User login
Urinary symptoms often unaddressed in MS
ORLANDO – Lower urinary tract symptoms in patients with multiple sclerosis are common, bothersome, and often undiscussed and untreated, according to a national patient survey.
The online survey was completed by a convenience sample of 1,052 MS patients recruited through the National Multiple Sclerosis Society and other patient advocacy organizations. Fully 88% of the respondents indicated they have lower urinary tract symptoms involving bladder dysfunction and urinary incontinence. The source of these common symptoms in patients with MS is increased contractile activity of the bladder’s detrusor muscle.
The most common lower urinary tract symptom reported by survey respondents was terminal dribble upon voiding, which affected 65% of patients. The next most common symptoms were urinary urgency, experienced by 62%, and incomplete emptying, cited by 61%, Kristin M. Khalaf, Pharm.D., reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Fifty-three percent of patients reported having urgency urinary incontinence, and 45% complained of stress urinary incontinence.
The lower urinary tract symptom that patients found most bothersome was urgency; indeed, one-third of the overall study population indicated they were bothered "quite a bit" or "a great deal" by this problem. Twenty-nine percent of respondents stated they were bothered at least quite a bit by urgency incontinence, added Dr. Khalaf of Allergan, Irvine, Calif.
Only one-third of the 922 MS patients with lower urinary tract symptoms had discussed their symptoms with a health care provider during the past year. When they did speak with a professional, 74% of the time it was with their neurologist. Fifty-two percent spoke to their primary care physician about their problem within the last year.
Among the 42% of survey respondents who indicated they were bothered at least quite a bit by urinary incontinence, 46% hadn’t discussed the problem with a health care provider within the past year, and 35% had never received any form of treatment for it.
Among patients who had ever discussed their lower urinary tract symptoms with a physician or other health care professional, 38% reported currently treating their problem via pelvic exercises or bladder training, 23% were using an oral anticholinergic agent, 4% were taking herbal medicines for their symptoms, 3% were receiving botulinum toxin type A (onabotulinumtoxinA) injections, and 2% had a neural stimulation device.
Most patients currently receiving treatment for their lower urinary tract symptoms pronounced themselves very or somewhat satisfied with their therapy.
The survey was funded by Allergan.
ORLANDO – Lower urinary tract symptoms in patients with multiple sclerosis are common, bothersome, and often undiscussed and untreated, according to a national patient survey.
The online survey was completed by a convenience sample of 1,052 MS patients recruited through the National Multiple Sclerosis Society and other patient advocacy organizations. Fully 88% of the respondents indicated they have lower urinary tract symptoms involving bladder dysfunction and urinary incontinence. The source of these common symptoms in patients with MS is increased contractile activity of the bladder’s detrusor muscle.
The most common lower urinary tract symptom reported by survey respondents was terminal dribble upon voiding, which affected 65% of patients. The next most common symptoms were urinary urgency, experienced by 62%, and incomplete emptying, cited by 61%, Kristin M. Khalaf, Pharm.D., reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Fifty-three percent of patients reported having urgency urinary incontinence, and 45% complained of stress urinary incontinence.
The lower urinary tract symptom that patients found most bothersome was urgency; indeed, one-third of the overall study population indicated they were bothered "quite a bit" or "a great deal" by this problem. Twenty-nine percent of respondents stated they were bothered at least quite a bit by urgency incontinence, added Dr. Khalaf of Allergan, Irvine, Calif.
Only one-third of the 922 MS patients with lower urinary tract symptoms had discussed their symptoms with a health care provider during the past year. When they did speak with a professional, 74% of the time it was with their neurologist. Fifty-two percent spoke to their primary care physician about their problem within the last year.
Among the 42% of survey respondents who indicated they were bothered at least quite a bit by urinary incontinence, 46% hadn’t discussed the problem with a health care provider within the past year, and 35% had never received any form of treatment for it.
Among patients who had ever discussed their lower urinary tract symptoms with a physician or other health care professional, 38% reported currently treating their problem via pelvic exercises or bladder training, 23% were using an oral anticholinergic agent, 4% were taking herbal medicines for their symptoms, 3% were receiving botulinum toxin type A (onabotulinumtoxinA) injections, and 2% had a neural stimulation device.
Most patients currently receiving treatment for their lower urinary tract symptoms pronounced themselves very or somewhat satisfied with their therapy.
The survey was funded by Allergan.
ORLANDO – Lower urinary tract symptoms in patients with multiple sclerosis are common, bothersome, and often undiscussed and untreated, according to a national patient survey.
The online survey was completed by a convenience sample of 1,052 MS patients recruited through the National Multiple Sclerosis Society and other patient advocacy organizations. Fully 88% of the respondents indicated they have lower urinary tract symptoms involving bladder dysfunction and urinary incontinence. The source of these common symptoms in patients with MS is increased contractile activity of the bladder’s detrusor muscle.
The most common lower urinary tract symptom reported by survey respondents was terminal dribble upon voiding, which affected 65% of patients. The next most common symptoms were urinary urgency, experienced by 62%, and incomplete emptying, cited by 61%, Kristin M. Khalaf, Pharm.D., reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Fifty-three percent of patients reported having urgency urinary incontinence, and 45% complained of stress urinary incontinence.
The lower urinary tract symptom that patients found most bothersome was urgency; indeed, one-third of the overall study population indicated they were bothered "quite a bit" or "a great deal" by this problem. Twenty-nine percent of respondents stated they were bothered at least quite a bit by urgency incontinence, added Dr. Khalaf of Allergan, Irvine, Calif.
Only one-third of the 922 MS patients with lower urinary tract symptoms had discussed their symptoms with a health care provider during the past year. When they did speak with a professional, 74% of the time it was with their neurologist. Fifty-two percent spoke to their primary care physician about their problem within the last year.
Among the 42% of survey respondents who indicated they were bothered at least quite a bit by urinary incontinence, 46% hadn’t discussed the problem with a health care provider within the past year, and 35% had never received any form of treatment for it.
Among patients who had ever discussed their lower urinary tract symptoms with a physician or other health care professional, 38% reported currently treating their problem via pelvic exercises or bladder training, 23% were using an oral anticholinergic agent, 4% were taking herbal medicines for their symptoms, 3% were receiving botulinum toxin type A (onabotulinumtoxinA) injections, and 2% had a neural stimulation device.
Most patients currently receiving treatment for their lower urinary tract symptoms pronounced themselves very or somewhat satisfied with their therapy.
The survey was funded by Allergan.
AT THE CMSC/ACTRIMS ANNUAL MEETING
Major Finding: Eighty-eight percent of patients with multiple sclerosis who responded to an online survey indicated they experience lower urinary tract symptoms, but only one-third of those with such symptoms reported having discussed the matter with a physician or other health professional within the past year.
Data Source: The survey included 1,052 respondents, a convenience sample recruited from several patient advocacy organizations.
Disclosures: The study was sponsored by Allergan. The presenter is a company employee.
BRUISE CONTROL: Warfarin bests heparin bridging
DENVER – Uninterrupted warfarin therapy during pacemaker or implantable cardioverter-defibrillator surgery in patients at high thromboembolic risk proved superior to the guideline-recommended practice of discontinuing warfarin and bridging with heparin, according to a large, multicenter, randomized clinical trial.
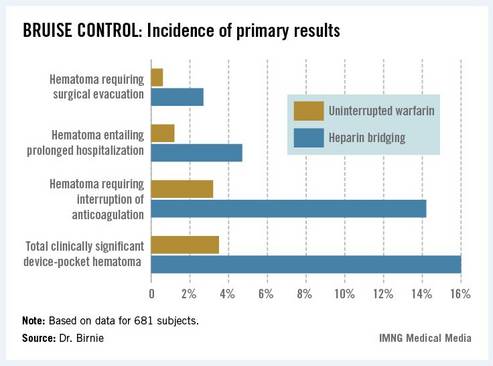
The primary outcome in the 17-center, 681-patient BRUISE CONTROL (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial) was the incidence of clinically significant device-pocket hematoma. The rate was 16% in patients randomized to heparin bridging, compared with 3.5% with uninterrupted warfarin, Dr. David H. Birnie reported at the annual meeting of the Heart Rhythm Society.
These results are clearly practice changing. Heparin bridging has been the standard of care. It is recommended in this common clinical scenario in all of the major guidelines, but that’s bound to change as a result of BRUISE CONTROL, predicted Dr. Birnie of the University of Ottawa Heart Institute.
"This trial was a home run. It was unequivocally positive," he commented. "For sure, our clinical practice changed as soon as we saw those results."
Device-pocket hematoma is a "very nasty" complication of cardiac device surgery, Dr. Birnie noted. It is quite painful, can cause device infection, and is difficult to treat. Clinically significant device-pocket hematoma was defined in this trial as a hematoma resulting in prolonged hospitalization for an additional day or more, or interruption of oral anticoagulation for at least 24 hours, and/or requiring additional surgery. All three components of the primary endpoint were significantly less frequent in the uninterrupted warfarin group (see chart).
Performing device surgery in patients on uninterrupted warfarin with a median international normalized ratio (INR) of 2.3 was not associated with any increase in major perioperative bleeding or other surgical or thromboembolic complications. And patient satisfaction surveys indicated subjects greatly preferred having their procedure without stopping their warfarin.
BRUISE CONTROL was planned as a definitive 1,000-patient clinical trial. However, the Data and Safety Monitoring Board halted the study after an interim analysis involving the first 681 subjects.
Of note, this was a study restricted to patients at high stroke risk – greater than 5% annually – as defined by the presence of atrial fibrillation and a CHADS2 score of 3 or more or the presence of a mechanical heart valve.
Dr. Birnie said that although it seems counterintuitive to have less bleeding when cardiac device surgery is performed on a fully anticoagulated patient, as occurred in BRUISE CONTROL, he and his coinvestigators have an explanatory hypothesis: "When bleeding occurs in a fully anticoagulated patient, the operator can readily see it and address it. On the contrary, with bridging you get hemostasis at the time of surgery, but you may have missed a tiny little thing, and then 24 hours later when you start up your heparin bridging again, that’s when the bleeding occurs. It’s a physiologically plausible explanation," he said.
In a multivariate analysis, neither the use of pressure dressings, nor the use of sandbags, nor injection of antibleeding agents into the device pocket before closure had any significant impact on the incidence of clinically significant hematomas. Only three factors did: uninterrupted warfarin therapy, which reduced the risk by 84%; aspirin therapy, which doubled the risk; and the presence of diabetes mellitus, which was inexplicably associated with a 52% reduction in hematoma risk.
Each year roughly 1.6 million people worldwide undergo pacemaker or implantable cardioverter-defibrillator (ICD) implantation. Up to one-third of them are on long-term oral anticoagulation, most commonly with warfarin.
Dr. Birnie stressed that the BRUISE CONTROL findings have no relevance to patients on one of the new oral anticoagulants.
"The whole risk/benefit ratio of the new agents is completely different from warfarin. Their onset and offset of action is hours as opposed to the 5 days for warfarin," he noted.
With that point in mind, he and his coinvestigators have just started the BRUISE CONTROL-2 trial, which is examining whether it’s better to stop the new agents around the time of device surgery or continue the medication uninterrupted.
Simultaneous with Dr. Birnie’s presentation of the BRUISE CONTROL data in Denver, the study results were published online (N. Engl. J. Med. 2013 May 9 [doi: 10.1056/NEJMoa1302946]).
BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
DENVER – Uninterrupted warfarin therapy during pacemaker or implantable cardioverter-defibrillator surgery in patients at high thromboembolic risk proved superior to the guideline-recommended practice of discontinuing warfarin and bridging with heparin, according to a large, multicenter, randomized clinical trial.
The primary outcome in the 17-center, 681-patient BRUISE CONTROL (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial) was the incidence of clinically significant device-pocket hematoma. The rate was 16% in patients randomized to heparin bridging, compared with 3.5% with uninterrupted warfarin, Dr. David H. Birnie reported at the annual meeting of the Heart Rhythm Society.
These results are clearly practice changing. Heparin bridging has been the standard of care. It is recommended in this common clinical scenario in all of the major guidelines, but that’s bound to change as a result of BRUISE CONTROL, predicted Dr. Birnie of the University of Ottawa Heart Institute.
"This trial was a home run. It was unequivocally positive," he commented. "For sure, our clinical practice changed as soon as we saw those results."
Device-pocket hematoma is a "very nasty" complication of cardiac device surgery, Dr. Birnie noted. It is quite painful, can cause device infection, and is difficult to treat. Clinically significant device-pocket hematoma was defined in this trial as a hematoma resulting in prolonged hospitalization for an additional day or more, or interruption of oral anticoagulation for at least 24 hours, and/or requiring additional surgery. All three components of the primary endpoint were significantly less frequent in the uninterrupted warfarin group (see chart).
Performing device surgery in patients on uninterrupted warfarin with a median international normalized ratio (INR) of 2.3 was not associated with any increase in major perioperative bleeding or other surgical or thromboembolic complications. And patient satisfaction surveys indicated subjects greatly preferred having their procedure without stopping their warfarin.
BRUISE CONTROL was planned as a definitive 1,000-patient clinical trial. However, the Data and Safety Monitoring Board halted the study after an interim analysis involving the first 681 subjects.
Of note, this was a study restricted to patients at high stroke risk – greater than 5% annually – as defined by the presence of atrial fibrillation and a CHADS2 score of 3 or more or the presence of a mechanical heart valve.
Dr. Birnie said that although it seems counterintuitive to have less bleeding when cardiac device surgery is performed on a fully anticoagulated patient, as occurred in BRUISE CONTROL, he and his coinvestigators have an explanatory hypothesis: "When bleeding occurs in a fully anticoagulated patient, the operator can readily see it and address it. On the contrary, with bridging you get hemostasis at the time of surgery, but you may have missed a tiny little thing, and then 24 hours later when you start up your heparin bridging again, that’s when the bleeding occurs. It’s a physiologically plausible explanation," he said.
