User login
Erythematous Plaques on a Tattoo
The Diagnosis: Epidermodysplasia Verruciformis
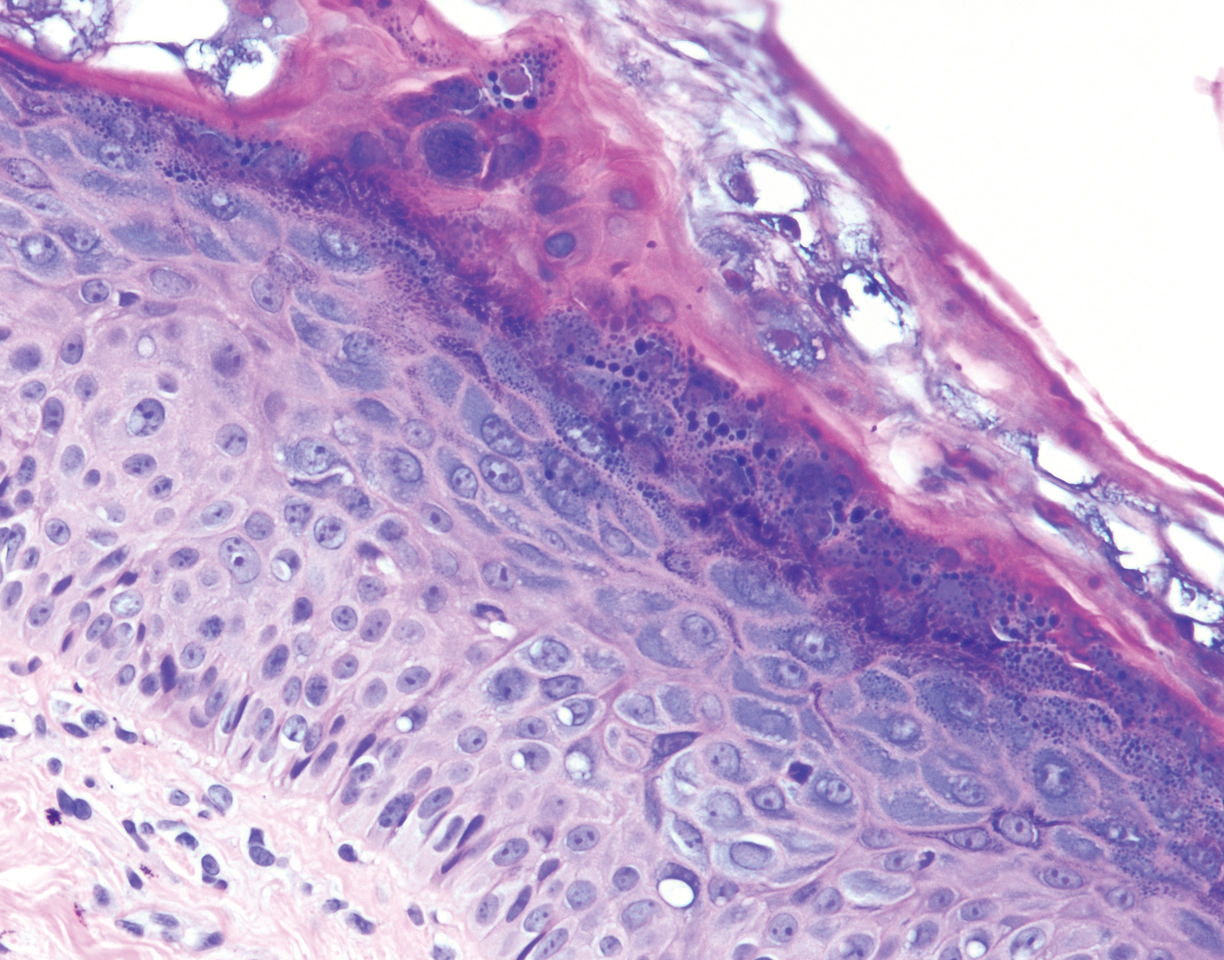
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
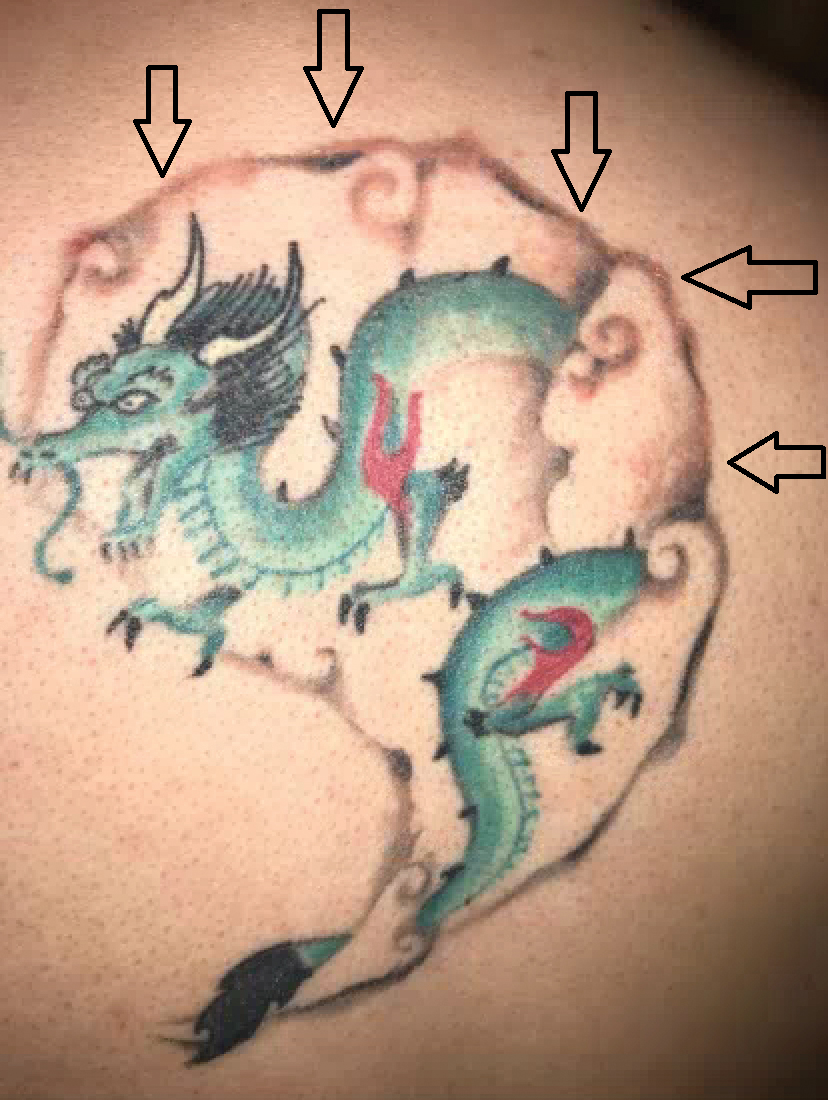
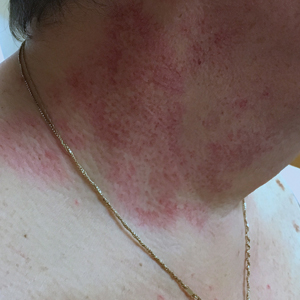
A 29-year-old man presented with increased redness, dryness, and pruritus at the periphery of a tattoo (arrows) on the upper back of 4 months' duration. He was diagnosed with human immunodeficiency virus 8 months prior to presentation and had a history of cystic fibrosis, eczema, and genital molluscum contagiosum. Laboratory analysis 1 month prior revealed a CD4 count of 42 cells/mm3 (reference range, 500-1200 cells/mm3), and the viral load was 2388 copies/mL (reference range, 20-10,000,000 copies/mL). Physical examination revealed multiple erythematous, eczematous, linear plaques along the dark gray lines of the tattoo. A 1.1.2 ×0.7.2 ×0.1-cm shave biopsy specimen was obtained. After the biopsy, tretinoin cream 0.1% and betamethasone dipropionate ointment 0.05% were prescribed to be alternately applied on the tattoo lesions until resolution.
Patient Questionnaire to Reduce Anxiety Prior to Full-Body Skin Examination
To the Editor:
A thorough full-body skin examination (FBSE) is an integral component of a dermatologic encounter and helps identify potentially malignant and high-risk lesions, particularly in areas that are difficult for the patient to visualize.1 Despite these benefits, many patients experience discomfort and anxiety about this examination because it involves sensitive anatomical areas. The true psychological impact of an FBSE is not clearly understood; however, research into improving patient comfort in these circumstances can have a broad positive impact.2 The purpose of this pilot study was to establish patients’ willingness to complete a pre-encounter questionnaire that defines their FBSE preferences as well as to identify the anatomical areas that are of most concern.
This study was approved by the University of Kansas institutional review board as nonhuman subjects research. A pre-encounter questionnaire that included information about the benefits of FBSEs was administered to 34 patients, allowing them to identify anatomic locations that they wanted to exclude from the FBSE.
Following the patient visit (in which the identified anatomical locations were excluded), patients were given a brief exit survey that asked about (1) their preference for a pre-encounter FBSE questionnaire and (2) the impact of the questionnaire on their anxiety level throughout the encounter. Preference for asking was surveyed using a 10-point scale (10=strong preference for the pre-encounter survey; 1=strong preference against the pre-encounter survey). Change in anxiety was surveyed using a 10-point scale (10=strong reduction in anxiety after the pre-encounter survey; 1=strong increase in anxiety after the pre-encounter survey). Statistical analysis was performed using 2-tailed unpaired t tests, with P<.05 considered statistically significant.
Twenty female and 14 male patients were enrolled (mean age, 53 years)(Table). The most commonly excluded anatomical location on the pre-encounter survey was the genitals, followed by the buttocks, breasts/chest, legs, feet, and abdomen (Table); 10 (71%) male and 13 (65%) female respondents did not exclude any component of the FBSE.
