User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Printable Guide on the Rosacea Patient Journey
The Rosacea Patient Journey: A Novel Approach to Conceptualizing Patient Experiences
Rosacea patients experience symptoms ranging from flushing to persistent acnelike rashes that can cause low self-esteem and anxiety, leading to social and professional isolation.1 Although it is estimated that 16 million individuals in the United States have rosacea, only 10% seek treatment.2,3 The motivation for patients to seek and adhere to treatment is not well characterized.
A patient journey is a map of the steps a patient takes as he/she progresses through different segments of the disease from diagnosis to management, including all the influences that can push him/her toward or away from certain decisions. The patient journey model provides a structure for understanding key issues in rosacea management, including barriers to successful treatment outcomes.
The patient journey model progresses from development of disease and diagnosis to treatment and disease management (Figure). We sought to examine each step of the rosacea patient journey to better understand key patient care boundaries faced by rosacea patients. We assessed the current literature regarding each step of the patient experience and identified areas of the patient journey with limited research.
Click here to view the figure as a PDF to print for future reference.
Researching the Patient Experience
A PubMed search of articles indexed for MEDLINE as well as a search of the National Rosacea Society Web site (http://www.rosacea.org) were conducted to identify articles and materials that quantitatively or qualitatively described rosacea patient experiences. Search terms included rosacea, rosacea patient experience, rosacea treatment, rosacea adherence, and rosacea quality of life. A Google search also was conducted using the same terms to obtain current news articles online. Current literature pertaining to the patient journey was summarized.
To create a model for the rosacea patient journey, we refined a rheumatoid arthritis patient journey map4 and included the critical components of the journey for rosacea patients. We organized the journey into stages, including prediagnosis, diagnosis, treatment, adherence, and management. We first explored what occurs prior to diagnosis, which includes the patient’s symptoms before visiting a physician. We then examined the process of diagnosis and the implementation of a treatment plan. Treatment adherence was then explored, ending with the ways patients self-manage their disease beyond the physician’s office.
Prediagnosis: What Motivates Patients to Seek Treatment
Rosacea can present with many symptoms that may lead patients to see a physician, including facial erythema and telangiectases, papules and pustules, phymatous changes, and ocular manifestations.5 The most common concern is temporary facial flushing, followed by persistent redness, then bumps and pimples.6 Many patients seek treatment after persistent facial flushing and an intolerable burning sensation. Some middle-aged patients decide to see a dermatologist for the first time when they break out in acne lesions after a history of clear skin. Others seek treatment because they can no longer tolerate the pain and embarrassment associated with their symptoms. However, patients who seek treatment only account for a small proportion of patients with rosacea, as only 10% of patients seek conventional medical treatment.7 Furthermore, symptomatic patients on average wait 7 months to 5 years before receiving a diagnosis.8,9
Care often is delayed or not pursued because many rosacea symptoms are mild when they first appear and may not initially bother the patient. Patients may not think anything of their symptoms and dismiss them as either acne vulgaris or sunburn. Due to the relapsing and remitting nature of the disease course, patients may feel their symptoms will resolve. Of patients diagnosed with rosacea, only one-half have heard of the condition prior to diagnosis,8 which can largely be attributed to lack of patient education on the signs and symptoms of rosacea, a concern that prompted the National Rosacea Society to designate the month of April as rosacea awareness month.5
With sales of antiredness facial care products growing 35% from 2002 to 2007, accounting for an increase of $300 million in revenue, patients also may be turning to over-the-counter products first.10 Men with rosacea tend to present with more severe symptoms such as rhinophyma, which may be due to their desire to wait until their symptoms reached more advanced stages of disease before seeking medical help.5
Diagnosis of Rosacea
After the patient decides that his/her symptoms are unusual, severe, or intolerable enough to seek treatment, the issues of access to dermatologic care and receiving the correct diagnosis come into play. Accessing dermatologic care can be difficult, as appointments may be hard to obtain, and even if the patient is able to get an appointment, it could be many weeks later.11 For some rosacea patients, the anxiety of waiting for their appointment prompts them to seek support and advice from online message boards (eg, http://www.rosacea-support.org). The long wait for appointments may be attributed to the increased demand for dermatologists for cosmetic procedures.12 Additionally, disparities according to insurance type can contribute to difficulties procuring an appointment. In one study, privately insured dermatology patients demonstrated a 91% acceptance rate and shorter wait times for appointments compared to publicly insured patients who were limited to a 29.8% acceptance rate and longer wait times.11 Many patients then are left to wait for an appointment with a dermatologist or instead turn to a primary care physician. Of patients diagnosed with rosacea in one study (N=2847), the majority of patients were seen by a dermatologist (79%), while the other patients were diagnosed by a family physician (14%) or other types of physicians such as internists and ophthalmologists (7%).6
The diagnosis of rosacea usually is not a major hurdle for dermatologists, but misdiagnoses can sometimes occur. The Rosacea Research & Development Institute compiled multiple patient anecdotes describing the struggles of finally reaching the correct diagnosis of rosacea; however, no estimates as to the frequency of misdiagnoses was estimated.13 Even with an accurate diagnosis of rosacea, correct classification of the 4 types of rosacea (ie, erythematotelangiectatic, papulopustular, phymatous, ocular) is necessary to avoid incorrect treatment recommendations. For example, patients with flushing often cannot tolerate topical medications in contrast to patients with the papulopustular subtype who benefit from them.14 In the meantime, the patients who are misdiagnosed may be met with frustration, as treatment was either delayed or incorrectly prescribed.
Although there are limited data regarding patient reactions after receiving a diagnosis of rosacea, it can be assumed that patients would be hopeful that diagnosis would lead to correct treatment. In a 2008 article in The New York Times, a rosacea patient was described as feeling relieved to be diagnosed with rosacea because it was an explanation for the development of pimples on the cheeks in her late 40s.10
Implementation of a Treatment Plan
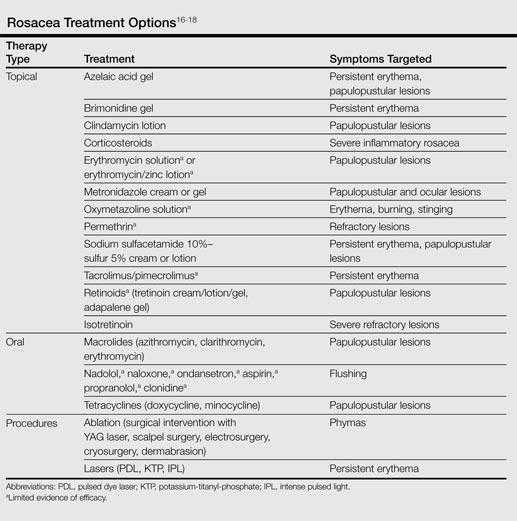
After recognizing the symptoms and receiving a correct diagnosis, the next step in the patient journey is treatment. Long-term management of incurable conditions such as rosacea is difficult. The main goals of treatment are to relieve symptoms, improve appearance, delay progression to advanced stages, and maintain remission.15 There are only a few reliable clinical trials regarding therapies for rosacea, so treatment has mostly relied on clinical experience (Table). The efficacy and safety of many older treatments has not been assessed.15 Mainstays of treatment include both topical agents and oral medications. The use of topical metronidazole, oral tetracycline, and oral isotretinoin have been found to improve both skin lesions and quality of life.18 Initially, a combination of a topical and an oral medication may be used for at least the first 12 weeks, and improvement is usually gradual, taking many weeks to become evident.15 Long-term treatment with topical medications often is required for maintenance, which can last another 6 months or more.19,20
Besides using pharmacologic therapies, some patients also may choose to undergo various procedures. The most common procedure is laser therapy, followed by dermabrasion, chemical peels, hot loop electrocoagulation, and surgical sculpting or plastic surgery.6 The use of these adjunct therapies may suggest impatience from the patient for improvement; it also indicates the lengths patients will go to and willingness to pay for improvement of symptoms.
Along with medication, patients are recommended to make changes to their skin care regimen and lifestyle. Rosacea patients typically have sensitive skin that may include symptoms such as dryness, scaling, stinging, burning, and pruritus.16 Skin care recommendations for rosacea patients include using a gentle cleanser and regularly applying sunscreen.5 Issues with physical appearance can be addressed with the use of cosmetic products such as green-tinted makeup to conceal skin lesions.21 Remission can be maintained by identifying certain triggers (eg, red wine, spicy foods, extreme temperatures, prolonged sun exposure, vigorous exercise) that can cause flare-ups.15 The most common trigger is sun exposure, making photoprotection an important component of the rosacea patient’s skin care regimen.6
Adherence
With a diagnosis and treatment plan in effect, the patient journey reaches the stage of treatment adherence, which should include ongoing education about the condition. Self-reported statistics from rosacea patients indicated that 28% of patients took time off from their treatment regimen,6 but actual nonadherence rates likely are higher. The most commonly reported reason for poor treatment adherence among rosacea patients was the impression that the symptoms had resolved or were adequately controlled.6 Treatment also must be affordable. In a national survey of rosacea patients, 24% of 427 patients receiving pharmacologic therapy planned on switching medications because of cost, and 17% of 769 patients discontinued medications due to co-pay/insurance issues.6 Other reasons cited for discontinuation of treatment included patient perception that symptoms were not that serious, co-pay/insurance issues, ineffectiveness of the medication, and side effects.6 Adherence to topical medications is lower than oral medications due to the time and inconvenience required for application.22 For some patients, topical medications may be too messy, have a strange odor, or stain clothing.
It is promising that most rosacea patients have reported the intent to continue using pharmacologic agents because the medication prevented worsening of their symptoms.6 However, there are still patients who switch or discontinue therapies without physician direction. These patients often cite that they desire more information at the time of diagnosis, particularly related to causes of flare-ups, physical symptoms to expect, drug treatment options, makeup to cover up visible symptoms, surgical or laser treatment options, psychological symptoms, patient support groups, and counseling options.6
Management
The last part of the journey is disease management, which occurs when the patient learns how to control his/her symptoms long-term. Important factors contributing to long-term control of rosacea flares are medication adherence and avoiding lifestyle triggers.23,24 Through the other stages of the journey, the patient has learned which treatments work and which factors may lead to exacerbation of symptoms.
Educating Patients on the Journey
The patient journey is a concept that can be applied to any disease state and brings to light roadblocks that patients may face from the initial diagnosis to successful disease management. Rosacea patients are faced with confusing and aggravating symptoms that can cause anxiety and may lead them to seek treatment from a physician. Facial flushing and phymatous changes of the nose can be mistaken for alcohol abuse, leading rosacea to be a socially stigmatizing disease.15 Because rosacea involves mostly the facial skin, it can disrupt social and professional interactions, leading to quality-of-life effects such as difficulty functioning on a day-to-day basis, which can be detrimental because patients usually are aged 30 to 50 years and may be perceived based on their appearance in the workforce.3 A lack of confidence, low self-esteem, embarrassment, and anxiety can even lead to serious psychiatric conditions such as depression and body dysmorphic disorder.25 Because the severity of rosacea increases over time, it is important to educate patients about seeking early treatment; therefore, understanding and awareness of rosacea symptoms are necessary to prompt patients to see a medical professional to either confirm or refute the diagnosis.
Rosacea is a clinical diagnosis that relies on patterns of primary and secondary features, as outlined in a 2002 report by the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea.5 Even with this consensus grading system, it appears that additional fine-tuning of the criteria is needed in the disease definition. Importantly, because much of the pathogenesis and progression of rosacea is still not completely understood, there is no laboratory benchmark test that can be utilized for correct diagnosis.14 Moreover, many of the clinical manifestations of rosacea are shared with other conditions, and patients may present with different symptoms or varying combinations.26
Treatment of rosacea is multifactorial and behavioral, as patients must not only be able to obtain and adhere to oral and topical regimens and possible procedures but also avoid various lifestyle and environmental triggers and learn to cope with emotional distress caused by their symptoms. Although patients who discontinue use of medications appear to be in the minority, education is still needed to stress the chronic nature of rosacea and the importance of the continuation of treatment. Collaboration between the physician and patient is needed to determine why a certain medication may not be effective and explore other treatment options. Treatment ineffectiveness could be due to incorrect use of the product, failure to use an adjunct skin care regimen, or inability to control rosacea triggers. Adequate early follow-up also is needed to maximize patient adherence to treatment.27 Working together with the patient to develop a treatment plan that can be followed is necessary for long-term control of rosacea symptoms.
There is little information on how to address the psychological needs of patients, but patients can find support from various avenues. For instance, the National Rosacea Society, a large advocacy group, produces newsletters and educational materials for both physicians and patients.28,29 There also are online support groups for rosacea patients that have thousands of members who exchange stories and provide words of encouragement. Although there are not many face-to-face support groups, physicians may consider developing live support groups for their rosacea patients. As patients achieve the later stages of the rosacea patient journey, they hopefully will have controlled their symptoms by following a treatment regimen and learning to adapt to a new life of successful disease management.
Many aspects of the rosacea patient journey have yet to be explored. It is uncertain how long patients with symptoms of rosacea wait before seeking treatment, what methods they use to control their rosacea before they receive a prescribed treatment or physician recommendations, and how they react to their diagnosis. It also is unknown how many rosacea patients receive an initial misdiagnosis of another condition and which physicians typically make the misdiagnosis. We also need to know more about the role of psychological issues in addressing patient adherence to treatment. Similarly, what role do support groups such as online forums play on adherence? There is a need for more patient education and awareness of rosacea.
Conclusion
Patients may be relieved that rosacea is not a life-threatening condition, but they may be disappointed that there is no cure for rosacea. As the patient and dermatologist work together to find an appropriate treatment plan, identify certain triggers, and modify the skin care routine, the patient can become disciplined in controlling rosacea symptoms. Ultimately, with the alleviation of visible symptoms, the patient’s quality of life also can improve. Better understanding of the rosacea patient perspective can lead to a more efficient health care system, improved patient care, and better patient satisfaction.
1. Baldwin HE. Systemic therapy for rosacea. Skin Therapy Lett. 2007;12:1-5, 9.
2. Drake L. Rosacea now estimated to affect at least 16 million Americans. Rosacea Review. Winter 2010. http://www.rosacea.org/rr/2010/winter/article_1.php. Accessed December 11, 2014.
3. Rosacea as an inflammatory disease: an expert interview with Brian Berman, MD, PhD. Medscape Web site. http://www.medscape.org/viewarticle/722156. Published May 27, 2010. Accessed December 11, 2014.
4. HealthEd Group, Inc. Rheumatoid arthritis patient journey map. http://visual.ly/rheumatoid-arthritis-patient-journey-map. Accessed December 19, 2014.
5. Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Elewski BE. Results of a national rosacea patient survey: common issues that concern rosacea sufferers. J Drugs Dermatol. 2009;8:120-123.
7. Del Rosso J. Management of rosacea in the United States: analysis based on recent prescribing patterns and insurance claims. J Am Acad Dermatol. 2008;58:AB13.
8. New survey reveals first impressions may not always be rosy for people with the widespread skin condition rosacea. Medical News Today Web site. http://www.medicalnew today.com/releases/185491.php. Updated April 15, 2010. Accessed December 12, 2014.
9. Shear NH, Levine C. Needs survey of Canadian rosacea patients. J Cutan Med Surg. 1999;3:178-181.
10. Sweeney C. In a perfect world, rosacea remains a problem. New York Times. April 24, 2008. http://www.nytimes.com/2008/04/24/fashion/24SKIN.html?pagewanted=all. Accessed December 12, 2014.
11. Alghothani L, Jacks SK, Vander HA, et al. Disparities in access to dermatologic care according to insurance type. Arch Dermatol. 2012;148:956-957.
12. Resneck J Jr. Too few or too many dermatologists? difficulties in assessing optimal workforce size. Arch Dermatol. 2001;137:1295-1301.
13. Rosacea Research & Development Institute Web site. http://irosacea.org/misdiagnosed_rosacea.html. Accessed December 19, 2014.
14. Crawford GH, Pelle MT, James WD. Rosacea: I. etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
15. Elewski BE, Draelos Z, Dreno B, et al. Rosacea—global diversity and optimized outcome: proposed international consensus from the Rosacea International Expert Group. J Eur Acad Dermatol Venereol. 2011;25:188-200.
16. Del Rosso JQ, Baldwin H, Webster G. American Acne & Rosacea Society rosacea medical management guidelines. J Drugs Dermatol. 2008;7:531-533.
17. Fowler J Jr, Jackson M, Moore A, et al. Efficacy and safety of once-daily topical brimonidine tartrate gel 0.5% for the treatment of moderate to severe facial erythema of rosacea: results of two randomized, double-blind, and vehicle-controlled pivotal studies. J Drugs Dermatol. 2013;12:650-656.
18. Aksoy B, Altaykan-Hapa A, Egemen D, et al. The impact of rosacea on quality of life: effects of demographic and clinical characteristics and various treatment modalities. Br J Dermatol. 2010;163:719-725.
19. Dahl MV, Katz HI, Krueger GG, et al. Topical metronidazole maintains remissions of rosacea. Arch Dermatol. 1998;134:679-683.
20. Thiboutot DM, Fleischer AB, Del Rosso JQ, et al. A multicenter study of topical azelaic acid 15% gel in combination with oral doxycycline as initial therapy and azelaic acid 15% gel as maintenance monotherapy. J Drugs Dermatol. 2009;8:639-648.
21. Boehncke WH, Ochsendorf F, Paeslack I, et al. Decorative cosmetics improve the quality of life in patients with disfiguring skin diseases. Eur J Dermatol. 2002;12:577-580.
22. Jackson JM, Pelle M. Topical rosacea therapy: the importance of vehicles for efficacy, tolerability and compliance. J Drugs Dermatol. 2011;10:627-633.
23. Wolf JE Jr. Medication adherence: a key factor in effective management of rosacea. Adv Ther. 2001;18:272-281.
24. Managing rosacea. National Rosacea Society Web site. http://www.rosacea.org/patients/materials/managing/lifestyle.php. Accessed December 19, 2014.
25. van Zuuren EJ, Fedorowicz Z. Lack of ‘appropriately assessed’ patient-reported outcomes in randomized controlled trials assessing the effectiveness of interventions for rosacea. Br J Dermatol. 2013;168:442-444.
26. Del Rosso JQ. Advances in understanding and managing rosacea: part 2: the central role, evaluation, and medical management of diffuse and persistent facial erythema of rosacea. J Clin Aesthet Dermatol. 2012;5:26-36.
27. Davis SA, Lin HC, Yu CH, et al. Underuse of early follow-up visits: a missed opportunity to improve patients’ adherence. 2014;13:833-836.
28. If you have rosacea, you’re not alone. National Rosacea Society Web site. http://www.rosacea.org/patients/index.php. Accessed December 19, 2014.
29. Tools for the professional. National Rosacea Society Web site. http://www.rosacea.org/physicians/index.php. Accessed December 19, 2014.
Rosacea patients experience symptoms ranging from flushing to persistent acnelike rashes that can cause low self-esteem and anxiety, leading to social and professional isolation.1 Although it is estimated that 16 million individuals in the United States have rosacea, only 10% seek treatment.2,3 The motivation for patients to seek and adhere to treatment is not well characterized.
A patient journey is a map of the steps a patient takes as he/she progresses through different segments of the disease from diagnosis to management, including all the influences that can push him/her toward or away from certain decisions. The patient journey model provides a structure for understanding key issues in rosacea management, including barriers to successful treatment outcomes.
The patient journey model progresses from development of disease and diagnosis to treatment and disease management (Figure). We sought to examine each step of the rosacea patient journey to better understand key patient care boundaries faced by rosacea patients. We assessed the current literature regarding each step of the patient experience and identified areas of the patient journey with limited research.
Click here to view the figure as a PDF to print for future reference.
Researching the Patient Experience
A PubMed search of articles indexed for MEDLINE as well as a search of the National Rosacea Society Web site (http://www.rosacea.org) were conducted to identify articles and materials that quantitatively or qualitatively described rosacea patient experiences. Search terms included rosacea, rosacea patient experience, rosacea treatment, rosacea adherence, and rosacea quality of life. A Google search also was conducted using the same terms to obtain current news articles online. Current literature pertaining to the patient journey was summarized.
To create a model for the rosacea patient journey, we refined a rheumatoid arthritis patient journey map4 and included the critical components of the journey for rosacea patients. We organized the journey into stages, including prediagnosis, diagnosis, treatment, adherence, and management. We first explored what occurs prior to diagnosis, which includes the patient’s symptoms before visiting a physician. We then examined the process of diagnosis and the implementation of a treatment plan. Treatment adherence was then explored, ending with the ways patients self-manage their disease beyond the physician’s office.
Prediagnosis: What Motivates Patients to Seek Treatment
Rosacea can present with many symptoms that may lead patients to see a physician, including facial erythema and telangiectases, papules and pustules, phymatous changes, and ocular manifestations.5 The most common concern is temporary facial flushing, followed by persistent redness, then bumps and pimples.6 Many patients seek treatment after persistent facial flushing and an intolerable burning sensation. Some middle-aged patients decide to see a dermatologist for the first time when they break out in acne lesions after a history of clear skin. Others seek treatment because they can no longer tolerate the pain and embarrassment associated with their symptoms. However, patients who seek treatment only account for a small proportion of patients with rosacea, as only 10% of patients seek conventional medical treatment.7 Furthermore, symptomatic patients on average wait 7 months to 5 years before receiving a diagnosis.8,9
Care often is delayed or not pursued because many rosacea symptoms are mild when they first appear and may not initially bother the patient. Patients may not think anything of their symptoms and dismiss them as either acne vulgaris or sunburn. Due to the relapsing and remitting nature of the disease course, patients may feel their symptoms will resolve. Of patients diagnosed with rosacea, only one-half have heard of the condition prior to diagnosis,8 which can largely be attributed to lack of patient education on the signs and symptoms of rosacea, a concern that prompted the National Rosacea Society to designate the month of April as rosacea awareness month.5
With sales of antiredness facial care products growing 35% from 2002 to 2007, accounting for an increase of $300 million in revenue, patients also may be turning to over-the-counter products first.10 Men with rosacea tend to present with more severe symptoms such as rhinophyma, which may be due to their desire to wait until their symptoms reached more advanced stages of disease before seeking medical help.5
Diagnosis of Rosacea
After the patient decides that his/her symptoms are unusual, severe, or intolerable enough to seek treatment, the issues of access to dermatologic care and receiving the correct diagnosis come into play. Accessing dermatologic care can be difficult, as appointments may be hard to obtain, and even if the patient is able to get an appointment, it could be many weeks later.11 For some rosacea patients, the anxiety of waiting for their appointment prompts them to seek support and advice from online message boards (eg, http://www.rosacea-support.org). The long wait for appointments may be attributed to the increased demand for dermatologists for cosmetic procedures.12 Additionally, disparities according to insurance type can contribute to difficulties procuring an appointment. In one study, privately insured dermatology patients demonstrated a 91% acceptance rate and shorter wait times for appointments compared to publicly insured patients who were limited to a 29.8% acceptance rate and longer wait times.11 Many patients then are left to wait for an appointment with a dermatologist or instead turn to a primary care physician. Of patients diagnosed with rosacea in one study (N=2847), the majority of patients were seen by a dermatologist (79%), while the other patients were diagnosed by a family physician (14%) or other types of physicians such as internists and ophthalmologists (7%).6
The diagnosis of rosacea usually is not a major hurdle for dermatologists, but misdiagnoses can sometimes occur. The Rosacea Research & Development Institute compiled multiple patient anecdotes describing the struggles of finally reaching the correct diagnosis of rosacea; however, no estimates as to the frequency of misdiagnoses was estimated.13 Even with an accurate diagnosis of rosacea, correct classification of the 4 types of rosacea (ie, erythematotelangiectatic, papulopustular, phymatous, ocular) is necessary to avoid incorrect treatment recommendations. For example, patients with flushing often cannot tolerate topical medications in contrast to patients with the papulopustular subtype who benefit from them.14 In the meantime, the patients who are misdiagnosed may be met with frustration, as treatment was either delayed or incorrectly prescribed.
