User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Topical Therapy for Acne in Women: Is There a Role for Clindamycin Phosphate–Benzoyl Peroxide Gel?
The management of acne vulgaris (AV) in women has been the subject of considerable attention over the last few years. It has become increasingly recognized that a greater number of patient encounters in dermatology offices involve women with AV who are beyond their adolescent years. Overall, it is estimated that up to approximately 22% of women in the United States are affected by AV, with approximately half of women in their 20s and one-third of women in their 30s reporting some degree of AV.1-4 Among women, the disease shows no predilection for certain skin types or ethnicities, can start during the preteenaged or adolescent years, can persist or recur in adulthood (persistent acne, 75%), or can start in adulthood (late-onset acne, 25%) in females with minimal or no history of AV occurring earlier in life.3,5-7 In the subpopulation of adult women, AV occurs at a time when many expect to be far beyond this “teenage affliction.” Women who are affected commonly express feeling embarrassed and frustrated.5-8
Most of the emphasis in the literature and in presentations at dermatology meetings regarding the management of AV in adult women has focused on excluding underlying disorders that cause excess androgens (eg, polycystic ovary syndrome, congenital adrenal hyperplasia, tumors, exogenous sources) as well as the use of systemic therapies such as oral contraceptives (OCs) and spironolactone.5-7,9,10 Little attention has been given to the selection of topical therapies in this patient population, especially with regard to evidence from clinical studies. To date, results from published study analyses using topical agents specifically for adult females with facial AV have only included adapalene gel 0.3% applied once daily and dapsone gel 5% applied twice daily.11-13 Both agents have been evaluated in subset analysis comparisons of outcomes in women aged 18 years and older versus adolescents aged 12 to 17 years based on data from 12-week phase 3 pivotal trials.14-16
Are there clinically relevant differences between AV in adult versus adolescent females?
Although much has been written about AV in women, epidemiology, demographics, assessment of clinical presentation, and correlation of clinical presentation with excess androgens have not been emphasized,1-3,5-10,17 likely due to marketing campaigns that emphasize AV as a disorder that predominantly affects teenagers as well as the focus on optimal use of oral spironolactone and/or OCs in the management of AV in the adult female population. Attention to spironolactone use is important because it is not approved by the US Food and Drug Administration for the treatment of AV. Spironolactone carries certain black-box warnings that may not be clinically relevant in all patients but still require attention. It also is associated with risks if taken during pregnancy, and it is a potassium-sparing diuretic with potential for hyperkalemia, especially in patients with reduced renal function or those who are taking potassium supplements or certain other medications.6,9,11,17 Use of OCs to treat AV also is not without potential risks, with specific warnings and relative contraindications reported, especially in relation to increased risks for cardiac complications, stroke, and thromboembolism.6,9,11,12 Because adult females are in a different stage of life than teenagers, there are defined psychosocial and medical considerations in managing AV in these patients compared with adolescents.5-8,17 Importantly for both clinicians and patients, addressing these differences and considerations can have a major impact on whether or not women with AV experience successful treatment outcomes.1-3,5,6,8,10,13 Skin color and ethnicity also can affect the psychosocial and physical factors that influence the overall management of adult female patients with AV, including selection of therapies and handling long-term visible sequelae that occur in some AV patients, such as dyschromia (eg, persistent or postinflammatory erythema or hyperpigmentation) and acne scarring.5-8,13,17-20
Psychosocial Considerations
With regard to psychosocial, emotional, and attitudinal considerations in women with AV, common findings include concern or frustration regarding the presence of AV beyond adolescence; anxiety; symptoms of depression; decreased self-confidence; increased self-consciousness, especially during public interactions or intimate situations; and interference with steady concentration at work or school.5,6,8,13 Long-term complications of AV, such as dyschromia and acne scarring, are more likely to be encountered in adult patients, especially if they had AV as a teenager, with women reporting that they remain conscious of these adverse sequelae.8 It is estimated that approximately three-fourths of women with AV also had AV as teenagers; therefore, most of them have already used many over-the-counter and prescription therapies and are likely to want treatments that are newer, well-tolerated, safe, and known to be effective in adult women.8,16,17 Convenience and simplicity are vital components of treatment selection and regimen design, as many women with AV frequently face time constraints in their daily routines due to family, social, employment, and home-related demands and responsibilities.6-8,17
Medical Considerations
It is apparent from reports in the literature as well as from clinical experience that some women with AV present with a U-shaped pattern of involvement on the face,5-7,10,13,17 which refers to the presence of predominantly inflammatory papules (many of them deep) and some nodules on the lower face, jawline, and anterolateral neck region, with comedones often sparse or absent.5-7 It often is perceived and may be true that women who present with this pattern of distribution are more androgen sensitive despite having normal serum androgen levels or in some cases exhibit detectable excess androgens (eg, in the setting of polycystic ovary syndrome) and may be more likely to respond to hormonal therapies (eg, spironolactone, OCs) than those with mixed facial AV (ie, multiple comedonal and inflammatory acne lesions, not limited to a U-shaped pattern, similar to adolescent AV), but data are limited to support differentiation between the U-shaped pattern group and the conventional mixed facial AV group.5-7,17 Adult and adolescent females in both groups sometimes report perimenstrual flares and frequent persistent papular AV that tends to concentrate on the perioral and chin area.
It is also important to consider that the current literature suggests approximately three-fourths of women with AV report that they also had AV as a teenager, with many indicating the same clinical pattern of AV and approximately one-third reporting AV that is more severe in adulthood than adolescence.5-8,17 The available literature on topical and oral therapies used to treat AV in both adolescent and adult females predominantly focuses on inclusion of both inflammatory and noninflammatory (comedonal) facial AV lesions, does not specifically address or include the U-shaped pattern of AV in adult women for inclusion in studies that evaluate efficacy in this subgroup, and does not include AV involving the neck region and below the jawline margin as part of any study protocols and/or discussions about therapy.5-7,9-12,17,21-26 Involvement of the neck and lower jawline is common in women presenting with the U-shaped pattern of AV, and available studies only evaluate AV involving the face and do not include AV lesions present below the jawline margin. As a result, there is a considerable need for well-designed studies with laboratory assessments to include or exclude underlying detectable excess androgens and to assess the efficacy, tolerability, and safety of specific therapeutic agents both alone and in combination in adult women who present with a U-shaped pattern of AV.17
Other medical considerations that can influence treatment selection and are more likely to be present in adult versus adolescent females include underlying chronic medical disorders; concomitant medications that may interact with other oral agents; potential for pregnancy; age, particularly when prescribing OCs; and the potential desire to stop taking OCs if already used over a prolonged period.6,7
Age-Related Differentiation of Female Subgroups With AV
The age-based dividing line that defines AV in adults versus adolescent females has been described in the literature; however, the basis for published definitions of female subgroups with AV is not well-supported by strong scientific evidence.1-3,5-7,17 The conventional dividing line that was originally selected to define adult females with AV was 25 years of age or older; persistent acne is present both during adolescence and at or after 25 years of age, while late-onset acne is described as AV that first presents at 25 years of age or older.3,5-7
More recently, a range of 18 years or older has been used to classify adult female AV and a range of 12 to 17 years for adolescent female AV in subset analyses that evaluated treatment outcomes in both patient populations from phase 3 pivotal trials completed with adapalene gel 0.3% applied once daily and dapsone gel 5% applied twice daily.14-16 These subanalyses included participants with facial AV that was predominantly moderate in severity, mandated specific lesion count ranges for both comedonal and inflammatory lesions, and included only facial AV that was above the mandibular (jawline) margin.15,16,21,26 Therefore, patients with AV presenting in a U-shaped pattern with involvement below the jawline and on the neck were not included in these study analyses, as these patients were excluded from the phase 3 trials on which the analyses were based. The outcomes of these analyses apply to treatment in women who present with both inflammatory and noninflammatory facial AV lesions, which supports the observation that AV in this patient population is not always predominantly inflammatory and does not always present in a U-shaped distribution.14-16 In fact, a U-shaped pattern of distribution appears to be less common in women with AV than a mixed inflammatory and comedonal distribution that involves the face more diffusely, though more data are needed from well-designed and large-scale epidemiologic and demographic studies.5,14,17
Are there data available on the use of benzoyl peroxide with or without a topical antibiotic in women with AV?
There is a conspicuous absence of prospective clinical trials and retrospective analyses evaluating the specific use of individual AV therapies in adult females, with a particular lack of studies with topical agents (eg, benzoyl peroxide [BP]).14 Subset analyses have been completed for adapalene gel 0.3% and dapsone gel 5%.15,16 Additionally, an age-based subset analysis in females with facial AV also has been completed with clindamycin phosphate (CP) 1.2%–BP 2.5% gel once daily, with data presented but not yet fully published.14
Two identical phase 3, double-blind, randomized, 12-week, 4-arm trials compared treatment outcomes in groups treated with an aqueous-based combination gel formulation containing BP 2.5% and CP 1.2% (n=797), active monad gels (BP [n=809] or CP [n=812]), or vehicle gel (n=395), all applied once daily in patients with facial AV.22 Participants were 12 years or older (mean age range, 19.1–19.6 years; age range, 12.1–70.2 years), were of either gender (approximately 50% split in each study arm), and presented with moderate (approximately 80% of participants) or severe AV (approximately 20% of participants) at baseline. The entry criteria for lesion types and number of lesions were 17 to 40 inflammatory lesions (ie, papules, pustules, <2 nodules)(range of mean number of lesions, 25.8–26.4) and 20 to 100 noninflammatory lesions (ie, closed comedones, open comedones)(range of mean number of lesions, 44.0–47.4). Participant demographics included white (73.9%–77.5%), black/African American (16.1%–20.4%), and Asian (2.1%–3.3%), with the remaining participants distributed among a variety of other ethnic groups such as Native Hawaiian/Native Pacific Islander and Native American Indian/Native Alaskan (collectively <5% in each study arm). Therefore, approximately 1 of every 4 patients had skin of color, which provided good diversity of patients considering the large study size (N=2813). Data analysis included dichotomization of participants by severity rating (moderate or severe based on evaluator global severity score) and skin phototype (Fitzpatrick skin types I–III or IV–VI).22
The pooled results from both studies completed at 68 investigative sites demonstrated that CP 1.2%–BP 2.5% gel was superior in efficacy to each individual monad and to the vehicle in inflammatory, noninflammatory, and total lesion reductions as early as week 4 (P<.001) and at week 12, which was the study end point (P<.001), with superiority also demonstrated in achieving treatment success (defined as a >2 grade improvement according to the evaluator global severity score) compared to the 3 other study arms (P<.001).22 Subject assessments also were consistent with outcomes noted by the investigators. Cutaneous tolerability was favorable and comparable in all 4 study arms with less than 1% of participants discontinuing treatment due to adverse events.22
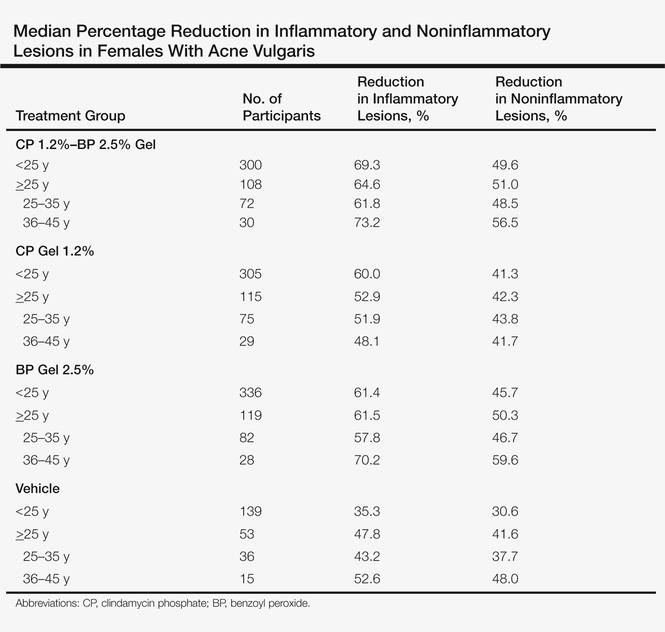
A subset analysis of the data from the phase 3 pivotal trials with CP 1.2%–BP 2.5% gel was completed to compare reductions in both inflammatory and noninflammatory lesions in female participants who were younger than 25 years and 25 years of age or older in all 4 study arms. This information has been presented14,17 but has not been previously published. Based on the overall results reported in the phase 3 studies, there were no differentiations in skin tolerability or safety based on participant age, gender, or skin type.22 The subanalysis included a total of 1080 females who were younger than 25 years and 395 females who were 25 years of age or older. The lesion reduction outcomes of this subanalysis are presented in the Table. Statistical analyses of the results among these age groups in the 4 study arms were not completed because the objective was to determine if there were any major or obvious differences in reduction of AV lesions based on the conventional dividing line of 25 years of age in adult women as compared to adolescent females treated with CP 1.2%–BP 2.5% gel. In addition, the large difference in numbers of female participants between the 2 age groups (>25 years of age, n=395; <25 years of age, n=1080) at least partially confounds both statistical and observational analysis. Among the women who were 25 years of age or older who were included in the subanalysis, 67.0% and 25.8% were between the ages of 25 to 35 years and 36 to 45 years, respectively. Based on the outcomes reported in the phase 3 trials and in this subgroup analysis, CP 1.2%–BP 2.5% gel applied once daily over a 12-week period appeared overall to be comparably effective in females regardless of age and with no apparent adverse events regarding differences in skin tolerability or safety.14,22 One observation that was noted was the possible trend of greater reduction in both lesion types in women older than 35 years versus younger females with the use of the combination gel or BP alone; however, the number of female participants who were older than 35 years of age was substantially less (n=102) than those who were 35 years of age or younger (n=1345), thus precluding support for any definitive conclusions about this possible trend.22
How can CP 1.2%–BP 2.5% gel be incorporated into a treatment regimen for women with facial AV?
The incorporation of CP 1.2%–BP 2.5% gel into a treatment regimen for women with facial AV is similar to the general use of BP-containing formulations in the overall management of AV.9,14,27,28 Because women with AV commonly present with facial inflammatory lesions and many also with facial comedones, CP 1.2%–BP 2.5% gel is best used once daily in the morning in combination with a topical retinoid in the evening,9,27 which can be achieved with use of CP 1.2%–BP 2.5% gel in the morning and a topical retinoid (ie, tretinoin, adapalene, tazarotene) in the evening or CP 1.2%–tretinoin 0.025% gel in the evening. It is important to note that cutaneous irritation may be more likely if neck lesions are present; the potential for bleaching of colored fabric by BP also is a practical concern.28 In addition, CP 1.2%–BP 2.5% gel may also be used in combination with topical dapsone, but both products should be applied separately at different times of the day to avoid temporary orange discoloration of the skin, which appears to be an uncommon side effect but remains a possibility based on the product information for dapsone gel 5% with regard to its concomitant use with BP.29,30
1. Perkins AC, Maglione J, Hillebrand GG, et al. Acne vulgaris in women: prevalence across the life span. J Womens Health. 2012;21:223-230.
2. Zeichner J. Evaluating and treating the adult female patient with acne. J Drugs Dermatol. 2013;12:1416-1427.
3. Goulden V, Stables GI, Cunliffe WJ. Prevalence of facial acne in adults. J Am Acad Dermatol. 1999;41:577-580.
4. Collier CN, Harper J, Cafardi JA, et al. The prevalence of acne in adults 20 years and older. J Am Acad Dermatol. 2008;58:56-59.
5. Dreno B, Layton A, Zouboulis CC, et al. Adult female acne: a new paradigm. J Eur Acad Dermatol Venereol. 2013;27:1063-1070.
6. Kim GK, Del Rosso JQ. Oral spironolactone in post-teenage female patients with acne vulgaris: practical considerations for the clinician based on current data and clinical experience. J Clin Aesthet Dermatol. 2012;5:37-50.
7. Kim GK, Michaels BB. Post-adolescent acne in women: more common and more clinical considerations. J Drugs Dermatol. 2012;11:708-713.
8. Tanghetti EA, Kawata AK, Daniels SR, et al. Understanding the burden of adult female acne. J Clin Aesthet Dermatol. 2014;7:22-30.
9. Gollnick H, Cunliffe W, Berson D, et al. Management of acne: a report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49(suppl 1):1-37.
10. Thiboutot DM. Endocrinological evaluation and hormonal therapy for women with difficult acne. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):57-61.
11. Sawaya ME, Samani N. Antiandrogens and androgen receptors. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelphia, PA: Saunders-Elsevier; 2012:361-374.
12. Harper JC. Should dermatologists prescribe hormonal contraceptives for acne? Dermatol Ther. 2009;22:452-457.
13. Preneau S, Dreno B. Female acne: a different subtype of teenager acne? J Eur Acad Dermatol Venereol. 2012;26:277-282.
14. Del Rosso JQ, Zeichner JA. What’s new in the medicine cabinet?: a panoramic review of clinically relevant information for the busy dermatologist. J Clin Aesthet Dermatol. 2014;7:26-30.
15. Berson D, Alexis A. Adapalene 0.3% for the treatment of acne in women. J Clin Aesthet Dermatol. 2013;6:32-35.
16. Del Rosso JQ, Kircik L, Gallagher C. Facing up to adult women with acne vulgaris: an analysis of pivotal trial data on dapsone 5% gel in the adult female population. Poster presented at: Fall Clinical Dermatology; October 2013; Las Vegas, NV.
17. Del Rosso JQ. Management of acne with oral spironolactone. Presented at: American Academy of Dermatology Summer Meeting; August 2013; Boston, MA.
18. Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
19. Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
20. Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasian, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
21. Draelos ZD, Carter E, Maloney JM, et al. Two randomized studies demonstrate the efficacy and safety of dapsone gel, 5% for the treatment of acne vulgaris [published online ahead of print January 17, 2007]. J Am Acad Dermatol. 2007;56:439.e1-439.e10.
22. Thiboutot D, Zaenglein A, Weiss J, et al. An aqueous gel fixed combination of clindamycin phosphate 1.2% and benzoyl peroxide 2.5% for the once-daily treatment of moderate to severe acne vulgaris: assessment of efficacy and safety in 2813 patients. J Am Acad Dermatol. 2008;59:792-800.
23. Schlessinger J, Menter A, Gold M, et al. Clinical safety and efficacy studies of a novel formulation combining
1.2% clindamycin phosphate and 0.025% tretinoin for the treatment of acne vulgaris. J Drugs Dermatol. 2007;6:607-615.
24. Fleischer AB Jr, Dinehart S, Stough D, et al. Safety and efficacy of a new extended-release formulation of minocycline. Cutis. 2006;78(suppl 4):21-31.
25. Gollnick HP, Draelos Z, Glenn MJ, et al. Adapalene-benzoyl peroxide, a unique fixed-dose combination topical gel for the treatment of acne vulgaris: a transatlantic, randomized, double-blind, controlled study in 1670 patients. Br J Dermatol. 2009;161:1180-1189.
26. Thiboutot D, Arsonnaud S, Soto P. Efficacy and tolerability of adapalene 0.3% gel compared to tazarotene 0.1% gel in the treatment of acne vulgaris. J Drugs Dermatol. 2008;7(suppl 6):3-10.
27. Zeichner JA. Optimizing topical combination therapy for the treatment of acne vulgaris. J Drugs Dermatol. 2012;11:313-317.
28. Tanghetti EA, Popp KF. A current review of topical benzoyl peroxide: new perspectives on formulation and utilization. Dermatol Clin. 2009;27:17-24.
29. Fleischer AB, Shalita A, Eichenfield LF. Dapsone gel 5% in combination with adapalene gel 0.1%, benzoyl peroxide gel 4% or moisturizer for the treatment of acne vulgaris: a 12-week, randomized, double-blind study. J Drugs Dermatol. 2010;9:33-40.
30. Aczone (dapsone gel 5%) [package insert]. Irvine, CA: Allergan, Inc; 2013.
The management of acne vulgaris (AV) in women has been the subject of considerable attention over the last few years. It has become increasingly recognized that a greater number of patient encounters in dermatology offices involve women with AV who are beyond their adolescent years. Overall, it is estimated that up to approximately 22% of women in the United States are affected by AV, with approximately half of women in their 20s and one-third of women in their 30s reporting some degree of AV.1-4 Among women, the disease shows no predilection for certain skin types or ethnicities, can start during the preteenaged or adolescent years, can persist or recur in adulthood (persistent acne, 75%), or can start in adulthood (late-onset acne, 25%) in females with minimal or no history of AV occurring earlier in life.3,5-7 In the subpopulation of adult women, AV occurs at a time when many expect to be far beyond this “teenage affliction.” Women who are affected commonly express feeling embarrassed and frustrated.5-8
Most of the emphasis in the literature and in presentations at dermatology meetings regarding the management of AV in adult women has focused on excluding underlying disorders that cause excess androgens (eg, polycystic ovary syndrome, congenital adrenal hyperplasia, tumors, exogenous sources) as well as the use of systemic therapies such as oral contraceptives (OCs) and spironolactone.5-7,9,10 Little attention has been given to the selection of topical therapies in this patient population, especially with regard to evidence from clinical studies. To date, results from published study analyses using topical agents specifically for adult females with facial AV have only included adapalene gel 0.3% applied once daily and dapsone gel 5% applied twice daily.11-13 Both agents have been evaluated in subset analysis comparisons of outcomes in women aged 18 years and older versus adolescents aged 12 to 17 years based on data from 12-week phase 3 pivotal trials.14-16
Are there clinically relevant differences between AV in adult versus adolescent females?
