User login
Empagliflozin improves cardiovascular risk factors in T2DM
CHICAGO – Twenty-four weeks of treatment with empagliflozin in patients with type 2 diabetes achieved clinically meaningful improvements in cardiovascular risk factors as well as in glycemic control, according to pooled data from four pivotal phase III clinical trials.
Empagliflozin is a sodium glucose cotransporter 2 (SGLT2) inhibitor. This is a novel emerging class of oral diabetic medications that work by boosting urinary glucose excretion. Canagliflozin (Invokana), the first-in-class SGLT2 inhibitor, received Food and Drug Administration marketing approval in March for treatment of type 2 diabetes. Empagliflozin’s application for marketing approval is now under review by the FDA and the European Medicines Agency, Dr. Thomas Hach noted at the annual scientific sessions of the American Diabetes Association.
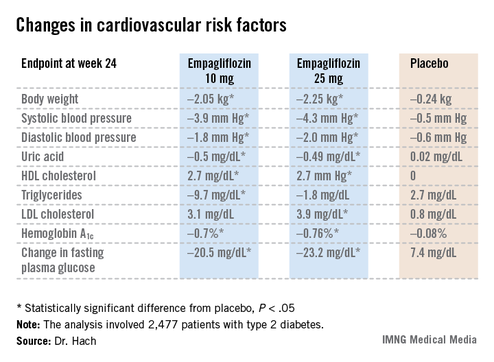

Empagliflozin resulted in significant, clinically meaningful improvements in blood pressure, body weight, uric acid, HDL cholesterol, hemoglobin A1c, and fasting plasma glucose, along with a modest, albeit unwelcome, increase in LDL cholesterol.
The improvement in blood pressure was particularly noteworthy. Among patients with uncontrolled blood pressure at baseline – that is, blood pressures of 130/80 mm Hg or higher – 33.3% of those who received empagliflozin at 10 mg/day and 32.2% on empagliflozin at 25 mg/day had controlled blood pressure at week 24. That was true of only 18.6% of placebo-treated controls, according to Dr. Hach of Boehringer Ingelheim Pharma in Ingelheim, Germany.
The impact of empagliflozin on cardiovascular events in patients with type 2 diabetes is under study in the ongoing EMPA-REG OUTCOME cardiovascular outcomes trial.
Empagliflozin is being jointly developed by Boehringer Ingelheim and Eli Lilly.
*Correction, 7/10/2013: An earlier version of the graphic accompanying this story incorrectly reported the change in uric acid.
CHICAGO – Twenty-four weeks of treatment with empagliflozin in patients with type 2 diabetes achieved clinically meaningful improvements in cardiovascular risk factors as well as in glycemic control, according to pooled data from four pivotal phase III clinical trials.
Empagliflozin is a sodium glucose cotransporter 2 (SGLT2) inhibitor. This is a novel emerging class of oral diabetic medications that work by boosting urinary glucose excretion. Canagliflozin (Invokana), the first-in-class SGLT2 inhibitor, received Food and Drug Administration marketing approval in March for treatment of type 2 diabetes. Empagliflozin’s application for marketing approval is now under review by the FDA and the European Medicines Agency, Dr. Thomas Hach noted at the annual scientific sessions of the American Diabetes Association.
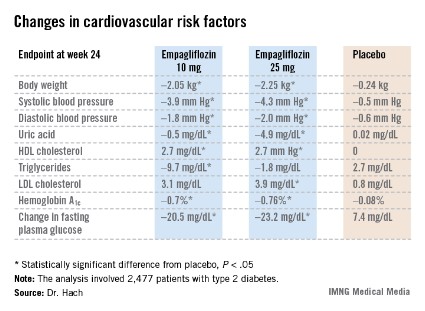
Empagliflozin resulted in significant, clinically meaningful improvements in blood pressure, body weight, uric acid, HDL cholesterol, hemoglobin A1c, and fasting plasma glucose, along with a modest, albeit unwelcome, increase in LDL cholesterol.
The improvement in blood pressure was particularly noteworthy. Among patients with uncontrolled blood pressure at baseline – that is, blood pressures of 130/80 mm Hg or higher – 33.3% of those who received empagliflozin at 10 mg/day and 32.2% on empagliflozin at 25 mg/day had controlled blood pressure at week 24. That was true of only 18.6% of placebo-treated controls, according to Dr. Hach of Boehringer Ingelheim Pharma in Ingelheim, Germany.
The impact of empagliflozin on cardiovascular events in patients with type 2 diabetes is under study in the ongoing EMPA-REG OUTCOME cardiovascular outcomes trial.
Empagliflozin is being jointly developed by Boehringer Ingelheim and Eli Lilly.
*Correction, 7/10/2013: An earlier version of the graphic accompanying this story incorrectly reported the change in uric acid.
CHICAGO – Twenty-four weeks of treatment with empagliflozin in patients with type 2 diabetes achieved clinically meaningful improvements in cardiovascular risk factors as well as in glycemic control, according to pooled data from four pivotal phase III clinical trials.
Empagliflozin is a sodium glucose cotransporter 2 (SGLT2) inhibitor. This is a novel emerging class of oral diabetic medications that work by boosting urinary glucose excretion. Canagliflozin (Invokana), the first-in-class SGLT2 inhibitor, received Food and Drug Administration marketing approval in March for treatment of type 2 diabetes. Empagliflozin’s application for marketing approval is now under review by the FDA and the European Medicines Agency, Dr. Thomas Hach noted at the annual scientific sessions of the American Diabetes Association.
Empagliflozin resulted in significant, clinically meaningful improvements in blood pressure, body weight, uric acid, HDL cholesterol, hemoglobin A1c, and fasting plasma glucose, along with a modest, albeit unwelcome, increase in LDL cholesterol.
The improvement in blood pressure was particularly noteworthy. Among patients with uncontrolled blood pressure at baseline – that is, blood pressures of 130/80 mm Hg or higher – 33.3% of those who received empagliflozin at 10 mg/day and 32.2% on empagliflozin at 25 mg/day had controlled blood pressure at week 24. That was true of only 18.6% of placebo-treated controls, according to Dr. Hach of Boehringer Ingelheim Pharma in Ingelheim, Germany.
The impact of empagliflozin on cardiovascular events in patients with type 2 diabetes is under study in the ongoing EMPA-REG OUTCOME cardiovascular outcomes trial.
Empagliflozin is being jointly developed by Boehringer Ingelheim and Eli Lilly.
*Correction, 7/10/2013: An earlier version of the graphic accompanying this story incorrectly reported the change in uric acid.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major Finding: One-third of type 2 diabetes patients with uncontrolled hypertension at baseline lowered their blood pressure to below 130/80 mm Hg after 24 weeks on empagliflozin.
Data Source: A pooled analysis of data from four randomized, placebo-controlled pivotal phase III clinical trials of empagliflozin in 2,477 patients with type 2 diabetes.
Disclosures: Empagliflozin is being jointly developed by Boehringer Ingelheim and Eli Lilly. Dr. Hach is an employee of Boehringer Ingelheim.
PML risk stratification tool could affect natalizumab usage
ORLANDO – The anti-JC virus antibody index appears to be an important new tool in further delineating the risk of developing progressive multifocal leukoencephalopathy in natalizumab-treated multiple sclerosis patients, according to Dr. Patricia K. Coyle.
Indeed, progressive multifocal leukoencephalopathy (PML) risk stratification could very well transform natalizumab (Tysabri) from a second-line agent for relapsing forms of MS to first-line therapy, she predicted at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.

After all, it’s widely accepted that natalizumab is the most effective of all the Food and Drug Administration–approved treatments for MS. And adherence is a nonissue, since the medication has to be given monthly at an infusion center. Natalizumab is clearly the preferred agent for patients with highly active disease, those with a poor prognosis, and African Americans, because the databases demonstrate they respond so well to it.
"The only thing holding back natalizumab from first-line treatment status is the PML risk. But there’s afoot the possibility of considering natalizumab as a first-line agent by risk-stratifying patients," explained Dr. Coyle, professor of neurology and director of the MS comprehensive care center at Stony Brook (N.Y.) Medical Center.
It’s increasingly clear that an individual’s risk level can be refined on the basis of three factors: anti-JC virus antibody status, natalizumab treatment duration, and a history of prior immunosuppressive therapy.
The JC virus is a ubiquitous DNA polyomavirus shed in the urine of 25% of normal individuals. Up to 70% of the general population is seropositive. It’s ordinarily a benign virus, yet it’s a requirement for developing PML.
An analysis of large clinical trial databases indicates that anti-JC virus antibody–negative MS patients on natalizumab have a reassuringly low risk of developing PML: roughly 1 in 10,000. The good news is that antibody-positive patients with a low antibody index appear to have a similarly low risk.
In an analysis presented at the CMSC/ACTRIMS meeting by Dr. Tatiana Plavina and Dr. Meena Subramanyam of Biogen Idec, Weston, Mass., the investigators estimated the risk of PML in anti-JC virus antibody–positive patients with an antibody index of 0.9 or less at 1 in 10,000 patients during their first 24 months on natalizumab, inching up to 3 per 10,000 with 25-40 months of use, and 4 per 10,000 with 49-72 months of use.
In contrast, the PML risk in anti-JC virus antibody–positive patients with an antibody index above 1.5 was 10 per 10,000 during the first 24 months, 81 per 10,000 with 25-48 months on natalizumab, and 85 per 10,000 with 49-72 months. These risk estimates were based on analysis of close to 6,000 blood samples from 51 natalizumab users who developed PML and 2,242 others who didn’t.
"This needs to be confirmed in other large databases. If this holds up, measuring the antibody index is going to be something we’ll routinely be doing," Dr. Coyle commented.
In fact, she’s already doing it in her own practice. She now checks the anti-JC virus antibody status of her antibody-negative patients on natalizumab every 3 months.
"The goal is to identify PML at an early asymptomatic stage," she noted.
If a patient turns positive, she obtains the antibody index and uses it to guide patient management. The antibody index reflects antibody titers but is a more reliable measure. The antibody index is easily measured using a commercially available kit. Typically, the index appears to persist at the same level – low or high risk – for an extended period.
An important caveat about the anti-JC virus antibody index as a risk stratification tool is that it doesn’t work if a patient has had prior immunosuppressive therapy of any type, even if only for a relatively brief time.
"All bets are off if you have prior immunosuppression. You can’t use the risk figures. This speaks to the importance of carefully staging your therapies," according to the neurologist.
Although the risk of PML appears to rise after about 2 years on natalizumab, in her view there’s nothing magic about that time line. In an anti-JC virus antibody–positive patient with a low antibody index who wants to continue on natalizumab after being fully informed of the risks as understood today, Dr. Coyle is willing to do so. Under those circumstances she suggests backing off from infusions every 4 weeks to a 6- to 8-week schedule, with MRIs every 4 months instead of 6.
"There are no data to show backing off in this way reduces PML risk, but I think it’s rational," she explained.
Natalizumab is such an effective agent that if a patient with MS experiences breakthrough disease activity on the drug, it’s time to check for the possible presence of neutralizing antibodies to natalizumab.
"We have very good data that if somebody has formed persistent neutralizing antibodies, the drug is not working. And there’s no point in putting a patient at any extra risk for a drug that is not working," Dr. Coyle said.
A significant infusion reaction is another indicator that neutralizing antibodies may be present, she added.
What does the future hold for natalizumab?
Dr. Coyle predicted that in the short term the drug will see increasing use as physicians become comfortable with risk stratification as a means of identifying the portion of the MS population at low risk for PML. But she forecast that several years from now this highly effective medication will have fallen by the wayside.
"I think the era of natalizumab is ending," she said. "In my opinion it’s going to be supplanted by anti-CD20 agents in the next couple of years – drugs with equivalent efficacy and greater safety."
She reported having received honoraria from Biogen Idec and eight other pharmaceutical companies.
ORLANDO – The anti-JC virus antibody index appears to be an important new tool in further delineating the risk of developing progressive multifocal leukoencephalopathy in natalizumab-treated multiple sclerosis patients, according to Dr. Patricia K. Coyle.
Indeed, progressive multifocal leukoencephalopathy (PML) risk stratification could very well transform natalizumab (Tysabri) from a second-line agent for relapsing forms of MS to first-line therapy, she predicted at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
After all, it’s widely accepted that natalizumab is the most effective of all the Food and Drug Administration–approved treatments for MS. And adherence is a nonissue, since the medication has to be given monthly at an infusion center. Natalizumab is clearly the preferred agent for patients with highly active disease, those with a poor prognosis, and African Americans, because the databases demonstrate they respond so well to it.
"The only thing holding back natalizumab from first-line treatment status is the PML risk. But there’s afoot the possibility of considering natalizumab as a first-line agent by risk-stratifying patients," explained Dr. Coyle, professor of neurology and director of the MS comprehensive care center at Stony Brook (N.Y.) Medical Center.
It’s increasingly clear that an individual’s risk level can be refined on the basis of three factors: anti-JC virus antibody status, natalizumab treatment duration, and a history of prior immunosuppressive therapy.
The JC virus is a ubiquitous DNA polyomavirus shed in the urine of 25% of normal individuals. Up to 70% of the general population is seropositive. It’s ordinarily a benign virus, yet it’s a requirement for developing PML.
An analysis of large clinical trial databases indicates that anti-JC virus antibody–negative MS patients on natalizumab have a reassuringly low risk of developing PML: roughly 1 in 10,000. The good news is that antibody-positive patients with a low antibody index appear to have a similarly low risk.
In an analysis presented at the CMSC/ACTRIMS meeting by Dr. Tatiana Plavina and Dr. Meena Subramanyam of Biogen Idec, Weston, Mass., the investigators estimated the risk of PML in anti-JC virus antibody–positive patients with an antibody index of 0.9 or less at 1 in 10,000 patients during their first 24 months on natalizumab, inching up to 3 per 10,000 with 25-40 months of use, and 4 per 10,000 with 49-72 months of use.
In contrast, the PML risk in anti-JC virus antibody–positive patients with an antibody index above 1.5 was 10 per 10,000 during the first 24 months, 81 per 10,000 with 25-48 months on natalizumab, and 85 per 10,000 with 49-72 months. These risk estimates were based on analysis of close to 6,000 blood samples from 51 natalizumab users who developed PML and 2,242 others who didn’t.
"This needs to be confirmed in other large databases. If this holds up, measuring the antibody index is going to be something we’ll routinely be doing," Dr. Coyle commented.
In fact, she’s already doing it in her own practice. She now checks the anti-JC virus antibody status of her antibody-negative patients on natalizumab every 3 months.
"The goal is to identify PML at an early asymptomatic stage," she noted.
If a patient turns positive, she obtains the antibody index and uses it to guide patient management. The antibody index reflects antibody titers but is a more reliable measure. The antibody index is easily measured using a commercially available kit. Typically, the index appears to persist at the same level – low or high risk – for an extended period.
An important caveat about the anti-JC virus antibody index as a risk stratification tool is that it doesn’t work if a patient has had prior immunosuppressive therapy of any type, even if only for a relatively brief time.
"All bets are off if you have prior immunosuppression. You can’t use the risk figures. This speaks to the importance of carefully staging your therapies," according to the neurologist.
Although the risk of PML appears to rise after about 2 years on natalizumab, in her view there’s nothing magic about that time line. In an anti-JC virus antibody–positive patient with a low antibody index who wants to continue on natalizumab after being fully informed of the risks as understood today, Dr. Coyle is willing to do so. Under those circumstances she suggests backing off from infusions every 4 weeks to a 6- to 8-week schedule, with MRIs every 4 months instead of 6.
"There are no data to show backing off in this way reduces PML risk, but I think it’s rational," she explained.
Natalizumab is such an effective agent that if a patient with MS experiences breakthrough disease activity on the drug, it’s time to check for the possible presence of neutralizing antibodies to natalizumab.
"We have very good data that if somebody has formed persistent neutralizing antibodies, the drug is not working. And there’s no point in putting a patient at any extra risk for a drug that is not working," Dr. Coyle said.
A significant infusion reaction is another indicator that neutralizing antibodies may be present, she added.
What does the future hold for natalizumab?
Dr. Coyle predicted that in the short term the drug will see increasing use as physicians become comfortable with risk stratification as a means of identifying the portion of the MS population at low risk for PML. But she forecast that several years from now this highly effective medication will have fallen by the wayside.
"I think the era of natalizumab is ending," she said. "In my opinion it’s going to be supplanted by anti-CD20 agents in the next couple of years – drugs with equivalent efficacy and greater safety."
She reported having received honoraria from Biogen Idec and eight other pharmaceutical companies.
ORLANDO – The anti-JC virus antibody index appears to be an important new tool in further delineating the risk of developing progressive multifocal leukoencephalopathy in natalizumab-treated multiple sclerosis patients, according to Dr. Patricia K. Coyle.
Indeed, progressive multifocal leukoencephalopathy (PML) risk stratification could very well transform natalizumab (Tysabri) from a second-line agent for relapsing forms of MS to first-line therapy, she predicted at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
After all, it’s widely accepted that natalizumab is the most effective of all the Food and Drug Administration–approved treatments for MS. And adherence is a nonissue, since the medication has to be given monthly at an infusion center. Natalizumab is clearly the preferred agent for patients with highly active disease, those with a poor prognosis, and African Americans, because the databases demonstrate they respond so well to it.
"The only thing holding back natalizumab from first-line treatment status is the PML risk. But there’s afoot the possibility of considering natalizumab as a first-line agent by risk-stratifying patients," explained Dr. Coyle, professor of neurology and director of the MS comprehensive care center at Stony Brook (N.Y.) Medical Center.
It’s increasingly clear that an individual’s risk level can be refined on the basis of three factors: anti-JC virus antibody status, natalizumab treatment duration, and a history of prior immunosuppressive therapy.
The JC virus is a ubiquitous DNA polyomavirus shed in the urine of 25% of normal individuals. Up to 70% of the general population is seropositive. It’s ordinarily a benign virus, yet it’s a requirement for developing PML.
An analysis of large clinical trial databases indicates that anti-JC virus antibody–negative MS patients on natalizumab have a reassuringly low risk of developing PML: roughly 1 in 10,000. The good news is that antibody-positive patients with a low antibody index appear to have a similarly low risk.
In an analysis presented at the CMSC/ACTRIMS meeting by Dr. Tatiana Plavina and Dr. Meena Subramanyam of Biogen Idec, Weston, Mass., the investigators estimated the risk of PML in anti-JC virus antibody–positive patients with an antibody index of 0.9 or less at 1 in 10,000 patients during their first 24 months on natalizumab, inching up to 3 per 10,000 with 25-40 months of use, and 4 per 10,000 with 49-72 months of use.
In contrast, the PML risk in anti-JC virus antibody–positive patients with an antibody index above 1.5 was 10 per 10,000 during the first 24 months, 81 per 10,000 with 25-48 months on natalizumab, and 85 per 10,000 with 49-72 months. These risk estimates were based on analysis of close to 6,000 blood samples from 51 natalizumab users who developed PML and 2,242 others who didn’t.
"This needs to be confirmed in other large databases. If this holds up, measuring the antibody index is going to be something we’ll routinely be doing," Dr. Coyle commented.
In fact, she’s already doing it in her own practice. She now checks the anti-JC virus antibody status of her antibody-negative patients on natalizumab every 3 months.
"The goal is to identify PML at an early asymptomatic stage," she noted.
If a patient turns positive, she obtains the antibody index and uses it to guide patient management. The antibody index reflects antibody titers but is a more reliable measure. The antibody index is easily measured using a commercially available kit. Typically, the index appears to persist at the same level – low or high risk – for an extended period.
An important caveat about the anti-JC virus antibody index as a risk stratification tool is that it doesn’t work if a patient has had prior immunosuppressive therapy of any type, even if only for a relatively brief time.
"All bets are off if you have prior immunosuppression. You can’t use the risk figures. This speaks to the importance of carefully staging your therapies," according to the neurologist.
Although the risk of PML appears to rise after about 2 years on natalizumab, in her view there’s nothing magic about that time line. In an anti-JC virus antibody–positive patient with a low antibody index who wants to continue on natalizumab after being fully informed of the risks as understood today, Dr. Coyle is willing to do so. Under those circumstances she suggests backing off from infusions every 4 weeks to a 6- to 8-week schedule, with MRIs every 4 months instead of 6.
"There are no data to show backing off in this way reduces PML risk, but I think it’s rational," she explained.
Natalizumab is such an effective agent that if a patient with MS experiences breakthrough disease activity on the drug, it’s time to check for the possible presence of neutralizing antibodies to natalizumab.
"We have very good data that if somebody has formed persistent neutralizing antibodies, the drug is not working. And there’s no point in putting a patient at any extra risk for a drug that is not working," Dr. Coyle said.
A significant infusion reaction is another indicator that neutralizing antibodies may be present, she added.
What does the future hold for natalizumab?
Dr. Coyle predicted that in the short term the drug will see increasing use as physicians become comfortable with risk stratification as a means of identifying the portion of the MS population at low risk for PML. But she forecast that several years from now this highly effective medication will have fallen by the wayside.
"I think the era of natalizumab is ending," she said. "In my opinion it’s going to be supplanted by anti-CD20 agents in the next couple of years – drugs with equivalent efficacy and greater safety."
She reported having received honoraria from Biogen Idec and eight other pharmaceutical companies.
EXPERT ANALYSIS FROM THE CMSC/ACTRIMS ANNUAL MEETING

