User login
2018-2019 flu season starts in earnest
National flu activity moved solidly into above-average territory during the week ending Dec. 15, as Colorado and Georgia took the lead with the highest activity levels in the country, according to the Centers for Disease Control and Prevention.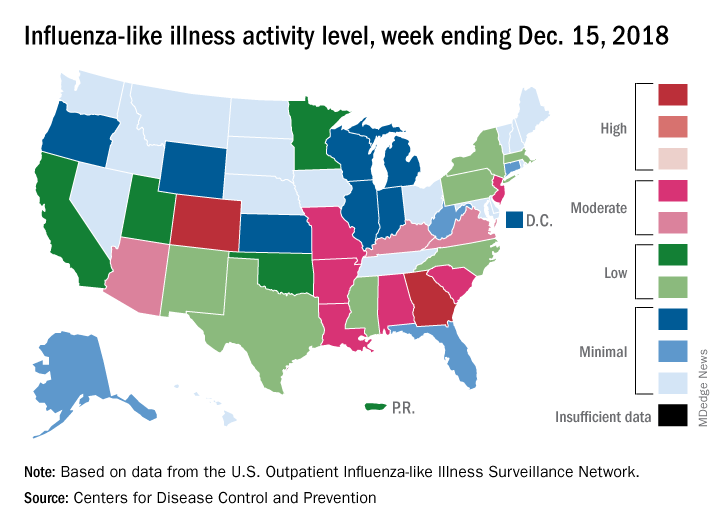
The proportion of outpatient visits for influenza-like illness (ILI) was 2.7% for the week, which was up from 2.3% the previous week and above the national baseline of 2.2%, the CDC reported. ILI is defined “as fever (temperature of 100°F [37.8°C] or greater) and cough and/or sore throat.”
Colorado and Georgia both reported ILI activity of 10 on the CDC’s 1-10 scale, making them the only states in the “high” range (8-10). Nine states and New York City had activity levels in the “moderate” range (6-7), Puerto Rico and 11 states were in the “low” range (4-5), and 28 states and the District of Columbia were in the “minimal” range (1-3), the CDC said.
During the comparable period of last year’s high-severity flu season, which ultimately resulted in 900,000 flu-related hospitalizations and 80,000 deaths (185 pediatric), nine states were already at level 10. For the 2018-2019 season so far, there have been seven ILI-related pediatric deaths, CDC data show.
National flu activity moved solidly into above-average territory during the week ending Dec. 15, as Colorado and Georgia took the lead with the highest activity levels in the country, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 2.7% for the week, which was up from 2.3% the previous week and above the national baseline of 2.2%, the CDC reported. ILI is defined “as fever (temperature of 100°F [37.8°C] or greater) and cough and/or sore throat.”
Colorado and Georgia both reported ILI activity of 10 on the CDC’s 1-10 scale, making them the only states in the “high” range (8-10). Nine states and New York City had activity levels in the “moderate” range (6-7), Puerto Rico and 11 states were in the “low” range (4-5), and 28 states and the District of Columbia were in the “minimal” range (1-3), the CDC said.
During the comparable period of last year’s high-severity flu season, which ultimately resulted in 900,000 flu-related hospitalizations and 80,000 deaths (185 pediatric), nine states were already at level 10. For the 2018-2019 season so far, there have been seven ILI-related pediatric deaths, CDC data show.
National flu activity moved solidly into above-average territory during the week ending Dec. 15, as Colorado and Georgia took the lead with the highest activity levels in the country, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 2.7% for the week, which was up from 2.3% the previous week and above the national baseline of 2.2%, the CDC reported. ILI is defined “as fever (temperature of 100°F [37.8°C] or greater) and cough and/or sore throat.”
Colorado and Georgia both reported ILI activity of 10 on the CDC’s 1-10 scale, making them the only states in the “high” range (8-10). Nine states and New York City had activity levels in the “moderate” range (6-7), Puerto Rico and 11 states were in the “low” range (4-5), and 28 states and the District of Columbia were in the “minimal” range (1-3), the CDC said.
During the comparable period of last year’s high-severity flu season, which ultimately resulted in 900,000 flu-related hospitalizations and 80,000 deaths (185 pediatric), nine states were already at level 10. For the 2018-2019 season so far, there have been seven ILI-related pediatric deaths, CDC data show.
Duodenoscopes contain more bacteria than expected
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.

