User login
Abortion measures continue to fall
Three important national measures of abortion dropped by at least 19% from 2006 to 2015, according to the Centers for Disease Control and Prevention.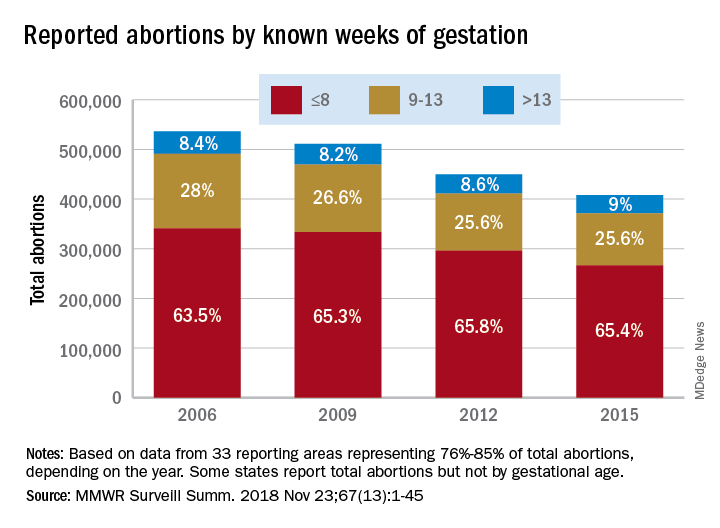
Surgical and medical abortions reported to the CDC dropped by 24%, going from almost 843,000 in 2006 to 638,000 in 2015, with declines occurring every year, Tara C. Jatlaoui, MD, and her associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
Over that same time period, the abortion rate fell from 15.9 per 1,000 women aged 15-44 years to 11.8 per 1,000 – a decline of 26%. Abortion ratio – the number of abortions per 1,000 live births within a given population – declined by 19%, dropping from 233 abortions per 1,000 live births in 2006 to 188 abortions per 1,000 live births in 2015, the investigators reported. The findings were based on data from 49 areas that continuously reported over the study period (excludes California, Maryland, and New Hampshire but includes the District of Columbia and New York City).
Abortion rates were highest for women aged 20-29 years for the study period, and this age group accounted for the largest share among the 44 reporting areas that provided data by maternal age each year. Those under age 15 years had the largest drops by age in total number of abortions (40%) and abortion rate (58%) but had the highest, by far, abortion ratio for each year of the study (700 per 1,000 live births in that age group in 2015. The abortion ratio for 15- to 19-year-olds was 289 per 1,000 live births).
The percentage of abortions performed before 14 weeks’ gestation changed little, going from 91.5% in 2006 to 91% in 2015, but “a shift occurred toward earlier gestational ages,” they noted. The percentage of abortions performed before or at 8 weeks increased 3% and those done at 9-13 weeks dropped almost 9% among the 33 areas that reported gestational age every year. Abortions done after 13 weeks gestation represented 9% of all abortions during the study period, with an increase of 7% occurring from 2006 to 2015, Dr. Jatlaoui and her associates said.
Removing barriers such as cost, “insufficient provider reimbursement and training, inadequate client-centered counseling, lack of youth-friendly services, and low client awareness ... might help improve contraceptive use, potentially reducing the number of unintended pregnancies and the number of abortions performed in the United States,” the researchers wrote.
SOURCE: Jatlaoui TC et al. MMWR Surveill Summ. 2018 Nov 23(13):1-45.
Three important national measures of abortion dropped by at least 19% from 2006 to 2015, according to the Centers for Disease Control and Prevention.
Surgical and medical abortions reported to the CDC dropped by 24%, going from almost 843,000 in 2006 to 638,000 in 2015, with declines occurring every year, Tara C. Jatlaoui, MD, and her associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
Over that same time period, the abortion rate fell from 15.9 per 1,000 women aged 15-44 years to 11.8 per 1,000 – a decline of 26%. Abortion ratio – the number of abortions per 1,000 live births within a given population – declined by 19%, dropping from 233 abortions per 1,000 live births in 2006 to 188 abortions per 1,000 live births in 2015, the investigators reported. The findings were based on data from 49 areas that continuously reported over the study period (excludes California, Maryland, and New Hampshire but includes the District of Columbia and New York City).
Abortion rates were highest for women aged 20-29 years for the study period, and this age group accounted for the largest share among the 44 reporting areas that provided data by maternal age each year. Those under age 15 years had the largest drops by age in total number of abortions (40%) and abortion rate (58%) but had the highest, by far, abortion ratio for each year of the study (700 per 1,000 live births in that age group in 2015. The abortion ratio for 15- to 19-year-olds was 289 per 1,000 live births).
