User login
Vyvanse shows promise for binge-eating disorder
HOLLYWOOD, FLA. – Not a single medication is approved for treatment of binge-eating disorder, but that could change if the favorable results of an ongoing phase III randomized trial of lisdexamfetamine mirror those of a recently completed phase II study.
Binge-eating disorder (BED) is the most common eating disorder. It is characterized by excessive food intake accompanied by psychological distress, but without the purging or fasting that are the hallmarks of bulimia nervosa and anorexia nervosa, respectively. Patients with BED are often severely obese, depressed, and have metabolic disorders.

BED is "gaining increasing recognition as a very significant public health problem, but it remains underdiagnosed," Dr. Susan L. McElroy observed at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
She presented the results of a 31-site, randomized, double-blind, placebo-controlled clinical trial of lisdexamfetamine (Vyvanse) in 213 patients with moderate or severe BED as defined by DSM-IV-TR. The study, in which participants recorded their binge-eating episodes in a daily diary, entailed a 2-week baseline period, 11 weeks of double-blind therapy, and a 1-week follow-up phase.
Patients were assigned to placebo or to lisdexamfetamine at 30, 50, or 70 mg/day. This was a forced-dose titration study. Everyone assigned to lisdexamfetamine started at 30 mg/day. After week 1, patients assigned to 50 or 70 mg/day increased their daily dose by 20 mg/wk until reaching their target.
Subjects assigned to lisdexamfetamine at 50 mg/day had a mean baseline of 4.54 binge-eating days/wk. By week 11 on the drug, this had improved to 0.31 days/wk.
This was a significantly better result than in placebo-treated controls, who went from 4.29 binge-eating days per week at baseline to 1.13 per week after 11 weeks. Patients assigned to lisdexamfetamine at 70 mg/day did best of all, improving from 4.47 binge-eating days/wk at baseline to 0.11 after 11 weeks.
In addition, 67% of patients in the 70-mg/day group had been free of any binge-eating episodes for 1 week at week 11, compared with 56% of subjects on lisdexamfetamine at 50 mg/day and 34% on placebo. These differences were statistically significant.
This was a dose-ranging study, and indeed a linear dose-response relationship was found. The 30-mg/day dose was not more effective than placebo. Using statistical analysis, the minimum effective dose of lisdexamfetamine for treatment of BED was estimated at 34 mg/day, said Dr. McElroy, chief research officer at the Lindner Center of HOPE in Mason, Ohio, and professor of psychiatry and behavioral neuroscience at the University of Cincinnati.
Lisdexamfetamine is a d-amphetamine prodrug that inhibits dopamine reuptake and stimulates release of monoamine neurotransmitters. The therapeutic rationale for its use in BED lies in the observation that BED is associated with abnormal signaling by the dopamine and norepinephrine neurotransmitter systems, she explained.
The drug’s approved indication is for treatment of attention-deficit/hyperactivity disorder, for which the recommended starting dose is 30 mg/day, with adjustments up to 70 mg/day permitted.
The side effects noted in the phase II BED study were typical of those seen when lisdexamfetamine is prescribed for ADHD.
This study was sponsored by Shire. Dr. McElroy is a consultant to or a member of the scientific advisory boards of Shire and a half-dozen other pharmaceutical companies.
Dr. Susan L. McElroy, Vyvanse
HOLLYWOOD, FLA. – Not a single medication is approved for treatment of binge-eating disorder, but that could change if the favorable results of an ongoing phase III randomized trial of lisdexamfetamine mirror those of a recently completed phase II study.
Binge-eating disorder (BED) is the most common eating disorder. It is characterized by excessive food intake accompanied by psychological distress, but without the purging or fasting that are the hallmarks of bulimia nervosa and anorexia nervosa, respectively. Patients with BED are often severely obese, depressed, and have metabolic disorders.
BED is "gaining increasing recognition as a very significant public health problem, but it remains underdiagnosed," Dr. Susan L. McElroy observed at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
She presented the results of a 31-site, randomized, double-blind, placebo-controlled clinical trial of lisdexamfetamine (Vyvanse) in 213 patients with moderate or severe BED as defined by DSM-IV-TR. The study, in which participants recorded their binge-eating episodes in a daily diary, entailed a 2-week baseline period, 11 weeks of double-blind therapy, and a 1-week follow-up phase.
Patients were assigned to placebo or to lisdexamfetamine at 30, 50, or 70 mg/day. This was a forced-dose titration study. Everyone assigned to lisdexamfetamine started at 30 mg/day. After week 1, patients assigned to 50 or 70 mg/day increased their daily dose by 20 mg/wk until reaching their target.
Subjects assigned to lisdexamfetamine at 50 mg/day had a mean baseline of 4.54 binge-eating days/wk. By week 11 on the drug, this had improved to 0.31 days/wk.
This was a significantly better result than in placebo-treated controls, who went from 4.29 binge-eating days per week at baseline to 1.13 per week after 11 weeks. Patients assigned to lisdexamfetamine at 70 mg/day did best of all, improving from 4.47 binge-eating days/wk at baseline to 0.11 after 11 weeks.
In addition, 67% of patients in the 70-mg/day group had been free of any binge-eating episodes for 1 week at week 11, compared with 56% of subjects on lisdexamfetamine at 50 mg/day and 34% on placebo. These differences were statistically significant.
This was a dose-ranging study, and indeed a linear dose-response relationship was found. The 30-mg/day dose was not more effective than placebo. Using statistical analysis, the minimum effective dose of lisdexamfetamine for treatment of BED was estimated at 34 mg/day, said Dr. McElroy, chief research officer at the Lindner Center of HOPE in Mason, Ohio, and professor of psychiatry and behavioral neuroscience at the University of Cincinnati.
Lisdexamfetamine is a d-amphetamine prodrug that inhibits dopamine reuptake and stimulates release of monoamine neurotransmitters. The therapeutic rationale for its use in BED lies in the observation that BED is associated with abnormal signaling by the dopamine and norepinephrine neurotransmitter systems, she explained.
The drug’s approved indication is for treatment of attention-deficit/hyperactivity disorder, for which the recommended starting dose is 30 mg/day, with adjustments up to 70 mg/day permitted.
The side effects noted in the phase II BED study were typical of those seen when lisdexamfetamine is prescribed for ADHD.
This study was sponsored by Shire. Dr. McElroy is a consultant to or a member of the scientific advisory boards of Shire and a half-dozen other pharmaceutical companies.
HOLLYWOOD, FLA. – Not a single medication is approved for treatment of binge-eating disorder, but that could change if the favorable results of an ongoing phase III randomized trial of lisdexamfetamine mirror those of a recently completed phase II study.
Binge-eating disorder (BED) is the most common eating disorder. It is characterized by excessive food intake accompanied by psychological distress, but without the purging or fasting that are the hallmarks of bulimia nervosa and anorexia nervosa, respectively. Patients with BED are often severely obese, depressed, and have metabolic disorders.
BED is "gaining increasing recognition as a very significant public health problem, but it remains underdiagnosed," Dr. Susan L. McElroy observed at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
She presented the results of a 31-site, randomized, double-blind, placebo-controlled clinical trial of lisdexamfetamine (Vyvanse) in 213 patients with moderate or severe BED as defined by DSM-IV-TR. The study, in which participants recorded their binge-eating episodes in a daily diary, entailed a 2-week baseline period, 11 weeks of double-blind therapy, and a 1-week follow-up phase.
Patients were assigned to placebo or to lisdexamfetamine at 30, 50, or 70 mg/day. This was a forced-dose titration study. Everyone assigned to lisdexamfetamine started at 30 mg/day. After week 1, patients assigned to 50 or 70 mg/day increased their daily dose by 20 mg/wk until reaching their target.
Subjects assigned to lisdexamfetamine at 50 mg/day had a mean baseline of 4.54 binge-eating days/wk. By week 11 on the drug, this had improved to 0.31 days/wk.
This was a significantly better result than in placebo-treated controls, who went from 4.29 binge-eating days per week at baseline to 1.13 per week after 11 weeks. Patients assigned to lisdexamfetamine at 70 mg/day did best of all, improving from 4.47 binge-eating days/wk at baseline to 0.11 after 11 weeks.
In addition, 67% of patients in the 70-mg/day group had been free of any binge-eating episodes for 1 week at week 11, compared with 56% of subjects on lisdexamfetamine at 50 mg/day and 34% on placebo. These differences were statistically significant.
This was a dose-ranging study, and indeed a linear dose-response relationship was found. The 30-mg/day dose was not more effective than placebo. Using statistical analysis, the minimum effective dose of lisdexamfetamine for treatment of BED was estimated at 34 mg/day, said Dr. McElroy, chief research officer at the Lindner Center of HOPE in Mason, Ohio, and professor of psychiatry and behavioral neuroscience at the University of Cincinnati.
Lisdexamfetamine is a d-amphetamine prodrug that inhibits dopamine reuptake and stimulates release of monoamine neurotransmitters. The therapeutic rationale for its use in BED lies in the observation that BED is associated with abnormal signaling by the dopamine and norepinephrine neurotransmitter systems, she explained.
The drug’s approved indication is for treatment of attention-deficit/hyperactivity disorder, for which the recommended starting dose is 30 mg/day, with adjustments up to 70 mg/day permitted.
The side effects noted in the phase II BED study were typical of those seen when lisdexamfetamine is prescribed for ADHD.
This study was sponsored by Shire. Dr. McElroy is a consultant to or a member of the scientific advisory boards of Shire and a half-dozen other pharmaceutical companies.
Dr. Susan L. McElroy, Vyvanse
Dr. Susan L. McElroy, Vyvanse
AT THE NCDEU MEETING
Major finding: Patients with moderate or severe binge-eating disorder showed a dose-dependent reduction in binge-eating episodes in response to lisdexamfetamine dimesylate. Binge-eating days per week in those assigned to the top dose of 70 mg/day dropped from a mean of 4.47 at baseline to 0.11 after 11 weeks.
Data source: Randomized, double-blind, placebo-controlled, multicenter phase II study involving 213 patients.
Disclosures: The study was sponsored by Shire, which markets lisdexamfetamine for treatment of ADHD. Dr. McElroy is a consultant to the company.

