User login
FDA approves elotuzumab with pom/dex in refractory myeloma
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.

Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
FDA approves lorlatinib as second line for ALK-positive advanced NSCLC
The Food and Drug Administration has granted accelerated approval to lorlatinib for patients with anaplastic lymphoma kinase (ALK)–positive metastatic non–small cell lung cancer (NSCLC) whose disease has progressed on crizotinib and at least one other ALK inhibitor for metastatic disease or whose disease has progressed on alectinib or ceritinib as the first ALK inhibitor therapy for metastatic disease.
Approval of the next-generation ALK inhibitor was based on an overall response rate of 48% – with 4% complete and 44% partial – in a subgroup of 215 patients with ALK-positive metastatic NSCLC enrolled in a nonrandomized, phase 2 trial, the FDA said in a press announcement. All patients had been previously treated with one or more ALK kinase inhibitors.
The median response duration was 12.5 months (95% confidence interval, 8.4-23.7) and the intracranial overall response rate in 89 patients with measurable lesions in the CNS was 60% (95% CI, 49-70) with 21% complete and 38% partial responses.
Common adverse reactions in patients receiving lorlatinib were edema, peripheral neuropathy, cognitive effects, dyspnea, fatigue, weight gain, arthralgia, mood effects, and diarrhea. The most common laboratory abnormalities were hypercholesterolemia and hypertriglyceridemia, the FDA said.
The recommended dose of lorlatinib, to be marketed as Lorbrena by Pfizer, is 100 mg orally once daily.
The Food and Drug Administration has granted accelerated approval to lorlatinib for patients with anaplastic lymphoma kinase (ALK)–positive metastatic non–small cell lung cancer (NSCLC) whose disease has progressed on crizotinib and at least one other ALK inhibitor for metastatic disease or whose disease has progressed on alectinib or ceritinib as the first ALK inhibitor therapy for metastatic disease.
Approval of the next-generation ALK inhibitor was based on an overall response rate of 48% – with 4% complete and 44% partial – in a subgroup of 215 patients with ALK-positive metastatic NSCLC enrolled in a nonrandomized, phase 2 trial, the FDA said in a press announcement. All patients had been previously treated with one or more ALK kinase inhibitors.
The median response duration was 12.5 months (95% confidence interval, 8.4-23.7) and the intracranial overall response rate in 89 patients with measurable lesions in the CNS was 60% (95% CI, 49-70) with 21% complete and 38% partial responses.
Common adverse reactions in patients receiving lorlatinib were edema, peripheral neuropathy, cognitive effects, dyspnea, fatigue, weight gain, arthralgia, mood effects, and diarrhea. The most common laboratory abnormalities were hypercholesterolemia and hypertriglyceridemia, the FDA said.
The recommended dose of lorlatinib, to be marketed as Lorbrena by Pfizer, is 100 mg orally once daily.
The Food and Drug Administration has granted accelerated approval to lorlatinib for patients with anaplastic lymphoma kinase (ALK)–positive metastatic non–small cell lung cancer (NSCLC) whose disease has progressed on crizotinib and at least one other ALK inhibitor for metastatic disease or whose disease has progressed on alectinib or ceritinib as the first ALK inhibitor therapy for metastatic disease.
Approval of the next-generation ALK inhibitor was based on an overall response rate of 48% – with 4% complete and 44% partial – in a subgroup of 215 patients with ALK-positive metastatic NSCLC enrolled in a nonrandomized, phase 2 trial, the FDA said in a press announcement. All patients had been previously treated with one or more ALK kinase inhibitors.
The median response duration was 12.5 months (95% confidence interval, 8.4-23.7) and the intracranial overall response rate in 89 patients with measurable lesions in the CNS was 60% (95% CI, 49-70) with 21% complete and 38% partial responses.
Common adverse reactions in patients receiving lorlatinib were edema, peripheral neuropathy, cognitive effects, dyspnea, fatigue, weight gain, arthralgia, mood effects, and diarrhea. The most common laboratory abnormalities were hypercholesterolemia and hypertriglyceridemia, the FDA said.
The recommended dose of lorlatinib, to be marketed as Lorbrena by Pfizer, is 100 mg orally once daily.
FDA approves sufentanil for adults with acute pain
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.

Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.
Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.
Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
FDA expands approval of pembrolizumab in NSCLC
The Food and Drug Administration .
The drug is now approved for use in combination with carboplatin and either paclitaxel or nanoparticle albumin–bound (nab) paclitaxel for the first-line treatment of NSCLC, regardless of PD-L1 expression status.
This makes pembrolizumab the first anti-PD-1 therapy approved in the first-line setting both as monotherapy and in combination treatment for certain patients with metastatic NSCLC. All appropriate patients with metastatic squamous NSCLC or metastatic nonsquamous NSCLC and no EGFR or ALK mutations are now eligible to receive pembrolizumab-based treatment first-line.
The FDA’s approval is based on results from the phase 3 KEYNOTE-407 trial. This randomized, double-blind study enrolled patients with metastatic squamous NSCLC, regardless of tumor PD-L1 expression status, who had received no prior systemic treatment for metastatic disease.
Patients in the pembrolizumab arm (n = 278) received pembrolizumab and carboplatin every 3 weeks for four cycles, plus paclitaxel every 3 weeks for four cycles or nab-paclitaxel on days 1, 8, and 15 of every 3-week cycle for four cycles, followed by pembrolizumab every 3 weeks.
Patients in the control arm (n = 281) received the same regimen of carboplatin and paclitaxel/nab-paclitaxel, but placebo instead of pembrolizumab.