Parenteral drugs for MS: What's ahead
ORLANDO – In the expanding matrix of disease-modifying therapeutic options for multiple sclerosis, some "old companions" among the parenteral agents are likely headed by the wayside, Dr. Patricia K. Coyle predicted at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Her plenary address, which highlighted parenteral agents now in the developmental pipeline, also touched on what’s likely to become of the current parenterals.

Here’s her expert analysis:
Interferon-betas and glatiramer acetate: These old standbys are Food and Drug Administration–approved for relapsing forms of MS and for clinically isolated syndrome. Their advantages: they’re fairly well tolerated and their safety profile, thoroughly established over the past 2 decades, holds absolutely no surprises. The big downside is that they are needle-injectable agents.
"I don’t think the market for interferon-betas and glatiramer acetate (Copaxone) is going to disappear, but I think it’s going to slowly shrink because now we have oral options. The biggest impact is going to be on treatment-naive, newly diagnosed patients. People are going to opt for an oral agent rather than an injectable," predicted Dr. Coyle, professor of neurology, vice chair for clinical affairs, and director of the MS Comprehensive Care Center at Stony Brook (N.Y.) Medical Center.
"Also, it’s beginning to become a very crowded market among the interferon-betas. It’s likely you’re going to see one interferon-beta emerge as the dominant option in a shrinking market," she continued.
Natalizumab (Tysabri): This highly effective agent has been dogged by its associated risk of progressive multifocal leukoencephalopathy (PML), but could actually see increased use over the next several years as a consequence of emerging risk-stratification methods that identify a subset of MS patients at low PML risk on natalizumab. But once the novel anti-CD20 monoclonal antibodies reach the marketplace (see ocrelizumab and daclizumab, below), the natalizumab era will be over, in Dr. Coyle’s view.
Mitoxantrone: A true induction agent with sustained efficacy lasting after treatment is stopped. This drug is FDA-approved for all forms of MS except for primary progressive MS. But the associated cardiotoxicity and leukemia risks dictate lifetime monitoring for a drug that can only be given 10 or 11 times. That’s a deal breaker.
"As far as I can tell, mitoxantrone is not being used for MS at all in the U.S. any longer," according to the neurologist.
Rituximab (Rituxan): This IgG1-anti-CD20 chimeric monoclonal antibody rapidly depletes B cells for 4-12 months. Eventually, naive B cells return preferentially over memory B cells. Rituximab is already marketed for non-Hodgkin lymphoma and refractory rheumatoid arthritis. Its manufacturer doesn’t intend to seek an indication for MS; however, earlier highly promising phase II MS studies (N. Engl. J. Med. 2008;358:676-88; Ann. Neurol. 2009;66:460-71) have spurred intensive development of novel anti-CD20 monoclonal antibodies targeting this disease.
Off label, some neurologists are using 1,000 mg of rituximab intravenously followed 15 days later by 500 mg, with dosing repeated every 6 months, she said.
Turning to the parenteral agents of the future, Dr. Coyle said the first up is likely to be:
Alemtuzumab: This agent, which knocks out T cells, is now under FDA review for possible marketing approval based upon impressive phase III results.
Alemtuzumab is a true induction agent. It is given on 8 days over the course of 2 years, and never again. The effects appear to last for several years after the last dose. It is not, however, a cure for MS, Dr. Coyle stressed.
Pegylated interferon beta-1a: The first-year results of the ongoing 2-year phase III ADVANCE trial showed self-administered subcutaneous dosing every 2 weeks to be more effective than every 4 weeks. Will this become the dominant interferon-beta in the marketplace? Way too early to tell, in Dr. Coyle’s view. She also noted the possibility that generic interferon-betas may become available in the United States.
Glatiramer acetate 40 mg: This drug is given subcutaneously three times per week, rather than daily as with the familiar 20-mg formulation. In the GALA (Glatiramer Acetate Low Frequency Administration Study) trial, glatiramer acetate 40 mg had an annualized relapse rate 34% lower than placebo, as well as a 34% reduction in new or relapsing T2 lesions on brain MRI.
Ocrelizumab: The furthest along in development of the novel anti-CD20 agents, this humanized monoclonal antibody is currently in three phase III clinical trials: two for relapsing MS and another for primary progressive MS.
In a phase II study, various doses of ocrelizumab showed 73%-80% reductions in relapses and 89%-96% suppression of MRI contrast lesions.
"The data with ocrelizumab looks great. I think, personally, it could potentially replace natalizumab, depending upon the safety," Dr. Coyle said.
Development of ocrelizumab as a treatment for rheumatoid arthritis and lupus was discontinued because of concerns about an increase in opportunistic infections.
"We’ve not really seen that in the MS cohort so far. It’s a slight background concern. So far, ocrelizumab seems pretty safe," according to the neurologist.
Daclizumab (Zenapax): This subcutaneously administered agent is a humanized IgG1 monoclonal antibody against CD25 and is already on the market for the prevention of organ transplant rejection. It is in the ongoing DECIDE study, a phase III MS clinical trial with intramuscular interferon beta-1a as the comparator. Earlier studies raised some concerns regarding safety and tolerability.
A particularly interesting feature of daclizumab is that it appears to have a biomarker predictive of efficacy: an increase in CD56 bright natural killer cells. Biomarkers of efficacy are desperately needed for MS therapies.
Miscellaneous parenterals: These include other anti-CD20 monoclonal antibodies, among them ofatumumab (Arzerra), already marketed for treatment of refractory chronic lymphocytic leukemia; secukinumab, an anti-interleukin-17 monoclonal antibody that showed efficacy in terms of MRI lesions in a small study; an anti-LINGO-1 monoclonal antibody aimed at stimulating myelin repair; and stem cell therapies that are early in development.
Dr. Coyle predicted more efficacy biomarkers are coming, and noted that neuroprotection and CNS restoration are areas wide open for drug development. Her final prediction regarding parenteral therapies for MS: "A high-efficacy monoclonal antibody, if it’s safe and convenient, could seize the market. The safety will be the critical issue."
She reported having received honoraria from Acorda, Accordant, Bayer, Biogen Idec, EMD Serono, Genzyme/Sanofi Aventis, Novartis, Roche, and Tera.
ORLANDO – In the expanding matrix of disease-modifying therapeutic options for multiple sclerosis, some "old companions" among the parenteral agents are likely headed by the wayside, Dr. Patricia K. Coyle predicted at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Her plenary address, which highlighted parenteral agents now in the developmental pipeline, also touched on what’s likely to become of the current parenterals.
Here’s her expert analysis:
Interferon-betas and glatiramer acetate: These old standbys are Food and Drug Administration–approved for relapsing forms of MS and for clinically isolated syndrome. Their advantages: they’re fairly well tolerated and their safety profile, thoroughly established over the past 2 decades, holds absolutely no surprises. The big downside is that they are needle-injectable agents.
"I don’t think the market for interferon-betas and glatiramer acetate (Copaxone) is going to disappear, but I think it’s going to slowly shrink because now we have oral options. The biggest impact is going to be on treatment-naive, newly diagnosed patients. People are going to opt for an oral agent rather than an injectable," predicted Dr. Coyle, professor of neurology, vice chair for clinical affairs, and director of the MS Comprehensive Care Center at Stony Brook (N.Y.) Medical Center.
"Also, it’s beginning to become a very crowded market among the interferon-betas. It’s likely you’re going to see one interferon-beta emerge as the dominant option in a shrinking market," she continued.
Natalizumab (Tysabri): This highly effective agent has been dogged by its associated risk of progressive multifocal leukoencephalopathy (PML), but could actually see increased use over the next several years as a consequence of emerging risk-stratification methods that identify a subset of MS patients at low PML risk on natalizumab. But once the novel anti-CD20 monoclonal antibodies reach the marketplace (see ocrelizumab and daclizumab, below), the natalizumab era will be over, in Dr. Coyle’s view.
Mitoxantrone: A true induction agent with sustained efficacy lasting after treatment is stopped. This drug is FDA-approved for all forms of MS except for primary progressive MS. But the associated cardiotoxicity and leukemia risks dictate lifetime monitoring for a drug that can only be given 10 or 11 times. That’s a deal breaker.
"As far as I can tell, mitoxantrone is not being used for MS at all in the U.S. any longer," according to the neurologist.
Rituximab (Rituxan): This IgG1-anti-CD20 chimeric monoclonal antibody rapidly depletes B cells for 4-12 months. Eventually, naive B cells return preferentially over memory B cells. Rituximab is already marketed for non-Hodgkin lymphoma and refractory rheumatoid arthritis. Its manufacturer doesn’t intend to seek an indication for MS; however, earlier highly promising phase II MS studies (N. Engl. J. Med. 2008;358:676-88; Ann. Neurol. 2009;66:460-71) have spurred intensive development of novel anti-CD20 monoclonal antibodies targeting this disease.
Off label, some neurologists are using 1,000 mg of rituximab intravenously followed 15 days later by 500 mg, with dosing repeated every 6 months, she said.
Turning to the parenteral agents of the future, Dr. Coyle said the first up is likely to be:
Alemtuzumab: This agent, which knocks out T cells, is now under FDA review for possible marketing approval based upon impressive phase III results.
Alemtuzumab is a true induction agent. It is given on 8 days over the course of 2 years, and never again. The effects appear to last for several years after the last dose. It is not, however, a cure for MS, Dr. Coyle stressed.
Pegylated interferon beta-1a: The first-year results of the ongoing 2-year phase III ADVANCE trial showed self-administered subcutaneous dosing every 2 weeks to be more effective than every 4 weeks. Will this become the dominant interferon-beta in the marketplace? Way too early to tell, in Dr. Coyle’s view. She also noted the possibility that generic interferon-betas may become available in the United States.
Glatiramer acetate 40 mg: This drug is given subcutaneously three times per week, rather than daily as with the familiar 20-mg formulation. In the GALA (Glatiramer Acetate Low Frequency Administration Study) trial, glatiramer acetate 40 mg had an annualized relapse rate 34% lower than placebo, as well as a 34% reduction in new or relapsing T2 lesions on brain MRI.
Ocrelizumab: The furthest along in development of the novel anti-CD20 agents, this humanized monoclonal antibody is currently in three phase III clinical trials: two for relapsing MS and another for primary progressive MS.
In a phase II study, various doses of ocrelizumab showed 73%-80% reductions in relapses and 89%-96% suppression of MRI contrast lesions.
"The data with ocrelizumab looks great. I think, personally, it could potentially replace natalizumab, depending upon the safety," Dr. Coyle said.
Development of ocrelizumab as a treatment for rheumatoid arthritis and lupus was discontinued because of concerns about an increase in opportunistic infections.
"We’ve not really seen that in the MS cohort so far. It’s a slight background concern. So far, ocrelizumab seems pretty safe," according to the neurologist.
Daclizumab (Zenapax): This subcutaneously administered agent is a humanized IgG1 monoclonal antibody against CD25 and is already on the market for the prevention of organ transplant rejection. It is in the ongoing DECIDE study, a phase III MS clinical trial with intramuscular interferon beta-1a as the comparator. Earlier studies raised some concerns regarding safety and tolerability.
A particularly interesting feature of daclizumab is that it appears to have a biomarker predictive of efficacy: an increase in CD56 bright natural killer cells. Biomarkers of efficacy are desperately needed for MS therapies.
Miscellaneous parenterals: These include other anti-CD20 monoclonal antibodies, among them ofatumumab (Arzerra), already marketed for treatment of refractory chronic lymphocytic leukemia; secukinumab, an anti-interleukin-17 monoclonal antibody that showed efficacy in terms of MRI lesions in a small study; an anti-LINGO-1 monoclonal antibody aimed at stimulating myelin repair; and stem cell therapies that are early in development.
Dr. Coyle predicted more efficacy biomarkers are coming, and noted that neuroprotection and CNS restoration are areas wide open for drug development. Her final prediction regarding parenteral therapies for MS: "A high-efficacy monoclonal antibody, if it’s safe and convenient, could seize the market. The safety will be the critical issue."
She reported having received honoraria from Acorda, Accordant, Bayer, Biogen Idec, EMD Serono, Genzyme/Sanofi Aventis, Novartis, Roche, and Tera.
ORLANDO – In the expanding matrix of disease-modifying therapeutic options for multiple sclerosis, some "old companions" among the parenteral agents are likely headed by the wayside, Dr. Patricia K. Coyle predicted at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Her plenary address, which highlighted parenteral agents now in the developmental pipeline, also touched on what’s likely to become of the current parenterals.
Here’s her expert analysis:
Interferon-betas and glatiramer acetate: These old standbys are Food and Drug Administration–approved for relapsing forms of MS and for clinically isolated syndrome. Their advantages: they’re fairly well tolerated and their safety profile, thoroughly established over the past 2 decades, holds absolutely no surprises. The big downside is that they are needle-injectable agents.
"I don’t think the market for interferon-betas and glatiramer acetate (Copaxone) is going to disappear, but I think it’s going to slowly shrink because now we have oral options. The biggest impact is going to be on treatment-naive, newly diagnosed patients. People are going to opt for an oral agent rather than an injectable," predicted Dr. Coyle, professor of neurology, vice chair for clinical affairs, and director of the MS Comprehensive Care Center at Stony Brook (N.Y.) Medical Center.
"Also, it’s beginning to become a very crowded market among the interferon-betas. It’s likely you’re going to see one interferon-beta emerge as the dominant option in a shrinking market," she continued.
Natalizumab (Tysabri): This highly effective agent has been dogged by its associated risk of progressive multifocal leukoencephalopathy (PML), but could actually see increased use over the next several years as a consequence of emerging risk-stratification methods that identify a subset of MS patients at low PML risk on natalizumab. But once the novel anti-CD20 monoclonal antibodies reach the marketplace (see ocrelizumab and daclizumab, below), the natalizumab era will be over, in Dr. Coyle’s view.
Mitoxantrone: A true induction agent with sustained efficacy lasting after treatment is stopped. This drug is FDA-approved for all forms of MS except for primary progressive MS. But the associated cardiotoxicity and leukemia risks dictate lifetime monitoring for a drug that can only be given 10 or 11 times. That’s a deal breaker.
"As far as I can tell, mitoxantrone is not being used for MS at all in the U.S. any longer," according to the neurologist.
Rituximab (Rituxan): This IgG1-anti-CD20 chimeric monoclonal antibody rapidly depletes B cells for 4-12 months. Eventually, naive B cells return preferentially over memory B cells. Rituximab is already marketed for non-Hodgkin lymphoma and refractory rheumatoid arthritis. Its manufacturer doesn’t intend to seek an indication for MS; however, earlier highly promising phase II MS studies (N. Engl. J. Med. 2008;358:676-88; Ann. Neurol. 2009;66:460-71) have spurred intensive development of novel anti-CD20 monoclonal antibodies targeting this disease.
Off label, some neurologists are using 1,000 mg of rituximab intravenously followed 15 days later by 500 mg, with dosing repeated every 6 months, she said.
Turning to the parenteral agents of the future, Dr. Coyle said the first up is likely to be:
Alemtuzumab: This agent, which knocks out T cells, is now under FDA review for possible marketing approval based upon impressive phase III results.
Alemtuzumab is a true induction agent. It is given on 8 days over the course of 2 years, and never again. The effects appear to last for several years after the last dose. It is not, however, a cure for MS, Dr. Coyle stressed.
Pegylated interferon beta-1a: The first-year results of the ongoing 2-year phase III ADVANCE trial showed self-administered subcutaneous dosing every 2 weeks to be more effective than every 4 weeks. Will this become the dominant interferon-beta in the marketplace? Way too early to tell, in Dr. Coyle’s view. She also noted the possibility that generic interferon-betas may become available in the United States.
Glatiramer acetate 40 mg: This drug is given subcutaneously three times per week, rather than daily as with the familiar 20-mg formulation. In the GALA (Glatiramer Acetate Low Frequency Administration Study) trial, glatiramer acetate 40 mg had an annualized relapse rate 34% lower than placebo, as well as a 34% reduction in new or relapsing T2 lesions on brain MRI.
Ocrelizumab: The furthest along in development of the novel anti-CD20 agents, this humanized monoclonal antibody is currently in three phase III clinical trials: two for relapsing MS and another for primary progressive MS.
In a phase II study, various doses of ocrelizumab showed 73%-80% reductions in relapses and 89%-96% suppression of MRI contrast lesions.
"The data with ocrelizumab looks great. I think, personally, it could potentially replace natalizumab, depending upon the safety," Dr. Coyle said.
Development of ocrelizumab as a treatment for rheumatoid arthritis and lupus was discontinued because of concerns about an increase in opportunistic infections.
"We’ve not really seen that in the MS cohort so far. It’s a slight background concern. So far, ocrelizumab seems pretty safe," according to the neurologist.
Daclizumab (Zenapax): This subcutaneously administered agent is a humanized IgG1 monoclonal antibody against CD25 and is already on the market for the prevention of organ transplant rejection. It is in the ongoing DECIDE study, a phase III MS clinical trial with intramuscular interferon beta-1a as the comparator. Earlier studies raised some concerns regarding safety and tolerability.
A particularly interesting feature of daclizumab is that it appears to have a biomarker predictive of efficacy: an increase in CD56 bright natural killer cells. Biomarkers of efficacy are desperately needed for MS therapies.
Miscellaneous parenterals: These include other anti-CD20 monoclonal antibodies, among them ofatumumab (Arzerra), already marketed for treatment of refractory chronic lymphocytic leukemia; secukinumab, an anti-interleukin-17 monoclonal antibody that showed efficacy in terms of MRI lesions in a small study; an anti-LINGO-1 monoclonal antibody aimed at stimulating myelin repair; and stem cell therapies that are early in development.
Dr. Coyle predicted more efficacy biomarkers are coming, and noted that neuroprotection and CNS restoration are areas wide open for drug development. Her final prediction regarding parenteral therapies for MS: "A high-efficacy monoclonal antibody, if it’s safe and convenient, could seize the market. The safety will be the critical issue."
She reported having received honoraria from Acorda, Accordant, Bayer, Biogen Idec, EMD Serono, Genzyme/Sanofi Aventis, Novartis, Roche, and Tera.
EXPERT ANALYSIS FROM THE CMSC/ACTRIMS ANNUAL MEETING