Although there are limited data regarding patient reactions after receiving a diagnosis of rosacea, it can be assumed that patients would be hopeful that diagnosis would lead to correct treatment. In a 2008 article in The New York Times, a rosacea patient was described as feeling relieved to be diagnosed with rosacea because it was an explanation for the development of pimples on the cheeks in her late 40s.10
Implementation of a Treatment Plan
After recognizing the symptoms and receiving a correct diagnosis, the next step in the patient journey is treatment. Long-term management of incurable conditions such as rosacea is difficult. The main goals of treatment are to relieve symptoms, improve appearance, delay progression to advanced stages, and maintain remission.15 There are only a few reliable clinical trials regarding therapies for rosacea, so treatment has mostly relied on clinical experience (Table). The efficacy and safety of many older treatments has not been assessed.15 Mainstays of treatment include both topical agents and oral medications. The use of topical metronidazole, oral tetracycline, and oral isotretinoin have been found to improve both skin lesions and quality of life.18 Initially, a combination of a topical and an oral medication may be used for at least the first 12 weeks, and improvement is usually gradual, taking many weeks to become evident.15 Long-term treatment with topical medications often is required for maintenance, which can last another 6 months or more.19,20
Besides using pharmacologic therapies, some patients also may choose to undergo various procedures. The most common procedure is laser therapy, followed by dermabrasion, chemical peels, hot loop electrocoagulation, and surgical sculpting or plastic surgery.6 The use of these adjunct therapies may suggest impatience from the patient for improvement; it also indicates the lengths patients will go to and willingness to pay for improvement of symptoms.
Along with medication, patients are recommended to make changes to their skin care regimen and lifestyle. Rosacea patients typically have sensitive skin that may include symptoms such as dryness, scaling, stinging, burning, and pruritus.16 Skin care recommendations for rosacea patients include using a gentle cleanser and regularly applying sunscreen.5 Issues with physical appearance can be addressed with the use of cosmetic products such as green-tinted makeup to conceal skin lesions.21 Remission can be maintained by identifying certain triggers (eg, red wine, spicy foods, extreme temperatures, prolonged sun exposure, vigorous exercise) that can cause flare-ups.15 The most common trigger is sun exposure, making photoprotection an important component of the rosacea patient’s skin care regimen.6
Adherence
With a diagnosis and treatment plan in effect, the patient journey reaches the stage of treatment adherence, which should include ongoing education about the condition. Self-reported statistics from rosacea patients indicated that 28% of patients took time off from their treatment regimen,6 but actual nonadherence rates likely are higher. The most commonly reported reason for poor treatment adherence among rosacea patients was the impression that the symptoms had resolved or were adequately controlled.6 Treatment also must be affordable. In a national survey of rosacea patients, 24% of 427 patients receiving pharmacologic therapy planned on switching medications because of cost, and 17% of 769 patients discontinued medications due to co-pay/insurance issues.6 Other reasons cited for discontinuation of treatment included patient perception that symptoms were not that serious, co-pay/insurance issues, ineffectiveness of the medication, and side effects.6 Adherence to topical medications is lower than oral medications due to the time and inconvenience required for application.22 For some patients, topical medications may be too messy, have a strange odor, or stain clothing.
It is promising that most rosacea patients have reported the intent to continue using pharmacologic agents because the medication prevented worsening of their symptoms.6 However, there are still patients who switch or discontinue therapies without physician direction. These patients often cite that they desire more information at the time of diagnosis, particularly related to causes of flare-ups, physical symptoms to expect, drug treatment options, makeup to cover up visible symptoms, surgical or laser treatment options, psychological symptoms, patient support groups, and counseling options.6
Management
The last part of the journey is disease management, which occurs when the patient learns how to control his/her symptoms long-term. Important factors contributing to long-term control of rosacea flares are medication adherence and avoiding lifestyle triggers.23,24 Through the other stages of the journey, the patient has learned which treatments work and which factors may lead to exacerbation of symptoms.
Educating Patients on the Journey
The patient journey is a concept that can be applied to any disease state and brings to light roadblocks that patients may face from the initial diagnosis to successful disease management. Rosacea patients are faced with confusing and aggravating symptoms that can cause anxiety and may lead them to seek treatment from a physician. Facial flushing and phymatous changes of the nose can be mistaken for alcohol abuse, leading rosacea to be a socially stigmatizing disease.15 Because rosacea involves mostly the facial skin, it can disrupt social and professional interactions, leading to quality-of-life effects such as difficulty functioning on a day-to-day basis, which can be detrimental because patients usually are aged 30 to 50 years and may be perceived based on their appearance in the workforce.3 A lack of confidence, low self-esteem, embarrassment, and anxiety can even lead to serious psychiatric conditions such as depression and body dysmorphic disorder.25 Because the severity of rosacea increases over time, it is important to educate patients about seeking early treatment; therefore, understanding and awareness of rosacea symptoms are necessary to prompt patients to see a medical professional to either confirm or refute the diagnosis.
Rosacea is a clinical diagnosis that relies on patterns of primary and secondary features, as outlined in a 2002 report by the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea.5 Even with this consensus grading system, it appears that additional fine-tuning of the criteria is needed in the disease definition. Importantly, because much of the pathogenesis and progression of rosacea is still not completely understood, there is no laboratory benchmark test that can be utilized for correct diagnosis.14 Moreover, many of the clinical manifestations of rosacea are shared with other conditions, and patients may present with different symptoms or varying combinations.26
Treatment of rosacea is multifactorial and behavioral, as patients must not only be able to obtain and adhere to oral and topical regimens and possible procedures but also avoid various lifestyle and environmental triggers and learn to cope with emotional distress caused by their symptoms. Although patients who discontinue use of medications appear to be in the minority, education is still needed to stress the chronic nature of rosacea and the importance of the continuation of treatment. Collaboration between the physician and patient is needed to determine why a certain medication may not be effective and explore other treatment options. Treatment ineffectiveness could be due to incorrect use of the product, failure to use an adjunct skin care regimen, or inability to control rosacea triggers. Adequate early follow-up also is needed to maximize patient adherence to treatment.27 Working together with the patient to develop a treatment plan that can be followed is necessary for long-term control of rosacea symptoms.
There is little information on how to address the psychological needs of patients, but patients can find support from various avenues. For instance, the National Rosacea Society, a large advocacy group, produces newsletters and educational materials for both physicians and patients.28,29 There also are online support groups for rosacea patients that have thousands of members who exchange stories and provide words of encouragement. Although there are not many face-to-face support groups, physicians may consider developing live support groups for their rosacea patients. As patients achieve the later stages of the rosacea patient journey, they hopefully will have controlled their symptoms by following a treatment regimen and learning to adapt to a new life of successful disease management.
Many aspects of the rosacea patient journey have yet to be explored. It is uncertain how long patients with symptoms of rosacea wait before seeking treatment, what methods they use to control their rosacea before they receive a prescribed treatment or physician recommendations, and how they react to their diagnosis. It also is unknown how many rosacea patients receive an initial misdiagnosis of another condition and which physicians typically make the misdiagnosis. We also need to know more about the role of psychological issues in addressing patient adherence to treatment. Similarly, what role do support groups such as online forums play on adherence? There is a need for more patient education and awareness of rosacea.
Conclusion
Patients may be relieved that rosacea is not a life-threatening condition, but they may be disappointed that there is no cure for rosacea. As the patient and dermatologist work together to find an appropriate treatment plan, identify certain triggers, and modify the skin care routine, the patient can become disciplined in controlling rosacea symptoms. Ultimately, with the alleviation of visible symptoms, the patient’s quality of life also can improve. Better understanding of the rosacea patient perspective can lead to a more efficient health care system, improved patient care, and better patient satisfaction.
Rosacea patients experience symptoms ranging from flushing to persistent acnelike rashes that can cause low self-esteem and anxiety, leading to social and professional isolation.1 Although it is estimated that 16 million individuals in the United States have rosacea, only 10% seek treatment.2,3 The motivation for patients to seek and adhere to treatment is not well characterized.
A patient journey is a map of the steps a patient takes as he/she progresses through different segments of the disease from diagnosis to management, including all the influences that can push him/her toward or away from certain decisions. The patient journey model provides a structure for understanding key issues in rosacea management, including barriers to successful treatment outcomes.
The patient journey model progresses from development of disease and diagnosis to treatment and disease management (Figure). We sought to examine each step of the rosacea patient journey to better understand key patient care boundaries faced by rosacea patients. We assessed the current literature regarding each step of the patient experience and identified areas of the patient journey with limited research.
Click here to view the figure as a PDF to print for future reference.
Researching the Patient Experience
A PubMed search of articles indexed for MEDLINE as well as a search of the National Rosacea Society Web site (http://www.rosacea.org) were conducted to identify articles and materials that quantitatively or qualitatively described rosacea patient experiences. Search terms included rosacea, rosacea patient experience, rosacea treatment, rosacea adherence, and rosacea quality of life. A Google search also was conducted using the same terms to obtain current news articles online. Current literature pertaining to the patient journey was summarized.
To create a model for the rosacea patient journey, we refined a rheumatoid arthritis patient journey map4 and included the critical components of the journey for rosacea patients. We organized the journey into stages, including prediagnosis, diagnosis, treatment, adherence, and management. We first explored what occurs prior to diagnosis, which includes the patient’s symptoms before visiting a physician. We then examined the process of diagnosis and the implementation of a treatment plan. Treatment adherence was then explored, ending with the ways patients self-manage their disease beyond the physician’s office.
Prediagnosis: What Motivates Patients to Seek Treatment
Rosacea can present with many symptoms that may lead patients to see a physician, including facial erythema and telangiectases, papules and pustules, phymatous changes, and ocular manifestations.5 The most common concern is temporary facial flushing, followed by persistent redness, then bumps and pimples.6 Many patients seek treatment after persistent facial flushing and an intolerable burning sensation. Some middle-aged patients decide to see a dermatologist for the first time when they break out in acne lesions after a history of clear skin. Others seek treatment because they can no longer tolerate the pain and embarrassment associated with their symptoms. However, patients who seek treatment only account for a small proportion of patients with rosacea, as only 10% of patients seek conventional medical treatment.7 Furthermore, symptomatic patients on average wait 7 months to 5 years before receiving a diagnosis.8,9
Care often is delayed or not pursued because many rosacea symptoms are mild when they first appear and may not initially bother the patient. Patients may not think anything of their symptoms and dismiss them as either acne vulgaris or sunburn. Due to the relapsing and remitting nature of the disease course, patients may feel their symptoms will resolve. Of patients diagnosed with rosacea, only one-half have heard of the condition prior to diagnosis,8 which can largely be attributed to lack of patient education on the signs and symptoms of rosacea, a concern that prompted the National Rosacea Society to designate the month of April as rosacea awareness month.5
With sales of antiredness facial care products growing 35% from 2002 to 2007, accounting for an increase of $300 million in revenue, patients also may be turning to over-the-counter products first.10 Men with rosacea tend to present with more severe symptoms such as rhinophyma, which may be due to their desire to wait until their symptoms reached more advanced stages of disease before seeking medical help.5
Diagnosis of Rosacea
After the patient decides that his/her symptoms are unusual, severe, or intolerable enough to seek treatment, the issues of access to dermatologic care and receiving the correct diagnosis come into play. Accessing dermatologic care can be difficult, as appointments may be hard to obtain, and even if the patient is able to get an appointment, it could be many weeks later.11 For some rosacea patients, the anxiety of waiting for their appointment prompts them to seek support and advice from online message boards (eg, http://www.rosacea-support.org). The long wait for appointments may be attributed to the increased demand for dermatologists for cosmetic procedures.12 Additionally, disparities according to insurance type can contribute to difficulties procuring an appointment. In one study, privately insured dermatology patients demonstrated a 91% acceptance rate and shorter wait times for appointments compared to publicly insured patients who were limited to a 29.8% acceptance rate and longer wait times.11 Many patients then are left to wait for an appointment with a dermatologist or instead turn to a primary care physician. Of patients diagnosed with rosacea in one study (N=2847), the majority of patients were seen by a dermatologist (79%), while the other patients were diagnosed by a family physician (14%) or other types of physicians such as internists and ophthalmologists (7%).6
The diagnosis of rosacea usually is not a major hurdle for dermatologists, but misdiagnoses can sometimes occur. The Rosacea Research & Development Institute compiled multiple patient anecdotes describing the struggles of finally reaching the correct diagnosis of rosacea; however, no estimates as to the frequency of misdiagnoses was estimated.13 Even with an accurate diagnosis of rosacea, correct classification of the 4 types of rosacea (ie, erythematotelangiectatic, papulopustular, phymatous, ocular) is necessary to avoid incorrect treatment recommendations. For example, patients with flushing often cannot tolerate topical medications in contrast to patients with the papulopustular subtype who benefit from them.14 In the meantime, the patients who are misdiagnosed may be met with frustration, as treatment was either delayed or incorrectly prescribed.
Although there are limited data regarding patient reactions after receiving a diagnosis of rosacea, it can be assumed that patients would be hopeful that diagnosis would lead to correct treatment. In a 2008 article in The New York Times, a rosacea patient was described as feeling relieved to be diagnosed with rosacea because it was an explanation for the development of pimples on the cheeks in her late 40s.10
Implementation of a Treatment Plan
After recognizing the symptoms and receiving a correct diagnosis, the next step in the patient journey is treatment. Long-term management of incurable conditions such as rosacea is difficult. The main goals of treatment are to relieve symptoms, improve appearance, delay progression to advanced stages, and maintain remission.15 There are only a few reliable clinical trials regarding therapies for rosacea, so treatment has mostly relied on clinical experience (Table). The efficacy and safety of many older treatments has not been assessed.15 Mainstays of treatment include both topical agents and oral medications. The use of topical metronidazole, oral tetracycline, and oral isotretinoin have been found to improve both skin lesions and quality of life.18 Initially, a combination of a topical and an oral medication may be used for at least the first 12 weeks, and improvement is usually gradual, taking many weeks to become evident.15 Long-term treatment with topical medications often is required for maintenance, which can last another 6 months or more.19,20
Besides using pharmacologic therapies, some patients also may choose to undergo various procedures. The most common procedure is laser therapy, followed by dermabrasion, chemical peels, hot loop electrocoagulation, and surgical sculpting or plastic surgery.6 The use of these adjunct therapies may suggest impatience from the patient for improvement; it also indicates the lengths patients will go to and willingness to pay for improvement of symptoms.
Along with medication, patients are recommended to make changes to their skin care regimen and lifestyle. Rosacea patients typically have sensitive skin that may include symptoms such as dryness, scaling, stinging, burning, and pruritus.16 Skin care recommendations for rosacea patients include using a gentle cleanser and regularly applying sunscreen.5 Issues with physical appearance can be addressed with the use of cosmetic products such as green-tinted makeup to conceal skin lesions.21 Remission can be maintained by identifying certain triggers (eg, red wine, spicy foods, extreme temperatures, prolonged sun exposure, vigorous exercise) that can cause flare-ups.15 The most common trigger is sun exposure, making photoprotection an important component of the rosacea patient’s skin care regimen.6
Adherence
With a diagnosis and treatment plan in effect, the patient journey reaches the stage of treatment adherence, which should include ongoing education about the condition. Self-reported statistics from rosacea patients indicated that 28% of patients took time off from their treatment regimen,6 but actual nonadherence rates likely are higher. The most commonly reported reason for poor treatment adherence among rosacea patients was the impression that the symptoms had resolved or were adequately controlled.6 Treatment also must be affordable. In a national survey of rosacea patients, 24% of 427 patients receiving pharmacologic therapy planned on switching medications because of cost, and 17% of 769 patients discontinued medications due to co-pay/insurance issues.6 Other reasons cited for discontinuation of treatment included patient perception that symptoms were not that serious, co-pay/insurance issues, ineffectiveness of the medication, and side effects.6 Adherence to topical medications is lower than oral medications due to the time and inconvenience required for application.22 For some patients, topical medications may be too messy, have a strange odor, or stain clothing.
It is promising that most rosacea patients have reported the intent to continue using pharmacologic agents because the medication prevented worsening of their symptoms.6 However, there are still patients who switch or discontinue therapies without physician direction. These patients often cite that they desire more information at the time of diagnosis, particularly related to causes of flare-ups, physical symptoms to expect, drug treatment options, makeup to cover up visible symptoms, surgical or laser treatment options, psychological symptoms, patient support groups, and counseling options.6
Management
The last part of the journey is disease management, which occurs when the patient learns how to control his/her symptoms long-term. Important factors contributing to long-term control of rosacea flares are medication adherence and avoiding lifestyle triggers.23,24 Through the other stages of the journey, the patient has learned which treatments work and which factors may lead to exacerbation of symptoms.
Educating Patients on the Journey
The patient journey is a concept that can be applied to any disease state and brings to light roadblocks that patients may face from the initial diagnosis to successful disease management. Rosacea patients are faced with confusing and aggravating symptoms that can cause anxiety and may lead them to seek treatment from a physician. Facial flushing and phymatous changes of the nose can be mistaken for alcohol abuse, leading rosacea to be a socially stigmatizing disease.15 Because rosacea involves mostly the facial skin, it can disrupt social and professional interactions, leading to quality-of-life effects such as difficulty functioning on a day-to-day basis, which can be detrimental because patients usually are aged 30 to 50 years and may be perceived based on their appearance in the workforce.3 A lack of confidence, low self-esteem, embarrassment, and anxiety can even lead to serious psychiatric conditions such as depression and body dysmorphic disorder.25 Because the severity of rosacea increases over time, it is important to educate patients about seeking early treatment; therefore, understanding and awareness of rosacea symptoms are necessary to prompt patients to see a medical professional to either confirm or refute the diagnosis.
Rosacea is a clinical diagnosis that relies on patterns of primary and secondary features, as outlined in a 2002 report by the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea.5 Even with this consensus grading system, it appears that additional fine-tuning of the criteria is needed in the disease definition. Importantly, because much of the pathogenesis and progression of rosacea is still not completely understood, there is no laboratory benchmark test that can be utilized for correct diagnosis.14 Moreover, many of the clinical manifestations of rosacea are shared with other conditions, and patients may present with different symptoms or varying combinations.26
Treatment of rosacea is multifactorial and behavioral, as patients must not only be able to obtain and adhere to oral and topical regimens and possible procedures but also avoid various lifestyle and environmental triggers and learn to cope with emotional distress caused by their symptoms. Although patients who discontinue use of medications appear to be in the minority, education is still needed to stress the chronic nature of rosacea and the importance of the continuation of treatment. Collaboration between the physician and patient is needed to determine why a certain medication may not be effective and explore other treatment options. Treatment ineffectiveness could be due to incorrect use of the product, failure to use an adjunct skin care regimen, or inability to control rosacea triggers. Adequate early follow-up also is needed to maximize patient adherence to treatment.27 Working together with the patient to develop a treatment plan that can be followed is necessary for long-term control of rosacea symptoms.
There is little information on how to address the psychological needs of patients, but patients can find support from various avenues. For instance, the National Rosacea Society, a large advocacy group, produces newsletters and educational materials for both physicians and patients.28,29 There also are online support groups for rosacea patients that have thousands of members who exchange stories and provide words of encouragement. Although there are not many face-to-face support groups, physicians may consider developing live support groups for their rosacea patients. As patients achieve the later stages of the rosacea patient journey, they hopefully will have controlled their symptoms by following a treatment regimen and learning to adapt to a new life of successful disease management.
Many aspects of the rosacea patient journey have yet to be explored. It is uncertain how long patients with symptoms of rosacea wait before seeking treatment, what methods they use to control their rosacea before they receive a prescribed treatment or physician recommendations, and how they react to their diagnosis. It also is unknown how many rosacea patients receive an initial misdiagnosis of another condition and which physicians typically make the misdiagnosis. We also need to know more about the role of psychological issues in addressing patient adherence to treatment. Similarly, what role do support groups such as online forums play on adherence? There is a need for more patient education and awareness of rosacea.
Conclusion
Patients may be relieved that rosacea is not a life-threatening condition, but they may be disappointed that there is no cure for rosacea. As the patient and dermatologist work together to find an appropriate treatment plan, identify certain triggers, and modify the skin care routine, the patient can become disciplined in controlling rosacea symptoms. Ultimately, with the alleviation of visible symptoms, the patient’s quality of life also can improve. Better understanding of the rosacea patient perspective can lead to a more efficient health care system, improved patient care, and better patient satisfaction.
1. Baldwin HE. Systemic therapy for rosacea. Skin Therapy Lett. 2007;12:1-5, 9.
2. Drake L. Rosacea now estimated to affect at least 16 million Americans. Rosacea Review. Winter 2010. http://www.rosacea.org/rr/2010/winter/article_1.php. Accessed December 11, 2014.
3. Rosacea as an inflammatory disease: an expert interview with Brian Berman, MD, PhD. Medscape Web site. http://www.medscape.org/viewarticle/722156. Published May 27, 2010. Accessed December 11, 2014.
4. HealthEd Group, Inc. Rheumatoid arthritis patient journey map. http://visual.ly/rheumatoid-arthritis-patient-journey-map. Accessed December 19, 2014.
5. Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Elewski BE. Results of a national rosacea patient survey: common issues that concern rosacea sufferers. J Drugs Dermatol. 2009;8:120-123.
7. Del Rosso J. Management of rosacea in the United States: analysis based on recent prescribing patterns and insurance claims. J Am Acad Dermatol. 2008;58:AB13.
8. New survey reveals first impressions may not always be rosy for people with the widespread skin condition rosacea. Medical News Today Web site. http://www.medicalnew today.com/releases/185491.php. Updated April 15, 2010. Accessed December 12, 2014.
9. Shear NH, Levine C. Needs survey of Canadian rosacea patients. J Cutan Med Surg. 1999;3:178-181.
10. Sweeney C. In a perfect world, rosacea remains a problem. New York Times. April 24, 2008. http://www.nytimes.com/2008/04/24/fashion/24SKIN.html?pagewanted=all. Accessed December 12, 2014.
11. Alghothani L, Jacks SK, Vander HA, et al. Disparities in access to dermatologic care according to insurance type. Arch Dermatol. 2012;148:956-957.
12. Resneck J Jr. Too few or too many dermatologists? difficulties in assessing optimal workforce size. Arch Dermatol. 2001;137:1295-1301.
13. Rosacea Research & Development Institute Web site. http://irosacea.org/misdiagnosed_rosacea.html. Accessed December 19, 2014.
14. Crawford GH, Pelle MT, James WD. Rosacea: I. etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
15. Elewski BE, Draelos Z, Dreno B, et al. Rosacea—global diversity and optimized outcome: proposed international consensus from the Rosacea International Expert Group. J Eur Acad Dermatol Venereol. 2011;25:188-200.
16. Del Rosso JQ, Baldwin H, Webster G. American Acne & Rosacea Society rosacea medical management guidelines. J Drugs Dermatol. 2008;7:531-533.
17. Fowler J Jr, Jackson M, Moore A, et al. Efficacy and safety of once-daily topical brimonidine tartrate gel 0.5% for the treatment of moderate to severe facial erythema of rosacea: results of two randomized, double-blind, and vehicle-controlled pivotal studies. J Drugs Dermatol. 2013;12:650-656.
18. Aksoy B, Altaykan-Hapa A, Egemen D, et al. The impact of rosacea on quality of life: effects of demographic and clinical characteristics and various treatment modalities. Br J Dermatol. 2010;163:719-725.
19. Dahl MV, Katz HI, Krueger GG, et al. Topical metronidazole maintains remissions of rosacea. Arch Dermatol. 1998;134:679-683.
20. Thiboutot DM, Fleischer AB, Del Rosso JQ, et al. A multicenter study of topical azelaic acid 15% gel in combination with oral doxycycline as initial therapy and azelaic acid 15% gel as maintenance monotherapy. J Drugs Dermatol. 2009;8:639-648.
21. Boehncke WH, Ochsendorf F, Paeslack I, et al. Decorative cosmetics improve the quality of life in patients with disfiguring skin diseases. Eur J Dermatol. 2002;12:577-580.