“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
The AGA Center for GI Innovation and Technology supports innovation and the development of new technology in gastroenterology, hepatology, nutrition and obesity by guiding medical device and therapeutics innovators through the technology development and adoption process. Learn more at www.gastro.org/CGIT
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
The AGA Center for GI Innovation and Technology supports innovation and the development of new technology in gastroenterology, hepatology, nutrition and obesity by guiding medical device and therapeutics innovators through the technology development and adoption process. Learn more at www.gastro.org/CGIT
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
The AGA Center for GI Innovation and Technology supports innovation and the development of new technology in gastroenterology, hepatology, nutrition and obesity by guiding medical device and therapeutics innovators through the technology development and adoption process. Learn more at www.gastro.org/CGIT
2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018

CDC: Acute flaccid myelitis on the decline for 2018
, according to the Centers for Disease Control and Prevention.
![]()
Through Nov. 30, 134 cases of AFM in 33 states have been confirmed out of the 299 reported to the CDC. That represents “an increase of 18 confirmed cases from the previous week, but most of the latest confirmed AFM cases occurred in September and October,” the CDC reported Dec. 3.
There has been a pattern of increased AFM cases every other year for the previous 4 years: 120 cases in 2014, 22 cases in 2015, 149 cases in 2016, and 33 cases in 2017. “Most cases are reported between August and October, and a marked reduction in cases is seen in November. That pattern appears to be repeating in 2018 because states have reported fewer [persons under investigation] over the past couple of weeks. CDC expects this decline to continue,” the statement said.
The 16 confirmed cases in Texas are the most for any state this year, followed by Colorado with 15; Ohio with 10; and Illinois, New Jersey, and Washington with 9 each. California and Florida have not had any confirmed cases as of Nov. 30. Since 2014, over 90% of all confirmed AFM cases have occurred in children, the CDC noted.
More information on AFM is available at a CDC website for health care professionals.
, according to the Centers for Disease Control and Prevention.
![]()
Through Nov. 30, 134 cases of AFM in 33 states have been confirmed out of the 299 reported to the CDC. That represents “an increase of 18 confirmed cases from the previous week, but most of the latest confirmed AFM cases occurred in September and October,” the CDC reported Dec. 3.
There has been a pattern of increased AFM cases every other year for the previous 4 years: 120 cases in 2014, 22 cases in 2015, 149 cases in 2016, and 33 cases in 2017. “Most cases are reported between August and October, and a marked reduction in cases is seen in November. That pattern appears to be repeating in 2018 because states have reported fewer [persons under investigation] over the past couple of weeks. CDC expects this decline to continue,” the statement said.
The 16 confirmed cases in Texas are the most for any state this year, followed by Colorado with 15; Ohio with 10; and Illinois, New Jersey, and Washington with 9 each. California and Florida have not had any confirmed cases as of Nov. 30. Since 2014, over 90% of all confirmed AFM cases have occurred in children, the CDC noted.
More information on AFM is available at a CDC website for health care professionals.
, according to the Centers for Disease Control and Prevention.
![]()
Through Nov. 30, 134 cases of AFM in 33 states have been confirmed out of the 299 reported to the CDC. That represents “an increase of 18 confirmed cases from the previous week, but most of the latest confirmed AFM cases occurred in September and October,” the CDC reported Dec. 3.
There has been a pattern of increased AFM cases every other year for the previous 4 years: 120 cases in 2014, 22 cases in 2015, 149 cases in 2016, and 33 cases in 2017. “Most cases are reported between August and October, and a marked reduction in cases is seen in November. That pattern appears to be repeating in 2018 because states have reported fewer [persons under investigation] over the past couple of weeks. CDC expects this decline to continue,” the statement said.
The 16 confirmed cases in Texas are the most for any state this year, followed by Colorado with 15; Ohio with 10; and Illinois, New Jersey, and Washington with 9 each. California and Florida have not had any confirmed cases as of Nov. 30. Since 2014, over 90% of all confirmed AFM cases have occurred in children, the CDC noted.
More information on AFM is available at a CDC website for health care professionals.
FDA warns of serious side effect of AML treatment
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
Infant mortality generally unchanged in 2016
Infant mortality in the United Sates dropped very slightly from 2015 to 2016 and has not changed significantly since 2011, according to the National Center for Health Statistics.
