User login
Shock-Less trial improves physicians' ICD programming
DENVER – The rate of adherence to evidence-based implantable cardioverter-defibrillator programming strategies known to reduce the rate of unnecessary shocks climbed significantly in a large, prospective study in which physicians received detailed reports on their own performance and how it stacked up to that of others.
The Shock-Less study was a real-world international study involving 4,131 ICD recipients and their physicians at 118 sites. The improved adherence to evidence-based programming achieved through the use of the individualized, multipage therapy programming reports (TPRs) translated into a highly significant 27% reduction in the risk of all-cause ICD shocks during follow-up, Dr. Marc T. Silver reported at the annual meeting of the Heart Rhythm Society.
This is welcome news for patients. It means less shock-related morbidity and potentially less mortality, said Dr. Silver of WakeMed in Raleigh, N.C.
The study included 2,693 patients who received their Medtronic primary or secondary prevention ICD before a center received its first Shock-Less report and 1,438 implanted after the report. A total of 265 all-cause shocks occurred in the "before" group, 116 in the "after" cohort.
In a multivariate logistic regression analysis adjusted for factors known to affect shock rates, including patient age, smoking status, New York Heart Association functional class, and atrial fibrillation, patients in the "after" group had a 27% reduction in the relative risk of both appropriate and inappropriate shocks (P = .002).
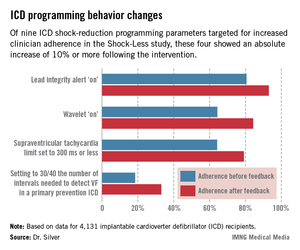
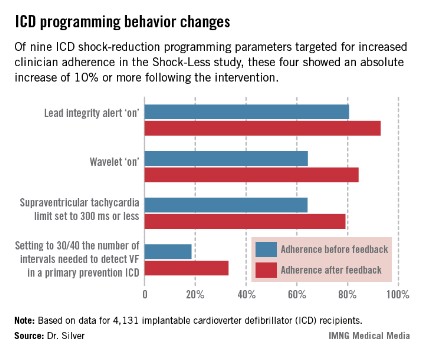
The TPRs provided ICD centers and their individual physicians with detailed feedback on rates of adherence to nine evidence-based programming settings that help reduce shocks. Most ICDs don’t arrive from the manufacturer with these settings in place. Some of the changes in programming were quite impressive (see graphic), including a near-doubling of the rate of primary prevention ICDs programmed to 30/40 as the number of intervals to detect ventricular fibrillation; this rate improved from 18.5% to 33.1%.
Dr. Silver offered personal testimony as to the power of the TPRs as a behavior-modification tool.

"Having received TPRs myself, it is a character-building experience. If you can get hold of information like this on your own practices, I guarantee you will leave a little smaller afterwards, like I did. Bigger in some way, smaller in others," he said.
Yet there remains a clear opportunity for further improvement in physician performance, Dr. Silver added.
"Achieving numbers in the 33% range for adherence to 30/40 [the number of intervals needed to detect ventricular fibrillation] is not what I think many of us would hope for one day," he observed.
Along those lines, audience member Dr. Thomas F. Deering of the Piedmont Heart Institute, Atlanta, commented that while the changes in physician behavior achieved through the Shock-Less project were significant, they were not sweeping in magnitude. Given the negative clinical consequences that result from lack of adherence to evidence-based programming, isn’t it time to request that the device industry change the default settings on their ICDs in accord with the evidence-based guidelines?, he asked.
"As someone who’s become very interested in physician behavior," Dr. Silver replied, "I regret to say that changing nominal settings on the devices may be the best way to move our profession forward. I say that with some degree of regret, but I think that’s the truth."
Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
DENVER – The rate of adherence to evidence-based implantable cardioverter-defibrillator programming strategies known to reduce the rate of unnecessary shocks climbed significantly in a large, prospective study in which physicians received detailed reports on their own performance and how it stacked up to that of others.
The Shock-Less study was a real-world international study involving 4,131 ICD recipients and their physicians at 118 sites. The improved adherence to evidence-based programming achieved through the use of the individualized, multipage therapy programming reports (TPRs) translated into a highly significant 27% reduction in the risk of all-cause ICD shocks during follow-up, Dr. Marc T. Silver reported at the annual meeting of the Heart Rhythm Society.
This is welcome news for patients. It means less shock-related morbidity and potentially less mortality, said Dr. Silver of WakeMed in Raleigh, N.C.
The study included 2,693 patients who received their Medtronic primary or secondary prevention ICD before a center received its first Shock-Less report and 1,438 implanted after the report. A total of 265 all-cause shocks occurred in the "before" group, 116 in the "after" cohort.
In a multivariate logistic regression analysis adjusted for factors known to affect shock rates, including patient age, smoking status, New York Heart Association functional class, and atrial fibrillation, patients in the "after" group had a 27% reduction in the relative risk of both appropriate and inappropriate shocks (P = .002).
The TPRs provided ICD centers and their individual physicians with detailed feedback on rates of adherence to nine evidence-based programming settings that help reduce shocks. Most ICDs don’t arrive from the manufacturer with these settings in place. Some of the changes in programming were quite impressive (see graphic), including a near-doubling of the rate of primary prevention ICDs programmed to 30/40 as the number of intervals to detect ventricular fibrillation; this rate improved from 18.5% to 33.1%.
Dr. Silver offered personal testimony as to the power of the TPRs as a behavior-modification tool.
"Having received TPRs myself, it is a character-building experience. If you can get hold of information like this on your own practices, I guarantee you will leave a little smaller afterwards, like I did. Bigger in some way, smaller in others," he said.
Yet there remains a clear opportunity for further improvement in physician performance, Dr. Silver added.
"Achieving numbers in the 33% range for adherence to 30/40 [the number of intervals needed to detect ventricular fibrillation] is not what I think many of us would hope for one day," he observed.
Along those lines, audience member Dr. Thomas F. Deering of the Piedmont Heart Institute, Atlanta, commented that while the changes in physician behavior achieved through the Shock-Less project were significant, they were not sweeping in magnitude. Given the negative clinical consequences that result from lack of adherence to evidence-based programming, isn’t it time to request that the device industry change the default settings on their ICDs in accord with the evidence-based guidelines?, he asked.
"As someone who’s become very interested in physician behavior," Dr. Silver replied, "I regret to say that changing nominal settings on the devices may be the best way to move our profession forward. I say that with some degree of regret, but I think that’s the truth."
Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
DENVER – The rate of adherence to evidence-based implantable cardioverter-defibrillator programming strategies known to reduce the rate of unnecessary shocks climbed significantly in a large, prospective study in which physicians received detailed reports on their own performance and how it stacked up to that of others.
The Shock-Less study was a real-world international study involving 4,131 ICD recipients and their physicians at 118 sites. The improved adherence to evidence-based programming achieved through the use of the individualized, multipage therapy programming reports (TPRs) translated into a highly significant 27% reduction in the risk of all-cause ICD shocks during follow-up, Dr. Marc T. Silver reported at the annual meeting of the Heart Rhythm Society.
This is welcome news for patients. It means less shock-related morbidity and potentially less mortality, said Dr. Silver of WakeMed in Raleigh, N.C.
The study included 2,693 patients who received their Medtronic primary or secondary prevention ICD before a center received its first Shock-Less report and 1,438 implanted after the report. A total of 265 all-cause shocks occurred in the "before" group, 116 in the "after" cohort.
In a multivariate logistic regression analysis adjusted for factors known to affect shock rates, including patient age, smoking status, New York Heart Association functional class, and atrial fibrillation, patients in the "after" group had a 27% reduction in the relative risk of both appropriate and inappropriate shocks (P = .002).
The TPRs provided ICD centers and their individual physicians with detailed feedback on rates of adherence to nine evidence-based programming settings that help reduce shocks. Most ICDs don’t arrive from the manufacturer with these settings in place. Some of the changes in programming were quite impressive (see graphic), including a near-doubling of the rate of primary prevention ICDs programmed to 30/40 as the number of intervals to detect ventricular fibrillation; this rate improved from 18.5% to 33.1%.
Dr. Silver offered personal testimony as to the power of the TPRs as a behavior-modification tool.
"Having received TPRs myself, it is a character-building experience. If you can get hold of information like this on your own practices, I guarantee you will leave a little smaller afterwards, like I did. Bigger in some way, smaller in others," he said.
Yet there remains a clear opportunity for further improvement in physician performance, Dr. Silver added.
"Achieving numbers in the 33% range for adherence to 30/40 [the number of intervals needed to detect ventricular fibrillation] is not what I think many of us would hope for one day," he observed.
Along those lines, audience member Dr. Thomas F. Deering of the Piedmont Heart Institute, Atlanta, commented that while the changes in physician behavior achieved through the Shock-Less project were significant, they were not sweeping in magnitude. Given the negative clinical consequences that result from lack of adherence to evidence-based programming, isn’t it time to request that the device industry change the default settings on their ICDs in accord with the evidence-based guidelines?, he asked.
"As someone who’s become very interested in physician behavior," Dr. Silver replied, "I regret to say that changing nominal settings on the devices may be the best way to move our profession forward. I say that with some degree of regret, but I think that’s the truth."
Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
AT HEART RHYTHM 2013
Major Finding: A 27% reduction in shocks from ICDs ensued after physicians received detailed, structured reports on their rates of adherence to evidence-based shock-reduction programming strategies.
Data Source: Shock-Less is an international prospective cohort study involving 4,131 ICD recipients and their physicians at 118 centers.
Disclosures: Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
Biomarkers predict response to cardiac resynchronization therapy
DENVER – Levels of troponin T and brain natriuretic peptide prior to implantation of a cardiac resynchronization device were strongly predictive of the risk of death or hospitalization for heart failure during the first year of device therapy in the BENEFIT study.
BENEFIT was a 1-year observational study undertaken to determine whether markers could predict which candidates for cardiac resynchronization therapy (CRT) were least likely to benefit from the costly device. At present, roughly 30% of patients who appropriately receive a CRT device do not respond to the treatment.
"We are all at this point frustrated with the persistent portion of CRT recipients who do not respond to therapy," BENEFIT principal investigator Dr. Alaa A. Shalaby said in presenting the study findings at the annual meeting of the Heart Rhythm Society. "We think our results suggest CRT should be offered earlier in the course of the disease"

