User login
Novel intranasal antidepressant shows results after 1 week
HOLLYWOOD, FLA. – An investigational intranasal spray antidepressant known for now as PH10 shows early promise in addressing two major unmet needs in the treatment of major depressive disorder: faster-acting drugs with novel mechanisms of action.
PH10 showed a large antidepressant effect in a small phase II study after just 1 week, when the first scheduled assessment took place. Future studies will look for an antidepressant effect even sooner, perhaps as early as day 1 of treatment, Dr. Michael R. Liebowitz said at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
PH10 is a proprietary pherine. The pherines are a class of intranasally administered psychoactive therapeutic agents that bind locally on nasal chemosensory receptors and trigger responses in the hypothalamus, amygdala, prefrontal cortex, and hippocampus. They have an excellent safety and tolerability profile, are effective in nanogram quantities, and do not circulate systemically in the blood. Instead, they initiate neural impulses that follow defined pathways in order to directly affect brain function, explained Dr. Liebowitz, professor of clinical psychiatry at Columbia University, New York.
He presented an 8-week, phase II, double-blind, single-site pilot randomized trial involving 30 patients with major depressive disorder. None had treatment-resistant depression. The participants were randomized to two self-administered inhalations in each nostril twice daily at a dose of 3.2 mcg/day of PH10 from a metered-dose spray device, or a high-dose group receiving 6.4 mcg/day, or placebo spray.
Baseline scores on the 17-item Hamilton Rating Scale for Depression (HAM-D) were in the low to mid 20s. After 8 weeks of treatment, mean HAM-D scores dropped by 10.9 points in the placebo-treated controls, 16.3 points in the low-dose PH10 group, and 17.8 points in the high-dose arm, said Dr. Liebowitz, also managing director and founder of the Medical Research Network.
Particularly intriguing were the results after just 1 week: a 4.2-point drop with placebo, compared with decreases of 8.4 and 10.1 points, respectively, in the low- and high-dose PH10 arms.
"The effect sizes are pretty substantial," Dr. Liebowitz noted. He cited the Cohen’s d value of 1.01 for the comparison between high-dose PH10 and placebo, indicative of a large effect size; and a Cohen’s d of 0.71, indicative of a moderate to large effect size, for low-dose PH10 vs. placebo.
Baseline scores on the patient self-rated Quality of Life Enjoyment and Satisfaction Questionnaire (QLESQ) averaged 40 but improved by a mean of 20.3 points in the high-dose PH10 group, 15.3 points in the low-dose group, and 10.1 points in controls.
Remission as defined by a final HAM-D score of 7 or less occurred in 80% of the low-dose PH10 group in this small study, in 60% of those on high-dose therapy, and 20% on placebo.
The most common side effects reported in patients on PH10 were daytime sleepiness, nasal irritation, and headache. Three patients on high-dose PH10 reported an increase in appetite, as did one patient in the low-dose arm and two patients on placebo. No significant changes in body weight were seen in the 8-week study.
As was frequently noted at the NCDEU meeting, depression is the mental illness with the largest prescription drug market in the United States. The need for antidepressants with new mechanisms of action is underscored by the observation that 50%-70% of patients do not experience remission on selective serotonin reuptake inhibitor/selective norepinephrine reuptake inhibitor therapy.
Pherin Pharmaceuticals, which is developing PH10 as a treatment for depression, also has a handful of other intranasal pherines in its pipeline. Furthest along is aloradine, now in phase III clinical trials for social anxiety disorder. Meanwhile, phase II studies have been completed for an intranasal spray used to treat premenstrual dysphoric disorder called PH80-PMD, and phase III studies for this product are gearing up. Salubrin-HF is currently in phase II studies for menopausal hot flashes. And PH15 is in preclinical testing as a cognitive enhancement agent.
Dr. Liebowitz serves as a consultant to and holds stock options in Pherin Pharmaceuticals. He receives research grants from more than 20 pharmaceutical companies.
HOLLYWOOD, FLA. – An investigational intranasal spray antidepressant known for now as PH10 shows early promise in addressing two major unmet needs in the treatment of major depressive disorder: faster-acting drugs with novel mechanisms of action.
PH10 showed a large antidepressant effect in a small phase II study after just 1 week, when the first scheduled assessment took place. Future studies will look for an antidepressant effect even sooner, perhaps as early as day 1 of treatment, Dr. Michael R. Liebowitz said at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
PH10 is a proprietary pherine. The pherines are a class of intranasally administered psychoactive therapeutic agents that bind locally on nasal chemosensory receptors and trigger responses in the hypothalamus, amygdala, prefrontal cortex, and hippocampus. They have an excellent safety and tolerability profile, are effective in nanogram quantities, and do not circulate systemically in the blood. Instead, they initiate neural impulses that follow defined pathways in order to directly affect brain function, explained Dr. Liebowitz, professor of clinical psychiatry at Columbia University, New York.
He presented an 8-week, phase II, double-blind, single-site pilot randomized trial involving 30 patients with major depressive disorder. None had treatment-resistant depression. The participants were randomized to two self-administered inhalations in each nostril twice daily at a dose of 3.2 mcg/day of PH10 from a metered-dose spray device, or a high-dose group receiving 6.4 mcg/day, or placebo spray.
Baseline scores on the 17-item Hamilton Rating Scale for Depression (HAM-D) were in the low to mid 20s. After 8 weeks of treatment, mean HAM-D scores dropped by 10.9 points in the placebo-treated controls, 16.3 points in the low-dose PH10 group, and 17.8 points in the high-dose arm, said Dr. Liebowitz, also managing director and founder of the Medical Research Network.
Particularly intriguing were the results after just 1 week: a 4.2-point drop with placebo, compared with decreases of 8.4 and 10.1 points, respectively, in the low- and high-dose PH10 arms.
"The effect sizes are pretty substantial," Dr. Liebowitz noted. He cited the Cohen’s d value of 1.01 for the comparison between high-dose PH10 and placebo, indicative of a large effect size; and a Cohen’s d of 0.71, indicative of a moderate to large effect size, for low-dose PH10 vs. placebo.
Baseline scores on the patient self-rated Quality of Life Enjoyment and Satisfaction Questionnaire (QLESQ) averaged 40 but improved by a mean of 20.3 points in the high-dose PH10 group, 15.3 points in the low-dose group, and 10.1 points in controls.
Remission as defined by a final HAM-D score of 7 or less occurred in 80% of the low-dose PH10 group in this small study, in 60% of those on high-dose therapy, and 20% on placebo.
The most common side effects reported in patients on PH10 were daytime sleepiness, nasal irritation, and headache. Three patients on high-dose PH10 reported an increase in appetite, as did one patient in the low-dose arm and two patients on placebo. No significant changes in body weight were seen in the 8-week study.
As was frequently noted at the NCDEU meeting, depression is the mental illness with the largest prescription drug market in the United States. The need for antidepressants with new mechanisms of action is underscored by the observation that 50%-70% of patients do not experience remission on selective serotonin reuptake inhibitor/selective norepinephrine reuptake inhibitor therapy.
Pherin Pharmaceuticals, which is developing PH10 as a treatment for depression, also has a handful of other intranasal pherines in its pipeline. Furthest along is aloradine, now in phase III clinical trials for social anxiety disorder. Meanwhile, phase II studies have been completed for an intranasal spray used to treat premenstrual dysphoric disorder called PH80-PMD, and phase III studies for this product are gearing up. Salubrin-HF is currently in phase II studies for menopausal hot flashes. And PH15 is in preclinical testing as a cognitive enhancement agent.
Dr. Liebowitz serves as a consultant to and holds stock options in Pherin Pharmaceuticals. He receives research grants from more than 20 pharmaceutical companies.
HOLLYWOOD, FLA. – An investigational intranasal spray antidepressant known for now as PH10 shows early promise in addressing two major unmet needs in the treatment of major depressive disorder: faster-acting drugs with novel mechanisms of action.
PH10 showed a large antidepressant effect in a small phase II study after just 1 week, when the first scheduled assessment took place. Future studies will look for an antidepressant effect even sooner, perhaps as early as day 1 of treatment, Dr. Michael R. Liebowitz said at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
PH10 is a proprietary pherine. The pherines are a class of intranasally administered psychoactive therapeutic agents that bind locally on nasal chemosensory receptors and trigger responses in the hypothalamus, amygdala, prefrontal cortex, and hippocampus. They have an excellent safety and tolerability profile, are effective in nanogram quantities, and do not circulate systemically in the blood. Instead, they initiate neural impulses that follow defined pathways in order to directly affect brain function, explained Dr. Liebowitz, professor of clinical psychiatry at Columbia University, New York.
He presented an 8-week, phase II, double-blind, single-site pilot randomized trial involving 30 patients with major depressive disorder. None had treatment-resistant depression. The participants were randomized to two self-administered inhalations in each nostril twice daily at a dose of 3.2 mcg/day of PH10 from a metered-dose spray device, or a high-dose group receiving 6.4 mcg/day, or placebo spray.
Baseline scores on the 17-item Hamilton Rating Scale for Depression (HAM-D) were in the low to mid 20s. After 8 weeks of treatment, mean HAM-D scores dropped by 10.9 points in the placebo-treated controls, 16.3 points in the low-dose PH10 group, and 17.8 points in the high-dose arm, said Dr. Liebowitz, also managing director and founder of the Medical Research Network.
Particularly intriguing were the results after just 1 week: a 4.2-point drop with placebo, compared with decreases of 8.4 and 10.1 points, respectively, in the low- and high-dose PH10 arms.
"The effect sizes are pretty substantial," Dr. Liebowitz noted. He cited the Cohen’s d value of 1.01 for the comparison between high-dose PH10 and placebo, indicative of a large effect size; and a Cohen’s d of 0.71, indicative of a moderate to large effect size, for low-dose PH10 vs. placebo.
Baseline scores on the patient self-rated Quality of Life Enjoyment and Satisfaction Questionnaire (QLESQ) averaged 40 but improved by a mean of 20.3 points in the high-dose PH10 group, 15.3 points in the low-dose group, and 10.1 points in controls.
Remission as defined by a final HAM-D score of 7 or less occurred in 80% of the low-dose PH10 group in this small study, in 60% of those on high-dose therapy, and 20% on placebo.
The most common side effects reported in patients on PH10 were daytime sleepiness, nasal irritation, and headache. Three patients on high-dose PH10 reported an increase in appetite, as did one patient in the low-dose arm and two patients on placebo. No significant changes in body weight were seen in the 8-week study.
As was frequently noted at the NCDEU meeting, depression is the mental illness with the largest prescription drug market in the United States. The need for antidepressants with new mechanisms of action is underscored by the observation that 50%-70% of patients do not experience remission on selective serotonin reuptake inhibitor/selective norepinephrine reuptake inhibitor therapy.
Pherin Pharmaceuticals, which is developing PH10 as a treatment for depression, also has a handful of other intranasal pherines in its pipeline. Furthest along is aloradine, now in phase III clinical trials for social anxiety disorder. Meanwhile, phase II studies have been completed for an intranasal spray used to treat premenstrual dysphoric disorder called PH80-PMD, and phase III studies for this product are gearing up. Salubrin-HF is currently in phase II studies for menopausal hot flashes. And PH15 is in preclinical testing as a cognitive enhancement agent.
Dr. Liebowitz serves as a consultant to and holds stock options in Pherin Pharmaceuticals. He receives research grants from more than 20 pharmaceutical companies.
AT THE NCDEU MEETING
Major finding: A self-administered intranasal spray antidepressant resulted in a mean 10.1-point reduction in scores on the Hamilton Rating Scale for Depression after just 1 week of high-dose therapy, compared to an 8.4-point decrease in patient on the low-dose regimen and 4.2 points in placebo-treated controls.
Data source: An 8-week, double-blind, single-center, phase II study involving 30 patients with major depressive disorder.
Disclosures: The clinical trial was sponsored by Pherin Pharmaceuticals. The presenter is a consultant to and holds stock options in the company.
Novel procognitive drug promising in schizophrenia, Alzheimer's
HOLLYWOOD, FLA. – A novel procognitive medication has entered pivotal phase III, randomized clinical trials in patients with chronic schizophrenia, with a separate phase III trial due to start later this year in Alzheimer’s disease.
The drug, known for now as EVP-6124, is an oral alpha-7 nicotinic partial agonist. The alpha-7 nicotinic receptor is an acetylcholine receptor highly localized to the cortical and hippocampal regions of the brain. EVP-6124 acts as a coagonist in combination with acetylcholine to activate the receptor and enhance cognition, Dr. Ilise Lombardo explained at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
In a phase IIa randomized, double-blind placebo-controlled study in patients with stable chronic schizophrenia who remained on their atypical antipsychotic therapy, EVP-6124 normalized three EEG biomarkers for cognitive processing: p50, p300, and MMN, or mismatch negativity. This encouraging finding, coupled with clinical evidence of a procognitive effect on the CogState computerized test of cognition, led to a subsequent 319-patient, 12-week, double-blind phase IIb study.