Canagliflozin bests sitagliptin for type 2 DM in comparative trial
CHICAGO – Canagliflozin outperformed sitagliptin for the treatment of patients with type 2 diabetes in the first randomized head-to-head comparison of drugs representing the two different classes of oral antidiabetic medications.
Canagliflozin (Invokana) received approval from the Food and Drug Administration in March as the first in a new class of medications for type 2 diabetes known as sodium glucose co-transporter 2 (SGLT2) inhibitors. SGLT2 is responsible for most glucose reabsorption in the kidney, so its inhibition boosts urinary glucose excretion. Other SGLT2 inhibitors are coming through the developmental pipeline.
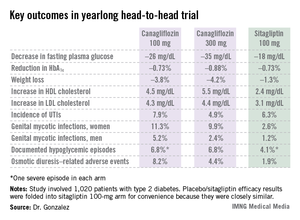
In a phase III, double-blind, 52-week clinical trial, canagliflozin improved glycemic control, lowered body weight, and reduced blood pressure to a significantly greater extent than did sitagliptin (Januvia), a dipeptidyl peptidase-4 (DPP-4) inhibitor, Dr. Fernando Lavalle Gonzalez reported at the annual scientific sessions of the American Diabetes Association.
The multicenter four-armed study included 1,020 type 2 patients with diabetes who had inadequate glycemic control on metformin monotherapy. They were randomized to canagliflozin at 100 or 300 mg/day, sitagliptin at 100 mg/day, or 26 weeks of placebo followed by 26 weeks on sitagliptin at 100 mg/day. All participants continued on metformin.
Reductions in fasting plasma glucose at 52 weeks averaged 26.2 mg/dL in the lower-dose canagliflozin group and 35.2 mg/dL in the higher-dose group, both significantly greater than the 17.7-mg/dL decrease in patients on sitagliptin.
In addition to the better outcomes seen with canagliflozin in terms of the major efficacy endpoints involving glycemic control, weight loss, and blood pressure, the SGLT2 inhibitor resulted in a bigger boost in HDL cholesterol (see chart). On the downside, both canagliflozin and sitagliptin resulted in modest increases in low-density lipoprotein (LDL), noted Dr. Gonzalez, an endocrinologist at the Autonomous University of Nuevo Leon in Monterrey, Mexico.
Canagliflozin was associated with significantly higher rates of genital mycotic infections in both men and women, as well as more adverse events related to osmotic diuresis. The infections were symptomatic, easily diagnosed, and readily treated with topical or oral antifungals. The adverse events related to increased urination were typically mild. These side effects led to very few study discontinuations, according to Dr. Gonzalez.