There was a significant improvement in overall response rate, progression-free survival, and overall survival in patients who received pembrolizumab.
The overall response rate was 58% in the pembrolizumab arm and 35% in the placebo arm (P = .0008). The median duration of response was 7.2 months and 4.9 months, respectively.
The median progression-free survival was 6.4 months in the pembrolizumab arm and 4.8 months in the placebo arm (P less than .0001). The median overall survival was 15.9 months and 11.3 months, respectively (P = .0017).
Safety data are available for the first 203 patients treated on the trial, 101 of them in the pembrolizumab arm.
Fifteen percent of patients discontinued pembrolizumab because of adverse events (AEs), and 43% of patients on pembrolizumab experienced AEs leading to dose interruption.
The most common AEs leading to dose interruption in the pembrolizumab arm were thrombocytopenia, neutropenia, anemia, asthenia, and diarrhea. The most frequent serious AEs in the pembrolizumab arm were febrile neutropenia, pneumonia, and urinary tract infection.
Additional details on this trial are available in the prescribing information, which can be found on the Keytruda website.
The Food and Drug Administration .
The drug is now approved for use in combination with carboplatin and either paclitaxel or nanoparticle albumin–bound (nab) paclitaxel for the first-line treatment of NSCLC, regardless of PD-L1 expression status.
This makes pembrolizumab the first anti-PD-1 therapy approved in the first-line setting both as monotherapy and in combination treatment for certain patients with metastatic NSCLC. All appropriate patients with metastatic squamous NSCLC or metastatic nonsquamous NSCLC and no EGFR or ALK mutations are now eligible to receive pembrolizumab-based treatment first-line.
The FDA’s approval is based on results from the phase 3 KEYNOTE-407 trial. This randomized, double-blind study enrolled patients with metastatic squamous NSCLC, regardless of tumor PD-L1 expression status, who had received no prior systemic treatment for metastatic disease.
Patients in the pembrolizumab arm (n = 278) received pembrolizumab and carboplatin every 3 weeks for four cycles, plus paclitaxel every 3 weeks for four cycles or nab-paclitaxel on days 1, 8, and 15 of every 3-week cycle for four cycles, followed by pembrolizumab every 3 weeks.
Patients in the control arm (n = 281) received the same regimen of carboplatin and paclitaxel/nab-paclitaxel, but placebo instead of pembrolizumab.
There was a significant improvement in overall response rate, progression-free survival, and overall survival in patients who received pembrolizumab.
The overall response rate was 58% in the pembrolizumab arm and 35% in the placebo arm (P = .0008). The median duration of response was 7.2 months and 4.9 months, respectively.
The median progression-free survival was 6.4 months in the pembrolizumab arm and 4.8 months in the placebo arm (P less than .0001). The median overall survival was 15.9 months and 11.3 months, respectively (P = .0017).
Safety data are available for the first 203 patients treated on the trial, 101 of them in the pembrolizumab arm.
Fifteen percent of patients discontinued pembrolizumab because of adverse events (AEs), and 43% of patients on pembrolizumab experienced AEs leading to dose interruption.
The most common AEs leading to dose interruption in the pembrolizumab arm were thrombocytopenia, neutropenia, anemia, asthenia, and diarrhea. The most frequent serious AEs in the pembrolizumab arm were febrile neutropenia, pneumonia, and urinary tract infection.
Additional details on this trial are available in the prescribing information, which can be found on the Keytruda website.
The Food and Drug Administration .
The drug is now approved for use in combination with carboplatin and either paclitaxel or nanoparticle albumin–bound (nab) paclitaxel for the first-line treatment of NSCLC, regardless of PD-L1 expression status.
This makes pembrolizumab the first anti-PD-1 therapy approved in the first-line setting both as monotherapy and in combination treatment for certain patients with metastatic NSCLC. All appropriate patients with metastatic squamous NSCLC or metastatic nonsquamous NSCLC and no EGFR or ALK mutations are now eligible to receive pembrolizumab-based treatment first-line.
The FDA’s approval is based on results from the phase 3 KEYNOTE-407 trial. This randomized, double-blind study enrolled patients with metastatic squamous NSCLC, regardless of tumor PD-L1 expression status, who had received no prior systemic treatment for metastatic disease.
Patients in the pembrolizumab arm (n = 278) received pembrolizumab and carboplatin every 3 weeks for four cycles, plus paclitaxel every 3 weeks for four cycles or nab-paclitaxel on days 1, 8, and 15 of every 3-week cycle for four cycles, followed by pembrolizumab every 3 weeks.
Patients in the control arm (n = 281) received the same regimen of carboplatin and paclitaxel/nab-paclitaxel, but placebo instead of pembrolizumab.
There was a significant improvement in overall response rate, progression-free survival, and overall survival in patients who received pembrolizumab.
The overall response rate was 58% in the pembrolizumab arm and 35% in the placebo arm (P = .0008). The median duration of response was 7.2 months and 4.9 months, respectively.
The median progression-free survival was 6.4 months in the pembrolizumab arm and 4.8 months in the placebo arm (P less than .0001). The median overall survival was 15.9 months and 11.3 months, respectively (P = .0017).
Safety data are available for the first 203 patients treated on the trial, 101 of them in the pembrolizumab arm.
Fifteen percent of patients discontinued pembrolizumab because of adverse events (AEs), and 43% of patients on pembrolizumab experienced AEs leading to dose interruption.