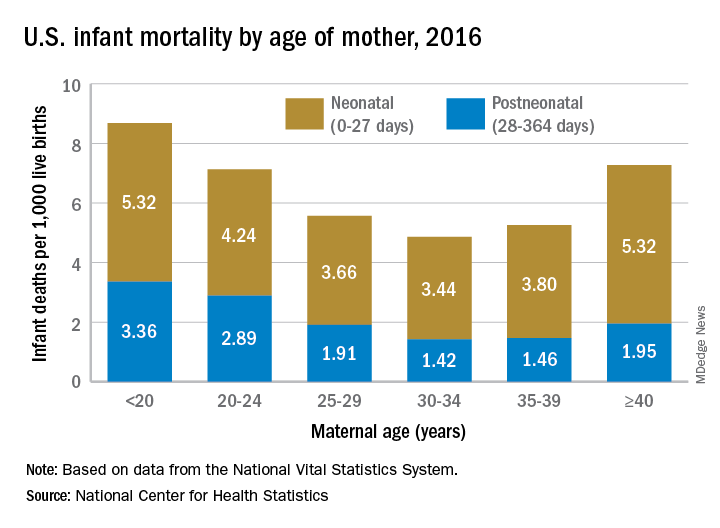
Overall infant mortality was 5.87 per 1,000 live births in 2016, which was not significantly less than the 2015 rate of 5.90 per 1,000 or the rate of 6.07 per 1,000 recorded in 2011, the NCHS said in a recent Data Brief. The rate for 2016 works out to 3.88 per 1,000 for the neonatal period (0-27 days) and 1.99 per 1,000 during the postneonatal period (28-364 days).
The rate was lowest for mothers aged 30-34 years (4.86 per 1,000) and highest for those under 20 years (8.69). All overall rates by maternal age were significantly different from each other, except for those of mothers aged 20-24 years (7.13) and those aged 40 years and over (7.27). Neonatal mortality was highest for the under-20 group and the 40-and-over group at 5.32 per 1,000, with the difference between them coming during the postneonatal period: 3.36 for those under 20 and 1.95 for the 40-and-overs, the NCHS investigators reported based on data from the National Vital Statistics System.
The leading cause of death during the neonatal period in 2016 was low birth weight at 98 per 1,000 live births, with congenital malformations second at 86 per 1,000. The leading cause of death in the postneonatal period was congenital malformations at 36 per 1,000, followed by sudden infant death syndrome (35 per 1,000), unintentional injuries (27 per 1,000), diseases of the circulatory system (9 per 1,000), and homicide (6 per 1,000), they added.
Infant mortality in the United Sates dropped very slightly from 2015 to 2016 and has not changed significantly since 2011, according to the National Center for Health Statistics.
Overall infant mortality was 5.87 per 1,000 live births in 2016, which was not significantly less than the 2015 rate of 5.90 per 1,000 or the rate of 6.07 per 1,000 recorded in 2011, the NCHS said in a recent Data Brief. The rate for 2016 works out to 3.88 per 1,000 for the neonatal period (0-27 days) and 1.99 per 1,000 during the postneonatal period (28-364 days).
The rate was lowest for mothers aged 30-34 years (4.86 per 1,000) and highest for those under 20 years (8.69). All overall rates by maternal age were significantly different from each other, except for those of mothers aged 20-24 years (7.13) and those aged 40 years and over (7.27). Neonatal mortality was highest for the under-20 group and the 40-and-over group at 5.32 per 1,000, with the difference between them coming during the postneonatal period: 3.36 for those under 20 and 1.95 for the 40-and-overs, the NCHS investigators reported based on data from the National Vital Statistics System.
The leading cause of death during the neonatal period in 2016 was low birth weight at 98 per 1,000 live births, with congenital malformations second at 86 per 1,000. The leading cause of death in the postneonatal period was congenital malformations at 36 per 1,000, followed by sudden infant death syndrome (35 per 1,000), unintentional injuries (27 per 1,000), diseases of the circulatory system (9 per 1,000), and homicide (6 per 1,000), they added.
Infant mortality in the United Sates dropped very slightly from 2015 to 2016 and has not changed significantly since 2011, according to the National Center for Health Statistics.
Overall infant mortality was 5.87 per 1,000 live births in 2016, which was not significantly less than the 2015 rate of 5.90 per 1,000 or the rate of 6.07 per 1,000 recorded in 2011, the NCHS said in a recent Data Brief. The rate for 2016 works out to 3.88 per 1,000 for the neonatal period (0-27 days) and 1.99 per 1,000 during the postneonatal period (28-364 days).
