cardiovascular death, MI, and stroke in patients with chronic coronary artery disease or peripheral artery disease.
This is the sixth indication for rivaroxaban, a factor Xa inhibitor that was first approved in 2011. It is also the first indication for cardiovascular prevention for any factor Xa inhibitor. Others on the U.S. market are apixaban (Eliquis), edoxaban (Savaysa), and betrixaban (Bevyxxa).
COMPASS was presented at the 2017 annual congress of the European Society of Cardiology. At that time, Eugene Braunwald, MD, of Harvard Medical School and Brigham and Women’s Hospital in Boston, commented that the trial produced “unambiguous results that should change guidelines and the management of stable coronary artery disease.” He added that the results are “an important step for thrombocardiology.”
when taken with aspirin, Janssen Pharmaceuticals announced on October 11.
The Food and Drug Administration’s approval was based on a review of the 27,000-patient COMPASS trial, which showed last year that a low dosage of rivaroxaban (Xarelto) plus aspirin reduced the combined rate of cardiovascular disease events by 24% in patients with coronary artery disease and by 28% in participants with peripheral artery disease, compared with aspirin alone. (N Engl J Med. 2017 Oct 5;377[14]:1319-30)
The flip side to the reduction in COMPASS’s combined primary endpoint was a 51% increase in major bleeding. However, that bump did not translate to increases in fatal bleeds, intracerebral bleeds, or bleeding in other critical organs.
COMPASS (Cardiovascular Outcomes for People Using Anticoagulation Strategies) studied two dosages of rivaroxaban, 2.5 mg and 5 mg twice daily, and it was the lower dosage that did the trick. Until this approval, that formulation wasn’t available; Janssen announced the coming of the 2.5-mg pill in its release.
The new prescribing information states specifically that Xarelto 2.5 mg is indicated, in combination with aspirin, to reduce the risk of major cardiovascular events, cardiovascular death, MI, and stroke in patients with chronic coronary artery disease or peripheral artery disease.
This is the sixth indication for rivaroxaban, a factor Xa inhibitor that was first approved in 2011. It is also the first indication for cardiovascular prevention for any factor Xa inhibitor. Others on the U.S. market are apixaban (Eliquis), edoxaban (Savaysa), and betrixaban (Bevyxxa).
COMPASS was presented at the 2017 annual congress of the European Society of Cardiology. At that time, Eugene Braunwald, MD, of Harvard Medical School and Brigham and Women’s Hospital in Boston, commented that the trial produced “unambiguous results that should change guidelines and the management of stable coronary artery disease.” He added that the results are “an important step for thrombocardiology.”
Anxiety and depression widespread among arthritis patients
Adults with arthritis are almost twice as likely to have symptoms of anxiety than depression, but the depressed patients are more likely to receive treatment, according to the Centers for Disease Control and Prevention.
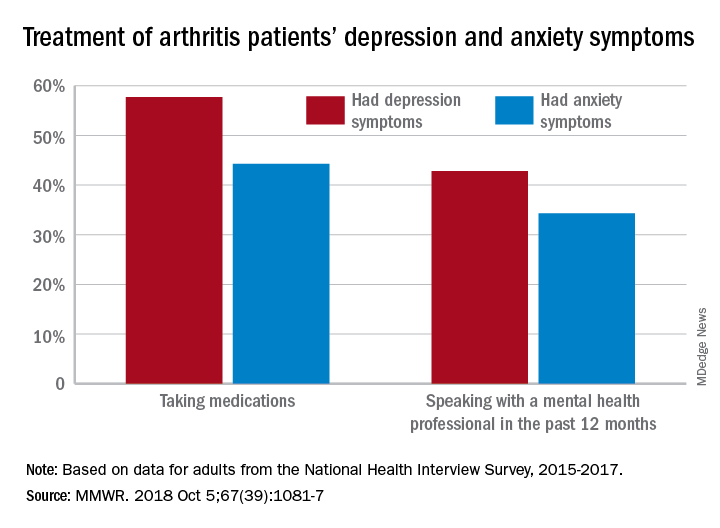
During 2015-2017, the prevalence of anxiety symptoms was 22.5% in adults with arthritis, compared with 12.1% for depression symptoms. Treatment of those symptoms, however, was another story: 57.7% of arthritis patients with depression symptoms were taking medications, versus 44.3% of those with anxiety symptoms, and 42.8% of those with symptoms of depression reported seeing a mental health professional the past 12 months, compared with 34.3% of adults with anxiety, Dana Guglielmo, MPH, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and her associates reported in the Morbidity and Mortality Weekly Report.