The phase IIb trial showed that adding once-daily EVP-6124 to standard antipsychotic medication resulted in statistically significant and clinically meaningful benefits compared with placebo in terms of global cognitive function on the CogState test as well as on the Positive and Negative Syndrome Scale (PANSS) cognitive impairment domain, the PANSS negative symptoms domain, and the Measurement and Treatment Research to Improve Cognition in Schizophrenia Consensus Cognitive Battery (MCCB). The drug had no effect on PANSS positive symptoms.
EVP-6124 also resulted in significant improvement in clinical function as measured on the interview-based Schizophrenia Cognition Rating Scale (SCoRS). That’s a key finding, because the Food and Drug Administration now requires evidence of improved clinical function as a condition for approval of any new treatment for schizophrenia, added Dr. Lombardo, vice president for clinical research at EnVivo Pharmaceuticals, which is developing the drug.
There are currently no approved treatments for the cognitive or negative symptoms of schizophrenia, making this a major unmet need in mental health care.
EVP-6124 proved safe and well tolerated in phase II testing. There were fewer adverse events and study dropouts than with placebo.
Based on these encouraging results, two phase III studies are underway. Each of the 26-week studies will randomize roughly 700 patients with schizophrenia. The MCCB and SCoRS tests will be the coprimary endpoints.
The recently completed double-blind, placebo-controlled, 24-week phase IIb study in Alzheimer’s disease involved 409 randomized patients. A dose-response effect was seen. The 0.3-mg dose of EVP-6124 was not significantly more effective than placebo. The 1.0-mg dose showed intermediate efficacy. And the 2.0-mg dose showed statistically significant benefits across the board, compared with placebo.
Moreover, these benefits were clinically meaningful in size. For example, the effect size for EVP-6124 at 2.0 mg/day was a solid 0.39 for the primary endpoint, which was improvement on the Alzheimer’s Disease Assessment Scale, 13-item subscale (ADAS-cog-13), Dr. Lombardo observed.
The top dose also resulted in significant improvement on numerous secondary endpoints, including the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB), the Controlled Oral Word Association Test (COWAT), the Mini-Mental State Examination, and the ADAS-cog-11.
HOLLYWOOD, FLA. – A novel procognitive medication has entered pivotal phase III, randomized clinical trials in patients with chronic schizophrenia, with a separate phase III trial due to start later this year in Alzheimer’s disease.
The drug, known for now as EVP-6124, is an oral alpha-7 nicotinic partial agonist. The alpha-7 nicotinic receptor is an acetylcholine receptor highly localized to the cortical and hippocampal regions of the brain. EVP-6124 acts as a coagonist in combination with acetylcholine to activate the receptor and enhance cognition, Dr. Ilise Lombardo explained at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
In a phase IIa randomized, double-blind placebo-controlled study in patients with stable chronic schizophrenia who remained on their atypical antipsychotic therapy, EVP-6124 normalized three EEG biomarkers for cognitive processing: p50, p300, and MMN, or mismatch negativity. This encouraging finding, coupled with clinical evidence of a procognitive effect on the CogState computerized test of cognition, led to a subsequent 319-patient, 12-week, double-blind phase IIb study.
The phase IIb trial showed that adding once-daily EVP-6124 to standard antipsychotic medication resulted in statistically significant and clinically meaningful benefits compared with placebo in terms of global cognitive function on the CogState test as well as on the Positive and Negative Syndrome Scale (PANSS) cognitive impairment domain, the PANSS negative symptoms domain, and the Measurement and Treatment Research to Improve Cognition in Schizophrenia Consensus Cognitive Battery (MCCB). The drug had no effect on PANSS positive symptoms.
EVP-6124 also resulted in significant improvement in clinical function as measured on the interview-based Schizophrenia Cognition Rating Scale (SCoRS). That’s a key finding, because the Food and Drug Administration now requires evidence of improved clinical function as a condition for approval of any new treatment for schizophrenia, added Dr. Lombardo, vice president for clinical research at EnVivo Pharmaceuticals, which is developing the drug.
There are currently no approved treatments for the cognitive or negative symptoms of schizophrenia, making this a major unmet need in mental health care.
EVP-6124 proved safe and well tolerated in phase II testing. There were fewer adverse events and study dropouts than with placebo.
Based on these encouraging results, two phase III studies are underway. Each of the 26-week studies will randomize roughly 700 patients with schizophrenia. The MCCB and SCoRS tests will be the coprimary endpoints.
The recently completed double-blind, placebo-controlled, 24-week phase IIb study in Alzheimer’s disease involved 409 randomized patients. A dose-response effect was seen. The 0.3-mg dose of EVP-6124 was not significantly more effective than placebo. The 1.0-mg dose showed intermediate efficacy. And the 2.0-mg dose showed statistically significant benefits across the board, compared with placebo.
Moreover, these benefits were clinically meaningful in size. For example, the effect size for EVP-6124 at 2.0 mg/day was a solid 0.39 for the primary endpoint, which was improvement on the Alzheimer’s Disease Assessment Scale, 13-item subscale (ADAS-cog-13), Dr. Lombardo observed.
The top dose also resulted in significant improvement on numerous secondary endpoints, including the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB), the Controlled Oral Word Association Test (COWAT), the Mini-Mental State Examination, and the ADAS-cog-11.
HOLLYWOOD, FLA. – A novel procognitive medication has entered pivotal phase III, randomized clinical trials in patients with chronic schizophrenia, with a separate phase III trial due to start later this year in Alzheimer’s disease.
The drug, known for now as EVP-6124, is an oral alpha-7 nicotinic partial agonist. The alpha-7 nicotinic receptor is an acetylcholine receptor highly localized to the cortical and hippocampal regions of the brain. EVP-6124 acts as a coagonist in combination with acetylcholine to activate the receptor and enhance cognition, Dr. Ilise Lombardo explained at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
In a phase IIa randomized, double-blind placebo-controlled study in patients with stable chronic schizophrenia who remained on their atypical antipsychotic therapy, EVP-6124 normalized three EEG biomarkers for cognitive processing: p50, p300, and MMN, or mismatch negativity. This encouraging finding, coupled with clinical evidence of a procognitive effect on the CogState computerized test of cognition, led to a subsequent 319-patient, 12-week, double-blind phase IIb study.
The phase IIb trial showed that adding once-daily EVP-6124 to standard antipsychotic medication resulted in statistically significant and clinically meaningful benefits compared with placebo in terms of global cognitive function on the CogState test as well as on the Positive and Negative Syndrome Scale (PANSS) cognitive impairment domain, the PANSS negative symptoms domain, and the Measurement and Treatment Research to Improve Cognition in Schizophrenia Consensus Cognitive Battery (MCCB). The drug had no effect on PANSS positive symptoms.
EVP-6124 also resulted in significant improvement in clinical function as measured on the interview-based Schizophrenia Cognition Rating Scale (SCoRS). That’s a key finding, because the Food and Drug Administration now requires evidence of improved clinical function as a condition for approval of any new treatment for schizophrenia, added Dr. Lombardo, vice president for clinical research at EnVivo Pharmaceuticals, which is developing the drug.
There are currently no approved treatments for the cognitive or negative symptoms of schizophrenia, making this a major unmet need in mental health care.
EVP-6124 proved safe and well tolerated in phase II testing. There were fewer adverse events and study dropouts than with placebo.
Based on these encouraging results, two phase III studies are underway. Each of the 26-week studies will randomize roughly 700 patients with schizophrenia. The MCCB and SCoRS tests will be the coprimary endpoints.
The recently completed double-blind, placebo-controlled, 24-week phase IIb study in Alzheimer’s disease involved 409 randomized patients. A dose-response effect was seen. The 0.3-mg dose of EVP-6124 was not significantly more effective than placebo. The 1.0-mg dose showed intermediate efficacy. And the 2.0-mg dose showed statistically significant benefits across the board, compared with placebo.
Moreover, these benefits were clinically meaningful in size. For example, the effect size for EVP-6124 at 2.0 mg/day was a solid 0.39 for the primary endpoint, which was improvement on the Alzheimer’s Disease Assessment Scale, 13-item subscale (ADAS-cog-13), Dr. Lombardo observed.
The top dose also resulted in significant improvement on numerous secondary endpoints, including the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB), the Controlled Oral Word Association Test (COWAT), the Mini-Mental State Examination, and the ADAS-cog-11.
EXPERT ANALYSIS FROM THE NCDEU MEETING

TMS may bring remission in bipolar depression
HOLLYWOOD, FLA. – Transcranial magnetic stimulation shows promise in highly treatment-resistant bipolar depression, a small observational study suggested.
Most studies of transcranial magnetic stimulation (TMS) have focused on treatment of major depressive disorder, the indication for which this noninvasive device therapy has Food and Drug Administration approval. TMS also is under study for the treatment of headaches, as well as for improvement of the negative symptoms of schizophrenia.
Yet little work has been done on TMS for bipolar depression, even though a pressing need exists for new treatment options for this often highly disruptive mood disorder. Antidepressant medications are often ineffective or can trigger a switch to a manic or hypomanic episode, Dr. William S. Gilmer noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

He reported on 10 patients with bipolar II disorder and 4 who met criteria for bipolar disorder not otherwise specified. All had complicated nonpsychotic depression. All were refractory to and/or intolerant of multiple antidepressant agents during their current episode; indeed, the patients had previously been on a mean of 6.4 antidepressant drugs during this episode, which had already lasted more than 18 months in 9 of 14 cases. Four patients were previous nonresponders to ECT. None of the subjects had bipolar activation symptoms such as grandiosity, nocturnal alertness, or agitation at baseline, according to Dr. Gilmer, associate professor of clinical psychiatry and behavioral sciences at Northwestern University, Chicago.
After an extended period during which he attempted to optimize the participants’ medications, including reducing benzodiazepines and increasing mood-stabilizing drugs if necessary, all patients underwent high-frequency TMS at 10-Hz over the left dorsolateral prefrontal cortex 5 days per week according to the standard procedure using the NeuroStar device marketed by Neuronetics.
Nine of 14 patients achieved a clinically meaningful antidepressant response to TMS, as defined by at least a 50% reduction in scores on the Quick Inventory of Depressive Symptomatology (QIDS-16 SR) from an initial mean baseline of 18.9. A mean of 24.6 sessions was required. Four patients achieved and maintained remission, as defined by a QIDS-16 SR score below 6; this required a mean of 24.8 TMS sessions, followed by a taper phase.
Two of the four previous nonresponders to ECT achieved full remission on TMS, and a third had a significant response.
Although there were no study dropouts, seven patients experienced bipolar activation symptoms during TMS therapy that required drug therapy. Of note, four of five TMS nonresponders experienced clinically significant activation symptoms, compared with just three of nine TMS responders. Thus, the emergence of activation symptoms during TMS may turn out to be a predictor of poor outcome. This is a possibility warranting further study in controlled trials aimed at establishing optimal TMS treatment parameters in bipolar depression, the psychiatrist suggested.
Dr. Gilmer is on the speakers’ bureau for Neuronetics.
HOLLYWOOD, FLA. – Transcranial magnetic stimulation shows promise in highly treatment-resistant bipolar depression, a small observational study suggested.
Most studies of transcranial magnetic stimulation (TMS) have focused on treatment of major depressive disorder, the indication for which this noninvasive device therapy has Food and Drug Administration approval. TMS also is under study for the treatment of headaches, as well as for improvement of the negative symptoms of schizophrenia.
Yet little work has been done on TMS for bipolar depression, even though a pressing need exists for new treatment options for this often highly disruptive mood disorder. Antidepressant medications are often ineffective or can trigger a switch to a manic or hypomanic episode, Dr. William S. Gilmer noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
He reported on 10 patients with bipolar II disorder and 4 who met criteria for bipolar disorder not otherwise specified. All had complicated nonpsychotic depression. All were refractory to and/or intolerant of multiple antidepressant agents during their current episode; indeed, the patients had previously been on a mean of 6.4 antidepressant drugs during this episode, which had already lasted more than 18 months in 9 of 14 cases. Four patients were previous nonresponders to ECT. None of the subjects had bipolar activation symptoms such as grandiosity, nocturnal alertness, or agitation at baseline, according to Dr. Gilmer, associate professor of clinical psychiatry and behavioral sciences at Northwestern University, Chicago.
After an extended period during which he attempted to optimize the participants’ medications, including reducing benzodiazepines and increasing mood-stabilizing drugs if necessary, all patients underwent high-frequency TMS at 10-Hz over the left dorsolateral prefrontal cortex 5 days per week according to the standard procedure using the NeuroStar device marketed by Neuronetics.