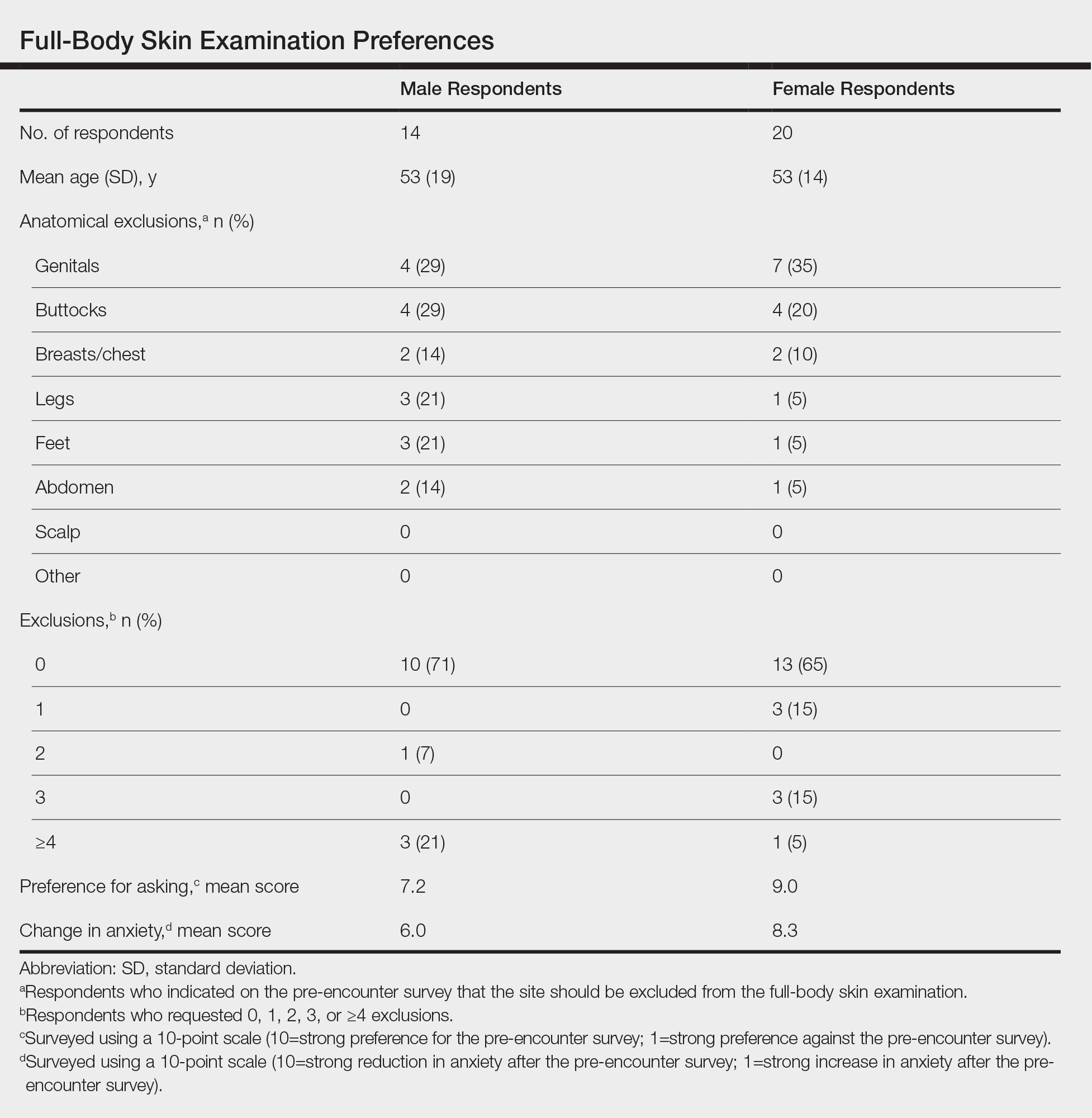
After the provider visit, females had a higher preference for the pre-encounter survey (mean score, 9.0) compared to males (mean score, 7.2; P=.021). Similarly, females had reduced anxiety about the office visit after survey administration compared to males (mean score, 8.3 vs 6.0; P=.001)(Table).
The results of our pilot study showed that a brief pre-encounter questionnaire may reduce the distress associated with an FBSE. Our survey took less than 1 minute to complete and served as a useful guide to direct the provider during the FBSE. Moreover, recognizing that patients do not want certain anatomic locations examined can serve as an opportunity for the dermatologist to provide helpful home skin check instructions and recommendations.
The small sample size was a limitation of this study. Future studies can assess with greater precision the clear benefits of a pre-encounter survey as well as the benefits or drawbacks of a survey compared to other modalities that are aimed at reducing patient anxiety about the FBSE, such as having the physician directly ask the patient about areas to avoid during the examination.
A pre-encounter survey about the FBSE can serve as an efficient means of determining patient preference and reducing self-reported anxiety about the visit.
- Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
- Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
To the Editor:
A thorough full-body skin examination (FBSE) is an integral component of a dermatologic encounter and helps identify potentially malignant and high-risk lesions, particularly in areas that are difficult for the patient to visualize.1 Despite these benefits, many patients experience discomfort and anxiety about this examination because it involves sensitive anatomical areas. The true psychological impact of an FBSE is not clearly understood; however, research into improving patient comfort in these circumstances can have a broad positive impact.2 The purpose of this pilot study was to establish patients’ willingness to complete a pre-encounter questionnaire that defines their FBSE preferences as well as to identify the anatomical areas that are of most concern.
This study was approved by the University of Kansas institutional review board as nonhuman subjects research. A pre-encounter questionnaire that included information about the benefits of FBSEs was administered to 34 patients, allowing them to identify anatomic locations that they wanted to exclude from the FBSE.
Following the patient visit (in which the identified anatomical locations were excluded), patients were given a brief exit survey that asked about (1) their preference for a pre-encounter FBSE questionnaire and (2) the impact of the questionnaire on their anxiety level throughout the encounter. Preference for asking was surveyed using a 10-point scale (10=strong preference for the pre-encounter survey; 1=strong preference against the pre-encounter survey). Change in anxiety was surveyed using a 10-point scale (10=strong reduction in anxiety after the pre-encounter survey; 1=strong increase in anxiety after the pre-encounter survey). Statistical analysis was performed using 2-tailed unpaired t tests, with P<.05 considered statistically significant.
Twenty female and 14 male patients were enrolled (mean age, 53 years)(Table). The most commonly excluded anatomical location on the pre-encounter survey was the genitals, followed by the buttocks, breasts/chest, legs, feet, and abdomen (Table); 10 (71%) male and 13 (65%) female respondents did not exclude any component of the FBSE.
After the provider visit, females had a higher preference for the pre-encounter survey (mean score, 9.0) compared to males (mean score, 7.2; P=.021). Similarly, females had reduced anxiety about the office visit after survey administration compared to males (mean score, 8.3 vs 6.0; P=.001)(Table).
The results of our pilot study showed that a brief pre-encounter questionnaire may reduce the distress associated with an FBSE. Our survey took less than 1 minute to complete and served as a useful guide to direct the provider during the FBSE. Moreover, recognizing that patients do not want certain anatomic locations examined can serve as an opportunity for the dermatologist to provide helpful home skin check instructions and recommendations.
The small sample size was a limitation of this study. Future studies can assess with greater precision the clear benefits of a pre-encounter survey as well as the benefits or drawbacks of a survey compared to other modalities that are aimed at reducing patient anxiety about the FBSE, such as having the physician directly ask the patient about areas to avoid during the examination.
A pre-encounter survey about the FBSE can serve as an efficient means of determining patient preference and reducing self-reported anxiety about the visit.
To the Editor:
A thorough full-body skin examination (FBSE) is an integral component of a dermatologic encounter and helps identify potentially malignant and high-risk lesions, particularly in areas that are difficult for the patient to visualize.1 Despite these benefits, many patients experience discomfort and anxiety about this examination because it involves sensitive anatomical areas. The true psychological impact of an FBSE is not clearly understood; however, research into improving patient comfort in these circumstances can have a broad positive impact.2 The purpose of this pilot study was to establish patients’ willingness to complete a pre-encounter questionnaire that defines their FBSE preferences as well as to identify the anatomical areas that are of most concern.
This study was approved by the University of Kansas institutional review board as nonhuman subjects research. A pre-encounter questionnaire that included information about the benefits of FBSEs was administered to 34 patients, allowing them to identify anatomic locations that they wanted to exclude from the FBSE.
Following the patient visit (in which the identified anatomical locations were excluded), patients were given a brief exit survey that asked about (1) their preference for a pre-encounter FBSE questionnaire and (2) the impact of the questionnaire on their anxiety level throughout the encounter. Preference for asking was surveyed using a 10-point scale (10=strong preference for the pre-encounter survey; 1=strong preference against the pre-encounter survey). Change in anxiety was surveyed using a 10-point scale (10=strong reduction in anxiety after the pre-encounter survey; 1=strong increase in anxiety after the pre-encounter survey). Statistical analysis was performed using 2-tailed unpaired t tests, with P<.05 considered statistically significant.
Twenty female and 14 male patients were enrolled (mean age, 53 years)(Table). The most commonly excluded anatomical location on the pre-encounter survey was the genitals, followed by the buttocks, breasts/chest, legs, feet, and abdomen (Table); 10 (71%) male and 13 (65%) female respondents did not exclude any component of the FBSE.
After the provider visit, females had a higher preference for the pre-encounter survey (mean score, 9.0) compared to males (mean score, 7.2; P=.021). Similarly, females had reduced anxiety about the office visit after survey administration compared to males (mean score, 8.3 vs 6.0; P=.001)(Table).
The results of our pilot study showed that a brief pre-encounter questionnaire may reduce the distress associated with an FBSE. Our survey took less than 1 minute to complete and served as a useful guide to direct the provider during the FBSE. Moreover, recognizing that patients do not want certain anatomic locations examined can serve as an opportunity for the dermatologist to provide helpful home skin check instructions and recommendations.