Novel drug in pipeline for comorbid schizophrenia, substance abuse
HOLLYWOOD, FLA. – A novel agent early in development for the treatment of schizophrenia aims for an efficacy trifecta: the well-established potent antipsychotic benefits of olanzapine, but without the associated substantial weight gain, and with expanded utility in patients with comorbid substance abuse, according to Dr. Bernard L. Silverman.
The drug, known for now as ALKS 3831, is a fixed oral combination of olanzapine plus the opioid modulator ALKS 33, a centrally acting mu antagonist far more potent and bioavailable than naltrexone.
The appeal of this investigational medication stems from the fact that roughly 50% of all patients with schizophrenia have a comorbid substance abuse disorder. Substance abuse is the leading cause of medication noncompliance in schizophrenia. It is associated with more severe schizophrenia symptoms, an increased risk of relapse, more frequent and lengthier hospitalizations, and more violent episodes. A drug that simultaneously addresses both conditions in these dual-diagnosis patients would be most welcome, Dr. Silverman said at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"ALKS 33 has potential applicability in many reward disorders, including alcohol use disorders. There are no predicted adverse effects on olanzapine’s safety or efficacy, as there is no drug-drug pharmacokinetic interaction between the two, and their neurotransmitter profiles are distinct," explained Dr. Silverman of Alkermes, Waltham, Mass., which is developing the olanzapine/ALKS 33 combination.
He noted that in a phase II, company-sponsored, 12-week randomized clinical trial involving 406 patients with alcohol dependence in which comorbid schizophrenia was an exclusion criterion, a 41% reduction in heavy drinking days relative to placebo was seen in patients on ALKS 33 at 10 mg/day. A heavy drinking day was defined as five or more alcoholic drinks in a day for men and at least four in women.
"Extending such reductions in heavy drinking to subjects with schizophrenia would be expected to provide significant therapeutic benefit to dual-diagnosis patients," Dr. Silverman said.
He noted that in the landmark Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, sponsored by the National Institute of Mental Health, olanzapine demonstrated efficacy superior to that of quetiapine, risperidone, ziprasidone, and perphenazine. However, olanzapine also had the highest dropout rate because of weight gain and metabolic side effects (N. Engl. J. Med. 2005;353:1209-23).
With animal studies showing diminished olanzapine-related weight gain with concomitant ALKS 33, Dr. Silverman and his coinvestigators conducted a phase I proof-of-concept study in 106 healthy young men with a baseline body mass index of 18-25 kg/m2. Again, schizophrenia was a contraindication to study participation. Subjects were randomized to 3 weeks of olanzapine (Zyprexa) at 10 mg/day, olanzapine plus ALKS 33 at 5 mg/day, ALKS 33 alone, or placebo.
The group on olanzapine alone showed rapid weight gain, averaging 3.4 kg in just 3 weeks. In contrast, subjects on the olanzapine/ALKS 33 combination averaged a weight gain of 2.5 kg, nearly a full kilo less (P = .014). Participants on ALKS 33 alone and those on placebo had a similarly minimal change in body weight.
Adverse events were those typically seen with olanzapine therapy in schizophrenia. The most common were orthostatic hypotension and sleepiness, each of which was present in about 20% of subjects on olanzapine alone or the combination.
Moving beyond this proof-of-concept study, the investigators plan to conduct trials using olanzapine in combination with higher doses of ALKS 33 for longer time periods, and in patients with schizophrenia.
HOLLYWOOD, FLA. – A novel agent early in development for the treatment of schizophrenia aims for an efficacy trifecta: the well-established potent antipsychotic benefits of olanzapine, but without the associated substantial weight gain, and with expanded utility in patients with comorbid substance abuse, according to Dr. Bernard L. Silverman.
The drug, known for now as ALKS 3831, is a fixed oral combination of olanzapine plus the opioid modulator ALKS 33, a centrally acting mu antagonist far more potent and bioavailable than naltrexone.
The appeal of this investigational medication stems from the fact that roughly 50% of all patients with schizophrenia have a comorbid substance abuse disorder. Substance abuse is the leading cause of medication noncompliance in schizophrenia. It is associated with more severe schizophrenia symptoms, an increased risk of relapse, more frequent and lengthier hospitalizations, and more violent episodes. A drug that simultaneously addresses both conditions in these dual-diagnosis patients would be most welcome, Dr. Silverman said at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"ALKS 33 has potential applicability in many reward disorders, including alcohol use disorders. There are no predicted adverse effects on olanzapine’s safety or efficacy, as there is no drug-drug pharmacokinetic interaction between the two, and their neurotransmitter profiles are distinct," explained Dr. Silverman of Alkermes, Waltham, Mass., which is developing the olanzapine/ALKS 33 combination.
He noted that in a phase II, company-sponsored, 12-week randomized clinical trial involving 406 patients with alcohol dependence in which comorbid schizophrenia was an exclusion criterion, a 41% reduction in heavy drinking days relative to placebo was seen in patients on ALKS 33 at 10 mg/day. A heavy drinking day was defined as five or more alcoholic drinks in a day for men and at least four in women.
"Extending such reductions in heavy drinking to subjects with schizophrenia would be expected to provide significant therapeutic benefit to dual-diagnosis patients," Dr. Silverman said.
He noted that in the landmark Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, sponsored by the National Institute of Mental Health, olanzapine demonstrated efficacy superior to that of quetiapine, risperidone, ziprasidone, and perphenazine. However, olanzapine also had the highest dropout rate because of weight gain and metabolic side effects (N. Engl. J. Med. 2005;353:1209-23).
With animal studies showing diminished olanzapine-related weight gain with concomitant ALKS 33, Dr. Silverman and his coinvestigators conducted a phase I proof-of-concept study in 106 healthy young men with a baseline body mass index of 18-25 kg/m2. Again, schizophrenia was a contraindication to study participation. Subjects were randomized to 3 weeks of olanzapine (Zyprexa) at 10 mg/day, olanzapine plus ALKS 33 at 5 mg/day, ALKS 33 alone, or placebo.
The group on olanzapine alone showed rapid weight gain, averaging 3.4 kg in just 3 weeks. In contrast, subjects on the olanzapine/ALKS 33 combination averaged a weight gain of 2.5 kg, nearly a full kilo less (P = .014). Participants on ALKS 33 alone and those on placebo had a similarly minimal change in body weight.
Adverse events were those typically seen with olanzapine therapy in schizophrenia. The most common were orthostatic hypotension and sleepiness, each of which was present in about 20% of subjects on olanzapine alone or the combination.
Moving beyond this proof-of-concept study, the investigators plan to conduct trials using olanzapine in combination with higher doses of ALKS 33 for longer time periods, and in patients with schizophrenia.
HOLLYWOOD, FLA. – A novel agent early in development for the treatment of schizophrenia aims for an efficacy trifecta: the well-established potent antipsychotic benefits of olanzapine, but without the associated substantial weight gain, and with expanded utility in patients with comorbid substance abuse, according to Dr. Bernard L. Silverman.
The drug, known for now as ALKS 3831, is a fixed oral combination of olanzapine plus the opioid modulator ALKS 33, a centrally acting mu antagonist far more potent and bioavailable than naltrexone.
The appeal of this investigational medication stems from the fact that roughly 50% of all patients with schizophrenia have a comorbid substance abuse disorder. Substance abuse is the leading cause of medication noncompliance in schizophrenia. It is associated with more severe schizophrenia symptoms, an increased risk of relapse, more frequent and lengthier hospitalizations, and more violent episodes. A drug that simultaneously addresses both conditions in these dual-diagnosis patients would be most welcome, Dr. Silverman said at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"ALKS 33 has potential applicability in many reward disorders, including alcohol use disorders. There are no predicted adverse effects on olanzapine’s safety or efficacy, as there is no drug-drug pharmacokinetic interaction between the two, and their neurotransmitter profiles are distinct," explained Dr. Silverman of Alkermes, Waltham, Mass., which is developing the olanzapine/ALKS 33 combination.
He noted that in a phase II, company-sponsored, 12-week randomized clinical trial involving 406 patients with alcohol dependence in which comorbid schizophrenia was an exclusion criterion, a 41% reduction in heavy drinking days relative to placebo was seen in patients on ALKS 33 at 10 mg/day. A heavy drinking day was defined as five or more alcoholic drinks in a day for men and at least four in women.
"Extending such reductions in heavy drinking to subjects with schizophrenia would be expected to provide significant therapeutic benefit to dual-diagnosis patients," Dr. Silverman said.
He noted that in the landmark Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, sponsored by the National Institute of Mental Health, olanzapine demonstrated efficacy superior to that of quetiapine, risperidone, ziprasidone, and perphenazine. However, olanzapine also had the highest dropout rate because of weight gain and metabolic side effects (N. Engl. J. Med. 2005;353:1209-23).
With animal studies showing diminished olanzapine-related weight gain with concomitant ALKS 33, Dr. Silverman and his coinvestigators conducted a phase I proof-of-concept study in 106 healthy young men with a baseline body mass index of 18-25 kg/m2. Again, schizophrenia was a contraindication to study participation. Subjects were randomized to 3 weeks of olanzapine (Zyprexa) at 10 mg/day, olanzapine plus ALKS 33 at 5 mg/day, ALKS 33 alone, or placebo.
The group on olanzapine alone showed rapid weight gain, averaging 3.4 kg in just 3 weeks. In contrast, subjects on the olanzapine/ALKS 33 combination averaged a weight gain of 2.5 kg, nearly a full kilo less (P = .014). Participants on ALKS 33 alone and those on placebo had a similarly minimal change in body weight.
Adverse events were those typically seen with olanzapine therapy in schizophrenia. The most common were orthostatic hypotension and sleepiness, each of which was present in about 20% of subjects on olanzapine alone or the combination.
Moving beyond this proof-of-concept study, the investigators plan to conduct trials using olanzapine in combination with higher doses of ALKS 33 for longer time periods, and in patients with schizophrenia.
AT THE NCDEU MEETING
Major finding: Subjects randomized to 3 weeks of ALKS 3831 – an investigational combination of olanzapine plus the potent opioid modulator ALKS 33 – gained significantly less weight than those assigned to olanzapine alone.
Data source: A phase I proof-of-concept study in which 106 healthy, lean young men without schizophrenia were assigned to 3 weeks of olanzapine alone, ALKS 3831, ALKS 33 alone, or placebo.
Disclosures: The study was sponsored by Alkermes, which is developing ALKS 3831. The presenter is a full-time company employee.
Wearable defibrillator vest useful as bridge to ICD
DENVER – The LifeVest wearable automatic defibrillator provides a safe and highly effective bridging strategy while physicians decide whether a patient should get an implantable cardioverter defibrillator, according to findings from the WEARIT-II registry.
Experience gleaned from the first 882 of a planned 3,000 patients to be enrolled in this prospective, real-world registry indicates the defibrillator vest consistently recognizes and safely terminates life-threatening arrhythmias while avoiding unnecessary shocks for non–life-threatening arrhythmic events, Dr. Ilan Goldenberg reported at the annual meeting of the Heart Rhythm Society.