BENEFIT included 267 CRT recipients at 33 centers. Patients were placed into one of three predefined risk categories based upon baseline levels of brain natriuretic peptide (BNP) and troponin T (TnT). On the basis os prior studies, high BNP was defined as 440 pg/mL or greater and a detectible TnT level was 0.01 ng/mL or greater. The low-risk group had an undetectable TnT and a BNP below 440 pg/mL. The intermediate-risk group had either an elevated BNP or TnT. The high-risk group had elevated BNP and TnT, reported Dr. Shalaby of the University of Pittsburgh.
The median baseline BNP in the study population was 198 pg/mL.
The three groups were similar in terms of baseline characteristics, including the proportion of patients with ischemic cardiomyopathy. Based on BNP and TnT results, 59% of patients were in the low-risk category, 33% had intermediate risk, and 8% were deemed high risk. The intermediate-risk group was evenly divided between patients who qualified on the basis of a high BNP and those with a detectable TnT.
During 12 months of follow-up there were 11 deaths and 19 heart failure hospitalizations. The incidence of either endpoint was 7% in the low-risk group, 15% in the intermediate-risk group, and 30% in the high-risk cohort.
After the researchers adjusted the results for age, left ventricular ejection fraction, QRS duration, and NYHA class, the risk of death or heart failure hospitalization was 2.5-fold greater in the group with intermediate-level baseline biomarkers as in the low-risk group. The high-biomarker group had a 7.3-fold increased risk, compared with the low-biomarker cohort. Both of these differences were statistically significant.
Changes in the biomarker levels after 6 and 12 months of CRT will be the subject of a future BENEFIT analysis, he noted.
The BENEFIT study was supported by St. Jude Medical. Dr. Shalaby reported having no conflicts of interest.
DENVER – Levels of troponin T and brain natriuretic peptide prior to implantation of a cardiac resynchronization device were strongly predictive of the risk of death or hospitalization for heart failure during the first year of device therapy in the BENEFIT study.
BENEFIT was a 1-year observational study undertaken to determine whether markers could predict which candidates for cardiac resynchronization therapy (CRT) were least likely to benefit from the costly device. At present, roughly 30% of patients who appropriately receive a CRT device do not respond to the treatment.
"We are all at this point frustrated with the persistent portion of CRT recipients who do not respond to therapy," BENEFIT principal investigator Dr. Alaa A. Shalaby said in presenting the study findings at the annual meeting of the Heart Rhythm Society. "We think our results suggest CRT should be offered earlier in the course of the disease"
BENEFIT included 267 CRT recipients at 33 centers. Patients were placed into one of three predefined risk categories based upon baseline levels of brain natriuretic peptide (BNP) and troponin T (TnT). On the basis os prior studies, high BNP was defined as 440 pg/mL or greater and a detectible TnT level was 0.01 ng/mL or greater. The low-risk group had an undetectable TnT and a BNP below 440 pg/mL. The intermediate-risk group had either an elevated BNP or TnT. The high-risk group had elevated BNP and TnT, reported Dr. Shalaby of the University of Pittsburgh.
The median baseline BNP in the study population was 198 pg/mL.
The three groups were similar in terms of baseline characteristics, including the proportion of patients with ischemic cardiomyopathy. Based on BNP and TnT results, 59% of patients were in the low-risk category, 33% had intermediate risk, and 8% were deemed high risk. The intermediate-risk group was evenly divided between patients who qualified on the basis of a high BNP and those with a detectable TnT.
During 12 months of follow-up there were 11 deaths and 19 heart failure hospitalizations. The incidence of either endpoint was 7% in the low-risk group, 15% in the intermediate-risk group, and 30% in the high-risk cohort.
After the researchers adjusted the results for age, left ventricular ejection fraction, QRS duration, and NYHA class, the risk of death or heart failure hospitalization was 2.5-fold greater in the group with intermediate-level baseline biomarkers as in the low-risk group. The high-biomarker group had a 7.3-fold increased risk, compared with the low-biomarker cohort. Both of these differences were statistically significant.
Changes in the biomarker levels after 6 and 12 months of CRT will be the subject of a future BENEFIT analysis, he noted.
The BENEFIT study was supported by St. Jude Medical. Dr. Shalaby reported having no conflicts of interest.
DENVER – Levels of troponin T and brain natriuretic peptide prior to implantation of a cardiac resynchronization device were strongly predictive of the risk of death or hospitalization for heart failure during the first year of device therapy in the BENEFIT study.
BENEFIT was a 1-year observational study undertaken to determine whether markers could predict which candidates for cardiac resynchronization therapy (CRT) were least likely to benefit from the costly device. At present, roughly 30% of patients who appropriately receive a CRT device do not respond to the treatment.
"We are all at this point frustrated with the persistent portion of CRT recipients who do not respond to therapy," BENEFIT principal investigator Dr. Alaa A. Shalaby said in presenting the study findings at the annual meeting of the Heart Rhythm Society. "We think our results suggest CRT should be offered earlier in the course of the disease"
BENEFIT included 267 CRT recipients at 33 centers. Patients were placed into one of three predefined risk categories based upon baseline levels of brain natriuretic peptide (BNP) and troponin T (TnT). On the basis os prior studies, high BNP was defined as 440 pg/mL or greater and a detectible TnT level was 0.01 ng/mL or greater. The low-risk group had an undetectable TnT and a BNP below 440 pg/mL. The intermediate-risk group had either an elevated BNP or TnT. The high-risk group had elevated BNP and TnT, reported Dr. Shalaby of the University of Pittsburgh.
The median baseline BNP in the study population was 198 pg/mL.
The three groups were similar in terms of baseline characteristics, including the proportion of patients with ischemic cardiomyopathy. Based on BNP and TnT results, 59% of patients were in the low-risk category, 33% had intermediate risk, and 8% were deemed high risk. The intermediate-risk group was evenly divided between patients who qualified on the basis of a high BNP and those with a detectable TnT.
During 12 months of follow-up there were 11 deaths and 19 heart failure hospitalizations. The incidence of either endpoint was 7% in the low-risk group, 15% in the intermediate-risk group, and 30% in the high-risk cohort.
After the researchers adjusted the results for age, left ventricular ejection fraction, QRS duration, and NYHA class, the risk of death or heart failure hospitalization was 2.5-fold greater in the group with intermediate-level baseline biomarkers as in the low-risk group. The high-biomarker group had a 7.3-fold increased risk, compared with the low-biomarker cohort. Both of these differences were statistically significant.
Changes in the biomarker levels after 6 and 12 months of CRT will be the subject of a future BENEFIT analysis, he noted.
The BENEFIT study was supported by St. Jude Medical. Dr. Shalaby reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major finding: Cardiac resynchronization therapy device recipients with a preimplantation brain natriuretic peptide level of at least 440 pg/mL and a detectable troponin T level were 7.3-fold more likely to die or be hospitalized for heart failure during the first 12 months after device implantation than were those with an undetectable troponin T level and a lower brain natriuretic peptide level.
Data source: The BENEFIT study was a prospective, 33-center study in which 267 patients who received a CRT device were followed for 1 year.
Disclosures: BENEFIT was supported by St. Jude Medical. The presenter reported having no conflicts of interest.

Optim ICD leads show tiny failure rate - so far
DENVER – Failure rates for the St. Jude Medical Riata ST Optim and Durata implantable cardioverter-defibrillator leads were reassuringly low in an independent analysis of three large prospective registries.
The study was undertaken at company expense because St. Jude Medical’s earlier-generation, silicone-wrapped Riata and Riata ST ICD leads were so failure-prone, that they were recalled several years ago. Their replacements – the subject of the new study – are insulated with Optim, a proprietary silicone/polyurethane copolymer with exceptional mechanical strength, flexibility, and abrasion resistance, according to Dr. John A. Cairns, who presented the registry study analysis at the annual meeting of the Heart Rhythm Society.

The prospective, St. Jude Medical–sponsored OPTIMUM, SJ4, and SCORE registries totaled 11,005 Durata and Riata ST Optim leads implanted in 10,820 patients. Median follow-up to date in the study, conducted independently under the direction of the Population Health Research Institute at McMaster University in Hamilton, Ont., is 3.0 years. Sixty-four percent of the leads remain under active follow-up, according to Dr. Cairns, who chairs the study steering committee and is professor of medicine at the University of British Columbia, Vancouver, as well as president-elect of the Canadian Academy of Health Sciences.
Participation in the registries entails patient follow-up every 6 months, with device interrogation encouraged. Participating physicians are supposed to return inactivated leads to St. Jude Medical for detailed analysis.
To date, the all-cause mechanical failure rate in the combined analysis is 0.35%, with a 0.22% conductor fracture rate, an insulation abrasion rate of 0.07%, and no cases of externalized conductors.
A life-table analysis was used to derive the 99.4% estimated 5-year rate of freedom from all-cause mechanical failure. The 5-year freedom from conductor fracture was 99.6%, with a 99.9% freedom from insulation abrasion and 100% freedom from externalized conductors.

Dr. Cairns said the company registries have been rigorously conducted, with central reporting and formal definitions of adverse events. Those strengths, coupled with the independent outside analysis, make the resultant findings more "in the realm of clinical trials rather than so much of the anecdotal information that’s out there," he added.
Session cochair John D. Day said in an interview that he considered the data "great news for patients with those leads" and was "cautiously optimistic" regarding the long-term performance of the ICD leads. He noted, however, that as of this analysis only 551 leads had actually reached the 5-year follow-up point. Many thousands more will do so in the next several years, at which point definitive conclusions can be drawn, according to Dr. Day, director of heart rhythm specialists at Intermountain Healthcare, Salt Lake City.
Dr. Cairns reported serving as a consultant to St. Jude Medical.
DENVER – Failure rates for the St. Jude Medical Riata ST Optim and Durata implantable cardioverter-defibrillator leads were reassuringly low in an independent analysis of three large prospective registries.
The study was undertaken at company expense because St. Jude Medical’s earlier-generation, silicone-wrapped Riata and Riata ST ICD leads were so failure-prone, that they were recalled several years ago. Their replacements – the subject of the new study – are insulated with Optim, a proprietary silicone/polyurethane copolymer with exceptional mechanical strength, flexibility, and abrasion resistance, according to Dr. John A. Cairns, who presented the registry study analysis at the annual meeting of the Heart Rhythm Society.
The prospective, St. Jude Medical–sponsored OPTIMUM, SJ4, and SCORE registries totaled 11,005 Durata and Riata ST Optim leads implanted in 10,820 patients. Median follow-up to date in the study, conducted independently under the direction of the Population Health Research Institute at McMaster University in Hamilton, Ont., is 3.0 years. Sixty-four percent of the leads remain under active follow-up, according to Dr. Cairns, who chairs the study steering committee and is professor of medicine at the University of British Columbia, Vancouver, as well as president-elect of the Canadian Academy of Health Sciences.
Participation in the registries entails patient follow-up every 6 months, with device interrogation encouraged. Participating physicians are supposed to return inactivated leads to St. Jude Medical for detailed analysis.
To date, the all-cause mechanical failure rate in the combined analysis is 0.35%, with a 0.22% conductor fracture rate, an insulation abrasion rate of 0.07%, and no cases of externalized conductors.
A life-table analysis was used to derive the 99.4% estimated 5-year rate of freedom from all-cause mechanical failure. The 5-year freedom from conductor fracture was 99.6%, with a 99.9% freedom from insulation abrasion and 100% freedom from externalized conductors.
Dr. Cairns said the company registries have been rigorously conducted, with central reporting and formal definitions of adverse events. Those strengths, coupled with the independent outside analysis, make the resultant findings more "in the realm of clinical trials rather than so much of the anecdotal information that’s out there," he added.
Session cochair John D. Day said in an interview that he considered the data "great news for patients with those leads" and was "cautiously optimistic" regarding the long-term performance of the ICD leads. He noted, however, that as of this analysis only 551 leads had actually reached the 5-year follow-up point. Many thousands more will do so in the next several years, at which point definitive conclusions can be drawn, according to Dr. Day, director of heart rhythm specialists at Intermountain Healthcare, Salt Lake City.
Dr. Cairns reported serving as a consultant to St. Jude Medical.
DENVER – Failure rates for the St. Jude Medical Riata ST Optim and Durata implantable cardioverter-defibrillator leads were reassuringly low in an independent analysis of three large prospective registries.
The study was undertaken at company expense because St. Jude Medical’s earlier-generation, silicone-wrapped Riata and Riata ST ICD leads were so failure-prone, that they were recalled several years ago. Their replacements – the subject of the new study – are insulated with Optim, a proprietary silicone/polyurethane copolymer with exceptional mechanical strength, flexibility, and abrasion resistance, according to Dr. John A. Cairns, who presented the registry study analysis at the annual meeting of the Heart Rhythm Society.
The prospective, St. Jude Medical–sponsored OPTIMUM, SJ4, and SCORE registries totaled 11,005 Durata and Riata ST Optim leads implanted in 10,820 patients. Median follow-up to date in the study, conducted independently under the direction of the Population Health Research Institute at McMaster University in Hamilton, Ont., is 3.0 years. Sixty-four percent of the leads remain under active follow-up, according to Dr. Cairns, who chairs the study steering committee and is professor of medicine at the University of British Columbia, Vancouver, as well as president-elect of the Canadian Academy of Health Sciences.
Participation in the registries entails patient follow-up every 6 months, with device interrogation encouraged. Participating physicians are supposed to return inactivated leads to St. Jude Medical for detailed analysis.
To date, the all-cause mechanical failure rate in the combined analysis is 0.35%, with a 0.22% conductor fracture rate, an insulation abrasion rate of 0.07%, and no cases of externalized conductors.
A life-table analysis was used to derive the 99.4% estimated 5-year rate of freedom from all-cause mechanical failure. The 5-year freedom from conductor fracture was 99.6%, with a 99.9% freedom from insulation abrasion and 100% freedom from externalized conductors.
Dr. Cairns said the company registries have been rigorously conducted, with central reporting and formal definitions of adverse events. Those strengths, coupled with the independent outside analysis, make the resultant findings more "in the realm of clinical trials rather than so much of the anecdotal information that’s out there," he added.
Session cochair John D. Day said in an interview that he considered the data "great news for patients with those leads" and was "cautiously optimistic" regarding the long-term performance of the ICD leads. He noted, however, that as of this analysis only 551 leads had actually reached the 5-year follow-up point. Many thousands more will do so in the next several years, at which point definitive conclusions can be drawn, according to Dr. Day, director of heart rhythm specialists at Intermountain Healthcare, Salt Lake City.
Dr. Cairns reported serving as a consultant to St. Jude Medical.
AT HEART RHYTHM 2013
Major finding: The 5-year rate of freedom from mechanical failure for St. Jude Medical’s Durata and Riata ST Optim ICD leads was 99.4% in a large study featuring outside analysis.
Data source: The study included 11,005 Durata or Riata ST Optim leads implanted in more than 10,000 patients with a median 3 years’ follow-up in three prospective registries.
Disclosures: The study was sponsored by St. Jude Medical and independently conducted by the Population Health Research Institute at McMaster University. Dr. Cairns reported serving as a consultant to St. Jude Medical.

