User login
Mitchel is a reporter for MDedge based in the Philadelphia area. He started with the company in 1992, when it was International Medical News Group (IMNG), and has since covered a range of medical specialties. Mitchel trained as a virologist at Roswell Park Memorial Institute in Buffalo, and then worked briefly as a researcher at Boston Children's Hospital before pivoting to journalism as a AAAS Mass Media Fellow in 1980. His first reporting job was with Science Digest magazine, and from the mid-1980s to early-1990s he was a reporter with Medical World News. @mitchelzoler
AMPLIFY: Apixaban beat warfarin on safety in acute VTE
AMSTERDAM – In patients with acute venous thromboembolism, 6 months of treatment with the oral-anticoagulant apixaban was as effective as was standard therapy with subcutaneous enoxaparin for a week followed by oral warfarin, and apixaban caused significantly fewer major bleeding complications in a randomized, multicenter trial with more than 5,000 patients.
But in addition to apixaban’s sterling individual performance in this pivotal trial, which seems to clear the way for the drug to eventually receive a labeled indication for acute venous thromboembolism, the results also appeared to further anoint the new, oral anticoagulant roster of dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) as the thrombotic-disease troika to be reckoned with, the newcomers whose time has come.

Ever since rivaroxaban became the first of the trio to gain acute VTE labeling, last November, physicians who manage patients with acute VTE had to wrestle with the question of how to integrate this option into their practices. The new findings on apixaban suggest that physicians will soon have to think about deciding between rivaroxaban and apixaban for this indication. And since recent results from other major trials also established dabigatran as the equal of warfarin for efficacy when treating acute VTE patients and with superior safety, dabigatran’s entry into acute VTE management seems imminent (N. Engl. J. Med. 2013;368:709-18).
Propelling this new anticoagulant era are the indications of efficacy that’s equivalent with heparin, but safer, and with far easier drug delivery as the need for anticoagulation clinics and regular measurement of international normalized ratio (INR) is eliminated by all three new drugs.
"An oral regimen without laboratory monitoring will simplify therapy," Dr. Giancarlo Agnelli noted when he presented the new apixaban findings at the congress of the International Society on Thrombosis and Haemostasis. Concurrently with his report at the meeting, the results were published online (N. Engl. J. Med. 2013;doi:10.1056/nejmoa1302507).
"I think the argument is overwhelming" to use one of the new drugs instead of warfarin. "They are oral drugs where you do not need a blood draw every 2 or 3 weeks, they are a lot easier to use, and they are at least as good as warfarin and at least as safe," said Dr. Frits R. Rosendaal, professor of clinical epidemiology in hemostasis and thrombosis at Leiden (The Netherlands) University.
The Apixaban for the Initial Management of Pulmonary Embolism and Deep-Vein Thrombosis as First-Line Therapy (AMPLIFY) trial randomized 5,400 acute VTE patients at 358 centers in 28 countries. Patients received either apixaban starting with a 10-mg b.i.d. dosage for 7 days, followed by a dosage of 5 mg b.i.d. for 6 months, or enoxaparin at a dosage of 1 mg/kg every 12 hours for a median of 7 days followed by warfarin for 6 months with a target INR of 2.0-3.0.
The study’s primary efficacy endpoint was the combined rate of recurrent, symptomatic VTE or death related to VTE. This occurred in 59 of 2,609 patients (2.3%) who received apixaban, and in 71 of 2,635 (2.7%) patients who received enoxaparin followed by warfarin. These results met the study’s prespecified criterion for apixaban’s noninferiority to standard treatment reported Dr. Agnelli, professor of medicine at the University of Perugia, Italy.
Major bleeding events occurred in 15 of 2,676 (0.6%) patients on apixaban and in 49 of 2,689 (1.8%) patients on enoxaparin and warfarin, a statistically significant difference. A composite safety outcome of major bleeds plus clinically relevant nonmajor bleeds occurred in 4.3% of the apixaban patients and in 9.7% of the patients on standard therapy, a statistically significant difference. Aside from bleeding events, the rates of all other adverse events were similar in the two treatment arms.
The trial was sponsored by Pfizer and Bristol-Myers Squibb, which market apixaban (Eliquis). Dr. Agnelli disclosed ties to Pfizer, Boehringer Ingelheim, Sanofi, Daiichi Sankyo, and Bayer. Dr. Rosendaal said that he had no disclosures.
The new anticoagulant era for patients with acute venous thromboembolism began late last year, when the Food and Drug Administration gave rivaroxaban this new labeled indication. Each physician who enters this new era, by opting to prescribe rivaroxaban instead of warfarin, needs to carefully think through which types of patients the evidence supports treating.
Clinicians who manage patients with acute venous thromboembolism (VTE) need to decide how to bring rivaroxaban into their practices, and eventually they will need to make similar decisions about dabigatran and apixaban once those drugs receive an acute VTE indication.

|
|
A few weeks after rivaroxaban’s approval for acute VTE, I got together with my colleagues at the University of Vermont in Burlington who manage these patients to draw up a framework for how we’ll decide which patients should be treated with rivaroxaban and which ones we should still treat with warfarin. I included the six main elements of that framework in an editorial I wrote about the AMPLIFY results reported by Dr. Agnelli (N. Engl. J. Med. 2013;doi:10.1056/nejme1307413).
One factor we urged clinicians to keep in mind was that the new-anticoagulant trials use selected patients. For example, the AMPLIFY trial excluded patients with provoked VTE because they are usually not treated for 6 months, the treatment duration studied in the trial. The new drugs are not suitable for every patient; patients on dialysis shouldn’t get them because these agents all involve renal clearance. Very old patients, those aged beyond 80 years, are an uncertain group because the trials have so far included few such patients, but on the other hand, eliminating the need for regular anticoagulation monitoring may make the new drugs more suitable for some elderly patients. Patients with cancer are another uncertain subgroup because of their low representation in the trials. My colleagues and I have for the time being decided not to use rivaroxaban on cancer patients with acute VTE because we judged the current evidence base too limited.
Rivaroxaban, apixaban, and dabigatran each have their own unique list of drug interactions and contraindications. None of them has as many caveats as warfarin, but it’s still essential to be familiar with each drug’s label and which clinical situations to avoid.
Another issue is how likely a patient will be to adhere to the prescribed regimen. A limitation of the new drugs is their short half-life. A patient just needs to miss a couple of doses and the anticoagulation effect starts to fade. In contrast, warfarin’s effect lingers much longer; a patient on a stable warfarin regimen can go several days without treatment before the international normalized ratio begins to fall out of the target range. Plus, patients on the new drugs provide physicians with no compliance feedback from monitoring results. That means patients should only be started on a new oral anticoagulant if they understand this limitation and the need for commitment to full compliance. There are also issues of cost and insurance coverage with the new drugs. Some patients have insurers that require preapproval for treatment of VTE with rivaroxaban, but then the payers deny the preapproval when we request it.
Warfarin has been the top anticoagulant for the past 60 years, much longer than my entire career in medicine. Physicians now face the challenge of applying the results from carefully controlled studies of the new anticoagulants – where selected cohorts of patients received their care from a small number of anticoagulation management experts – to a much broader and more diverse spectrum of patients in a variety of clinical settings. How exactly that gets done needs careful consideration.
Dr. Mary Cushman is a hematologist and professor of medicine at the University of Vermont in Burlington. She made these comments in an interview. She said that she had no disclosures.
The new anticoagulant era for patients with acute venous thromboembolism began late last year, when the Food and Drug Administration gave rivaroxaban this new labeled indication. Each physician who enters this new era, by opting to prescribe rivaroxaban instead of warfarin, needs to carefully think through which types of patients the evidence supports treating.
Clinicians who manage patients with acute venous thromboembolism (VTE) need to decide how to bring rivaroxaban into their practices, and eventually they will need to make similar decisions about dabigatran and apixaban once those drugs receive an acute VTE indication.
|
|
|
A few weeks after rivaroxaban’s approval for acute VTE, I got together with my colleagues at the University of Vermont in Burlington who manage these patients to draw up a framework for how we’ll decide which patients should be treated with rivaroxaban and which ones we should still treat with warfarin. I included the six main elements of that framework in an editorial I wrote about the AMPLIFY results reported by Dr. Agnelli (N. Engl. J. Med. 2013;doi:10.1056/nejme1307413).
One factor we urged clinicians to keep in mind was that the new-anticoagulant trials use selected patients. For example, the AMPLIFY trial excluded patients with provoked VTE because they are usually not treated for 6 months, the treatment duration studied in the trial. The new drugs are not suitable for every patient; patients on dialysis shouldn’t get them because these agents all involve renal clearance. Very old patients, those aged beyond 80 years, are an uncertain group because the trials have so far included few such patients, but on the other hand, eliminating the need for regular anticoagulation monitoring may make the new drugs more suitable for some elderly patients. Patients with cancer are another uncertain subgroup because of their low representation in the trials. My colleagues and I have for the time being decided not to use rivaroxaban on cancer patients with acute VTE because we judged the current evidence base too limited.
Rivaroxaban, apixaban, and dabigatran each have their own unique list of drug interactions and contraindications. None of them has as many caveats as warfarin, but it’s still essential to be familiar with each drug’s label and which clinical situations to avoid.
Another issue is how likely a patient will be to adhere to the prescribed regimen. A limitation of the new drugs is their short half-life. A patient just needs to miss a couple of doses and the anticoagulation effect starts to fade. In contrast, warfarin’s effect lingers much longer; a patient on a stable warfarin regimen can go several days without treatment before the international normalized ratio begins to fall out of the target range. Plus, patients on the new drugs provide physicians with no compliance feedback from monitoring results. That means patients should only be started on a new oral anticoagulant if they understand this limitation and the need for commitment to full compliance. There are also issues of cost and insurance coverage with the new drugs. Some patients have insurers that require preapproval for treatment of VTE with rivaroxaban, but then the payers deny the preapproval when we request it.
Warfarin has been the top anticoagulant for the past 60 years, much longer than my entire career in medicine. Physicians now face the challenge of applying the results from carefully controlled studies of the new anticoagulants – where selected cohorts of patients received their care from a small number of anticoagulation management experts – to a much broader and more diverse spectrum of patients in a variety of clinical settings. How exactly that gets done needs careful consideration.
Dr. Mary Cushman is a hematologist and professor of medicine at the University of Vermont in Burlington. She made these comments in an interview. She said that she had no disclosures.
The new anticoagulant era for patients with acute venous thromboembolism began late last year, when the Food and Drug Administration gave rivaroxaban this new labeled indication. Each physician who enters this new era, by opting to prescribe rivaroxaban instead of warfarin, needs to carefully think through which types of patients the evidence supports treating.
Clinicians who manage patients with acute venous thromboembolism (VTE) need to decide how to bring rivaroxaban into their practices, and eventually they will need to make similar decisions about dabigatran and apixaban once those drugs receive an acute VTE indication.
|
|
|
A few weeks after rivaroxaban’s approval for acute VTE, I got together with my colleagues at the University of Vermont in Burlington who manage these patients to draw up a framework for how we’ll decide which patients should be treated with rivaroxaban and which ones we should still treat with warfarin. I included the six main elements of that framework in an editorial I wrote about the AMPLIFY results reported by Dr. Agnelli (N. Engl. J. Med. 2013;doi:10.1056/nejme1307413).
One factor we urged clinicians to keep in mind was that the new-anticoagulant trials use selected patients. For example, the AMPLIFY trial excluded patients with provoked VTE because they are usually not treated for 6 months, the treatment duration studied in the trial. The new drugs are not suitable for every patient; patients on dialysis shouldn’t get them because these agents all involve renal clearance. Very old patients, those aged beyond 80 years, are an uncertain group because the trials have so far included few such patients, but on the other hand, eliminating the need for regular anticoagulation monitoring may make the new drugs more suitable for some elderly patients. Patients with cancer are another uncertain subgroup because of their low representation in the trials. My colleagues and I have for the time being decided not to use rivaroxaban on cancer patients with acute VTE because we judged the current evidence base too limited.
Rivaroxaban, apixaban, and dabigatran each have their own unique list of drug interactions and contraindications. None of them has as many caveats as warfarin, but it’s still essential to be familiar with each drug’s label and which clinical situations to avoid.
Another issue is how likely a patient will be to adhere to the prescribed regimen. A limitation of the new drugs is their short half-life. A patient just needs to miss a couple of doses and the anticoagulation effect starts to fade. In contrast, warfarin’s effect lingers much longer; a patient on a stable warfarin regimen can go several days without treatment before the international normalized ratio begins to fall out of the target range. Plus, patients on the new drugs provide physicians with no compliance feedback from monitoring results. That means patients should only be started on a new oral anticoagulant if they understand this limitation and the need for commitment to full compliance. There are also issues of cost and insurance coverage with the new drugs. Some patients have insurers that require preapproval for treatment of VTE with rivaroxaban, but then the payers deny the preapproval when we request it.
Warfarin has been the top anticoagulant for the past 60 years, much longer than my entire career in medicine. Physicians now face the challenge of applying the results from carefully controlled studies of the new anticoagulants – where selected cohorts of patients received their care from a small number of anticoagulation management experts – to a much broader and more diverse spectrum of patients in a variety of clinical settings. How exactly that gets done needs careful consideration.
Dr. Mary Cushman is a hematologist and professor of medicine at the University of Vermont in Burlington. She made these comments in an interview. She said that she had no disclosures.
AMSTERDAM – In patients with acute venous thromboembolism, 6 months of treatment with the oral-anticoagulant apixaban was as effective as was standard therapy with subcutaneous enoxaparin for a week followed by oral warfarin, and apixaban caused significantly fewer major bleeding complications in a randomized, multicenter trial with more than 5,000 patients.
But in addition to apixaban’s sterling individual performance in this pivotal trial, which seems to clear the way for the drug to eventually receive a labeled indication for acute venous thromboembolism, the results also appeared to further anoint the new, oral anticoagulant roster of dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) as the thrombotic-disease troika to be reckoned with, the newcomers whose time has come.
Ever since rivaroxaban became the first of the trio to gain acute VTE labeling, last November, physicians who manage patients with acute VTE had to wrestle with the question of how to integrate this option into their practices. The new findings on apixaban suggest that physicians will soon have to think about deciding between rivaroxaban and apixaban for this indication. And since recent results from other major trials also established dabigatran as the equal of warfarin for efficacy when treating acute VTE patients and with superior safety, dabigatran’s entry into acute VTE management seems imminent (N. Engl. J. Med. 2013;368:709-18).
Propelling this new anticoagulant era are the indications of efficacy that’s equivalent with heparin, but safer, and with far easier drug delivery as the need for anticoagulation clinics and regular measurement of international normalized ratio (INR) is eliminated by all three new drugs.
"An oral regimen without laboratory monitoring will simplify therapy," Dr. Giancarlo Agnelli noted when he presented the new apixaban findings at the congress of the International Society on Thrombosis and Haemostasis. Concurrently with his report at the meeting, the results were published online (N. Engl. J. Med. 2013;doi:10.1056/nejmoa1302507).
"I think the argument is overwhelming" to use one of the new drugs instead of warfarin. "They are oral drugs where you do not need a blood draw every 2 or 3 weeks, they are a lot easier to use, and they are at least as good as warfarin and at least as safe," said Dr. Frits R. Rosendaal, professor of clinical epidemiology in hemostasis and thrombosis at Leiden (The Netherlands) University.
The Apixaban for the Initial Management of Pulmonary Embolism and Deep-Vein Thrombosis as First-Line Therapy (AMPLIFY) trial randomized 5,400 acute VTE patients at 358 centers in 28 countries. Patients received either apixaban starting with a 10-mg b.i.d. dosage for 7 days, followed by a dosage of 5 mg b.i.d. for 6 months, or enoxaparin at a dosage of 1 mg/kg every 12 hours for a median of 7 days followed by warfarin for 6 months with a target INR of 2.0-3.0.
The study’s primary efficacy endpoint was the combined rate of recurrent, symptomatic VTE or death related to VTE. This occurred in 59 of 2,609 patients (2.3%) who received apixaban, and in 71 of 2,635 (2.7%) patients who received enoxaparin followed by warfarin. These results met the study’s prespecified criterion for apixaban’s noninferiority to standard treatment reported Dr. Agnelli, professor of medicine at the University of Perugia, Italy.
Major bleeding events occurred in 15 of 2,676 (0.6%) patients on apixaban and in 49 of 2,689 (1.8%) patients on enoxaparin and warfarin, a statistically significant difference. A composite safety outcome of major bleeds plus clinically relevant nonmajor bleeds occurred in 4.3% of the apixaban patients and in 9.7% of the patients on standard therapy, a statistically significant difference. Aside from bleeding events, the rates of all other adverse events were similar in the two treatment arms.
The trial was sponsored by Pfizer and Bristol-Myers Squibb, which market apixaban (Eliquis). Dr. Agnelli disclosed ties to Pfizer, Boehringer Ingelheim, Sanofi, Daiichi Sankyo, and Bayer. Dr. Rosendaal said that he had no disclosures.
AMSTERDAM – In patients with acute venous thromboembolism, 6 months of treatment with the oral-anticoagulant apixaban was as effective as was standard therapy with subcutaneous enoxaparin for a week followed by oral warfarin, and apixaban caused significantly fewer major bleeding complications in a randomized, multicenter trial with more than 5,000 patients.
But in addition to apixaban’s sterling individual performance in this pivotal trial, which seems to clear the way for the drug to eventually receive a labeled indication for acute venous thromboembolism, the results also appeared to further anoint the new, oral anticoagulant roster of dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) as the thrombotic-disease troika to be reckoned with, the newcomers whose time has come.
Ever since rivaroxaban became the first of the trio to gain acute VTE labeling, last November, physicians who manage patients with acute VTE had to wrestle with the question of how to integrate this option into their practices. The new findings on apixaban suggest that physicians will soon have to think about deciding between rivaroxaban and apixaban for this indication. And since recent results from other major trials also established dabigatran as the equal of warfarin for efficacy when treating acute VTE patients and with superior safety, dabigatran’s entry into acute VTE management seems imminent (N. Engl. J. Med. 2013;368:709-18).
Propelling this new anticoagulant era are the indications of efficacy that’s equivalent with heparin, but safer, and with far easier drug delivery as the need for anticoagulation clinics and regular measurement of international normalized ratio (INR) is eliminated by all three new drugs.
"An oral regimen without laboratory monitoring will simplify therapy," Dr. Giancarlo Agnelli noted when he presented the new apixaban findings at the congress of the International Society on Thrombosis and Haemostasis. Concurrently with his report at the meeting, the results were published online (N. Engl. J. Med. 2013;doi:10.1056/nejmoa1302507).
"I think the argument is overwhelming" to use one of the new drugs instead of warfarin. "They are oral drugs where you do not need a blood draw every 2 or 3 weeks, they are a lot easier to use, and they are at least as good as warfarin and at least as safe," said Dr. Frits R. Rosendaal, professor of clinical epidemiology in hemostasis and thrombosis at Leiden (The Netherlands) University.
The Apixaban for the Initial Management of Pulmonary Embolism and Deep-Vein Thrombosis as First-Line Therapy (AMPLIFY) trial randomized 5,400 acute VTE patients at 358 centers in 28 countries. Patients received either apixaban starting with a 10-mg b.i.d. dosage for 7 days, followed by a dosage of 5 mg b.i.d. for 6 months, or enoxaparin at a dosage of 1 mg/kg every 12 hours for a median of 7 days followed by warfarin for 6 months with a target INR of 2.0-3.0.
The study’s primary efficacy endpoint was the combined rate of recurrent, symptomatic VTE or death related to VTE. This occurred in 59 of 2,609 patients (2.3%) who received apixaban, and in 71 of 2,635 (2.7%) patients who received enoxaparin followed by warfarin. These results met the study’s prespecified criterion for apixaban’s noninferiority to standard treatment reported Dr. Agnelli, professor of medicine at the University of Perugia, Italy.
Major bleeding events occurred in 15 of 2,676 (0.6%) patients on apixaban and in 49 of 2,689 (1.8%) patients on enoxaparin and warfarin, a statistically significant difference. A composite safety outcome of major bleeds plus clinically relevant nonmajor bleeds occurred in 4.3% of the apixaban patients and in 9.7% of the patients on standard therapy, a statistically significant difference. Aside from bleeding events, the rates of all other adverse events were similar in the two treatment arms.
The trial was sponsored by Pfizer and Bristol-Myers Squibb, which market apixaban (Eliquis). Dr. Agnelli disclosed ties to Pfizer, Boehringer Ingelheim, Sanofi, Daiichi Sankyo, and Bayer. Dr. Rosendaal said that he had no disclosures.
AT THE 2013 ISTH CONGRESS
Major finding: Major bleeds occurred in 0.6% of patients on apixaban and 1.8% of patients on warfarin, with similar efficacy.
Data source: The AMPLIFY study, which randomized 5,400 patients with acute venous thromboembolism to treatment with apixaban or enoxaparin followed by warfarin for 6 months.
Disclosures: The trial was sponsored by Pfizer and Bristol-Myers Squibb, which market apixaban (Eliquis). Dr. Agnelli disclosed ties to Pfizer, Boehringer Ingelheim, Sanofi, Daiichi Sankyo, and Bayer. Dr. Rosendaal said that he had no disclosures.