The most common AEs leading to dose interruption in the pembrolizumab arm were thrombocytopenia, neutropenia, anemia, asthenia, and diarrhea. The most frequent serious AEs in the pembrolizumab arm were febrile neutropenia, pneumonia, and urinary tract infection.
Additional details on this trial are available in the prescribing information, which can be found on the Keytruda website.
FDA approves Xyrem to treat children with narcolepsy
The Food and Drug Administration has cleared Xyrem (sodium oxybate) oral solution to treat cataplexy and excessive daytime sleepiness in patients ages 7-17 with narcolepsy.
The central nervous system depressant previously had been approved to treat cataplexy in adults with narcolepsy.
The current approval was granted by the FDA under a Priority Review designation. Xyrem also received the FDA’s Orphan Drug designation, which is intended to encourage the development of drugs for rare diseases.
The agency noted in a press release, however, that the drug would continue to be available only through risk evaluation mitigation strategy (REMS) programs because of “the risk of serious outcomes resulting from inappropriate prescribing, misuse, abuse and diversion.” Xyrem either alone or in combination with other CNS depressants may be associated with reactions including seizure, respiratory depression, decreases in the level of consciousness, coma, and death, the FDA said.
The most common adverse reactions in pediatric patients were enuresis, nausea, headache, vomiting, weight decrease, decreased appetite, and dizziness.
For more information on prescribing Xyrem for pediatric patients, see the revised labeling information on the FDA website.
The Food and Drug Administration has cleared Xyrem (sodium oxybate) oral solution to treat cataplexy and excessive daytime sleepiness in patients ages 7-17 with narcolepsy.
The central nervous system depressant previously had been approved to treat cataplexy in adults with narcolepsy.
The current approval was granted by the FDA under a Priority Review designation. Xyrem also received the FDA’s Orphan Drug designation, which is intended to encourage the development of drugs for rare diseases.
The agency noted in a press release, however, that the drug would continue to be available only through risk evaluation mitigation strategy (REMS) programs because of “the risk of serious outcomes resulting from inappropriate prescribing, misuse, abuse and diversion.” Xyrem either alone or in combination with other CNS depressants may be associated with reactions including seizure, respiratory depression, decreases in the level of consciousness, coma, and death, the FDA said.
The most common adverse reactions in pediatric patients were enuresis, nausea, headache, vomiting, weight decrease, decreased appetite, and dizziness.
For more information on prescribing Xyrem for pediatric patients, see the revised labeling information on the FDA website.
The Food and Drug Administration has cleared Xyrem (sodium oxybate) oral solution to treat cataplexy and excessive daytime sleepiness in patients ages 7-17 with narcolepsy.
The central nervous system depressant previously had been approved to treat cataplexy in adults with narcolepsy.
The current approval was granted by the FDA under a Priority Review designation. Xyrem also received the FDA’s Orphan Drug designation, which is intended to encourage the development of drugs for rare diseases.
The agency noted in a press release, however, that the drug would continue to be available only through risk evaluation mitigation strategy (REMS) programs because of “the risk of serious outcomes resulting from inappropriate prescribing, misuse, abuse and diversion.” Xyrem either alone or in combination with other CNS depressants may be associated with reactions including seizure, respiratory depression, decreases in the level of consciousness, coma, and death, the FDA said.
The most common adverse reactions in pediatric patients were enuresis, nausea, headache, vomiting, weight decrease, decreased appetite, and dizziness.
For more information on prescribing Xyrem for pediatric patients, see the revised labeling information on the FDA website.
Ruxolitinib receives priority review for acute GVHD
The Food and Drug Administration has accepted the JAK1/JAK2 inhibitor ruxolitinib (Jakafi) for priority review.
Incyte is seeking approval for ruxolitinib as a treatment for patients with acute graft-versus-host disease (GVHD) who have had an inadequate response to corticosteroids.
“If approved, ruxolitinib will be the first and only treatment available in the U.S. for patients with acute GVHD who have not responded adequately to corticosteroid therapy,” Steven Stein, MD, chief medical officer at Incyte, said in a statement.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The designation generally means that the agency will act on the application within 6 months, rather than 10 months.
In addition to priority review, the FDA previously granted ruxolitinib breakthrough therapy and orphan drug designations.
The application is based on data from the ongoing, phase 2 REACH1 trial (NCT02953678), which is evaluating ruxolitinib in combination with corticosteroids in patients who have steroid-refractory acute GVHD.
Incyte announced top-line results from REACH1 in June, reporting on outcomes in 71 patients.
The study’s primary endpoint – overall response rate at day 28 – was met. Ruxolitinib produced an overall response rate of 55% at that time. However, 73% of patients responded to ruxolitinib at some point during the trial. Incyte said the most common treatment-emergent adverse events were anemia, thrombocytopenia, and neutropenia.
The Food and Drug Administration has accepted the JAK1/JAK2 inhibitor ruxolitinib (Jakafi) for priority review.
Incyte is seeking approval for ruxolitinib as a treatment for patients with acute graft-versus-host disease (GVHD) who have had an inadequate response to corticosteroids.
“If approved, ruxolitinib will be the first and only treatment available in the U.S. for patients with acute GVHD who have not responded adequately to corticosteroid therapy,” Steven Stein, MD, chief medical officer at Incyte, said in a statement.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The designation generally means that the agency will act on the application within 6 months, rather than 10 months.
In addition to priority review, the FDA previously granted ruxolitinib breakthrough therapy and orphan drug designations.
The application is based on data from the ongoing, phase 2 REACH1 trial (NCT02953678), which is evaluating ruxolitinib in combination with corticosteroids in patients who have steroid-refractory acute GVHD.