Although much has been written about AV in women, epidemiology, demographics, assessment of clinical presentation, and correlation of clinical presentation with excess androgens have not been emphasized,1-3,5-10,17 likely due to marketing campaigns that emphasize AV as a disorder that predominantly affects teenagers as well as the focus on optimal use of oral spironolactone and/or OCs in the management of AV in the adult female population. Attention to spironolactone use is important because it is not approved by the US Food and Drug Administration for the treatment of AV. Spironolactone carries certain black-box warnings that may not be clinically relevant in all patients but still require attention. It also is associated with risks if taken during pregnancy, and it is a potassium-sparing diuretic with potential for hyperkalemia, especially in patients with reduced renal function or those who are taking potassium supplements or certain other medications.6,9,11,17 Use of OCs to treat AV also is not without potential risks, with specific warnings and relative contraindications reported, especially in relation to increased risks for cardiac complications, stroke, and thromboembolism.6,9,11,12 Because adult females are in a different stage of life than teenagers, there are defined psychosocial and medical considerations in managing AV in these patients compared with adolescents.5-8,17 Importantly for both clinicians and patients, addressing these differences and considerations can have a major impact on whether or not women with AV experience successful treatment outcomes.1-3,5,6,8,10,13 Skin color and ethnicity also can affect the psychosocial and physical factors that influence the overall management of adult female patients with AV, including selection of therapies and handling long-term visible sequelae that occur in some AV patients, such as dyschromia (eg, persistent or postinflammatory erythema or hyperpigmentation) and acne scarring.5-8,13,17-20
Psychosocial Considerations
With regard to psychosocial, emotional, and attitudinal considerations in women with AV, common findings include concern or frustration regarding the presence of AV beyond adolescence; anxiety; symptoms of depression; decreased self-confidence; increased self-consciousness, especially during public interactions or intimate situations; and interference with steady concentration at work or school.5,6,8,13 Long-term complications of AV, such as dyschromia and acne scarring, are more likely to be encountered in adult patients, especially if they had AV as a teenager, with women reporting that they remain conscious of these adverse sequelae.8 It is estimated that approximately three-fourths of women with AV also had AV as teenagers; therefore, most of them have already used many over-the-counter and prescription therapies and are likely to want treatments that are newer, well-tolerated, safe, and known to be effective in adult women.8,16,17 Convenience and simplicity are vital components of treatment selection and regimen design, as many women with AV frequently face time constraints in their daily routines due to family, social, employment, and home-related demands and responsibilities.6-8,17
Medical Considerations
It is apparent from reports in the literature as well as from clinical experience that some women with AV present with a U-shaped pattern of involvement on the face,5-7,10,13,17 which refers to the presence of predominantly inflammatory papules (many of them deep) and some nodules on the lower face, jawline, and anterolateral neck region, with comedones often sparse or absent.5-7 It often is perceived and may be true that women who present with this pattern of distribution are more androgen sensitive despite having normal serum androgen levels or in some cases exhibit detectable excess androgens (eg, in the setting of polycystic ovary syndrome) and may be more likely to respond to hormonal therapies (eg, spironolactone, OCs) than those with mixed facial AV (ie, multiple comedonal and inflammatory acne lesions, not limited to a U-shaped pattern, similar to adolescent AV), but data are limited to support differentiation between the U-shaped pattern group and the conventional mixed facial AV group.5-7,17 Adult and adolescent females in both groups sometimes report perimenstrual flares and frequent persistent papular AV that tends to concentrate on the perioral and chin area.
It is also important to consider that the current literature suggests approximately three-fourths of women with AV report that they also had AV as a teenager, with many indicating the same clinical pattern of AV and approximately one-third reporting AV that is more severe in adulthood than adolescence.5-8,17 The available literature on topical and oral therapies used to treat AV in both adolescent and adult females predominantly focuses on inclusion of both inflammatory and noninflammatory (comedonal) facial AV lesions, does not specifically address or include the U-shaped pattern of AV in adult women for inclusion in studies that evaluate efficacy in this subgroup, and does not include AV involving the neck region and below the jawline margin as part of any study protocols and/or discussions about therapy.5-7,9-12,17,21-26 Involvement of the neck and lower jawline is common in women presenting with the U-shaped pattern of AV, and available studies only evaluate AV involving the face and do not include AV lesions present below the jawline margin. As a result, there is a considerable need for well-designed studies with laboratory assessments to include or exclude underlying detectable excess androgens and to assess the efficacy, tolerability, and safety of specific therapeutic agents both alone and in combination in adult women who present with a U-shaped pattern of AV.17
Other medical considerations that can influence treatment selection and are more likely to be present in adult versus adolescent females include underlying chronic medical disorders; concomitant medications that may interact with other oral agents; potential for pregnancy; age, particularly when prescribing OCs; and the potential desire to stop taking OCs if already used over a prolonged period.6,7
Age-Related Differentiation of Female Subgroups With AV
The age-based dividing line that defines AV in adults versus adolescent females has been described in the literature; however, the basis for published definitions of female subgroups with AV is not well-supported by strong scientific evidence.1-3,5-7,17 The conventional dividing line that was originally selected to define adult females with AV was 25 years of age or older; persistent acne is present both during adolescence and at or after 25 years of age, while late-onset acne is described as AV that first presents at 25 years of age or older.3,5-7
More recently, a range of 18 years or older has been used to classify adult female AV and a range of 12 to 17 years for adolescent female AV in subset analyses that evaluated treatment outcomes in both patient populations from phase 3 pivotal trials completed with adapalene gel 0.3% applied once daily and dapsone gel 5% applied twice daily.14-16 These subanalyses included participants with facial AV that was predominantly moderate in severity, mandated specific lesion count ranges for both comedonal and inflammatory lesions, and included only facial AV that was above the mandibular (jawline) margin.15,16,21,26 Therefore, patients with AV presenting in a U-shaped pattern with involvement below the jawline and on the neck were not included in these study analyses, as these patients were excluded from the phase 3 trials on which the analyses were based. The outcomes of these analyses apply to treatment in women who present with both inflammatory and noninflammatory facial AV lesions, which supports the observation that AV in this patient population is not always predominantly inflammatory and does not always present in a U-shaped distribution.14-16 In fact, a U-shaped pattern of distribution appears to be less common in women with AV than a mixed inflammatory and comedonal distribution that involves the face more diffusely, though more data are needed from well-designed and large-scale epidemiologic and demographic studies.5,14,17
Are there data available on the use of benzoyl peroxide with or without a topical antibiotic in women with AV?
There is a conspicuous absence of prospective clinical trials and retrospective analyses evaluating the specific use of individual AV therapies in adult females, with a particular lack of studies with topical agents (eg, benzoyl peroxide [BP]).14 Subset analyses have been completed for adapalene gel 0.3% and dapsone gel 5%.15,16 Additionally, an age-based subset analysis in females with facial AV also has been completed with clindamycin phosphate (CP) 1.2%–BP 2.5% gel once daily, with data presented but not yet fully published.14
Two identical phase 3, double-blind, randomized, 12-week, 4-arm trials compared treatment outcomes in groups treated with an aqueous-based combination gel formulation containing BP 2.5% and CP 1.2% (n=797), active monad gels (BP [n=809] or CP [n=812]), or vehicle gel (n=395), all applied once daily in patients with facial AV.22 Participants were 12 years or older (mean age range, 19.1–19.6 years; age range, 12.1–70.2 years), were of either gender (approximately 50% split in each study arm), and presented with moderate (approximately 80% of participants) or severe AV (approximately 20% of participants) at baseline. The entry criteria for lesion types and number of lesions were 17 to 40 inflammatory lesions (ie, papules, pustules, <2 nodules)(range of mean number of lesions, 25.8–26.4) and 20 to 100 noninflammatory lesions (ie, closed comedones, open comedones)(range of mean number of lesions, 44.0–47.4). Participant demographics included white (73.9%–77.5%), black/African American (16.1%–20.4%), and Asian (2.1%–3.3%), with the remaining participants distributed among a variety of other ethnic groups such as Native Hawaiian/Native Pacific Islander and Native American Indian/Native Alaskan (collectively <5% in each study arm). Therefore, approximately 1 of every 4 patients had skin of color, which provided good diversity of patients considering the large study size (N=2813). Data analysis included dichotomization of participants by severity rating (moderate or severe based on evaluator global severity score) and skin phototype (Fitzpatrick skin types I–III or IV–VI).22
The pooled results from both studies completed at 68 investigative sites demonstrated that CP 1.2%–BP 2.5% gel was superior in efficacy to each individual monad and to the vehicle in inflammatory, noninflammatory, and total lesion reductions as early as week 4 (P<.001) and at week 12, which was the study end point (P<.001), with superiority also demonstrated in achieving treatment success (defined as a >2 grade improvement according to the evaluator global severity score) compared to the 3 other study arms (P<.001).22 Subject assessments also were consistent with outcomes noted by the investigators. Cutaneous tolerability was favorable and comparable in all 4 study arms with less than 1% of participants discontinuing treatment due to adverse events.22
A subset analysis of the data from the phase 3 pivotal trials with CP 1.2%–BP 2.5% gel was completed to compare reductions in both inflammatory and noninflammatory lesions in female participants who were younger than 25 years and 25 years of age or older in all 4 study arms. This information has been presented14,17 but has not been previously published. Based on the overall results reported in the phase 3 studies, there were no differentiations in skin tolerability or safety based on participant age, gender, or skin type.22 The subanalysis included a total of 1080 females who were younger than 25 years and 395 females who were 25 years of age or older. The lesion reduction outcomes of this subanalysis are presented in the Table. Statistical analyses of the results among these age groups in the 4 study arms were not completed because the objective was to determine if there were any major or obvious differences in reduction of AV lesions based on the conventional dividing line of 25 years of age in adult women as compared to adolescent females treated with CP 1.2%–BP 2.5% gel. In addition, the large difference in numbers of female participants between the 2 age groups (>25 years of age, n=395; <25 years of age, n=1080) at least partially confounds both statistical and observational analysis. Among the women who were 25 years of age or older who were included in the subanalysis, 67.0% and 25.8% were between the ages of 25 to 35 years and 36 to 45 years, respectively. Based on the outcomes reported in the phase 3 trials and in this subgroup analysis, CP 1.2%–BP 2.5% gel applied once daily over a 12-week period appeared overall to be comparably effective in females regardless of age and with no apparent adverse events regarding differences in skin tolerability or safety.14,22 One observation that was noted was the possible trend of greater reduction in both lesion types in women older than 35 years versus younger females with the use of the combination gel or BP alone; however, the number of female participants who were older than 35 years of age was substantially less (n=102) than those who were 35 years of age or younger (n=1345), thus precluding support for any definitive conclusions about this possible trend.22
How can CP 1.2%–BP 2.5% gel be incorporated into a treatment regimen for women with facial AV?
The incorporation of CP 1.2%–BP 2.5% gel into a treatment regimen for women with facial AV is similar to the general use of BP-containing formulations in the overall management of AV.9,14,27,28 Because women with AV commonly present with facial inflammatory lesions and many also with facial comedones, CP 1.2%–BP 2.5% gel is best used once daily in the morning in combination with a topical retinoid in the evening,9,27 which can be achieved with use of CP 1.2%–BP 2.5% gel in the morning and a topical retinoid (ie, tretinoin, adapalene, tazarotene) in the evening or CP 1.2%–tretinoin 0.025% gel in the evening. It is important to note that cutaneous irritation may be more likely if neck lesions are present; the potential for bleaching of colored fabric by BP also is a practical concern.28 In addition, CP 1.2%–BP 2.5% gel may also be used in combination with topical dapsone, but both products should be applied separately at different times of the day to avoid temporary orange discoloration of the skin, which appears to be an uncommon side effect but remains a possibility based on the product information for dapsone gel 5% with regard to its concomitant use with BP.29,30
The management of acne vulgaris (AV) in women has been the subject of considerable attention over the last few years. It has become increasingly recognized that a greater number of patient encounters in dermatology offices involve women with AV who are beyond their adolescent years. Overall, it is estimated that up to approximately 22% of women in the United States are affected by AV, with approximately half of women in their 20s and one-third of women in their 30s reporting some degree of AV.1-4 Among women, the disease shows no predilection for certain skin types or ethnicities, can start during the preteenaged or adolescent years, can persist or recur in adulthood (persistent acne, 75%), or can start in adulthood (late-onset acne, 25%) in females with minimal or no history of AV occurring earlier in life.3,5-7 In the subpopulation of adult women, AV occurs at a time when many expect to be far beyond this “teenage affliction.” Women who are affected commonly express feeling embarrassed and frustrated.5-8
Most of the emphasis in the literature and in presentations at dermatology meetings regarding the management of AV in adult women has focused on excluding underlying disorders that cause excess androgens (eg, polycystic ovary syndrome, congenital adrenal hyperplasia, tumors, exogenous sources) as well as the use of systemic therapies such as oral contraceptives (OCs) and spironolactone.5-7,9,10 Little attention has been given to the selection of topical therapies in this patient population, especially with regard to evidence from clinical studies. To date, results from published study analyses using topical agents specifically for adult females with facial AV have only included adapalene gel 0.3% applied once daily and dapsone gel 5% applied twice daily.11-13 Both agents have been evaluated in subset analysis comparisons of outcomes in women aged 18 years and older versus adolescents aged 12 to 17 years based on data from 12-week phase 3 pivotal trials.14-16
Are there clinically relevant differences between AV in adult versus adolescent females?
Although much has been written about AV in women, epidemiology, demographics, assessment of clinical presentation, and correlation of clinical presentation with excess androgens have not been emphasized,1-3,5-10,17 likely due to marketing campaigns that emphasize AV as a disorder that predominantly affects teenagers as well as the focus on optimal use of oral spironolactone and/or OCs in the management of AV in the adult female population. Attention to spironolactone use is important because it is not approved by the US Food and Drug Administration for the treatment of AV. Spironolactone carries certain black-box warnings that may not be clinically relevant in all patients but still require attention. It also is associated with risks if taken during pregnancy, and it is a potassium-sparing diuretic with potential for hyperkalemia, especially in patients with reduced renal function or those who are taking potassium supplements or certain other medications.6,9,11,17 Use of OCs to treat AV also is not without potential risks, with specific warnings and relative contraindications reported, especially in relation to increased risks for cardiac complications, stroke, and thromboembolism.6,9,11,12 Because adult females are in a different stage of life than teenagers, there are defined psychosocial and medical considerations in managing AV in these patients compared with adolescents.5-8,17 Importantly for both clinicians and patients, addressing these differences and considerations can have a major impact on whether or not women with AV experience successful treatment outcomes.1-3,5,6,8,10,13 Skin color and ethnicity also can affect the psychosocial and physical factors that influence the overall management of adult female patients with AV, including selection of therapies and handling long-term visible sequelae that occur in some AV patients, such as dyschromia (eg, persistent or postinflammatory erythema or hyperpigmentation) and acne scarring.5-8,13,17-20
Psychosocial Considerations
With regard to psychosocial, emotional, and attitudinal considerations in women with AV, common findings include concern or frustration regarding the presence of AV beyond adolescence; anxiety; symptoms of depression; decreased self-confidence; increased self-consciousness, especially during public interactions or intimate situations; and interference with steady concentration at work or school.5,6,8,13 Long-term complications of AV, such as dyschromia and acne scarring, are more likely to be encountered in adult patients, especially if they had AV as a teenager, with women reporting that they remain conscious of these adverse sequelae.8 It is estimated that approximately three-fourths of women with AV also had AV as teenagers; therefore, most of them have already used many over-the-counter and prescription therapies and are likely to want treatments that are newer, well-tolerated, safe, and known to be effective in adult women.8,16,17 Convenience and simplicity are vital components of treatment selection and regimen design, as many women with AV frequently face time constraints in their daily routines due to family, social, employment, and home-related demands and responsibilities.6-8,17
Medical Considerations
It is apparent from reports in the literature as well as from clinical experience that some women with AV present with a U-shaped pattern of involvement on the face,5-7,10,13,17 which refers to the presence of predominantly inflammatory papules (many of them deep) and some nodules on the lower face, jawline, and anterolateral neck region, with comedones often sparse or absent.5-7 It often is perceived and may be true that women who present with this pattern of distribution are more androgen sensitive despite having normal serum androgen levels or in some cases exhibit detectable excess androgens (eg, in the setting of polycystic ovary syndrome) and may be more likely to respond to hormonal therapies (eg, spironolactone, OCs) than those with mixed facial AV (ie, multiple comedonal and inflammatory acne lesions, not limited to a U-shaped pattern, similar to adolescent AV), but data are limited to support differentiation between the U-shaped pattern group and the conventional mixed facial AV group.5-7,17 Adult and adolescent females in both groups sometimes report perimenstrual flares and frequent persistent papular AV that tends to concentrate on the perioral and chin area.
It is also important to consider that the current literature suggests approximately three-fourths of women with AV report that they also had AV as a teenager, with many indicating the same clinical pattern of AV and approximately one-third reporting AV that is more severe in adulthood than adolescence.5-8,17 The available literature on topical and oral therapies used to treat AV in both adolescent and adult females predominantly focuses on inclusion of both inflammatory and noninflammatory (comedonal) facial AV lesions, does not specifically address or include the U-shaped pattern of AV in adult women for inclusion in studies that evaluate efficacy in this subgroup, and does not include AV involving the neck region and below the jawline margin as part of any study protocols and/or discussions about therapy.5-7,9-12,17,21-26 Involvement of the neck and lower jawline is common in women presenting with the U-shaped pattern of AV, and available studies only evaluate AV involving the face and do not include AV lesions present below the jawline margin. As a result, there is a considerable need for well-designed studies with laboratory assessments to include or exclude underlying detectable excess androgens and to assess the efficacy, tolerability, and safety of specific therapeutic agents both alone and in combination in adult women who present with a U-shaped pattern of AV.17
Other medical considerations that can influence treatment selection and are more likely to be present in adult versus adolescent females include underlying chronic medical disorders; concomitant medications that may interact with other oral agents; potential for pregnancy; age, particularly when prescribing OCs; and the potential desire to stop taking OCs if already used over a prolonged period.6,7
Age-Related Differentiation of Female Subgroups With AV
The age-based dividing line that defines AV in adults versus adolescent females has been described in the literature; however, the basis for published definitions of female subgroups with AV is not well-supported by strong scientific evidence.1-3,5-7,17 The conventional dividing line that was originally selected to define adult females with AV was 25 years of age or older; persistent acne is present both during adolescence and at or after 25 years of age, while late-onset acne is described as AV that first presents at 25 years of age or older.3,5-7
More recently, a range of 18 years or older has been used to classify adult female AV and a range of 12 to 17 years for adolescent female AV in subset analyses that evaluated treatment outcomes in both patient populations from phase 3 pivotal trials completed with adapalene gel 0.3% applied once daily and dapsone gel 5% applied twice daily.14-16 These subanalyses included participants with facial AV that was predominantly moderate in severity, mandated specific lesion count ranges for both comedonal and inflammatory lesions, and included only facial AV that was above the mandibular (jawline) margin.15,16,21,26 Therefore, patients with AV presenting in a U-shaped pattern with involvement below the jawline and on the neck were not included in these study analyses, as these patients were excluded from the phase 3 trials on which the analyses were based. The outcomes of these analyses apply to treatment in women who present with both inflammatory and noninflammatory facial AV lesions, which supports the observation that AV in this patient population is not always predominantly inflammatory and does not always present in a U-shaped distribution.14-16 In fact, a U-shaped pattern of distribution appears to be less common in women with AV than a mixed inflammatory and comedonal distribution that involves the face more diffusely, though more data are needed from well-designed and large-scale epidemiologic and demographic studies.5,14,17
Are there data available on the use of benzoyl peroxide with or without a topical antibiotic in women with AV?
There is a conspicuous absence of prospective clinical trials and retrospective analyses evaluating the specific use of individual AV therapies in adult females, with a particular lack of studies with topical agents (eg, benzoyl peroxide [BP]).14 Subset analyses have been completed for adapalene gel 0.3% and dapsone gel 5%.15,16 Additionally, an age-based subset analysis in females with facial AV also has been completed with clindamycin phosphate (CP) 1.2%–BP 2.5% gel once daily, with data presented but not yet fully published.14
Two identical phase 3, double-blind, randomized, 12-week, 4-arm trials compared treatment outcomes in groups treated with an aqueous-based combination gel formulation containing BP 2.5% and CP 1.2% (n=797), active monad gels (BP [n=809] or CP [n=812]), or vehicle gel (n=395), all applied once daily in patients with facial AV.22 Participants were 12 years or older (mean age range, 19.1–19.6 years; age range, 12.1–70.2 years), were of either gender (approximately 50% split in each study arm), and presented with moderate (approximately 80% of participants) or severe AV (approximately 20% of participants) at baseline. The entry criteria for lesion types and number of lesions were 17 to 40 inflammatory lesions (ie, papules, pustules, <2 nodules)(range of mean number of lesions, 25.8–26.4) and 20 to 100 noninflammatory lesions (ie, closed comedones, open comedones)(range of mean number of lesions, 44.0–47.4). Participant demographics included white (73.9%–77.5%), black/African American (16.1%–20.4%), and Asian (2.1%–3.3%), with the remaining participants distributed among a variety of other ethnic groups such as Native Hawaiian/Native Pacific Islander and Native American Indian/Native Alaskan (collectively <5% in each study arm). Therefore, approximately 1 of every 4 patients had skin of color, which provided good diversity of patients considering the large study size (N=2813). Data analysis included dichotomization of participants by severity rating (moderate or severe based on evaluator global severity score) and skin phototype (Fitzpatrick skin types I–III or IV–VI).22
The pooled results from both studies completed at 68 investigative sites demonstrated that CP 1.2%–BP 2.5% gel was superior in efficacy to each individual monad and to the vehicle in inflammatory, noninflammatory, and total lesion reductions as early as week 4 (P<.001) and at week 12, which was the study end point (P<.001), with superiority also demonstrated in achieving treatment success (defined as a >2 grade improvement according to the evaluator global severity score) compared to the 3 other study arms (P<.001).22 Subject assessments also were consistent with outcomes noted by the investigators. Cutaneous tolerability was favorable and comparable in all 4 study arms with less than 1% of participants discontinuing treatment due to adverse events.22
A subset analysis of the data from the phase 3 pivotal trials with CP 1.2%–BP 2.5% gel was completed to compare reductions in both inflammatory and noninflammatory lesions in female participants who were younger than 25 years and 25 years of age or older in all 4 study arms. This information has been presented14,17 but has not been previously published. Based on the overall results reported in the phase 3 studies, there were no differentiations in skin tolerability or safety based on participant age, gender, or skin type.22 The subanalysis included a total of 1080 females who were younger than 25 years and 395 females who were 25 years of age or older. The lesion reduction outcomes of this subanalysis are presented in the Table. Statistical analyses of the results among these age groups in the 4 study arms were not completed because the objective was to determine if there were any major or obvious differences in reduction of AV lesions based on the conventional dividing line of 25 years of age in adult women as compared to adolescent females treated with CP 1.2%–BP 2.5% gel. In addition, the large difference in numbers of female participants between the 2 age groups (>25 years of age, n=395; <25 years of age, n=1080) at least partially confounds both statistical and observational analysis. Among the women who were 25 years of age or older who were included in the subanalysis, 67.0% and 25.8% were between the ages of 25 to 35 years and 36 to 45 years, respectively. Based on the outcomes reported in the phase 3 trials and in this subgroup analysis, CP 1.2%–BP 2.5% gel applied once daily over a 12-week period appeared overall to be comparably effective in females regardless of age and with no apparent adverse events regarding differences in skin tolerability or safety.14,22 One observation that was noted was the possible trend of greater reduction in both lesion types in women older than 35 years versus younger females with the use of the combination gel or BP alone; however, the number of female participants who were older than 35 years of age was substantially less (n=102) than those who were 35 years of age or younger (n=1345), thus precluding support for any definitive conclusions about this possible trend.22
How can CP 1.2%–BP 2.5% gel be incorporated into a treatment regimen for women with facial AV?