Prevalences of anxiety and depression symptoms varied considerably by sociodemographic characteristic during 2015-2017. Anxiety and depression were both more common in those aged 18-44 years (28.3% and 13.7%, respectively) than in those aged over 65 (9.7% and 6.2%), and women with arthritis were more likely than were men to experience symptoms of anxiety (26.9% vs. 16%) and depression (14% vs. 9.2%), the investigators said, based on data from the National Health Interview Survey.
Among racial/ethnic groups, the prevalence of anxiety was highest for whites (23.9%) and lowest for Asians (10.6%), who also had lowest depression symptom prevalence at 3.3%, with American Indians/Alaska Natives highest at 15.4%. Adults categorized as other/multiple race, however, were highest in both cases at 32.3% for anxiety and 17.4% for depression, Ms. Guglielmo and her associates said.
The overall prevalence of anxiety and depression symptoms in patients with arthritis was much higher than in those without arthritis – 10.7% for anxiety and 4.7% for depression – which “suggests that all adults with arthritis would benefit from mental health screening,” they noted.
, and encouraging physical activity, which is an effective nonpharmacologic strategy that can help reduce the symptoms of anxiety and depression, improve arthritis symptoms, and promote better quality of life,” the investigators wrote.
SOURCE: Guglielmo D et al. MMWR. 2018 Oct 5;67(39):1081-7.
Adults with arthritis are almost twice as likely to have symptoms of anxiety than depression, but the depressed patients are more likely to receive treatment, according to the Centers for Disease Control and Prevention.
During 2015-2017, the prevalence of anxiety symptoms was 22.5% in adults with arthritis, compared with 12.1% for depression symptoms. Treatment of those symptoms, however, was another story: 57.7% of arthritis patients with depression symptoms were taking medications, versus 44.3% of those with anxiety symptoms, and 42.8% of those with symptoms of depression reported seeing a mental health professional the past 12 months, compared with 34.3% of adults with anxiety, Dana Guglielmo, MPH, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and her associates reported in the Morbidity and Mortality Weekly Report.
Prevalences of anxiety and depression symptoms varied considerably by sociodemographic characteristic during 2015-2017. Anxiety and depression were both more common in those aged 18-44 years (28.3% and 13.7%, respectively) than in those aged over 65 (9.7% and 6.2%), and women with arthritis were more likely than were men to experience symptoms of anxiety (26.9% vs. 16%) and depression (14% vs. 9.2%), the investigators said, based on data from the National Health Interview Survey.
Among racial/ethnic groups, the prevalence of anxiety was highest for whites (23.9%) and lowest for Asians (10.6%), who also had lowest depression symptom prevalence at 3.3%, with American Indians/Alaska Natives highest at 15.4%. Adults categorized as other/multiple race, however, were highest in both cases at 32.3% for anxiety and 17.4% for depression, Ms. Guglielmo and her associates said.
The overall prevalence of anxiety and depression symptoms in patients with arthritis was much higher than in those without arthritis – 10.7% for anxiety and 4.7% for depression – which “suggests that all adults with arthritis would benefit from mental health screening,” they noted.
, and encouraging physical activity, which is an effective nonpharmacologic strategy that can help reduce the symptoms of anxiety and depression, improve arthritis symptoms, and promote better quality of life,” the investigators wrote.
SOURCE: Guglielmo D et al. MMWR. 2018 Oct 5;67(39):1081-7.
