User login
Functional medicine offers another approach to treating psychiatric illness
The shortage of psychiatrists, other mental health clinicians, and primary care physicians who treat patients with mental illness is a profound problem in the United States and around the world. What would happen to those trends if psychiatrists incorporated a functional medicine approach to treating patients?

In functional medicine, we look for underlying causes, physiological damage that results from those causes, clinical body system imbalances, and ultimately, symptoms that patients are experiencing. By addressing the root causes of chronic problems, treating physiological damage, and creating balance in body systems, psychiatrists and other physicians can help our patients achieve optimal health.
For example, a functional medicine approach to treating a child with ADHD might focus on encouraging behavioral changes such as improving sleep hygiene,1 increasing hydration,2 changing nutrition, or prescribing adjunctive meditation rather than medication alone. A functional medicine approach to Alzheimer’s prevention, for example, could include “prescribing” an increase in the amount of regular physical exercise.3 In other words, functional medicine uses a different lens to prevent, arrest, and in some cases, reverse certain diseases.
Medicine has long recognized the links between inflammation and chronic illness. Autoimmune conditions, asthma, heart disease, stroke, diabetes, obesity, peripheral neuropathy, thyroid problems, joint pain, and cancer all are chronic inflammatory diseases. Because inflammation affects the brain, it has been theorized and is being investigated that psychiatric disorders such as depression, schizophrenia, anxiety, panic attacks, dementia, and autism might result.4,5,6,7
Besides the brain, the GI tract is the only organ system that has its own nervous system, which is called the enteric nervous system, or ENS. The ENS functions independently from the central nervous system, and transmits important messages to and from the brain. When one feels stressed, the brain communicates to the hormonal system and floods the body with stress hormones, such as cortisol, which by themselves, can cause increased intestinal permeability. In addition, the gut produces its own neurotransmitters that affect the brain. In fact, every class of neurotransmitter found in the brain also is found in the GI tract. For example, serotonin is an important neurotransmitter for feeling happy and optimistic. Ninety-five percent of the body’s serotonin is produced in the gut. It is produced from 5-HTP, which is derived from tryptophan. However, in the presence of inflammation in the body, tryptophan is converted into kynurenate and quinolate. Both cause fatigue, and quinolate causes neurotoxicity. The subsequent depletion of serotonin produces symptoms of depression. Problems in the gut can lead to problems in the brain and the whole body.
Other problems affecting patients are tied to toxins in the environment. The air we breathe, food we eat, water we drink, and clothing we wear all are sources of toxins. Toxins include biotoxins, dioxine, phthalates, PCBs, and heavy metals, such as mercury, lead, cadmium, aluminum. About 2,000 new chemicals have been introduced into our environment each year since the 1940s, and it is estimated that we are exposed to more than 80,000 chemicals on a regular basis.8
The Environmental Working Group, a nonprofit organization dedicated to educating the public about the environment, has estimated that average babies are born with 287 chemicals in their body, 217 of which are neurotoxins.9 As children grow up, their body accumulates more toxins. According to the Centers for Disease Control and Prevention, every American has hundreds of neurotoxins in their bodies right now.
As we become more aware of the many changes in our environment, functional medicine brings a new way of thinking about and looking at chronic disease. As physicians, we can continue treating symptoms, and we should. But we can look deeper and ask ourselves what has changed in our lives that has caused such a decline in human mental and physical health. I urge psychiatrists to help lead the way.
Dr. Gaitour, a physiatrist, trained at NYU Langone Medical Center in New York. She is a functional medicine practitioner.
References
1. Peppers KH et al. J Pediatr Health Care. 2016 Nov-Dec;30(6):e43-8.
2. Martin EB and PG Hammerness. ADHD, stimulant medication, and dehydration. CHADD.org. 2014 Aug.
3. Guitar NA et al. Ageing Res Rev. 2018 Nov;47:159-67.
4. Mørch RH et al. Acta Psychiatr Scand. 2017 Oct;136(4):400-8.
5. Dooley LN et al. Neurosci Biobehav Rev. 2018 Nov;94:219-37.
6. Yang L et al. Brain Behav Immun. 2016 Aug;56:352-62.
7. Doenyas C. Neuroscience. 2018 Mar 15;374:271-86.
8. PBS News Hour. 2016 Jun 22.
9. Houlihan J. Environmental Working Group. 2005 Jul 14.
The shortage of psychiatrists, other mental health clinicians, and primary care physicians who treat patients with mental illness is a profound problem in the United States and around the world. What would happen to those trends if psychiatrists incorporated a functional medicine approach to treating patients?
In functional medicine, we look for underlying causes, physiological damage that results from those causes, clinical body system imbalances, and ultimately, symptoms that patients are experiencing. By addressing the root causes of chronic problems, treating physiological damage, and creating balance in body systems, psychiatrists and other physicians can help our patients achieve optimal health.
For example, a functional medicine approach to treating a child with ADHD might focus on encouraging behavioral changes such as improving sleep hygiene,1 increasing hydration,2 changing nutrition, or prescribing adjunctive meditation rather than medication alone. A functional medicine approach to Alzheimer’s prevention, for example, could include “prescribing” an increase in the amount of regular physical exercise.3 In other words, functional medicine uses a different lens to prevent, arrest, and in some cases, reverse certain diseases.
Medicine has long recognized the links between inflammation and chronic illness. Autoimmune conditions, asthma, heart disease, stroke, diabetes, obesity, peripheral neuropathy, thyroid problems, joint pain, and cancer all are chronic inflammatory diseases. Because inflammation affects the brain, it has been theorized and is being investigated that psychiatric disorders such as depression, schizophrenia, anxiety, panic attacks, dementia, and autism might result.4,5,6,7
Besides the brain, the GI tract is the only organ system that has its own nervous system, which is called the enteric nervous system, or ENS. The ENS functions independently from the central nervous system, and transmits important messages to and from the brain. When one feels stressed, the brain communicates to the hormonal system and floods the body with stress hormones, such as cortisol, which by themselves, can cause increased intestinal permeability. In addition, the gut produces its own neurotransmitters that affect the brain. In fact, every class of neurotransmitter found in the brain also is found in the GI tract. For example, serotonin is an important neurotransmitter for feeling happy and optimistic. Ninety-five percent of the body’s serotonin is produced in the gut. It is produced from 5-HTP, which is derived from tryptophan. However, in the presence of inflammation in the body, tryptophan is converted into kynurenate and quinolate. Both cause fatigue, and quinolate causes neurotoxicity. The subsequent depletion of serotonin produces symptoms of depression. Problems in the gut can lead to problems in the brain and the whole body.
Other problems affecting patients are tied to toxins in the environment. The air we breathe, food we eat, water we drink, and clothing we wear all are sources of toxins. Toxins include biotoxins, dioxine, phthalates, PCBs, and heavy metals, such as mercury, lead, cadmium, aluminum. About 2,000 new chemicals have been introduced into our environment each year since the 1940s, and it is estimated that we are exposed to more than 80,000 chemicals on a regular basis.8
The Environmental Working Group, a nonprofit organization dedicated to educating the public about the environment, has estimated that average babies are born with 287 chemicals in their body, 217 of which are neurotoxins.9 As children grow up, their body accumulates more toxins. According to the Centers for Disease Control and Prevention, every American has hundreds of neurotoxins in their bodies right now.
As we become more aware of the many changes in our environment, functional medicine brings a new way of thinking about and looking at chronic disease. As physicians, we can continue treating symptoms, and we should. But we can look deeper and ask ourselves what has changed in our lives that has caused such a decline in human mental and physical health. I urge psychiatrists to help lead the way.
Dr. Gaitour, a physiatrist, trained at NYU Langone Medical Center in New York. She is a functional medicine practitioner.
References
1. Peppers KH et al. J Pediatr Health Care. 2016 Nov-Dec;30(6):e43-8.
2. Martin EB and PG Hammerness. ADHD, stimulant medication, and dehydration. CHADD.org. 2014 Aug.
3. Guitar NA et al. Ageing Res Rev. 2018 Nov;47:159-67.
4. Mørch RH et al. Acta Psychiatr Scand. 2017 Oct;136(4):400-8.
5. Dooley LN et al. Neurosci Biobehav Rev. 2018 Nov;94:219-37.
6. Yang L et al. Brain Behav Immun. 2016 Aug;56:352-62.
7. Doenyas C. Neuroscience. 2018 Mar 15;374:271-86.
8. PBS News Hour. 2016 Jun 22.
9. Houlihan J. Environmental Working Group. 2005 Jul 14.
The shortage of psychiatrists, other mental health clinicians, and primary care physicians who treat patients with mental illness is a profound problem in the United States and around the world. What would happen to those trends if psychiatrists incorporated a functional medicine approach to treating patients?
In functional medicine, we look for underlying causes, physiological damage that results from those causes, clinical body system imbalances, and ultimately, symptoms that patients are experiencing. By addressing the root causes of chronic problems, treating physiological damage, and creating balance in body systems, psychiatrists and other physicians can help our patients achieve optimal health.
For example, a functional medicine approach to treating a child with ADHD might focus on encouraging behavioral changes such as improving sleep hygiene,1 increasing hydration,2 changing nutrition, or prescribing adjunctive meditation rather than medication alone. A functional medicine approach to Alzheimer’s prevention, for example, could include “prescribing” an increase in the amount of regular physical exercise.3 In other words, functional medicine uses a different lens to prevent, arrest, and in some cases, reverse certain diseases.
Medicine has long recognized the links between inflammation and chronic illness. Autoimmune conditions, asthma, heart disease, stroke, diabetes, obesity, peripheral neuropathy, thyroid problems, joint pain, and cancer all are chronic inflammatory diseases. Because inflammation affects the brain, it has been theorized and is being investigated that psychiatric disorders such as depression, schizophrenia, anxiety, panic attacks, dementia, and autism might result.4,5,6,7
Besides the brain, the GI tract is the only organ system that has its own nervous system, which is called the enteric nervous system, or ENS. The ENS functions independently from the central nervous system, and transmits important messages to and from the brain. When one feels stressed, the brain communicates to the hormonal system and floods the body with stress hormones, such as cortisol, which by themselves, can cause increased intestinal permeability. In addition, the gut produces its own neurotransmitters that affect the brain. In fact, every class of neurotransmitter found in the brain also is found in the GI tract. For example, serotonin is an important neurotransmitter for feeling happy and optimistic. Ninety-five percent of the body’s serotonin is produced in the gut. It is produced from 5-HTP, which is derived from tryptophan. However, in the presence of inflammation in the body, tryptophan is converted into kynurenate and quinolate. Both cause fatigue, and quinolate causes neurotoxicity. The subsequent depletion of serotonin produces symptoms of depression. Problems in the gut can lead to problems in the brain and the whole body.
Other problems affecting patients are tied to toxins in the environment. The air we breathe, food we eat, water we drink, and clothing we wear all are sources of toxins. Toxins include biotoxins, dioxine, phthalates, PCBs, and heavy metals, such as mercury, lead, cadmium, aluminum. About 2,000 new chemicals have been introduced into our environment each year since the 1940s, and it is estimated that we are exposed to more than 80,000 chemicals on a regular basis.8
The Environmental Working Group, a nonprofit organization dedicated to educating the public about the environment, has estimated that average babies are born with 287 chemicals in their body, 217 of which are neurotoxins.9 As children grow up, their body accumulates more toxins. According to the Centers for Disease Control and Prevention, every American has hundreds of neurotoxins in their bodies right now.
As we become more aware of the many changes in our environment, functional medicine brings a new way of thinking about and looking at chronic disease. As physicians, we can continue treating symptoms, and we should. But we can look deeper and ask ourselves what has changed in our lives that has caused such a decline in human mental and physical health. I urge psychiatrists to help lead the way.
Dr. Gaitour, a physiatrist, trained at NYU Langone Medical Center in New York. She is a functional medicine practitioner.
References
1. Peppers KH et al. J Pediatr Health Care. 2016 Nov-Dec;30(6):e43-8.
2. Martin EB and PG Hammerness. ADHD, stimulant medication, and dehydration. CHADD.org. 2014 Aug.
3. Guitar NA et al. Ageing Res Rev. 2018 Nov;47:159-67.
4. Mørch RH et al. Acta Psychiatr Scand. 2017 Oct;136(4):400-8.
5. Dooley LN et al. Neurosci Biobehav Rev. 2018 Nov;94:219-37.
6. Yang L et al. Brain Behav Immun. 2016 Aug;56:352-62.
7. Doenyas C. Neuroscience. 2018 Mar 15;374:271-86.
8. PBS News Hour. 2016 Jun 22.
9. Houlihan J. Environmental Working Group. 2005 Jul 14.
I have seen the future
Many patients have seen their long-term physicians retire. When I ask how they like their new doctors, they say: “She’s okay, I guess. Quite efficient. Seems thorough. But it’s not the same. It’s just business. Nothing personal.”