Indeed, the inappropriate shock rate in WEARIT-II was a mere 0.3%, far lower than with contemporary implantable cardioverter defibrillator (ICD) therapy, noted Dr. Goldenberg of the University of Rochester (N.Y.).
This bridging strategy deserves broad use in high-risk populations, he said. The WEARIT-II data show that bridging is particularly useful as a means of protecting patients with a transient arrhythmia risk as well as those whose long-term arrhythmia risk is undefined and requires further evaluation. Examples include patients who are post MI, or have new-onset heart failure with a depressed left ventricular ejection fraction (LVEF) of 35% or less, have recently undergone coronary revascularization, or are undergoing detailed evaluation of a possible inherited or congenital arrhythmic disorder.
Guidelines state that an ICD is indicated for primary prevention of sudden cardiac death in patients with an LVEF of 35% or less, but not within 40 days following an MI, or within 3 months after diagnosis of heart failure, or within 90 days following coronary revascularization. The reason for these mandatory delays is that many patients will experience improvement in LVEF in response to medical therapy such that they no longer qualify for ICD implantation. But if their physician is concerned about their arrhythmia risk during that waiting period, the wearable defibrillator is an excellent solution, Dr. Goldenberg continued.
There is a clear need for more selective prescription of ICDs for primary prevention of cardiac arrest. This was already evident more than a decade ago, when the MADIT-II (Multicenter Automatic Defibrillator Trial–II) demonstrated that only one-third of patients received appropriate ICD therapy during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-83). More recently, the MADIT-Reduce Inappropriate Therapy trial reported that participating ICD recipients had a low appropriate shock rate of 3 shocks per 100 patient-years (N. Engl. J. Med. 2012;367:2275-83) . The wearable defibrillator bridging strategy offers a means of safely being more selective in ICD placement. Patients whose risk isn’t yet clearly defined can in effect have a nonpermanent trial run of up to 6 months’ duration using the LifeVest, the electrophysiologist explained.
The LifeVest is commercially available and routinely covered by insurers. It’s a thin, lightweight vest designed to be worn under clothes. It is attached to a waist battery pack. The LifeVest provides continuous heart rhythm monitoring and automatic defibrillation upon detection of a potentially fatal ventricular arrhythmia. The device has an override button that enables a patient experiencing a sustained ventricular tachyarrhythmia (VT) to allow the event to terminate spontaneously, thereby avoiding unnecessary shocks. Should the patient pass out while the VT continues, the LifeVest will deliver shock therapy.
The 882 patients in the WEAR-IT registry had a mean LVEF of 25%. A total of 771 patients had acquired heart conditions, most commonly nonischemic or ischemic cardiomyopathy. The other 111 patients had inherited or congenital conditions, such as long QT syndrome. Patients wore the LifeVest for an average of 81 days and for a mean of 21 hours daily.
Appropriate LifeVest shock therapy that terminated life-threatening fast VT or ventricular fibrillation occurred at a rate of 9 events per 100 patient-years. Sustained VT that was allowed to spontaneously terminate as a result of the patient’s use of the device’s override button occurred at a rate of 27 events per 100 patient-years.
The device also detected nonsustained VT at a rate of 47 events per 100 patient-years, atrial arrhythmias and other supraventricular tachycardias at 64 events per 100 patient-years, and asystole at 3 events per 100 patient-years.
Four deaths occurred. Three of those happened when the patient was not wearing the vest; the fourth was caused by asystole.
Upon ending their use of the LifeVest, 41% of patients did not receive an ICD because their LVEF improved. Moreover, the arrhythmias detected by the LifeVest affected patient disposition: 80% of patients who received an appropriate shock from the vest got an ICD, as did fewer than 40% of those with no arrhythmias detected during vest wear.
Patients within 40 days post MI or 90 days post revascularization had the highest arrhythmic event rates among those with acquired heart conditions; however, the event rate was even higher among those with inherited or congenital conditions.
Dr. Michael R. Gold commented that in his experience, another important group of candidates for the wearable defibrillator are arrhythmia-prone patients who develop a cardiac device infection requiring device removal.
"You’re worried about that patient yet you can’t implant another device because it may take weeks or months to clear the infection," noted Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
Dr. Goldenberg reported having received research grants from Zoll Medical, which sponsors the WEARIT-II registry and markets the LifeVest, as well as from Boston Scientific.
DENVER – The LifeVest wearable automatic defibrillator provides a safe and highly effective bridging strategy while physicians decide whether a patient should get an implantable cardioverter defibrillator, according to findings from the WEARIT-II registry.
Experience gleaned from the first 882 of a planned 3,000 patients to be enrolled in this prospective, real-world registry indicates the defibrillator vest consistently recognizes and safely terminates life-threatening arrhythmias while avoiding unnecessary shocks for non–life-threatening arrhythmic events, Dr. Ilan Goldenberg reported at the annual meeting of the Heart Rhythm Society.
Indeed, the inappropriate shock rate in WEARIT-II was a mere 0.3%, far lower than with contemporary implantable cardioverter defibrillator (ICD) therapy, noted Dr. Goldenberg of the University of Rochester (N.Y.).
This bridging strategy deserves broad use in high-risk populations, he said. The WEARIT-II data show that bridging is particularly useful as a means of protecting patients with a transient arrhythmia risk as well as those whose long-term arrhythmia risk is undefined and requires further evaluation. Examples include patients who are post MI, or have new-onset heart failure with a depressed left ventricular ejection fraction (LVEF) of 35% or less, have recently undergone coronary revascularization, or are undergoing detailed evaluation of a possible inherited or congenital arrhythmic disorder.
Guidelines state that an ICD is indicated for primary prevention of sudden cardiac death in patients with an LVEF of 35% or less, but not within 40 days following an MI, or within 3 months after diagnosis of heart failure, or within 90 days following coronary revascularization. The reason for these mandatory delays is that many patients will experience improvement in LVEF in response to medical therapy such that they no longer qualify for ICD implantation. But if their physician is concerned about their arrhythmia risk during that waiting period, the wearable defibrillator is an excellent solution, Dr. Goldenberg continued.
There is a clear need for more selective prescription of ICDs for primary prevention of cardiac arrest. This was already evident more than a decade ago, when the MADIT-II (Multicenter Automatic Defibrillator Trial–II) demonstrated that only one-third of patients received appropriate ICD therapy during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-83). More recently, the MADIT-Reduce Inappropriate Therapy trial reported that participating ICD recipients had a low appropriate shock rate of 3 shocks per 100 patient-years (N. Engl. J. Med. 2012;367:2275-83) . The wearable defibrillator bridging strategy offers a means of safely being more selective in ICD placement. Patients whose risk isn’t yet clearly defined can in effect have a nonpermanent trial run of up to 6 months’ duration using the LifeVest, the electrophysiologist explained.
The LifeVest is commercially available and routinely covered by insurers. It’s a thin, lightweight vest designed to be worn under clothes. It is attached to a waist battery pack. The LifeVest provides continuous heart rhythm monitoring and automatic defibrillation upon detection of a potentially fatal ventricular arrhythmia. The device has an override button that enables a patient experiencing a sustained ventricular tachyarrhythmia (VT) to allow the event to terminate spontaneously, thereby avoiding unnecessary shocks. Should the patient pass out while the VT continues, the LifeVest will deliver shock therapy.
The 882 patients in the WEAR-IT registry had a mean LVEF of 25%. A total of 771 patients had acquired heart conditions, most commonly nonischemic or ischemic cardiomyopathy. The other 111 patients had inherited or congenital conditions, such as long QT syndrome. Patients wore the LifeVest for an average of 81 days and for a mean of 21 hours daily.
Appropriate LifeVest shock therapy that terminated life-threatening fast VT or ventricular fibrillation occurred at a rate of 9 events per 100 patient-years. Sustained VT that was allowed to spontaneously terminate as a result of the patient’s use of the device’s override button occurred at a rate of 27 events per 100 patient-years.
The device also detected nonsustained VT at a rate of 47 events per 100 patient-years, atrial arrhythmias and other supraventricular tachycardias at 64 events per 100 patient-years, and asystole at 3 events per 100 patient-years.
Four deaths occurred. Three of those happened when the patient was not wearing the vest; the fourth was caused by asystole.
Upon ending their use of the LifeVest, 41% of patients did not receive an ICD because their LVEF improved. Moreover, the arrhythmias detected by the LifeVest affected patient disposition: 80% of patients who received an appropriate shock from the vest got an ICD, as did fewer than 40% of those with no arrhythmias detected during vest wear.
Patients within 40 days post MI or 90 days post revascularization had the highest arrhythmic event rates among those with acquired heart conditions; however, the event rate was even higher among those with inherited or congenital conditions.
Dr. Michael R. Gold commented that in his experience, another important group of candidates for the wearable defibrillator are arrhythmia-prone patients who develop a cardiac device infection requiring device removal.
"You’re worried about that patient yet you can’t implant another device because it may take weeks or months to clear the infection," noted Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
Dr. Goldenberg reported having received research grants from Zoll Medical, which sponsors the WEARIT-II registry and markets the LifeVest, as well as from Boston Scientific.
DENVER – The LifeVest wearable automatic defibrillator provides a safe and highly effective bridging strategy while physicians decide whether a patient should get an implantable cardioverter defibrillator, according to findings from the WEARIT-II registry.
Experience gleaned from the first 882 of a planned 3,000 patients to be enrolled in this prospective, real-world registry indicates the defibrillator vest consistently recognizes and safely terminates life-threatening arrhythmias while avoiding unnecessary shocks for non–life-threatening arrhythmic events, Dr. Ilan Goldenberg reported at the annual meeting of the Heart Rhythm Society.
Indeed, the inappropriate shock rate in WEARIT-II was a mere 0.3%, far lower than with contemporary implantable cardioverter defibrillator (ICD) therapy, noted Dr. Goldenberg of the University of Rochester (N.Y.).
This bridging strategy deserves broad use in high-risk populations, he said. The WEARIT-II data show that bridging is particularly useful as a means of protecting patients with a transient arrhythmia risk as well as those whose long-term arrhythmia risk is undefined and requires further evaluation. Examples include patients who are post MI, or have new-onset heart failure with a depressed left ventricular ejection fraction (LVEF) of 35% or less, have recently undergone coronary revascularization, or are undergoing detailed evaluation of a possible inherited or congenital arrhythmic disorder.
Guidelines state that an ICD is indicated for primary prevention of sudden cardiac death in patients with an LVEF of 35% or less, but not within 40 days following an MI, or within 3 months after diagnosis of heart failure, or within 90 days following coronary revascularization. The reason for these mandatory delays is that many patients will experience improvement in LVEF in response to medical therapy such that they no longer qualify for ICD implantation. But if their physician is concerned about their arrhythmia risk during that waiting period, the wearable defibrillator is an excellent solution, Dr. Goldenberg continued.
There is a clear need for more selective prescription of ICDs for primary prevention of cardiac arrest. This was already evident more than a decade ago, when the MADIT-II (Multicenter Automatic Defibrillator Trial–II) demonstrated that only one-third of patients received appropriate ICD therapy during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-83). More recently, the MADIT-Reduce Inappropriate Therapy trial reported that participating ICD recipients had a low appropriate shock rate of 3 shocks per 100 patient-years (N. Engl. J. Med. 2012;367:2275-83) . The wearable defibrillator bridging strategy offers a means of safely being more selective in ICD placement. Patients whose risk isn’t yet clearly defined can in effect have a nonpermanent trial run of up to 6 months’ duration using the LifeVest, the electrophysiologist explained.
The LifeVest is commercially available and routinely covered by insurers. It’s a thin, lightweight vest designed to be worn under clothes. It is attached to a waist battery pack. The LifeVest provides continuous heart rhythm monitoring and automatic defibrillation upon detection of a potentially fatal ventricular arrhythmia. The device has an override button that enables a patient experiencing a sustained ventricular tachyarrhythmia (VT) to allow the event to terminate spontaneously, thereby avoiding unnecessary shocks. Should the patient pass out while the VT continues, the LifeVest will deliver shock therapy.
The 882 patients in the WEAR-IT registry had a mean LVEF of 25%. A total of 771 patients had acquired heart conditions, most commonly nonischemic or ischemic cardiomyopathy. The other 111 patients had inherited or congenital conditions, such as long QT syndrome. Patients wore the LifeVest for an average of 81 days and for a mean of 21 hours daily.
Appropriate LifeVest shock therapy that terminated life-threatening fast VT or ventricular fibrillation occurred at a rate of 9 events per 100 patient-years. Sustained VT that was allowed to spontaneously terminate as a result of the patient’s use of the device’s override button occurred at a rate of 27 events per 100 patient-years.
The device also detected nonsustained VT at a rate of 47 events per 100 patient-years, atrial arrhythmias and other supraventricular tachycardias at 64 events per 100 patient-years, and asystole at 3 events per 100 patient-years.
Four deaths occurred. Three of those happened when the patient was not wearing the vest; the fourth was caused by asystole.
Upon ending their use of the LifeVest, 41% of patients did not receive an ICD because their LVEF improved. Moreover, the arrhythmias detected by the LifeVest affected patient disposition: 80% of patients who received an appropriate shock from the vest got an ICD, as did fewer than 40% of those with no arrhythmias detected during vest wear.
Patients within 40 days post MI or 90 days post revascularization had the highest arrhythmic event rates among those with acquired heart conditions; however, the event rate was even higher among those with inherited or congenital conditions.
Dr. Michael R. Gold commented that in his experience, another important group of candidates for the wearable defibrillator are arrhythmia-prone patients who develop a cardiac device infection requiring device removal.
"You’re worried about that patient yet you can’t implant another device because it may take weeks or months to clear the infection," noted Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
Dr. Goldenberg reported having received research grants from Zoll Medical, which sponsors the WEARIT-II registry and markets the LifeVest, as well as from Boston Scientific.
AT HEART RHYTHM 2013
Major finding: During an average of 81 days using the LifeVest wearable automatic defibrillator, patients experienced appropriate device shocks to terminate potentially fatal ventricular tachyarrhythmia/ventricular fibrillation at a rate of 9 events per 100 person-years while appropriately avoiding shocks for sustained VT with spontaneous termination at a rate of 27 events per 100 person-years. Only 0.3% of 882 vest users experienced an inappropriate shock.
Data source: The WEARIT-II registry, which to date includes 882 patients who have been prescribed the LifeVest wearable defibrillator.
Disclosures: The registry is sponsored by Zoll Medical, which markets the LifeVest. The presenter said he has received research grants from the company.

Dabigatran post-AF ablation may be riskier than warfarin
DENVER – Using dabigatran rather than warfarin for periprocedural anticoagulation in patients undergoing radiofrequency ablation for atrial fibrillation may entail a small but statistically increased risk of stroke or transient ischemic attack, according to a meta-analysis of 10 observational cohort studies.
Guidelines recommend periprocedural anticoagulation for at least 2 months post ablation. Warfarin continues to be the most widely used agent for this purpose, but new oral alternatives are attracting a great deal of interest from physicians and patients.

The meta-analysis, which included 1,501 patients on periprocedural dabigatran (Pradaxa) and 2,356 on warfarin, had dual primary end points. One was stroke or TIA, which occurred in 0.7% of the dabigatran group, compared with 0.2% of those on warfarin – a statistically significant difference (P = .0007), Dr. Benjamin A. Steinberg reported at the annual meeting of the Heart Rhythm Society.
The co-primary end point, major bleeding, was recorded in 1.6% of the dabigatran group, with a closely similar 1.7% incidence in the warfarin group, added Dr. Steinberg of Duke University in Durham, N.C.
Rates of cardiac tamponade, a secondary end point, were also similar: 1.1% with dabigatran and 0.9% with warfarin.
Although dabigatran is approved for the prevention of stroke or systemic embolism in patients with AF, a definitive determination of its safety and effectiveness in the setting of AF ablation therapy will require randomized trials, Dr. Steinberg observed.
Such trials would, of necessity, have to be quite large. The data from this meta-analysis suggest that, for every 200 patients undergoing AF ablation under the protection of dabigatran rather than warfarin, one additional case of stroke or TIA would occur.
The meta-analysis was supported in part by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.
DENVER – Using dabigatran rather than warfarin for periprocedural anticoagulation in patients undergoing radiofrequency ablation for atrial fibrillation may entail a small but statistically increased risk of stroke or transient ischemic attack, according to a meta-analysis of 10 observational cohort studies.
Guidelines recommend periprocedural anticoagulation for at least 2 months post ablation. Warfarin continues to be the most widely used agent for this purpose, but new oral alternatives are attracting a great deal of interest from physicians and patients.
The meta-analysis, which included 1,501 patients on periprocedural dabigatran (Pradaxa) and 2,356 on warfarin, had dual primary end points. One was stroke or TIA, which occurred in 0.7% of the dabigatran group, compared with 0.2% of those on warfarin – a statistically significant difference (P = .0007), Dr. Benjamin A. Steinberg reported at the annual meeting of the Heart Rhythm Society.
The co-primary end point, major bleeding, was recorded in 1.6% of the dabigatran group, with a closely similar 1.7% incidence in the warfarin group, added Dr. Steinberg of Duke University in Durham, N.C.
Rates of cardiac tamponade, a secondary end point, were also similar: 1.1% with dabigatran and 0.9% with warfarin.
Although dabigatran is approved for the prevention of stroke or systemic embolism in patients with AF, a definitive determination of its safety and effectiveness in the setting of AF ablation therapy will require randomized trials, Dr. Steinberg observed.
Such trials would, of necessity, have to be quite large. The data from this meta-analysis suggest that, for every 200 patients undergoing AF ablation under the protection of dabigatran rather than warfarin, one additional case of stroke or TIA would occur.
The meta-analysis was supported in part by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.
DENVER – Using dabigatran rather than warfarin for periprocedural anticoagulation in patients undergoing radiofrequency ablation for atrial fibrillation may entail a small but statistically increased risk of stroke or transient ischemic attack, according to a meta-analysis of 10 observational cohort studies.
Guidelines recommend periprocedural anticoagulation for at least 2 months post ablation. Warfarin continues to be the most widely used agent for this purpose, but new oral alternatives are attracting a great deal of interest from physicians and patients.
The meta-analysis, which included 1,501 patients on periprocedural dabigatran (Pradaxa) and 2,356 on warfarin, had dual primary end points. One was stroke or TIA, which occurred in 0.7% of the dabigatran group, compared with 0.2% of those on warfarin – a statistically significant difference (P = .0007), Dr. Benjamin A. Steinberg reported at the annual meeting of the Heart Rhythm Society.
The co-primary end point, major bleeding, was recorded in 1.6% of the dabigatran group, with a closely similar 1.7% incidence in the warfarin group, added Dr. Steinberg of Duke University in Durham, N.C.
Rates of cardiac tamponade, a secondary end point, were also similar: 1.1% with dabigatran and 0.9% with warfarin.
Although dabigatran is approved for the prevention of stroke or systemic embolism in patients with AF, a definitive determination of its safety and effectiveness in the setting of AF ablation therapy will require randomized trials, Dr. Steinberg observed.
Such trials would, of necessity, have to be quite large. The data from this meta-analysis suggest that, for every 200 patients undergoing AF ablation under the protection of dabigatran rather than warfarin, one additional case of stroke or TIA would occur.
The meta-analysis was supported in part by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major finding: The incidence of stroke or transient ischemic attack in patients on dabigatran for periprocedural anticoagulation in conjunction with radiofrequency ablation for atrial fibrillation was 0.7%, statistically greater than the 0.2% in patients on warfarin.
Data source: A meta-analysis of 10 observational cohort studies totaling 1,501 AF ablation patients on dabigatran and 2,356 on warfarin.
Disclosures: The meta-analysis was funded by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.

Cardiac implantable device infection rate is falling
DENVER – Whether cardiac electronic devices are implanted in an inpatient or outpatient setting doesn’t affect device infection rates, according to a national analysis of Medicare data for 1997-2010.
Outpatient implantations are on the upswing. From 1997 to 2010, the proportion of pacemaker implantations performed in Medicare patients on an outpatient basis climbed from 8.6% to 29%. In addition, the proportion of pacemaker revision procedures done in outpatient settings nearly doubled, from 37% to 72%, Dr. Arnold J. Greenspon reported at the annual meeting of the Heart Rhythm Society.