The incorporation of CP 1.2%–BP 2.5% gel into a treatment regimen for women with facial AV is similar to the general use of BP-containing formulations in the overall management of AV.9,14,27,28 Because women with AV commonly present with facial inflammatory lesions and many also with facial comedones, CP 1.2%–BP 2.5% gel is best used once daily in the morning in combination with a topical retinoid in the evening,9,27 which can be achieved with use of CP 1.2%–BP 2.5% gel in the morning and a topical retinoid (ie, tretinoin, adapalene, tazarotene) in the evening or CP 1.2%–tretinoin 0.025% gel in the evening. It is important to note that cutaneous irritation may be more likely if neck lesions are present; the potential for bleaching of colored fabric by BP also is a practical concern.28 In addition, CP 1.2%–BP 2.5% gel may also be used in combination with topical dapsone, but both products should be applied separately at different times of the day to avoid temporary orange discoloration of the skin, which appears to be an uncommon side effect but remains a possibility based on the product information for dapsone gel 5% with regard to its concomitant use with BP.29,30
1. Perkins AC, Maglione J, Hillebrand GG, et al. Acne vulgaris in women: prevalence across the life span. J Womens Health. 2012;21:223-230.
2. Zeichner J. Evaluating and treating the adult female patient with acne. J Drugs Dermatol. 2013;12:1416-1427.
3. Goulden V, Stables GI, Cunliffe WJ. Prevalence of facial acne in adults. J Am Acad Dermatol. 1999;41:577-580.
4. Collier CN, Harper J, Cafardi JA, et al. The prevalence of acne in adults 20 years and older. J Am Acad Dermatol. 2008;58:56-59.
5. Dreno B, Layton A, Zouboulis CC, et al. Adult female acne: a new paradigm. J Eur Acad Dermatol Venereol. 2013;27:1063-1070.
6. Kim GK, Del Rosso JQ. Oral spironolactone in post-teenage female patients with acne vulgaris: practical considerations for the clinician based on current data and clinical experience. J Clin Aesthet Dermatol. 2012;5:37-50.
7. Kim GK, Michaels BB. Post-adolescent acne in women: more common and more clinical considerations. J Drugs Dermatol. 2012;11:708-713.
8. Tanghetti EA, Kawata AK, Daniels SR, et al. Understanding the burden of adult female acne. J Clin Aesthet Dermatol. 2014;7:22-30.
9. Gollnick H, Cunliffe W, Berson D, et al. Management of acne: a report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49(suppl 1):1-37.
10. Thiboutot DM. Endocrinological evaluation and hormonal therapy for women with difficult acne. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):57-61.
11. Sawaya ME, Samani N. Antiandrogens and androgen receptors. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelphia, PA: Saunders-Elsevier; 2012:361-374.
12. Harper JC. Should dermatologists prescribe hormonal contraceptives for acne? Dermatol Ther. 2009;22:452-457.
13. Preneau S, Dreno B. Female acne: a different subtype of teenager acne? J Eur Acad Dermatol Venereol. 2012;26:277-282.
14. Del Rosso JQ, Zeichner JA. What’s new in the medicine cabinet?: a panoramic review of clinically relevant information for the busy dermatologist. J Clin Aesthet Dermatol. 2014;7:26-30.
15. Berson D, Alexis A. Adapalene 0.3% for the treatment of acne in women. J Clin Aesthet Dermatol. 2013;6:32-35.
16. Del Rosso JQ, Kircik L, Gallagher C. Facing up to adult women with acne vulgaris: an analysis of pivotal trial data on dapsone 5% gel in the adult female population. Poster presented at: Fall Clinical Dermatology; October 2013; Las Vegas, NV.
17. Del Rosso JQ. Management of acne with oral spironolactone. Presented at: American Academy of Dermatology Summer Meeting; August 2013; Boston, MA.
18. Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
19. Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
20. Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasian, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
21. Draelos ZD, Carter E, Maloney JM, et al. Two randomized studies demonstrate the efficacy and safety of dapsone gel, 5% for the treatment of acne vulgaris [published online ahead of print January 17, 2007]. J Am Acad Dermatol. 2007;56:439.e1-439.e10.
22. Thiboutot D, Zaenglein A, Weiss J, et al. An aqueous gel fixed combination of clindamycin phosphate 1.2% and benzoyl peroxide 2.5% for the once-daily treatment of moderate to severe acne vulgaris: assessment of efficacy and safety in 2813 patients. J Am Acad Dermatol. 2008;59:792-800.
23. Schlessinger J, Menter A, Gold M, et al. Clinical safety and efficacy studies of a novel formulation combining
1.2% clindamycin phosphate and 0.025% tretinoin for the treatment of acne vulgaris. J Drugs Dermatol. 2007;6:607-615.
24. Fleischer AB Jr, Dinehart S, Stough D, et al. Safety and efficacy of a new extended-release formulation of minocycline. Cutis. 2006;78(suppl 4):21-31.
25. Gollnick HP, Draelos Z, Glenn MJ, et al. Adapalene-benzoyl peroxide, a unique fixed-dose combination topical gel for the treatment of acne vulgaris: a transatlantic, randomized, double-blind, controlled study in 1670 patients. Br J Dermatol. 2009;161:1180-1189.
26. Thiboutot D, Arsonnaud S, Soto P. Efficacy and tolerability of adapalene 0.3% gel compared to tazarotene 0.1% gel in the treatment of acne vulgaris. J Drugs Dermatol. 2008;7(suppl 6):3-10.
27. Zeichner JA. Optimizing topical combination therapy for the treatment of acne vulgaris. J Drugs Dermatol. 2012;11:313-317.
28. Tanghetti EA, Popp KF. A current review of topical benzoyl peroxide: new perspectives on formulation and utilization. Dermatol Clin. 2009;27:17-24.
29. Fleischer AB, Shalita A, Eichenfield LF. Dapsone gel 5% in combination with adapalene gel 0.1%, benzoyl peroxide gel 4% or moisturizer for the treatment of acne vulgaris: a 12-week, randomized, double-blind study. J Drugs Dermatol. 2010;9:33-40.
30. Aczone (dapsone gel 5%) [package insert]. Irvine, CA: Allergan, Inc; 2013.
1. Perkins AC, Maglione J, Hillebrand GG, et al. Acne vulgaris in women: prevalence across the life span. J Womens Health. 2012;21:223-230.
2. Zeichner J. Evaluating and treating the adult female patient with acne. J Drugs Dermatol. 2013;12:1416-1427.
3. Goulden V, Stables GI, Cunliffe WJ. Prevalence of facial acne in adults. J Am Acad Dermatol. 1999;41:577-580.
4. Collier CN, Harper J, Cafardi JA, et al. The prevalence of acne in adults 20 years and older. J Am Acad Dermatol. 2008;58:56-59.
5. Dreno B, Layton A, Zouboulis CC, et al. Adult female acne: a new paradigm. J Eur Acad Dermatol Venereol. 2013;27:1063-1070.
6. Kim GK, Del Rosso JQ. Oral spironolactone in post-teenage female patients with acne vulgaris: practical considerations for the clinician based on current data and clinical experience. J Clin Aesthet Dermatol. 2012;5:37-50.
7. Kim GK, Michaels BB. Post-adolescent acne in women: more common and more clinical considerations. J Drugs Dermatol. 2012;11:708-713.
8. Tanghetti EA, Kawata AK, Daniels SR, et al. Understanding the burden of adult female acne. J Clin Aesthet Dermatol. 2014;7:22-30.
9. Gollnick H, Cunliffe W, Berson D, et al. Management of acne: a report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49(suppl 1):1-37.
10. Thiboutot DM. Endocrinological evaluation and hormonal therapy for women with difficult acne. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):57-61.
11. Sawaya ME, Samani N. Antiandrogens and androgen receptors. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelphia, PA: Saunders-Elsevier; 2012:361-374.
12. Harper JC. Should dermatologists prescribe hormonal contraceptives for acne? Dermatol Ther. 2009;22:452-457.
13. Preneau S, Dreno B. Female acne: a different subtype of teenager acne? J Eur Acad Dermatol Venereol. 2012;26:277-282.
14. Del Rosso JQ, Zeichner JA. What’s new in the medicine cabinet?: a panoramic review of clinically relevant information for the busy dermatologist. J Clin Aesthet Dermatol. 2014;7:26-30.
15. Berson D, Alexis A. Adapalene 0.3% for the treatment of acne in women. J Clin Aesthet Dermatol. 2013;6:32-35.
16. Del Rosso JQ, Kircik L, Gallagher C. Facing up to adult women with acne vulgaris: an analysis of pivotal trial data on dapsone 5% gel in the adult female population. Poster presented at: Fall Clinical Dermatology; October 2013; Las Vegas, NV.
17. Del Rosso JQ. Management of acne with oral spironolactone. Presented at: American Academy of Dermatology Summer Meeting; August 2013; Boston, MA.
18. Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
19. Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
20. Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasian, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
21. Draelos ZD, Carter E, Maloney JM, et al. Two randomized studies demonstrate the efficacy and safety of dapsone gel, 5% for the treatment of acne vulgaris [published online ahead of print January 17, 2007]. J Am Acad Dermatol. 2007;56:439.e1-439.e10.
22. Thiboutot D, Zaenglein A, Weiss J, et al. An aqueous gel fixed combination of clindamycin phosphate 1.2% and benzoyl peroxide 2.5% for the once-daily treatment of moderate to severe acne vulgaris: assessment of efficacy and safety in 2813 patients. J Am Acad Dermatol. 2008;59:792-800.
23. Schlessinger J, Menter A, Gold M, et al. Clinical safety and efficacy studies of a novel formulation combining
1.2% clindamycin phosphate and 0.025% tretinoin for the treatment of acne vulgaris. J Drugs Dermatol. 2007;6:607-615.
24. Fleischer AB Jr, Dinehart S, Stough D, et al. Safety and efficacy of a new extended-release formulation of minocycline. Cutis. 2006;78(suppl 4):21-31.
25. Gollnick HP, Draelos Z, Glenn MJ, et al. Adapalene-benzoyl peroxide, a unique fixed-dose combination topical gel for the treatment of acne vulgaris: a transatlantic, randomized, double-blind, controlled study in 1670 patients. Br J Dermatol. 2009;161:1180-1189.
26. Thiboutot D, Arsonnaud S, Soto P. Efficacy and tolerability of adapalene 0.3% gel compared to tazarotene 0.1% gel in the treatment of acne vulgaris. J Drugs Dermatol. 2008;7(suppl 6):3-10.
27. Zeichner JA. Optimizing topical combination therapy for the treatment of acne vulgaris. J Drugs Dermatol. 2012;11:313-317.
28. Tanghetti EA, Popp KF. A current review of topical benzoyl peroxide: new perspectives on formulation and utilization. Dermatol Clin. 2009;27:17-24.
29. Fleischer AB, Shalita A, Eichenfield LF. Dapsone gel 5% in combination with adapalene gel 0.1%, benzoyl peroxide gel 4% or moisturizer for the treatment of acne vulgaris: a 12-week, randomized, double-blind study. J Drugs Dermatol. 2010;9:33-40.
30. Aczone (dapsone gel 5%) [package insert]. Irvine, CA: Allergan, Inc; 2013.
Practice Points
- Adult women with acne vulgaris (AV) can present with a U-shaped pattern of predominantly inflammatory papules, pustules, and nodules involving the lower face, jawline region, and anterior and lateral neck; however, many women present with mixed comedonal and inflammatory acne involving the face more diffusely with a presentation similar to what is typically seen in adolescent AV.
- Topical therapy for adult women with acne has been evaluated in subanalyses of data from phase 3 studies with good efficacy and favorable tolerability shown with adapalene gel 0.3% once daily, dapsone gel 5% twice daily, and clindamycin phosphate 1.2%–benzoyl peroxide (BP) 2.5% gel once daily. The latter agent requires cautious use below the jawline margin, as BP can bleach colored fabric of clothing such as shirts, blouses, and sweaters. These topical agents may be used in combination based on the clinical situation, including with systemic therapies for AV.
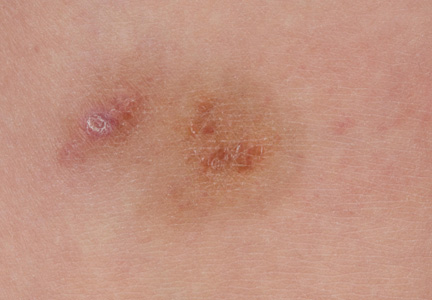
Lipidized Dermatofibroma
Lipidized dermatofibromas most commonly are found on the ankles, which has led some authors to refer to these lesions as ankle-type fibrous histiocytomas.1 Compared to ordinary dermatofibromas, patients with lipidized dermatofibromas tend to be older, most commonly presenting in the fifth or sixth decades of life, and are predominantly male. Lipidized dermatofibromas typically present as well-circumscribed solitary nodules in the dermis. Characteristic features include numerous xanthomatous cells dissected by distinctive hyalinized wiry collagen fibers (Figures 1 and 2).1 Xanthomatous cells can be round, polygonal, or stellate in shape. These characteristic features in combination with others of dermatofibromas (eg, epidermal acanthosis [Figure 1]) fulfill the criteria for diagnosis of a lipidized dermatofibroma. Additionally, lipidized dermatofibromas tend to be larger than ordinary dermatofibromas, which typically are less than 2 cm in diameter.1
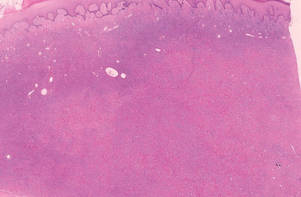 |
Figure 1. Lipidized dermatofibromas are characterized by classic epidermal features of dermatofibromas, such as acanthosis, along with numerous foam cells and extensive stromal hyalinization (H&E, original magnification ×1.5). |
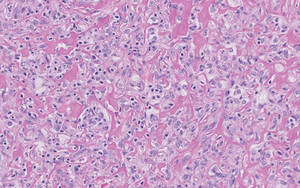 |
Figure 2. Higher-power view of a lipidized dermatofibroma shows the characteristic irregular dissection of hyalinized wiry collagen fibers between the xanthomatous cells (H&E, original magnification ×20). |
Eruptive xanthomas are characterized by a lacelike infiltrate of extravascular lipid deposits between collagen bundles (Figure 3).2 Granular cell tumors are composed of sheets and/or nests of large cells with abundant eosinophilic cytoplasm and may be confused with lipidized dermatofibromas, as they also may induce overlying pseudoepitheliomatous hyperplasia3; however, on closer examination of the cells, the cytoplasm is found to be granular (Figure 4), which contrasts the finely vacuolated cytoplasm of xanthomatous cells found in lipidized dermatofibromas. Giant lysosomal granules (eg, pustulo-ovoid bodies of Milian) are present in some cases.2 Of note, an unusual variant of dermatofibroma exists that features prominent granular cells.4
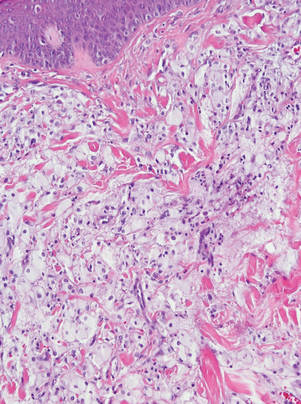 |
Figure 3. Lacelike deposition of extravascular lipid deposits is seen infiltrating between collagen bundles in an eruptive xanthoma (H&E, original magnification ×20). |
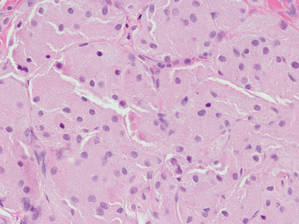 |
Figure 4. An abundant eosinophilic, finely granular cytoplasm is characteristic of granular cell tumor (H&E, original magnification ×40). |
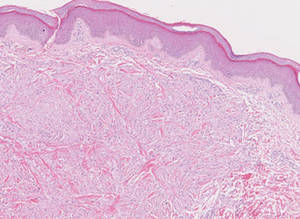
Tuberous xanthomas most commonly occur around the pressure areas, such as the knees, elbows, and buttocks. Foam cells are a main feature of tuberous xanthomas and are arranged in large aggregates throughout the dermis.2 Tuberous xanthomas lack Touton giant cells or inflammatory cells. Older lesions tend to develop substantial fibrosis (Figure 5). Although foam cells can be present in older lesions, they are never as conspicuous as those found in other xanthomas.
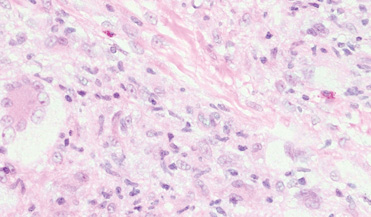
Xanthogranulomas commonly occur on the head and neck. Findings noted on low magnification include a well-circumscribed exophytic nodule and an epidermal collarette, which help to easily distinguish xanthogranulomas from lipidized dermatofibromas. Additionally, the presence of a more prominent inflammatory infiltrate, which often includes eosinophils, as well as multinucleated Touton giant cells (Figure 6) and histiocytes with more eosinophilic and less xanthomatous cytoplasm can help distinguish between the lesions.1,5 Notably, Touton giant cells also can be seen in lipidized dermatofibromas,1 but the presence of unique features such as distinctive stromal hyalinization are clues to the correct diagnosis of a lipidized dermatofibroma.
- Iwata J, Fletcher CD. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol. 2000;22:126-134.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Elsevier Health Sciences; 2009.
- Elston DM, Ferringer T. Dermatopathology. Philadelphia, PA: Saunders Elsevier; 2009.
- Yogesh TL, Sowmya SV. Granules in granular cell lesions of the head and neck: a review. ISRN Pathol. 2011;2011:10.
- Fujita Y, Tsunemi Y, Kadono T, et al. Lipidized fibrous histiocytoma on the left condyle of the tibia. Int J Dermatol. 2011;50:634-636.
Lipidized dermatofibromas most commonly are found on the ankles, which has led some authors to refer to these lesions as ankle-type fibrous histiocytomas.1 Compared to ordinary dermatofibromas, patients with lipidized dermatofibromas tend to be older, most commonly presenting in the fifth or sixth decades of life, and are predominantly male. Lipidized dermatofibromas typically present as well-circumscribed solitary nodules in the dermis. Characteristic features include numerous xanthomatous cells dissected by distinctive hyalinized wiry collagen fibers (Figures 1 and 2).1 Xanthomatous cells can be round, polygonal, or stellate in shape. These characteristic features in combination with others of dermatofibromas (eg, epidermal acanthosis [Figure 1]) fulfill the criteria for diagnosis of a lipidized dermatofibroma. Additionally, lipidized dermatofibromas tend to be larger than ordinary dermatofibromas, which typically are less than 2 cm in diameter.1
|
Figure 1. Lipidized dermatofibromas are characterized by classic epidermal features of dermatofibromas, such as acanthosis, along with numerous foam cells and extensive stromal hyalinization (H&E, original magnification ×1.5). |
|
Figure 2. Higher-power view of a lipidized dermatofibroma shows the characteristic irregular dissection of hyalinized wiry collagen fibers between the xanthomatous cells (H&E, original magnification ×20). |
Eruptive xanthomas are characterized by a lacelike infiltrate of extravascular lipid deposits between collagen bundles (Figure 3).2 Granular cell tumors are composed of sheets and/or nests of large cells with abundant eosinophilic cytoplasm and may be confused with lipidized dermatofibromas, as they also may induce overlying pseudoepitheliomatous hyperplasia3; however, on closer examination of the cells, the cytoplasm is found to be granular (Figure 4), which contrasts the finely vacuolated cytoplasm of xanthomatous cells found in lipidized dermatofibromas. Giant lysosomal granules (eg, pustulo-ovoid bodies of Milian) are present in some cases.2 Of note, an unusual variant of dermatofibroma exists that features prominent granular cells.4
|
Figure 3. Lacelike deposition of extravascular lipid deposits is seen infiltrating between collagen bundles in an eruptive xanthoma (H&E, original magnification ×20). |
|
Figure 4. An abundant eosinophilic, finely granular cytoplasm is characteristic of granular cell tumor (H&E, original magnification ×40). |
Tuberous xanthomas most commonly occur around the pressure areas, such as the knees, elbows, and buttocks. Foam cells are a main feature of tuberous xanthomas and are arranged in large aggregates throughout the dermis.2 Tuberous xanthomas lack Touton giant cells or inflammatory cells. Older lesions tend to develop substantial fibrosis (Figure 5). Although foam cells can be present in older lesions, they are never as conspicuous as those found in other xanthomas.
Xanthogranulomas commonly occur on the head and neck. Findings noted on low magnification include a well-circumscribed exophytic nodule and an epidermal collarette, which help to easily distinguish xanthogranulomas from lipidized dermatofibromas. Additionally, the presence of a more prominent inflammatory infiltrate, which often includes eosinophils, as well as multinucleated Touton giant cells (Figure 6) and histiocytes with more eosinophilic and less xanthomatous cytoplasm can help distinguish between the lesions.1,5 Notably, Touton giant cells also can be seen in lipidized dermatofibromas,1 but the presence of unique features such as distinctive stromal hyalinization are clues to the correct diagnosis of a lipidized dermatofibroma.