Sometimes you have to look backward to look forward. So it’s perhaps fitting that I glimpsed the future at my last colonoscopy.
In recent years, I’ve had such procedures at a local suburban surgicenter. Easy access, plenty of parking.
The woman who checks me in is all business. She scans my insurance cards and hands me a clipboard with a medical history form. Have I ever had cancer? A hernia? Am I pregnant? I wonder whether anyone reads these.
A different young woman brings me inside, the first of many new faces. Their roles are murky.
In a curtained cubby, yet another staff person asks me to pack my clothes in a plastic bag and put on a johnny. Then an older man enters, initiating furious multitasking. A different nursing assistant asks me to confirm my name and date of birth, then inserts an intravenous line in one arm, while the old doctor hands me an anesthesia consent form to sign with the other hand. I check many answers very fast, ignore the small-print boilerplate, and sign.
I am handed two more consent forms to sign, one from each side. The staff makes no pretense of explaining them or even telling me what they are for, and I make none of reading them.
They depart, replaced by still another person, who rolls me into the next room. He confirms my name and date of birth, and which procedure I am there for. The purpose of these multiple checks is clear, along with dispiriting depersonalization. One could mitigate this with some light banter, but no one bothers. No time.
My physician – whom I actually know – enters, says hello, and exchanges pleasantries. The last guy asks me to turn onto my left side. Intravenous sedation flows into my veins. The rest is silence.
Sometime later I wake up, greeted by another staff person. She asks if I am okay and offers me water or juice and saltines. Noting her Boston Red Sox sweatshirt, I say, “Great game last night,” but she does not know what I am talking about. She cares only for football and plans to fly to Nashville, Tenn., to watch her favorites.
Curtains are closed, and I am asked to dress. Another assistant directs me to a chair, where I will await my ride home. Through I try to walk alone, she takes my arm. “We assist everyone,” she explains.
As the sedation wears off, I observe. All around me I see movement, brisk and purposeful. Staff members crisscross before me from all angles, striding from one task to another, from prep room A to cubby D, walking with or pushing patients from procedure room M to holding area 8H. No one I’ve just met recognizes me, or acknowledges having met me before.
At last, the final staff member approaches. She flashes a kind smile as she takes my arm to walk me to the door. I take this for a personal touch, until she explains that she must make sure I don’t fall and that I get into the right car. As we pass, no one in the waiting room, neither staff nor patients, takes any notice.
My wife is outside, idling in the correct car. She’s brought coffee and a chocolate croissant, which – almost – makes last night’s prep worthwhile. She confirms neither my name nor date of birth.
Altogether, I have been in and out in 90 minutes. In the car, I peruse the handout that had been given to me as I exited. Drinking my coffee, I read the postcare instructions and enjoy its full-color pictures. Seldom has my cecum looked more radiant.
In “The Checklist Manifesto,” Atul Gawande described the outcome improvement that systematized practice can achieve. Data analysis confirms the measurably superior efficacy of such a method.
As for me, I feel like output from one of today’s cataract factories: like a car just extruded from an automated wash, with a photo on its front seat of the shiny, Simonized hubcaps included with the premium service package.
Just business, though. Nothing personal.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Many patients have seen their long-term physicians retire. When I ask how they like their new doctors, they say: “She’s okay, I guess. Quite efficient. Seems thorough. But it’s not the same. It’s just business. Nothing personal.”
Sometimes you have to look backward to look forward. So it’s perhaps fitting that I glimpsed the future at my last colonoscopy.
In recent years, I’ve had such procedures at a local suburban surgicenter. Easy access, plenty of parking.
The woman who checks me in is all business. She scans my insurance cards and hands me a clipboard with a medical history form. Have I ever had cancer? A hernia? Am I pregnant? I wonder whether anyone reads these.
A different young woman brings me inside, the first of many new faces. Their roles are murky.
In a curtained cubby, yet another staff person asks me to pack my clothes in a plastic bag and put on a johnny. Then an older man enters, initiating furious multitasking. A different nursing assistant asks me to confirm my name and date of birth, then inserts an intravenous line in one arm, while the old doctor hands me an anesthesia consent form to sign with the other hand. I check many answers very fast, ignore the small-print boilerplate, and sign.
I am handed two more consent forms to sign, one from each side. The staff makes no pretense of explaining them or even telling me what they are for, and I make none of reading them.
They depart, replaced by still another person, who rolls me into the next room. He confirms my name and date of birth, and which procedure I am there for. The purpose of these multiple checks is clear, along with dispiriting depersonalization. One could mitigate this with some light banter, but no one bothers. No time.
My physician – whom I actually know – enters, says hello, and exchanges pleasantries. The last guy asks me to turn onto my left side. Intravenous sedation flows into my veins. The rest is silence.
Sometime later I wake up, greeted by another staff person. She asks if I am okay and offers me water or juice and saltines. Noting her Boston Red Sox sweatshirt, I say, “Great game last night,” but she does not know what I am talking about. She cares only for football and plans to fly to Nashville, Tenn., to watch her favorites.
Curtains are closed, and I am asked to dress. Another assistant directs me to a chair, where I will await my ride home. Through I try to walk alone, she takes my arm. “We assist everyone,” she explains.
As the sedation wears off, I observe. All around me I see movement, brisk and purposeful. Staff members crisscross before me from all angles, striding from one task to another, from prep room A to cubby D, walking with or pushing patients from procedure room M to holding area 8H. No one I’ve just met recognizes me, or acknowledges having met me before.
At last, the final staff member approaches. She flashes a kind smile as she takes my arm to walk me to the door. I take this for a personal touch, until she explains that she must make sure I don’t fall and that I get into the right car. As we pass, no one in the waiting room, neither staff nor patients, takes any notice.
My wife is outside, idling in the correct car. She’s brought coffee and a chocolate croissant, which – almost – makes last night’s prep worthwhile. She confirms neither my name nor date of birth.
Altogether, I have been in and out in 90 minutes. In the car, I peruse the handout that had been given to me as I exited. Drinking my coffee, I read the postcare instructions and enjoy its full-color pictures. Seldom has my cecum looked more radiant.
In “The Checklist Manifesto,” Atul Gawande described the outcome improvement that systematized practice can achieve. Data analysis confirms the measurably superior efficacy of such a method.
As for me, I feel like output from one of today’s cataract factories: like a car just extruded from an automated wash, with a photo on its front seat of the shiny, Simonized hubcaps included with the premium service package.
Just business, though. Nothing personal.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Many patients have seen their long-term physicians retire. When I ask how they like their new doctors, they say: “She’s okay, I guess. Quite efficient. Seems thorough. But it’s not the same. It’s just business. Nothing personal.”
Sometimes you have to look backward to look forward. So it’s perhaps fitting that I glimpsed the future at my last colonoscopy.
In recent years, I’ve had such procedures at a local suburban surgicenter. Easy access, plenty of parking.
The woman who checks me in is all business. She scans my insurance cards and hands me a clipboard with a medical history form. Have I ever had cancer? A hernia? Am I pregnant? I wonder whether anyone reads these.
A different young woman brings me inside, the first of many new faces. Their roles are murky.
In a curtained cubby, yet another staff person asks me to pack my clothes in a plastic bag and put on a johnny. Then an older man enters, initiating furious multitasking. A different nursing assistant asks me to confirm my name and date of birth, then inserts an intravenous line in one arm, while the old doctor hands me an anesthesia consent form to sign with the other hand. I check many answers very fast, ignore the small-print boilerplate, and sign.
I am handed two more consent forms to sign, one from each side. The staff makes no pretense of explaining them or even telling me what they are for, and I make none of reading them.
They depart, replaced by still another person, who rolls me into the next room. He confirms my name and date of birth, and which procedure I am there for. The purpose of these multiple checks is clear, along with dispiriting depersonalization. One could mitigate this with some light banter, but no one bothers. No time.
My physician – whom I actually know – enters, says hello, and exchanges pleasantries. The last guy asks me to turn onto my left side. Intravenous sedation flows into my veins. The rest is silence.
Sometime later I wake up, greeted by another staff person. She asks if I am okay and offers me water or juice and saltines. Noting her Boston Red Sox sweatshirt, I say, “Great game last night,” but she does not know what I am talking about. She cares only for football and plans to fly to Nashville, Tenn., to watch her favorites.
Curtains are closed, and I am asked to dress. Another assistant directs me to a chair, where I will await my ride home. Through I try to walk alone, she takes my arm. “We assist everyone,” she explains.
As the sedation wears off, I observe. All around me I see movement, brisk and purposeful. Staff members crisscross before me from all angles, striding from one task to another, from prep room A to cubby D, walking with or pushing patients from procedure room M to holding area 8H. No one I’ve just met recognizes me, or acknowledges having met me before.
At last, the final staff member approaches. She flashes a kind smile as she takes my arm to walk me to the door. I take this for a personal touch, until she explains that she must make sure I don’t fall and that I get into the right car. As we pass, no one in the waiting room, neither staff nor patients, takes any notice.
My wife is outside, idling in the correct car. She’s brought coffee and a chocolate croissant, which – almost – makes last night’s prep worthwhile. She confirms neither my name nor date of birth.
Altogether, I have been in and out in 90 minutes. In the car, I peruse the handout that had been given to me as I exited. Drinking my coffee, I read the postcare instructions and enjoy its full-color pictures. Seldom has my cecum looked more radiant.
In “The Checklist Manifesto,” Atul Gawande described the outcome improvement that systematized practice can achieve. Data analysis confirms the measurably superior efficacy of such a method.
As for me, I feel like output from one of today’s cataract factories: like a car just extruded from an automated wash, with a photo on its front seat of the shiny, Simonized hubcaps included with the premium service package.
Just business, though. Nothing personal.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Snapshots of an oncologist
It’s 6:30 on a Friday night, and I am triaging three admissions to the leukemia service at once. The call from the ED about you makes me pause. I recognize your name – you were my patient a few years before. At the time, you were undergoing chemotherapy for acute myeloid leukemia, and I cared for you during the aftermath. I now pull up your chart and fill in the gaps of the last 2 years. You got into remission and received a bone marrow transplant. For 2 years, you were cured. But today, you are back. The ED has picked up an abundance of blasts – cancer cells – in your blood. I walk to your ED gurney slowly, thinking of how to tell you this. You recognize me, too. And I can see in your eyes that you already know. “I am so sorry this is happening,” I say.

You are here for your third cycle of chemotherapy. It’s a standard check-in. The first cycle was tolerable, the second cycle was rough, and now you are exhausted. You wonder if it’s normal to be so beat up from this. You ask how much nausea is too much nausea. But your hair didn’t fall out – isn’t that strange? Is it a sure thing that it will? And, by the way, is there anything to prevent the neuropathy? You wiggle your fingers as if to emphasize the point. We go through each of your symptoms and strategize ways to make this cycle better than the last. “OK,” you conclude triumphantly. “I got this!”
It’s your 1-month follow-up and it’s time to pivot. After you were diagnosed with an aggressive triple-negative breast cancer, you met with a medical oncologist and a surgeon. Chemotherapy first, they agreed. The chemo would shrink the tumor, they said, so that it could all be scooped out with surgery. The medications were rough, but you knew it was for the best. But now it’s been two cycles and the lump in your breast is getting bigger not smaller. I ask if I may draw on your skin, promising I’ll wash it off. I gently trace the mass in pen and pull out a tape measure. Yes. It is bigger. I listen to your heart and hear it racing. “What now?” you ask.
When you saw your doctor for bloating and were told it’s not gas, actually, but stage 4 cancer, you didn’t cry. You didn’t deny it. You prepared. You called your lawyer and made a will. You contacted your job and planned for retirement. You organized your things so your children wouldn’t have to. Your oncologist recommended palliative chemotherapy as it could give you some more good days. The best case scenario would be 1 year. That was 2½ years ago. You still like to be prepared, you tell me, but that’s on the back burner now. You are busy, after all – your feet still ache from dancing all night in heels at your niece’s wedding last weekend. I pull up your latest PET scan and we look together: Again, wonderfully, everything appears stable. “See you in 3 months,” I say.
You called three times to move up this appointment because you didn’t know if you’d be alive this long. You want a second opinion. When your kidney cancer grew after surgery, two immunotherapy drugs, and a chemotherapy pill, the latest setback has been fevers up to 104 ° with drenching night sweats. They found a deep infection gnawing around the edges of your tumor, and antibiotics aren’t touching it. The only chance to stop the cancer is more chemotherapy, but that could make the infection worse and lead to a rapid demise. You can’t decide. Today, in the exam room, you are sweating. Your temperature is 101 °. Your partner is trying to keep it together, but the crumpled tissues in her hand give it away. She looks at me earnestly: “What would you do if this were your family member?”