VTE risk-prediction formula gains validation
AMSTERDAM – A simple formula for calculating the risk faced by acutely ill, hospitalized patients for venous thromboembolism was validated in a case-control study with more than 400 patients.
This VTE risk-calculator formula "is the first [risk-assessment model (RAM)] to be validated on a large scale in hospitalized medical patients," Charles E. Mahan, Pharm.D., said at the congress of the International Society on Thrombosis and Haemostasis.

"Applying this RAM could spare 20%-30% of these patients from getting unnecessary prophylaxis" with an anticoagulant, said Dr. Mahan, director of outcomes research at the New Mexico Heart Institute in Albuquerque.
He cautioned that the new evidence he presented still needs to be published, and a prospective test of the risk formula should also be done, but the new findings give this risk-scoring method a leg up over the several other risk-assessment methods that are out there.
"This gives us some information that we can comfortably use," Dr. Mahan said in an interview. Other formulas for estimating VTE risk in patients hospitalized for medical reasons include the Padua Prediction Score (J. Thromb. Haemost. 2010;8:2450-7), but the RAM tested by Dr. Mahan now "has the best evidence base for hospitalized, acutely ill patients."

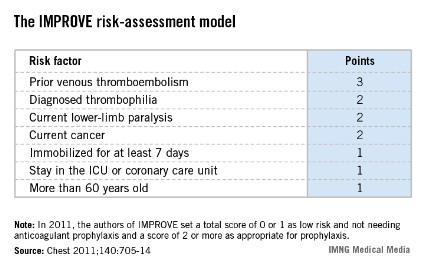
The validation used the risk formula developed by the IMPROVE (International Medical Prevention Registry in Venous Thromboembolism) study, which included more than 15,000 medical patients seen at 52 hospitals in 12 countries (Chest 2011;140:705-14).
The IMPROVE RAM includes seven risk factors that each score from 1 to 3 points (see box). A history of VTE scores 3 points; immobilization for a week or more, intensive care unit stay, and age over 60 each score 1 point; and three other factors each score 2 points.
The validation cohort came from the more than 130,000 patients aged 18 years or older who were hospitalized for at least 3 days during 2005-2011 at a McMaster University–affiliated hospital in Hamilton, Ont. After excluding pregnancies, patients with recent surgery, and patients with VTE at the time of admission, the investigators identified 139 patients who developed VTE within 90 days of hospital admission and matched them with 278 patients who did not develop a VTE as controls. Matching was by gender, hospital, and date of admission.
The IMPROVE RAM showed "good" discrimination in the validation cohort, Dr. Mahan reported. The incidence of VTE during the 90 days following hospitalization in the validation cohort was 0.20% in patients with low scores, 0 or 1; 1.04% in patients with moderate scores, 2 or 3; and 4.15% in those with high scores, 4 or greater. By comparison, in the first IMPROVE cohort the VTE rates for 90 days were 0.45% in patients with low scores, 1.30% in those with moderate scores, and 4.74% in those with high scores.
Receiver-operator characteristic curve analysis showed that in the new cohort, the IMPROVE formula could account for about 77% of the variability in VTE incidence, performance that was also similar to that of the derivation cohort. But the formula failed to predict a VTE in several patients: 26 patients (19%) who had a VTE during follow-up had an IMPROVE score of 0 or 1 at the time of their hospitalization.
In 2011, the IMPROVE authors suggested that clinicians apply VTE prophylaxis to patients who scored 2 points or higher on the risk formula, which represented 31% of the more than 15,000 patients in the derivation cohort. In the validation cohort, 37% of the patients had a score of 2 or more. The new data suggest that a score of 3 or more may be an even better cutoff for starting VTE prophylaxis with heparin or low-molecular-weight heparin, Dr. Mahan said, but he added that this needs more analysis.
The new data showed that a score cut-point of 2 had a sensitivity of 81% and a specificity of 60%, whereas a cut-point of 3 had a sensitivity of 63% and a specificity of 78%.
"We’re still working on the cut-point. I’m sure that a score of 0 or 1 needs no prophylaxis," but deciding between 2 and 3 points will take more time, he said. "In the United States especially, we are over-prophylaxing," giving anticoagulant prophylaxis to "a large group of U.S. hospital patients who don’t need it. Some hospitals do blanket prophylaxis" for virtually all patients hospitalized for medical reasons, Dr. Mahan said.
"We want to identify patients who are not at risk so they don’t get prophylaxis."
Dr. Mahan said that he has been a consultant to or speaker for several drug companies including Janssen, Sanofi-Aventis, Boehringer Ingelheim, Bristol-Myers Squibb, and Pfizer.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
This research addresses quality, safety, and cost for our patients in hospital medicine. We have recognized that not all inpatients require VTE prevention, and this work offers a validated approach to assist with appropriate prophylaxis.

|
| Dr. Steven B. Deitelzweig |
It will be important to see if this can be incorporated into our workflow via electronic health records. This is exactly where we need assistance to prevent one of the leading causes of death in our hospitalized patients in a cost-effective manner.
We still need to better define the risk score, but this is definitely an advance.
Dr. Steven B. Deitelzweig is chair of the department of hospital medicine, Ochsner Health System, New Orleans.
This research addresses quality, safety, and cost for our patients in hospital medicine. We have recognized that not all inpatients require VTE prevention, and this work offers a validated approach to assist with appropriate prophylaxis.
|
|
| Dr. Steven B. Deitelzweig |
It will be important to see if this can be incorporated into our workflow via electronic health records. This is exactly where we need assistance to prevent one of the leading causes of death in our hospitalized patients in a cost-effective manner.
We still need to better define the risk score, but this is definitely an advance.
Dr. Steven B. Deitelzweig is chair of the department of hospital medicine, Ochsner Health System, New Orleans.
This research addresses quality, safety, and cost for our patients in hospital medicine. We have recognized that not all inpatients require VTE prevention, and this work offers a validated approach to assist with appropriate prophylaxis.
|
|
| Dr. Steven B. Deitelzweig |
It will be important to see if this can be incorporated into our workflow via electronic health records. This is exactly where we need assistance to prevent one of the leading causes of death in our hospitalized patients in a cost-effective manner.
We still need to better define the risk score, but this is definitely an advance.
Dr. Steven B. Deitelzweig is chair of the department of hospital medicine, Ochsner Health System, New Orleans.
AMSTERDAM – A simple formula for calculating the risk faced by acutely ill, hospitalized patients for venous thromboembolism was validated in a case-control study with more than 400 patients.
This VTE risk-calculator formula "is the first [risk-assessment model (RAM)] to be validated on a large scale in hospitalized medical patients," Charles E. Mahan, Pharm.D., said at the congress of the International Society on Thrombosis and Haemostasis.
"Applying this RAM could spare 20%-30% of these patients from getting unnecessary prophylaxis" with an anticoagulant, said Dr. Mahan, director of outcomes research at the New Mexico Heart Institute in Albuquerque.
He cautioned that the new evidence he presented still needs to be published, and a prospective test of the risk formula should also be done, but the new findings give this risk-scoring method a leg up over the several other risk-assessment methods that are out there.
"This gives us some information that we can comfortably use," Dr. Mahan said in an interview. Other formulas for estimating VTE risk in patients hospitalized for medical reasons include the Padua Prediction Score (J. Thromb. Haemost. 2010;8:2450-7), but the RAM tested by Dr. Mahan now "has the best evidence base for hospitalized, acutely ill patients."
The validation used the risk formula developed by the IMPROVE (International Medical Prevention Registry in Venous Thromboembolism) study, which included more than 15,000 medical patients seen at 52 hospitals in 12 countries (Chest 2011;140:705-14).
The IMPROVE RAM includes seven risk factors that each score from 1 to 3 points (see box). A history of VTE scores 3 points; immobilization for a week or more, intensive care unit stay, and age over 60 each score 1 point; and three other factors each score 2 points.
The validation cohort came from the more than 130,000 patients aged 18 years or older who were hospitalized for at least 3 days during 2005-2011 at a McMaster University–affiliated hospital in Hamilton, Ont. After excluding pregnancies, patients with recent surgery, and patients with VTE at the time of admission, the investigators identified 139 patients who developed VTE within 90 days of hospital admission and matched them with 278 patients who did not develop a VTE as controls. Matching was by gender, hospital, and date of admission.
The IMPROVE RAM showed "good" discrimination in the validation cohort, Dr. Mahan reported. The incidence of VTE during the 90 days following hospitalization in the validation cohort was 0.20% in patients with low scores, 0 or 1; 1.04% in patients with moderate scores, 2 or 3; and 4.15% in those with high scores, 4 or greater. By comparison, in the first IMPROVE cohort the VTE rates for 90 days were 0.45% in patients with low scores, 1.30% in those with moderate scores, and 4.74% in those with high scores.
Receiver-operator characteristic curve analysis showed that in the new cohort, the IMPROVE formula could account for about 77% of the variability in VTE incidence, performance that was also similar to that of the derivation cohort. But the formula failed to predict a VTE in several patients: 26 patients (19%) who had a VTE during follow-up had an IMPROVE score of 0 or 1 at the time of their hospitalization.
In 2011, the IMPROVE authors suggested that clinicians apply VTE prophylaxis to patients who scored 2 points or higher on the risk formula, which represented 31% of the more than 15,000 patients in the derivation cohort. In the validation cohort, 37% of the patients had a score of 2 or more. The new data suggest that a score of 3 or more may be an even better cutoff for starting VTE prophylaxis with heparin or low-molecular-weight heparin, Dr. Mahan said, but he added that this needs more analysis.
The new data showed that a score cut-point of 2 had a sensitivity of 81% and a specificity of 60%, whereas a cut-point of 3 had a sensitivity of 63% and a specificity of 78%.
"We’re still working on the cut-point. I’m sure that a score of 0 or 1 needs no prophylaxis," but deciding between 2 and 3 points will take more time, he said. "In the United States especially, we are over-prophylaxing," giving anticoagulant prophylaxis to "a large group of U.S. hospital patients who don’t need it. Some hospitals do blanket prophylaxis" for virtually all patients hospitalized for medical reasons, Dr. Mahan said.
"We want to identify patients who are not at risk so they don’t get prophylaxis."
Dr. Mahan said that he has been a consultant to or speaker for several drug companies including Janssen, Sanofi-Aventis, Boehringer Ingelheim, Bristol-Myers Squibb, and Pfizer.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
AMSTERDAM – A simple formula for calculating the risk faced by acutely ill, hospitalized patients for venous thromboembolism was validated in a case-control study with more than 400 patients.
This VTE risk-calculator formula "is the first [risk-assessment model (RAM)] to be validated on a large scale in hospitalized medical patients," Charles E. Mahan, Pharm.D., said at the congress of the International Society on Thrombosis and Haemostasis.
"Applying this RAM could spare 20%-30% of these patients from getting unnecessary prophylaxis" with an anticoagulant, said Dr. Mahan, director of outcomes research at the New Mexico Heart Institute in Albuquerque.
He cautioned that the new evidence he presented still needs to be published, and a prospective test of the risk formula should also be done, but the new findings give this risk-scoring method a leg up over the several other risk-assessment methods that are out there.
"This gives us some information that we can comfortably use," Dr. Mahan said in an interview. Other formulas for estimating VTE risk in patients hospitalized for medical reasons include the Padua Prediction Score (J. Thromb. Haemost. 2010;8:2450-7), but the RAM tested by Dr. Mahan now "has the best evidence base for hospitalized, acutely ill patients."
The validation used the risk formula developed by the IMPROVE (International Medical Prevention Registry in Venous Thromboembolism) study, which included more than 15,000 medical patients seen at 52 hospitals in 12 countries (Chest 2011;140:705-14).
The IMPROVE RAM includes seven risk factors that each score from 1 to 3 points (see box). A history of VTE scores 3 points; immobilization for a week or more, intensive care unit stay, and age over 60 each score 1 point; and three other factors each score 2 points.
The validation cohort came from the more than 130,000 patients aged 18 years or older who were hospitalized for at least 3 days during 2005-2011 at a McMaster University–affiliated hospital in Hamilton, Ont. After excluding pregnancies, patients with recent surgery, and patients with VTE at the time of admission, the investigators identified 139 patients who developed VTE within 90 days of hospital admission and matched them with 278 patients who did not develop a VTE as controls. Matching was by gender, hospital, and date of admission.
The IMPROVE RAM showed "good" discrimination in the validation cohort, Dr. Mahan reported. The incidence of VTE during the 90 days following hospitalization in the validation cohort was 0.20% in patients with low scores, 0 or 1; 1.04% in patients with moderate scores, 2 or 3; and 4.15% in those with high scores, 4 or greater. By comparison, in the first IMPROVE cohort the VTE rates for 90 days were 0.45% in patients with low scores, 1.30% in those with moderate scores, and 4.74% in those with high scores.
Receiver-operator characteristic curve analysis showed that in the new cohort, the IMPROVE formula could account for about 77% of the variability in VTE incidence, performance that was also similar to that of the derivation cohort. But the formula failed to predict a VTE in several patients: 26 patients (19%) who had a VTE during follow-up had an IMPROVE score of 0 or 1 at the time of their hospitalization.
In 2011, the IMPROVE authors suggested that clinicians apply VTE prophylaxis to patients who scored 2 points or higher on the risk formula, which represented 31% of the more than 15,000 patients in the derivation cohort. In the validation cohort, 37% of the patients had a score of 2 or more. The new data suggest that a score of 3 or more may be an even better cutoff for starting VTE prophylaxis with heparin or low-molecular-weight heparin, Dr. Mahan said, but he added that this needs more analysis.
The new data showed that a score cut-point of 2 had a sensitivity of 81% and a specificity of 60%, whereas a cut-point of 3 had a sensitivity of 63% and a specificity of 78%.
"We’re still working on the cut-point. I’m sure that a score of 0 or 1 needs no prophylaxis," but deciding between 2 and 3 points will take more time, he said. "In the United States especially, we are over-prophylaxing," giving anticoagulant prophylaxis to "a large group of U.S. hospital patients who don’t need it. Some hospitals do blanket prophylaxis" for virtually all patients hospitalized for medical reasons, Dr. Mahan said.
"We want to identify patients who are not at risk so they don’t get prophylaxis."
Dr. Mahan said that he has been a consultant to or speaker for several drug companies including Janssen, Sanofi-Aventis, Boehringer Ingelheim, Bristol-Myers Squibb, and Pfizer.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
AT THE 2013 ISTH CONGRESS
Orbital atherectomy shown safe, effective in coronaries
PARIS – An atherectomy device already on the U.S. market for treating calcified lesions in peripheral arteries showed safety and efficacy for treating severely calcified coronary arteries in preparation for stenting in a pivotal, multicenter study with 443 patients.
"We believe that the orbital atherectomy system is a unique technology that appears to address an unmet need in this difficult-to-treat patient population," Dr. Jeffrey W. Chambers said at the annual meeting of the European Association of Percutaneous Cardiovascular Intervention.