He said mechanistic studies have identified two factors as being responsible for the clinically meaningful reductions in blood pressure seen with canagliflozin in this study, which amounted to a mean 3.5 mm Hg decrease in systolic blood pressure at 100 mg/day and a 4.7 mm Hg reduction at 300 mg/day. It appears that about half of the blood pressure reduction is due to the negative salt and water balance, on the order of 750-1,000 cc, occurring in the first 3-4 days of treatment, and the other half is related to the weight loss accompanying canagliflozin therapy.
Session chair Dr. Ralph A. DeFronzo, who is involved in SGLT2 inhibitor research, said a widespread misconception exists that the medications are associated with an increased risk of urinary tract infections.
"It’s very commonly stated that this class of drugs is associated with an increase in UTIs [urinary tract infections]. In fact, if you look at the data rather than what’s said, this really in my opinion doesn’t hold up. You can see it in this study, where there’s no significant difference between the groups," said Dr. DeFronzo, professor of medicine and director of the diabetes division at the University of Texas Health Science Center, San Antonio.
He also put into perspective the increase in LDL seen with canagliflozin.
"Looking across the studies, with canagliflozin at the 100-mg dose the rise in LDL is about 4 mg/dL, and it’s about 8 mg/dL at the 300-mg dose. So if you’re truly treating your patients to goal and they’re at an LDL of 70 mg/dL, in the worst case scenario you’d be going from 70 to 78 mg/dL. I think many of us here in the audience probably wouldn’t even increase the dose of a statin, but if you did up it by one dose, you’d get the LDL back down to 70," he said.
Dr. DeFronzo reported serving on advisory panels for Janssen, which markets canagliflozin, as well as for Amylin Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Novo Nordisk, and Takeda. Dr. Gonzalez is on speakers bureaus and advisory panels for Janssen and numerous other companies.
Invokana, Food and Drug Administration, FDA approval, SGLT2,
CHICAGO – Canagliflozin outperformed sitagliptin for the treatment of patients with type 2 diabetes in the first randomized head-to-head comparison of drugs representing the two different classes of oral antidiabetic medications.
Canagliflozin (Invokana) received approval from the Food and Drug Administration in March as the first in a new class of medications for type 2 diabetes known as sodium glucose co-transporter 2 (SGLT2) inhibitors. SGLT2 is responsible for most glucose reabsorption in the kidney, so its inhibition boosts urinary glucose excretion. Other SGLT2 inhibitors are coming through the developmental pipeline.
In a phase III, double-blind, 52-week clinical trial, canagliflozin improved glycemic control, lowered body weight, and reduced blood pressure to a significantly greater extent than did sitagliptin (Januvia), a dipeptidyl peptidase-4 (DPP-4) inhibitor, Dr. Fernando Lavalle Gonzalez reported at the annual scientific sessions of the American Diabetes Association.
The multicenter four-armed study included 1,020 type 2 patients with diabetes who had inadequate glycemic control on metformin monotherapy. They were randomized to canagliflozin at 100 or 300 mg/day, sitagliptin at 100 mg/day, or 26 weeks of placebo followed by 26 weeks on sitagliptin at 100 mg/day. All participants continued on metformin.
Reductions in fasting plasma glucose at 52 weeks averaged 26.2 mg/dL in the lower-dose canagliflozin group and 35.2 mg/dL in the higher-dose group, both significantly greater than the 17.7-mg/dL decrease in patients on sitagliptin.
In addition to the better outcomes seen with canagliflozin in terms of the major efficacy endpoints involving glycemic control, weight loss, and blood pressure, the SGLT2 inhibitor resulted in a bigger boost in HDL cholesterol (see chart). On the downside, both canagliflozin and sitagliptin resulted in modest increases in low-density lipoprotein (LDL), noted Dr. Gonzalez, an endocrinologist at the Autonomous University of Nuevo Leon in Monterrey, Mexico.
Canagliflozin was associated with significantly higher rates of genital mycotic infections in both men and women, as well as more adverse events related to osmotic diuresis. The infections were symptomatic, easily diagnosed, and readily treated with topical or oral antifungals. The adverse events related to increased urination were typically mild. These side effects led to very few study discontinuations, according to Dr. Gonzalez.
He said mechanistic studies have identified two factors as being responsible for the clinically meaningful reductions in blood pressure seen with canagliflozin in this study, which amounted to a mean 3.5 mm Hg decrease in systolic blood pressure at 100 mg/day and a 4.7 mm Hg reduction at 300 mg/day. It appears that about half of the blood pressure reduction is due to the negative salt and water balance, on the order of 750-1,000 cc, occurring in the first 3-4 days of treatment, and the other half is related to the weight loss accompanying canagliflozin therapy.
Session chair Dr. Ralph A. DeFronzo, who is involved in SGLT2 inhibitor research, said a widespread misconception exists that the medications are associated with an increased risk of urinary tract infections.
"It’s very commonly stated that this class of drugs is associated with an increase in UTIs [urinary tract infections]. In fact, if you look at the data rather than what’s said, this really in my opinion doesn’t hold up. You can see it in this study, where there’s no significant difference between the groups," said Dr. DeFronzo, professor of medicine and director of the diabetes division at the University of Texas Health Science Center, San Antonio.
He also put into perspective the increase in LDL seen with canagliflozin.
"Looking across the studies, with canagliflozin at the 100-mg dose the rise in LDL is about 4 mg/dL, and it’s about 8 mg/dL at the 300-mg dose. So if you’re truly treating your patients to goal and they’re at an LDL of 70 mg/dL, in the worst case scenario you’d be going from 70 to 78 mg/dL. I think many of us here in the audience probably wouldn’t even increase the dose of a statin, but if you did up it by one dose, you’d get the LDL back down to 70," he said.
Dr. DeFronzo reported serving on advisory panels for Janssen, which markets canagliflozin, as well as for Amylin Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Novo Nordisk, and Takeda. Dr. Gonzalez is on speakers bureaus and advisory panels for Janssen and numerous other companies.
CHICAGO – Canagliflozin outperformed sitagliptin for the treatment of patients with type 2 diabetes in the first randomized head-to-head comparison of drugs representing the two different classes of oral antidiabetic medications.
Canagliflozin (Invokana) received approval from the Food and Drug Administration in March as the first in a new class of medications for type 2 diabetes known as sodium glucose co-transporter 2 (SGLT2) inhibitors. SGLT2 is responsible for most glucose reabsorption in the kidney, so its inhibition boosts urinary glucose excretion. Other SGLT2 inhibitors are coming through the developmental pipeline.
In a phase III, double-blind, 52-week clinical trial, canagliflozin improved glycemic control, lowered body weight, and reduced blood pressure to a significantly greater extent than did sitagliptin (Januvia), a dipeptidyl peptidase-4 (DPP-4) inhibitor, Dr. Fernando Lavalle Gonzalez reported at the annual scientific sessions of the American Diabetes Association.
The multicenter four-armed study included 1,020 type 2 patients with diabetes who had inadequate glycemic control on metformin monotherapy. They were randomized to canagliflozin at 100 or 300 mg/day, sitagliptin at 100 mg/day, or 26 weeks of placebo followed by 26 weeks on sitagliptin at 100 mg/day. All participants continued on metformin.
Reductions in fasting plasma glucose at 52 weeks averaged 26.2 mg/dL in the lower-dose canagliflozin group and 35.2 mg/dL in the higher-dose group, both significantly greater than the 17.7-mg/dL decrease in patients on sitagliptin.
In addition to the better outcomes seen with canagliflozin in terms of the major efficacy endpoints involving glycemic control, weight loss, and blood pressure, the SGLT2 inhibitor resulted in a bigger boost in HDL cholesterol (see chart). On the downside, both canagliflozin and sitagliptin resulted in modest increases in low-density lipoprotein (LDL), noted Dr. Gonzalez, an endocrinologist at the Autonomous University of Nuevo Leon in Monterrey, Mexico.
Canagliflozin was associated with significantly higher rates of genital mycotic infections in both men and women, as well as more adverse events related to osmotic diuresis. The infections were symptomatic, easily diagnosed, and readily treated with topical or oral antifungals. The adverse events related to increased urination were typically mild. These side effects led to very few study discontinuations, according to Dr. Gonzalez.
He said mechanistic studies have identified two factors as being responsible for the clinically meaningful reductions in blood pressure seen with canagliflozin in this study, which amounted to a mean 3.5 mm Hg decrease in systolic blood pressure at 100 mg/day and a 4.7 mm Hg reduction at 300 mg/day. It appears that about half of the blood pressure reduction is due to the negative salt and water balance, on the order of 750-1,000 cc, occurring in the first 3-4 days of treatment, and the other half is related to the weight loss accompanying canagliflozin therapy.
Session chair Dr. Ralph A. DeFronzo, who is involved in SGLT2 inhibitor research, said a widespread misconception exists that the medications are associated with an increased risk of urinary tract infections.
"It’s very commonly stated that this class of drugs is associated with an increase in UTIs [urinary tract infections]. In fact, if you look at the data rather than what’s said, this really in my opinion doesn’t hold up. You can see it in this study, where there’s no significant difference between the groups," said Dr. DeFronzo, professor of medicine and director of the diabetes division at the University of Texas Health Science Center, San Antonio.
He also put into perspective the increase in LDL seen with canagliflozin.
"Looking across the studies, with canagliflozin at the 100-mg dose the rise in LDL is about 4 mg/dL, and it’s about 8 mg/dL at the 300-mg dose. So if you’re truly treating your patients to goal and they’re at an LDL of 70 mg/dL, in the worst case scenario you’d be going from 70 to 78 mg/dL. I think many of us here in the audience probably wouldn’t even increase the dose of a statin, but if you did up it by one dose, you’d get the LDL back down to 70," he said.
Dr. DeFronzo reported serving on advisory panels for Janssen, which markets canagliflozin, as well as for Amylin Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Novo Nordisk, and Takeda. Dr. Gonzalez is on speakers bureaus and advisory panels for Janssen and numerous other companies.
Invokana, Food and Drug Administration, FDA approval, SGLT2,
Invokana, Food and Drug Administration, FDA approval, SGLT2,
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major finding: Patients with type 2 diabetes averaged a 26.2-mg/dL reduction from baseline in fasting plasma glucose after 52 weeks on canagliflozin at 100 mg/day and a 35.2-mg/dL decrease on the sodium glucose transporter 2 inhibitor at 300 mg/day, both superior to the 17.7-mg/dL reduction seen with sitagliptin.
Data source: A 52-week, double-blind, multicenter, phase III randomized trial involving 1,020 patients with type 2 diabetes who had inadequate glycemic control with metformin monotherapy.
Disclosures: Dr. DeFronzo reported serving on advisory panels for Janssen, which markets canagliflozin, as well as for Amylin Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Novo Nordisk, and Takeda. Dr. Gonzalez is on speakers bureaus and advisory panels for Janssen and numerous other companies.
Investigational albiglutide bests sitagliptin in head-to-head trial
CHICAGO – Once-weekly, fixed-dose albiglutide achieved superior glycemic control compared with dose-adjusted sitagliptin in patients with type 2 diabetes who had inadequate glycemic control and varying degrees of renal impairment in a phase III trial.
Albiglutide is an investigational glucagonlike peptide–1 (GLP-1) agonist now under review at both the U.S. Food and Drug Administration and the European Medicines Agency as a potential new treatment for type 2 diabetes. It is a large molecule cleared by the reticuloendothelial system rather than by the kidney, so its metabolism is not affected by the renal dysfunction that is common in patients with type 2 diabetes.
"Albiglutide will be a useful treatment addition for patients with renal impairment and type 2 diabetes. Current treatments for such patients are limited. Several antidiabetic medications are contraindicated in this population, and many others require dose adjustment," Dr. Lawrence A. Leiter observed in presenting the trial results at the annual scientific sessions of the American Diabetes Association.
The 52-week double-blind study involved 495 patients with type 2 diabetes whose hyperglycemia wasn’t adequately controlled by diet and exercise or oral antidiabetic medications. Renal impairment was mild in slightly over half of the participants, moderate in 41%, and severe in the remainder.
Patients were randomized to self-administered subcutaneous injection of albiglutide at 30 mg once a week or the oral dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin, dose-adjusted on the basis of the degree of renal impairment as specified in the product labeling. Thirty-five percent of patients in the albiglutide group had their daily dose uptitrated from 30 mg to 50 mg owing to insufficient response to the initial dose.
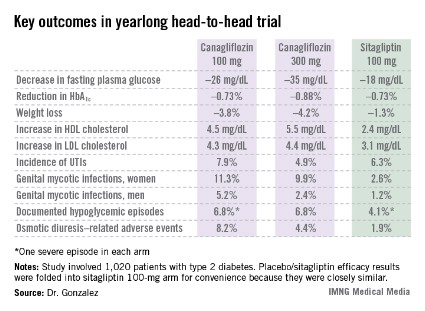
The primary endpoint was the change in mean hemoglobin A1c from baseline to week 26. The HbA1c dropped by 0.8% in the albiglutide group, significantly better than the 0.5% decrease with sitagliptin (P = .0003). The advantage favoring the investigational agent was seen in all three renal impairment severity groups, reported Dr. Leiter, professor of medicine and nutritional sciences at the University of Toronto and president of the Canadian Society of Endocrinology and Metabolism.
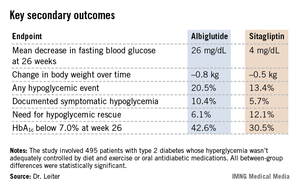
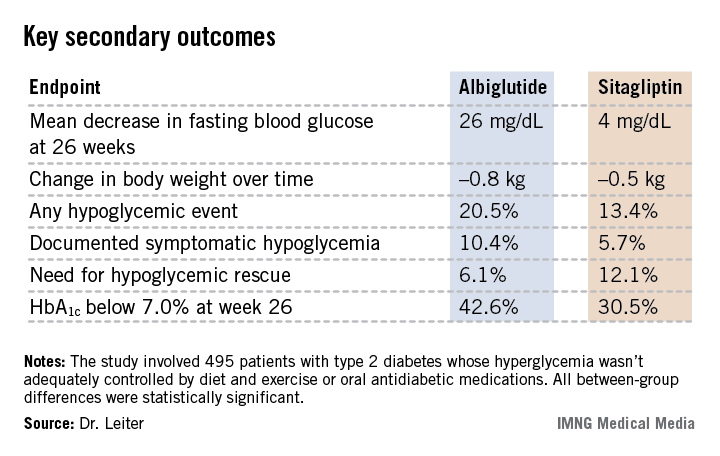
Albiglutide-treated patients also fared significantly better in terms of multiple key secondary endpoints (see chart). For example, 42.6% of them achieved a clinically meaningful HbA1c response by 26 weeks, defined as an HbA1c below 7.0%, compared with 30.5% of patients on sitagliptin.