You teach about this disease in your classes and never thought it would happen to you. It started simply enough – you were bruising. Your joints ached. Small things; odd things. The ER doctor cleverly noticed that some numbers were off in your blood counts and sent you to a hematology-oncology doctor, who then cleverly ordered a molecular blood test. It was a long shot. He didn’t really expect it to come back with chronic myeloid leukemia. But there it is, and here we are. You return to talk about treatment options. You understand in detail the biology of how they work. What you don’t know is which is best for you. I go through the four choices and unpleasant effects of each. Muscle aches; diarrhea; risk of bleeding; twice a day dosing tied to mealtimes. “Is there an Option 5?” you wonder.

You have been in the hospital for 34 days, but who’s counting? You are. Because it has been Thirty. Four. Days. You knew the chemotherapy would suppress your blood counts. Now you know what “impaired immune system” really means. You had the bloodstream bacterial infection, requiring 2 days in the ICU. You had the invasive fungus growing in your lungs. The nurses post a calendar on your wall and kindly fill it in every day with your white blood cell count so you don’t have to ask. For days, it’s the same. Your bag stays packed – “just in case,” you explain. Your spouse diligently keeps your children – 2 and 4 years old – away, as kids are notorious germ factories. Then one Sunday morning and – finally! “Put me on speakerphone,” you tell your spouse. “Daddy is coming home!”
One of the most precious parts of hematology and oncology is the relationships. You are there not just for one difficult moment, but for the journey. I await getting to help you over the years to come. For now, I will settle for snapshots.
Dr. Yurkiewicz is a fellow in hematology and oncology at Stanford (Calif.) University. Follow her on Twitter @ilanayurkiewicz and listen to her each week on the Blood & Cancer podcast.
It’s 6:30 on a Friday night, and I am triaging three admissions to the leukemia service at once. The call from the ED about you makes me pause. I recognize your name – you were my patient a few years before. At the time, you were undergoing chemotherapy for acute myeloid leukemia, and I cared for you during the aftermath. I now pull up your chart and fill in the gaps of the last 2 years. You got into remission and received a bone marrow transplant. For 2 years, you were cured. But today, you are back. The ED has picked up an abundance of blasts – cancer cells – in your blood. I walk to your ED gurney slowly, thinking of how to tell you this. You recognize me, too. And I can see in your eyes that you already know. “I am so sorry this is happening,” I say.
You are here for your third cycle of chemotherapy. It’s a standard check-in. The first cycle was tolerable, the second cycle was rough, and now you are exhausted. You wonder if it’s normal to be so beat up from this. You ask how much nausea is too much nausea. But your hair didn’t fall out – isn’t that strange? Is it a sure thing that it will? And, by the way, is there anything to prevent the neuropathy? You wiggle your fingers as if to emphasize the point. We go through each of your symptoms and strategize ways to make this cycle better than the last. “OK,” you conclude triumphantly. “I got this!”
It’s your 1-month follow-up and it’s time to pivot. After you were diagnosed with an aggressive triple-negative breast cancer, you met with a medical oncologist and a surgeon. Chemotherapy first, they agreed. The chemo would shrink the tumor, they said, so that it could all be scooped out with surgery. The medications were rough, but you knew it was for the best. But now it’s been two cycles and the lump in your breast is getting bigger not smaller. I ask if I may draw on your skin, promising I’ll wash it off. I gently trace the mass in pen and pull out a tape measure. Yes. It is bigger. I listen to your heart and hear it racing. “What now?” you ask.
When you saw your doctor for bloating and were told it’s not gas, actually, but stage 4 cancer, you didn’t cry. You didn’t deny it. You prepared. You called your lawyer and made a will. You contacted your job and planned for retirement. You organized your things so your children wouldn’t have to. Your oncologist recommended palliative chemotherapy as it could give you some more good days. The best case scenario would be 1 year. That was 2½ years ago. You still like to be prepared, you tell me, but that’s on the back burner now. You are busy, after all – your feet still ache from dancing all night in heels at your niece’s wedding last weekend. I pull up your latest PET scan and we look together: Again, wonderfully, everything appears stable. “See you in 3 months,” I say.
You called three times to move up this appointment because you didn’t know if you’d be alive this long. You want a second opinion. When your kidney cancer grew after surgery, two immunotherapy drugs, and a chemotherapy pill, the latest setback has been fevers up to 104 ° with drenching night sweats. They found a deep infection gnawing around the edges of your tumor, and antibiotics aren’t touching it. The only chance to stop the cancer is more chemotherapy, but that could make the infection worse and lead to a rapid demise. You can’t decide. Today, in the exam room, you are sweating. Your temperature is 101 °. Your partner is trying to keep it together, but the crumpled tissues in her hand give it away. She looks at me earnestly: “What would you do if this were your family member?”
You teach about this disease in your classes and never thought it would happen to you. It started simply enough – you were bruising. Your joints ached. Small things; odd things. The ER doctor cleverly noticed that some numbers were off in your blood counts and sent you to a hematology-oncology doctor, who then cleverly ordered a molecular blood test. It was a long shot. He didn’t really expect it to come back with chronic myeloid leukemia. But there it is, and here we are. You return to talk about treatment options. You understand in detail the biology of how they work. What you don’t know is which is best for you. I go through the four choices and unpleasant effects of each. Muscle aches; diarrhea; risk of bleeding; twice a day dosing tied to mealtimes. “Is there an Option 5?” you wonder.
You have been in the hospital for 34 days, but who’s counting? You are. Because it has been Thirty. Four. Days. You knew the chemotherapy would suppress your blood counts. Now you know what “impaired immune system” really means. You had the bloodstream bacterial infection, requiring 2 days in the ICU. You had the invasive fungus growing in your lungs. The nurses post a calendar on your wall and kindly fill it in every day with your white blood cell count so you don’t have to ask. For days, it’s the same. Your bag stays packed – “just in case,” you explain. Your spouse diligently keeps your children – 2 and 4 years old – away, as kids are notorious germ factories. Then one Sunday morning and – finally! “Put me on speakerphone,” you tell your spouse. “Daddy is coming home!”
One of the most precious parts of hematology and oncology is the relationships. You are there not just for one difficult moment, but for the journey. I await getting to help you over the years to come. For now, I will settle for snapshots.
Dr. Yurkiewicz is a fellow in hematology and oncology at Stanford (Calif.) University. Follow her on Twitter @ilanayurkiewicz and listen to her each week on the Blood & Cancer podcast.
It’s 6:30 on a Friday night, and I am triaging three admissions to the leukemia service at once. The call from the ED about you makes me pause. I recognize your name – you were my patient a few years before. At the time, you were undergoing chemotherapy for acute myeloid leukemia, and I cared for you during the aftermath. I now pull up your chart and fill in the gaps of the last 2 years. You got into remission and received a bone marrow transplant. For 2 years, you were cured. But today, you are back. The ED has picked up an abundance of blasts – cancer cells – in your blood. I walk to your ED gurney slowly, thinking of how to tell you this. You recognize me, too. And I can see in your eyes that you already know. “I am so sorry this is happening,” I say.
You are here for your third cycle of chemotherapy. It’s a standard check-in. The first cycle was tolerable, the second cycle was rough, and now you are exhausted. You wonder if it’s normal to be so beat up from this. You ask how much nausea is too much nausea. But your hair didn’t fall out – isn’t that strange? Is it a sure thing that it will? And, by the way, is there anything to prevent the neuropathy? You wiggle your fingers as if to emphasize the point. We go through each of your symptoms and strategize ways to make this cycle better than the last. “OK,” you conclude triumphantly. “I got this!”
It’s your 1-month follow-up and it’s time to pivot. After you were diagnosed with an aggressive triple-negative breast cancer, you met with a medical oncologist and a surgeon. Chemotherapy first, they agreed. The chemo would shrink the tumor, they said, so that it could all be scooped out with surgery. The medications were rough, but you knew it was for the best. But now it’s been two cycles and the lump in your breast is getting bigger not smaller. I ask if I may draw on your skin, promising I’ll wash it off. I gently trace the mass in pen and pull out a tape measure. Yes. It is bigger. I listen to your heart and hear it racing. “What now?” you ask.
When you saw your doctor for bloating and were told it’s not gas, actually, but stage 4 cancer, you didn’t cry. You didn’t deny it. You prepared. You called your lawyer and made a will. You contacted your job and planned for retirement. You organized your things so your children wouldn’t have to. Your oncologist recommended palliative chemotherapy as it could give you some more good days. The best case scenario would be 1 year. That was 2½ years ago. You still like to be prepared, you tell me, but that’s on the back burner now. You are busy, after all – your feet still ache from dancing all night in heels at your niece’s wedding last weekend. I pull up your latest PET scan and we look together: Again, wonderfully, everything appears stable. “See you in 3 months,” I say.
You called three times to move up this appointment because you didn’t know if you’d be alive this long. You want a second opinion. When your kidney cancer grew after surgery, two immunotherapy drugs, and a chemotherapy pill, the latest setback has been fevers up to 104 ° with drenching night sweats. They found a deep infection gnawing around the edges of your tumor, and antibiotics aren’t touching it. The only chance to stop the cancer is more chemotherapy, but that could make the infection worse and lead to a rapid demise. You can’t decide. Today, in the exam room, you are sweating. Your temperature is 101 °. Your partner is trying to keep it together, but the crumpled tissues in her hand give it away. She looks at me earnestly: “What would you do if this were your family member?”
You teach about this disease in your classes and never thought it would happen to you. It started simply enough – you were bruising. Your joints ached. Small things; odd things. The ER doctor cleverly noticed that some numbers were off in your blood counts and sent you to a hematology-oncology doctor, who then cleverly ordered a molecular blood test. It was a long shot. He didn’t really expect it to come back with chronic myeloid leukemia. But there it is, and here we are. You return to talk about treatment options. You understand in detail the biology of how they work. What you don’t know is which is best for you. I go through the four choices and unpleasant effects of each. Muscle aches; diarrhea; risk of bleeding; twice a day dosing tied to mealtimes. “Is there an Option 5?” you wonder.
You have been in the hospital for 34 days, but who’s counting? You are. Because it has been Thirty. Four. Days. You knew the chemotherapy would suppress your blood counts. Now you know what “impaired immune system” really means. You had the bloodstream bacterial infection, requiring 2 days in the ICU. You had the invasive fungus growing in your lungs. The nurses post a calendar on your wall and kindly fill it in every day with your white blood cell count so you don’t have to ask. For days, it’s the same. Your bag stays packed – “just in case,” you explain. Your spouse diligently keeps your children – 2 and 4 years old – away, as kids are notorious germ factories. Then one Sunday morning and – finally! “Put me on speakerphone,” you tell your spouse. “Daddy is coming home!”
One of the most precious parts of hematology and oncology is the relationships. You are there not just for one difficult moment, but for the journey. I await getting to help you over the years to come. For now, I will settle for snapshots.
Dr. Yurkiewicz is a fellow in hematology and oncology at Stanford (Calif.) University. Follow her on Twitter @ilanayurkiewicz and listen to her each week on the Blood & Cancer podcast.
Food as therapy and toxin
I return to write the Editor’s comments after missing last month because I joined over 700,000 Americans who, this year, will undergo knee replacement surgery.
This month, we feature a couple articles from the 2019 James W. Freston Conference (an annual AGA event that highlights cutting-edge science). Jim was the 89th AGA President (1995) and this conference is a fitting legacy. This year’s topic was “Food at the intersection of gut health and disease.” As usual, the Freston Conference attracted international experts and interested clinicians who want to understand how current research will alter our clinical care in the near future.

Our front-page articles are fascinating. One highlights new advances in the management of celiac disease. Although the only current treatment that reverses intestinal immunological damage is adoption of a gluten-free diet, there is demand for alternative treatments including medical therapies targeting specific steps in the celiac damage pathway. While none are ready for wide-spread adoption, research will continue. Patient self-management with gluten detection-devices were also discussed.
Advances in the genetics of Crohn’s disease are being published at an accelerating rate. This month we highlight an article about how gene expression analysis can predict response to a Crohn’s flare. Evidence-based therapy for inflammatory bowel disease is complex, so clinicians need to stay current. Each year, the premier IBD educational venue is co-produced by the AGA and the Crohn’s & Colitis Foundation. The 2020 Crohn’s and Colitis Congress will be held in Austin, Texas January 23-25. Learn more at: https://www.crohnscolitiscongress.org.
Finally, I want to highlight an article about the risk of venous thromboembolism (VTE) during and after an IBD flare. This risk is underappreciated by many treating physicians but it is real and can be life-threatening. Gastroenterologists must be knowledgeable about current guidelines for VTE in IBD patients (see Gastroenterology 2014;146:835-48).
John I. Allen, MD, MBA, AGAF
Editor in Chief
I return to write the Editor’s comments after missing last month because I joined over 700,000 Americans who, this year, will undergo knee replacement surgery.