The percentage of abortions performed before 14 weeks’ gestation changed little, going from 91.5% in 2006 to 91% in 2015, but “a shift occurred toward earlier gestational ages,” they noted. The percentage of abortions performed before or at 8 weeks increased 3% and those done at 9-13 weeks dropped almost 9% among the 33 areas that reported gestational age every year. Abortions done after 13 weeks gestation represented 9% of all abortions during the study period, with an increase of 7% occurring from 2006 to 2015, Dr. Jatlaoui and her associates said.
Removing barriers such as cost, “insufficient provider reimbursement and training, inadequate client-centered counseling, lack of youth-friendly services, and low client awareness ... might help improve contraceptive use, potentially reducing the number of unintended pregnancies and the number of abortions performed in the United States,” the researchers wrote.
SOURCE: Jatlaoui TC et al. MMWR Surveill Summ. 2018 Nov 23(13):1-45.
Three important national measures of abortion dropped by at least 19% from 2006 to 2015, according to the Centers for Disease Control and Prevention.
Surgical and medical abortions reported to the CDC dropped by 24%, going from almost 843,000 in 2006 to 638,000 in 2015, with declines occurring every year, Tara C. Jatlaoui, MD, and her associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
Over that same time period, the abortion rate fell from 15.9 per 1,000 women aged 15-44 years to 11.8 per 1,000 – a decline of 26%. Abortion ratio – the number of abortions per 1,000 live births within a given population – declined by 19%, dropping from 233 abortions per 1,000 live births in 2006 to 188 abortions per 1,000 live births in 2015, the investigators reported. The findings were based on data from 49 areas that continuously reported over the study period (excludes California, Maryland, and New Hampshire but includes the District of Columbia and New York City).
Abortion rates were highest for women aged 20-29 years for the study period, and this age group accounted for the largest share among the 44 reporting areas that provided data by maternal age each year. Those under age 15 years had the largest drops by age in total number of abortions (40%) and abortion rate (58%) but had the highest, by far, abortion ratio for each year of the study (700 per 1,000 live births in that age group in 2015. The abortion ratio for 15- to 19-year-olds was 289 per 1,000 live births).
The percentage of abortions performed before 14 weeks’ gestation changed little, going from 91.5% in 2006 to 91% in 2015, but “a shift occurred toward earlier gestational ages,” they noted. The percentage of abortions performed before or at 8 weeks increased 3% and those done at 9-13 weeks dropped almost 9% among the 33 areas that reported gestational age every year. Abortions done after 13 weeks gestation represented 9% of all abortions during the study period, with an increase of 7% occurring from 2006 to 2015, Dr. Jatlaoui and her associates said.
Removing barriers such as cost, “insufficient provider reimbursement and training, inadequate client-centered counseling, lack of youth-friendly services, and low client awareness ... might help improve contraceptive use, potentially reducing the number of unintended pregnancies and the number of abortions performed in the United States,” the researchers wrote.
SOURCE: Jatlaoui TC et al. MMWR Surveill Summ. 2018 Nov 23(13):1-45.
FROM MMWR SURVEILLANCE SUMMARIES
FDA approves emapalumab for primary HLH
The (HLH).

Emapalumab, an interferon gamma-blocking antibody, is approved to treat patients of all ages (newborn and older) with primary HLH who have refractory, recurrent, or progressive disease or who cannot tolerate conventional HLH therapy. Emapalumab is the first treatment to be FDA approved for primary HLH, and it is expected to be available in the United States in the first quarter of 2019. The FDA previously granted emapalumab priority review, breakthrough therapy designation, orphan drug designation, and rare pediatric disease designation. The FDA’s approval of emapalumab is based on results from a phase 2/3 trial (NCT01818492).
The trial included 34 patients, 27 of whom had refractory, recurrent, or progressive disease or could not tolerate conventional HLH therapy. Patients received emapalumab in combination with dexamethasone. At the end of treatment, 63% (17/27) of patients had achieved a response, which was defined as complete response (n = 7), partial response (n=8), or HLH improvement (n = 2). A total of 70% (n=19) of patients went on to hematopoietic stem cell transplant. The most common adverse events were infections (56%), hypertension (41%), infusion-related reactions (27%), and pyrexia (24%).
Results also are scheduled to be presented at the 2018 annual meeting of the American Society of Hematology in the late-breaker abstract session (Abstract LBA-6).
Emapalumab was developed by Novimmune SA. Sobi acquired global rights to the drug in August 2018.
The (HLH).
