User login
TB in 2017: Good news and bad news
according to the Centers for Disease Control and Prevention.
Those new lows – TB incidence of 2.8 per 100,000 persons and 9,093 new cases – continue a downward trend that started in 1993, but the current rate of decline is much lower than the threshold needed to eliminate TB by the year 2100, Rebekah J. Stewart and her associates at the CDC’s Division of Tuberculosis Elimination, Atlanta, wrote in the Morbidity and Mortality Weekly Report.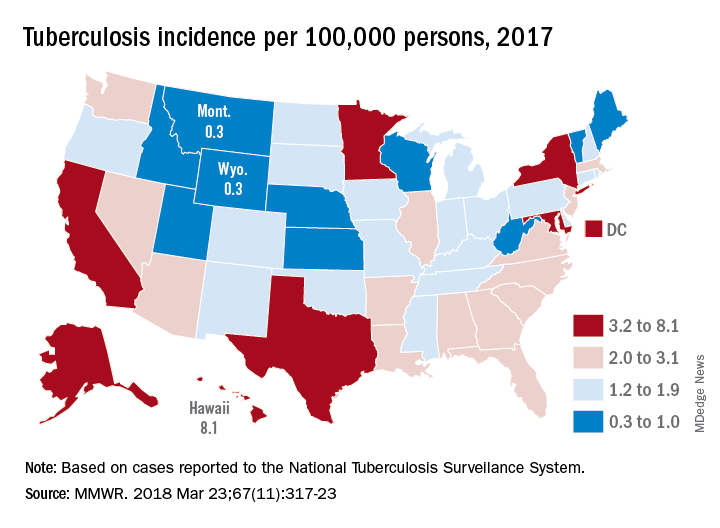
Geographically, at least, the states with populations at the highest risk are Hawaii, which had a TB incidence of 8.1 per 100,000 persons in 2017, and Alaska, with an incidence of 7.0 per 100,000. California and the District of Columbia were next, each with an incidence of 5.2. The states with the lowest rates were Montana and Wyoming at 0.3 per 100,000, the investigators reported, based on data from the National Tuberculosis Surveillance System as of Feb. 12, 2018.
Groups most affected by TB include persons housed in congregate settings – homeless shelters, long-term care facilities, and correctional facilities – and those from countries that have high TB prevalence. Overall incidence for non–U.S. born residents was 14.6 per 100,000 in 2017, compared with 1.0 for the native born, with large discrepancies seen between U.S. and non–U.S. born blacks (2.8 vs. 22.0), native Hawaiian/Pacific Islanders (6.5 vs. 21.0), and Asians (2.0 vs. 27.0), Ms. Stewart and her associates said.
“Increased support of global TB elimination efforts would help to reduce global … prevalence, thereby indirectly reducing the incidence of reactivation TB in the United States among non–U.S. born persons from higher-prevalence countries,” they wrote.
The issue of global action on TB was addressed by the Forum of International Respiratory Societies in a statement recognizing World TB Day (March 24). “TB is the world’s most common infectious disease killer, yet is identifiable, treatable and preventable; what is missing is the political will to dedicate the resources necessary to eradicate it, once and for all,” said Dean E. Schraufnagel, MD, the organization’s executive director.
SOURCE: Stewart RJ et al. MMWR 2018 Mar 23;67(11):317-23.
according to the Centers for Disease Control and Prevention.
Those new lows – TB incidence of 2.8 per 100,000 persons and 9,093 new cases – continue a downward trend that started in 1993, but the current rate of decline is much lower than the threshold needed to eliminate TB by the year 2100, Rebekah J. Stewart and her associates at the CDC’s Division of Tuberculosis Elimination, Atlanta, wrote in the Morbidity and Mortality Weekly Report.
Geographically, at least, the states with populations at the highest risk are Hawaii, which had a TB incidence of 8.1 per 100,000 persons in 2017, and Alaska, with an incidence of 7.0 per 100,000. California and the District of Columbia were next, each with an incidence of 5.2. The states with the lowest rates were Montana and Wyoming at 0.3 per 100,000, the investigators reported, based on data from the National Tuberculosis Surveillance System as of Feb. 12, 2018.
Groups most affected by TB include persons housed in congregate settings – homeless shelters, long-term care facilities, and correctional facilities – and those from countries that have high TB prevalence. Overall incidence for non–U.S. born residents was 14.6 per 100,000 in 2017, compared with 1.0 for the native born, with large discrepancies seen between U.S. and non–U.S. born blacks (2.8 vs. 22.0), native Hawaiian/Pacific Islanders (6.5 vs. 21.0), and Asians (2.0 vs. 27.0), Ms. Stewart and her associates said.
“Increased support of global TB elimination efforts would help to reduce global … prevalence, thereby indirectly reducing the incidence of reactivation TB in the United States among non–U.S. born persons from higher-prevalence countries,” they wrote.
The issue of global action on TB was addressed by the Forum of International Respiratory Societies in a statement recognizing World TB Day (March 24). “TB is the world’s most common infectious disease killer, yet is identifiable, treatable and preventable; what is missing is the political will to dedicate the resources necessary to eradicate it, once and for all,” said Dean E. Schraufnagel, MD, the organization’s executive director.
SOURCE: Stewart RJ et al. MMWR 2018 Mar 23;67(11):317-23.
according to the Centers for Disease Control and Prevention.