Similarly, outpatient installation of implantable cardioverter defibrillators (ICDs) accounted for just 6.2% of all ICD implantations in 1997, but 36% in 2010. The proportion of ICD revision procedures performed on an outpatient basis rose from 31% to 69% during this time period, added Dr. Greenspon, professor of medicine and director of the cardiac electrophysiology laboratory at Thomas Jefferson University, Philadelphia.
Device infection rates following primary implantation have been waning. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010. Pacemaker infections are less frequent: The rate was 0.9% in 1997, falling to just 0.2% in 2010.
However, the risk of deep infection warranting inpatient revision surgery has increased over time. The annual number of patients undergoing inpatient device removal on the basis of ICD diagnosis codes for deep infection or sepsis increased two- to -threefold during 1997-2010.
Risk factors for infection identified in this study included advanced age, renal failure and other medical comorbidities, and an increasing number of cardiac device procedures. Indeed, the pacemaker and ICD infection rates climbed to 14% and 9%, respectively, in patients who had undergone five device procedures.
This study was funded by Medtronic. Dr. Greenspon reported serving as a consultant to Medtronic, Boston Scientific, and St. Jude Medical.
DENVER – Whether cardiac electronic devices are implanted in an inpatient or outpatient setting doesn’t affect device infection rates, according to a national analysis of Medicare data for 1997-2010.
Outpatient implantations are on the upswing. From 1997 to 2010, the proportion of pacemaker implantations performed in Medicare patients on an outpatient basis climbed from 8.6% to 29%. In addition, the proportion of pacemaker revision procedures done in outpatient settings nearly doubled, from 37% to 72%, Dr. Arnold J. Greenspon reported at the annual meeting of the Heart Rhythm Society.
Similarly, outpatient installation of implantable cardioverter defibrillators (ICDs) accounted for just 6.2% of all ICD implantations in 1997, but 36% in 2010. The proportion of ICD revision procedures performed on an outpatient basis rose from 31% to 69% during this time period, added Dr. Greenspon, professor of medicine and director of the cardiac electrophysiology laboratory at Thomas Jefferson University, Philadelphia.
Device infection rates following primary implantation have been waning. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010. Pacemaker infections are less frequent: The rate was 0.9% in 1997, falling to just 0.2% in 2010.
However, the risk of deep infection warranting inpatient revision surgery has increased over time. The annual number of patients undergoing inpatient device removal on the basis of ICD diagnosis codes for deep infection or sepsis increased two- to -threefold during 1997-2010.
Risk factors for infection identified in this study included advanced age, renal failure and other medical comorbidities, and an increasing number of cardiac device procedures. Indeed, the pacemaker and ICD infection rates climbed to 14% and 9%, respectively, in patients who had undergone five device procedures.
This study was funded by Medtronic. Dr. Greenspon reported serving as a consultant to Medtronic, Boston Scientific, and St. Jude Medical.
DENVER – Whether cardiac electronic devices are implanted in an inpatient or outpatient setting doesn’t affect device infection rates, according to a national analysis of Medicare data for 1997-2010.
Outpatient implantations are on the upswing. From 1997 to 2010, the proportion of pacemaker implantations performed in Medicare patients on an outpatient basis climbed from 8.6% to 29%. In addition, the proportion of pacemaker revision procedures done in outpatient settings nearly doubled, from 37% to 72%, Dr. Arnold J. Greenspon reported at the annual meeting of the Heart Rhythm Society.
Similarly, outpatient installation of implantable cardioverter defibrillators (ICDs) accounted for just 6.2% of all ICD implantations in 1997, but 36% in 2010. The proportion of ICD revision procedures performed on an outpatient basis rose from 31% to 69% during this time period, added Dr. Greenspon, professor of medicine and director of the cardiac electrophysiology laboratory at Thomas Jefferson University, Philadelphia.
Device infection rates following primary implantation have been waning. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010. Pacemaker infections are less frequent: The rate was 0.9% in 1997, falling to just 0.2% in 2010.
However, the risk of deep infection warranting inpatient revision surgery has increased over time. The annual number of patients undergoing inpatient device removal on the basis of ICD diagnosis codes for deep infection or sepsis increased two- to -threefold during 1997-2010.
Risk factors for infection identified in this study included advanced age, renal failure and other medical comorbidities, and an increasing number of cardiac device procedures. Indeed, the pacemaker and ICD infection rates climbed to 14% and 9%, respectively, in patients who had undergone five device procedures.
This study was funded by Medtronic. Dr. Greenspon reported serving as a consultant to Medtronic, Boston Scientific, and St. Jude Medical.
AT HEART RHYTHM 2013
Major finding: Annual infection rates following primary pacemaker implantation declined nationally from 0.9% in 1997 to just 0.2% in 2010. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010.
Data source: An analysis of the Medicare database covering 1997-2010.
Disclosures: The study was sponsored by Medtronic. The presenter is a consultant to Medtronic and other medical device manufacturers.

Leadless cardiac pacemaker draws plaudits
DENVER – A first-in-man study has demonstrated that a novel leadless cardiac pacemaker is easy to implant percutaneously in the right ventricle, is readily retrievable when required, and performs like a conventional single-chamber permanent VVIR pacemaker.
"This is a relatively small feasibility study, but I think this new leadless pacemaker has the possibility of effecting a real paradigm shift in how we think about pacing. We’re going to be able to eliminate what has been the weak link in the pacemaker system: the lead," Dr. Vivek Reddy predicted in presenting the results of the LEADLESS study at the annual meeting of the Heart Rhythm Society.
More than 4.4 million people worldwide have a cardiac pacemaker. More than 65,000 of them develop chronic problems related to the pacemaker leads, including lead dislodgement, infection, lead failure, and hematoma. In one major study, the overall complication rate over the life of conventional pacemakers was close to 8%.

These complications come at considerable cost. A lead infection can easily add $50,000 to the cost of care, a lead revision $16,000, noted Dr. Reddy of Mount Sinai School of Medicine, New York.
The leadless pacemaker is about the size of a triple-A battery. Vascular access for its placement is gained in standard fashion through the femoral vein using a steerable 18F catheter. The device is typically placed in the right ventricular apex and fixed in place by a single turn of an attached helix. The single-chamber pacemaker uses low-power electronics and has an estimated battery life of 8-17 years, depending upon how much of the time it’s actively pacing. The pacemaker senses right ventricular blood temperature, using it as a guide to boost pacing rate in response to increased metabolic demand.
In the LEADLESS study, 32 of 33 patients at three European centers underwent successful pacemaker implantation. The procedure took roughly 30 minutes initially, but times came down with greater operator experience. The most common indication for a permanent single-chamber pacemaker among study participants was chronic atrial fibrillation with second- or third-degree atrioventricular heart block.
The pacemaker is designed so the interventionalist can quickly determine if the device has been placed in an optimal location; if not, it can be repositioned during the same procedure before being finally locked into place. Twenty-three of the 32 patients did not require any device repositioning.

During 3 months of study follow-up, two patients required device retrieval. In one, the leadless pacemaker had migrated through an open patent foramen ovale into the left ventricle. The other patient developed indications warranting an implantable cardioverter-defibrillator. Device retrieval was a straightforward matter and took 6 and 13 minutes, respectively, using a basket catheter.
The one serious adverse event in the study involved a patient who experienced cardiac perforation and tamponade during pacemaker implantation. Five days after an uncomplicated surgical repair, the patient had a large right-sided stroke and died.
Dr. Reddy said the leadless pacemaker will be commercialized in Europe later this year. Planning is underway for a large, multicenter U.S. trial that will probably start next year. And Nanostim, the company developing the device, is also working on a leadless atrial pacemaker that will be able to communicate with the right ventricular pacemaker in order to provide multichamber cardiac pacing.
Dr. Reddy’s presentation met with an enthusiastic response.
"This is fascinating, incredible technology," declared Dr. Andrea M. Russo of Cooper University Health Care in Camden, N.J.
"The idea of being able to put a pacemaker into someone without having leads and without having a device under the skin is probably the next phase of what we’d hope to do with cardiac pacing therapy," Dr. Michael R. Gold observed.
"I think most of us, if we were to make predictions about what’s in store 10 years from now, we’d be hoping that we’d no longer use leads, which are the component that fails most frequently in any device. If we can get rid of the leads, put these little pacemakers anywhere we want in the heart, and get them to cross-talk and communicate with each other, that’s very exciting," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
DENVER – A first-in-man study has demonstrated that a novel leadless cardiac pacemaker is easy to implant percutaneously in the right ventricle, is readily retrievable when required, and performs like a conventional single-chamber permanent VVIR pacemaker.
"This is a relatively small feasibility study, but I think this new leadless pacemaker has the possibility of effecting a real paradigm shift in how we think about pacing. We’re going to be able to eliminate what has been the weak link in the pacemaker system: the lead," Dr. Vivek Reddy predicted in presenting the results of the LEADLESS study at the annual meeting of the Heart Rhythm Society.
More than 4.4 million people worldwide have a cardiac pacemaker. More than 65,000 of them develop chronic problems related to the pacemaker leads, including lead dislodgement, infection, lead failure, and hematoma. In one major study, the overall complication rate over the life of conventional pacemakers was close to 8%.
These complications come at considerable cost. A lead infection can easily add $50,000 to the cost of care, a lead revision $16,000, noted Dr. Reddy of Mount Sinai School of Medicine, New York.
The leadless pacemaker is about the size of a triple-A battery. Vascular access for its placement is gained in standard fashion through the femoral vein using a steerable 18F catheter. The device is typically placed in the right ventricular apex and fixed in place by a single turn of an attached helix. The single-chamber pacemaker uses low-power electronics and has an estimated battery life of 8-17 years, depending upon how much of the time it’s actively pacing. The pacemaker senses right ventricular blood temperature, using it as a guide to boost pacing rate in response to increased metabolic demand.
In the LEADLESS study, 32 of 33 patients at three European centers underwent successful pacemaker implantation. The procedure took roughly 30 minutes initially, but times came down with greater operator experience. The most common indication for a permanent single-chamber pacemaker among study participants was chronic atrial fibrillation with second- or third-degree atrioventricular heart block.
The pacemaker is designed so the interventionalist can quickly determine if the device has been placed in an optimal location; if not, it can be repositioned during the same procedure before being finally locked into place. Twenty-three of the 32 patients did not require any device repositioning.
During 3 months of study follow-up, two patients required device retrieval. In one, the leadless pacemaker had migrated through an open patent foramen ovale into the left ventricle. The other patient developed indications warranting an implantable cardioverter-defibrillator. Device retrieval was a straightforward matter and took 6 and 13 minutes, respectively, using a basket catheter.
The one serious adverse event in the study involved a patient who experienced cardiac perforation and tamponade during pacemaker implantation. Five days after an uncomplicated surgical repair, the patient had a large right-sided stroke and died.
Dr. Reddy said the leadless pacemaker will be commercialized in Europe later this year. Planning is underway for a large, multicenter U.S. trial that will probably start next year. And Nanostim, the company developing the device, is also working on a leadless atrial pacemaker that will be able to communicate with the right ventricular pacemaker in order to provide multichamber cardiac pacing.
Dr. Reddy’s presentation met with an enthusiastic response.
"This is fascinating, incredible technology," declared Dr. Andrea M. Russo of Cooper University Health Care in Camden, N.J.
"The idea of being able to put a pacemaker into someone without having leads and without having a device under the skin is probably the next phase of what we’d hope to do with cardiac pacing therapy," Dr. Michael R. Gold observed.
"I think most of us, if we were to make predictions about what’s in store 10 years from now, we’d be hoping that we’d no longer use leads, which are the component that fails most frequently in any device. If we can get rid of the leads, put these little pacemakers anywhere we want in the heart, and get them to cross-talk and communicate with each other, that’s very exciting," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
DENVER – A first-in-man study has demonstrated that a novel leadless cardiac pacemaker is easy to implant percutaneously in the right ventricle, is readily retrievable when required, and performs like a conventional single-chamber permanent VVIR pacemaker.
"This is a relatively small feasibility study, but I think this new leadless pacemaker has the possibility of effecting a real paradigm shift in how we think about pacing. We’re going to be able to eliminate what has been the weak link in the pacemaker system: the lead," Dr. Vivek Reddy predicted in presenting the results of the LEADLESS study at the annual meeting of the Heart Rhythm Society.
More than 4.4 million people worldwide have a cardiac pacemaker. More than 65,000 of them develop chronic problems related to the pacemaker leads, including lead dislodgement, infection, lead failure, and hematoma. In one major study, the overall complication rate over the life of conventional pacemakers was close to 8%.
These complications come at considerable cost. A lead infection can easily add $50,000 to the cost of care, a lead revision $16,000, noted Dr. Reddy of Mount Sinai School of Medicine, New York.
The leadless pacemaker is about the size of a triple-A battery. Vascular access for its placement is gained in standard fashion through the femoral vein using a steerable 18F catheter. The device is typically placed in the right ventricular apex and fixed in place by a single turn of an attached helix. The single-chamber pacemaker uses low-power electronics and has an estimated battery life of 8-17 years, depending upon how much of the time it’s actively pacing. The pacemaker senses right ventricular blood temperature, using it as a guide to boost pacing rate in response to increased metabolic demand.
In the LEADLESS study, 32 of 33 patients at three European centers underwent successful pacemaker implantation. The procedure took roughly 30 minutes initially, but times came down with greater operator experience. The most common indication for a permanent single-chamber pacemaker among study participants was chronic atrial fibrillation with second- or third-degree atrioventricular heart block.
The pacemaker is designed so the interventionalist can quickly determine if the device has been placed in an optimal location; if not, it can be repositioned during the same procedure before being finally locked into place. Twenty-three of the 32 patients did not require any device repositioning.
During 3 months of study follow-up, two patients required device retrieval. In one, the leadless pacemaker had migrated through an open patent foramen ovale into the left ventricle. The other patient developed indications warranting an implantable cardioverter-defibrillator. Device retrieval was a straightforward matter and took 6 and 13 minutes, respectively, using a basket catheter.
The one serious adverse event in the study involved a patient who experienced cardiac perforation and tamponade during pacemaker implantation. Five days after an uncomplicated surgical repair, the patient had a large right-sided stroke and died.
Dr. Reddy said the leadless pacemaker will be commercialized in Europe later this year. Planning is underway for a large, multicenter U.S. trial that will probably start next year. And Nanostim, the company developing the device, is also working on a leadless atrial pacemaker that will be able to communicate with the right ventricular pacemaker in order to provide multichamber cardiac pacing.
Dr. Reddy’s presentation met with an enthusiastic response.
"This is fascinating, incredible technology," declared Dr. Andrea M. Russo of Cooper University Health Care in Camden, N.J.
"The idea of being able to put a pacemaker into someone without having leads and without having a device under the skin is probably the next phase of what we’d hope to do with cardiac pacing therapy," Dr. Michael R. Gold observed.
"I think most of us, if we were to make predictions about what’s in store 10 years from now, we’d be hoping that we’d no longer use leads, which are the component that fails most frequently in any device. If we can get rid of the leads, put these little pacemakers anywhere we want in the heart, and get them to cross-talk and communicate with each other, that’s very exciting," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
AT HEART RHYTHM 2013
Major finding: A leadless cardiac pacemaker the size of a triple-A battery was successfully implanted in 32 of 33 candidates for permanent single-chamber pacing. The device performed well during 3 months of follow-up.
Data source: The first-in-man, prospective, single-arm, nonrandomized study conducted at three European centers.
Disclosures: The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.