Lipidized dermatofibromas most commonly are found on the ankles, which has led some authors to refer to these lesions as ankle-type fibrous histiocytomas.1 Compared to ordinary dermatofibromas, patients with lipidized dermatofibromas tend to be older, most commonly presenting in the fifth or sixth decades of life, and are predominantly male. Lipidized dermatofibromas typically present as well-circumscribed solitary nodules in the dermis. Characteristic features include numerous xanthomatous cells dissected by distinctive hyalinized wiry collagen fibers (Figures 1 and 2).1 Xanthomatous cells can be round, polygonal, or stellate in shape. These characteristic features in combination with others of dermatofibromas (eg, epidermal acanthosis [Figure 1]) fulfill the criteria for diagnosis of a lipidized dermatofibroma. Additionally, lipidized dermatofibromas tend to be larger than ordinary dermatofibromas, which typically are less than 2 cm in diameter.1
|
Figure 1. Lipidized dermatofibromas are characterized by classic epidermal features of dermatofibromas, such as acanthosis, along with numerous foam cells and extensive stromal hyalinization (H&E, original magnification ×1.5). |
|
Figure 2. Higher-power view of a lipidized dermatofibroma shows the characteristic irregular dissection of hyalinized wiry collagen fibers between the xanthomatous cells (H&E, original magnification ×20). |
Eruptive xanthomas are characterized by a lacelike infiltrate of extravascular lipid deposits between collagen bundles (Figure 3).2 Granular cell tumors are composed of sheets and/or nests of large cells with abundant eosinophilic cytoplasm and may be confused with lipidized dermatofibromas, as they also may induce overlying pseudoepitheliomatous hyperplasia3; however, on closer examination of the cells, the cytoplasm is found to be granular (Figure 4), which contrasts the finely vacuolated cytoplasm of xanthomatous cells found in lipidized dermatofibromas. Giant lysosomal granules (eg, pustulo-ovoid bodies of Milian) are present in some cases.2 Of note, an unusual variant of dermatofibroma exists that features prominent granular cells.4
|
Figure 3. Lacelike deposition of extravascular lipid deposits is seen infiltrating between collagen bundles in an eruptive xanthoma (H&E, original magnification ×20). |
|
Figure 4. An abundant eosinophilic, finely granular cytoplasm is characteristic of granular cell tumor (H&E, original magnification ×40). |
Tuberous xanthomas most commonly occur around the pressure areas, such as the knees, elbows, and buttocks. Foam cells are a main feature of tuberous xanthomas and are arranged in large aggregates throughout the dermis.2 Tuberous xanthomas lack Touton giant cells or inflammatory cells. Older lesions tend to develop substantial fibrosis (Figure 5). Although foam cells can be present in older lesions, they are never as conspicuous as those found in other xanthomas.
Xanthogranulomas commonly occur on the head and neck. Findings noted on low magnification include a well-circumscribed exophytic nodule and an epidermal collarette, which help to easily distinguish xanthogranulomas from lipidized dermatofibromas. Additionally, the presence of a more prominent inflammatory infiltrate, which often includes eosinophils, as well as multinucleated Touton giant cells (Figure 6) and histiocytes with more eosinophilic and less xanthomatous cytoplasm can help distinguish between the lesions.1,5 Notably, Touton giant cells also can be seen in lipidized dermatofibromas,1 but the presence of unique features such as distinctive stromal hyalinization are clues to the correct diagnosis of a lipidized dermatofibroma.
- Iwata J, Fletcher CD. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol. 2000;22:126-134.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Elsevier Health Sciences; 2009.
- Elston DM, Ferringer T. Dermatopathology. Philadelphia, PA: Saunders Elsevier; 2009.
- Yogesh TL, Sowmya SV. Granules in granular cell lesions of the head and neck: a review. ISRN Pathol. 2011;2011:10.
- Fujita Y, Tsunemi Y, Kadono T, et al. Lipidized fibrous histiocytoma on the left condyle of the tibia. Int J Dermatol. 2011;50:634-636.
- Iwata J, Fletcher CD. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol. 2000;22:126-134.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Elsevier Health Sciences; 2009.
- Elston DM, Ferringer T. Dermatopathology. Philadelphia, PA: Saunders Elsevier; 2009.
- Yogesh TL, Sowmya SV. Granules in granular cell lesions of the head and neck: a review. ISRN Pathol. 2011;2011:10.
- Fujita Y, Tsunemi Y, Kadono T, et al. Lipidized fibrous histiocytoma on the left condyle of the tibia. Int J Dermatol. 2011;50:634-636.
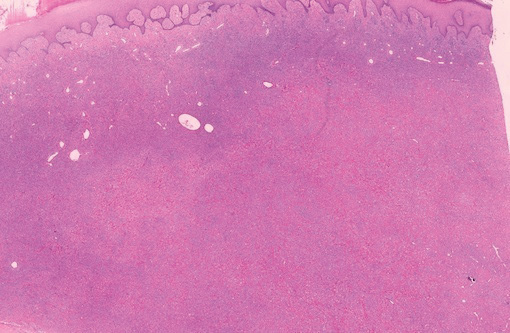
What Is Your Diagnosis? Rhino-orbital-cerebral Mucormycosis
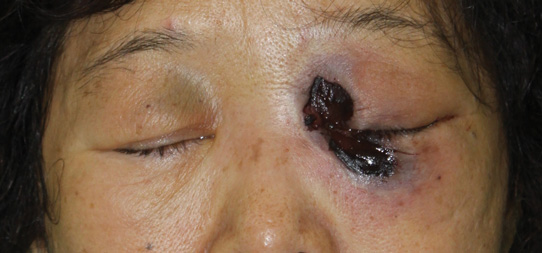
A 56-year-old woman presented with painful, erythematous to violaceous patches with necrosis of the left eye and periorbital area of 1 day’s duration. She reported headaches and periorbital pain in the 3 weeks prior to presentation. She was being treated for hypertension, type 2 diabetes mellitus, and end-stage renal disease. The patient denied prior trauma to the area.
The Diagnosis: Rhino-orbital-cerebral Mucormycosis
Cutaneous examination revealed a dusky, erythematous to violaceous patch with a necrotic center involving the left eye and periorbital area (Figure 1). The differential diagnosis included herpes zoster, cellulitis, and fungal infection. We obtained patient consent for a punch biopsy. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (Figure 2). Magnetic resonance imaging of the orbit and head showed involvement of the periorbital soft tissues; the ethmoidal, sphenoidal, and maxillary sinuses; and the left medial temporal lobe. The patient was started on an empirical antifungal treatment of amphotericin B deoxycholate 50 mg daily but died 4 days later due to multiorgan failure.
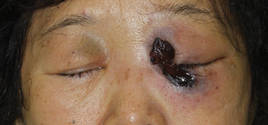 |
| Figure 1. A dusky, erythematous to violaceous patch with a necrotic center on the left eye and periorbital area. |
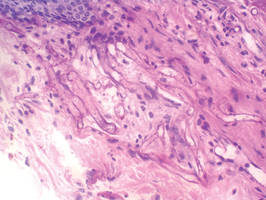 |
| Figure 2. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (periodic acid–Schiff, original magnification ×400). |
Mucormycosis is a rare but fatal infection that may rapidly progress.1 Risk factors include defects in host defense such as malignancy, immunodeficiency from bone marrow or solid organ transplantation, diabetes mellitus, malnutrition, abnormal metabolic states, and deferoxamine use.1,2 Rhino-orbital-cerebral mucormycosis usually starts with eye or facial pain and unilateral facial swelling.3,4 Visual impairment, fever, and mental status changes may follow.1,3,4 Skin findings may progress from erythema to violaceous color changes and lastly to a black necrotic eschar resulting from tissue infarction.5
Radiologic imaging may be helpful but rarely is diagnostic in mucormycosis, and reliable serologic tests are lacking.1 Therefore, suspicion of mucormycosis based on clinical and histopathologic factors followed by immediate initiation of empirical antifungal treatment is critical. The key factors in treating mucormycosis include early diagnosis, correction of underlying risk factors, prompt antifungal therapy, and surgical debridement.1 Amphotericin B deoxycholate and its lipid derivatives (eg, amphotericin B lipid complex, liposomal amphotericin B) are the standard antifungal agents used in the treatment of mucormycosis.6,7 Posaconazole is an extended-spectrum triazole with in vitro activity against Mucorales. Posaconazole may be useful as salvage therapy; however, strong clinical evidence to support its role as a primary therapeutic agent is lacking in the literature.6,7 Blood vessel thrombosis and tissue necrosis can result in poor penetration of antifungal agents to the infection site; therefore, surgical debridement also may be critical for complete eradication of the disease.6 Confirmative diagnosis of mucormycosis can be made based on histopathologic findings.
Our case highlights the importance of clinician awareness of the typical presentation of rhino-orbital-cerebral mucormycosis to ensure prompt diagnosis and initiation of immediate treatment of this possibly fatal infection.
1. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18:556-569.
2. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope. 1982;92:1140-1143.
3. Peterson KL, Wang M, Canalis RF, et al. Rhinocerebral mucormycosis: evolution of the disease and treatment options. Laryngoscope. 1997;107:855-862.
4. Khor BS, Lee MH, Leu HS, et al. Rhinocerebral mucormycosis in Taiwan. J Microbiol Immunol Infect. 2003;36:266-269.
5. Petrikkos G, Skiada A, Sambatakou H, et al. Mucormycosis: ten-year experience at a tertiary-care center in Greece. Eur J Clin Microbiol Infect Dis. 2003;22:753-756.
6. Spellberg B, Ibrahim AS. Recent advances in the treatment of mucormycosis. Curr Infect Dis Rep. 2010;12:423-429.
7. Enoch DA, Aliyu SH, Sule O, et al. Posaconazole for the treatment of mucormycosis. Int J Antimicrob Agents. 2011;38:465-473.
A 56-year-old woman presented with painful, erythematous to violaceous patches with necrosis of the left eye and periorbital area of 1 day’s duration. She reported headaches and periorbital pain in the 3 weeks prior to presentation. She was being treated for hypertension, type 2 diabetes mellitus, and end-stage renal disease. The patient denied prior trauma to the area.
The Diagnosis: Rhino-orbital-cerebral Mucormycosis
Cutaneous examination revealed a dusky, erythematous to violaceous patch with a necrotic center involving the left eye and periorbital area (Figure 1). The differential diagnosis included herpes zoster, cellulitis, and fungal infection. We obtained patient consent for a punch biopsy. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (Figure 2). Magnetic resonance imaging of the orbit and head showed involvement of the periorbital soft tissues; the ethmoidal, sphenoidal, and maxillary sinuses; and the left medial temporal lobe. The patient was started on an empirical antifungal treatment of amphotericin B deoxycholate 50 mg daily but died 4 days later due to multiorgan failure.
|
| Figure 1. A dusky, erythematous to violaceous patch with a necrotic center on the left eye and periorbital area. |
|
| Figure 2. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (periodic acid–Schiff, original magnification ×400). |
Mucormycosis is a rare but fatal infection that may rapidly progress.1 Risk factors include defects in host defense such as malignancy, immunodeficiency from bone marrow or solid organ transplantation, diabetes mellitus, malnutrition, abnormal metabolic states, and deferoxamine use.1,2 Rhino-orbital-cerebral mucormycosis usually starts with eye or facial pain and unilateral facial swelling.3,4 Visual impairment, fever, and mental status changes may follow.1,3,4 Skin findings may progress from erythema to violaceous color changes and lastly to a black necrotic eschar resulting from tissue infarction.5
Radiologic imaging may be helpful but rarely is diagnostic in mucormycosis, and reliable serologic tests are lacking.1 Therefore, suspicion of mucormycosis based on clinical and histopathologic factors followed by immediate initiation of empirical antifungal treatment is critical. The key factors in treating mucormycosis include early diagnosis, correction of underlying risk factors, prompt antifungal therapy, and surgical debridement.1 Amphotericin B deoxycholate and its lipid derivatives (eg, amphotericin B lipid complex, liposomal amphotericin B) are the standard antifungal agents used in the treatment of mucormycosis.6,7 Posaconazole is an extended-spectrum triazole with in vitro activity against Mucorales. Posaconazole may be useful as salvage therapy; however, strong clinical evidence to support its role as a primary therapeutic agent is lacking in the literature.6,7 Blood vessel thrombosis and tissue necrosis can result in poor penetration of antifungal agents to the infection site; therefore, surgical debridement also may be critical for complete eradication of the disease.6 Confirmative diagnosis of mucormycosis can be made based on histopathologic findings.
Our case highlights the importance of clinician awareness of the typical presentation of rhino-orbital-cerebral mucormycosis to ensure prompt diagnosis and initiation of immediate treatment of this possibly fatal infection.
A 56-year-old woman presented with painful, erythematous to violaceous patches with necrosis of the left eye and periorbital area of 1 day’s duration. She reported headaches and periorbital pain in the 3 weeks prior to presentation. She was being treated for hypertension, type 2 diabetes mellitus, and end-stage renal disease. The patient denied prior trauma to the area.
The Diagnosis: Rhino-orbital-cerebral Mucormycosis
Cutaneous examination revealed a dusky, erythematous to violaceous patch with a necrotic center involving the left eye and periorbital area (Figure 1). The differential diagnosis included herpes zoster, cellulitis, and fungal infection. We obtained patient consent for a punch biopsy. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (Figure 2). Magnetic resonance imaging of the orbit and head showed involvement of the periorbital soft tissues; the ethmoidal, sphenoidal, and maxillary sinuses; and the left medial temporal lobe. The patient was started on an empirical antifungal treatment of amphotericin B deoxycholate 50 mg daily but died 4 days later due to multiorgan failure.
|
| Figure 1. A dusky, erythematous to violaceous patch with a necrotic center on the left eye and periorbital area. |
|
| Figure 2. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (periodic acid–Schiff, original magnification ×400). |
Mucormycosis is a rare but fatal infection that may rapidly progress.1 Risk factors include defects in host defense such as malignancy, immunodeficiency from bone marrow or solid organ transplantation, diabetes mellitus, malnutrition, abnormal metabolic states, and deferoxamine use.1,2 Rhino-orbital-cerebral mucormycosis usually starts with eye or facial pain and unilateral facial swelling.3,4 Visual impairment, fever, and mental status changes may follow.1,3,4 Skin findings may progress from erythema to violaceous color changes and lastly to a black necrotic eschar resulting from tissue infarction.5
Radiologic imaging may be helpful but rarely is diagnostic in mucormycosis, and reliable serologic tests are lacking.1 Therefore, suspicion of mucormycosis based on clinical and histopathologic factors followed by immediate initiation of empirical antifungal treatment is critical. The key factors in treating mucormycosis include early diagnosis, correction of underlying risk factors, prompt antifungal therapy, and surgical debridement.1 Amphotericin B deoxycholate and its lipid derivatives (eg, amphotericin B lipid complex, liposomal amphotericin B) are the standard antifungal agents used in the treatment of mucormycosis.6,7 Posaconazole is an extended-spectrum triazole with in vitro activity against Mucorales. Posaconazole may be useful as salvage therapy; however, strong clinical evidence to support its role as a primary therapeutic agent is lacking in the literature.6,7 Blood vessel thrombosis and tissue necrosis can result in poor penetration of antifungal agents to the infection site; therefore, surgical debridement also may be critical for complete eradication of the disease.6 Confirmative diagnosis of mucormycosis can be made based on histopathologic findings.
Our case highlights the importance of clinician awareness of the typical presentation of rhino-orbital-cerebral mucormycosis to ensure prompt diagnosis and initiation of immediate treatment of this possibly fatal infection.
1. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18:556-569.
2. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope. 1982;92:1140-1143.
3. Peterson KL, Wang M, Canalis RF, et al. Rhinocerebral mucormycosis: evolution of the disease and treatment options. Laryngoscope. 1997;107:855-862.
4. Khor BS, Lee MH, Leu HS, et al. Rhinocerebral mucormycosis in Taiwan. J Microbiol Immunol Infect. 2003;36:266-269.
5. Petrikkos G, Skiada A, Sambatakou H, et al. Mucormycosis: ten-year experience at a tertiary-care center in Greece. Eur J Clin Microbiol Infect Dis. 2003;22:753-756.
6. Spellberg B, Ibrahim AS. Recent advances in the treatment of mucormycosis. Curr Infect Dis Rep. 2010;12:423-429.
7. Enoch DA, Aliyu SH, Sule O, et al. Posaconazole for the treatment of mucormycosis. Int J Antimicrob Agents. 2011;38:465-473.
1. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18:556-569.
2. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope. 1982;92:1140-1143.
3. Peterson KL, Wang M, Canalis RF, et al. Rhinocerebral mucormycosis: evolution of the disease and treatment options. Laryngoscope. 1997;107:855-862.
4. Khor BS, Lee MH, Leu HS, et al. Rhinocerebral mucormycosis in Taiwan. J Microbiol Immunol Infect. 2003;36:266-269.
5. Petrikkos G, Skiada A, Sambatakou H, et al. Mucormycosis: ten-year experience at a tertiary-care center in Greece. Eur J Clin Microbiol Infect Dis. 2003;22:753-756.
6. Spellberg B, Ibrahim AS. Recent advances in the treatment of mucormycosis. Curr Infect Dis Rep. 2010;12:423-429.
7. Enoch DA, Aliyu SH, Sule O, et al. Posaconazole for the treatment of mucormycosis. Int J Antimicrob Agents. 2011;38:465-473.
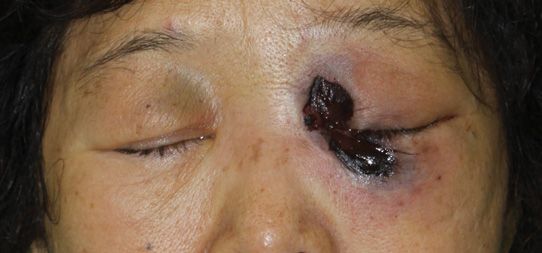
ICD-10 Codes: More Specificity With More Characters
As I have mentioned in prior columns, the key to success with International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) coding will be additional specificity, which will come in the form of more characters to communicate with payors and statisticians regarding the services that have been provided during the office visit. The addition of characters will permit more information about the visit to be delivered in the code.
Some of these additional characters will be required to submit the code for processing. Specifically look out for codes that communicate accidents or injury. Some common circumstances for dermatologists will be lacerations, abrasions, laceration repairs, and burns. The structure of the ICD-10-CM codes is outlined below to explain circumstances in which the increased number of characters will be most important for dermatologists.
The ICD-10-CM codes will be 3 to 7 characters long.1 The first 3 characters are the general categories. For example, L70 is the 3-character category for acne and L40 is the category for psoriasis. The next 3 characters (ie, characters 4–6) correspond to the related etiology (ie, the cause, set of causes, manner of causation of a disease or condition), anatomic site, severity, and other vital clinical details.1
Take the case of a burn on the hand. With the International Classification of Diseases, Ninth Revision, a first-degree burn on the back of the hand is coded with 5 characters (944.16).2 According to the ICD-10-CM coding system, the code for an initial visit for a first-degree burn on the back of the hand would include 7 characters (T23.169A).1 There will be times when the sixth character may not be necessary; in these instances, an X must be placed in this position as a placeholder, but the absence of a sixth character does not negate the need for a seventh. Thankfully, the seventh character can only be 1 of 3 letters. As illustrated above in the code for a first-degree burn on the back of the hand, the A designates an initial encounter; a D would designate a follow-up visit for this burn, and an S would represent a sequela from the initial burn, such as a postinflammatory change, which would have its own code.1
Conclusion
To be reimbursed, appropriate ICD-10-CM codes must be used. Be sure to master the structure of these codes before the October 1, 2015, compliance deadline; plan now for a training and testing period.
1. Centers for Medicare & Medicaid Services. ICD-10-CM Tabular List of Diseases and Injuries. http://www.cms.gov/Medicare/Coding/ICD10/downloads/6_I10tab2010.pdf. Published 2010. Accessed September 18, 2014.
2. ICD-9 code lookup. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/medicare-coverage-database/staticpages/icd-9-code-lookup.aspx. Accessed September 18, 2014.
As I have mentioned in prior columns, the key to success with International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) coding will be additional specificity, which will come in the form of more characters to communicate with payors and statisticians regarding the services that have been provided during the office visit. The addition of characters will permit more information about the visit to be delivered in the code.
Some of these additional characters will be required to submit the code for processing. Specifically look out for codes that communicate accidents or injury. Some common circumstances for dermatologists will be lacerations, abrasions, laceration repairs, and burns. The structure of the ICD-10-CM codes is outlined below to explain circumstances in which the increased number of characters will be most important for dermatologists.
The ICD-10-CM codes will be 3 to 7 characters long.1 The first 3 characters are the general categories. For example, L70 is the 3-character category for acne and L40 is the category for psoriasis. The next 3 characters (ie, characters 4–6) correspond to the related etiology (ie, the cause, set of causes, manner of causation of a disease or condition), anatomic site, severity, and other vital clinical details.1
Take the case of a burn on the hand. With the International Classification of Diseases, Ninth Revision, a first-degree burn on the back of the hand is coded with 5 characters (944.16).2 According to the ICD-10-CM coding system, the code for an initial visit for a first-degree burn on the back of the hand would include 7 characters (T23.169A).1 There will be times when the sixth character may not be necessary; in these instances, an X must be placed in this position as a placeholder, but the absence of a sixth character does not negate the need for a seventh. Thankfully, the seventh character can only be 1 of 3 letters. As illustrated above in the code for a first-degree burn on the back of the hand, the A designates an initial encounter; a D would designate a follow-up visit for this burn, and an S would represent a sequela from the initial burn, such as a postinflammatory change, which would have its own code.1
Conclusion
To be reimbursed, appropriate ICD-10-CM codes must be used. Be sure to master the structure of these codes before the October 1, 2015, compliance deadline; plan now for a training and testing period.
As I have mentioned in prior columns, the key to success with International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) coding will be additional specificity, which will come in the form of more characters to communicate with payors and statisticians regarding the services that have been provided during the office visit. The addition of characters will permit more information about the visit to be delivered in the code.
Some of these additional characters will be required to submit the code for processing. Specifically look out for codes that communicate accidents or injury. Some common circumstances for dermatologists will be lacerations, abrasions, laceration repairs, and burns. The structure of the ICD-10-CM codes is outlined below to explain circumstances in which the increased number of characters will be most important for dermatologists.
The ICD-10-CM codes will be 3 to 7 characters long.1 The first 3 characters are the general categories. For example, L70 is the 3-character category for acne and L40 is the category for psoriasis. The next 3 characters (ie, characters 4–6) correspond to the related etiology (ie, the cause, set of causes, manner of causation of a disease or condition), anatomic site, severity, and other vital clinical details.1
Take the case of a burn on the hand. With the International Classification of Diseases, Ninth Revision, a first-degree burn on the back of the hand is coded with 5 characters (944.16).2 According to the ICD-10-CM coding system, the code for an initial visit for a first-degree burn on the back of the hand would include 7 characters (T23.169A).1 There will be times when the sixth character may not be necessary; in these instances, an X must be placed in this position as a placeholder, but the absence of a sixth character does not negate the need for a seventh. Thankfully, the seventh character can only be 1 of 3 letters. As illustrated above in the code for a first-degree burn on the back of the hand, the A designates an initial encounter; a D would designate a follow-up visit for this burn, and an S would represent a sequela from the initial burn, such as a postinflammatory change, which would have its own code.1
Conclusion
To be reimbursed, appropriate ICD-10-CM codes must be used. Be sure to master the structure of these codes before the October 1, 2015, compliance deadline; plan now for a training and testing period.
1. Centers for Medicare & Medicaid Services. ICD-10-CM Tabular List of Diseases and Injuries. http://www.cms.gov/Medicare/Coding/ICD10/downloads/6_I10tab2010.pdf. Published 2010. Accessed September 18, 2014.
2. ICD-9 code lookup. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/medicare-coverage-database/staticpages/icd-9-code-lookup.aspx. Accessed September 18, 2014.