Adults with arthritis are almost twice as likely to have symptoms of anxiety than depression, but the depressed patients are more likely to receive treatment, according to the Centers for Disease Control and Prevention.
During 2015-2017, the prevalence of anxiety symptoms was 22.5% in adults with arthritis, compared with 12.1% for depression symptoms. Treatment of those symptoms, however, was another story: 57.7% of arthritis patients with depression symptoms were taking medications, versus 44.3% of those with anxiety symptoms, and 42.8% of those with symptoms of depression reported seeing a mental health professional the past 12 months, compared with 34.3% of adults with anxiety, Dana Guglielmo, MPH, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and her associates reported in the Morbidity and Mortality Weekly Report.
Prevalences of anxiety and depression symptoms varied considerably by sociodemographic characteristic during 2015-2017. Anxiety and depression were both more common in those aged 18-44 years (28.3% and 13.7%, respectively) than in those aged over 65 (9.7% and 6.2%), and women with arthritis were more likely than were men to experience symptoms of anxiety (26.9% vs. 16%) and depression (14% vs. 9.2%), the investigators said, based on data from the National Health Interview Survey.
Among racial/ethnic groups, the prevalence of anxiety was highest for whites (23.9%) and lowest for Asians (10.6%), who also had lowest depression symptom prevalence at 3.3%, with American Indians/Alaska Natives highest at 15.4%. Adults categorized as other/multiple race, however, were highest in both cases at 32.3% for anxiety and 17.4% for depression, Ms. Guglielmo and her associates said.
The overall prevalence of anxiety and depression symptoms in patients with arthritis was much higher than in those without arthritis – 10.7% for anxiety and 4.7% for depression – which “suggests that all adults with arthritis would benefit from mental health screening,” they noted.
, and encouraging physical activity, which is an effective nonpharmacologic strategy that can help reduce the symptoms of anxiety and depression, improve arthritis symptoms, and promote better quality of life,” the investigators wrote.
SOURCE: Guglielmo D et al. MMWR. 2018 Oct 5;67(39):1081-7.
FROM MMWR
FDA approves emicizumab for hemophilia A without inhibitors
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for subcutaneous prophylactic treatment in hemophilia A without factor VIII inhibitors.
Genentech announced the new approval on Oct. 4 of this year. In 2017, the bispecific antibody, which targets both factors IXa and X, was approved for patients as young as newborns who had factor VIII inhibitors; the latest approval allows it to be used for patients without inhibitors as well.
The approval is based on a pair of trials. HAVEN 3 (NCT02847637) is a phase 3 trial in which investigators looked at emicizumab prophylaxis weekly or every other week, versus on-demand factor VIII treatment in patients without inhibitors. The study included 152 patients aged 12 years and older who were previously treated with factor VIII therapy.
Compared with patients not receiving prophylactic treatments, those receiving weekly doses had a 96% reduction in treated bleeds, and those receiving doses every other week saw a 97% reduction. Investigators also found that 55.6% of patients treated every week, 60% of those treated every other week, and 0% of those treated with no prophylaxis experienced zero bleeds; similarly, 91.7%, 94.3%, and 5.6% experienced three or fewer bleeds.
The single-arm HAVEN 4 (NCT03020160) trial evaluated dosing patients every 4 weeks among 48 patients aged 12 years and older, with or without inhibitors, and results showed that even that dosing regimen could lead to a clinically meaningful control of bleeds: 56.1% had no bleeds, and 90.2% had three or fewer bleeds.
The most common adverse reactions were joint pain, headache, and injection-site reaction. When emicizumab-kxwh is used with activated prothrombin complex concentrate, there’s a risk of thrombotic microangiopathy and thrombotic events. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for subcutaneous prophylactic treatment in hemophilia A without factor VIII inhibitors.
Genentech announced the new approval on Oct. 4 of this year. In 2017, the bispecific antibody, which targets both factors IXa and X, was approved for patients as young as newborns who had factor VIII inhibitors; the latest approval allows it to be used for patients without inhibitors as well.