Based on the data collected in the new study, Cardiovascular Systems, the company that markets the orbital atherectomy system (OAS) Diamondback 360° applied to the Food and Drug Administration in March for approval to add severe coronary artery calcification as an indication for the device. The device received FDA approval for treating peripheral arteries in 2007.
The study met its primary safety and efficacy endpoints "by a significant margin," said Dr. Chambers, director of the cardiac catheterization laboratory at the Metropolitan Heart and Vascular Institute of Allina Mercy Hospital in Minneapolis. OAS treatment resulted in a 10% rate of major adverse coronary events during 30 days following treatment, significantly less than the 17% prespecified performance goal. The procedural success rate of 89% also significantly surpassed the 82% prespecified efficacy performance goal.
"This study is very important and clinically relevant," commented Dr. William Wijns, codirector of the cardiovascular center at OLV Hospital in Aalst, Belgium, who cochaired the session where Dr. Chambers gave his report.
The ORBIT II (Evaluate the Safety and Efficacy of OAS in Treating Severely Calcified Coronary Lesions) trial enrolled 443 patients at 49 U.S. sites. All participants had severely calcified coronary lesions that were at least 15 mm long. The patients averaged 71 years old, and two-thirds were men. The mean lesion length was almost 19 mm, with an average stenosis rate of 84%.
Patients had a cumulative 7% rate of severe angiographic complications following OAS treatment, which included type C dissections, perforations, abrupt closures, or slow or no reflow. The only significant clinical factor associated with the occurrence of one of these complications was lesion length: The longer the lesion, the more likely a complication. The complication rates for OAS seen in this study were similar to or less than the rates reported from past studies of rotational atherectomy for treating coronary arteries, Dr. Chambers said.
Safety was defined as the combined rate of cardiac death, myocardial infarction, or need for target vessel or lesion revascularization during the 30 days after treatment. The most common of these adverse events was non–Q-wave myocardial infarction, which occurred in nearly 9% of patients. Procedural success was defined as successful stent delivery, which occurred in almost 98% of patients, minus the rate of major adverse coronary events during hospitalization.
ORBIT II’s original design called for comparing OAS with rotational atherectomy, but that plan was abandoned because rotational atherectomy devices are not approved for treating severely calcified lesions, Dr. Chambers said. "The FDA felt that no other devices do this," which led to the uncontrolled study design.
The OAS uses a special guide wire that allows direct crossing of "almost all lesions," he said. The system works by removing calcium to improve arterial compliance and allow full stent expansion; the goal of OAS treatment is not debulking or plaque removal, Dr. Chambers said. The microparticles produced during the procedure are "smaller than with rotational ablation," small enough to safely pass through capillaries.
"There is a relatively easy learning curve of a couple of cases if you have used rotational ablation," he said. "The device has a leading edge that guides you into the lesion, acting as a pathfinder. The OAS is designed to engage the hard components of plaque and flex the soft components out of the way."
The ORBIT II study was sponsored by Cardiovascular Systems, which markets the OAS. Dr. Chambers is a consultant to Cardiovascular Systems and Boston Scientific. Dr. Wijns said he has received honoraria and research grants from several drug and device companies, but he had no disclosures related to Cardiovascular Systems; he has received honoraria and research grants from Boston Scientific, which markets a rotational ablation device.
On Twitter @mitchelzoler
PARIS – An atherectomy device already on the U.S. market for treating calcified lesions in peripheral arteries showed safety and efficacy for treating severely calcified coronary arteries in preparation for stenting in a pivotal, multicenter study with 443 patients.
"We believe that the orbital atherectomy system is a unique technology that appears to address an unmet need in this difficult-to-treat patient population," Dr. Jeffrey W. Chambers said at the annual meeting of the European Association of Percutaneous Cardiovascular Intervention.
Based on the data collected in the new study, Cardiovascular Systems, the company that markets the orbital atherectomy system (OAS) Diamondback 360° applied to the Food and Drug Administration in March for approval to add severe coronary artery calcification as an indication for the device. The device received FDA approval for treating peripheral arteries in 2007.
The study met its primary safety and efficacy endpoints "by a significant margin," said Dr. Chambers, director of the cardiac catheterization laboratory at the Metropolitan Heart and Vascular Institute of Allina Mercy Hospital in Minneapolis. OAS treatment resulted in a 10% rate of major adverse coronary events during 30 days following treatment, significantly less than the 17% prespecified performance goal. The procedural success rate of 89% also significantly surpassed the 82% prespecified efficacy performance goal.
"This study is very important and clinically relevant," commented Dr. William Wijns, codirector of the cardiovascular center at OLV Hospital in Aalst, Belgium, who cochaired the session where Dr. Chambers gave his report.
The ORBIT II (Evaluate the Safety and Efficacy of OAS in Treating Severely Calcified Coronary Lesions) trial enrolled 443 patients at 49 U.S. sites. All participants had severely calcified coronary lesions that were at least 15 mm long. The patients averaged 71 years old, and two-thirds were men. The mean lesion length was almost 19 mm, with an average stenosis rate of 84%.
Patients had a cumulative 7% rate of severe angiographic complications following OAS treatment, which included type C dissections, perforations, abrupt closures, or slow or no reflow. The only significant clinical factor associated with the occurrence of one of these complications was lesion length: The longer the lesion, the more likely a complication. The complication rates for OAS seen in this study were similar to or less than the rates reported from past studies of rotational atherectomy for treating coronary arteries, Dr. Chambers said.
Safety was defined as the combined rate of cardiac death, myocardial infarction, or need for target vessel or lesion revascularization during the 30 days after treatment. The most common of these adverse events was non–Q-wave myocardial infarction, which occurred in nearly 9% of patients. Procedural success was defined as successful stent delivery, which occurred in almost 98% of patients, minus the rate of major adverse coronary events during hospitalization.
ORBIT II’s original design called for comparing OAS with rotational atherectomy, but that plan was abandoned because rotational atherectomy devices are not approved for treating severely calcified lesions, Dr. Chambers said. "The FDA felt that no other devices do this," which led to the uncontrolled study design.
The OAS uses a special guide wire that allows direct crossing of "almost all lesions," he said. The system works by removing calcium to improve arterial compliance and allow full stent expansion; the goal of OAS treatment is not debulking or plaque removal, Dr. Chambers said. The microparticles produced during the procedure are "smaller than with rotational ablation," small enough to safely pass through capillaries.
"There is a relatively easy learning curve of a couple of cases if you have used rotational ablation," he said. "The device has a leading edge that guides you into the lesion, acting as a pathfinder. The OAS is designed to engage the hard components of plaque and flex the soft components out of the way."
The ORBIT II study was sponsored by Cardiovascular Systems, which markets the OAS. Dr. Chambers is a consultant to Cardiovascular Systems and Boston Scientific. Dr. Wijns said he has received honoraria and research grants from several drug and device companies, but he had no disclosures related to Cardiovascular Systems; he has received honoraria and research grants from Boston Scientific, which markets a rotational ablation device.
On Twitter @mitchelzoler
PARIS – An atherectomy device already on the U.S. market for treating calcified lesions in peripheral arteries showed safety and efficacy for treating severely calcified coronary arteries in preparation for stenting in a pivotal, multicenter study with 443 patients.
"We believe that the orbital atherectomy system is a unique technology that appears to address an unmet need in this difficult-to-treat patient population," Dr. Jeffrey W. Chambers said at the annual meeting of the European Association of Percutaneous Cardiovascular Intervention.
Based on the data collected in the new study, Cardiovascular Systems, the company that markets the orbital atherectomy system (OAS) Diamondback 360° applied to the Food and Drug Administration in March for approval to add severe coronary artery calcification as an indication for the device. The device received FDA approval for treating peripheral arteries in 2007.
The study met its primary safety and efficacy endpoints "by a significant margin," said Dr. Chambers, director of the cardiac catheterization laboratory at the Metropolitan Heart and Vascular Institute of Allina Mercy Hospital in Minneapolis. OAS treatment resulted in a 10% rate of major adverse coronary events during 30 days following treatment, significantly less than the 17% prespecified performance goal. The procedural success rate of 89% also significantly surpassed the 82% prespecified efficacy performance goal.
"This study is very important and clinically relevant," commented Dr. William Wijns, codirector of the cardiovascular center at OLV Hospital in Aalst, Belgium, who cochaired the session where Dr. Chambers gave his report.
The ORBIT II (Evaluate the Safety and Efficacy of OAS in Treating Severely Calcified Coronary Lesions) trial enrolled 443 patients at 49 U.S. sites. All participants had severely calcified coronary lesions that were at least 15 mm long. The patients averaged 71 years old, and two-thirds were men. The mean lesion length was almost 19 mm, with an average stenosis rate of 84%.
Patients had a cumulative 7% rate of severe angiographic complications following OAS treatment, which included type C dissections, perforations, abrupt closures, or slow or no reflow. The only significant clinical factor associated with the occurrence of one of these complications was lesion length: The longer the lesion, the more likely a complication. The complication rates for OAS seen in this study were similar to or less than the rates reported from past studies of rotational atherectomy for treating coronary arteries, Dr. Chambers said.
Safety was defined as the combined rate of cardiac death, myocardial infarction, or need for target vessel or lesion revascularization during the 30 days after treatment. The most common of these adverse events was non–Q-wave myocardial infarction, which occurred in nearly 9% of patients. Procedural success was defined as successful stent delivery, which occurred in almost 98% of patients, minus the rate of major adverse coronary events during hospitalization.
ORBIT II’s original design called for comparing OAS with rotational atherectomy, but that plan was abandoned because rotational atherectomy devices are not approved for treating severely calcified lesions, Dr. Chambers said. "The FDA felt that no other devices do this," which led to the uncontrolled study design.
The OAS uses a special guide wire that allows direct crossing of "almost all lesions," he said. The system works by removing calcium to improve arterial compliance and allow full stent expansion; the goal of OAS treatment is not debulking or plaque removal, Dr. Chambers said. The microparticles produced during the procedure are "smaller than with rotational ablation," small enough to safely pass through capillaries.
"There is a relatively easy learning curve of a couple of cases if you have used rotational ablation," he said. "The device has a leading edge that guides you into the lesion, acting as a pathfinder. The OAS is designed to engage the hard components of plaque and flex the soft components out of the way."
The ORBIT II study was sponsored by Cardiovascular Systems, which markets the OAS. Dr. Chambers is a consultant to Cardiovascular Systems and Boston Scientific. Dr. Wijns said he has received honoraria and research grants from several drug and device companies, but he had no disclosures related to Cardiovascular Systems; he has received honoraria and research grants from Boston Scientific, which markets a rotational ablation device.
On Twitter @mitchelzoler
AT EUROPCR 2013
Major finding: Orbital atherectomy had an 89% procedural success rate and a 10% rate of major adverse coronary events during 30 days after treatment.
Data source: The ORBIT II study, an uncontrolled, open-label study of orbital atherectomy in 443 patients with severely calcified coronary arteries at 49 U.S. sites.
Disclosures: The ORBIT II study was sponsored by Cardiovascular Systems, which markets the orbital atherectomy system. Dr. Chambers said he is a consultant to Cardiovascular Systems and Boston Scientific. Dr. Wijns said he has received honoraria and research grants from several drug and device companies, but he had no disclosures related to Cardiovascular Systems; he has received honoraria and research grants from Boston Scientific, which markets a rotational ablation device.

Evidence grows for TNF inhibitors in spondyloarthritis
MADRID – Tumor necrosis factor inhibitors are further solidifying their position as the go-to drug class for patients with spondyloarthritis who fail to adequately respond to treatment with nonsteroidal anti-inflammatory drugs.
Results from a series of reports at the annual European Congress of Rheumatology gave further support for the safety and efficacy of tumor necrosis factor (TNF) inhibitors for treating axial spondyloarthritis (SpA), and another report at the meeting provided some of the first evidence for efficacy of the TNF inhibitor class in patients with the less-studied variant, peripheral SpA.

TNF inhibitors "work well for symptoms, and are the gold standard for treating active axial SpA," said Dr. Philip J. Mease, a rheumatologist at Swedish Medical Center in Seattle. He reported evidence for the efficacy of a TNF inhibitor in patients with peripheral SpA without psoriatic involvement, a form of SpA that he said is increasingly being diagnosed after it was first defined a few years ago. The study that Dr. Mease reported on was the first to use the diagnostic criteria for peripheral SpA published by the Assessment of Spondyloarthritis International Society (ASAS) in 2011 (Ann. Rheum. Dis. 2011;70:25-31). Although several TNF inhibitors now have labeling for treating axial SpA and psoriatic arthritis, none currently have U.S. approval for treating peripheral SpA.
The ABILITY-2 (Study of Adalimumab in Subjects With Peripheral Spondyloarthritis) study enrolled patients in the United States, Canada, and several European countries. Patients either had an inadequate response to at least two different nonsteroidal anti-inflammatory drugs (NSAIDs) or were intolerant of or had contraindications for these drugs. Study participants received either 40 mg of adalimumab (Humira) subcutaneously every other week or placebo for 12 weeks.
The study’s primary endpoint was the percentage of patients achieving the peripheral SpA response criteria 40 at 12 weeks, a composite endpoint that requires at least a 40% improvement on each of three measures: patient global assessment of disease activity; patient global assessment of disease pain; and swollen and tender joint count, enthesitis count, or dactylitis count.
The rate of patients fulfilling the primary endpoint was 39% in 84 patients treated with adalimumab and 20% in 81 patients on placebo, a significant difference. Treatment with adalimumab also was linked to "substantial" and statistically significant improvements after 12 weeks in physical function, health-related quality of life, and work productivity, Dr. Mease reported.
Reports on using TNF inhibitors to treat axial SpA at the congress included results from the first randomized, controlled, phase III trial of a TNF inhibitor to enroll patients from the full range of axial SpA, including roughly equal numbers of patients with ankylosing spondylitis and patients diagnosed with axial SpA but without radiographic changes. The phase III RAPID-axSpA (Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Efficacy and Safety of Certolizumab Pegol in Subjects with Active Axial Spondyloarthritis) trial included 325 patients enrolled at 104 sites in the United States and several other countries. The study enrolled patients who had an elevated blood level of C-reactive protein, evidence of sacroiliitis on an MRI scan, or both, and had failed to adequately respond to treatment with at least one NSAID.
Researchers randomized patients to receive either 200 mg of certolizumab pegol (Cimzia) subcutaneously every 2 weeks, 400 mg of certolizumab pegol subcutaneously every 4 weeks, or placebo. All patients randomized to receive certolizumab pegol began with three subcutaneous loading doses of 400 mg administered at the study’s start and after 2 and 4 weeks. Currently, certolizumab pegol has no labeling for treating patients with axial SpA, unlike several other TNF inhibitors such as adalimumab and etanercept (Enbrel).