In a multivariate analysis, neither the use of pressure dressings, nor the use of sandbags, nor injection of antibleeding agents into the device pocket before closure had any significant impact on the incidence of clinically significant hematomas. Only three factors did: uninterrupted warfarin therapy, which reduced the risk by 84%; aspirin therapy, which doubled the risk; and the presence of diabetes mellitus, which was inexplicably associated with a 52% reduction in hematoma risk.
Each year roughly 1.6 million people worldwide undergo pacemaker or implantable cardioverter-defibrillator (ICD) implantation. Up to one-third of them are on long-term oral anticoagulation, most commonly with warfarin.
Dr. Birnie stressed that the BRUISE CONTROL findings have no relevance to patients on one of the new oral anticoagulants.
"The whole risk/benefit ratio of the new agents is completely different from warfarin. Their onset and offset of action is hours as opposed to the 5 days for warfarin," he noted.
With that point in mind, he and his coinvestigators have just started the BRUISE CONTROL-2 trial, which is examining whether it’s better to stop the new agents around the time of device surgery or continue the medication uninterrupted.
Simultaneous with Dr. Birnie’s presentation of the BRUISE CONTROL data in Denver, the study results were published online (N. Engl. J. Med. 2013 May 9 [doi: 10.1056/NEJMoa1302946]).
BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
DENVER – Uninterrupted warfarin therapy during pacemaker or implantable cardioverter-defibrillator surgery in patients at high thromboembolic risk proved superior to the guideline-recommended practice of discontinuing warfarin and bridging with heparin, according to a large, multicenter, randomized clinical trial.
The primary outcome in the 17-center, 681-patient BRUISE CONTROL (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial) was the incidence of clinically significant device-pocket hematoma. The rate was 16% in patients randomized to heparin bridging, compared with 3.5% with uninterrupted warfarin, Dr. David H. Birnie reported at the annual meeting of the Heart Rhythm Society.
These results are clearly practice changing. Heparin bridging has been the standard of care. It is recommended in this common clinical scenario in all of the major guidelines, but that’s bound to change as a result of BRUISE CONTROL, predicted Dr. Birnie of the University of Ottawa Heart Institute.
"This trial was a home run. It was unequivocally positive," he commented. "For sure, our clinical practice changed as soon as we saw those results."
Device-pocket hematoma is a "very nasty" complication of cardiac device surgery, Dr. Birnie noted. It is quite painful, can cause device infection, and is difficult to treat. Clinically significant device-pocket hematoma was defined in this trial as a hematoma resulting in prolonged hospitalization for an additional day or more, or interruption of oral anticoagulation for at least 24 hours, and/or requiring additional surgery. All three components of the primary endpoint were significantly less frequent in the uninterrupted warfarin group (see chart).
Performing device surgery in patients on uninterrupted warfarin with a median international normalized ratio (INR) of 2.3 was not associated with any increase in major perioperative bleeding or other surgical or thromboembolic complications. And patient satisfaction surveys indicated subjects greatly preferred having their procedure without stopping their warfarin.
BRUISE CONTROL was planned as a definitive 1,000-patient clinical trial. However, the Data and Safety Monitoring Board halted the study after an interim analysis involving the first 681 subjects.
Of note, this was a study restricted to patients at high stroke risk – greater than 5% annually – as defined by the presence of atrial fibrillation and a CHADS2 score of 3 or more or the presence of a mechanical heart valve.
Dr. Birnie said that although it seems counterintuitive to have less bleeding when cardiac device surgery is performed on a fully anticoagulated patient, as occurred in BRUISE CONTROL, he and his coinvestigators have an explanatory hypothesis: "When bleeding occurs in a fully anticoagulated patient, the operator can readily see it and address it. On the contrary, with bridging you get hemostasis at the time of surgery, but you may have missed a tiny little thing, and then 24 hours later when you start up your heparin bridging again, that’s when the bleeding occurs. It’s a physiologically plausible explanation," he said.
In a multivariate analysis, neither the use of pressure dressings, nor the use of sandbags, nor injection of antibleeding agents into the device pocket before closure had any significant impact on the incidence of clinically significant hematomas. Only three factors did: uninterrupted warfarin therapy, which reduced the risk by 84%; aspirin therapy, which doubled the risk; and the presence of diabetes mellitus, which was inexplicably associated with a 52% reduction in hematoma risk.
Each year roughly 1.6 million people worldwide undergo pacemaker or implantable cardioverter-defibrillator (ICD) implantation. Up to one-third of them are on long-term oral anticoagulation, most commonly with warfarin.
Dr. Birnie stressed that the BRUISE CONTROL findings have no relevance to patients on one of the new oral anticoagulants.
"The whole risk/benefit ratio of the new agents is completely different from warfarin. Their onset and offset of action is hours as opposed to the 5 days for warfarin," he noted.
With that point in mind, he and his coinvestigators have just started the BRUISE CONTROL-2 trial, which is examining whether it’s better to stop the new agents around the time of device surgery or continue the medication uninterrupted.
Simultaneous with Dr. Birnie’s presentation of the BRUISE CONTROL data in Denver, the study results were published online (N. Engl. J. Med. 2013 May 9 [doi: 10.1056/NEJMoa1302946]).
BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
Major finding: The incidence of clinically significant device-pocket hematoma in patients at high thromboembolic risk who underwent pacemaker or implantable cardioverter-defibrillator surgery was 3.5% if they remained on full-dose warfarin, compared with 16% with the guideline-recommended, standard-of-care practice of interrupting warfarin in favor of heparin bridging.
Data source: BRUISE CONTROL was a 681-patient, 17-center randomized clinical trial.
Disclosures: BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
Study: Reablate, don't medicate, after failed AF ablation
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. The question of what to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo ablation as well (see graphic).
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group.
Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachy-arrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication because of intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. The question of what to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo ablation as well (see graphic).
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group.
Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachy-arrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication because of intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. The question of what to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo ablation as well (see graphic).
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group.
Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachy-arrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication because of intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
Major Finding: At 3 years after a first ablation procedure had failed for symptomatic paroxysmal atrial fibrillation, 12% of those randomized to drug therapy and 58% of those in the redo-ablation group were free of atrial tachyarrhythmias.
Data Source: A randomized, prospective, multicenter clinical trial involving 154 patients whose atrial arrhythmia status was monitored via implantable loop recorder.
Disclosures: The presenter reported having no conflicts of interest.
Evidence of delayed cancer detection in MS patients
ORLANDO – Patients with multiple sclerosis are at significantly reduced risk of being diagnosed with cancer, compared with the general population, but delayed cancer detection – that is, diagnostic neglect – appears to be a contributing factor, a study from British Columbia has shown.
"Diagnostic neglect is unlikely to account for the entire reduced cancer risk that we’re seeing, but I think it could have major implications for the health, well-being, and longevity of people with multiple sclerosis," said Helen Tremlett, Ph.D, a neuroepidemiologist at the University of British Columbia, Vancouver.
In the population-based Malignancy and Multiple Sclerosis (MaMS) study, she and her coinvestigators linked data from the British Columbia MS registry with the provincial cancer registry. The study included 6,820 MS patients who visited a British Columbia MS clinic in 1980-2004. Most had never been exposed to an immunomodulatory therapy. They had a collective 110,666 person-years of follow-up. Their cancer incidence over time was compared with that of the age-, sex-, and calendar year–matched general population of British Columbia.

The standardized incidence ratio for all cancers in the MS cohort was 0.86, meaning MS patients had a highly significant overall 14% reduction in the risk of being diagnosed with cancer. The risk reduction was particularly striking for colorectal cancer: Patients with MS were 44% less likely than controls to be diagnosed with this malignancy.
The cancer risk reductions were similar in men and women with MS, and in those with relapsing-remitting as compared with primary progressive MS, Dr. Tremlett reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Of note, the cancer risk reduction identified in the MaMS study was consistent with an earlier meta-analysis of five studies by other investigators around the world, who concluded that the risk of being diagnosed with cancer was 8% lower in MS patients than controls, a statistically significant difference (J. Neurol. Neurosurg. Psychiatry 2010;81:1413-4).
Unlike the other researchers, however, Dr. Tremlett and her coworkers also looked at tumor size at the time of diagnosis for the four most common cancers: breast, prostate, lung, and colorectal cancer. They found a consistent pattern: MS patients diagnosed with cancer had a lower-than-expected rate of the smaller T1 and T2 tumors than in the matched general population with cancer, and a greater-than-expected rate of T3 and T4 tumors.
The most likely explanation for the observed larger-than-expected tumor size is that when MS patients report, say, a new sense of fatigue, it may be ascribed to their MS, whereas if a patient from the general population had the same complaint, a battery of tests would be ordered, she said.
The MS patients’ 14% reduction in the risk of being diagnosed with cancer had an impressive P value. In contrast, the association between MS and larger tumor size, while statistically significant, had a less than stellar P value of .04. For this reason, Dr. Tremlett said she doesn’t believe diagnostic neglect is the entire explanation for the MS patients’ reduction in cancer incidence. Other possible contributing factors worthy of further study are that MS patients’ hypervigilant immune system renders them less vulnerable to cancer growth, or that MS and cancer share a common and as-yet-unidentified genetic predisposition, or that upon receiving the diagnosis of MS, affected patients adopt a healthier lifestyle.
Dr. Tremlett stressed the importance of being on the lookout for cancer and other aging-related comorbidities, especially now that patients with MS are living longer. In a recent study of 6,917 MS patients in British Columbia, she and her coinvestigators determined that the median survival age was 78.6 years for women and 74.3 years for men. Those are some of the longest life spans ever reported, but still about 6 years less than expected for the general British Columbia population.
Median survival from disease onset was markedly longer for patients with relapsing-onset MS than primary progressive MS: 49.7 years compared with 32.5 years. However, the two groups lived to about the same age.
Patients with primary progressive MS had a 1.52-fold greater relative mortality risk than did those with relapsing-onset MS. Women with primary progressive MS had a 1.55-fold survival disadvantage compared with men with primary progressive disease (J. Neurol. Neurosurg. Psychiatry 2012;83:61-6).
The MaMS study is sponsored by the Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada. Dr. Tremlett reported having no relevant financial disclosures.
ORLANDO – Patients with multiple sclerosis are at significantly reduced risk of being diagnosed with cancer, compared with the general population, but delayed cancer detection – that is, diagnostic neglect – appears to be a contributing factor, a study from British Columbia has shown.
"Diagnostic neglect is unlikely to account for the entire reduced cancer risk that we’re seeing, but I think it could have major implications for the health, well-being, and longevity of people with multiple sclerosis," said Helen Tremlett, Ph.D, a neuroepidemiologist at the University of British Columbia, Vancouver.
In the population-based Malignancy and Multiple Sclerosis (MaMS) study, she and her coinvestigators linked data from the British Columbia MS registry with the provincial cancer registry. The study included 6,820 MS patients who visited a British Columbia MS clinic in 1980-2004. Most had never been exposed to an immunomodulatory therapy. They had a collective 110,666 person-years of follow-up. Their cancer incidence over time was compared with that of the age-, sex-, and calendar year–matched general population of British Columbia.
The standardized incidence ratio for all cancers in the MS cohort was 0.86, meaning MS patients had a highly significant overall 14% reduction in the risk of being diagnosed with cancer. The risk reduction was particularly striking for colorectal cancer: Patients with MS were 44% less likely than controls to be diagnosed with this malignancy.
The cancer risk reductions were similar in men and women with MS, and in those with relapsing-remitting as compared with primary progressive MS, Dr. Tremlett reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Of note, the cancer risk reduction identified in the MaMS study was consistent with an earlier meta-analysis of five studies by other investigators around the world, who concluded that the risk of being diagnosed with cancer was 8% lower in MS patients than controls, a statistically significant difference (J. Neurol. Neurosurg. Psychiatry 2010;81:1413-4).
Unlike the other researchers, however, Dr. Tremlett and her coworkers also looked at tumor size at the time of diagnosis for the four most common cancers: breast, prostate, lung, and colorectal cancer. They found a consistent pattern: MS patients diagnosed with cancer had a lower-than-expected rate of the smaller T1 and T2 tumors than in the matched general population with cancer, and a greater-than-expected rate of T3 and T4 tumors.
The most likely explanation for the observed larger-than-expected tumor size is that when MS patients report, say, a new sense of fatigue, it may be ascribed to their MS, whereas if a patient from the general population had the same complaint, a battery of tests would be ordered, she said.
The MS patients’ 14% reduction in the risk of being diagnosed with cancer had an impressive P value. In contrast, the association between MS and larger tumor size, while statistically significant, had a less than stellar P value of .04. For this reason, Dr. Tremlett said she doesn’t believe diagnostic neglect is the entire explanation for the MS patients’ reduction in cancer incidence. Other possible contributing factors worthy of further study are that MS patients’ hypervigilant immune system renders them less vulnerable to cancer growth, or that MS and cancer share a common and as-yet-unidentified genetic predisposition, or that upon receiving the diagnosis of MS, affected patients adopt a healthier lifestyle.
Dr. Tremlett stressed the importance of being on the lookout for cancer and other aging-related comorbidities, especially now that patients with MS are living longer. In a recent study of 6,917 MS patients in British Columbia, she and her coinvestigators determined that the median survival age was 78.6 years for women and 74.3 years for men. Those are some of the longest life spans ever reported, but still about 6 years less than expected for the general British Columbia population.
Median survival from disease onset was markedly longer for patients with relapsing-onset MS than primary progressive MS: 49.7 years compared with 32.5 years. However, the two groups lived to about the same age.