Emapalumab, an interferon gamma-blocking antibody, is approved to treat patients of all ages (newborn and older) with primary HLH who have refractory, recurrent, or progressive disease or who cannot tolerate conventional HLH therapy. Emapalumab is the first treatment to be FDA approved for primary HLH, and it is expected to be available in the United States in the first quarter of 2019. The FDA previously granted emapalumab priority review, breakthrough therapy designation, orphan drug designation, and rare pediatric disease designation. The FDA’s approval of emapalumab is based on results from a phase 2/3 trial (NCT01818492).
The trial included 34 patients, 27 of whom had refractory, recurrent, or progressive disease or could not tolerate conventional HLH therapy. Patients received emapalumab in combination with dexamethasone. At the end of treatment, 63% (17/27) of patients had achieved a response, which was defined as complete response (n = 7), partial response (n=8), or HLH improvement (n = 2). A total of 70% (n=19) of patients went on to hematopoietic stem cell transplant. The most common adverse events were infections (56%), hypertension (41%), infusion-related reactions (27%), and pyrexia (24%).
Results also are scheduled to be presented at the 2018 annual meeting of the American Society of Hematology in the late-breaker abstract session (Abstract LBA-6).
Emapalumab was developed by Novimmune SA. Sobi acquired global rights to the drug in August 2018.
The (HLH).
Emapalumab, an interferon gamma-blocking antibody, is approved to treat patients of all ages (newborn and older) with primary HLH who have refractory, recurrent, or progressive disease or who cannot tolerate conventional HLH therapy. Emapalumab is the first treatment to be FDA approved for primary HLH, and it is expected to be available in the United States in the first quarter of 2019. The FDA previously granted emapalumab priority review, breakthrough therapy designation, orphan drug designation, and rare pediatric disease designation. The FDA’s approval of emapalumab is based on results from a phase 2/3 trial (NCT01818492).
The trial included 34 patients, 27 of whom had refractory, recurrent, or progressive disease or could not tolerate conventional HLH therapy. Patients received emapalumab in combination with dexamethasone. At the end of treatment, 63% (17/27) of patients had achieved a response, which was defined as complete response (n = 7), partial response (n=8), or HLH improvement (n = 2). A total of 70% (n=19) of patients went on to hematopoietic stem cell transplant. The most common adverse events were infections (56%), hypertension (41%), infusion-related reactions (27%), and pyrexia (24%).
Results also are scheduled to be presented at the 2018 annual meeting of the American Society of Hematology in the late-breaker abstract session (Abstract LBA-6).
Emapalumab was developed by Novimmune SA. Sobi acquired global rights to the drug in August 2018.
FDA expands approval of brentuximab vedotin to PTCL
The , marking the first FDA approval of a treatment for newly-diagnosed PTCL.
The drug, which is marketed by Seattle Genetics as Adcetris, is a monoclonal antibody that binds to CD30 protein found on some cancer cells.
It was previously approved for adult patients with untreated stage III or IV classical Hodgkin lymphoma (cHL), cHL after relapse, cHL after stem cell transplant in patients at high risk for relapse or progression, systemic anaplastic large cell lymphoma (ALCL) after other treatments fail, and primary cutaneous ALCL or CD30-expressing mycosis fungoides after other treatments fail.
The expanded approval, which followed the granting of Priority Review and Breakthrough Therapy designations for the supplemental Biologic License Application, was made using the FDA’s new Real-Time Oncology Review pilot program (RTOR). This program allows for data review and communication with a sponsor prior to official application submission with the goal of speeding up the review process.
The brentuximab vedotin approval now extends to previously untreated systemic ALCL and other CD30-expressing PTCLs in combination with chemotherapy.
Approval was based on the ECHELON-2 clinical trial involving 452 patients, which demonstrated improved progression-free survival (PFS) in patients with certain types of PTCL who were treated first-line with either brentuximab vedotin plus chemotherapy with cyclophosphamide, doxorubicin, prednisone (CHP), or standard chemotherapy with CHP and vincristine (CHOP). Median PFS was 48 months vs. 21 months in the groups, respectively (hazard ratio, 0.71).

The FDA advises health care providers to “monitor patients for infusion reactions, life-threatening allergic reactions (anaphylaxis), neuropathy, fever, gastrointestinal complications, and infections,” according to a press release announcing the approval, which also states that patients should be monitored for tumor lysis syndrome, serious skin reactions, pulmonary toxicity, and hepatotoxicity.
The drug may cause harm to a developing fetus or newborn and should not be used in women who are pregnant or breastfeeding. A Boxed Warning regarding risk of progressive multifocal leukoencephalopathy is also included in the prescribing information.
The current standard of care for initial treatment of PTCL is multiagent chemotherapy – a treatment that “has not significantly changed in decades and is too often unsuccessful in leading to long-term remissions, underscoring the need for new treatments, ” Steven Horwitz, MD, of Memorial Sloan Kettering Cancer Center, New York, said in a statement issued by Seattle Genetics.