This month, we feature a couple articles from the 2019 James W. Freston Conference (an annual AGA event that highlights cutting-edge science). Jim was the 89th AGA President (1995) and this conference is a fitting legacy. This year’s topic was “Food at the intersection of gut health and disease.” As usual, the Freston Conference attracted international experts and interested clinicians who want to understand how current research will alter our clinical care in the near future.
Our front-page articles are fascinating. One highlights new advances in the management of celiac disease. Although the only current treatment that reverses intestinal immunological damage is adoption of a gluten-free diet, there is demand for alternative treatments including medical therapies targeting specific steps in the celiac damage pathway. While none are ready for wide-spread adoption, research will continue. Patient self-management with gluten detection-devices were also discussed.
Advances in the genetics of Crohn’s disease are being published at an accelerating rate. This month we highlight an article about how gene expression analysis can predict response to a Crohn’s flare. Evidence-based therapy for inflammatory bowel disease is complex, so clinicians need to stay current. Each year, the premier IBD educational venue is co-produced by the AGA and the Crohn’s & Colitis Foundation. The 2020 Crohn’s and Colitis Congress will be held in Austin, Texas January 23-25. Learn more at: https://www.crohnscolitiscongress.org.
Finally, I want to highlight an article about the risk of venous thromboembolism (VTE) during and after an IBD flare. This risk is underappreciated by many treating physicians but it is real and can be life-threatening. Gastroenterologists must be knowledgeable about current guidelines for VTE in IBD patients (see Gastroenterology 2014;146:835-48).
John I. Allen, MD, MBA, AGAF
Editor in Chief
I return to write the Editor’s comments after missing last month because I joined over 700,000 Americans who, this year, will undergo knee replacement surgery.
This month, we feature a couple articles from the 2019 James W. Freston Conference (an annual AGA event that highlights cutting-edge science). Jim was the 89th AGA President (1995) and this conference is a fitting legacy. This year’s topic was “Food at the intersection of gut health and disease.” As usual, the Freston Conference attracted international experts and interested clinicians who want to understand how current research will alter our clinical care in the near future.
Our front-page articles are fascinating. One highlights new advances in the management of celiac disease. Although the only current treatment that reverses intestinal immunological damage is adoption of a gluten-free diet, there is demand for alternative treatments including medical therapies targeting specific steps in the celiac damage pathway. While none are ready for wide-spread adoption, research will continue. Patient self-management with gluten detection-devices were also discussed.
Advances in the genetics of Crohn’s disease are being published at an accelerating rate. This month we highlight an article about how gene expression analysis can predict response to a Crohn’s flare. Evidence-based therapy for inflammatory bowel disease is complex, so clinicians need to stay current. Each year, the premier IBD educational venue is co-produced by the AGA and the Crohn’s & Colitis Foundation. The 2020 Crohn’s and Colitis Congress will be held in Austin, Texas January 23-25. Learn more at: https://www.crohnscolitiscongress.org.
Finally, I want to highlight an article about the risk of venous thromboembolism (VTE) during and after an IBD flare. This risk is underappreciated by many treating physicians but it is real and can be life-threatening. Gastroenterologists must be knowledgeable about current guidelines for VTE in IBD patients (see Gastroenterology 2014;146:835-48).
John I. Allen, MD, MBA, AGAF
Editor in Chief
Transcervical ablation of symptomatic uterine fibroids under US guidance
On Aug. 29, 2019, the first commercial case utilizing the Sonata system to transcervically ablate symptomatic uterine fibroids under ultrasound guidance was performed at Stamford (Conn.) Hospital. This truly minimally invasive new treatment expands our options in the surgical management of uterine fibroids.
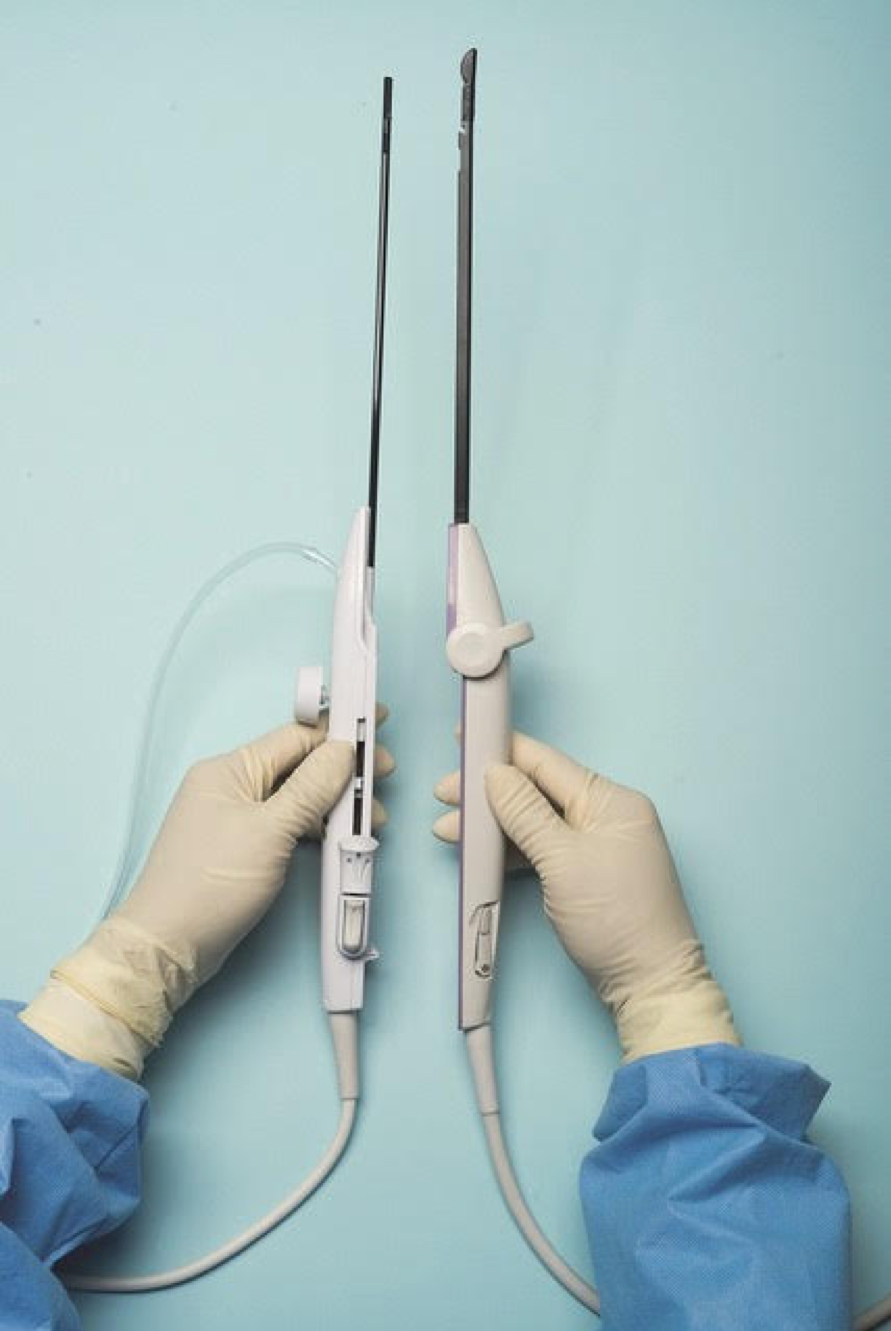
Uterine fibroids are the most common benign tumors of the reproductive tract. It has been estimated that nearly half of the 70%-80% of women who develop fibroids during their reproductive years are symptomatic. Given that some patients present with fertility concerns, it also has been estimated that at least one in three women with fibroids have symptoms such as heavy bleeding (menorrhagia) and bulk symptoms, pain (dyspareunia, dysmenorrhea, noncyclic pain), and increased urinary frequency.
Fibroids are the most common cause of hysterectomy in the United States, with 240,000 (40% of 600,000) performed annually, yet research shows that many women are interested in minimally invasive options and in uterine conservation. In a 2013 national survey published in the American Journal of Obstetrics and Gynecology, 79% of women expressed an interest in minimally invasive approaches for fibroid treatment, and over 50% reported a desire for uterine conservation.1
Both myomectomy and uterine artery embolization are uterine-sparing procedures. However, uterine artery embolization should not be performed in a woman interested in pregnancy. Moreover, there are reports of ovarian reserve issues when the procedure is performed in women in their later reproductive years.
Depending on the technique performed, women undergoing hysteroscopic myomectomy are at risk of fluid overload, hyponatremia, gas-related embolism, and postoperative adhesions. The suture requirements of a laparoscopic myomectomy make this approach an often-difficult one to master, even with robotic assistance. It also requires intubation and potentially places the patient at risk for bleeding and infection. Furthermore, long-term risks include adhesions and the need for C-section with pregnancy.
The impact of uterine fibroids on patients’ lives and their desire for uterine conservation has spurred growing interest in the use of radiofrequency (RF) energy to ablate uterine fibroids. In a 2018 systematic review of nonresective treatments for uterine fibroids published in the International Journal of Hyperthermia, investigators found that the pooled fibroid volume reductions at 6 months after RF ablation and uterine artery embolization were 70% and 54%, respectively.2
The first commercially available system utilizing RF frequency to shrink fibrosis – Acessa – involves laparoscopy, and thus requires abdominal incisions. In August 2018, the Sonata system (Gynesonics: Redwood, Calif.) received Food and Drug Administration clearance after having received European CE-Mark approval in 2010 (for the original device, the VizAblate) and in 2014 (for the next-generation device, the Sonata).
The technology
For a complete description of transcervical, intrauterine sonography–guided radiofrequency ablation of uterine fibroids, one can refer to the excellent outline by David Toub, MD, in Current Obstetrics and Gynecology Reports.3 Basically, the Sonata system allows for real-time, image-guided treatment through the use of a reusable intrauterine ultrasound (IUUS) probe, a single-use RF ablation (RFA) handpiece, and graphical guidance software for diagnosis and targeting.
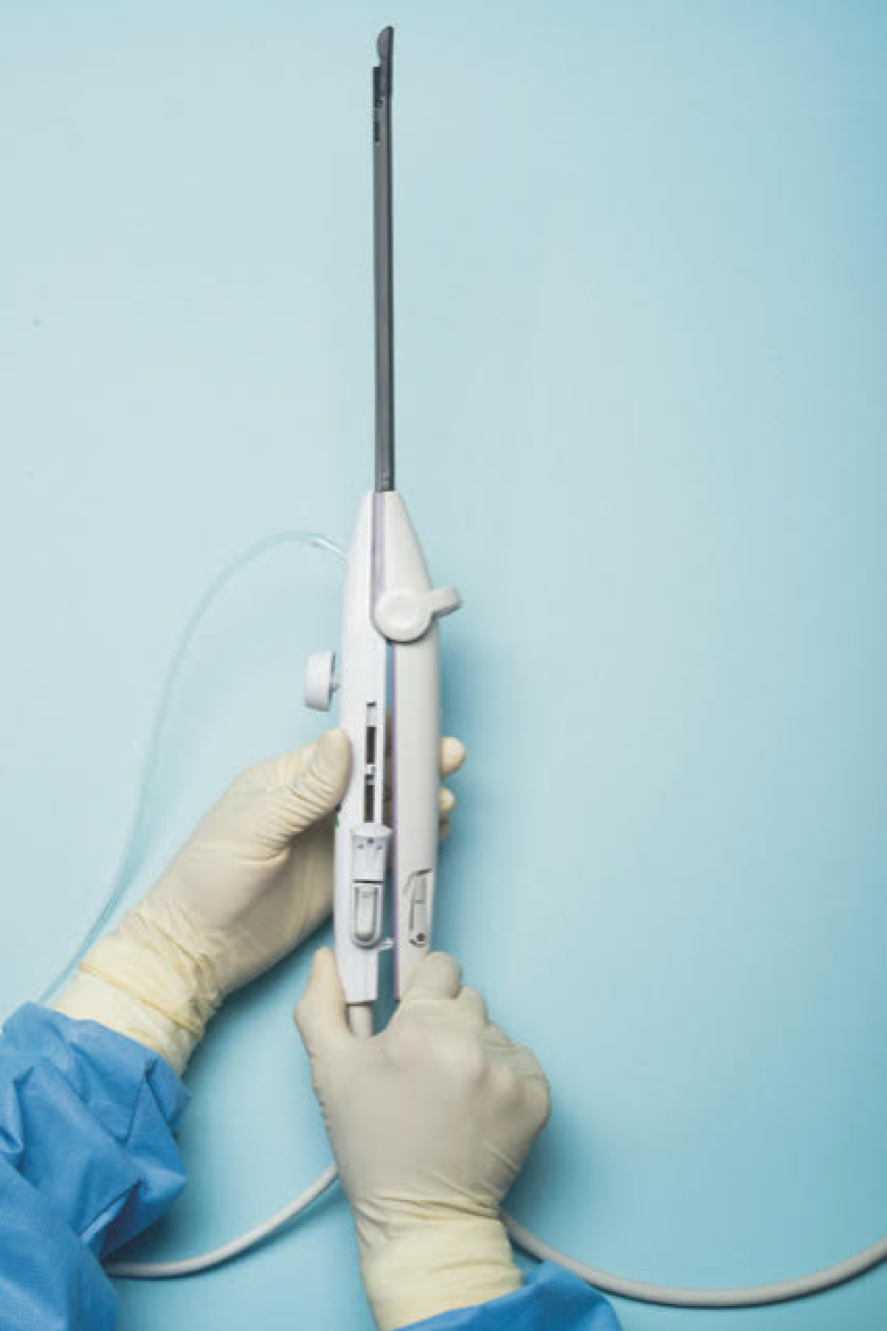
Initially, the IUUS probe enables identification of fibroids from within the uterine cavity, then guides deployment of an introducer and needle electrode into the targeted fibroid(s). The probe image is curvilinear, penetrates more than 9 cm, and provides a 90-degree field of view.