Incyte announced top-line results from REACH1 in June, reporting on outcomes in 71 patients.
The study’s primary endpoint – overall response rate at day 28 – was met. Ruxolitinib produced an overall response rate of 55% at that time. However, 73% of patients responded to ruxolitinib at some point during the trial. Incyte said the most common treatment-emergent adverse events were anemia, thrombocytopenia, and neutropenia.
The Food and Drug Administration has accepted the JAK1/JAK2 inhibitor ruxolitinib (Jakafi) for priority review.
Incyte is seeking approval for ruxolitinib as a treatment for patients with acute graft-versus-host disease (GVHD) who have had an inadequate response to corticosteroids.
“If approved, ruxolitinib will be the first and only treatment available in the U.S. for patients with acute GVHD who have not responded adequately to corticosteroid therapy,” Steven Stein, MD, chief medical officer at Incyte, said in a statement.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The designation generally means that the agency will act on the application within 6 months, rather than 10 months.
In addition to priority review, the FDA previously granted ruxolitinib breakthrough therapy and orphan drug designations.
The application is based on data from the ongoing, phase 2 REACH1 trial (NCT02953678), which is evaluating ruxolitinib in combination with corticosteroids in patients who have steroid-refractory acute GVHD.
Incyte announced top-line results from REACH1 in June, reporting on outcomes in 71 patients.
The study’s primary endpoint – overall response rate at day 28 – was met. Ruxolitinib produced an overall response rate of 55% at that time. However, 73% of patients responded to ruxolitinib at some point during the trial. Incyte said the most common treatment-emergent adverse events were anemia, thrombocytopenia, and neutropenia.
OMS721 gains orphan designation for HSCT-associated thrombotic microangiopathy
The Food and Drug Administration has granted OMS721 orphan designation for the treatment of hematopoietic stem cell transplant–associated thrombotic microangiopathy (HSCT-TMA).
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The FDA previously granted OMS721 breakthrough therapy designation for HSCT-TMA and orphan designation for the prevention of complement-mediated TMA, including HSCT-TMA.
Omeros, the company developing OMS721, has established a compassionate use program for OMS721, which is active in the United States and Europe.
Phase 3 clinical programs are in progress for OMS721 in atypical hemolytic uremic syndrome, immunoglobulin A nephropathy, and HSCT-TMA. Two phase 2 trials of OMS721 – one in TMA and one in immunoglobulin A nephropathy – are ongoing.
Omeros announced results from the phase 2 TMA trial (NCT02222545) in February. The study includes adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post transplant. Patients receive weekly OMS721 treatments for 4-8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 HSCT-TMA patients had been treated.
These patients had a significantly longer median overall survival at 347 days, compared with historical controls at 21 days (P less than .0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P = .017). The mean lactate dehydrogenase decreased from 591 U/L to 250 U/L (P less than .001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P = .003).
The most commonly reported adverse events were diarrhea and neutropenia. Four deaths occurred during the study. One of these – attributable to acute renal and respiratory failure – was considered possibly related to OMS721.
The Food and Drug Administration has granted OMS721 orphan designation for the treatment of hematopoietic stem cell transplant–associated thrombotic microangiopathy (HSCT-TMA).
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The FDA previously granted OMS721 breakthrough therapy designation for HSCT-TMA and orphan designation for the prevention of complement-mediated TMA, including HSCT-TMA.
Omeros, the company developing OMS721, has established a compassionate use program for OMS721, which is active in the United States and Europe.
Phase 3 clinical programs are in progress for OMS721 in atypical hemolytic uremic syndrome, immunoglobulin A nephropathy, and HSCT-TMA. Two phase 2 trials of OMS721 – one in TMA and one in immunoglobulin A nephropathy – are ongoing.
Omeros announced results from the phase 2 TMA trial (NCT02222545) in February. The study includes adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post transplant. Patients receive weekly OMS721 treatments for 4-8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 HSCT-TMA patients had been treated.
These patients had a significantly longer median overall survival at 347 days, compared with historical controls at 21 days (P less than .0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P = .017). The mean lactate dehydrogenase decreased from 591 U/L to 250 U/L (P less than .001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P = .003).
The most commonly reported adverse events were diarrhea and neutropenia. Four deaths occurred during the study. One of these – attributable to acute renal and respiratory failure – was considered possibly related to OMS721.
The Food and Drug Administration has granted OMS721 orphan designation for the treatment of hematopoietic stem cell transplant–associated thrombotic microangiopathy (HSCT-TMA).
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The FDA previously granted OMS721 breakthrough therapy designation for HSCT-TMA and orphan designation for the prevention of complement-mediated TMA, including HSCT-TMA.
Omeros, the company developing OMS721, has established a compassionate use program for OMS721, which is active in the United States and Europe.
Phase 3 clinical programs are in progress for OMS721 in atypical hemolytic uremic syndrome, immunoglobulin A nephropathy, and HSCT-TMA. Two phase 2 trials of OMS721 – one in TMA and one in immunoglobulin A nephropathy – are ongoing.
Omeros announced results from the phase 2 TMA trial (NCT02222545) in February. The study includes adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post transplant. Patients receive weekly OMS721 treatments for 4-8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 HSCT-TMA patients had been treated.
These patients had a significantly longer median overall survival at 347 days, compared with historical controls at 21 days (P less than .0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P = .017). The mean lactate dehydrogenase decreased from 591 U/L to 250 U/L (P less than .001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P = .003).