“With this approval, clinicians have the opportunity to transform the way newly diagnosed CD30-expressing PTCL patients are treated,” Dr. Horwitz said.
The ECHELON-2 data will be presented at the American Society of Hematology annual meeting in San Diego on Monday, Dec. 3, 2018.
The , marking the first FDA approval of a treatment for newly-diagnosed PTCL.
The drug, which is marketed by Seattle Genetics as Adcetris, is a monoclonal antibody that binds to CD30 protein found on some cancer cells.
It was previously approved for adult patients with untreated stage III or IV classical Hodgkin lymphoma (cHL), cHL after relapse, cHL after stem cell transplant in patients at high risk for relapse or progression, systemic anaplastic large cell lymphoma (ALCL) after other treatments fail, and primary cutaneous ALCL or CD30-expressing mycosis fungoides after other treatments fail.
The expanded approval, which followed the granting of Priority Review and Breakthrough Therapy designations for the supplemental Biologic License Application, was made using the FDA’s new Real-Time Oncology Review pilot program (RTOR). This program allows for data review and communication with a sponsor prior to official application submission with the goal of speeding up the review process.
The brentuximab vedotin approval now extends to previously untreated systemic ALCL and other CD30-expressing PTCLs in combination with chemotherapy.
Approval was based on the ECHELON-2 clinical trial involving 452 patients, which demonstrated improved progression-free survival (PFS) in patients with certain types of PTCL who were treated first-line with either brentuximab vedotin plus chemotherapy with cyclophosphamide, doxorubicin, prednisone (CHP), or standard chemotherapy with CHP and vincristine (CHOP). Median PFS was 48 months vs. 21 months in the groups, respectively (hazard ratio, 0.71).
The FDA advises health care providers to “monitor patients for infusion reactions, life-threatening allergic reactions (anaphylaxis), neuropathy, fever, gastrointestinal complications, and infections,” according to a press release announcing the approval, which also states that patients should be monitored for tumor lysis syndrome, serious skin reactions, pulmonary toxicity, and hepatotoxicity.
The drug may cause harm to a developing fetus or newborn and should not be used in women who are pregnant or breastfeeding. A Boxed Warning regarding risk of progressive multifocal leukoencephalopathy is also included in the prescribing information.
The current standard of care for initial treatment of PTCL is multiagent chemotherapy – a treatment that “has not significantly changed in decades and is too often unsuccessful in leading to long-term remissions, underscoring the need for new treatments, ” Steven Horwitz, MD, of Memorial Sloan Kettering Cancer Center, New York, said in a statement issued by Seattle Genetics.
“With this approval, clinicians have the opportunity to transform the way newly diagnosed CD30-expressing PTCL patients are treated,” Dr. Horwitz said.
The ECHELON-2 data will be presented at the American Society of Hematology annual meeting in San Diego on Monday, Dec. 3, 2018.
The , marking the first FDA approval of a treatment for newly-diagnosed PTCL.
The drug, which is marketed by Seattle Genetics as Adcetris, is a monoclonal antibody that binds to CD30 protein found on some cancer cells.
It was previously approved for adult patients with untreated stage III or IV classical Hodgkin lymphoma (cHL), cHL after relapse, cHL after stem cell transplant in patients at high risk for relapse or progression, systemic anaplastic large cell lymphoma (ALCL) after other treatments fail, and primary cutaneous ALCL or CD30-expressing mycosis fungoides after other treatments fail.
The expanded approval, which followed the granting of Priority Review and Breakthrough Therapy designations for the supplemental Biologic License Application, was made using the FDA’s new Real-Time Oncology Review pilot program (RTOR). This program allows for data review and communication with a sponsor prior to official application submission with the goal of speeding up the review process.
The brentuximab vedotin approval now extends to previously untreated systemic ALCL and other CD30-expressing PTCLs in combination with chemotherapy.
Approval was based on the ECHELON-2 clinical trial involving 452 patients, which demonstrated improved progression-free survival (PFS) in patients with certain types of PTCL who were treated first-line with either brentuximab vedotin plus chemotherapy with cyclophosphamide, doxorubicin, prednisone (CHP), or standard chemotherapy with CHP and vincristine (CHOP). Median PFS was 48 months vs. 21 months in the groups, respectively (hazard ratio, 0.71).
The FDA advises health care providers to “monitor patients for infusion reactions, life-threatening allergic reactions (anaphylaxis), neuropathy, fever, gastrointestinal complications, and infections,” according to a press release announcing the approval, which also states that patients should be monitored for tumor lysis syndrome, serious skin reactions, pulmonary toxicity, and hepatotoxicity.