The study’s primary endpoint was the percentage of patients achieving an ASAS 20 response after 12 weeks of treatment, which requires at least a 20% improvement in at least three of these four criteria: patient global assessment, spinal pain assessment, function, and inflammation. This endpoint was reached by 38% of the 107 placebo patients, 58% of the 111 patients who received certolizumab pegol every 2 weeks, and 64% of those who received certolizumab pegol every 4 weeks, showing statistically significant differences in favor of the active treatment, reported Dr. Robert B.M. Landewé at the congress.
Response rates were similar among the patients with ankylosing spondylitis and those with no radiographic pathology. After 24 weeks of treatment, the rate of ASAS 20 responders fell to 29% of the placebo patients, compared with increases to 67% of patients receiving certolizumab pegol every 2 weeks and to 70% in those getting the drug every 4 weeks.
Dr. Landewé, who is a professor of rheumatology at the Academic Medical Center in Amsterdam, also presented results for several other secondary measures of response. One of these, the Ankylosing Spondylitis Disease Activity Score (ASDAS), showed that inactive disease developed after 24 weeks of treatment in 4% of the placebo-treated patients, 30% of the patients who received certolizumab pegol every 2 weeks, and 31% of patients who received the drug every 4 weeks. The results also showed no new signals of adverse effects, compared with several prior pivotal trials of certolizumab pegol.
Another set of measures in the same study focused on the impact of 24 weeks of treatment on work and household productivity and participation in social activities. Among the 69% of patients in the study who were employed, treatment with either dosage of certolizumab pegol was associated with an average of 10 more productive days of paid work per patient, compared with placebo, reported Dr. Désirée van der Heijde, professor of rheumatology at Leiden University in the Netherlands. During the 24 weeks of treatment, the active regimens also resulted in an added 13-17 days of productive household work and an average of about 10 added days of social or leisure activities, compared with placebo-treated patients.
Results from a third study reported at the meeting included outcomes from patients with axial SpA and objective evidence of inflammation at entry who remained on treatment with adalimumab during 2 years of follow-up in the ABILITY-1 study. This trial’s primary-endpoint results, which were recently published (Ann. Rheum. Dis. 2013;72:815-22), showed that 40 mg of adalimumab administered every other week was significantly better than placebo for reducing disease activity after 12 weeks of treatment. The new results came from 107 patients who remained in the study and received 104 weeks of adalimumab treatment.
After 2 years, 66% of patients showed ASAS 40 responses, and 44% had inactive disease based on their ASDAS, reported Dr. Joachim Sieper, professor and chief of rheumatology at Charité University Hospital in Berlin. Most of the patients in remission at 104 weeks had also been in remission after 52 and 80 weeks of treatment. In addition, the 2-year data showed no new safety concerns, compared with several other prior reports of long-term treatment with adalimumab, he said.
The ABILITY-1 and ABILITY-2 trials were sponsored by AbbVie, which markets adalimumab. Dr. Mease has been a consultant to and has received research support from AbbVie and other companies. Dr. Sieper has been a consultant to and has received research support from Abbott (from which AbbVie was created) as well as Merck, Pfizer, and UCB. The RAPID-axSpA trial was sponsored by UCB, which markets certolizumab. Dr. Landewé has been a consultant to and has received research support from UCB and other companies. Dr. van der Heijde has been a consultant to and has received grant support from UCB and other companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
MADRID – Tumor necrosis factor inhibitors are further solidifying their position as the go-to drug class for patients with spondyloarthritis who fail to adequately respond to treatment with nonsteroidal anti-inflammatory drugs.
Results from a series of reports at the annual European Congress of Rheumatology gave further support for the safety and efficacy of tumor necrosis factor (TNF) inhibitors for treating axial spondyloarthritis (SpA), and another report at the meeting provided some of the first evidence for efficacy of the TNF inhibitor class in patients with the less-studied variant, peripheral SpA.
TNF inhibitors "work well for symptoms, and are the gold standard for treating active axial SpA," said Dr. Philip J. Mease, a rheumatologist at Swedish Medical Center in Seattle. He reported evidence for the efficacy of a TNF inhibitor in patients with peripheral SpA without psoriatic involvement, a form of SpA that he said is increasingly being diagnosed after it was first defined a few years ago. The study that Dr. Mease reported on was the first to use the diagnostic criteria for peripheral SpA published by the Assessment of Spondyloarthritis International Society (ASAS) in 2011 (Ann. Rheum. Dis. 2011;70:25-31). Although several TNF inhibitors now have labeling for treating axial SpA and psoriatic arthritis, none currently have U.S. approval for treating peripheral SpA.
The ABILITY-2 (Study of Adalimumab in Subjects With Peripheral Spondyloarthritis) study enrolled patients in the United States, Canada, and several European countries. Patients either had an inadequate response to at least two different nonsteroidal anti-inflammatory drugs (NSAIDs) or were intolerant of or had contraindications for these drugs. Study participants received either 40 mg of adalimumab (Humira) subcutaneously every other week or placebo for 12 weeks.
The study’s primary endpoint was the percentage of patients achieving the peripheral SpA response criteria 40 at 12 weeks, a composite endpoint that requires at least a 40% improvement on each of three measures: patient global assessment of disease activity; patient global assessment of disease pain; and swollen and tender joint count, enthesitis count, or dactylitis count.
The rate of patients fulfilling the primary endpoint was 39% in 84 patients treated with adalimumab and 20% in 81 patients on placebo, a significant difference. Treatment with adalimumab also was linked to "substantial" and statistically significant improvements after 12 weeks in physical function, health-related quality of life, and work productivity, Dr. Mease reported.
Reports on using TNF inhibitors to treat axial SpA at the congress included results from the first randomized, controlled, phase III trial of a TNF inhibitor to enroll patients from the full range of axial SpA, including roughly equal numbers of patients with ankylosing spondylitis and patients diagnosed with axial SpA but without radiographic changes. The phase III RAPID-axSpA (Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Efficacy and Safety of Certolizumab Pegol in Subjects with Active Axial Spondyloarthritis) trial included 325 patients enrolled at 104 sites in the United States and several other countries. The study enrolled patients who had an elevated blood level of C-reactive protein, evidence of sacroiliitis on an MRI scan, or both, and had failed to adequately respond to treatment with at least one NSAID.
Researchers randomized patients to receive either 200 mg of certolizumab pegol (Cimzia) subcutaneously every 2 weeks, 400 mg of certolizumab pegol subcutaneously every 4 weeks, or placebo. All patients randomized to receive certolizumab pegol began with three subcutaneous loading doses of 400 mg administered at the study’s start and after 2 and 4 weeks. Currently, certolizumab pegol has no labeling for treating patients with axial SpA, unlike several other TNF inhibitors such as adalimumab and etanercept (Enbrel).
The study’s primary endpoint was the percentage of patients achieving an ASAS 20 response after 12 weeks of treatment, which requires at least a 20% improvement in at least three of these four criteria: patient global assessment, spinal pain assessment, function, and inflammation. This endpoint was reached by 38% of the 107 placebo patients, 58% of the 111 patients who received certolizumab pegol every 2 weeks, and 64% of those who received certolizumab pegol every 4 weeks, showing statistically significant differences in favor of the active treatment, reported Dr. Robert B.M. Landewé at the congress.
Response rates were similar among the patients with ankylosing spondylitis and those with no radiographic pathology. After 24 weeks of treatment, the rate of ASAS 20 responders fell to 29% of the placebo patients, compared with increases to 67% of patients receiving certolizumab pegol every 2 weeks and to 70% in those getting the drug every 4 weeks.
Dr. Landewé, who is a professor of rheumatology at the Academic Medical Center in Amsterdam, also presented results for several other secondary measures of response. One of these, the Ankylosing Spondylitis Disease Activity Score (ASDAS), showed that inactive disease developed after 24 weeks of treatment in 4% of the placebo-treated patients, 30% of the patients who received certolizumab pegol every 2 weeks, and 31% of patients who received the drug every 4 weeks. The results also showed no new signals of adverse effects, compared with several prior pivotal trials of certolizumab pegol.
Another set of measures in the same study focused on the impact of 24 weeks of treatment on work and household productivity and participation in social activities. Among the 69% of patients in the study who were employed, treatment with either dosage of certolizumab pegol was associated with an average of 10 more productive days of paid work per patient, compared with placebo, reported Dr. Désirée van der Heijde, professor of rheumatology at Leiden University in the Netherlands. During the 24 weeks of treatment, the active regimens also resulted in an added 13-17 days of productive household work and an average of about 10 added days of social or leisure activities, compared with placebo-treated patients.
Results from a third study reported at the meeting included outcomes from patients with axial SpA and objective evidence of inflammation at entry who remained on treatment with adalimumab during 2 years of follow-up in the ABILITY-1 study. This trial’s primary-endpoint results, which were recently published (Ann. Rheum. Dis. 2013;72:815-22), showed that 40 mg of adalimumab administered every other week was significantly better than placebo for reducing disease activity after 12 weeks of treatment. The new results came from 107 patients who remained in the study and received 104 weeks of adalimumab treatment.
After 2 years, 66% of patients showed ASAS 40 responses, and 44% had inactive disease based on their ASDAS, reported Dr. Joachim Sieper, professor and chief of rheumatology at Charité University Hospital in Berlin. Most of the patients in remission at 104 weeks had also been in remission after 52 and 80 weeks of treatment. In addition, the 2-year data showed no new safety concerns, compared with several other prior reports of long-term treatment with adalimumab, he said.
The ABILITY-1 and ABILITY-2 trials were sponsored by AbbVie, which markets adalimumab. Dr. Mease has been a consultant to and has received research support from AbbVie and other companies. Dr. Sieper has been a consultant to and has received research support from Abbott (from which AbbVie was created) as well as Merck, Pfizer, and UCB. The RAPID-axSpA trial was sponsored by UCB, which markets certolizumab. Dr. Landewé has been a consultant to and has received research support from UCB and other companies. Dr. van der Heijde has been a consultant to and has received grant support from UCB and other companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
MADRID – Tumor necrosis factor inhibitors are further solidifying their position as the go-to drug class for patients with spondyloarthritis who fail to adequately respond to treatment with nonsteroidal anti-inflammatory drugs.
Results from a series of reports at the annual European Congress of Rheumatology gave further support for the safety and efficacy of tumor necrosis factor (TNF) inhibitors for treating axial spondyloarthritis (SpA), and another report at the meeting provided some of the first evidence for efficacy of the TNF inhibitor class in patients with the less-studied variant, peripheral SpA.
TNF inhibitors "work well for symptoms, and are the gold standard for treating active axial SpA," said Dr. Philip J. Mease, a rheumatologist at Swedish Medical Center in Seattle. He reported evidence for the efficacy of a TNF inhibitor in patients with peripheral SpA without psoriatic involvement, a form of SpA that he said is increasingly being diagnosed after it was first defined a few years ago. The study that Dr. Mease reported on was the first to use the diagnostic criteria for peripheral SpA published by the Assessment of Spondyloarthritis International Society (ASAS) in 2011 (Ann. Rheum. Dis. 2011;70:25-31). Although several TNF inhibitors now have labeling for treating axial SpA and psoriatic arthritis, none currently have U.S. approval for treating peripheral SpA.
The ABILITY-2 (Study of Adalimumab in Subjects With Peripheral Spondyloarthritis) study enrolled patients in the United States, Canada, and several European countries. Patients either had an inadequate response to at least two different nonsteroidal anti-inflammatory drugs (NSAIDs) or were intolerant of or had contraindications for these drugs. Study participants received either 40 mg of adalimumab (Humira) subcutaneously every other week or placebo for 12 weeks.
The study’s primary endpoint was the percentage of patients achieving the peripheral SpA response criteria 40 at 12 weeks, a composite endpoint that requires at least a 40% improvement on each of three measures: patient global assessment of disease activity; patient global assessment of disease pain; and swollen and tender joint count, enthesitis count, or dactylitis count.
The rate of patients fulfilling the primary endpoint was 39% in 84 patients treated with adalimumab and 20% in 81 patients on placebo, a significant difference. Treatment with adalimumab also was linked to "substantial" and statistically significant improvements after 12 weeks in physical function, health-related quality of life, and work productivity, Dr. Mease reported.
Reports on using TNF inhibitors to treat axial SpA at the congress included results from the first randomized, controlled, phase III trial of a TNF inhibitor to enroll patients from the full range of axial SpA, including roughly equal numbers of patients with ankylosing spondylitis and patients diagnosed with axial SpA but without radiographic changes. The phase III RAPID-axSpA (Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Efficacy and Safety of Certolizumab Pegol in Subjects with Active Axial Spondyloarthritis) trial included 325 patients enrolled at 104 sites in the United States and several other countries. The study enrolled patients who had an elevated blood level of C-reactive protein, evidence of sacroiliitis on an MRI scan, or both, and had failed to adequately respond to treatment with at least one NSAID.
Researchers randomized patients to receive either 200 mg of certolizumab pegol (Cimzia) subcutaneously every 2 weeks, 400 mg of certolizumab pegol subcutaneously every 4 weeks, or placebo. All patients randomized to receive certolizumab pegol began with three subcutaneous loading doses of 400 mg administered at the study’s start and after 2 and 4 weeks. Currently, certolizumab pegol has no labeling for treating patients with axial SpA, unlike several other TNF inhibitors such as adalimumab and etanercept (Enbrel).
The study’s primary endpoint was the percentage of patients achieving an ASAS 20 response after 12 weeks of treatment, which requires at least a 20% improvement in at least three of these four criteria: patient global assessment, spinal pain assessment, function, and inflammation. This endpoint was reached by 38% of the 107 placebo patients, 58% of the 111 patients who received certolizumab pegol every 2 weeks, and 64% of those who received certolizumab pegol every 4 weeks, showing statistically significant differences in favor of the active treatment, reported Dr. Robert B.M. Landewé at the congress.
Response rates were similar among the patients with ankylosing spondylitis and those with no radiographic pathology. After 24 weeks of treatment, the rate of ASAS 20 responders fell to 29% of the placebo patients, compared with increases to 67% of patients receiving certolizumab pegol every 2 weeks and to 70% in those getting the drug every 4 weeks.
Dr. Landewé, who is a professor of rheumatology at the Academic Medical Center in Amsterdam, also presented results for several other secondary measures of response. One of these, the Ankylosing Spondylitis Disease Activity Score (ASDAS), showed that inactive disease developed after 24 weeks of treatment in 4% of the placebo-treated patients, 30% of the patients who received certolizumab pegol every 2 weeks, and 31% of patients who received the drug every 4 weeks. The results also showed no new signals of adverse effects, compared with several prior pivotal trials of certolizumab pegol.
Another set of measures in the same study focused on the impact of 24 weeks of treatment on work and household productivity and participation in social activities. Among the 69% of patients in the study who were employed, treatment with either dosage of certolizumab pegol was associated with an average of 10 more productive days of paid work per patient, compared with placebo, reported Dr. Désirée van der Heijde, professor of rheumatology at Leiden University in the Netherlands. During the 24 weeks of treatment, the active regimens also resulted in an added 13-17 days of productive household work and an average of about 10 added days of social or leisure activities, compared with placebo-treated patients.
Results from a third study reported at the meeting included outcomes from patients with axial SpA and objective evidence of inflammation at entry who remained on treatment with adalimumab during 2 years of follow-up in the ABILITY-1 study. This trial’s primary-endpoint results, which were recently published (Ann. Rheum. Dis. 2013;72:815-22), showed that 40 mg of adalimumab administered every other week was significantly better than placebo for reducing disease activity after 12 weeks of treatment. The new results came from 107 patients who remained in the study and received 104 weeks of adalimumab treatment.
After 2 years, 66% of patients showed ASAS 40 responses, and 44% had inactive disease based on their ASDAS, reported Dr. Joachim Sieper, professor and chief of rheumatology at Charité University Hospital in Berlin. Most of the patients in remission at 104 weeks had also been in remission after 52 and 80 weeks of treatment. In addition, the 2-year data showed no new safety concerns, compared with several other prior reports of long-term treatment with adalimumab, he said.
The ABILITY-1 and ABILITY-2 trials were sponsored by AbbVie, which markets adalimumab. Dr. Mease has been a consultant to and has received research support from AbbVie and other companies. Dr. Sieper has been a consultant to and has received research support from Abbott (from which AbbVie was created) as well as Merck, Pfizer, and UCB. The RAPID-axSpA trial was sponsored by UCB, which markets certolizumab. Dr. Landewé has been a consultant to and has received research support from UCB and other companies. Dr. van der Heijde has been a consultant to and has received grant support from UCB and other companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AT THE EULAR CONGRESS 2013

Twice daily regimen eases aspirin resistance in diabetes
PARIS – Twice-daily dosing with aspirin worked substantially better for blocking platelet aggregation than did once-daily treatment with the same total dosage, in patients with diabetes and coronary artery disease in a single-center crossover study with 92 patients.
Patients with diabetes are often less responsive to aspirin treatment for secondary prevention of cardiovascular events than are patients without diabetes, apparently because of faster platelet turnover in patients with diabetes, Dr. Jean-Guillaume Dillinger said at the annual meeting of the European Association for Percutaneous Cardiovascular Interventions. Because aspirin levels sufficient to dampen platelet aggregation persist only for about 2 hours following a dose, faster platelet turnover leads to fewer platelets exposed to an adequate aspirin level, increased aspirin resistance, and greater thrombotic potential.

"The results of our study offer a new option to overcome biological aspirin resistance in patients with diabetes," said Dr. Dillinger, a cardiologist at Lariboisière Hospital in Paris. But Dr. Dillinger also cautioned that "large, prospective trials are needed to test if aspirin twice daily can decrease the rate of atherothrombotic events without increasing side effects such as gastrointestinal toxicity." Another potential drawback of twice-daily dosing is that patient compliance with the regimen may be reduced.
Dr. Dillinger also stressed that his study specifically enrolled patients with diabetes with at least one additional risk factor for loss of aspirin efficacy: They were either smokers or had elevated inflammatory markers, and hence, "the results cannot be extrapolated to the whole population of patients with diabetes and coronary artery disease." But he estimated that more than half of patients with diabetes and coronary artery disease (CAD) likely have at least one of these increased-risk factors.
"The results from Dr. Dillinger’s pharmacodynamic study show that a twice-daily regimen may offer greater antiplatelet effects than once daily and are in line with the results from prior investigations," commented Dr. Dominick J. Angiolillo, director of the Cardiovascular Research and Thrombosis Research Center at the University of Florida in Jacksonville. "The data support that [a twice-daily] regimen can be an effective strategy to improve response profiles, but this was a pharmacodynamic study and no conclusions can be made from the results on its clinical efficacy of safety. Clinical studies are warranted to support a change in practice," he said in an interview.