Nine of 14 patients achieved a clinically meaningful antidepressant response to TMS, as defined by at least a 50% reduction in scores on the Quick Inventory of Depressive Symptomatology (QIDS-16 SR) from an initial mean baseline of 18.9. A mean of 24.6 sessions was required. Four patients achieved and maintained remission, as defined by a QIDS-16 SR score below 6; this required a mean of 24.8 TMS sessions, followed by a taper phase.
Two of the four previous nonresponders to ECT achieved full remission on TMS, and a third had a significant response.
Although there were no study dropouts, seven patients experienced bipolar activation symptoms during TMS therapy that required drug therapy. Of note, four of five TMS nonresponders experienced clinically significant activation symptoms, compared with just three of nine TMS responders. Thus, the emergence of activation symptoms during TMS may turn out to be a predictor of poor outcome. This is a possibility warranting further study in controlled trials aimed at establishing optimal TMS treatment parameters in bipolar depression, the psychiatrist suggested.
Dr. Gilmer is on the speakers’ bureau for Neuronetics.
HOLLYWOOD, FLA. – Transcranial magnetic stimulation shows promise in highly treatment-resistant bipolar depression, a small observational study suggested.
Most studies of transcranial magnetic stimulation (TMS) have focused on treatment of major depressive disorder, the indication for which this noninvasive device therapy has Food and Drug Administration approval. TMS also is under study for the treatment of headaches, as well as for improvement of the negative symptoms of schizophrenia.
Yet little work has been done on TMS for bipolar depression, even though a pressing need exists for new treatment options for this often highly disruptive mood disorder. Antidepressant medications are often ineffective or can trigger a switch to a manic or hypomanic episode, Dr. William S. Gilmer noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
He reported on 10 patients with bipolar II disorder and 4 who met criteria for bipolar disorder not otherwise specified. All had complicated nonpsychotic depression. All were refractory to and/or intolerant of multiple antidepressant agents during their current episode; indeed, the patients had previously been on a mean of 6.4 antidepressant drugs during this episode, which had already lasted more than 18 months in 9 of 14 cases. Four patients were previous nonresponders to ECT. None of the subjects had bipolar activation symptoms such as grandiosity, nocturnal alertness, or agitation at baseline, according to Dr. Gilmer, associate professor of clinical psychiatry and behavioral sciences at Northwestern University, Chicago.
After an extended period during which he attempted to optimize the participants’ medications, including reducing benzodiazepines and increasing mood-stabilizing drugs if necessary, all patients underwent high-frequency TMS at 10-Hz over the left dorsolateral prefrontal cortex 5 days per week according to the standard procedure using the NeuroStar device marketed by Neuronetics.
Nine of 14 patients achieved a clinically meaningful antidepressant response to TMS, as defined by at least a 50% reduction in scores on the Quick Inventory of Depressive Symptomatology (QIDS-16 SR) from an initial mean baseline of 18.9. A mean of 24.6 sessions was required. Four patients achieved and maintained remission, as defined by a QIDS-16 SR score below 6; this required a mean of 24.8 TMS sessions, followed by a taper phase.
Two of the four previous nonresponders to ECT achieved full remission on TMS, and a third had a significant response.
Although there were no study dropouts, seven patients experienced bipolar activation symptoms during TMS therapy that required drug therapy. Of note, four of five TMS nonresponders experienced clinically significant activation symptoms, compared with just three of nine TMS responders. Thus, the emergence of activation symptoms during TMS may turn out to be a predictor of poor outcome. This is a possibility warranting further study in controlled trials aimed at establishing optimal TMS treatment parameters in bipolar depression, the psychiatrist suggested.
Dr. Gilmer is on the speakers’ bureau for Neuronetics.
AT THE NCDEU MEETING
Major finding: Nine of 14 patients with highly treatment-resistant bipolar depression achieved a clinically meaningful antidepressant response to transcranial magnetic stimulation, including 4 who reached and maintained remission. Two of the four in remission were prior nonresponders to ECT.
Data source: This observational study was a naturalistic case series with no control group. Patients had been unresponsive to and/or intolerant of a mean of 6.4 antidepressant medications during their current episode of bipolar depression, which had been ongoing for more than 18 months in nine cases.
Disclosures: Dr. Gilmer is on the speakers’ bureau for Neuronetics, which markets the transcranial magnetic stimulation device used in the investigation.

Lurasidone produces less daytime sleepiness than quetiapine XR
HOLLYWOOD, FLA. – Lurasidone resulted in significantly less daytime sleepiness and improved cognitive performance, compared with extended-release quetiapine in a long-term, double-blind, head-to-head comparative trial in patients with schizophrenia.
The 6-month, double-blind study involved 207 patients on lurasidone (Latuda) flexibly dosed at 40-160 mg once daily in the evening and 85 patients on extended-release quetiapine (Seroquel XR) at 200-800 mg/day.
One of the two major endpoints was change in daytime alertness as measured by the validated Epworth Sleepiness Scale. From a mean baseline ESS of 5.9, scores in the lurasidone group improved to a mean of 4.4. This was a significantly greater improvement in daytime sleepiness than with quetiapine, where scores went from 6.48 to 5.9, Philip D. Harvey, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The other major endpoint was change over time in cognitive performance as assessed using the computerized CogState schizophrenia battery. From a mean baseline composite Z-score of –3.9, scores in the lurasidone group improved by roughly 1.5 points, a threefold greater gain than in patients on quetiapine, added Dr. Harvey, professor of psychiatry and behavioral sciences, and chief of the division of psychology at the University of Miami.
Although it seems logical that daytime sleepiness would impair cognition and interfere with everyday functional capacity, in a multivariate analysis, the superior cognitive performance at month 6 in the lurasidone group was independent of the antipsychotic agent’s effect upon sleepiness.
Lurasidone has greater affinity for serotonin 5HT7 and 5HT1A receptors than other second-generation antipsychotic agents. These receptors are abundant in areas of the brain involved in sleep, mood regulation, and memory. These pharmacologic attributes provide a biologic basis in support of the improvements in cognitive performance and daytime sleepiness seen in the head-to-head trial.
Dr. Harvey is a consultant to Sunovion Pharmaceuticals, which sponsored the comparative trial, as well as to a half-dozen other pharmaceutical companies.
HOLLYWOOD, FLA. – Lurasidone resulted in significantly less daytime sleepiness and improved cognitive performance, compared with extended-release quetiapine in a long-term, double-blind, head-to-head comparative trial in patients with schizophrenia.
The 6-month, double-blind study involved 207 patients on lurasidone (Latuda) flexibly dosed at 40-160 mg once daily in the evening and 85 patients on extended-release quetiapine (Seroquel XR) at 200-800 mg/day.
One of the two major endpoints was change in daytime alertness as measured by the validated Epworth Sleepiness Scale. From a mean baseline ESS of 5.9, scores in the lurasidone group improved to a mean of 4.4. This was a significantly greater improvement in daytime sleepiness than with quetiapine, where scores went from 6.48 to 5.9, Philip D. Harvey, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The other major endpoint was change over time in cognitive performance as assessed using the computerized CogState schizophrenia battery. From a mean baseline composite Z-score of –3.9, scores in the lurasidone group improved by roughly 1.5 points, a threefold greater gain than in patients on quetiapine, added Dr. Harvey, professor of psychiatry and behavioral sciences, and chief of the division of psychology at the University of Miami.
Although it seems logical that daytime sleepiness would impair cognition and interfere with everyday functional capacity, in a multivariate analysis, the superior cognitive performance at month 6 in the lurasidone group was independent of the antipsychotic agent’s effect upon sleepiness.
Lurasidone has greater affinity for serotonin 5HT7 and 5HT1A receptors than other second-generation antipsychotic agents. These receptors are abundant in areas of the brain involved in sleep, mood regulation, and memory. These pharmacologic attributes provide a biologic basis in support of the improvements in cognitive performance and daytime sleepiness seen in the head-to-head trial.
Dr. Harvey is a consultant to Sunovion Pharmaceuticals, which sponsored the comparative trial, as well as to a half-dozen other pharmaceutical companies.
HOLLYWOOD, FLA. – Lurasidone resulted in significantly less daytime sleepiness and improved cognitive performance, compared with extended-release quetiapine in a long-term, double-blind, head-to-head comparative trial in patients with schizophrenia.
The 6-month, double-blind study involved 207 patients on lurasidone (Latuda) flexibly dosed at 40-160 mg once daily in the evening and 85 patients on extended-release quetiapine (Seroquel XR) at 200-800 mg/day.
One of the two major endpoints was change in daytime alertness as measured by the validated Epworth Sleepiness Scale. From a mean baseline ESS of 5.9, scores in the lurasidone group improved to a mean of 4.4. This was a significantly greater improvement in daytime sleepiness than with quetiapine, where scores went from 6.48 to 5.9, Philip D. Harvey, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The other major endpoint was change over time in cognitive performance as assessed using the computerized CogState schizophrenia battery. From a mean baseline composite Z-score of –3.9, scores in the lurasidone group improved by roughly 1.5 points, a threefold greater gain than in patients on quetiapine, added Dr. Harvey, professor of psychiatry and behavioral sciences, and chief of the division of psychology at the University of Miami.
Although it seems logical that daytime sleepiness would impair cognition and interfere with everyday functional capacity, in a multivariate analysis, the superior cognitive performance at month 6 in the lurasidone group was independent of the antipsychotic agent’s effect upon sleepiness.
Lurasidone has greater affinity for serotonin 5HT7 and 5HT1A receptors than other second-generation antipsychotic agents. These receptors are abundant in areas of the brain involved in sleep, mood regulation, and memory. These pharmacologic attributes provide a biologic basis in support of the improvements in cognitive performance and daytime sleepiness seen in the head-to-head trial.
Dr. Harvey is a consultant to Sunovion Pharmaceuticals, which sponsored the comparative trial, as well as to a half-dozen other pharmaceutical companies.
AT THE NCDEU MEETING
Major finding: Patients with schizophrenia who were randomized to 6 months of double-blind lurasidone experienced significantly less daytime sleepiness than did those on extended-release quetiapine as reflected in their mean 1.5-point improvement over baseline on the Epworth Sleepiness Scale as compared with a 0.58-point improvement with quetiapine XR.
Data source: This was a randomized double-blind clinical trial involving 207 patients with schizophrenia assigned to lurasidone and 85 others placed on extended-release quetiapine.
Disclosures: The study was sponsored by Sunovion Pharmaceuticals, which markets lurasidone. The presenter is a consultant to the company.
Duloxetine proves beneficial in elderly GAD
HOLLYWOOD, FLA. – Duloxetine proved safe and effective for the treatment of generalized anxiety disorder in the elderly in a phase IV clinical trial restricted to patients aged 65 and up.
This study fills a major gap in the evidence base for duloxetine (Cymbalta). Generalized anxiety disorder (GAD) is one of the most common psychiatric disorders in the elderly, with a prevalence estimated at up to 7%. Yet prior studies of duloxetine for GAD largely excluded the elderly. Indeed, the product labeling states prominently that premarketing studies did not include enough patients over age 65 to determine whether they respond differently than younger subjects.
With the new evidence provided by the phase IV study, the answer to that question is now in: Duloxetine proved significantly more effective than placebo in seniors. Moreover, the discontinuation rate because of adverse events did not differ significantly between the duloxetine and placebo groups, Karla J. Alaka reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The study included 291 patients aged 65 or older (mean age, 72) who met DSM-IV-TR criteria for GAD. They were randomized to 10 weeks of double-blind treatment with duloxetine dosed at 30-120 mg/day or placebo. Throughout the study, roughly one-third of the duloxetine group remained on 30 mg/day, another third bumped up to 60 mg/day, one-quarter increased to 90 mg/day, and the remainder eventually received 120 mg/day.
Elderly patients with an additional Axis I diagnosis plus GAD were not eligible for the trial. However, patients with comorbid medical illnesses were not excluded from participation so long as those conditions were stable and not expected to result in hospitalization within the next 6 months. Fully 83% of subjects had one or more preexisting medical conditions for which they took concomitant medication during the study. Geriatricians and other physicians have become increasingly vocal about the absence of quality safety and efficacy data for many widely prescribed drugs in the elderly, where issues such as polypharmacy, drug-drug interactions, and slowed drug metabolism become key considerations.
The primary efficacy measure in the study was a change in the Hamilton Anxiety Scale (HAS) total score from baseline to week 10. From a mean baseline HAS score of 24.6, the duloxetine group averaged a 15.9-point decrease, significantly better than the 11.7-point drop with placebo, according to Ms. Alaka of Eli Lilly, Indianapolis.