The RFA handpiece contains the introducer and needle electrode array. It snaps together with the IUUS probe to form and integrate into a single treatment device that contains all controls needed to place and size the ablation. Mechanical stops and lockouts within the RFA handpiece further enhance proper localization and sizing of the ablation.
The system’s graphical guidance software, also known as the SMART Guide, is a real-time graphical overlay on the ultrasound display, which enables one to visually select deployment length, width, and position of the ablation guides. In so doing, the mechanical stops for the introducer and needle electrodes are determined prior to their insertion into the targeted fibroid(s). This was validated in more than 4,000 ablations in bovine muscle and human-extirpated uteri, as well as in vivo at time of laparotomy.
By displaying the ellipsoidal region where the ablation will take place (ablation zone) along with a surrounding ellipsoid (thermal safety border) where tissue temperature will be elevated, the SMART Guide provides a safer and more accurate understanding of the ablation than if it showed only the ablation zone.

As with transabdominal or transvaginal sonography, the serosa will appear hyperechoic at the time of intrauterine ultrasound. By using the SMART Guide, the ablation is sized and positioned to encompass as much of the fibroid as possible while maintaining thermal energy within the uterine serosal margin. Once the desired ablation size has been selected, and safe placement of the needle electrodes is confirmed by rotating the IUUS probe in multiple planes, therapeutic RF energy is delivered to the fibroid; the fixed treatment cycle is dependent on ablation size.
The system will modulate power (up to 150W) to keep temperature at the tips of the needle electrode at 105° C. Moreover, the time of energy delivery at the temperature of 105° – 2-7 minutes – is automatically set based on ablation size, which is a continuum up to 4 cm wide and up to 5 cm long. Multiple ablations may be utilized in a particularly large fibroid.
Unlike hysteroscopic myomectomy, only a small amount of hypotonic solution is instilled within the uterine cavity to enhance acoustic coupling. Furthermore, the treatment device (RFA handpiece and IUUS probe) is only 8.3 mm in diameter. This requires Hegar dilatation of the cervix to 9.
The procedure
After administering anesthesia (regional or sedation), dispersive electrode pads are placed on the anterior thighs. After the cervix is dilated to Hegar dilatation of 9, the treatment device is inserted transcervically into the uterine cavity and the fibroid(s) are identified with the ultrasound probe. The physician plans and optimizes the ablation by sizing and aligning the graphical overlay targeting guide (the SMART Guide) over the live image. Once the size and location of the ablation are set, the trocar-tipped introducer is advanced into the fibroid. After ensuring the guide is within the serosal boundary, the needle electrodes are deployed.
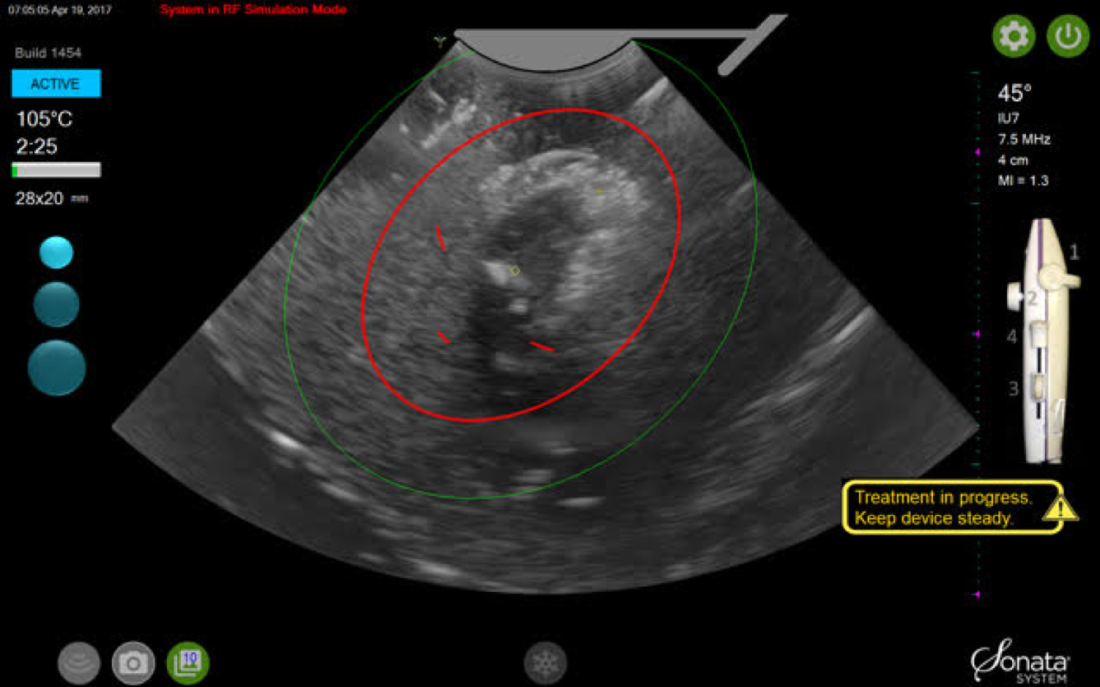
A second visual safety check is completed, and the delivery of RF energy is initiated using a footswitch control. The time of energy delivery is determined based on the size of the desired ablation, up to 7 minutes for the largest ablation size (5 cm x 4 cm). The targeting and treatment steps are repeated as required to treat additional fibroids. Once the treatment is completed, the needle electrodes and introducer are retracted, and the treatment device removed.
Study results and the future
The 12-month safety and effectiveness data for ultrasound-guided transcervical ablation of uterine fibroids were reported in January 2019 in Obstetrics & Gynecology.4 Women enrolled in the prospective, multicenter, single-arm, interventional trial had 1-10 fibroids – the International Federation of Gynecology and Obstetrics (FIGO) types 1, 2, 3, 4, and 2-5 (pedunculated fibroids excluded) – with diameters of 1-5 centimeters. Patients also were required to have at least one fibroid indenting or impinging on the endometrial cavity (FIGO type 1, 2, 3, or 2-5).
Upon study entry, the pictorial assessment blood loss was required to be 150-500 cc. The study included 147 patients. Both coprimary endpoints were satisfied at 12 months; that is, 65% of patients experienced a 50% or greater reduction in menstrual bleeding, and 99% were free from surgical intervention at 1 year.
The mean pictorial blood loss decreased by 39%, 48%, and 51% at 3, 6, and 12 months respectively. Moreover, 95% of the study population experienced some reduction in menstrual bleeding at 12 months. There also were mean improvements in symptom severity and health-related quality-of-life parameters. Mean maximal fibroid volume reduction per patient was 62%.
More than half of the patients returned to normal activity within 1 day, 96% of patients reported symptom improvement at 12 months, and 97% expressed satisfaction with the procedure and results at 12 months. There were no device-related adverse events.

I am the lead author for the 2-year follow-up study utilizing transcervical RFA of symptomatic uterine fibroids, which currently is in press. Suffice it to say, the quality-of-life data, symptom improvement, and lower rate of surgical reintervention all are significant and compelling. Ultimately, I believe Sonata will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.
Dr. Miller is a clinical associate professor at the University of Illinois in Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. Dr. Miller disclosed that he is a consultant for Gynesonics and holds a stock option agreement with the company.
References
1. Am J Obstet Gynecol. 2013 Oct;209(4):319.e1-319.e20.
2. Int J Hyperthermia. 2019;36(1):295-301.
3. Curr Obstet Gynecol Rep. 2017; 6(1): 67-73.
4. Obstet Gynecol. 2019 Jan;133(1):13-22.
On Aug. 29, 2019, the first commercial case utilizing the Sonata system to transcervically ablate symptomatic uterine fibroids under ultrasound guidance was performed at Stamford (Conn.) Hospital. This truly minimally invasive new treatment expands our options in the surgical management of uterine fibroids.
Uterine fibroids are the most common benign tumors of the reproductive tract. It has been estimated that nearly half of the 70%-80% of women who develop fibroids during their reproductive years are symptomatic. Given that some patients present with fertility concerns, it also has been estimated that at least one in three women with fibroids have symptoms such as heavy bleeding (menorrhagia) and bulk symptoms, pain (dyspareunia, dysmenorrhea, noncyclic pain), and increased urinary frequency.
Fibroids are the most common cause of hysterectomy in the United States, with 240,000 (40% of 600,000) performed annually, yet research shows that many women are interested in minimally invasive options and in uterine conservation. In a 2013 national survey published in the American Journal of Obstetrics and Gynecology, 79% of women expressed an interest in minimally invasive approaches for fibroid treatment, and over 50% reported a desire for uterine conservation.1
Both myomectomy and uterine artery embolization are uterine-sparing procedures. However, uterine artery embolization should not be performed in a woman interested in pregnancy. Moreover, there are reports of ovarian reserve issues when the procedure is performed in women in their later reproductive years.
Depending on the technique performed, women undergoing hysteroscopic myomectomy are at risk of fluid overload, hyponatremia, gas-related embolism, and postoperative adhesions. The suture requirements of a laparoscopic myomectomy make this approach an often-difficult one to master, even with robotic assistance. It also requires intubation and potentially places the patient at risk for bleeding and infection. Furthermore, long-term risks include adhesions and the need for C-section with pregnancy.
The impact of uterine fibroids on patients’ lives and their desire for uterine conservation has spurred growing interest in the use of radiofrequency (RF) energy to ablate uterine fibroids. In a 2018 systematic review of nonresective treatments for uterine fibroids published in the International Journal of Hyperthermia, investigators found that the pooled fibroid volume reductions at 6 months after RF ablation and uterine artery embolization were 70% and 54%, respectively.2
The first commercially available system utilizing RF frequency to shrink fibrosis – Acessa – involves laparoscopy, and thus requires abdominal incisions. In August 2018, the Sonata system (Gynesonics: Redwood, Calif.) received Food and Drug Administration clearance after having received European CE-Mark approval in 2010 (for the original device, the VizAblate) and in 2014 (for the next-generation device, the Sonata).
The technology
For a complete description of transcervical, intrauterine sonography–guided radiofrequency ablation of uterine fibroids, one can refer to the excellent outline by David Toub, MD, in Current Obstetrics and Gynecology Reports.3 Basically, the Sonata system allows for real-time, image-guided treatment through the use of a reusable intrauterine ultrasound (IUUS) probe, a single-use RF ablation (RFA) handpiece, and graphical guidance software for diagnosis and targeting.
Initially, the IUUS probe enables identification of fibroids from within the uterine cavity, then guides deployment of an introducer and needle electrode into the targeted fibroid(s). The probe image is curvilinear, penetrates more than 9 cm, and provides a 90-degree field of view.
The RFA handpiece contains the introducer and needle electrode array. It snaps together with the IUUS probe to form and integrate into a single treatment device that contains all controls needed to place and size the ablation. Mechanical stops and lockouts within the RFA handpiece further enhance proper localization and sizing of the ablation.
The system’s graphical guidance software, also known as the SMART Guide, is a real-time graphical overlay on the ultrasound display, which enables one to visually select deployment length, width, and position of the ablation guides. In so doing, the mechanical stops for the introducer and needle electrodes are determined prior to their insertion into the targeted fibroid(s). This was validated in more than 4,000 ablations in bovine muscle and human-extirpated uteri, as well as in vivo at time of laparotomy.
By displaying the ellipsoidal region where the ablation will take place (ablation zone) along with a surrounding ellipsoid (thermal safety border) where tissue temperature will be elevated, the SMART Guide provides a safer and more accurate understanding of the ablation than if it showed only the ablation zone.