22. Jackson JM, Pelle M. Topical rosacea therapy: the importance of vehicles for efficacy, tolerability and compliance. J Drugs Dermatol. 2011;10:627-633.
23. Wolf JE Jr. Medication adherence: a key factor in effective management of rosacea. Adv Ther. 2001;18:272-281.
24. Managing rosacea. National Rosacea Society Web site. http://www.rosacea.org/patients/materials/managing/lifestyle.php. Accessed December 19, 2014.
25. van Zuuren EJ, Fedorowicz Z. Lack of ‘appropriately assessed’ patient-reported outcomes in randomized controlled trials assessing the effectiveness of interventions for rosacea. Br J Dermatol. 2013;168:442-444.
26. Del Rosso JQ. Advances in understanding and managing rosacea: part 2: the central role, evaluation, and medical management of diffuse and persistent facial erythema of rosacea. J Clin Aesthet Dermatol. 2012;5:26-36.
27. Davis SA, Lin HC, Yu CH, et al. Underuse of early follow-up visits: a missed opportunity to improve patients’ adherence. 2014;13:833-836.
28. If you have rosacea, you’re not alone. National Rosacea Society Web site. http://www.rosacea.org/patients/index.php. Accessed December 19, 2014.
29. Tools for the professional. National Rosacea Society Web site. http://www.rosacea.org/physicians/index.php. Accessed December 19, 2014.
1. Baldwin HE. Systemic therapy for rosacea. Skin Therapy Lett. 2007;12:1-5, 9.
2. Drake L. Rosacea now estimated to affect at least 16 million Americans. Rosacea Review. Winter 2010. http://www.rosacea.org/rr/2010/winter/article_1.php. Accessed December 11, 2014.
3. Rosacea as an inflammatory disease: an expert interview with Brian Berman, MD, PhD. Medscape Web site. http://www.medscape.org/viewarticle/722156. Published May 27, 2010. Accessed December 11, 2014.
4. HealthEd Group, Inc. Rheumatoid arthritis patient journey map. http://visual.ly/rheumatoid-arthritis-patient-journey-map. Accessed December 19, 2014.
5. Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Elewski BE. Results of a national rosacea patient survey: common issues that concern rosacea sufferers. J Drugs Dermatol. 2009;8:120-123.
7. Del Rosso J. Management of rosacea in the United States: analysis based on recent prescribing patterns and insurance claims. J Am Acad Dermatol. 2008;58:AB13.
8. New survey reveals first impressions may not always be rosy for people with the widespread skin condition rosacea. Medical News Today Web site. http://www.medicalnew today.com/releases/185491.php. Updated April 15, 2010. Accessed December 12, 2014.
9. Shear NH, Levine C. Needs survey of Canadian rosacea patients. J Cutan Med Surg. 1999;3:178-181.
10. Sweeney C. In a perfect world, rosacea remains a problem. New York Times. April 24, 2008. http://www.nytimes.com/2008/04/24/fashion/24SKIN.html?pagewanted=all. Accessed December 12, 2014.
11. Alghothani L, Jacks SK, Vander HA, et al. Disparities in access to dermatologic care according to insurance type. Arch Dermatol. 2012;148:956-957.
12. Resneck J Jr. Too few or too many dermatologists? difficulties in assessing optimal workforce size. Arch Dermatol. 2001;137:1295-1301.
13. Rosacea Research & Development Institute Web site. http://irosacea.org/misdiagnosed_rosacea.html. Accessed December 19, 2014.
14. Crawford GH, Pelle MT, James WD. Rosacea: I. etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
15. Elewski BE, Draelos Z, Dreno B, et al. Rosacea—global diversity and optimized outcome: proposed international consensus from the Rosacea International Expert Group. J Eur Acad Dermatol Venereol. 2011;25:188-200.
16. Del Rosso JQ, Baldwin H, Webster G. American Acne & Rosacea Society rosacea medical management guidelines. J Drugs Dermatol. 2008;7:531-533.
17. Fowler J Jr, Jackson M, Moore A, et al. Efficacy and safety of once-daily topical brimonidine tartrate gel 0.5% for the treatment of moderate to severe facial erythema of rosacea: results of two randomized, double-blind, and vehicle-controlled pivotal studies. J Drugs Dermatol. 2013;12:650-656.
18. Aksoy B, Altaykan-Hapa A, Egemen D, et al. The impact of rosacea on quality of life: effects of demographic and clinical characteristics and various treatment modalities. Br J Dermatol. 2010;163:719-725.
19. Dahl MV, Katz HI, Krueger GG, et al. Topical metronidazole maintains remissions of rosacea. Arch Dermatol. 1998;134:679-683.
20. Thiboutot DM, Fleischer AB, Del Rosso JQ, et al. A multicenter study of topical azelaic acid 15% gel in combination with oral doxycycline as initial therapy and azelaic acid 15% gel as maintenance monotherapy. J Drugs Dermatol. 2009;8:639-648.
21. Boehncke WH, Ochsendorf F, Paeslack I, et al. Decorative cosmetics improve the quality of life in patients with disfiguring skin diseases. Eur J Dermatol. 2002;12:577-580.
22. Jackson JM, Pelle M. Topical rosacea therapy: the importance of vehicles for efficacy, tolerability and compliance. J Drugs Dermatol. 2011;10:627-633.
23. Wolf JE Jr. Medication adherence: a key factor in effective management of rosacea. Adv Ther. 2001;18:272-281.
24. Managing rosacea. National Rosacea Society Web site. http://www.rosacea.org/patients/materials/managing/lifestyle.php. Accessed December 19, 2014.
25. van Zuuren EJ, Fedorowicz Z. Lack of ‘appropriately assessed’ patient-reported outcomes in randomized controlled trials assessing the effectiveness of interventions for rosacea. Br J Dermatol. 2013;168:442-444.
26. Del Rosso JQ. Advances in understanding and managing rosacea: part 2: the central role, evaluation, and medical management of diffuse and persistent facial erythema of rosacea. J Clin Aesthet Dermatol. 2012;5:26-36.
27. Davis SA, Lin HC, Yu CH, et al. Underuse of early follow-up visits: a missed opportunity to improve patients’ adherence. 2014;13:833-836.
28. If you have rosacea, you’re not alone. National Rosacea Society Web site. http://www.rosacea.org/patients/index.php. Accessed December 19, 2014.
29. Tools for the professional. National Rosacea Society Web site. http://www.rosacea.org/physicians/index.php. Accessed December 19, 2014.
Practice Points
- For patients who are emotionally distressed by their rosacea and who lack a social support network, several rosacea-focused online support systems are available.
- An early follow-up visit to evaluate newly prescribed treatments can positively influence disease management.

Solitary Morphea Profunda Following Trauma Sustained in an Automobile Accident
Case Report
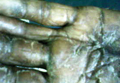
A 50-year-old white woman presented to our clinic for evaluation of what she described as a “very hard red line” on the right upper arm. The lesion had developed suddenly overnight. Several months prior to presentation the patient sustained trauma to the same area in a car accident and she thought the lesion might be related to the resulting nerve damage. Initially she presented to her primary care physician who used ultrasonography of the area to rule out muscle or bone involvement. The patient presented to our dermatology clinic 2 months later with an 18×4-cm, brownish, rectangular, sclerotic, bound-down, hypertrophic plaque that started on the right mid forearm and extended to the right shoulder (Figure 1). Her medical history was notable for high blood pressure, which was controlled with valsartan.
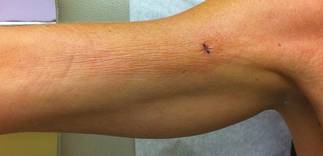
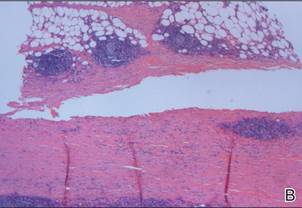
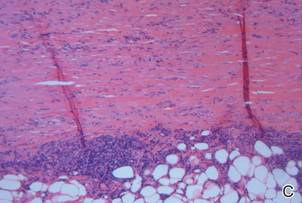
A review of systems was unremarkable. Physical examination revealed a well-developed, well-nourished woman. Examination of the right arm revealed no motion restriction (muscle strength, 5/5) and no pain; however, she described a burning sensation at the site of the lesion. She reported no allergies. A 4-mm punch biopsy was performed and laboratory tests were ordered including an antinuclear antibody (ANA) test with reflex, double-stranded DNA test, DNA antitopoisomerase antibodies test, and Lyme titers (IgM and IgG). Initially, the patient was treated with calcipotriene 0.005%–betamethasone dipropionate 0.064% ointment twice daily; she also was treated empirically for Lyme disease with doxycycline 50 mg twice daily. All laboratory tests were within reference range, and a punch biopsy revealed markedly thickened fibrous septa within the subcutaneous fat. At the edge of the septa there were nodular aggregates of lymphocytes. Due to clinical presentation, laboratory data, and histopathology, solitary morphea profunda (SMP) was diagnosed.
Following histopathologic examination (Figure 2), the patient was instructed to continue treatment with calcipotriene–betamethasone dipropionate as well as doxycycline. A trial of prednisone and/or hydroxychloroquine also was considered pending her response to the initial treatment. At approximately 1-month follow-up, remarkable improvement of the lesion was noted.
Comment
|
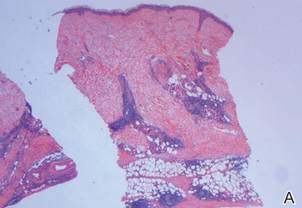
There is limited literature available about the diagnosis and treatment of SMP. Our case prompted us to further examine the data to emphasize the necessity of greater research surrounding SMP.
Classification of SMP
Morphea is a localized form of scleroderma, an inflammatory disease that primarily affects the dermis but can extend down to the bone and also can limit motion. There are several types of morphea that are classified according to the extent, depth, and distribution of the lesions, including plaque, generalized, bullous, linear (including morphea en coup de sabre), guttate, nodular, and deep morphea.1,2 Other subtypes have been described including subcutaneous morphea, eosinophilic fasciitis (EF), pansclerotic morphea, and morphea profunda.3 Linear and deep morphea are characterized by involvement of the deep dermis, subcutaneous tissue, fascia, and/or superficial muscle.2,4
In 1981, Su and Person5 first described morphea profunda (MP). In their study, 22 of 23 patients presented with generalized MP. One patient developed a single lesion,5 which ultimately was classified as SMP by Whittaker et al6 in 1989.
Epidemiology
Morphea profunda occurs more frequently in females than in males, with sclerosis manifesting over a period of several months.7 In 2004, Azad et al4 suggested that only 9 cases of SMP had been reported in the literature. Although there is insignificant data to determine the epidemiology of SMP, the authors concluded that it most commonly affects middle-aged individuals with equal sex distribution.4 The single plaque in patients with SMP most commonly presents on the shoulder, back, or neck or in the paraspinal area.
Etiology
Because of the limited amount of literature on MP, a definitive etiology is unknown, but investigators have cited many possible causes. Genetic, autoimmune, hormonal, traumatic,8 vaccination,2,8 radiation,9 viral, neurogenic, and vascular factors all have been implicated,10 as well as infectious agents such as Borrelia burgdorferi in the United States,11,12Borrelia afzelii in Europe,2 and Borrelia garinii in Japan.2 Because our patient experienced a traumatic episode several months prior to presentation, it is important to investigate trauma as a likely etiology. Furthermore, traumatic events have been reported in 23% of children with linear morphea.13
Diagnostic Studies
Morphea profunda is diagnosed clinically and skin biopsy can be used for confirmation. Biopsy requires deep excision down to the muscle, which can aid in determining if the fascia is incorporated. Elevated levels of IgG and IgM have been detected in deep and linear morphea and are known to correlate with disease activity and the development of joint contractures in linear morphea.2 Serum procollagen type I has been considered by some as a useful indicator of disease severity.14 Elevated serum levels of antifibrillin-1 antibodies also have been demonstrated in patients with localized scleroderma (LS).15 Radiography and magnetic resonance imaging can be used for monitoring and analyzing lesion depth. Furthermore, magnetic resonance imaging can be used to differentiate MP from EF.2
The presence of ANAs in LS is controversial. According to Nguyen et al,2 ANAs are present in approximately 46% to 80% of patients with morphea, with a higher prevalence in patients with generalized, linear, and deep subtypes. However, Savoia et al16 found that patients with morphea typically do not present with ANAs; rather ANAs usually are found in patients with EF.
Pathogenesis
After the inflammatory phase in LS, fibrillar collagen types I and III accumulate, causing dermal fibrosis. The extracellular matrix increases due to the activation of connective tissue growth factor, transforming growth factor β (TGF-β), TGF-β receptors, IL-4, and several other cytokines.17 The TGF-β receptors combine with the connective tissue growth factor released by fibroblasts to create an autocrine production loop that causes fibroblast and matrix production.17 As the inflammation progresses to sclerosis, the CD34 count decreases.18
Physical Findings
In patients with MP, lesions manifest as thickened taut skin with deep, solitary, and sclerotic indurated plaques. Clinically, plaques are mildly inflamed, hyperpigmented, symmetric, and somewhat ill defined, and the skin feels thickened and bound to the underlying fascia and muscle. Plaques usually are smooth and shiny, but areas of both dermal and subcutaneous atrophy may be present, particularly in chronic lesions.19 Morphea profunda also can be described as having a cobblestone or pseudocellulite appearance. The groove sign is used to describe a depression along the course of a vein and/or between muscle groups. Both clinical presentations may manifest later in the course of disease.2
Histopathology
Su and Person5 described 3 main characteristics of MP that stand out histopathologically. First, there is thickening and hyalinization of collagen bundles in the deep dermis, subcutis, and fascia that are prominent between the junction of the dermis and subcutaneous fat. There also are fewer sebaceous glands and hair follicles. Second, MP presents with an increased inflammatory cell infiltrate composed mainly of lymphocytes located around small blood vessels and the interstitium. In some patients, the lymphocytes consist predominantly of collections of plasma cells. Third, MP contains deposits of mucin in deep portions of the dermis with occasional eosinophils and mast cells. The presence of eosinophils allows EF to be a part of this spectrum and to be included as a differential diagnosis.5 Eosinophilic fasciitis has a similar presentation to MP because the fibrosis affects the dermis, subcutaneous fat, and underlying structures.20 Although EF presents with the histopathologic characteristic of fascial fibrosis, a clear distinction between EF and morphea has not been established in the literature. Some authors classify EF as a variant of morphea, whereas others consider it as its own entity. We believe EF is its own entity. Eosinophilic fasciitis can be distinguished from morphea because 60% to 80% of patients with EF have peripheral eosinophilia and 20% to 70% of patients with EF have hypergammaglobulinemia. Additionally, morphea does not present as symmetrically or abruptly as EF.21
Treatment
To date, there is conflicting literature regarding the treatment regimen for MP. There is controversy regarding whether MP responds to corticosteroids.19 Different treatment regimens have been discussed for LS, but there is a lack of reports specifically describing therapies for MP and SMP. Because MP and SMP fall under the umbrella of LS, many investigators have reported using the following treatment regimens for patients with MP and SMP: bosentan,22 D-penicillamine,23 phototherapy,24-26 retinoids,26 oral steroids,27 methotrexate,27-29 vitamin D3 (oral calcitriol),30,31 cyclosporine,32 mycophenolate mofetil,33 and extracorporeal photochemotherapy.34
Falanga and Medsger23 reported 64% (7/11) treatment success with D-penicillamine in patients who exhibited severe LS. Psoralen plus UVA,24 methoxsalen, and UVA1 therapy are widely used in the treatment of LS.25 Kreuter et al25 advocated for phototherapy as the first approach in the management of LS after reporting improvement in all participants in their study (N=64), 2 participants with deep morphea while the rest exhibited other forms of morphea. Ozdemir et al26 proposed that retinoic acid combined with psoralen plus UVA is a good treatment choice for plaque-type LS; however, UVA only has the ability to target the epidermis and dermis, which may not be useful for deep forms of morphea.
Several studies have shown positive results in patients treated with methylprednisolone combined with low-dose methotrexate sodium.27-29 Kreuter et al30 and Elst et al31 proposed that calcitriol is effective in treating LS, whereas Hulshof et al35 indicated that it is not. It should be noted that none of these studies specifically mentioned MP. Martini et al33 demonstrated success with mycophenolate mofetil in the treatment of 10 LS patients who were resistant to methotrexate sodium and corticosteroids. Although none of the participants in the study had MP, 2 patients had disabling pansclerotic morphea, 3 had generalized morphea, and 5 had linear scleroderma (morphea en coup de sabre) affecting the limbs (n=2) and face (n=3).33 Because there is no established therapy or consensus for the treatment of MP, we have found success in starting with corticosteroids and then trying alternative therapies.
Prognosis
Morphea has transitioned into systemic scleroderma in a small number of reported cases.10,16,20,36 Therefore, patient follow-up is imperative to consistently identify systemic evolution. Although visceral complications are rare in the setting of LS, associated clinical findings have been reported, including arthralgia, arthritis, contractures, and carpal tunnel syndrome, as well as pulmonary, esophageal, and cardiac abnormalities.7,34
Conclusion
The morphologic features observed in our patient appear to correspond most closely to the type of lesion described by Su and Person5 and Whittaker et al.6 Although our case was clinically difficult to distinguish from linear morphea, the histology suggested SMP over other causes. If our patient’s SMP progressed to the joints, physical therapy would be needed to maintain range of motion and function of the extremities,2 and mandatory long-term follow-up would be required due to the risk for relapse after discontinuation of therapy. Our case highlights the inherent difficulties in the treatment of MP. Due to limited reports of SMP and MP in the literature as well as the conflicting views regarding effective and appropriate treatment options, additional investigation of these conditions and therapeutic options are necessary to further understand this debilitating condition.
1. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
2. Nguyen JV, Werth VP, Fett N. Morphea treatment & management. Medscape Web site. http://emedicine.medscape.com/article/1065782-treatment. Updated July 21, 2014. Accessed December 16, 2104.
3. Melani L, Cardinali C, Giomi B, et al. Case study: periodic follow-up is necessary in morphea profunda to identify systemic evolution. Skinmed. 2005;4:188-190.
4. Azad J, Dawn G, Shaffrali FC, et al. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29:25-27.
5. Su WP, Person JR. Morphea profunda. a new concept and a histopathologic study of 23 cases. Am J Dermatopathol. 1981;3:251-260.
6. Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol. 1989;120:431-440.
7. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
8. Torrelo A, Suárez J, Colmenero I, et al. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23:484-487.
9. Kreft B, Wohlrab J, Radant K, et al. Unrecognized radiation-induced localized scleroderma: a cause of postoperative wound-healing disorder [published online ahead of print June 22, 2009]. Clin Exp Dermatol. 2009;34:e383-e384.
10. Braun-Falco O, Plewig G, Wolff HH, et al, eds. Dermatology. Berlin, Germany: Springer; 2002.
11. Prinz JC, Kutasi Z, Weisenseel P, et al. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? results of a cohort analysis and presentation of three cases [published online ahead of print November 20, 2008]. J Am Acad Dermatol. 2009;60:248-255.
12. Wojas-Pelc A, Wielowieyska-Szybińska D, Kiełtyka A. Presence of the antinuclear antibodies and antibodies to Borrelia burgdorferi among patients with morphea en plaque, deep linear scleroderma and atrophoderma Pasini-Pierini [in Polish]. Przegl Lek. 2002;59:898-902.
13. Falanga V, Medsger TA Jr, Reichlin M, et al. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104:849-857.
14. Kikuchi K, Sato S, Kadono T, et al. Serum concentration of procollagen type I carboxyterminal propeptide in localized scleroderma. Arch Dermatol. 1994;130:1269-1272.
15. Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrilin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656-2659.
16. Savoia P, Zaccagna A, Bernengo MG. Guess what? inflammatory disseminated morphea profunda. Eur J Dermatol. 1999;9:654-656.
17. Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106:729-733.
18. Gilmour TK, Wilkinson B, Breit SN, et al. Analysis of dendritic cell populations using a revised histological staging of morphoea. Br J Dermatol. 2000;143:1183-1192.
19. Sayama K, Chen M, Shiraishi S, et al. Morphea profunda. Int J Dermatol. 1991;30:873-875.
20. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
21. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47:29-35.
22. Roldan R, Morote G, Castro Mdel C, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol. 2006;33:2538-2540.
23. Falanga V, Medsger TA Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990;126:609-612.
24. Breuckmann F, Gambichler T, Altmeyer P, et al. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol. 2004;4:11.
25. Kreuter A, Hyun J, Stücker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma [published online ahead of print January 30, 2006]. J Am Acad Dermatol. 2006;54:440-447.
26. Ozdemir M, Engin B, Toy H, et al. Treatment of plaque-type localized scleroderma with retinoic acid and ultraviolet A plus the photosensitizer psoralen: a case series. J Eur Acad Dermatol Venereol. 2008;22:519-521.
27. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847-852.
28. Kroft EB, Creemers MC, van den Hoogen FH, et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study [published online ahead of print February 4, 2009]. Br J Dermatol. 2009;160:1075-1082.
29. Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155:1013-1020.
30. Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241-245.
31. Elst EF, Van Suijlekom-Smit LW, Oranje AP. Treatment of linear scleroderma with oral 1,25-dihydroxy vitamin D3 (calcitriol) in seven children. Pediatr Dermatol. 1999;16:53-58.
32. Crespo MP, Mas IB, Díaz JM, et al. Rapid response to cyclosporine and maintenance with methotrexate in linear scleroderma in a young girl. Pediatr Dermatol. 2009;26:118-120.
33. Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil [published online ahead of print August 27, 2009]. Rheumatology (Oxford). 2009;48:1410-1413.
34. Neustadter JH, Samarin F, Carlson KR, et al. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol. 2009;145:127-130.
35. Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017-1023.
36. Toledano C, Rabhi S, Kettaneh A, et al. Localized scleroderma: a series of 52 patients [published online ahead of print September 17, 2008]. Eur J Intern Med. 2009;20:331-336.
Case Report
A 50-year-old white woman presented to our clinic for evaluation of what she described as a “very hard red line” on the right upper arm. The lesion had developed suddenly overnight. Several months prior to presentation the patient sustained trauma to the same area in a car accident and she thought the lesion might be related to the resulting nerve damage. Initially she presented to her primary care physician who used ultrasonography of the area to rule out muscle or bone involvement. The patient presented to our dermatology clinic 2 months later with an 18×4-cm, brownish, rectangular, sclerotic, bound-down, hypertrophic plaque that started on the right mid forearm and extended to the right shoulder (Figure 1). Her medical history was notable for high blood pressure, which was controlled with valsartan.
A review of systems was unremarkable. Physical examination revealed a well-developed, well-nourished woman. Examination of the right arm revealed no motion restriction (muscle strength, 5/5) and no pain; however, she described a burning sensation at the site of the lesion. She reported no allergies. A 4-mm punch biopsy was performed and laboratory tests were ordered including an antinuclear antibody (ANA) test with reflex, double-stranded DNA test, DNA antitopoisomerase antibodies test, and Lyme titers (IgM and IgG). Initially, the patient was treated with calcipotriene 0.005%–betamethasone dipropionate 0.064% ointment twice daily; she also was treated empirically for Lyme disease with doxycycline 50 mg twice daily. All laboratory tests were within reference range, and a punch biopsy revealed markedly thickened fibrous septa within the subcutaneous fat. At the edge of the septa there were nodular aggregates of lymphocytes. Due to clinical presentation, laboratory data, and histopathology, solitary morphea profunda (SMP) was diagnosed.
Following histopathologic examination (Figure 2), the patient was instructed to continue treatment with calcipotriene–betamethasone dipropionate as well as doxycycline. A trial of prednisone and/or hydroxychloroquine also was considered pending her response to the initial treatment. At approximately 1-month follow-up, remarkable improvement of the lesion was noted.
Comment
|
There is limited literature available about the diagnosis and treatment of SMP. Our case prompted us to further examine the data to emphasize the necessity of greater research surrounding SMP.