1. Centers for Medicare & Medicaid Services. ICD-10-CM Tabular List of Diseases and Injuries. http://www.cms.gov/Medicare/Coding/ICD10/downloads/6_I10tab2010.pdf. Published 2010. Accessed September 18, 2014.
2. ICD-9 code lookup. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/medicare-coverage-database/staticpages/icd-9-code-lookup.aspx. Accessed September 18, 2014.
Practice Points
- The addition of characters in International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) codes will permit more information about the visit to be communicated to payors and statisticians.
- Codes with ICD-10-CM will be 3 to 7 characters long and indicate the disease category, followed by the related etiology, anatomic site, severity, and other vital clinical details. The last character is 1 of 3 letters to indicate if the visit is an initial encounter, subsequent encounter, or sequela.
Private Practice Will Survive But Patient Billing Will Not
For many years I have advised physicians that aggressive management of accounts receivable is the key to financial health for any private practice. In the current health care reform climate, it has become more important than ever. A crucial step toward proper management of accounts receivable in the age of the Patient Protection and Affordable Care Act is minimization, if not outright elimination, of patient billing, which is a hallowed yet obsolete tradition in private practice. Billing, in effect, is extending free credit to patients, and independent physicians can no longer afford it.
Some physicians of a traditional bent cling to the idea that accepting credit cards or even asking for payment at the time of service smacks of “storekeeping.” They feel more comfortable billing patients for outstanding balances but complain that their bills often are ignored; with each passing day following treatment in your office, the likelihood decreases that a patient will pay the bill.
Patient billing also is expensive. When you total the costs of materials, postage, and staff labor, each bill can cost anywhere from $2 to $10 or more. Every minute the office staff spends producing and mailing bills is time not spent on more productive work. Billing services are an alternative, but they also are expensive, and those bills get ignored too. Requiring immediate payment may seem distasteful to some physicians, but for physicians who wish to keep their office private and independent, it is rapidly becoming the only viable option.
Health Savings Accounts
Private practices will need to become increasingly flexible in how they accept payments as the population continues to age. This flexibility becomes increasingly important as more and more patients rely on health savings accounts (HSAs). Enrollment in these specialized, tax-deductible, tax-free accounts has increased 10-fold over the last decade.1 Private practice physicians will want to accommodate for HSAs as much as possible.
A few credit companies are already promoting cards to finance HSAs and other private-pay portions of health care expenses, such as The HELPcard (www.helpcard.com). Major credit card companies also have begun to appreciate this largely untapped segment of potential business for them. Soon you may begin receiving help from them in setting up creative payment plans for your patients. Some financial institutions have even begun creating medical credit and debit cards called health benefit cards that are designed specifically for use at physicians’ offices.2
Credit and Debit Cards
Credit and debit cards eliminate many of the problems associated with patient billing. They allow you to collect more fees at the time of service while you still have the patient’s attention and the service you provided is still appreciated.
Charging to a credit or debit card also reduces the chances of a balance owed falling through the cracks, getting lost in the mail, or getting embezzled, and it cannot bounce so it is better than a check. Card payments also can improve your practice’s cash flow, which is always a welcome benefit. Additionally, if a patient is delinquent in paying a credit card bill, it is the credit card company’s problem, not yours.
Credit cards also offer more payment flexibility for patients. In the case of a large balance, offer your patient the option of charging all of the services to a credit card, which he/she can then pay off in affordable monthly installments. Your practice will get reimbursed in full, even as the patient is paying it off slowly, and the patient is able to pay off the debt at a pace that makes sense for his/her finances.
Payment Policies
Beyond simply accepting credit cards, the next step is one that every hotel, rental car agency, and many other businesses have used for years: Retain a card number in each patient’s file, and bill balances as they come in.
Every new patient in my office receives a letter at his/her first visit explaining our policy: We will keep a credit card number on file and use it to bill any outstanding balances after third parties pay their portion. At the bottom of the letter is a brief statement of consent for the patient to sign, along with a place to write the credit card number and expiration date. This policy also comes in handy for patients who claim to have come to the office without cash, a checkbook, credit cards, or any other method of payment. In such situations, my office manager can say, “No problem, we have your credit card information on file!”
Do patients object to this policy? Some do, mostly older patients. But when we explain that we are doing nothing different than hotels do at check-in and that this policy also will work to their advantage by decreasing the number of bills they receive and checks they must write, most come around. Make it an option at first if you wish; then, when everyone is accustomed, you can make it a mandatory policy. My office manager has the authority to make exceptions on a case-by-case basis when necessary.
Do patients worry about confidentiality or unauthorized use? Most individuals do not worry when they use a credit card at a restaurant, hotel, or the Internet. Guard your patients’ financial information as carefully as their medical information. If you have electronic health records, the patient’s credit card number can go in the medical chart. Otherwise use a separate portable filing system that can be locked up each night.
Does this policy work? In only 1 year, my total accounts receivable dropped by nearly 50%; after another year they stabilized at 30% to 35% of prior levels and have remained there ever since, which was a source of consternation for our new accountant who we hired shortly thereafter. “Something must be wrong,” he said nervously after his first look at our books. “Accounts receivable totals are never that low in a medical office with your level of volume.” His eyes widened as I explained our system. “Why doesn’t every private practice do that?” he asked. Why, indeed.
Final Thoughts
The business of health care delivery is currently being rocked at its foundations, as I have been detailing in this column. Without considerable adaptation to these fundamental changes, a private practice can do little more than survive, and even that will take luck. A crucial component of adaptation involves doing more of what we do best, treating patients. Leave the business of extending credit to the banks and credit card companies.
1. Stroud M. Making the most of that shiny new HSA. Reuters. April 19, 2012. http://www.reuters.com/article/2012/04/19/us-healthcare-savings-idU BRE83I0ZI20120419. Accessed September 16, 2014.
2. Prater C. Is there a health care debit card in your future? CreditCards.com Web site. http://www.credit cards.com/credit-card-news/payment-cards-health-care-expenses-1271.php. Published April 14, 2009. Accessed September 16, 2014.
For many years I have advised physicians that aggressive management of accounts receivable is the key to financial health for any private practice. In the current health care reform climate, it has become more important than ever. A crucial step toward proper management of accounts receivable in the age of the Patient Protection and Affordable Care Act is minimization, if not outright elimination, of patient billing, which is a hallowed yet obsolete tradition in private practice. Billing, in effect, is extending free credit to patients, and independent physicians can no longer afford it.
Some physicians of a traditional bent cling to the idea that accepting credit cards or even asking for payment at the time of service smacks of “storekeeping.” They feel more comfortable billing patients for outstanding balances but complain that their bills often are ignored; with each passing day following treatment in your office, the likelihood decreases that a patient will pay the bill.
Patient billing also is expensive. When you total the costs of materials, postage, and staff labor, each bill can cost anywhere from $2 to $10 or more. Every minute the office staff spends producing and mailing bills is time not spent on more productive work. Billing services are an alternative, but they also are expensive, and those bills get ignored too. Requiring immediate payment may seem distasteful to some physicians, but for physicians who wish to keep their office private and independent, it is rapidly becoming the only viable option.
Health Savings Accounts
Private practices will need to become increasingly flexible in how they accept payments as the population continues to age. This flexibility becomes increasingly important as more and more patients rely on health savings accounts (HSAs). Enrollment in these specialized, tax-deductible, tax-free accounts has increased 10-fold over the last decade.1 Private practice physicians will want to accommodate for HSAs as much as possible.
A few credit companies are already promoting cards to finance HSAs and other private-pay portions of health care expenses, such as The HELPcard (www.helpcard.com). Major credit card companies also have begun to appreciate this largely untapped segment of potential business for them. Soon you may begin receiving help from them in setting up creative payment plans for your patients. Some financial institutions have even begun creating medical credit and debit cards called health benefit cards that are designed specifically for use at physicians’ offices.2
Credit and Debit Cards
Credit and debit cards eliminate many of the problems associated with patient billing. They allow you to collect more fees at the time of service while you still have the patient’s attention and the service you provided is still appreciated.
Charging to a credit or debit card also reduces the chances of a balance owed falling through the cracks, getting lost in the mail, or getting embezzled, and it cannot bounce so it is better than a check. Card payments also can improve your practice’s cash flow, which is always a welcome benefit. Additionally, if a patient is delinquent in paying a credit card bill, it is the credit card company’s problem, not yours.
Credit cards also offer more payment flexibility for patients. In the case of a large balance, offer your patient the option of charging all of the services to a credit card, which he/she can then pay off in affordable monthly installments. Your practice will get reimbursed in full, even as the patient is paying it off slowly, and the patient is able to pay off the debt at a pace that makes sense for his/her finances.
Payment Policies
Beyond simply accepting credit cards, the next step is one that every hotel, rental car agency, and many other businesses have used for years: Retain a card number in each patient’s file, and bill balances as they come in.
Every new patient in my office receives a letter at his/her first visit explaining our policy: We will keep a credit card number on file and use it to bill any outstanding balances after third parties pay their portion. At the bottom of the letter is a brief statement of consent for the patient to sign, along with a place to write the credit card number and expiration date. This policy also comes in handy for patients who claim to have come to the office without cash, a checkbook, credit cards, or any other method of payment. In such situations, my office manager can say, “No problem, we have your credit card information on file!”
Do patients object to this policy? Some do, mostly older patients. But when we explain that we are doing nothing different than hotels do at check-in and that this policy also will work to their advantage by decreasing the number of bills they receive and checks they must write, most come around. Make it an option at first if you wish; then, when everyone is accustomed, you can make it a mandatory policy. My office manager has the authority to make exceptions on a case-by-case basis when necessary.
Do patients worry about confidentiality or unauthorized use? Most individuals do not worry when they use a credit card at a restaurant, hotel, or the Internet. Guard your patients’ financial information as carefully as their medical information. If you have electronic health records, the patient’s credit card number can go in the medical chart. Otherwise use a separate portable filing system that can be locked up each night.
Does this policy work? In only 1 year, my total accounts receivable dropped by nearly 50%; after another year they stabilized at 30% to 35% of prior levels and have remained there ever since, which was a source of consternation for our new accountant who we hired shortly thereafter. “Something must be wrong,” he said nervously after his first look at our books. “Accounts receivable totals are never that low in a medical office with your level of volume.” His eyes widened as I explained our system. “Why doesn’t every private practice do that?” he asked. Why, indeed.
Final Thoughts
The business of health care delivery is currently being rocked at its foundations, as I have been detailing in this column. Without considerable adaptation to these fundamental changes, a private practice can do little more than survive, and even that will take luck. A crucial component of adaptation involves doing more of what we do best, treating patients. Leave the business of extending credit to the banks and credit card companies.
For many years I have advised physicians that aggressive management of accounts receivable is the key to financial health for any private practice. In the current health care reform climate, it has become more important than ever. A crucial step toward proper management of accounts receivable in the age of the Patient Protection and Affordable Care Act is minimization, if not outright elimination, of patient billing, which is a hallowed yet obsolete tradition in private practice. Billing, in effect, is extending free credit to patients, and independent physicians can no longer afford it.
Some physicians of a traditional bent cling to the idea that accepting credit cards or even asking for payment at the time of service smacks of “storekeeping.” They feel more comfortable billing patients for outstanding balances but complain that their bills often are ignored; with each passing day following treatment in your office, the likelihood decreases that a patient will pay the bill.
Patient billing also is expensive. When you total the costs of materials, postage, and staff labor, each bill can cost anywhere from $2 to $10 or more. Every minute the office staff spends producing and mailing bills is time not spent on more productive work. Billing services are an alternative, but they also are expensive, and those bills get ignored too. Requiring immediate payment may seem distasteful to some physicians, but for physicians who wish to keep their office private and independent, it is rapidly becoming the only viable option.
Health Savings Accounts
Private practices will need to become increasingly flexible in how they accept payments as the population continues to age. This flexibility becomes increasingly important as more and more patients rely on health savings accounts (HSAs). Enrollment in these specialized, tax-deductible, tax-free accounts has increased 10-fold over the last decade.1 Private practice physicians will want to accommodate for HSAs as much as possible.
A few credit companies are already promoting cards to finance HSAs and other private-pay portions of health care expenses, such as The HELPcard (www.helpcard.com). Major credit card companies also have begun to appreciate this largely untapped segment of potential business for them. Soon you may begin receiving help from them in setting up creative payment plans for your patients. Some financial institutions have even begun creating medical credit and debit cards called health benefit cards that are designed specifically for use at physicians’ offices.2
Credit and Debit Cards
Credit and debit cards eliminate many of the problems associated with patient billing. They allow you to collect more fees at the time of service while you still have the patient’s attention and the service you provided is still appreciated.
Charging to a credit or debit card also reduces the chances of a balance owed falling through the cracks, getting lost in the mail, or getting embezzled, and it cannot bounce so it is better than a check. Card payments also can improve your practice’s cash flow, which is always a welcome benefit. Additionally, if a patient is delinquent in paying a credit card bill, it is the credit card company’s problem, not yours.
Credit cards also offer more payment flexibility for patients. In the case of a large balance, offer your patient the option of charging all of the services to a credit card, which he/she can then pay off in affordable monthly installments. Your practice will get reimbursed in full, even as the patient is paying it off slowly, and the patient is able to pay off the debt at a pace that makes sense for his/her finances.
Payment Policies
Beyond simply accepting credit cards, the next step is one that every hotel, rental car agency, and many other businesses have used for years: Retain a card number in each patient’s file, and bill balances as they come in.
Every new patient in my office receives a letter at his/her first visit explaining our policy: We will keep a credit card number on file and use it to bill any outstanding balances after third parties pay their portion. At the bottom of the letter is a brief statement of consent for the patient to sign, along with a place to write the credit card number and expiration date. This policy also comes in handy for patients who claim to have come to the office without cash, a checkbook, credit cards, or any other method of payment. In such situations, my office manager can say, “No problem, we have your credit card information on file!”
Do patients object to this policy? Some do, mostly older patients. But when we explain that we are doing nothing different than hotels do at check-in and that this policy also will work to their advantage by decreasing the number of bills they receive and checks they must write, most come around. Make it an option at first if you wish; then, when everyone is accustomed, you can make it a mandatory policy. My office manager has the authority to make exceptions on a case-by-case basis when necessary.
Do patients worry about confidentiality or unauthorized use? Most individuals do not worry when they use a credit card at a restaurant, hotel, or the Internet. Guard your patients’ financial information as carefully as their medical information. If you have electronic health records, the patient’s credit card number can go in the medical chart. Otherwise use a separate portable filing system that can be locked up each night.
Does this policy work? In only 1 year, my total accounts receivable dropped by nearly 50%; after another year they stabilized at 30% to 35% of prior levels and have remained there ever since, which was a source of consternation for our new accountant who we hired shortly thereafter. “Something must be wrong,” he said nervously after his first look at our books. “Accounts receivable totals are never that low in a medical office with your level of volume.” His eyes widened as I explained our system. “Why doesn’t every private practice do that?” he asked. Why, indeed.
Final Thoughts
The business of health care delivery is currently being rocked at its foundations, as I have been detailing in this column. Without considerable adaptation to these fundamental changes, a private practice can do little more than survive, and even that will take luck. A crucial component of adaptation involves doing more of what we do best, treating patients. Leave the business of extending credit to the banks and credit card companies.
1. Stroud M. Making the most of that shiny new HSA. Reuters. April 19, 2012. http://www.reuters.com/article/2012/04/19/us-healthcare-savings-idU BRE83I0ZI20120419. Accessed September 16, 2014.
2. Prater C. Is there a health care debit card in your future? CreditCards.com Web site. http://www.credit cards.com/credit-card-news/payment-cards-health-care-expenses-1271.php. Published April 14, 2009. Accessed September 16, 2014.
1. Stroud M. Making the most of that shiny new HSA. Reuters. April 19, 2012. http://www.reuters.com/article/2012/04/19/us-healthcare-savings-idU BRE83I0ZI20120419. Accessed September 16, 2014.
2. Prater C. Is there a health care debit card in your future? CreditCards.com Web site. http://www.credit cards.com/credit-card-news/payment-cards-health-care-expenses-1271.php. Published April 14, 2009. Accessed September 16, 2014.
Practice Points
- Aggressive management of accounts receivable is the key to the financial health of any private practice. Physicians must become increasingly flexible in how they accept payments as the population continues to age.
- Consider requiring patients to supply a credit card or debit card to bill for outstanding balances after third parties pay their portion.
- Accommodate health savings accounts and health benefit cards.

Challenges Facing Our Specialty
The health care environment is changing rapidly and the smart dermatologist will stay informed and respond proactively. Our strength lies in our unity and identity as dermatologists. There is strength in numbers, and for us to thrive, all dermatologists should be members of the American Academy of Dermatology (AAD) and the American Medical Association. These memberships ensure that we have a seat at the table when important decisions are being made. If you have let your membership lapse, I strongly encourage you to join. Our representation as a specialty depends on the number of members we have in each of these societies. The AAD provides many ways to stay informed, including member-to-member communications, Dermatology World, and special communications from the AAD president. Member alerts will let you know when critical action is required to affect pending legislation that impacts our specialty. Stay informed and respond when called upon.
Dermatologists face unprecedented challenges that pose a very real threat to patient access to high-quality care by a board-certified dermatologist and the future of private practice, including limited provider networks, challenges to fair reimbursement, and bad audit policies. Limited provider networks may represent the single greatest threat to the independent practice of medicine in the United States. Recent actions by payors have unenrolled large numbers of providers. In some cases, dermatologists have found that 20% of their patients became “out of network” overnight. Higher patient co-pays and difficulty with reimbursement may follow, limiting a patient’s ability to continue to see his/her physician. Challenges to fair reimbursement abound and tiered payments are becoming commonplace, with the criteria for tiering often driven by economics rather than quality. Medical necessity auditors have inappropriately used the ABCD public education tool for melanoma, applying it to medical records and ruling biopsies positive for melanoma as “not medically necessary” because the ABCDs were not documented in the physician’s note. In other cases, biopsies positive for skin cancer were ruled “not medically necessary” because of “lack of documentation of signs and symptoms.” Melanomas rarely itch, and the ABCD tool was designed for laypeople. Ignorance and lack of understanding of the care we provide jeopardizes patient access to care.
Even bigger challenges loom. Where will dermatology fit into the big picture as national health care priorities focus on large public health issues such as hypertension, diabetes, obesity, and depression? Dermatologists play a critical role in reducing the burden of skin cancer, preventing both death and morbidity, but most policymakers do not understand the critical services we provide. Individual physicians have a limited ability to respond to these challenges, and our state and subspecialty societies have limited resources to fight these battles. Over the last 2 years, the AAD has responded by transforming a good state affairs office into a superbly effective and nimble group of highly talented individuals with expertise in advocacy, law, and health policy. Our new Strategic Alliance Liaison Committee is designed to coordinate the efforts of patient advocacy groups and dermatology societies to help ensure an effective response. If your state or subspecialty society is not actively engaged with the AAD’s state affairs office, it is time to contact them.
It is critical that dermatologists project a unified voice. Dermatology is a small specialty, representing less than 2% of physicians, but we have always been successful in projecting a voice much larger than our numbers. Unity is key to our success. This past year, the AAD established a rapid response checklist to ensure that all critical steps fall into place when responding to a rapidly evolving critical issue, including coordination with key patient advocacy groups and other key dermatological societies such as the American Society of Dermatologic Surgery, the Mohs College, Mohs Society, American Society of Dermatopathology, the American Osteopathic College of Dermatology, and many others. There are many payment and scope of practice issues that are difficult for us to present without appearing self-serving, but these very same messages can succeed when the focus is on patient safety, quality of care, and patient access. Patient advocacy groups are our best allies because they fight for patient rights to timely and effective care for diseases of the skin.
Change is occurring quickly and there is a lot of work to be done. Key priorities fundamental to the future of our specialty include ensuring effective advocacy, establishing how dermatologists fit into new payment and care delivery models, obtaining the data we need to demonstrate the unique value dermatologists bring to patient care and the health care system, enhancing the image of our specialty, and optimizing our support of state and local dermatological societies as they confront a growing range of issues.
We are privileged to practice a specialty that can provide patients with dramatic improvements in health and quality of life. We give back in so many ways, such as volunteering to help underserved populations overseas or at home. We have raised public awareness of the threat of melanoma. The Canadian Dermatology Association turned Niagara Falls orange on Melanoma Monday this year to raise skin cancer awareness; well done! Every one of us who helps support our patient advocacy efforts or the continued success of Camp Discovery (http://www.aad.org/dermatology-a-to-z/for-kids/camp-discovery) enhances the image of our specialty. Each time you see a hospital consultation, volunteer in the community, or squeeze in a patient who cannot pay at the end of a long day, you do more than help an individual; you help ensure the very future of our specialty.
To face the challenges ahead, we must stick together and project a unified voice. Stay informed! If you do not regularly read Dermatology World and the AAD’s member-to-member alerts, you are missing a lot. Our future depends on each one of us working together for our patients and our specialty.
The health care environment is changing rapidly and the smart dermatologist will stay informed and respond proactively. Our strength lies in our unity and identity as dermatologists. There is strength in numbers, and for us to thrive, all dermatologists should be members of the American Academy of Dermatology (AAD) and the American Medical Association. These memberships ensure that we have a seat at the table when important decisions are being made. If you have let your membership lapse, I strongly encourage you to join. Our representation as a specialty depends on the number of members we have in each of these societies. The AAD provides many ways to stay informed, including member-to-member communications, Dermatology World, and special communications from the AAD president. Member alerts will let you know when critical action is required to affect pending legislation that impacts our specialty. Stay informed and respond when called upon.
Dermatologists face unprecedented challenges that pose a very real threat to patient access to high-quality care by a board-certified dermatologist and the future of private practice, including limited provider networks, challenges to fair reimbursement, and bad audit policies. Limited provider networks may represent the single greatest threat to the independent practice of medicine in the United States. Recent actions by payors have unenrolled large numbers of providers. In some cases, dermatologists have found that 20% of their patients became “out of network” overnight. Higher patient co-pays and difficulty with reimbursement may follow, limiting a patient’s ability to continue to see his/her physician. Challenges to fair reimbursement abound and tiered payments are becoming commonplace, with the criteria for tiering often driven by economics rather than quality. Medical necessity auditors have inappropriately used the ABCD public education tool for melanoma, applying it to medical records and ruling biopsies positive for melanoma as “not medically necessary” because the ABCDs were not documented in the physician’s note. In other cases, biopsies positive for skin cancer were ruled “not medically necessary” because of “lack of documentation of signs and symptoms.” Melanomas rarely itch, and the ABCD tool was designed for laypeople. Ignorance and lack of understanding of the care we provide jeopardizes patient access to care.