The approval is based on a pair of trials. HAVEN 3 (NCT02847637) is a phase 3 trial in which investigators looked at emicizumab prophylaxis weekly or every other week, versus on-demand factor VIII treatment in patients without inhibitors. The study included 152 patients aged 12 years and older who were previously treated with factor VIII therapy.
Compared with patients not receiving prophylactic treatments, those receiving weekly doses had a 96% reduction in treated bleeds, and those receiving doses every other week saw a 97% reduction. Investigators also found that 55.6% of patients treated every week, 60% of those treated every other week, and 0% of those treated with no prophylaxis experienced zero bleeds; similarly, 91.7%, 94.3%, and 5.6% experienced three or fewer bleeds.
The single-arm HAVEN 4 (NCT03020160) trial evaluated dosing patients every 4 weeks among 48 patients aged 12 years and older, with or without inhibitors, and results showed that even that dosing regimen could lead to a clinically meaningful control of bleeds: 56.1% had no bleeds, and 90.2% had three or fewer bleeds.
The most common adverse reactions were joint pain, headache, and injection-site reaction. When emicizumab-kxwh is used with activated prothrombin complex concentrate, there’s a risk of thrombotic microangiopathy and thrombotic events. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for subcutaneous prophylactic treatment in hemophilia A without factor VIII inhibitors.
Genentech announced the new approval on Oct. 4 of this year. In 2017, the bispecific antibody, which targets both factors IXa and X, was approved for patients as young as newborns who had factor VIII inhibitors; the latest approval allows it to be used for patients without inhibitors as well.
The approval is based on a pair of trials. HAVEN 3 (NCT02847637) is a phase 3 trial in which investigators looked at emicizumab prophylaxis weekly or every other week, versus on-demand factor VIII treatment in patients without inhibitors. The study included 152 patients aged 12 years and older who were previously treated with factor VIII therapy.
Compared with patients not receiving prophylactic treatments, those receiving weekly doses had a 96% reduction in treated bleeds, and those receiving doses every other week saw a 97% reduction. Investigators also found that 55.6% of patients treated every week, 60% of those treated every other week, and 0% of those treated with no prophylaxis experienced zero bleeds; similarly, 91.7%, 94.3%, and 5.6% experienced three or fewer bleeds.
The single-arm HAVEN 4 (NCT03020160) trial evaluated dosing patients every 4 weeks among 48 patients aged 12 years and older, with or without inhibitors, and results showed that even that dosing regimen could lead to a clinically meaningful control of bleeds: 56.1% had no bleeds, and 90.2% had three or fewer bleeds.
The most common adverse reactions were joint pain, headache, and injection-site reaction. When emicizumab-kxwh is used with activated prothrombin complex concentrate, there’s a risk of thrombotic microangiopathy and thrombotic events. Full prescribing information can be found on the FDA website.
FDA approves omadacycline for pneumonia and skin infections
The, for treating community-acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI) in adults, the manufacturer, Paratek, announced in a press release.
The company expects that omadacycline will be available in the first quarter of 2019. Administered once-daily in either oral or IV formulations, the antibiotic was effective and well tolerated across multiple trials, which altogether included almost 2,000 patients, according to Paratek. As part of the approval, the company has agreed to conduct postmarketing studies, specifically, more studies in CABP and in pediatric populations. “To reduce the development of drug-resistant bacteria and maintain the effectiveness of Nuzyra and other antibacterial drugs, Nuzyra should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria,” according to a statement in the indications section of the prescribing information.
Omadacycline is contraindicated for patients with a known hypersensitivity to the drug or any members of the tetracycline class of antibacterial drugs; hypersensitivity reactions have been observed, so use should be discontinued if one is suspected. Use of this drug during later stages of pregnancy can lead to irreversible discoloration of the infant’s teeth and inhibition of bone growth; it should also not be used during breastfeeding.