The most commonly reported adverse events were diarrhea and neutropenia. Four deaths occurred during the study. One of these – attributable to acute renal and respiratory failure – was considered possibly related to OMS721.
Endocrine Society raises concerns about FDA’s “safe” classification of bisphenol A in food containers
An initial report from the Endocrine Society has raised concerns about bisphenol A use in products such as food and drink containers, toys, and medical devices, citing recent data that show the synthetic compound is linked to reproductive, behavioral, and metabolic disorders.

Although the Food and Drug Administration classifies bisphenol A (BPA) as safe to use in food containers, there have been hundreds of studies tying BPA to health problems such as “neurological outcomes, abnormal metabolism, reproductive effects as well as growth and development effects,” according to Laura N. Vandenberg, PhD, an Endocrine Society spokesperson, who spoke at a press briefing held on Oct. 23. Dr. Vandenberg explained the FDA’s 2014 position on BPA safety comes from a small subset of publicly available data, but these are not all the data on BPA, as some academic data are still under review.
The Endocrine Society recently held the news conference because they are concerned the FDA has “jumped the gun” before all the research has been published. “Even considering the fact that the data that have been presented by FDA have been interpreted by FDA as suggesting that BPA is safe, scientists still disagree,” Dr. Vandenberg said.

However, the Endocrine Society noted there is an issue with the current literature, which can be used to interpret and report different results. Heather Patisaul, PhD, cited a joint report from the Food and Agriculture Organization of the United Nations and World Health Organization, as well as a report from the National Toxicology Program (NTP), to illustrate this problem. Both reports expressed concern about BPA safety but took different approaches and a wider viewpoint, and came to different conclusions, she said.
“These two documents both concluded that there was some concern about bisphenol A and behavior, but they identified there was a big problem with trying to pool all this literature together because the experimental protocols were different, the animals were different, the dosing was different,” she said. “It was not a very harmonious literature.”
To combat this issue, the National Institute of Environmental Health Sciences and the FDA have funded the CLARITY-BPA (Consortium Linking Academic and Regulatory Insights on BPA Toxicity) study. Dr. Patisaul said CLARITY-BPA is “the most ambitious project that’s ever been done” to study the health effects of a chemical, bringing together scientists from academic institutions, the NTP, and the FDA to help create data for risk assessment.
“The goal was to create this culture of partnership and communication between the agencies that have to make these decisions about safety and the scientists who are producing the data that’s trying to inform those assessments,” Dr. Patisaul said.
Dr. Vandenberg and Dr. Patisaul presented results from the CLARITY-BPA Core Study, which studied the effects of continuous doses of BPA in rats starting from 6 days of pregnancy; after birth, the rat offspring were fed doses of BPA for 1 year and 2 years. A second group of rats in a stop-dose group were fed BPA from early development, where the mothers were fed BPA at day 6 of pregnancy and the offspring fed BPA from birth until puberty (21 days) and followed for 1 year or 2 years. The researchers also examined 2.5 mcg/kg, 25 mcg/kg, 250 mcg/kg, 2,500 mcg/kg, and 25,000 mcg/kg doses of BPA exposure as well as continuous ethinyl estradiol exposure as a positive control.
In the FDA Core Study, there was a significantly increased incidence of mammary adenocarcinoma in the stop dose group and inflammation of the dorsal and lateral lobes of the prostate in the continuous dose group at a dose of 2.5 mcg/kg. In addition, kidney nephropathy and increased body weight in female rats in the continuous group were also seen at the 2.5 mcg/kg dose, Dr. Vandenberg noted.
“I think one of the reasons why FDA is dismissing those low-dose effects is that there’s an expectation with increasing dose, there should be an increase in an effect,” Dr. Vandenberg said.
In the low-dose range, BPA could be acting as a hormone such as estrogen, but also could be acting through other hormone receptors or as a toxicant at the high-dose range, she explained.
Dr. Patisaul also presented results of BPA-related effects on the brain and behavior in the existing literature from the TEDX Low-dose Bisphenol A project, which is a comparison of 391 in vivo and in vitro studies of BPA prior to 2009. The results showed brain and behavior was “heavily impacted” by BPA, as were organ systems such as the heart, which supports the results from the CLARITY-BPA study, Dr. Patisaul noted.
“When you think about reproducibility in the broadest sense, and you look at the effects that the FDA found at low dose, you look at the effects the CLARITY investigators found at low dose, and you go back and look at the existing literature, you see a very clear picture of BPA-produced effects on brain and behavior, female reproductive systems, and the cardiovascular system,” she said.
Dr. Patisaul is a study investigator for CLARITY-BPA.
An initial report from the Endocrine Society has raised concerns about bisphenol A use in products such as food and drink containers, toys, and medical devices, citing recent data that show the synthetic compound is linked to reproductive, behavioral, and metabolic disorders.
Although the Food and Drug Administration classifies bisphenol A (BPA) as safe to use in food containers, there have been hundreds of studies tying BPA to health problems such as “neurological outcomes, abnormal metabolism, reproductive effects as well as growth and development effects,” according to Laura N. Vandenberg, PhD, an Endocrine Society spokesperson, who spoke at a press briefing held on Oct. 23. Dr. Vandenberg explained the FDA’s 2014 position on BPA safety comes from a small subset of publicly available data, but these are not all the data on BPA, as some academic data are still under review.
The Endocrine Society recently held the news conference because they are concerned the FDA has “jumped the gun” before all the research has been published. “Even considering the fact that the data that have been presented by FDA have been interpreted by FDA as suggesting that BPA is safe, scientists still disagree,” Dr. Vandenberg said.