Both drugs were well tolerated. The rate of adverse events leading to study withdrawal was 6.4% with albiglutide and 8.1% with sitagliptin. Both drugs had impressively low rates of nausea and vomiting; typically, less than 1% of patients per week reported either symptom.
Session chair Dr. Julio Rosenstock called this study noteworthy for several reasons. It is the first study of a GLP-1 agonist in patients with renal insufficiency; head-to-head comparative randomized trials in diabetes are rare. And the low-single-digit rates of nausea and vomiting seen with albiglutide in this study are "remarkable." Other GLP-1 agonists are typically associated with nausea and vomiting rates in the 15%-20% range or even higher, noted Dr. Rosenstock, director of the Dallas Diabetes and Endocrine Center at Medical City.
Dr. Leiter agreed, offering two potential explanations for the rock-bottom nausea and vomiting rates. One is that the once-weekly injection results in a very gradual increase in drug levels. Also, as a large molecule, albiglutide probably has less central action, including less activity at the central nervous system’s nausea and vomiting centers.
Other large, completed phase III trials have shown benefits for albiglutide in additional, common clinical scenarios, including yearlong studies of albiglutide as add-on therapy in patients with type 2 diabetes not controlled on pioglitazone and metformin, albiglutide monotherapy in drug-naive type 2 diabetic patients, a comparison of the novel GLP-1 agonist to insulin glargine in patients with type 2 diabetes, and albiglutide versus pioglitazone as add-on therapy in patients on background metformin and glimepiride.
Albiglutide is being developed by GlaxoSmithKline. Dr. Leiter has received research grants from and served as a consultant to GSK and other companies.
CHICAGO – Once-weekly, fixed-dose albiglutide achieved superior glycemic control compared with dose-adjusted sitagliptin in patients with type 2 diabetes who had inadequate glycemic control and varying degrees of renal impairment in a phase III trial.
Albiglutide is an investigational glucagonlike peptide–1 (GLP-1) agonist now under review at both the U.S. Food and Drug Administration and the European Medicines Agency as a potential new treatment for type 2 diabetes. It is a large molecule cleared by the reticuloendothelial system rather than by the kidney, so its metabolism is not affected by the renal dysfunction that is common in patients with type 2 diabetes.
"Albiglutide will be a useful treatment addition for patients with renal impairment and type 2 diabetes. Current treatments for such patients are limited. Several antidiabetic medications are contraindicated in this population, and many others require dose adjustment," Dr. Lawrence A. Leiter observed in presenting the trial results at the annual scientific sessions of the American Diabetes Association.
The 52-week double-blind study involved 495 patients with type 2 diabetes whose hyperglycemia wasn’t adequately controlled by diet and exercise or oral antidiabetic medications. Renal impairment was mild in slightly over half of the participants, moderate in 41%, and severe in the remainder.
Patients were randomized to self-administered subcutaneous injection of albiglutide at 30 mg once a week or the oral dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin, dose-adjusted on the basis of the degree of renal impairment as specified in the product labeling. Thirty-five percent of patients in the albiglutide group had their daily dose uptitrated from 30 mg to 50 mg owing to insufficient response to the initial dose.
The primary endpoint was the change in mean hemoglobin A1c from baseline to week 26. The HbA1c dropped by 0.8% in the albiglutide group, significantly better than the 0.5% decrease with sitagliptin (P = .0003). The advantage favoring the investigational agent was seen in all three renal impairment severity groups, reported Dr. Leiter, professor of medicine and nutritional sciences at the University of Toronto and president of the Canadian Society of Endocrinology and Metabolism.
Albiglutide-treated patients also fared significantly better in terms of multiple key secondary endpoints (see chart). For example, 42.6% of them achieved a clinically meaningful HbA1c response by 26 weeks, defined as an HbA1c below 7.0%, compared with 30.5% of patients on sitagliptin.
Both drugs were well tolerated. The rate of adverse events leading to study withdrawal was 6.4% with albiglutide and 8.1% with sitagliptin. Both drugs had impressively low rates of nausea and vomiting; typically, less than 1% of patients per week reported either symptom.
Session chair Dr. Julio Rosenstock called this study noteworthy for several reasons. It is the first study of a GLP-1 agonist in patients with renal insufficiency; head-to-head comparative randomized trials in diabetes are rare. And the low-single-digit rates of nausea and vomiting seen with albiglutide in this study are "remarkable." Other GLP-1 agonists are typically associated with nausea and vomiting rates in the 15%-20% range or even higher, noted Dr. Rosenstock, director of the Dallas Diabetes and Endocrine Center at Medical City.
Dr. Leiter agreed, offering two potential explanations for the rock-bottom nausea and vomiting rates. One is that the once-weekly injection results in a very gradual increase in drug levels. Also, as a large molecule, albiglutide probably has less central action, including less activity at the central nervous system’s nausea and vomiting centers.
Other large, completed phase III trials have shown benefits for albiglutide in additional, common clinical scenarios, including yearlong studies of albiglutide as add-on therapy in patients with type 2 diabetes not controlled on pioglitazone and metformin, albiglutide monotherapy in drug-naive type 2 diabetic patients, a comparison of the novel GLP-1 agonist to insulin glargine in patients with type 2 diabetes, and albiglutide versus pioglitazone as add-on therapy in patients on background metformin and glimepiride.
Albiglutide is being developed by GlaxoSmithKline. Dr. Leiter has received research grants from and served as a consultant to GSK and other companies.
CHICAGO – Once-weekly, fixed-dose albiglutide achieved superior glycemic control compared with dose-adjusted sitagliptin in patients with type 2 diabetes who had inadequate glycemic control and varying degrees of renal impairment in a phase III trial.
Albiglutide is an investigational glucagonlike peptide–1 (GLP-1) agonist now under review at both the U.S. Food and Drug Administration and the European Medicines Agency as a potential new treatment for type 2 diabetes. It is a large molecule cleared by the reticuloendothelial system rather than by the kidney, so its metabolism is not affected by the renal dysfunction that is common in patients with type 2 diabetes.
"Albiglutide will be a useful treatment addition for patients with renal impairment and type 2 diabetes. Current treatments for such patients are limited. Several antidiabetic medications are contraindicated in this population, and many others require dose adjustment," Dr. Lawrence A. Leiter observed in presenting the trial results at the annual scientific sessions of the American Diabetes Association.
The 52-week double-blind study involved 495 patients with type 2 diabetes whose hyperglycemia wasn’t adequately controlled by diet and exercise or oral antidiabetic medications. Renal impairment was mild in slightly over half of the participants, moderate in 41%, and severe in the remainder.
Patients were randomized to self-administered subcutaneous injection of albiglutide at 30 mg once a week or the oral dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin, dose-adjusted on the basis of the degree of renal impairment as specified in the product labeling. Thirty-five percent of patients in the albiglutide group had their daily dose uptitrated from 30 mg to 50 mg owing to insufficient response to the initial dose.
The primary endpoint was the change in mean hemoglobin A1c from baseline to week 26. The HbA1c dropped by 0.8% in the albiglutide group, significantly better than the 0.5% decrease with sitagliptin (P = .0003). The advantage favoring the investigational agent was seen in all three renal impairment severity groups, reported Dr. Leiter, professor of medicine and nutritional sciences at the University of Toronto and president of the Canadian Society of Endocrinology and Metabolism.
Albiglutide-treated patients also fared significantly better in terms of multiple key secondary endpoints (see chart). For example, 42.6% of them achieved a clinically meaningful HbA1c response by 26 weeks, defined as an HbA1c below 7.0%, compared with 30.5% of patients on sitagliptin.
Both drugs were well tolerated. The rate of adverse events leading to study withdrawal was 6.4% with albiglutide and 8.1% with sitagliptin. Both drugs had impressively low rates of nausea and vomiting; typically, less than 1% of patients per week reported either symptom.
Session chair Dr. Julio Rosenstock called this study noteworthy for several reasons. It is the first study of a GLP-1 agonist in patients with renal insufficiency; head-to-head comparative randomized trials in diabetes are rare. And the low-single-digit rates of nausea and vomiting seen with albiglutide in this study are "remarkable." Other GLP-1 agonists are typically associated with nausea and vomiting rates in the 15%-20% range or even higher, noted Dr. Rosenstock, director of the Dallas Diabetes and Endocrine Center at Medical City.
Dr. Leiter agreed, offering two potential explanations for the rock-bottom nausea and vomiting rates. One is that the once-weekly injection results in a very gradual increase in drug levels. Also, as a large molecule, albiglutide probably has less central action, including less activity at the central nervous system’s nausea and vomiting centers.
Other large, completed phase III trials have shown benefits for albiglutide in additional, common clinical scenarios, including yearlong studies of albiglutide as add-on therapy in patients with type 2 diabetes not controlled on pioglitazone and metformin, albiglutide monotherapy in drug-naive type 2 diabetic patients, a comparison of the novel GLP-1 agonist to insulin glargine in patients with type 2 diabetes, and albiglutide versus pioglitazone as add-on therapy in patients on background metformin and glimepiride.
Albiglutide is being developed by GlaxoSmithKline. Dr. Leiter has received research grants from and served as a consultant to GSK and other companies.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major finding: Mean HbA1c levels dropped by 0.8% after 26 weeks on albiglutide in renally impaired type 2 diabetic patients, a significantly better result than the mean 0.5% decrease with sitagliptin. Patients on albiglutide also fared significantly better in terms of numerous secondary endpoints.
Data source: A phase III, randomized, double-blind trial in 495 patients with type 2 diabetes and varying degrees of renal insufficiency and baseline inadequate glycemic control assigned to either the novel once-weekly GLP-1 agonist albiglutide or sitagliptin.
Disclosures: Albiglutide is being developed by GlaxoSmithKline. Dr. Leiter has received research grants from and served as a consultant to GSK and other companies.
Alemtuzumab shows 'wow' factor in highly-active MS
ORLANDO – Alemtuzumab is far more effective than is high-dose interferon beta-1a in treating patients with highly active relapsing-remitting multiple sclerosis despite prior therapy, according to a new subgroup analysis of the CARE-MS II trial.
Twenty-four percent of alemtuzumab-treated patients in the subgroup with highly-active disease at baseline remained entirely free of demonstrable MS disease activity – both clinically and by MRI – throughout the 2-year randomized trial. In contrast, none of the patients on subcutaneous interferon beta-1a (Rebif) achieved that high standard, Dr. Stephen Krieger reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.

"That’s an unusual result to see in any of our trials and in any subgroups: zero percent of patients achieving an efficacy goal with one of our approved and highly efficacious agents. I think this demonstrates the high disease activity of these patients, the efficacy gradient between alemtuzumab and interferon, and it also highlights the unmet need for patients with highly-active disease," said Dr. Krieger of Mount Sinai Medical Center, New York.
The CARE-MS II subanalysis also strongly suggests alemtuzumab may provide an important new treatment option for this group of severely affected patients, he added.
Alemtuzumab is a humanized monoclonal antibody to CD52, a protein present on the surface of mature lymphocytes but not on the stem cells from which they originate. Alemtuzumab is approved as Campath for treatment of B-cell chronic lymphocytic leukemia. Genzyme has applied to the Food and Drug Administration and European regulators for a new indication under the trade name Lemtrada for treatment of MS.
CARE-MS II (Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis) was a 2-year, rater-blinded, multicenter, international, phase III clinical trial in which 667 patients with relapsing-remitting MS who had experienced two or more relapses despite treatment during the 2 years immediately prior to enrollment. Participants were randomized 2:1 to alemtuzumab or interferon beta-1a. Alemtuzumab at 12 mg was administered intravenously on 5 consecutive days at the start of the study and again on 3 consecutive days at the 1-year mark, for a total of just 8 doses in 2 years. In contrast, subcutaneous interferon beta-1a at 44 mcg was given three times weekly throughout the study.
The overall results, now published (Lancet 2012;380:1829-39), showed the alemtuzumab group had reductions of 49% in the risk of relapse and 42% in the risk of sustained accumulation of disability, compared with the interferon group.
Dr. Krieger presented a new analysis restricted to the 145 CARE-MS II participants with the most highly active disease as defined by two or more relapses in the year prior to randomization as well as the presence of at least one gadolinium-enhancing lesion on baseline MRI.
"Basically, we’re taking the most active patients from the trial of the more active patients who had already experienced disease activity despite prior therapy," the neurologist explained.
The annualized relapse rate in these patients with highly active disease was 0.33 events with alemtuzumab and .65 events with interferon, for a 51% reduction in favor of the investigational agent. Sixty-four percent of the alemtuzumab group and 36% on interferon remained relapse free during the 2-year study.
Freedom from clinical disease activity was defined as being relapse free as well as free of sustained accumulation of disability, meaning an absence of at least a 1-point increase over baseline on the Expanded Disability Status Scale lasting for at least 6 months. Clinical disease activity–free status for the full 2 years of follow-up was attained by 61% of the alemtuzumab group, compared with 33% of patients on interferon. The adjusted odds ratio was 3.18, meaning patients with highly active disease were more than three times more likely to remain free of clinical disease activity throughout the study if they were on alemtuzumab rather than interferon.
Turning to the serial MRI results, Dr. Krieger noted that 40% of the alemtuzumab group remained free of disease activity by MRI, meaning no new gadolinium-enhancing lesions and no new or enlarging T2 lesions. Only 7.5% of patients with highly active disease assigned to interferon therapy remained free of MRI evidence of disease activity during the 2-year study. Patients on alemtuzumab were an adjusted ninefold more likely to be free of MRI activity than were those on interferon.
The post hoc composite endpoint of freedom from demonstrable MS disease activity required 2 years of freedom from relapse, freedom from MRI activity, and freedom from sustained accumulation of disability. This was the outcome achieved by 24% of patients on alemtuzumab and 0% of those on interferon.
The safety data in the subgroup with highly active disease reflected the findings in the overall CARE-MS II population. Infections occurred in 84% of highly active patients on alemtuzumab and 64% on interferon. Thyroid disorders occurred in 15.5% on alemtuzumab, compared with 2.4% on interferon. Adverse events leading to study discontinuation were seen in 1% on alemtuzumab, compared with 2.4% on interferon.
CARE-MS II was sponsored by Genzyme. Dr. Krieger is a consultant to Genzyme and half a dozen other pharmaceutical companies.
ORLANDO – Alemtuzumab is far more effective than is high-dose interferon beta-1a in treating patients with highly active relapsing-remitting multiple sclerosis despite prior therapy, according to a new subgroup analysis of the CARE-MS II trial.
Twenty-four percent of alemtuzumab-treated patients in the subgroup with highly-active disease at baseline remained entirely free of demonstrable MS disease activity – both clinically and by MRI – throughout the 2-year randomized trial. In contrast, none of the patients on subcutaneous interferon beta-1a (Rebif) achieved that high standard, Dr. Stephen Krieger reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"That’s an unusual result to see in any of our trials and in any subgroups: zero percent of patients achieving an efficacy goal with one of our approved and highly efficacious agents. I think this demonstrates the high disease activity of these patients, the efficacy gradient between alemtuzumab and interferon, and it also highlights the unmet need for patients with highly-active disease," said Dr. Krieger of Mount Sinai Medical Center, New York.
The CARE-MS II subanalysis also strongly suggests alemtuzumab may provide an important new treatment option for this group of severely affected patients, he added.
Alemtuzumab is a humanized monoclonal antibody to CD52, a protein present on the surface of mature lymphocytes but not on the stem cells from which they originate. Alemtuzumab is approved as Campath for treatment of B-cell chronic lymphocytic leukemia. Genzyme has applied to the Food and Drug Administration and European regulators for a new indication under the trade name Lemtrada for treatment of MS.
CARE-MS II (Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis) was a 2-year, rater-blinded, multicenter, international, phase III clinical trial in which 667 patients with relapsing-remitting MS who had experienced two or more relapses despite treatment during the 2 years immediately prior to enrollment. Participants were randomized 2:1 to alemtuzumab or interferon beta-1a. Alemtuzumab at 12 mg was administered intravenously on 5 consecutive days at the start of the study and again on 3 consecutive days at the 1-year mark, for a total of just 8 doses in 2 years. In contrast, subcutaneous interferon beta-1a at 44 mcg was given three times weekly throughout the study.
The overall results, now published (Lancet 2012;380:1829-39), showed the alemtuzumab group had reductions of 49% in the risk of relapse and 42% in the risk of sustained accumulation of disability, compared with the interferon group.
Dr. Krieger presented a new analysis restricted to the 145 CARE-MS II participants with the most highly active disease as defined by two or more relapses in the year prior to randomization as well as the presence of at least one gadolinium-enhancing lesion on baseline MRI.
"Basically, we’re taking the most active patients from the trial of the more active patients who had already experienced disease activity despite prior therapy," the neurologist explained.
The annualized relapse rate in these patients with highly active disease was 0.33 events with alemtuzumab and .65 events with interferon, for a 51% reduction in favor of the investigational agent. Sixty-four percent of the alemtuzumab group and 36% on interferon remained relapse free during the 2-year study.
Freedom from clinical disease activity was defined as being relapse free as well as free of sustained accumulation of disability, meaning an absence of at least a 1-point increase over baseline on the Expanded Disability Status Scale lasting for at least 6 months. Clinical disease activity–free status for the full 2 years of follow-up was attained by 61% of the alemtuzumab group, compared with 33% of patients on interferon. The adjusted odds ratio was 3.18, meaning patients with highly active disease were more than three times more likely to remain free of clinical disease activity throughout the study if they were on alemtuzumab rather than interferon.
Turning to the serial MRI results, Dr. Krieger noted that 40% of the alemtuzumab group remained free of disease activity by MRI, meaning no new gadolinium-enhancing lesions and no new or enlarging T2 lesions. Only 7.5% of patients with highly active disease assigned to interferon therapy remained free of MRI evidence of disease activity during the 2-year study. Patients on alemtuzumab were an adjusted ninefold more likely to be free of MRI activity than were those on interferon.
The post hoc composite endpoint of freedom from demonstrable MS disease activity required 2 years of freedom from relapse, freedom from MRI activity, and freedom from sustained accumulation of disability. This was the outcome achieved by 24% of patients on alemtuzumab and 0% of those on interferon.
The safety data in the subgroup with highly active disease reflected the findings in the overall CARE-MS II population. Infections occurred in 84% of highly active patients on alemtuzumab and 64% on interferon. Thyroid disorders occurred in 15.5% on alemtuzumab, compared with 2.4% on interferon. Adverse events leading to study discontinuation were seen in 1% on alemtuzumab, compared with 2.4% on interferon.
CARE-MS II was sponsored by Genzyme. Dr. Krieger is a consultant to Genzyme and half a dozen other pharmaceutical companies.
ORLANDO – Alemtuzumab is far more effective than is high-dose interferon beta-1a in treating patients with highly active relapsing-remitting multiple sclerosis despite prior therapy, according to a new subgroup analysis of the CARE-MS II trial.
Twenty-four percent of alemtuzumab-treated patients in the subgroup with highly-active disease at baseline remained entirely free of demonstrable MS disease activity – both clinically and by MRI – throughout the 2-year randomized trial. In contrast, none of the patients on subcutaneous interferon beta-1a (Rebif) achieved that high standard, Dr. Stephen Krieger reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"That’s an unusual result to see in any of our trials and in any subgroups: zero percent of patients achieving an efficacy goal with one of our approved and highly efficacious agents. I think this demonstrates the high disease activity of these patients, the efficacy gradient between alemtuzumab and interferon, and it also highlights the unmet need for patients with highly-active disease," said Dr. Krieger of Mount Sinai Medical Center, New York.
The CARE-MS II subanalysis also strongly suggests alemtuzumab may provide an important new treatment option for this group of severely affected patients, he added.
Alemtuzumab is a humanized monoclonal antibody to CD52, a protein present on the surface of mature lymphocytes but not on the stem cells from which they originate. Alemtuzumab is approved as Campath for treatment of B-cell chronic lymphocytic leukemia. Genzyme has applied to the Food and Drug Administration and European regulators for a new indication under the trade name Lemtrada for treatment of MS.
CARE-MS II (Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis) was a 2-year, rater-blinded, multicenter, international, phase III clinical trial in which 667 patients with relapsing-remitting MS who had experienced two or more relapses despite treatment during the 2 years immediately prior to enrollment. Participants were randomized 2:1 to alemtuzumab or interferon beta-1a. Alemtuzumab at 12 mg was administered intravenously on 5 consecutive days at the start of the study and again on 3 consecutive days at the 1-year mark, for a total of just 8 doses in 2 years. In contrast, subcutaneous interferon beta-1a at 44 mcg was given three times weekly throughout the study.
The overall results, now published (Lancet 2012;380:1829-39), showed the alemtuzumab group had reductions of 49% in the risk of relapse and 42% in the risk of sustained accumulation of disability, compared with the interferon group.
Dr. Krieger presented a new analysis restricted to the 145 CARE-MS II participants with the most highly active disease as defined by two or more relapses in the year prior to randomization as well as the presence of at least one gadolinium-enhancing lesion on baseline MRI.
"Basically, we’re taking the most active patients from the trial of the more active patients who had already experienced disease activity despite prior therapy," the neurologist explained.
The annualized relapse rate in these patients with highly active disease was 0.33 events with alemtuzumab and .65 events with interferon, for a 51% reduction in favor of the investigational agent. Sixty-four percent of the alemtuzumab group and 36% on interferon remained relapse free during the 2-year study.
Freedom from clinical disease activity was defined as being relapse free as well as free of sustained accumulation of disability, meaning an absence of at least a 1-point increase over baseline on the Expanded Disability Status Scale lasting for at least 6 months. Clinical disease activity–free status for the full 2 years of follow-up was attained by 61% of the alemtuzumab group, compared with 33% of patients on interferon. The adjusted odds ratio was 3.18, meaning patients with highly active disease were more than three times more likely to remain free of clinical disease activity throughout the study if they were on alemtuzumab rather than interferon.
Turning to the serial MRI results, Dr. Krieger noted that 40% of the alemtuzumab group remained free of disease activity by MRI, meaning no new gadolinium-enhancing lesions and no new or enlarging T2 lesions. Only 7.5% of patients with highly active disease assigned to interferon therapy remained free of MRI evidence of disease activity during the 2-year study. Patients on alemtuzumab were an adjusted ninefold more likely to be free of MRI activity than were those on interferon.
The post hoc composite endpoint of freedom from demonstrable MS disease activity required 2 years of freedom from relapse, freedom from MRI activity, and freedom from sustained accumulation of disability. This was the outcome achieved by 24% of patients on alemtuzumab and 0% of those on interferon.
The safety data in the subgroup with highly active disease reflected the findings in the overall CARE-MS II population. Infections occurred in 84% of highly active patients on alemtuzumab and 64% on interferon. Thyroid disorders occurred in 15.5% on alemtuzumab, compared with 2.4% on interferon. Adverse events leading to study discontinuation were seen in 1% on alemtuzumab, compared with 2.4% on interferon.
CARE-MS II was sponsored by Genzyme. Dr. Krieger is a consultant to Genzyme and half a dozen other pharmaceutical companies.
AT THE CMSC/ACTRIMS ANNUAL MEETING
Major Finding: Twenty-four percent of patients with highly active relapsing-remitting multiple sclerosis placed on the investigational agent alemtuzumab remained free from clinical relapse, sustained accumulation of disability, and MRI evidence of disease activity during 2 years of follow-up, compared with 0% of those randomized to standard therapy with subcutaneous high-dose interferon beta-1a.
Data Source: An analysis of the 145 participants in the phase III randomized CARE-MS II study who had the most highly active disease.
Disclosures: The CARE-MS II trial and this new subgroup analysis were sponsored by Genzyme. The presenter is a consultant to the company.