Memantine delays driving impairment in mild Alzheimer's
HOLLYWOOD, FLA. – Treatment with memantine appears to delay progressive driving impairment in patients with mild Alzheimer’s disease, according to a year-long, double-blind, albeit small, randomized controlled trial.
"For many patients with Alzheimer’s disease, driving cessation is a major life-changing event that negatively impacts them and their caregivers. Until highly effective disease-altering treatments are available, there is a need to develop therapies that can prolong independent functioning," Dr. Peter J. Holland said in explaining the rationale for the driving skills study he presented at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

He chose memantine for the study for a couple of reasons. Preclinical evidence suggests the drug might have neuroprotective properties. And, in his experience, it’s a very well-tolerated drug.
There are a number of large controlled studies in which the dropout rate from adverse events was higher in the placebo group than with memantine, observed Dr. Holland, a psychiatrist and research professor of biomedical science at Florida Atlantic University, Boca Raton.
He reported on 60 otherwise healthy subjects with mild Alzheimer’s disease who were randomized to 12 months of double-blind memantine titrated to a target of 20 mg/day or placebo. All participants were still driving. The primary study endpoint was their score on the DriveABLE On Road Evaluation at 12 months. This structured, behind-the-wheel road test measures driving-related neurocognitive performance.
All 13 subjects on memantine either improved or maintained their baseline driving ability at 12 months, as did only 9 of 12 on placebo, a statistically significant difference (P = .05).
Asked about the high dropout rate, Dr. Holland noted that it’s a real challenge to keep older patients with Alzheimer’s disease in a study lasting a full year.
"The P value is there. I’d like to see the study replicated. If this holds true, it’s such a benign medication – it’s so well tolerated – that if it has a chance of being helpful, I think it’s worth further study," Dr. Holland added.
Memantine’s approved indication is in treating patients with moderate to-severe Alzheimer’s disease, so this application of the drug to delay driving impairment in patients with mild Alzheimer’s disease is off-label use.
In addition to the DriveABLE On Road test, study participants completed a battery of neurocognitive assessments at baseline, 6, and 12 months. These assessments were chosen because they test skills necessary for safe driving, including executive function, selective attention, and visuospatial abilities.
Thus far, Dr. Holland has determined that the memantine-treated group performed significantly better than controls at 12 months on the Rey-Osterrieth Complex Figure Test. In contrast, no significant difference between the two groups was noted on the Trail Making Part B test. As the study was only recently completed, the investigator is still analyzing the results on the Useful Field of View, Motor Free Visual Perception Test, and Alzheimer's Disease Assessment Scale-Cognitive test.
An estimated 5.2 million Americans aged 65 years or older have Alzheimer’s disease. And according to the American Automobile Association, in another dozen years, fully one-quarter of all U.S. drivers will be 65 years or older. A medication that slows driving impairment would have a large clinical impact, Dr. Holland noted.
His investigator-initiated study was funded by a research grant from Forest Pharmaceuticals.
HOLLYWOOD, FLA. – Treatment with memantine appears to delay progressive driving impairment in patients with mild Alzheimer’s disease, according to a year-long, double-blind, albeit small, randomized controlled trial.
"For many patients with Alzheimer’s disease, driving cessation is a major life-changing event that negatively impacts them and their caregivers. Until highly effective disease-altering treatments are available, there is a need to develop therapies that can prolong independent functioning," Dr. Peter J. Holland said in explaining the rationale for the driving skills study he presented at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
He chose memantine for the study for a couple of reasons. Preclinical evidence suggests the drug might have neuroprotective properties. And, in his experience, it’s a very well-tolerated drug.
There are a number of large controlled studies in which the dropout rate from adverse events was higher in the placebo group than with memantine, observed Dr. Holland, a psychiatrist and research professor of biomedical science at Florida Atlantic University, Boca Raton.
He reported on 60 otherwise healthy subjects with mild Alzheimer’s disease who were randomized to 12 months of double-blind memantine titrated to a target of 20 mg/day or placebo. All participants were still driving. The primary study endpoint was their score on the DriveABLE On Road Evaluation at 12 months. This structured, behind-the-wheel road test measures driving-related neurocognitive performance.
All 13 subjects on memantine either improved or maintained their baseline driving ability at 12 months, as did only 9 of 12 on placebo, a statistically significant difference (P = .05).
Asked about the high dropout rate, Dr. Holland noted that it’s a real challenge to keep older patients with Alzheimer’s disease in a study lasting a full year.
"The P value is there. I’d like to see the study replicated. If this holds true, it’s such a benign medication – it’s so well tolerated – that if it has a chance of being helpful, I think it’s worth further study," Dr. Holland added.
Memantine’s approved indication is in treating patients with moderate to-severe Alzheimer’s disease, so this application of the drug to delay driving impairment in patients with mild Alzheimer’s disease is off-label use.
In addition to the DriveABLE On Road test, study participants completed a battery of neurocognitive assessments at baseline, 6, and 12 months. These assessments were chosen because they test skills necessary for safe driving, including executive function, selective attention, and visuospatial abilities.
Thus far, Dr. Holland has determined that the memantine-treated group performed significantly better than controls at 12 months on the Rey-Osterrieth Complex Figure Test. In contrast, no significant difference between the two groups was noted on the Trail Making Part B test. As the study was only recently completed, the investigator is still analyzing the results on the Useful Field of View, Motor Free Visual Perception Test, and Alzheimer's Disease Assessment Scale-Cognitive test.
An estimated 5.2 million Americans aged 65 years or older have Alzheimer’s disease. And according to the American Automobile Association, in another dozen years, fully one-quarter of all U.S. drivers will be 65 years or older. A medication that slows driving impairment would have a large clinical impact, Dr. Holland noted.
His investigator-initiated study was funded by a research grant from Forest Pharmaceuticals.
HOLLYWOOD, FLA. – Treatment with memantine appears to delay progressive driving impairment in patients with mild Alzheimer’s disease, according to a year-long, double-blind, albeit small, randomized controlled trial.
"For many patients with Alzheimer’s disease, driving cessation is a major life-changing event that negatively impacts them and their caregivers. Until highly effective disease-altering treatments are available, there is a need to develop therapies that can prolong independent functioning," Dr. Peter J. Holland said in explaining the rationale for the driving skills study he presented at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
He chose memantine for the study for a couple of reasons. Preclinical evidence suggests the drug might have neuroprotective properties. And, in his experience, it’s a very well-tolerated drug.
There are a number of large controlled studies in which the dropout rate from adverse events was higher in the placebo group than with memantine, observed Dr. Holland, a psychiatrist and research professor of biomedical science at Florida Atlantic University, Boca Raton.
He reported on 60 otherwise healthy subjects with mild Alzheimer’s disease who were randomized to 12 months of double-blind memantine titrated to a target of 20 mg/day or placebo. All participants were still driving. The primary study endpoint was their score on the DriveABLE On Road Evaluation at 12 months. This structured, behind-the-wheel road test measures driving-related neurocognitive performance.
All 13 subjects on memantine either improved or maintained their baseline driving ability at 12 months, as did only 9 of 12 on placebo, a statistically significant difference (P = .05).
Asked about the high dropout rate, Dr. Holland noted that it’s a real challenge to keep older patients with Alzheimer’s disease in a study lasting a full year.
"The P value is there. I’d like to see the study replicated. If this holds true, it’s such a benign medication – it’s so well tolerated – that if it has a chance of being helpful, I think it’s worth further study," Dr. Holland added.
Memantine’s approved indication is in treating patients with moderate to-severe Alzheimer’s disease, so this application of the drug to delay driving impairment in patients with mild Alzheimer’s disease is off-label use.
In addition to the DriveABLE On Road test, study participants completed a battery of neurocognitive assessments at baseline, 6, and 12 months. These assessments were chosen because they test skills necessary for safe driving, including executive function, selective attention, and visuospatial abilities.
Thus far, Dr. Holland has determined that the memantine-treated group performed significantly better than controls at 12 months on the Rey-Osterrieth Complex Figure Test. In contrast, no significant difference between the two groups was noted on the Trail Making Part B test. As the study was only recently completed, the investigator is still analyzing the results on the Useful Field of View, Motor Free Visual Perception Test, and Alzheimer's Disease Assessment Scale-Cognitive test.
An estimated 5.2 million Americans aged 65 years or older have Alzheimer’s disease. And according to the American Automobile Association, in another dozen years, fully one-quarter of all U.S. drivers will be 65 years or older. A medication that slows driving impairment would have a large clinical impact, Dr. Holland noted.
His investigator-initiated study was funded by a research grant from Forest Pharmaceuticals.
AT THE NCDEU MEETING
Major finding: One hundred percent of patients with mild Alzheimer’s disease passed a structured on-the-road driving test after 12 months on memantine, compared to 75% on placebo in a small double-blind randomized trial.
Data source: A randomized, double-blind, prospective, 12-month study involving 60 patients with mild Alzheimer’s disease.
Disclosures: The study was funded by an investigator-initiated research grant from Forest Pharmaceuticals.

Treating adult ADHD improves parenting performance
HOLLYWOOD, FLA. – Pharmacologic treatment of parental attention-deficit/hyperactivity disorder might provide a novel means of improving parenting skills while reducing inappropriate behaviors in their unmedicated children with the disorder, according to Dr. James G. Waxmonsky.
In a structured study conducted by formally trained evaluators in a university family behavioral sciences center, treatment of parental ADHD with lisdexamfetamine dimesylate (Vyvanse) not only resulted in the expected reduction in parental ADHD symptoms, but was also associated with improved parenting performance and more harmonious child behavior in the laboratory setting, he reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Parents on ADHD medication gradually increased their use of praise over time by fivefold, while those on placebo did not change their use of praise. The parents on lisdexamfetamine dimesylate (LDX) also were significantly more verbally responsive to their child. There was less negative talk. Parents issued fewer commands, as well.