However, the Endocrine Society noted there is an issue with the current literature, which can be used to interpret and report different results. Heather Patisaul, PhD, cited a joint report from the Food and Agriculture Organization of the United Nations and World Health Organization, as well as a report from the National Toxicology Program (NTP), to illustrate this problem. Both reports expressed concern about BPA safety but took different approaches and a wider viewpoint, and came to different conclusions, she said.
“These two documents both concluded that there was some concern about bisphenol A and behavior, but they identified there was a big problem with trying to pool all this literature together because the experimental protocols were different, the animals were different, the dosing was different,” she said. “It was not a very harmonious literature.”
To combat this issue, the National Institute of Environmental Health Sciences and the FDA have funded the CLARITY-BPA (Consortium Linking Academic and Regulatory Insights on BPA Toxicity) study. Dr. Patisaul said CLARITY-BPA is “the most ambitious project that’s ever been done” to study the health effects of a chemical, bringing together scientists from academic institutions, the NTP, and the FDA to help create data for risk assessment.
“The goal was to create this culture of partnership and communication between the agencies that have to make these decisions about safety and the scientists who are producing the data that’s trying to inform those assessments,” Dr. Patisaul said.
Dr. Vandenberg and Dr. Patisaul presented results from the CLARITY-BPA Core Study, which studied the effects of continuous doses of BPA in rats starting from 6 days of pregnancy; after birth, the rat offspring were fed doses of BPA for 1 year and 2 years. A second group of rats in a stop-dose group were fed BPA from early development, where the mothers were fed BPA at day 6 of pregnancy and the offspring fed BPA from birth until puberty (21 days) and followed for 1 year or 2 years. The researchers also examined 2.5 mcg/kg, 25 mcg/kg, 250 mcg/kg, 2,500 mcg/kg, and 25,000 mcg/kg doses of BPA exposure as well as continuous ethinyl estradiol exposure as a positive control.
In the FDA Core Study, there was a significantly increased incidence of mammary adenocarcinoma in the stop dose group and inflammation of the dorsal and lateral lobes of the prostate in the continuous dose group at a dose of 2.5 mcg/kg. In addition, kidney nephropathy and increased body weight in female rats in the continuous group were also seen at the 2.5 mcg/kg dose, Dr. Vandenberg noted.
“I think one of the reasons why FDA is dismissing those low-dose effects is that there’s an expectation with increasing dose, there should be an increase in an effect,” Dr. Vandenberg said.
In the low-dose range, BPA could be acting as a hormone such as estrogen, but also could be acting through other hormone receptors or as a toxicant at the high-dose range, she explained.
Dr. Patisaul also presented results of BPA-related effects on the brain and behavior in the existing literature from the TEDX Low-dose Bisphenol A project, which is a comparison of 391 in vivo and in vitro studies of BPA prior to 2009. The results showed brain and behavior was “heavily impacted” by BPA, as were organ systems such as the heart, which supports the results from the CLARITY-BPA study, Dr. Patisaul noted.
“When you think about reproducibility in the broadest sense, and you look at the effects that the FDA found at low dose, you look at the effects the CLARITY investigators found at low dose, and you go back and look at the existing literature, you see a very clear picture of BPA-produced effects on brain and behavior, female reproductive systems, and the cardiovascular system,” she said.
Dr. Patisaul is a study investigator for CLARITY-BPA.
An initial report from the Endocrine Society has raised concerns about bisphenol A use in products such as food and drink containers, toys, and medical devices, citing recent data that show the synthetic compound is linked to reproductive, behavioral, and metabolic disorders.
Although the Food and Drug Administration classifies bisphenol A (BPA) as safe to use in food containers, there have been hundreds of studies tying BPA to health problems such as “neurological outcomes, abnormal metabolism, reproductive effects as well as growth and development effects,” according to Laura N. Vandenberg, PhD, an Endocrine Society spokesperson, who spoke at a press briefing held on Oct. 23. Dr. Vandenberg explained the FDA’s 2014 position on BPA safety comes from a small subset of publicly available data, but these are not all the data on BPA, as some academic data are still under review.
The Endocrine Society recently held the news conference because they are concerned the FDA has “jumped the gun” before all the research has been published. “Even considering the fact that the data that have been presented by FDA have been interpreted by FDA as suggesting that BPA is safe, scientists still disagree,” Dr. Vandenberg said.
However, the Endocrine Society noted there is an issue with the current literature, which can be used to interpret and report different results. Heather Patisaul, PhD, cited a joint report from the Food and Agriculture Organization of the United Nations and World Health Organization, as well as a report from the National Toxicology Program (NTP), to illustrate this problem. Both reports expressed concern about BPA safety but took different approaches and a wider viewpoint, and came to different conclusions, she said.
“These two documents both concluded that there was some concern about bisphenol A and behavior, but they identified there was a big problem with trying to pool all this literature together because the experimental protocols were different, the animals were different, the dosing was different,” she said. “It was not a very harmonious literature.”
To combat this issue, the National Institute of Environmental Health Sciences and the FDA have funded the CLARITY-BPA (Consortium Linking Academic and Regulatory Insights on BPA Toxicity) study. Dr. Patisaul said CLARITY-BPA is “the most ambitious project that’s ever been done” to study the health effects of a chemical, bringing together scientists from academic institutions, the NTP, and the FDA to help create data for risk assessment.