The small sample size was a limitation of this study. Future studies can assess with greater precision the clear benefits of a pre-encounter survey as well as the benefits or drawbacks of a survey compared to other modalities that are aimed at reducing patient anxiety about the FBSE, such as having the physician directly ask the patient about areas to avoid during the examination.
A pre-encounter survey about the FBSE can serve as an efficient means of determining patient preference and reducing self-reported anxiety about the visit.
- Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
- Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
- Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
- Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
Practice Points
- Full-body skin examination (FBSE) is an assessment that requires examination of sensitive body areas, any of which can be seen as intrusive by certain patients.
- A pre-encounter survey on the FBSE can offer an efficient means by which to determine patient preference and reduce visit-associated anxiety.
Pruritic Papules on the Face and Chest
The Diagnosis: Eosinophilic Folliculitis
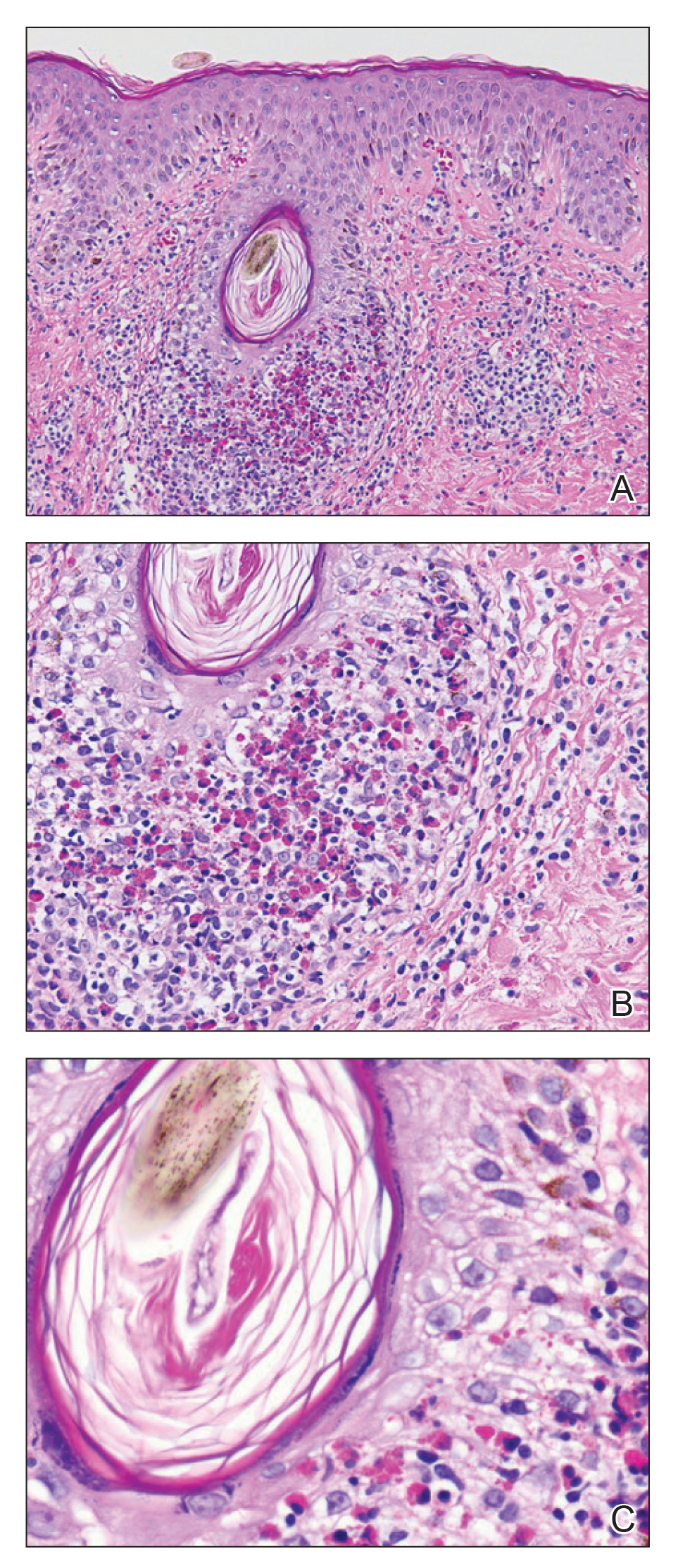
A shave biopsy specimen of an intact pustule on the left side of the chest was obtained. Histopathologic examination revealed follicular inflammation with copious eosinophils (Figure, A and B). Based on the histopathology and clinical presentation, a diagnosis of human immunodeficiency virus (HIV)-associated eosinophilic folliculitis (EF) was made.
The patient was started on triamcinolone ointment 0.1% twice daily to active lesions, oral cetirizine 10 mg in the morning, and oral hydroxyzine 25 mg at bedtime. Laboratory evaluation at the time of diagnosis showed eosinophilia with a peripheral blood eosinophil count of 0.5 K/μL (reference range, 0.03–0.48 K/μL).
Human immunodeficiency virus-associated EF is a pruritic follicular eruption that occurs in HIV-positive individuals with advanced disease. Clinically, it is characterized by intermittent, urticarial, red or flesh-colored, 2- to 5-mm papules with sparse pustules involving the head, neck, arms, and upper trunk.1,2 The cardinal clinical feature of the disorder is intense pruritus, with overlying crusts and excoriations present on physical examination.3
Patients usually have a CD4 count of less than 250 cells/mm3.2,3 Patients with HIV can develop an exacerbation of EF in the first 3 to 6 months after initiating antiretroviral therapy. This clinical pattern is believed to be due to the reconstituted immune system and increased circulation of inflammatory cells.4 Peripheral eosinophilia and elevated serum IgE levels are found in 25% to 50% of patients with HIV-associated EF.2,3
Clinically, the differential diagnosis of intensely pruritic papules with excoriations should include scabies.3 Other diagnoses to consider include opportunistic infections and papular urticaria.5 Acne vulgaris and Demodex folliculitis also may present with lesions similar to HIV-associated EF; however, these lesions tend not to be as intensely pruritic.1,5
The etiology of HIV-associated EF is unknown.3 One proposed mechanism involves a hypersensitivity reaction to Pityrosporum or Demodex mite fragments, as evidenced by studies that found fragments of these microorganisms in biopsied lesions of HIV-associated EF.3,6 In our patient's histopathology, it was noted that the afflicted hair follicle held a single Demodex mite (Figure, C).
The histopathology is characterized by a perifollicular inflammatory infiltrate of eosinophils and CD8+ lymphocytes with areas of sebaceous lysis.3,6 Spongiosis of the follicular epithelium is seen in early lesions of HIV-associated EF.6
The first-line treatment of HIV-associated EF includes antiretroviral therapy with topical steroids and antihistamines. Human immunodeficiency virus-associated EF improves as CD4 helper T-cell counts rise above 250 cells/mm3 with continued antiretroviral therapy, though it initially can cause a flare of the condition.4 High-potency steroids and antihistamines are added during this period to treat the severe pruritus.1,7 In particular, daily cetirizine has been shown to be effective, which may be due to its ability to block eosinophil migration in addition to H1-receptor antagonist properties.3,7
Various alternative therapies have been described in case reports and case series; however, there have been no controlled studies comparing therapies. Phototherapy with UVB light 3 times weekly for 3 to 6 weeks has been effective and curative in recalcitrant cases.7 Other frequently used treatments include oral metronidazole, oral itraconazole, and permethrin cream 5%. The effectiveness of the latter 2 treatments is believed to be related to the proposed role of Pityrosporum and Demodex in the pathogenesis.3
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his help compiling the histopathological images and their diagnostic descriptions.
- Parker SR, Parker DC, McCall CO. Eosinophilic folliculitis in HIV-infected women: case series and review. Am J Clin Dermatol. 2006;7:193-200.
- Rosenthal D, LeBoit PE, Klumpp L, et al. Human immunodeficiency virus-associated eosinophilic folliculitis. a unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206-209.
- Fearfield LA, Rowe A, Francis N, et al. Itchy folliculitis and human immunodeficiency virus infection: clinicopathological and immunological features, pathogenesis, and treatment. Br J Dermatol. 1999;141:3-11.
- Rajendran PM, Dolev JC, Heaphy MR, et al. Eosinophilic folliculitis: before and after the introduction of antiretroviral therapy. Arch Dermatol. 2005;141:1227-1231.
- Nervi SJ, Schwartz RA, Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- McCalmont TH, Altemus D, Maurer T, et al. Eosinophilic folliculitis: the histological spectrum. Am J Dermatopathol. 1995;17:439-446.
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197.
The Diagnosis: Eosinophilic Folliculitis
A shave biopsy specimen of an intact pustule on the left side of the chest was obtained. Histopathologic examination revealed follicular inflammation with copious eosinophils (Figure, A and B). Based on the histopathology and clinical presentation, a diagnosis of human immunodeficiency virus (HIV)-associated eosinophilic folliculitis (EF) was made.
The patient was started on triamcinolone ointment 0.1% twice daily to active lesions, oral cetirizine 10 mg in the morning, and oral hydroxyzine 25 mg at bedtime. Laboratory evaluation at the time of diagnosis showed eosinophilia with a peripheral blood eosinophil count of 0.5 K/μL (reference range, 0.03–0.48 K/μL).
Human immunodeficiency virus-associated EF is a pruritic follicular eruption that occurs in HIV-positive individuals with advanced disease. Clinically, it is characterized by intermittent, urticarial, red or flesh-colored, 2- to 5-mm papules with sparse pustules involving the head, neck, arms, and upper trunk.1,2 The cardinal clinical feature of the disorder is intense pruritus, with overlying crusts and excoriations present on physical examination.3
Patients usually have a CD4 count of less than 250 cells/mm3.2,3 Patients with HIV can develop an exacerbation of EF in the first 3 to 6 months after initiating antiretroviral therapy. This clinical pattern is believed to be due to the reconstituted immune system and increased circulation of inflammatory cells.4 Peripheral eosinophilia and elevated serum IgE levels are found in 25% to 50% of patients with HIV-associated EF.2,3
Clinically, the differential diagnosis of intensely pruritic papules with excoriations should include scabies.3 Other diagnoses to consider include opportunistic infections and papular urticaria.5 Acne vulgaris and Demodex folliculitis also may present with lesions similar to HIV-associated EF; however, these lesions tend not to be as intensely pruritic.1,5
The etiology of HIV-associated EF is unknown.3 One proposed mechanism involves a hypersensitivity reaction to Pityrosporum or Demodex mite fragments, as evidenced by studies that found fragments of these microorganisms in biopsied lesions of HIV-associated EF.3,6 In our patient's histopathology, it was noted that the afflicted hair follicle held a single Demodex mite (Figure, C).