Even bigger challenges loom. Where will dermatology fit into the big picture as national health care priorities focus on large public health issues such as hypertension, diabetes, obesity, and depression? Dermatologists play a critical role in reducing the burden of skin cancer, preventing both death and morbidity, but most policymakers do not understand the critical services we provide. Individual physicians have a limited ability to respond to these challenges, and our state and subspecialty societies have limited resources to fight these battles. Over the last 2 years, the AAD has responded by transforming a good state affairs office into a superbly effective and nimble group of highly talented individuals with expertise in advocacy, law, and health policy. Our new Strategic Alliance Liaison Committee is designed to coordinate the efforts of patient advocacy groups and dermatology societies to help ensure an effective response. If your state or subspecialty society is not actively engaged with the AAD’s state affairs office, it is time to contact them.
It is critical that dermatologists project a unified voice. Dermatology is a small specialty, representing less than 2% of physicians, but we have always been successful in projecting a voice much larger than our numbers. Unity is key to our success. This past year, the AAD established a rapid response checklist to ensure that all critical steps fall into place when responding to a rapidly evolving critical issue, including coordination with key patient advocacy groups and other key dermatological societies such as the American Society of Dermatologic Surgery, the Mohs College, Mohs Society, American Society of Dermatopathology, the American Osteopathic College of Dermatology, and many others. There are many payment and scope of practice issues that are difficult for us to present without appearing self-serving, but these very same messages can succeed when the focus is on patient safety, quality of care, and patient access. Patient advocacy groups are our best allies because they fight for patient rights to timely and effective care for diseases of the skin.
Change is occurring quickly and there is a lot of work to be done. Key priorities fundamental to the future of our specialty include ensuring effective advocacy, establishing how dermatologists fit into new payment and care delivery models, obtaining the data we need to demonstrate the unique value dermatologists bring to patient care and the health care system, enhancing the image of our specialty, and optimizing our support of state and local dermatological societies as they confront a growing range of issues.
We are privileged to practice a specialty that can provide patients with dramatic improvements in health and quality of life. We give back in so many ways, such as volunteering to help underserved populations overseas or at home. We have raised public awareness of the threat of melanoma. The Canadian Dermatology Association turned Niagara Falls orange on Melanoma Monday this year to raise skin cancer awareness; well done! Every one of us who helps support our patient advocacy efforts or the continued success of Camp Discovery (http://www.aad.org/dermatology-a-to-z/for-kids/camp-discovery) enhances the image of our specialty. Each time you see a hospital consultation, volunteer in the community, or squeeze in a patient who cannot pay at the end of a long day, you do more than help an individual; you help ensure the very future of our specialty.
To face the challenges ahead, we must stick together and project a unified voice. Stay informed! If you do not regularly read Dermatology World and the AAD’s member-to-member alerts, you are missing a lot. Our future depends on each one of us working together for our patients and our specialty.
The health care environment is changing rapidly and the smart dermatologist will stay informed and respond proactively. Our strength lies in our unity and identity as dermatologists. There is strength in numbers, and for us to thrive, all dermatologists should be members of the American Academy of Dermatology (AAD) and the American Medical Association. These memberships ensure that we have a seat at the table when important decisions are being made. If you have let your membership lapse, I strongly encourage you to join. Our representation as a specialty depends on the number of members we have in each of these societies. The AAD provides many ways to stay informed, including member-to-member communications, Dermatology World, and special communications from the AAD president. Member alerts will let you know when critical action is required to affect pending legislation that impacts our specialty. Stay informed and respond when called upon.
Dermatologists face unprecedented challenges that pose a very real threat to patient access to high-quality care by a board-certified dermatologist and the future of private practice, including limited provider networks, challenges to fair reimbursement, and bad audit policies. Limited provider networks may represent the single greatest threat to the independent practice of medicine in the United States. Recent actions by payors have unenrolled large numbers of providers. In some cases, dermatologists have found that 20% of their patients became “out of network” overnight. Higher patient co-pays and difficulty with reimbursement may follow, limiting a patient’s ability to continue to see his/her physician. Challenges to fair reimbursement abound and tiered payments are becoming commonplace, with the criteria for tiering often driven by economics rather than quality. Medical necessity auditors have inappropriately used the ABCD public education tool for melanoma, applying it to medical records and ruling biopsies positive for melanoma as “not medically necessary” because the ABCDs were not documented in the physician’s note. In other cases, biopsies positive for skin cancer were ruled “not medically necessary” because of “lack of documentation of signs and symptoms.” Melanomas rarely itch, and the ABCD tool was designed for laypeople. Ignorance and lack of understanding of the care we provide jeopardizes patient access to care.
Even bigger challenges loom. Where will dermatology fit into the big picture as national health care priorities focus on large public health issues such as hypertension, diabetes, obesity, and depression? Dermatologists play a critical role in reducing the burden of skin cancer, preventing both death and morbidity, but most policymakers do not understand the critical services we provide. Individual physicians have a limited ability to respond to these challenges, and our state and subspecialty societies have limited resources to fight these battles. Over the last 2 years, the AAD has responded by transforming a good state affairs office into a superbly effective and nimble group of highly talented individuals with expertise in advocacy, law, and health policy. Our new Strategic Alliance Liaison Committee is designed to coordinate the efforts of patient advocacy groups and dermatology societies to help ensure an effective response. If your state or subspecialty society is not actively engaged with the AAD’s state affairs office, it is time to contact them.
It is critical that dermatologists project a unified voice. Dermatology is a small specialty, representing less than 2% of physicians, but we have always been successful in projecting a voice much larger than our numbers. Unity is key to our success. This past year, the AAD established a rapid response checklist to ensure that all critical steps fall into place when responding to a rapidly evolving critical issue, including coordination with key patient advocacy groups and other key dermatological societies such as the American Society of Dermatologic Surgery, the Mohs College, Mohs Society, American Society of Dermatopathology, the American Osteopathic College of Dermatology, and many others. There are many payment and scope of practice issues that are difficult for us to present without appearing self-serving, but these very same messages can succeed when the focus is on patient safety, quality of care, and patient access. Patient advocacy groups are our best allies because they fight for patient rights to timely and effective care for diseases of the skin.
Change is occurring quickly and there is a lot of work to be done. Key priorities fundamental to the future of our specialty include ensuring effective advocacy, establishing how dermatologists fit into new payment and care delivery models, obtaining the data we need to demonstrate the unique value dermatologists bring to patient care and the health care system, enhancing the image of our specialty, and optimizing our support of state and local dermatological societies as they confront a growing range of issues.
We are privileged to practice a specialty that can provide patients with dramatic improvements in health and quality of life. We give back in so many ways, such as volunteering to help underserved populations overseas or at home. We have raised public awareness of the threat of melanoma. The Canadian Dermatology Association turned Niagara Falls orange on Melanoma Monday this year to raise skin cancer awareness; well done! Every one of us who helps support our patient advocacy efforts or the continued success of Camp Discovery (http://www.aad.org/dermatology-a-to-z/for-kids/camp-discovery) enhances the image of our specialty. Each time you see a hospital consultation, volunteer in the community, or squeeze in a patient who cannot pay at the end of a long day, you do more than help an individual; you help ensure the very future of our specialty.
To face the challenges ahead, we must stick together and project a unified voice. Stay informed! If you do not regularly read Dermatology World and the AAD’s member-to-member alerts, you are missing a lot. Our future depends on each one of us working together for our patients and our specialty.
Pemphigus Vulgaris in Pregnancy
Pemphigus vulgaris (PV) is a rare autoimmune bullous dermatosis that has not shown a predilection toward a particular race or sex.1 Autoantibodies for desmoglein 1 and desmoglein 3, members of the cadherin family that are involved in cellular adhesion, have been linked to the pathogenesis of PV.2 These autoantibodies play a role in the loss of cell-to-cell adhesion in the basal and suprabasal layers of the deep epidermis while cellular adhesion in the superficial epidermis remains intact, leading to the clinical presentation of epidermal blistering and ulcerations most commonly found on the scalp, face, groin, and axillae. Diagnosis typically is made based on skin biopsy and confirmed by direct immunofluorescence. Histologically, PV displays acantholysis and suprabasal cleft formation. Immunofluorescence may show IgG antibodies against the PV antigen in the epidermis.3 Once a diagnosis has been made, treatment typically consists of systemic steroids, as the use of steroids has had great effect in preventing infections, sepsis, and fatality that were once associated with PV.4 Mortality rates associated with PV have decreased to 10% to 15% with systemic steroids from a mortality rate as high as 70% in the presteroid era.1,5 Treatment of PV during pregnancy, as in our patient, requires obstetric and pediatric consultations before therapy is initiated. Use of corticosteroids during pregnancy can be potentially dangerous to the fetus, particularly if high doses are necessary to control maternal disease.6,7
Case Report
A 34-year-old pregnant woman at 6 weeks’ gestation presented with widespread blistering dermatitis and associated burning and pruritus. Her obstetrical history was gravida 3, para 2. The patient reported a “rash” on the scalp that had developed 9 months prior. She had been treated as an outpatient at an outside institution with topical antibiotics and antifungal medications, yet the dermatitis progressed. Three weeks prior to hospitalization, the rash was present on the skin and mucosal surfaces, including the groin, chest, face, hard palate, buccal mucosa, lips (Figure 1), and back (Figure 2). Nontender bullae ruptured after 3 days, releasing clear, yellow, serous fluid with associated burning and pruritus. The bullae were hemorrhagic and erythematous at the base.
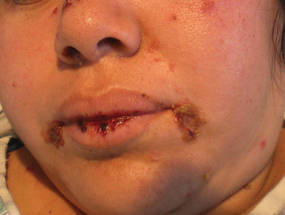 |
| Figure 1. Facial involvement with bullae, crusted hemorrhagic lesions, and eschar in a 34-year-old pregnant woman. |
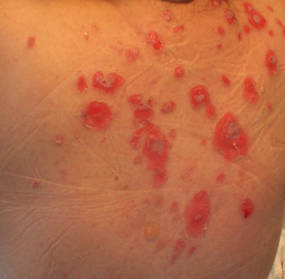 |
| Figure 2. Involvement of the back with bullae in various stages. Some bullae were intact while others newly erupted. |
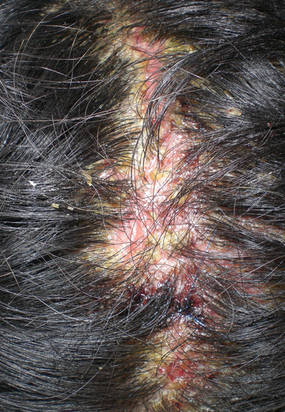 |
| Figure 3. Superinfected and flaking scalp. |
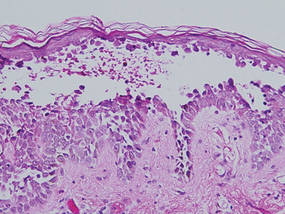 |
| Figure 4. Biopsy revealed suprabasal acantholysis with a tombstone effect of residual basal cells (H&E, original magnification ×200). |
At the current presentation, the patient had several excoriated 1- to 2-cm oval denudations; some were crusted with eschar. Nikolsky sign was negative. Multiple confluent bullous lesions had erupted on the entire scalp with a thick, impetiginous, yellow crust. She had a wet, boggy, foul-smelling, superinfected scalp that was mildly tender to touch with flaking tissue debris (Figure 3). A white blood cell count was 13.2×109/L (reference range, 4.5–11.0×109/L) with 5% eosinophils (reference range, 2.7%). The differential diagnosis included bullous impetigo, pemphigoid, Stevens-Johnson syndrome, dermatitis herpetiformis, and pemphigus vulgaris.
Biopsies of the scalp and back were taken and showed suprabasal acantholysis with a tombstone effect of residual basal cells standing up on the basement membrane without the characteristic acantholysis into skin appendages (Figure 4). The acantholytic cells in the bullous chamber did not round up as in Hailey-Hailey disease nor was there the dyskeratosis of Grover disease. Direct immunofluorescence on an elbow punch biopsy found diffuse 1+ intercellular IgG in the epidermis and diffuse 1+ basal intercellular C3, and was negative for IgA, IgM, and C1q, thus confirming a diagnosis of PV.
The patient was started on prednisone 20 mg once daily. An increase to prednisone 60 mg led to initial improvement of symptoms, but there was a relapse after several days, which is typical of PV in pregnancy,7 prompting the dose to be increased to 120 mg. Following alleviation of symptoms, the dose was later tapered back to 60 mg. No lesions were present at discharge or for 2.5 months thereafter, as the prednisone was tapered from 60 to 45 mg daily after discharge.
On follow-up, the patient’s PV was well controlled, but the prednisone dose was back up to 60 mg daily because of 2 new skin lesions that had developed since her last visit 2.5 months prior. Ultrasonography showed no fetal abnormalities as the pregnancy progressed to 28 weeks’ gestation. The patient developed hypertension and went into premature labor due to placenta previa. The neonate showed no skin lesions or anomalies while in the neonatal intensive care unit. The mother’s prednisone dose was tapered from 60 to 20 mg daily while the white blood cell count was 7.1×109/L with 2% eosinophils and a new scalp lesion appeared. Seven months after her initial discharge from the hospital for the dermatologic condition, she was no longer nursing and azathioprine was added to prednisone 60 mg daily.
Comment
Pemphigus vulgaris is associated with infertility in its active phase; therefore, PV during pregnancy is rare.8 Pregnancy may exacerbate PV, which has been a similar finding in other well-documented autoimmune diseases.7 One review of PV in pregnancy reported that 11 of 49 patients (22%) experienced an exacerbation of the disease.8 This finding pre-sents 2 problems: (1) severe active disease during pregnancy with high antibody titers has been shown to heighten risk for morbidity and mortality for the fetus, and (2) a patient with active PV during pregnancy may require systemic therapy with doses high enough to subdue the disease. The presence of PV was a challenge throughout our patient’s pregnancy. Transient skin lesions may occasionally appear in the neonate and seem to have an increased association with severe active PV in the mother; however, neonatal PV also has been present in mild cases in the mother.7 These lesions are secondary to passive transplacental transfer of PV antibodies but do not have long-lasting clinical implications because of an antibody’s brief half-life.9 The lesions either spontaneously resolve or can be treated with a topical corticosteroid.
Treatment with high-dose systemic corticosteroids or immunosuppressants can be problematic because of the risks posed to the fetus, especially if the mother must be treated when the embryo is particularly susceptible (eg, during organogenesis).10 If a woman with known PV is planning to become pregnant, it is recommended to first control and suppress the disease so that therapy can be minimal during the pregnancy. It also is recommended to use aggressive topical therapy if possible to control PV in a pregnant woman.8 This option would not have been efficacious in our patient because of her severe widespread disease.
Prednisone is considered one of the first-line treatments of PV and has been historically successful as a treatment for pregnant patients with PV if maintained at a low dosage. Prednisone, similar to other corticosteroids, can cross the placental barrier and can increase the chance of premature birth, infection, and mortality in high doses.7 Similar to prednisone, azathioprine is not recommended during pregnancy, but if use is necessary, it is suggested to keep the dose low to prevent fetal harm.11 Inadequate treatment and control of PV can be life threatening to the patient because of the severe infection that may ensue; thus it is necessary for the health of the patient and fetus to suppress the PV. One alternative to treatment with steroids and immunosuppressants is plasma exchange, which has been successful in the clinical context of pregnancy.12 The cons of plasma exchange are repeat procedures, the need to give the patient more immunosuppressants to prevent a rejection, and the return of the autoantibody.7
Several studies have evaluated the safety and efficacy of rituximab in the treatment of refractory PV. Multiple case reports state that both 1 and 2 courses of intravenous rituximab therapy at a dosage of 375 mg per square meter of body surface area affected once weekly for 4 weeks proved to be useful in clinical improvement for patients with refractory disease.13,14 Studies are currently underway to look at the effects of rituximab on pregnancy and the fetus. Preliminary findings show neonates may have B-cell abnormalities initially yet recover fully without infectious complications or sequelae.15 Rituximab currently is a pregnancy category C drug, and women are counseled to avoid pregnancy for at least 12 months after rituximab exposure and use contraception while actively taking the drug.16
Conclusion
Contrary to traditional thinking, PV itself may be associated with poor neonatal outcome, including prematurity and fetal death. These complications seem to be restricted to pregnancies with clinically severe PV.7 Our patient decided to progress with her pregnancy despite the potential risk to the fetus from the disease and treatment. Ultimately, the infant was delivered prematurely but was free of disease.
1. Fainaru O, Mashiach R, Kupferminc M, et al. Pemphigus vulgaris in pregnancy: a case report and review of literature. Hum Reprod. 2000;15:1195-1197.
2. Joly P, Gilbert D, Thomine E, et al. Identification of a new antibody population directed against a desmosomal plaque antigen in pemphigus vulgaris and pemphigus foliaceus. J Invest Dermatol. 1997;108:469-475.
3. Daniel Y, Shenhav M, Botchan A, et al. Pregnancy associated with pemphigus. Br J Obstet Gynecol. 1995;102:667-669.
4. Ruach M, Ohel G, Rahav D, et al. Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv. 1995;50:755-760.
5. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on clinical course of pemphigus vulgaris. J Am Acad Dermatol. 1996;34:645-652.
6. Goldberg NS, DeFeo C, Kirshenbaum N. Pemphigus and pregnancy: risk factors and recommendations. J Am Acad Dermatol. 1993;28(5, pt 2):877-879.
7. Lehman JS, Mueller KK, Schraith DF. Do safe and effective treatment options exist for patients with active pemphigus vulgaris who plan conception and pregnancy? Arch Dermatol. 2008;144:783-785.
8. Kardos M, Levine D, Gurcan H, et al. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739-749.
9. Fenniche S, Benmously R, Marrak H, et al. Neonatal pemphigus vulgaris in an infant born to a mother with pemphigus vulgaris in remission. Pediatr Dermatol. 2006;23:124-127.
10. Kalayciyan A, Engin B, Serdaroglu S, et al. A retrospective analysis of patients with pemphigus vulgaris associated with pregnancy. Br J Dermatol. 2002;147:396-397.
11. Hup JM, Bruinsma RA, Boersma ER, et al. Neonatal pemphigus vulgaris: transplacental transmission of antibodies. Pediatr Dermatol. 1986;3:468-472.
12. Piontek JO, Borberg H, Sollberg S, et al. Severe exacerbation of pemphigus vulgaris in pregnancy: successful treatment with plasma exchange. Br J Dermatol. 2000;143:455-456.
13. Faurschou A, Gniadecki R. Two courses of rituximab (anti-CD20 monoclonal antibody) for recalcitrant pemphigus vulgaris. Int J Dermatol. 2008;47:292-294.
14. Marzano AV, Fanoni D, Venegoni L, et al. Treatment of refractory pemphigus with the anti-CD20 monoclonal antibody (rituximab). Dermatology. 2007;214:310-318.
15. Braunstein I, Werth V. Treatment of dermatologic connective tissue disease and autoimmune blistering disorders in pregnancy. Dermatol Ther. 2013;26:354-363.
16. Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
Pemphigus vulgaris (PV) is a rare autoimmune bullous dermatosis that has not shown a predilection toward a particular race or sex.1 Autoantibodies for desmoglein 1 and desmoglein 3, members of the cadherin family that are involved in cellular adhesion, have been linked to the pathogenesis of PV.2 These autoantibodies play a role in the loss of cell-to-cell adhesion in the basal and suprabasal layers of the deep epidermis while cellular adhesion in the superficial epidermis remains intact, leading to the clinical presentation of epidermal blistering and ulcerations most commonly found on the scalp, face, groin, and axillae. Diagnosis typically is made based on skin biopsy and confirmed by direct immunofluorescence. Histologically, PV displays acantholysis and suprabasal cleft formation. Immunofluorescence may show IgG antibodies against the PV antigen in the epidermis.3 Once a diagnosis has been made, treatment typically consists of systemic steroids, as the use of steroids has had great effect in preventing infections, sepsis, and fatality that were once associated with PV.4 Mortality rates associated with PV have decreased to 10% to 15% with systemic steroids from a mortality rate as high as 70% in the presteroid era.1,5 Treatment of PV during pregnancy, as in our patient, requires obstetric and pediatric consultations before therapy is initiated. Use of corticosteroids during pregnancy can be potentially dangerous to the fetus, particularly if high doses are necessary to control maternal disease.6,7
Case Report
A 34-year-old pregnant woman at 6 weeks’ gestation presented with widespread blistering dermatitis and associated burning and pruritus. Her obstetrical history was gravida 3, para 2. The patient reported a “rash” on the scalp that had developed 9 months prior. She had been treated as an outpatient at an outside institution with topical antibiotics and antifungal medications, yet the dermatitis progressed. Three weeks prior to hospitalization, the rash was present on the skin and mucosal surfaces, including the groin, chest, face, hard palate, buccal mucosa, lips (Figure 1), and back (Figure 2). Nontender bullae ruptured after 3 days, releasing clear, yellow, serous fluid with associated burning and pruritus. The bullae were hemorrhagic and erythematous at the base.
|
| Figure 1. Facial involvement with bullae, crusted hemorrhagic lesions, and eschar in a 34-year-old pregnant woman. |
|
| Figure 2. Involvement of the back with bullae in various stages. Some bullae were intact while others newly erupted. |
|
| Figure 3. Superinfected and flaking scalp. |
|
| Figure 4. Biopsy revealed suprabasal acantholysis with a tombstone effect of residual basal cells (H&E, original magnification ×200). |
At the current presentation, the patient had several excoriated 1- to 2-cm oval denudations; some were crusted with eschar. Nikolsky sign was negative. Multiple confluent bullous lesions had erupted on the entire scalp with a thick, impetiginous, yellow crust. She had a wet, boggy, foul-smelling, superinfected scalp that was mildly tender to touch with flaking tissue debris (Figure 3). A white blood cell count was 13.2×109/L (reference range, 4.5–11.0×109/L) with 5% eosinophils (reference range, 2.7%). The differential diagnosis included bullous impetigo, pemphigoid, Stevens-Johnson syndrome, dermatitis herpetiformis, and pemphigus vulgaris.
Biopsies of the scalp and back were taken and showed suprabasal acantholysis with a tombstone effect of residual basal cells standing up on the basement membrane without the characteristic acantholysis into skin appendages (Figure 4). The acantholytic cells in the bullous chamber did not round up as in Hailey-Hailey disease nor was there the dyskeratosis of Grover disease. Direct immunofluorescence on an elbow punch biopsy found diffuse 1+ intercellular IgG in the epidermis and diffuse 1+ basal intercellular C3, and was negative for IgA, IgM, and C1q, thus confirming a diagnosis of PV.