Patients with primary progressive MS had a 1.52-fold greater relative mortality risk than did those with relapsing-onset MS. Women with primary progressive MS had a 1.55-fold survival disadvantage compared with men with primary progressive disease (J. Neurol. Neurosurg. Psychiatry 2012;83:61-6).
The MaMS study is sponsored by the Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada. Dr. Tremlett reported having no relevant financial disclosures.
ORLANDO – Patients with multiple sclerosis are at significantly reduced risk of being diagnosed with cancer, compared with the general population, but delayed cancer detection – that is, diagnostic neglect – appears to be a contributing factor, a study from British Columbia has shown.
"Diagnostic neglect is unlikely to account for the entire reduced cancer risk that we’re seeing, but I think it could have major implications for the health, well-being, and longevity of people with multiple sclerosis," said Helen Tremlett, Ph.D, a neuroepidemiologist at the University of British Columbia, Vancouver.
In the population-based Malignancy and Multiple Sclerosis (MaMS) study, she and her coinvestigators linked data from the British Columbia MS registry with the provincial cancer registry. The study included 6,820 MS patients who visited a British Columbia MS clinic in 1980-2004. Most had never been exposed to an immunomodulatory therapy. They had a collective 110,666 person-years of follow-up. Their cancer incidence over time was compared with that of the age-, sex-, and calendar year–matched general population of British Columbia.
The standardized incidence ratio for all cancers in the MS cohort was 0.86, meaning MS patients had a highly significant overall 14% reduction in the risk of being diagnosed with cancer. The risk reduction was particularly striking for colorectal cancer: Patients with MS were 44% less likely than controls to be diagnosed with this malignancy.
The cancer risk reductions were similar in men and women with MS, and in those with relapsing-remitting as compared with primary progressive MS, Dr. Tremlett reported at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Of note, the cancer risk reduction identified in the MaMS study was consistent with an earlier meta-analysis of five studies by other investigators around the world, who concluded that the risk of being diagnosed with cancer was 8% lower in MS patients than controls, a statistically significant difference (J. Neurol. Neurosurg. Psychiatry 2010;81:1413-4).
Unlike the other researchers, however, Dr. Tremlett and her coworkers also looked at tumor size at the time of diagnosis for the four most common cancers: breast, prostate, lung, and colorectal cancer. They found a consistent pattern: MS patients diagnosed with cancer had a lower-than-expected rate of the smaller T1 and T2 tumors than in the matched general population with cancer, and a greater-than-expected rate of T3 and T4 tumors.
The most likely explanation for the observed larger-than-expected tumor size is that when MS patients report, say, a new sense of fatigue, it may be ascribed to their MS, whereas if a patient from the general population had the same complaint, a battery of tests would be ordered, she said.
The MS patients’ 14% reduction in the risk of being diagnosed with cancer had an impressive P value. In contrast, the association between MS and larger tumor size, while statistically significant, had a less than stellar P value of .04. For this reason, Dr. Tremlett said she doesn’t believe diagnostic neglect is the entire explanation for the MS patients’ reduction in cancer incidence. Other possible contributing factors worthy of further study are that MS patients’ hypervigilant immune system renders them less vulnerable to cancer growth, or that MS and cancer share a common and as-yet-unidentified genetic predisposition, or that upon receiving the diagnosis of MS, affected patients adopt a healthier lifestyle.
Dr. Tremlett stressed the importance of being on the lookout for cancer and other aging-related comorbidities, especially now that patients with MS are living longer. In a recent study of 6,917 MS patients in British Columbia, she and her coinvestigators determined that the median survival age was 78.6 years for women and 74.3 years for men. Those are some of the longest life spans ever reported, but still about 6 years less than expected for the general British Columbia population.
Median survival from disease onset was markedly longer for patients with relapsing-onset MS than primary progressive MS: 49.7 years compared with 32.5 years. However, the two groups lived to about the same age.
Patients with primary progressive MS had a 1.52-fold greater relative mortality risk than did those with relapsing-onset MS. Women with primary progressive MS had a 1.55-fold survival disadvantage compared with men with primary progressive disease (J. Neurol. Neurosurg. Psychiatry 2012;83:61-6).
The MaMS study is sponsored by the Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada. Dr. Tremlett reported having no relevant financial disclosures.
AT THE CMSC/ACTRIMS ANNUAL MEETING
Major Finding: Patients with multiple sclerosis in British Columbia had a highly significant 14% reduction in the risk of being diagnosed with cancer, compared with the matched general population. But they also had a larger-than-expected tumor size, suggestive of delayed cancer diagnosis.
Data Source: The population-based Malignancy and Multiple Sclerosis study, which includes 6,820 MS patients with 110,666 person-years of follow-up.
Disclosures: The MaMS study is sponsored by the Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada. The presenter reported having no relevant financial disclosures.

Novel antipsychotic shows early promise
HOLLYWOOD, FLA. – A novel oral antipsychotic, RP5063, displayed broad safety and efficacy for the treatment of schizophrenia and schizoaffective disorder in a phase II study.
RP5063 is a dopamine-serotonin system stabilizer. The agent is a potent partial agonist at the dopamine D2, D3, and D4 receptors and the serotonin 5-HT1A and 5-HT2A receptors, as well as an antagonist at the serotonin 5-HT6 and 5-HT7 receptors. Several of those sites have never been targeted by other medications, Dr. Marc Cantillon noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Sixty-one percent of the drug’s metabolism is by the CYP3A4 pathway, the rest by the CYP2D6 pathway, an arrangement that provides a potential for low drug-drug interaction.

The agent was designed to provide safe, less side effect laden, and more broadly effective alternatives to current atypical antipsychotics. RP5063 has minimal effects on off-target receptors, such as the histamine receptor, which are responsible for the common side effects of atypical antipsychotics that often result in poor treatment adherence, Dr. Cantillon explained.
He presented the results of the REFRESH trial, a 4-week, double-blind study of 234 patients with acute exacerbation of schizophrenia or schizoaffective disorder in the United States, Europe, and Asia. The patients were randomized 3:3:3:2:1 to once-daily RP5063 at 15, 30, or 50 mg/day, placebo, or aripiprazole (Abilify) at 15 mg/day.
Andreasen schizophrenia remission criteria (Am. J. Psychiatry 2005;162:441-9) were met by 34% of patients on RP5063 at 15 mg, 30% at 30 mg, and 46% at 50 mg, all significantly better than the 22% rate with placebo. The aripiprazole group was too small to include in the results comparison. Efficacy in the RP5063 group became significantly better than placebo within the first week and continued to steadily improve throughout the 4-week investigation, reported Dr. Cantillon, a psychiatrist and geriatrician in Livingston, N.J., and chief medical officer at Reviva Pharmaceuticals, which is developing RP5063.
Efficacy was defined as at least a 20% improvement over baseline on the Positive and Negative Syndrome Scale (PANSS) plus a 2-point improvement on the Clinical Global Impression Severity (CGI-S) scale; 46% of patients assigned to RP5063 at 15 mg/day met this bar, as did 32% of those on RP5063 at 30 mg/day and 33% on 50 mg/day, compared with 19% of placebo-treated controls.
"These remission efficacy levels within such a short study place RP5063 among the robust antipsychotics," Dr. Cantillon said.
While comparisons between different placebo-controlled randomized trials must be taken with a grain of salt, he said, the effect sizes for changes in PANSS scores with the three doses of RP5063 used in the REFRESH study are considerably bigger than in published meta-analyses of placebo-controlled trials of amisulpride, aripiprazole, quetiapine, olanzapine, or risperidone (Arch. Gen. Psychiatry 2003;60:553-64 and Mol. Psychiatry 2009;14:429-47).
On the safety front, 4 weeks of RP5063 resulted in no differences compared with placebo in terms of body weight, lipids, or blood glucose, suggesting the drug may produce fewer of the metabolic problems common of current atypical antipsychotics, noted Dr. Cantillon. Also, there were no significant differences between active treatment and control patients in terms of movement side effects or ECG changes. Serum prolactin levels declined by nearly 50% in all RP5063 treatment arms, then climbed back after the study ended.
Based upon the findings of REFRESH, a large phase III clinical trial of RP5063 for schizophrenia and schizoaffective disorder will start later this year.
"We’ll probably be including a lower dose for use in pediatric and geriatric populations in phase III, since 15 mg performed so well in REFRESH," Dr. Cantillon said.
Because of its favorable balance of agonism and antagonism of key dopaminergic and serotonergic receptors and minimal off-target effects, RP5063 is also under development for the treatment of major depressive disorder, bipolar disorder, Tourette syndrome, autism, attention-deficit/hyperactivity disorder, and psychosis in Alzheimer’s and Parkinson’s disease.
The REFRESH trial was funded by Reviva Pharmaceuticals.
HOLLYWOOD, FLA. – A novel oral antipsychotic, RP5063, displayed broad safety and efficacy for the treatment of schizophrenia and schizoaffective disorder in a phase II study.
RP5063 is a dopamine-serotonin system stabilizer. The agent is a potent partial agonist at the dopamine D2, D3, and D4 receptors and the serotonin 5-HT1A and 5-HT2A receptors, as well as an antagonist at the serotonin 5-HT6 and 5-HT7 receptors. Several of those sites have never been targeted by other medications, Dr. Marc Cantillon noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Sixty-one percent of the drug’s metabolism is by the CYP3A4 pathway, the rest by the CYP2D6 pathway, an arrangement that provides a potential for low drug-drug interaction.
The agent was designed to provide safe, less side effect laden, and more broadly effective alternatives to current atypical antipsychotics. RP5063 has minimal effects on off-target receptors, such as the histamine receptor, which are responsible for the common side effects of atypical antipsychotics that often result in poor treatment adherence, Dr. Cantillon explained.
He presented the results of the REFRESH trial, a 4-week, double-blind study of 234 patients with acute exacerbation of schizophrenia or schizoaffective disorder in the United States, Europe, and Asia. The patients were randomized 3:3:3:2:1 to once-daily RP5063 at 15, 30, or 50 mg/day, placebo, or aripiprazole (Abilify) at 15 mg/day.
Andreasen schizophrenia remission criteria (Am. J. Psychiatry 2005;162:441-9) were met by 34% of patients on RP5063 at 15 mg, 30% at 30 mg, and 46% at 50 mg, all significantly better than the 22% rate with placebo. The aripiprazole group was too small to include in the results comparison. Efficacy in the RP5063 group became significantly better than placebo within the first week and continued to steadily improve throughout the 4-week investigation, reported Dr. Cantillon, a psychiatrist and geriatrician in Livingston, N.J., and chief medical officer at Reviva Pharmaceuticals, which is developing RP5063.
Efficacy was defined as at least a 20% improvement over baseline on the Positive and Negative Syndrome Scale (PANSS) plus a 2-point improvement on the Clinical Global Impression Severity (CGI-S) scale; 46% of patients assigned to RP5063 at 15 mg/day met this bar, as did 32% of those on RP5063 at 30 mg/day and 33% on 50 mg/day, compared with 19% of placebo-treated controls.
"These remission efficacy levels within such a short study place RP5063 among the robust antipsychotics," Dr. Cantillon said.
While comparisons between different placebo-controlled randomized trials must be taken with a grain of salt, he said, the effect sizes for changes in PANSS scores with the three doses of RP5063 used in the REFRESH study are considerably bigger than in published meta-analyses of placebo-controlled trials of amisulpride, aripiprazole, quetiapine, olanzapine, or risperidone (Arch. Gen. Psychiatry 2003;60:553-64 and Mol. Psychiatry 2009;14:429-47).
On the safety front, 4 weeks of RP5063 resulted in no differences compared with placebo in terms of body weight, lipids, or blood glucose, suggesting the drug may produce fewer of the metabolic problems common of current atypical antipsychotics, noted Dr. Cantillon. Also, there were no significant differences between active treatment and control patients in terms of movement side effects or ECG changes. Serum prolactin levels declined by nearly 50% in all RP5063 treatment arms, then climbed back after the study ended.
Based upon the findings of REFRESH, a large phase III clinical trial of RP5063 for schizophrenia and schizoaffective disorder will start later this year.
"We’ll probably be including a lower dose for use in pediatric and geriatric populations in phase III, since 15 mg performed so well in REFRESH," Dr. Cantillon said.
Because of its favorable balance of agonism and antagonism of key dopaminergic and serotonergic receptors and minimal off-target effects, RP5063 is also under development for the treatment of major depressive disorder, bipolar disorder, Tourette syndrome, autism, attention-deficit/hyperactivity disorder, and psychosis in Alzheimer’s and Parkinson’s disease.
The REFRESH trial was funded by Reviva Pharmaceuticals.
HOLLYWOOD, FLA. – A novel oral antipsychotic, RP5063, displayed broad safety and efficacy for the treatment of schizophrenia and schizoaffective disorder in a phase II study.
RP5063 is a dopamine-serotonin system stabilizer. The agent is a potent partial agonist at the dopamine D2, D3, and D4 receptors and the serotonin 5-HT1A and 5-HT2A receptors, as well as an antagonist at the serotonin 5-HT6 and 5-HT7 receptors. Several of those sites have never been targeted by other medications, Dr. Marc Cantillon noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Sixty-one percent of the drug’s metabolism is by the CYP3A4 pathway, the rest by the CYP2D6 pathway, an arrangement that provides a potential for low drug-drug interaction.
The agent was designed to provide safe, less side effect laden, and more broadly effective alternatives to current atypical antipsychotics. RP5063 has minimal effects on off-target receptors, such as the histamine receptor, which are responsible for the common side effects of atypical antipsychotics that often result in poor treatment adherence, Dr. Cantillon explained.