Classification of SMP
Morphea is a localized form of scleroderma, an inflammatory disease that primarily affects the dermis but can extend down to the bone and also can limit motion. There are several types of morphea that are classified according to the extent, depth, and distribution of the lesions, including plaque, generalized, bullous, linear (including morphea en coup de sabre), guttate, nodular, and deep morphea.1,2 Other subtypes have been described including subcutaneous morphea, eosinophilic fasciitis (EF), pansclerotic morphea, and morphea profunda.3 Linear and deep morphea are characterized by involvement of the deep dermis, subcutaneous tissue, fascia, and/or superficial muscle.2,4
In 1981, Su and Person5 first described morphea profunda (MP). In their study, 22 of 23 patients presented with generalized MP. One patient developed a single lesion,5 which ultimately was classified as SMP by Whittaker et al6 in 1989.
Epidemiology
Morphea profunda occurs more frequently in females than in males, with sclerosis manifesting over a period of several months.7 In 2004, Azad et al4 suggested that only 9 cases of SMP had been reported in the literature. Although there is insignificant data to determine the epidemiology of SMP, the authors concluded that it most commonly affects middle-aged individuals with equal sex distribution.4 The single plaque in patients with SMP most commonly presents on the shoulder, back, or neck or in the paraspinal area.
Etiology
Because of the limited amount of literature on MP, a definitive etiology is unknown, but investigators have cited many possible causes. Genetic, autoimmune, hormonal, traumatic,8 vaccination,2,8 radiation,9 viral, neurogenic, and vascular factors all have been implicated,10 as well as infectious agents such as Borrelia burgdorferi in the United States,11,12Borrelia afzelii in Europe,2 and Borrelia garinii in Japan.2 Because our patient experienced a traumatic episode several months prior to presentation, it is important to investigate trauma as a likely etiology. Furthermore, traumatic events have been reported in 23% of children with linear morphea.13
Diagnostic Studies
Morphea profunda is diagnosed clinically and skin biopsy can be used for confirmation. Biopsy requires deep excision down to the muscle, which can aid in determining if the fascia is incorporated. Elevated levels of IgG and IgM have been detected in deep and linear morphea and are known to correlate with disease activity and the development of joint contractures in linear morphea.2 Serum procollagen type I has been considered by some as a useful indicator of disease severity.14 Elevated serum levels of antifibrillin-1 antibodies also have been demonstrated in patients with localized scleroderma (LS).15 Radiography and magnetic resonance imaging can be used for monitoring and analyzing lesion depth. Furthermore, magnetic resonance imaging can be used to differentiate MP from EF.2
The presence of ANAs in LS is controversial. According to Nguyen et al,2 ANAs are present in approximately 46% to 80% of patients with morphea, with a higher prevalence in patients with generalized, linear, and deep subtypes. However, Savoia et al16 found that patients with morphea typically do not present with ANAs; rather ANAs usually are found in patients with EF.
Pathogenesis
After the inflammatory phase in LS, fibrillar collagen types I and III accumulate, causing dermal fibrosis. The extracellular matrix increases due to the activation of connective tissue growth factor, transforming growth factor β (TGF-β), TGF-β receptors, IL-4, and several other cytokines.17 The TGF-β receptors combine with the connective tissue growth factor released by fibroblasts to create an autocrine production loop that causes fibroblast and matrix production.17 As the inflammation progresses to sclerosis, the CD34 count decreases.18
Physical Findings
In patients with MP, lesions manifest as thickened taut skin with deep, solitary, and sclerotic indurated plaques. Clinically, plaques are mildly inflamed, hyperpigmented, symmetric, and somewhat ill defined, and the skin feels thickened and bound to the underlying fascia and muscle. Plaques usually are smooth and shiny, but areas of both dermal and subcutaneous atrophy may be present, particularly in chronic lesions.19 Morphea profunda also can be described as having a cobblestone or pseudocellulite appearance. The groove sign is used to describe a depression along the course of a vein and/or between muscle groups. Both clinical presentations may manifest later in the course of disease.2
Histopathology
Su and Person5 described 3 main characteristics of MP that stand out histopathologically. First, there is thickening and hyalinization of collagen bundles in the deep dermis, subcutis, and fascia that are prominent between the junction of the dermis and subcutaneous fat. There also are fewer sebaceous glands and hair follicles. Second, MP presents with an increased inflammatory cell infiltrate composed mainly of lymphocytes located around small blood vessels and the interstitium. In some patients, the lymphocytes consist predominantly of collections of plasma cells. Third, MP contains deposits of mucin in deep portions of the dermis with occasional eosinophils and mast cells. The presence of eosinophils allows EF to be a part of this spectrum and to be included as a differential diagnosis.5 Eosinophilic fasciitis has a similar presentation to MP because the fibrosis affects the dermis, subcutaneous fat, and underlying structures.20 Although EF presents with the histopathologic characteristic of fascial fibrosis, a clear distinction between EF and morphea has not been established in the literature. Some authors classify EF as a variant of morphea, whereas others consider it as its own entity. We believe EF is its own entity. Eosinophilic fasciitis can be distinguished from morphea because 60% to 80% of patients with EF have peripheral eosinophilia and 20% to 70% of patients with EF have hypergammaglobulinemia. Additionally, morphea does not present as symmetrically or abruptly as EF.21
Treatment
To date, there is conflicting literature regarding the treatment regimen for MP. There is controversy regarding whether MP responds to corticosteroids.19 Different treatment regimens have been discussed for LS, but there is a lack of reports specifically describing therapies for MP and SMP. Because MP and SMP fall under the umbrella of LS, many investigators have reported using the following treatment regimens for patients with MP and SMP: bosentan,22 D-penicillamine,23 phototherapy,24-26 retinoids,26 oral steroids,27 methotrexate,27-29 vitamin D3 (oral calcitriol),30,31 cyclosporine,32 mycophenolate mofetil,33 and extracorporeal photochemotherapy.34
Falanga and Medsger23 reported 64% (7/11) treatment success with D-penicillamine in patients who exhibited severe LS. Psoralen plus UVA,24 methoxsalen, and UVA1 therapy are widely used in the treatment of LS.25 Kreuter et al25 advocated for phototherapy as the first approach in the management of LS after reporting improvement in all participants in their study (N=64), 2 participants with deep morphea while the rest exhibited other forms of morphea. Ozdemir et al26 proposed that retinoic acid combined with psoralen plus UVA is a good treatment choice for plaque-type LS; however, UVA only has the ability to target the epidermis and dermis, which may not be useful for deep forms of morphea.
Several studies have shown positive results in patients treated with methylprednisolone combined with low-dose methotrexate sodium.27-29 Kreuter et al30 and Elst et al31 proposed that calcitriol is effective in treating LS, whereas Hulshof et al35 indicated that it is not. It should be noted that none of these studies specifically mentioned MP. Martini et al33 demonstrated success with mycophenolate mofetil in the treatment of 10 LS patients who were resistant to methotrexate sodium and corticosteroids. Although none of the participants in the study had MP, 2 patients had disabling pansclerotic morphea, 3 had generalized morphea, and 5 had linear scleroderma (morphea en coup de sabre) affecting the limbs (n=2) and face (n=3).33 Because there is no established therapy or consensus for the treatment of MP, we have found success in starting with corticosteroids and then trying alternative therapies.
Prognosis
Morphea has transitioned into systemic scleroderma in a small number of reported cases.10,16,20,36 Therefore, patient follow-up is imperative to consistently identify systemic evolution. Although visceral complications are rare in the setting of LS, associated clinical findings have been reported, including arthralgia, arthritis, contractures, and carpal tunnel syndrome, as well as pulmonary, esophageal, and cardiac abnormalities.7,34
Conclusion
The morphologic features observed in our patient appear to correspond most closely to the type of lesion described by Su and Person5 and Whittaker et al.6 Although our case was clinically difficult to distinguish from linear morphea, the histology suggested SMP over other causes. If our patient’s SMP progressed to the joints, physical therapy would be needed to maintain range of motion and function of the extremities,2 and mandatory long-term follow-up would be required due to the risk for relapse after discontinuation of therapy. Our case highlights the inherent difficulties in the treatment of MP. Due to limited reports of SMP and MP in the literature as well as the conflicting views regarding effective and appropriate treatment options, additional investigation of these conditions and therapeutic options are necessary to further understand this debilitating condition.
Case Report
A 50-year-old white woman presented to our clinic for evaluation of what she described as a “very hard red line” on the right upper arm. The lesion had developed suddenly overnight. Several months prior to presentation the patient sustained trauma to the same area in a car accident and she thought the lesion might be related to the resulting nerve damage. Initially she presented to her primary care physician who used ultrasonography of the area to rule out muscle or bone involvement. The patient presented to our dermatology clinic 2 months later with an 18×4-cm, brownish, rectangular, sclerotic, bound-down, hypertrophic plaque that started on the right mid forearm and extended to the right shoulder (Figure 1). Her medical history was notable for high blood pressure, which was controlled with valsartan.
A review of systems was unremarkable. Physical examination revealed a well-developed, well-nourished woman. Examination of the right arm revealed no motion restriction (muscle strength, 5/5) and no pain; however, she described a burning sensation at the site of the lesion. She reported no allergies. A 4-mm punch biopsy was performed and laboratory tests were ordered including an antinuclear antibody (ANA) test with reflex, double-stranded DNA test, DNA antitopoisomerase antibodies test, and Lyme titers (IgM and IgG). Initially, the patient was treated with calcipotriene 0.005%–betamethasone dipropionate 0.064% ointment twice daily; she also was treated empirically for Lyme disease with doxycycline 50 mg twice daily. All laboratory tests were within reference range, and a punch biopsy revealed markedly thickened fibrous septa within the subcutaneous fat. At the edge of the septa there were nodular aggregates of lymphocytes. Due to clinical presentation, laboratory data, and histopathology, solitary morphea profunda (SMP) was diagnosed.
Following histopathologic examination (Figure 2), the patient was instructed to continue treatment with calcipotriene–betamethasone dipropionate as well as doxycycline. A trial of prednisone and/or hydroxychloroquine also was considered pending her response to the initial treatment. At approximately 1-month follow-up, remarkable improvement of the lesion was noted.
Comment
|
There is limited literature available about the diagnosis and treatment of SMP. Our case prompted us to further examine the data to emphasize the necessity of greater research surrounding SMP.
Classification of SMP
Morphea is a localized form of scleroderma, an inflammatory disease that primarily affects the dermis but can extend down to the bone and also can limit motion. There are several types of morphea that are classified according to the extent, depth, and distribution of the lesions, including plaque, generalized, bullous, linear (including morphea en coup de sabre), guttate, nodular, and deep morphea.1,2 Other subtypes have been described including subcutaneous morphea, eosinophilic fasciitis (EF), pansclerotic morphea, and morphea profunda.3 Linear and deep morphea are characterized by involvement of the deep dermis, subcutaneous tissue, fascia, and/or superficial muscle.2,4
In 1981, Su and Person5 first described morphea profunda (MP). In their study, 22 of 23 patients presented with generalized MP. One patient developed a single lesion,5 which ultimately was classified as SMP by Whittaker et al6 in 1989.
Epidemiology
Morphea profunda occurs more frequently in females than in males, with sclerosis manifesting over a period of several months.7 In 2004, Azad et al4 suggested that only 9 cases of SMP had been reported in the literature. Although there is insignificant data to determine the epidemiology of SMP, the authors concluded that it most commonly affects middle-aged individuals with equal sex distribution.4 The single plaque in patients with SMP most commonly presents on the shoulder, back, or neck or in the paraspinal area.
Etiology
Because of the limited amount of literature on MP, a definitive etiology is unknown, but investigators have cited many possible causes. Genetic, autoimmune, hormonal, traumatic,8 vaccination,2,8 radiation,9 viral, neurogenic, and vascular factors all have been implicated,10 as well as infectious agents such as Borrelia burgdorferi in the United States,11,12Borrelia afzelii in Europe,2 and Borrelia garinii in Japan.2 Because our patient experienced a traumatic episode several months prior to presentation, it is important to investigate trauma as a likely etiology. Furthermore, traumatic events have been reported in 23% of children with linear morphea.13
Diagnostic Studies
Morphea profunda is diagnosed clinically and skin biopsy can be used for confirmation. Biopsy requires deep excision down to the muscle, which can aid in determining if the fascia is incorporated. Elevated levels of IgG and IgM have been detected in deep and linear morphea and are known to correlate with disease activity and the development of joint contractures in linear morphea.2 Serum procollagen type I has been considered by some as a useful indicator of disease severity.14 Elevated serum levels of antifibrillin-1 antibodies also have been demonstrated in patients with localized scleroderma (LS).15 Radiography and magnetic resonance imaging can be used for monitoring and analyzing lesion depth. Furthermore, magnetic resonance imaging can be used to differentiate MP from EF.2
The presence of ANAs in LS is controversial. According to Nguyen et al,2 ANAs are present in approximately 46% to 80% of patients with morphea, with a higher prevalence in patients with generalized, linear, and deep subtypes. However, Savoia et al16 found that patients with morphea typically do not present with ANAs; rather ANAs usually are found in patients with EF.
Pathogenesis
After the inflammatory phase in LS, fibrillar collagen types I and III accumulate, causing dermal fibrosis. The extracellular matrix increases due to the activation of connective tissue growth factor, transforming growth factor β (TGF-β), TGF-β receptors, IL-4, and several other cytokines.17 The TGF-β receptors combine with the connective tissue growth factor released by fibroblasts to create an autocrine production loop that causes fibroblast and matrix production.17 As the inflammation progresses to sclerosis, the CD34 count decreases.18
Physical Findings
In patients with MP, lesions manifest as thickened taut skin with deep, solitary, and sclerotic indurated plaques. Clinically, plaques are mildly inflamed, hyperpigmented, symmetric, and somewhat ill defined, and the skin feels thickened and bound to the underlying fascia and muscle. Plaques usually are smooth and shiny, but areas of both dermal and subcutaneous atrophy may be present, particularly in chronic lesions.19 Morphea profunda also can be described as having a cobblestone or pseudocellulite appearance. The groove sign is used to describe a depression along the course of a vein and/or between muscle groups. Both clinical presentations may manifest later in the course of disease.2
Histopathology
Su and Person5 described 3 main characteristics of MP that stand out histopathologically. First, there is thickening and hyalinization of collagen bundles in the deep dermis, subcutis, and fascia that are prominent between the junction of the dermis and subcutaneous fat. There also are fewer sebaceous glands and hair follicles. Second, MP presents with an increased inflammatory cell infiltrate composed mainly of lymphocytes located around small blood vessels and the interstitium. In some patients, the lymphocytes consist predominantly of collections of plasma cells. Third, MP contains deposits of mucin in deep portions of the dermis with occasional eosinophils and mast cells. The presence of eosinophils allows EF to be a part of this spectrum and to be included as a differential diagnosis.5 Eosinophilic fasciitis has a similar presentation to MP because the fibrosis affects the dermis, subcutaneous fat, and underlying structures.20 Although EF presents with the histopathologic characteristic of fascial fibrosis, a clear distinction between EF and morphea has not been established in the literature. Some authors classify EF as a variant of morphea, whereas others consider it as its own entity. We believe EF is its own entity. Eosinophilic fasciitis can be distinguished from morphea because 60% to 80% of patients with EF have peripheral eosinophilia and 20% to 70% of patients with EF have hypergammaglobulinemia. Additionally, morphea does not present as symmetrically or abruptly as EF.21
Treatment
To date, there is conflicting literature regarding the treatment regimen for MP. There is controversy regarding whether MP responds to corticosteroids.19 Different treatment regimens have been discussed for LS, but there is a lack of reports specifically describing therapies for MP and SMP. Because MP and SMP fall under the umbrella of LS, many investigators have reported using the following treatment regimens for patients with MP and SMP: bosentan,22 D-penicillamine,23 phototherapy,24-26 retinoids,26 oral steroids,27 methotrexate,27-29 vitamin D3 (oral calcitriol),30,31 cyclosporine,32 mycophenolate mofetil,33 and extracorporeal photochemotherapy.34
Falanga and Medsger23 reported 64% (7/11) treatment success with D-penicillamine in patients who exhibited severe LS. Psoralen plus UVA,24 methoxsalen, and UVA1 therapy are widely used in the treatment of LS.25 Kreuter et al25 advocated for phototherapy as the first approach in the management of LS after reporting improvement in all participants in their study (N=64), 2 participants with deep morphea while the rest exhibited other forms of morphea. Ozdemir et al26 proposed that retinoic acid combined with psoralen plus UVA is a good treatment choice for plaque-type LS; however, UVA only has the ability to target the epidermis and dermis, which may not be useful for deep forms of morphea.
Several studies have shown positive results in patients treated with methylprednisolone combined with low-dose methotrexate sodium.27-29 Kreuter et al30 and Elst et al31 proposed that calcitriol is effective in treating LS, whereas Hulshof et al35 indicated that it is not. It should be noted that none of these studies specifically mentioned MP. Martini et al33 demonstrated success with mycophenolate mofetil in the treatment of 10 LS patients who were resistant to methotrexate sodium and corticosteroids. Although none of the participants in the study had MP, 2 patients had disabling pansclerotic morphea, 3 had generalized morphea, and 5 had linear scleroderma (morphea en coup de sabre) affecting the limbs (n=2) and face (n=3).33 Because there is no established therapy or consensus for the treatment of MP, we have found success in starting with corticosteroids and then trying alternative therapies.
Prognosis
Morphea has transitioned into systemic scleroderma in a small number of reported cases.10,16,20,36 Therefore, patient follow-up is imperative to consistently identify systemic evolution. Although visceral complications are rare in the setting of LS, associated clinical findings have been reported, including arthralgia, arthritis, contractures, and carpal tunnel syndrome, as well as pulmonary, esophageal, and cardiac abnormalities.7,34
Conclusion
The morphologic features observed in our patient appear to correspond most closely to the type of lesion described by Su and Person5 and Whittaker et al.6 Although our case was clinically difficult to distinguish from linear morphea, the histology suggested SMP over other causes. If our patient’s SMP progressed to the joints, physical therapy would be needed to maintain range of motion and function of the extremities,2 and mandatory long-term follow-up would be required due to the risk for relapse after discontinuation of therapy. Our case highlights the inherent difficulties in the treatment of MP. Due to limited reports of SMP and MP in the literature as well as the conflicting views regarding effective and appropriate treatment options, additional investigation of these conditions and therapeutic options are necessary to further understand this debilitating condition.
1. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
2. Nguyen JV, Werth VP, Fett N. Morphea treatment & management. Medscape Web site. http://emedicine.medscape.com/article/1065782-treatment. Updated July 21, 2014. Accessed December 16, 2104.
3. Melani L, Cardinali C, Giomi B, et al. Case study: periodic follow-up is necessary in morphea profunda to identify systemic evolution. Skinmed. 2005;4:188-190.
4. Azad J, Dawn G, Shaffrali FC, et al. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29:25-27.
5. Su WP, Person JR. Morphea profunda. a new concept and a histopathologic study of 23 cases. Am J Dermatopathol. 1981;3:251-260.
6. Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol. 1989;120:431-440.
7. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
8. Torrelo A, Suárez J, Colmenero I, et al. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23:484-487.
9. Kreft B, Wohlrab J, Radant K, et al. Unrecognized radiation-induced localized scleroderma: a cause of postoperative wound-healing disorder [published online ahead of print June 22, 2009]. Clin Exp Dermatol. 2009;34:e383-e384.
10. Braun-Falco O, Plewig G, Wolff HH, et al, eds. Dermatology. Berlin, Germany: Springer; 2002.
11. Prinz JC, Kutasi Z, Weisenseel P, et al. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? results of a cohort analysis and presentation of three cases [published online ahead of print November 20, 2008]. J Am Acad Dermatol. 2009;60:248-255.
12. Wojas-Pelc A, Wielowieyska-Szybińska D, Kiełtyka A. Presence of the antinuclear antibodies and antibodies to Borrelia burgdorferi among patients with morphea en plaque, deep linear scleroderma and atrophoderma Pasini-Pierini [in Polish]. Przegl Lek. 2002;59:898-902.
13. Falanga V, Medsger TA Jr, Reichlin M, et al. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104:849-857.
14. Kikuchi K, Sato S, Kadono T, et al. Serum concentration of procollagen type I carboxyterminal propeptide in localized scleroderma. Arch Dermatol. 1994;130:1269-1272.
15. Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrilin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656-2659.
16. Savoia P, Zaccagna A, Bernengo MG. Guess what? inflammatory disseminated morphea profunda. Eur J Dermatol. 1999;9:654-656.
17. Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106:729-733.
18. Gilmour TK, Wilkinson B, Breit SN, et al. Analysis of dendritic cell populations using a revised histological staging of morphoea. Br J Dermatol. 2000;143:1183-1192.
19. Sayama K, Chen M, Shiraishi S, et al. Morphea profunda. Int J Dermatol. 1991;30:873-875.
20. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
21. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47:29-35.
22. Roldan R, Morote G, Castro Mdel C, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol. 2006;33:2538-2540.
23. Falanga V, Medsger TA Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990;126:609-612.
24. Breuckmann F, Gambichler T, Altmeyer P, et al. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol. 2004;4:11.
25. Kreuter A, Hyun J, Stücker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma [published online ahead of print January 30, 2006]. J Am Acad Dermatol. 2006;54:440-447.
26. Ozdemir M, Engin B, Toy H, et al. Treatment of plaque-type localized scleroderma with retinoic acid and ultraviolet A plus the photosensitizer psoralen: a case series. J Eur Acad Dermatol Venereol. 2008;22:519-521.
27. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847-852.
28. Kroft EB, Creemers MC, van den Hoogen FH, et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study [published online ahead of print February 4, 2009]. Br J Dermatol. 2009;160:1075-1082.
29. Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155:1013-1020.
30. Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241-245.
31. Elst EF, Van Suijlekom-Smit LW, Oranje AP. Treatment of linear scleroderma with oral 1,25-dihydroxy vitamin D3 (calcitriol) in seven children. Pediatr Dermatol. 1999;16:53-58.
32. Crespo MP, Mas IB, Díaz JM, et al. Rapid response to cyclosporine and maintenance with methotrexate in linear scleroderma in a young girl. Pediatr Dermatol. 2009;26:118-120.
33. Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil [published online ahead of print August 27, 2009]. Rheumatology (Oxford). 2009;48:1410-1413.
34. Neustadter JH, Samarin F, Carlson KR, et al. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol. 2009;145:127-130.
35. Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017-1023.
36. Toledano C, Rabhi S, Kettaneh A, et al. Localized scleroderma: a series of 52 patients [published online ahead of print September 17, 2008]. Eur J Intern Med. 2009;20:331-336.
1. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
2. Nguyen JV, Werth VP, Fett N. Morphea treatment & management. Medscape Web site. http://emedicine.medscape.com/article/1065782-treatment. Updated July 21, 2014. Accessed December 16, 2104.
3. Melani L, Cardinali C, Giomi B, et al. Case study: periodic follow-up is necessary in morphea profunda to identify systemic evolution. Skinmed. 2005;4:188-190.
4. Azad J, Dawn G, Shaffrali FC, et al. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29:25-27.
5. Su WP, Person JR. Morphea profunda. a new concept and a histopathologic study of 23 cases. Am J Dermatopathol. 1981;3:251-260.
6. Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol. 1989;120:431-440.
7. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
8. Torrelo A, Suárez J, Colmenero I, et al. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23:484-487.
9. Kreft B, Wohlrab J, Radant K, et al. Unrecognized radiation-induced localized scleroderma: a cause of postoperative wound-healing disorder [published online ahead of print June 22, 2009]. Clin Exp Dermatol. 2009;34:e383-e384.
10. Braun-Falco O, Plewig G, Wolff HH, et al, eds. Dermatology. Berlin, Germany: Springer; 2002.
11. Prinz JC, Kutasi Z, Weisenseel P, et al. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? results of a cohort analysis and presentation of three cases [published online ahead of print November 20, 2008]. J Am Acad Dermatol. 2009;60:248-255.