Promising autism drugs progress through pipeline
HOLLYWOOD, FLA. – Arbaclofen, intranasal oxytocin, and D-cycloserine for the treatment of core deficits in autism spectrum disorders are all moving forward to more advanced clinical trials on the basis of encouraging studies highlighted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Arbaclofen, also known as R-baclofen and STX209, is an oral selective GABA-B agonist to be studied in a large phase III trial on the strength of encouraging improvements in social function seen in a 150-patient phase II study. Intranasal oxytocin showed improvements in selected aspects of social functioning as well as in irritability in a 25-patient feasibility study that cleared the way for the ongoing 300-patient SOARS-B study, sponsored by the National Institutes of Health.

D-cycloserine is earlier in development. The first placebo-controlled study is now being planned based upon favorable results on measures of social communication in an uncontrolled 20-patient pilot study.
This all comes as most welcome news. At present, the sole drugs approved for treatment of autism spectrum disorders (ASD) are risperidone (Risperdal) and aripiprazole (Abilify). They merely target the irritability associated with ASD rather than the core clinical features involving social deficits and repetitive behaviors. Meanwhile, the ASD caseload has been growing relentlessly for years. Indeed, the latest Centers for Disease Control and Prevention estimate of the ASD prevalence is 1 in 88 among U.S. children born in 2000 (MMWR 2012;61:1-19).
Arbaclofen
Dr. Paul P. Wang presented findings from a 12-week, double-blind, phase II study involving 150 subjects aged 5-21 years with autism disorder or Asperger’s disorder who were randomized to arbaclofen titrated to a maximum dose of 30 mg/day or placebo. The primary endpoint was improvement on the Aberrant Behavior Checklist–Lethargy/Social Withdrawal subscale. Such improvement was seen in the arbaclofen group, but also to a similar extent in the placebo arm. Dr. Wang was philosophical about the negative result.
"We are very early, obviously, in drug development for ASD. There is no Hamilton Autism Scale, no PANSS Autism Scale. It’s not entirely clear what scales are best psychometrically as well as being clinically valid. This is going to be a large challenge in all of the trials you’re likely to hear about in the next couple of years," predicted Dr. Wang, a behavioral pediatrician who is vice president for clinical development at Seaside Therapeutics Inc., Cambridge, Mass.
Discussant Dr. James McCracken concurred and added some advice for Dr. Wang and others searching for new ASD therapies.
"The real challenge in this area is our traditional overreliance on parent-reported observations of child behavior. It just sets us up for placebo effects that will kill any drug signal to be found. You have to work really hard to develop other measurement strategies that don’t depend so centrally on parent and caregiver report," said Dr. McCracken, professor of child psychiatry, and director of the division of child and adolescent psychiatry at the University of California, Los Angeles.
"You may be wondering, why are these people even working in this area? ASD is complicated, and the measurements are kind of lame. The secret is there are some great targets in ASD. This is a field ripe for treatment discovery," he said.

Dr. Wang said that fortunately, a post hoc analysis of a key secondary endpoint – the Vineland Adaptive Behavior Scales socialization score – showed an impressive improvement in response to arbaclofen among the 96 patients whose serial Vineland assessments were conducted by the same trained clinician and caregiver, in accord with the study protocol. Indeed, the arbaclofen group showed a mean 7.2-point gain, significantly better than the 1.8-point improvement with placebo.
Moreover, in the roughly half of study participants with an IQ of 70 or more, this effect was even more pronounced: an average gain of more than 9-points over the course of the study. To put this benefit in perspective, the mean score on the Vineland socialization measure in the general population is 100, and 1 standard deviation is 15 points.
"From a regulatory perspective and the narrow viewpoint of regulatory approval, this is a negative study. But I believe that from a scientific point of view, this is an extremely interesting study, and clinically potentially very important. We found what we believe to be a signal in the social-communicative domain that is the core impairment in autism," said Dr. Wang.
An efficacy signal also was noted in irritability and sensory symptoms, he added.
Eight patients in the arbaclofen group and two on placebo dropped out of the study because of adverse events, mostly behavioral. The only serious adverse event in the arbaclofen group involved one patient who developed suicidal thoughts on the drug, which disappeared promptly upon drug discontinuation; however, the same thing happened to one patient in the control group.
Arbaclofen is the active isomer of racemic baclofen, which was approved by the Food and Drug Administration in 1977 for treating spasticity in multiple sclerosis patients aged 12 and up. Baclofen also is widely used in the treatment of spasticity in cerebral palsy patients, many of whom are much younger than age 12. Its safety is well established. Baclofen is a strongly sedating drug; roughly one-third of cerebral palsy patients have to discontinue it for that reason. Yet only 8% of arbaclofen-treated patients in the phase II study reported any sedation.
Intranasal oxytocin
Oxytocin levels have been shown to be decreased in children with ASD; oxytocin-related genes are associated with increased risk for ASD; and patients with ASD have increased methylation of the oxytocin receptor. Therein lies the therapeutic rationale for exogenous oxytocin therapy.
Oxytocin is being used by thousands of patients as a complementary and alternative therapy for ASD, with numerous anecdotal reports of good benefit and tolerability but no published data. Internet pop-up ads tout oxytocin as "the love hormone." Alternative medicine providers use a host of different preparations and doses, typically with no monitoring of vital signs or documentation of outcomes. It’s not a healthy situation.
"We felt like it was time to act now," noted Dr. Linmarie Sikich, a psychiatrist at the University of North Carolina, Chapel Hill.

She and her coinvestigators therefore obtained a grant from Autism Speaks to conduct a phase II feasibility study in which 25 children with autistic disorder by DSM-IV criteria were randomized to 8 weeks of double-blind, twice-daily intranasal oxytocin flexibly dosed at 4-64 IU daily or placebo, followed by open-label oxytocin for all for an additional 8 weeks, then 3 months off-treatment before final follow-up. Eleven children were nonverbal, and 14 had fluent speech.
Oxytocin therapy was associated with improvement in multiple measures of social functioning, compared to placebo. Moreover, the benefits remained stable, and in some cases, increased during the 3 months after the end of treatment. Even the low-functioning, nonverbal young children were able to tolerate intranasal oxytocin.
Typical parental reports were that their child was more engaged at school, more compliant at home, more tolerant of peers, and better able to recognize the feelings of others, Dr. Sikich said.
The phase III SOARS-B study entails 6 months of double-blind intranasal oxytocin or placebo, then 6 months of open-label oxytocin for all, followed by 6 months off-therapy. Potential biomarkers of response as well as genetic samples are being obtained.
"We think the core social symptoms that are our target aren’t going to change in 8 weeks," she said "We think seeing meaningful clinical change is going to take more time."
Oxytocin is off patent. It lacks profit potential. As a result, pharmaceutical industry interest in developing it as an ASD therapy is nil. So Dr. Sikich and her academic colleagues are trying to do so, going so far as to arrange for intranasal oxytocin to be compounded in the United States to avoid the unpredictable shipment delays from European suppliers that occurred in the phase II study. She described working with the Food and Drug Administration in this effort as an eye-opening experience fraught with numbing hassles, bureaucratic indecision, and delays.
"Collaborating with pharma is a lot easier. I was not at all prepared for all of the things we’ve had to do that pharma typically does, and all of the associated costs," she said.
D-cycloserine
An antibiotic used as a second-line treatment for tuberculosis, D-cycloserine is a partial glycine agonist that binds with glutamate at the NMDA receptor to promote calcium conductance and normalize NMDA neurotransmission.
Based upon favorable animal studies, Dr. Maria R. Urbano and her colleagues conducted a 10-week uncontrolled, randomized, double-blind trial comparing D-cycloserine once-daily at 50 mg or once-weekly at 50 mg for 8 weeks in 20 patients with ASD. Participants were 14-25 years old, and all had an IQ above 70. Dr. Urbano chose this age range deliberately.