As with transabdominal or transvaginal sonography, the serosa will appear hyperechoic at the time of intrauterine ultrasound. By using the SMART Guide, the ablation is sized and positioned to encompass as much of the fibroid as possible while maintaining thermal energy within the uterine serosal margin. Once the desired ablation size has been selected, and safe placement of the needle electrodes is confirmed by rotating the IUUS probe in multiple planes, therapeutic RF energy is delivered to the fibroid; the fixed treatment cycle is dependent on ablation size.
The system will modulate power (up to 150W) to keep temperature at the tips of the needle electrode at 105° C. Moreover, the time of energy delivery at the temperature of 105° – 2-7 minutes – is automatically set based on ablation size, which is a continuum up to 4 cm wide and up to 5 cm long. Multiple ablations may be utilized in a particularly large fibroid.
Unlike hysteroscopic myomectomy, only a small amount of hypotonic solution is instilled within the uterine cavity to enhance acoustic coupling. Furthermore, the treatment device (RFA handpiece and IUUS probe) is only 8.3 mm in diameter. This requires Hegar dilatation of the cervix to 9.
The procedure
After administering anesthesia (regional or sedation), dispersive electrode pads are placed on the anterior thighs. After the cervix is dilated to Hegar dilatation of 9, the treatment device is inserted transcervically into the uterine cavity and the fibroid(s) are identified with the ultrasound probe. The physician plans and optimizes the ablation by sizing and aligning the graphical overlay targeting guide (the SMART Guide) over the live image. Once the size and location of the ablation are set, the trocar-tipped introducer is advanced into the fibroid. After ensuring the guide is within the serosal boundary, the needle electrodes are deployed.
A second visual safety check is completed, and the delivery of RF energy is initiated using a footswitch control. The time of energy delivery is determined based on the size of the desired ablation, up to 7 minutes for the largest ablation size (5 cm x 4 cm). The targeting and treatment steps are repeated as required to treat additional fibroids. Once the treatment is completed, the needle electrodes and introducer are retracted, and the treatment device removed.
Study results and the future
The 12-month safety and effectiveness data for ultrasound-guided transcervical ablation of uterine fibroids were reported in January 2019 in Obstetrics & Gynecology.4 Women enrolled in the prospective, multicenter, single-arm, interventional trial had 1-10 fibroids – the International Federation of Gynecology and Obstetrics (FIGO) types 1, 2, 3, 4, and 2-5 (pedunculated fibroids excluded) – with diameters of 1-5 centimeters. Patients also were required to have at least one fibroid indenting or impinging on the endometrial cavity (FIGO type 1, 2, 3, or 2-5).
Upon study entry, the pictorial assessment blood loss was required to be 150-500 cc. The study included 147 patients. Both coprimary endpoints were satisfied at 12 months; that is, 65% of patients experienced a 50% or greater reduction in menstrual bleeding, and 99% were free from surgical intervention at 1 year.
The mean pictorial blood loss decreased by 39%, 48%, and 51% at 3, 6, and 12 months respectively. Moreover, 95% of the study population experienced some reduction in menstrual bleeding at 12 months. There also were mean improvements in symptom severity and health-related quality-of-life parameters. Mean maximal fibroid volume reduction per patient was 62%.
More than half of the patients returned to normal activity within 1 day, 96% of patients reported symptom improvement at 12 months, and 97% expressed satisfaction with the procedure and results at 12 months. There were no device-related adverse events.
I am the lead author for the 2-year follow-up study utilizing transcervical RFA of symptomatic uterine fibroids, which currently is in press. Suffice it to say, the quality-of-life data, symptom improvement, and lower rate of surgical reintervention all are significant and compelling. Ultimately, I believe Sonata will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.
Dr. Miller is a clinical associate professor at the University of Illinois in Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. Dr. Miller disclosed that he is a consultant for Gynesonics and holds a stock option agreement with the company.
References
1. Am J Obstet Gynecol. 2013 Oct;209(4):319.e1-319.e20.
2. Int J Hyperthermia. 2019;36(1):295-301.
3. Curr Obstet Gynecol Rep. 2017; 6(1): 67-73.
4. Obstet Gynecol. 2019 Jan;133(1):13-22.
On Aug. 29, 2019, the first commercial case utilizing the Sonata system to transcervically ablate symptomatic uterine fibroids under ultrasound guidance was performed at Stamford (Conn.) Hospital. This truly minimally invasive new treatment expands our options in the surgical management of uterine fibroids.
Uterine fibroids are the most common benign tumors of the reproductive tract. It has been estimated that nearly half of the 70%-80% of women who develop fibroids during their reproductive years are symptomatic. Given that some patients present with fertility concerns, it also has been estimated that at least one in three women with fibroids have symptoms such as heavy bleeding (menorrhagia) and bulk symptoms, pain (dyspareunia, dysmenorrhea, noncyclic pain), and increased urinary frequency.
Fibroids are the most common cause of hysterectomy in the United States, with 240,000 (40% of 600,000) performed annually, yet research shows that many women are interested in minimally invasive options and in uterine conservation. In a 2013 national survey published in the American Journal of Obstetrics and Gynecology, 79% of women expressed an interest in minimally invasive approaches for fibroid treatment, and over 50% reported a desire for uterine conservation.1
Both myomectomy and uterine artery embolization are uterine-sparing procedures. However, uterine artery embolization should not be performed in a woman interested in pregnancy. Moreover, there are reports of ovarian reserve issues when the procedure is performed in women in their later reproductive years.
Depending on the technique performed, women undergoing hysteroscopic myomectomy are at risk of fluid overload, hyponatremia, gas-related embolism, and postoperative adhesions. The suture requirements of a laparoscopic myomectomy make this approach an often-difficult one to master, even with robotic assistance. It also requires intubation and potentially places the patient at risk for bleeding and infection. Furthermore, long-term risks include adhesions and the need for C-section with pregnancy.
The impact of uterine fibroids on patients’ lives and their desire for uterine conservation has spurred growing interest in the use of radiofrequency (RF) energy to ablate uterine fibroids. In a 2018 systematic review of nonresective treatments for uterine fibroids published in the International Journal of Hyperthermia, investigators found that the pooled fibroid volume reductions at 6 months after RF ablation and uterine artery embolization were 70% and 54%, respectively.2
The first commercially available system utilizing RF frequency to shrink fibrosis – Acessa – involves laparoscopy, and thus requires abdominal incisions. In August 2018, the Sonata system (Gynesonics: Redwood, Calif.) received Food and Drug Administration clearance after having received European CE-Mark approval in 2010 (for the original device, the VizAblate) and in 2014 (for the next-generation device, the Sonata).
The technology
For a complete description of transcervical, intrauterine sonography–guided radiofrequency ablation of uterine fibroids, one can refer to the excellent outline by David Toub, MD, in Current Obstetrics and Gynecology Reports.3 Basically, the Sonata system allows for real-time, image-guided treatment through the use of a reusable intrauterine ultrasound (IUUS) probe, a single-use RF ablation (RFA) handpiece, and graphical guidance software for diagnosis and targeting.
Initially, the IUUS probe enables identification of fibroids from within the uterine cavity, then guides deployment of an introducer and needle electrode into the targeted fibroid(s). The probe image is curvilinear, penetrates more than 9 cm, and provides a 90-degree field of view.
The RFA handpiece contains the introducer and needle electrode array. It snaps together with the IUUS probe to form and integrate into a single treatment device that contains all controls needed to place and size the ablation. Mechanical stops and lockouts within the RFA handpiece further enhance proper localization and sizing of the ablation.
The system’s graphical guidance software, also known as the SMART Guide, is a real-time graphical overlay on the ultrasound display, which enables one to visually select deployment length, width, and position of the ablation guides. In so doing, the mechanical stops for the introducer and needle electrodes are determined prior to their insertion into the targeted fibroid(s). This was validated in more than 4,000 ablations in bovine muscle and human-extirpated uteri, as well as in vivo at time of laparotomy.
By displaying the ellipsoidal region where the ablation will take place (ablation zone) along with a surrounding ellipsoid (thermal safety border) where tissue temperature will be elevated, the SMART Guide provides a safer and more accurate understanding of the ablation than if it showed only the ablation zone.
As with transabdominal or transvaginal sonography, the serosa will appear hyperechoic at the time of intrauterine ultrasound. By using the SMART Guide, the ablation is sized and positioned to encompass as much of the fibroid as possible while maintaining thermal energy within the uterine serosal margin. Once the desired ablation size has been selected, and safe placement of the needle electrodes is confirmed by rotating the IUUS probe in multiple planes, therapeutic RF energy is delivered to the fibroid; the fixed treatment cycle is dependent on ablation size.
The system will modulate power (up to 150W) to keep temperature at the tips of the needle electrode at 105° C. Moreover, the time of energy delivery at the temperature of 105° – 2-7 minutes – is automatically set based on ablation size, which is a continuum up to 4 cm wide and up to 5 cm long. Multiple ablations may be utilized in a particularly large fibroid.
Unlike hysteroscopic myomectomy, only a small amount of hypotonic solution is instilled within the uterine cavity to enhance acoustic coupling. Furthermore, the treatment device (RFA handpiece and IUUS probe) is only 8.3 mm in diameter. This requires Hegar dilatation of the cervix to 9.
The procedure
After administering anesthesia (regional or sedation), dispersive electrode pads are placed on the anterior thighs. After the cervix is dilated to Hegar dilatation of 9, the treatment device is inserted transcervically into the uterine cavity and the fibroid(s) are identified with the ultrasound probe. The physician plans and optimizes the ablation by sizing and aligning the graphical overlay targeting guide (the SMART Guide) over the live image. Once the size and location of the ablation are set, the trocar-tipped introducer is advanced into the fibroid. After ensuring the guide is within the serosal boundary, the needle electrodes are deployed.
A second visual safety check is completed, and the delivery of RF energy is initiated using a footswitch control. The time of energy delivery is determined based on the size of the desired ablation, up to 7 minutes for the largest ablation size (5 cm x 4 cm). The targeting and treatment steps are repeated as required to treat additional fibroids. Once the treatment is completed, the needle electrodes and introducer are retracted, and the treatment device removed.
Study results and the future
The 12-month safety and effectiveness data for ultrasound-guided transcervical ablation of uterine fibroids were reported in January 2019 in Obstetrics & Gynecology.4 Women enrolled in the prospective, multicenter, single-arm, interventional trial had 1-10 fibroids – the International Federation of Gynecology and Obstetrics (FIGO) types 1, 2, 3, 4, and 2-5 (pedunculated fibroids excluded) – with diameters of 1-5 centimeters. Patients also were required to have at least one fibroid indenting or impinging on the endometrial cavity (FIGO type 1, 2, 3, or 2-5).
Upon study entry, the pictorial assessment blood loss was required to be 150-500 cc. The study included 147 patients. Both coprimary endpoints were satisfied at 12 months; that is, 65% of patients experienced a 50% or greater reduction in menstrual bleeding, and 99% were free from surgical intervention at 1 year.
The mean pictorial blood loss decreased by 39%, 48%, and 51% at 3, 6, and 12 months respectively. Moreover, 95% of the study population experienced some reduction in menstrual bleeding at 12 months. There also were mean improvements in symptom severity and health-related quality-of-life parameters. Mean maximal fibroid volume reduction per patient was 62%.
More than half of the patients returned to normal activity within 1 day, 96% of patients reported symptom improvement at 12 months, and 97% expressed satisfaction with the procedure and results at 12 months. There were no device-related adverse events.
I am the lead author for the 2-year follow-up study utilizing transcervical RFA of symptomatic uterine fibroids, which currently is in press. Suffice it to say, the quality-of-life data, symptom improvement, and lower rate of surgical reintervention all are significant and compelling. Ultimately, I believe Sonata will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.
Dr. Miller is a clinical associate professor at the University of Illinois in Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. Dr. Miller disclosed that he is a consultant for Gynesonics and holds a stock option agreement with the company.
References
1. Am J Obstet Gynecol. 2013 Oct;209(4):319.e1-319.e20.
2. Int J Hyperthermia. 2019;36(1):295-301.
3. Curr Obstet Gynecol Rep. 2017; 6(1): 67-73.
4. Obstet Gynecol. 2019 Jan;133(1):13-22.
Treating uterine fibroids
Uterine fibroids are the most common benign tumor in women originating from the smooth muscles of the myometrium. While some women are asymptomatic, others experience pelvic pain, pressure, and abnormal uterine bleeding. Uterine fibroids also are associated with gastrointestinal disturbances; urinary problems; infertility; and obstetrical complications including miscarriages, preterm delivery, and cesarean sections.