The HAS response rate, defined by at least a 50% reduction in the HAS total score, was 75% in duloxetine-treated patients, compared with 56% in controls. The Hamilton Anxiety Scale remission rate, a more stringent endpoint requiring a week 10 total score of 10 or less, was achieved by 62% of the duloxetine group and 40% on placebo.
Duloxetine-treated patients also outperformed controls in terms of several other secondary endpoints. For example, the Sheehan Disability Scale global function impairment score in the duloxetine group improved by a mean of 7.6 points from a baseline score of 13.7, compared with a 4.3-point improvement with placebo.
Study discontinuation because of treatment-emergent adverse events occurred in 9.9% of elderly patients on duloxetine and 10.7% on placebo. The only adverse event that was significantly more common in the active treatment group was dry mouth, which occurred in 7% on duloxetine, compared with 1% of controls. Duloxetine was not associated with any clinically significant changes in laboratory findings, ECG parameters, or body weight.
The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
HOLLYWOOD, FLA. – Duloxetine proved safe and effective for the treatment of generalized anxiety disorder in the elderly in a phase IV clinical trial restricted to patients aged 65 and up.
This study fills a major gap in the evidence base for duloxetine (Cymbalta). Generalized anxiety disorder (GAD) is one of the most common psychiatric disorders in the elderly, with a prevalence estimated at up to 7%. Yet prior studies of duloxetine for GAD largely excluded the elderly. Indeed, the product labeling states prominently that premarketing studies did not include enough patients over age 65 to determine whether they respond differently than younger subjects.
With the new evidence provided by the phase IV study, the answer to that question is now in: Duloxetine proved significantly more effective than placebo in seniors. Moreover, the discontinuation rate because of adverse events did not differ significantly between the duloxetine and placebo groups, Karla J. Alaka reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The study included 291 patients aged 65 or older (mean age, 72) who met DSM-IV-TR criteria for GAD. They were randomized to 10 weeks of double-blind treatment with duloxetine dosed at 30-120 mg/day or placebo. Throughout the study, roughly one-third of the duloxetine group remained on 30 mg/day, another third bumped up to 60 mg/day, one-quarter increased to 90 mg/day, and the remainder eventually received 120 mg/day.
Elderly patients with an additional Axis I diagnosis plus GAD were not eligible for the trial. However, patients with comorbid medical illnesses were not excluded from participation so long as those conditions were stable and not expected to result in hospitalization within the next 6 months. Fully 83% of subjects had one or more preexisting medical conditions for which they took concomitant medication during the study. Geriatricians and other physicians have become increasingly vocal about the absence of quality safety and efficacy data for many widely prescribed drugs in the elderly, where issues such as polypharmacy, drug-drug interactions, and slowed drug metabolism become key considerations.
The primary efficacy measure in the study was a change in the Hamilton Anxiety Scale (HAS) total score from baseline to week 10. From a mean baseline HAS score of 24.6, the duloxetine group averaged a 15.9-point decrease, significantly better than the 11.7-point drop with placebo, according to Ms. Alaka of Eli Lilly, Indianapolis.
The HAS response rate, defined by at least a 50% reduction in the HAS total score, was 75% in duloxetine-treated patients, compared with 56% in controls. The Hamilton Anxiety Scale remission rate, a more stringent endpoint requiring a week 10 total score of 10 or less, was achieved by 62% of the duloxetine group and 40% on placebo.
Duloxetine-treated patients also outperformed controls in terms of several other secondary endpoints. For example, the Sheehan Disability Scale global function impairment score in the duloxetine group improved by a mean of 7.6 points from a baseline score of 13.7, compared with a 4.3-point improvement with placebo.
Study discontinuation because of treatment-emergent adverse events occurred in 9.9% of elderly patients on duloxetine and 10.7% on placebo. The only adverse event that was significantly more common in the active treatment group was dry mouth, which occurred in 7% on duloxetine, compared with 1% of controls. Duloxetine was not associated with any clinically significant changes in laboratory findings, ECG parameters, or body weight.
The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
HOLLYWOOD, FLA. – Duloxetine proved safe and effective for the treatment of generalized anxiety disorder in the elderly in a phase IV clinical trial restricted to patients aged 65 and up.
This study fills a major gap in the evidence base for duloxetine (Cymbalta). Generalized anxiety disorder (GAD) is one of the most common psychiatric disorders in the elderly, with a prevalence estimated at up to 7%. Yet prior studies of duloxetine for GAD largely excluded the elderly. Indeed, the product labeling states prominently that premarketing studies did not include enough patients over age 65 to determine whether they respond differently than younger subjects.
With the new evidence provided by the phase IV study, the answer to that question is now in: Duloxetine proved significantly more effective than placebo in seniors. Moreover, the discontinuation rate because of adverse events did not differ significantly between the duloxetine and placebo groups, Karla J. Alaka reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The study included 291 patients aged 65 or older (mean age, 72) who met DSM-IV-TR criteria for GAD. They were randomized to 10 weeks of double-blind treatment with duloxetine dosed at 30-120 mg/day or placebo. Throughout the study, roughly one-third of the duloxetine group remained on 30 mg/day, another third bumped up to 60 mg/day, one-quarter increased to 90 mg/day, and the remainder eventually received 120 mg/day.
Elderly patients with an additional Axis I diagnosis plus GAD were not eligible for the trial. However, patients with comorbid medical illnesses were not excluded from participation so long as those conditions were stable and not expected to result in hospitalization within the next 6 months. Fully 83% of subjects had one or more preexisting medical conditions for which they took concomitant medication during the study. Geriatricians and other physicians have become increasingly vocal about the absence of quality safety and efficacy data for many widely prescribed drugs in the elderly, where issues such as polypharmacy, drug-drug interactions, and slowed drug metabolism become key considerations.
The primary efficacy measure in the study was a change in the Hamilton Anxiety Scale (HAS) total score from baseline to week 10. From a mean baseline HAS score of 24.6, the duloxetine group averaged a 15.9-point decrease, significantly better than the 11.7-point drop with placebo, according to Ms. Alaka of Eli Lilly, Indianapolis.
The HAS response rate, defined by at least a 50% reduction in the HAS total score, was 75% in duloxetine-treated patients, compared with 56% in controls. The Hamilton Anxiety Scale remission rate, a more stringent endpoint requiring a week 10 total score of 10 or less, was achieved by 62% of the duloxetine group and 40% on placebo.
Duloxetine-treated patients also outperformed controls in terms of several other secondary endpoints. For example, the Sheehan Disability Scale global function impairment score in the duloxetine group improved by a mean of 7.6 points from a baseline score of 13.7, compared with a 4.3-point improvement with placebo.
Study discontinuation because of treatment-emergent adverse events occurred in 9.9% of elderly patients on duloxetine and 10.7% on placebo. The only adverse event that was significantly more common in the active treatment group was dry mouth, which occurred in 7% on duloxetine, compared with 1% of controls. Duloxetine was not associated with any clinically significant changes in laboratory findings, ECG parameters, or body weight.
The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
AT THE NCDEU MEETING
Major finding: The first-ever randomized clinical trial of duloxetine for generalized anxiety disorder to include a substantial number of patients aged 65 and older showed a 62% remission rate with duloxetine, compared with 40% with placebo.
Data source: This 10-week, randomized, double-blind phase IV clinical trial included 291 patients with generalized anxiety disorder, all aged 65 or older.
Disclosures: The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
In MS, 44% of excess mortality is potentially preventable
ORLANDO – Sepsis, pulmonary infections, aspiration, and ischemic heart disease accounted for the bulk of excess mortality seen in patients with multiple sclerosis, compared with the matched general population, in a large U.S. study.
"Increased awareness of these potentially fatal conditions for patients with MS can improve patient care by increasing physician vigilance and facilitating early intervention," Dr. Michael J. Corwin said at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Because MS is a chronic, progressive, incurable disease, the death certificates of MS patients often list MS as the underlying cause of death. Accepting that at face value would mean nearly two-thirds of excess mortality seen in the study was because of advanced MS, said Dr. Corwin. But with the use of a novel cause-of-death algorithm, he and his colleagues were able to identify key intermediate and potentially interruptible steps on the pathway from end-stage MS to death.
He presented a retrospective matched cohort study of 30,402 patients with MS and 89,818 matched non-MS comparators, all drawn from a large, national, U.S. commercial health-plan database. The mortality rate was doubled among the MS population: 5.2%, compared with 2.6% in controls. This translated to a death rate of 899 per 100,000 person-years in the MS group and 446 per 100,000 person-years in the control group, reported Dr. Corwin, an associate professor of pediatrics and epidemiology at Boston University’s Schools of Medicine and Public Health.
The study entailed detailed analysis of the death records of 1,579 MS patients and 2,332 matched controls. Because of the notorious lack of uniformity in the way death certificate data are recorded, it can be difficult to determine the immediate cause of death from these records, noted Dr. Corwin. Therefore, he and his coinvestigators developed the algorithm for doing so.
Specifically, the algorithm showed that infection accounted for 21% of the excess mortality in the MS population, cardiovascular disease 13%, and pulmonary disease 10%. Collectively, these three causes of death accounted for 44% of the excess mortality. Another 29% of the excess remained attributable to late-stage MS because of the lack of additional death record data that might have allowed a more specific categorization.
Delving more deeply into the principal causes of death, the investigators determined that sepsis was the No. 1 contributor to the excess mortality seen in the MS population. It accounted for 45 of the 453 excess deaths per 100,000 person-years. Sepsis was followed by pulmonary infection at 41, pulmonary aspiration at 27, and ischemic heart disease at 17.
The prominent role of pulmonary infections in contributing to excess mortality in patients with MS seen in this study confirms a finding from the 21-year follow-up of the landmark, multicenter, interferon beta-1b randomized treatment trial (BMJ Open 2012 Nov 30;2(6). pii: e001972 [doi: 10.1136/bmjopen-2012-001972]). Respiratory diseases were also the top cause of excess mortality found among MS patients in a recent, large, British, national population-based registry (Eur. J. Neurol. 2012;19:1007-14).
Dr. Corwin is a principal in Care-Safe, a consulting firm contracted by Bayer HealthCare Pharmaceuticals to conduct the U.S. study.
ORLANDO – Sepsis, pulmonary infections, aspiration, and ischemic heart disease accounted for the bulk of excess mortality seen in patients with multiple sclerosis, compared with the matched general population, in a large U.S. study.
"Increased awareness of these potentially fatal conditions for patients with MS can improve patient care by increasing physician vigilance and facilitating early intervention," Dr. Michael J. Corwin said at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Because MS is a chronic, progressive, incurable disease, the death certificates of MS patients often list MS as the underlying cause of death. Accepting that at face value would mean nearly two-thirds of excess mortality seen in the study was because of advanced MS, said Dr. Corwin. But with the use of a novel cause-of-death algorithm, he and his colleagues were able to identify key intermediate and potentially interruptible steps on the pathway from end-stage MS to death.
He presented a retrospective matched cohort study of 30,402 patients with MS and 89,818 matched non-MS comparators, all drawn from a large, national, U.S. commercial health-plan database. The mortality rate was doubled among the MS population: 5.2%, compared with 2.6% in controls. This translated to a death rate of 899 per 100,000 person-years in the MS group and 446 per 100,000 person-years in the control group, reported Dr. Corwin, an associate professor of pediatrics and epidemiology at Boston University’s Schools of Medicine and Public Health.
The study entailed detailed analysis of the death records of 1,579 MS patients and 2,332 matched controls. Because of the notorious lack of uniformity in the way death certificate data are recorded, it can be difficult to determine the immediate cause of death from these records, noted Dr. Corwin. Therefore, he and his coinvestigators developed the algorithm for doing so.
Specifically, the algorithm showed that infection accounted for 21% of the excess mortality in the MS population, cardiovascular disease 13%, and pulmonary disease 10%. Collectively, these three causes of death accounted for 44% of the excess mortality. Another 29% of the excess remained attributable to late-stage MS because of the lack of additional death record data that might have allowed a more specific categorization.
Delving more deeply into the principal causes of death, the investigators determined that sepsis was the No. 1 contributor to the excess mortality seen in the MS population. It accounted for 45 of the 453 excess deaths per 100,000 person-years. Sepsis was followed by pulmonary infection at 41, pulmonary aspiration at 27, and ischemic heart disease at 17.
The prominent role of pulmonary infections in contributing to excess mortality in patients with MS seen in this study confirms a finding from the 21-year follow-up of the landmark, multicenter, interferon beta-1b randomized treatment trial (BMJ Open 2012 Nov 30;2(6). pii: e001972 [doi: 10.1136/bmjopen-2012-001972]). Respiratory diseases were also the top cause of excess mortality found among MS patients in a recent, large, British, national population-based registry (Eur. J. Neurol. 2012;19:1007-14).
Dr. Corwin is a principal in Care-Safe, a consulting firm contracted by Bayer HealthCare Pharmaceuticals to conduct the U.S. study.