"I’m particularly interested in older adolescents and young adults with ASD, because in this group it’s imperative to try to interact socially as they try to launch from their families. There are too many of my patients who are sitting at home at age 20 or 21 doing nothing," observed Dr. Urbano, a psychiatrist and codirector of the ASD program at Eastern Virginia Medical School, Norfolk.
Ten of the 20 patients were deemed clinical responders based upon at least a 16-point (1 standard deviation) improvement in scores on the Social Responsiveness Scale. The mean improvement over the course of the study was 17.9 points, with no differences in the rate or magnitude of response between the two dosing arms. Since the efficacy was the same, once-weekly dosing will be used in the placebo-controlled trial now being planned. Once-weekly dosing might lessen the risk of side effects or tachyphylaxis, she noted.
Dr. Sikich and Dr. Urbano reported having no relevant financial interests. Dr. McCracken is a consultant to Roche and Seaside.
HOLLYWOOD, FLA. – Arbaclofen, intranasal oxytocin, and D-cycloserine for the treatment of core deficits in autism spectrum disorders are all moving forward to more advanced clinical trials on the basis of encouraging studies highlighted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Arbaclofen, also known as R-baclofen and STX209, is an oral selective GABA-B agonist to be studied in a large phase III trial on the strength of encouraging improvements in social function seen in a 150-patient phase II study. Intranasal oxytocin showed improvements in selected aspects of social functioning as well as in irritability in a 25-patient feasibility study that cleared the way for the ongoing 300-patient SOARS-B study, sponsored by the National Institutes of Health.
D-cycloserine is earlier in development. The first placebo-controlled study is now being planned based upon favorable results on measures of social communication in an uncontrolled 20-patient pilot study.
This all comes as most welcome news. At present, the sole drugs approved for treatment of autism spectrum disorders (ASD) are risperidone (Risperdal) and aripiprazole (Abilify). They merely target the irritability associated with ASD rather than the core clinical features involving social deficits and repetitive behaviors. Meanwhile, the ASD caseload has been growing relentlessly for years. Indeed, the latest Centers for Disease Control and Prevention estimate of the ASD prevalence is 1 in 88 among U.S. children born in 2000 (MMWR 2012;61:1-19).
Arbaclofen
Dr. Paul P. Wang presented findings from a 12-week, double-blind, phase II study involving 150 subjects aged 5-21 years with autism disorder or Asperger’s disorder who were randomized to arbaclofen titrated to a maximum dose of 30 mg/day or placebo. The primary endpoint was improvement on the Aberrant Behavior Checklist–Lethargy/Social Withdrawal subscale. Such improvement was seen in the arbaclofen group, but also to a similar extent in the placebo arm. Dr. Wang was philosophical about the negative result.
"We are very early, obviously, in drug development for ASD. There is no Hamilton Autism Scale, no PANSS Autism Scale. It’s not entirely clear what scales are best psychometrically as well as being clinically valid. This is going to be a large challenge in all of the trials you’re likely to hear about in the next couple of years," predicted Dr. Wang, a behavioral pediatrician who is vice president for clinical development at Seaside Therapeutics Inc., Cambridge, Mass.
Discussant Dr. James McCracken concurred and added some advice for Dr. Wang and others searching for new ASD therapies.
"The real challenge in this area is our traditional overreliance on parent-reported observations of child behavior. It just sets us up for placebo effects that will kill any drug signal to be found. You have to work really hard to develop other measurement strategies that don’t depend so centrally on parent and caregiver report," said Dr. McCracken, professor of child psychiatry, and director of the division of child and adolescent psychiatry at the University of California, Los Angeles.
"You may be wondering, why are these people even working in this area? ASD is complicated, and the measurements are kind of lame. The secret is there are some great targets in ASD. This is a field ripe for treatment discovery," he said.
Dr. Wang said that fortunately, a post hoc analysis of a key secondary endpoint – the Vineland Adaptive Behavior Scales socialization score – showed an impressive improvement in response to arbaclofen among the 96 patients whose serial Vineland assessments were conducted by the same trained clinician and caregiver, in accord with the study protocol. Indeed, the arbaclofen group showed a mean 7.2-point gain, significantly better than the 1.8-point improvement with placebo.
Moreover, in the roughly half of study participants with an IQ of 70 or more, this effect was even more pronounced: an average gain of more than 9-points over the course of the study. To put this benefit in perspective, the mean score on the Vineland socialization measure in the general population is 100, and 1 standard deviation is 15 points.
"From a regulatory perspective and the narrow viewpoint of regulatory approval, this is a negative study. But I believe that from a scientific point of view, this is an extremely interesting study, and clinically potentially very important. We found what we believe to be a signal in the social-communicative domain that is the core impairment in autism," said Dr. Wang.
An efficacy signal also was noted in irritability and sensory symptoms, he added.
Eight patients in the arbaclofen group and two on placebo dropped out of the study because of adverse events, mostly behavioral. The only serious adverse event in the arbaclofen group involved one patient who developed suicidal thoughts on the drug, which disappeared promptly upon drug discontinuation; however, the same thing happened to one patient in the control group.
Arbaclofen is the active isomer of racemic baclofen, which was approved by the Food and Drug Administration in 1977 for treating spasticity in multiple sclerosis patients aged 12 and up. Baclofen also is widely used in the treatment of spasticity in cerebral palsy patients, many of whom are much younger than age 12. Its safety is well established. Baclofen is a strongly sedating drug; roughly one-third of cerebral palsy patients have to discontinue it for that reason. Yet only 8% of arbaclofen-treated patients in the phase II study reported any sedation.
Intranasal oxytocin
Oxytocin levels have been shown to be decreased in children with ASD; oxytocin-related genes are associated with increased risk for ASD; and patients with ASD have increased methylation of the oxytocin receptor. Therein lies the therapeutic rationale for exogenous oxytocin therapy.
Oxytocin is being used by thousands of patients as a complementary and alternative therapy for ASD, with numerous anecdotal reports of good benefit and tolerability but no published data. Internet pop-up ads tout oxytocin as "the love hormone." Alternative medicine providers use a host of different preparations and doses, typically with no monitoring of vital signs or documentation of outcomes. It’s not a healthy situation.
"We felt like it was time to act now," noted Dr. Linmarie Sikich, a psychiatrist at the University of North Carolina, Chapel Hill.
She and her coinvestigators therefore obtained a grant from Autism Speaks to conduct a phase II feasibility study in which 25 children with autistic disorder by DSM-IV criteria were randomized to 8 weeks of double-blind, twice-daily intranasal oxytocin flexibly dosed at 4-64 IU daily or placebo, followed by open-label oxytocin for all for an additional 8 weeks, then 3 months off-treatment before final follow-up. Eleven children were nonverbal, and 14 had fluent speech.
Oxytocin therapy was associated with improvement in multiple measures of social functioning, compared to placebo. Moreover, the benefits remained stable, and in some cases, increased during the 3 months after the end of treatment. Even the low-functioning, nonverbal young children were able to tolerate intranasal oxytocin.
Typical parental reports were that their child was more engaged at school, more compliant at home, more tolerant of peers, and better able to recognize the feelings of others, Dr. Sikich said.
The phase III SOARS-B study entails 6 months of double-blind intranasal oxytocin or placebo, then 6 months of open-label oxytocin for all, followed by 6 months off-therapy. Potential biomarkers of response as well as genetic samples are being obtained.
"We think the core social symptoms that are our target aren’t going to change in 8 weeks," she said "We think seeing meaningful clinical change is going to take more time."
Oxytocin is off patent. It lacks profit potential. As a result, pharmaceutical industry interest in developing it as an ASD therapy is nil. So Dr. Sikich and her academic colleagues are trying to do so, going so far as to arrange for intranasal oxytocin to be compounded in the United States to avoid the unpredictable shipment delays from European suppliers that occurred in the phase II study. She described working with the Food and Drug Administration in this effort as an eye-opening experience fraught with numbing hassles, bureaucratic indecision, and delays.
"Collaborating with pharma is a lot easier. I was not at all prepared for all of the things we’ve had to do that pharma typically does, and all of the associated costs," she said.
D-cycloserine
An antibiotic used as a second-line treatment for tuberculosis, D-cycloserine is a partial glycine agonist that binds with glutamate at the NMDA receptor to promote calcium conductance and normalize NMDA neurotransmission.
Based upon favorable animal studies, Dr. Maria R. Urbano and her colleagues conducted a 10-week uncontrolled, randomized, double-blind trial comparing D-cycloserine once-daily at 50 mg or once-weekly at 50 mg for 8 weeks in 20 patients with ASD. Participants were 14-25 years old, and all had an IQ above 70. Dr. Urbano chose this age range deliberately.
"I’m particularly interested in older adolescents and young adults with ASD, because in this group it’s imperative to try to interact socially as they try to launch from their families. There are too many of my patients who are sitting at home at age 20 or 21 doing nothing," observed Dr. Urbano, a psychiatrist and codirector of the ASD program at Eastern Virginia Medical School, Norfolk.
Ten of the 20 patients were deemed clinical responders based upon at least a 16-point (1 standard deviation) improvement in scores on the Social Responsiveness Scale. The mean improvement over the course of the study was 17.9 points, with no differences in the rate or magnitude of response between the two dosing arms. Since the efficacy was the same, once-weekly dosing will be used in the placebo-controlled trial now being planned. Once-weekly dosing might lessen the risk of side effects or tachyphylaxis, she noted.
Dr. Sikich and Dr. Urbano reported having no relevant financial interests. Dr. McCracken is a consultant to Roche and Seaside.
HOLLYWOOD, FLA. – Arbaclofen, intranasal oxytocin, and D-cycloserine for the treatment of core deficits in autism spectrum disorders are all moving forward to more advanced clinical trials on the basis of encouraging studies highlighted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Arbaclofen, also known as R-baclofen and STX209, is an oral selective GABA-B agonist to be studied in a large phase III trial on the strength of encouraging improvements in social function seen in a 150-patient phase II study. Intranasal oxytocin showed improvements in selected aspects of social functioning as well as in irritability in a 25-patient feasibility study that cleared the way for the ongoing 300-patient SOARS-B study, sponsored by the National Institutes of Health.
D-cycloserine is earlier in development. The first placebo-controlled study is now being planned based upon favorable results on measures of social communication in an uncontrolled 20-patient pilot study.
This all comes as most welcome news. At present, the sole drugs approved for treatment of autism spectrum disorders (ASD) are risperidone (Risperdal) and aripiprazole (Abilify). They merely target the irritability associated with ASD rather than the core clinical features involving social deficits and repetitive behaviors. Meanwhile, the ASD caseload has been growing relentlessly for years. Indeed, the latest Centers for Disease Control and Prevention estimate of the ASD prevalence is 1 in 88 among U.S. children born in 2000 (MMWR 2012;61:1-19).
Arbaclofen
Dr. Paul P. Wang presented findings from a 12-week, double-blind, phase II study involving 150 subjects aged 5-21 years with autism disorder or Asperger’s disorder who were randomized to arbaclofen titrated to a maximum dose of 30 mg/day or placebo. The primary endpoint was improvement on the Aberrant Behavior Checklist–Lethargy/Social Withdrawal subscale. Such improvement was seen in the arbaclofen group, but also to a similar extent in the placebo arm. Dr. Wang was philosophical about the negative result.
"We are very early, obviously, in drug development for ASD. There is no Hamilton Autism Scale, no PANSS Autism Scale. It’s not entirely clear what scales are best psychometrically as well as being clinically valid. This is going to be a large challenge in all of the trials you’re likely to hear about in the next couple of years," predicted Dr. Wang, a behavioral pediatrician who is vice president for clinical development at Seaside Therapeutics Inc., Cambridge, Mass.
Discussant Dr. James McCracken concurred and added some advice for Dr. Wang and others searching for new ASD therapies.
"The real challenge in this area is our traditional overreliance on parent-reported observations of child behavior. It just sets us up for placebo effects that will kill any drug signal to be found. You have to work really hard to develop other measurement strategies that don’t depend so centrally on parent and caregiver report," said Dr. McCracken, professor of child psychiatry, and director of the division of child and adolescent psychiatry at the University of California, Los Angeles.
"You may be wondering, why are these people even working in this area? ASD is complicated, and the measurements are kind of lame. The secret is there are some great targets in ASD. This is a field ripe for treatment discovery," he said.
Dr. Wang said that fortunately, a post hoc analysis of a key secondary endpoint – the Vineland Adaptive Behavior Scales socialization score – showed an impressive improvement in response to arbaclofen among the 96 patients whose serial Vineland assessments were conducted by the same trained clinician and caregiver, in accord with the study protocol. Indeed, the arbaclofen group showed a mean 7.2-point gain, significantly better than the 1.8-point improvement with placebo.
Moreover, in the roughly half of study participants with an IQ of 70 or more, this effect was even more pronounced: an average gain of more than 9-points over the course of the study. To put this benefit in perspective, the mean score on the Vineland socialization measure in the general population is 100, and 1 standard deviation is 15 points.
"From a regulatory perspective and the narrow viewpoint of regulatory approval, this is a negative study. But I believe that from a scientific point of view, this is an extremely interesting study, and clinically potentially very important. We found what we believe to be a signal in the social-communicative domain that is the core impairment in autism," said Dr. Wang.
An efficacy signal also was noted in irritability and sensory symptoms, he added.
Eight patients in the arbaclofen group and two on placebo dropped out of the study because of adverse events, mostly behavioral. The only serious adverse event in the arbaclofen group involved one patient who developed suicidal thoughts on the drug, which disappeared promptly upon drug discontinuation; however, the same thing happened to one patient in the control group.
Arbaclofen is the active isomer of racemic baclofen, which was approved by the Food and Drug Administration in 1977 for treating spasticity in multiple sclerosis patients aged 12 and up. Baclofen also is widely used in the treatment of spasticity in cerebral palsy patients, many of whom are much younger than age 12. Its safety is well established. Baclofen is a strongly sedating drug; roughly one-third of cerebral palsy patients have to discontinue it for that reason. Yet only 8% of arbaclofen-treated patients in the phase II study reported any sedation.
Intranasal oxytocin
Oxytocin levels have been shown to be decreased in children with ASD; oxytocin-related genes are associated with increased risk for ASD; and patients with ASD have increased methylation of the oxytocin receptor. Therein lies the therapeutic rationale for exogenous oxytocin therapy.
Oxytocin is being used by thousands of patients as a complementary and alternative therapy for ASD, with numerous anecdotal reports of good benefit and tolerability but no published data. Internet pop-up ads tout oxytocin as "the love hormone." Alternative medicine providers use a host of different preparations and doses, typically with no monitoring of vital signs or documentation of outcomes. It’s not a healthy situation.
"We felt like it was time to act now," noted Dr. Linmarie Sikich, a psychiatrist at the University of North Carolina, Chapel Hill.
She and her coinvestigators therefore obtained a grant from Autism Speaks to conduct a phase II feasibility study in which 25 children with autistic disorder by DSM-IV criteria were randomized to 8 weeks of double-blind, twice-daily intranasal oxytocin flexibly dosed at 4-64 IU daily or placebo, followed by open-label oxytocin for all for an additional 8 weeks, then 3 months off-treatment before final follow-up. Eleven children were nonverbal, and 14 had fluent speech.
Oxytocin therapy was associated with improvement in multiple measures of social functioning, compared to placebo. Moreover, the benefits remained stable, and in some cases, increased during the 3 months after the end of treatment. Even the low-functioning, nonverbal young children were able to tolerate intranasal oxytocin.
Typical parental reports were that their child was more engaged at school, more compliant at home, more tolerant of peers, and better able to recognize the feelings of others, Dr. Sikich said.
The phase III SOARS-B study entails 6 months of double-blind intranasal oxytocin or placebo, then 6 months of open-label oxytocin for all, followed by 6 months off-therapy. Potential biomarkers of response as well as genetic samples are being obtained.
"We think the core social symptoms that are our target aren’t going to change in 8 weeks," she said "We think seeing meaningful clinical change is going to take more time."
Oxytocin is off patent. It lacks profit potential. As a result, pharmaceutical industry interest in developing it as an ASD therapy is nil. So Dr. Sikich and her academic colleagues are trying to do so, going so far as to arrange for intranasal oxytocin to be compounded in the United States to avoid the unpredictable shipment delays from European suppliers that occurred in the phase II study. She described working with the Food and Drug Administration in this effort as an eye-opening experience fraught with numbing hassles, bureaucratic indecision, and delays.
"Collaborating with pharma is a lot easier. I was not at all prepared for all of the things we’ve had to do that pharma typically does, and all of the associated costs," she said.
D-cycloserine
An antibiotic used as a second-line treatment for tuberculosis, D-cycloserine is a partial glycine agonist that binds with glutamate at the NMDA receptor to promote calcium conductance and normalize NMDA neurotransmission.
Based upon favorable animal studies, Dr. Maria R. Urbano and her colleagues conducted a 10-week uncontrolled, randomized, double-blind trial comparing D-cycloserine once-daily at 50 mg or once-weekly at 50 mg for 8 weeks in 20 patients with ASD. Participants were 14-25 years old, and all had an IQ above 70. Dr. Urbano chose this age range deliberately.
"I’m particularly interested in older adolescents and young adults with ASD, because in this group it’s imperative to try to interact socially as they try to launch from their families. There are too many of my patients who are sitting at home at age 20 or 21 doing nothing," observed Dr. Urbano, a psychiatrist and codirector of the ASD program at Eastern Virginia Medical School, Norfolk.
Ten of the 20 patients were deemed clinical responders based upon at least a 16-point (1 standard deviation) improvement in scores on the Social Responsiveness Scale. The mean improvement over the course of the study was 17.9 points, with no differences in the rate or magnitude of response between the two dosing arms. Since the efficacy was the same, once-weekly dosing will be used in the placebo-controlled trial now being planned. Once-weekly dosing might lessen the risk of side effects or tachyphylaxis, she noted.
Dr. Sikich and Dr. Urbano reported having no relevant financial interests. Dr. McCracken is a consultant to Roche and Seaside.
EXPERT ANALYSIS FROM THE NCDEU MEETING