Those new lows – TB incidence of 2.8 per 100,000 persons and 9,093 new cases – continue a downward trend that started in 1993, but the current rate of decline is much lower than the threshold needed to eliminate TB by the year 2100, Rebekah J. Stewart and her associates at the CDC’s Division of Tuberculosis Elimination, Atlanta, wrote in the Morbidity and Mortality Weekly Report.
Geographically, at least, the states with populations at the highest risk are Hawaii, which had a TB incidence of 8.1 per 100,000 persons in 2017, and Alaska, with an incidence of 7.0 per 100,000. California and the District of Columbia were next, each with an incidence of 5.2. The states with the lowest rates were Montana and Wyoming at 0.3 per 100,000, the investigators reported, based on data from the National Tuberculosis Surveillance System as of Feb. 12, 2018.
Groups most affected by TB include persons housed in congregate settings – homeless shelters, long-term care facilities, and correctional facilities – and those from countries that have high TB prevalence. Overall incidence for non–U.S. born residents was 14.6 per 100,000 in 2017, compared with 1.0 for the native born, with large discrepancies seen between U.S. and non–U.S. born blacks (2.8 vs. 22.0), native Hawaiian/Pacific Islanders (6.5 vs. 21.0), and Asians (2.0 vs. 27.0), Ms. Stewart and her associates said.
“Increased support of global TB elimination efforts would help to reduce global … prevalence, thereby indirectly reducing the incidence of reactivation TB in the United States among non–U.S. born persons from higher-prevalence countries,” they wrote.
The issue of global action on TB was addressed by the Forum of International Respiratory Societies in a statement recognizing World TB Day (March 24). “TB is the world’s most common infectious disease killer, yet is identifiable, treatable and preventable; what is missing is the political will to dedicate the resources necessary to eradicate it, once and for all,” said Dean E. Schraufnagel, MD, the organization’s executive director.
SOURCE: Stewart RJ et al. MMWR 2018 Mar 23;67(11):317-23.
FROM MMWR
FDA approves certolizumab label update for pregnancy, breastfeeding
The manufacturer of certolizumab pegol, UCB, announced March 22 that the Food and Drug Administration approved a label update to the biologic that includes pharmacokinetic data showing negligible to low transfer of the biologic through the placenta and minimal mother-to-infant transfer from breast milk.

In the CRIB study, certolizumab levels were below the lower limit of quantification (defined as 0.032 mcg/mL) in 13 out of 15 infant blood samples at birth and in all samples at weeks 4 and 8. No anticertolizumab antibodies were detected in mothers, umbilical cords, or infants.
In the CRADLE study, 56% of 137 breast milk samples from 17 mothers had no measurable certolizumab, and the remaining samples showed minimal levels of the biologic. No serious adverse reactions were noted in the 17 infants in the study.
“It is well recognized that women with chronic inflammatory disease face uncertainty during motherhood given the lack of information on treatment during pregnancy and breastfeeding. Many women with chronic inflammatory disease discontinue their biologic treatment during pregnancy, often when they need disease control the most,” said CRADLE lead study author Megan E. B. Clowse, MD, of Duke University, Durham, N.C., in a press release issued by UCB. “These data for Cimzia provide important information to empower women and healthcare providers making decisions about treatment during pregnancy and breastfeeding.”
UCB said that limited data from an ongoing pregnancy registry regarding the use of certolizumab in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes.
The manufacturer of certolizumab pegol, UCB, announced March 22 that the Food and Drug Administration approved a label update to the biologic that includes pharmacokinetic data showing negligible to low transfer of the biologic through the placenta and minimal mother-to-infant transfer from breast milk.
In the CRIB study, certolizumab levels were below the lower limit of quantification (defined as 0.032 mcg/mL) in 13 out of 15 infant blood samples at birth and in all samples at weeks 4 and 8. No anticertolizumab antibodies were detected in mothers, umbilical cords, or infants.
In the CRADLE study, 56% of 137 breast milk samples from 17 mothers had no measurable certolizumab, and the remaining samples showed minimal levels of the biologic. No serious adverse reactions were noted in the 17 infants in the study.
“It is well recognized that women with chronic inflammatory disease face uncertainty during motherhood given the lack of information on treatment during pregnancy and breastfeeding. Many women with chronic inflammatory disease discontinue their biologic treatment during pregnancy, often when they need disease control the most,” said CRADLE lead study author Megan E. B. Clowse, MD, of Duke University, Durham, N.C., in a press release issued by UCB. “These data for Cimzia provide important information to empower women and healthcare providers making decisions about treatment during pregnancy and breastfeeding.”
UCB said that limited data from an ongoing pregnancy registry regarding the use of certolizumab in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes.
The manufacturer of certolizumab pegol, UCB, announced March 22 that the Food and Drug Administration approved a label update to the biologic that includes pharmacokinetic data showing negligible to low transfer of the biologic through the placenta and minimal mother-to-infant transfer from breast milk.
In the CRIB study, certolizumab levels were below the lower limit of quantification (defined as 0.032 mcg/mL) in 13 out of 15 infant blood samples at birth and in all samples at weeks 4 and 8. No anticertolizumab antibodies were detected in mothers, umbilical cords, or infants.
In the CRADLE study, 56% of 137 breast milk samples from 17 mothers had no measurable certolizumab, and the remaining samples showed minimal levels of the biologic. No serious adverse reactions were noted in the 17 infants in the study.
