User login
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
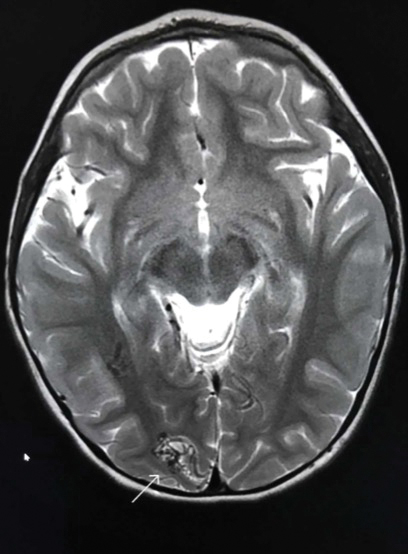
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
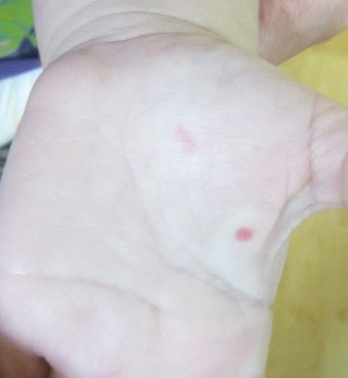
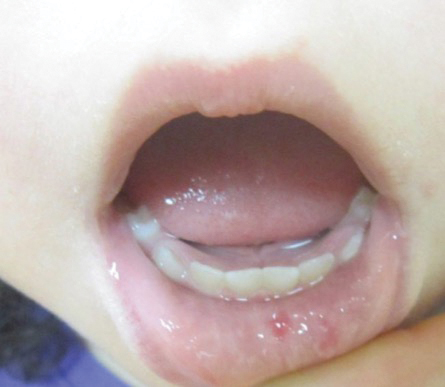
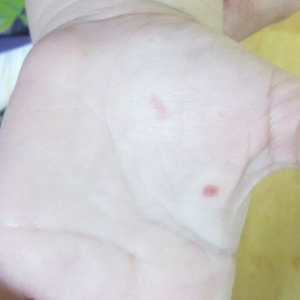
A 2-year-old girl presented with an erythematous macule on the left nasal sidewall that had been present since birth as well as other similar-appearing macules that had slowly evolved over the last 2 years. The patient was born via normal spontaneous vaginal delivery to healthy parents. She had 2 healthy siblings. Her parents reported that she was otherwise growing and developing normally. The patient had no history of epistaxis, and there was no family history of vascular anomalies. Physical examination revealed 2- to 6-mm vascular macules that blanched with pressure and filled quickly thereafter on the left nasal sidewall, upper (top) and lower extremities, and trunk. Some macules were surrounded by blanched halos. Several 1-mm red macules also were noted on the exterior and interior of the mucosal lower lip (bottom).