“The goal was to create this culture of partnership and communication between the agencies that have to make these decisions about safety and the scientists who are producing the data that’s trying to inform those assessments,” Dr. Patisaul said.
Dr. Vandenberg and Dr. Patisaul presented results from the CLARITY-BPA Core Study, which studied the effects of continuous doses of BPA in rats starting from 6 days of pregnancy; after birth, the rat offspring were fed doses of BPA for 1 year and 2 years. A second group of rats in a stop-dose group were fed BPA from early development, where the mothers were fed BPA at day 6 of pregnancy and the offspring fed BPA from birth until puberty (21 days) and followed for 1 year or 2 years. The researchers also examined 2.5 mcg/kg, 25 mcg/kg, 250 mcg/kg, 2,500 mcg/kg, and 25,000 mcg/kg doses of BPA exposure as well as continuous ethinyl estradiol exposure as a positive control.
In the FDA Core Study, there was a significantly increased incidence of mammary adenocarcinoma in the stop dose group and inflammation of the dorsal and lateral lobes of the prostate in the continuous dose group at a dose of 2.5 mcg/kg. In addition, kidney nephropathy and increased body weight in female rats in the continuous group were also seen at the 2.5 mcg/kg dose, Dr. Vandenberg noted.
“I think one of the reasons why FDA is dismissing those low-dose effects is that there’s an expectation with increasing dose, there should be an increase in an effect,” Dr. Vandenberg said.
In the low-dose range, BPA could be acting as a hormone such as estrogen, but also could be acting through other hormone receptors or as a toxicant at the high-dose range, she explained.
Dr. Patisaul also presented results of BPA-related effects on the brain and behavior in the existing literature from the TEDX Low-dose Bisphenol A project, which is a comparison of 391 in vivo and in vitro studies of BPA prior to 2009. The results showed brain and behavior was “heavily impacted” by BPA, as were organ systems such as the heart, which supports the results from the CLARITY-BPA study, Dr. Patisaul noted.
“When you think about reproducibility in the broadest sense, and you look at the effects that the FDA found at low dose, you look at the effects the CLARITY investigators found at low dose, and you go back and look at the existing literature, you see a very clear picture of BPA-produced effects on brain and behavior, female reproductive systems, and the cardiovascular system,” she said.
Dr. Patisaul is a study investigator for CLARITY-BPA.
Key clinical point: Despite claims from the Food and Drug Administration, results from the CLARITY-BPA (Consortium Linking Academic and Regulatory Insights on BPA Toxicity) Core Study show serious effects in humans of bisphenol A at low doses.
Major finding: Research from CLARITY-BPA has shown brain and behavior, female reproduction, and organ systems such as the heart can be adversely affected by bisphenol A even at low doses.
Study details: An initial report from the CLARITY-BPA Core Study.
Disclosures: Dr. Patisaul is a study investigator for CLARITY-BPA.
FDA clears Abbott’s Influenza A & B 2, Strep A 2 assays
The Food and Drug Administration has cleared Abbott Laboratories’ next-generation Influenza A & B 2 and Strep A 2 molecular assays for point-of-care testing.
The Influenza A & B 2 assay can detect and differentiate influenza A and B in 13 minutes, with a call-out of positive results at 5 minutes. It can be stored at room temperature, simplifying storage and ordering. The Strep A 2 assay detects group A streptococcus bacterial nucleic acid in 6 minutes, with a call-out of positive results at 2 minutes. Both will be the fastest tests currently on the market in their respective fields, according to a corporate press release.
The assays will be available in a variety of inpatient and outpatient settings, particularly in locations where patients commonly access health care services, such as EDs, physician offices, walk-in clinics, and urgent care centers. This will allow health care providers to make a fast, informed diagnosis and provide appropriate treatment within the span of a single patient visit.
“The ability to obtain early call outs for positive test results with molecular accuracy in as little as 5 minutes for influenza and 2 minutes for strep A is a game-changing development that allows prompt treatment decisions at the point of care. Rapid testing may also help reduce improper antibiotic usage, which can occur when treatment is based exclusively on a patient’s symptoms, and contributes to antibiotic resistance,” Gregory J. Berry, PhD, director of molecular diagnostics at Northwell Health Laboratories in Lake Success, N.Y., said in the press release.
Find the full press release on the Abbott Laboratories website.
The Food and Drug Administration has cleared Abbott Laboratories’ next-generation Influenza A & B 2 and Strep A 2 molecular assays for point-of-care testing.
The Influenza A & B 2 assay can detect and differentiate influenza A and B in 13 minutes, with a call-out of positive results at 5 minutes. It can be stored at room temperature, simplifying storage and ordering. The Strep A 2 assay detects group A streptococcus bacterial nucleic acid in 6 minutes, with a call-out of positive results at 2 minutes. Both will be the fastest tests currently on the market in their respective fields, according to a corporate press release.
The assays will be available in a variety of inpatient and outpatient settings, particularly in locations where patients commonly access health care services, such as EDs, physician offices, walk-in clinics, and urgent care centers. This will allow health care providers to make a fast, informed diagnosis and provide appropriate treatment within the span of a single patient visit.
“The ability to obtain early call outs for positive test results with molecular accuracy in as little as 5 minutes for influenza and 2 minutes for strep A is a game-changing development that allows prompt treatment decisions at the point of care. Rapid testing may also help reduce improper antibiotic usage, which can occur when treatment is based exclusively on a patient’s symptoms, and contributes to antibiotic resistance,” Gregory J. Berry, PhD, director of molecular diagnostics at Northwell Health Laboratories in Lake Success, N.Y., said in the press release.