Peginterferon beta-1a shows promise in relapsing-remitting MS
ORLANDO – An investigational pegylated formulation of subcutaneous interferon beta-1a appears to offer the efficacy and safety of currently available interferon therapy for relapsing-remitting multiple sclerosis with the advantage of less frequent injections, a study has shown.
In the phase III, double-blind ADVANCE trial, peginterferon beta-1a at 125 mcg self-administered subcutaneously every 2 weeks for 1 year consistently provided statistically significant advantages over placebo for all clinical and radiologic outcomes, Dr. Peter A. Calabresi reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Peginterferon beta-1a dosed every 4 weeks was less effective than every 2 weeks. It bested placebo on some but not all endpoints, added Dr. Calabresi, professor of neurology and director of the multiple sclerosis center at Johns Hopkins University, Baltimore.

The ADVANCE trial involved 1,516 randomized patients with relapsing-remitting MS, 86% of whom had a baseline Expanded Disability Status Scale (EDSS) score of less than 4.
The primary endpoint was the annualized relapse rate at 1 year: 0.397 with placebo; 0.2888 with peginterferon beta-1a every 4 weeks, for a 27.5% reduction compared with placebo; and 0.256 with biweekly therapy, for a 35.6% reduction. The 1-year incidence of relapse was reduced by 26% with monthly dosing and by 39% with biweekly dosing of peginterferon beta-1a, compared with placebo.
A key secondary outcome measure, disability progression during year 1 as defined by at least a 1-point increase in the EDSS sustained for 12 weeks or more, was reduced by 38% in both peginterferon beta-1a arms, compared with placebo.
In terms of MRI outcomes reflecting disease activity, the mean number of new or enlarging T2 lesions at 1 year was 10.9 with placebo, 7.9 with peginterferon every 4 weeks, and 3.6 – a 67% reduction compared with placebo – with biweekly therapy. The mean number of gadolinium-enhancing MRI lesions was 1.4 with placebo, 0.9 with peginterferon every 4 weeks, and 0.2 with biweekly peginterferon, an 86% reduction compared with placebo.
The incidence of interferon-neutralizing antibodies believed capable of interfering with the effect of the drug was reassuringly low at less than 1% in all three study arms at 1 year.
The adverse events profile of peginterferon beta-1a would be familiar to physicians who prescribe other interferons: injection site reactions, flulike illness, and headaches, for example – all typically rated mild and lasting less than 48 hours, Dr. Calabresi continued.
One audience member, noting that peginterferon beta-1a every 2 weeks was clearly more effective than every 4 weeks, asked why once-weekly injections weren’t studied. Dr. Calabresi replied that the every-2-week regimen appears to provide a reduction in the relapse rate that’s roughly equivalent to that achieved with standard nonpegylated interferon beta-1a given more frequently. For patients who’ve done well on interferon therapy for years, the idea of switching from Avonex given intramuscularly once weekly or Rebif given subcutaneously three times per week to an every 2-week dosing regimen that provides the efficacy and safety they’re accustomed to is likely to be very attractive, he added.
Pegylation is a strategy for increasing the half-life and pharmacodynamic effects of a drug. Less frequent dosing means greater convenience for patients and possibly improved adherence as well. Ninety-one percent of the placebo group completed year 1 of ADVANCE, as did 88% on peginterferon beta-1a given every 4 weeks and 86% on biweekly dosing.
The ADVANCE study is now in its second and final year, during which patients continue on the same peginterferon dosing schedule they had in year 1, while patients formerly on placebo are randomized to a year on one of the two peginterferon dosing regimens. After the 2 years of ADVANCE are completed, there will be a 2-year extension study called ATTAIN.
The ADVANCE trial is sponsored by Biogen Idec. Dr. Calabresi reported receiving grant support from that company and serving as a consultant to Vaccinex, Abbott, and Vertex.
ORLANDO – An investigational pegylated formulation of subcutaneous interferon beta-1a appears to offer the efficacy and safety of currently available interferon therapy for relapsing-remitting multiple sclerosis with the advantage of less frequent injections, a study has shown.
In the phase III, double-blind ADVANCE trial, peginterferon beta-1a at 125 mcg self-administered subcutaneously every 2 weeks for 1 year consistently provided statistically significant advantages over placebo for all clinical and radiologic outcomes, Dr. Peter A. Calabresi reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Peginterferon beta-1a dosed every 4 weeks was less effective than every 2 weeks. It bested placebo on some but not all endpoints, added Dr. Calabresi, professor of neurology and director of the multiple sclerosis center at Johns Hopkins University, Baltimore.
The ADVANCE trial involved 1,516 randomized patients with relapsing-remitting MS, 86% of whom had a baseline Expanded Disability Status Scale (EDSS) score of less than 4.
The primary endpoint was the annualized relapse rate at 1 year: 0.397 with placebo; 0.2888 with peginterferon beta-1a every 4 weeks, for a 27.5% reduction compared with placebo; and 0.256 with biweekly therapy, for a 35.6% reduction. The 1-year incidence of relapse was reduced by 26% with monthly dosing and by 39% with biweekly dosing of peginterferon beta-1a, compared with placebo.
A key secondary outcome measure, disability progression during year 1 as defined by at least a 1-point increase in the EDSS sustained for 12 weeks or more, was reduced by 38% in both peginterferon beta-1a arms, compared with placebo.
In terms of MRI outcomes reflecting disease activity, the mean number of new or enlarging T2 lesions at 1 year was 10.9 with placebo, 7.9 with peginterferon every 4 weeks, and 3.6 – a 67% reduction compared with placebo – with biweekly therapy. The mean number of gadolinium-enhancing MRI lesions was 1.4 with placebo, 0.9 with peginterferon every 4 weeks, and 0.2 with biweekly peginterferon, an 86% reduction compared with placebo.
The incidence of interferon-neutralizing antibodies believed capable of interfering with the effect of the drug was reassuringly low at less than 1% in all three study arms at 1 year.
The adverse events profile of peginterferon beta-1a would be familiar to physicians who prescribe other interferons: injection site reactions, flulike illness, and headaches, for example – all typically rated mild and lasting less than 48 hours, Dr. Calabresi continued.
One audience member, noting that peginterferon beta-1a every 2 weeks was clearly more effective than every 4 weeks, asked why once-weekly injections weren’t studied. Dr. Calabresi replied that the every-2-week regimen appears to provide a reduction in the relapse rate that’s roughly equivalent to that achieved with standard nonpegylated interferon beta-1a given more frequently. For patients who’ve done well on interferon therapy for years, the idea of switching from Avonex given intramuscularly once weekly or Rebif given subcutaneously three times per week to an every 2-week dosing regimen that provides the efficacy and safety they’re accustomed to is likely to be very attractive, he added.
Pegylation is a strategy for increasing the half-life and pharmacodynamic effects of a drug. Less frequent dosing means greater convenience for patients and possibly improved adherence as well. Ninety-one percent of the placebo group completed year 1 of ADVANCE, as did 88% on peginterferon beta-1a given every 4 weeks and 86% on biweekly dosing.
The ADVANCE study is now in its second and final year, during which patients continue on the same peginterferon dosing schedule they had in year 1, while patients formerly on placebo are randomized to a year on one of the two peginterferon dosing regimens. After the 2 years of ADVANCE are completed, there will be a 2-year extension study called ATTAIN.
The ADVANCE trial is sponsored by Biogen Idec. Dr. Calabresi reported receiving grant support from that company and serving as a consultant to Vaccinex, Abbott, and Vertex.
ORLANDO – An investigational pegylated formulation of subcutaneous interferon beta-1a appears to offer the efficacy and safety of currently available interferon therapy for relapsing-remitting multiple sclerosis with the advantage of less frequent injections, a study has shown.
In the phase III, double-blind ADVANCE trial, peginterferon beta-1a at 125 mcg self-administered subcutaneously every 2 weeks for 1 year consistently provided statistically significant advantages over placebo for all clinical and radiologic outcomes, Dr. Peter A. Calabresi reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Peginterferon beta-1a dosed every 4 weeks was less effective than every 2 weeks. It bested placebo on some but not all endpoints, added Dr. Calabresi, professor of neurology and director of the multiple sclerosis center at Johns Hopkins University, Baltimore.
The ADVANCE trial involved 1,516 randomized patients with relapsing-remitting MS, 86% of whom had a baseline Expanded Disability Status Scale (EDSS) score of less than 4.
The primary endpoint was the annualized relapse rate at 1 year: 0.397 with placebo; 0.2888 with peginterferon beta-1a every 4 weeks, for a 27.5% reduction compared with placebo; and 0.256 with biweekly therapy, for a 35.6% reduction. The 1-year incidence of relapse was reduced by 26% with monthly dosing and by 39% with biweekly dosing of peginterferon beta-1a, compared with placebo.
A key secondary outcome measure, disability progression during year 1 as defined by at least a 1-point increase in the EDSS sustained for 12 weeks or more, was reduced by 38% in both peginterferon beta-1a arms, compared with placebo.
In terms of MRI outcomes reflecting disease activity, the mean number of new or enlarging T2 lesions at 1 year was 10.9 with placebo, 7.9 with peginterferon every 4 weeks, and 3.6 – a 67% reduction compared with placebo – with biweekly therapy. The mean number of gadolinium-enhancing MRI lesions was 1.4 with placebo, 0.9 with peginterferon every 4 weeks, and 0.2 with biweekly peginterferon, an 86% reduction compared with placebo.
The incidence of interferon-neutralizing antibodies believed capable of interfering with the effect of the drug was reassuringly low at less than 1% in all three study arms at 1 year.
The adverse events profile of peginterferon beta-1a would be familiar to physicians who prescribe other interferons: injection site reactions, flulike illness, and headaches, for example – all typically rated mild and lasting less than 48 hours, Dr. Calabresi continued.
One audience member, noting that peginterferon beta-1a every 2 weeks was clearly more effective than every 4 weeks, asked why once-weekly injections weren’t studied. Dr. Calabresi replied that the every-2-week regimen appears to provide a reduction in the relapse rate that’s roughly equivalent to that achieved with standard nonpegylated interferon beta-1a given more frequently. For patients who’ve done well on interferon therapy for years, the idea of switching from Avonex given intramuscularly once weekly or Rebif given subcutaneously three times per week to an every 2-week dosing regimen that provides the efficacy and safety they’re accustomed to is likely to be very attractive, he added.
Pegylation is a strategy for increasing the half-life and pharmacodynamic effects of a drug. Less frequent dosing means greater convenience for patients and possibly improved adherence as well. Ninety-one percent of the placebo group completed year 1 of ADVANCE, as did 88% on peginterferon beta-1a given every 4 weeks and 86% on biweekly dosing.
The ADVANCE study is now in its second and final year, during which patients continue on the same peginterferon dosing schedule they had in year 1, while patients formerly on placebo are randomized to a year on one of the two peginterferon dosing regimens. After the 2 years of ADVANCE are completed, there will be a 2-year extension study called ATTAIN.
The ADVANCE trial is sponsored by Biogen Idec. Dr. Calabresi reported receiving grant support from that company and serving as a consultant to Vaccinex, Abbott, and Vertex.
AT THE CMSC/ACTRIMS ANNUAL MEETING
Major Finding: Patients on investigational pegylated interferon beta-1a self-administered by subcutaneous injection once every 2 weeks had an annualized relapse rate at 1 year of 0.256, a 35.6% reduction compared with placebo. Injections once every 4 weeks resulted in an annualized relapse rate at 1 year of 0.2888.
Data Source: A phase III, double-blind, multicenter clinical trial (ADVANCE) including 1,516 patients with relapsing-remitting MS randomized to 1 year of placebo or subcutaneous pegylated interferon beta-1a at 125 mcg self-administered either every 2 weeks or every 4 weeks.
Disclosures: The ADVANCE trial is sponsored by Biogen Idec. Dr. Calabresi reported receiving grant support from that company and serving as a consultant to Vaccinex, Abbott, and Vertex.