“It is well recognized that women with chronic inflammatory disease face uncertainty during motherhood given the lack of information on treatment during pregnancy and breastfeeding. Many women with chronic inflammatory disease discontinue their biologic treatment during pregnancy, often when they need disease control the most,” said CRADLE lead study author Megan E. B. Clowse, MD, of Duke University, Durham, N.C., in a press release issued by UCB. “These data for Cimzia provide important information to empower women and healthcare providers making decisions about treatment during pregnancy and breastfeeding.”
UCB said that limited data from an ongoing pregnancy registry regarding the use of certolizumab in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes.
FDA approves IL-23 antagonist for plaque psoriasis
in adults who are eligible for systemic therapy or phototherapy, according to a statement from Sun Pharma.
Tildrakizumab is administered at a dose of 100 mg, subcutaneously, at weeks 0 and 4, then every 12 weeks. Approval is based on data from two phase 3, identically designed clinical trials, reSURFACE1 and reSURFACE2. Both studies were multicenter, randomized, double-blind, and placebo controlled. In the studies, 926 patients received tildrakizumab (616 patients) or placebo (310 patients).
The effectiveness of tildrakizumab extended beyond 12 weeks, with 74% of patients achieving a PASI 75 at 28 weeks after three doses. This percentage grew to 84% at week 64 in patients who continued treatment. Similar results were observed with PGA scores, with 69% of patients who had a PGA score of 0 or 1 at 12 weeks maintaining that score at week 28.
Tildrakizumab has been associated with serious side effects, including serious allergic reactions including skin rash, swelling of the face and mouth, trouble breathing, and chest tightness. It may also increase patient susceptibility to infection. It is approved with a Medication Guide for patients, explaining the potential risks associated with treatment.
Tildrakizumab will be marketed as Ilumya.
Sun Pharma is working with the FDA on postapproval commitments, and once that has been completed, they will have a better idea of when it will become available, according to a spokesperson for the manufacturer. The cost is not yet available.
in adults who are eligible for systemic therapy or phototherapy, according to a statement from Sun Pharma.
Tildrakizumab is administered at a dose of 100 mg, subcutaneously, at weeks 0 and 4, then every 12 weeks. Approval is based on data from two phase 3, identically designed clinical trials, reSURFACE1 and reSURFACE2. Both studies were multicenter, randomized, double-blind, and placebo controlled. In the studies, 926 patients received tildrakizumab (616 patients) or placebo (310 patients).
The effectiveness of tildrakizumab extended beyond 12 weeks, with 74% of patients achieving a PASI 75 at 28 weeks after three doses. This percentage grew to 84% at week 64 in patients who continued treatment. Similar results were observed with PGA scores, with 69% of patients who had a PGA score of 0 or 1 at 12 weeks maintaining that score at week 28.
Tildrakizumab has been associated with serious side effects, including serious allergic reactions including skin rash, swelling of the face and mouth, trouble breathing, and chest tightness. It may also increase patient susceptibility to infection. It is approved with a Medication Guide for patients, explaining the potential risks associated with treatment.
Tildrakizumab will be marketed as Ilumya.
Sun Pharma is working with the FDA on postapproval commitments, and once that has been completed, they will have a better idea of when it will become available, according to a spokesperson for the manufacturer. The cost is not yet available.
in adults who are eligible for systemic therapy or phototherapy, according to a statement from Sun Pharma.
Tildrakizumab is administered at a dose of 100 mg, subcutaneously, at weeks 0 and 4, then every 12 weeks. Approval is based on data from two phase 3, identically designed clinical trials, reSURFACE1 and reSURFACE2. Both studies were multicenter, randomized, double-blind, and placebo controlled. In the studies, 926 patients received tildrakizumab (616 patients) or placebo (310 patients).
The effectiveness of tildrakizumab extended beyond 12 weeks, with 74% of patients achieving a PASI 75 at 28 weeks after three doses. This percentage grew to 84% at week 64 in patients who continued treatment. Similar results were observed with PGA scores, with 69% of patients who had a PGA score of 0 or 1 at 12 weeks maintaining that score at week 28.
Tildrakizumab has been associated with serious side effects, including serious allergic reactions including skin rash, swelling of the face and mouth, trouble breathing, and chest tightness. It may also increase patient susceptibility to infection. It is approved with a Medication Guide for patients, explaining the potential risks associated with treatment.
Tildrakizumab will be marketed as Ilumya.
Sun Pharma is working with the FDA on postapproval commitments, and once that has been completed, they will have a better idea of when it will become available, according to a spokesperson for the manufacturer. The cost is not yet available.
FDA updates breast implant–associated lymphoma cases, risk
(BIA-ALCL), including nine deaths.
This figure includes all medical device reports received by the agency between 2011 and September 2017. The FDA recently provided an update on ALCL linked to breast implants and an estimate of lifetime risk of developing ALCL.
Based on available medical literature, the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000, according to the update.
Of the 272 reports with data on surface type, 242 were textured implants and 30 were smooth implants. In addition, 413 reports include information on the implant fill type: 234 used silicone gel and 179 were saline filled.
“The FDA has been closely tracking the relationship between breast implants and a rare type of non-Hodgkin’s lymphoma since we first identified this possible association. We’ve been working to gather additional information to better characterize and quantify the risk so that patients and providers can have more informed discussions about breast implants,” said Binita Ashar, MD, director of the division of surgical devices in the FDA’s Center for Devices and Radiological Health. “As part of that effort, we are working to update and enhance the information we have on this association, including updating the total number of known cases of BIA-ALCL and the lifetime risk of developing BIA-ALCL as reported in medical literature.”