He presented the results of the REFRESH trial, a 4-week, double-blind study of 234 patients with acute exacerbation of schizophrenia or schizoaffective disorder in the United States, Europe, and Asia. The patients were randomized 3:3:3:2:1 to once-daily RP5063 at 15, 30, or 50 mg/day, placebo, or aripiprazole (Abilify) at 15 mg/day.
Andreasen schizophrenia remission criteria (Am. J. Psychiatry 2005;162:441-9) were met by 34% of patients on RP5063 at 15 mg, 30% at 30 mg, and 46% at 50 mg, all significantly better than the 22% rate with placebo. The aripiprazole group was too small to include in the results comparison. Efficacy in the RP5063 group became significantly better than placebo within the first week and continued to steadily improve throughout the 4-week investigation, reported Dr. Cantillon, a psychiatrist and geriatrician in Livingston, N.J., and chief medical officer at Reviva Pharmaceuticals, which is developing RP5063.
Efficacy was defined as at least a 20% improvement over baseline on the Positive and Negative Syndrome Scale (PANSS) plus a 2-point improvement on the Clinical Global Impression Severity (CGI-S) scale; 46% of patients assigned to RP5063 at 15 mg/day met this bar, as did 32% of those on RP5063 at 30 mg/day and 33% on 50 mg/day, compared with 19% of placebo-treated controls.
"These remission efficacy levels within such a short study place RP5063 among the robust antipsychotics," Dr. Cantillon said.
While comparisons between different placebo-controlled randomized trials must be taken with a grain of salt, he said, the effect sizes for changes in PANSS scores with the three doses of RP5063 used in the REFRESH study are considerably bigger than in published meta-analyses of placebo-controlled trials of amisulpride, aripiprazole, quetiapine, olanzapine, or risperidone (Arch. Gen. Psychiatry 2003;60:553-64 and Mol. Psychiatry 2009;14:429-47).
On the safety front, 4 weeks of RP5063 resulted in no differences compared with placebo in terms of body weight, lipids, or blood glucose, suggesting the drug may produce fewer of the metabolic problems common of current atypical antipsychotics, noted Dr. Cantillon. Also, there were no significant differences between active treatment and control patients in terms of movement side effects or ECG changes. Serum prolactin levels declined by nearly 50% in all RP5063 treatment arms, then climbed back after the study ended.
Based upon the findings of REFRESH, a large phase III clinical trial of RP5063 for schizophrenia and schizoaffective disorder will start later this year.
"We’ll probably be including a lower dose for use in pediatric and geriatric populations in phase III, since 15 mg performed so well in REFRESH," Dr. Cantillon said.
Because of its favorable balance of agonism and antagonism of key dopaminergic and serotonergic receptors and minimal off-target effects, RP5063 is also under development for the treatment of major depressive disorder, bipolar disorder, Tourette syndrome, autism, attention-deficit/hyperactivity disorder, and psychosis in Alzheimer’s and Parkinson’s disease.
The REFRESH trial was funded by Reviva Pharmaceuticals.
AT THE NCDEU MEETING
Major Finding: Forty-five percent of patients with schizophrenia or schizoaffective disorder randomized to RP5063 responded with at least a 2-point reduction from baseline on the CGI-S scale plus a 20% improvement on PANSS, compared with 19% on placebo.
Data Source: The phase-II REFRESH study, a 4-week, double-blind trial of 234 patients with acute exacerbation of schizophrenia or schizoaffective disorder who were randomized to RP5063 at one of three daily doses, aripiprazole at 15 mg/day, or placebo.
Disclosures: REFRESH was funded by Reviva Pharmaceuticals. Dr. Cantillon is the company’s chief medical officer.

Recommendations outline how to improve dimethyl fumarate tolerability
ORLANDO – The flushing frequently reported in conjunction with oral dimethyl fumarate therapy for relapsing forms of multiple sclerosis is greatly reduced by aspirin pretreatment, Dr. J. Theodore Phillips said at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
In contrast, slowed titration of dimethyl fumarate (Tecfidera) does not diminish the gastrointestinal adverse effects which are the other common side effect encountered during the first month or two of therapy, added Dr. Phillips, a neurologist in the multiple sclerosis research program at the Baylor Institute for Immunology Research, Dallas.

Dr. Phillips was part of an expert consensus panel which presented recommendations for maximizing the tolerability of dimethyl fumarate, approved earlier this year as the third oral agent for treatment of MS.
In an interview, he said the recommendations are largely based upon expert opinion rather than being rigorously evidence-based guidelines. After the pivotal phase III DEFINE and CONFIRM clinical trials were completed, he and four other study leaders decided to poll the investigators who had enrolled at least 10 patients in the trials as to how they managed the flushing and GI upset problems which arose. The flushing and GI side effects were reported by 36% and 42%, respectively, of study patients randomized to the drug. The incidence decreased after the first month. These side effects were generally rated by investigators as mild-to-moderate in nature. Flushing resulted in study dropout in 2.5% of patients, while another 4.3% discontinued due to GI adverse events.
Thirty of the 84 invited clinical investigators completed the questionnaire. Meanwhile, investigators at Biogen Idec, which markets Tecfidera, conducted their own randomized, double-blind, phase IIIb study in 172 healthy volunteers, the results of which have been incorporated into the expert panel’s recommendations. Participants in the 8-week trial were randomized to one of four treatment arms: dimethyl fumarate titrated in standard fashion over 1 week plus 325 mg of non–enteric-coated aspirin taken 30 minutes beforehand during weeks 1-4, replaced by aspirin placebo in weeks 5-8; dimethyl fumarate plus aspirin placebo during weeks 1-4; dimethyl fumarate slow-titrated over the course of 3 weeks; and double placebo.
Roughly 80% of subjects on dimethyl fumarate without aspirin experienced flushing events, self-assessed as mild-to-moderate. In contrast, while subjects were on both dimethyl fumarate and aspirin, their flushing frequency and severity were similar to participants on double-placebo.
Slow titration of dimethyl fumarate had no impact on GI symptoms or flushing frequency or severity.
Given that slow titration of dimethyl fumarate proved ineffective in reducing GI symptoms in the phase IIIb study, the expert panel’s recommendations for managing nausea/vomiting or abdominal pain were to take the drug with food and consider prescribing a proton pump inhibitor or H2 receptor antagonist. Metoclopramide or domperidone is another recommended option for those with nausea/vomiting. For patients who experience medication-related diarrhea, the panel advised loperamide or other standard antidiarrheal agents.
"Vasocutaneous flushing and GI upset in association with dosing of Tecfidera could for obvious reasons affect a person’s enthusiasm for going on," Dr. Phillips observed. "The main thing is for the physician to set expectations by up-front acknowledging these issues as part of the risk/benefit discussion prior to initiating the drug. Tell the patient that if those side effects were to happen, we’ve got game plans to deal with them."
The investigator survey that formed the basis for the expert panel recommendations was funded by Biogen Idec. Dr. Phillips is on the company’s medical advisory board. He has also received honoraria from Avanir, Genzyme, Novartis, and Teva.
ORLANDO – The flushing frequently reported in conjunction with oral dimethyl fumarate therapy for relapsing forms of multiple sclerosis is greatly reduced by aspirin pretreatment, Dr. J. Theodore Phillips said at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
In contrast, slowed titration of dimethyl fumarate (Tecfidera) does not diminish the gastrointestinal adverse effects which are the other common side effect encountered during the first month or two of therapy, added Dr. Phillips, a neurologist in the multiple sclerosis research program at the Baylor Institute for Immunology Research, Dallas.
Dr. Phillips was part of an expert consensus panel which presented recommendations for maximizing the tolerability of dimethyl fumarate, approved earlier this year as the third oral agent for treatment of MS.
In an interview, he said the recommendations are largely based upon expert opinion rather than being rigorously evidence-based guidelines. After the pivotal phase III DEFINE and CONFIRM clinical trials were completed, he and four other study leaders decided to poll the investigators who had enrolled at least 10 patients in the trials as to how they managed the flushing and GI upset problems which arose. The flushing and GI side effects were reported by 36% and 42%, respectively, of study patients randomized to the drug. The incidence decreased after the first month. These side effects were generally rated by investigators as mild-to-moderate in nature. Flushing resulted in study dropout in 2.5% of patients, while another 4.3% discontinued due to GI adverse events.
Thirty of the 84 invited clinical investigators completed the questionnaire. Meanwhile, investigators at Biogen Idec, which markets Tecfidera, conducted their own randomized, double-blind, phase IIIb study in 172 healthy volunteers, the results of which have been incorporated into the expert panel’s recommendations. Participants in the 8-week trial were randomized to one of four treatment arms: dimethyl fumarate titrated in standard fashion over 1 week plus 325 mg of non–enteric-coated aspirin taken 30 minutes beforehand during weeks 1-4, replaced by aspirin placebo in weeks 5-8; dimethyl fumarate plus aspirin placebo during weeks 1-4; dimethyl fumarate slow-titrated over the course of 3 weeks; and double placebo.
Roughly 80% of subjects on dimethyl fumarate without aspirin experienced flushing events, self-assessed as mild-to-moderate. In contrast, while subjects were on both dimethyl fumarate and aspirin, their flushing frequency and severity were similar to participants on double-placebo.
Slow titration of dimethyl fumarate had no impact on GI symptoms or flushing frequency or severity.
Given that slow titration of dimethyl fumarate proved ineffective in reducing GI symptoms in the phase IIIb study, the expert panel’s recommendations for managing nausea/vomiting or abdominal pain were to take the drug with food and consider prescribing a proton pump inhibitor or H2 receptor antagonist. Metoclopramide or domperidone is another recommended option for those with nausea/vomiting. For patients who experience medication-related diarrhea, the panel advised loperamide or other standard antidiarrheal agents.
"Vasocutaneous flushing and GI upset in association with dosing of Tecfidera could for obvious reasons affect a person’s enthusiasm for going on," Dr. Phillips observed. "The main thing is for the physician to set expectations by up-front acknowledging these issues as part of the risk/benefit discussion prior to initiating the drug. Tell the patient that if those side effects were to happen, we’ve got game plans to deal with them."
The investigator survey that formed the basis for the expert panel recommendations was funded by Biogen Idec. Dr. Phillips is on the company’s medical advisory board. He has also received honoraria from Avanir, Genzyme, Novartis, and Teva.
ORLANDO – The flushing frequently reported in conjunction with oral dimethyl fumarate therapy for relapsing forms of multiple sclerosis is greatly reduced by aspirin pretreatment, Dr. J. Theodore Phillips said at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
In contrast, slowed titration of dimethyl fumarate (Tecfidera) does not diminish the gastrointestinal adverse effects which are the other common side effect encountered during the first month or two of therapy, added Dr. Phillips, a neurologist in the multiple sclerosis research program at the Baylor Institute for Immunology Research, Dallas.
Dr. Phillips was part of an expert consensus panel which presented recommendations for maximizing the tolerability of dimethyl fumarate, approved earlier this year as the third oral agent for treatment of MS.
In an interview, he said the recommendations are largely based upon expert opinion rather than being rigorously evidence-based guidelines. After the pivotal phase III DEFINE and CONFIRM clinical trials were completed, he and four other study leaders decided to poll the investigators who had enrolled at least 10 patients in the trials as to how they managed the flushing and GI upset problems which arose. The flushing and GI side effects were reported by 36% and 42%, respectively, of study patients randomized to the drug. The incidence decreased after the first month. These side effects were generally rated by investigators as mild-to-moderate in nature. Flushing resulted in study dropout in 2.5% of patients, while another 4.3% discontinued due to GI adverse events.
Thirty of the 84 invited clinical investigators completed the questionnaire. Meanwhile, investigators at Biogen Idec, which markets Tecfidera, conducted their own randomized, double-blind, phase IIIb study in 172 healthy volunteers, the results of which have been incorporated into the expert panel’s recommendations. Participants in the 8-week trial were randomized to one of four treatment arms: dimethyl fumarate titrated in standard fashion over 1 week plus 325 mg of non–enteric-coated aspirin taken 30 minutes beforehand during weeks 1-4, replaced by aspirin placebo in weeks 5-8; dimethyl fumarate plus aspirin placebo during weeks 1-4; dimethyl fumarate slow-titrated over the course of 3 weeks; and double placebo.
Roughly 80% of subjects on dimethyl fumarate without aspirin experienced flushing events, self-assessed as mild-to-moderate. In contrast, while subjects were on both dimethyl fumarate and aspirin, their flushing frequency and severity were similar to participants on double-placebo.
Slow titration of dimethyl fumarate had no impact on GI symptoms or flushing frequency or severity.
Given that slow titration of dimethyl fumarate proved ineffective in reducing GI symptoms in the phase IIIb study, the expert panel’s recommendations for managing nausea/vomiting or abdominal pain were to take the drug with food and consider prescribing a proton pump inhibitor or H2 receptor antagonist. Metoclopramide or domperidone is another recommended option for those with nausea/vomiting. For patients who experience medication-related diarrhea, the panel advised loperamide or other standard antidiarrheal agents.
"Vasocutaneous flushing and GI upset in association with dosing of Tecfidera could for obvious reasons affect a person’s enthusiasm for going on," Dr. Phillips observed. "The main thing is for the physician to set expectations by up-front acknowledging these issues as part of the risk/benefit discussion prior to initiating the drug. Tell the patient that if those side effects were to happen, we’ve got game plans to deal with them."
The investigator survey that formed the basis for the expert panel recommendations was funded by Biogen Idec. Dr. Phillips is on the company’s medical advisory board. He has also received honoraria from Avanir, Genzyme, Novartis, and Teva.
AT THE CMSC/ACTRIMS ANNUAL MEETING

Oral sitagliptin promising for inpatient glycemic management in type 2 diabetes
CHICAGO – Once-daily oral sitagliptin appears to be safe and effective for the management of hyperglycemia in patients with type 2 diabetes hospitalized on general medicine or surgery wards, according to an industry-sponsored pilot study.