The drug may cause harm to a developing fetus or newborn and should not be used in women who are pregnant or breastfeeding. A Boxed Warning regarding risk of progressive multifocal leukoencephalopathy is also included in the prescribing information.
The current standard of care for initial treatment of PTCL is multiagent chemotherapy – a treatment that “has not significantly changed in decades and is too often unsuccessful in leading to long-term remissions, underscoring the need for new treatments, ” Steven Horwitz, MD, of Memorial Sloan Kettering Cancer Center, New York, said in a statement issued by Seattle Genetics.
“With this approval, clinicians have the opportunity to transform the way newly diagnosed CD30-expressing PTCL patients are treated,” Dr. Horwitz said.
The ECHELON-2 data will be presented at the American Society of Hematology annual meeting in San Diego on Monday, Dec. 3, 2018.
FDA clears new blood typing, screening instrument
The Food and Drug Administration has granted marketing clearance for the immunohematology instrument NEO Iris.
NEO Iris is a fully automated blood bank instrument designed for the mid- to high-volume laboratory, according to Immucor, the company marketing the device. Immucor says NEO Iris provides the highest type and screen throughput on the market – up to 60 types and screens per hour.
NEO Iris performs ABO/Rh D typing, weak D testing, donor confirmation, cytomegalovirus screening, immunoglobulin G direct antiglobulin test and crossmatch, and antibody identification and screening.
The workflow management tool on Neo Iris has STAT priority and allows operators to run tests in any order at any time, according to Immucor.
The company says NEO Iris can hold up to 224 samples, and “modules can pipette, incubate, centrifuge, and read simultaneously.” NEO Iris integrates with Immucor’s data management software, ImmuLINK, to aggregate test results and produce reports with complete testing history.
The Food and Drug Administration has granted marketing clearance for the immunohematology instrument NEO Iris.
NEO Iris is a fully automated blood bank instrument designed for the mid- to high-volume laboratory, according to Immucor, the company marketing the device. Immucor says NEO Iris provides the highest type and screen throughput on the market – up to 60 types and screens per hour.
NEO Iris performs ABO/Rh D typing, weak D testing, donor confirmation, cytomegalovirus screening, immunoglobulin G direct antiglobulin test and crossmatch, and antibody identification and screening.
The workflow management tool on Neo Iris has STAT priority and allows operators to run tests in any order at any time, according to Immucor.
The company says NEO Iris can hold up to 224 samples, and “modules can pipette, incubate, centrifuge, and read simultaneously.” NEO Iris integrates with Immucor’s data management software, ImmuLINK, to aggregate test results and produce reports with complete testing history.
The Food and Drug Administration has granted marketing clearance for the immunohematology instrument NEO Iris.
NEO Iris is a fully automated blood bank instrument designed for the mid- to high-volume laboratory, according to Immucor, the company marketing the device. Immucor says NEO Iris provides the highest type and screen throughput on the market – up to 60 types and screens per hour.
NEO Iris performs ABO/Rh D typing, weak D testing, donor confirmation, cytomegalovirus screening, immunoglobulin G direct antiglobulin test and crossmatch, and antibody identification and screening.
The workflow management tool on Neo Iris has STAT priority and allows operators to run tests in any order at any time, according to Immucor.
The company says NEO Iris can hold up to 224 samples, and “modules can pipette, incubate, centrifuge, and read simultaneously.” NEO Iris integrates with Immucor’s data management software, ImmuLINK, to aggregate test results and produce reports with complete testing history.
FDA approves Yupelri for COPD maintenance therapy
The Food and Drug Administration has approved Yupelri (revefenacin) for maintenance therapy of patients with chronic obstructive pulmonary disease (COPD).

Revefenacin is a long-acting muscarinic antagonist aimed at improving the lung function of patients with COPD. Yupelri is an inhalation solution administered once daily through a standard jet nebulizer.
The most common adverse events associated with Yupelri are cough, nasopharyngitis, upper respiratory tract infection, headache, and back pain. Patients receiving other anticholinergic-containing drugs or OATP1B1 and OATP1B3 inhibitors should not receive Yupelri.
“Patients should also be alert for signs and symptoms of acute narrow-angle glaucoma [e.g., eye pain or discomfort, blurred vision, visual changes]. Patients should consult a healthcare professional immediately if any of these signs or symptoms develop,” the FDA said in the press release.
The expanded label for Yupelri can be found on the FDA website.
The Food and Drug Administration has approved Yupelri (revefenacin) for maintenance therapy of patients with chronic obstructive pulmonary disease (COPD).
Revefenacin is a long-acting muscarinic antagonist aimed at improving the lung function of patients with COPD. Yupelri is an inhalation solution administered once daily through a standard jet nebulizer.