The patient was started on prednisone 20 mg once daily. An increase to prednisone 60 mg led to initial improvement of symptoms, but there was a relapse after several days, which is typical of PV in pregnancy,7 prompting the dose to be increased to 120 mg. Following alleviation of symptoms, the dose was later tapered back to 60 mg. No lesions were present at discharge or for 2.5 months thereafter, as the prednisone was tapered from 60 to 45 mg daily after discharge.
On follow-up, the patient’s PV was well controlled, but the prednisone dose was back up to 60 mg daily because of 2 new skin lesions that had developed since her last visit 2.5 months prior. Ultrasonography showed no fetal abnormalities as the pregnancy progressed to 28 weeks’ gestation. The patient developed hypertension and went into premature labor due to placenta previa. The neonate showed no skin lesions or anomalies while in the neonatal intensive care unit. The mother’s prednisone dose was tapered from 60 to 20 mg daily while the white blood cell count was 7.1×109/L with 2% eosinophils and a new scalp lesion appeared. Seven months after her initial discharge from the hospital for the dermatologic condition, she was no longer nursing and azathioprine was added to prednisone 60 mg daily.
Comment
Pemphigus vulgaris is associated with infertility in its active phase; therefore, PV during pregnancy is rare.8 Pregnancy may exacerbate PV, which has been a similar finding in other well-documented autoimmune diseases.7 One review of PV in pregnancy reported that 11 of 49 patients (22%) experienced an exacerbation of the disease.8 This finding pre-sents 2 problems: (1) severe active disease during pregnancy with high antibody titers has been shown to heighten risk for morbidity and mortality for the fetus, and (2) a patient with active PV during pregnancy may require systemic therapy with doses high enough to subdue the disease. The presence of PV was a challenge throughout our patient’s pregnancy. Transient skin lesions may occasionally appear in the neonate and seem to have an increased association with severe active PV in the mother; however, neonatal PV also has been present in mild cases in the mother.7 These lesions are secondary to passive transplacental transfer of PV antibodies but do not have long-lasting clinical implications because of an antibody’s brief half-life.9 The lesions either spontaneously resolve or can be treated with a topical corticosteroid.
Treatment with high-dose systemic corticosteroids or immunosuppressants can be problematic because of the risks posed to the fetus, especially if the mother must be treated when the embryo is particularly susceptible (eg, during organogenesis).10 If a woman with known PV is planning to become pregnant, it is recommended to first control and suppress the disease so that therapy can be minimal during the pregnancy. It also is recommended to use aggressive topical therapy if possible to control PV in a pregnant woman.8 This option would not have been efficacious in our patient because of her severe widespread disease.
Prednisone is considered one of the first-line treatments of PV and has been historically successful as a treatment for pregnant patients with PV if maintained at a low dosage. Prednisone, similar to other corticosteroids, can cross the placental barrier and can increase the chance of premature birth, infection, and mortality in high doses.7 Similar to prednisone, azathioprine is not recommended during pregnancy, but if use is necessary, it is suggested to keep the dose low to prevent fetal harm.11 Inadequate treatment and control of PV can be life threatening to the patient because of the severe infection that may ensue; thus it is necessary for the health of the patient and fetus to suppress the PV. One alternative to treatment with steroids and immunosuppressants is plasma exchange, which has been successful in the clinical context of pregnancy.12 The cons of plasma exchange are repeat procedures, the need to give the patient more immunosuppressants to prevent a rejection, and the return of the autoantibody.7
Several studies have evaluated the safety and efficacy of rituximab in the treatment of refractory PV. Multiple case reports state that both 1 and 2 courses of intravenous rituximab therapy at a dosage of 375 mg per square meter of body surface area affected once weekly for 4 weeks proved to be useful in clinical improvement for patients with refractory disease.13,14 Studies are currently underway to look at the effects of rituximab on pregnancy and the fetus. Preliminary findings show neonates may have B-cell abnormalities initially yet recover fully without infectious complications or sequelae.15 Rituximab currently is a pregnancy category C drug, and women are counseled to avoid pregnancy for at least 12 months after rituximab exposure and use contraception while actively taking the drug.16
Conclusion
Contrary to traditional thinking, PV itself may be associated with poor neonatal outcome, including prematurity and fetal death. These complications seem to be restricted to pregnancies with clinically severe PV.7 Our patient decided to progress with her pregnancy despite the potential risk to the fetus from the disease and treatment. Ultimately, the infant was delivered prematurely but was free of disease.
Pemphigus vulgaris (PV) is a rare autoimmune bullous dermatosis that has not shown a predilection toward a particular race or sex.1 Autoantibodies for desmoglein 1 and desmoglein 3, members of the cadherin family that are involved in cellular adhesion, have been linked to the pathogenesis of PV.2 These autoantibodies play a role in the loss of cell-to-cell adhesion in the basal and suprabasal layers of the deep epidermis while cellular adhesion in the superficial epidermis remains intact, leading to the clinical presentation of epidermal blistering and ulcerations most commonly found on the scalp, face, groin, and axillae. Diagnosis typically is made based on skin biopsy and confirmed by direct immunofluorescence. Histologically, PV displays acantholysis and suprabasal cleft formation. Immunofluorescence may show IgG antibodies against the PV antigen in the epidermis.3 Once a diagnosis has been made, treatment typically consists of systemic steroids, as the use of steroids has had great effect in preventing infections, sepsis, and fatality that were once associated with PV.4 Mortality rates associated with PV have decreased to 10% to 15% with systemic steroids from a mortality rate as high as 70% in the presteroid era.1,5 Treatment of PV during pregnancy, as in our patient, requires obstetric and pediatric consultations before therapy is initiated. Use of corticosteroids during pregnancy can be potentially dangerous to the fetus, particularly if high doses are necessary to control maternal disease.6,7
Case Report
A 34-year-old pregnant woman at 6 weeks’ gestation presented with widespread blistering dermatitis and associated burning and pruritus. Her obstetrical history was gravida 3, para 2. The patient reported a “rash” on the scalp that had developed 9 months prior. She had been treated as an outpatient at an outside institution with topical antibiotics and antifungal medications, yet the dermatitis progressed. Three weeks prior to hospitalization, the rash was present on the skin and mucosal surfaces, including the groin, chest, face, hard palate, buccal mucosa, lips (Figure 1), and back (Figure 2). Nontender bullae ruptured after 3 days, releasing clear, yellow, serous fluid with associated burning and pruritus. The bullae were hemorrhagic and erythematous at the base.
|
| Figure 1. Facial involvement with bullae, crusted hemorrhagic lesions, and eschar in a 34-year-old pregnant woman. |
|
| Figure 2. Involvement of the back with bullae in various stages. Some bullae were intact while others newly erupted. |
|
| Figure 3. Superinfected and flaking scalp. |
|
| Figure 4. Biopsy revealed suprabasal acantholysis with a tombstone effect of residual basal cells (H&E, original magnification ×200). |
At the current presentation, the patient had several excoriated 1- to 2-cm oval denudations; some were crusted with eschar. Nikolsky sign was negative. Multiple confluent bullous lesions had erupted on the entire scalp with a thick, impetiginous, yellow crust. She had a wet, boggy, foul-smelling, superinfected scalp that was mildly tender to touch with flaking tissue debris (Figure 3). A white blood cell count was 13.2×109/L (reference range, 4.5–11.0×109/L) with 5% eosinophils (reference range, 2.7%). The differential diagnosis included bullous impetigo, pemphigoid, Stevens-Johnson syndrome, dermatitis herpetiformis, and pemphigus vulgaris.
Biopsies of the scalp and back were taken and showed suprabasal acantholysis with a tombstone effect of residual basal cells standing up on the basement membrane without the characteristic acantholysis into skin appendages (Figure 4). The acantholytic cells in the bullous chamber did not round up as in Hailey-Hailey disease nor was there the dyskeratosis of Grover disease. Direct immunofluorescence on an elbow punch biopsy found diffuse 1+ intercellular IgG in the epidermis and diffuse 1+ basal intercellular C3, and was negative for IgA, IgM, and C1q, thus confirming a diagnosis of PV.
The patient was started on prednisone 20 mg once daily. An increase to prednisone 60 mg led to initial improvement of symptoms, but there was a relapse after several days, which is typical of PV in pregnancy,7 prompting the dose to be increased to 120 mg. Following alleviation of symptoms, the dose was later tapered back to 60 mg. No lesions were present at discharge or for 2.5 months thereafter, as the prednisone was tapered from 60 to 45 mg daily after discharge.
On follow-up, the patient’s PV was well controlled, but the prednisone dose was back up to 60 mg daily because of 2 new skin lesions that had developed since her last visit 2.5 months prior. Ultrasonography showed no fetal abnormalities as the pregnancy progressed to 28 weeks’ gestation. The patient developed hypertension and went into premature labor due to placenta previa. The neonate showed no skin lesions or anomalies while in the neonatal intensive care unit. The mother’s prednisone dose was tapered from 60 to 20 mg daily while the white blood cell count was 7.1×109/L with 2% eosinophils and a new scalp lesion appeared. Seven months after her initial discharge from the hospital for the dermatologic condition, she was no longer nursing and azathioprine was added to prednisone 60 mg daily.
Comment
Pemphigus vulgaris is associated with infertility in its active phase; therefore, PV during pregnancy is rare.8 Pregnancy may exacerbate PV, which has been a similar finding in other well-documented autoimmune diseases.7 One review of PV in pregnancy reported that 11 of 49 patients (22%) experienced an exacerbation of the disease.8 This finding pre-sents 2 problems: (1) severe active disease during pregnancy with high antibody titers has been shown to heighten risk for morbidity and mortality for the fetus, and (2) a patient with active PV during pregnancy may require systemic therapy with doses high enough to subdue the disease. The presence of PV was a challenge throughout our patient’s pregnancy. Transient skin lesions may occasionally appear in the neonate and seem to have an increased association with severe active PV in the mother; however, neonatal PV also has been present in mild cases in the mother.7 These lesions are secondary to passive transplacental transfer of PV antibodies but do not have long-lasting clinical implications because of an antibody’s brief half-life.9 The lesions either spontaneously resolve or can be treated with a topical corticosteroid.
Treatment with high-dose systemic corticosteroids or immunosuppressants can be problematic because of the risks posed to the fetus, especially if the mother must be treated when the embryo is particularly susceptible (eg, during organogenesis).10 If a woman with known PV is planning to become pregnant, it is recommended to first control and suppress the disease so that therapy can be minimal during the pregnancy. It also is recommended to use aggressive topical therapy if possible to control PV in a pregnant woman.8 This option would not have been efficacious in our patient because of her severe widespread disease.
Prednisone is considered one of the first-line treatments of PV and has been historically successful as a treatment for pregnant patients with PV if maintained at a low dosage. Prednisone, similar to other corticosteroids, can cross the placental barrier and can increase the chance of premature birth, infection, and mortality in high doses.7 Similar to prednisone, azathioprine is not recommended during pregnancy, but if use is necessary, it is suggested to keep the dose low to prevent fetal harm.11 Inadequate treatment and control of PV can be life threatening to the patient because of the severe infection that may ensue; thus it is necessary for the health of the patient and fetus to suppress the PV. One alternative to treatment with steroids and immunosuppressants is plasma exchange, which has been successful in the clinical context of pregnancy.12 The cons of plasma exchange are repeat procedures, the need to give the patient more immunosuppressants to prevent a rejection, and the return of the autoantibody.7
Several studies have evaluated the safety and efficacy of rituximab in the treatment of refractory PV. Multiple case reports state that both 1 and 2 courses of intravenous rituximab therapy at a dosage of 375 mg per square meter of body surface area affected once weekly for 4 weeks proved to be useful in clinical improvement for patients with refractory disease.13,14 Studies are currently underway to look at the effects of rituximab on pregnancy and the fetus. Preliminary findings show neonates may have B-cell abnormalities initially yet recover fully without infectious complications or sequelae.15 Rituximab currently is a pregnancy category C drug, and women are counseled to avoid pregnancy for at least 12 months after rituximab exposure and use contraception while actively taking the drug.16
Conclusion
Contrary to traditional thinking, PV itself may be associated with poor neonatal outcome, including prematurity and fetal death. These complications seem to be restricted to pregnancies with clinically severe PV.7 Our patient decided to progress with her pregnancy despite the potential risk to the fetus from the disease and treatment. Ultimately, the infant was delivered prematurely but was free of disease.
1. Fainaru O, Mashiach R, Kupferminc M, et al. Pemphigus vulgaris in pregnancy: a case report and review of literature. Hum Reprod. 2000;15:1195-1197.
2. Joly P, Gilbert D, Thomine E, et al. Identification of a new antibody population directed against a desmosomal plaque antigen in pemphigus vulgaris and pemphigus foliaceus. J Invest Dermatol. 1997;108:469-475.
3. Daniel Y, Shenhav M, Botchan A, et al. Pregnancy associated with pemphigus. Br J Obstet Gynecol. 1995;102:667-669.
4. Ruach M, Ohel G, Rahav D, et al. Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv. 1995;50:755-760.
5. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on clinical course of pemphigus vulgaris. J Am Acad Dermatol. 1996;34:645-652.
6. Goldberg NS, DeFeo C, Kirshenbaum N. Pemphigus and pregnancy: risk factors and recommendations. J Am Acad Dermatol. 1993;28(5, pt 2):877-879.
7. Lehman JS, Mueller KK, Schraith DF. Do safe and effective treatment options exist for patients with active pemphigus vulgaris who plan conception and pregnancy? Arch Dermatol. 2008;144:783-785.
8. Kardos M, Levine D, Gurcan H, et al. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739-749.
9. Fenniche S, Benmously R, Marrak H, et al. Neonatal pemphigus vulgaris in an infant born to a mother with pemphigus vulgaris in remission. Pediatr Dermatol. 2006;23:124-127.
10. Kalayciyan A, Engin B, Serdaroglu S, et al. A retrospective analysis of patients with pemphigus vulgaris associated with pregnancy. Br J Dermatol. 2002;147:396-397.
11. Hup JM, Bruinsma RA, Boersma ER, et al. Neonatal pemphigus vulgaris: transplacental transmission of antibodies. Pediatr Dermatol. 1986;3:468-472.
12. Piontek JO, Borberg H, Sollberg S, et al. Severe exacerbation of pemphigus vulgaris in pregnancy: successful treatment with plasma exchange. Br J Dermatol. 2000;143:455-456.
13. Faurschou A, Gniadecki R. Two courses of rituximab (anti-CD20 monoclonal antibody) for recalcitrant pemphigus vulgaris. Int J Dermatol. 2008;47:292-294.
14. Marzano AV, Fanoni D, Venegoni L, et al. Treatment of refractory pemphigus with the anti-CD20 monoclonal antibody (rituximab). Dermatology. 2007;214:310-318.
15. Braunstein I, Werth V. Treatment of dermatologic connective tissue disease and autoimmune blistering disorders in pregnancy. Dermatol Ther. 2013;26:354-363.
16. Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
1. Fainaru O, Mashiach R, Kupferminc M, et al. Pemphigus vulgaris in pregnancy: a case report and review of literature. Hum Reprod. 2000;15:1195-1197.
2. Joly P, Gilbert D, Thomine E, et al. Identification of a new antibody population directed against a desmosomal plaque antigen in pemphigus vulgaris and pemphigus foliaceus. J Invest Dermatol. 1997;108:469-475.
3. Daniel Y, Shenhav M, Botchan A, et al. Pregnancy associated with pemphigus. Br J Obstet Gynecol. 1995;102:667-669.
4. Ruach M, Ohel G, Rahav D, et al. Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv. 1995;50:755-760.
5. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on clinical course of pemphigus vulgaris. J Am Acad Dermatol. 1996;34:645-652.
6. Goldberg NS, DeFeo C, Kirshenbaum N. Pemphigus and pregnancy: risk factors and recommendations. J Am Acad Dermatol. 1993;28(5, pt 2):877-879.
7. Lehman JS, Mueller KK, Schraith DF. Do safe and effective treatment options exist for patients with active pemphigus vulgaris who plan conception and pregnancy? Arch Dermatol. 2008;144:783-785.
8. Kardos M, Levine D, Gurcan H, et al. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739-749.
9. Fenniche S, Benmously R, Marrak H, et al. Neonatal pemphigus vulgaris in an infant born to a mother with pemphigus vulgaris in remission. Pediatr Dermatol. 2006;23:124-127.
10. Kalayciyan A, Engin B, Serdaroglu S, et al. A retrospective analysis of patients with pemphigus vulgaris associated with pregnancy. Br J Dermatol. 2002;147:396-397.
11. Hup JM, Bruinsma RA, Boersma ER, et al. Neonatal pemphigus vulgaris: transplacental transmission of antibodies. Pediatr Dermatol. 1986;3:468-472.
12. Piontek JO, Borberg H, Sollberg S, et al. Severe exacerbation of pemphigus vulgaris in pregnancy: successful treatment with plasma exchange. Br J Dermatol. 2000;143:455-456.
13. Faurschou A, Gniadecki R. Two courses of rituximab (anti-CD20 monoclonal antibody) for recalcitrant pemphigus vulgaris. Int J Dermatol. 2008;47:292-294.
14. Marzano AV, Fanoni D, Venegoni L, et al. Treatment of refractory pemphigus with the anti-CD20 monoclonal antibody (rituximab). Dermatology. 2007;214:310-318.
15. Braunstein I, Werth V. Treatment of dermatologic connective tissue disease and autoimmune blistering disorders in pregnancy. Dermatol Ther. 2013;26:354-363.
16. Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
Practice Points
- Early diagnosis and appropriate treatment of pemphigus vulgaris in pregnancy is paramount in protecting the health of the mother and fetus.
- Management of autoimmune diseases during pregnancy continues to present numerous challenges for physicians due to the pathology of the diseases as well as the sensitive nature of pregnancy and lack of robust data in this patient population.
Cosmetic Corner: Dermatologists Weigh in on OTC Pigment Control Products
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top OTC pigment control products. Consideration must be given to:
- Even Better
Clinique Laboratories, LLC
“It also is useful as prevention and offers many different options.”—Antonella Tosti, MD, Miami, Florida
“These OTC products have good clinical data to support use for hyperpigmentation. Patients tell me that they feel good on their skin and aren’t irritating.”—Gary Goldenberg, MD, New York, New York
- Lumixyl Brightening System
Envy Medical, Inc
“A great option for patients who may be experiencing modest issues with pigmentation. Use of a retinoid with this product also may enhance its efficacy.”—Joel Schlessinger, MD, Omaha, Nebraska
- Lytera Skin Brightening Complex
SkinMedica
“With key ingredients such as hexylresorcinol, retinol, and niacinamide, it has been clinically shown to lighten dark patches in its trials as well as adding luminosity to the skin.”—Anthony Rossi, MD, New York, New York
Recommended by Elizabeth K. Hale, MD, New York, New York
- Pigmentclar Serum
La-Roche Posay Laboratoire Dermatologique
“It attacks pigment production at every stage.”—Whitney Bowe, MD, Brooklyn, New York
Cutis invites readers to send us their recommendations. Mineral makeup, eyelash enhancers, and facial scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to cutis@frontlinemedcom.com.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top OTC pigment control products. Consideration must be given to:
- Even Better
Clinique Laboratories, LLC
“It also is useful as prevention and offers many different options.”—Antonella Tosti, MD, Miami, Florida
“These OTC products have good clinical data to support use for hyperpigmentation. Patients tell me that they feel good on their skin and aren’t irritating.”—Gary Goldenberg, MD, New York, New York
- Lumixyl Brightening System
Envy Medical, Inc
“A great option for patients who may be experiencing modest issues with pigmentation. Use of a retinoid with this product also may enhance its efficacy.”—Joel Schlessinger, MD, Omaha, Nebraska
- Lytera Skin Brightening Complex
SkinMedica
“With key ingredients such as hexylresorcinol, retinol, and niacinamide, it has been clinically shown to lighten dark patches in its trials as well as adding luminosity to the skin.”—Anthony Rossi, MD, New York, New York
Recommended by Elizabeth K. Hale, MD, New York, New York
- Pigmentclar Serum
La-Roche Posay Laboratoire Dermatologique
“It attacks pigment production at every stage.”—Whitney Bowe, MD, Brooklyn, New York
Cutis invites readers to send us their recommendations. Mineral makeup, eyelash enhancers, and facial scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to cutis@frontlinemedcom.com.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top OTC pigment control products. Consideration must be given to:
- Even Better
Clinique Laboratories, LLC
“It also is useful as prevention and offers many different options.”—Antonella Tosti, MD, Miami, Florida
“These OTC products have good clinical data to support use for hyperpigmentation. Patients tell me that they feel good on their skin and aren’t irritating.”—Gary Goldenberg, MD, New York, New York
- Lumixyl Brightening System
Envy Medical, Inc
“A great option for patients who may be experiencing modest issues with pigmentation. Use of a retinoid with this product also may enhance its efficacy.”—Joel Schlessinger, MD, Omaha, Nebraska
- Lytera Skin Brightening Complex
SkinMedica
“With key ingredients such as hexylresorcinol, retinol, and niacinamide, it has been clinically shown to lighten dark patches in its trials as well as adding luminosity to the skin.”—Anthony Rossi, MD, New York, New York
Recommended by Elizabeth K. Hale, MD, New York, New York
- Pigmentclar Serum
La-Roche Posay Laboratoire Dermatologique
“It attacks pigment production at every stage.”—Whitney Bowe, MD, Brooklyn, New York
Cutis invites readers to send us their recommendations. Mineral makeup, eyelash enhancers, and facial scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to cutis@frontlinemedcom.com.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
Unilateral Generalized Keratosis Pilaris Following Pregnancy
Keratosis pilaris (KP), also known as lichen planopilaris, is a common inherited disorder characterized by the presence of small folliculocentric keratotic papules that may have surrounding erythema, which gives the skin a stippled appearance resembling gooseflesh.1 Keratosis pilaris has a predilection for the extensor surfaces of the upper arms, thighs, and buttocks; however, a generalized presentation may occur.1-3 Patients usually are asymptomatic with concerns regarding cosmetic appearance. A number of diseases are associated with KP, such as KP atrophicans, erythromelanosis follicularis faciei et colli, and ichthyosis vulgaris. Treatment of KP mainly focuses on avoiding skin dryness and adding keratolytic agents when necessary.1 We report the case of a 29-year-old woman who presented with unilateral generalized KP in the second month of her second pregnancy.