The histopathology is characterized by a perifollicular inflammatory infiltrate of eosinophils and CD8+ lymphocytes with areas of sebaceous lysis.3,6 Spongiosis of the follicular epithelium is seen in early lesions of HIV-associated EF.6
The first-line treatment of HIV-associated EF includes antiretroviral therapy with topical steroids and antihistamines. Human immunodeficiency virus-associated EF improves as CD4 helper T-cell counts rise above 250 cells/mm3 with continued antiretroviral therapy, though it initially can cause a flare of the condition.4 High-potency steroids and antihistamines are added during this period to treat the severe pruritus.1,7 In particular, daily cetirizine has been shown to be effective, which may be due to its ability to block eosinophil migration in addition to H1-receptor antagonist properties.3,7
Various alternative therapies have been described in case reports and case series; however, there have been no controlled studies comparing therapies. Phototherapy with UVB light 3 times weekly for 3 to 6 weeks has been effective and curative in recalcitrant cases.7 Other frequently used treatments include oral metronidazole, oral itraconazole, and permethrin cream 5%. The effectiveness of the latter 2 treatments is believed to be related to the proposed role of Pityrosporum and Demodex in the pathogenesis.3
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his help compiling the histopathological images and their diagnostic descriptions.
The Diagnosis: Eosinophilic Folliculitis
A shave biopsy specimen of an intact pustule on the left side of the chest was obtained. Histopathologic examination revealed follicular inflammation with copious eosinophils (Figure, A and B). Based on the histopathology and clinical presentation, a diagnosis of human immunodeficiency virus (HIV)-associated eosinophilic folliculitis (EF) was made.
The patient was started on triamcinolone ointment 0.1% twice daily to active lesions, oral cetirizine 10 mg in the morning, and oral hydroxyzine 25 mg at bedtime. Laboratory evaluation at the time of diagnosis showed eosinophilia with a peripheral blood eosinophil count of 0.5 K/μL (reference range, 0.03–0.48 K/μL).
Human immunodeficiency virus-associated EF is a pruritic follicular eruption that occurs in HIV-positive individuals with advanced disease. Clinically, it is characterized by intermittent, urticarial, red or flesh-colored, 2- to 5-mm papules with sparse pustules involving the head, neck, arms, and upper trunk.1,2 The cardinal clinical feature of the disorder is intense pruritus, with overlying crusts and excoriations present on physical examination.3
Patients usually have a CD4 count of less than 250 cells/mm3.2,3 Patients with HIV can develop an exacerbation of EF in the first 3 to 6 months after initiating antiretroviral therapy. This clinical pattern is believed to be due to the reconstituted immune system and increased circulation of inflammatory cells.4 Peripheral eosinophilia and elevated serum IgE levels are found in 25% to 50% of patients with HIV-associated EF.2,3
Clinically, the differential diagnosis of intensely pruritic papules with excoriations should include scabies.3 Other diagnoses to consider include opportunistic infections and papular urticaria.5 Acne vulgaris and Demodex folliculitis also may present with lesions similar to HIV-associated EF; however, these lesions tend not to be as intensely pruritic.1,5
The etiology of HIV-associated EF is unknown.3 One proposed mechanism involves a hypersensitivity reaction to Pityrosporum or Demodex mite fragments, as evidenced by studies that found fragments of these microorganisms in biopsied lesions of HIV-associated EF.3,6 In our patient's histopathology, it was noted that the afflicted hair follicle held a single Demodex mite (Figure, C).
The histopathology is characterized by a perifollicular inflammatory infiltrate of eosinophils and CD8+ lymphocytes with areas of sebaceous lysis.3,6 Spongiosis of the follicular epithelium is seen in early lesions of HIV-associated EF.6
The first-line treatment of HIV-associated EF includes antiretroviral therapy with topical steroids and antihistamines. Human immunodeficiency virus-associated EF improves as CD4 helper T-cell counts rise above 250 cells/mm3 with continued antiretroviral therapy, though it initially can cause a flare of the condition.4 High-potency steroids and antihistamines are added during this period to treat the severe pruritus.1,7 In particular, daily cetirizine has been shown to be effective, which may be due to its ability to block eosinophil migration in addition to H1-receptor antagonist properties.3,7
Various alternative therapies have been described in case reports and case series; however, there have been no controlled studies comparing therapies. Phototherapy with UVB light 3 times weekly for 3 to 6 weeks has been effective and curative in recalcitrant cases.7 Other frequently used treatments include oral metronidazole, oral itraconazole, and permethrin cream 5%. The effectiveness of the latter 2 treatments is believed to be related to the proposed role of Pityrosporum and Demodex in the pathogenesis.3
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his help compiling the histopathological images and their diagnostic descriptions.
- Parker SR, Parker DC, McCall CO. Eosinophilic folliculitis in HIV-infected women: case series and review. Am J Clin Dermatol. 2006;7:193-200.
- Rosenthal D, LeBoit PE, Klumpp L, et al. Human immunodeficiency virus-associated eosinophilic folliculitis. a unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206-209.
- Fearfield LA, Rowe A, Francis N, et al. Itchy folliculitis and human immunodeficiency virus infection: clinicopathological and immunological features, pathogenesis, and treatment. Br J Dermatol. 1999;141:3-11.
- Rajendran PM, Dolev JC, Heaphy MR, et al. Eosinophilic folliculitis: before and after the introduction of antiretroviral therapy. Arch Dermatol. 2005;141:1227-1231.
- Nervi SJ, Schwartz RA, Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- McCalmont TH, Altemus D, Maurer T, et al. Eosinophilic folliculitis: the histological spectrum. Am J Dermatopathol. 1995;17:439-446.
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197.
- Parker SR, Parker DC, McCall CO. Eosinophilic folliculitis in HIV-infected women: case series and review. Am J Clin Dermatol. 2006;7:193-200.
- Rosenthal D, LeBoit PE, Klumpp L, et al. Human immunodeficiency virus-associated eosinophilic folliculitis. a unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206-209.
- Fearfield LA, Rowe A, Francis N, et al. Itchy folliculitis and human immunodeficiency virus infection: clinicopathological and immunological features, pathogenesis, and treatment. Br J Dermatol. 1999;141:3-11.
- Rajendran PM, Dolev JC, Heaphy MR, et al. Eosinophilic folliculitis: before and after the introduction of antiretroviral therapy. Arch Dermatol. 2005;141:1227-1231.
- Nervi SJ, Schwartz RA, Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- McCalmont TH, Altemus D, Maurer T, et al. Eosinophilic folliculitis: the histological spectrum. Am J Dermatopathol. 1995;17:439-446.
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197.

A 31-year-old man presented with a severely pruritic rash of 2 weeks' duration. Physical examination revealed numerous urticarial papules and rare erythematous pustules over the face (top), upper chest (bottom), and proximal arms; most lesions were excoriated. Additionally, there were numerous hyperpigmented papules with central hypopigmentation on the upper chest and arms. The lower half of the body was spared. His medical history was notable for human immunodeficiency virus/AIDS with a prior episode of Pneumocystis pneumonia. He had been noncompliant with antiretroviral therapy for the last 2 years but restarted therapy 3 weeks prior to presentation. Laboratory test results revealed a CD4 cell count of 13 cells/mm3 (reference range, 500-1500 cells/mm3) with a viral load of 179 copies/mL (reference range, undetectable).