The most common adverse events associated with Yupelri are cough, nasopharyngitis, upper respiratory tract infection, headache, and back pain. Patients receiving other anticholinergic-containing drugs or OATP1B1 and OATP1B3 inhibitors should not receive Yupelri.
“Patients should also be alert for signs and symptoms of acute narrow-angle glaucoma [e.g., eye pain or discomfort, blurred vision, visual changes]. Patients should consult a healthcare professional immediately if any of these signs or symptoms develop,” the FDA said in the press release.
The expanded label for Yupelri can be found on the FDA website.
The Food and Drug Administration has approved Yupelri (revefenacin) for maintenance therapy of patients with chronic obstructive pulmonary disease (COPD).
Revefenacin is a long-acting muscarinic antagonist aimed at improving the lung function of patients with COPD. Yupelri is an inhalation solution administered once daily through a standard jet nebulizer.
The most common adverse events associated with Yupelri are cough, nasopharyngitis, upper respiratory tract infection, headache, and back pain. Patients receiving other anticholinergic-containing drugs or OATP1B1 and OATP1B3 inhibitors should not receive Yupelri.
“Patients should also be alert for signs and symptoms of acute narrow-angle glaucoma [e.g., eye pain or discomfort, blurred vision, visual changes]. Patients should consult a healthcare professional immediately if any of these signs or symptoms develop,” the FDA said in the press release.
The expanded label for Yupelri can be found on the FDA website.
P-BCMA-101 gains FDA regenerative medicine designation
(MM), has received the regenerative medicine advanced therapy (RMAT) designation from the Food and Drug Administration.
P-BCMA-101 modifies patients’ T cells using a nonviral DNA modification system known as piggyBac. The modified T cells target cells expressing B-cell maturation antigen (BCMA), which is expressed on essentially all MM cells.
Early results from the phase 1 clinical trial of P-BCMA-101 were recently reported at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics, the company developing P-BCMA-101.
Initial results of the trial (NCT03288493) included data on 11 patients with heavily pretreated MM. Patients were a median age of 60, and 73% were high risk. They had a median of six prior therapies.
Patients received conditioning treatment with fludarabine and cyclophosphamide for 3 days prior to receiving P-BCMA-101. They then received one of three doses of CAR T cells – 51×106 (n=3), 152×106 (n=7), or 430×106 (n=1).
The investigators observed no dose-limiting toxicities. Adverse events included neutropenia in eight patients and thrombocytopenia in five.
One patient may have had cytokine release syndrome, but the condition resolved without drug intervention. And investigators observed no neurotoxicity.
Seven of ten patients evaluable for response by International Myeloma Working Group criteria achieved at least a partial response, including very good partial responses and stringent complete response.
The eleventh patient has oligosecretory disease and was only evaluable by PET, which indicated a near-complete response.
Poseida expects to have additional data to report by the end of the year, according to Dr. Ostertag. The study is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.RMAT designation is intended to expedite development and review of regenerative medicines that are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition.
Preliminary evidence must indicate that the therapy has the potential to address unmet medical needs for the disease or condition. RMAT designation includes all the benefits of fast track and breakthrough therapy designations, including early interactions with the FDA.
(MM), has received the regenerative medicine advanced therapy (RMAT) designation from the Food and Drug Administration.
P-BCMA-101 modifies patients’ T cells using a nonviral DNA modification system known as piggyBac. The modified T cells target cells expressing B-cell maturation antigen (BCMA), which is expressed on essentially all MM cells.
Early results from the phase 1 clinical trial of P-BCMA-101 were recently reported at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics, the company developing P-BCMA-101.
Initial results of the trial (NCT03288493) included data on 11 patients with heavily pretreated MM. Patients were a median age of 60, and 73% were high risk. They had a median of six prior therapies.
Patients received conditioning treatment with fludarabine and cyclophosphamide for 3 days prior to receiving P-BCMA-101. They then received one of three doses of CAR T cells – 51×106 (n=3), 152×106 (n=7), or 430×106 (n=1).
The investigators observed no dose-limiting toxicities. Adverse events included neutropenia in eight patients and thrombocytopenia in five.
One patient may have had cytokine release syndrome, but the condition resolved without drug intervention. And investigators observed no neurotoxicity.
Seven of ten patients evaluable for response by International Myeloma Working Group criteria achieved at least a partial response, including very good partial responses and stringent complete response.
The eleventh patient has oligosecretory disease and was only evaluable by PET, which indicated a near-complete response.
Poseida expects to have additional data to report by the end of the year, according to Dr. Ostertag. The study is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.RMAT designation is intended to expedite development and review of regenerative medicines that are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition.