The rate was lowest for mothers aged 30-34 years (4.86 per 1,000) and highest for those under 20 years (8.69). All overall rates by maternal age were significantly different from each other, except for those of mothers aged 20-24 years (7.13) and those aged 40 years and over (7.27). Neonatal mortality was highest for the under-20 group and the 40-and-over group at 5.32 per 1,000, with the difference between them coming during the postneonatal period: 3.36 for those under 20 and 1.95 for the 40-and-overs, the NCHS investigators reported based on data from the National Vital Statistics System.
The leading cause of death during the neonatal period in 2016 was low birth weight at 98 per 1,000 live births, with congenital malformations second at 86 per 1,000. The leading cause of death in the postneonatal period was congenital malformations at 36 per 1,000, followed by sudden infant death syndrome (35 per 1,000), unintentional injuries (27 per 1,000), diseases of the circulatory system (9 per 1,000), and homicide (6 per 1,000), they added.
FDA approves gilteritinib for AML with FLT3 mutation
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
FDA approves rituximab biosimilar for lymphoma
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
Temixys plus other antiretrovirals approved for HIV-1
The Food and Drug Administration has approved the combination of lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) known as Temixys for treatment of HIV-1 when used with other antiretrovirals. The approval is for adult and pediatric patients with HIV-1 who weigh at least 35 kg.
The approval is based on data through 144 weeks in a double-blind, active-controlled, multicenter trial in 600 antiretroviral-naive patients. The trial compared TDF/3TC plus efavirenz (EFV) with 3TC/EFV plus stavudine (d4T). The results showed similar responses at 144 weeks between both groups: 62% of patients taking TDF/3TC/EFV and 58% of patients taking d4T/3TC/EFV achieved and maintained fewer than 50 copies/mL of HIV-1 RNA.
The most common adverse events include headache, pain, depression, rash, and diarrhea. Prior to initiating treatment, patients should be tested for hepatitis B virus because there have been reports of 3TC-resistant strains of hepatitis B virus associated with treatment of HIV-1 with 3TC-containing regimens in coinfected patients. Patients should also be tested for estimated creatinine clearance, urine glucose, and urine protein because TDF/3TC is not recommended for patients with renal impairment.
The full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved the combination of lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) known as Temixys for treatment of HIV-1 when used with other antiretrovirals. The approval is for adult and pediatric patients with HIV-1 who weigh at least 35 kg.
The approval is based on data through 144 weeks in a double-blind, active-controlled, multicenter trial in 600 antiretroviral-naive patients. The trial compared TDF/3TC plus efavirenz (EFV) with 3TC/EFV plus stavudine (d4T). The results showed similar responses at 144 weeks between both groups: 62% of patients taking TDF/3TC/EFV and 58% of patients taking d4T/3TC/EFV achieved and maintained fewer than 50 copies/mL of HIV-1 RNA.
The most common adverse events include headache, pain, depression, rash, and diarrhea. Prior to initiating treatment, patients should be tested for hepatitis B virus because there have been reports of 3TC-resistant strains of hepatitis B virus associated with treatment of HIV-1 with 3TC-containing regimens in coinfected patients. Patients should also be tested for estimated creatinine clearance, urine glucose, and urine protein because TDF/3TC is not recommended for patients with renal impairment.
The full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved the combination of lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) known as Temixys for treatment of HIV-1 when used with other antiretrovirals. The approval is for adult and pediatric patients with HIV-1 who weigh at least 35 kg.
The approval is based on data through 144 weeks in a double-blind, active-controlled, multicenter trial in 600 antiretroviral-naive patients. The trial compared TDF/3TC plus efavirenz (EFV) with 3TC/EFV plus stavudine (d4T). The results showed similar responses at 144 weeks between both groups: 62% of patients taking TDF/3TC/EFV and 58% of patients taking d4T/3TC/EFV achieved and maintained fewer than 50 copies/mL of HIV-1 RNA.
The most common adverse events include headache, pain, depression, rash, and diarrhea. Prior to initiating treatment, patients should be tested for hepatitis B virus because there have been reports of 3TC-resistant strains of hepatitis B virus associated with treatment of HIV-1 with 3TC-containing regimens in coinfected patients. Patients should also be tested for estimated creatinine clearance, urine glucose, and urine protein because TDF/3TC is not recommended for patients with renal impairment.
The full prescribing information can be found on the FDA website.
Venetoclax gets accelerated approval in older AML patients
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.