Even before the treated adults’ parenting performance began to show objective improvement, their children displayed a reduction in inappropriate and oppositional behaviors during joint homework assignments and other observed interactions.
"Most effects emerged several weeks into treatment with the optimal dose. The strength of effects on parenting behaviors paralleled that seen in trials of established parent training programs, while the degree of improvements in children’s oppositional behaviors matched that seen with stimulant medication," according to Dr. Waxmonsky, a child and adolescent psychiatrist serving as medical director of the center for children and families at Florida International University, Miami.
He presented a study involving 38 parents who met full DSM-IV criteria for ADHD and who also had a 5- to 15-year-old child with ADHD. The parents were started on LDX at 30 mg/day, titrated over the next several weeks to a maximum of 70 mg/day or until occurrence of at least a 30% reduction in scores on the ADHD Rating Scale. The mean optimized drug dose was 50 mg/day, with an average 58% reduction in ADHD Rating Scale score severity.
Once the medication was optimized, the parent and child were assessed using the Dyadic Parent-Child Interaction Coding System in the behavior laboratory on two occasions 1 week apart. In these sessions, the parent and child collaborated on a simulated homework task as well as an age-appropriate nonacademic task such as joint play or planning a family activity. In the first session, the blinded parent was on medication; for the second session, on placebo. The child with ADHD remained unmedicated throughout the study.
Significant improvement in parental ADHD symptoms was noted after just 1 week on the optimal medication dose. But in the initial pair of evaluations, the first of which was conducted when parents had been on their optimal drug dose for just 1-2 weeks, there was no significant difference between parenting behaviors when the parents were on and off medication.
In contrast to the lack of change in parenting behaviors during this initial study phase, significant reductions in the children’s inappropriate behavior during the homework task was documented when the parent was on LDX versus placebo. One plausible explanation for this observation is that a reduction in parental ADHD symptoms triggers improved child behavior. Then, as parents note improvements in their child’s behavior, they might respond by engaging in more positive parenting behaviors, in turn fostering further improvement over time in their child’s behavior. That would account for what was seen later in the study, Dr. Waxmonsky said.
In phase II of the study, the blinded parents were randomized to 8 weeks of optimized medication or placebo, followed by another evaluation of parent-child interaction in the behavior lab. This time, striking improvements in parenting performance were evident in the adults on extended duration of optimized medication compared with those on placebo. Moreover, the improvements in the behavior of the children whose parents were on LDX compared with that of the kids whose parents were on placebo were even larger than those seen in the first phase of the study. For example, children whose parents had been on optimized LDX for 8 weeks showed a fourfold greater reduction in inappropriate and oppositional behaviors than did children of parents on placebo.
The chief limitation of this in-depth study, Dr. Waxmonsky, is its small size. Almost none of the participating parents had ever been on anti-ADHD medication before; 10 of the 38 dropped out because of medication side effects, most prominently appetite loss, insomnia, and headaches.
The study was funded by a research grant from Shire Pharmaceuticals.
HOLLYWOOD, FLA. – Pharmacologic treatment of parental attention-deficit/hyperactivity disorder might provide a novel means of improving parenting skills while reducing inappropriate behaviors in their unmedicated children with the disorder, according to Dr. James G. Waxmonsky.
In a structured study conducted by formally trained evaluators in a university family behavioral sciences center, treatment of parental ADHD with lisdexamfetamine dimesylate (Vyvanse) not only resulted in the expected reduction in parental ADHD symptoms, but was also associated with improved parenting performance and more harmonious child behavior in the laboratory setting, he reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Parents on ADHD medication gradually increased their use of praise over time by fivefold, while those on placebo did not change their use of praise. The parents on lisdexamfetamine dimesylate (LDX) also were significantly more verbally responsive to their child. There was less negative talk. Parents issued fewer commands, as well.
Even before the treated adults’ parenting performance began to show objective improvement, their children displayed a reduction in inappropriate and oppositional behaviors during joint homework assignments and other observed interactions.
"Most effects emerged several weeks into treatment with the optimal dose. The strength of effects on parenting behaviors paralleled that seen in trials of established parent training programs, while the degree of improvements in children’s oppositional behaviors matched that seen with stimulant medication," according to Dr. Waxmonsky, a child and adolescent psychiatrist serving as medical director of the center for children and families at Florida International University, Miami.
He presented a study involving 38 parents who met full DSM-IV criteria for ADHD and who also had a 5- to 15-year-old child with ADHD. The parents were started on LDX at 30 mg/day, titrated over the next several weeks to a maximum of 70 mg/day or until occurrence of at least a 30% reduction in scores on the ADHD Rating Scale. The mean optimized drug dose was 50 mg/day, with an average 58% reduction in ADHD Rating Scale score severity.
Once the medication was optimized, the parent and child were assessed using the Dyadic Parent-Child Interaction Coding System in the behavior laboratory on two occasions 1 week apart. In these sessions, the parent and child collaborated on a simulated homework task as well as an age-appropriate nonacademic task such as joint play or planning a family activity. In the first session, the blinded parent was on medication; for the second session, on placebo. The child with ADHD remained unmedicated throughout the study.
Significant improvement in parental ADHD symptoms was noted after just 1 week on the optimal medication dose. But in the initial pair of evaluations, the first of which was conducted when parents had been on their optimal drug dose for just 1-2 weeks, there was no significant difference between parenting behaviors when the parents were on and off medication.
In contrast to the lack of change in parenting behaviors during this initial study phase, significant reductions in the children’s inappropriate behavior during the homework task was documented when the parent was on LDX versus placebo. One plausible explanation for this observation is that a reduction in parental ADHD symptoms triggers improved child behavior. Then, as parents note improvements in their child’s behavior, they might respond by engaging in more positive parenting behaviors, in turn fostering further improvement over time in their child’s behavior. That would account for what was seen later in the study, Dr. Waxmonsky said.
In phase II of the study, the blinded parents were randomized to 8 weeks of optimized medication or placebo, followed by another evaluation of parent-child interaction in the behavior lab. This time, striking improvements in parenting performance were evident in the adults on extended duration of optimized medication compared with those on placebo. Moreover, the improvements in the behavior of the children whose parents were on LDX compared with that of the kids whose parents were on placebo were even larger than those seen in the first phase of the study. For example, children whose parents had been on optimized LDX for 8 weeks showed a fourfold greater reduction in inappropriate and oppositional behaviors than did children of parents on placebo.
The chief limitation of this in-depth study, Dr. Waxmonsky, is its small size. Almost none of the participating parents had ever been on anti-ADHD medication before; 10 of the 38 dropped out because of medication side effects, most prominently appetite loss, insomnia, and headaches.
The study was funded by a research grant from Shire Pharmaceuticals.
HOLLYWOOD, FLA. – Pharmacologic treatment of parental attention-deficit/hyperactivity disorder might provide a novel means of improving parenting skills while reducing inappropriate behaviors in their unmedicated children with the disorder, according to Dr. James G. Waxmonsky.
In a structured study conducted by formally trained evaluators in a university family behavioral sciences center, treatment of parental ADHD with lisdexamfetamine dimesylate (Vyvanse) not only resulted in the expected reduction in parental ADHD symptoms, but was also associated with improved parenting performance and more harmonious child behavior in the laboratory setting, he reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Parents on ADHD medication gradually increased their use of praise over time by fivefold, while those on placebo did not change their use of praise. The parents on lisdexamfetamine dimesylate (LDX) also were significantly more verbally responsive to their child. There was less negative talk. Parents issued fewer commands, as well.
Even before the treated adults’ parenting performance began to show objective improvement, their children displayed a reduction in inappropriate and oppositional behaviors during joint homework assignments and other observed interactions.
"Most effects emerged several weeks into treatment with the optimal dose. The strength of effects on parenting behaviors paralleled that seen in trials of established parent training programs, while the degree of improvements in children’s oppositional behaviors matched that seen with stimulant medication," according to Dr. Waxmonsky, a child and adolescent psychiatrist serving as medical director of the center for children and families at Florida International University, Miami.
He presented a study involving 38 parents who met full DSM-IV criteria for ADHD and who also had a 5- to 15-year-old child with ADHD. The parents were started on LDX at 30 mg/day, titrated over the next several weeks to a maximum of 70 mg/day or until occurrence of at least a 30% reduction in scores on the ADHD Rating Scale. The mean optimized drug dose was 50 mg/day, with an average 58% reduction in ADHD Rating Scale score severity.
Once the medication was optimized, the parent and child were assessed using the Dyadic Parent-Child Interaction Coding System in the behavior laboratory on two occasions 1 week apart. In these sessions, the parent and child collaborated on a simulated homework task as well as an age-appropriate nonacademic task such as joint play or planning a family activity. In the first session, the blinded parent was on medication; for the second session, on placebo. The child with ADHD remained unmedicated throughout the study.
Significant improvement in parental ADHD symptoms was noted after just 1 week on the optimal medication dose. But in the initial pair of evaluations, the first of which was conducted when parents had been on their optimal drug dose for just 1-2 weeks, there was no significant difference between parenting behaviors when the parents were on and off medication.
In contrast to the lack of change in parenting behaviors during this initial study phase, significant reductions in the children’s inappropriate behavior during the homework task was documented when the parent was on LDX versus placebo. One plausible explanation for this observation is that a reduction in parental ADHD symptoms triggers improved child behavior. Then, as parents note improvements in their child’s behavior, they might respond by engaging in more positive parenting behaviors, in turn fostering further improvement over time in their child’s behavior. That would account for what was seen later in the study, Dr. Waxmonsky said.
In phase II of the study, the blinded parents were randomized to 8 weeks of optimized medication or placebo, followed by another evaluation of parent-child interaction in the behavior lab. This time, striking improvements in parenting performance were evident in the adults on extended duration of optimized medication compared with those on placebo. Moreover, the improvements in the behavior of the children whose parents were on LDX compared with that of the kids whose parents were on placebo were even larger than those seen in the first phase of the study. For example, children whose parents had been on optimized LDX for 8 weeks showed a fourfold greater reduction in inappropriate and oppositional behaviors than did children of parents on placebo.
The chief limitation of this in-depth study, Dr. Waxmonsky, is its small size. Almost none of the participating parents had ever been on anti-ADHD medication before; 10 of the 38 dropped out because of medication side effects, most prominently appetite loss, insomnia, and headaches.
The study was funded by a research grant from Shire Pharmaceuticals.
AT THE NCDEU MEETING
Major finding: Parenting performance improved when adults with ADHD underwent pharmacologic treatment. Marked improvements also were documented in the behavior of their children, who also had ADHD but remained unmedicated.
Data source: A study conducted in a university behavior lab involving 38 parents with ADHD, each with a 5- to 15-year-old child who also had ADHD.
Disclosures: The study was funded by a research grant from Shire Pharmaceuticals.
Novel contact force catheter advances AF ablation
DENVER – An investigational atrial fibrillation ablation catheter that provides the interventionalist with real-time measurement of contact force with the beating heart wall is safe and appears to improve 12-month success rates, according to the SMART-AF trial.
The principle underlying the development of this novel ablation catheter is that the durability of the therapeutic lesion created during AF ablation depends upon the amount of contact between the radiofrequency electrode and target tissue. Conventional catheters require the operator to assess this mainly visually, a hit-or-miss proposition. In contrast, the new catheter uses contact force sensing technology to provide the operator with quantitative feedback as to whether adequate tissue contact is occurring. The Biosense Webster ThermoCool SmartTouch contact force catheter is integrated with a standard 3-D navigation system, so the operator can see the data on the monitor during the procedure, Dr. Andrea Natale explained in presenting the SMART-AF results at the annual meeting of the Heart Rhythm Society.

The primary efficacy endpoint was freedom from documented AF, atrial flutter, and atrial tachycardia episodes through 12 months of follow-up, not including the standard 3-month blanking period immediately post ablation. Seventy-two percent of the 122 patients with symptomatic paroxysmal AF included in the efficacy analysis remained recurrence free at 12 months.
More impressively, the success rate climbed to 84% in cases where the interventionalist maintained contact force in the therapeutic range more than 82% of the time, while dropping to 61% when the contact force time was 82% or less, reported Dr. Natale, executive medical director of the Texas Cardiac Arrhythmia Institute, Austin.
While therapeutic efficacy proved to be a function of time spent in the therapeutic contact force range, adverse events were not, he noted.
The rate of serious adverse events occurring within 7 days of ablation was 9.9% among the 161 patients included in the SMART-AF safety analysis. The efficacy analysis included 39 fewer patients than the safety analysis. Those first 39 patients comprised investigators’ learning curve in the multicenter, prospective, single-arm study.
An independent safety committee deemed the single case of pericardial effusion that occurred in SMART-AF to be the only device-related adverse event. In addition, the committee judged the four cases of cardiac tamponade and the single case of pericarditis to be possibly device related. No strokes, MIs, or thromboembolisms occurred in the study.
The Food and Drug Administration allowed SMART-AF to use historical controls from earlier clinical trials utilizing a similar Biosense Webster ablation catheter which lacked contact force sensing. The required endpoints in SMART-AF were a 12-month freedom from recurrence greater than 50% and an early adverse event rate less than 16.6%, both of which were met.
SMART-AF investigators selected individual contact force target ranges based upon their early experience with the technology along with patient characteristics. To date, it hasn’t been possible to zero in on a fixed quantity of contact force that predicts treatment success regardless of who is doing the procedure.
All patients underwent circumferential pulmonary vein isolation, with additional radiofrequency lesions placed outside the pulmonary vein ostia as warranted.
The contact force sensing catheter utilizes a 7.5 F shaft with an 8 F electrode. Dr. Natale said it’s his anecdotal impression that the catheter results in reduced fluoroscopy and procedure times, but they weren’t measured in the study. What patients and physicians really care about, he added, is therapeutic success.
Asked if he thought ablation catheters using contact force sensing technology, if eventually approved by the FDA, would become the standard of care, he replied that he believes so, as long as the price is reasonable.
Dr. Michael R. Gold, who wasn’t involved in the SMART-AF study, commented that he found the results "very exciting."
"Our ability – using our fingers 3 feet away from a catheter – to know whether we’re making the right amount of contact is limited. Often, we worry, is it too little or too much? Experts like Dr. Natale do it wonderfully, but for the rest of us, it’s a challenge to try to do these ablations. So the concept of being able to know how much force you’re creating, and to optimize it while minimizing the risk of complications [such as] pushing too hard and poking a hole in the heart, is clearly the Holy Grail we’ve been trying to achieve. This is the first step in that direction," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the Heart and Vascular Center at the Medical University of South Carolina, Charleston.
Dr. Natale serves as a consultant to Biosense Webster, the sponsor of the SMART-AF study, as well as to other medical device companies.
DENVER – An investigational atrial fibrillation ablation catheter that provides the interventionalist with real-time measurement of contact force with the beating heart wall is safe and appears to improve 12-month success rates, according to the SMART-AF trial.
The principle underlying the development of this novel ablation catheter is that the durability of the therapeutic lesion created during AF ablation depends upon the amount of contact between the radiofrequency electrode and target tissue. Conventional catheters require the operator to assess this mainly visually, a hit-or-miss proposition. In contrast, the new catheter uses contact force sensing technology to provide the operator with quantitative feedback as to whether adequate tissue contact is occurring. The Biosense Webster ThermoCool SmartTouch contact force catheter is integrated with a standard 3-D navigation system, so the operator can see the data on the monitor during the procedure, Dr. Andrea Natale explained in presenting the SMART-AF results at the annual meeting of the Heart Rhythm Society.
The primary efficacy endpoint was freedom from documented AF, atrial flutter, and atrial tachycardia episodes through 12 months of follow-up, not including the standard 3-month blanking period immediately post ablation. Seventy-two percent of the 122 patients with symptomatic paroxysmal AF included in the efficacy analysis remained recurrence free at 12 months.
More impressively, the success rate climbed to 84% in cases where the interventionalist maintained contact force in the therapeutic range more than 82% of the time, while dropping to 61% when the contact force time was 82% or less, reported Dr. Natale, executive medical director of the Texas Cardiac Arrhythmia Institute, Austin.
While therapeutic efficacy proved to be a function of time spent in the therapeutic contact force range, adverse events were not, he noted.
The rate of serious adverse events occurring within 7 days of ablation was 9.9% among the 161 patients included in the SMART-AF safety analysis. The efficacy analysis included 39 fewer patients than the safety analysis. Those first 39 patients comprised investigators’ learning curve in the multicenter, prospective, single-arm study.
An independent safety committee deemed the single case of pericardial effusion that occurred in SMART-AF to be the only device-related adverse event. In addition, the committee judged the four cases of cardiac tamponade and the single case of pericarditis to be possibly device related. No strokes, MIs, or thromboembolisms occurred in the study.
The Food and Drug Administration allowed SMART-AF to use historical controls from earlier clinical trials utilizing a similar Biosense Webster ablation catheter which lacked contact force sensing. The required endpoints in SMART-AF were a 12-month freedom from recurrence greater than 50% and an early adverse event rate less than 16.6%, both of which were met.
SMART-AF investigators selected individual contact force target ranges based upon their early experience with the technology along with patient characteristics. To date, it hasn’t been possible to zero in on a fixed quantity of contact force that predicts treatment success regardless of who is doing the procedure.
All patients underwent circumferential pulmonary vein isolation, with additional radiofrequency lesions placed outside the pulmonary vein ostia as warranted.
The contact force sensing catheter utilizes a 7.5 F shaft with an 8 F electrode. Dr. Natale said it’s his anecdotal impression that the catheter results in reduced fluoroscopy and procedure times, but they weren’t measured in the study. What patients and physicians really care about, he added, is therapeutic success.
Asked if he thought ablation catheters using contact force sensing technology, if eventually approved by the FDA, would become the standard of care, he replied that he believes so, as long as the price is reasonable.
Dr. Michael R. Gold, who wasn’t involved in the SMART-AF study, commented that he found the results "very exciting."
"Our ability – using our fingers 3 feet away from a catheter – to know whether we’re making the right amount of contact is limited. Often, we worry, is it too little or too much? Experts like Dr. Natale do it wonderfully, but for the rest of us, it’s a challenge to try to do these ablations. So the concept of being able to know how much force you’re creating, and to optimize it while minimizing the risk of complications [such as] pushing too hard and poking a hole in the heart, is clearly the Holy Grail we’ve been trying to achieve. This is the first step in that direction," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the Heart and Vascular Center at the Medical University of South Carolina, Charleston.
Dr. Natale serves as a consultant to Biosense Webster, the sponsor of the SMART-AF study, as well as to other medical device companies.
DENVER – An investigational atrial fibrillation ablation catheter that provides the interventionalist with real-time measurement of contact force with the beating heart wall is safe and appears to improve 12-month success rates, according to the SMART-AF trial.
The principle underlying the development of this novel ablation catheter is that the durability of the therapeutic lesion created during AF ablation depends upon the amount of contact between the radiofrequency electrode and target tissue. Conventional catheters require the operator to assess this mainly visually, a hit-or-miss proposition. In contrast, the new catheter uses contact force sensing technology to provide the operator with quantitative feedback as to whether adequate tissue contact is occurring. The Biosense Webster ThermoCool SmartTouch contact force catheter is integrated with a standard 3-D navigation system, so the operator can see the data on the monitor during the procedure, Dr. Andrea Natale explained in presenting the SMART-AF results at the annual meeting of the Heart Rhythm Society.
The primary efficacy endpoint was freedom from documented AF, atrial flutter, and atrial tachycardia episodes through 12 months of follow-up, not including the standard 3-month blanking period immediately post ablation. Seventy-two percent of the 122 patients with symptomatic paroxysmal AF included in the efficacy analysis remained recurrence free at 12 months.
More impressively, the success rate climbed to 84% in cases where the interventionalist maintained contact force in the therapeutic range more than 82% of the time, while dropping to 61% when the contact force time was 82% or less, reported Dr. Natale, executive medical director of the Texas Cardiac Arrhythmia Institute, Austin.
While therapeutic efficacy proved to be a function of time spent in the therapeutic contact force range, adverse events were not, he noted.
The rate of serious adverse events occurring within 7 days of ablation was 9.9% among the 161 patients included in the SMART-AF safety analysis. The efficacy analysis included 39 fewer patients than the safety analysis. Those first 39 patients comprised investigators’ learning curve in the multicenter, prospective, single-arm study.
An independent safety committee deemed the single case of pericardial effusion that occurred in SMART-AF to be the only device-related adverse event. In addition, the committee judged the four cases of cardiac tamponade and the single case of pericarditis to be possibly device related. No strokes, MIs, or thromboembolisms occurred in the study.
The Food and Drug Administration allowed SMART-AF to use historical controls from earlier clinical trials utilizing a similar Biosense Webster ablation catheter which lacked contact force sensing. The required endpoints in SMART-AF were a 12-month freedom from recurrence greater than 50% and an early adverse event rate less than 16.6%, both of which were met.
SMART-AF investigators selected individual contact force target ranges based upon their early experience with the technology along with patient characteristics. To date, it hasn’t been possible to zero in on a fixed quantity of contact force that predicts treatment success regardless of who is doing the procedure.
All patients underwent circumferential pulmonary vein isolation, with additional radiofrequency lesions placed outside the pulmonary vein ostia as warranted.
The contact force sensing catheter utilizes a 7.5 F shaft with an 8 F electrode. Dr. Natale said it’s his anecdotal impression that the catheter results in reduced fluoroscopy and procedure times, but they weren’t measured in the study. What patients and physicians really care about, he added, is therapeutic success.
Asked if he thought ablation catheters using contact force sensing technology, if eventually approved by the FDA, would become the standard of care, he replied that he believes so, as long as the price is reasonable.
Dr. Michael R. Gold, who wasn’t involved in the SMART-AF study, commented that he found the results "very exciting."
"Our ability – using our fingers 3 feet away from a catheter – to know whether we’re making the right amount of contact is limited. Often, we worry, is it too little or too much? Experts like Dr. Natale do it wonderfully, but for the rest of us, it’s a challenge to try to do these ablations. So the concept of being able to know how much force you’re creating, and to optimize it while minimizing the risk of complications [such as] pushing too hard and poking a hole in the heart, is clearly the Holy Grail we’ve been trying to achieve. This is the first step in that direction," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the Heart and Vascular Center at the Medical University of South Carolina, Charleston.
Dr. Natale serves as a consultant to Biosense Webster, the sponsor of the SMART-AF study, as well as to other medical device companies.
AT HEART RHYTHM 2013
Major finding: Patients undergoing ablation of paroxysmal atrial fibrillation using an ablation catheter featuring contact force sensing technology had an impressive 84% rate of freedom from recurrence at 12 months if the operator kept contact force with the target tissue within the desired range more than 82% of the time.
Data source: SMART-AF, a multicenter, prospective, nonrandomized study with 161 evaluated patients.
Disclosures: SMART-AF was sponsored by Biosense Webster. The presenter serves as a consultant to the company.