The possible association between breast implants and the development of anaplastic large cell lymphoma (ALCL) was first identified in 2011. At that time, there were not enough cases of to determine what factors increased a patient’s risk of developing the disease. As more information became available, the World Health Organization designated BIA-ALCL as a T-cell lymphoma that can develop following breast implants.
(BIA-ALCL), including nine deaths.
This figure includes all medical device reports received by the agency between 2011 and September 2017. The FDA recently provided an update on ALCL linked to breast implants and an estimate of lifetime risk of developing ALCL.
Based on available medical literature, the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000, according to the update.
Of the 272 reports with data on surface type, 242 were textured implants and 30 were smooth implants. In addition, 413 reports include information on the implant fill type: 234 used silicone gel and 179 were saline filled.
“The FDA has been closely tracking the relationship between breast implants and a rare type of non-Hodgkin’s lymphoma since we first identified this possible association. We’ve been working to gather additional information to better characterize and quantify the risk so that patients and providers can have more informed discussions about breast implants,” said Binita Ashar, MD, director of the division of surgical devices in the FDA’s Center for Devices and Radiological Health. “As part of that effort, we are working to update and enhance the information we have on this association, including updating the total number of known cases of BIA-ALCL and the lifetime risk of developing BIA-ALCL as reported in medical literature.”
The possible association between breast implants and the development of anaplastic large cell lymphoma (ALCL) was first identified in 2011. At that time, there were not enough cases of to determine what factors increased a patient’s risk of developing the disease. As more information became available, the World Health Organization designated BIA-ALCL as a T-cell lymphoma that can develop following breast implants.
(BIA-ALCL), including nine deaths.
This figure includes all medical device reports received by the agency between 2011 and September 2017. The FDA recently provided an update on ALCL linked to breast implants and an estimate of lifetime risk of developing ALCL.
Based on available medical literature, the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000, according to the update.
Of the 272 reports with data on surface type, 242 were textured implants and 30 were smooth implants. In addition, 413 reports include information on the implant fill type: 234 used silicone gel and 179 were saline filled.
“The FDA has been closely tracking the relationship between breast implants and a rare type of non-Hodgkin’s lymphoma since we first identified this possible association. We’ve been working to gather additional information to better characterize and quantify the risk so that patients and providers can have more informed discussions about breast implants,” said Binita Ashar, MD, director of the division of surgical devices in the FDA’s Center for Devices and Radiological Health. “As part of that effort, we are working to update and enhance the information we have on this association, including updating the total number of known cases of BIA-ALCL and the lifetime risk of developing BIA-ALCL as reported in medical literature.”
The possible association between breast implants and the development of anaplastic large cell lymphoma (ALCL) was first identified in 2011. At that time, there were not enough cases of to determine what factors increased a patient’s risk of developing the disease. As more information became available, the World Health Organization designated BIA-ALCL as a T-cell lymphoma that can develop following breast implants.
FDA wants data on role of flavored tobacco products in youth initiation
The Food and Drug Administration is seeking data on the role that flavors, including menthol, in tobacco products play in the initiation, use, and cessation of tobacco products, with an emphasis on how flavoring impacts young people.
“In the spirit of our commitment to preventing kids from using tobacco, we are taking a closer look at flavors in tobacco products to better understand their level of impact on youth initiation,” FDA Commissioner Scott Gottlieb, MD, said in statement. It is important “that we also explore how flavors, under a properly regulated framework that protects youth, may also be helping some currently addicted adult cigarette smokers switch to certain noncombustible forms of tobacco products.”
“Youth consistently report product flavoring as a leading reason for using tobacco products,” Dr. Gottlieb noted. “In fact, there is evidence indicating that youth tobacco users who reported their first tobacco was flavored had a higher prevalence of current tobacco product use, compared to youth whose product was not flavored.”
The advance notice calls for information across a number of areas, including the role of flavors other than tobacco in tobacco products; flavors and initiation and patterns of tobacco product use, particularly among youths and young adults; and flavors and cessation, dual-use, and relapse among current and former tobacco product users.
It also is seeking comment on whether standards should be set on tobacco flavoring, including whether there should a prohibition or restriction on flavors and to which types of products these standards should apply. The notice specifically asks about menthol and its role in cigarette initiation and whether limitations on menthol could lead to use of other tobacco products.
“Because almost 90% of adult smokers started smoking by the age of 18, it’s imperative we look at new ways we can ensure that kids don’t progress from experimentation to regular use,” Commissioner Gottlieb said.
The American Heart Association called the action “long overdue.”
“We encourage the FDA to quickly move beyond information gathering and develop a strong flavoring product standard,” CEO Nancy Brown said in a statement. “There is already clear evidence that flavored tobacco products, including menthol, harm the public health. To make it worse, fruit- and candy-flavored e-cigarettes, cigars, and other tobacco products are highly attractive to kids and make it more likely that they will take up this addiction.”
The action comes less than a week after FDA published an advance notice seeking information comments on reducing nicotine levels in cigarettes to help combat nicotine addiction.
The advance notice will be published March 21 in the Federal Register; comments will be accepted at www.regulations.gov for 90 days.
The Food and Drug Administration is seeking data on the role that flavors, including menthol, in tobacco products play in the initiation, use, and cessation of tobacco products, with an emphasis on how flavoring impacts young people.
“In the spirit of our commitment to preventing kids from using tobacco, we are taking a closer look at flavors in tobacco products to better understand their level of impact on youth initiation,” FDA Commissioner Scott Gottlieb, MD, said in statement. It is important “that we also explore how flavors, under a properly regulated framework that protects youth, may also be helping some currently addicted adult cigarette smokers switch to certain noncombustible forms of tobacco products.”