Preliminary evidence must indicate that the therapy has the potential to address unmet medical needs for the disease or condition. RMAT designation includes all the benefits of fast track and breakthrough therapy designations, including early interactions with the FDA.
(MM), has received the regenerative medicine advanced therapy (RMAT) designation from the Food and Drug Administration.
P-BCMA-101 modifies patients’ T cells using a nonviral DNA modification system known as piggyBac. The modified T cells target cells expressing B-cell maturation antigen (BCMA), which is expressed on essentially all MM cells.
Early results from the phase 1 clinical trial of P-BCMA-101 were recently reported at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics, the company developing P-BCMA-101.
Initial results of the trial (NCT03288493) included data on 11 patients with heavily pretreated MM. Patients were a median age of 60, and 73% were high risk. They had a median of six prior therapies.
Patients received conditioning treatment with fludarabine and cyclophosphamide for 3 days prior to receiving P-BCMA-101. They then received one of three doses of CAR T cells – 51×106 (n=3), 152×106 (n=7), or 430×106 (n=1).
The investigators observed no dose-limiting toxicities. Adverse events included neutropenia in eight patients and thrombocytopenia in five.
One patient may have had cytokine release syndrome, but the condition resolved without drug intervention. And investigators observed no neurotoxicity.
Seven of ten patients evaluable for response by International Myeloma Working Group criteria achieved at least a partial response, including very good partial responses and stringent complete response.
The eleventh patient has oligosecretory disease and was only evaluable by PET, which indicated a near-complete response.
Poseida expects to have additional data to report by the end of the year, according to Dr. Ostertag. The study is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.RMAT designation is intended to expedite development and review of regenerative medicines that are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition.
Preliminary evidence must indicate that the therapy has the potential to address unmet medical needs for the disease or condition. RMAT designation includes all the benefits of fast track and breakthrough therapy designations, including early interactions with the FDA.
FDA authorizes emergency use of rapid fingerstick test for Ebola
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
FDA puts selinexor on fast track for DLBCL
The Food and Drug Administration has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and who are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor (CAR) T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (Abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export compound being developed by Karyopharm Therapeutics.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs. Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL,” Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm, said in a statement.
In October, the FDA accepted an NDA for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
The Food and Drug Administration has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and who are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor (CAR) T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (Abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export compound being developed by Karyopharm Therapeutics.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs. Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL,” Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm, said in a statement.
In October, the FDA accepted an NDA for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
The Food and Drug Administration has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and who are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor (CAR) T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (Abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export compound being developed by Karyopharm Therapeutics.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs. Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL,” Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm, said in a statement.
In October, the FDA accepted an NDA for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
Portable hematology analyzer gets FDA nod
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
Cigarette smoking at lowest level ever
“This new all-time low in cigarette smoking among U.S. adults is a tremendous public health accomplishment, and it demonstrates the importance of continued proven strategies to reduce smoking,” CDC Director Robert Redfield said in a written statement.
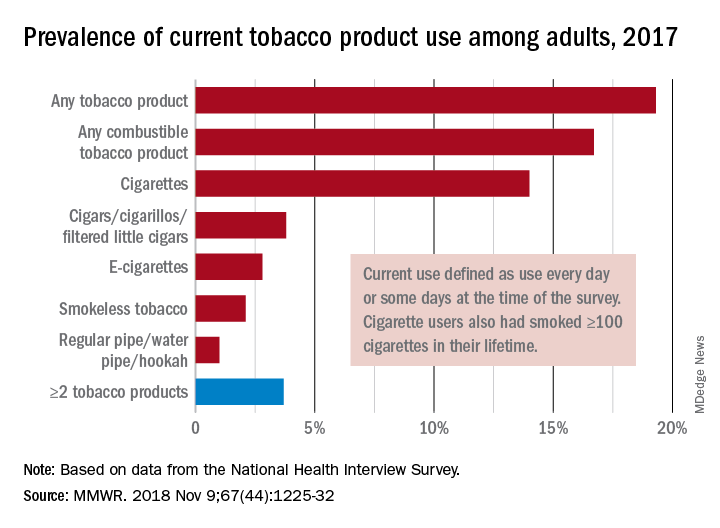
In 2017, 19.3% of adults aged 18 years and older – approximately 47.4 million Americans – reported current use of some type of tobacco product, and current use of combustible tobacco was 16.7%, Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and her associates reported in the Morbidity and Mortality Weekly Report. Current use was defined as use every day or some days, with an added requirement of at least 100 cigarettes in a lifetime added for cigarette smokers.
Data from the National Health Interview Survey showed that from 2016 to 2017, current use declined for any tobacco product, any combustible tobacco product, cigarettes, smokeless tobacco, and the combination of two or more tobacco products. The most common combination in 2017 was cigarettes and e-cigarettes, which was reported by 30.1% of the 9 million adults who used more than one product, Dr. Wang and her associates said.