The first successful abdominal myomectomy was described in 1845 but the procedure quickly fell out of favor because of unacceptably high mortality rates. Myomectomies require special skills and, at times, are associated with bleeding resulting in massive transfusions or sometimes unwanted hysterectomies. In 1922, Victor Bonney developed a uterine artery clamp which significantly decreased bleeding associated with morbidity and mortality.1
The latter part of the 20th century belonged to the minimally invasive surgery (MIS) evolution. Currently, video- or robotic-assisted laparoscopic myomectomies are increasingly employed in fertility-sparing surgery. In 2014, electromechanical morcellators came under scrutiny with concerns about iatrogenic dissemination of both benign and malignant tissues. A media storm ensued, resulting in the 2014 Food and Drug Administration black-box warning, and electromechanical morcellators were pulled from shelves. Data are being collected to quantify and understand these risks more clearly.
While exposing patients to even a small risk of dissemination of an occult uterine malignancy is unwise, MIS should not be abandoned altogether given its advantages to patients.2 Most recently, the American College of Obstetricians and Gynecologists concluded that, although abdominal hysterectomy or myomectomy may reduce the chance of spreading undiagnosed leiomyosarcoma cells, it is associated with increased morbidity, compared with noninvasive approaches, and ob.gyns. should engage in open decision-making processes and explain nonsurgical options with patients.3
The author of this Master Class, Dr. Charles Miller, a world-renowned MIS surgeon, will enlighten readers on the latest development in noninvasive treatment of symptomatic patients. The Sonata system, a promising transcervical (and thus incisionless) treatment modality utilizing intrauterine sonography–guided radiofrequency ablation for uterine fibroids which does not require general anesthesia or hospitalization. He believes that Sonata “will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.”
Dr. Miller is on the editorial advisory boards of numerous academic journals and serves as the editor of the award-winning Master Class in Gynecologic Surgery column. For this installment, he has stepped into the role of guest author. Dr. Miller has received numerous awards for his educational contributions and was recently granted the distinct honor of taking the lead in the March 28, 2020 Worldwide EndoMarch–Chicago. It is my pleasure to take part in this introduction.
Dr. Nezhat is director of minimally invasive surgery and robotics as well as the medical director of training and education at Northside Hospital, both in Atlanta. He is fellowship director at Atlanta Center for Special Minimally Invasive Surgery & Reproductive Medicine. Dr. Nezhat also is an adjunct professor of gynecology and obstetrics at Emory University, Atlanta, and is past president of the Society of Reproductive Surgeons and the AAGL. He reported that he has no disclosures relevant to this Master Class. Email him at obnews@mdedge.com.
References
1. BJOG. 2018 Apr;125(5):586.
2. JAMA Oncol. 2015;1(1):78-9.
3. Obstet Gynecol. 2019 Mar;133(3):e238-48.
Uterine fibroids are the most common benign tumor in women originating from the smooth muscles of the myometrium. While some women are asymptomatic, others experience pelvic pain, pressure, and abnormal uterine bleeding. Uterine fibroids also are associated with gastrointestinal disturbances; urinary problems; infertility; and obstetrical complications including miscarriages, preterm delivery, and cesarean sections.
The first successful abdominal myomectomy was described in 1845 but the procedure quickly fell out of favor because of unacceptably high mortality rates. Myomectomies require special skills and, at times, are associated with bleeding resulting in massive transfusions or sometimes unwanted hysterectomies. In 1922, Victor Bonney developed a uterine artery clamp which significantly decreased bleeding associated with morbidity and mortality.1
The latter part of the 20th century belonged to the minimally invasive surgery (MIS) evolution. Currently, video- or robotic-assisted laparoscopic myomectomies are increasingly employed in fertility-sparing surgery. In 2014, electromechanical morcellators came under scrutiny with concerns about iatrogenic dissemination of both benign and malignant tissues. A media storm ensued, resulting in the 2014 Food and Drug Administration black-box warning, and electromechanical morcellators were pulled from shelves. Data are being collected to quantify and understand these risks more clearly.
While exposing patients to even a small risk of dissemination of an occult uterine malignancy is unwise, MIS should not be abandoned altogether given its advantages to patients.2 Most recently, the American College of Obstetricians and Gynecologists concluded that, although abdominal hysterectomy or myomectomy may reduce the chance of spreading undiagnosed leiomyosarcoma cells, it is associated with increased morbidity, compared with noninvasive approaches, and ob.gyns. should engage in open decision-making processes and explain nonsurgical options with patients.3
The author of this Master Class, Dr. Charles Miller, a world-renowned MIS surgeon, will enlighten readers on the latest development in noninvasive treatment of symptomatic patients. The Sonata system, a promising transcervical (and thus incisionless) treatment modality utilizing intrauterine sonography–guided radiofrequency ablation for uterine fibroids which does not require general anesthesia or hospitalization. He believes that Sonata “will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.”
Dr. Miller is on the editorial advisory boards of numerous academic journals and serves as the editor of the award-winning Master Class in Gynecologic Surgery column. For this installment, he has stepped into the role of guest author. Dr. Miller has received numerous awards for his educational contributions and was recently granted the distinct honor of taking the lead in the March 28, 2020 Worldwide EndoMarch–Chicago. It is my pleasure to take part in this introduction.
Dr. Nezhat is director of minimally invasive surgery and robotics as well as the medical director of training and education at Northside Hospital, both in Atlanta. He is fellowship director at Atlanta Center for Special Minimally Invasive Surgery & Reproductive Medicine. Dr. Nezhat also is an adjunct professor of gynecology and obstetrics at Emory University, Atlanta, and is past president of the Society of Reproductive Surgeons and the AAGL. He reported that he has no disclosures relevant to this Master Class. Email him at obnews@mdedge.com.
References
1. BJOG. 2018 Apr;125(5):586.
2. JAMA Oncol. 2015;1(1):78-9.
3. Obstet Gynecol. 2019 Mar;133(3):e238-48.
Uterine fibroids are the most common benign tumor in women originating from the smooth muscles of the myometrium. While some women are asymptomatic, others experience pelvic pain, pressure, and abnormal uterine bleeding. Uterine fibroids also are associated with gastrointestinal disturbances; urinary problems; infertility; and obstetrical complications including miscarriages, preterm delivery, and cesarean sections.
The first successful abdominal myomectomy was described in 1845 but the procedure quickly fell out of favor because of unacceptably high mortality rates. Myomectomies require special skills and, at times, are associated with bleeding resulting in massive transfusions or sometimes unwanted hysterectomies. In 1922, Victor Bonney developed a uterine artery clamp which significantly decreased bleeding associated with morbidity and mortality.1
The latter part of the 20th century belonged to the minimally invasive surgery (MIS) evolution. Currently, video- or robotic-assisted laparoscopic myomectomies are increasingly employed in fertility-sparing surgery. In 2014, electromechanical morcellators came under scrutiny with concerns about iatrogenic dissemination of both benign and malignant tissues. A media storm ensued, resulting in the 2014 Food and Drug Administration black-box warning, and electromechanical morcellators were pulled from shelves. Data are being collected to quantify and understand these risks more clearly.
While exposing patients to even a small risk of dissemination of an occult uterine malignancy is unwise, MIS should not be abandoned altogether given its advantages to patients.2 Most recently, the American College of Obstetricians and Gynecologists concluded that, although abdominal hysterectomy or myomectomy may reduce the chance of spreading undiagnosed leiomyosarcoma cells, it is associated with increased morbidity, compared with noninvasive approaches, and ob.gyns. should engage in open decision-making processes and explain nonsurgical options with patients.3
The author of this Master Class, Dr. Charles Miller, a world-renowned MIS surgeon, will enlighten readers on the latest development in noninvasive treatment of symptomatic patients. The Sonata system, a promising transcervical (and thus incisionless) treatment modality utilizing intrauterine sonography–guided radiofrequency ablation for uterine fibroids which does not require general anesthesia or hospitalization. He believes that Sonata “will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.”
Dr. Miller is on the editorial advisory boards of numerous academic journals and serves as the editor of the award-winning Master Class in Gynecologic Surgery column. For this installment, he has stepped into the role of guest author. Dr. Miller has received numerous awards for his educational contributions and was recently granted the distinct honor of taking the lead in the March 28, 2020 Worldwide EndoMarch–Chicago. It is my pleasure to take part in this introduction.
Dr. Nezhat is director of minimally invasive surgery and robotics as well as the medical director of training and education at Northside Hospital, both in Atlanta. He is fellowship director at Atlanta Center for Special Minimally Invasive Surgery & Reproductive Medicine. Dr. Nezhat also is an adjunct professor of gynecology and obstetrics at Emory University, Atlanta, and is past president of the Society of Reproductive Surgeons and the AAGL. He reported that he has no disclosures relevant to this Master Class. Email him at obnews@mdedge.com.
References
1. BJOG. 2018 Apr;125(5):586.
2. JAMA Oncol. 2015;1(1):78-9.
3. Obstet Gynecol. 2019 Mar;133(3):e238-48.
Preemptive pacifier promotion
I recently encountered an article aimed at parents who were struggling with what to do about their child’s persistent attachment to his pacifier (“How to Ditch the Pacifier,” by Anna Nowogrodski, New York Times, 2019 Sept. 16). For the most part, the author presented a sampling of sound advice from pediatricians and other health experts.

Most children will abandon their pacifiers at a time that is consistent with their developmental stage. Pacifiers seldom do any permanent damage, although they aren’t terribly appealing to look at when hanging out of a toddling toddler’s mouth. Parents were urged to be patient and consistent and were told that allowing the gooey thing to self-destruct often works, as does accelerating the process with a razor blade. Enlisting the aid of the Pacifier Fairy was suggested, but I’m not so sure that would work terribly well.
As I finished perusing the article, I couldn’t help think of how this vexing issue of pacifier removal can be avoided if parents follow a simple rule when they first introduced a pacifier to their child. If experienced parents think back to when they first resorted to using the pacifier, it wasn’t because the plastic and rubber gadget was a family heirloom that had been passed down from generation to generation like an engraved silver spoon. It wasn’t because the dentist told them that children who use pacifiers are less likely to need braces on their teeth. Nor was it a rumor filtered down from speech therapists that pacifiers improve articulation.
Parents reach for a pacifier in hopes that it will help their child will fall asleep. I think most parents of older children agree that at the beginning the pacifier was first and foremost a sleep aid. But here is where the critical oversight occurs: If you give your children pacifiers when you want them to go to sleep, why not simply add the stipulation of where you would like them to go to sleep as well?

Most parents prefer that their children sleep in their own space. We can argue of whether that should be in a side sleeper or their own crib, but most parents don’t want their 3-year-olds sleeping in their bed. Nor do they want their children sleeping on the couch in the living room with them while they watch a movie at 10:30 at night. And as pediatricians, we prefer that children not sleep with their necks flexed in a car seat or baby rocker, particularly if they’re a preemie.
Augmenting the primary association between sleep and the pacifier by adding a place has several important advantages. It gives parents more control of where their children will sleep or, more importantly, where they won’t be sleeping. It helps transitions to nonhome sleeping places like day care and long trips to grandma’s house go more smoothly.
Even more importantly, the crib/pacifier association helps parents who have had trouble reading their children’s cues. If they want a pacifier, it means they are tired and want to go to where the pacifier lives: bed
Finally, maintaining the link between sleeping and the pacifier promotes a more natural weaning process than going cold turkey or hiring the Pacifier Fairy. As naps disappear, the pacifier gradually become a less obvious accessory in the child’s life. However, it may linger in the background as a reminder of when the child needs some restorative sleep.
Of course, helping parents to think clearly enough to create and enforce a simple rule long enough to forge a healthy association when they are sleep deprived themselves is just another one of those challenges we must accept as concerned primary care pediatricians.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Is My Child Overtired? The Sleep Solution for Raising Happier, Healthier Children.” Email him at pdnews@mdedge.com.
I recently encountered an article aimed at parents who were struggling with what to do about their child’s persistent attachment to his pacifier (“How to Ditch the Pacifier,” by Anna Nowogrodski, New York Times, 2019 Sept. 16). For the most part, the author presented a sampling of sound advice from pediatricians and other health experts.
Most children will abandon their pacifiers at a time that is consistent with their developmental stage. Pacifiers seldom do any permanent damage, although they aren’t terribly appealing to look at when hanging out of a toddling toddler’s mouth. Parents were urged to be patient and consistent and were told that allowing the gooey thing to self-destruct often works, as does accelerating the process with a razor blade. Enlisting the aid of the Pacifier Fairy was suggested, but I’m not so sure that would work terribly well.
As I finished perusing the article, I couldn’t help think of how this vexing issue of pacifier removal can be avoided if parents follow a simple rule when they first introduced a pacifier to their child. If experienced parents think back to when they first resorted to using the pacifier, it wasn’t because the plastic and rubber gadget was a family heirloom that had been passed down from generation to generation like an engraved silver spoon. It wasn’t because the dentist told them that children who use pacifiers are less likely to need braces on their teeth. Nor was it a rumor filtered down from speech therapists that pacifiers improve articulation.