"Our results suggest that many hospitalized patients with type 2 diabetes could be treated with sitagliptin plus corrective rapid-acting insulin as needed. Patients with persistent hyperglycemia can be treated with sitagliptin and a low dose of glargine once daily or with basal-bolus insulin," Dr. Roma Gianchandani said at the annual scientific sessions of the American Diabetes Association.
Sitagliptin (Januvia) is an attractive alternative to current guideline-recommended basal-bolus insulin regimens, which are labor intensive, require multiple daily injections, and entail a significant risk of hypoglycemia, said Dr. Gianchandani of the University of Michigan, Ann Arbor.

She presented a randomized, open-label study of 82 noncritically ill patients with type 2 diabetes hospitalized on general medicine or surgery wards. Their mean hospital length of stay was 6.5 days. Because there have been no previous studies on the use of a dipeptidyl peptidase–4 (DPP-4) inhibitor such as sitagliptin in this context, entry criteria were stringent. Patients were enrolled if they had early-stage type 2 diabetes as reflected in a prehospital treatment regimen involving diet and oral antidiabetic agents or a low total daily insulin dose of up to 0.4 units/kg per day.
Participants were randomized to one of three treatment arms: sitagliptin at 100 mg once daily provided their creatinine clearance exceeded 50 mL/min, or 50 mg/day if their creatinine clearance was 30-50 mL/min; sitagliptin plus once-daily insulin glargine at 0.2 units/kg per day if their admission blood glucose value was 140-200 mg/dL, and glargine at 0.25 units/kg per day if it was 201-400 mg/dL; or a standard basal-bolus insulin regimen at a total daily insulin dose of 0.44 units/kg per day if their admission blood glucose was 140-200 mg/dL and 0.5 units/kg per day if it was 201-400 mg/dL. In the basal-bolus group, half of the total daily dose was given as glargine once daily and half as lispro in three equal doses before meals or every 6 hours.
Patients in all three study arms received a supplemental dose of lispro before meals if their premeal blood glucose exceeded 140 mg/dL.
A key study finding was that glycemic control improved similarly in all three study groups. After the first day of treatment there were no significant differences in mean daily blood glucose. Nor did the percentage of blood glucose readings falling within the target range of 70-140 mg/dL differ significantly: It was 36% in the sitagliptin-only group and 43% in the other two treatment arms. The percentage of blood glucose readings above the cutoffs of 140 and 200 mg/dL was likewise similar in all three treatment groups. The rate of treatment failure, defined as three or more consecutive blood glucose readings greater than 240 mg/dL or a mean daily blood glucose above that level, was 11% in the sitagliptin-alone group, 10% with sitagliptin plus basal insulin, and 8% with basal-bolus therapy.
The rate of hypoglycemia below 70 mg/dL was statistically similar in all three groups: 4% with sitagliptin alone, 7% with sitagliptin and basal insulin, and 8% with basal-bolus insulin.
Patients in the sitagliptin group averaged 1.8 insulin injections per day after day 1 of their hospital stay, significantly less than the 2.5 per day in the basal-bolus therapy group. Patients on sitagliptin plus basal insulin averaged 1.3 injections per day. Total insulin per day averaged 11.5 units in the sitagliptin group, 28.2 units in the sitagliptin plus basal insulin–treated patients, and 39.6 units in the basal-bolus group.
There were no between-group differences in complication rates or length of stay, Dr. Gianchandani continued.
One audience member said the generalizability of the study findings is limited by the small number of patients and stringent entry criteria. He estimated that 80% of his own type 2 diabetic patients hospitalized on general medicine or surgery wards wouldn’t have qualified for the trial.
Dr. Gianchandani responded that based on the favorable results of this pilot study, a larger study with a broader range of patients is being planned.
Another audience member, Dr. Stanley Schwartz of Ardmore, Pa., rose to enthusiastically declare, "I think this is actually a seminal study." He noted that nearly half of the patients in the sitagliptin-alone group never required any supplemental insulin injections during their hospital stay.
"Think about it: 40%-50% of these patients with early diabetes who may have otherwise required low doses of insulin didn’t need insulin because they were on sitagliptin alone. I think this is a superb study and should be pursued further," he said.
"We’re very excited about it, too," Dr. Gianchandani replied.
The study was sponsored by Merck, which markets sitagliptin. Dr. Gianchandani reported having no financial conflicts.
CHICAGO – Once-daily oral sitagliptin appears to be safe and effective for the management of hyperglycemia in patients with type 2 diabetes hospitalized on general medicine or surgery wards, according to an industry-sponsored pilot study.
"Our results suggest that many hospitalized patients with type 2 diabetes could be treated with sitagliptin plus corrective rapid-acting insulin as needed. Patients with persistent hyperglycemia can be treated with sitagliptin and a low dose of glargine once daily or with basal-bolus insulin," Dr. Roma Gianchandani said at the annual scientific sessions of the American Diabetes Association.
Sitagliptin (Januvia) is an attractive alternative to current guideline-recommended basal-bolus insulin regimens, which are labor intensive, require multiple daily injections, and entail a significant risk of hypoglycemia, said Dr. Gianchandani of the University of Michigan, Ann Arbor.
She presented a randomized, open-label study of 82 noncritically ill patients with type 2 diabetes hospitalized on general medicine or surgery wards. Their mean hospital length of stay was 6.5 days. Because there have been no previous studies on the use of a dipeptidyl peptidase–4 (DPP-4) inhibitor such as sitagliptin in this context, entry criteria were stringent. Patients were enrolled if they had early-stage type 2 diabetes as reflected in a prehospital treatment regimen involving diet and oral antidiabetic agents or a low total daily insulin dose of up to 0.4 units/kg per day.
Participants were randomized to one of three treatment arms: sitagliptin at 100 mg once daily provided their creatinine clearance exceeded 50 mL/min, or 50 mg/day if their creatinine clearance was 30-50 mL/min; sitagliptin plus once-daily insulin glargine at 0.2 units/kg per day if their admission blood glucose value was 140-200 mg/dL, and glargine at 0.25 units/kg per day if it was 201-400 mg/dL; or a standard basal-bolus insulin regimen at a total daily insulin dose of 0.44 units/kg per day if their admission blood glucose was 140-200 mg/dL and 0.5 units/kg per day if it was 201-400 mg/dL. In the basal-bolus group, half of the total daily dose was given as glargine once daily and half as lispro in three equal doses before meals or every 6 hours.
Patients in all three study arms received a supplemental dose of lispro before meals if their premeal blood glucose exceeded 140 mg/dL.
A key study finding was that glycemic control improved similarly in all three study groups. After the first day of treatment there were no significant differences in mean daily blood glucose. Nor did the percentage of blood glucose readings falling within the target range of 70-140 mg/dL differ significantly: It was 36% in the sitagliptin-only group and 43% in the other two treatment arms. The percentage of blood glucose readings above the cutoffs of 140 and 200 mg/dL was likewise similar in all three treatment groups. The rate of treatment failure, defined as three or more consecutive blood glucose readings greater than 240 mg/dL or a mean daily blood glucose above that level, was 11% in the sitagliptin-alone group, 10% with sitagliptin plus basal insulin, and 8% with basal-bolus therapy.
The rate of hypoglycemia below 70 mg/dL was statistically similar in all three groups: 4% with sitagliptin alone, 7% with sitagliptin and basal insulin, and 8% with basal-bolus insulin.
Patients in the sitagliptin group averaged 1.8 insulin injections per day after day 1 of their hospital stay, significantly less than the 2.5 per day in the basal-bolus therapy group. Patients on sitagliptin plus basal insulin averaged 1.3 injections per day. Total insulin per day averaged 11.5 units in the sitagliptin group, 28.2 units in the sitagliptin plus basal insulin–treated patients, and 39.6 units in the basal-bolus group.
There were no between-group differences in complication rates or length of stay, Dr. Gianchandani continued.
One audience member said the generalizability of the study findings is limited by the small number of patients and stringent entry criteria. He estimated that 80% of his own type 2 diabetic patients hospitalized on general medicine or surgery wards wouldn’t have qualified for the trial.
Dr. Gianchandani responded that based on the favorable results of this pilot study, a larger study with a broader range of patients is being planned.
Another audience member, Dr. Stanley Schwartz of Ardmore, Pa., rose to enthusiastically declare, "I think this is actually a seminal study." He noted that nearly half of the patients in the sitagliptin-alone group never required any supplemental insulin injections during their hospital stay.
"Think about it: 40%-50% of these patients with early diabetes who may have otherwise required low doses of insulin didn’t need insulin because they were on sitagliptin alone. I think this is a superb study and should be pursued further," he said.
"We’re very excited about it, too," Dr. Gianchandani replied.
The study was sponsored by Merck, which markets sitagliptin. Dr. Gianchandani reported having no financial conflicts.
CHICAGO – Once-daily oral sitagliptin appears to be safe and effective for the management of hyperglycemia in patients with type 2 diabetes hospitalized on general medicine or surgery wards, according to an industry-sponsored pilot study.
"Our results suggest that many hospitalized patients with type 2 diabetes could be treated with sitagliptin plus corrective rapid-acting insulin as needed. Patients with persistent hyperglycemia can be treated with sitagliptin and a low dose of glargine once daily or with basal-bolus insulin," Dr. Roma Gianchandani said at the annual scientific sessions of the American Diabetes Association.
Sitagliptin (Januvia) is an attractive alternative to current guideline-recommended basal-bolus insulin regimens, which are labor intensive, require multiple daily injections, and entail a significant risk of hypoglycemia, said Dr. Gianchandani of the University of Michigan, Ann Arbor.
She presented a randomized, open-label study of 82 noncritically ill patients with type 2 diabetes hospitalized on general medicine or surgery wards. Their mean hospital length of stay was 6.5 days. Because there have been no previous studies on the use of a dipeptidyl peptidase–4 (DPP-4) inhibitor such as sitagliptin in this context, entry criteria were stringent. Patients were enrolled if they had early-stage type 2 diabetes as reflected in a prehospital treatment regimen involving diet and oral antidiabetic agents or a low total daily insulin dose of up to 0.4 units/kg per day.
Participants were randomized to one of three treatment arms: sitagliptin at 100 mg once daily provided their creatinine clearance exceeded 50 mL/min, or 50 mg/day if their creatinine clearance was 30-50 mL/min; sitagliptin plus once-daily insulin glargine at 0.2 units/kg per day if their admission blood glucose value was 140-200 mg/dL, and glargine at 0.25 units/kg per day if it was 201-400 mg/dL; or a standard basal-bolus insulin regimen at a total daily insulin dose of 0.44 units/kg per day if their admission blood glucose was 140-200 mg/dL and 0.5 units/kg per day if it was 201-400 mg/dL. In the basal-bolus group, half of the total daily dose was given as glargine once daily and half as lispro in three equal doses before meals or every 6 hours.
Patients in all three study arms received a supplemental dose of lispro before meals if their premeal blood glucose exceeded 140 mg/dL.
A key study finding was that glycemic control improved similarly in all three study groups. After the first day of treatment there were no significant differences in mean daily blood glucose. Nor did the percentage of blood glucose readings falling within the target range of 70-140 mg/dL differ significantly: It was 36% in the sitagliptin-only group and 43% in the other two treatment arms. The percentage of blood glucose readings above the cutoffs of 140 and 200 mg/dL was likewise similar in all three treatment groups. The rate of treatment failure, defined as three or more consecutive blood glucose readings greater than 240 mg/dL or a mean daily blood glucose above that level, was 11% in the sitagliptin-alone group, 10% with sitagliptin plus basal insulin, and 8% with basal-bolus therapy.
The rate of hypoglycemia below 70 mg/dL was statistically similar in all three groups: 4% with sitagliptin alone, 7% with sitagliptin and basal insulin, and 8% with basal-bolus insulin.
Patients in the sitagliptin group averaged 1.8 insulin injections per day after day 1 of their hospital stay, significantly less than the 2.5 per day in the basal-bolus therapy group. Patients on sitagliptin plus basal insulin averaged 1.3 injections per day. Total insulin per day averaged 11.5 units in the sitagliptin group, 28.2 units in the sitagliptin plus basal insulin–treated patients, and 39.6 units in the basal-bolus group.
There were no between-group differences in complication rates or length of stay, Dr. Gianchandani continued.
One audience member said the generalizability of the study findings is limited by the small number of patients and stringent entry criteria. He estimated that 80% of his own type 2 diabetic patients hospitalized on general medicine or surgery wards wouldn’t have qualified for the trial.
Dr. Gianchandani responded that based on the favorable results of this pilot study, a larger study with a broader range of patients is being planned.
Another audience member, Dr. Stanley Schwartz of Ardmore, Pa., rose to enthusiastically declare, "I think this is actually a seminal study." He noted that nearly half of the patients in the sitagliptin-alone group never required any supplemental insulin injections during their hospital stay.
"Think about it: 40%-50% of these patients with early diabetes who may have otherwise required low doses of insulin didn’t need insulin because they were on sitagliptin alone. I think this is a superb study and should be pursued further," he said.
"We’re very excited about it, too," Dr. Gianchandani replied.
The study was sponsored by Merck, which markets sitagliptin. Dr. Gianchandani reported having no financial conflicts.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major finding: Glycemic control was statistically as good with once-daily oral sitagliptin as with guideline-recommended multi-injection basal-bolus insulin (36% and 43%, respectively).
Data source: A randomized, prospective, open-label pilot study involving 82 type 2 diabetic patients hospitalized on general medicine or surgery wards.
Disclosures: The study was sponsored by Merck, which markets sitagliptin. Dr. Gianchandani reported having no financial conflicts.