Tense Bullae on the Hands
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
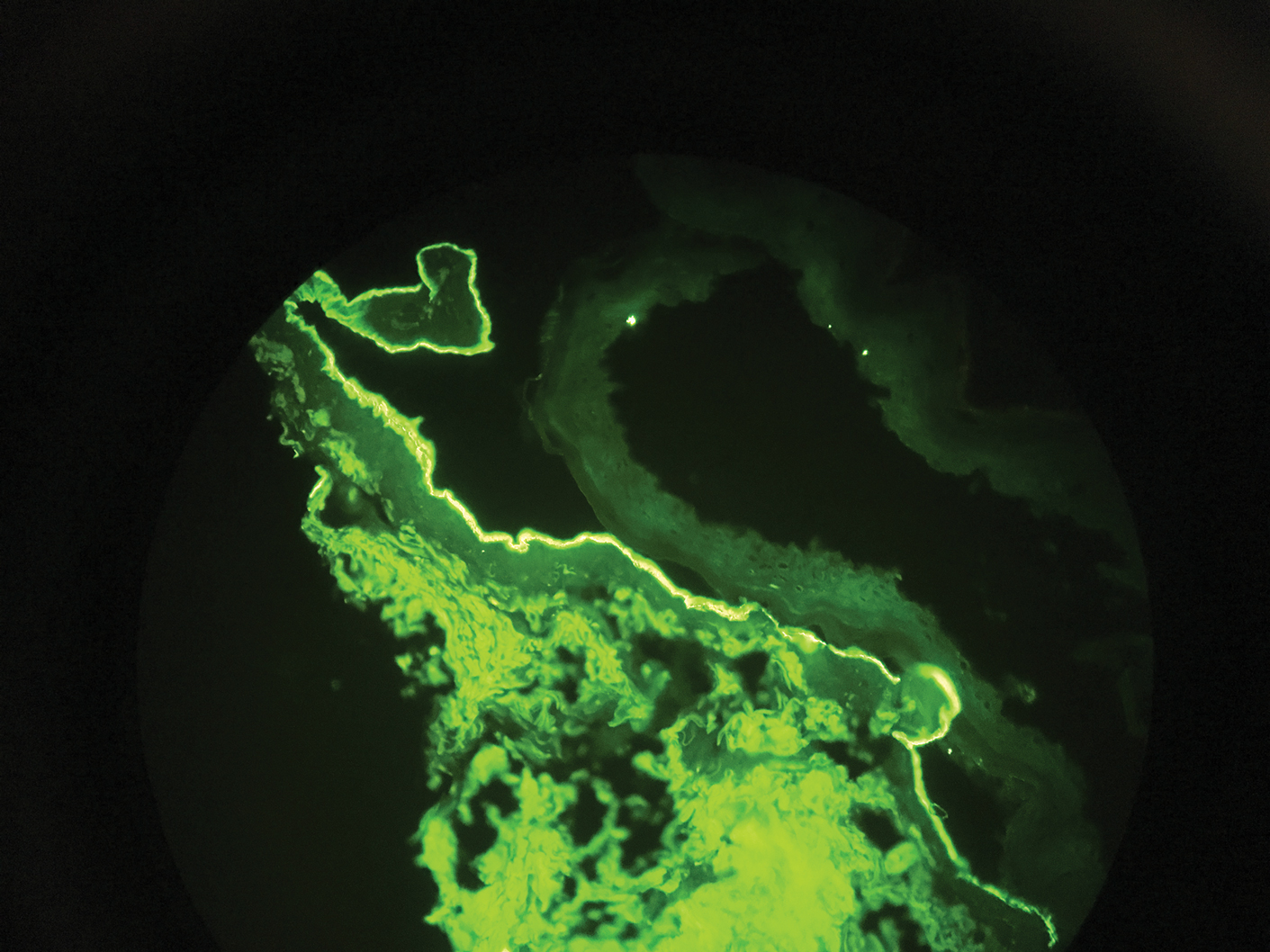
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
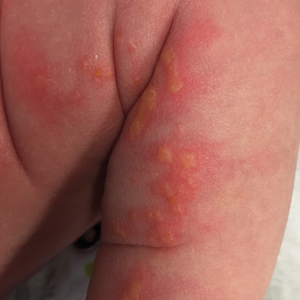
A 75-year-old man presented to our clinic with nonpainful, nonpruritic, tense bullae and erosions on the dorsal aspects of the hands and extensor surfaces of the elbows of 1 month's duration. The patient also had erythematous erosions and crusted papules on the left cheek and surrounding the left eye. He denied any new medications, history of liver or kidney disease, or history of hepatitis or human immunodeficiency virus. There were no obvious exacerbating factors, including exposure to sunlight. Direct immunofluorescence using a salt-split preparation was performed on a biopsy of the perilesional skin.
Unilateral Vesicular Eruption in a Neonate
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.

A 4-day-old female neonate presented to the dermatology clinic with a vesicular eruption on the left leg of 1 day's duration. The eruption was asymptomatic without any extracutaneous findings. This term infant was born without complication, and the mother denied any symptoms consistent with herpes simplex virus infection. Physical examination revealed yellow-red vesicles on an erythematous base in a blaschkoid distribution on the left leg. The rest of the examination was unremarkable. Herpes simplex virus polymerase chain reaction testing was negative.
Smooth Papules on the Left Hand
The Diagnosis: Adult Colloid Milium
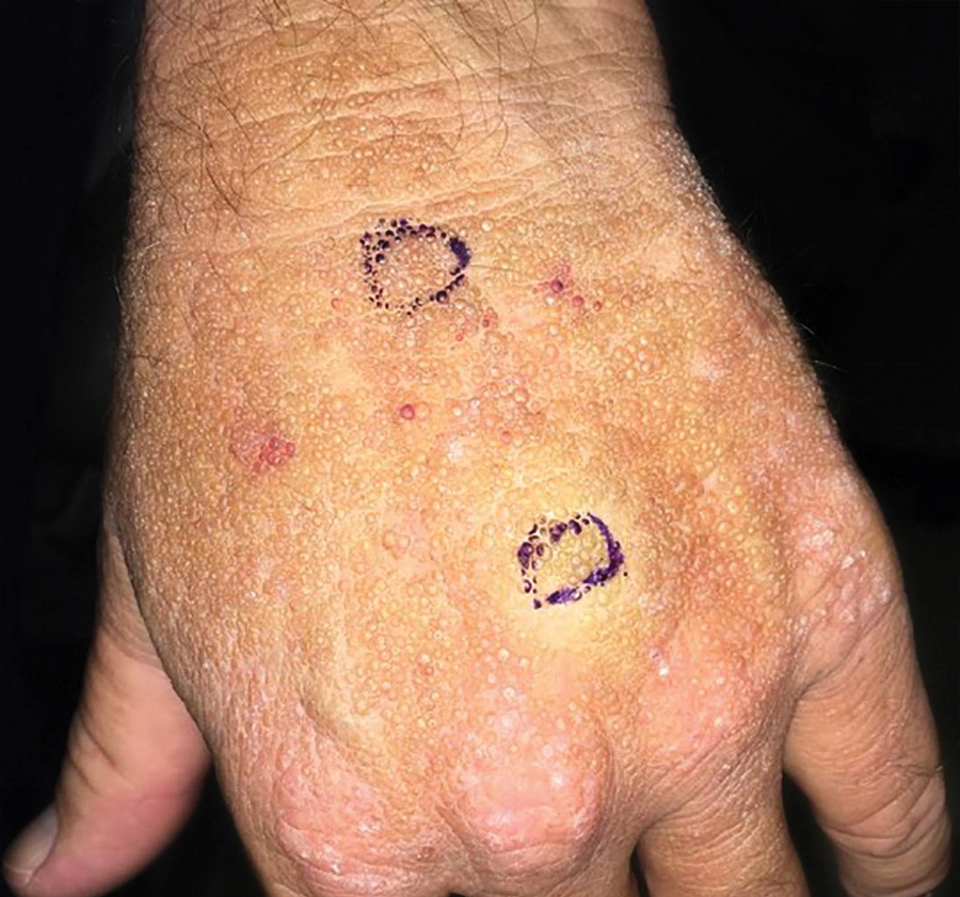
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).

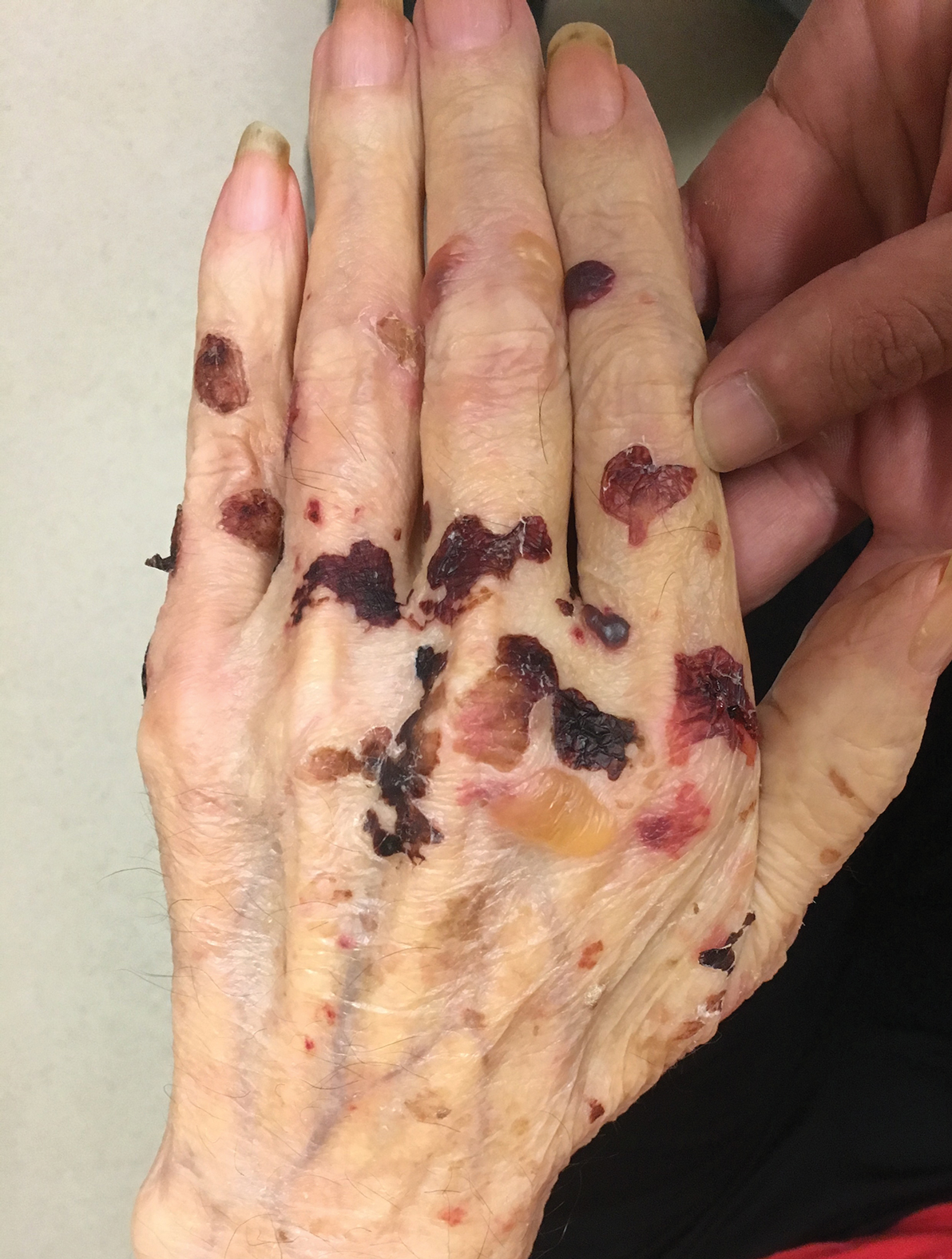
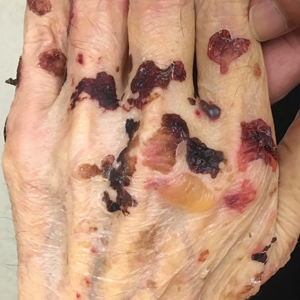
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
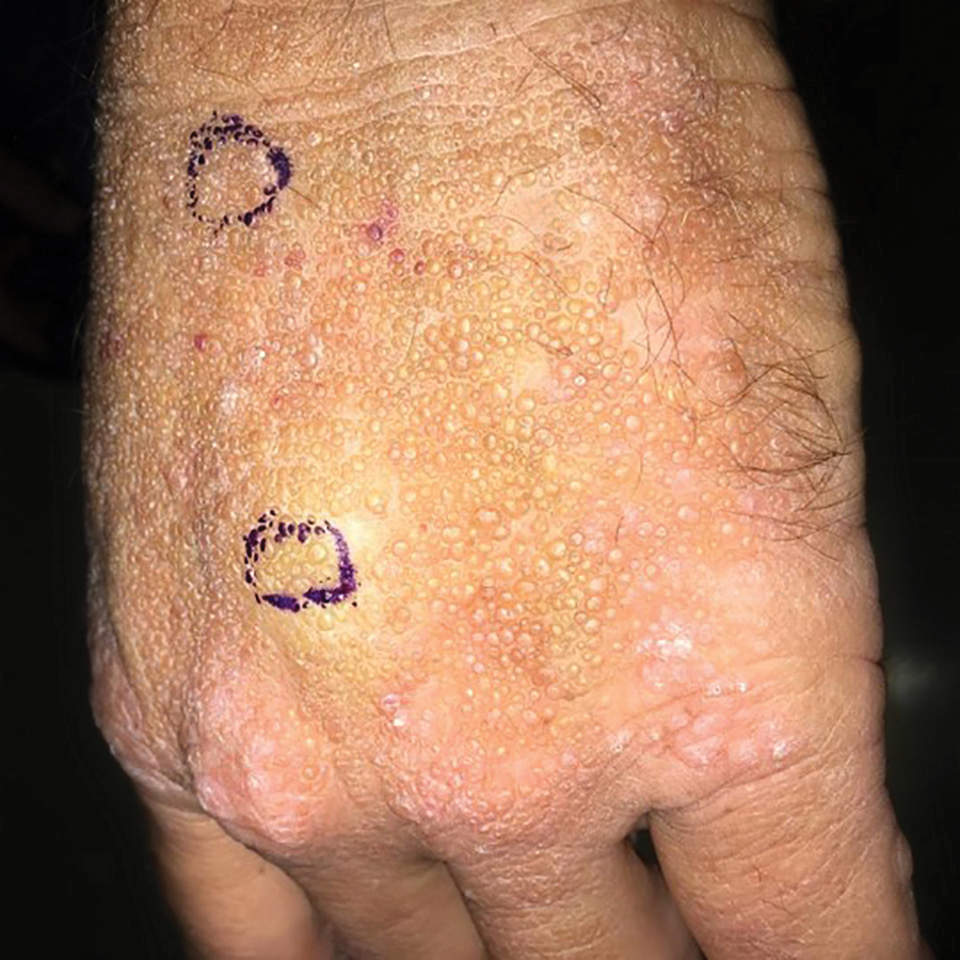
A 41-year-old man presented to the outpatient dermatology clinic with multiple smooth papules on the left hand of 7 years' duration. The papules had been steadily increasing in number, and the patient reported that they were frequently symptomatic with a burning itching sensation. Physical examination revealed multiple 1- to 3-mm, dome-shaped, translucent to flesh-colored papules on the left hand with a few scattered bright red papules. No similar lesions were present on the right hand or elsewhere on the body. He had a history of hypertension but was otherwise healthy with no other chronic medical conditions.
Numerous Flesh-Colored Nodules on the Trunk
The Diagnosis: Steatocystoma Multiplex
The punch biopsy of an abdominal lesion demonstrated a folded cyst wall with a wavy eosinophilic cuticle (Figure), characteristics consistent with steatocystoma multiplex (SM).
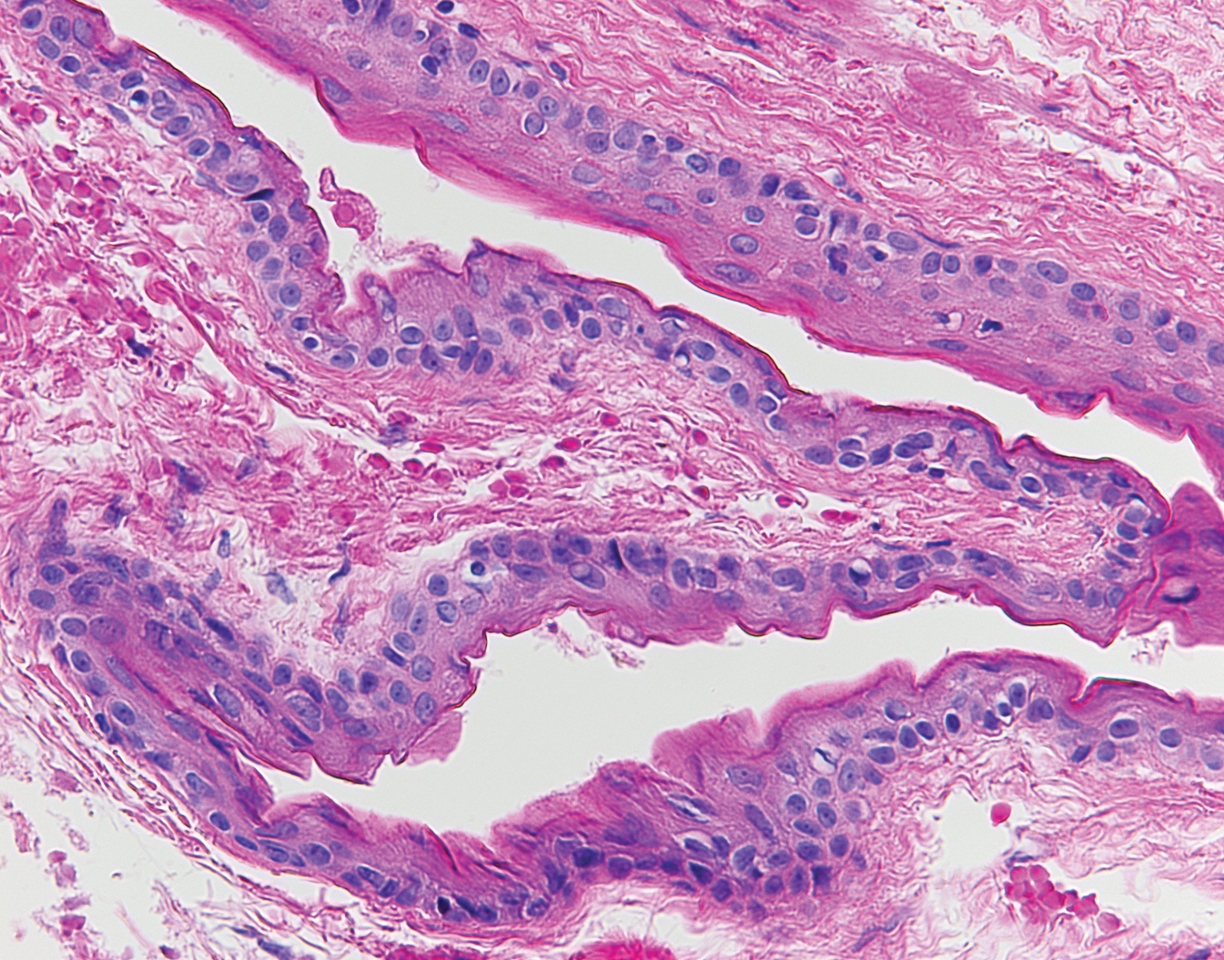