“Youth consistently report product flavoring as a leading reason for using tobacco products,” Dr. Gottlieb noted. “In fact, there is evidence indicating that youth tobacco users who reported their first tobacco was flavored had a higher prevalence of current tobacco product use, compared to youth whose product was not flavored.”
The advance notice calls for information across a number of areas, including the role of flavors other than tobacco in tobacco products; flavors and initiation and patterns of tobacco product use, particularly among youths and young adults; and flavors and cessation, dual-use, and relapse among current and former tobacco product users.
It also is seeking comment on whether standards should be set on tobacco flavoring, including whether there should a prohibition or restriction on flavors and to which types of products these standards should apply. The notice specifically asks about menthol and its role in cigarette initiation and whether limitations on menthol could lead to use of other tobacco products.
“Because almost 90% of adult smokers started smoking by the age of 18, it’s imperative we look at new ways we can ensure that kids don’t progress from experimentation to regular use,” Commissioner Gottlieb said.
The American Heart Association called the action “long overdue.”
“We encourage the FDA to quickly move beyond information gathering and develop a strong flavoring product standard,” CEO Nancy Brown said in a statement. “There is already clear evidence that flavored tobacco products, including menthol, harm the public health. To make it worse, fruit- and candy-flavored e-cigarettes, cigars, and other tobacco products are highly attractive to kids and make it more likely that they will take up this addiction.”
The action comes less than a week after FDA published an advance notice seeking information comments on reducing nicotine levels in cigarettes to help combat nicotine addiction.
The advance notice will be published March 21 in the Federal Register; comments will be accepted at www.regulations.gov for 90 days.
The Food and Drug Administration is seeking data on the role that flavors, including menthol, in tobacco products play in the initiation, use, and cessation of tobacco products, with an emphasis on how flavoring impacts young people.
“In the spirit of our commitment to preventing kids from using tobacco, we are taking a closer look at flavors in tobacco products to better understand their level of impact on youth initiation,” FDA Commissioner Scott Gottlieb, MD, said in statement. It is important “that we also explore how flavors, under a properly regulated framework that protects youth, may also be helping some currently addicted adult cigarette smokers switch to certain noncombustible forms of tobacco products.”
“Youth consistently report product flavoring as a leading reason for using tobacco products,” Dr. Gottlieb noted. “In fact, there is evidence indicating that youth tobacco users who reported their first tobacco was flavored had a higher prevalence of current tobacco product use, compared to youth whose product was not flavored.”
The advance notice calls for information across a number of areas, including the role of flavors other than tobacco in tobacco products; flavors and initiation and patterns of tobacco product use, particularly among youths and young adults; and flavors and cessation, dual-use, and relapse among current and former tobacco product users.
It also is seeking comment on whether standards should be set on tobacco flavoring, including whether there should a prohibition or restriction on flavors and to which types of products these standards should apply. The notice specifically asks about menthol and its role in cigarette initiation and whether limitations on menthol could lead to use of other tobacco products.
“Because almost 90% of adult smokers started smoking by the age of 18, it’s imperative we look at new ways we can ensure that kids don’t progress from experimentation to regular use,” Commissioner Gottlieb said.
The American Heart Association called the action “long overdue.”
“We encourage the FDA to quickly move beyond information gathering and develop a strong flavoring product standard,” CEO Nancy Brown said in a statement. “There is already clear evidence that flavored tobacco products, including menthol, harm the public health. To make it worse, fruit- and candy-flavored e-cigarettes, cigars, and other tobacco products are highly attractive to kids and make it more likely that they will take up this addiction.”
The action comes less than a week after FDA published an advance notice seeking information comments on reducing nicotine levels in cigarettes to help combat nicotine addiction.
The advance notice will be published March 21 in the Federal Register; comments will be accepted at www.regulations.gov for 90 days.
Arthritis limits physical activity the most in the South
“Physical activity is a proven strategy for managing arthritis symptoms,” but 35% of Americans with arthritis do not participate in any such activities or exercise, according to investigators who analyzed data from a national survey of more than 440,000 adults.
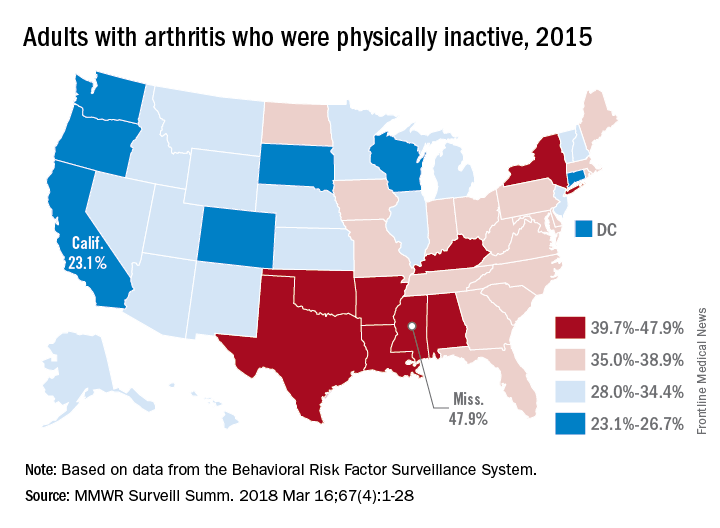
The low rates of inactivity in the western half of the country were topped by California’s 23.1% and South Dakota’s 23.4%, with Oregon (24.0%) and Wisconsin (24.6%) not too far behind, Dr. Barbour and his associates said based on data for 441,456 adults aged 18 years and older who were interviewed for the Behavioral Risk Factor Surveillance System.