Investigational basal insulin has less nocturnal hypoglycemia
CHICAGO – A new longer-acting basal insulin glargine formulation was as effective as the familiar insulin glargine 100 U/mL/Lantus in controlling glycemia while producing a 21% lower rate of severe nocturnal hypoglycemic events in the phase III EDITION I trial.
The investigational formulation contains insulin glargine 300 U/mL and is known as U300. Like insulin glargine U100, the investigational agent is a once-daily, long-acting insulin analog. But U300 has an even flatter, longer, and more stable pharmacodynamic profile, which translates into a lower risk of hypoglycemia, as shown in the EDITION I trial, principal investigator Dr. Matthew C. Riddle explained at the annual scientific sessions of the American Diabetes Association.

EDITION I included 807 adults with type 2 diabetes who were on an established treatment with at least 42 U/day of basal glargine or NPH along with mealtime insulin with or without metformin. Their mean duration of diabetes was 15.8 years, and their mean body mass index was 36.6 kg/m2. They were randomized to 6 months of open-label insulin glargine U300 or insulin glargine U100 once daily in the evening while continuing on their mealtime insulin. The basal insulin was titrated to achieve a fasting plasma glucose of 80-100 mg/dL.
The primary efficacy endpoint was change in hemoglobin A1c from baseline through month 6. The reductions were the same in both groups: a mean decrease of 0.83%.
The chief secondary endpoint was the occurrence of one or more episodes of severe and/or confirmed nocturnal hypoglycemia to a level of 70 mg/dL or less during months 3-6. The rate was 36.1% in the U300 group, compared with 46% with U100, for a highly significant 21% risk reduction, said Dr. Riddle, professor of medicine at the Oregon Health and Science Center, Portland.
Any nocturnal hypoglycemia during the 6-month study occurred in 45.3% of patients on U300 vs. 59.7% on U100, for a 24% relative risk reduction, he added.
EDITION I is the first in a large worldwide series of phase III trials of the investigational insulin. The recently completed EDITION II trial replicated the EDITION I findings blood glucose reductions similar to those seen in patients taking Lantus, with fewer serious nocturnal hypoglycemic events – in 811 type 2 diabetic patients with earlier-stage disease as evidenced by the enrollment requirement that they be established on basal insulin and oral antidiabetic therapy. EDITION III is ongoing in insulin-naive type 2 diabetic patients, EDITION IV focuses on patients with type 1 diabetes, and EDITION JP I and II are ongoing in Japan. These trials are designed to assess the relative performance of U100 and the investigational insulin in a range of common clinical scenarios.
"The question is what proportion of patients is going to have a clinically relevant difference in success with the new formulation versus the old one. I don’t know the answer to that yet," Dr. Riddle said in an interview. "U100 has been a really successful insulin, and the likelihood of it going away is really, really low. We’ll probably end up dividing the basal insulin patients into a good many continuing on Lantus U100, and a good many others using newer, longer-acting insulins, U300 being one, but not the only one. When affordability is an issue, taking any insulin is better than not taking it at all."
Dr. Riddle receives research grant support from and is paid consultant to several pharmaceutical companies, among them Sanofi, which sponsors the EDITION trials.
CHICAGO – A new longer-acting basal insulin glargine formulation was as effective as the familiar insulin glargine 100 U/mL/Lantus in controlling glycemia while producing a 21% lower rate of severe nocturnal hypoglycemic events in the phase III EDITION I trial.
The investigational formulation contains insulin glargine 300 U/mL and is known as U300. Like insulin glargine U100, the investigational agent is a once-daily, long-acting insulin analog. But U300 has an even flatter, longer, and more stable pharmacodynamic profile, which translates into a lower risk of hypoglycemia, as shown in the EDITION I trial, principal investigator Dr. Matthew C. Riddle explained at the annual scientific sessions of the American Diabetes Association.
EDITION I included 807 adults with type 2 diabetes who were on an established treatment with at least 42 U/day of basal glargine or NPH along with mealtime insulin with or without metformin. Their mean duration of diabetes was 15.8 years, and their mean body mass index was 36.6 kg/m2. They were randomized to 6 months of open-label insulin glargine U300 or insulin glargine U100 once daily in the evening while continuing on their mealtime insulin. The basal insulin was titrated to achieve a fasting plasma glucose of 80-100 mg/dL.
The primary efficacy endpoint was change in hemoglobin A1c from baseline through month 6. The reductions were the same in both groups: a mean decrease of 0.83%.
The chief secondary endpoint was the occurrence of one or more episodes of severe and/or confirmed nocturnal hypoglycemia to a level of 70 mg/dL or less during months 3-6. The rate was 36.1% in the U300 group, compared with 46% with U100, for a highly significant 21% risk reduction, said Dr. Riddle, professor of medicine at the Oregon Health and Science Center, Portland.
Any nocturnal hypoglycemia during the 6-month study occurred in 45.3% of patients on U300 vs. 59.7% on U100, for a 24% relative risk reduction, he added.
EDITION I is the first in a large worldwide series of phase III trials of the investigational insulin. The recently completed EDITION II trial replicated the EDITION I findings blood glucose reductions similar to those seen in patients taking Lantus, with fewer serious nocturnal hypoglycemic events – in 811 type 2 diabetic patients with earlier-stage disease as evidenced by the enrollment requirement that they be established on basal insulin and oral antidiabetic therapy. EDITION III is ongoing in insulin-naive type 2 diabetic patients, EDITION IV focuses on patients with type 1 diabetes, and EDITION JP I and II are ongoing in Japan. These trials are designed to assess the relative performance of U100 and the investigational insulin in a range of common clinical scenarios.
"The question is what proportion of patients is going to have a clinically relevant difference in success with the new formulation versus the old one. I don’t know the answer to that yet," Dr. Riddle said in an interview. "U100 has been a really successful insulin, and the likelihood of it going away is really, really low. We’ll probably end up dividing the basal insulin patients into a good many continuing on Lantus U100, and a good many others using newer, longer-acting insulins, U300 being one, but not the only one. When affordability is an issue, taking any insulin is better than not taking it at all."
Dr. Riddle receives research grant support from and is paid consultant to several pharmaceutical companies, among them Sanofi, which sponsors the EDITION trials.
CHICAGO – A new longer-acting basal insulin glargine formulation was as effective as the familiar insulin glargine 100 U/mL/Lantus in controlling glycemia while producing a 21% lower rate of severe nocturnal hypoglycemic events in the phase III EDITION I trial.
The investigational formulation contains insulin glargine 300 U/mL and is known as U300. Like insulin glargine U100, the investigational agent is a once-daily, long-acting insulin analog. But U300 has an even flatter, longer, and more stable pharmacodynamic profile, which translates into a lower risk of hypoglycemia, as shown in the EDITION I trial, principal investigator Dr. Matthew C. Riddle explained at the annual scientific sessions of the American Diabetes Association.
EDITION I included 807 adults with type 2 diabetes who were on an established treatment with at least 42 U/day of basal glargine or NPH along with mealtime insulin with or without metformin. Their mean duration of diabetes was 15.8 years, and their mean body mass index was 36.6 kg/m2. They were randomized to 6 months of open-label insulin glargine U300 or insulin glargine U100 once daily in the evening while continuing on their mealtime insulin. The basal insulin was titrated to achieve a fasting plasma glucose of 80-100 mg/dL.
The primary efficacy endpoint was change in hemoglobin A1c from baseline through month 6. The reductions were the same in both groups: a mean decrease of 0.83%.
The chief secondary endpoint was the occurrence of one or more episodes of severe and/or confirmed nocturnal hypoglycemia to a level of 70 mg/dL or less during months 3-6. The rate was 36.1% in the U300 group, compared with 46% with U100, for a highly significant 21% risk reduction, said Dr. Riddle, professor of medicine at the Oregon Health and Science Center, Portland.
Any nocturnal hypoglycemia during the 6-month study occurred in 45.3% of patients on U300 vs. 59.7% on U100, for a 24% relative risk reduction, he added.
EDITION I is the first in a large worldwide series of phase III trials of the investigational insulin. The recently completed EDITION II trial replicated the EDITION I findings blood glucose reductions similar to those seen in patients taking Lantus, with fewer serious nocturnal hypoglycemic events – in 811 type 2 diabetic patients with earlier-stage disease as evidenced by the enrollment requirement that they be established on basal insulin and oral antidiabetic therapy. EDITION III is ongoing in insulin-naive type 2 diabetic patients, EDITION IV focuses on patients with type 1 diabetes, and EDITION JP I and II are ongoing in Japan. These trials are designed to assess the relative performance of U100 and the investigational insulin in a range of common clinical scenarios.
"The question is what proportion of patients is going to have a clinically relevant difference in success with the new formulation versus the old one. I don’t know the answer to that yet," Dr. Riddle said in an interview. "U100 has been a really successful insulin, and the likelihood of it going away is really, really low. We’ll probably end up dividing the basal insulin patients into a good many continuing on Lantus U100, and a good many others using newer, longer-acting insulins, U300 being one, but not the only one. When affordability is an issue, taking any insulin is better than not taking it at all."
Dr. Riddle receives research grant support from and is paid consultant to several pharmaceutical companies, among them Sanofi, which sponsors the EDITION trials.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major Finding: An investigational longer-acting insulin glargine formulation resulted in equivalent glucose control with a 21% reduction in the risk of severe nocturnal hypoglycemic events, compared with insulin glargine 100 U/mL (Lantus) in type 2 diabetes patients.
Data Source: EDITION I, a 6-month, open-label, phase III randomized trial in 807 patients with type 2 diabetes on mealtime and basal insulin with or without oral therapy.
Disclosures: The EDITION I trial was sponsored by Sanofi. The presenter is a consultant to Sanofi and several other pharmaceutical companies.

Initial triple-drug therapy best in new T2DM
CHICAGO – Starting patients with newly diagnosed type 2 diabetes on the combination of metformin, pioglitazone, and exenatide from the get-go proved superior to standard guideline-recommended sequential add-on therapy with metformin, a sulfonylurea, and basal insulin in a 2-year randomized trial.
The triple-therapy group achieved significantly greater and more durable reductions in hemoglobin A1c with less risk of hypoglycemia than patients on the conventional treatment strategy. They also experienced weight loss rather than the weight gain seen with conventional management, Dr. Muhammad A. Abdul-Ghani reported at the annual scientific sessions of the American Diabetes Association.