Watchman device hits home run in PROTECT AF trial
DENVER – The investigational Watchman left atrial appendage closure device for stroke prevention in patients with atrial fibrillation proved markedly superior to warfarin in the 4-year follow-up of the randomized PROTECT AF trial.
Watchman recipients demonstrated a 40% reduction in the primary efficacy endpoint – a composite of stroke, systemic embolism, or cardiovascular death – compared with the warfarin-treated controls (95% confidence interval, 0.38-0.97; P = .03).
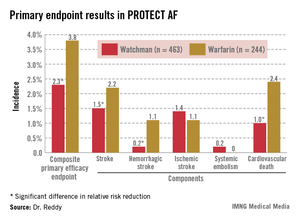
The Watchman group also showed a statistically significant 34% reduction in risk of all-cause mortality, compared with the 4.8% incidence in the warfarin arm (95% CI, 0.45-0.98; P = .04).
"This is something that’s really extraordinary. I wasn’t expecting to see a statistically significant decrease in all-cause mortality," Dr. Vivek Reddy said in presenting the new PROTECT AF data at the annual meeting of the Heart Rhythm Society.
The reduced all-cause mortality rate in the Watchman group resulted from fewer deaths due to cardiovascular-, pulmonary disease–, and hemorrhagic stroke–related causes.
In an earlier interim study analysis with 2.3 years and roughly 1,600 person-years of follow-up, the Watchman was merely statistically noninferior to warfarin in terms of efficacy (Circulation 2013;127:720-9). But in the updated analysis, with a mean follow-up of 45 months and 2,621 person-years, the device therapy has evolved in statistical parlance from noninferior to superior.
"The amount of benefit hasn’t changed. It’s been about a 40% difference throughout most of the trial. What’s changed is that with more follow-up the confidence intervals have narrowed, increasing the degree of certainty that the number is really true," according to Dr. Reddy of Mount Sinai School of Medicine, New York.
The Watchman’s superiority in terms of the primary efficacy endpoint was driven by an 85% reduction in the risk of hemorrhagic stroke and a 60% decrease in cardiovascular death compared with the warfarin group (see graphic). Importantly, the Watchman’s efficacy advantage was preserved in all prespecified patient subgroups, including the participants at greatest stroke risk: the roughly 20% with a baseline prior stroke or transient ischemic attack (TIA).
The Watchman is designed as a replacement for warfarin, a stroke prophylaxis drug whose numerous shortcomings are well known. The device is implanted in the left atrial appendage in a catheter procedure which entails an atrial puncture. Upon deployment, the Watchman excludes the left atrial appendage from the systemic circulation. The benefits seen in PROTECT AF and other Watchman studies confirm the notion that the left atrial appendage is critical to the pathogenesis of stroke in atrial fibrillation (AF), Dr. Reddy observed.
The 707 PROTECT AF participants at 59 centers were split fairly evenly between those with paroxysmal, persistent, and permanent AF. Subjects were randomized two-to-one to the Watchman or conventional warfarin therapy. Subjects in the warfarin group spent an average of 70% of their time with drug levels in therapeutic range, a considerably higher figure than studies would suggest is the norm in everyday clinical practice.

The composite primary safety endpoint occurred in a similar proportion of Watchman recipients and controls. However, the nature and timing of the adverse events differed in the two groups.
The most common adverse event in the Watchman group was serious pericardial effusion, which occurred in 4.8% of patients. Two-thirds of affected patients were managed via percutaneous intervention; the rest underwent surgery. The incidence of pericardial effusion decreased with greater operator experience: The rate was 6.3% in the first half of the study and 3.7% afterwards. Of note, in more recent studies begun since procedural refinements were introduced, the pericardial effusion rate has been lower than in PROTECT AF.
The chief adverse event in the warfarin group was, not surprisingly, major bleeding, which occurred in 4.1% of subjects, compared with a 0.6% rate in Watchman recipients. Unlike adverse events in the Watchman group, which were mostly procedure related, complications in the warfarin group were spread out over the full course of follow-up.
Dr. Reddy considers the PROTECT AF results broadly applicable even in this era of new oral anticoagulants.
"Despite the availability of novel anticoagulants, warfarin is still the most widely prescribed agent for stroke prevention," he noted.
Asked if he thought the Food and Drug Administration is likely to approve the Watchman based upon the new evidence from PROTECT AF as well as other studies, Dr. Reddy said it’s difficult to predict the agency’s actions but he certainly considers the device approvable.
If and when the Watchman is approved, he added, the first population where it is likely to see widespread use is in AF patients for whom warfarin is contraindicated. It would also be logical to consider the device in patients with a relative contraindication to warfarin, such as those on dialysis, at increased risk for falls, and elderly patients on polypharmacy; indeed, those are patient groups in which any anticoagulant is problematic. Head-to-head comparative studies of the Watchman and the novel anticoagulants would be welcome but are unlikely to occur anytime soon, Dr. Reddy said.
A fifth and final year of follow-up is scheduled for the PROTECT AF study.
The trial is sponsored by Boston Scientific. Dr. Reddy reported serving as a consultant to or receiving research grant support from Boston Scientific, Coherex Medical, and St. Jude Medical.
DENVER – The investigational Watchman left atrial appendage closure device for stroke prevention in patients with atrial fibrillation proved markedly superior to warfarin in the 4-year follow-up of the randomized PROTECT AF trial.
Watchman recipients demonstrated a 40% reduction in the primary efficacy endpoint – a composite of stroke, systemic embolism, or cardiovascular death – compared with the warfarin-treated controls (95% confidence interval, 0.38-0.97; P = .03).
The Watchman group also showed a statistically significant 34% reduction in risk of all-cause mortality, compared with the 4.8% incidence in the warfarin arm (95% CI, 0.45-0.98; P = .04).
"This is something that’s really extraordinary. I wasn’t expecting to see a statistically significant decrease in all-cause mortality," Dr. Vivek Reddy said in presenting the new PROTECT AF data at the annual meeting of the Heart Rhythm Society.
The reduced all-cause mortality rate in the Watchman group resulted from fewer deaths due to cardiovascular-, pulmonary disease–, and hemorrhagic stroke–related causes.
In an earlier interim study analysis with 2.3 years and roughly 1,600 person-years of follow-up, the Watchman was merely statistically noninferior to warfarin in terms of efficacy (Circulation 2013;127:720-9). But in the updated analysis, with a mean follow-up of 45 months and 2,621 person-years, the device therapy has evolved in statistical parlance from noninferior to superior.
"The amount of benefit hasn’t changed. It’s been about a 40% difference throughout most of the trial. What’s changed is that with more follow-up the confidence intervals have narrowed, increasing the degree of certainty that the number is really true," according to Dr. Reddy of Mount Sinai School of Medicine, New York.
The Watchman’s superiority in terms of the primary efficacy endpoint was driven by an 85% reduction in the risk of hemorrhagic stroke and a 60% decrease in cardiovascular death compared with the warfarin group (see graphic). Importantly, the Watchman’s efficacy advantage was preserved in all prespecified patient subgroups, including the participants at greatest stroke risk: the roughly 20% with a baseline prior stroke or transient ischemic attack (TIA).
The Watchman is designed as a replacement for warfarin, a stroke prophylaxis drug whose numerous shortcomings are well known. The device is implanted in the left atrial appendage in a catheter procedure which entails an atrial puncture. Upon deployment, the Watchman excludes the left atrial appendage from the systemic circulation. The benefits seen in PROTECT AF and other Watchman studies confirm the notion that the left atrial appendage is critical to the pathogenesis of stroke in atrial fibrillation (AF), Dr. Reddy observed.
The 707 PROTECT AF participants at 59 centers were split fairly evenly between those with paroxysmal, persistent, and permanent AF. Subjects were randomized two-to-one to the Watchman or conventional warfarin therapy. Subjects in the warfarin group spent an average of 70% of their time with drug levels in therapeutic range, a considerably higher figure than studies would suggest is the norm in everyday clinical practice.
The composite primary safety endpoint occurred in a similar proportion of Watchman recipients and controls. However, the nature and timing of the adverse events differed in the two groups.
The most common adverse event in the Watchman group was serious pericardial effusion, which occurred in 4.8% of patients. Two-thirds of affected patients were managed via percutaneous intervention; the rest underwent surgery. The incidence of pericardial effusion decreased with greater operator experience: The rate was 6.3% in the first half of the study and 3.7% afterwards. Of note, in more recent studies begun since procedural refinements were introduced, the pericardial effusion rate has been lower than in PROTECT AF.
The chief adverse event in the warfarin group was, not surprisingly, major bleeding, which occurred in 4.1% of subjects, compared with a 0.6% rate in Watchman recipients. Unlike adverse events in the Watchman group, which were mostly procedure related, complications in the warfarin group were spread out over the full course of follow-up.
Dr. Reddy considers the PROTECT AF results broadly applicable even in this era of new oral anticoagulants.
"Despite the availability of novel anticoagulants, warfarin is still the most widely prescribed agent for stroke prevention," he noted.
Asked if he thought the Food and Drug Administration is likely to approve the Watchman based upon the new evidence from PROTECT AF as well as other studies, Dr. Reddy said it’s difficult to predict the agency’s actions but he certainly considers the device approvable.
If and when the Watchman is approved, he added, the first population where it is likely to see widespread use is in AF patients for whom warfarin is contraindicated. It would also be logical to consider the device in patients with a relative contraindication to warfarin, such as those on dialysis, at increased risk for falls, and elderly patients on polypharmacy; indeed, those are patient groups in which any anticoagulant is problematic. Head-to-head comparative studies of the Watchman and the novel anticoagulants would be welcome but are unlikely to occur anytime soon, Dr. Reddy said.
A fifth and final year of follow-up is scheduled for the PROTECT AF study.
The trial is sponsored by Boston Scientific. Dr. Reddy reported serving as a consultant to or receiving research grant support from Boston Scientific, Coherex Medical, and St. Jude Medical.
DENVER – The investigational Watchman left atrial appendage closure device for stroke prevention in patients with atrial fibrillation proved markedly superior to warfarin in the 4-year follow-up of the randomized PROTECT AF trial.
Watchman recipients demonstrated a 40% reduction in the primary efficacy endpoint – a composite of stroke, systemic embolism, or cardiovascular death – compared with the warfarin-treated controls (95% confidence interval, 0.38-0.97; P = .03).
The Watchman group also showed a statistically significant 34% reduction in risk of all-cause mortality, compared with the 4.8% incidence in the warfarin arm (95% CI, 0.45-0.98; P = .04).
"This is something that’s really extraordinary. I wasn’t expecting to see a statistically significant decrease in all-cause mortality," Dr. Vivek Reddy said in presenting the new PROTECT AF data at the annual meeting of the Heart Rhythm Society.
The reduced all-cause mortality rate in the Watchman group resulted from fewer deaths due to cardiovascular-, pulmonary disease–, and hemorrhagic stroke–related causes.
In an earlier interim study analysis with 2.3 years and roughly 1,600 person-years of follow-up, the Watchman was merely statistically noninferior to warfarin in terms of efficacy (Circulation 2013;127:720-9). But in the updated analysis, with a mean follow-up of 45 months and 2,621 person-years, the device therapy has evolved in statistical parlance from noninferior to superior.
"The amount of benefit hasn’t changed. It’s been about a 40% difference throughout most of the trial. What’s changed is that with more follow-up the confidence intervals have narrowed, increasing the degree of certainty that the number is really true," according to Dr. Reddy of Mount Sinai School of Medicine, New York.
The Watchman’s superiority in terms of the primary efficacy endpoint was driven by an 85% reduction in the risk of hemorrhagic stroke and a 60% decrease in cardiovascular death compared with the warfarin group (see graphic). Importantly, the Watchman’s efficacy advantage was preserved in all prespecified patient subgroups, including the participants at greatest stroke risk: the roughly 20% with a baseline prior stroke or transient ischemic attack (TIA).
The Watchman is designed as a replacement for warfarin, a stroke prophylaxis drug whose numerous shortcomings are well known. The device is implanted in the left atrial appendage in a catheter procedure which entails an atrial puncture. Upon deployment, the Watchman excludes the left atrial appendage from the systemic circulation. The benefits seen in PROTECT AF and other Watchman studies confirm the notion that the left atrial appendage is critical to the pathogenesis of stroke in atrial fibrillation (AF), Dr. Reddy observed.
The 707 PROTECT AF participants at 59 centers were split fairly evenly between those with paroxysmal, persistent, and permanent AF. Subjects were randomized two-to-one to the Watchman or conventional warfarin therapy. Subjects in the warfarin group spent an average of 70% of their time with drug levels in therapeutic range, a considerably higher figure than studies would suggest is the norm in everyday clinical practice.
The composite primary safety endpoint occurred in a similar proportion of Watchman recipients and controls. However, the nature and timing of the adverse events differed in the two groups.
The most common adverse event in the Watchman group was serious pericardial effusion, which occurred in 4.8% of patients. Two-thirds of affected patients were managed via percutaneous intervention; the rest underwent surgery. The incidence of pericardial effusion decreased with greater operator experience: The rate was 6.3% in the first half of the study and 3.7% afterwards. Of note, in more recent studies begun since procedural refinements were introduced, the pericardial effusion rate has been lower than in PROTECT AF.
The chief adverse event in the warfarin group was, not surprisingly, major bleeding, which occurred in 4.1% of subjects, compared with a 0.6% rate in Watchman recipients. Unlike adverse events in the Watchman group, which were mostly procedure related, complications in the warfarin group were spread out over the full course of follow-up.
Dr. Reddy considers the PROTECT AF results broadly applicable even in this era of new oral anticoagulants.
"Despite the availability of novel anticoagulants, warfarin is still the most widely prescribed agent for stroke prevention," he noted.
Asked if he thought the Food and Drug Administration is likely to approve the Watchman based upon the new evidence from PROTECT AF as well as other studies, Dr. Reddy said it’s difficult to predict the agency’s actions but he certainly considers the device approvable.
If and when the Watchman is approved, he added, the first population where it is likely to see widespread use is in AF patients for whom warfarin is contraindicated. It would also be logical to consider the device in patients with a relative contraindication to warfarin, such as those on dialysis, at increased risk for falls, and elderly patients on polypharmacy; indeed, those are patient groups in which any anticoagulant is problematic. Head-to-head comparative studies of the Watchman and the novel anticoagulants would be welcome but are unlikely to occur anytime soon, Dr. Reddy said.
A fifth and final year of follow-up is scheduled for the PROTECT AF study.
The trial is sponsored by Boston Scientific. Dr. Reddy reported serving as a consultant to or receiving research grant support from Boston Scientific, Coherex Medical, and St. Jude Medical.
AT HEART RHYTHM 2013
Major finding: Patients with atrial fibrillation who received the Watchman left atrial appendage closure device had a 40% lower risk of the combined endpoint of stroke, systemic embolism, or cardiovascular death than those randomized to warfarin. They also had a 34% lower risk of all-cause mortality.
Data source: This analysis of the prospective, multicenter PROTECT AF trial included 707 randomized patients followed for a mean of 45 months.
Disclosures: PROTECT AF is sponsored by Boston Scientific. Dr. Reddy reported serving as a consultant to or receiving research grant support from Boston Scientific, Coherex Medical, and St. Jude Medical.
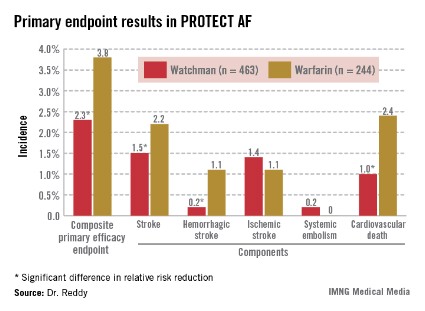
Renal dysfunction improved after AF ablation
DENVER – Catheter ablation of atrial fibrillation in patients with comorbid moderate chronic kidney disease may have a side benefit: improved renal function, according to Dr. Nazem W. Akoum.
Among 81 patients with stage 3 or 4 chronic kidney disease (CKD) who underwent successful AF ablation, the mean estimated glomerular filtration rate (eGFR) improved significantly, from 46.7 mL/min per 1.73 m2 at baseline to 53 mL/min per 1.73 m2 at follow-up 3 months post procedure, Dr. Akoum reported at the annual meeting of the Heart Rhythm Society.