Dr. Dillinger and his associates enrolled 92 patients with diabetes and angiography-confirmed CAD who were already on aspirin treatment. All patients had been first diagnosed with diabetes after they were 30 years old, and they had not received insulin as their initial diabetes treatment unless their treatment had begun during an episode of acute coronary syndrome. In addition, all enrolled patients had at least one of these additional risk factors: current smoker, a high-sensitivity C-reaction protein (hsCRP) level of more than 4 mg/L, a fibrinogen level of more than 4 g/L, or a platelet count of more than 270,000/mm3.
The patients averaged 64 years old, 85% were men, and they entered the study an average of 12 years following their diabetes diagnosis. Of the 92 patients, 48% had one high-risk marker, 30% had two, 15% had three, and 7% had all four of the high-risk markers. Forty-eight (52%) of the patients entered the study on a daily clopidogrel regimen, and that treatment continued.
Patients were randomized to receive either 150 mg aspirin once daily taken every morning for 7-14 days before blood sampling, or 75 mg of aspirin taken twice daily, once in the morning and once in the evening for 7-14 days before their blood was drawn. Following the blood draw, they immediately switched to the alternative regimen that again continued for 7-14 days before the researchers took a second blood sample.
They assessed aspirin resistance by measuring platelet aggregation of each blood specimen using light transmission aggregometry. Specimens that produced a maximum aggregation intensity of at least 20% were considered to indicate aspirin resistance.
Testing revealed aspirin resistance in 39 (42%) patients when the study group received aspirin once daily, and in 16 patients (17%) when they were on the twice-daily regimen, a statistically significant difference.
When the 39 patients who showed aspirin resistance on the once-daily dosage were switched to twice-daily, 24 became aspirin responders on the twice-daily schedule. In contrast, among the 16 patients who were aspirin resistant on twice-daily treatment, 1 patient became responsive on the once-daily regimen, Dr. Dillinger reported.
The researchers also applied a second measure of platelet function – global platelet reactivity – using a platelet function analyzer. By this measure, they found that 38 (41%) of patients had normal platelet reactivity with once-daily aspirin, compared with 27 (29%) when patients were on twice-daily treatment. Within the subgroup of 27 patients who were resistant on either of the two regimens but not the other, 19 (70%) became responders when switched to the bid regimen, with 8 (30%) becoming responders when switched to once daily, a statistically significant difference between the two regimens.
Clopidogrel treatment had no discernable impact on the links between dosing frequency and aspirin’s antiplatelet effects.
A multivariate analysis of factors potentially affecting aspirin’s antiplatelet effect found that twice-daily treatment linked with a statistically significant 79% cut in the likelihood of aspirin resistance, compared with once daily. The only other parameter measured with a significant impact on aspirin resistance in this analysis was level of hsCRP. Every 1 mg/L rise in hsCRP linked with a 15% increased rate of resistance.
Dr. Dillinger also reported these findings in a published article (Am. Heart J. 2012;164:600-6)
Dr. Dillinger had no disclosures. Dr. Angiolillo said that he has received honoraria from several drug and device companies including Bristol Myers Squibb, Sanofi-Aventis, Eli Lilly, and others. He has also received research grants from several companies
On Twitter @mitchelzoler
PARIS – Twice-daily dosing with aspirin worked substantially better for blocking platelet aggregation than did once-daily treatment with the same total dosage, in patients with diabetes and coronary artery disease in a single-center crossover study with 92 patients.
Patients with diabetes are often less responsive to aspirin treatment for secondary prevention of cardiovascular events than are patients without diabetes, apparently because of faster platelet turnover in patients with diabetes, Dr. Jean-Guillaume Dillinger said at the annual meeting of the European Association for Percutaneous Cardiovascular Interventions. Because aspirin levels sufficient to dampen platelet aggregation persist only for about 2 hours following a dose, faster platelet turnover leads to fewer platelets exposed to an adequate aspirin level, increased aspirin resistance, and greater thrombotic potential.
"The results of our study offer a new option to overcome biological aspirin resistance in patients with diabetes," said Dr. Dillinger, a cardiologist at Lariboisière Hospital in Paris. But Dr. Dillinger also cautioned that "large, prospective trials are needed to test if aspirin twice daily can decrease the rate of atherothrombotic events without increasing side effects such as gastrointestinal toxicity." Another potential drawback of twice-daily dosing is that patient compliance with the regimen may be reduced.
Dr. Dillinger also stressed that his study specifically enrolled patients with diabetes with at least one additional risk factor for loss of aspirin efficacy: They were either smokers or had elevated inflammatory markers, and hence, "the results cannot be extrapolated to the whole population of patients with diabetes and coronary artery disease." But he estimated that more than half of patients with diabetes and coronary artery disease (CAD) likely have at least one of these increased-risk factors.
"The results from Dr. Dillinger’s pharmacodynamic study show that a twice-daily regimen may offer greater antiplatelet effects than once daily and are in line with the results from prior investigations," commented Dr. Dominick J. Angiolillo, director of the Cardiovascular Research and Thrombosis Research Center at the University of Florida in Jacksonville. "The data support that [a twice-daily] regimen can be an effective strategy to improve response profiles, but this was a pharmacodynamic study and no conclusions can be made from the results on its clinical efficacy of safety. Clinical studies are warranted to support a change in practice," he said in an interview.
Dr. Dillinger and his associates enrolled 92 patients with diabetes and angiography-confirmed CAD who were already on aspirin treatment. All patients had been first diagnosed with diabetes after they were 30 years old, and they had not received insulin as their initial diabetes treatment unless their treatment had begun during an episode of acute coronary syndrome. In addition, all enrolled patients had at least one of these additional risk factors: current smoker, a high-sensitivity C-reaction protein (hsCRP) level of more than 4 mg/L, a fibrinogen level of more than 4 g/L, or a platelet count of more than 270,000/mm3.
The patients averaged 64 years old, 85% were men, and they entered the study an average of 12 years following their diabetes diagnosis. Of the 92 patients, 48% had one high-risk marker, 30% had two, 15% had three, and 7% had all four of the high-risk markers. Forty-eight (52%) of the patients entered the study on a daily clopidogrel regimen, and that treatment continued.
Patients were randomized to receive either 150 mg aspirin once daily taken every morning for 7-14 days before blood sampling, or 75 mg of aspirin taken twice daily, once in the morning and once in the evening for 7-14 days before their blood was drawn. Following the blood draw, they immediately switched to the alternative regimen that again continued for 7-14 days before the researchers took a second blood sample.
They assessed aspirin resistance by measuring platelet aggregation of each blood specimen using light transmission aggregometry. Specimens that produced a maximum aggregation intensity of at least 20% were considered to indicate aspirin resistance.
Testing revealed aspirin resistance in 39 (42%) patients when the study group received aspirin once daily, and in 16 patients (17%) when they were on the twice-daily regimen, a statistically significant difference.
When the 39 patients who showed aspirin resistance on the once-daily dosage were switched to twice-daily, 24 became aspirin responders on the twice-daily schedule. In contrast, among the 16 patients who were aspirin resistant on twice-daily treatment, 1 patient became responsive on the once-daily regimen, Dr. Dillinger reported.
The researchers also applied a second measure of platelet function – global platelet reactivity – using a platelet function analyzer. By this measure, they found that 38 (41%) of patients had normal platelet reactivity with once-daily aspirin, compared with 27 (29%) when patients were on twice-daily treatment. Within the subgroup of 27 patients who were resistant on either of the two regimens but not the other, 19 (70%) became responders when switched to the bid regimen, with 8 (30%) becoming responders when switched to once daily, a statistically significant difference between the two regimens.
Clopidogrel treatment had no discernable impact on the links between dosing frequency and aspirin’s antiplatelet effects.
A multivariate analysis of factors potentially affecting aspirin’s antiplatelet effect found that twice-daily treatment linked with a statistically significant 79% cut in the likelihood of aspirin resistance, compared with once daily. The only other parameter measured with a significant impact on aspirin resistance in this analysis was level of hsCRP. Every 1 mg/L rise in hsCRP linked with a 15% increased rate of resistance.
Dr. Dillinger also reported these findings in a published article (Am. Heart J. 2012;164:600-6)
Dr. Dillinger had no disclosures. Dr. Angiolillo said that he has received honoraria from several drug and device companies including Bristol Myers Squibb, Sanofi-Aventis, Eli Lilly, and others. He has also received research grants from several companies
On Twitter @mitchelzoler
PARIS – Twice-daily dosing with aspirin worked substantially better for blocking platelet aggregation than did once-daily treatment with the same total dosage, in patients with diabetes and coronary artery disease in a single-center crossover study with 92 patients.
Patients with diabetes are often less responsive to aspirin treatment for secondary prevention of cardiovascular events than are patients without diabetes, apparently because of faster platelet turnover in patients with diabetes, Dr. Jean-Guillaume Dillinger said at the annual meeting of the European Association for Percutaneous Cardiovascular Interventions. Because aspirin levels sufficient to dampen platelet aggregation persist only for about 2 hours following a dose, faster platelet turnover leads to fewer platelets exposed to an adequate aspirin level, increased aspirin resistance, and greater thrombotic potential.
"The results of our study offer a new option to overcome biological aspirin resistance in patients with diabetes," said Dr. Dillinger, a cardiologist at Lariboisière Hospital in Paris. But Dr. Dillinger also cautioned that "large, prospective trials are needed to test if aspirin twice daily can decrease the rate of atherothrombotic events without increasing side effects such as gastrointestinal toxicity." Another potential drawback of twice-daily dosing is that patient compliance with the regimen may be reduced.
Dr. Dillinger also stressed that his study specifically enrolled patients with diabetes with at least one additional risk factor for loss of aspirin efficacy: They were either smokers or had elevated inflammatory markers, and hence, "the results cannot be extrapolated to the whole population of patients with diabetes and coronary artery disease." But he estimated that more than half of patients with diabetes and coronary artery disease (CAD) likely have at least one of these increased-risk factors.
"The results from Dr. Dillinger’s pharmacodynamic study show that a twice-daily regimen may offer greater antiplatelet effects than once daily and are in line with the results from prior investigations," commented Dr. Dominick J. Angiolillo, director of the Cardiovascular Research and Thrombosis Research Center at the University of Florida in Jacksonville. "The data support that [a twice-daily] regimen can be an effective strategy to improve response profiles, but this was a pharmacodynamic study and no conclusions can be made from the results on its clinical efficacy of safety. Clinical studies are warranted to support a change in practice," he said in an interview.
Dr. Dillinger and his associates enrolled 92 patients with diabetes and angiography-confirmed CAD who were already on aspirin treatment. All patients had been first diagnosed with diabetes after they were 30 years old, and they had not received insulin as their initial diabetes treatment unless their treatment had begun during an episode of acute coronary syndrome. In addition, all enrolled patients had at least one of these additional risk factors: current smoker, a high-sensitivity C-reaction protein (hsCRP) level of more than 4 mg/L, a fibrinogen level of more than 4 g/L, or a platelet count of more than 270,000/mm3.
The patients averaged 64 years old, 85% were men, and they entered the study an average of 12 years following their diabetes diagnosis. Of the 92 patients, 48% had one high-risk marker, 30% had two, 15% had three, and 7% had all four of the high-risk markers. Forty-eight (52%) of the patients entered the study on a daily clopidogrel regimen, and that treatment continued.
Patients were randomized to receive either 150 mg aspirin once daily taken every morning for 7-14 days before blood sampling, or 75 mg of aspirin taken twice daily, once in the morning and once in the evening for 7-14 days before their blood was drawn. Following the blood draw, they immediately switched to the alternative regimen that again continued for 7-14 days before the researchers took a second blood sample.
They assessed aspirin resistance by measuring platelet aggregation of each blood specimen using light transmission aggregometry. Specimens that produced a maximum aggregation intensity of at least 20% were considered to indicate aspirin resistance.
Testing revealed aspirin resistance in 39 (42%) patients when the study group received aspirin once daily, and in 16 patients (17%) when they were on the twice-daily regimen, a statistically significant difference.
When the 39 patients who showed aspirin resistance on the once-daily dosage were switched to twice-daily, 24 became aspirin responders on the twice-daily schedule. In contrast, among the 16 patients who were aspirin resistant on twice-daily treatment, 1 patient became responsive on the once-daily regimen, Dr. Dillinger reported.
The researchers also applied a second measure of platelet function – global platelet reactivity – using a platelet function analyzer. By this measure, they found that 38 (41%) of patients had normal platelet reactivity with once-daily aspirin, compared with 27 (29%) when patients were on twice-daily treatment. Within the subgroup of 27 patients who were resistant on either of the two regimens but not the other, 19 (70%) became responders when switched to the bid regimen, with 8 (30%) becoming responders when switched to once daily, a statistically significant difference between the two regimens.
Clopidogrel treatment had no discernable impact on the links between dosing frequency and aspirin’s antiplatelet effects.
A multivariate analysis of factors potentially affecting aspirin’s antiplatelet effect found that twice-daily treatment linked with a statistically significant 79% cut in the likelihood of aspirin resistance, compared with once daily. The only other parameter measured with a significant impact on aspirin resistance in this analysis was level of hsCRP. Every 1 mg/L rise in hsCRP linked with a 15% increased rate of resistance.
Dr. Dillinger also reported these findings in a published article (Am. Heart J. 2012;164:600-6)
Dr. Dillinger had no disclosures. Dr. Angiolillo said that he has received honoraria from several drug and device companies including Bristol Myers Squibb, Sanofi-Aventis, Eli Lilly, and others. He has also received research grants from several companies
On Twitter @mitchelzoler
AT EUROPCR 2013
Major finding: Aspirin resistance occurred in 42% of patients receiving 150 mg aspirin once daily and in 17% on 75 mg twice daily.
Data source: A single-center crossover study of 92 patients with diabetes and coronary artery disease.
Disclosures: Dr. Dillinger had no disclosures. Dr. Angiolillo said that he has received honoraria from several drug and device companies including Bristol Myers Squibb, Sanofi-Aventis, Eli Lilly, and others. He has also received research grants from several companies.

Renal denervation benefits moderate-hypertension patients
PARIS – Renal denervation may help lower blood pressure in patients with moderate hypertension that is resistant to drug treatment, a study has shown.
In a pilot, uncontrolled study with 54 patients at one German center, renal denervation was safe and reasonably effective for patients with treatment-resistant hypertension and an average baseline blood pressure of 151/82 mm Hg.
The patients in the study had persistent office blood pressure measures of at least 140/90 mm Hg but less than 160/100 mm Hg, despite stable treatment with at least three antihypertensive drugs, including one diuretic. To enter the study, patients also needed 24-hour blood pressure monitoring showing consistent blood pressure levels of at least 130/80 mm Hg.

A total of 51% of the patients reached their target blood pressure of less than 140/90 mm Hg at 6 months after renal denervation treatment, Dr. Roland E. Schmieder said at the annual meeting of the European Association of Percutaneous Cardiovascular Intervention.
The percentage of patients reaching their goal pressure in the new study surpassed the outcome reported in the Symplicity HTN-2 trial, which showed that 39% of patients with severe treatment-resistant hypertension reached their goal pressure 6 months after renal denervation (Lancet 2010;376:1903-9).
Patients in the new study had their blood pressure fall by substantially less than in the Symplicity HTN-2 study, by an average of 12.5/7.5 mm Hg after 6 months in the new study, compared with 32/12 mm Hg in the renal denervation arm of Symplicity HTN-2. However, the smaller reduction was expected because patients in the new study entered treatment with much lower blood pressures than did patients in the older trial.
"The lower the starting blood pressure, the less the response" to renal denervation, explained Dr. Schmieder, professor and head of the clinical research center of hypertension and vascular medicine at University Hospital in Erlangen, Germany. Several years of study on the effect of renal denervation clearly established this relationship, he noted.
The new study enrolled patients with a mean age of 64 years who all underwent standard renal denervation treatment using the Symplicity Flex system. The procedures produced no complications, and in addition to reducing office blood pressure measurements, the treatment produced an average reduction in heart rate of 4 bpm after 6 months. The reported changes in blood pressure occurred even though 37% of the patients in the study reduced their antihypertensive treatment regimen, despite instructions from their physicians to continue their usual treatment dosages. Moreover, none of the patients received an increase in their hypertension treatment medications during follow-up, Dr. Schmieder said.
"Our data are encouraging" for using renal denervation to treat moderate treatment-resistant hypertension, he concluded. Researchers will soon start a multicenter, controlled study to more definitively assess the safety and efficacy of renal denervation in patients with resistant hypertension with systolic blood pressures of 140-159 mm Hg, Dr. Schmieder said.
The study received no commercial support. Dr. Schmieder said he has been a consultant to and has received research support from Medtronic, the company that markets the Symplicity renal denervation systems.
On Twitter @mitchelzoler
PARIS – Renal denervation may help lower blood pressure in patients with moderate hypertension that is resistant to drug treatment, a study has shown.
In a pilot, uncontrolled study with 54 patients at one German center, renal denervation was safe and reasonably effective for patients with treatment-resistant hypertension and an average baseline blood pressure of 151/82 mm Hg.
The patients in the study had persistent office blood pressure measures of at least 140/90 mm Hg but less than 160/100 mm Hg, despite stable treatment with at least three antihypertensive drugs, including one diuretic. To enter the study, patients also needed 24-hour blood pressure monitoring showing consistent blood pressure levels of at least 130/80 mm Hg.
A total of 51% of the patients reached their target blood pressure of less than 140/90 mm Hg at 6 months after renal denervation treatment, Dr. Roland E. Schmieder said at the annual meeting of the European Association of Percutaneous Cardiovascular Intervention.
The percentage of patients reaching their goal pressure in the new study surpassed the outcome reported in the Symplicity HTN-2 trial, which showed that 39% of patients with severe treatment-resistant hypertension reached their goal pressure 6 months after renal denervation (Lancet 2010;376:1903-9).
Patients in the new study had their blood pressure fall by substantially less than in the Symplicity HTN-2 study, by an average of 12.5/7.5 mm Hg after 6 months in the new study, compared with 32/12 mm Hg in the renal denervation arm of Symplicity HTN-2. However, the smaller reduction was expected because patients in the new study entered treatment with much lower blood pressures than did patients in the older trial.
"The lower the starting blood pressure, the less the response" to renal denervation, explained Dr. Schmieder, professor and head of the clinical research center of hypertension and vascular medicine at University Hospital in Erlangen, Germany. Several years of study on the effect of renal denervation clearly established this relationship, he noted.
The new study enrolled patients with a mean age of 64 years who all underwent standard renal denervation treatment using the Symplicity Flex system. The procedures produced no complications, and in addition to reducing office blood pressure measurements, the treatment produced an average reduction in heart rate of 4 bpm after 6 months. The reported changes in blood pressure occurred even though 37% of the patients in the study reduced their antihypertensive treatment regimen, despite instructions from their physicians to continue their usual treatment dosages. Moreover, none of the patients received an increase in their hypertension treatment medications during follow-up, Dr. Schmieder said.
"Our data are encouraging" for using renal denervation to treat moderate treatment-resistant hypertension, he concluded. Researchers will soon start a multicenter, controlled study to more definitively assess the safety and efficacy of renal denervation in patients with resistant hypertension with systolic blood pressures of 140-159 mm Hg, Dr. Schmieder said.
The study received no commercial support. Dr. Schmieder said he has been a consultant to and has received research support from Medtronic, the company that markets the Symplicity renal denervation systems.
On Twitter @mitchelzoler
PARIS – Renal denervation may help lower blood pressure in patients with moderate hypertension that is resistant to drug treatment, a study has shown.
In a pilot, uncontrolled study with 54 patients at one German center, renal denervation was safe and reasonably effective for patients with treatment-resistant hypertension and an average baseline blood pressure of 151/82 mm Hg.
The patients in the study had persistent office blood pressure measures of at least 140/90 mm Hg but less than 160/100 mm Hg, despite stable treatment with at least three antihypertensive drugs, including one diuretic. To enter the study, patients also needed 24-hour blood pressure monitoring showing consistent blood pressure levels of at least 130/80 mm Hg.
A total of 51% of the patients reached their target blood pressure of less than 140/90 mm Hg at 6 months after renal denervation treatment, Dr. Roland E. Schmieder said at the annual meeting of the European Association of Percutaneous Cardiovascular Intervention.
The percentage of patients reaching their goal pressure in the new study surpassed the outcome reported in the Symplicity HTN-2 trial, which showed that 39% of patients with severe treatment-resistant hypertension reached their goal pressure 6 months after renal denervation (Lancet 2010;376:1903-9).
Patients in the new study had their blood pressure fall by substantially less than in the Symplicity HTN-2 study, by an average of 12.5/7.5 mm Hg after 6 months in the new study, compared with 32/12 mm Hg in the renal denervation arm of Symplicity HTN-2. However, the smaller reduction was expected because patients in the new study entered treatment with much lower blood pressures than did patients in the older trial.
"The lower the starting blood pressure, the less the response" to renal denervation, explained Dr. Schmieder, professor and head of the clinical research center of hypertension and vascular medicine at University Hospital in Erlangen, Germany. Several years of study on the effect of renal denervation clearly established this relationship, he noted.
The new study enrolled patients with a mean age of 64 years who all underwent standard renal denervation treatment using the Symplicity Flex system. The procedures produced no complications, and in addition to reducing office blood pressure measurements, the treatment produced an average reduction in heart rate of 4 bpm after 6 months. The reported changes in blood pressure occurred even though 37% of the patients in the study reduced their antihypertensive treatment regimen, despite instructions from their physicians to continue their usual treatment dosages. Moreover, none of the patients received an increase in their hypertension treatment medications during follow-up, Dr. Schmieder said.
"Our data are encouraging" for using renal denervation to treat moderate treatment-resistant hypertension, he concluded. Researchers will soon start a multicenter, controlled study to more definitively assess the safety and efficacy of renal denervation in patients with resistant hypertension with systolic blood pressures of 140-159 mm Hg, Dr. Schmieder said.
The study received no commercial support. Dr. Schmieder said he has been a consultant to and has received research support from Medtronic, the company that markets the Symplicity renal denervation systems.
On Twitter @mitchelzoler
AT EUROPCR 2013
Major finding: Renal denervation led to attainment of goal blood pressure in 51% of patients with moderate, treatment-resistant hypertension.
Data source: A single-center uncontrolled study with 54 patients.
Disclosures: The study received no commercial support. Dr. Schmieder said he has been a consultant to and has received research support from Medtronic, the company that markets the Symplicity renal denervation systems.

Cardiac rehabilitation benefits elderly heart failure patients
ROME – A multiweek program of cardiac rehabilitation is as beneficial in elderly patients with chronic heart failure as it is in younger heart failure patients, according to a review of 243 patients at one Belgium center.
"Although they have lower exercise capacity at baseline, older patients have at least as much benefit from an exercise program as younger patients with chronic heart failure," Ms. Sofie Pardaens* reported in a poster at the annual meeting of the European Association for Cardiovascular Prevention and Rehabilitation.
A second analysis by Ms. Pardaens, a researcher at Ghent (Belgium) University, and her associates, reported in a separate poster, showed that a prolonged cardiac rehabilitation program was as effective in patients recently discharged from a heart failure hospitalization as it was in patients following cardiac surgery or after an acute coronary syndrome (ACS) event.