Overall prevalence rates show that arthritis has the greatest effect in southern states, which, in addition to high inactivity, had more arthritis-attributable severe joint pain, more arthritis-attributable social participation restriction, and less leisure-time walking among adults with arthritis. This information, the investigators suggested, may help public health professionals “to better understand and target evidence-based nonpharmaceutical interventions, such as arthritis self-management education and physical activity.”
SOURCE: Barbour KE et al. MMWR Surveill Summ. 2018 Mar 16;67(4):1-28.
“Physical activity is a proven strategy for managing arthritis symptoms,” but 35% of Americans with arthritis do not participate in any such activities or exercise, according to investigators who analyzed data from a national survey of more than 440,000 adults.
The low rates of inactivity in the western half of the country were topped by California’s 23.1% and South Dakota’s 23.4%, with Oregon (24.0%) and Wisconsin (24.6%) not too far behind, Dr. Barbour and his associates said based on data for 441,456 adults aged 18 years and older who were interviewed for the Behavioral Risk Factor Surveillance System.
Overall prevalence rates show that arthritis has the greatest effect in southern states, which, in addition to high inactivity, had more arthritis-attributable severe joint pain, more arthritis-attributable social participation restriction, and less leisure-time walking among adults with arthritis. This information, the investigators suggested, may help public health professionals “to better understand and target evidence-based nonpharmaceutical interventions, such as arthritis self-management education and physical activity.”
SOURCE: Barbour KE et al. MMWR Surveill Summ. 2018 Mar 16;67(4):1-28.
“Physical activity is a proven strategy for managing arthritis symptoms,” but 35% of Americans with arthritis do not participate in any such activities or exercise, according to investigators who analyzed data from a national survey of more than 440,000 adults.
The low rates of inactivity in the western half of the country were topped by California’s 23.1% and South Dakota’s 23.4%, with Oregon (24.0%) and Wisconsin (24.6%) not too far behind, Dr. Barbour and his associates said based on data for 441,456 adults aged 18 years and older who were interviewed for the Behavioral Risk Factor Surveillance System.
Overall prevalence rates show that arthritis has the greatest effect in southern states, which, in addition to high inactivity, had more arthritis-attributable severe joint pain, more arthritis-attributable social participation restriction, and less leisure-time walking among adults with arthritis. This information, the investigators suggested, may help public health professionals “to better understand and target evidence-based nonpharmaceutical interventions, such as arthritis self-management education and physical activity.”
SOURCE: Barbour KE et al. MMWR Surveill Summ. 2018 Mar 16;67(4):1-28.
FROM MMWR SURVEILLANCE SUMMARIES
FDA meeting on medical devices for sleep apnea scheduled
In a statement sent to members, CHEST invited all to attend this open meeting, which will be held at the FDA White Oak Campus in Silver Spring, Md.![]()
In a statement sent to members, CHEST invited all to attend this open meeting, which will be held at the FDA White Oak Campus in Silver Spring, Md.![]()
In a statement sent to members, CHEST invited all to attend this open meeting, which will be held at the FDA White Oak Campus in Silver Spring, Md.![]()
Top-selling drugs going to patients with diabetes
, and more than half of the 20 biggest-selling drugs for the year are regularly prescribed to patients with diabetes, according to the Agency for Healthcare Research and Quality.
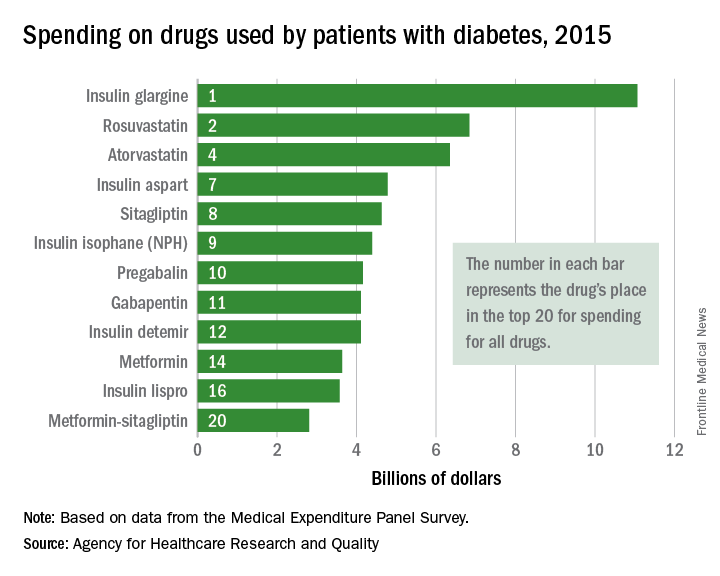
Drugs used by patients with diabetes also took up half of the next 10 spots in the list: gabapentin was 11th, insulin detemir was 12th, metformin was 14th, insulin lispro was 16th, and metformin-sitagliptin was 20th, according to the MEPS data.
The drugs in the MEPS top 10 for 2015 – the most recent year for which data are available – that are not commonly prescribed for diabetes were the asthma/chronic obstructive pulmonary drug fluticasone-salmeterol (third at $6.7 billion), the gastroesophageal reflux disease drug esomeprazole (fifth at $5.3 billion), and aripiprazole (sixth at $5.2 billion), which is used to treat schizophrenia and bipolar disorder.