Because omadacycline is structurally similar to tetracycline class drugs, some adverse reactions to those drugs may be seen with this one, such as photosensitivity, pseudotumor cerebri, and antianabolic action. Adverse reactions known to have an association with omadacycline include nausea, vomiting, hypertension, insomnia, diarrhea, constipation, and increases of alanine aminotransferase, aspartate aminotransferase, and/or gamma-glutamyl transferase.
Drug interactions may occur with anticoagulants, so dosage of those drugs may need to be reduced while treating with omadacycline. Antacids also are believed to have a drug interaction – specifically, impairing absorption of omadacycline
The, for treating community-acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI) in adults, the manufacturer, Paratek, announced in a press release.
The company expects that omadacycline will be available in the first quarter of 2019. Administered once-daily in either oral or IV formulations, the antibiotic was effective and well tolerated across multiple trials, which altogether included almost 2,000 patients, according to Paratek. As part of the approval, the company has agreed to conduct postmarketing studies, specifically, more studies in CABP and in pediatric populations. “To reduce the development of drug-resistant bacteria and maintain the effectiveness of Nuzyra and other antibacterial drugs, Nuzyra should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria,” according to a statement in the indications section of the prescribing information.
Omadacycline is contraindicated for patients with a known hypersensitivity to the drug or any members of the tetracycline class of antibacterial drugs; hypersensitivity reactions have been observed, so use should be discontinued if one is suspected. Use of this drug during later stages of pregnancy can lead to irreversible discoloration of the infant’s teeth and inhibition of bone growth; it should also not be used during breastfeeding.
Because omadacycline is structurally similar to tetracycline class drugs, some adverse reactions to those drugs may be seen with this one, such as photosensitivity, pseudotumor cerebri, and antianabolic action. Adverse reactions known to have an association with omadacycline include nausea, vomiting, hypertension, insomnia, diarrhea, constipation, and increases of alanine aminotransferase, aspartate aminotransferase, and/or gamma-glutamyl transferase.
Drug interactions may occur with anticoagulants, so dosage of those drugs may need to be reduced while treating with omadacycline. Antacids also are believed to have a drug interaction – specifically, impairing absorption of omadacycline
The, for treating community-acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI) in adults, the manufacturer, Paratek, announced in a press release.
The company expects that omadacycline will be available in the first quarter of 2019. Administered once-daily in either oral or IV formulations, the antibiotic was effective and well tolerated across multiple trials, which altogether included almost 2,000 patients, according to Paratek. As part of the approval, the company has agreed to conduct postmarketing studies, specifically, more studies in CABP and in pediatric populations. “To reduce the development of drug-resistant bacteria and maintain the effectiveness of Nuzyra and other antibacterial drugs, Nuzyra should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria,” according to a statement in the indications section of the prescribing information.
Omadacycline is contraindicated for patients with a known hypersensitivity to the drug or any members of the tetracycline class of antibacterial drugs; hypersensitivity reactions have been observed, so use should be discontinued if one is suspected. Use of this drug during later stages of pregnancy can lead to irreversible discoloration of the infant’s teeth and inhibition of bone growth; it should also not be used during breastfeeding.
Because omadacycline is structurally similar to tetracycline class drugs, some adverse reactions to those drugs may be seen with this one, such as photosensitivity, pseudotumor cerebri, and antianabolic action. Adverse reactions known to have an association with omadacycline include nausea, vomiting, hypertension, insomnia, diarrhea, constipation, and increases of alanine aminotransferase, aspartate aminotransferase, and/or gamma-glutamyl transferase.
Drug interactions may occur with anticoagulants, so dosage of those drugs may need to be reduced while treating with omadacycline. Antacids also are believed to have a drug interaction – specifically, impairing absorption of omadacycline
FDA approves Arikayce for MAC lung diseases
that has been caused by members of the Mycobacterium avium complex and is refractory to other treatments.