In contrast, the eGFR in 382 patients with stage 1 or 2 CKD who underwent AF ablation didn’t change significantly, from a baseline mean of 103.6 mL/min per 1.73 m2, added Dr. Akoum, an electrophysiologist at the University of Utah, Salt Lake City.
Patients with AF and stage 5 or 6 CKD were excluded from participation in this study. Given the requirement for preablation quantification of the extent of atrial fibrosis via delayed-enhancement MRI with gadolinium contrast, there were safety concerns regarding exposing patients with advanced CKD to gadolinium.
Patients with stage 1 or 2 CKD averaged 15.8% left atrial fibrosis, while stage 3 or 4 patients averaged 19.1%.
The improvement in eGFR noted in the group with stage 3 or 4 CKD following restoration of sinus rhythm appeared to be independent of left ventricular systolic performance or medication effects. The postablation regularization of ventricular activation and the resultant improvement in endothelial function may have contributed to the welcome renal side benefit, Dr. Akoum said.
He reported having no relevant financial conflicts.
DENVER – Catheter ablation of atrial fibrillation in patients with comorbid moderate chronic kidney disease may have a side benefit: improved renal function, according to Dr. Nazem W. Akoum.
Among 81 patients with stage 3 or 4 chronic kidney disease (CKD) who underwent successful AF ablation, the mean estimated glomerular filtration rate (eGFR) improved significantly, from 46.7 mL/min per 1.73 m2 at baseline to 53 mL/min per 1.73 m2 at follow-up 3 months post procedure, Dr. Akoum reported at the annual meeting of the Heart Rhythm Society.
In contrast, the eGFR in 382 patients with stage 1 or 2 CKD who underwent AF ablation didn’t change significantly, from a baseline mean of 103.6 mL/min per 1.73 m2, added Dr. Akoum, an electrophysiologist at the University of Utah, Salt Lake City.
Patients with AF and stage 5 or 6 CKD were excluded from participation in this study. Given the requirement for preablation quantification of the extent of atrial fibrosis via delayed-enhancement MRI with gadolinium contrast, there were safety concerns regarding exposing patients with advanced CKD to gadolinium.
Patients with stage 1 or 2 CKD averaged 15.8% left atrial fibrosis, while stage 3 or 4 patients averaged 19.1%.
The improvement in eGFR noted in the group with stage 3 or 4 CKD following restoration of sinus rhythm appeared to be independent of left ventricular systolic performance or medication effects. The postablation regularization of ventricular activation and the resultant improvement in endothelial function may have contributed to the welcome renal side benefit, Dr. Akoum said.
He reported having no relevant financial conflicts.
DENVER – Catheter ablation of atrial fibrillation in patients with comorbid moderate chronic kidney disease may have a side benefit: improved renal function, according to Dr. Nazem W. Akoum.
Among 81 patients with stage 3 or 4 chronic kidney disease (CKD) who underwent successful AF ablation, the mean estimated glomerular filtration rate (eGFR) improved significantly, from 46.7 mL/min per 1.73 m2 at baseline to 53 mL/min per 1.73 m2 at follow-up 3 months post procedure, Dr. Akoum reported at the annual meeting of the Heart Rhythm Society.
In contrast, the eGFR in 382 patients with stage 1 or 2 CKD who underwent AF ablation didn’t change significantly, from a baseline mean of 103.6 mL/min per 1.73 m2, added Dr. Akoum, an electrophysiologist at the University of Utah, Salt Lake City.
Patients with AF and stage 5 or 6 CKD were excluded from participation in this study. Given the requirement for preablation quantification of the extent of atrial fibrosis via delayed-enhancement MRI with gadolinium contrast, there were safety concerns regarding exposing patients with advanced CKD to gadolinium.
Patients with stage 1 or 2 CKD averaged 15.8% left atrial fibrosis, while stage 3 or 4 patients averaged 19.1%.
The improvement in eGFR noted in the group with stage 3 or 4 CKD following restoration of sinus rhythm appeared to be independent of left ventricular systolic performance or medication effects. The postablation regularization of ventricular activation and the resultant improvement in endothelial function may have contributed to the welcome renal side benefit, Dr. Akoum said.
He reported having no relevant financial conflicts.
AT HEART RHYTHM 2013
Major finding: Patients with stage 3 or 4 chronic kidney disease who underwent catheter ablation of atrial fibrillation showed significant improvement in renal function 3 months post procedure, with their estimated glomerular filtration rate improving from a baseline mean of 46.7 mL/min per 1.73 m2 to 53 mL/min per 1.73 m2.
Data source: A prospective study of 463 patients with stage 1-4 chronic kidney disease undergoing AF ablation.
Disclosures: Dr. Nazem W. Akoum reported having no relevant financial conflicts.

Antipsychotics linked to sudden cardiac death risk
DENVER – Both the second-generation as well as the first-generation antipsychotic agents proved independently associated with greater than threefold increased risks of sudden cardiac death, according to results from a large, population-based study.
However, schizophrenia per se was not linked to an increased risk, Dr. Audrey Uy-Evanado reported at the annual meeting of the Heart Rhythm Society.
She presented data from the ongoing landmark Oregon Sudden Unexplained Death Study involving 1,544 documented sudden cardiac deaths (SCDs) and 774 matched controls.
The prevalence of diagnosed schizophrenia among patients with SCD was 2.5%, significantly greater than the 0.5% figure in controls. At the time of SCD, 8.2% of subjects were on an antipsychotic agent, compared with 1.9% of controls.
Second-generation, or "atypical," antipsychotic agents such as risperidone (Risperdal) and olanzapine (Zyprexa) were being used by 6.3% of the SCD cohort and 1.3% of controls. First-generation antipsychotic agents such as haloperidol, fluphenazine, and chlorpromazine were used by 2.6% of the SCD group, compared with 0.5% of controls. All of these between-group differences were highly significant.
Third-generation antipsychotic agents such as aripiprazole (Abilify) were so rarely used – only 0.1% of patients were on a third-generation agent – that it wasn’t possible to draw any conclusions about SCD risk, according to Dr. Uy-Evanado of Cedars-Sinai Medical Center in Los Angeles.
In terms of cardiovascular risk factors, hypertension and obesity were significantly more prevalent in the control group – but the SCD cohort had increased rates of diabetes, chronic kidney disease, and severe left ventricular dysfunction.
Aside from schizophrenia, rates of other psychiatric disorders didn’t differ between the two groups.
In a multivariate logistic regression analysis adjusted for age, gender, and comorbidities, having schizophrenia was not associated with an increased risk of SCD. However, patients on a first-generation antipsychotic agent had a 3.76-fold increased risk of SCD (P = .0002), and those on a second-generation antipsychotic drug had a 3.3-fold increased risk.
This study was undertaken as a follow-up to an earlier report from the Oregon Sudden Unexplained Death Study, which broadly identified antipsychotic drug therapy as being associated with SCD. That observation raised the question as to whether the risk is limited to specific classes of antipsychotic agents. The answer appears to be no.
The Oregon Sudden Unexplained Death Study is funded by Oregon Health and Science University and the Centers for Disease Control and Prevention. Dr. Uy-Evanado reported having no conflicts of interest.
DENVER – Both the second-generation as well as the first-generation antipsychotic agents proved independently associated with greater than threefold increased risks of sudden cardiac death, according to results from a large, population-based study.
However, schizophrenia per se was not linked to an increased risk, Dr. Audrey Uy-Evanado reported at the annual meeting of the Heart Rhythm Society.
She presented data from the ongoing landmark Oregon Sudden Unexplained Death Study involving 1,544 documented sudden cardiac deaths (SCDs) and 774 matched controls.
The prevalence of diagnosed schizophrenia among patients with SCD was 2.5%, significantly greater than the 0.5% figure in controls. At the time of SCD, 8.2% of subjects were on an antipsychotic agent, compared with 1.9% of controls.
Second-generation, or "atypical," antipsychotic agents such as risperidone (Risperdal) and olanzapine (Zyprexa) were being used by 6.3% of the SCD cohort and 1.3% of controls. First-generation antipsychotic agents such as haloperidol, fluphenazine, and chlorpromazine were used by 2.6% of the SCD group, compared with 0.5% of controls. All of these between-group differences were highly significant.
Third-generation antipsychotic agents such as aripiprazole (Abilify) were so rarely used – only 0.1% of patients were on a third-generation agent – that it wasn’t possible to draw any conclusions about SCD risk, according to Dr. Uy-Evanado of Cedars-Sinai Medical Center in Los Angeles.
In terms of cardiovascular risk factors, hypertension and obesity were significantly more prevalent in the control group – but the SCD cohort had increased rates of diabetes, chronic kidney disease, and severe left ventricular dysfunction.
Aside from schizophrenia, rates of other psychiatric disorders didn’t differ between the two groups.
In a multivariate logistic regression analysis adjusted for age, gender, and comorbidities, having schizophrenia was not associated with an increased risk of SCD. However, patients on a first-generation antipsychotic agent had a 3.76-fold increased risk of SCD (P = .0002), and those on a second-generation antipsychotic drug had a 3.3-fold increased risk.
This study was undertaken as a follow-up to an earlier report from the Oregon Sudden Unexplained Death Study, which broadly identified antipsychotic drug therapy as being associated with SCD. That observation raised the question as to whether the risk is limited to specific classes of antipsychotic agents. The answer appears to be no.
The Oregon Sudden Unexplained Death Study is funded by Oregon Health and Science University and the Centers for Disease Control and Prevention. Dr. Uy-Evanado reported having no conflicts of interest.
DENVER – Both the second-generation as well as the first-generation antipsychotic agents proved independently associated with greater than threefold increased risks of sudden cardiac death, according to results from a large, population-based study.
However, schizophrenia per se was not linked to an increased risk, Dr. Audrey Uy-Evanado reported at the annual meeting of the Heart Rhythm Society.
She presented data from the ongoing landmark Oregon Sudden Unexplained Death Study involving 1,544 documented sudden cardiac deaths (SCDs) and 774 matched controls.
The prevalence of diagnosed schizophrenia among patients with SCD was 2.5%, significantly greater than the 0.5% figure in controls. At the time of SCD, 8.2% of subjects were on an antipsychotic agent, compared with 1.9% of controls.
Second-generation, or "atypical," antipsychotic agents such as risperidone (Risperdal) and olanzapine (Zyprexa) were being used by 6.3% of the SCD cohort and 1.3% of controls. First-generation antipsychotic agents such as haloperidol, fluphenazine, and chlorpromazine were used by 2.6% of the SCD group, compared with 0.5% of controls. All of these between-group differences were highly significant.
Third-generation antipsychotic agents such as aripiprazole (Abilify) were so rarely used – only 0.1% of patients were on a third-generation agent – that it wasn’t possible to draw any conclusions about SCD risk, according to Dr. Uy-Evanado of Cedars-Sinai Medical Center in Los Angeles.
In terms of cardiovascular risk factors, hypertension and obesity were significantly more prevalent in the control group – but the SCD cohort had increased rates of diabetes, chronic kidney disease, and severe left ventricular dysfunction.
Aside from schizophrenia, rates of other psychiatric disorders didn’t differ between the two groups.
In a multivariate logistic regression analysis adjusted for age, gender, and comorbidities, having schizophrenia was not associated with an increased risk of SCD. However, patients on a first-generation antipsychotic agent had a 3.76-fold increased risk of SCD (P = .0002), and those on a second-generation antipsychotic drug had a 3.3-fold increased risk.
This study was undertaken as a follow-up to an earlier report from the Oregon Sudden Unexplained Death Study, which broadly identified antipsychotic drug therapy as being associated with SCD. That observation raised the question as to whether the risk is limited to specific classes of antipsychotic agents. The answer appears to be no.
The Oregon Sudden Unexplained Death Study is funded by Oregon Health and Science University and the Centers for Disease Control and Prevention. Dr. Uy-Evanado reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major finding: Use of a first-generation antipsychotic agent was independently associated with a 3.76-fold increased risk of sudden cardiac death in a large, ongoing community-based study. Use of a second-generation antipsychotic drug was associated with a 3.31-fold increased risk.
Data source: This analysis from the Oregon Sudden Unexplained Death Study involved 1,544 documented cases of sudden cardiac death and 774 controls.
Disclosures: The Oregon Sudden Unexplained Death Study is sponsored by Oregon Health and Science University and the Centers for Disease Control and Prevention. The presenter reported having no conflicts of interest.