12. Wojas-Pelc A, Wielowieyska-Szybińska D, Kiełtyka A. Presence of the antinuclear antibodies and antibodies to Borrelia burgdorferi among patients with morphea en plaque, deep linear scleroderma and atrophoderma Pasini-Pierini [in Polish]. Przegl Lek. 2002;59:898-902.
13. Falanga V, Medsger TA Jr, Reichlin M, et al. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104:849-857.
14. Kikuchi K, Sato S, Kadono T, et al. Serum concentration of procollagen type I carboxyterminal propeptide in localized scleroderma. Arch Dermatol. 1994;130:1269-1272.
15. Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrilin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656-2659.
16. Savoia P, Zaccagna A, Bernengo MG. Guess what? inflammatory disseminated morphea profunda. Eur J Dermatol. 1999;9:654-656.
17. Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106:729-733.
18. Gilmour TK, Wilkinson B, Breit SN, et al. Analysis of dendritic cell populations using a revised histological staging of morphoea. Br J Dermatol. 2000;143:1183-1192.
19. Sayama K, Chen M, Shiraishi S, et al. Morphea profunda. Int J Dermatol. 1991;30:873-875.
20. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
21. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47:29-35.
22. Roldan R, Morote G, Castro Mdel C, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol. 2006;33:2538-2540.
23. Falanga V, Medsger TA Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990;126:609-612.
24. Breuckmann F, Gambichler T, Altmeyer P, et al. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol. 2004;4:11.
25. Kreuter A, Hyun J, Stücker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma [published online ahead of print January 30, 2006]. J Am Acad Dermatol. 2006;54:440-447.
26. Ozdemir M, Engin B, Toy H, et al. Treatment of plaque-type localized scleroderma with retinoic acid and ultraviolet A plus the photosensitizer psoralen: a case series. J Eur Acad Dermatol Venereol. 2008;22:519-521.
27. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847-852.
28. Kroft EB, Creemers MC, van den Hoogen FH, et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study [published online ahead of print February 4, 2009]. Br J Dermatol. 2009;160:1075-1082.
29. Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155:1013-1020.
30. Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241-245.
31. Elst EF, Van Suijlekom-Smit LW, Oranje AP. Treatment of linear scleroderma with oral 1,25-dihydroxy vitamin D3 (calcitriol) in seven children. Pediatr Dermatol. 1999;16:53-58.
32. Crespo MP, Mas IB, Díaz JM, et al. Rapid response to cyclosporine and maintenance with methotrexate in linear scleroderma in a young girl. Pediatr Dermatol. 2009;26:118-120.
33. Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil [published online ahead of print August 27, 2009]. Rheumatology (Oxford). 2009;48:1410-1413.
34. Neustadter JH, Samarin F, Carlson KR, et al. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol. 2009;145:127-130.
35. Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017-1023.
36. Toledano C, Rabhi S, Kettaneh A, et al. Localized scleroderma: a series of 52 patients [published online ahead of print September 17, 2008]. Eur J Intern Med. 2009;20:331-336.
Practice Points
- Localized trauma to the skin may be an inciting event to trigger morphea.
- Morphea is a clinical diagnosis but should be confirmed through biopsy to differentiate it from other similar entities.
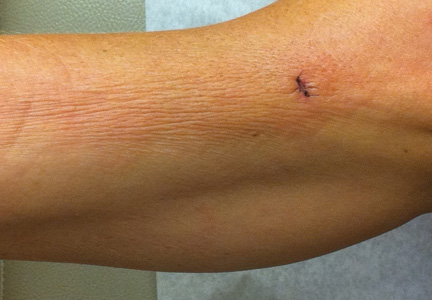
Cold Panniculitis: Delayed Onset in an Adult
The panniculitides can be a complex dermatologic entity for both dermatologists and dermatopathologists. The history, clinical examination, and histology need to be correlated to arrive at a differential diagnosis that will ultimately provide a diagnosis for the subcutaneous lesions. Panniculitis is an inflammation of the subcutaneous adipose tissue and can be associated with systemic diseases. According to Peters and Su,1 “Anatomic location of lesions, presence or absence of ulceration, occurrence of lipoatrophy, history of trauma, association with immunologic or metabolic disorders, and age of the patient are important clinical data to consider in conjunction with the microscopic features.” The panniculitides histologic differences may be subtle because they all include septal and lobular components, but one is usually more dominant in leading to a diagnosis along with the clinical findings.2
Cold panniculitis is a form of traumatic panniculitis. We present a unique case of this condition that was caused by use of a cold therapy unit following surgery to relieve pain.
Case Report
A 37-year-old woman presented for a routine postoperative visit 15 days following arthroscopic repair of a superior labrum anterior posterior tear in the left shoulder with a single suture anchor. The patient reported a rash that had developed 10 days postoperatively on the left upper arm. The rash started as red dots that progressively became larger, painful, and warm to the touch. The rash did not spread anywhere else on the patient’s body, and she denied fever, chills, and pruritus. She had tried using diphenhydramine without relief. The only new medication the patient had started prior to the eruption was oxycodone, which was initiated immediately following surgery. Prior to surgery, the entire left upper extremity including the shoulder had been prepared with a preoperative surgical skin antiseptic. There were no visible signs of the antiseptic on the skin at the time of presentation. The patient reported that she had applied a cold therapy unit to the left upper arm over her clothing for 1 hour every night since surgery. The cold therapy unit frequently is used to help decrease postoperative pain, swelling, inflammation, and narcotic use following surgical procedures.
Physical examination revealed multiple well-defined, erythematous, tender, indurated, warm nodules on the lateral aspect of the left upper arm (Figure 1). No other areas of eruption were noted on the body, and there was no swelling of the left elbow, forearm, wrist, or hand. The left upper extremity demonstrated intact sensation, rapid capillary refill, and a palpable radial pulse. Her weight was 230.1 lb with a body mass index of 35.
|
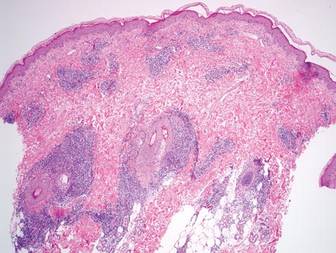
A 5-mm punch biopsy from a nodule on the left upper arm was performed, and pathology demonstrated vacuolar interface changes with patchy parakeratosis, spongiosis, and dyskeratosis on staining with hematoxylin and eosin. Pandermal and subcutaneous perivascular, periadnexal, and mild interstitial lymphohistiocytic infiltrate with occasional neutrophils and eosinophils were noted (Figure 2). The inflammation extended to the subcutaneous fat involving both septae and lobules with a primarily lobular distribution.
Clinical and pathologic correlation was required to arrive at a definitive diagnosis of cold panniculitis. The epidermal and dermal changes were consistent with a pernio or chilblains type of insult, and the septal and lobular panniculitis was indicative of cold panniculitis. The patient was advised to discontinue use of the cold therapy device as well as any other form of icing of the left shoulder or arm. She continued the oxycodone for pain control. Four weeks postoperatively, only desquamation remained where the nodules had previously appeared, which also eventually resolved.
Comment
Infants and small children are more predisposed to cold panniculitis than adults. In their 2008 review, Quesada-Cortés et al3 found the first report of cold panniculitis by Hochsinger in 1902 in a German pediatric journal, followed by reports from Lemez in 1928 and Haxthausen in 1941, which subsequently described similar cases in infants. Adult cases were not reported until 1963 by Solomon and Beerman4 and then in 1980 by Beacham et al.5
Etiologies for children have included popsicles, ice packs applied to the face to control supraventricular tachycardia or to the lower extremities after vaccinations, and cold weather exposure.6 The chemical composition of fat tissue plays a role in pediatric patients. According to Quesada-Cortés et al,3 subcutaneous fat in newborns is rich in saturated oils such as palmitic and stearic acids that have a higher solidification point. A small decrease in an infant’s temperature may result in crystallization of fat. The subcutaneous fat tends to become more unsaturated with aging with more oleic acid, and the solidification temperature diminishes.7
Cryoglobulins and cold agglutinins have not been demonstrated to be a cause of cold panniculitis in infants.7 Severe cold exposure or predisposition to certain conditions such as cryofibrinogenemia may occur in some adult patients. Gender does not seem to be a factor in children; however, in adults, women tend to be more predisposed to cold panniculitis secondary to obesity and participation in activities such as cycling, motorcycling, or horseback riding in cold conditions.3
On clinical examination, cold panniculitis features erythematous, firm, tender nodules on the cheeks and chin in infants and small children.2 These areas often are exposed to cold weather or wind because they typically are not covered with protective clothing.3 Nodules generally occur 1 to 3 days following exposure to cold and usually resolve spontaneously within 2 weeks.8 Popsicle panniculitis is characterized by a reddish discoloration on both cheeks 1 or 2 days after sucking on popsicles or ice cubes. This reaction can be reproduced in a half day by applying an ice cube to the volar forearm for 2 minutes, which can help diagnose and differentiate this subset of cold panniuculits.3 The red area in cold panniculitis eventually turns purple, becomes less indurated, and fades in approximately 3 months, but occasionally residual hyperpigmentation will last for a few months. Ice packs used as treatment of congenital cardiac arrhythmias in some cardiac surgeries and as surface cooling for management of birth asphyxia can produce a similar physical presentation.3
Equestrian panniculitis is characterized by erythematous, violaceous, tender plaques on the upper lateral thighs of young females who participate in horseback riding in the winter while wearing tight-fitting pants.2,5 These plaques typically occur within several hours and over the next week become painful, violaceous, and indurated or develop red nodules or plaques that can ulcerate or become crusted.3 These lesions usually will spontaneously resolve within 3 weeks, but new areas may occur again during the winter on further exposure with occasional persistent hyperpigmentation. These areas usually disappear at the end of winter with warmer weather or when horseback riding is discontinued. Perniosis also needs to be considered in the differential diagnosis due to the location and appearance of the lesions.3
It is important to obtain the correct specimen for biopsy. According to Peters and Su,1 a deep excisional biopsy that includes multiple fat lobules in addition to dermis and epidermis is critical. On histology, cold panniculitis usually demonstrates a primarily lobular inflammation. There typically is a superficial and deep perivascular lymphocytic infiltrate in the papillary dermis with edema noted in the connective tissue around the eccrine glands that can appear similar to perniosis on histopathology.9 Deposition of mucin, focal panniculitis surrounded by fatty tissue without inflammatory changes within the same field, and fat necrosis with pseudocysts and numerous lipophages also are characteristic features of cold panniculitis.10 Needlelike clefts are not present in cold panniculitis but appear in subcutaneous fat necrosis of the newborn.1
Different treatments have been tried, but no substantial impact on the rate of dissipation of the lesions has been noted. The plaques slowly resolve without scarring over 2 to 3 weeks if the cold source is removed.2 Application of a heating pad to the affected area has been used with limited success. Vasodilators such as nifedipine have been used but have not been found to be effective.3 Antihistamines also have failed to control the lesions.11
Treatment of cold panniculitis is based on the prevention of further insult versus trying to cure the condition. Avoidance of cold and wind exposure as well as direct contact with ice are key methods in preventing cold panniculitis.
Our patient’s presentation of this condition was unique. Although cold panniculitis lesions usually develop 1 to 3 days after cold exposure, our patient did not develop lesions until 10 days following surgery. The cold therapy unit used by our patient was evaluated in our office and also by the manufacturer and was found to be functioning normally with no defects. The late onset of the lesions was attributed to limited application of the cold therapy unit; our patient used it for only 1 hour every night, whereas application for 6 to 8 hours continuously is normally recommended. The lesions may have occurred sooner had the patient been using a solid ice pack versus the continuous cold circulating water of the cold therapy unit. Pathology was consistent with the patient’s history and physical examination indicating a diagnosis of cold panniculitis. The challenge of treatment was to alleviate the pain of the lesions as well as the postoperative shoulder pain without the aid of any form of cold therapy. The patient only needed a tincture of time, as the lesions resolved after 4 weeks. Patient education was provided on future prevention of this condition by avoiding exposure to cold or applying cold packs directly to the skin.
Acknowledgment
The authors thank the staff at the Office of Scientific Writing and Publication at the Marshfield Clinic Research Foundation, Wisconsin, for their editorial assistance in the preparation of this manuscript.
1. Peters MS, Su WP. Panniculitis. Dermatol Clin. 1992;10:37-57.
2. Patterson JW. Panniculitis. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008:1515-1530.
3. Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489.
4. Solomon LM, Beerman H. Cold panniculitis. Arch Dermatol. 1963;88:897-900.
5. Beacham BE, Cooper PH, Buchanan CS, et al. Equestrian cold panniculitis in women. Arch Dermatol. 1980;116:1025-1027.
6. Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421-433.
7. Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol. 1995;33(2, pt 2):383-385.
8. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
9. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
10. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22:530-549.
11. Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol. 1966;94:722-724.
The panniculitides can be a complex dermatologic entity for both dermatologists and dermatopathologists. The history, clinical examination, and histology need to be correlated to arrive at a differential diagnosis that will ultimately provide a diagnosis for the subcutaneous lesions. Panniculitis is an inflammation of the subcutaneous adipose tissue and can be associated with systemic diseases. According to Peters and Su,1 “Anatomic location of lesions, presence or absence of ulceration, occurrence of lipoatrophy, history of trauma, association with immunologic or metabolic disorders, and age of the patient are important clinical data to consider in conjunction with the microscopic features.” The panniculitides histologic differences may be subtle because they all include septal and lobular components, but one is usually more dominant in leading to a diagnosis along with the clinical findings.2
Cold panniculitis is a form of traumatic panniculitis. We present a unique case of this condition that was caused by use of a cold therapy unit following surgery to relieve pain.
Case Report
A 37-year-old woman presented for a routine postoperative visit 15 days following arthroscopic repair of a superior labrum anterior posterior tear in the left shoulder with a single suture anchor. The patient reported a rash that had developed 10 days postoperatively on the left upper arm. The rash started as red dots that progressively became larger, painful, and warm to the touch. The rash did not spread anywhere else on the patient’s body, and she denied fever, chills, and pruritus. She had tried using diphenhydramine without relief. The only new medication the patient had started prior to the eruption was oxycodone, which was initiated immediately following surgery. Prior to surgery, the entire left upper extremity including the shoulder had been prepared with a preoperative surgical skin antiseptic. There were no visible signs of the antiseptic on the skin at the time of presentation. The patient reported that she had applied a cold therapy unit to the left upper arm over her clothing for 1 hour every night since surgery. The cold therapy unit frequently is used to help decrease postoperative pain, swelling, inflammation, and narcotic use following surgical procedures.
Physical examination revealed multiple well-defined, erythematous, tender, indurated, warm nodules on the lateral aspect of the left upper arm (Figure 1). No other areas of eruption were noted on the body, and there was no swelling of the left elbow, forearm, wrist, or hand. The left upper extremity demonstrated intact sensation, rapid capillary refill, and a palpable radial pulse. Her weight was 230.1 lb with a body mass index of 35.
|
A 5-mm punch biopsy from a nodule on the left upper arm was performed, and pathology demonstrated vacuolar interface changes with patchy parakeratosis, spongiosis, and dyskeratosis on staining with hematoxylin and eosin. Pandermal and subcutaneous perivascular, periadnexal, and mild interstitial lymphohistiocytic infiltrate with occasional neutrophils and eosinophils were noted (Figure 2). The inflammation extended to the subcutaneous fat involving both septae and lobules with a primarily lobular distribution.
Clinical and pathologic correlation was required to arrive at a definitive diagnosis of cold panniculitis. The epidermal and dermal changes were consistent with a pernio or chilblains type of insult, and the septal and lobular panniculitis was indicative of cold panniculitis. The patient was advised to discontinue use of the cold therapy device as well as any other form of icing of the left shoulder or arm. She continued the oxycodone for pain control. Four weeks postoperatively, only desquamation remained where the nodules had previously appeared, which also eventually resolved.
Comment
Infants and small children are more predisposed to cold panniculitis than adults. In their 2008 review, Quesada-Cortés et al3 found the first report of cold panniculitis by Hochsinger in 1902 in a German pediatric journal, followed by reports from Lemez in 1928 and Haxthausen in 1941, which subsequently described similar cases in infants. Adult cases were not reported until 1963 by Solomon and Beerman4 and then in 1980 by Beacham et al.5
Etiologies for children have included popsicles, ice packs applied to the face to control supraventricular tachycardia or to the lower extremities after vaccinations, and cold weather exposure.6 The chemical composition of fat tissue plays a role in pediatric patients. According to Quesada-Cortés et al,3 subcutaneous fat in newborns is rich in saturated oils such as palmitic and stearic acids that have a higher solidification point. A small decrease in an infant’s temperature may result in crystallization of fat. The subcutaneous fat tends to become more unsaturated with aging with more oleic acid, and the solidification temperature diminishes.7
Cryoglobulins and cold agglutinins have not been demonstrated to be a cause of cold panniculitis in infants.7 Severe cold exposure or predisposition to certain conditions such as cryofibrinogenemia may occur in some adult patients. Gender does not seem to be a factor in children; however, in adults, women tend to be more predisposed to cold panniculitis secondary to obesity and participation in activities such as cycling, motorcycling, or horseback riding in cold conditions.3
On clinical examination, cold panniculitis features erythematous, firm, tender nodules on the cheeks and chin in infants and small children.2 These areas often are exposed to cold weather or wind because they typically are not covered with protective clothing.3 Nodules generally occur 1 to 3 days following exposure to cold and usually resolve spontaneously within 2 weeks.8 Popsicle panniculitis is characterized by a reddish discoloration on both cheeks 1 or 2 days after sucking on popsicles or ice cubes. This reaction can be reproduced in a half day by applying an ice cube to the volar forearm for 2 minutes, which can help diagnose and differentiate this subset of cold panniuculits.3 The red area in cold panniculitis eventually turns purple, becomes less indurated, and fades in approximately 3 months, but occasionally residual hyperpigmentation will last for a few months. Ice packs used as treatment of congenital cardiac arrhythmias in some cardiac surgeries and as surface cooling for management of birth asphyxia can produce a similar physical presentation.3
Equestrian panniculitis is characterized by erythematous, violaceous, tender plaques on the upper lateral thighs of young females who participate in horseback riding in the winter while wearing tight-fitting pants.2,5 These plaques typically occur within several hours and over the next week become painful, violaceous, and indurated or develop red nodules or plaques that can ulcerate or become crusted.3 These lesions usually will spontaneously resolve within 3 weeks, but new areas may occur again during the winter on further exposure with occasional persistent hyperpigmentation. These areas usually disappear at the end of winter with warmer weather or when horseback riding is discontinued. Perniosis also needs to be considered in the differential diagnosis due to the location and appearance of the lesions.3
It is important to obtain the correct specimen for biopsy. According to Peters and Su,1 a deep excisional biopsy that includes multiple fat lobules in addition to dermis and epidermis is critical. On histology, cold panniculitis usually demonstrates a primarily lobular inflammation. There typically is a superficial and deep perivascular lymphocytic infiltrate in the papillary dermis with edema noted in the connective tissue around the eccrine glands that can appear similar to perniosis on histopathology.9 Deposition of mucin, focal panniculitis surrounded by fatty tissue without inflammatory changes within the same field, and fat necrosis with pseudocysts and numerous lipophages also are characteristic features of cold panniculitis.10 Needlelike clefts are not present in cold panniculitis but appear in subcutaneous fat necrosis of the newborn.1
Different treatments have been tried, but no substantial impact on the rate of dissipation of the lesions has been noted. The plaques slowly resolve without scarring over 2 to 3 weeks if the cold source is removed.2 Application of a heating pad to the affected area has been used with limited success. Vasodilators such as nifedipine have been used but have not been found to be effective.3 Antihistamines also have failed to control the lesions.11
Treatment of cold panniculitis is based on the prevention of further insult versus trying to cure the condition. Avoidance of cold and wind exposure as well as direct contact with ice are key methods in preventing cold panniculitis.
Our patient’s presentation of this condition was unique. Although cold panniculitis lesions usually develop 1 to 3 days after cold exposure, our patient did not develop lesions until 10 days following surgery. The cold therapy unit used by our patient was evaluated in our office and also by the manufacturer and was found to be functioning normally with no defects. The late onset of the lesions was attributed to limited application of the cold therapy unit; our patient used it for only 1 hour every night, whereas application for 6 to 8 hours continuously is normally recommended. The lesions may have occurred sooner had the patient been using a solid ice pack versus the continuous cold circulating water of the cold therapy unit. Pathology was consistent with the patient’s history and physical examination indicating a diagnosis of cold panniculitis. The challenge of treatment was to alleviate the pain of the lesions as well as the postoperative shoulder pain without the aid of any form of cold therapy. The patient only needed a tincture of time, as the lesions resolved after 4 weeks. Patient education was provided on future prevention of this condition by avoiding exposure to cold or applying cold packs directly to the skin.
Acknowledgment
The authors thank the staff at the Office of Scientific Writing and Publication at the Marshfield Clinic Research Foundation, Wisconsin, for their editorial assistance in the preparation of this manuscript.
The panniculitides can be a complex dermatologic entity for both dermatologists and dermatopathologists. The history, clinical examination, and histology need to be correlated to arrive at a differential diagnosis that will ultimately provide a diagnosis for the subcutaneous lesions. Panniculitis is an inflammation of the subcutaneous adipose tissue and can be associated with systemic diseases. According to Peters and Su,1 “Anatomic location of lesions, presence or absence of ulceration, occurrence of lipoatrophy, history of trauma, association with immunologic or metabolic disorders, and age of the patient are important clinical data to consider in conjunction with the microscopic features.” The panniculitides histologic differences may be subtle because they all include septal and lobular components, but one is usually more dominant in leading to a diagnosis along with the clinical findings.2
Cold panniculitis is a form of traumatic panniculitis. We present a unique case of this condition that was caused by use of a cold therapy unit following surgery to relieve pain.
Case Report
A 37-year-old woman presented for a routine postoperative visit 15 days following arthroscopic repair of a superior labrum anterior posterior tear in the left shoulder with a single suture anchor. The patient reported a rash that had developed 10 days postoperatively on the left upper arm. The rash started as red dots that progressively became larger, painful, and warm to the touch. The rash did not spread anywhere else on the patient’s body, and she denied fever, chills, and pruritus. She had tried using diphenhydramine without relief. The only new medication the patient had started prior to the eruption was oxycodone, which was initiated immediately following surgery. Prior to surgery, the entire left upper extremity including the shoulder had been prepared with a preoperative surgical skin antiseptic. There were no visible signs of the antiseptic on the skin at the time of presentation. The patient reported that she had applied a cold therapy unit to the left upper arm over her clothing for 1 hour every night since surgery. The cold therapy unit frequently is used to help decrease postoperative pain, swelling, inflammation, and narcotic use following surgical procedures.
Physical examination revealed multiple well-defined, erythematous, tender, indurated, warm nodules on the lateral aspect of the left upper arm (Figure 1). No other areas of eruption were noted on the body, and there was no swelling of the left elbow, forearm, wrist, or hand. The left upper extremity demonstrated intact sensation, rapid capillary refill, and a palpable radial pulse. Her weight was 230.1 lb with a body mass index of 35.
|
A 5-mm punch biopsy from a nodule on the left upper arm was performed, and pathology demonstrated vacuolar interface changes with patchy parakeratosis, spongiosis, and dyskeratosis on staining with hematoxylin and eosin. Pandermal and subcutaneous perivascular, periadnexal, and mild interstitial lymphohistiocytic infiltrate with occasional neutrophils and eosinophils were noted (Figure 2). The inflammation extended to the subcutaneous fat involving both septae and lobules with a primarily lobular distribution.
Clinical and pathologic correlation was required to arrive at a definitive diagnosis of cold panniculitis. The epidermal and dermal changes were consistent with a pernio or chilblains type of insult, and the septal and lobular panniculitis was indicative of cold panniculitis. The patient was advised to discontinue use of the cold therapy device as well as any other form of icing of the left shoulder or arm. She continued the oxycodone for pain control. Four weeks postoperatively, only desquamation remained where the nodules had previously appeared, which also eventually resolved.