Find the full press release on the Abbott Laboratories website.
The Food and Drug Administration has cleared Abbott Laboratories’ next-generation Influenza A & B 2 and Strep A 2 molecular assays for point-of-care testing.
The Influenza A & B 2 assay can detect and differentiate influenza A and B in 13 minutes, with a call-out of positive results at 5 minutes. It can be stored at room temperature, simplifying storage and ordering. The Strep A 2 assay detects group A streptococcus bacterial nucleic acid in 6 minutes, with a call-out of positive results at 2 minutes. Both will be the fastest tests currently on the market in their respective fields, according to a corporate press release.
The assays will be available in a variety of inpatient and outpatient settings, particularly in locations where patients commonly access health care services, such as EDs, physician offices, walk-in clinics, and urgent care centers. This will allow health care providers to make a fast, informed diagnosis and provide appropriate treatment within the span of a single patient visit.
“The ability to obtain early call outs for positive test results with molecular accuracy in as little as 5 minutes for influenza and 2 minutes for strep A is a game-changing development that allows prompt treatment decisions at the point of care. Rapid testing may also help reduce improper antibiotic usage, which can occur when treatment is based exclusively on a patient’s symptoms, and contributes to antibiotic resistance,” Gregory J. Berry, PhD, director of molecular diagnostics at Northwell Health Laboratories in Lake Success, N.Y., said in the press release.
Find the full press release on the Abbott Laboratories website.
Drug overdose deaths down since late 2017
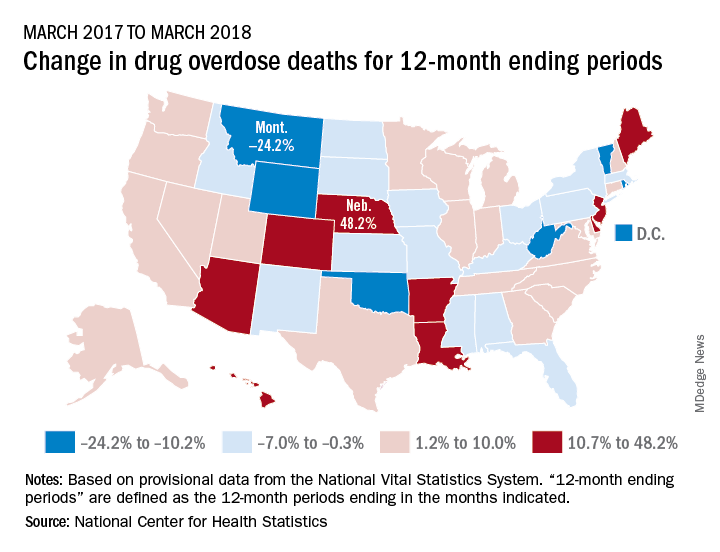
Longer-term data, however, show an increase over the year from March 2017 to March 2018, as the short-term decrease was not enough to overcome the previous year’s increase. The provisional 12-month ending count – deaths during the 12-month period ending in the month indicated – went from 66,859 in March 2017 to 68,690 in March 2018, an increase of 2.7%, the NCHS reported.
That year-long increase was not spread evenly among the states. Nebraska’s 12-month ending count jumped over 48% from March 2017 to March 2018, more than twice as much as second-place Hawaii’s 20.9%. Montana had the largest drop over that year, –24.2%, with Wyoming next at –20.7% and the District of Columbia third at –14.8%, data from the National Vital Statistics System show.
“Provisional drug overdose death data are often incomplete,” the NCHS noted, “and the degree of completeness varies by jurisdiction and 12-month ending period. Consequently, the numbers of drug overdose deaths are underestimated, based on provisional data relative to final data and are subject to random variation.”
Longer-term data, however, show an increase over the year from March 2017 to March 2018, as the short-term decrease was not enough to overcome the previous year’s increase. The provisional 12-month ending count – deaths during the 12-month period ending in the month indicated – went from 66,859 in March 2017 to 68,690 in March 2018, an increase of 2.7%, the NCHS reported.
That year-long increase was not spread evenly among the states. Nebraska’s 12-month ending count jumped over 48% from March 2017 to March 2018, more than twice as much as second-place Hawaii’s 20.9%. Montana had the largest drop over that year, –24.2%, with Wyoming next at –20.7% and the District of Columbia third at –14.8%, data from the National Vital Statistics System show.
“Provisional drug overdose death data are often incomplete,” the NCHS noted, “and the degree of completeness varies by jurisdiction and 12-month ending period. Consequently, the numbers of drug overdose deaths are underestimated, based on provisional data relative to final data and are subject to random variation.”
Longer-term data, however, show an increase over the year from March 2017 to March 2018, as the short-term decrease was not enough to overcome the previous year’s increase. The provisional 12-month ending count – deaths during the 12-month period ending in the month indicated – went from 66,859 in March 2017 to 68,690 in March 2018, an increase of 2.7%, the NCHS reported.
That year-long increase was not spread evenly among the states. Nebraska’s 12-month ending count jumped over 48% from March 2017 to March 2018, more than twice as much as second-place Hawaii’s 20.9%. Montana had the largest drop over that year, –24.2%, with Wyoming next at –20.7% and the District of Columbia third at –14.8%, data from the National Vital Statistics System show.
“Provisional drug overdose death data are often incomplete,” the NCHS noted, “and the degree of completeness varies by jurisdiction and 12-month ending period. Consequently, the numbers of drug overdose deaths are underestimated, based on provisional data relative to final data and are subject to random variation.”