User login
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
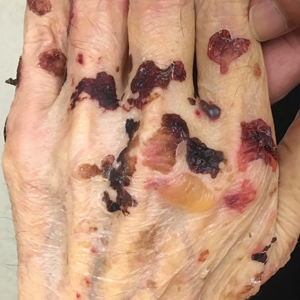
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
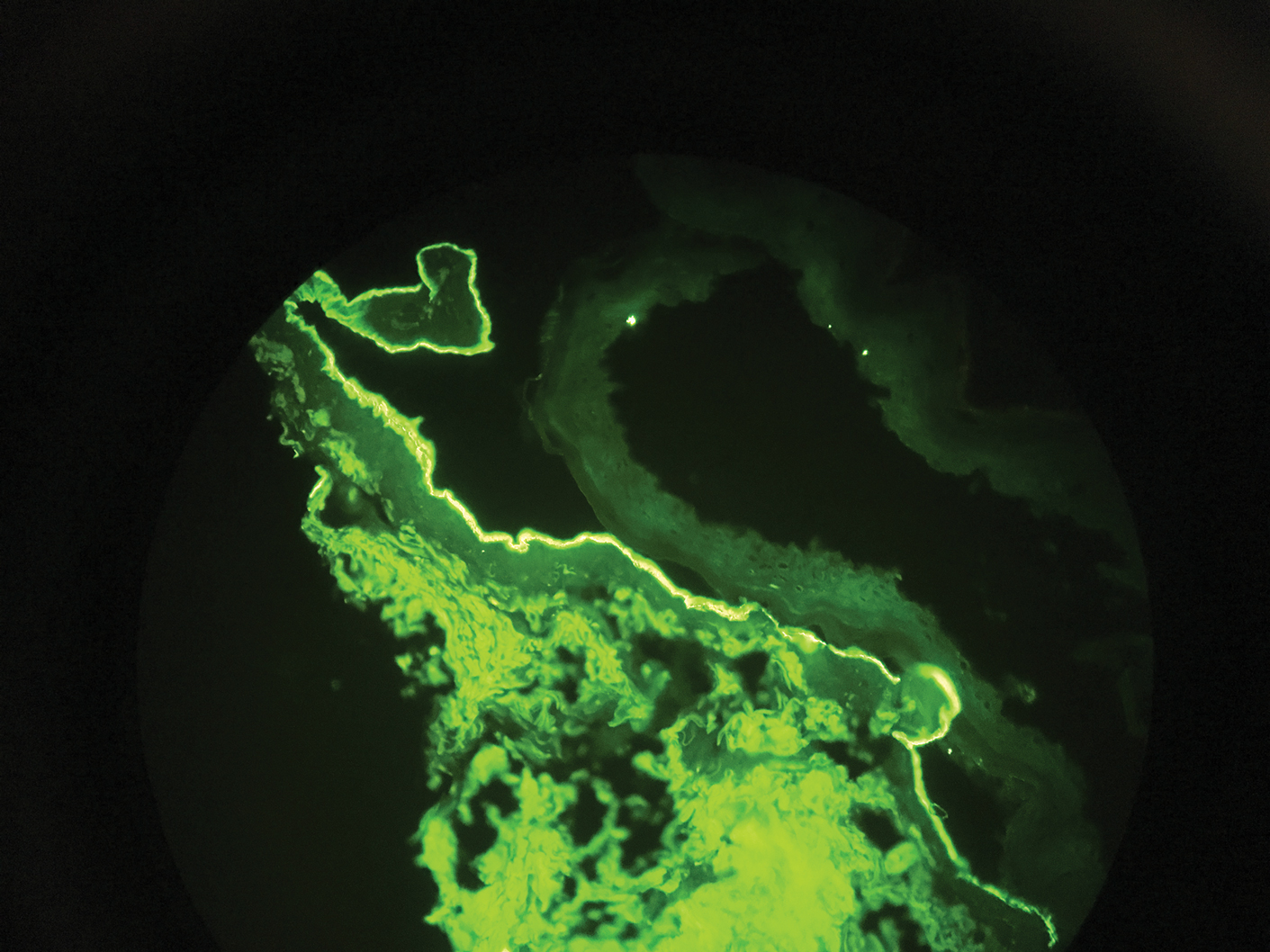
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
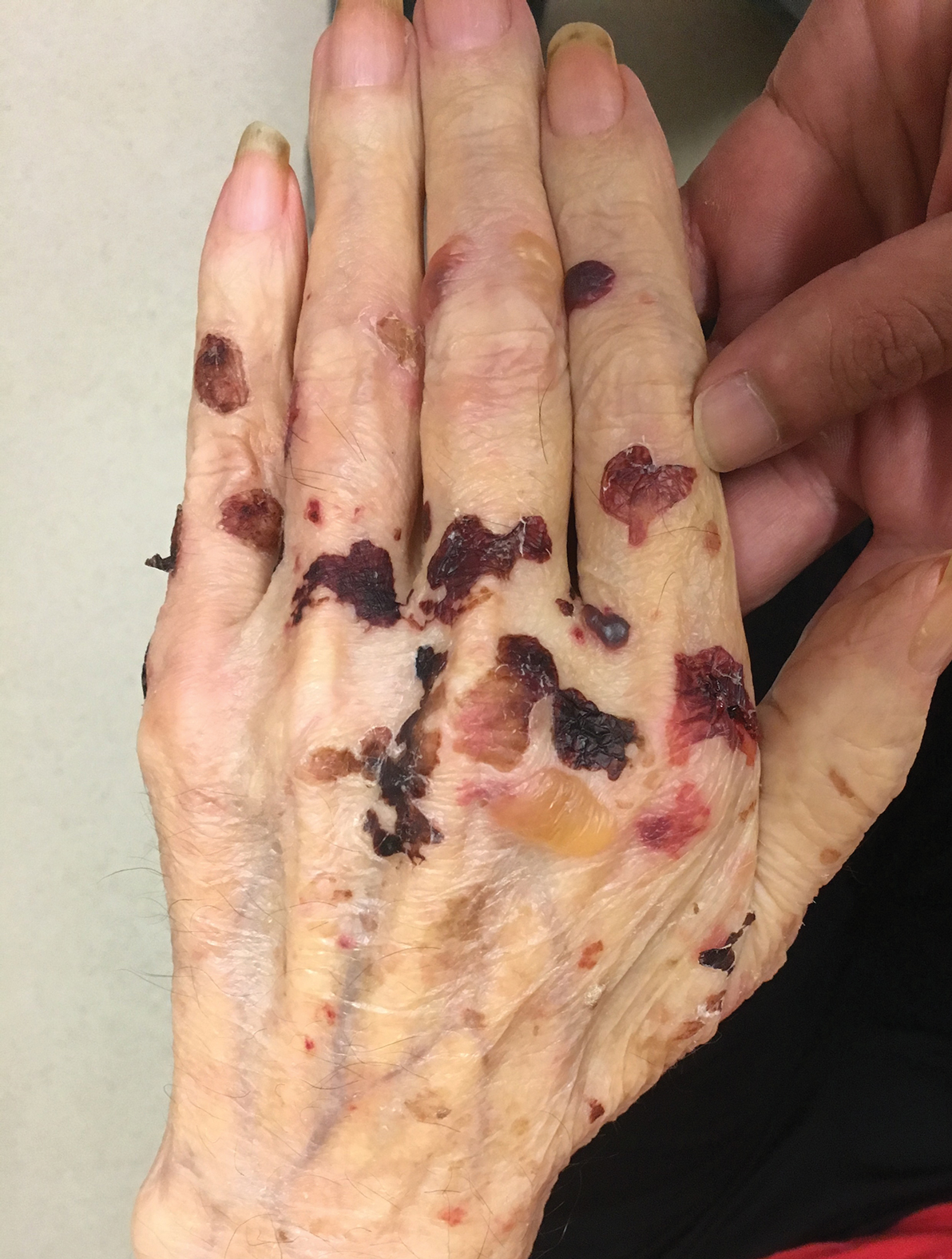
A 75-year-old man presented to our clinic with nonpainful, nonpruritic, tense bullae and erosions on the dorsal aspects of the hands and extensor surfaces of the elbows of 1 month's duration. The patient also had erythematous erosions and crusted papules on the left cheek and surrounding the left eye. He denied any new medications, history of liver or kidney disease, or history of hepatitis or human immunodeficiency virus. There were no obvious exacerbating factors, including exposure to sunlight. Direct immunofluorescence using a salt-split preparation was performed on a biopsy of the perilesional skin.