Comment
Infants and small children are more predisposed to cold panniculitis than adults. In their 2008 review, Quesada-Cortés et al3 found the first report of cold panniculitis by Hochsinger in 1902 in a German pediatric journal, followed by reports from Lemez in 1928 and Haxthausen in 1941, which subsequently described similar cases in infants. Adult cases were not reported until 1963 by Solomon and Beerman4 and then in 1980 by Beacham et al.5
Etiologies for children have included popsicles, ice packs applied to the face to control supraventricular tachycardia or to the lower extremities after vaccinations, and cold weather exposure.6 The chemical composition of fat tissue plays a role in pediatric patients. According to Quesada-Cortés et al,3 subcutaneous fat in newborns is rich in saturated oils such as palmitic and stearic acids that have a higher solidification point. A small decrease in an infant’s temperature may result in crystallization of fat. The subcutaneous fat tends to become more unsaturated with aging with more oleic acid, and the solidification temperature diminishes.7
Cryoglobulins and cold agglutinins have not been demonstrated to be a cause of cold panniculitis in infants.7 Severe cold exposure or predisposition to certain conditions such as cryofibrinogenemia may occur in some adult patients. Gender does not seem to be a factor in children; however, in adults, women tend to be more predisposed to cold panniculitis secondary to obesity and participation in activities such as cycling, motorcycling, or horseback riding in cold conditions.3
On clinical examination, cold panniculitis features erythematous, firm, tender nodules on the cheeks and chin in infants and small children.2 These areas often are exposed to cold weather or wind because they typically are not covered with protective clothing.3 Nodules generally occur 1 to 3 days following exposure to cold and usually resolve spontaneously within 2 weeks.8 Popsicle panniculitis is characterized by a reddish discoloration on both cheeks 1 or 2 days after sucking on popsicles or ice cubes. This reaction can be reproduced in a half day by applying an ice cube to the volar forearm for 2 minutes, which can help diagnose and differentiate this subset of cold panniuculits.3 The red area in cold panniculitis eventually turns purple, becomes less indurated, and fades in approximately 3 months, but occasionally residual hyperpigmentation will last for a few months. Ice packs used as treatment of congenital cardiac arrhythmias in some cardiac surgeries and as surface cooling for management of birth asphyxia can produce a similar physical presentation.3
Equestrian panniculitis is characterized by erythematous, violaceous, tender plaques on the upper lateral thighs of young females who participate in horseback riding in the winter while wearing tight-fitting pants.2,5 These plaques typically occur within several hours and over the next week become painful, violaceous, and indurated or develop red nodules or plaques that can ulcerate or become crusted.3 These lesions usually will spontaneously resolve within 3 weeks, but new areas may occur again during the winter on further exposure with occasional persistent hyperpigmentation. These areas usually disappear at the end of winter with warmer weather or when horseback riding is discontinued. Perniosis also needs to be considered in the differential diagnosis due to the location and appearance of the lesions.3
It is important to obtain the correct specimen for biopsy. According to Peters and Su,1 a deep excisional biopsy that includes multiple fat lobules in addition to dermis and epidermis is critical. On histology, cold panniculitis usually demonstrates a primarily lobular inflammation. There typically is a superficial and deep perivascular lymphocytic infiltrate in the papillary dermis with edema noted in the connective tissue around the eccrine glands that can appear similar to perniosis on histopathology.9 Deposition of mucin, focal panniculitis surrounded by fatty tissue without inflammatory changes within the same field, and fat necrosis with pseudocysts and numerous lipophages also are characteristic features of cold panniculitis.10 Needlelike clefts are not present in cold panniculitis but appear in subcutaneous fat necrosis of the newborn.1
Different treatments have been tried, but no substantial impact on the rate of dissipation of the lesions has been noted. The plaques slowly resolve without scarring over 2 to 3 weeks if the cold source is removed.2 Application of a heating pad to the affected area has been used with limited success. Vasodilators such as nifedipine have been used but have not been found to be effective.3 Antihistamines also have failed to control the lesions.11
Treatment of cold panniculitis is based on the prevention of further insult versus trying to cure the condition. Avoidance of cold and wind exposure as well as direct contact with ice are key methods in preventing cold panniculitis.
Our patient’s presentation of this condition was unique. Although cold panniculitis lesions usually develop 1 to 3 days after cold exposure, our patient did not develop lesions until 10 days following surgery. The cold therapy unit used by our patient was evaluated in our office and also by the manufacturer and was found to be functioning normally with no defects. The late onset of the lesions was attributed to limited application of the cold therapy unit; our patient used it for only 1 hour every night, whereas application for 6 to 8 hours continuously is normally recommended. The lesions may have occurred sooner had the patient been using a solid ice pack versus the continuous cold circulating water of the cold therapy unit. Pathology was consistent with the patient’s history and physical examination indicating a diagnosis of cold panniculitis. The challenge of treatment was to alleviate the pain of the lesions as well as the postoperative shoulder pain without the aid of any form of cold therapy. The patient only needed a tincture of time, as the lesions resolved after 4 weeks. Patient education was provided on future prevention of this condition by avoiding exposure to cold or applying cold packs directly to the skin.
Acknowledgment
The authors thank the staff at the Office of Scientific Writing and Publication at the Marshfield Clinic Research Foundation, Wisconsin, for their editorial assistance in the preparation of this manuscript.
1. Peters MS, Su WP. Panniculitis. Dermatol Clin. 1992;10:37-57.
2. Patterson JW. Panniculitis. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008:1515-1530.
3. Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489.
4. Solomon LM, Beerman H. Cold panniculitis. Arch Dermatol. 1963;88:897-900.
5. Beacham BE, Cooper PH, Buchanan CS, et al. Equestrian cold panniculitis in women. Arch Dermatol. 1980;116:1025-1027.
6. Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421-433.
7. Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol. 1995;33(2, pt 2):383-385.
8. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
9. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
10. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22:530-549.
11. Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol. 1966;94:722-724.
1. Peters MS, Su WP. Panniculitis. Dermatol Clin. 1992;10:37-57.
2. Patterson JW. Panniculitis. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008:1515-1530.
3. Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489.
4. Solomon LM, Beerman H. Cold panniculitis. Arch Dermatol. 1963;88:897-900.
5. Beacham BE, Cooper PH, Buchanan CS, et al. Equestrian cold panniculitis in women. Arch Dermatol. 1980;116:1025-1027.
6. Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421-433.
7. Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol. 1995;33(2, pt 2):383-385.
8. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
9. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
10. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22:530-549.
11. Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol. 1966;94:722-724.
Practice Points
- Cold panniculitis is a form of traumatic panniculitis.
- Cold panniculitis often appears on the cheeks and chin, areas that are exposed to cold weather or wind because they are not covered with protective clothing, in infants and small children.
- Treatment of cold panniculitis is based on the prevention of further insult.
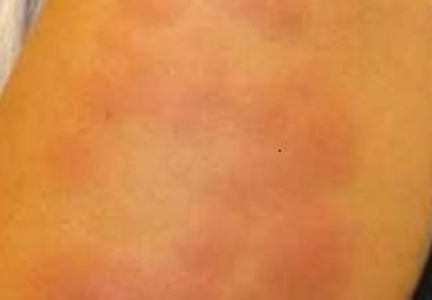
Autosomal-Dominant Familial Angiolipomatosis
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
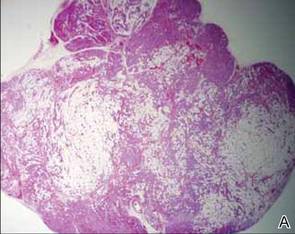
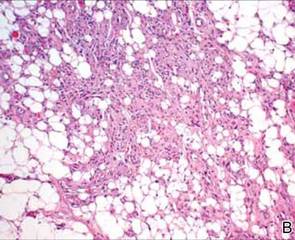
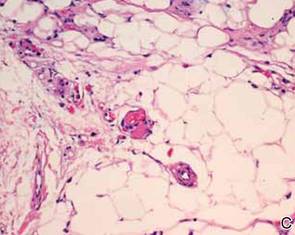
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Practice Points
- Dermatologists should be familiar with the clinical and histological features of angiolipomas along with their potential inheritance patterns.
- Familial angiolipomatosis is a rare condition characterized by multiple angiolipomas that has been described as having an autosomal-recessive transmission pattern. Autosomal-dominant inheritance also may occur, as illustrated in the current case report.
- Awareness of the autosomal-dominant form of this entity is important to prevent its misdiagnosis as
neurofibromatosis type I, which has a similar family history and clinical presentation.
Incidence and Epidemiology of Onychomycosis in Patients Visiting a Tertiary Care Hospital in India
Onychomycosis is a chronic fungal infection of the nails. Dermatophytes are the most common etiologic agents, but yeasts and nondermatophyte molds also constitute a substantial number of cases.1 An accumulation of debris under distorted, deformed, thickened, and discolored nails, particularly with ragged and furrowed edges, strongly suggests tinea unguium.2 Candidal onychomycosis (CO) lacks gross distortion and accumulated detritus and mainly affects fingernails.3 Nondermatophytic molds cause 1.5% to 6% of cases of onychomycosis, mostly seen in toenails of elderly individuals with a history of trauma.4 Onychomycosis affects 5.5% of the world population5 and represents 20% to 40% of all onychopathies and approximately 30% of cutaneous mycotic infections.6
The incidence of onychomycosis ranges from 0.5% to 5% in the general population in India.7 The incidence is particularly high in warm humid climates such as India.8 Researchers have found certain habits of the population in the Indian subcontinent (eg, walking with bare feet, wearing ill-fitting shoes, nail-biting [eg, onychophagia], working with chemicals) to be contributing factors for onychomycosis.9 Several studies have shown that the prevalence of onychomycosis increases with age, possibly due to poor peripheral circulation, diabetes mellitus, repeated nail trauma, prolonged exposure to pathogenic fungi, suboptimal immune function, inactivity, or inability to trim the toenails and care for the feet.10 Nail infection is a cosmetic problem with serious physical and psychological morbidity and also serves as the fungal reservoir for skin infections. Besides destruction and disfigurement of the nail plate, onychomycosis can lead to self-consciousness and impairment of daily functioning.11
Nail dystrophy occurs secondary to various systemic disorders or can be associated with other dermatologic conditions. Nail discoloration and other onychia should be differentiated from onychomycosis by classifying nail lesions as distal lateral subungual onychomycosis, proximal subungual onychomycosis (PSO), CO, white superficial onychomycosis (WSO), and total dystrophic onychomycosis.12 Laboratory investigation is necessary to accurately differentiate between fungal infections and other skin diseases before starting treatment. Our hospital-based study sought to determine the incidence and epidemiology of onychomycosis with an analysis of 134 participants with clinically suspected onychomycosis. We evaluated prevalence based on age, sex, and occupation, as well as the most common pathogens.
Materials and Methods
Study Design and Participants
The study population consisted of 134 patients with clinically suspected onychomycosis who visited the dermatology department at the Veer Chandra Singh Garhwali Government Institute of Medical Sciences and Research Institute in Uttarakhand, India (October 2010 to October 2011). A thorough history was obtained and a detailed examination of the distorted nails was conducted in the microbiology laboratory. Patient history and demographic factors such as age, sex, occupation, and related history of risk factors for onychomycosis were recorded pro forma. Some of the details such as itching, family history of fungal infection, and prior cutaneous infections were recorded. Patients who were undergoing treatment with systemic or topical antifungal agents in the 4 weeks preceding the study period were excluded to rule out false-negative cases and to avoid the influence of antifungal agents on the disease course.
Assessments
Two samples were taken from each patient on different days. Participants were divided into 4 groups based on occupation: farmer, housewife, student, and other (eg, clerk, shopkeeper, painter). Clinical presentation of discoloration, onycholysis, subungual hyperkeratosis, and nail thickening affecting the distal and/or lateral nail plate was defined as distal lateral subungual onychomycosis; discoloration and onycholysis affecting the proximal part of the nail was defined as PSO; association with paronychia and distal and lateral onycholysis was defined as CO; white opaque patches on the nail surface were defined as WSO; and end-stage nail disease was defined as total dystrophic onychomycosis.
Prior to sampling, the nails were cleaned with a 70% alcohol solution. Nail clippings were obtained using presterilized nail clippers and a blunt no. 15 scalpel blade and were placed on sterilized black paper. Each nail sample was divided into 2 parts: one for direct microscopy and one for culture. Nail clippings were subjected to microscopic examination after clearing in 20% potassium hydroxide solution. The slides were examined for fungal hyphae, arthrospores, yeasts, and pseudohyphal forms. Culture was done with Emmons modification of Sabouraud dextrose agar (incubated at 27°C for molds and 37°C for yeasts) as well as with 0.4% chloramphenicol and 5% cycloheximide (incubated at 27°C). Culture tubes were examined daily for the first week and on alternate days thereafter for 4 weeks of incubation.
Dermatophytes were identified based on the colony morphology, growth rate, texture, border, and pigmentation in the obverse and reverse of culture media and microscopic examination using lactophenol cotton blue tease mount. Yeast colonies were identified microscopically with Gram stain, and species were identified by germ tube, carbohydrate assimilation, and fermentation tests.13 Nondermatophyte molds were identified by colony morphology, microscopic examination, and slide culture. Molds were considered as pathogens in the presence of the following criteria: (1) absence of other fungal growth in the same culture tube; (2) presence of mold growth in all 3 samples; and (3) presence of filaments identified on direct examination.
Results
Of 134 clinically suspected cases of onychomycosis, 78 (58.2%) were from fingernails and 56 (41.8%) from toenails. Clinical diagnosis was confirmed in 96 (71.6%) cases by both fungal culture and direct microscopy but was confirmed by direct microscopy alone in only 76 (56.7%) cases. False-negative results were found in 23.9% (32/134) of participants with direct microscopy and 9.0% (12/134) with fungal cultures. The results of direct microscopy and fungal culture are outlined in Table 1. The study included 78 (58.2%) males and 56 (41.8%) females with a mean age of 44 years. Highest prevalence (47.8%) was seen in participants older than 40 years and lowest prevalence (11.9%) in participants younger than 20 years. In total, 32.8% of participants were farmers, 31.3% were housewives, 14.9% were students, and 20.9% performed other occupations. Disease history at the time of first presentation varied from 1 month to more than 2 years; 33.6% of participants had a 1- to 6-month history of disease, while only 3.7% had a disease history of less than 1 month at presentation. The demographic data are further outlined in Table 2.


Distal lateral subungual onychomycosis was the most prevalent clinical pattern found in 66 (49.3%) participants; fungal isolates were found in 60 of these participants. The next most prevalent clinical pattern was PSO, which was found in 34 (25.4%) participants, 12 showing fungal growth. A clinical pattern of CO was noted in 28 (20.9%) participants, 22 showing fungal growth; WSO was noted in 10 (7.5%) participants, 2 showing fungal growth.
Of 96 culture-positive cases, dermatophytes were the most common pathogens isolated in 56 (58.3%) participants, followed by Candida species in 28 (29.2%) participants. Nondermatophyte molds were isolated in 12 (12.5%) participants. The various dermatophytes, Candida species, and nondermatophyte molds that were isolated on fungal culture are outlined in Table 3. Of the 96 participants with positive fungal cultures, 30 (31.2%) were farmers working with soil, 28 (29.2%) were housewives associated with wet work, 16 (16.7%) were students associated with increased physical exercise from extracurricular activity, and 22 (22.9%) were in other occupations (Table 4).
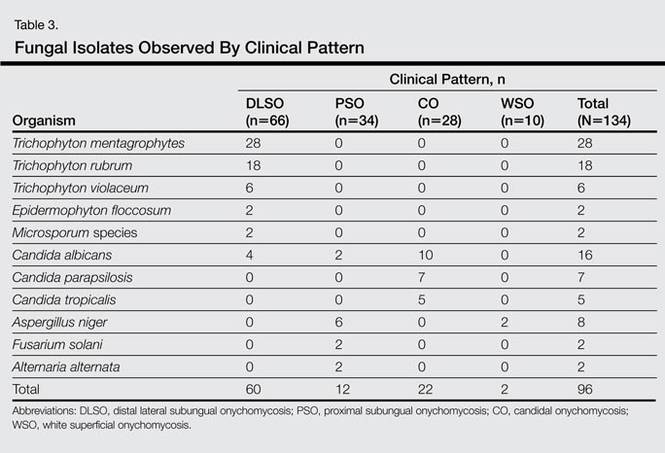
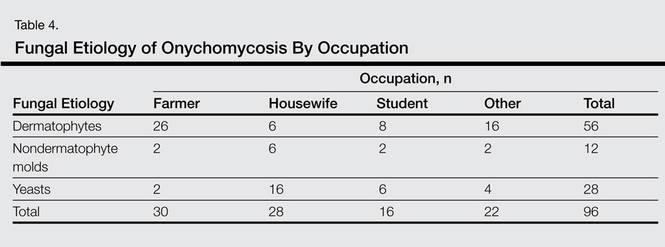
Comment
The term onychomycosis is derived from onyx, the Greek word for nail, and mykes, the Greek word for fungus. Onychomycosis is a chronic mycotic infection of the fingernails and toenails that can have a serious impact on patients’ quality of life. The fungi known to cause onychomycosis vary among geographic areas, primarily due to differences in climate.14 The isolation rate of onychomycosis in our hospital-based study was 71.6%, which is in accordance with various studies in India and abroad, including 60% in Karnataka, India5; 82.3% in Sikkim, India6; and 86.9% in Turkey.1 However, other studies have shown lower isolation rates of 39.5% in Central Delhi, India,15 and 37.6% in Himachal Pradesh, India.16 Some patients with onychomycosis may not seek medical attention, which may explain the difference in the prevalence of onychomycosis observed worldwide.17 The prevalence of onychomycosis by age also varies. In our study, participants older than 40 years showed the highest prevalence (47.8%), which is in accordance with other studies from India18 and abroad.19,20 In contrast, some Indian studies15,21,22 have reported a higher prevalence in younger adults (ie, 21–30 years), which may be attributed to greater self-consciousness about nail discoloration and disfigurement as well as increased physical activity and different shoe-wearing habits. A higher prevalence in older adults, as observed in our study as well some other studies,19,21 may be due to poor peripheral circulation, diabetes mellitus, repeated nail trauma, longer exposure to pathogenic fungi, suboptimal immune function, inactivity, and poor hygiene.10
In our study, suspected onychomycosis was more common in males (58.2%) than in females (41.8%). These results are in accordance with many of the studies in the worldwide literature.1,10,11,15,16,23-25 A higher isolation rate in males worldwide may be due to common use of occlusive footwear, more exposure to outdoor conditions, and increased physical activity, leading to an increased likelihood of trauma. The importance of trauma to the nails as a predisposing factor for onychomycosis is well established.24 In our study, the majority of males wore shoes regardless of occupation. Perspiration of the feet when wearing socks and/or shoes can generate a warm moist environment that promotes the growth of fungi and predisposes patients to onychomycosis. Similar observations have been reported by other investigators.21,22,25,26
The incidence of onychomycosis was almost evenly distributed among farmers, housewives, and the miscellaneous group, whereas a high isolation rate was noted among students. Of 20 students included in our study, onychomycosis was confirmed in 16, which may be related to an increased use of synthetic sports shoes and socks that retain sweat as well as vigorous physical activity frequently resulting in nail injuries among this patient population.11 Younger patients may be more conscious of their appearance and therefore may be more likely to seek treatment. Similar observations have been reported by other researchers.15,21,22
In our study, dermatophytes were the most commonly found pathogens (58.3%), which is comparable to other studies.15,18,22Trichophyton mentagrophytes was the most frequently isolated dermatophyte from cultures, which was in concordance with a study from Delhi.15 In some studies,18,20,22Trichophyton rubrum has been reported as the most prevalent dermatophyte, but we identified Trichophyton rubrum in only 18 participants, which can be attributed to variations in epidemiology based on geographic region. Nondermatophyte molds were isolated in 12.5% of participants, with Aspergillus niger being the most common isolate found in 8 cases. Other isolated species were Alternaria alternata and Fusarium solani found in 2 cases each. Aspergillus niger has been reported in worldwide studies as an important cause of onychomycosis.15,18,19,21,22
In 28 cases (29.2%) involving Candida species, Candida albicans, Candida parapsilosis, and Candida tropicalis were the most common pathogens, respectively, which is in accordance with many studies.15,20-22,25 In 28 cases of CO, females (n=16) were affected more than males (n=12). All of the females were housewives and C albicans was predominantly isolated from the fingernails. Household responsibilities involving kitchen work (eg, cutting and peeling vegetables, washing utensils, cleaning the house/laundry) may chronically expose housewives to moist environments and make them more prone to injury, thus facilitating easy entry of fungal agents.
Distal lateral subungual onychomycosis was the most prevalent clinical type found (n=66), which is comparable to other reports.20,22,25 Proximal subungual onychomycosis was the second most common type; however, a greater incidence has been reported by some researchers,23,24 while others have reported a lower incidence.20,21 Candidial onychomycosis and WSO were not common in our study, and PSO was not associated with any immunodeficiency disease, as reported by other researchers.15,20
Of 134 suspected cases of onychomycosis, 71.6% were confirmed by both direct microscopy and fungal culture, but only 56.7% were confirmed by direct microscopy alone. If we had relied on microscopy with potassium hydroxide only, we would have missed 23.9% of cases. Therefore, nail scrapings should always be subjected to fungal culture as well as direct microscopy, as both are necessary for accurate diagnosis and treatment of onychomycosis. If onychomycosis is not successfully treated, it can act as a reservoir of fungal infection affecting other parts of the body with the potential to pass infection on to others.
Conclusion
Clinical examination alone is not sufficient for diagnosing onychomycosis14,18,20; in many cases of suspected onychomycosis with nail changes, mycologic examination does not confirm fungal infection. In our study, only 71.6% of participants with nail changes proved to be of fungal etiology. Other researchers from different geographic locations have reported similar results with lower incidence (eg, 39.5%,15 37.6%,16 51.7%,18 45.3%21) of fungal etiology in such cases. Therefore, both clinical and mycologic examinations are important for establishing the diagnosis and selecting the most suitable antifungal agent, which is possible only if the underlying pathogen is correctly identified.
1. Yenişehirli G, Bulut Y, Sezer E, et al. Onychomycosis infections in the Middle Black Sea Region, Turkey. Int J Dermatol. 2009;48:956-959.
2. Kouskoukis CE, Scher RK, Ackerman AB. What histologic finding distinguishes onychomycosis and psoriasis? Am J Dermatopathol. 1983;5:501-503.
3. Rippon JW. Medical mycology. In: Wonsiewicz M, ed. The Pathogenic Fungi and the Pathogenic Actinomycetes. 3rd ed. Philadelphia, PA: WB Saunders; 1988:169-275.
4. Greer DL. Evolving role of nondermatophytes in onychomycosis. Int J Dermatol. 1995;34:521-524.
5. Murray SC, Dawber RP. Onychomycosis of toenails: orthopaedic and podiatric considerations. Australas J Dermatol. 2002;43:105-112.
6. Achten G, Wanet-Rouard J. Onychomycoses in the laboratory. Mykosen Suppl. 1978;1:125-127.
7. Sobhanadri C, Rao DT, Babu KS. Clinical and mycological study of superficial fungal infections at Government General Hospital: guntur and their response to treatment with hamycin, dermostatin and dermamycin. Indian J Dermatol Venereol. 1970;36:209-214.
8. Jain S, Sehgal VN. Commentary: onychomycosis: an epidemio-etiologic perspective. Int J Dermatol. 2000;39:100-103.
9. Sehgal VN, Aggarwal AK, Srivastava G, et al. Onychomycosis: a 3 year clinicomycologic hospital-based study. Skinmed. 2007;6:11-17.
10. Elewski BE, Charif MA. Prevalence of onychomycosis in patients attending a dermatology clinic in northeastern Ohio for the other conditions. Arch Dermatol. 1997;133:1172-1173.