Also known as eruptive steatocystoma, SM consists of numerous flesh-colored, dome-shaped papules and nodules that most commonly arise during adolescence, with a median age of onset of 26 years.1 These hamartomatous nevoid malformations arise in areas with well-developed pilosebaceous units, such as the upper extremities, neck, axillae, and trunk.1,2 They occur less commonly on the scalp, face, and acral surfaces.2-5 The lesions range in size from 2 to 30 mm6 and usually are asymptomatic.1 Occasionally, steatocystomas become tender or can rupture.7
Steatocystoma multiplex may arise sporadically or may be inherited in an autosomal-dominant fashion. Mutations in exon 1 of the keratin 17 gene, KRT17, have been identified in autosomal-dominant SM.6,8 KRT17 mutations also are responsible for pachyonychia congenita type 2, which is associated with SM.9 Some patients with pachyonychia congenita type 2 who have prominent SM and mild nail findings may be misdiagnosed as having pure SM.2
The histopathologic features of SM were described in a study by Cho and colleagues1 of 64 patients. Steatocystomas have cyst walls that may be either intricately folded or round/oval, comprised of an average of 4.9 epithelial cell layers. In most cases, the cyst wall contains sebaceous lobules. In all cases, an acellular eosinophilic cuticle was present, and no granular layer was seen. Few vellus hairs may be observed in the cystic cavity.1
The differential diagnosis of SM includes eruptive vellus hair cysts, lipomas, Muir-Torre syndrome, and Gardner syndrome. Some have suggested that eruptive vellus hair cysts and SM exist on a disease spectrum because of their similar clinical presentation.10 In contrast to SM, however, eruptive vellus hair cysts originate in the infundibulum of the hair shaft rather than the sebaceous duct, and more numerous vellus hair shafts are seen on histopathology.1
Various treatment modalities have been described, including isotretinoin for inflamed lesions,11 cryotherapy for noninflamed lesions,11 aspiration of lesions smallerthan 1 cm,12 and electrocautery combined with topical retinoids.13 Laser treatment has been described, with a 1450-nm diode laser used to target the abnormal sebaceous glands and a 1550-nm fractionated erbium-doped fiber laser used to target the dermal cysts.14 Carbon dioxide lasers also may be used to open the cyst for drainage.15 Surgical excision or mini-incision also may be performed.16,17
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his assistance with interpretation of the dermatopathology in this case.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Rollins T, Levin RM, Heymann WR. Acral steatocystoma multiplex.J Am Acad Dermatol. 2000;43(2, pt 2):396-399.