Acute antipsychotics do not worsen glycemia in psychiatric inpatients
CHICAGO – Treatment with antipsychotic agents in acute psychiatric inpatient units is not associated with increased blood glucose levels, according to a multicenter study.
"This finding is contrary to what many of us may have previously considered," Dr. Dawn D. Smiley observed in presenting the study findings at the annual scientific sessions of the American Diabetes Association.

A well-recognized association exists between the use of antipsychotic agents and an increased prevalence of diabetes and hyperglycemia, although it remains unclear whether the relationship is causal. The association is especially strong for the atypical or second-generation antipsychotics, which account for more than 90% of all antipsychotic use and are among the top-selling prescription drugs in the United States. Indeed, the Food and Drug Administration has for a decade required the product labeling for all atypical antipsychotics to include a warning about the risks of hyperglycemia and diabetes.
But most data on antipsychotic-associated diabetes come from long-term studies, which show that diabetes, when it occurs, most often does so within the first 6 months on the medication. Little is known about the acute impact of antipsychotic agents on blood glucose levels and clinical outcomes in patients admitted to acute psychiatric units for stays averaging a few weeks. This information gap provided the rationale for Dr. Smiley’s study.
She presented a chart review of 1,212 patients admitted to three acute inpatient psychiatric units in inner-city Atlanta during 2008. The mean age of the patients was 57 years, they had a mean body mass index of 27 kg/m2, and the mean daily blood glucose concentration was 122 mg/dL during their stay. Twenty-one percent of the patients had a history of diabetes, and 6% had new hyperglycemia at admission. The most common primary diagnoses on admission were suicidal ideation in 28% of patients, a primary psychotic disorder in 27%, and depression in 16%.
Thirty-seven percent of patients had no prior experience with antipsychotic agents but were newly started on an antipsychotic during their acute inpatient stay. An additional 38% had been on antipsychotic therapy prior to admission and continued on it while hospitalized. Twenty-five percent of patients had never been treated with antipsychotic agents and were not during their hospitalization. Fifty-two percent of patients were treated with atypical antipsychotic agents during their inpatient stay, 17% received both atypical and typical antipsychotics, and 6% got typical antipsychotics only, explained Dr. Smiley, an endocrinologist at Emory University, Atlanta.
She sought answers to three research questions:
• Does prior exposure to antipsychotic therapy affect inpatient blood glucose levels or clinical outcome measures? Mean psychiatric inpatient length of stay was significantly shorter for patients never exposed to antipsychotics, at 9.5 days, compared with 14.2 days in patients who continued on their previous antipsychotic regimen and 13.7 days in antipsychotic-naive patients started on an antipsychotic for the first time. The most likely explanation for the longer stays in patients on antipsychotic therapy was the need to monitor them for side effects, according to Dr. Smiley.
Importantly, mean daily blood glucose concentrations during psychiatric hospitalization did not differ between patients never exposed to antipsychotic agents, those started on an antipsychotic for the first time during their hospitalization, and patients continued on their previous antipsychotics.
• Does the type of antipsychotic agent used during the acute inpatient admission affect blood glucose levels or clinical outcomes?
Mean daily blood glucose levels were similar regardless of whether patients were on atypical antipsychotics, typical antipsychotics, or both. The mean hemoglobin A1c level was 7.3% in all three groups of antipsychotic-treated patients. However, the rate of medical complications during psychiatric hospitalization was significantly higher among patients on atypical agents, at 23%, compared with 17% in patients on typical antipsychotics and 13% in those on both.
Three percent of patients on atypical antipsychotics-only required transfer to a medical unit for treatment of a complication that arose during psychiatric hospitalization. That was also true for 3% of patients on combined atypical/typical antipsychotic therapy. In contrast, none of the relatively few patients on typical antipsychotics-only required transfer to a medical unit. It’s worth noting that all atypical antipsychotics are not alike in terms of the strength of their association with diabetes. Most antipsychotic-treated patients in this study got olanzapine (Zyprexa) or clozapine (Clozaril). Newer, less diabetogenic atypical agents were less frequently used in this largely indigent population.
• Does a history of diabetes or new hyperglycemia at admission affect outcome measures? Not unexpectedly, patients with a history of diabetes prior to admission had a significantly higher rate of medical complications during their psychiatric inpatient stay: a 28% rate, compared with 22% in patients with no history of diabetes but with hyperglycemia at admission and 20% in patients with neither metabolic abnormality. The most common complication in patients with a history of diabetes was urinary tract infection.
Again unsurprisingly, patients with a history of diabetes had a significantly higher rate of transfer to a medical unit because of medical complications: 5%, compared with no transfers in patients with new hyperglycemia at admission and a 1% transfer rate in patients with neither metabolic abnormality.
Dr. Smiley reported that her research funding came from the National Institutes of Health, Sanofi-Aventis, and Merck.
CHICAGO – Treatment with antipsychotic agents in acute psychiatric inpatient units is not associated with increased blood glucose levels, according to a multicenter study.
"This finding is contrary to what many of us may have previously considered," Dr. Dawn D. Smiley observed in presenting the study findings at the annual scientific sessions of the American Diabetes Association.
A well-recognized association exists between the use of antipsychotic agents and an increased prevalence of diabetes and hyperglycemia, although it remains unclear whether the relationship is causal. The association is especially strong for the atypical or second-generation antipsychotics, which account for more than 90% of all antipsychotic use and are among the top-selling prescription drugs in the United States. Indeed, the Food and Drug Administration has for a decade required the product labeling for all atypical antipsychotics to include a warning about the risks of hyperglycemia and diabetes.
But most data on antipsychotic-associated diabetes come from long-term studies, which show that diabetes, when it occurs, most often does so within the first 6 months on the medication. Little is known about the acute impact of antipsychotic agents on blood glucose levels and clinical outcomes in patients admitted to acute psychiatric units for stays averaging a few weeks. This information gap provided the rationale for Dr. Smiley’s study.
She presented a chart review of 1,212 patients admitted to three acute inpatient psychiatric units in inner-city Atlanta during 2008. The mean age of the patients was 57 years, they had a mean body mass index of 27 kg/m2, and the mean daily blood glucose concentration was 122 mg/dL during their stay. Twenty-one percent of the patients had a history of diabetes, and 6% had new hyperglycemia at admission. The most common primary diagnoses on admission were suicidal ideation in 28% of patients, a primary psychotic disorder in 27%, and depression in 16%.
Thirty-seven percent of patients had no prior experience with antipsychotic agents but were newly started on an antipsychotic during their acute inpatient stay. An additional 38% had been on antipsychotic therapy prior to admission and continued on it while hospitalized. Twenty-five percent of patients had never been treated with antipsychotic agents and were not during their hospitalization. Fifty-two percent of patients were treated with atypical antipsychotic agents during their inpatient stay, 17% received both atypical and typical antipsychotics, and 6% got typical antipsychotics only, explained Dr. Smiley, an endocrinologist at Emory University, Atlanta.
She sought answers to three research questions:
• Does prior exposure to antipsychotic therapy affect inpatient blood glucose levels or clinical outcome measures? Mean psychiatric inpatient length of stay was significantly shorter for patients never exposed to antipsychotics, at 9.5 days, compared with 14.2 days in patients who continued on their previous antipsychotic regimen and 13.7 days in antipsychotic-naive patients started on an antipsychotic for the first time. The most likely explanation for the longer stays in patients on antipsychotic therapy was the need to monitor them for side effects, according to Dr. Smiley.
Importantly, mean daily blood glucose concentrations during psychiatric hospitalization did not differ between patients never exposed to antipsychotic agents, those started on an antipsychotic for the first time during their hospitalization, and patients continued on their previous antipsychotics.
• Does the type of antipsychotic agent used during the acute inpatient admission affect blood glucose levels or clinical outcomes?
Mean daily blood glucose levels were similar regardless of whether patients were on atypical antipsychotics, typical antipsychotics, or both. The mean hemoglobin A1c level was 7.3% in all three groups of antipsychotic-treated patients. However, the rate of medical complications during psychiatric hospitalization was significantly higher among patients on atypical agents, at 23%, compared with 17% in patients on typical antipsychotics and 13% in those on both.
Three percent of patients on atypical antipsychotics-only required transfer to a medical unit for treatment of a complication that arose during psychiatric hospitalization. That was also true for 3% of patients on combined atypical/typical antipsychotic therapy. In contrast, none of the relatively few patients on typical antipsychotics-only required transfer to a medical unit. It’s worth noting that all atypical antipsychotics are not alike in terms of the strength of their association with diabetes. Most antipsychotic-treated patients in this study got olanzapine (Zyprexa) or clozapine (Clozaril). Newer, less diabetogenic atypical agents were less frequently used in this largely indigent population.
• Does a history of diabetes or new hyperglycemia at admission affect outcome measures? Not unexpectedly, patients with a history of diabetes prior to admission had a significantly higher rate of medical complications during their psychiatric inpatient stay: a 28% rate, compared with 22% in patients with no history of diabetes but with hyperglycemia at admission and 20% in patients with neither metabolic abnormality. The most common complication in patients with a history of diabetes was urinary tract infection.
Again unsurprisingly, patients with a history of diabetes had a significantly higher rate of transfer to a medical unit because of medical complications: 5%, compared with no transfers in patients with new hyperglycemia at admission and a 1% transfer rate in patients with neither metabolic abnormality.
Dr. Smiley reported that her research funding came from the National Institutes of Health, Sanofi-Aventis, and Merck.
CHICAGO – Treatment with antipsychotic agents in acute psychiatric inpatient units is not associated with increased blood glucose levels, according to a multicenter study.
"This finding is contrary to what many of us may have previously considered," Dr. Dawn D. Smiley observed in presenting the study findings at the annual scientific sessions of the American Diabetes Association.
A well-recognized association exists between the use of antipsychotic agents and an increased prevalence of diabetes and hyperglycemia, although it remains unclear whether the relationship is causal. The association is especially strong for the atypical or second-generation antipsychotics, which account for more than 90% of all antipsychotic use and are among the top-selling prescription drugs in the United States. Indeed, the Food and Drug Administration has for a decade required the product labeling for all atypical antipsychotics to include a warning about the risks of hyperglycemia and diabetes.
But most data on antipsychotic-associated diabetes come from long-term studies, which show that diabetes, when it occurs, most often does so within the first 6 months on the medication. Little is known about the acute impact of antipsychotic agents on blood glucose levels and clinical outcomes in patients admitted to acute psychiatric units for stays averaging a few weeks. This information gap provided the rationale for Dr. Smiley’s study.
She presented a chart review of 1,212 patients admitted to three acute inpatient psychiatric units in inner-city Atlanta during 2008. The mean age of the patients was 57 years, they had a mean body mass index of 27 kg/m2, and the mean daily blood glucose concentration was 122 mg/dL during their stay. Twenty-one percent of the patients had a history of diabetes, and 6% had new hyperglycemia at admission. The most common primary diagnoses on admission were suicidal ideation in 28% of patients, a primary psychotic disorder in 27%, and depression in 16%.
Thirty-seven percent of patients had no prior experience with antipsychotic agents but were newly started on an antipsychotic during their acute inpatient stay. An additional 38% had been on antipsychotic therapy prior to admission and continued on it while hospitalized. Twenty-five percent of patients had never been treated with antipsychotic agents and were not during their hospitalization. Fifty-two percent of patients were treated with atypical antipsychotic agents during their inpatient stay, 17% received both atypical and typical antipsychotics, and 6% got typical antipsychotics only, explained Dr. Smiley, an endocrinologist at Emory University, Atlanta.
She sought answers to three research questions:
• Does prior exposure to antipsychotic therapy affect inpatient blood glucose levels or clinical outcome measures? Mean psychiatric inpatient length of stay was significantly shorter for patients never exposed to antipsychotics, at 9.5 days, compared with 14.2 days in patients who continued on their previous antipsychotic regimen and 13.7 days in antipsychotic-naive patients started on an antipsychotic for the first time. The most likely explanation for the longer stays in patients on antipsychotic therapy was the need to monitor them for side effects, according to Dr. Smiley.
Importantly, mean daily blood glucose concentrations during psychiatric hospitalization did not differ between patients never exposed to antipsychotic agents, those started on an antipsychotic for the first time during their hospitalization, and patients continued on their previous antipsychotics.
• Does the type of antipsychotic agent used during the acute inpatient admission affect blood glucose levels or clinical outcomes?
Mean daily blood glucose levels were similar regardless of whether patients were on atypical antipsychotics, typical antipsychotics, or both. The mean hemoglobin A1c level was 7.3% in all three groups of antipsychotic-treated patients. However, the rate of medical complications during psychiatric hospitalization was significantly higher among patients on atypical agents, at 23%, compared with 17% in patients on typical antipsychotics and 13% in those on both.
Three percent of patients on atypical antipsychotics-only required transfer to a medical unit for treatment of a complication that arose during psychiatric hospitalization. That was also true for 3% of patients on combined atypical/typical antipsychotic therapy. In contrast, none of the relatively few patients on typical antipsychotics-only required transfer to a medical unit. It’s worth noting that all atypical antipsychotics are not alike in terms of the strength of their association with diabetes. Most antipsychotic-treated patients in this study got olanzapine (Zyprexa) or clozapine (Clozaril). Newer, less diabetogenic atypical agents were less frequently used in this largely indigent population.
• Does a history of diabetes or new hyperglycemia at admission affect outcome measures? Not unexpectedly, patients with a history of diabetes prior to admission had a significantly higher rate of medical complications during their psychiatric inpatient stay: a 28% rate, compared with 22% in patients with no history of diabetes but with hyperglycemia at admission and 20% in patients with neither metabolic abnormality. The most common complication in patients with a history of diabetes was urinary tract infection.
Again unsurprisingly, patients with a history of diabetes had a significantly higher rate of transfer to a medical unit because of medical complications: 5%, compared with no transfers in patients with new hyperglycemia at admission and a 1% transfer rate in patients with neither metabolic abnormality.