11. Scher RK. Onychomycosis is more than a cosmetic problem. Br J Dermatol. 1994;130(suppl 43):S15.
12. Godoy-Martinez PG, Nunes FG, Tomimori-Yamashita J, et al. Onychomycosis in São Paulo, Brazil [published online ahead of print May 8, 2009]. Mycopathologia. 2009;168:111-116.
13. Larone DH. Medically Important Fungi: A Guide to Identification. 4th ed. Washington, DC: American Society for Microbiology Press; 2002.
14. Sehgal VN, Srivastava G, Dogra S, et al. Onychomycosis: an Asian perspective. Skinmed. 2010;8:37-45.
15. Sanjiv A, Shalini M, Charoo H. Etiological agents of onychomycosis from a tertiary care hospital in Central Delhi, India. Indian J Fund Appl Life Science. 2011;1:11-14.
16. Gupta M, Sharma NL, Kanga AK, et al. Onychomycosis: clinic-mycologic study of 130 patients from Himachal Pradesh, India. Indian J Dermatol Venereol Leprol. 2007;73:389-392.
17. Eleweski BE. Diagnostic techniques for confirming onychomycosis. J Am Acad Dermatol. 1996;35(3, pt 2):S6-S9.
18. Das NK, Ghosh P, Das S, et al. A study on the etiological agent and clinico-mycological correlation of fingernail onychomycosis in eastern India. Indian J Dermatol. 2008;53:75-79.
19. Bassiri-Jahromi S, Khaksar AA. Nondermatophytic moulds as a causative agent of onychomycosis in Tehran. Indian J Dermatol. 2010;55:140-143.
20. Bokhari MA, Hussain I, Jahangir M, et al. Onychomycosis in Lahore, Pakistan. Int J Dermatol. 1999;38:591-595.
21. Jesudanam TM, Rao GR, Lakshmi DJ, et al. Onychomycosis: a significant medical problem. Indian J Dermatol Venereol Leprol. 2002;68:326-329.
22. Ahmad M, Gupta S, Gupte S. A clinico-mycological study of onychomycosis. EDOJ. 2010;6:1-9.
23. Vinod S, Grover S, Dash K, et al. A clinico-mycological evaluation of onychomycosis. Indian J Dermatol Venereol Leprol. 2000;66:238-240.
24. Veer P, Patwardhan NS, Damle AS. Study of onychomycosis: prevailing fungi and pattern of infection. Indian J Med Microbiol. 2007;25:53-56.
25. Garg A, Venkatesh V, Singh M, et al. Onychomycosis in central India: a clinicoetiologic correlation. Int J Dermatol. 2004;43:498-502.
26. Adhikari L, Das Gupta A, Pal R, et al. Clinico-etiologic correlates of onychomycosis in Sikkim. Indian J Pathol Microbiol. 2009;52:194-197.
Onychomycosis is a chronic fungal infection of the nails. Dermatophytes are the most common etiologic agents, but yeasts and nondermatophyte molds also constitute a substantial number of cases.1 An accumulation of debris under distorted, deformed, thickened, and discolored nails, particularly with ragged and furrowed edges, strongly suggests tinea unguium.2 Candidal onychomycosis (CO) lacks gross distortion and accumulated detritus and mainly affects fingernails.3 Nondermatophytic molds cause 1.5% to 6% of cases of onychomycosis, mostly seen in toenails of elderly individuals with a history of trauma.4 Onychomycosis affects 5.5% of the world population5 and represents 20% to 40% of all onychopathies and approximately 30% of cutaneous mycotic infections.6
The incidence of onychomycosis ranges from 0.5% to 5% in the general population in India.7 The incidence is particularly high in warm humid climates such as India.8 Researchers have found certain habits of the population in the Indian subcontinent (eg, walking with bare feet, wearing ill-fitting shoes, nail-biting [eg, onychophagia], working with chemicals) to be contributing factors for onychomycosis.9 Several studies have shown that the prevalence of onychomycosis increases with age, possibly due to poor peripheral circulation, diabetes mellitus, repeated nail trauma, prolonged exposure to pathogenic fungi, suboptimal immune function, inactivity, or inability to trim the toenails and care for the feet.10 Nail infection is a cosmetic problem with serious physical and psychological morbidity and also serves as the fungal reservoir for skin infections. Besides destruction and disfigurement of the nail plate, onychomycosis can lead to self-consciousness and impairment of daily functioning.11
Nail dystrophy occurs secondary to various systemic disorders or can be associated with other dermatologic conditions. Nail discoloration and other onychia should be differentiated from onychomycosis by classifying nail lesions as distal lateral subungual onychomycosis, proximal subungual onychomycosis (PSO), CO, white superficial onychomycosis (WSO), and total dystrophic onychomycosis.12 Laboratory investigation is necessary to accurately differentiate between fungal infections and other skin diseases before starting treatment. Our hospital-based study sought to determine the incidence and epidemiology of onychomycosis with an analysis of 134 participants with clinically suspected onychomycosis. We evaluated prevalence based on age, sex, and occupation, as well as the most common pathogens.
Materials and Methods
Study Design and Participants
The study population consisted of 134 patients with clinically suspected onychomycosis who visited the dermatology department at the Veer Chandra Singh Garhwali Government Institute of Medical Sciences and Research Institute in Uttarakhand, India (October 2010 to October 2011). A thorough history was obtained and a detailed examination of the distorted nails was conducted in the microbiology laboratory. Patient history and demographic factors such as age, sex, occupation, and related history of risk factors for onychomycosis were recorded pro forma. Some of the details such as itching, family history of fungal infection, and prior cutaneous infections were recorded. Patients who were undergoing treatment with systemic or topical antifungal agents in the 4 weeks preceding the study period were excluded to rule out false-negative cases and to avoid the influence of antifungal agents on the disease course.
Assessments
Two samples were taken from each patient on different days. Participants were divided into 4 groups based on occupation: farmer, housewife, student, and other (eg, clerk, shopkeeper, painter). Clinical presentation of discoloration, onycholysis, subungual hyperkeratosis, and nail thickening affecting the distal and/or lateral nail plate was defined as distal lateral subungual onychomycosis; discoloration and onycholysis affecting the proximal part of the nail was defined as PSO; association with paronychia and distal and lateral onycholysis was defined as CO; white opaque patches on the nail surface were defined as WSO; and end-stage nail disease was defined as total dystrophic onychomycosis.
Prior to sampling, the nails were cleaned with a 70% alcohol solution. Nail clippings were obtained using presterilized nail clippers and a blunt no. 15 scalpel blade and were placed on sterilized black paper. Each nail sample was divided into 2 parts: one for direct microscopy and one for culture. Nail clippings were subjected to microscopic examination after clearing in 20% potassium hydroxide solution. The slides were examined for fungal hyphae, arthrospores, yeasts, and pseudohyphal forms. Culture was done with Emmons modification of Sabouraud dextrose agar (incubated at 27°C for molds and 37°C for yeasts) as well as with 0.4% chloramphenicol and 5% cycloheximide (incubated at 27°C). Culture tubes were examined daily for the first week and on alternate days thereafter for 4 weeks of incubation.
Dermatophytes were identified based on the colony morphology, growth rate, texture, border, and pigmentation in the obverse and reverse of culture media and microscopic examination using lactophenol cotton blue tease mount. Yeast colonies were identified microscopically with Gram stain, and species were identified by germ tube, carbohydrate assimilation, and fermentation tests.13 Nondermatophyte molds were identified by colony morphology, microscopic examination, and slide culture. Molds were considered as pathogens in the presence of the following criteria: (1) absence of other fungal growth in the same culture tube; (2) presence of mold growth in all 3 samples; and (3) presence of filaments identified on direct examination.
Results
Of 134 clinically suspected cases of onychomycosis, 78 (58.2%) were from fingernails and 56 (41.8%) from toenails. Clinical diagnosis was confirmed in 96 (71.6%) cases by both fungal culture and direct microscopy but was confirmed by direct microscopy alone in only 76 (56.7%) cases. False-negative results were found in 23.9% (32/134) of participants with direct microscopy and 9.0% (12/134) with fungal cultures. The results of direct microscopy and fungal culture are outlined in Table 1. The study included 78 (58.2%) males and 56 (41.8%) females with a mean age of 44 years. Highest prevalence (47.8%) was seen in participants older than 40 years and lowest prevalence (11.9%) in participants younger than 20 years. In total, 32.8% of participants were farmers, 31.3% were housewives, 14.9% were students, and 20.9% performed other occupations. Disease history at the time of first presentation varied from 1 month to more than 2 years; 33.6% of participants had a 1- to 6-month history of disease, while only 3.7% had a disease history of less than 1 month at presentation. The demographic data are further outlined in Table 2.
Distal lateral subungual onychomycosis was the most prevalent clinical pattern found in 66 (49.3%) participants; fungal isolates were found in 60 of these participants. The next most prevalent clinical pattern was PSO, which was found in 34 (25.4%) participants, 12 showing fungal growth. A clinical pattern of CO was noted in 28 (20.9%) participants, 22 showing fungal growth; WSO was noted in 10 (7.5%) participants, 2 showing fungal growth.
Of 96 culture-positive cases, dermatophytes were the most common pathogens isolated in 56 (58.3%) participants, followed by Candida species in 28 (29.2%) participants. Nondermatophyte molds were isolated in 12 (12.5%) participants. The various dermatophytes, Candida species, and nondermatophyte molds that were isolated on fungal culture are outlined in Table 3. Of the 96 participants with positive fungal cultures, 30 (31.2%) were farmers working with soil, 28 (29.2%) were housewives associated with wet work, 16 (16.7%) were students associated with increased physical exercise from extracurricular activity, and 22 (22.9%) were in other occupations (Table 4).
Comment
The term onychomycosis is derived from onyx, the Greek word for nail, and mykes, the Greek word for fungus. Onychomycosis is a chronic mycotic infection of the fingernails and toenails that can have a serious impact on patients’ quality of life. The fungi known to cause onychomycosis vary among geographic areas, primarily due to differences in climate.14 The isolation rate of onychomycosis in our hospital-based study was 71.6%, which is in accordance with various studies in India and abroad, including 60% in Karnataka, India5; 82.3% in Sikkim, India6; and 86.9% in Turkey.1 However, other studies have shown lower isolation rates of 39.5% in Central Delhi, India,15 and 37.6% in Himachal Pradesh, India.16 Some patients with onychomycosis may not seek medical attention, which may explain the difference in the prevalence of onychomycosis observed worldwide.17 The prevalence of onychomycosis by age also varies. In our study, participants older than 40 years showed the highest prevalence (47.8%), which is in accordance with other studies from India18 and abroad.19,20 In contrast, some Indian studies15,21,22 have reported a higher prevalence in younger adults (ie, 21–30 years), which may be attributed to greater self-consciousness about nail discoloration and disfigurement as well as increased physical activity and different shoe-wearing habits. A higher prevalence in older adults, as observed in our study as well some other studies,19,21 may be due to poor peripheral circulation, diabetes mellitus, repeated nail trauma, longer exposure to pathogenic fungi, suboptimal immune function, inactivity, and poor hygiene.10
In our study, suspected onychomycosis was more common in males (58.2%) than in females (41.8%). These results are in accordance with many of the studies in the worldwide literature.1,10,11,15,16,23-25 A higher isolation rate in males worldwide may be due to common use of occlusive footwear, more exposure to outdoor conditions, and increased physical activity, leading to an increased likelihood of trauma. The importance of trauma to the nails as a predisposing factor for onychomycosis is well established.24 In our study, the majority of males wore shoes regardless of occupation. Perspiration of the feet when wearing socks and/or shoes can generate a warm moist environment that promotes the growth of fungi and predisposes patients to onychomycosis. Similar observations have been reported by other investigators.21,22,25,26
The incidence of onychomycosis was almost evenly distributed among farmers, housewives, and the miscellaneous group, whereas a high isolation rate was noted among students. Of 20 students included in our study, onychomycosis was confirmed in 16, which may be related to an increased use of synthetic sports shoes and socks that retain sweat as well as vigorous physical activity frequently resulting in nail injuries among this patient population.11 Younger patients may be more conscious of their appearance and therefore may be more likely to seek treatment. Similar observations have been reported by other researchers.15,21,22
In our study, dermatophytes were the most commonly found pathogens (58.3%), which is comparable to other studies.15,18,22Trichophyton mentagrophytes was the most frequently isolated dermatophyte from cultures, which was in concordance with a study from Delhi.15 In some studies,18,20,22Trichophyton rubrum has been reported as the most prevalent dermatophyte, but we identified Trichophyton rubrum in only 18 participants, which can be attributed to variations in epidemiology based on geographic region. Nondermatophyte molds were isolated in 12.5% of participants, with Aspergillus niger being the most common isolate found in 8 cases. Other isolated species were Alternaria alternata and Fusarium solani found in 2 cases each. Aspergillus niger has been reported in worldwide studies as an important cause of onychomycosis.15,18,19,21,22
In 28 cases (29.2%) involving Candida species, Candida albicans, Candida parapsilosis, and Candida tropicalis were the most common pathogens, respectively, which is in accordance with many studies.15,20-22,25 In 28 cases of CO, females (n=16) were affected more than males (n=12). All of the females were housewives and C albicans was predominantly isolated from the fingernails. Household responsibilities involving kitchen work (eg, cutting and peeling vegetables, washing utensils, cleaning the house/laundry) may chronically expose housewives to moist environments and make them more prone to injury, thus facilitating easy entry of fungal agents.
Distal lateral subungual onychomycosis was the most prevalent clinical type found (n=66), which is comparable to other reports.20,22,25 Proximal subungual onychomycosis was the second most common type; however, a greater incidence has been reported by some researchers,23,24 while others have reported a lower incidence.20,21 Candidial onychomycosis and WSO were not common in our study, and PSO was not associated with any immunodeficiency disease, as reported by other researchers.15,20
Of 134 suspected cases of onychomycosis, 71.6% were confirmed by both direct microscopy and fungal culture, but only 56.7% were confirmed by direct microscopy alone. If we had relied on microscopy with potassium hydroxide only, we would have missed 23.9% of cases. Therefore, nail scrapings should always be subjected to fungal culture as well as direct microscopy, as both are necessary for accurate diagnosis and treatment of onychomycosis. If onychomycosis is not successfully treated, it can act as a reservoir of fungal infection affecting other parts of the body with the potential to pass infection on to others.
Conclusion
Clinical examination alone is not sufficient for diagnosing onychomycosis14,18,20; in many cases of suspected onychomycosis with nail changes, mycologic examination does not confirm fungal infection. In our study, only 71.6% of participants with nail changes proved to be of fungal etiology. Other researchers from different geographic locations have reported similar results with lower incidence (eg, 39.5%,15 37.6%,16 51.7%,18 45.3%21) of fungal etiology in such cases. Therefore, both clinical and mycologic examinations are important for establishing the diagnosis and selecting the most suitable antifungal agent, which is possible only if the underlying pathogen is correctly identified.
Onychomycosis is a chronic fungal infection of the nails. Dermatophytes are the most common etiologic agents, but yeasts and nondermatophyte molds also constitute a substantial number of cases.1 An accumulation of debris under distorted, deformed, thickened, and discolored nails, particularly with ragged and furrowed edges, strongly suggests tinea unguium.2 Candidal onychomycosis (CO) lacks gross distortion and accumulated detritus and mainly affects fingernails.3 Nondermatophytic molds cause 1.5% to 6% of cases of onychomycosis, mostly seen in toenails of elderly individuals with a history of trauma.4 Onychomycosis affects 5.5% of the world population5 and represents 20% to 40% of all onychopathies and approximately 30% of cutaneous mycotic infections.6
The incidence of onychomycosis ranges from 0.5% to 5% in the general population in India.7 The incidence is particularly high in warm humid climates such as India.8 Researchers have found certain habits of the population in the Indian subcontinent (eg, walking with bare feet, wearing ill-fitting shoes, nail-biting [eg, onychophagia], working with chemicals) to be contributing factors for onychomycosis.9 Several studies have shown that the prevalence of onychomycosis increases with age, possibly due to poor peripheral circulation, diabetes mellitus, repeated nail trauma, prolonged exposure to pathogenic fungi, suboptimal immune function, inactivity, or inability to trim the toenails and care for the feet.10 Nail infection is a cosmetic problem with serious physical and psychological morbidity and also serves as the fungal reservoir for skin infections. Besides destruction and disfigurement of the nail plate, onychomycosis can lead to self-consciousness and impairment of daily functioning.11
Nail dystrophy occurs secondary to various systemic disorders or can be associated with other dermatologic conditions. Nail discoloration and other onychia should be differentiated from onychomycosis by classifying nail lesions as distal lateral subungual onychomycosis, proximal subungual onychomycosis (PSO), CO, white superficial onychomycosis (WSO), and total dystrophic onychomycosis.12 Laboratory investigation is necessary to accurately differentiate between fungal infections and other skin diseases before starting treatment. Our hospital-based study sought to determine the incidence and epidemiology of onychomycosis with an analysis of 134 participants with clinically suspected onychomycosis. We evaluated prevalence based on age, sex, and occupation, as well as the most common pathogens.
Materials and Methods
Study Design and Participants
The study population consisted of 134 patients with clinically suspected onychomycosis who visited the dermatology department at the Veer Chandra Singh Garhwali Government Institute of Medical Sciences and Research Institute in Uttarakhand, India (October 2010 to October 2011). A thorough history was obtained and a detailed examination of the distorted nails was conducted in the microbiology laboratory. Patient history and demographic factors such as age, sex, occupation, and related history of risk factors for onychomycosis were recorded pro forma. Some of the details such as itching, family history of fungal infection, and prior cutaneous infections were recorded. Patients who were undergoing treatment with systemic or topical antifungal agents in the 4 weeks preceding the study period were excluded to rule out false-negative cases and to avoid the influence of antifungal agents on the disease course.
Assessments
Two samples were taken from each patient on different days. Participants were divided into 4 groups based on occupation: farmer, housewife, student, and other (eg, clerk, shopkeeper, painter). Clinical presentation of discoloration, onycholysis, subungual hyperkeratosis, and nail thickening affecting the distal and/or lateral nail plate was defined as distal lateral subungual onychomycosis; discoloration and onycholysis affecting the proximal part of the nail was defined as PSO; association with paronychia and distal and lateral onycholysis was defined as CO; white opaque patches on the nail surface were defined as WSO; and end-stage nail disease was defined as total dystrophic onychomycosis.
Prior to sampling, the nails were cleaned with a 70% alcohol solution. Nail clippings were obtained using presterilized nail clippers and a blunt no. 15 scalpel blade and were placed on sterilized black paper. Each nail sample was divided into 2 parts: one for direct microscopy and one for culture. Nail clippings were subjected to microscopic examination after clearing in 20% potassium hydroxide solution. The slides were examined for fungal hyphae, arthrospores, yeasts, and pseudohyphal forms. Culture was done with Emmons modification of Sabouraud dextrose agar (incubated at 27°C for molds and 37°C for yeasts) as well as with 0.4% chloramphenicol and 5% cycloheximide (incubated at 27°C). Culture tubes were examined daily for the first week and on alternate days thereafter for 4 weeks of incubation.
Dermatophytes were identified based on the colony morphology, growth rate, texture, border, and pigmentation in the obverse and reverse of culture media and microscopic examination using lactophenol cotton blue tease mount. Yeast colonies were identified microscopically with Gram stain, and species were identified by germ tube, carbohydrate assimilation, and fermentation tests.13 Nondermatophyte molds were identified by colony morphology, microscopic examination, and slide culture. Molds were considered as pathogens in the presence of the following criteria: (1) absence of other fungal growth in the same culture tube; (2) presence of mold growth in all 3 samples; and (3) presence of filaments identified on direct examination.
Results
Of 134 clinically suspected cases of onychomycosis, 78 (58.2%) were from fingernails and 56 (41.8%) from toenails. Clinical diagnosis was confirmed in 96 (71.6%) cases by both fungal culture and direct microscopy but was confirmed by direct microscopy alone in only 76 (56.7%) cases. False-negative results were found in 23.9% (32/134) of participants with direct microscopy and 9.0% (12/134) with fungal cultures. The results of direct microscopy and fungal culture are outlined in Table 1. The study included 78 (58.2%) males and 56 (41.8%) females with a mean age of 44 years. Highest prevalence (47.8%) was seen in participants older than 40 years and lowest prevalence (11.9%) in participants younger than 20 years. In total, 32.8% of participants were farmers, 31.3% were housewives, 14.9% were students, and 20.9% performed other occupations. Disease history at the time of first presentation varied from 1 month to more than 2 years; 33.6% of participants had a 1- to 6-month history of disease, while only 3.7% had a disease history of less than 1 month at presentation. The demographic data are further outlined in Table 2.
Distal lateral subungual onychomycosis was the most prevalent clinical pattern found in 66 (49.3%) participants; fungal isolates were found in 60 of these participants. The next most prevalent clinical pattern was PSO, which was found in 34 (25.4%) participants, 12 showing fungal growth. A clinical pattern of CO was noted in 28 (20.9%) participants, 22 showing fungal growth; WSO was noted in 10 (7.5%) participants, 2 showing fungal growth.
Of 96 culture-positive cases, dermatophytes were the most common pathogens isolated in 56 (58.3%) participants, followed by Candida species in 28 (29.2%) participants. Nondermatophyte molds were isolated in 12 (12.5%) participants. The various dermatophytes, Candida species, and nondermatophyte molds that were isolated on fungal culture are outlined in Table 3. Of the 96 participants with positive fungal cultures, 30 (31.2%) were farmers working with soil, 28 (29.2%) were housewives associated with wet work, 16 (16.7%) were students associated with increased physical exercise from extracurricular activity, and 22 (22.9%) were in other occupations (Table 4).
Comment
The term onychomycosis is derived from onyx, the Greek word for nail, and mykes, the Greek word for fungus. Onychomycosis is a chronic mycotic infection of the fingernails and toenails that can have a serious impact on patients’ quality of life. The fungi known to cause onychomycosis vary among geographic areas, primarily due to differences in climate.14 The isolation rate of onychomycosis in our hospital-based study was 71.6%, which is in accordance with various studies in India and abroad, including 60% in Karnataka, India5; 82.3% in Sikkim, India6; and 86.9% in Turkey.1 However, other studies have shown lower isolation rates of 39.5% in Central Delhi, India,15 and 37.6% in Himachal Pradesh, India.16 Some patients with onychomycosis may not seek medical attention, which may explain the difference in the prevalence of onychomycosis observed worldwide.17 The prevalence of onychomycosis by age also varies. In our study, participants older than 40 years showed the highest prevalence (47.8%), which is in accordance with other studies from India18 and abroad.19,20 In contrast, some Indian studies15,21,22 have reported a higher prevalence in younger adults (ie, 21–30 years), which may be attributed to greater self-consciousness about nail discoloration and disfigurement as well as increased physical activity and different shoe-wearing habits. A higher prevalence in older adults, as observed in our study as well some other studies,19,21 may be due to poor peripheral circulation, diabetes mellitus, repeated nail trauma, longer exposure to pathogenic fungi, suboptimal immune function, inactivity, and poor hygiene.10
In our study, suspected onychomycosis was more common in males (58.2%) than in females (41.8%). These results are in accordance with many of the studies in the worldwide literature.1,10,11,15,16,23-25 A higher isolation rate in males worldwide may be due to common use of occlusive footwear, more exposure to outdoor conditions, and increased physical activity, leading to an increased likelihood of trauma. The importance of trauma to the nails as a predisposing factor for onychomycosis is well established.24 In our study, the majority of males wore shoes regardless of occupation. Perspiration of the feet when wearing socks and/or shoes can generate a warm moist environment that promotes the growth of fungi and predisposes patients to onychomycosis. Similar observations have been reported by other investigators.21,22,25,26
The incidence of onychomycosis was almost evenly distributed among farmers, housewives, and the miscellaneous group, whereas a high isolation rate was noted among students. Of 20 students included in our study, onychomycosis was confirmed in 16, which may be related to an increased use of synthetic sports shoes and socks that retain sweat as well as vigorous physical activity frequently resulting in nail injuries among this patient population.11 Younger patients may be more conscious of their appearance and therefore may be more likely to seek treatment. Similar observations have been reported by other researchers.15,21,22
In our study, dermatophytes were the most commonly found pathogens (58.3%), which is comparable to other studies.15,18,22Trichophyton mentagrophytes was the most frequently isolated dermatophyte from cultures, which was in concordance with a study from Delhi.15 In some studies,18,20,22Trichophyton rubrum has been reported as the most prevalent dermatophyte, but we identified Trichophyton rubrum in only 18 participants, which can be attributed to variations in epidemiology based on geographic region. Nondermatophyte molds were isolated in 12.5% of participants, with Aspergillus niger being the most common isolate found in 8 cases. Other isolated species were Alternaria alternata and Fusarium solani found in 2 cases each. Aspergillus niger has been reported in worldwide studies as an important cause of onychomycosis.15,18,19,21,22
In 28 cases (29.2%) involving Candida species, Candida albicans, Candida parapsilosis, and Candida tropicalis were the most common pathogens, respectively, which is in accordance with many studies.15,20-22,25 In 28 cases of CO, females (n=16) were affected more than males (n=12). All of the females were housewives and C albicans was predominantly isolated from the fingernails. Household responsibilities involving kitchen work (eg, cutting and peeling vegetables, washing utensils, cleaning the house/laundry) may chronically expose housewives to moist environments and make them more prone to injury, thus facilitating easy entry of fungal agents.