Case Report
A 29-year-old woman presented in the second month of pregnancy with follicular hyperkeratotic papules on the right side of the face, neck, trunk, buttocks, arms, and legs with mild pruritus (Figure 1). According to the patient, when she found out she was pregnant, she discovered several reddish gray papules on the right cheek and dorsal aspect of the right hand that rapidly spread to the neck and trunk but remained restricted to the right side of the body. She occasionally experienced mild pruritus.
 | 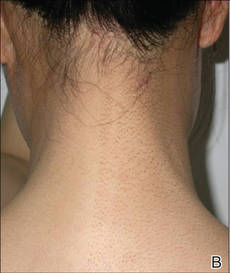 |
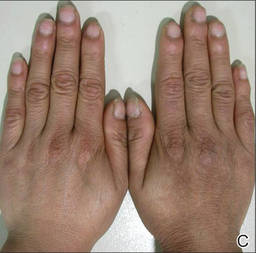 | 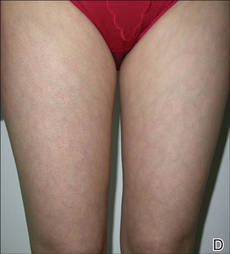 |
Figure 1. Several reddish gray follicular papules formed red patches on the right side of the face (A). Numerous small hyperpigmented follicular papules were scattered on the right side of the neck; the left side was devoid of lesions, revealing a sharp distinct demarcation line between the involved and uninvolved skin (B). Reddish gray or pigmented lesions were distributed on the hands and thighs but were restricted to the right side (C and D). | |
Physical examination revealed numerous small reddish or hyperpigmented follicular papules scattered on the right side of the body, both in the flexor and extensor aspects. The distribution of the lesions did not follow the lines of Blaschko. Strangely, the left side was devoid of lesions, and a sharp distinct line of demarcation separated the involved and uninvolved sides. Physical examination of the scalp, hair, nails, and oral cavity was normal with no atrophy or scarring. The patient’s first pregnancy was aborted in the third month of pregnancy (14 months prior to presentation), but no skin lesions were reported at that time. The patient’s medical history was otherwise unremarkable, and there was no known family history of similar skin lesions. Histopathologic examination of a biopsy taken from a lesion on the right side of the neck showed hyperpigmentation of the basal layer, follicular and interfollicular hyperkeratosis, dilated hair follicles containing keratinous plugs, dermal vascular dilatation and congestion, and mild perivascular inflammatory infiltration (Figure 2).
Based on the clinical and pathologic findings, a diagnosis of unilateral generalized KP was made. No treatment was administered due to the pregnancy. The patient’s skin condition continued to worsen throughout her pregnancy with more widespread and hyperpigmented lesions, but they remained unilateral. The patient showed no obvious improvement 4 months following delivery, and no treatment was administered because the patient was breastfeeding.
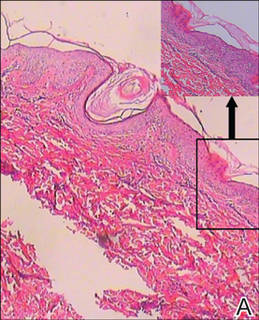 |
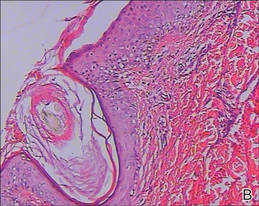 |
| Figure 2. Histopathologic examination revealed hyperpigmentation of the basal layer, dilated hair follicles containing keratinous plugs, dermal vascular dilatation and congestion, and mild perivascular inflammatory infiltration, which suggested typical keratosis pilaris (A and B)(H&E, original magnifications ×40 [inset, original magnification ×100] and ×100). |
Comment
Keratosis pilaris is a common hyperkeratotic condition that typically presents as flesh-colored follicular papules surrounded by varying degrees of perifollicular erythema. Occasionally, inflammatory acneform pustules and papules may appear.1,2 Keratosis pilaris usually presents between the ages of 2 and 3 years, flourishes until 20 years of age, and usually subsides in adulthood.4 Xerotic or atopic individuals are prone to develop KP.2 Other conditions associated with KP include ichthyosis follicularis, atrichia with papular lesions, cardiofasciocutaneous syndrome, ectodermal dysplasia with corkscrew hairs, and keratitis-ichthyosis-deafness syndrome.5 Although its etiology is uncertain, KP is thought to be inherited in an autosomal-dominant fashion.
Histologically, the lesions result from the formation of an orthokeratotic plug, which may contain twisted hairs, that blocks and dilates the orifice and upper portion of the follicular infundibulum. Mild perivascular mononuclear cell infiltrates usually are present in the adjacent dermis.5 Most cases of KP involve lesions that are bilaterally distributed. One report described a 5-year-old girl with lesions that were limited to the right cheek. They were diagnosed as atrophoderma vermiculata, a form of KP atrophicans, which is a less common variant of KP.6 Another report described a 2-year-old girl who developed KP 7 months after birth, which was the only case we found in the English-language literature, according to a PubMed search of articles indexed for MEDLINE using the search terms keratosis pilaris, hemicorporal, and pregnancy, with near-total involvement of one side of the body. The authors believed this peculiar manifestation was probably caused by an inherited segmental anomaly from a postzygotic genetic mutation.7
Our patient’s condition was unusual because the generalized lesions were unilaterally distributed and they occurred early in the pregnancy. Some studies have indicated that hormonal influences may play a role in the development of KP.3,8 Our case also demonstrates KP as a condition in which the onset and severity of the disorder may be associated with the hormonal changes in pregnancy. Additionally, it is worth noting that the lesions presented during the patient’s second pregnancy, but there were no lesions reported during her first pregnancy 14 months prior, which was aborted in the third month. We cannot explain this phenomenon and cannot predict if the lesions will recur in future pregnancies.
Conclusion
We suggest that both a genetic mutation and pregnancy-induced hormonal changes played possible roles in the development and progress of unilateral generalized KP in our patient. Follow-up is necessary in these patients and further work needs to be done to reveal the underlying mechanism.
1. Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
2. Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: WB Saunders; 2000.
3. Jackson JB, Touma SC, Norton AB. Keratosis pilaris in pregnancy: an unrecognized dematosis of pregnancy? W V Med J. 2004;100:26-28.
4. Poskitt L, Wilkinson JD. Natural history of keratosis pilaris. Br J Dermatol. 1994;130:711-713.
5. Griffiths WAD, Judge MR, Leigh IM. Disorders of keratinisation. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. 6th ed. Oxford, MA: Blackwell Scientific; 1998.
6. Nico MM, Valente NY, Sotto MN. Folliculitis ulerythematosa reticulata (atrophoderma vermiculata): early detection of a case with unilateral lesions. Pediatr Dermatol. 1998;15:285-286.
7. Ehsani A, Namazi MR, Barikbin B, et al. Unilaterally generalized keratosis pilaris. J Eur Acad Dermatol Venereol. 2003;17:361-362.
8. Barth JH, Wojnarowska F, Dawber RP. Is keratosis pilaris another androgen-dependent dermatosis? Clin Exp Dermatol. 1988;13:240-241.
Keratosis pilaris (KP), also known as lichen planopilaris, is a common inherited disorder characterized by the presence of small folliculocentric keratotic papules that may have surrounding erythema, which gives the skin a stippled appearance resembling gooseflesh.1 Keratosis pilaris has a predilection for the extensor surfaces of the upper arms, thighs, and buttocks; however, a generalized presentation may occur.1-3 Patients usually are asymptomatic with concerns regarding cosmetic appearance. A number of diseases are associated with KP, such as KP atrophicans, erythromelanosis follicularis faciei et colli, and ichthyosis vulgaris. Treatment of KP mainly focuses on avoiding skin dryness and adding keratolytic agents when necessary.1 We report the case of a 29-year-old woman who presented with unilateral generalized KP in the second month of her second pregnancy.
Case Report
A 29-year-old woman presented in the second month of pregnancy with follicular hyperkeratotic papules on the right side of the face, neck, trunk, buttocks, arms, and legs with mild pruritus (Figure 1). According to the patient, when she found out she was pregnant, she discovered several reddish gray papules on the right cheek and dorsal aspect of the right hand that rapidly spread to the neck and trunk but remained restricted to the right side of the body. She occasionally experienced mild pruritus.
| |
| |
Figure 1. Several reddish gray follicular papules formed red patches on the right side of the face (A). Numerous small hyperpigmented follicular papules were scattered on the right side of the neck; the left side was devoid of lesions, revealing a sharp distinct demarcation line between the involved and uninvolved skin (B). Reddish gray or pigmented lesions were distributed on the hands and thighs but were restricted to the right side (C and D). | |
Physical examination revealed numerous small reddish or hyperpigmented follicular papules scattered on the right side of the body, both in the flexor and extensor aspects. The distribution of the lesions did not follow the lines of Blaschko. Strangely, the left side was devoid of lesions, and a sharp distinct line of demarcation separated the involved and uninvolved sides. Physical examination of the scalp, hair, nails, and oral cavity was normal with no atrophy or scarring. The patient’s first pregnancy was aborted in the third month of pregnancy (14 months prior to presentation), but no skin lesions were reported at that time. The patient’s medical history was otherwise unremarkable, and there was no known family history of similar skin lesions. Histopathologic examination of a biopsy taken from a lesion on the right side of the neck showed hyperpigmentation of the basal layer, follicular and interfollicular hyperkeratosis, dilated hair follicles containing keratinous plugs, dermal vascular dilatation and congestion, and mild perivascular inflammatory infiltration (Figure 2).
Based on the clinical and pathologic findings, a diagnosis of unilateral generalized KP was made. No treatment was administered due to the pregnancy. The patient’s skin condition continued to worsen throughout her pregnancy with more widespread and hyperpigmented lesions, but they remained unilateral. The patient showed no obvious improvement 4 months following delivery, and no treatment was administered because the patient was breastfeeding.
|
|
| Figure 2. Histopathologic examination revealed hyperpigmentation of the basal layer, dilated hair follicles containing keratinous plugs, dermal vascular dilatation and congestion, and mild perivascular inflammatory infiltration, which suggested typical keratosis pilaris (A and B)(H&E, original magnifications ×40 [inset, original magnification ×100] and ×100). |
Comment
Keratosis pilaris is a common hyperkeratotic condition that typically presents as flesh-colored follicular papules surrounded by varying degrees of perifollicular erythema. Occasionally, inflammatory acneform pustules and papules may appear.1,2 Keratosis pilaris usually presents between the ages of 2 and 3 years, flourishes until 20 years of age, and usually subsides in adulthood.4 Xerotic or atopic individuals are prone to develop KP.2 Other conditions associated with KP include ichthyosis follicularis, atrichia with papular lesions, cardiofasciocutaneous syndrome, ectodermal dysplasia with corkscrew hairs, and keratitis-ichthyosis-deafness syndrome.5 Although its etiology is uncertain, KP is thought to be inherited in an autosomal-dominant fashion.
Histologically, the lesions result from the formation of an orthokeratotic plug, which may contain twisted hairs, that blocks and dilates the orifice and upper portion of the follicular infundibulum. Mild perivascular mononuclear cell infiltrates usually are present in the adjacent dermis.5 Most cases of KP involve lesions that are bilaterally distributed. One report described a 5-year-old girl with lesions that were limited to the right cheek. They were diagnosed as atrophoderma vermiculata, a form of KP atrophicans, which is a less common variant of KP.6 Another report described a 2-year-old girl who developed KP 7 months after birth, which was the only case we found in the English-language literature, according to a PubMed search of articles indexed for MEDLINE using the search terms keratosis pilaris, hemicorporal, and pregnancy, with near-total involvement of one side of the body. The authors believed this peculiar manifestation was probably caused by an inherited segmental anomaly from a postzygotic genetic mutation.7
Our patient’s condition was unusual because the generalized lesions were unilaterally distributed and they occurred early in the pregnancy. Some studies have indicated that hormonal influences may play a role in the development of KP.3,8 Our case also demonstrates KP as a condition in which the onset and severity of the disorder may be associated with the hormonal changes in pregnancy. Additionally, it is worth noting that the lesions presented during the patient’s second pregnancy, but there were no lesions reported during her first pregnancy 14 months prior, which was aborted in the third month. We cannot explain this phenomenon and cannot predict if the lesions will recur in future pregnancies.
Conclusion
We suggest that both a genetic mutation and pregnancy-induced hormonal changes played possible roles in the development and progress of unilateral generalized KP in our patient. Follow-up is necessary in these patients and further work needs to be done to reveal the underlying mechanism.
Keratosis pilaris (KP), also known as lichen planopilaris, is a common inherited disorder characterized by the presence of small folliculocentric keratotic papules that may have surrounding erythema, which gives the skin a stippled appearance resembling gooseflesh.1 Keratosis pilaris has a predilection for the extensor surfaces of the upper arms, thighs, and buttocks; however, a generalized presentation may occur.1-3 Patients usually are asymptomatic with concerns regarding cosmetic appearance. A number of diseases are associated with KP, such as KP atrophicans, erythromelanosis follicularis faciei et colli, and ichthyosis vulgaris. Treatment of KP mainly focuses on avoiding skin dryness and adding keratolytic agents when necessary.1 We report the case of a 29-year-old woman who presented with unilateral generalized KP in the second month of her second pregnancy.
Case Report
A 29-year-old woman presented in the second month of pregnancy with follicular hyperkeratotic papules on the right side of the face, neck, trunk, buttocks, arms, and legs with mild pruritus (Figure 1). According to the patient, when she found out she was pregnant, she discovered several reddish gray papules on the right cheek and dorsal aspect of the right hand that rapidly spread to the neck and trunk but remained restricted to the right side of the body. She occasionally experienced mild pruritus.
| |
| |
Figure 1. Several reddish gray follicular papules formed red patches on the right side of the face (A). Numerous small hyperpigmented follicular papules were scattered on the right side of the neck; the left side was devoid of lesions, revealing a sharp distinct demarcation line between the involved and uninvolved skin (B). Reddish gray or pigmented lesions were distributed on the hands and thighs but were restricted to the right side (C and D). | |
Physical examination revealed numerous small reddish or hyperpigmented follicular papules scattered on the right side of the body, both in the flexor and extensor aspects. The distribution of the lesions did not follow the lines of Blaschko. Strangely, the left side was devoid of lesions, and a sharp distinct line of demarcation separated the involved and uninvolved sides. Physical examination of the scalp, hair, nails, and oral cavity was normal with no atrophy or scarring. The patient’s first pregnancy was aborted in the third month of pregnancy (14 months prior to presentation), but no skin lesions were reported at that time. The patient’s medical history was otherwise unremarkable, and there was no known family history of similar skin lesions. Histopathologic examination of a biopsy taken from a lesion on the right side of the neck showed hyperpigmentation of the basal layer, follicular and interfollicular hyperkeratosis, dilated hair follicles containing keratinous plugs, dermal vascular dilatation and congestion, and mild perivascular inflammatory infiltration (Figure 2).
Based on the clinical and pathologic findings, a diagnosis of unilateral generalized KP was made. No treatment was administered due to the pregnancy. The patient’s skin condition continued to worsen throughout her pregnancy with more widespread and hyperpigmented lesions, but they remained unilateral. The patient showed no obvious improvement 4 months following delivery, and no treatment was administered because the patient was breastfeeding.
|
|
| Figure 2. Histopathologic examination revealed hyperpigmentation of the basal layer, dilated hair follicles containing keratinous plugs, dermal vascular dilatation and congestion, and mild perivascular inflammatory infiltration, which suggested typical keratosis pilaris (A and B)(H&E, original magnifications ×40 [inset, original magnification ×100] and ×100). |
Comment
Keratosis pilaris is a common hyperkeratotic condition that typically presents as flesh-colored follicular papules surrounded by varying degrees of perifollicular erythema. Occasionally, inflammatory acneform pustules and papules may appear.1,2 Keratosis pilaris usually presents between the ages of 2 and 3 years, flourishes until 20 years of age, and usually subsides in adulthood.4 Xerotic or atopic individuals are prone to develop KP.2 Other conditions associated with KP include ichthyosis follicularis, atrichia with papular lesions, cardiofasciocutaneous syndrome, ectodermal dysplasia with corkscrew hairs, and keratitis-ichthyosis-deafness syndrome.5 Although its etiology is uncertain, KP is thought to be inherited in an autosomal-dominant fashion.
Histologically, the lesions result from the formation of an orthokeratotic plug, which may contain twisted hairs, that blocks and dilates the orifice and upper portion of the follicular infundibulum. Mild perivascular mononuclear cell infiltrates usually are present in the adjacent dermis.5 Most cases of KP involve lesions that are bilaterally distributed. One report described a 5-year-old girl with lesions that were limited to the right cheek. They were diagnosed as atrophoderma vermiculata, a form of KP atrophicans, which is a less common variant of KP.6 Another report described a 2-year-old girl who developed KP 7 months after birth, which was the only case we found in the English-language literature, according to a PubMed search of articles indexed for MEDLINE using the search terms keratosis pilaris, hemicorporal, and pregnancy, with near-total involvement of one side of the body. The authors believed this peculiar manifestation was probably caused by an inherited segmental anomaly from a postzygotic genetic mutation.7
Our patient’s condition was unusual because the generalized lesions were unilaterally distributed and they occurred early in the pregnancy. Some studies have indicated that hormonal influences may play a role in the development of KP.3,8 Our case also demonstrates KP as a condition in which the onset and severity of the disorder may be associated with the hormonal changes in pregnancy. Additionally, it is worth noting that the lesions presented during the patient’s second pregnancy, but there were no lesions reported during her first pregnancy 14 months prior, which was aborted in the third month. We cannot explain this phenomenon and cannot predict if the lesions will recur in future pregnancies.
Conclusion
We suggest that both a genetic mutation and pregnancy-induced hormonal changes played possible roles in the development and progress of unilateral generalized KP in our patient. Follow-up is necessary in these patients and further work needs to be done to reveal the underlying mechanism.
1. Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
2. Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: WB Saunders; 2000.
3. Jackson JB, Touma SC, Norton AB. Keratosis pilaris in pregnancy: an unrecognized dematosis of pregnancy? W V Med J. 2004;100:26-28.
4. Poskitt L, Wilkinson JD. Natural history of keratosis pilaris. Br J Dermatol. 1994;130:711-713.
5. Griffiths WAD, Judge MR, Leigh IM. Disorders of keratinisation. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. 6th ed. Oxford, MA: Blackwell Scientific; 1998.
6. Nico MM, Valente NY, Sotto MN. Folliculitis ulerythematosa reticulata (atrophoderma vermiculata): early detection of a case with unilateral lesions. Pediatr Dermatol. 1998;15:285-286.
7. Ehsani A, Namazi MR, Barikbin B, et al. Unilaterally generalized keratosis pilaris. J Eur Acad Dermatol Venereol. 2003;17:361-362.
8. Barth JH, Wojnarowska F, Dawber RP. Is keratosis pilaris another androgen-dependent dermatosis? Clin Exp Dermatol. 1988;13:240-241.
1. Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
2. Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: WB Saunders; 2000.
3. Jackson JB, Touma SC, Norton AB. Keratosis pilaris in pregnancy: an unrecognized dematosis of pregnancy? W V Med J. 2004;100:26-28.
4. Poskitt L, Wilkinson JD. Natural history of keratosis pilaris. Br J Dermatol. 1994;130:711-713.
5. Griffiths WAD, Judge MR, Leigh IM. Disorders of keratinisation. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. 6th ed. Oxford, MA: Blackwell Scientific; 1998.
6. Nico MM, Valente NY, Sotto MN. Folliculitis ulerythematosa reticulata (atrophoderma vermiculata): early detection of a case with unilateral lesions. Pediatr Dermatol. 1998;15:285-286.
7. Ehsani A, Namazi MR, Barikbin B, et al. Unilaterally generalized keratosis pilaris. J Eur Acad Dermatol Venereol. 2003;17:361-362.
8. Barth JH, Wojnarowska F, Dawber RP. Is keratosis pilaris another androgen-dependent dermatosis? Clin Exp Dermatol. 1988;13:240-241.
Practice Points
- Keratosis pilaris (KP) is a common disorder, with a genetic background and hormonal changes playing possible roles in its development. It also may be associated with a number of diseases.
- The skin lesions of KP usually are bilaterally distributed, either in a generalized or localized distribution. Unilateral lesions are rare. The underlying mechanism of unilateral generalized KP remains unknown.
Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
 |  |  | ||
 | 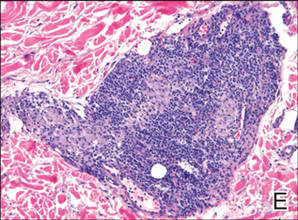 |  | ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
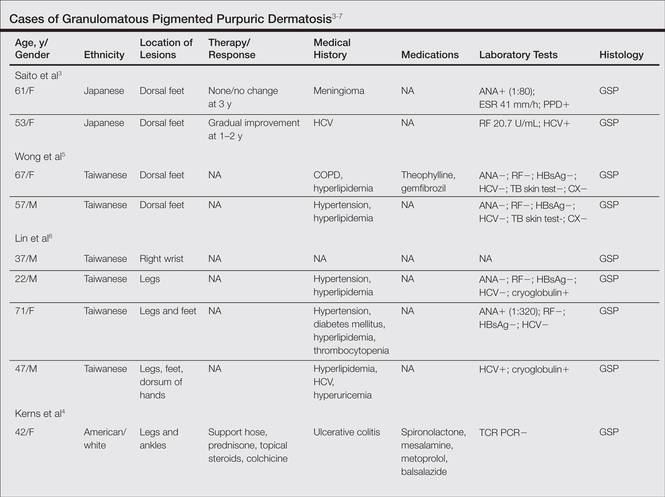
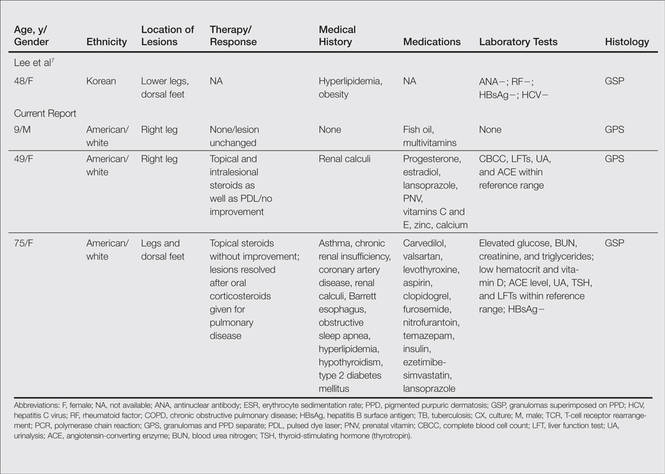
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
| | | ||
| | | ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
| | | ||
| | | ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Practice Points
- Consider a punch biopsy when sampling suspected inflammatory dermatoses, such as pigmented purpuric dermatosis, to allow deeper sampling.
- Provide all clinical details to the dermatopathologist to assist with clinicopathologic correlation and diagnostic accuracy.