In a randomized, controlled trial, patients with refractory M. avium complex infections were assigned to receive either Arikayce plus a multidrug antibacterial regimen or just the antibacterial regimen. By 6 months, sputum cultures for 29% of those treated with the combination had shown no mycobacterial growth for 3 consecutive months, whereas this was only true for the cultures for 9% of patients on the multidrug antibacterial regimen alone.
The Arikayce prescribing information includes a boxed warning regarding the increased risk of respiratory conditions, including hypersensitivity pneumonitis, bronchospasm, exacerbation of underlying lung disease, and hemoptysis, some of which have proven serious enough to lead to hospitalization. Other side effects include dysphonia, cough, musculoskeletal pain, nausea, and fatigue.
According to the press announcement from the FDA, this is the first approval under the Limited Population Pathway for Antibacterial and Antifungal Drugs, which was set up by Congress “to advance development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need.” It does so by allowing a more streamlined clinical development program that may involve smaller, shorter, or fewer clinical trials.
More information can be found in the full press announcement.
that has been caused by members of the Mycobacterium avium complex and is refractory to other treatments.
In a randomized, controlled trial, patients with refractory M. avium complex infections were assigned to receive either Arikayce plus a multidrug antibacterial regimen or just the antibacterial regimen. By 6 months, sputum cultures for 29% of those treated with the combination had shown no mycobacterial growth for 3 consecutive months, whereas this was only true for the cultures for 9% of patients on the multidrug antibacterial regimen alone.
The Arikayce prescribing information includes a boxed warning regarding the increased risk of respiratory conditions, including hypersensitivity pneumonitis, bronchospasm, exacerbation of underlying lung disease, and hemoptysis, some of which have proven serious enough to lead to hospitalization. Other side effects include dysphonia, cough, musculoskeletal pain, nausea, and fatigue.
According to the press announcement from the FDA, this is the first approval under the Limited Population Pathway for Antibacterial and Antifungal Drugs, which was set up by Congress “to advance development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need.” It does so by allowing a more streamlined clinical development program that may involve smaller, shorter, or fewer clinical trials.
More information can be found in the full press announcement.
that has been caused by members of the Mycobacterium avium complex and is refractory to other treatments.
In a randomized, controlled trial, patients with refractory M. avium complex infections were assigned to receive either Arikayce plus a multidrug antibacterial regimen or just the antibacterial regimen. By 6 months, sputum cultures for 29% of those treated with the combination had shown no mycobacterial growth for 3 consecutive months, whereas this was only true for the cultures for 9% of patients on the multidrug antibacterial regimen alone.
The Arikayce prescribing information includes a boxed warning regarding the increased risk of respiratory conditions, including hypersensitivity pneumonitis, bronchospasm, exacerbation of underlying lung disease, and hemoptysis, some of which have proven serious enough to lead to hospitalization. Other side effects include dysphonia, cough, musculoskeletal pain, nausea, and fatigue.
According to the press announcement from the FDA, this is the first approval under the Limited Population Pathway for Antibacterial and Antifungal Drugs, which was set up by Congress “to advance development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need.” It does so by allowing a more streamlined clinical development program that may involve smaller, shorter, or fewer clinical trials.
More information can be found in the full press announcement.
FDA approves cemiplimab for advanced cutaneous squamous cell carcinoma
The U.S. Food and Drug Administration has approved cemiplimab-rwlc (Libtayo) for the treatment of metastatic or locally advanced cutaneous squamous cell carcinoma, the agency announced in a press release.
The approval was granted based on data from two open-label clinical trials involving a total of 108 patients: the phase 2 EMPOWER-CSCC-1 trial (NCT02760498) and two expansion cohorts from an open-label, nonrandomized phase 1 trial.
These trials, which included 75 patients with metastatic disease and 33 with locally advanced disease, found an overall response rate of 47.2%, and most of those patients still showed ongoing responses at the time of data analysis. Among patients with metastatic disease, 5% had a complete response, according to a press release from the manufacturer, Sanofi.