Their assessment of cardiac rehabilitation relative to a patient’s age included 243 patients who participated in a rehabilitation program at the University of Ghent, who had chronic heart failure, and who had an amino-terminal pro-B-type natriuretic peptide value of at least 400 pg/mL, a level very suggestive of heart failure (Circulation 2011;123:2015-9). The group included 43 patients (18%) who were at least 75 years old (average, 78 years) and 68 patients younger than 60 (average, 51 years), with the remaining 132 patients evenly distributed across the range of 60-74 years old.
All participants had just been hospitalized, for an ACS event, cardiac surgery, or heart failure.
The hospital-based rehabilitation program combined aerobic and strength training, and was designed to bring a patient’s heart rate to his anaerobic threshold during each session. Sessions occurred two or three times a week, and the full program included 45 sessions over a period of 4-5 months. The patients studied averaged 34 sessions each; patients aged 75 or older averaged 32 sessions each, while those younger than 60 averaged 35 sessions each.
The researchers measured peak exercise capacity using cardiopulmonary exercise testing at baseline and at the end of the rehabilitation session sequence, and found that the 16% average level of improvement among patients at least 75 years old closely matched the average 19% improvement among the patients younger than 60, and the 17% average improvement among everyone else, Ms. Pardaens and her associates reported. All age groups also showed similar improvements in their average ventilatory equivalence ratio, as well as their average 6-minute walk distance; however, the 29% average increased distance among patients younger than 60 years significantly exceeded the 19% average increase among those aged 75 or older.
The group’s second analysis focused on the 371 patients who underwent cardiac rehabilitation at the University of Ghent during January 2010 through May 2012 from among the 1,253 patients hospitalized during this period for an acute coronary syndrome event, cardiac surgery, or heart failure. In this pool of more than 1,000 patients who were potentially eligible to participate, only 30% actually enrolled in the rehabilitation program. The cardiac rehabilitation program again involved two to three sessions per week, with a goal for patients to complete 45 sessions within 5 months.
The sign-up rate for rehabilitation lagged even more among the 428 patients from the larger group whose index hospitalization had been for heart failure, with 37 of the acute heart failure patients (9%) actually engaging in rehabilitation. Rehabilitation participation was highest, a 56% rate, among the 358 patients who had been hospitalized for cardiac surgery, with a 28% uptake rate among 467 patients who had an ACS event.
Despite the low, 9% uptake of cardiac rehabilitation in heart failure patients, their benefit from participation closely tracked the benefit seen in surgery and ACS patients. Improvement in peak exercise capacity over baseline at the end of rehabilitation averaged 19% in the heart failure patients, 17% in the ACS patients, and 24% in the surgery patients, differences that were not statistically significant, reported Ms. Pardaens. All three subgroups also had similar average improvements in their 6-minute walk distance, which rose by an average of 21% in the heart failure patients and by averages of 27% and 28% in the other two subgroups.
Based on the efficacy but low usage of cardiac rehabilitation, future research should examine ways to boost its use by heart failure patients, concluded Ms. Pardaens.
Ms. Pardaens and her associates said they had no relevant financial disclosures.
On Twitter @mitchelzoler
*Correction 6/28/13: An earlier version of this article incorrectly reported researcher Sofie Pardaens' title. She is currently studying for her PhD in the department of internal medicine at Ghent University.
Cardiac rehabilitation is undoubtedly an essential component of the contemporary treatment of patients with coronary disease and heart failure.
Exercise training has the potential to act as a catalyst for promoting other aspects of rehabilitation, including risk factor modification through therapeutic lifestyle changes and optimization of psychosocial support. Similarly, among patients who are elderly, such outcome measures may include the achievement of functional independence, the prevention of premature disability, and a reduction in the need for custodial care.
Despite limited data, older patients have shown improvement in their exercise tolerance comparable to that of younger patients participating in equivalent exercise programs. In addition, the safety of exercise within cardiac rehabilitation programs is well accepted and established.
Dr. Jun Chiong is associate professor of medicine at Loma Linda (Calif.) University Medical Center. He is on the on the advisory board of CHEST Physician.
Cardiac rehabilitation is undoubtedly an essential component of the contemporary treatment of patients with coronary disease and heart failure.
Exercise training has the potential to act as a catalyst for promoting other aspects of rehabilitation, including risk factor modification through therapeutic lifestyle changes and optimization of psychosocial support. Similarly, among patients who are elderly, such outcome measures may include the achievement of functional independence, the prevention of premature disability, and a reduction in the need for custodial care.
Despite limited data, older patients have shown improvement in their exercise tolerance comparable to that of younger patients participating in equivalent exercise programs. In addition, the safety of exercise within cardiac rehabilitation programs is well accepted and established.
Dr. Jun Chiong is associate professor of medicine at Loma Linda (Calif.) University Medical Center. He is on the on the advisory board of CHEST Physician.
Cardiac rehabilitation is undoubtedly an essential component of the contemporary treatment of patients with coronary disease and heart failure.
Exercise training has the potential to act as a catalyst for promoting other aspects of rehabilitation, including risk factor modification through therapeutic lifestyle changes and optimization of psychosocial support. Similarly, among patients who are elderly, such outcome measures may include the achievement of functional independence, the prevention of premature disability, and a reduction in the need for custodial care.
Despite limited data, older patients have shown improvement in their exercise tolerance comparable to that of younger patients participating in equivalent exercise programs. In addition, the safety of exercise within cardiac rehabilitation programs is well accepted and established.
Dr. Jun Chiong is associate professor of medicine at Loma Linda (Calif.) University Medical Center. He is on the on the advisory board of CHEST Physician.
ROME – A multiweek program of cardiac rehabilitation is as beneficial in elderly patients with chronic heart failure as it is in younger heart failure patients, according to a review of 243 patients at one Belgium center.
"Although they have lower exercise capacity at baseline, older patients have at least as much benefit from an exercise program as younger patients with chronic heart failure," Ms. Sofie Pardaens* reported in a poster at the annual meeting of the European Association for Cardiovascular Prevention and Rehabilitation.
A second analysis by Ms. Pardaens, a researcher at Ghent (Belgium) University, and her associates, reported in a separate poster, showed that a prolonged cardiac rehabilitation program was as effective in patients recently discharged from a heart failure hospitalization as it was in patients following cardiac surgery or after an acute coronary syndrome (ACS) event.
Their assessment of cardiac rehabilitation relative to a patient’s age included 243 patients who participated in a rehabilitation program at the University of Ghent, who had chronic heart failure, and who had an amino-terminal pro-B-type natriuretic peptide value of at least 400 pg/mL, a level very suggestive of heart failure (Circulation 2011;123:2015-9). The group included 43 patients (18%) who were at least 75 years old (average, 78 years) and 68 patients younger than 60 (average, 51 years), with the remaining 132 patients evenly distributed across the range of 60-74 years old.
All participants had just been hospitalized, for an ACS event, cardiac surgery, or heart failure.
The hospital-based rehabilitation program combined aerobic and strength training, and was designed to bring a patient’s heart rate to his anaerobic threshold during each session. Sessions occurred two or three times a week, and the full program included 45 sessions over a period of 4-5 months. The patients studied averaged 34 sessions each; patients aged 75 or older averaged 32 sessions each, while those younger than 60 averaged 35 sessions each.
The researchers measured peak exercise capacity using cardiopulmonary exercise testing at baseline and at the end of the rehabilitation session sequence, and found that the 16% average level of improvement among patients at least 75 years old closely matched the average 19% improvement among the patients younger than 60, and the 17% average improvement among everyone else, Ms. Pardaens and her associates reported. All age groups also showed similar improvements in their average ventilatory equivalence ratio, as well as their average 6-minute walk distance; however, the 29% average increased distance among patients younger than 60 years significantly exceeded the 19% average increase among those aged 75 or older.
The group’s second analysis focused on the 371 patients who underwent cardiac rehabilitation at the University of Ghent during January 2010 through May 2012 from among the 1,253 patients hospitalized during this period for an acute coronary syndrome event, cardiac surgery, or heart failure. In this pool of more than 1,000 patients who were potentially eligible to participate, only 30% actually enrolled in the rehabilitation program. The cardiac rehabilitation program again involved two to three sessions per week, with a goal for patients to complete 45 sessions within 5 months.
The sign-up rate for rehabilitation lagged even more among the 428 patients from the larger group whose index hospitalization had been for heart failure, with 37 of the acute heart failure patients (9%) actually engaging in rehabilitation. Rehabilitation participation was highest, a 56% rate, among the 358 patients who had been hospitalized for cardiac surgery, with a 28% uptake rate among 467 patients who had an ACS event.
Despite the low, 9% uptake of cardiac rehabilitation in heart failure patients, their benefit from participation closely tracked the benefit seen in surgery and ACS patients. Improvement in peak exercise capacity over baseline at the end of rehabilitation averaged 19% in the heart failure patients, 17% in the ACS patients, and 24% in the surgery patients, differences that were not statistically significant, reported Ms. Pardaens. All three subgroups also had similar average improvements in their 6-minute walk distance, which rose by an average of 21% in the heart failure patients and by averages of 27% and 28% in the other two subgroups.
Based on the efficacy but low usage of cardiac rehabilitation, future research should examine ways to boost its use by heart failure patients, concluded Ms. Pardaens.
Ms. Pardaens and her associates said they had no relevant financial disclosures.
On Twitter @mitchelzoler
*Correction 6/28/13: An earlier version of this article incorrectly reported researcher Sofie Pardaens' title. She is currently studying for her PhD in the department of internal medicine at Ghent University.
ROME – A multiweek program of cardiac rehabilitation is as beneficial in elderly patients with chronic heart failure as it is in younger heart failure patients, according to a review of 243 patients at one Belgium center.
"Although they have lower exercise capacity at baseline, older patients have at least as much benefit from an exercise program as younger patients with chronic heart failure," Ms. Sofie Pardaens* reported in a poster at the annual meeting of the European Association for Cardiovascular Prevention and Rehabilitation.
A second analysis by Ms. Pardaens, a researcher at Ghent (Belgium) University, and her associates, reported in a separate poster, showed that a prolonged cardiac rehabilitation program was as effective in patients recently discharged from a heart failure hospitalization as it was in patients following cardiac surgery or after an acute coronary syndrome (ACS) event.
Their assessment of cardiac rehabilitation relative to a patient’s age included 243 patients who participated in a rehabilitation program at the University of Ghent, who had chronic heart failure, and who had an amino-terminal pro-B-type natriuretic peptide value of at least 400 pg/mL, a level very suggestive of heart failure (Circulation 2011;123:2015-9). The group included 43 patients (18%) who were at least 75 years old (average, 78 years) and 68 patients younger than 60 (average, 51 years), with the remaining 132 patients evenly distributed across the range of 60-74 years old.
All participants had just been hospitalized, for an ACS event, cardiac surgery, or heart failure.
The hospital-based rehabilitation program combined aerobic and strength training, and was designed to bring a patient’s heart rate to his anaerobic threshold during each session. Sessions occurred two or three times a week, and the full program included 45 sessions over a period of 4-5 months. The patients studied averaged 34 sessions each; patients aged 75 or older averaged 32 sessions each, while those younger than 60 averaged 35 sessions each.
The researchers measured peak exercise capacity using cardiopulmonary exercise testing at baseline and at the end of the rehabilitation session sequence, and found that the 16% average level of improvement among patients at least 75 years old closely matched the average 19% improvement among the patients younger than 60, and the 17% average improvement among everyone else, Ms. Pardaens and her associates reported. All age groups also showed similar improvements in their average ventilatory equivalence ratio, as well as their average 6-minute walk distance; however, the 29% average increased distance among patients younger than 60 years significantly exceeded the 19% average increase among those aged 75 or older.
The group’s second analysis focused on the 371 patients who underwent cardiac rehabilitation at the University of Ghent during January 2010 through May 2012 from among the 1,253 patients hospitalized during this period for an acute coronary syndrome event, cardiac surgery, or heart failure. In this pool of more than 1,000 patients who were potentially eligible to participate, only 30% actually enrolled in the rehabilitation program. The cardiac rehabilitation program again involved two to three sessions per week, with a goal for patients to complete 45 sessions within 5 months.
The sign-up rate for rehabilitation lagged even more among the 428 patients from the larger group whose index hospitalization had been for heart failure, with 37 of the acute heart failure patients (9%) actually engaging in rehabilitation. Rehabilitation participation was highest, a 56% rate, among the 358 patients who had been hospitalized for cardiac surgery, with a 28% uptake rate among 467 patients who had an ACS event.
Despite the low, 9% uptake of cardiac rehabilitation in heart failure patients, their benefit from participation closely tracked the benefit seen in surgery and ACS patients. Improvement in peak exercise capacity over baseline at the end of rehabilitation averaged 19% in the heart failure patients, 17% in the ACS patients, and 24% in the surgery patients, differences that were not statistically significant, reported Ms. Pardaens. All three subgroups also had similar average improvements in their 6-minute walk distance, which rose by an average of 21% in the heart failure patients and by averages of 27% and 28% in the other two subgroups.
Based on the efficacy but low usage of cardiac rehabilitation, future research should examine ways to boost its use by heart failure patients, concluded Ms. Pardaens.
Ms. Pardaens and her associates said they had no relevant financial disclosures.
On Twitter @mitchelzoler
*Correction 6/28/13: An earlier version of this article incorrectly reported researcher Sofie Pardaens' title. She is currently studying for her PhD in the department of internal medicine at Ghent University.
AT EUROPREVENT 2013
Major finding: Cardiac rehabilitation boosted exercise capacity by an average 16% in elderly patients and an average 19% in younger patients.
Data source: Data came from a review of 243 patients with chronic heart failure who participated in cardiac rehabilitation at one Belgium center.
Disclosures: Dr. Pardaens and her associates said they had no relevant financial disclosures.

Annual pulmonary hypertension screening recommended for systemic sclerosis
MADRID – Patients with systemic sclerosis should undergo annual screening for pulmonary arterial hypertension using a combination of transthoracic echocardiography and pulmonary function tests, an international expert panel said.
These are the first evidence- and consensus-based recommendations for pulmonary arterial hypertension (PAH) screening in patients with systemic sclerosis, and the panel also called for screening patients with mixed or other connective tissue diseases with scleroderma features. "Our hope is that these recommendations will lead to earlier detection of PAH in connective tissue diseases and improve patient outcomes," Dr. Dinesh Khanna said while presenting the screening recommendations at the annual European Congress of Rheumatology.