- Setoyama M, Mizoguchi S, Usuki K, et al. Steatocystoma multiplex: a case with unusual clinical and histological manifestation. Am J Dermatopathol. 1997;19:89-92.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273-278.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sato K, Shibuya K, Taguchi H, et al. Aspiration therapy in steatocystoma multiplex. Arch Dermatol. 1993;129:35-37.
- Papakonstantinou E, Franke I, Gollnick H. Facial steatocystoma multiplex combined with eruptive vellus hair cysts: a hybrid? J Eur Acad Dermatol Venereol. 2015;29:2051-2053.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7, pt 1):1104-1106.
- Rossi R, Cappugi P, Battini M, et al. CO2 laser therapy in a case of steatocystoma multiplex with prominent nodules on the face and neck. Int J Dermatol. 2003;42:302-304.
- Schmook T, Burg G, Hafner J. Surgical pearl: mini-incisions for the extraction of steatocystoma multiplex. J Am Acad Dermatol. 2001;44:1041-1042.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
The Diagnosis: Steatocystoma Multiplex
The punch biopsy of an abdominal lesion demonstrated a folded cyst wall with a wavy eosinophilic cuticle (Figure), characteristics consistent with steatocystoma multiplex (SM).
Also known as eruptive steatocystoma, SM consists of numerous flesh-colored, dome-shaped papules and nodules that most commonly arise during adolescence, with a median age of onset of 26 years.1 These hamartomatous nevoid malformations arise in areas with well-developed pilosebaceous units, such as the upper extremities, neck, axillae, and trunk.1,2 They occur less commonly on the scalp, face, and acral surfaces.2-5 The lesions range in size from 2 to 30 mm6 and usually are asymptomatic.1 Occasionally, steatocystomas become tender or can rupture.7
Steatocystoma multiplex may arise sporadically or may be inherited in an autosomal-dominant fashion. Mutations in exon 1 of the keratin 17 gene, KRT17, have been identified in autosomal-dominant SM.6,8 KRT17 mutations also are responsible for pachyonychia congenita type 2, which is associated with SM.9 Some patients with pachyonychia congenita type 2 who have prominent SM and mild nail findings may be misdiagnosed as having pure SM.2
The histopathologic features of SM were described in a study by Cho and colleagues1 of 64 patients. Steatocystomas have cyst walls that may be either intricately folded or round/oval, comprised of an average of 4.9 epithelial cell layers. In most cases, the cyst wall contains sebaceous lobules. In all cases, an acellular eosinophilic cuticle was present, and no granular layer was seen. Few vellus hairs may be observed in the cystic cavity.1
The differential diagnosis of SM includes eruptive vellus hair cysts, lipomas, Muir-Torre syndrome, and Gardner syndrome. Some have suggested that eruptive vellus hair cysts and SM exist on a disease spectrum because of their similar clinical presentation.10 In contrast to SM, however, eruptive vellus hair cysts originate in the infundibulum of the hair shaft rather than the sebaceous duct, and more numerous vellus hair shafts are seen on histopathology.1
Various treatment modalities have been described, including isotretinoin for inflamed lesions,11 cryotherapy for noninflamed lesions,11 aspiration of lesions smallerthan 1 cm,12 and electrocautery combined with topical retinoids.13 Laser treatment has been described, with a 1450-nm diode laser used to target the abnormal sebaceous glands and a 1550-nm fractionated erbium-doped fiber laser used to target the dermal cysts.14 Carbon dioxide lasers also may be used to open the cyst for drainage.15 Surgical excision or mini-incision also may be performed.16,17
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his assistance with interpretation of the dermatopathology in this case.
The Diagnosis: Steatocystoma Multiplex
The punch biopsy of an abdominal lesion demonstrated a folded cyst wall with a wavy eosinophilic cuticle (Figure), characteristics consistent with steatocystoma multiplex (SM).
Also known as eruptive steatocystoma, SM consists of numerous flesh-colored, dome-shaped papules and nodules that most commonly arise during adolescence, with a median age of onset of 26 years.1 These hamartomatous nevoid malformations arise in areas with well-developed pilosebaceous units, such as the upper extremities, neck, axillae, and trunk.1,2 They occur less commonly on the scalp, face, and acral surfaces.2-5 The lesions range in size from 2 to 30 mm6 and usually are asymptomatic.1 Occasionally, steatocystomas become tender or can rupture.7
Steatocystoma multiplex may arise sporadically or may be inherited in an autosomal-dominant fashion. Mutations in exon 1 of the keratin 17 gene, KRT17, have been identified in autosomal-dominant SM.6,8 KRT17 mutations also are responsible for pachyonychia congenita type 2, which is associated with SM.9 Some patients with pachyonychia congenita type 2 who have prominent SM and mild nail findings may be misdiagnosed as having pure SM.2
The histopathologic features of SM were described in a study by Cho and colleagues1 of 64 patients. Steatocystomas have cyst walls that may be either intricately folded or round/oval, comprised of an average of 4.9 epithelial cell layers. In most cases, the cyst wall contains sebaceous lobules. In all cases, an acellular eosinophilic cuticle was present, and no granular layer was seen. Few vellus hairs may be observed in the cystic cavity.1
The differential diagnosis of SM includes eruptive vellus hair cysts, lipomas, Muir-Torre syndrome, and Gardner syndrome. Some have suggested that eruptive vellus hair cysts and SM exist on a disease spectrum because of their similar clinical presentation.10 In contrast to SM, however, eruptive vellus hair cysts originate in the infundibulum of the hair shaft rather than the sebaceous duct, and more numerous vellus hair shafts are seen on histopathology.1
Various treatment modalities have been described, including isotretinoin for inflamed lesions,11 cryotherapy for noninflamed lesions,11 aspiration of lesions smallerthan 1 cm,12 and electrocautery combined with topical retinoids.13 Laser treatment has been described, with a 1450-nm diode laser used to target the abnormal sebaceous glands and a 1550-nm fractionated erbium-doped fiber laser used to target the dermal cysts.14 Carbon dioxide lasers also may be used to open the cyst for drainage.15 Surgical excision or mini-incision also may be performed.16,17
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his assistance with interpretation of the dermatopathology in this case.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Rollins T, Levin RM, Heymann WR. Acral steatocystoma multiplex.J Am Acad Dermatol. 2000;43(2, pt 2):396-399.
- Setoyama M, Mizoguchi S, Usuki K, et al. Steatocystoma multiplex: a case with unusual clinical and histological manifestation. Am J Dermatopathol. 1997;19:89-92.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273-278.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sato K, Shibuya K, Taguchi H, et al. Aspiration therapy in steatocystoma multiplex. Arch Dermatol. 1993;129:35-37.
- Papakonstantinou E, Franke I, Gollnick H. Facial steatocystoma multiplex combined with eruptive vellus hair cysts: a hybrid? J Eur Acad Dermatol Venereol. 2015;29:2051-2053.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7, pt 1):1104-1106.
- Rossi R, Cappugi P, Battini M, et al. CO2 laser therapy in a case of steatocystoma multiplex with prominent nodules on the face and neck. Int J Dermatol. 2003;42:302-304.
- Schmook T, Burg G, Hafner J. Surgical pearl: mini-incisions for the extraction of steatocystoma multiplex. J Am Acad Dermatol. 2001;44:1041-1042.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Rollins T, Levin RM, Heymann WR. Acral steatocystoma multiplex.J Am Acad Dermatol. 2000;43(2, pt 2):396-399.
- Setoyama M, Mizoguchi S, Usuki K, et al. Steatocystoma multiplex: a case with unusual clinical and histological manifestation. Am J Dermatopathol. 1997;19:89-92.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273-278.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sato K, Shibuya K, Taguchi H, et al. Aspiration therapy in steatocystoma multiplex. Arch Dermatol. 1993;129:35-37.
- Papakonstantinou E, Franke I, Gollnick H. Facial steatocystoma multiplex combined with eruptive vellus hair cysts: a hybrid? J Eur Acad Dermatol Venereol. 2015;29:2051-2053.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7, pt 1):1104-1106.
- Rossi R, Cappugi P, Battini M, et al. CO2 laser therapy in a case of steatocystoma multiplex with prominent nodules on the face and neck. Int J Dermatol. 2003;42:302-304.
- Schmook T, Burg G, Hafner J. Surgical pearl: mini-incisions for the extraction of steatocystoma multiplex. J Am Acad Dermatol. 2001;44:1041-1042.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.

A 33-year-old woman presented with numerous firm, noncompressible, flesh-colored nodules that measured 3 to 4 mm and were distributed across the abdomen, chest, back, and neck. The lesions had been present for approximately 10 years. The patient denied any lesion-associated pain, itching, or bleeding, and there was no family history of similar lesions. A punch biopsy of a lesion on the central abdomen was obtained.
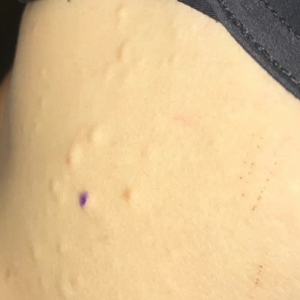
Painful and Pruritic Erosions on the Back
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
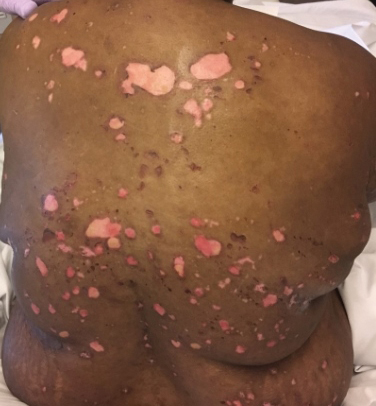
A 51-year-old black woman presented to the dermatology clinic with painful and pruritic erosions on the back, abdomen, neck, and arms of approximately 2 months' duration. The lesions started on the back and spread in a cephalocaudal manner. The patient denied any new changes in medication. Physical examination revealed large erosions with mild weeping of serosanguineous fluid on the back, abdomen, neck, and upper extremities. A few tense bullae were present on the dorsal aspect of the right hand. She had experienced a similar flare approximately 1.5 years prior to the current presentation. At that time, 2 shave biopsies from vesiculobullous lesions on the right side of the neck were sent for hematoxylin and eosin staining and direct immunofluorescence. Biopsy results showed a subepidermal blister that extended along the course of the hair follicle and was associated with an infiltrate of neutrophilic granulocytes that also extended along the course of the hair follicle. Direct immunofluorescence showed IgG and C3 deposition in the basement membrane zone extending along the floor of the blister where the epidermis was separated from the dermis.