Vortioxetine effective in treatment-resistant depression
HOLLYWOOD, FLA. – The investigational antidepressant vortioxetine achieved a 55% remission rate in a large, double-blind study of patients with major depressive disorder switched to the drug after an inadequate response to one of a half-dozen approved selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors.
"I think it’s quite impressive to see a remission rate on the order of 55% in this difficult-to-treat population," Dr. Marianne Dragheim observed in presenting the study findings at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

After all, in the landmark NIMH-sponsored sequential treatment alternatives to relieve depression, or STAR*D trial, in which citalopram nonresponders were switched to second-line treatment with sustained-release bupropion, extended-release venlafaxine, or sertraline, the mean remission rate after 12-14 weeks on the backup medication was just 31% (N. Engl. J. Med. 2006;354:1231-42), noted Dr. Dragheim of H. Lundbeck A/S, Valby, Denmark.
Vortioxetine is a first of its kind multimodal antidepressant. It is an inhibitor of the 5-hydroxytryptamine (5-HT) or serotonin transporter. In addition, vortioxetine is a 5-HT3, 5-HT7, and 5-HT10 receptor antagonist, a 5-HT1A receptor agonist, and a 5-HT1B receptor partial agonist.
Vortioxetine is currently under review at the Food and Drug Administration for possible marketing approval. The evidence submitted to the FDA includes seven positive studies, including four phase III randomized trials presented in May at the annual meeting of the American Psychiatric Association in San Francisco. The switching study Dr. Dragheim presented at the NCDEU meeting was completed so recently that it was not included in the FDA’s data package.
She reported on 501 European patients with major depressive disorder who responded inadequately to at least 6 weeks of monotherapy with citalopram, escitalopram, sertraline, paroxetine, venlafaxine, or duloxetine at approved doses. The study participants, all of whom wanted to change their antidepressant because of inadequate response, were randomized to 12 weeks of double-blind treatment with flexibly dosed vortioxetine at 10-20 mg/day or agomelatine at 25-50 mg/day. Agomelatine is approved as an antidepressant in the European Union and elsewhere in the world, but not in the United States.
The primary study endpoint was the change in scores on the Montgomery-Åsberg Depression Rating Scale (MADRS) between baseline and week 8. From a baseline MADRS total score of 29, the vortioxetine group improved by a mean of 2.2 more points than the agomelatine group, a statistically significant difference.
In addition, the vortioxetine-treated patients fared significantly better in terms of numerous secondary endpoints. They had a mean 1.9-point greater improvement than did the agomelatine group at 8 weeks in the Hamilton Anxiety Rating Scale total score, a 0.3-point greater improvement on the Clinical Global Impression-Severity, and a 2.2-point bigger improvement on the Sheehan Disability Scale.
The week-8 response rate as reflected in at least a 50% improvement from baseline in MADRS score was 61.5% in the vortioxetine group and 47.3% with agomelatine.
Remission as defined by a MADRS score of 10 or less occurred in 40.5% of the vortioxetine group and 29.5% of the agomelatine group at week 8, and in 55.2% vs. 39.4%, respectively, at week 12 (P = .0002).
Study withdrawal because of adverse events occurred in 5.9% of the vortioxetine group, compared with 9.5% of patients on agomelatine. Nausea, reported by 16% of patients on vortioxetine, was the only side effect more common in patients on that drug; however, less than 1% of subjects on vortioxetine left the study because of nausea.
Vortioxetine is being developed jointly by H. Lundbeck and Takeda Pharmaceutical Co.Dr. Dragheim is a company employee.
HOLLYWOOD, FLA. – The investigational antidepressant vortioxetine achieved a 55% remission rate in a large, double-blind study of patients with major depressive disorder switched to the drug after an inadequate response to one of a half-dozen approved selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors.
"I think it’s quite impressive to see a remission rate on the order of 55% in this difficult-to-treat population," Dr. Marianne Dragheim observed in presenting the study findings at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
After all, in the landmark NIMH-sponsored sequential treatment alternatives to relieve depression, or STAR*D trial, in which citalopram nonresponders were switched to second-line treatment with sustained-release bupropion, extended-release venlafaxine, or sertraline, the mean remission rate after 12-14 weeks on the backup medication was just 31% (N. Engl. J. Med. 2006;354:1231-42), noted Dr. Dragheim of H. Lundbeck A/S, Valby, Denmark.
Vortioxetine is a first of its kind multimodal antidepressant. It is an inhibitor of the 5-hydroxytryptamine (5-HT) or serotonin transporter. In addition, vortioxetine is a 5-HT3, 5-HT7, and 5-HT10 receptor antagonist, a 5-HT1A receptor agonist, and a 5-HT1B receptor partial agonist.
Vortioxetine is currently under review at the Food and Drug Administration for possible marketing approval. The evidence submitted to the FDA includes seven positive studies, including four phase III randomized trials presented in May at the annual meeting of the American Psychiatric Association in San Francisco. The switching study Dr. Dragheim presented at the NCDEU meeting was completed so recently that it was not included in the FDA’s data package.
She reported on 501 European patients with major depressive disorder who responded inadequately to at least 6 weeks of monotherapy with citalopram, escitalopram, sertraline, paroxetine, venlafaxine, or duloxetine at approved doses. The study participants, all of whom wanted to change their antidepressant because of inadequate response, were randomized to 12 weeks of double-blind treatment with flexibly dosed vortioxetine at 10-20 mg/day or agomelatine at 25-50 mg/day. Agomelatine is approved as an antidepressant in the European Union and elsewhere in the world, but not in the United States.
The primary study endpoint was the change in scores on the Montgomery-Åsberg Depression Rating Scale (MADRS) between baseline and week 8. From a baseline MADRS total score of 29, the vortioxetine group improved by a mean of 2.2 more points than the agomelatine group, a statistically significant difference.
In addition, the vortioxetine-treated patients fared significantly better in terms of numerous secondary endpoints. They had a mean 1.9-point greater improvement than did the agomelatine group at 8 weeks in the Hamilton Anxiety Rating Scale total score, a 0.3-point greater improvement on the Clinical Global Impression-Severity, and a 2.2-point bigger improvement on the Sheehan Disability Scale.
The week-8 response rate as reflected in at least a 50% improvement from baseline in MADRS score was 61.5% in the vortioxetine group and 47.3% with agomelatine.
Remission as defined by a MADRS score of 10 or less occurred in 40.5% of the vortioxetine group and 29.5% of the agomelatine group at week 8, and in 55.2% vs. 39.4%, respectively, at week 12 (P = .0002).
Study withdrawal because of adverse events occurred in 5.9% of the vortioxetine group, compared with 9.5% of patients on agomelatine. Nausea, reported by 16% of patients on vortioxetine, was the only side effect more common in patients on that drug; however, less than 1% of subjects on vortioxetine left the study because of nausea.
Vortioxetine is being developed jointly by H. Lundbeck and Takeda Pharmaceutical Co.Dr. Dragheim is a company employee.
HOLLYWOOD, FLA. – The investigational antidepressant vortioxetine achieved a 55% remission rate in a large, double-blind study of patients with major depressive disorder switched to the drug after an inadequate response to one of a half-dozen approved selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors.
"I think it’s quite impressive to see a remission rate on the order of 55% in this difficult-to-treat population," Dr. Marianne Dragheim observed in presenting the study findings at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
After all, in the landmark NIMH-sponsored sequential treatment alternatives to relieve depression, or STAR*D trial, in which citalopram nonresponders were switched to second-line treatment with sustained-release bupropion, extended-release venlafaxine, or sertraline, the mean remission rate after 12-14 weeks on the backup medication was just 31% (N. Engl. J. Med. 2006;354:1231-42), noted Dr. Dragheim of H. Lundbeck A/S, Valby, Denmark.
Vortioxetine is a first of its kind multimodal antidepressant. It is an inhibitor of the 5-hydroxytryptamine (5-HT) or serotonin transporter. In addition, vortioxetine is a 5-HT3, 5-HT7, and 5-HT10 receptor antagonist, a 5-HT1A receptor agonist, and a 5-HT1B receptor partial agonist.
Vortioxetine is currently under review at the Food and Drug Administration for possible marketing approval. The evidence submitted to the FDA includes seven positive studies, including four phase III randomized trials presented in May at the annual meeting of the American Psychiatric Association in San Francisco. The switching study Dr. Dragheim presented at the NCDEU meeting was completed so recently that it was not included in the FDA’s data package.
She reported on 501 European patients with major depressive disorder who responded inadequately to at least 6 weeks of monotherapy with citalopram, escitalopram, sertraline, paroxetine, venlafaxine, or duloxetine at approved doses. The study participants, all of whom wanted to change their antidepressant because of inadequate response, were randomized to 12 weeks of double-blind treatment with flexibly dosed vortioxetine at 10-20 mg/day or agomelatine at 25-50 mg/day. Agomelatine is approved as an antidepressant in the European Union and elsewhere in the world, but not in the United States.
The primary study endpoint was the change in scores on the Montgomery-Åsberg Depression Rating Scale (MADRS) between baseline and week 8. From a baseline MADRS total score of 29, the vortioxetine group improved by a mean of 2.2 more points than the agomelatine group, a statistically significant difference.
In addition, the vortioxetine-treated patients fared significantly better in terms of numerous secondary endpoints. They had a mean 1.9-point greater improvement than did the agomelatine group at 8 weeks in the Hamilton Anxiety Rating Scale total score, a 0.3-point greater improvement on the Clinical Global Impression-Severity, and a 2.2-point bigger improvement on the Sheehan Disability Scale.
The week-8 response rate as reflected in at least a 50% improvement from baseline in MADRS score was 61.5% in the vortioxetine group and 47.3% with agomelatine.
Remission as defined by a MADRS score of 10 or less occurred in 40.5% of the vortioxetine group and 29.5% of the agomelatine group at week 8, and in 55.2% vs. 39.4%, respectively, at week 12 (P = .0002).
Study withdrawal because of adverse events occurred in 5.9% of the vortioxetine group, compared with 9.5% of patients on agomelatine. Nausea, reported by 16% of patients on vortioxetine, was the only side effect more common in patients on that drug; however, less than 1% of subjects on vortioxetine left the study because of nausea.
Vortioxetine is being developed jointly by H. Lundbeck and Takeda Pharmaceutical Co.Dr. Dragheim is a company employee.
AT THE NCDEU MEETING
Major finding: Patients with major depressive disorder who did not respond adequately to a course of selective serotonin reuptake inhibitor or selective norepinephrine reuptake inhibitor therapy and who were then switched to double-blind treatment with the novel antidepressant vortioxetine had a 55% remission rate after 8 weeks of therapy.
Data source: A 501-patient, multicenter, randomized comparator European study.
Disclosures: The study was sponsored by H. Lundbeck A/S. Dr. Dragheim is a company employee.