About 5%-15% of patients with systemic sclerosis develop PAH, and once PAH occurs, up to 30% of patients will die within 3 years, said Dr. Khanna, director of the scleroderma program at the University of Michigan, Ann Arbor.
"Despite having approved drugs available" to treat systemic sclerosis and other scleroderma-spectrum disorder connective tissue diseases, these treatments "have not had a huge impact on survival. The only thing we can offer patients is screening, followed by early diagnosis and treatment," Dr. Khanna said in an interview.
The new recommendations say that patients with a tricuspid regurgitant velocity measured by transthoracic echocardiography greater than 2.8 m/s require assessment for PAH by right heart catheterization. Right heart catheterization is also needed for patients with a tricuspid regurgitant velocity of 2.5-2.8 m/s if they also have signs or symptoms of PAH such as dyspnea, fatigue, chest pain, dizziness, loud pulmonary sound, or peripheral edema. Another echo finding that should trigger right heart catheterization regardless of signs or symptoms or tricuspid regurgitation is right atrial or ventricular enlargement.
The key measures on pulmonary function tests that trigger right heart catheterization is a forced vital capacity (FVC) to diffusion capacity of lungs for carbon monoxide (DLCO) ratio of more than 1.6, or a DLCO of less than 60% if either appears in the setting of PAH signs or symptoms. Alternatively, meeting either of these pulmonary criteria should lead to right heart catheterization regardless of signs and symptoms if the patient’s most recent blood level of N-terminal pro-brain natriuretic peptide (NT-ProBNP) was greater than twice the upper limit of normal.
The panel also said that patients should undergo right heart catheterization regardless of PAH signs and symptoms if they fulfill the screening algorithm developed for the DETECT study (Ann. Rheum. Dis. 2013 May 18 [doi:10.1036/annrheumdis-2013-203301]).
The panel recommended annual transthoracic echo and pulmonary function test screening, or more frequently if a patient shows new signs or symptoms. Measurement of NT-ProBNP should happen at baseline, and then be repeated if new signs or symptoms of PAH appear. They also recommended applying the full DETECT screening algorithm in patients diagnosed with systemic sclerosis or other scleroderma spectrum connective-tissue disease for more than 3 years and a DLCO that is less than 60%. Right heart catheterization is mandatory to definitively diagnose PAH, Dr. Khanna stressed. The panel also said screening is not needed in patients with mixed- or other connective tissue disorders who did not have scleroderma-like features.
In a separate report at the meeting Dr. Khanna and his associates assessed the ability of transthoracic echocardiography and pulmonary function tests to screen patients with PAH. They used data from 69 patients with PAH in two separate reported series that together had 347 patients with systemic sclerosis who underwent assessment for suspected PAH (J. Rheumatol. 2011;38:2172-9 and J. Rheumatol. 2010;37:2290-8).
The new, retrospective analysis showed that combining transthoracic echo and pulmonary-function test screens can have a negative predictive accuracy of 98% for correctly ruling out PAH in patients with systemic sclerosis, reported Dr. Heather Gladue, a rheumatology fellow at the University of Michigan.
The recommendations panel cautioned that its proposals should not substitute for individualized, direct assessment of each patient. The panel also noted that the cost effectiveness of its recommendations had not yet been assessed. In addition to representatives from the University of Michigan, the task force included members from the University of California, Los Angeles; Massachusetts General Hospital, Boston; Stanford (Calif.) University; the University of Zurich; University Hospital in Lille, France; the University of Paris-South; McGill University, Montreal; Johns Hopkins University, Baltimore; and St. Joseph Hospital, Phoenix.
The task force was supported by the Scleroderma Foundation and the Pulmonary Hypertension Association. Dr. Khanna said that he has been a consultant to several drug companies including Actelion, Bayer, Genentech/Roche, Gilead, Merck, and DIGNA. Dr. Gladue said that she had no disclosures.
Twitter @mitchelzoler
MADRID – Patients with systemic sclerosis should undergo annual screening for pulmonary arterial hypertension using a combination of transthoracic echocardiography and pulmonary function tests, an international expert panel said.
These are the first evidence- and consensus-based recommendations for pulmonary arterial hypertension (PAH) screening in patients with systemic sclerosis, and the panel also called for screening patients with mixed or other connective tissue diseases with scleroderma features. "Our hope is that these recommendations will lead to earlier detection of PAH in connective tissue diseases and improve patient outcomes," Dr. Dinesh Khanna said while presenting the screening recommendations at the annual European Congress of Rheumatology.
About 5%-15% of patients with systemic sclerosis develop PAH, and once PAH occurs, up to 30% of patients will die within 3 years, said Dr. Khanna, director of the scleroderma program at the University of Michigan, Ann Arbor.
"Despite having approved drugs available" to treat systemic sclerosis and other scleroderma-spectrum disorder connective tissue diseases, these treatments "have not had a huge impact on survival. The only thing we can offer patients is screening, followed by early diagnosis and treatment," Dr. Khanna said in an interview.
The new recommendations say that patients with a tricuspid regurgitant velocity measured by transthoracic echocardiography greater than 2.8 m/s require assessment for PAH by right heart catheterization. Right heart catheterization is also needed for patients with a tricuspid regurgitant velocity of 2.5-2.8 m/s if they also have signs or symptoms of PAH such as dyspnea, fatigue, chest pain, dizziness, loud pulmonary sound, or peripheral edema. Another echo finding that should trigger right heart catheterization regardless of signs or symptoms or tricuspid regurgitation is right atrial or ventricular enlargement.
The key measures on pulmonary function tests that trigger right heart catheterization is a forced vital capacity (FVC) to diffusion capacity of lungs for carbon monoxide (DLCO) ratio of more than 1.6, or a DLCO of less than 60% if either appears in the setting of PAH signs or symptoms. Alternatively, meeting either of these pulmonary criteria should lead to right heart catheterization regardless of signs and symptoms if the patient’s most recent blood level of N-terminal pro-brain natriuretic peptide (NT-ProBNP) was greater than twice the upper limit of normal.
The panel also said that patients should undergo right heart catheterization regardless of PAH signs and symptoms if they fulfill the screening algorithm developed for the DETECT study (Ann. Rheum. Dis. 2013 May 18 [doi:10.1036/annrheumdis-2013-203301]).
The panel recommended annual transthoracic echo and pulmonary function test screening, or more frequently if a patient shows new signs or symptoms. Measurement of NT-ProBNP should happen at baseline, and then be repeated if new signs or symptoms of PAH appear. They also recommended applying the full DETECT screening algorithm in patients diagnosed with systemic sclerosis or other scleroderma spectrum connective-tissue disease for more than 3 years and a DLCO that is less than 60%. Right heart catheterization is mandatory to definitively diagnose PAH, Dr. Khanna stressed. The panel also said screening is not needed in patients with mixed- or other connective tissue disorders who did not have scleroderma-like features.
In a separate report at the meeting Dr. Khanna and his associates assessed the ability of transthoracic echocardiography and pulmonary function tests to screen patients with PAH. They used data from 69 patients with PAH in two separate reported series that together had 347 patients with systemic sclerosis who underwent assessment for suspected PAH (J. Rheumatol. 2011;38:2172-9 and J. Rheumatol. 2010;37:2290-8).
The new, retrospective analysis showed that combining transthoracic echo and pulmonary-function test screens can have a negative predictive accuracy of 98% for correctly ruling out PAH in patients with systemic sclerosis, reported Dr. Heather Gladue, a rheumatology fellow at the University of Michigan.
The recommendations panel cautioned that its proposals should not substitute for individualized, direct assessment of each patient. The panel also noted that the cost effectiveness of its recommendations had not yet been assessed. In addition to representatives from the University of Michigan, the task force included members from the University of California, Los Angeles; Massachusetts General Hospital, Boston; Stanford (Calif.) University; the University of Zurich; University Hospital in Lille, France; the University of Paris-South; McGill University, Montreal; Johns Hopkins University, Baltimore; and St. Joseph Hospital, Phoenix.
The task force was supported by the Scleroderma Foundation and the Pulmonary Hypertension Association. Dr. Khanna said that he has been a consultant to several drug companies including Actelion, Bayer, Genentech/Roche, Gilead, Merck, and DIGNA. Dr. Gladue said that she had no disclosures.
Twitter @mitchelzoler
MADRID – Patients with systemic sclerosis should undergo annual screening for pulmonary arterial hypertension using a combination of transthoracic echocardiography and pulmonary function tests, an international expert panel said.
These are the first evidence- and consensus-based recommendations for pulmonary arterial hypertension (PAH) screening in patients with systemic sclerosis, and the panel also called for screening patients with mixed or other connective tissue diseases with scleroderma features. "Our hope is that these recommendations will lead to earlier detection of PAH in connective tissue diseases and improve patient outcomes," Dr. Dinesh Khanna said while presenting the screening recommendations at the annual European Congress of Rheumatology.
About 5%-15% of patients with systemic sclerosis develop PAH, and once PAH occurs, up to 30% of patients will die within 3 years, said Dr. Khanna, director of the scleroderma program at the University of Michigan, Ann Arbor.
"Despite having approved drugs available" to treat systemic sclerosis and other scleroderma-spectrum disorder connective tissue diseases, these treatments "have not had a huge impact on survival. The only thing we can offer patients is screening, followed by early diagnosis and treatment," Dr. Khanna said in an interview.
The new recommendations say that patients with a tricuspid regurgitant velocity measured by transthoracic echocardiography greater than 2.8 m/s require assessment for PAH by right heart catheterization. Right heart catheterization is also needed for patients with a tricuspid regurgitant velocity of 2.5-2.8 m/s if they also have signs or symptoms of PAH such as dyspnea, fatigue, chest pain, dizziness, loud pulmonary sound, or peripheral edema. Another echo finding that should trigger right heart catheterization regardless of signs or symptoms or tricuspid regurgitation is right atrial or ventricular enlargement.
The key measures on pulmonary function tests that trigger right heart catheterization is a forced vital capacity (FVC) to diffusion capacity of lungs for carbon monoxide (DLCO) ratio of more than 1.6, or a DLCO of less than 60% if either appears in the setting of PAH signs or symptoms. Alternatively, meeting either of these pulmonary criteria should lead to right heart catheterization regardless of signs and symptoms if the patient’s most recent blood level of N-terminal pro-brain natriuretic peptide (NT-ProBNP) was greater than twice the upper limit of normal.
The panel also said that patients should undergo right heart catheterization regardless of PAH signs and symptoms if they fulfill the screening algorithm developed for the DETECT study (Ann. Rheum. Dis. 2013 May 18 [doi:10.1036/annrheumdis-2013-203301]).
The panel recommended annual transthoracic echo and pulmonary function test screening, or more frequently if a patient shows new signs or symptoms. Measurement of NT-ProBNP should happen at baseline, and then be repeated if new signs or symptoms of PAH appear. They also recommended applying the full DETECT screening algorithm in patients diagnosed with systemic sclerosis or other scleroderma spectrum connective-tissue disease for more than 3 years and a DLCO that is less than 60%. Right heart catheterization is mandatory to definitively diagnose PAH, Dr. Khanna stressed. The panel also said screening is not needed in patients with mixed- or other connective tissue disorders who did not have scleroderma-like features.
In a separate report at the meeting Dr. Khanna and his associates assessed the ability of transthoracic echocardiography and pulmonary function tests to screen patients with PAH. They used data from 69 patients with PAH in two separate reported series that together had 347 patients with systemic sclerosis who underwent assessment for suspected PAH (J. Rheumatol. 2011;38:2172-9 and J. Rheumatol. 2010;37:2290-8).
The new, retrospective analysis showed that combining transthoracic echo and pulmonary-function test screens can have a negative predictive accuracy of 98% for correctly ruling out PAH in patients with systemic sclerosis, reported Dr. Heather Gladue, a rheumatology fellow at the University of Michigan.
The recommendations panel cautioned that its proposals should not substitute for individualized, direct assessment of each patient. The panel also noted that the cost effectiveness of its recommendations had not yet been assessed. In addition to representatives from the University of Michigan, the task force included members from the University of California, Los Angeles; Massachusetts General Hospital, Boston; Stanford (Calif.) University; the University of Zurich; University Hospital in Lille, France; the University of Paris-South; McGill University, Montreal; Johns Hopkins University, Baltimore; and St. Joseph Hospital, Phoenix.
The task force was supported by the Scleroderma Foundation and the Pulmonary Hypertension Association. Dr. Khanna said that he has been a consultant to several drug companies including Actelion, Bayer, Genentech/Roche, Gilead, Merck, and DIGNA. Dr. Gladue said that she had no disclosures.
Twitter @mitchelzoler
AT THE EULAR CONGRESS 2013

Synthetic triple therapy matches anti-TNF therapy in RA
MADRID – When rheumatoid arthritis patients fail initial treatment with methotrexate monotherapy, are they better served by a less expensive step-up treatment or the one that may better slow their radiographic progression and produce faster responses?
That seems to be the choice between step-up from methotrexate monotherapy by adding synthetic disease-modifying antirheumatic drugs (DMARDs) such as sulfasalazine and hydroxychloroquine, or by adding a tumor necrosis factor (TNF) inhibitor such as etanercept.

The RACAT (Rheumatoid Arthritis: Comparison of Active Therapies) trial tested those two options in 353 rheumatoid arthritis (RA) patients at 36 U.S. or Canadian sites between July 2007 and December 2010. The study’s primary endpoint, improvement in average disease activity score in 28 joints (DAS28) from baseline to 48 weeks of treatment, was similar in both treatment arms, proving the noninferiority of the triple synthetic DMARD regimen against the etanercept, biological arm, Dr. James R. O’Dell reported in a poster at the annual European Congress of Rheumatology. The results also appeared online simultaneously with Dr. O’Dell’s poster presentation (N. Engl. J. Med. 2013 June 11 [doi: 10.1056/NEJMoal1303006]).
"We showed that starting first with a TNF inhibitor or first with triple therapy resulted in the same outcomes. But costs were not the same. We [successfully] treated another 30% of patients who did not need a biologic" with the synthetic triple therapy. "And in the patients where triple therapy doesn’t work, you can change them to a biologic and they have identical outcomes, clinically by DAS28 and radiographically," compared with patients who began on etanercept added to methotrexate from the start, he said. Dr. O’Dell is chief of rheumatology at the VA Medical Center in Omaha, Neb., and professor of medicine at the University of Nebraska.
"Nothing is lost for the patient; if they don’t do well on triple therapy they can switch to a biological. The data are persuasive and the economic case is easy to make. You absolutely ought to go with triple therapy," he concluded based on the study findings.
But "it is very difficult to get physicians to buy into this" strategy, he said in an interview. TNF inhibitors such as etanercept and other biological DMARDs are "seductive," Dr. O’Dell said, because they are effective, work quickly, and appear more "targeted" than synthetic DMARDs, and they are promoted by well-financed marketing campaigns.
Dr. O’Dell conceded that the study data showed a signal of more radiographic progression with triple synthetic DMARD treatment that could potentially, over time, accrue to more substantial differences. At 48 weeks after the onset of treatment, patients on triple therapy had an average 0.54-point increase (worsening) in their van der Heijde modified Sharp score, compared with an average 0.29-point rise in the patients who received etanercept, a 0.25-point average difference in favor of etanercept that did not reach statistical significance.
But this trend toward greater radiographic progression in patients on triple therapy was consistent with the statistically significant, roughly 1-point average additional increased radiographic progression with triple therapy, compared with patients on methotrexate plus etanercept, that was seen after 2 years of follow-up in the TEAR (Treatment of Early Aggressive Rheumatoid Arthritis) trial (Arthritis Rheum. 2012;64:2824-35).
The TEAR study, which enrolled patients with very early RA, included a subgroup with an inadequate response to methotrexate monotherapy, and in that subgroup, patients randomized to triple therapy and those randomized to etanercept plus methotrexate had similar clinical outcomes, consistent with the new study findings.

"It’s hard to say that 1 or 2 units on the Sharp score doesn’t matter much, even if you don’t see the difference for several years," commented Dr. Daniel Furst, professor of medicine and director of the Rheumatology Clinical Research Center at the University of California, Los Angeles.
"Triple therapy is effective clinically, but it doesn’t do so well for x-rays. There are relationships between a 1 or 2 Sharp score difference and long-term outcomes. Across the board with biologics, the difference is 1 or 2 Sharp units. If one’s philosophy is to hit the RA hard and stop it, then perhaps a biologic is better," Dr. Furst commented in an interview.
But another view was that triple therapy could play a useful and cost-effective role. The new findings, along with the TEAR results, make "initial treatment of early RA with triple therapy a reasonable approach," said Dr. Joel M. Kremer, a professor of medicine at Albany (N.Y.) Medical College and director of research at the Center for Rheumatology in Albany. The only clear downside is that if triple therapy doesn’t work, the patient loses time, but that’s true for every treatment option, he noted.
"We don’t usually have the opportunity to hear the data for generic drugs" as much as for brand-name formulations, Dr. Kremer said in an interview. "Will data like this substantially change prescribing patterns? Probably not, but what might happen is that insurers may look at these data and say that patients should fail triple therapy before starting a biologic. That would be a sea change" for rheumatology, he added.
"I have always used methotrexate first, usually in combination with hydroxychloroquine," Dr. Kremer said. He has not usually also prescribed sulfasalazine, but said he would consider adding it based on the new data.
The new study enrolled patients with a DAS28 score of 4.4 or higher despite at least 12 weeks of stable methotrexate therapy with a weekly dosage of 15-25 mg. The patients averaged about 57 years old. After the first 24 weeks on randomized treatment, patients who did not have a decrease in their DAS28 of at least 1.2 units switched to the alternate regimens. The primary outcome was change in DAS28 at week 48 according to initial treatment assignment; the researchers collected 48-week DAS28 scores from 309 enrolled patients. The mean change in DAS28 from baseline at 48 weeks was a 2.12-unit reduction in the triple-therapy patients and a 2.29-unit reduction in the etanercept patients, an average 0.17-unit difference between the two treatment arms that was not statistically significant and that fell within the study’s prespecified range for noninferiority for triple therapy.
A similar percentage of patients, 27%, in each of the treatment arms switched to the alternate therapy at 24 weeks due to lack of an adequate initial response. There were also no statistically significant differences between the treatment arms in their rate of American College of Rheumatology (ACR) 20 and 50 responses at both 24 and 48 weeks.