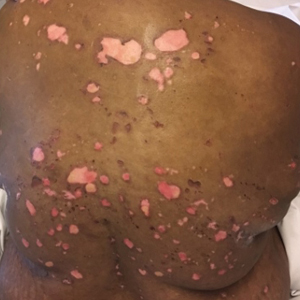
Annular Atrophic Plaques on the Forearm
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
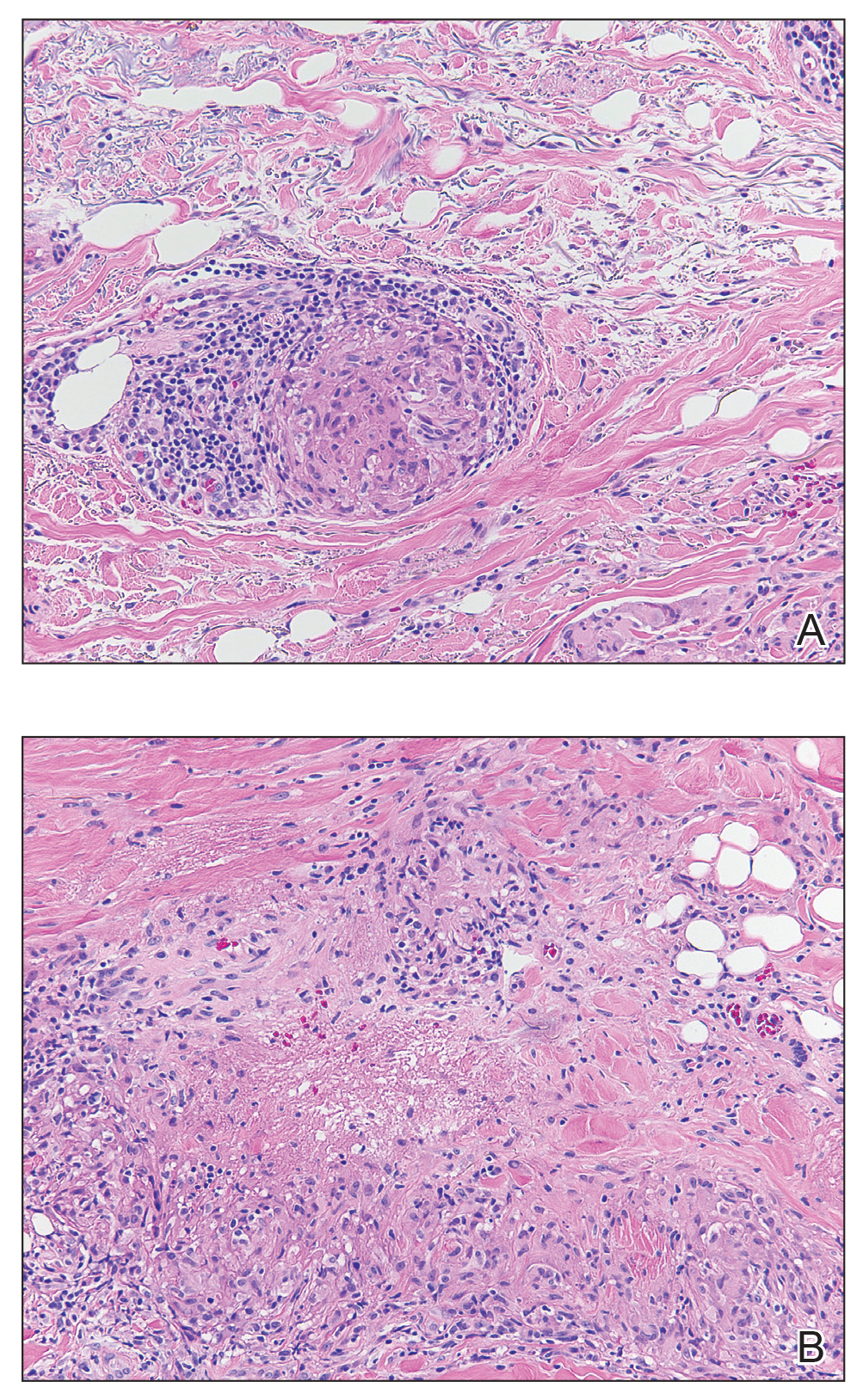
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
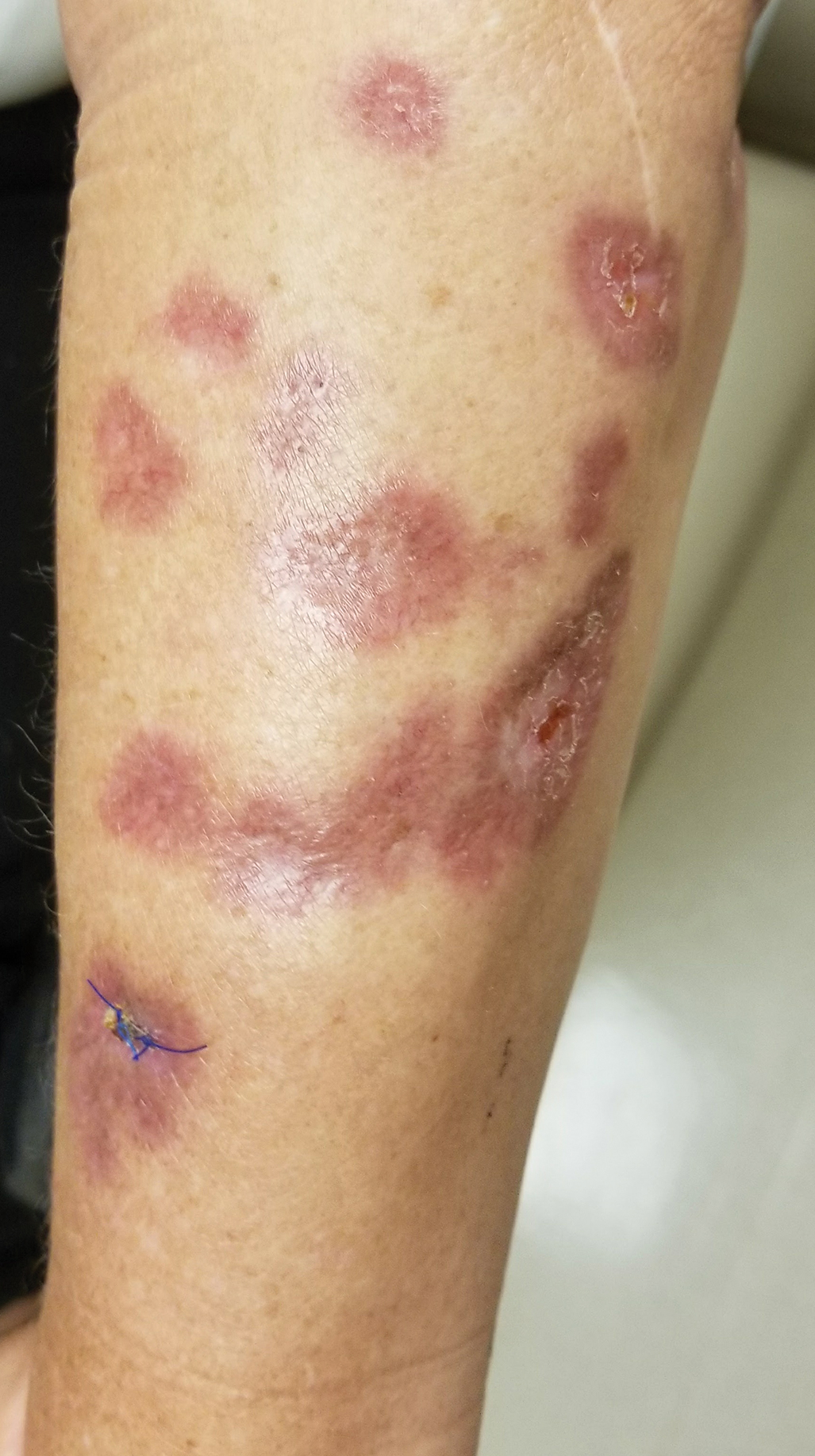
A 57-year-old woman presented with several lesions on the left extensor forearm of 10 years’ duration. A single annular indurated lesion with central atrophy initially developed near a prior surgical site. The lesions were pruritic with no associated pain or bleeding. Over 5 years, similar lesions had developed extending up the arm. No benefit was seen with low-potency topical steroid application. Biopsy for histopathologic examination was performed to confirm the diagnosis.
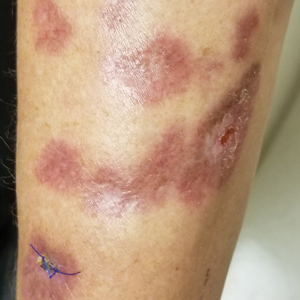
Telangiectatic Patch on the Neck
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
A 55-year-old woman presented to our clinic for a total-body skin examination and was noted to have a completely blanchable telangiectatic patch on the right side of the neck extending down onto the chest and breast. The patient reported that it had been present for 15 years and had slowly expanded in size. The lesion was asymptomatic. Pertinent medical history included cryptogenic cirrhosis of the liver, and she was undergoing a workup for a liver transplant.