Antibacterial envelope may limit cardiac device infections
DENVER – Encasing implantable cardioverter-defibrillators and cardiac resynchronization therapy devices in a proprietary antibiotic-impregnated envelope during implantation was associated with a low 90-day major infection rate in an interim analysis of a large prospective multicenter study.
The CITADEL/CENTURION study involves 1,000 patients who underwent ICD or CRT device replacement using the antibacterial envelope at 55 U.S. centers. A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient through 3 months of prospective follow-up, for an infection incidence of 0.1%, Dr. Charles Henrikson reported at the annual meeting of the Heart Rhythm Society.

CITADEL/CENTURION is not a randomized trial. Instead, the control group consists of 533 Canadian patients in a published study, all of whom underwent ICD or CRT replacement because of device advisories or recalls. The major device infection rate in this retrospective study (JAMA 2006;295:1907-11) during a mean 2.7 months following device replacement was 1.9%, or roughly 19-fold greater than in CITADEL/CENTURION, noted Dr. Henrikson of Oregon Health and Science University, Portland.
Device replacement is a known risk factor for major device infections, which carry a substantial toll in terms of morbidity, mortality, and expense.
The antibacterial envelope, known as AIGISRx, has received Food and Drug Administration approval and is commercially available. The antibacterial envelope is designed to prevent device migration as well as infections. "It changes the device from something that’s slick and slides right in into something with an incredibly high coefficient of friction, so you need to make a larger pocket to put it in. That’s why we’re paying a lot of attention to the hematoma rate," according to Dr. Henrikson.
The device consists of a nonresorbable polypropylene mesh coated with a resorbable polymer layer, which releases rifampin and minocycline for 7-10 days. These two antibiotics are active against the pathogens that cause most device infections, including both methicillin-sensitive and -resistant Staphylococcus aureus as well as S. epidermis.
The indication for device replacement in CITADEL/CENTURION was end of battery life in about three-quarters of patients, with device upgrades and lead revisions accounting for nearly all of the rest. Explantation because of device infection was a contraindication to study participation. All subjects were on perioperative prophylactic antibiotics, as were the historical controls.
The chief device-related complication recorded during the study was major pocket hematoma requiring a change in patient management, typically open drainage or transfusion. The complication occurred in 1.5% of patients, as compared to 2.25% of the historical controls.
Two of the 1,000 patients developed generalized rashes thought by independent reviewers to be possibly related to AIGISRx.
The plan is to monitor CITADEL/CENTURION participants for device infections and mechanical complications through 12 months, Dr. Henrikson explained. Only the 3-month data were available for presentation at a late-breaking clinical trials session during Heart Rhythm 2013.
CITADEL/CENTURION is sponsored by TYRX, which markets AIGISRx. Dr. Henrikson reported having received research support from the company.
DENVER – Encasing implantable cardioverter-defibrillators and cardiac resynchronization therapy devices in a proprietary antibiotic-impregnated envelope during implantation was associated with a low 90-day major infection rate in an interim analysis of a large prospective multicenter study.
The CITADEL/CENTURION study involves 1,000 patients who underwent ICD or CRT device replacement using the antibacterial envelope at 55 U.S. centers. A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient through 3 months of prospective follow-up, for an infection incidence of 0.1%, Dr. Charles Henrikson reported at the annual meeting of the Heart Rhythm Society.
CITADEL/CENTURION is not a randomized trial. Instead, the control group consists of 533 Canadian patients in a published study, all of whom underwent ICD or CRT replacement because of device advisories or recalls. The major device infection rate in this retrospective study (JAMA 2006;295:1907-11) during a mean 2.7 months following device replacement was 1.9%, or roughly 19-fold greater than in CITADEL/CENTURION, noted Dr. Henrikson of Oregon Health and Science University, Portland.
Device replacement is a known risk factor for major device infections, which carry a substantial toll in terms of morbidity, mortality, and expense.
The antibacterial envelope, known as AIGISRx, has received Food and Drug Administration approval and is commercially available. The antibacterial envelope is designed to prevent device migration as well as infections. "It changes the device from something that’s slick and slides right in into something with an incredibly high coefficient of friction, so you need to make a larger pocket to put it in. That’s why we’re paying a lot of attention to the hematoma rate," according to Dr. Henrikson.
The device consists of a nonresorbable polypropylene mesh coated with a resorbable polymer layer, which releases rifampin and minocycline for 7-10 days. These two antibiotics are active against the pathogens that cause most device infections, including both methicillin-sensitive and -resistant Staphylococcus aureus as well as S. epidermis.
The indication for device replacement in CITADEL/CENTURION was end of battery life in about three-quarters of patients, with device upgrades and lead revisions accounting for nearly all of the rest. Explantation because of device infection was a contraindication to study participation. All subjects were on perioperative prophylactic antibiotics, as were the historical controls.
The chief device-related complication recorded during the study was major pocket hematoma requiring a change in patient management, typically open drainage or transfusion. The complication occurred in 1.5% of patients, as compared to 2.25% of the historical controls.
Two of the 1,000 patients developed generalized rashes thought by independent reviewers to be possibly related to AIGISRx.
The plan is to monitor CITADEL/CENTURION participants for device infections and mechanical complications through 12 months, Dr. Henrikson explained. Only the 3-month data were available for presentation at a late-breaking clinical trials session during Heart Rhythm 2013.
CITADEL/CENTURION is sponsored by TYRX, which markets AIGISRx. Dr. Henrikson reported having received research support from the company.
DENVER – Encasing implantable cardioverter-defibrillators and cardiac resynchronization therapy devices in a proprietary antibiotic-impregnated envelope during implantation was associated with a low 90-day major infection rate in an interim analysis of a large prospective multicenter study.
The CITADEL/CENTURION study involves 1,000 patients who underwent ICD or CRT device replacement using the antibacterial envelope at 55 U.S. centers. A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient through 3 months of prospective follow-up, for an infection incidence of 0.1%, Dr. Charles Henrikson reported at the annual meeting of the Heart Rhythm Society.
CITADEL/CENTURION is not a randomized trial. Instead, the control group consists of 533 Canadian patients in a published study, all of whom underwent ICD or CRT replacement because of device advisories or recalls. The major device infection rate in this retrospective study (JAMA 2006;295:1907-11) during a mean 2.7 months following device replacement was 1.9%, or roughly 19-fold greater than in CITADEL/CENTURION, noted Dr. Henrikson of Oregon Health and Science University, Portland.
Device replacement is a known risk factor for major device infections, which carry a substantial toll in terms of morbidity, mortality, and expense.
The antibacterial envelope, known as AIGISRx, has received Food and Drug Administration approval and is commercially available. The antibacterial envelope is designed to prevent device migration as well as infections. "It changes the device from something that’s slick and slides right in into something with an incredibly high coefficient of friction, so you need to make a larger pocket to put it in. That’s why we’re paying a lot of attention to the hematoma rate," according to Dr. Henrikson.
The device consists of a nonresorbable polypropylene mesh coated with a resorbable polymer layer, which releases rifampin and minocycline for 7-10 days. These two antibiotics are active against the pathogens that cause most device infections, including both methicillin-sensitive and -resistant Staphylococcus aureus as well as S. epidermis.
The indication for device replacement in CITADEL/CENTURION was end of battery life in about three-quarters of patients, with device upgrades and lead revisions accounting for nearly all of the rest. Explantation because of device infection was a contraindication to study participation. All subjects were on perioperative prophylactic antibiotics, as were the historical controls.
The chief device-related complication recorded during the study was major pocket hematoma requiring a change in patient management, typically open drainage or transfusion. The complication occurred in 1.5% of patients, as compared to 2.25% of the historical controls.
Two of the 1,000 patients developed generalized rashes thought by independent reviewers to be possibly related to AIGISRx.
The plan is to monitor CITADEL/CENTURION participants for device infections and mechanical complications through 12 months, Dr. Henrikson explained. Only the 3-month data were available for presentation at a late-breaking clinical trials session during Heart Rhythm 2013.
CITADEL/CENTURION is sponsored by TYRX, which markets AIGISRx. Dr. Henrikson reported having received research support from the company.
AT HEART RHYTHM 2013
Major finding: A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient, for an infection incidence of 0.1%.
Data source: Three months of data from 1,000 patients in CITADEL/CENTURION study, an ongoing multicenter, prospective study.
Disclosures: The study is sponsored by TYRX, which markets the AIGISRx antibacterial envelope. The presenter reported receiving research support from the company.