ORLANDO – Sepsis, pulmonary infections, aspiration, and ischemic heart disease accounted for the bulk of excess mortality seen in patients with multiple sclerosis, compared with the matched general population, in a large U.S. study.
"Increased awareness of these potentially fatal conditions for patients with MS can improve patient care by increasing physician vigilance and facilitating early intervention," Dr. Michael J. Corwin said at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Because MS is a chronic, progressive, incurable disease, the death certificates of MS patients often list MS as the underlying cause of death. Accepting that at face value would mean nearly two-thirds of excess mortality seen in the study was because of advanced MS, said Dr. Corwin. But with the use of a novel cause-of-death algorithm, he and his colleagues were able to identify key intermediate and potentially interruptible steps on the pathway from end-stage MS to death.
He presented a retrospective matched cohort study of 30,402 patients with MS and 89,818 matched non-MS comparators, all drawn from a large, national, U.S. commercial health-plan database. The mortality rate was doubled among the MS population: 5.2%, compared with 2.6% in controls. This translated to a death rate of 899 per 100,000 person-years in the MS group and 446 per 100,000 person-years in the control group, reported Dr. Corwin, an associate professor of pediatrics and epidemiology at Boston University’s Schools of Medicine and Public Health.
The study entailed detailed analysis of the death records of 1,579 MS patients and 2,332 matched controls. Because of the notorious lack of uniformity in the way death certificate data are recorded, it can be difficult to determine the immediate cause of death from these records, noted Dr. Corwin. Therefore, he and his coinvestigators developed the algorithm for doing so.
Specifically, the algorithm showed that infection accounted for 21% of the excess mortality in the MS population, cardiovascular disease 13%, and pulmonary disease 10%. Collectively, these three causes of death accounted for 44% of the excess mortality. Another 29% of the excess remained attributable to late-stage MS because of the lack of additional death record data that might have allowed a more specific categorization.
Delving more deeply into the principal causes of death, the investigators determined that sepsis was the No. 1 contributor to the excess mortality seen in the MS population. It accounted for 45 of the 453 excess deaths per 100,000 person-years. Sepsis was followed by pulmonary infection at 41, pulmonary aspiration at 27, and ischemic heart disease at 17.
The prominent role of pulmonary infections in contributing to excess mortality in patients with MS seen in this study confirms a finding from the 21-year follow-up of the landmark, multicenter, interferon beta-1b randomized treatment trial (BMJ Open 2012 Nov 30;2(6). pii: e001972 [doi: 10.1136/bmjopen-2012-001972]). Respiratory diseases were also the top cause of excess mortality found among MS patients in a recent, large, British, national population-based registry (Eur. J. Neurol. 2012;19:1007-14).
Dr. Corwin is a principal in Care-Safe, a consulting firm contracted by Bayer HealthCare Pharmaceuticals to conduct the U.S. study.
AT THE CMSC/ACTRIMS ANNUAL MEETING
Major Finding: Fatal sepsis, pulmonary infection, ischemic heart disease, and pulmonary aspiration occurred significantly more often in MS patients than in controls, collectively accounting for 44% of the excess mortality in the MS population.
Data Source: A retrospective cohort study of the death records of 1,579 patients with MS and 2,332 matched controls.
Disclosures: Dr. Corwin is a principal in Care-Safe, a consulting firm contracted by Bayer HealthCare Pharmaceuticals to conduct the U.S. study.
Long-acting injectable antipsychotics provide better outcomes
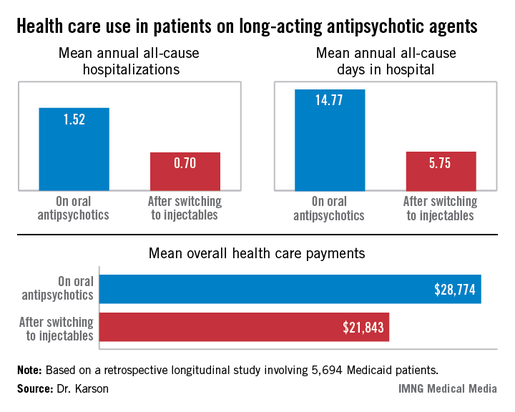
HOLLYWOOD, FLA. – Once patients were switched from oral antipsychotic agents to long-acting injectable ones, their annual all-cause hospitalization rate was cut by more than half in an observational study involving nearly 6,000 Medicaid patients with schizophrenia.
Moreover, the mean number of days per year spent in the hospital for any reason also dropped dramatically after the switch. Plus, the mean overall annual health care costs decreased by $6,901, Dr. Craig N. Karson reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"We may have an opportunity here to improve the management of schizophrenia, and in particular the cost management," said Dr. Karson, a psychiatrist in Wayne, Pa.
He presented a retrospective longitudinal study of 5,694 Medicaid patients with schizophrenia who switched from oral to long-acting injectable antipsychotics during 2005-2010. The data came from the Thomson Reuters MarketScan Research Medicaid Database.

Forty-four percent of patients were placed on long-acting injectable risperidone, 41% on haloperidol, and the rest on fluphenazine, which were the three antipsychotic agents available in depot form during the study period.
Roughly 80% of the all-cause hospitalizations in this study were for schizophrenia.
The switch from oral to long-acting injectable antipsychotic agents was associated with a striking reduction in overall health care resource use, including a decrease in the mean annual number of all-cause hospitalizations from 1.52 to 0.70 (see box).
In order to see whether patients’ duration on long-acting injectable antipsychotics was related to health care resource use, Dr. Karson and his coinvestigators divided the study population into two groups: the 2,856 patients who were short-term users of long-acting injectable therapy, defined as less than 180 days of treatment; and the 2,838 users of long-acting injectables for at least 180 days. The short-term users averaged 0.79 all-cause hospitalizations annually; the long-term users, 0.61. The mean annualized total days in the hospital was 6.56 for the short-term users, compared with 4.93 in longer-term users.
Several speakers argued that the MarketScan database has significant methodologic shortcomings, including selection bias, rendering it better suited as a basis for hypothesis generation than for drawing firm conclusions. Dr. Karson acknowledged that the database is limited in that it provides a convenience sample rather than a random sample of the Medicaid population, but he argued that the shear size of the study population overcomes that limitation.
"Ask yourself, how many treatment studies do we have in psychiatry with almost 6,000 patients? Very few," he said. "When you get into these kinds of patient numbers, which provide enormous statistical power, and then use a very hard endpoint like annual all-cause hospitalizations, I think it changes the game a little. I would argue that these patients are representative of the Medicaid population with serious mental illness, regardless."
Discussant Dr. Stephen R. Marder, who was not involved in the study, agreed that the data are compelling."
"The differences seen in your study were large and persuasive. And I thought the analysis of short- versus long-term use showing a dose-response effect added to the confidence in the results. Being on depot medication longer makes a difference," concluded Dr. Marder, director of the section on psychosis at the University of California, Los Angeles, Neuropsychiatric Institute.
"The problem lies with us as providers: We see long-acting injectable antipsychotics as the last resort, said Dr. Marder, also professor of psychiatry and behavioral sciences at UCLA. "We’re probably reluctant to do it because we see the infrastructure for providing the therapy is very poor in many places."

At the VA medical center where he works, for example, patients formerly had access to depot medication on a 24/7 basis, so that if they had a job, they could obtain treatment without missing work. That service isn’t available, anymore, Dr. Marder noted.
His hope, he added, is that thought leaders in psychiatry will be able to influence implementation of the Affordable Care Act so as to create a much better infrastructure for providing long-acting injectable antipsychotic therapy. Ideally, access to long-acting injectable antipsychotics should be available in places where psychosocial and rehabilitation services are located. "That’s what seems to work the best," according to the psychiatrist.
Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
HOLLYWOOD, FLA. – Once patients were switched from oral antipsychotic agents to long-acting injectable ones, their annual all-cause hospitalization rate was cut by more than half in an observational study involving nearly 6,000 Medicaid patients with schizophrenia.
Moreover, the mean number of days per year spent in the hospital for any reason also dropped dramatically after the switch. Plus, the mean overall annual health care costs decreased by $6,901, Dr. Craig N. Karson reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"We may have an opportunity here to improve the management of schizophrenia, and in particular the cost management," said Dr. Karson, a psychiatrist in Wayne, Pa.
He presented a retrospective longitudinal study of 5,694 Medicaid patients with schizophrenia who switched from oral to long-acting injectable antipsychotics during 2005-2010. The data came from the Thomson Reuters MarketScan Research Medicaid Database.
Forty-four percent of patients were placed on long-acting injectable risperidone, 41% on haloperidol, and the rest on fluphenazine, which were the three antipsychotic agents available in depot form during the study period.
Roughly 80% of the all-cause hospitalizations in this study were for schizophrenia.
The switch from oral to long-acting injectable antipsychotic agents was associated with a striking reduction in overall health care resource use, including a decrease in the mean annual number of all-cause hospitalizations from 1.52 to 0.70 (see box).
In order to see whether patients’ duration on long-acting injectable antipsychotics was related to health care resource use, Dr. Karson and his coinvestigators divided the study population into two groups: the 2,856 patients who were short-term users of long-acting injectable therapy, defined as less than 180 days of treatment; and the 2,838 users of long-acting injectables for at least 180 days. The short-term users averaged 0.79 all-cause hospitalizations annually; the long-term users, 0.61. The mean annualized total days in the hospital was 6.56 for the short-term users, compared with 4.93 in longer-term users.
Several speakers argued that the MarketScan database has significant methodologic shortcomings, including selection bias, rendering it better suited as a basis for hypothesis generation than for drawing firm conclusions. Dr. Karson acknowledged that the database is limited in that it provides a convenience sample rather than a random sample of the Medicaid population, but he argued that the shear size of the study population overcomes that limitation.
"Ask yourself, how many treatment studies do we have in psychiatry with almost 6,000 patients? Very few," he said. "When you get into these kinds of patient numbers, which provide enormous statistical power, and then use a very hard endpoint like annual all-cause hospitalizations, I think it changes the game a little. I would argue that these patients are representative of the Medicaid population with serious mental illness, regardless."
Discussant Dr. Stephen R. Marder, who was not involved in the study, agreed that the data are compelling."
"The differences seen in your study were large and persuasive. And I thought the analysis of short- versus long-term use showing a dose-response effect added to the confidence in the results. Being on depot medication longer makes a difference," concluded Dr. Marder, director of the section on psychosis at the University of California, Los Angeles, Neuropsychiatric Institute.
"The problem lies with us as providers: We see long-acting injectable antipsychotics as the last resort, said Dr. Marder, also professor of psychiatry and behavioral sciences at UCLA. "We’re probably reluctant to do it because we see the infrastructure for providing the therapy is very poor in many places."
At the VA medical center where he works, for example, patients formerly had access to depot medication on a 24/7 basis, so that if they had a job, they could obtain treatment without missing work. That service isn’t available, anymore, Dr. Marder noted.
His hope, he added, is that thought leaders in psychiatry will be able to influence implementation of the Affordable Care Act so as to create a much better infrastructure for providing long-acting injectable antipsychotic therapy. Ideally, access to long-acting injectable antipsychotics should be available in places where psychosocial and rehabilitation services are located. "That’s what seems to work the best," according to the psychiatrist.
Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
HOLLYWOOD, FLA. – Once patients were switched from oral antipsychotic agents to long-acting injectable ones, their annual all-cause hospitalization rate was cut by more than half in an observational study involving nearly 6,000 Medicaid patients with schizophrenia.
Moreover, the mean number of days per year spent in the hospital for any reason also dropped dramatically after the switch. Plus, the mean overall annual health care costs decreased by $6,901, Dr. Craig N. Karson reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"We may have an opportunity here to improve the management of schizophrenia, and in particular the cost management," said Dr. Karson, a psychiatrist in Wayne, Pa.
He presented a retrospective longitudinal study of 5,694 Medicaid patients with schizophrenia who switched from oral to long-acting injectable antipsychotics during 2005-2010. The data came from the Thomson Reuters MarketScan Research Medicaid Database.
Forty-four percent of patients were placed on long-acting injectable risperidone, 41% on haloperidol, and the rest on fluphenazine, which were the three antipsychotic agents available in depot form during the study period.
Roughly 80% of the all-cause hospitalizations in this study were for schizophrenia.
The switch from oral to long-acting injectable antipsychotic agents was associated with a striking reduction in overall health care resource use, including a decrease in the mean annual number of all-cause hospitalizations from 1.52 to 0.70 (see box).