, and more than half of the 20 biggest-selling drugs for the year are regularly prescribed to patients with diabetes, according to the Agency for Healthcare Research and Quality.
Drugs used by patients with diabetes also took up half of the next 10 spots in the list: gabapentin was 11th, insulin detemir was 12th, metformin was 14th, insulin lispro was 16th, and metformin-sitagliptin was 20th, according to the MEPS data.
The drugs in the MEPS top 10 for 2015 – the most recent year for which data are available – that are not commonly prescribed for diabetes were the asthma/chronic obstructive pulmonary drug fluticasone-salmeterol (third at $6.7 billion), the gastroesophageal reflux disease drug esomeprazole (fifth at $5.3 billion), and aripiprazole (sixth at $5.2 billion), which is used to treat schizophrenia and bipolar disorder.
, and more than half of the 20 biggest-selling drugs for the year are regularly prescribed to patients with diabetes, according to the Agency for Healthcare Research and Quality.
Drugs used by patients with diabetes also took up half of the next 10 spots in the list: gabapentin was 11th, insulin detemir was 12th, metformin was 14th, insulin lispro was 16th, and metformin-sitagliptin was 20th, according to the MEPS data.
The drugs in the MEPS top 10 for 2015 – the most recent year for which data are available – that are not commonly prescribed for diabetes were the asthma/chronic obstructive pulmonary drug fluticasone-salmeterol (third at $6.7 billion), the gastroesophageal reflux disease drug esomeprazole (fifth at $5.3 billion), and aripiprazole (sixth at $5.2 billion), which is used to treat schizophrenia and bipolar disorder.
FDA issues warning to all duodenoscope manufacturers
The Food and Drug Administration on March 9 issued warning letters to all three duodenoscope manufacturers for failing to comply with the requirements of federal law under which they were ordered to conduct postmarket surveillance studies to assess the effectiveness of reprocessing the devices.
The warning is part of an ongoing effort to prevent patient infections associated with the transmission of bacteria from contaminated duodenoscopes. The three manufacturers – Olympus, Fujifilm, and Pentax – are required to conduct studies to sample and culture reprocessed duodenoscopes that are in clinical use to learn more about issues that contribute to contamination, and to study human factors to determine how hospital staff who have had training are following the reprocessing instructions. In 2015, the FDA ordered the companies to conduct a postmarket surveillance study to determine whether health care facilities were able to properly clean and disinfect the devices.
Currently, the Olympus manufacturer has failed to start data collection, while both Pentax and Fujifilm have failed to provide sufficient data required for their respective studies to sample and culture reprocessed duodenoscopes that are in clinical use. In addition, Olympus and Pentax have not complied with requirements to assess how well staff members have followed the reprocessing instructions after the human factors studies and Fujifilm has been meeting its requirements for its human factors study only.
“The FDA has taken important steps to improve the reprocessing of duodenoscopes, and we’ve seen a reduction in reports of patient infections, but we need the required postmarket studies to determine whether these measures are being properly implemented in real-world clinical settings and whether we need to take additional action to further improve the safety of these devices,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in a press release. “We expect these device manufacturers to meet their study obligations to ensure patient safety.”
The companies have until March 24 to submit a plan that outlines how study milestones will be achieved. If the companies fail to respond to the warning letter, the FDA states that they may take additional action, such as seizure, injunction, and civil monetary penalties.
Read the full press release on the FDA’s website.
The Food and Drug Administration on March 9 issued warning letters to all three duodenoscope manufacturers for failing to comply with the requirements of federal law under which they were ordered to conduct postmarket surveillance studies to assess the effectiveness of reprocessing the devices.
The warning is part of an ongoing effort to prevent patient infections associated with the transmission of bacteria from contaminated duodenoscopes. The three manufacturers – Olympus, Fujifilm, and Pentax – are required to conduct studies to sample and culture reprocessed duodenoscopes that are in clinical use to learn more about issues that contribute to contamination, and to study human factors to determine how hospital staff who have had training are following the reprocessing instructions. In 2015, the FDA ordered the companies to conduct a postmarket surveillance study to determine whether health care facilities were able to properly clean and disinfect the devices.
Currently, the Olympus manufacturer has failed to start data collection, while both Pentax and Fujifilm have failed to provide sufficient data required for their respective studies to sample and culture reprocessed duodenoscopes that are in clinical use. In addition, Olympus and Pentax have not complied with requirements to assess how well staff members have followed the reprocessing instructions after the human factors studies and Fujifilm has been meeting its requirements for its human factors study only.
“The FDA has taken important steps to improve the reprocessing of duodenoscopes, and we’ve seen a reduction in reports of patient infections, but we need the required postmarket studies to determine whether these measures are being properly implemented in real-world clinical settings and whether we need to take additional action to further improve the safety of these devices,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in a press release. “We expect these device manufacturers to meet their study obligations to ensure patient safety.”
The companies have until March 24 to submit a plan that outlines how study milestones will be achieved. If the companies fail to respond to the warning letter, the FDA states that they may take additional action, such as seizure, injunction, and civil monetary penalties.
Read the full press release on the FDA’s website.
The Food and Drug Administration on March 9 issued warning letters to all three duodenoscope manufacturers for failing to comply with the requirements of federal law under which they were ordered to conduct postmarket surveillance studies to assess the effectiveness of reprocessing the devices.