Valacyclovir improves cognition in bipolar patients
HOLLYWOOD, FLA. – A 4-month course of the oral antiviral agent valacyclovir boosted cognition in herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment in a randomized, double-blind placebo-controlled clinical trial.
In this 60-patient study, 53% of participants assigned to valacyclovir (Valtrex) exhibited a clinically meaningful improvement in cognitive function, defined as at least a 10-point gain over baseline on the Repeatable Battery for the Assessment of Neurological Status (RBANS), compared with 14% in the placebo arm, Dr. Jennifer L. Payne reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

The impressive improvement in response to valacyclovir documented in this study lends support to the viral hypothesis of mental illness, she said. This hypothesis posits that infection with one of the herpes viruses can in genetically vulnerable individuals lead to major mental illness, including schizophrenia, bipolar disorder, and perhaps Alzheimer’s disease.
"The idea behind this is that episodic reactivation of the virus in the [central nervous system] by stress or other stimuli could trigger mood, cognitive, or psychotic symptoms. If you treat patients you see this all the time: A patient comes under some kind of stress and becomes psychiatrically ill. One of the thoughts is that [herpes simplex virus] could underlie some of that psychopathology," according to Dr. Payne, a psychiatrist and director of the women’s mood disorders center at Johns Hopkins University, Baltimore.
She credited Faith B. Dickerson, Ph.D., of the Sheppard Pratt Institute in Baltimore with earlier groundbreaking work identifying a link between herpes simplex virus type 1 (HSV-1) and cognitive dysfunction in bipolar disorder. Dr. Dickerson and her colleagues reported a roughly 20-fold increased risk for cognitive impairment on the RBANS in HSV-1–seropositive, compared with seronegative patients with bipolar disorder (Biol. Psychiatry 2004;55:588-93).
The RBANS is a paper and pencil test that takes roughly 25 minutes. The mean score in the general population is 100. One standard deviation below the mean is a score of 85. Upon retaking the test, 16% of the general population are able to improve their score by 10 points or more, a rate close to the placebo group’s performance in Dr. Payne’s bipolar disorder study. The RBANS has individual sections for attention, immediate memory, delayed memory, language, and visuospatial construction.
HSV-1 typically causes oral herpes lesions. It’s an extremely common infection. By middle age, roughly 70% of Americans have serologic antibody titers to HSV-1. The virus infects the CNS and often remains in a latent state for many years, punctuated by symptomatic recurrences.
Dr. Payne screened 106 bipolar disorder patients; 84 proved HSV-1 seropositive. The mean baseline RBANS score in the seropositive patients was 75, compared with 92 in the seronegative cohort. The observed association between HSV-1 serologic status and RBANS score remained significant in a multivariate logistic regression analysis after investigators controlled for education level.
The 60 study participants were bipolar disorder outpatients, with an average age of 43. Thirty-seven percent met diagnostic criteria for bipolar I disorder; the rest, for bipolar II disorder. All were required to have baseline cognitive impairment as reflected by an RBANS score of 85 or less. Their average baseline Montgomery-Asberg Depression Rating Scale (MADRS) score was 24, with a Young Mania Rating Scale (YMRS) score of 8. Patients remained on their usual psychiatric medications during the study.
Participants in the 4-month trial were evaluated every 2 weeks for a change in mood symptoms using the MADRS and YMRS. The results came as a surprise.
"Our hypothesis had been that cognitive improvement with valacyclovir would be associated with improvement in depression, but the MADRS scores didn’t change over time," according to Dr. Payne.
As is typical in months-long clinical trials conducted in patients with bipolar disorder, there was a high dropout rate. Mean RBANS scores in the 19 patients in the valacyclovir group who completed the study improved from a baseline of 67.6 to 77.7 at 4 months. The 22 study completers in the control group showed no significant change in scores over time.
Dr. Payne said that if these results are confirmed in another clinical trial – and she plans to conduct one including seropositive bipolar disorder patients who are not cognitively impaired – it would be practice changing.
"If these findings hold up, it would indicate that as clinicians, we need to be testing for HSV-1 and treating it in our patients," she said.
Her future plans also include studying HSV-1 antibody status, cognition, and the possible impact of antiviral therapy in patients with major depressive disorder.
Patients with bipolar disorder often complain and exhibit symptoms of cognitive dysfunction, particularly in the domains of attention, memory, and executive function. The dysfunction typically worsens during manic or depressive episodes, but it’s often still present when bipolar disorder patients are affectively neutral.
The randomized trial was funded by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.
HOLLYWOOD, FLA. – A 4-month course of the oral antiviral agent valacyclovir boosted cognition in herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment in a randomized, double-blind placebo-controlled clinical trial.
In this 60-patient study, 53% of participants assigned to valacyclovir (Valtrex) exhibited a clinically meaningful improvement in cognitive function, defined as at least a 10-point gain over baseline on the Repeatable Battery for the Assessment of Neurological Status (RBANS), compared with 14% in the placebo arm, Dr. Jennifer L. Payne reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The impressive improvement in response to valacyclovir documented in this study lends support to the viral hypothesis of mental illness, she said. This hypothesis posits that infection with one of the herpes viruses can in genetically vulnerable individuals lead to major mental illness, including schizophrenia, bipolar disorder, and perhaps Alzheimer’s disease.
"The idea behind this is that episodic reactivation of the virus in the [central nervous system] by stress or other stimuli could trigger mood, cognitive, or psychotic symptoms. If you treat patients you see this all the time: A patient comes under some kind of stress and becomes psychiatrically ill. One of the thoughts is that [herpes simplex virus] could underlie some of that psychopathology," according to Dr. Payne, a psychiatrist and director of the women’s mood disorders center at Johns Hopkins University, Baltimore.
She credited Faith B. Dickerson, Ph.D., of the Sheppard Pratt Institute in Baltimore with earlier groundbreaking work identifying a link between herpes simplex virus type 1 (HSV-1) and cognitive dysfunction in bipolar disorder. Dr. Dickerson and her colleagues reported a roughly 20-fold increased risk for cognitive impairment on the RBANS in HSV-1–seropositive, compared with seronegative patients with bipolar disorder (Biol. Psychiatry 2004;55:588-93).
The RBANS is a paper and pencil test that takes roughly 25 minutes. The mean score in the general population is 100. One standard deviation below the mean is a score of 85. Upon retaking the test, 16% of the general population are able to improve their score by 10 points or more, a rate close to the placebo group’s performance in Dr. Payne’s bipolar disorder study. The RBANS has individual sections for attention, immediate memory, delayed memory, language, and visuospatial construction.
HSV-1 typically causes oral herpes lesions. It’s an extremely common infection. By middle age, roughly 70% of Americans have serologic antibody titers to HSV-1. The virus infects the CNS and often remains in a latent state for many years, punctuated by symptomatic recurrences.
Dr. Payne screened 106 bipolar disorder patients; 84 proved HSV-1 seropositive. The mean baseline RBANS score in the seropositive patients was 75, compared with 92 in the seronegative cohort. The observed association between HSV-1 serologic status and RBANS score remained significant in a multivariate logistic regression analysis after investigators controlled for education level.
The 60 study participants were bipolar disorder outpatients, with an average age of 43. Thirty-seven percent met diagnostic criteria for bipolar I disorder; the rest, for bipolar II disorder. All were required to have baseline cognitive impairment as reflected by an RBANS score of 85 or less. Their average baseline Montgomery-Asberg Depression Rating Scale (MADRS) score was 24, with a Young Mania Rating Scale (YMRS) score of 8. Patients remained on their usual psychiatric medications during the study.
Participants in the 4-month trial were evaluated every 2 weeks for a change in mood symptoms using the MADRS and YMRS. The results came as a surprise.
"Our hypothesis had been that cognitive improvement with valacyclovir would be associated with improvement in depression, but the MADRS scores didn’t change over time," according to Dr. Payne.
As is typical in months-long clinical trials conducted in patients with bipolar disorder, there was a high dropout rate. Mean RBANS scores in the 19 patients in the valacyclovir group who completed the study improved from a baseline of 67.6 to 77.7 at 4 months. The 22 study completers in the control group showed no significant change in scores over time.
Dr. Payne said that if these results are confirmed in another clinical trial – and she plans to conduct one including seropositive bipolar disorder patients who are not cognitively impaired – it would be practice changing.
"If these findings hold up, it would indicate that as clinicians, we need to be testing for HSV-1 and treating it in our patients," she said.
Her future plans also include studying HSV-1 antibody status, cognition, and the possible impact of antiviral therapy in patients with major depressive disorder.
Patients with bipolar disorder often complain and exhibit symptoms of cognitive dysfunction, particularly in the domains of attention, memory, and executive function. The dysfunction typically worsens during manic or depressive episodes, but it’s often still present when bipolar disorder patients are affectively neutral.
The randomized trial was funded by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.
HOLLYWOOD, FLA. – A 4-month course of the oral antiviral agent valacyclovir boosted cognition in herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment in a randomized, double-blind placebo-controlled clinical trial.
In this 60-patient study, 53% of participants assigned to valacyclovir (Valtrex) exhibited a clinically meaningful improvement in cognitive function, defined as at least a 10-point gain over baseline on the Repeatable Battery for the Assessment of Neurological Status (RBANS), compared with 14% in the placebo arm, Dr. Jennifer L. Payne reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The impressive improvement in response to valacyclovir documented in this study lends support to the viral hypothesis of mental illness, she said. This hypothesis posits that infection with one of the herpes viruses can in genetically vulnerable individuals lead to major mental illness, including schizophrenia, bipolar disorder, and perhaps Alzheimer’s disease.
"The idea behind this is that episodic reactivation of the virus in the [central nervous system] by stress or other stimuli could trigger mood, cognitive, or psychotic symptoms. If you treat patients you see this all the time: A patient comes under some kind of stress and becomes psychiatrically ill. One of the thoughts is that [herpes simplex virus] could underlie some of that psychopathology," according to Dr. Payne, a psychiatrist and director of the women’s mood disorders center at Johns Hopkins University, Baltimore.
She credited Faith B. Dickerson, Ph.D., of the Sheppard Pratt Institute in Baltimore with earlier groundbreaking work identifying a link between herpes simplex virus type 1 (HSV-1) and cognitive dysfunction in bipolar disorder. Dr. Dickerson and her colleagues reported a roughly 20-fold increased risk for cognitive impairment on the RBANS in HSV-1–seropositive, compared with seronegative patients with bipolar disorder (Biol. Psychiatry 2004;55:588-93).
The RBANS is a paper and pencil test that takes roughly 25 minutes. The mean score in the general population is 100. One standard deviation below the mean is a score of 85. Upon retaking the test, 16% of the general population are able to improve their score by 10 points or more, a rate close to the placebo group’s performance in Dr. Payne’s bipolar disorder study. The RBANS has individual sections for attention, immediate memory, delayed memory, language, and visuospatial construction.
HSV-1 typically causes oral herpes lesions. It’s an extremely common infection. By middle age, roughly 70% of Americans have serologic antibody titers to HSV-1. The virus infects the CNS and often remains in a latent state for many years, punctuated by symptomatic recurrences.
Dr. Payne screened 106 bipolar disorder patients; 84 proved HSV-1 seropositive. The mean baseline RBANS score in the seropositive patients was 75, compared with 92 in the seronegative cohort. The observed association between HSV-1 serologic status and RBANS score remained significant in a multivariate logistic regression analysis after investigators controlled for education level.
The 60 study participants were bipolar disorder outpatients, with an average age of 43. Thirty-seven percent met diagnostic criteria for bipolar I disorder; the rest, for bipolar II disorder. All were required to have baseline cognitive impairment as reflected by an RBANS score of 85 or less. Their average baseline Montgomery-Asberg Depression Rating Scale (MADRS) score was 24, with a Young Mania Rating Scale (YMRS) score of 8. Patients remained on their usual psychiatric medications during the study.
Participants in the 4-month trial were evaluated every 2 weeks for a change in mood symptoms using the MADRS and YMRS. The results came as a surprise.
"Our hypothesis had been that cognitive improvement with valacyclovir would be associated with improvement in depression, but the MADRS scores didn’t change over time," according to Dr. Payne.
As is typical in months-long clinical trials conducted in patients with bipolar disorder, there was a high dropout rate. Mean RBANS scores in the 19 patients in the valacyclovir group who completed the study improved from a baseline of 67.6 to 77.7 at 4 months. The 22 study completers in the control group showed no significant change in scores over time.
Dr. Payne said that if these results are confirmed in another clinical trial – and she plans to conduct one including seropositive bipolar disorder patients who are not cognitively impaired – it would be practice changing.
"If these findings hold up, it would indicate that as clinicians, we need to be testing for HSV-1 and treating it in our patients," she said.
Her future plans also include studying HSV-1 antibody status, cognition, and the possible impact of antiviral therapy in patients with major depressive disorder.
Patients with bipolar disorder often complain and exhibit symptoms of cognitive dysfunction, particularly in the domains of attention, memory, and executive function. The dysfunction typically worsens during manic or depressive episodes, but it’s often still present when bipolar disorder patients are affectively neutral.
The randomized trial was funded by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.
AT THE NCDEU MEETING
Major finding: Fifty-three percent of herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment exhibited objective cognitive improvement after 4 months on valacyclovir, as did a mere 14% on placebo.
Data source: A 60-patient randomized, double-blind, placebo-controlled clinical trial.
Disclosures: The study was sponsored by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.