In order to see whether patients’ duration on long-acting injectable antipsychotics was related to health care resource use, Dr. Karson and his coinvestigators divided the study population into two groups: the 2,856 patients who were short-term users of long-acting injectable therapy, defined as less than 180 days of treatment; and the 2,838 users of long-acting injectables for at least 180 days. The short-term users averaged 0.79 all-cause hospitalizations annually; the long-term users, 0.61. The mean annualized total days in the hospital was 6.56 for the short-term users, compared with 4.93 in longer-term users.
Several speakers argued that the MarketScan database has significant methodologic shortcomings, including selection bias, rendering it better suited as a basis for hypothesis generation than for drawing firm conclusions. Dr. Karson acknowledged that the database is limited in that it provides a convenience sample rather than a random sample of the Medicaid population, but he argued that the shear size of the study population overcomes that limitation.
"Ask yourself, how many treatment studies do we have in psychiatry with almost 6,000 patients? Very few," he said. "When you get into these kinds of patient numbers, which provide enormous statistical power, and then use a very hard endpoint like annual all-cause hospitalizations, I think it changes the game a little. I would argue that these patients are representative of the Medicaid population with serious mental illness, regardless."
Discussant Dr. Stephen R. Marder, who was not involved in the study, agreed that the data are compelling."
"The differences seen in your study were large and persuasive. And I thought the analysis of short- versus long-term use showing a dose-response effect added to the confidence in the results. Being on depot medication longer makes a difference," concluded Dr. Marder, director of the section on psychosis at the University of California, Los Angeles, Neuropsychiatric Institute.
"The problem lies with us as providers: We see long-acting injectable antipsychotics as the last resort, said Dr. Marder, also professor of psychiatry and behavioral sciences at UCLA. "We’re probably reluctant to do it because we see the infrastructure for providing the therapy is very poor in many places."
At the VA medical center where he works, for example, patients formerly had access to depot medication on a 24/7 basis, so that if they had a job, they could obtain treatment without missing work. That service isn’t available, anymore, Dr. Marder noted.
His hope, he added, is that thought leaders in psychiatry will be able to influence implementation of the Affordable Care Act so as to create a much better infrastructure for providing long-acting injectable antipsychotic therapy. Ideally, access to long-acting injectable antipsychotics should be available in places where psychosocial and rehabilitation services are located. "That’s what seems to work the best," according to the psychiatrist.
Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
AT THE NCDEU MEETING
Major finding: Medicaid patients with schizophrenia had a mean 1.52 hospitalizations per year for any cause while they were on oral antipsychotic medication, dropping to 0.7 all-cause hospitalizations per year after they switched to long-acting injectable therapy.
Data source: A retrospective observational study involving 5,694 Medicaid patients with schizophrenia drawn from the Thomson Reuters MarketScan Research Medicaid Database.
Disclosures: Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
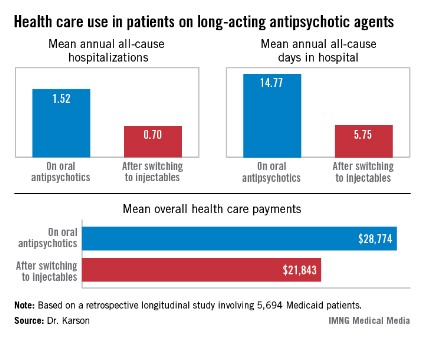
Reablate, don't medicate, after failed AF ablation
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. What to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo-ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo-ablation as well.
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group. Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachyarrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication due to intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. What to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo-ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo-ablation as well.
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group. Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachyarrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication due to intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. What to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo-ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo-ablation as well.
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group. Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachyarrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication due to intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major Finding: At 3 years after a first ablation procedure had failed for symptomatic paroxysmal atrial fibrillation, 12% of those randomized to drug therapy and 58% of those in the redo-ablation group were free of atrial tachyarrhythmias.
Data Source: A randomized, prospective, multicenter clinical trial involving 154 patients whose atrial arrhythmia status was monitored via implantable loop recorder.
Disclosures: The presenter reported having no conflicts of interest.
Esophagectomy cases have been rising steadily
INDIANAPOLIS - Transthoracic esophagectomy for esophageal cancer provides significantly lower in-hospital mortality and major morbidity rates than does transhiatal esophagectomy, according to an analysis of a large multiyear national database.
Further, in-hospital outcomes of esophagectomy didn?t differ significantly between high-volume centers ? in this study, defined as those doing 10 or more cases per year ? and low-volume centers, Dr. Mehraneh D. Jafari reported at the annual meeting of the American Surgical Association.
That finding was met with skepticism, and discussants were quick to argue that study limitations make it difficult to draw any meaningful conclusions from the data. For one thing, speakers contended that defining a high-volume center based upon an institutional threshold of 10 or more cases per year sets the bar far too low given that a single dedicated esophageal surgery specialist might easily perform 50 or more esophagectomies annually.
Dr. Jafari presented an analysis of 11,473 transthoracic and 3,717 transhiatal esophagectomies performed for esophageal cancer. The data came from the Nationwide Inpatient Sample (NIS) during 2001-2010. The NIS records data on in-hospital outcomes for a nationally representative sample composed of roughly 20% of the country?s hospital discharges each year.
The number of esophagectomies rose steadily by an average of 4% annually during the study years, reflecting the substantial national increase in cases of esophageal cancer. The growing case count, expected to reach an estimated 18,000 cases of esophageal cancer nationwide in 2013, has been attributed to rising rates of gastroesophageal reflux disease, Barrett?s esophagus, and obesity. Transthoracic esophagectomy, used in 76% of cases, remained the preferred operative strategy throughout the study years.
In-hospital outcomes were markedly better in patients who had transthoracic esophagectomy. After adjustment for potential confounding variables in a multivariate analysis, transhiatal esophagectomy recipients had a 67% increased risk of in-hospital mortality and a 39% greater risk of serious complications, including a 37% increased risk of pulmonary complications. However, anastomotic leak rates were similar with both operations, according to Dr. Jafari of the University of California, Irvine.
The referral rate to high-volume esophagectomy centers climbed steadily over time, rising from 22% of all cases in 2001 to 58% in 2010.
The 35 high-volume centers performed an average of 16 cases per year. In contrast, the 484 low-volume centers averaged 2 cases per year. In-hospital mortality among the 9,386 patients treated in low-volume centers averaged 7.6% compared with 4.3% for patients in high-volume centers. Overall in-hospital serious morbidity rates were greater in the low-volume centers as well: 47% versus 41%. While these raw differences were statistically significant, a risk-adjusted multivariate analysis found no significant outcome differences between low- and high-volume centers.
Discussant Dr. Michael J. Zinner noted that in an earlier study he and his coworkers showed that an institutional threshold of roughly 30 esophagectomies per year is required to discriminate between low- and high-volume centers in terms of in-hospital mortality. So why define high-volume centers as those doing a mere 10 cases per year? asked Dr. Zinner, chairman of the department of surgery at Brigham and Women?s Hospital and professor of surgery at Harvard Medical School, Boston.
"The problem here is if you establish 30 cases per year as the threshold for a high-volume center, I can tell you there are probably less than 20 centers in the whole U.S. capable of doing that volume. That?s a real issue, because then how are patients who live in a remote region going to get care at one of those centers?" replied Dr. Jafari?s senior coauthor Dr. Ninh T. Nguyen, professor and vice-chair of surgery at UC Irvine.
"I think instead we should try to lift all boats: Develop a national esophageal center network to identify the qualities reflective of better outcomes in the high-volume centers and introduce those factors at low-volume centers. This way we?re not impeding access to care for our patients," he continued.
Dr. Nguyen said that in-hospital surgical morbidity rates in the NIS need to be taken with a grain of salt, as the accuracy of coding for complications is "rather low." The development of minimally invasive techniques for intrathoracic anastomosis has transformed transthoracic esophagectomy into a procedure with an improved complication profile.
"I switched to transthoracic esophagectomy 5 years ago. One reason was development of the minimally invasive approach. As a result, we?re not scared of a chest anastomosis like we used to be. When patients undergoing transthoracic esophagectomy with thoracotomy had a leak in the chest they had a very high risk for mortality. That?s not the case anymore. We have not observed any mortality associated with a leak in the chest for many, many years now," he said.
The investigators reported having no conflicts of interest.
I have major problems with this study, stemming from inherent limitations in the Nationwide Inpatient Sample. It?s an administrative database set up chiefly to track costs, utilization, and length of stay. It contains no information at all on key clinical outcomes such as 30- and 90-day mortality, discharge disposition, or 30-day readmission rates.
| Dr. Luketich |
In addition, the accuracy of the quoted in-hospital morbidity rates is suspect, probably because data entry isn?t performed by trained researchers. For example, the 8% incidence of renal failure in esophagectomy patients cited in this study sounds too high to be right.
And there?s another major problem with this database: The superior outcomes reported for transthoracic esophagectomy recipients in this study fly in the face of earlier, well-conducted meta-analyses that reached the opposite conclusion. The most likely explanation for the discordant findings lies in the fact that the NIS doesn?t show whether a transthoracic esophagectomy was performed via open thoracotomy in the old-school manner or with an intrathoracic anastomosis created using contemporary minimally invasive techniques which, while complex, have been associated with better outcomes.
Dr. James D. Luketich is professor of surgery and chief of the Heart, Lung, and Esophageal Surgery Institute at the University of Pittsburgh. He was a designated discussant of the study at the meeting.
I have major problems with this study, stemming from inherent limitations in the Nationwide Inpatient Sample. It?s an administrative database set up chiefly to track costs, utilization, and length of stay. It contains no information at all on key clinical outcomes such as 30- and 90-day mortality, discharge disposition, or 30-day readmission rates.
| Dr. Luketich |
In addition, the accuracy of the quoted in-hospital morbidity rates is suspect, probably because data entry isn?t performed by trained researchers. For example, the 8% incidence of renal failure in esophagectomy patients cited in this study sounds too high to be right.
And there?s another major problem with this database: The superior outcomes reported for transthoracic esophagectomy recipients in this study fly in the face of earlier, well-conducted meta-analyses that reached the opposite conclusion. The most likely explanation for the discordant findings lies in the fact that the NIS doesn?t show whether a transthoracic esophagectomy was performed via open thoracotomy in the old-school manner or with an intrathoracic anastomosis created using contemporary minimally invasive techniques which, while complex, have been associated with better outcomes.
Dr. James D. Luketich is professor of surgery and chief of the Heart, Lung, and Esophageal Surgery Institute at the University of Pittsburgh. He was a designated discussant of the study at the meeting.
I have major problems with this study, stemming from inherent limitations in the Nationwide Inpatient Sample. It?s an administrative database set up chiefly to track costs, utilization, and length of stay. It contains no information at all on key clinical outcomes such as 30- and 90-day mortality, discharge disposition, or 30-day readmission rates.
| Dr. Luketich |
In addition, the accuracy of the quoted in-hospital morbidity rates is suspect, probably because data entry isn?t performed by trained researchers. For example, the 8% incidence of renal failure in esophagectomy patients cited in this study sounds too high to be right.
And there?s another major problem with this database: The superior outcomes reported for transthoracic esophagectomy recipients in this study fly in the face of earlier, well-conducted meta-analyses that reached the opposite conclusion. The most likely explanation for the discordant findings lies in the fact that the NIS doesn?t show whether a transthoracic esophagectomy was performed via open thoracotomy in the old-school manner or with an intrathoracic anastomosis created using contemporary minimally invasive techniques which, while complex, have been associated with better outcomes.
Dr. James D. Luketich is professor of surgery and chief of the Heart, Lung, and Esophageal Surgery Institute at the University of Pittsburgh. He was a designated discussant of the study at the meeting.
INDIANAPOLIS - Transthoracic esophagectomy for esophageal cancer provides significantly lower in-hospital mortality and major morbidity rates than does transhiatal esophagectomy, according to an analysis of a large multiyear national database.
Further, in-hospital outcomes of esophagectomy didn?t differ significantly between high-volume centers ? in this study, defined as those doing 10 or more cases per year ? and low-volume centers, Dr. Mehraneh D. Jafari reported at the annual meeting of the American Surgical Association.
That finding was met with skepticism, and discussants were quick to argue that study limitations make it difficult to draw any meaningful conclusions from the data. For one thing, speakers contended that defining a high-volume center based upon an institutional threshold of 10 or more cases per year sets the bar far too low given that a single dedicated esophageal surgery specialist might easily perform 50 or more esophagectomies annually.
Dr. Jafari presented an analysis of 11,473 transthoracic and 3,717 transhiatal esophagectomies performed for esophageal cancer. The data came from the Nationwide Inpatient Sample (NIS) during 2001-2010. The NIS records data on in-hospital outcomes for a nationally representative sample composed of roughly 20% of the country?s hospital discharges each year.