The observed differences in therapeutic effectiveness and safety validate the strategy behind initial triple therapy, namely that it’s better to target the insulin resistance and progressive beta cell failure that constitute the core metabolic defects responsible for hyperglycemia and type 2 diabetes instead of simply focusing on lowering plasma glucose, added Dr. Abdul-Ghani of the University of Texas Health Science Center, San Antonio.
"My take home message for you is when you treat people with type 2 diabetes, treat the disease – diabetes – not the symptom of hyperglycemia," he declared.
The open-label study comprised 155 patients who had been recently diagnosed with type 2 diabetes. Average age was 47 years, average body mass index was 30.5 kg/m2, and average baseline HbA1c was 8.6%. Those randomized to triple therapy started on metformin at 1,000 mg/day, pioglitazone at 15 mg/day, and exenatide at 5 mcg twice daily. After 1 month, the doses of all three drugs were doubled. If 3 months into the study a patient hadn’t achieved the goal of an HbA1c below 6.5%, pioglitazone was boosted to 45 mg/day.
The conventional treatment group started on metformin followed by sequential addition of glipizide and then basal insulin as needed in an effort to reach an HbA1c below 6.5%.
The primary outcome was the HbA1c difference between the two groups at 2 years. The mean HbA1c decreased from 8.6% at baseline to 6% in the triple-therapy group, significantly better than the 6.6% with conventional therapy. The median HbA1c was 5.8% with triple therapy compared to 6.4% with add-on therapy. In the triple therapy group, 60% had an HbA1c below 6% compared to 27% of controls. In the triple therapy, 92% met the ADA target of an HbA1c below 7% compared to 72% on conventional therapy.
Participants were seen in the clinic every 3 months. Treatment failure, defined as an HbA1c above 6.5% on two consecutive visits 3 months apart, occurred in 17% of the triple-therapy group and 42% on conventional therapy.
Even though the mean HbA1c was 0.5% lower in the triple-therapy group, their incidence of hypoglycemia over the course of 2 years was also significantly lower: 15% versus 46% in the conventional therapy group, the diabetologist continued.
Patients receiving triple therapy had a mean 1.2-kg weight loss over 24 months versus a 4.1-kg weight gain with conventional therapy.
In a multivariate regression analysis, triple-therapy was associated with an 84% reduction in the risk of treatment failure. The other protective factor was age: for each decade beyond age 40 years, a patient’s risk of treatment failure was reduced by 64%.
"We don’t know yet whether this is an issue of patient compliance with therapy or there is something in the pathophysiology of the disease that makes age protective against treatment failure," Dr. Abdul-Ghani said.
Metformin was selected for inclusion in the triple-drug regimen because it corrects insulin resistance in the liver, pioglitazone because it corrects insulin resistance in adipocytes and muscle along with preserving beta cell function, and exenatide because it enhances beta cell function, the physician explained.
Several audience members called the study controversial, noting that it was a single-center study with fewer than 200 patients, yet it aims to overturn current guideline-based clinical practice.
In an interview, Dr. Abdul-Ghani said he and his coinvestigators are trying to drum up support for a larger, multicenter follow-up study featuring microvascular complications as a primary endpoint.
"Every novel approach is controversial at first. But if the difference we’ve seen in A1c can be maintained for longer than 24 months it could result in a substantially reduced risk of microvascular complications. That would really be a game changer in terms of attitudes toward patient treatment," he observed.
He added that even in the absence of data showing a reduction in retinopathy and other microvascular complications, he would "absolutely" recommend that physicians start applying initial triple therapy in their patients with newly diagnosed type 2 diabetes.
"Our data are so strong: there is no increased risk, and so many benefits," according to Dr. Abdul-Ghani.
Both metformin and pioglitazone are available as low-cost generics, and with so many new GLP-1 agonists in the developmental pipeline, the cost of an older, twice-daily agent such as exenatide might come down, he said.
The study was funded by the American Diabetes Association with support from Amylin Pharmaceuticals and Takeda. Dr. Abdul-Ghani reported having no conflicts of interest.
CHICAGO – Starting patients with newly diagnosed type 2 diabetes on the combination of metformin, pioglitazone, and exenatide from the get-go proved superior to standard guideline-recommended sequential add-on therapy with metformin, a sulfonylurea, and basal insulin in a 2-year randomized trial.
The triple-therapy group achieved significantly greater and more durable reductions in hemoglobin A1c with less risk of hypoglycemia than patients on the conventional treatment strategy. They also experienced weight loss rather than the weight gain seen with conventional management, Dr. Muhammad A. Abdul-Ghani reported at the annual scientific sessions of the American Diabetes Association.
The observed differences in therapeutic effectiveness and safety validate the strategy behind initial triple therapy, namely that it’s better to target the insulin resistance and progressive beta cell failure that constitute the core metabolic defects responsible for hyperglycemia and type 2 diabetes instead of simply focusing on lowering plasma glucose, added Dr. Abdul-Ghani of the University of Texas Health Science Center, San Antonio.
"My take home message for you is when you treat people with type 2 diabetes, treat the disease – diabetes – not the symptom of hyperglycemia," he declared.
The open-label study comprised 155 patients who had been recently diagnosed with type 2 diabetes. Average age was 47 years, average body mass index was 30.5 kg/m2, and average baseline HbA1c was 8.6%. Those randomized to triple therapy started on metformin at 1,000 mg/day, pioglitazone at 15 mg/day, and exenatide at 5 mcg twice daily. After 1 month, the doses of all three drugs were doubled. If 3 months into the study a patient hadn’t achieved the goal of an HbA1c below 6.5%, pioglitazone was boosted to 45 mg/day.
The conventional treatment group started on metformin followed by sequential addition of glipizide and then basal insulin as needed in an effort to reach an HbA1c below 6.5%.
The primary outcome was the HbA1c difference between the two groups at 2 years. The mean HbA1c decreased from 8.6% at baseline to 6% in the triple-therapy group, significantly better than the 6.6% with conventional therapy. The median HbA1c was 5.8% with triple therapy compared to 6.4% with add-on therapy. In the triple therapy group, 60% had an HbA1c below 6% compared to 27% of controls. In the triple therapy, 92% met the ADA target of an HbA1c below 7% compared to 72% on conventional therapy.
Participants were seen in the clinic every 3 months. Treatment failure, defined as an HbA1c above 6.5% on two consecutive visits 3 months apart, occurred in 17% of the triple-therapy group and 42% on conventional therapy.
Even though the mean HbA1c was 0.5% lower in the triple-therapy group, their incidence of hypoglycemia over the course of 2 years was also significantly lower: 15% versus 46% in the conventional therapy group, the diabetologist continued.
Patients receiving triple therapy had a mean 1.2-kg weight loss over 24 months versus a 4.1-kg weight gain with conventional therapy.
In a multivariate regression analysis, triple-therapy was associated with an 84% reduction in the risk of treatment failure. The other protective factor was age: for each decade beyond age 40 years, a patient’s risk of treatment failure was reduced by 64%.
"We don’t know yet whether this is an issue of patient compliance with therapy or there is something in the pathophysiology of the disease that makes age protective against treatment failure," Dr. Abdul-Ghani said.
Metformin was selected for inclusion in the triple-drug regimen because it corrects insulin resistance in the liver, pioglitazone because it corrects insulin resistance in adipocytes and muscle along with preserving beta cell function, and exenatide because it enhances beta cell function, the physician explained.
Several audience members called the study controversial, noting that it was a single-center study with fewer than 200 patients, yet it aims to overturn current guideline-based clinical practice.
In an interview, Dr. Abdul-Ghani said he and his coinvestigators are trying to drum up support for a larger, multicenter follow-up study featuring microvascular complications as a primary endpoint.
"Every novel approach is controversial at first. But if the difference we’ve seen in A1c can be maintained for longer than 24 months it could result in a substantially reduced risk of microvascular complications. That would really be a game changer in terms of attitudes toward patient treatment," he observed.
He added that even in the absence of data showing a reduction in retinopathy and other microvascular complications, he would "absolutely" recommend that physicians start applying initial triple therapy in their patients with newly diagnosed type 2 diabetes.
"Our data are so strong: there is no increased risk, and so many benefits," according to Dr. Abdul-Ghani.
Both metformin and pioglitazone are available as low-cost generics, and with so many new GLP-1 agonists in the developmental pipeline, the cost of an older, twice-daily agent such as exenatide might come down, he said.
The study was funded by the American Diabetes Association with support from Amylin Pharmaceuticals and Takeda. Dr. Abdul-Ghani reported having no conflicts of interest.
CHICAGO – Starting patients with newly diagnosed type 2 diabetes on the combination of metformin, pioglitazone, and exenatide from the get-go proved superior to standard guideline-recommended sequential add-on therapy with metformin, a sulfonylurea, and basal insulin in a 2-year randomized trial.
The triple-therapy group achieved significantly greater and more durable reductions in hemoglobin A1c with less risk of hypoglycemia than patients on the conventional treatment strategy. They also experienced weight loss rather than the weight gain seen with conventional management, Dr. Muhammad A. Abdul-Ghani reported at the annual scientific sessions of the American Diabetes Association.
The observed differences in therapeutic effectiveness and safety validate the strategy behind initial triple therapy, namely that it’s better to target the insulin resistance and progressive beta cell failure that constitute the core metabolic defects responsible for hyperglycemia and type 2 diabetes instead of simply focusing on lowering plasma glucose, added Dr. Abdul-Ghani of the University of Texas Health Science Center, San Antonio.
"My take home message for you is when you treat people with type 2 diabetes, treat the disease – diabetes – not the symptom of hyperglycemia," he declared.
The open-label study comprised 155 patients who had been recently diagnosed with type 2 diabetes. Average age was 47 years, average body mass index was 30.5 kg/m2, and average baseline HbA1c was 8.6%. Those randomized to triple therapy started on metformin at 1,000 mg/day, pioglitazone at 15 mg/day, and exenatide at 5 mcg twice daily. After 1 month, the doses of all three drugs were doubled. If 3 months into the study a patient hadn’t achieved the goal of an HbA1c below 6.5%, pioglitazone was boosted to 45 mg/day.
The conventional treatment group started on metformin followed by sequential addition of glipizide and then basal insulin as needed in an effort to reach an HbA1c below 6.5%.
The primary outcome was the HbA1c difference between the two groups at 2 years. The mean HbA1c decreased from 8.6% at baseline to 6% in the triple-therapy group, significantly better than the 6.6% with conventional therapy. The median HbA1c was 5.8% with triple therapy compared to 6.4% with add-on therapy. In the triple therapy group, 60% had an HbA1c below 6% compared to 27% of controls. In the triple therapy, 92% met the ADA target of an HbA1c below 7% compared to 72% on conventional therapy.
Participants were seen in the clinic every 3 months. Treatment failure, defined as an HbA1c above 6.5% on two consecutive visits 3 months apart, occurred in 17% of the triple-therapy group and 42% on conventional therapy.
Even though the mean HbA1c was 0.5% lower in the triple-therapy group, their incidence of hypoglycemia over the course of 2 years was also significantly lower: 15% versus 46% in the conventional therapy group, the diabetologist continued.
Patients receiving triple therapy had a mean 1.2-kg weight loss over 24 months versus a 4.1-kg weight gain with conventional therapy.
In a multivariate regression analysis, triple-therapy was associated with an 84% reduction in the risk of treatment failure. The other protective factor was age: for each decade beyond age 40 years, a patient’s risk of treatment failure was reduced by 64%.
"We don’t know yet whether this is an issue of patient compliance with therapy or there is something in the pathophysiology of the disease that makes age protective against treatment failure," Dr. Abdul-Ghani said.
Metformin was selected for inclusion in the triple-drug regimen because it corrects insulin resistance in the liver, pioglitazone because it corrects insulin resistance in adipocytes and muscle along with preserving beta cell function, and exenatide because it enhances beta cell function, the physician explained.
Several audience members called the study controversial, noting that it was a single-center study with fewer than 200 patients, yet it aims to overturn current guideline-based clinical practice.
In an interview, Dr. Abdul-Ghani said he and his coinvestigators are trying to drum up support for a larger, multicenter follow-up study featuring microvascular complications as a primary endpoint.
"Every novel approach is controversial at first. But if the difference we’ve seen in A1c can be maintained for longer than 24 months it could result in a substantially reduced risk of microvascular complications. That would really be a game changer in terms of attitudes toward patient treatment," he observed.
He added that even in the absence of data showing a reduction in retinopathy and other microvascular complications, he would "absolutely" recommend that physicians start applying initial triple therapy in their patients with newly diagnosed type 2 diabetes.
"Our data are so strong: there is no increased risk, and so many benefits," according to Dr. Abdul-Ghani.
Both metformin and pioglitazone are available as low-cost generics, and with so many new GLP-1 agonists in the developmental pipeline, the cost of an older, twice-daily agent such as exenatide might come down, he said.
The study was funded by the American Diabetes Association with support from Amylin Pharmaceuticals and Takeda. Dr. Abdul-Ghani reported having no conflicts of interest.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major Finding: Patients with recently diagnosed type 2 diabetes who were managed from the outset with the triple-drug combination of metformin, pioglitazone, and exenatide went from a mean A1c of 8.6% at baseline to 6% at 2 years, significantly better than the 6.6% achieved with conventional sequential add-on therapy with metformin, a sulfonylurea, and basal insulin.
Data Source: A single-center, open-label, randomized trial involving 155 patients with newly diagnosed type 2 diabetes.
Disclosures: The study was funded by the American Diabetes Association. The presenter reported having no financial conflicts.

Selegiline transdermal system promising for social phobia
HOLLYWOOD, FLA. – Patients with social phobia responded favorably to treatment with the selegiline transdermal system, showing significant improvement in the anxiety component of the Liebowitz Social Anxiety Scale and other efficacy endpoints in a small phase II study.
Twenty patients participated in this 12-week, open-label, pilot study. They were treated with the selegiline transdermal system (Emsam) at a dose of 6 mg/24 hours. They demonstrated a significant mean 7.1-point drop from a baseline score of 17.6 on the avoidance component of the Liebowitz Social Anxiety Scale, as well as a 6.6-point drop on the scale’s anxiety component, a result that just missed statistical significance (P = .053), Kimberly Portland, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The selegiline transdermal system is approved for the treatment of major depression. Selegiline is an MAO-A and -B inhibitor. Its transdermal administration bypasses the GI tract and liver, thereby essentially eliminating the risks of hypertensive crisis and dietary interactions with tyramine-rich foods, which have demoted the traditional oral monoamine oxidase (MAO) inhibitors in antidepressant treatment hierarchies.
Oral MAO inhibitors previously have been shown in clinical trials to be at least as effective as cognitive-behavioral therapy or benzodiazepines in treating social phobia. So it seemed appropriate to examine the safety and efficacy of the selegiline transdermal system for this condition, which has an estimated prevalence in the United States of 5%-12%. The study was particularly timely because the selegiline transdermal system has dopaminergic effects, and recent evidence points to dopamine systems as playing a role in social phobia, explained Dr. Portland of Mylan Specialty, Basking Ridge, N.J., which markets Emsam.
In addition to the favorable results on the Liebowitz Social Anxiety Scale over the course of 12 weeks, participants also showed statistically significant improvements on the Social Life/Leisure Activities domain of the Sheehan Disability Scale, the Clinical Global Impression Severity of Illness Scale, and the Clinical Global Impression Change Scale.
Three patients dropped out of the study because of adverse events. The most common adverse events in the study were headache, reported by 25% of participants, and application site reactions, reported by 20%.
The major limitation of this pilot study is its small size, Dr. Portland noted. Further studies incorporating larger patient numbers, a double-blind format, and higher doses of the MAO inhibitor are planned.
HOLLYWOOD, FLA. – Patients with social phobia responded favorably to treatment with the selegiline transdermal system, showing significant improvement in the anxiety component of the Liebowitz Social Anxiety Scale and other efficacy endpoints in a small phase II study.
Twenty patients participated in this 12-week, open-label, pilot study. They were treated with the selegiline transdermal system (Emsam) at a dose of 6 mg/24 hours. They demonstrated a significant mean 7.1-point drop from a baseline score of 17.6 on the avoidance component of the Liebowitz Social Anxiety Scale, as well as a 6.6-point drop on the scale’s anxiety component, a result that just missed statistical significance (P = .053), Kimberly Portland, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The selegiline transdermal system is approved for the treatment of major depression. Selegiline is an MAO-A and -B inhibitor. Its transdermal administration bypasses the GI tract and liver, thereby essentially eliminating the risks of hypertensive crisis and dietary interactions with tyramine-rich foods, which have demoted the traditional oral monoamine oxidase (MAO) inhibitors in antidepressant treatment hierarchies.
Oral MAO inhibitors previously have been shown in clinical trials to be at least as effective as cognitive-behavioral therapy or benzodiazepines in treating social phobia. So it seemed appropriate to examine the safety and efficacy of the selegiline transdermal system for this condition, which has an estimated prevalence in the United States of 5%-12%. The study was particularly timely because the selegiline transdermal system has dopaminergic effects, and recent evidence points to dopamine systems as playing a role in social phobia, explained Dr. Portland of Mylan Specialty, Basking Ridge, N.J., which markets Emsam.
In addition to the favorable results on the Liebowitz Social Anxiety Scale over the course of 12 weeks, participants also showed statistically significant improvements on the Social Life/Leisure Activities domain of the Sheehan Disability Scale, the Clinical Global Impression Severity of Illness Scale, and the Clinical Global Impression Change Scale.
Three patients dropped out of the study because of adverse events. The most common adverse events in the study were headache, reported by 25% of participants, and application site reactions, reported by 20%.
The major limitation of this pilot study is its small size, Dr. Portland noted. Further studies incorporating larger patient numbers, a double-blind format, and higher doses of the MAO inhibitor are planned.
HOLLYWOOD, FLA. – Patients with social phobia responded favorably to treatment with the selegiline transdermal system, showing significant improvement in the anxiety component of the Liebowitz Social Anxiety Scale and other efficacy endpoints in a small phase II study.
Twenty patients participated in this 12-week, open-label, pilot study. They were treated with the selegiline transdermal system (Emsam) at a dose of 6 mg/24 hours. They demonstrated a significant mean 7.1-point drop from a baseline score of 17.6 on the avoidance component of the Liebowitz Social Anxiety Scale, as well as a 6.6-point drop on the scale’s anxiety component, a result that just missed statistical significance (P = .053), Kimberly Portland, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The selegiline transdermal system is approved for the treatment of major depression. Selegiline is an MAO-A and -B inhibitor. Its transdermal administration bypasses the GI tract and liver, thereby essentially eliminating the risks of hypertensive crisis and dietary interactions with tyramine-rich foods, which have demoted the traditional oral monoamine oxidase (MAO) inhibitors in antidepressant treatment hierarchies.
Oral MAO inhibitors previously have been shown in clinical trials to be at least as effective as cognitive-behavioral therapy or benzodiazepines in treating social phobia. So it seemed appropriate to examine the safety and efficacy of the selegiline transdermal system for this condition, which has an estimated prevalence in the United States of 5%-12%. The study was particularly timely because the selegiline transdermal system has dopaminergic effects, and recent evidence points to dopamine systems as playing a role in social phobia, explained Dr. Portland of Mylan Specialty, Basking Ridge, N.J., which markets Emsam.
In addition to the favorable results on the Liebowitz Social Anxiety Scale over the course of 12 weeks, participants also showed statistically significant improvements on the Social Life/Leisure Activities domain of the Sheehan Disability Scale, the Clinical Global Impression Severity of Illness Scale, and the Clinical Global Impression Change Scale.
Three patients dropped out of the study because of adverse events. The most common adverse events in the study were headache, reported by 25% of participants, and application site reactions, reported by 20%.
The major limitation of this pilot study is its small size, Dr. Portland noted. Further studies incorporating larger patient numbers, a double-blind format, and higher doses of the MAO inhibitor are planned.
AT THE NCDEU MEETING
Major finding: Patients with social phobia showed significant improvement on multiple efficacy measures in response to treatment with the selegiline transdermal system.
Data source: This was an open-label, 20-patient, 12-week phase II study.
Disclosures: The study was sponsored by Mylan Specialty, which markets the selegiline transdermal system for the treatment of major depression. The presenter is a company employee.