Prevalence of current tobacco use was higher among men than women (24.8% vs. 14.2%), and adults aged 25-44 years (22.5%) had the highest level by age, followed by those aged 45-64 years (21.3%), 18-24 years (18.3%), and 65 years or older (11%). Use by race/ethnicity was highest among American Indian/Alaska Natives (29.8%), with the Midwest putting up the highest prevalence by region at 23.5%, they said.
“Although cigarette smoking among U.S. adults has declined considerably, tobacco products have evolved in recent years to include various combustible, noncombustible, and electronic products,” Dr. Wang and her associates wrote. “Implementation of evidence-based tobacco control interventions that address the diversity of tobacco products used by U.S. adults, in coordination with regulation of tobacco product manufacturing, marketing, and sales, can reduce tobacco-related disease and death in the United States.”
SOURCE: Wang TW et al. MMWR. 2018 Nov 9;67[44]:1225-32.
“This new all-time low in cigarette smoking among U.S. adults is a tremendous public health accomplishment, and it demonstrates the importance of continued proven strategies to reduce smoking,” CDC Director Robert Redfield said in a written statement.
In 2017, 19.3% of adults aged 18 years and older – approximately 47.4 million Americans – reported current use of some type of tobacco product, and current use of combustible tobacco was 16.7%, Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and her associates reported in the Morbidity and Mortality Weekly Report. Current use was defined as use every day or some days, with an added requirement of at least 100 cigarettes in a lifetime added for cigarette smokers.
Data from the National Health Interview Survey showed that from 2016 to 2017, current use declined for any tobacco product, any combustible tobacco product, cigarettes, smokeless tobacco, and the combination of two or more tobacco products. The most common combination in 2017 was cigarettes and e-cigarettes, which was reported by 30.1% of the 9 million adults who used more than one product, Dr. Wang and her associates said.
Prevalence of current tobacco use was higher among men than women (24.8% vs. 14.2%), and adults aged 25-44 years (22.5%) had the highest level by age, followed by those aged 45-64 years (21.3%), 18-24 years (18.3%), and 65 years or older (11%). Use by race/ethnicity was highest among American Indian/Alaska Natives (29.8%), with the Midwest putting up the highest prevalence by region at 23.5%, they said.
“Although cigarette smoking among U.S. adults has declined considerably, tobacco products have evolved in recent years to include various combustible, noncombustible, and electronic products,” Dr. Wang and her associates wrote. “Implementation of evidence-based tobacco control interventions that address the diversity of tobacco products used by U.S. adults, in coordination with regulation of tobacco product manufacturing, marketing, and sales, can reduce tobacco-related disease and death in the United States.”
SOURCE: Wang TW et al. MMWR. 2018 Nov 9;67[44]:1225-32.
“This new all-time low in cigarette smoking among U.S. adults is a tremendous public health accomplishment, and it demonstrates the importance of continued proven strategies to reduce smoking,” CDC Director Robert Redfield said in a written statement.
In 2017, 19.3% of adults aged 18 years and older – approximately 47.4 million Americans – reported current use of some type of tobacco product, and current use of combustible tobacco was 16.7%, Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and her associates reported in the Morbidity and Mortality Weekly Report. Current use was defined as use every day or some days, with an added requirement of at least 100 cigarettes in a lifetime added for cigarette smokers.
Data from the National Health Interview Survey showed that from 2016 to 2017, current use declined for any tobacco product, any combustible tobacco product, cigarettes, smokeless tobacco, and the combination of two or more tobacco products. The most common combination in 2017 was cigarettes and e-cigarettes, which was reported by 30.1% of the 9 million adults who used more than one product, Dr. Wang and her associates said.
Prevalence of current tobacco use was higher among men than women (24.8% vs. 14.2%), and adults aged 25-44 years (22.5%) had the highest level by age, followed by those aged 45-64 years (21.3%), 18-24 years (18.3%), and 65 years or older (11%). Use by race/ethnicity was highest among American Indian/Alaska Natives (29.8%), with the Midwest putting up the highest prevalence by region at 23.5%, they said.
“Although cigarette smoking among U.S. adults has declined considerably, tobacco products have evolved in recent years to include various combustible, noncombustible, and electronic products,” Dr. Wang and her associates wrote. “Implementation of evidence-based tobacco control interventions that address the diversity of tobacco products used by U.S. adults, in coordination with regulation of tobacco product manufacturing, marketing, and sales, can reduce tobacco-related disease and death in the United States.”
SOURCE: Wang TW et al. MMWR. 2018 Nov 9;67[44]:1225-32.
FROM MMWR