The number of esophagectomies rose steadily by an average of 4% annually during the study years, reflecting the substantial national increase in cases of esophageal cancer. The growing case count, expected to reach an estimated 18,000 cases of esophageal cancer nationwide in 2013, has been attributed to rising rates of gastroesophageal reflux disease, Barrett?s esophagus, and obesity. Transthoracic esophagectomy, used in 76% of cases, remained the preferred operative strategy throughout the study years.
In-hospital outcomes were markedly better in patients who had transthoracic esophagectomy. After adjustment for potential confounding variables in a multivariate analysis, transhiatal esophagectomy recipients had a 67% increased risk of in-hospital mortality and a 39% greater risk of serious complications, including a 37% increased risk of pulmonary complications. However, anastomotic leak rates were similar with both operations, according to Dr. Jafari of the University of California, Irvine.
The referral rate to high-volume esophagectomy centers climbed steadily over time, rising from 22% of all cases in 2001 to 58% in 2010.
The 35 high-volume centers performed an average of 16 cases per year. In contrast, the 484 low-volume centers averaged 2 cases per year. In-hospital mortality among the 9,386 patients treated in low-volume centers averaged 7.6% compared with 4.3% for patients in high-volume centers. Overall in-hospital serious morbidity rates were greater in the low-volume centers as well: 47% versus 41%. While these raw differences were statistically significant, a risk-adjusted multivariate analysis found no significant outcome differences between low- and high-volume centers.
Discussant Dr. Michael J. Zinner noted that in an earlier study he and his coworkers showed that an institutional threshold of roughly 30 esophagectomies per year is required to discriminate between low- and high-volume centers in terms of in-hospital mortality. So why define high-volume centers as those doing a mere 10 cases per year? asked Dr. Zinner, chairman of the department of surgery at Brigham and Women?s Hospital and professor of surgery at Harvard Medical School, Boston.
"The problem here is if you establish 30 cases per year as the threshold for a high-volume center, I can tell you there are probably less than 20 centers in the whole U.S. capable of doing that volume. That?s a real issue, because then how are patients who live in a remote region going to get care at one of those centers?" replied Dr. Jafari?s senior coauthor Dr. Ninh T. Nguyen, professor and vice-chair of surgery at UC Irvine.
"I think instead we should try to lift all boats: Develop a national esophageal center network to identify the qualities reflective of better outcomes in the high-volume centers and introduce those factors at low-volume centers. This way we?re not impeding access to care for our patients," he continued.
Dr. Nguyen said that in-hospital surgical morbidity rates in the NIS need to be taken with a grain of salt, as the accuracy of coding for complications is "rather low." The development of minimally invasive techniques for intrathoracic anastomosis has transformed transthoracic esophagectomy into a procedure with an improved complication profile.
"I switched to transthoracic esophagectomy 5 years ago. One reason was development of the minimally invasive approach. As a result, we?re not scared of a chest anastomosis like we used to be. When patients undergoing transthoracic esophagectomy with thoracotomy had a leak in the chest they had a very high risk for mortality. That?s not the case anymore. We have not observed any mortality associated with a leak in the chest for many, many years now," he said.
The investigators reported having no conflicts of interest.
INDIANAPOLIS - Transthoracic esophagectomy for esophageal cancer provides significantly lower in-hospital mortality and major morbidity rates than does transhiatal esophagectomy, according to an analysis of a large multiyear national database.
Further, in-hospital outcomes of esophagectomy didn?t differ significantly between high-volume centers ? in this study, defined as those doing 10 or more cases per year ? and low-volume centers, Dr. Mehraneh D. Jafari reported at the annual meeting of the American Surgical Association.
That finding was met with skepticism, and discussants were quick to argue that study limitations make it difficult to draw any meaningful conclusions from the data. For one thing, speakers contended that defining a high-volume center based upon an institutional threshold of 10 or more cases per year sets the bar far too low given that a single dedicated esophageal surgery specialist might easily perform 50 or more esophagectomies annually.
Dr. Jafari presented an analysis of 11,473 transthoracic and 3,717 transhiatal esophagectomies performed for esophageal cancer. The data came from the Nationwide Inpatient Sample (NIS) during 2001-2010. The NIS records data on in-hospital outcomes for a nationally representative sample composed of roughly 20% of the country?s hospital discharges each year.
The number of esophagectomies rose steadily by an average of 4% annually during the study years, reflecting the substantial national increase in cases of esophageal cancer. The growing case count, expected to reach an estimated 18,000 cases of esophageal cancer nationwide in 2013, has been attributed to rising rates of gastroesophageal reflux disease, Barrett?s esophagus, and obesity. Transthoracic esophagectomy, used in 76% of cases, remained the preferred operative strategy throughout the study years.
In-hospital outcomes were markedly better in patients who had transthoracic esophagectomy. After adjustment for potential confounding variables in a multivariate analysis, transhiatal esophagectomy recipients had a 67% increased risk of in-hospital mortality and a 39% greater risk of serious complications, including a 37% increased risk of pulmonary complications. However, anastomotic leak rates were similar with both operations, according to Dr. Jafari of the University of California, Irvine.
The referral rate to high-volume esophagectomy centers climbed steadily over time, rising from 22% of all cases in 2001 to 58% in 2010.
The 35 high-volume centers performed an average of 16 cases per year. In contrast, the 484 low-volume centers averaged 2 cases per year. In-hospital mortality among the 9,386 patients treated in low-volume centers averaged 7.6% compared with 4.3% for patients in high-volume centers. Overall in-hospital serious morbidity rates were greater in the low-volume centers as well: 47% versus 41%. While these raw differences were statistically significant, a risk-adjusted multivariate analysis found no significant outcome differences between low- and high-volume centers.
Discussant Dr. Michael J. Zinner noted that in an earlier study he and his coworkers showed that an institutional threshold of roughly 30 esophagectomies per year is required to discriminate between low- and high-volume centers in terms of in-hospital mortality. So why define high-volume centers as those doing a mere 10 cases per year? asked Dr. Zinner, chairman of the department of surgery at Brigham and Women?s Hospital and professor of surgery at Harvard Medical School, Boston.
"The problem here is if you establish 30 cases per year as the threshold for a high-volume center, I can tell you there are probably less than 20 centers in the whole U.S. capable of doing that volume. That?s a real issue, because then how are patients who live in a remote region going to get care at one of those centers?" replied Dr. Jafari?s senior coauthor Dr. Ninh T. Nguyen, professor and vice-chair of surgery at UC Irvine.
"I think instead we should try to lift all boats: Develop a national esophageal center network to identify the qualities reflective of better outcomes in the high-volume centers and introduce those factors at low-volume centers. This way we?re not impeding access to care for our patients," he continued.
Dr. Nguyen said that in-hospital surgical morbidity rates in the NIS need to be taken with a grain of salt, as the accuracy of coding for complications is "rather low." The development of minimally invasive techniques for intrathoracic anastomosis has transformed transthoracic esophagectomy into a procedure with an improved complication profile.
"I switched to transthoracic esophagectomy 5 years ago. One reason was development of the minimally invasive approach. As a result, we?re not scared of a chest anastomosis like we used to be. When patients undergoing transthoracic esophagectomy with thoracotomy had a leak in the chest they had a very high risk for mortality. That?s not the case anymore. We have not observed any mortality associated with a leak in the chest for many, many years now," he said.
The investigators reported having no conflicts of interest.
At the ASA Annual Meeting
Major finding: In-hospital mortality occurred nationally in 5.8% of esophageal cancer patients who underwent transthoracic esophagectomy compared with 8.3% of transhiatal esophagectomy recipients.
Data source: A retrospective study of more than 15,000 patients who underwent esophagectomy for esophageal cancer during 2001-2010 and were Included in the Nationwide Inpatient Sample, a database sponsored by the Agency for Healthcare Research and Quality.
Disclosures: The study presenters reported having no financial conflicts.

Cariprazine performs well for schizophrenia, bipolar mania
HOLLYWOOD, FLA. – The novel oral antipsychotic agent cariprazine showed efficacy across the broad spectrum of schizophrenia manifestations – including negative symptoms – in a multicenter phase III clinical trial.
This is welcome news. Current antipsychotic agents are only modestly effective in treating the negative symptoms, mood disturbances, and neurocognitive dysfunction that so often hamper recovery in patients with schizophrenia. In contrast, the investigational agent cariprazine demonstrated efficacy in all these domains, Dr. John M. Kane reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Cariprazine is a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. As such, it would be expected to have broad efficacy across the spectrum of schizophrenia symptoms. This has been borne out, first in animal studies and now in this phase III randomized trial, noted Dr. Kane, chairman of psychiatry at Zucker Hillside Hospital in Glen Oaks, N.Y., and professor of psychiatry, neurology, and neuroscience at the Albert Einstein College of Medicine in New York.

Forest Pharmaceuticals filed an application with the Food and Drug Administration last fall for marketing approval of cariprazine in the treatment of schizophrenia as well as for treatment of bipolar mania. The company expects a decision from the regulatory agency before year’s end.
Dr. Kane reported on 446 patients with acute exacerbation of schizophrenia placed on 6 weeks of double-blind placebo or cariprazine at either 3-6 mg/day or 6-9 mg/day. Sixty-one percent of subjects completed the study. The mean daily dosage of cariprazine in the lower-dose study arm was 4.2 mg/day, versus 6.6 mg/day in the higher-dose arm.
The primary study endpoint was improvement in Positive and Negative Syndrome Scale (PANSS) total score. From a baseline mean PANSS score of 96, indicative of marked illness, patients in the lower-dose cariprazine group showed a placebo-subtracted net 6.8-point improvement. Those on higher-dose therapy had a net 9.9-point improvement. Both dosing strategies were significantly more effective than was placebo. Patients on cariprazine at the more effective dosing regimen of 6-9 mg/day showed a significant advantage over placebo starting at week 1.
To more closely examine the drug’s efficacy in domains other than positive symptoms such as delusions and hallucinations, Dr. Kane and his coinvestigators conducted a post hoc analysis focusing on cariprazine’s impact on each of the five PANSS-derived Marder symptom factor groups. These five groupings are negative symptoms, positive symptoms, uncontrolled hostility/excitement, disorganized thought, and anxiety/depression.
Cariprazine at 6-9 mg/day proved superior to placebo in all five Marder factor groupings. Cariprazine at 3-6 mg/day outperformed placebo in all but the Marder negative symptom factor grouping, where a trend favoring the drug fell short of statistical significance.
Of the 30 individual items that make up the PANSS, higher-dose cariprazine was significantly more effective than was placebo in 21. These included most positive symptom factor items as well as others where conventional antipsychotic agents typically don’t fare very well, such as emotional withdrawal, active social avoidance, poor rapport, anxiety, depression, tension, poor impulse control, hostility, excitement, uncooperativeness, disturbance of volition, and conceptual disorganization.
Cariprazine at 3-6 mg/day outperformed placebo on 11 of the 30 individual PANSS items.
Long-term cariprazine in bipolar I
Also at the NCDEU meeting, Dr. Terence A. Ketter presented a phase III, open-label, 16-week, study of flexibly dosed cariprazine in 402 patients with bipolar I disorder.
Dosing of cariprazine was permitted at 3-12 mg/day. The actual mean dose used in the study was 6.2 mg/day. The primary study endpoints involved safety and tolerability, since the drug already had demonstrated efficacy in three separate 3-week-long studies in acute mania.
This 16-week study was undertaken in recognition that the real challenge in bipolar I disorder is long-term management. Nearly half of all patients who respond to initial conventional therapies relapse within 2 years, mainly because of high rates of nonadherence, explained Dr. Ketter, professor of psychiatry and behavioral sciences and chief of the bipolar disorders clinic at Stanford (Calif.) University.
Just 33% of participants completed the 16-week study. Sixteen percent of subjects withdrew because of adverse events, roughly the same number as for protocol violations.
The major tolerability issue in the phase III trial was treatment-emergent akathisia, which occurred in 37% of participants. In addition, 7% experienced treatment-emergent extrapyramidal symptoms as defined by a Simpson-Angus Scale score of 3 or less at baseline rising to greater than 3 on treatment.
Ninety-eight percent of these cases of treatment-emergent akathisia or extrapyramidal symptoms were rated as mild to moderate in intensity. Five percent of subjects dropped out of the trial because of treatment-emergent akathisia, making this the No. 1 reason for discontinuation because of adverse events, followed by depression at 2%. Only 0.7% of subjects dropped out because of extrapyramidal symptoms, the same small number as quit because of worsening mania.
No clinically meaningful changes occurred in lipids, liver enzymes, ECG parameters, or prolactin levels. Systolic and diastolic blood pressure each rose modestly by less than 2 mm Hg. Weight gain was minimal: a mean increase of 0.9 kg over 16 weeks, with an accompanying mean 1.0-cm increase in waist circumference.