The results showed significant differences between the treatment arms after the first 24 weeks of treatment for higher-level responses. For example, the percentage of patients achieving an ACR 70 response was 5% in the triple-therapy patients and 16% in the etanercept patients, a statistically significant difference. The rate of patients with a DAS28 score of 2.6 points or less at 24 weeks was 13% in the triple-therapy patients and 22% in those on etanercept, a significant difference.
Based on findings like these, "I would start a biologic in a patient who failed high-dose methotrexate and had poor prognostic factors and highly active disease, because at 6 months etanercept had the edge," said Dr. Edward C. Keystone, director of the Centre for Arthritis and Autoimmune Disease at Mount Sinai Hospital in Toronto, and a coinvestigator on the new study. But for all the other patients, "why not start on triple therapy first if you can switch them later if needed and the patients do well?" he asked. "The important observation is that the same percentage of patients failed in each arm. That is a huge message."
The RACAT study received no commercial support. Dr. O’Dell said that he had no disclosures. Dr. Furst has been a consultant to or received grant support from Abbott, Amgen, Bristol-Myers Squibb, and other companies. Dr. Kremer has been a consultant to or received grant support from Pfizer, Abbott, Genentech, and other companies. Dr. Keystone said that he has been a consultant to or has received research grants from Amgen, Pfizer, Merck, and other companies.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
MADRID – When rheumatoid arthritis patients fail initial treatment with methotrexate monotherapy, are they better served by a less expensive step-up treatment or the one that may better slow their radiographic progression and produce faster responses?
That seems to be the choice between step-up from methotrexate monotherapy by adding synthetic disease-modifying antirheumatic drugs (DMARDs) such as sulfasalazine and hydroxychloroquine, or by adding a tumor necrosis factor (TNF) inhibitor such as etanercept.
The RACAT (Rheumatoid Arthritis: Comparison of Active Therapies) trial tested those two options in 353 rheumatoid arthritis (RA) patients at 36 U.S. or Canadian sites between July 2007 and December 2010. The study’s primary endpoint, improvement in average disease activity score in 28 joints (DAS28) from baseline to 48 weeks of treatment, was similar in both treatment arms, proving the noninferiority of the triple synthetic DMARD regimen against the etanercept, biological arm, Dr. James R. O’Dell reported in a poster at the annual European Congress of Rheumatology. The results also appeared online simultaneously with Dr. O’Dell’s poster presentation (N. Engl. J. Med. 2013 June 11 [doi: 10.1056/NEJMoal1303006]).
"We showed that starting first with a TNF inhibitor or first with triple therapy resulted in the same outcomes. But costs were not the same. We [successfully] treated another 30% of patients who did not need a biologic" with the synthetic triple therapy. "And in the patients where triple therapy doesn’t work, you can change them to a biologic and they have identical outcomes, clinically by DAS28 and radiographically," compared with patients who began on etanercept added to methotrexate from the start, he said. Dr. O’Dell is chief of rheumatology at the VA Medical Center in Omaha, Neb., and professor of medicine at the University of Nebraska.
"Nothing is lost for the patient; if they don’t do well on triple therapy they can switch to a biological. The data are persuasive and the economic case is easy to make. You absolutely ought to go with triple therapy," he concluded based on the study findings.
But "it is very difficult to get physicians to buy into this" strategy, he said in an interview. TNF inhibitors such as etanercept and other biological DMARDs are "seductive," Dr. O’Dell said, because they are effective, work quickly, and appear more "targeted" than synthetic DMARDs, and they are promoted by well-financed marketing campaigns.
Dr. O’Dell conceded that the study data showed a signal of more radiographic progression with triple synthetic DMARD treatment that could potentially, over time, accrue to more substantial differences. At 48 weeks after the onset of treatment, patients on triple therapy had an average 0.54-point increase (worsening) in their van der Heijde modified Sharp score, compared with an average 0.29-point rise in the patients who received etanercept, a 0.25-point average difference in favor of etanercept that did not reach statistical significance.
But this trend toward greater radiographic progression in patients on triple therapy was consistent with the statistically significant, roughly 1-point average additional increased radiographic progression with triple therapy, compared with patients on methotrexate plus etanercept, that was seen after 2 years of follow-up in the TEAR (Treatment of Early Aggressive Rheumatoid Arthritis) trial (Arthritis Rheum. 2012;64:2824-35).
The TEAR study, which enrolled patients with very early RA, included a subgroup with an inadequate response to methotrexate monotherapy, and in that subgroup, patients randomized to triple therapy and those randomized to etanercept plus methotrexate had similar clinical outcomes, consistent with the new study findings.
"It’s hard to say that 1 or 2 units on the Sharp score doesn’t matter much, even if you don’t see the difference for several years," commented Dr. Daniel Furst, professor of medicine and director of the Rheumatology Clinical Research Center at the University of California, Los Angeles.
"Triple therapy is effective clinically, but it doesn’t do so well for x-rays. There are relationships between a 1 or 2 Sharp score difference and long-term outcomes. Across the board with biologics, the difference is 1 or 2 Sharp units. If one’s philosophy is to hit the RA hard and stop it, then perhaps a biologic is better," Dr. Furst commented in an interview.
But another view was that triple therapy could play a useful and cost-effective role. The new findings, along with the TEAR results, make "initial treatment of early RA with triple therapy a reasonable approach," said Dr. Joel M. Kremer, a professor of medicine at Albany (N.Y.) Medical College and director of research at the Center for Rheumatology in Albany. The only clear downside is that if triple therapy doesn’t work, the patient loses time, but that’s true for every treatment option, he noted.
"We don’t usually have the opportunity to hear the data for generic drugs" as much as for brand-name formulations, Dr. Kremer said in an interview. "Will data like this substantially change prescribing patterns? Probably not, but what might happen is that insurers may look at these data and say that patients should fail triple therapy before starting a biologic. That would be a sea change" for rheumatology, he added.
"I have always used methotrexate first, usually in combination with hydroxychloroquine," Dr. Kremer said. He has not usually also prescribed sulfasalazine, but said he would consider adding it based on the new data.
The new study enrolled patients with a DAS28 score of 4.4 or higher despite at least 12 weeks of stable methotrexate therapy with a weekly dosage of 15-25 mg. The patients averaged about 57 years old. After the first 24 weeks on randomized treatment, patients who did not have a decrease in their DAS28 of at least 1.2 units switched to the alternate regimens. The primary outcome was change in DAS28 at week 48 according to initial treatment assignment; the researchers collected 48-week DAS28 scores from 309 enrolled patients. The mean change in DAS28 from baseline at 48 weeks was a 2.12-unit reduction in the triple-therapy patients and a 2.29-unit reduction in the etanercept patients, an average 0.17-unit difference between the two treatment arms that was not statistically significant and that fell within the study’s prespecified range for noninferiority for triple therapy.
A similar percentage of patients, 27%, in each of the treatment arms switched to the alternate therapy at 24 weeks due to lack of an adequate initial response. There were also no statistically significant differences between the treatment arms in their rate of American College of Rheumatology (ACR) 20 and 50 responses at both 24 and 48 weeks.
The results showed significant differences between the treatment arms after the first 24 weeks of treatment for higher-level responses. For example, the percentage of patients achieving an ACR 70 response was 5% in the triple-therapy patients and 16% in the etanercept patients, a statistically significant difference. The rate of patients with a DAS28 score of 2.6 points or less at 24 weeks was 13% in the triple-therapy patients and 22% in those on etanercept, a significant difference.
Based on findings like these, "I would start a biologic in a patient who failed high-dose methotrexate and had poor prognostic factors and highly active disease, because at 6 months etanercept had the edge," said Dr. Edward C. Keystone, director of the Centre for Arthritis and Autoimmune Disease at Mount Sinai Hospital in Toronto, and a coinvestigator on the new study. But for all the other patients, "why not start on triple therapy first if you can switch them later if needed and the patients do well?" he asked. "The important observation is that the same percentage of patients failed in each arm. That is a huge message."
The RACAT study received no commercial support. Dr. O’Dell said that he had no disclosures. Dr. Furst has been a consultant to or received grant support from Abbott, Amgen, Bristol-Myers Squibb, and other companies. Dr. Kremer has been a consultant to or received grant support from Pfizer, Abbott, Genentech, and other companies. Dr. Keystone said that he has been a consultant to or has received research grants from Amgen, Pfizer, Merck, and other companies.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
MADRID – When rheumatoid arthritis patients fail initial treatment with methotrexate monotherapy, are they better served by a less expensive step-up treatment or the one that may better slow their radiographic progression and produce faster responses?
That seems to be the choice between step-up from methotrexate monotherapy by adding synthetic disease-modifying antirheumatic drugs (DMARDs) such as sulfasalazine and hydroxychloroquine, or by adding a tumor necrosis factor (TNF) inhibitor such as etanercept.
The RACAT (Rheumatoid Arthritis: Comparison of Active Therapies) trial tested those two options in 353 rheumatoid arthritis (RA) patients at 36 U.S. or Canadian sites between July 2007 and December 2010. The study’s primary endpoint, improvement in average disease activity score in 28 joints (DAS28) from baseline to 48 weeks of treatment, was similar in both treatment arms, proving the noninferiority of the triple synthetic DMARD regimen against the etanercept, biological arm, Dr. James R. O’Dell reported in a poster at the annual European Congress of Rheumatology. The results also appeared online simultaneously with Dr. O’Dell’s poster presentation (N. Engl. J. Med. 2013 June 11 [doi: 10.1056/NEJMoal1303006]).
"We showed that starting first with a TNF inhibitor or first with triple therapy resulted in the same outcomes. But costs were not the same. We [successfully] treated another 30% of patients who did not need a biologic" with the synthetic triple therapy. "And in the patients where triple therapy doesn’t work, you can change them to a biologic and they have identical outcomes, clinically by DAS28 and radiographically," compared with patients who began on etanercept added to methotrexate from the start, he said. Dr. O’Dell is chief of rheumatology at the VA Medical Center in Omaha, Neb., and professor of medicine at the University of Nebraska.
"Nothing is lost for the patient; if they don’t do well on triple therapy they can switch to a biological. The data are persuasive and the economic case is easy to make. You absolutely ought to go with triple therapy," he concluded based on the study findings.
But "it is very difficult to get physicians to buy into this" strategy, he said in an interview. TNF inhibitors such as etanercept and other biological DMARDs are "seductive," Dr. O’Dell said, because they are effective, work quickly, and appear more "targeted" than synthetic DMARDs, and they are promoted by well-financed marketing campaigns.
Dr. O’Dell conceded that the study data showed a signal of more radiographic progression with triple synthetic DMARD treatment that could potentially, over time, accrue to more substantial differences. At 48 weeks after the onset of treatment, patients on triple therapy had an average 0.54-point increase (worsening) in their van der Heijde modified Sharp score, compared with an average 0.29-point rise in the patients who received etanercept, a 0.25-point average difference in favor of etanercept that did not reach statistical significance.
But this trend toward greater radiographic progression in patients on triple therapy was consistent with the statistically significant, roughly 1-point average additional increased radiographic progression with triple therapy, compared with patients on methotrexate plus etanercept, that was seen after 2 years of follow-up in the TEAR (Treatment of Early Aggressive Rheumatoid Arthritis) trial (Arthritis Rheum. 2012;64:2824-35).
The TEAR study, which enrolled patients with very early RA, included a subgroup with an inadequate response to methotrexate monotherapy, and in that subgroup, patients randomized to triple therapy and those randomized to etanercept plus methotrexate had similar clinical outcomes, consistent with the new study findings.
"It’s hard to say that 1 or 2 units on the Sharp score doesn’t matter much, even if you don’t see the difference for several years," commented Dr. Daniel Furst, professor of medicine and director of the Rheumatology Clinical Research Center at the University of California, Los Angeles.
"Triple therapy is effective clinically, but it doesn’t do so well for x-rays. There are relationships between a 1 or 2 Sharp score difference and long-term outcomes. Across the board with biologics, the difference is 1 or 2 Sharp units. If one’s philosophy is to hit the RA hard and stop it, then perhaps a biologic is better," Dr. Furst commented in an interview.
But another view was that triple therapy could play a useful and cost-effective role. The new findings, along with the TEAR results, make "initial treatment of early RA with triple therapy a reasonable approach," said Dr. Joel M. Kremer, a professor of medicine at Albany (N.Y.) Medical College and director of research at the Center for Rheumatology in Albany. The only clear downside is that if triple therapy doesn’t work, the patient loses time, but that’s true for every treatment option, he noted.
"We don’t usually have the opportunity to hear the data for generic drugs" as much as for brand-name formulations, Dr. Kremer said in an interview. "Will data like this substantially change prescribing patterns? Probably not, but what might happen is that insurers may look at these data and say that patients should fail triple therapy before starting a biologic. That would be a sea change" for rheumatology, he added.
"I have always used methotrexate first, usually in combination with hydroxychloroquine," Dr. Kremer said. He has not usually also prescribed sulfasalazine, but said he would consider adding it based on the new data.
The new study enrolled patients with a DAS28 score of 4.4 or higher despite at least 12 weeks of stable methotrexate therapy with a weekly dosage of 15-25 mg. The patients averaged about 57 years old. After the first 24 weeks on randomized treatment, patients who did not have a decrease in their DAS28 of at least 1.2 units switched to the alternate regimens. The primary outcome was change in DAS28 at week 48 according to initial treatment assignment; the researchers collected 48-week DAS28 scores from 309 enrolled patients. The mean change in DAS28 from baseline at 48 weeks was a 2.12-unit reduction in the triple-therapy patients and a 2.29-unit reduction in the etanercept patients, an average 0.17-unit difference between the two treatment arms that was not statistically significant and that fell within the study’s prespecified range for noninferiority for triple therapy.
A similar percentage of patients, 27%, in each of the treatment arms switched to the alternate therapy at 24 weeks due to lack of an adequate initial response. There were also no statistically significant differences between the treatment arms in their rate of American College of Rheumatology (ACR) 20 and 50 responses at both 24 and 48 weeks.
The results showed significant differences between the treatment arms after the first 24 weeks of treatment for higher-level responses. For example, the percentage of patients achieving an ACR 70 response was 5% in the triple-therapy patients and 16% in the etanercept patients, a statistically significant difference. The rate of patients with a DAS28 score of 2.6 points or less at 24 weeks was 13% in the triple-therapy patients and 22% in those on etanercept, a significant difference.
Based on findings like these, "I would start a biologic in a patient who failed high-dose methotrexate and had poor prognostic factors and highly active disease, because at 6 months etanercept had the edge," said Dr. Edward C. Keystone, director of the Centre for Arthritis and Autoimmune Disease at Mount Sinai Hospital in Toronto, and a coinvestigator on the new study. But for all the other patients, "why not start on triple therapy first if you can switch them later if needed and the patients do well?" he asked. "The important observation is that the same percentage of patients failed in each arm. That is a huge message."
The RACAT study received no commercial support. Dr. O’Dell said that he had no disclosures. Dr. Furst has been a consultant to or received grant support from Abbott, Amgen, Bristol-Myers Squibb, and other companies. Dr. Kremer has been a consultant to or received grant support from Pfizer, Abbott, Genentech, and other companies. Dr. Keystone said that he has been a consultant to or has received research grants from Amgen, Pfizer, Merck, and other companies.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
AT THE EULAR CONGRESS 2013
Major finding: After 48 weeks of treatment, patients randomized to triple therapy or to etanercept and methotrexate had similar 2-point drops from their baseline DAS28 score.
Data source: The RACAT (Rheumatoid Arthritis: Comparison of Active Therapies) trial, a multicenter, 48-week, randomized study with 353 rheumatoid arthritis patients.
Disclosures: The RACAT study received no commercial support. Dr. O’Dell said that he had no disclosures. Dr. Furst has been a consultant to or received grant support from Abbott, Amgen, Bristol-Myers Squibb, and other companies. Dr. Kremer has been a consultant to or received grant support from Pfizer, Abbott, Genentech, and other companies. Dr. Keystone said that he has been a consultant to or has received research grants from Amgen, Pfizer, Merck, and other companies.

New TAVR device cuts aortic regurgitation
PARIS – A next-generation system for transcatheter aortic valve replacement produced substantially less aortic regurgitation, compared with the historical performance of marketed, percutaneous valve-replacement systems in a pilot, multicenter, uncontrolled study with 60 patients.
At 30 days after transcatheter aortic valve replacement (TAVR) using the investigational Lotus valve system, 1 of 53 patients assessed had moderate aortic regurgitation and no patients had severe regurgitation, Dr. Ian T. Meredith said at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions. In addition, 10 patients (19%) had mild regurgitation, 5 patients (9%) had trace regurgitation, and 37 patients, 70%, had no regurgitation.

By contrast, results from one of the most recent TAVR studies reported using the Sapien valve systems now on the worldwide market in more than 400 patients (the Placement of Aortic Transcatheter Valves [PARTNER] II cohort B trial) showed a roughly 20% incidence of severe or moderate aortic regurgitation 30 days after TAVR, with about 40% of patients having mild regurgitation and fewer than 20% of patients with no regurgitation.
Dr. Meredith attributed the superior regurgitation performance with the new device to a key feature of the Lotus valve, an adaptive seal around the valve that he likened to "cling wrap" that fills most or all of the small spaces around the valve after it’s placed. This produces a tight seal and prevents regurgitation, said Dr. Meredith, professor and director of MonasHeart at Monash University in Victoria, Australia.
The other major feature of the new valve is that it is easily retrievable after deployment, allowing operators to reposition it if needed following initial deployment. In the 60 cases reported by Dr. Meredith operators took advantage of the retrievable feature in 16 cases: 12 times to reposition the valve, and in 4 cases, they completely removed the first valve and replaced it with a different-size (the valves used in the study came in two different sizes).
The REPRISE (Repositionable Percutaneous Replacement of Stenotic Aortic Valve Through Implantation of Lotus Valve System–Evaluation of Safety and Performance) II trial enrolled 60 patients with severe, calcific aortic stenosis at seven centers in Australia and France during October 2012-January 2013. The patients averaged 86 years of age, and their average Society of Thoracic Surgeons risk score was 6.4%. At baseline their average aortic valve gradient was 47.5 mm Hg.
Valves were placed successfully in all 60 patients. The study’s primary performance endpoint was to produce an average aortic valve gradient of 18 mm Hg or less measured 30 days after valve placement. Actual performance was an average aortic valve gradient of 11.3 mm Hg in 52 patients measured at 30 days. In addition, one patient died, two patients had a disabling stroke, three had a nondisabling stroke, and five had a life-threatening bleed. The overall 30-day safety composite-event rate was 19%.
Valve calcification is the main cause of regurgitation following TAVR, Dr. Meredith said in an interview. The calcium in the aortic annulus appears in various shapes and sizes and can be difficult to flatten so that a simple valve seal could still be subject to leaks. The adaptive seal of the Lotus valve is better able to stop leaks, he said.
The study was sponsored by Boston Scientific, which is developing the Lotus valve system. Dr. Meredith said that he has been a consultant to and has received honoraria from Boston Scientific and Medtronic.
On Twitter @mitchelzoler
PARIS – A next-generation system for transcatheter aortic valve replacement produced substantially less aortic regurgitation, compared with the historical performance of marketed, percutaneous valve-replacement systems in a pilot, multicenter, uncontrolled study with 60 patients.
At 30 days after transcatheter aortic valve replacement (TAVR) using the investigational Lotus valve system, 1 of 53 patients assessed had moderate aortic regurgitation and no patients had severe regurgitation, Dr. Ian T. Meredith said at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions. In addition, 10 patients (19%) had mild regurgitation, 5 patients (9%) had trace regurgitation, and 37 patients, 70%, had no regurgitation.
By contrast, results from one of the most recent TAVR studies reported using the Sapien valve systems now on the worldwide market in more than 400 patients (the Placement of Aortic Transcatheter Valves [PARTNER] II cohort B trial) showed a roughly 20% incidence of severe or moderate aortic regurgitation 30 days after TAVR, with about 40% of patients having mild regurgitation and fewer than 20% of patients with no regurgitation.
Dr. Meredith attributed the superior regurgitation performance with the new device to a key feature of the Lotus valve, an adaptive seal around the valve that he likened to "cling wrap" that fills most or all of the small spaces around the valve after it’s placed. This produces a tight seal and prevents regurgitation, said Dr. Meredith, professor and director of MonasHeart at Monash University in Victoria, Australia.
The other major feature of the new valve is that it is easily retrievable after deployment, allowing operators to reposition it if needed following initial deployment. In the 60 cases reported by Dr. Meredith operators took advantage of the retrievable feature in 16 cases: 12 times to reposition the valve, and in 4 cases, they completely removed the first valve and replaced it with a different-size (the valves used in the study came in two different sizes).
The REPRISE (Repositionable Percutaneous Replacement of Stenotic Aortic Valve Through Implantation of Lotus Valve System–Evaluation of Safety and Performance) II trial enrolled 60 patients with severe, calcific aortic stenosis at seven centers in Australia and France during October 2012-January 2013. The patients averaged 86 years of age, and their average Society of Thoracic Surgeons risk score was 6.4%. At baseline their average aortic valve gradient was 47.5 mm Hg.
Valves were placed successfully in all 60 patients. The study’s primary performance endpoint was to produce an average aortic valve gradient of 18 mm Hg or less measured 30 days after valve placement. Actual performance was an average aortic valve gradient of 11.3 mm Hg in 52 patients measured at 30 days. In addition, one patient died, two patients had a disabling stroke, three had a nondisabling stroke, and five had a life-threatening bleed. The overall 30-day safety composite-event rate was 19%.
Valve calcification is the main cause of regurgitation following TAVR, Dr. Meredith said in an interview. The calcium in the aortic annulus appears in various shapes and sizes and can be difficult to flatten so that a simple valve seal could still be subject to leaks. The adaptive seal of the Lotus valve is better able to stop leaks, he said.
The study was sponsored by Boston Scientific, which is developing the Lotus valve system. Dr. Meredith said that he has been a consultant to and has received honoraria from Boston Scientific and Medtronic.
On Twitter @mitchelzoler
PARIS – A next-generation system for transcatheter aortic valve replacement produced substantially less aortic regurgitation, compared with the historical performance of marketed, percutaneous valve-replacement systems in a pilot, multicenter, uncontrolled study with 60 patients.
At 30 days after transcatheter aortic valve replacement (TAVR) using the investigational Lotus valve system, 1 of 53 patients assessed had moderate aortic regurgitation and no patients had severe regurgitation, Dr. Ian T. Meredith said at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions. In addition, 10 patients (19%) had mild regurgitation, 5 patients (9%) had trace regurgitation, and 37 patients, 70%, had no regurgitation.
By contrast, results from one of the most recent TAVR studies reported using the Sapien valve systems now on the worldwide market in more than 400 patients (the Placement of Aortic Transcatheter Valves [PARTNER] II cohort B trial) showed a roughly 20% incidence of severe or moderate aortic regurgitation 30 days after TAVR, with about 40% of patients having mild regurgitation and fewer than 20% of patients with no regurgitation.
Dr. Meredith attributed the superior regurgitation performance with the new device to a key feature of the Lotus valve, an adaptive seal around the valve that he likened to "cling wrap" that fills most or all of the small spaces around the valve after it’s placed. This produces a tight seal and prevents regurgitation, said Dr. Meredith, professor and director of MonasHeart at Monash University in Victoria, Australia.
The other major feature of the new valve is that it is easily retrievable after deployment, allowing operators to reposition it if needed following initial deployment. In the 60 cases reported by Dr. Meredith operators took advantage of the retrievable feature in 16 cases: 12 times to reposition the valve, and in 4 cases, they completely removed the first valve and replaced it with a different-size (the valves used in the study came in two different sizes).
The REPRISE (Repositionable Percutaneous Replacement of Stenotic Aortic Valve Through Implantation of Lotus Valve System–Evaluation of Safety and Performance) II trial enrolled 60 patients with severe, calcific aortic stenosis at seven centers in Australia and France during October 2012-January 2013. The patients averaged 86 years of age, and their average Society of Thoracic Surgeons risk score was 6.4%. At baseline their average aortic valve gradient was 47.5 mm Hg.
Valves were placed successfully in all 60 patients. The study’s primary performance endpoint was to produce an average aortic valve gradient of 18 mm Hg or less measured 30 days after valve placement. Actual performance was an average aortic valve gradient of 11.3 mm Hg in 52 patients measured at 30 days. In addition, one patient died, two patients had a disabling stroke, three had a nondisabling stroke, and five had a life-threatening bleed. The overall 30-day safety composite-event rate was 19%.
Valve calcification is the main cause of regurgitation following TAVR, Dr. Meredith said in an interview. The calcium in the aortic annulus appears in various shapes and sizes and can be difficult to flatten so that a simple valve seal could still be subject to leaks. The adaptive seal of the Lotus valve is better able to stop leaks, he said.
The study was sponsored by Boston Scientific, which is developing the Lotus valve system. Dr. Meredith said that he has been a consultant to and has received honoraria from Boston Scientific and Medtronic.
On Twitter @mitchelzoler
AT EUROPCR 2013
Major finding: The Lotus valve had a 79% rate of no or trace aortic-valve regurgitation 30 days after transcatheter aortic valve replacement.
Data source: An uncontrolled, multicenter study with 60 patients.
Disclosures: The study was sponsored by Boston Scientific, which is developing the Lotus valve system. Dr. Meredith said that he has been a consultant to and has received honoraria from Boston Scientific and Medtronic.