The warning is part of an ongoing effort to prevent patient infections associated with the transmission of bacteria from contaminated duodenoscopes. The three manufacturers – Olympus, Fujifilm, and Pentax – are required to conduct studies to sample and culture reprocessed duodenoscopes that are in clinical use to learn more about issues that contribute to contamination, and to study human factors to determine how hospital staff who have had training are following the reprocessing instructions. In 2015, the FDA ordered the companies to conduct a postmarket surveillance study to determine whether health care facilities were able to properly clean and disinfect the devices.
Currently, the Olympus manufacturer has failed to start data collection, while both Pentax and Fujifilm have failed to provide sufficient data required for their respective studies to sample and culture reprocessed duodenoscopes that are in clinical use. In addition, Olympus and Pentax have not complied with requirements to assess how well staff members have followed the reprocessing instructions after the human factors studies and Fujifilm has been meeting its requirements for its human factors study only.
“The FDA has taken important steps to improve the reprocessing of duodenoscopes, and we’ve seen a reduction in reports of patient infections, but we need the required postmarket studies to determine whether these measures are being properly implemented in real-world clinical settings and whether we need to take additional action to further improve the safety of these devices,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in a press release. “We expect these device manufacturers to meet their study obligations to ensure patient safety.”
The companies have until March 24 to submit a plan that outlines how study milestones will be achieved. If the companies fail to respond to the warning letter, the FDA states that they may take additional action, such as seizure, injunction, and civil monetary penalties.
Read the full press release on the FDA’s website.
Flu activity continues to decline
The 2017-2018 flu season continued to loosen its grip on the country as both outpatient activity and pediatric deaths dropped during the week ending March 3, according to the Centers for Disease Control and Prevention.
After five consecutive weeks of double-digit pediatric deaths related to influenza-like illness (ILI), five deaths were reported for the week ending March 3, four of which occurred in previous weeks. The total for the 2017-2018 season is now 119, the CDC said in its weekly surveillance report.
The proportion of outpatient visits for ILI was 3.7% for the week, which is down from 4.9% the week before and less than half of the seasonal high of 7.5% that was recorded for the week of Feb. 3, CDC data show. The national baseline level of outpatient activity is 2.2%.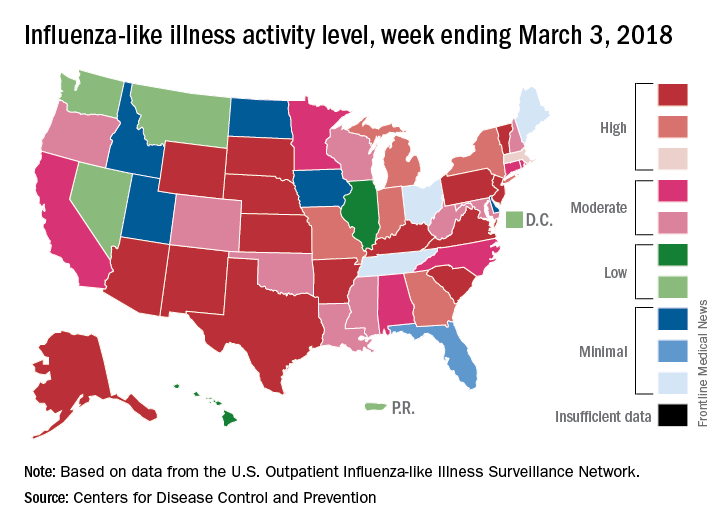
The cumulative hospitalization rate for the 2017-2018 flu season climbed from 84.2 the previous week to 86.3 per 100,000 population – well above the rate of 57.2 per 100,000 that was recorded for the corresponding week of the hospitalization-record-setting 2014-2015 season, FluView data show.
The 2017-2018 flu season continued to loosen its grip on the country as both outpatient activity and pediatric deaths dropped during the week ending March 3, according to the Centers for Disease Control and Prevention.
After five consecutive weeks of double-digit pediatric deaths related to influenza-like illness (ILI), five deaths were reported for the week ending March 3, four of which occurred in previous weeks. The total for the 2017-2018 season is now 119, the CDC said in its weekly surveillance report.
The proportion of outpatient visits for ILI was 3.7% for the week, which is down from 4.9% the week before and less than half of the seasonal high of 7.5% that was recorded for the week of Feb. 3, CDC data show. The national baseline level of outpatient activity is 2.2%.
The cumulative hospitalization rate for the 2017-2018 flu season climbed from 84.2 the previous week to 86.3 per 100,000 population – well above the rate of 57.2 per 100,000 that was recorded for the corresponding week of the hospitalization-record-setting 2014-2015 season, FluView data show.
The 2017-2018 flu season continued to loosen its grip on the country as both outpatient activity and pediatric deaths dropped during the week ending March 3, according to the Centers for Disease Control and Prevention.
After five consecutive weeks of double-digit pediatric deaths related to influenza-like illness (ILI), five deaths were reported for the week ending March 3, four of which occurred in previous weeks. The total for the 2017-2018 season is now 119, the CDC said in its weekly surveillance report.
The proportion of outpatient visits for ILI was 3.7% for the week, which is down from 4.9% the week before and less than half of the seasonal high of 7.5% that was recorded for the week of Feb. 3, CDC data show. The national baseline level of outpatient activity is 2.2%.
The cumulative hospitalization rate for the 2017-2018 flu season climbed from 84.2 the previous week to 86.3 per 100,000 population – well above the rate of 57.2 per 100,000 that was recorded for the corresponding week of the hospitalization-record-setting 2014-2015 season, FluView data show.