Distal lateral subungual onychomycosis was the most prevalent clinical type found (n=66), which is comparable to other reports.20,22,25 Proximal subungual onychomycosis was the second most common type; however, a greater incidence has been reported by some researchers,23,24 while others have reported a lower incidence.20,21 Candidial onychomycosis and WSO were not common in our study, and PSO was not associated with any immunodeficiency disease, as reported by other researchers.15,20
Of 134 suspected cases of onychomycosis, 71.6% were confirmed by both direct microscopy and fungal culture, but only 56.7% were confirmed by direct microscopy alone. If we had relied on microscopy with potassium hydroxide only, we would have missed 23.9% of cases. Therefore, nail scrapings should always be subjected to fungal culture as well as direct microscopy, as both are necessary for accurate diagnosis and treatment of onychomycosis. If onychomycosis is not successfully treated, it can act as a reservoir of fungal infection affecting other parts of the body with the potential to pass infection on to others.
Conclusion
Clinical examination alone is not sufficient for diagnosing onychomycosis14,18,20; in many cases of suspected onychomycosis with nail changes, mycologic examination does not confirm fungal infection. In our study, only 71.6% of participants with nail changes proved to be of fungal etiology. Other researchers from different geographic locations have reported similar results with lower incidence (eg, 39.5%,15 37.6%,16 51.7%,18 45.3%21) of fungal etiology in such cases. Therefore, both clinical and mycologic examinations are important for establishing the diagnosis and selecting the most suitable antifungal agent, which is possible only if the underlying pathogen is correctly identified.
1. Yenişehirli G, Bulut Y, Sezer E, et al. Onychomycosis infections in the Middle Black Sea Region, Turkey. Int J Dermatol. 2009;48:956-959.
2. Kouskoukis CE, Scher RK, Ackerman AB. What histologic finding distinguishes onychomycosis and psoriasis? Am J Dermatopathol. 1983;5:501-503.
3. Rippon JW. Medical mycology. In: Wonsiewicz M, ed. The Pathogenic Fungi and the Pathogenic Actinomycetes. 3rd ed. Philadelphia, PA: WB Saunders; 1988:169-275.
4. Greer DL. Evolving role of nondermatophytes in onychomycosis. Int J Dermatol. 1995;34:521-524.
5. Murray SC, Dawber RP. Onychomycosis of toenails: orthopaedic and podiatric considerations. Australas J Dermatol. 2002;43:105-112.
6. Achten G, Wanet-Rouard J. Onychomycoses in the laboratory. Mykosen Suppl. 1978;1:125-127.
7. Sobhanadri C, Rao DT, Babu KS. Clinical and mycological study of superficial fungal infections at Government General Hospital: guntur and their response to treatment with hamycin, dermostatin and dermamycin. Indian J Dermatol Venereol. 1970;36:209-214.
8. Jain S, Sehgal VN. Commentary: onychomycosis: an epidemio-etiologic perspective. Int J Dermatol. 2000;39:100-103.
9. Sehgal VN, Aggarwal AK, Srivastava G, et al. Onychomycosis: a 3 year clinicomycologic hospital-based study. Skinmed. 2007;6:11-17.
10. Elewski BE, Charif MA. Prevalence of onychomycosis in patients attending a dermatology clinic in northeastern Ohio for the other conditions. Arch Dermatol. 1997;133:1172-1173.
11. Scher RK. Onychomycosis is more than a cosmetic problem. Br J Dermatol. 1994;130(suppl 43):S15.
12. Godoy-Martinez PG, Nunes FG, Tomimori-Yamashita J, et al. Onychomycosis in São Paulo, Brazil [published online ahead of print May 8, 2009]. Mycopathologia. 2009;168:111-116.
13. Larone DH. Medically Important Fungi: A Guide to Identification. 4th ed. Washington, DC: American Society for Microbiology Press; 2002.
14. Sehgal VN, Srivastava G, Dogra S, et al. Onychomycosis: an Asian perspective. Skinmed. 2010;8:37-45.
15. Sanjiv A, Shalini M, Charoo H. Etiological agents of onychomycosis from a tertiary care hospital in Central Delhi, India. Indian J Fund Appl Life Science. 2011;1:11-14.
16. Gupta M, Sharma NL, Kanga AK, et al. Onychomycosis: clinic-mycologic study of 130 patients from Himachal Pradesh, India. Indian J Dermatol Venereol Leprol. 2007;73:389-392.
17. Eleweski BE. Diagnostic techniques for confirming onychomycosis. J Am Acad Dermatol. 1996;35(3, pt 2):S6-S9.
18. Das NK, Ghosh P, Das S, et al. A study on the etiological agent and clinico-mycological correlation of fingernail onychomycosis in eastern India. Indian J Dermatol. 2008;53:75-79.
19. Bassiri-Jahromi S, Khaksar AA. Nondermatophytic moulds as a causative agent of onychomycosis in Tehran. Indian J Dermatol. 2010;55:140-143.
20. Bokhari MA, Hussain I, Jahangir M, et al. Onychomycosis in Lahore, Pakistan. Int J Dermatol. 1999;38:591-595.
21. Jesudanam TM, Rao GR, Lakshmi DJ, et al. Onychomycosis: a significant medical problem. Indian J Dermatol Venereol Leprol. 2002;68:326-329.
22. Ahmad M, Gupta S, Gupte S. A clinico-mycological study of onychomycosis. EDOJ. 2010;6:1-9.
23. Vinod S, Grover S, Dash K, et al. A clinico-mycological evaluation of onychomycosis. Indian J Dermatol Venereol Leprol. 2000;66:238-240.
24. Veer P, Patwardhan NS, Damle AS. Study of onychomycosis: prevailing fungi and pattern of infection. Indian J Med Microbiol. 2007;25:53-56.
25. Garg A, Venkatesh V, Singh M, et al. Onychomycosis in central India: a clinicoetiologic correlation. Int J Dermatol. 2004;43:498-502.
26. Adhikari L, Das Gupta A, Pal R, et al. Clinico-etiologic correlates of onychomycosis in Sikkim. Indian J Pathol Microbiol. 2009;52:194-197.
1. Yenişehirli G, Bulut Y, Sezer E, et al. Onychomycosis infections in the Middle Black Sea Region, Turkey. Int J Dermatol. 2009;48:956-959.
2. Kouskoukis CE, Scher RK, Ackerman AB. What histologic finding distinguishes onychomycosis and psoriasis? Am J Dermatopathol. 1983;5:501-503.
3. Rippon JW. Medical mycology. In: Wonsiewicz M, ed. The Pathogenic Fungi and the Pathogenic Actinomycetes. 3rd ed. Philadelphia, PA: WB Saunders; 1988:169-275.
4. Greer DL. Evolving role of nondermatophytes in onychomycosis. Int J Dermatol. 1995;34:521-524.
5. Murray SC, Dawber RP. Onychomycosis of toenails: orthopaedic and podiatric considerations. Australas J Dermatol. 2002;43:105-112.
6. Achten G, Wanet-Rouard J. Onychomycoses in the laboratory. Mykosen Suppl. 1978;1:125-127.
7. Sobhanadri C, Rao DT, Babu KS. Clinical and mycological study of superficial fungal infections at Government General Hospital: guntur and their response to treatment with hamycin, dermostatin and dermamycin. Indian J Dermatol Venereol. 1970;36:209-214.
8. Jain S, Sehgal VN. Commentary: onychomycosis: an epidemio-etiologic perspective. Int J Dermatol. 2000;39:100-103.
9. Sehgal VN, Aggarwal AK, Srivastava G, et al. Onychomycosis: a 3 year clinicomycologic hospital-based study. Skinmed. 2007;6:11-17.
10. Elewski BE, Charif MA. Prevalence of onychomycosis in patients attending a dermatology clinic in northeastern Ohio for the other conditions. Arch Dermatol. 1997;133:1172-1173.
11. Scher RK. Onychomycosis is more than a cosmetic problem. Br J Dermatol. 1994;130(suppl 43):S15.
12. Godoy-Martinez PG, Nunes FG, Tomimori-Yamashita J, et al. Onychomycosis in São Paulo, Brazil [published online ahead of print May 8, 2009]. Mycopathologia. 2009;168:111-116.
13. Larone DH. Medically Important Fungi: A Guide to Identification. 4th ed. Washington, DC: American Society for Microbiology Press; 2002.
14. Sehgal VN, Srivastava G, Dogra S, et al. Onychomycosis: an Asian perspective. Skinmed. 2010;8:37-45.
15. Sanjiv A, Shalini M, Charoo H. Etiological agents of onychomycosis from a tertiary care hospital in Central Delhi, India. Indian J Fund Appl Life Science. 2011;1:11-14.
16. Gupta M, Sharma NL, Kanga AK, et al. Onychomycosis: clinic-mycologic study of 130 patients from Himachal Pradesh, India. Indian J Dermatol Venereol Leprol. 2007;73:389-392.
17. Eleweski BE. Diagnostic techniques for confirming onychomycosis. J Am Acad Dermatol. 1996;35(3, pt 2):S6-S9.
18. Das NK, Ghosh P, Das S, et al. A study on the etiological agent and clinico-mycological correlation of fingernail onychomycosis in eastern India. Indian J Dermatol. 2008;53:75-79.
19. Bassiri-Jahromi S, Khaksar AA. Nondermatophytic moulds as a causative agent of onychomycosis in Tehran. Indian J Dermatol. 2010;55:140-143.
20. Bokhari MA, Hussain I, Jahangir M, et al. Onychomycosis in Lahore, Pakistan. Int J Dermatol. 1999;38:591-595.
21. Jesudanam TM, Rao GR, Lakshmi DJ, et al. Onychomycosis: a significant medical problem. Indian J Dermatol Venereol Leprol. 2002;68:326-329.
22. Ahmad M, Gupta S, Gupte S. A clinico-mycological study of onychomycosis. EDOJ. 2010;6:1-9.
23. Vinod S, Grover S, Dash K, et al. A clinico-mycological evaluation of onychomycosis. Indian J Dermatol Venereol Leprol. 2000;66:238-240.
24. Veer P, Patwardhan NS, Damle AS. Study of onychomycosis: prevailing fungi and pattern of infection. Indian J Med Microbiol. 2007;25:53-56.
25. Garg A, Venkatesh V, Singh M, et al. Onychomycosis in central India: a clinicoetiologic correlation. Int J Dermatol. 2004;43:498-502.
26. Adhikari L, Das Gupta A, Pal R, et al. Clinico-etiologic correlates of onychomycosis in Sikkim. Indian J Pathol Microbiol. 2009;52:194-197.
Practice Points
- Onychomycosis is a chronic fungal infection of the nails and represents 20% to 40% of all onycho-pathies worldwide.
- Apart from dermatophytes as etiologic agents, nondermatophyte molds and yeasts also can contribute to the disease.
- Categorization of onychomycosis clinically as well as mycologically will surely ensure better patient care.
- Avoiding certain habits (eg, walking with bare feet, wearing ill-fitting shoes, onychophagia) can decrease disease incidence.
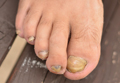
Cutaneous Metastasis of Gastric Adenocarcinoma at the Site of a Traumatic Ecchymosis
To the Editor:
In a recent Cutis® article, Cesaretti et al1 reported a case of cutaneous metastasis from primary gastric cancer that appeared on a resection scar 6 years after remission and without any relapse of the primary tumor. We report a case of a 68-year-old man who was referred to the dermatology clinic with a 15×20-cm nonpruritic, nonscaly, bruiselike lesion on the right forearm of 1 month’s duration. Approximately 1.5 years prior to presentation, the patient was diagnosed with gastric adenocarcinoma (stage IV: T4N3M1) with hepatic and lung metastasis. Following 6 months of chemotherapy with cisplatin and 5-fluorouracil, a positron emission tomography–computed tomography scan was performed and showed a reduction in metastasis but growth of the primitive tumor. After 1 year of chemotherapy, the new positron emission tomography–computed tomography scan showed no metastases. However, the primitive tumor had increased in size.
One month prior to presentation to the dermatology department, a traumatic blood sample on the right forearm left the patient with a persistent ecchymosis. The lesion was thought to be a healing ecchymosis and no biopsy was performed. One month later, the skin lesion had become much thicker and more erythematous (Figure) but not larger. A skin biopsy of this well-defined plaque was performed. Histologic examination showed neovascularization, proliferative epithelial cells, and cytokeratin markers AE1/AE3 and CK20, leading to the diagnosis of skin metastasis of the gastric adenocarcinoma. Chemotherapy was discontinued because of the patient’s altered general status and palliative care was given until he died the following month (2 months after presentation).
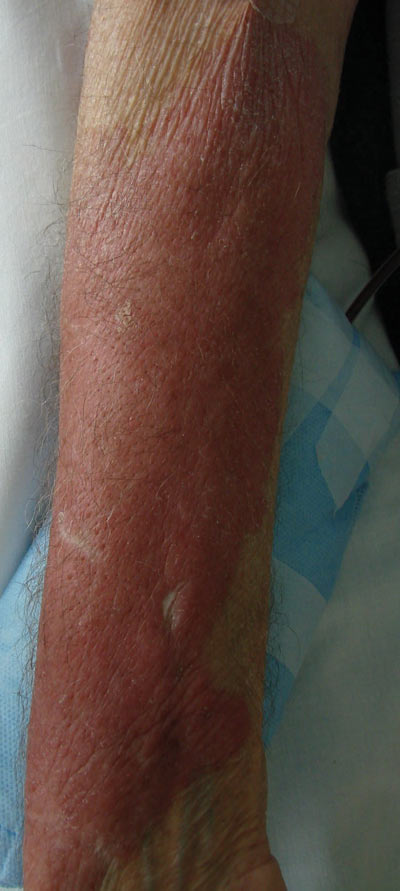
Iatrogenic dissemination of cancer cells has been described often on scars of tumor surgery,2 and in malignant melanoma, bruises and hematoma revealing preexisting metastases have been reported.3,4 Our report of secondary metastasis on an ecchymosis suggests that the traumatic blood sample performed before the development of the metastasis caused circulating tumor cells in the skin, which led to their local proliferation. The skin metastasis was the first sign of relapse and was followed by alteration of the general status and death.
Our patient is an example of the “soil and seed”5 hypothesis. Our case illustrates the abilities of tumor cells to colonize the skin under favorable conditions and emphasizes the importance of minimizing bleeding events and iatrogenic seeding of internal neoplasms in daily practice.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:e9-e13.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
- Pham-ledard A, Taieb A, Vergier B, et al. Metastatic cutaneous hematoma variant from melanoma: five cases. Bull Cancer. 2011;98:108-112.
- Connolly CM, Soldin M, Dawson A, et al. Metastatic malignant melanoma presenting with a bruise. Br J Plast Surg. 2003;56:725.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101.
To the Editor:
In a recent Cutis® article, Cesaretti et al1 reported a case of cutaneous metastasis from primary gastric cancer that appeared on a resection scar 6 years after remission and without any relapse of the primary tumor. We report a case of a 68-year-old man who was referred to the dermatology clinic with a 15×20-cm nonpruritic, nonscaly, bruiselike lesion on the right forearm of 1 month’s duration. Approximately 1.5 years prior to presentation, the patient was diagnosed with gastric adenocarcinoma (stage IV: T4N3M1) with hepatic and lung metastasis. Following 6 months of chemotherapy with cisplatin and 5-fluorouracil, a positron emission tomography–computed tomography scan was performed and showed a reduction in metastasis but growth of the primitive tumor. After 1 year of chemotherapy, the new positron emission tomography–computed tomography scan showed no metastases. However, the primitive tumor had increased in size.
One month prior to presentation to the dermatology department, a traumatic blood sample on the right forearm left the patient with a persistent ecchymosis. The lesion was thought to be a healing ecchymosis and no biopsy was performed. One month later, the skin lesion had become much thicker and more erythematous (Figure) but not larger. A skin biopsy of this well-defined plaque was performed. Histologic examination showed neovascularization, proliferative epithelial cells, and cytokeratin markers AE1/AE3 and CK20, leading to the diagnosis of skin metastasis of the gastric adenocarcinoma. Chemotherapy was discontinued because of the patient’s altered general status and palliative care was given until he died the following month (2 months after presentation).
Iatrogenic dissemination of cancer cells has been described often on scars of tumor surgery,2 and in malignant melanoma, bruises and hematoma revealing preexisting metastases have been reported.3,4 Our report of secondary metastasis on an ecchymosis suggests that the traumatic blood sample performed before the development of the metastasis caused circulating tumor cells in the skin, which led to their local proliferation. The skin metastasis was the first sign of relapse and was followed by alteration of the general status and death.
Our patient is an example of the “soil and seed”5 hypothesis. Our case illustrates the abilities of tumor cells to colonize the skin under favorable conditions and emphasizes the importance of minimizing bleeding events and iatrogenic seeding of internal neoplasms in daily practice.
To the Editor:
In a recent Cutis® article, Cesaretti et al1 reported a case of cutaneous metastasis from primary gastric cancer that appeared on a resection scar 6 years after remission and without any relapse of the primary tumor. We report a case of a 68-year-old man who was referred to the dermatology clinic with a 15×20-cm nonpruritic, nonscaly, bruiselike lesion on the right forearm of 1 month’s duration. Approximately 1.5 years prior to presentation, the patient was diagnosed with gastric adenocarcinoma (stage IV: T4N3M1) with hepatic and lung metastasis. Following 6 months of chemotherapy with cisplatin and 5-fluorouracil, a positron emission tomography–computed tomography scan was performed and showed a reduction in metastasis but growth of the primitive tumor. After 1 year of chemotherapy, the new positron emission tomography–computed tomography scan showed no metastases. However, the primitive tumor had increased in size.
One month prior to presentation to the dermatology department, a traumatic blood sample on the right forearm left the patient with a persistent ecchymosis. The lesion was thought to be a healing ecchymosis and no biopsy was performed. One month later, the skin lesion had become much thicker and more erythematous (Figure) but not larger. A skin biopsy of this well-defined plaque was performed. Histologic examination showed neovascularization, proliferative epithelial cells, and cytokeratin markers AE1/AE3 and CK20, leading to the diagnosis of skin metastasis of the gastric adenocarcinoma. Chemotherapy was discontinued because of the patient’s altered general status and palliative care was given until he died the following month (2 months after presentation).
Iatrogenic dissemination of cancer cells has been described often on scars of tumor surgery,2 and in malignant melanoma, bruises and hematoma revealing preexisting metastases have been reported.3,4 Our report of secondary metastasis on an ecchymosis suggests that the traumatic blood sample performed before the development of the metastasis caused circulating tumor cells in the skin, which led to their local proliferation. The skin metastasis was the first sign of relapse and was followed by alteration of the general status and death.
Our patient is an example of the “soil and seed”5 hypothesis. Our case illustrates the abilities of tumor cells to colonize the skin under favorable conditions and emphasizes the importance of minimizing bleeding events and iatrogenic seeding of internal neoplasms in daily practice.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:e9-e13.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
- Pham-ledard A, Taieb A, Vergier B, et al. Metastatic cutaneous hematoma variant from melanoma: five cases. Bull Cancer. 2011;98:108-112.
- Connolly CM, Soldin M, Dawson A, et al. Metastatic malignant melanoma presenting with a bruise. Br J Plast Surg. 2003;56:725.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:e9-e13.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
- Pham-ledard A, Taieb A, Vergier B, et al. Metastatic cutaneous hematoma variant from melanoma: five cases. Bull Cancer. 2011;98:108-112.
- Connolly CM, Soldin M, Dawson A, et al. Metastatic malignant melanoma presenting with a bruise. Br J Plast Surg. 2003;56:725.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101.
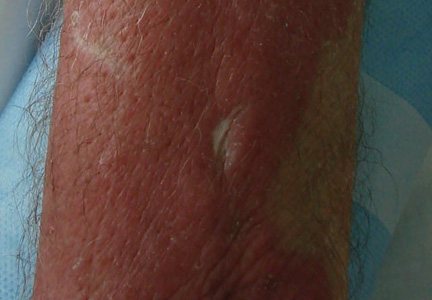
Paclitaxel-Associated Melanonychia
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
 |  | |
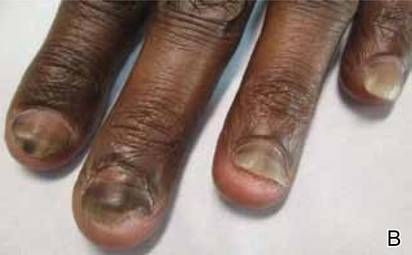 | 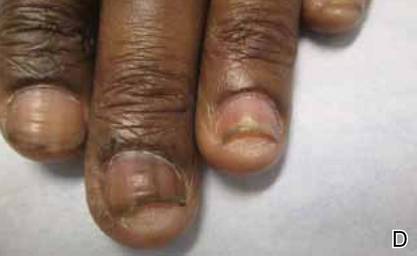 |
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
| | |
| |
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
| | |
| |
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
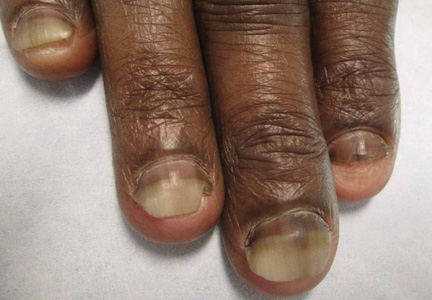
Verrucous Kaposi Sarcoma in an HIV-Positive Man
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
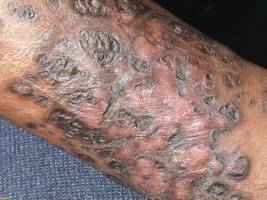
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
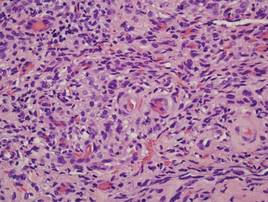
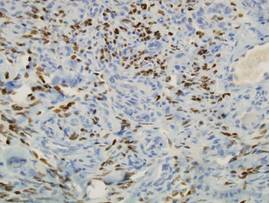
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
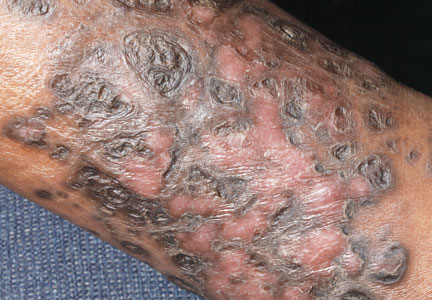
Aspergillus nidulans Causing Primary Cutaneous Aspergillosis in an Immunocompetent Patient
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.

The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.