Bremelanotide improves female sexual dysfunction
HOLLYWOOD, FLA. – Subcutaneous bremelanotide self-administered at home by premenopausal women with sexual dysfunction significantly boosted sexual arousal and desire and their number of satisfying sexual events, based on data from a phase IIb clinical trial.
The novel therapy proved effective both in women with hypoactive sexual desire disorder and in those with combined hypoactive sexual desire disorder/female sexual arousal disorder, among the most common forms of female sexual dysfunction, Dr. Anita H. Clayton noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Female sexual dysfunction is distressing, very common, and multifactorial, and there is at present no approved pharmacotherapy for these disorders, according to Dr. Clayton, professor of psychiatry and neurobehavioral sciences at the University of Virginia, Charlottesville. Thus, this represents an area of significant unmet medical need, she said.
Bremelanotide is a cyclic 7-amino-acid melanocortin peptide. It is a synthetic analog of the hormone alpha-melanocyte–stimulating hormone (alpha-MSH). It functions as a melanocortin-4 receptor agonist. Bremelanotide, like alpha-MSH, is thought to modulate brain pathways involved in sexual response, Dr. Clayton explained.
She reported data from a phase IIb randomized, double-blind, multicenter trial involving 327 women who met diagnostic criteria for hypoactive sexual desire disorder or combined hypoactive sexual desire disorder/female sexual arousal disorder. After receiving instruction in self-administration of subcutaneous injections, all participants underwent 4 weeks of single-blind placebo self-dosing at home on an as-needed basis. Then they were randomized to 12 weeks of double-blind home treatment with placebo or bremelanotide at 0.75 mg, 1.25 mg, or 1.75 mg in prefilled syringes. Participants were instructed to inject themselves approximately 45 minutes prior to sexual activity. They were not to exceed 1 dose per day, or 16 doses in a 4-week period.
The primary endpoint was change between the numbers of satisfying sexual events during the 28-day baseline period on placebo and during the final 28 days of the 12-week double-blind study period, using the Female Sexual Encounter Profile-Revised
The mean increase was 0.2 events in placebo-treated controls. Women randomized to 0.75 of bremelanotide didn’t fare significantly better than that.
However, women using bremelanotide at 1.25 mg had a mean 0.7-event increase from baseline, and those on 1.75 mg averaged a 0.8-event increase, both of which were significantly better than placebo.
Secondary endpoints were also positive, in dose-dependent fashion. The mean change over time in the Female Sexual Function Index total score, a validated measure of overall sexual functioning, was 1.88 for placebo, 3.6 in the pooled analysis of patients on bremelanotide at 1.25 or 1.75 mg, and 4.4 in those on 1.75 mg.
Similarly, the mean improvement on the FSDS-DAO (Female Sexual Distress Scale-Desire/Arousal/Orgasm) total score, an indicator of sexual dysfunction–related distress, was –6.8 for placebo, –11.1 for the pooled group on 1.25 or 1.75 mg of bremelanotide, and –13.1 for women on 1.75 mg.
These are clinically meaningful improvements, according to Dr. Clayton. Of note, the mean total score improvements, compared with baseline on these outcome measures, were still growing during the third and final month of double-blind treatment.
Also encouraging was the large percentage of women on bremelanotide whose scores reached thresholds indicative of normal levels of sexual function, she continued. For example, a Female Sexual Function Index total score greater than 26.5 was achieved in 42.7% of women on bremelanotide at 0.75 mg, 45.5% of those on 1.25 mg, and 49% on 1.75 mg, compared with 36.5% of placebo-treated controls. Moreover, a FSDS-DAO total score less than 18 was attained by 28.5% of women on placebo, 40.5% of those on bremelanotide at 0.75 mg, 45.6% on 1.25 mg, and 47.5% of those on 1.75 mg.
The drug therapy was safe and generally well tolerated. The most common bremelanotide-associated side effects were nausea, facial flushing, and headache, which affected 9%-24% of patients in dose-dependent fashion and were typically mild to moderate in nature.
Bremelanotide-treated patients averaged an increase in blood pressure of approximately 2 mm Hg, largely restricted to the first 4 hours after dosing. The number of patients forced to withdraw from the study based on predefined blood pressure change criteria was evenly distributed among the placebo and treatment groups, which was reassuring, Dr. Clayton said. Approximately 5 years ago, development of an intranasal formulation of bremelanotide for treatment of male erectile dysfunction as well as female sexual dysfunction was discontinued because of concerns about significant drug-induced hypertension. The current study, as well as other data, indicates that hypertension isn’t an issue with subcutaneous administration, she noted.
A definitive phase III clinical trial of at-home, self-administered subcutaneous bremelanotide for treatment of female sexual dysfunction is anticipated to start later this year.
Dr. Clayton reported receiving research support and consulting fees from Palatin Technologies, which is developing bremelanotide, as well as from other pharmaceutical companies.
HOLLYWOOD, FLA. – Subcutaneous bremelanotide self-administered at home by premenopausal women with sexual dysfunction significantly boosted sexual arousal and desire and their number of satisfying sexual events, based on data from a phase IIb clinical trial.
The novel therapy proved effective both in women with hypoactive sexual desire disorder and in those with combined hypoactive sexual desire disorder/female sexual arousal disorder, among the most common forms of female sexual dysfunction, Dr. Anita H. Clayton noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Female sexual dysfunction is distressing, very common, and multifactorial, and there is at present no approved pharmacotherapy for these disorders, according to Dr. Clayton, professor of psychiatry and neurobehavioral sciences at the University of Virginia, Charlottesville. Thus, this represents an area of significant unmet medical need, she said.
Bremelanotide is a cyclic 7-amino-acid melanocortin peptide. It is a synthetic analog of the hormone alpha-melanocyte–stimulating hormone (alpha-MSH). It functions as a melanocortin-4 receptor agonist. Bremelanotide, like alpha-MSH, is thought to modulate brain pathways involved in sexual response, Dr. Clayton explained.
She reported data from a phase IIb randomized, double-blind, multicenter trial involving 327 women who met diagnostic criteria for hypoactive sexual desire disorder or combined hypoactive sexual desire disorder/female sexual arousal disorder. After receiving instruction in self-administration of subcutaneous injections, all participants underwent 4 weeks of single-blind placebo self-dosing at home on an as-needed basis. Then they were randomized to 12 weeks of double-blind home treatment with placebo or bremelanotide at 0.75 mg, 1.25 mg, or 1.75 mg in prefilled syringes. Participants were instructed to inject themselves approximately 45 minutes prior to sexual activity. They were not to exceed 1 dose per day, or 16 doses in a 4-week period.
The primary endpoint was change between the numbers of satisfying sexual events during the 28-day baseline period on placebo and during the final 28 days of the 12-week double-blind study period, using the Female Sexual Encounter Profile-Revised
The mean increase was 0.2 events in placebo-treated controls. Women randomized to 0.75 of bremelanotide didn’t fare significantly better than that.
However, women using bremelanotide at 1.25 mg had a mean 0.7-event increase from baseline, and those on 1.75 mg averaged a 0.8-event increase, both of which were significantly better than placebo.
Secondary endpoints were also positive, in dose-dependent fashion. The mean change over time in the Female Sexual Function Index total score, a validated measure of overall sexual functioning, was 1.88 for placebo, 3.6 in the pooled analysis of patients on bremelanotide at 1.25 or 1.75 mg, and 4.4 in those on 1.75 mg.
Similarly, the mean improvement on the FSDS-DAO (Female Sexual Distress Scale-Desire/Arousal/Orgasm) total score, an indicator of sexual dysfunction–related distress, was –6.8 for placebo, –11.1 for the pooled group on 1.25 or 1.75 mg of bremelanotide, and –13.1 for women on 1.75 mg.
These are clinically meaningful improvements, according to Dr. Clayton. Of note, the mean total score improvements, compared with baseline on these outcome measures, were still growing during the third and final month of double-blind treatment.
Also encouraging was the large percentage of women on bremelanotide whose scores reached thresholds indicative of normal levels of sexual function, she continued. For example, a Female Sexual Function Index total score greater than 26.5 was achieved in 42.7% of women on bremelanotide at 0.75 mg, 45.5% of those on 1.25 mg, and 49% on 1.75 mg, compared with 36.5% of placebo-treated controls. Moreover, a FSDS-DAO total score less than 18 was attained by 28.5% of women on placebo, 40.5% of those on bremelanotide at 0.75 mg, 45.6% on 1.25 mg, and 47.5% of those on 1.75 mg.
The drug therapy was safe and generally well tolerated. The most common bremelanotide-associated side effects were nausea, facial flushing, and headache, which affected 9%-24% of patients in dose-dependent fashion and were typically mild to moderate in nature.
Bremelanotide-treated patients averaged an increase in blood pressure of approximately 2 mm Hg, largely restricted to the first 4 hours after dosing. The number of patients forced to withdraw from the study based on predefined blood pressure change criteria was evenly distributed among the placebo and treatment groups, which was reassuring, Dr. Clayton said. Approximately 5 years ago, development of an intranasal formulation of bremelanotide for treatment of male erectile dysfunction as well as female sexual dysfunction was discontinued because of concerns about significant drug-induced hypertension. The current study, as well as other data, indicates that hypertension isn’t an issue with subcutaneous administration, she noted.
A definitive phase III clinical trial of at-home, self-administered subcutaneous bremelanotide for treatment of female sexual dysfunction is anticipated to start later this year.
Dr. Clayton reported receiving research support and consulting fees from Palatin Technologies, which is developing bremelanotide, as well as from other pharmaceutical companies.
HOLLYWOOD, FLA. – Subcutaneous bremelanotide self-administered at home by premenopausal women with sexual dysfunction significantly boosted sexual arousal and desire and their number of satisfying sexual events, based on data from a phase IIb clinical trial.
The novel therapy proved effective both in women with hypoactive sexual desire disorder and in those with combined hypoactive sexual desire disorder/female sexual arousal disorder, among the most common forms of female sexual dysfunction, Dr. Anita H. Clayton noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Female sexual dysfunction is distressing, very common, and multifactorial, and there is at present no approved pharmacotherapy for these disorders, according to Dr. Clayton, professor of psychiatry and neurobehavioral sciences at the University of Virginia, Charlottesville. Thus, this represents an area of significant unmet medical need, she said.
Bremelanotide is a cyclic 7-amino-acid melanocortin peptide. It is a synthetic analog of the hormone alpha-melanocyte–stimulating hormone (alpha-MSH). It functions as a melanocortin-4 receptor agonist. Bremelanotide, like alpha-MSH, is thought to modulate brain pathways involved in sexual response, Dr. Clayton explained.
She reported data from a phase IIb randomized, double-blind, multicenter trial involving 327 women who met diagnostic criteria for hypoactive sexual desire disorder or combined hypoactive sexual desire disorder/female sexual arousal disorder. After receiving instruction in self-administration of subcutaneous injections, all participants underwent 4 weeks of single-blind placebo self-dosing at home on an as-needed basis. Then they were randomized to 12 weeks of double-blind home treatment with placebo or bremelanotide at 0.75 mg, 1.25 mg, or 1.75 mg in prefilled syringes. Participants were instructed to inject themselves approximately 45 minutes prior to sexual activity. They were not to exceed 1 dose per day, or 16 doses in a 4-week period.
The primary endpoint was change between the numbers of satisfying sexual events during the 28-day baseline period on placebo and during the final 28 days of the 12-week double-blind study period, using the Female Sexual Encounter Profile-Revised
The mean increase was 0.2 events in placebo-treated controls. Women randomized to 0.75 of bremelanotide didn’t fare significantly better than that.
However, women using bremelanotide at 1.25 mg had a mean 0.7-event increase from baseline, and those on 1.75 mg averaged a 0.8-event increase, both of which were significantly better than placebo.
Secondary endpoints were also positive, in dose-dependent fashion. The mean change over time in the Female Sexual Function Index total score, a validated measure of overall sexual functioning, was 1.88 for placebo, 3.6 in the pooled analysis of patients on bremelanotide at 1.25 or 1.75 mg, and 4.4 in those on 1.75 mg.
Similarly, the mean improvement on the FSDS-DAO (Female Sexual Distress Scale-Desire/Arousal/Orgasm) total score, an indicator of sexual dysfunction–related distress, was –6.8 for placebo, –11.1 for the pooled group on 1.25 or 1.75 mg of bremelanotide, and –13.1 for women on 1.75 mg.
These are clinically meaningful improvements, according to Dr. Clayton. Of note, the mean total score improvements, compared with baseline on these outcome measures, were still growing during the third and final month of double-blind treatment.
Also encouraging was the large percentage of women on bremelanotide whose scores reached thresholds indicative of normal levels of sexual function, she continued. For example, a Female Sexual Function Index total score greater than 26.5 was achieved in 42.7% of women on bremelanotide at 0.75 mg, 45.5% of those on 1.25 mg, and 49% on 1.75 mg, compared with 36.5% of placebo-treated controls. Moreover, a FSDS-DAO total score less than 18 was attained by 28.5% of women on placebo, 40.5% of those on bremelanotide at 0.75 mg, 45.6% on 1.25 mg, and 47.5% of those on 1.75 mg.
The drug therapy was safe and generally well tolerated. The most common bremelanotide-associated side effects were nausea, facial flushing, and headache, which affected 9%-24% of patients in dose-dependent fashion and were typically mild to moderate in nature.
Bremelanotide-treated patients averaged an increase in blood pressure of approximately 2 mm Hg, largely restricted to the first 4 hours after dosing. The number of patients forced to withdraw from the study based on predefined blood pressure change criteria was evenly distributed among the placebo and treatment groups, which was reassuring, Dr. Clayton said. Approximately 5 years ago, development of an intranasal formulation of bremelanotide for treatment of male erectile dysfunction as well as female sexual dysfunction was discontinued because of concerns about significant drug-induced hypertension. The current study, as well as other data, indicates that hypertension isn’t an issue with subcutaneous administration, she noted.
A definitive phase III clinical trial of at-home, self-administered subcutaneous bremelanotide for treatment of female sexual dysfunction is anticipated to start later this year.
Dr. Clayton reported receiving research support and consulting fees from Palatin Technologies, which is developing bremelanotide, as well as from other pharmaceutical companies.
AT THE NCDEU MEETING
Major finding: Self-administered subcutaneous injections of the novel agent bremelanotide at the top dose tested resulted in quadruple the number of satisfying sexual events, compared with placebo, in premenopausal women with female sexual dysfunction.
Data source: A multicenter, double-blind, phase IIb randomized trial involving 397 women with female sexual dysfunction, 327 of whom were evaluable.
Disclosures: The phase IIb study was sponsored by Palatin Technologies. The presenter has received research funding and consulting fees from this and other pharmaceutical companies.