Dr. Smiley reported that her research funding came from the National Institutes of Health, Sanofi-Aventis, and Merck.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major finding: Blood glucose levels during acute psychiatric hospitalizations did not differ between antipsychotic-naive patients newly placed on antipsychotic therapy during their inpatient stay, and patients never exposed to antipsychotic agents.
Data source: This was a chart review of 1,212 patients admitted to three acute inpatient psychiatric units located in Atlanta.
Disclosures: Dr. Smiley reported that her research funding came from the National Institutes of Health, Sanofi-Aventis, and Merck.

Lurasidone shows efficacy in bipolar depression
HOLLYWOOD, FLA. – Lurasidone has hit all of its primary and secondary efficacy endpoints in a phase III clinical trial for the treatment of bipolar I depression.
As a result, the drug, already marketed as Latuda for the treatment of schizophrenia, has been approved by the Food and Drug Administration for a requested expanded indication in treating bipolar I depression. The approval, which came July 1, was made for lurasidone as monotherapy and adjunctive therapy with lithium or valproate.
The phase III PREVAIL 2 (Program to Evaluate the Antidepressant Impact of Lurasidone) study was a 6-week, double-blind, placebo-controlled, multicenter clinical trial involving 505 patients with bipolar I depression. They were randomized to once-daily, flexibly dosed lurasidone at either 20-60 mg/day or 80-120 mg/day, or to placebo.
The primary efficacy endpoint was change from baseline through week 6 in scores on MADRS (the Montgomery-Åsberg Depression Rating Scale). From a mean baseline score of 30, both the lower- and higher-dose lurasidone groups averaged identical 15.4-point reductions, a significantly greater improvement than the 10.7-point decrease in placebo-treated controls, Dr. Antony D. Loebel reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
This pattern of closely similar efficacy in the lower- and higher-dose lurasidone groups, whose mean modal doses were 34.9 and 92.3 mg/day, respectively, was repeated for the other study endpoints, observed Dr. Loebel, executive vice president and chief medical officer at Sunovion Pharmaceuticals, Fort Lee, N.J.
For example, the Clinical Global Impression, Bipolar Severity depression score decreased from a baseline of 4.5 by a mean of 1.8 points in the lower-dose lurasidone group and 1.7 points in the higher-dose arm, significantly greater than the 1.1-point decline with placebo.
Similarly, 53% and 51% of the lower- and higher-dose lurasidone groups, respectively, were deemed treatment responders based upon at least a 50% reduction in MADRS scores, compared with 30% of controls.
The remission rate, defined as a final MADRS score of 12 or less, was 42% in the lower-dose lurasidone arm, 40% in patients on higher-dose therapy, and 25% with placebo.
All three patient groups showed similarly modest decreases over time in the Young Mania Rating Scale: a mean 1-point drop in the lower-dose lurasidone arm, a 0.7-point reduction with higher-dose therapy, and a 0.9-point reduction with placebo. That’s an important and reassuring finding, because attempts to treat bipolar mania using conventional antidepressants can result in a switch to mania. In this study, treatment-emergent mania occurred in just 1% of subjects on lower-dose lurasidone, 0% on higher-dose therapy, and 1% on placebo, Dr. Loebel continued.
Numerous other studies have established that roughly 90% of patients with bipolar depression experience severe functional impairment. Of note, PREVAIL 2 patients in the lower-dose lurasidone arm displayed a highly significant mean 9.5-point reduction from a baseline score of 20 on the Sheehan Disability Scale, and the higher-dose lurasidone group showed a 9.8-point decrease. In contrast, scores did not change over time in the control group.
In a similar vein, scores on the Quick Inventory of Depressive Symptomatology-Self-Report improved from a baseline of 33.5 by 19.3 and 19.8 points, respectively, in the lower- and higher-dose lurasidone arms, significantly better than the 12.8-point improvement with placebo, reported Dr. Loebel also of the department of psychiatry at New York University.
A total of 6%-7% of subjects in each study arm discontinued the trial because of adverse events. Nausea and akathisia were the two adverse events that were seen more frequently with lurasidone than placebo. No significant changes in lipids, body weight, or glycemic control were observed.
Lurasidone’s efficacy in bipolar depression is attributed to the drug’s unique pharmacodynamic profile. It is a more potent blocker of the serotonin 5HT7 receptor than are other atypical antipsychotics. It also is a strong antagonist of the dopamine D2 and 5-HT2A receptors, a moderate partial agonist at the 5HT1a receptor, and has a moderate antagonist effect at the alpha-2c receptor.
Before the approval, quetiapine (Seroquel) was the only drug approved as monotherapy for bipolar depression.
Sunovion Pharmaceuticals sponsored the phase III trial. Dr. Loebel is a company employee.
*This story was updated 7/3/2013.
HOLLYWOOD, FLA. – Lurasidone has hit all of its primary and secondary efficacy endpoints in a phase III clinical trial for the treatment of bipolar I depression.
As a result, the drug, already marketed as Latuda for the treatment of schizophrenia, has been approved by the Food and Drug Administration for a requested expanded indication in treating bipolar I depression. The approval, which came July 1, was made for lurasidone as monotherapy and adjunctive therapy with lithium or valproate.
The phase III PREVAIL 2 (Program to Evaluate the Antidepressant Impact of Lurasidone) study was a 6-week, double-blind, placebo-controlled, multicenter clinical trial involving 505 patients with bipolar I depression. They were randomized to once-daily, flexibly dosed lurasidone at either 20-60 mg/day or 80-120 mg/day, or to placebo.
The primary efficacy endpoint was change from baseline through week 6 in scores on MADRS (the Montgomery-Åsberg Depression Rating Scale). From a mean baseline score of 30, both the lower- and higher-dose lurasidone groups averaged identical 15.4-point reductions, a significantly greater improvement than the 10.7-point decrease in placebo-treated controls, Dr. Antony D. Loebel reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
This pattern of closely similar efficacy in the lower- and higher-dose lurasidone groups, whose mean modal doses were 34.9 and 92.3 mg/day, respectively, was repeated for the other study endpoints, observed Dr. Loebel, executive vice president and chief medical officer at Sunovion Pharmaceuticals, Fort Lee, N.J.
For example, the Clinical Global Impression, Bipolar Severity depression score decreased from a baseline of 4.5 by a mean of 1.8 points in the lower-dose lurasidone group and 1.7 points in the higher-dose arm, significantly greater than the 1.1-point decline with placebo.
Similarly, 53% and 51% of the lower- and higher-dose lurasidone groups, respectively, were deemed treatment responders based upon at least a 50% reduction in MADRS scores, compared with 30% of controls.
The remission rate, defined as a final MADRS score of 12 or less, was 42% in the lower-dose lurasidone arm, 40% in patients on higher-dose therapy, and 25% with placebo.
All three patient groups showed similarly modest decreases over time in the Young Mania Rating Scale: a mean 1-point drop in the lower-dose lurasidone arm, a 0.7-point reduction with higher-dose therapy, and a 0.9-point reduction with placebo. That’s an important and reassuring finding, because attempts to treat bipolar mania using conventional antidepressants can result in a switch to mania. In this study, treatment-emergent mania occurred in just 1% of subjects on lower-dose lurasidone, 0% on higher-dose therapy, and 1% on placebo, Dr. Loebel continued.
Numerous other studies have established that roughly 90% of patients with bipolar depression experience severe functional impairment. Of note, PREVAIL 2 patients in the lower-dose lurasidone arm displayed a highly significant mean 9.5-point reduction from a baseline score of 20 on the Sheehan Disability Scale, and the higher-dose lurasidone group showed a 9.8-point decrease. In contrast, scores did not change over time in the control group.
In a similar vein, scores on the Quick Inventory of Depressive Symptomatology-Self-Report improved from a baseline of 33.5 by 19.3 and 19.8 points, respectively, in the lower- and higher-dose lurasidone arms, significantly better than the 12.8-point improvement with placebo, reported Dr. Loebel also of the department of psychiatry at New York University.
A total of 6%-7% of subjects in each study arm discontinued the trial because of adverse events. Nausea and akathisia were the two adverse events that were seen more frequently with lurasidone than placebo. No significant changes in lipids, body weight, or glycemic control were observed.
Lurasidone’s efficacy in bipolar depression is attributed to the drug’s unique pharmacodynamic profile. It is a more potent blocker of the serotonin 5HT7 receptor than are other atypical antipsychotics. It also is a strong antagonist of the dopamine D2 and 5-HT2A receptors, a moderate partial agonist at the 5HT1a receptor, and has a moderate antagonist effect at the alpha-2c receptor.
Before the approval, quetiapine (Seroquel) was the only drug approved as monotherapy for bipolar depression.
Sunovion Pharmaceuticals sponsored the phase III trial. Dr. Loebel is a company employee.
*This story was updated 7/3/2013.
HOLLYWOOD, FLA. – Lurasidone has hit all of its primary and secondary efficacy endpoints in a phase III clinical trial for the treatment of bipolar I depression.
As a result, the drug, already marketed as Latuda for the treatment of schizophrenia, has been approved by the Food and Drug Administration for a requested expanded indication in treating bipolar I depression. The approval, which came July 1, was made for lurasidone as monotherapy and adjunctive therapy with lithium or valproate.
The phase III PREVAIL 2 (Program to Evaluate the Antidepressant Impact of Lurasidone) study was a 6-week, double-blind, placebo-controlled, multicenter clinical trial involving 505 patients with bipolar I depression. They were randomized to once-daily, flexibly dosed lurasidone at either 20-60 mg/day or 80-120 mg/day, or to placebo.
The primary efficacy endpoint was change from baseline through week 6 in scores on MADRS (the Montgomery-Åsberg Depression Rating Scale). From a mean baseline score of 30, both the lower- and higher-dose lurasidone groups averaged identical 15.4-point reductions, a significantly greater improvement than the 10.7-point decrease in placebo-treated controls, Dr. Antony D. Loebel reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
This pattern of closely similar efficacy in the lower- and higher-dose lurasidone groups, whose mean modal doses were 34.9 and 92.3 mg/day, respectively, was repeated for the other study endpoints, observed Dr. Loebel, executive vice president and chief medical officer at Sunovion Pharmaceuticals, Fort Lee, N.J.
For example, the Clinical Global Impression, Bipolar Severity depression score decreased from a baseline of 4.5 by a mean of 1.8 points in the lower-dose lurasidone group and 1.7 points in the higher-dose arm, significantly greater than the 1.1-point decline with placebo.
Similarly, 53% and 51% of the lower- and higher-dose lurasidone groups, respectively, were deemed treatment responders based upon at least a 50% reduction in MADRS scores, compared with 30% of controls.
The remission rate, defined as a final MADRS score of 12 or less, was 42% in the lower-dose lurasidone arm, 40% in patients on higher-dose therapy, and 25% with placebo.
All three patient groups showed similarly modest decreases over time in the Young Mania Rating Scale: a mean 1-point drop in the lower-dose lurasidone arm, a 0.7-point reduction with higher-dose therapy, and a 0.9-point reduction with placebo. That’s an important and reassuring finding, because attempts to treat bipolar mania using conventional antidepressants can result in a switch to mania. In this study, treatment-emergent mania occurred in just 1% of subjects on lower-dose lurasidone, 0% on higher-dose therapy, and 1% on placebo, Dr. Loebel continued.
Numerous other studies have established that roughly 90% of patients with bipolar depression experience severe functional impairment. Of note, PREVAIL 2 patients in the lower-dose lurasidone arm displayed a highly significant mean 9.5-point reduction from a baseline score of 20 on the Sheehan Disability Scale, and the higher-dose lurasidone group showed a 9.8-point decrease. In contrast, scores did not change over time in the control group.
In a similar vein, scores on the Quick Inventory of Depressive Symptomatology-Self-Report improved from a baseline of 33.5 by 19.3 and 19.8 points, respectively, in the lower- and higher-dose lurasidone arms, significantly better than the 12.8-point improvement with placebo, reported Dr. Loebel also of the department of psychiatry at New York University.
A total of 6%-7% of subjects in each study arm discontinued the trial because of adverse events. Nausea and akathisia were the two adverse events that were seen more frequently with lurasidone than placebo. No significant changes in lipids, body weight, or glycemic control were observed.
Lurasidone’s efficacy in bipolar depression is attributed to the drug’s unique pharmacodynamic profile. It is a more potent blocker of the serotonin 5HT7 receptor than are other atypical antipsychotics. It also is a strong antagonist of the dopamine D2 and 5-HT2A receptors, a moderate partial agonist at the 5HT1a receptor, and has a moderate antagonist effect at the alpha-2c receptor.
Before the approval, quetiapine (Seroquel) was the only drug approved as monotherapy for bipolar depression.
Sunovion Pharmaceuticals sponsored the phase III trial. Dr. Loebel is a company employee.
*This story was updated 7/3/2013.
AT THE NCDEU MEETING
Major finding: Patients with bipolar I depression responded to 6 weeks of lurasidone at either a lower or higher dose range with identical 15.4-point improvements from a baseline score of 30 on the Montgomery-Åsberg Depression Rating Scale, significantly better than the 10.7-point improvement with placebo.
Data source: PREVAIL 2 was a phase III, double-blind, 6-week randomized trial involving 505 patients with bipolar I depression.
Disclosures: The study was sponsored by Sunovion. The presenter is a company employee.
New metformin formulation broadens candidate population
CHICAGO – A novel delayed-release formulation of oral metformin holds the promise of making the drug work for the many patients with type 2 diabetes who are unable to take generic metformin because of renal insufficiency or GI intolerance.