While this was primarily a phase III safety and tolerability study, efficacy was also measured. At week 16, 63% of subjects met study criteria for disease remission as defined by a Young Mania Rating Scale (YMRS) total score of 12 or less, starting from a mean baseline YMRS score of 26.1. Most of the symptomatic improvement was attained within the first 3 weeks of treatment, with a mean 13.6-point reduction in the YMRS, modestly improved upon to a mean 15.2-point reduction by week 16.
These schizophrenia and bipolar I disorder studies were sponsored by Forest Pharmaceuticals. Dr. Kane reported serving as a consultant and/or having received honoraria for lectures from Forest and 17 other pharmaceutical companies. Dr. Ketter reported serving as a consultant to Forest and four other pharmaceutical companies as well as receiving lecture honoraria from GlaxoSmithKline and Otsuka.
HOLLYWOOD, FLA. – The novel oral antipsychotic agent cariprazine showed efficacy across the broad spectrum of schizophrenia manifestations – including negative symptoms – in a multicenter phase III clinical trial.
This is welcome news. Current antipsychotic agents are only modestly effective in treating the negative symptoms, mood disturbances, and neurocognitive dysfunction that so often hamper recovery in patients with schizophrenia. In contrast, the investigational agent cariprazine demonstrated efficacy in all these domains, Dr. John M. Kane reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Cariprazine is a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. As such, it would be expected to have broad efficacy across the spectrum of schizophrenia symptoms. This has been borne out, first in animal studies and now in this phase III randomized trial, noted Dr. Kane, chairman of psychiatry at Zucker Hillside Hospital in Glen Oaks, N.Y., and professor of psychiatry, neurology, and neuroscience at the Albert Einstein College of Medicine in New York.
Forest Pharmaceuticals filed an application with the Food and Drug Administration last fall for marketing approval of cariprazine in the treatment of schizophrenia as well as for treatment of bipolar mania. The company expects a decision from the regulatory agency before year’s end.
Dr. Kane reported on 446 patients with acute exacerbation of schizophrenia placed on 6 weeks of double-blind placebo or cariprazine at either 3-6 mg/day or 6-9 mg/day. Sixty-one percent of subjects completed the study. The mean daily dosage of cariprazine in the lower-dose study arm was 4.2 mg/day, versus 6.6 mg/day in the higher-dose arm.
The primary study endpoint was improvement in Positive and Negative Syndrome Scale (PANSS) total score. From a baseline mean PANSS score of 96, indicative of marked illness, patients in the lower-dose cariprazine group showed a placebo-subtracted net 6.8-point improvement. Those on higher-dose therapy had a net 9.9-point improvement. Both dosing strategies were significantly more effective than was placebo. Patients on cariprazine at the more effective dosing regimen of 6-9 mg/day showed a significant advantage over placebo starting at week 1.
To more closely examine the drug’s efficacy in domains other than positive symptoms such as delusions and hallucinations, Dr. Kane and his coinvestigators conducted a post hoc analysis focusing on cariprazine’s impact on each of the five PANSS-derived Marder symptom factor groups. These five groupings are negative symptoms, positive symptoms, uncontrolled hostility/excitement, disorganized thought, and anxiety/depression.
Cariprazine at 6-9 mg/day proved superior to placebo in all five Marder factor groupings. Cariprazine at 3-6 mg/day outperformed placebo in all but the Marder negative symptom factor grouping, where a trend favoring the drug fell short of statistical significance.
Of the 30 individual items that make up the PANSS, higher-dose cariprazine was significantly more effective than was placebo in 21. These included most positive symptom factor items as well as others where conventional antipsychotic agents typically don’t fare very well, such as emotional withdrawal, active social avoidance, poor rapport, anxiety, depression, tension, poor impulse control, hostility, excitement, uncooperativeness, disturbance of volition, and conceptual disorganization.
Cariprazine at 3-6 mg/day outperformed placebo on 11 of the 30 individual PANSS items.
Long-term cariprazine in bipolar I
Also at the NCDEU meeting, Dr. Terence A. Ketter presented a phase III, open-label, 16-week, study of flexibly dosed cariprazine in 402 patients with bipolar I disorder.
Dosing of cariprazine was permitted at 3-12 mg/day. The actual mean dose used in the study was 6.2 mg/day. The primary study endpoints involved safety and tolerability, since the drug already had demonstrated efficacy in three separate 3-week-long studies in acute mania.
This 16-week study was undertaken in recognition that the real challenge in bipolar I disorder is long-term management. Nearly half of all patients who respond to initial conventional therapies relapse within 2 years, mainly because of high rates of nonadherence, explained Dr. Ketter, professor of psychiatry and behavioral sciences and chief of the bipolar disorders clinic at Stanford (Calif.) University.
Just 33% of participants completed the 16-week study. Sixteen percent of subjects withdrew because of adverse events, roughly the same number as for protocol violations.
The major tolerability issue in the phase III trial was treatment-emergent akathisia, which occurred in 37% of participants. In addition, 7% experienced treatment-emergent extrapyramidal symptoms as defined by a Simpson-Angus Scale score of 3 or less at baseline rising to greater than 3 on treatment.
Ninety-eight percent of these cases of treatment-emergent akathisia or extrapyramidal symptoms were rated as mild to moderate in intensity. Five percent of subjects dropped out of the trial because of treatment-emergent akathisia, making this the No. 1 reason for discontinuation because of adverse events, followed by depression at 2%. Only 0.7% of subjects dropped out because of extrapyramidal symptoms, the same small number as quit because of worsening mania.
No clinically meaningful changes occurred in lipids, liver enzymes, ECG parameters, or prolactin levels. Systolic and diastolic blood pressure each rose modestly by less than 2 mm Hg. Weight gain was minimal: a mean increase of 0.9 kg over 16 weeks, with an accompanying mean 1.0-cm increase in waist circumference.
While this was primarily a phase III safety and tolerability study, efficacy was also measured. At week 16, 63% of subjects met study criteria for disease remission as defined by a Young Mania Rating Scale (YMRS) total score of 12 or less, starting from a mean baseline YMRS score of 26.1. Most of the symptomatic improvement was attained within the first 3 weeks of treatment, with a mean 13.6-point reduction in the YMRS, modestly improved upon to a mean 15.2-point reduction by week 16.
These schizophrenia and bipolar I disorder studies were sponsored by Forest Pharmaceuticals. Dr. Kane reported serving as a consultant and/or having received honoraria for lectures from Forest and 17 other pharmaceutical companies. Dr. Ketter reported serving as a consultant to Forest and four other pharmaceutical companies as well as receiving lecture honoraria from GlaxoSmithKline and Otsuka.
HOLLYWOOD, FLA. – The novel oral antipsychotic agent cariprazine showed efficacy across the broad spectrum of schizophrenia manifestations – including negative symptoms – in a multicenter phase III clinical trial.
This is welcome news. Current antipsychotic agents are only modestly effective in treating the negative symptoms, mood disturbances, and neurocognitive dysfunction that so often hamper recovery in patients with schizophrenia. In contrast, the investigational agent cariprazine demonstrated efficacy in all these domains, Dr. John M. Kane reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Cariprazine is a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. As such, it would be expected to have broad efficacy across the spectrum of schizophrenia symptoms. This has been borne out, first in animal studies and now in this phase III randomized trial, noted Dr. Kane, chairman of psychiatry at Zucker Hillside Hospital in Glen Oaks, N.Y., and professor of psychiatry, neurology, and neuroscience at the Albert Einstein College of Medicine in New York.
Forest Pharmaceuticals filed an application with the Food and Drug Administration last fall for marketing approval of cariprazine in the treatment of schizophrenia as well as for treatment of bipolar mania. The company expects a decision from the regulatory agency before year’s end.
Dr. Kane reported on 446 patients with acute exacerbation of schizophrenia placed on 6 weeks of double-blind placebo or cariprazine at either 3-6 mg/day or 6-9 mg/day. Sixty-one percent of subjects completed the study. The mean daily dosage of cariprazine in the lower-dose study arm was 4.2 mg/day, versus 6.6 mg/day in the higher-dose arm.
The primary study endpoint was improvement in Positive and Negative Syndrome Scale (PANSS) total score. From a baseline mean PANSS score of 96, indicative of marked illness, patients in the lower-dose cariprazine group showed a placebo-subtracted net 6.8-point improvement. Those on higher-dose therapy had a net 9.9-point improvement. Both dosing strategies were significantly more effective than was placebo. Patients on cariprazine at the more effective dosing regimen of 6-9 mg/day showed a significant advantage over placebo starting at week 1.
To more closely examine the drug’s efficacy in domains other than positive symptoms such as delusions and hallucinations, Dr. Kane and his coinvestigators conducted a post hoc analysis focusing on cariprazine’s impact on each of the five PANSS-derived Marder symptom factor groups. These five groupings are negative symptoms, positive symptoms, uncontrolled hostility/excitement, disorganized thought, and anxiety/depression.
Cariprazine at 6-9 mg/day proved superior to placebo in all five Marder factor groupings. Cariprazine at 3-6 mg/day outperformed placebo in all but the Marder negative symptom factor grouping, where a trend favoring the drug fell short of statistical significance.
Of the 30 individual items that make up the PANSS, higher-dose cariprazine was significantly more effective than was placebo in 21. These included most positive symptom factor items as well as others where conventional antipsychotic agents typically don’t fare very well, such as emotional withdrawal, active social avoidance, poor rapport, anxiety, depression, tension, poor impulse control, hostility, excitement, uncooperativeness, disturbance of volition, and conceptual disorganization.
Cariprazine at 3-6 mg/day outperformed placebo on 11 of the 30 individual PANSS items.
Long-term cariprazine in bipolar I
Also at the NCDEU meeting, Dr. Terence A. Ketter presented a phase III, open-label, 16-week, study of flexibly dosed cariprazine in 402 patients with bipolar I disorder.
Dosing of cariprazine was permitted at 3-12 mg/day. The actual mean dose used in the study was 6.2 mg/day. The primary study endpoints involved safety and tolerability, since the drug already had demonstrated efficacy in three separate 3-week-long studies in acute mania.
This 16-week study was undertaken in recognition that the real challenge in bipolar I disorder is long-term management. Nearly half of all patients who respond to initial conventional therapies relapse within 2 years, mainly because of high rates of nonadherence, explained Dr. Ketter, professor of psychiatry and behavioral sciences and chief of the bipolar disorders clinic at Stanford (Calif.) University.
Just 33% of participants completed the 16-week study. Sixteen percent of subjects withdrew because of adverse events, roughly the same number as for protocol violations.
The major tolerability issue in the phase III trial was treatment-emergent akathisia, which occurred in 37% of participants. In addition, 7% experienced treatment-emergent extrapyramidal symptoms as defined by a Simpson-Angus Scale score of 3 or less at baseline rising to greater than 3 on treatment.
Ninety-eight percent of these cases of treatment-emergent akathisia or extrapyramidal symptoms were rated as mild to moderate in intensity. Five percent of subjects dropped out of the trial because of treatment-emergent akathisia, making this the No. 1 reason for discontinuation because of adverse events, followed by depression at 2%. Only 0.7% of subjects dropped out because of extrapyramidal symptoms, the same small number as quit because of worsening mania.
No clinically meaningful changes occurred in lipids, liver enzymes, ECG parameters, or prolactin levels. Systolic and diastolic blood pressure each rose modestly by less than 2 mm Hg. Weight gain was minimal: a mean increase of 0.9 kg over 16 weeks, with an accompanying mean 1.0-cm increase in waist circumference.
While this was primarily a phase III safety and tolerability study, efficacy was also measured. At week 16, 63% of subjects met study criteria for disease remission as defined by a Young Mania Rating Scale (YMRS) total score of 12 or less, starting from a mean baseline YMRS score of 26.1. Most of the symptomatic improvement was attained within the first 3 weeks of treatment, with a mean 13.6-point reduction in the YMRS, modestly improved upon to a mean 15.2-point reduction by week 16.
These schizophrenia and bipolar I disorder studies were sponsored by Forest Pharmaceuticals. Dr. Kane reported serving as a consultant and/or having received honoraria for lectures from Forest and 17 other pharmaceutical companies. Dr. Ketter reported serving as a consultant to Forest and four other pharmaceutical companies as well as receiving lecture honoraria from GlaxoSmithKline and Otsuka.
AT THE NCDEU MEETING
Major finding: Schizophrenia study: Patients in the higher-dose cariprazine group showed a net 9.9-point improvement in Positive and Negative Syndrome Scale total score. Bipolar I study: Among those who took cariprazine, 63% met study criteria for disease remission as defined by a Young Mania Rating Scale total score of 12 or less.
Data source: Multicenter phase III trial of 446 patients with acute exacerbation of schizophrenia and a phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I.
Disclosures: Studies were funded by Forest Pharmaceuticals. Dr. Kane and Dr. Ketter reported serving as consultants to Forest.