This is the sixth FDA approval for a checkpoint inhibitor targeting the PD-1/PD-L1 pathway. The drug was evaluated under the FDA’s Priority Review program for drugs that represent significant improvements in the safety or effectiveness of treatments for serious conditions. Manufacturer Sanofi was granted Breakthrough Therapy designation for cemiplimab in 2017 for advanced cutaneous squamous cell carcinoma, and the drug is also being reviewed by the European Medicines Agency.
Cemiplimab is administered as a 350-mg intravenous therapy every 3 weeks – costing $9,100 per treatment – until the disease progresses or patients experience unacceptable toxicity, according to the manufacturer. The most common side effects include fatigue, rash and diarrhea, but more serious adverse events can include immune-mediated reactions such as pneumonitis, colitis, hepatitis, endocrinopathies, and skin and kidney problems.
The U.S. Food and Drug Administration has approved cemiplimab-rwlc (Libtayo) for the treatment of metastatic or locally advanced cutaneous squamous cell carcinoma, the agency announced in a press release.
The approval was granted based on data from two open-label clinical trials involving a total of 108 patients: the phase 2 EMPOWER-CSCC-1 trial (NCT02760498) and two expansion cohorts from an open-label, nonrandomized phase 1 trial.
These trials, which included 75 patients with metastatic disease and 33 with locally advanced disease, found an overall response rate of 47.2%, and most of those patients still showed ongoing responses at the time of data analysis. Among patients with metastatic disease, 5% had a complete response, according to a press release from the manufacturer, Sanofi.
This is the sixth FDA approval for a checkpoint inhibitor targeting the PD-1/PD-L1 pathway. The drug was evaluated under the FDA’s Priority Review program for drugs that represent significant improvements in the safety or effectiveness of treatments for serious conditions. Manufacturer Sanofi was granted Breakthrough Therapy designation for cemiplimab in 2017 for advanced cutaneous squamous cell carcinoma, and the drug is also being reviewed by the European Medicines Agency.
Cemiplimab is administered as a 350-mg intravenous therapy every 3 weeks – costing $9,100 per treatment – until the disease progresses or patients experience unacceptable toxicity, according to the manufacturer. The most common side effects include fatigue, rash and diarrhea, but more serious adverse events can include immune-mediated reactions such as pneumonitis, colitis, hepatitis, endocrinopathies, and skin and kidney problems.
The U.S. Food and Drug Administration has approved cemiplimab-rwlc (Libtayo) for the treatment of metastatic or locally advanced cutaneous squamous cell carcinoma, the agency announced in a press release.
The monoclonal antibody drug – a checkpoint inhibitor that blocks the PD-1 pathway – is the first treatment to be approved specifically for advanced cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.
The approval was granted based on data from two open-label clinical trials involving a total of 108 patients: the phase 2 EMPOWER-CSCC-1 trial (NCT02760498) and two expansion cohorts from an open-label, nonrandomized phase 1 trial.
These trials, which included 75 patients with metastatic disease and 33 with locally advanced disease, found an overall response rate of 47.2%, and most of those patients still showed ongoing responses at the time of data analysis. Among patients with metastatic disease, 5% had a complete response, according to a press release from the manufacturer, Sanofi.
This is the sixth FDA approval for a checkpoint inhibitor targeting the PD-1/PD-L1 pathway. The drug was evaluated under the FDA’s Priority Review program for drugs that represent significant improvements in the safety or effectiveness of treatments for serious conditions. Manufacturer Sanofi was granted Breakthrough Therapy designation for cemiplimab in 2017 for advanced cutaneous squamous cell carcinoma, and the drug is also being reviewed by the European Medicines Agency.
Cemiplimab is administered as a 350-mg intravenous therapy every 3 weeks – costing $9,100 per treatment – until the disease progresses or patients experience unacceptable toxicity, according to the manufacturer. The most common side effects include fatigue, rash and diarrhea, but more serious adverse events can include immune-mediated reactions such as pneumonitis, colitis, hepatitis, endocrinopathies, and skin and kidney problems.