The key to this development was the discovery that metformin’s primary site of action isn’t the circulation, it’s the lower bowel – specifically, the gut enteroendocrine L-cells. Metformin plasma exposure is not required for the drug’s glucose-lowering effects, Dr. Ralph A. DeFronzo asserted at the annual scientific sessions of the American Diabetes Association.

The investigational proprietary delayed-release formulation of metformin, known as NewMet, is designed to minimize the drug’s bioavailability. The delayed-release tablet bypasses the highly absorptive upper bowel, instead delivering the medication to the distal small intestine, where metformin is poorly absorbed into the circulation and enteroendocrine L-cells are concentrated. Metformin’s glucose-lowering effects are now thought to result from the drug’s stimulation of the L-cells to release gut hormones, including glucagon-like peptide-1 (GLP-1) and peptide YY, said Dr. DeFronzo, professor of medicine and director of the diabetes division at University of Texas Health Science Center at San Antonio.
He presented a proof-of-concept study demonstrating that metformin’s glucose-lowering effect is dissociated from plasma drug levels. The randomized, double-blind, crossover study involved 24 patients with type 2 diabetes. After being withdrawn from oral diabetes therapy for 2 weeks, they received in random order 5 days of commercially available, immediate-release metformin at 1,000 mg twice daily; delayed-release metformin at 1,000 mg twice daily; and delayed-release metformin at 500 mg twice daily. Each 5-day treatment period was separated by a 9- to 12-day washout.
Each of the two dosing regimens of delayed-release metformin proved as effective as conventional metformin. They reduced fasting and postprandial plasma glucose levels as well as the 10-hour glucose area under the curve to the same extent as that of immediate-release metformin, without changing the insulin area under the curve. In other words, 1,000 mg/day of delayed-release metformin was as effective as was standard full-dose conventional metformin at 2,000 mg/day. Yet plasma metformin exposure was reduced by 45% with delayed-release metformin at 1,000 mg twice daily and by 57% by the investigational agent at 500 mg twice daily. Both the conventional and delayed-release formulations of metformin increased GLP-1 and peptide YY levels to a similar extent.
Delayed-release metformin was better tolerated. Unlike conventional metformin, it did not cause any nausea or vomiting, since it was not yet released in the upper bowel. However, both immediate- and delayed-release metformin were associated with a 10%-15% incidence of diarrhea, a side effect of metformin in the lower bowel.
It appears that when a patient is on full-dose conventional metformin, about half of the dose is absorbed into the circulation from the stomach, while the other half continues downstream to the lower bowel, where it actually exerts its therapeutic effects, according to Dr. DeFronzo.
Conventional metformin is contraindicated in patients with renal insufficiency. That’s because once metformin is absorbed into the circulation, it gets cleared by the kidney. If renal function is impaired, the drug accumulates and can cause lactic acidosis due to metformin-induced liver toxicity.

Delayed-release metformin should sidestep this problem because the plasma levels are so much lower than with the conventional drug. That was suggested in a separate study presented at the ADA meeting by Mark Fineman, Ph.D., senior vice president for research and development at San Diego–based Elcelyx Therapeutics Inc., which is developing NewMet. He presented a model designed to predict plasma metformin concentrations that would result if type 2 diabetes patients with varying degrees of renal impairment were to take delayed-release metformin. The results suggested no increased risk of lactic acidosis at 1,000 or 2,000 mg/day. And a study is now underway to see if 800 or 600 mg once daily can provide the full effect seen at 500 mg twice daily in Dr. DeFronzo’s study, Dr. Fineman said in an interview.
Metformin is recommended in ADA treatment guidelines as the initial drug of choice for patients with type 2 diabetes. It is the most commonly prescribed medication for type 2 diabetes in the world. Yet roughly 40% of patients don’t take it due to renal insufficiency or GI intolerance. Moreover, many patients on metformin are unable to take full-dose therapy because of limited tolerability. These are the patient populations where delayed-release metformin could be a boon.
Dr. Fineman said Elcelyx is now in discussions with the FDA about the requirements for marketing approval, which are likely to be abbreviated since NewMet is a new formulation of an approved drug.
"We’ll need to do a phase III program to show that in non–renally impaired people delayed-release metformin works just as well as regular metformin, maybe better, and then we’ll do studies in renally impaired patients to show what the efficacy is," he said.
Dr. DeFronzo is a clinical adviser to Elcelyx.
CHICAGO – A novel delayed-release formulation of oral metformin holds the promise of making the drug work for the many patients with type 2 diabetes who are unable to take generic metformin because of renal insufficiency or GI intolerance.
The key to this development was the discovery that metformin’s primary site of action isn’t the circulation, it’s the lower bowel – specifically, the gut enteroendocrine L-cells. Metformin plasma exposure is not required for the drug’s glucose-lowering effects, Dr. Ralph A. DeFronzo asserted at the annual scientific sessions of the American Diabetes Association.
The investigational proprietary delayed-release formulation of metformin, known as NewMet, is designed to minimize the drug’s bioavailability. The delayed-release tablet bypasses the highly absorptive upper bowel, instead delivering the medication to the distal small intestine, where metformin is poorly absorbed into the circulation and enteroendocrine L-cells are concentrated. Metformin’s glucose-lowering effects are now thought to result from the drug’s stimulation of the L-cells to release gut hormones, including glucagon-like peptide-1 (GLP-1) and peptide YY, said Dr. DeFronzo, professor of medicine and director of the diabetes division at University of Texas Health Science Center at San Antonio.
He presented a proof-of-concept study demonstrating that metformin’s glucose-lowering effect is dissociated from plasma drug levels. The randomized, double-blind, crossover study involved 24 patients with type 2 diabetes. After being withdrawn from oral diabetes therapy for 2 weeks, they received in random order 5 days of commercially available, immediate-release metformin at 1,000 mg twice daily; delayed-release metformin at 1,000 mg twice daily; and delayed-release metformin at 500 mg twice daily. Each 5-day treatment period was separated by a 9- to 12-day washout.
Each of the two dosing regimens of delayed-release metformin proved as effective as conventional metformin. They reduced fasting and postprandial plasma glucose levels as well as the 10-hour glucose area under the curve to the same extent as that of immediate-release metformin, without changing the insulin area under the curve. In other words, 1,000 mg/day of delayed-release metformin was as effective as was standard full-dose conventional metformin at 2,000 mg/day. Yet plasma metformin exposure was reduced by 45% with delayed-release metformin at 1,000 mg twice daily and by 57% by the investigational agent at 500 mg twice daily. Both the conventional and delayed-release formulations of metformin increased GLP-1 and peptide YY levels to a similar extent.
Delayed-release metformin was better tolerated. Unlike conventional metformin, it did not cause any nausea or vomiting, since it was not yet released in the upper bowel. However, both immediate- and delayed-release metformin were associated with a 10%-15% incidence of diarrhea, a side effect of metformin in the lower bowel.
It appears that when a patient is on full-dose conventional metformin, about half of the dose is absorbed into the circulation from the stomach, while the other half continues downstream to the lower bowel, where it actually exerts its therapeutic effects, according to Dr. DeFronzo.
Conventional metformin is contraindicated in patients with renal insufficiency. That’s because once metformin is absorbed into the circulation, it gets cleared by the kidney. If renal function is impaired, the drug accumulates and can cause lactic acidosis due to metformin-induced liver toxicity.
Delayed-release metformin should sidestep this problem because the plasma levels are so much lower than with the conventional drug. That was suggested in a separate study presented at the ADA meeting by Mark Fineman, Ph.D., senior vice president for research and development at San Diego–based Elcelyx Therapeutics Inc., which is developing NewMet. He presented a model designed to predict plasma metformin concentrations that would result if type 2 diabetes patients with varying degrees of renal impairment were to take delayed-release metformin. The results suggested no increased risk of lactic acidosis at 1,000 or 2,000 mg/day. And a study is now underway to see if 800 or 600 mg once daily can provide the full effect seen at 500 mg twice daily in Dr. DeFronzo’s study, Dr. Fineman said in an interview.
Metformin is recommended in ADA treatment guidelines as the initial drug of choice for patients with type 2 diabetes. It is the most commonly prescribed medication for type 2 diabetes in the world. Yet roughly 40% of patients don’t take it due to renal insufficiency or GI intolerance. Moreover, many patients on metformin are unable to take full-dose therapy because of limited tolerability. These are the patient populations where delayed-release metformin could be a boon.
Dr. Fineman said Elcelyx is now in discussions with the FDA about the requirements for marketing approval, which are likely to be abbreviated since NewMet is a new formulation of an approved drug.
"We’ll need to do a phase III program to show that in non–renally impaired people delayed-release metformin works just as well as regular metformin, maybe better, and then we’ll do studies in renally impaired patients to show what the efficacy is," he said.
Dr. DeFronzo is a clinical adviser to Elcelyx.
CHICAGO – A novel delayed-release formulation of oral metformin holds the promise of making the drug work for the many patients with type 2 diabetes who are unable to take generic metformin because of renal insufficiency or GI intolerance.
The key to this development was the discovery that metformin’s primary site of action isn’t the circulation, it’s the lower bowel – specifically, the gut enteroendocrine L-cells. Metformin plasma exposure is not required for the drug’s glucose-lowering effects, Dr. Ralph A. DeFronzo asserted at the annual scientific sessions of the American Diabetes Association.
The investigational proprietary delayed-release formulation of metformin, known as NewMet, is designed to minimize the drug’s bioavailability. The delayed-release tablet bypasses the highly absorptive upper bowel, instead delivering the medication to the distal small intestine, where metformin is poorly absorbed into the circulation and enteroendocrine L-cells are concentrated. Metformin’s glucose-lowering effects are now thought to result from the drug’s stimulation of the L-cells to release gut hormones, including glucagon-like peptide-1 (GLP-1) and peptide YY, said Dr. DeFronzo, professor of medicine and director of the diabetes division at University of Texas Health Science Center at San Antonio.
He presented a proof-of-concept study demonstrating that metformin’s glucose-lowering effect is dissociated from plasma drug levels. The randomized, double-blind, crossover study involved 24 patients with type 2 diabetes. After being withdrawn from oral diabetes therapy for 2 weeks, they received in random order 5 days of commercially available, immediate-release metformin at 1,000 mg twice daily; delayed-release metformin at 1,000 mg twice daily; and delayed-release metformin at 500 mg twice daily. Each 5-day treatment period was separated by a 9- to 12-day washout.
Each of the two dosing regimens of delayed-release metformin proved as effective as conventional metformin. They reduced fasting and postprandial plasma glucose levels as well as the 10-hour glucose area under the curve to the same extent as that of immediate-release metformin, without changing the insulin area under the curve. In other words, 1,000 mg/day of delayed-release metformin was as effective as was standard full-dose conventional metformin at 2,000 mg/day. Yet plasma metformin exposure was reduced by 45% with delayed-release metformin at 1,000 mg twice daily and by 57% by the investigational agent at 500 mg twice daily. Both the conventional and delayed-release formulations of metformin increased GLP-1 and peptide YY levels to a similar extent.
Delayed-release metformin was better tolerated. Unlike conventional metformin, it did not cause any nausea or vomiting, since it was not yet released in the upper bowel. However, both immediate- and delayed-release metformin were associated with a 10%-15% incidence of diarrhea, a side effect of metformin in the lower bowel.
It appears that when a patient is on full-dose conventional metformin, about half of the dose is absorbed into the circulation from the stomach, while the other half continues downstream to the lower bowel, where it actually exerts its therapeutic effects, according to Dr. DeFronzo.
Conventional metformin is contraindicated in patients with renal insufficiency. That’s because once metformin is absorbed into the circulation, it gets cleared by the kidney. If renal function is impaired, the drug accumulates and can cause lactic acidosis due to metformin-induced liver toxicity.
Delayed-release metformin should sidestep this problem because the plasma levels are so much lower than with the conventional drug. That was suggested in a separate study presented at the ADA meeting by Mark Fineman, Ph.D., senior vice president for research and development at San Diego–based Elcelyx Therapeutics Inc., which is developing NewMet. He presented a model designed to predict plasma metformin concentrations that would result if type 2 diabetes patients with varying degrees of renal impairment were to take delayed-release metformin. The results suggested no increased risk of lactic acidosis at 1,000 or 2,000 mg/day. And a study is now underway to see if 800 or 600 mg once daily can provide the full effect seen at 500 mg twice daily in Dr. DeFronzo’s study, Dr. Fineman said in an interview.
Metformin is recommended in ADA treatment guidelines as the initial drug of choice for patients with type 2 diabetes. It is the most commonly prescribed medication for type 2 diabetes in the world. Yet roughly 40% of patients don’t take it due to renal insufficiency or GI intolerance. Moreover, many patients on metformin are unable to take full-dose therapy because of limited tolerability. These are the patient populations where delayed-release metformin could be a boon.
Dr. Fineman said Elcelyx is now in discussions with the FDA about the requirements for marketing approval, which are likely to be abbreviated since NewMet is a new formulation of an approved drug.
"We’ll need to do a phase III program to show that in non–renally impaired people delayed-release metformin works just as well as regular metformin, maybe better, and then we’ll do studies in renally impaired patients to show what the efficacy is," he said.
Dr. DeFronzo is a clinical adviser to Elcelyx.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Major Finding: A novel delayed-release formulation of metformin reduced fasting plasma glucose in patients with type 2 diabetes by 19.9 mg/dL at 1,000 mg twice daily and by 16.4 mg/dL at 500 mg twice daily, effect sizes similar to what was seen with commercially available immediate-release metformin at 1,000 mg twice daily.
Data Source: A proof-of-concept study demonstrating that metformin’s mechanism of lowering glucose is mediated by the gut rather than the circulation.
Disclosures: The study was sponsored by Elcelyx Therapeutics, which is developing the proprietary delayed-release metformin. Dr. DeFronzo is a clinical adviser to the company.