Parents reach for a pacifier in hopes that it will help their child will fall asleep. I think most parents of older children agree that at the beginning the pacifier was first and foremost a sleep aid. But here is where the critical oversight occurs: If you give your children pacifiers when you want them to go to sleep, why not simply add the stipulation of where you would like them to go to sleep as well?
Most parents prefer that their children sleep in their own space. We can argue of whether that should be in a side sleeper or their own crib, but most parents don’t want their 3-year-olds sleeping in their bed. Nor do they want their children sleeping on the couch in the living room with them while they watch a movie at 10:30 at night. And as pediatricians, we prefer that children not sleep with their necks flexed in a car seat or baby rocker, particularly if they’re a preemie.
Augmenting the primary association between sleep and the pacifier by adding a place has several important advantages. It gives parents more control of where their children will sleep or, more importantly, where they won’t be sleeping. It helps transitions to nonhome sleeping places like day care and long trips to grandma’s house go more smoothly.
Even more importantly, the crib/pacifier association helps parents who have had trouble reading their children’s cues. If they want a pacifier, it means they are tired and want to go to where the pacifier lives: bed
Finally, maintaining the link between sleeping and the pacifier promotes a more natural weaning process than going cold turkey or hiring the Pacifier Fairy. As naps disappear, the pacifier gradually become a less obvious accessory in the child’s life. However, it may linger in the background as a reminder of when the child needs some restorative sleep.
Of course, helping parents to think clearly enough to create and enforce a simple rule long enough to forge a healthy association when they are sleep deprived themselves is just another one of those challenges we must accept as concerned primary care pediatricians.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Is My Child Overtired? The Sleep Solution for Raising Happier, Healthier Children.” Email him at pdnews@mdedge.com.
I recently encountered an article aimed at parents who were struggling with what to do about their child’s persistent attachment to his pacifier (“How to Ditch the Pacifier,” by Anna Nowogrodski, New York Times, 2019 Sept. 16). For the most part, the author presented a sampling of sound advice from pediatricians and other health experts.
Most children will abandon their pacifiers at a time that is consistent with their developmental stage. Pacifiers seldom do any permanent damage, although they aren’t terribly appealing to look at when hanging out of a toddling toddler’s mouth. Parents were urged to be patient and consistent and were told that allowing the gooey thing to self-destruct often works, as does accelerating the process with a razor blade. Enlisting the aid of the Pacifier Fairy was suggested, but I’m not so sure that would work terribly well.
As I finished perusing the article, I couldn’t help think of how this vexing issue of pacifier removal can be avoided if parents follow a simple rule when they first introduced a pacifier to their child. If experienced parents think back to when they first resorted to using the pacifier, it wasn’t because the plastic and rubber gadget was a family heirloom that had been passed down from generation to generation like an engraved silver spoon. It wasn’t because the dentist told them that children who use pacifiers are less likely to need braces on their teeth. Nor was it a rumor filtered down from speech therapists that pacifiers improve articulation.
Parents reach for a pacifier in hopes that it will help their child will fall asleep. I think most parents of older children agree that at the beginning the pacifier was first and foremost a sleep aid. But here is where the critical oversight occurs: If you give your children pacifiers when you want them to go to sleep, why not simply add the stipulation of where you would like them to go to sleep as well?
Most parents prefer that their children sleep in their own space. We can argue of whether that should be in a side sleeper or their own crib, but most parents don’t want their 3-year-olds sleeping in their bed. Nor do they want their children sleeping on the couch in the living room with them while they watch a movie at 10:30 at night. And as pediatricians, we prefer that children not sleep with their necks flexed in a car seat or baby rocker, particularly if they’re a preemie.
Augmenting the primary association between sleep and the pacifier by adding a place has several important advantages. It gives parents more control of where their children will sleep or, more importantly, where they won’t be sleeping. It helps transitions to nonhome sleeping places like day care and long trips to grandma’s house go more smoothly.
Even more importantly, the crib/pacifier association helps parents who have had trouble reading their children’s cues. If they want a pacifier, it means they are tired and want to go to where the pacifier lives: bed
Finally, maintaining the link between sleeping and the pacifier promotes a more natural weaning process than going cold turkey or hiring the Pacifier Fairy. As naps disappear, the pacifier gradually become a less obvious accessory in the child’s life. However, it may linger in the background as a reminder of when the child needs some restorative sleep.
Of course, helping parents to think clearly enough to create and enforce a simple rule long enough to forge a healthy association when they are sleep deprived themselves is just another one of those challenges we must accept as concerned primary care pediatricians.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Is My Child Overtired? The Sleep Solution for Raising Happier, Healthier Children.” Email him at pdnews@mdedge.com.
Labeling of medication warnings
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.

The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
NSCLC: Predicting benefit from immunotherapy plus chemo
In this edition of “How I will treat my next patient,” I highlight three studies presented at the World Conference on Lung Cancer (WCLC 2019) regarding the use of tissue biomarkers to predict benefit when immune checkpoint inhibitors (ICIs) are combined with chemotherapy in the treatment of stage IV non–small cell lung cancer (NSCLC) patients.
TMB in nonsquamous NSCLC

Marina C. Garassino, MD, and colleagues examined tumor mutation burden (TMB) to predict benefit from pembrolizumab in the KEYNOTE-189 study. KEYNOTE-189 was a double-blind comparison of first-line chemotherapy plus either pembrolizumab or placebo in 616 patients with stage IV nonsquamous NSCLC who were randomized 2:1 to the treatment arms.
Overall, adding pembrolizumab to pemetrexed and platinum significantly improved overall survival (hazard ratio, 0.49), progression-free survival (HR, 0.52), and overall response rate (47.6% vs. 18.9%). Benefit was observed in all analyzed subgroups, including patients with programmed death-ligand 1 (PD-L1) of less than 1%, 1%-49%, and 50% or greater.
In 293 patients – less than 50% of the total participants in the trial – with evaluable TMB data, TMB as a continuous variable showed no significant association with overall survival, progression-free survival, or overall response rate. There was no cut point at which TMB predicted outcome from treatment.
Similarly, Corey Langer, MD, and colleagues presented an exploratory analysis of the randomized, phase 2 KEYNOTE-021 trial (open label, pembrolizumab plus chemotherapy in 70 stage IV nonsquamous NSCLC patients). There was no association between tissue TMB and overall survival, progression-free survival, or overall response rate. In patients with tissue TMB greater than 175 mutations/exome and less than 175 mutations/exome, the overall response rate was 71% and 61%, respectively.
Both presenters recommended that tissue TMB not yet be used in therapeutic decision making.
How these results influence clinical practice
TMB has been associated with response to ICIs, but there is little information regarding whether TMB predicts for response to chemotherapy, either given alone or with ICIs. Logistical issues have limited the clinical utility of TMB. There are a variety of methodologies to measure TMB and no consensus on the ideal cut point for defining benefit from ICI therapy.
While TMB remains a marker of interest, the two presentations at WCLC 2019 demonstrate that additional research is needed to define whether TMB needs to be combined with other markers in an algorithm or matrix to guide decision making or whether we should focus entirely on identifying better biomarkers of immunogenicity.
PD-L1 expression and overall survival
Federico Cappuzzo, MD, and colleagues reported a subset analysis of IMpower131, a randomized, phase 3 trial of chemotherapy plus or minus atezolizumab as first-line therapy in 1,021 patients with stage IV squamous NSCLC. Patients were randomized to arm A (atezolizumab plus carboplatin plus paclitaxel), arm B (atezolizumab plus carboplatin plus nab-paclitaxel) or arm C (carboplatin plus nab-paclitaxel). Investigator-assessed progression-free survival, reported at the 2018 annual meeting of the American Society of Clinical Oncology, showed a small (about 21 days), but statistically significant, improvement in median progression-free survival in arm B versus arm C. The progression-free survival benefit was seen in all PD-L1-positive subgroups. At WCLC 2019, he reported the final overall survival results of arms B versus C.
Median overall survival in the intent-to-treat population was 14.2 months with atezolizumab versus 13.5 months without it (HR, 0.88). Patients with high PD-L1 expression (14% and 13% of patients in the groups, respectively), experienced dramatic, clinically important improvement in overall survival with atezolizumab plus chemotherapy, compared with chemotherapy alone (median of 23.4 vs. 10.2 months; HR, 0.48).
In IMpower 131, PD-L1-high expression was defined as TC3 or IC3 – expression on greater than 50% of tumor cells or greater than 10% of immune cells. Patients were also categorized as PD-L1 positive (TC 1/2/3 or IC 1/2/3 – expression of PD-L1 on 1% or greater of tumor cells or immune cells) or PD-LI negative (TC 0 or IC 0 – expression on less than 1% of cells). The PD-L1-positive and negative subsets did not demonstrate improved overall survival with atezolizumab.
How these results influence clinical practice
As noted above, in NSCLC patients (regardless of histology), we need biomarkers that predict benefit from ICIs alone and additive benefit when ICIs are combined with other, potentially toxic therapies. In the subset analysis of IMpower 131, despite clinically relevant differences in overall survival for the “PD-L1-high” patients, the PD-L1-positive patients did not benefit, so PD-L1 tumor proportion score remains an imperfect biomarker.
To put this report in its proper context, it will be important to analyze the details of the final manuscript of IMpower 131, particularly the comparison of arms A plus B versus C and the proportion of arm C patients who ultimately received an ICI in the second- or later-line setting. In the meantime, clinicians will select their ICI and chemotherapy regimen of choice, utilizing PD-L1 expression as an “eyebrow raiser,” but not an exclusionary criteria – as they did prior to WCLC 2019.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I will treat my next patient,” I highlight three studies presented at the World Conference on Lung Cancer (WCLC 2019) regarding the use of tissue biomarkers to predict benefit when immune checkpoint inhibitors (ICIs) are combined with chemotherapy in the treatment of stage IV non–small cell lung cancer (NSCLC) patients.
TMB in nonsquamous NSCLC
Marina C. Garassino, MD, and colleagues examined tumor mutation burden (TMB) to predict benefit from pembrolizumab in the KEYNOTE-189 study. KEYNOTE-189 was a double-blind comparison of first-line chemotherapy plus either pembrolizumab or placebo in 616 patients with stage IV nonsquamous NSCLC who were randomized 2:1 to the treatment arms.
Overall, adding pembrolizumab to pemetrexed and platinum significantly improved overall survival (hazard ratio, 0.49), progression-free survival (HR, 0.52), and overall response rate (47.6% vs. 18.9%). Benefit was observed in all analyzed subgroups, including patients with programmed death-ligand 1 (PD-L1) of less than 1%, 1%-49%, and 50% or greater.
In 293 patients – less than 50% of the total participants in the trial – with evaluable TMB data, TMB as a continuous variable showed no significant association with overall survival, progression-free survival, or overall response rate. There was no cut point at which TMB predicted outcome from treatment.
Similarly, Corey Langer, MD, and colleagues presented an exploratory analysis of the randomized, phase 2 KEYNOTE-021 trial (open label, pembrolizumab plus chemotherapy in 70 stage IV nonsquamous NSCLC patients). There was no association between tissue TMB and overall survival, progression-free survival, or overall response rate. In patients with tissue TMB greater than 175 mutations/exome and less than 175 mutations/exome, the overall response rate was 71% and 61%, respectively.
Both presenters recommended that tissue TMB not yet be used in therapeutic decision making.
How these results influence clinical practice
TMB has been associated with response to ICIs, but there is little information regarding whether TMB predicts for response to chemotherapy, either given alone or with ICIs. Logistical issues have limited the clinical utility of TMB. There are a variety of methodologies to measure TMB and no consensus on the ideal cut point for defining benefit from ICI therapy.
While TMB remains a marker of interest, the two presentations at WCLC 2019 demonstrate that additional research is needed to define whether TMB needs to be combined with other markers in an algorithm or matrix to guide decision making or whether we should focus entirely on identifying better biomarkers of immunogenicity.
PD-L1 expression and overall survival
Federico Cappuzzo, MD, and colleagues reported a subset analysis of IMpower131, a randomized, phase 3 trial of chemotherapy plus or minus atezolizumab as first-line therapy in 1,021 patients with stage IV squamous NSCLC. Patients were randomized to arm A (atezolizumab plus carboplatin plus paclitaxel), arm B (atezolizumab plus carboplatin plus nab-paclitaxel) or arm C (carboplatin plus nab-paclitaxel). Investigator-assessed progression-free survival, reported at the 2018 annual meeting of the American Society of Clinical Oncology, showed a small (about 21 days), but statistically significant, improvement in median progression-free survival in arm B versus arm C. The progression-free survival benefit was seen in all PD-L1-positive subgroups. At WCLC 2019, he reported the final overall survival results of arms B versus C.
Median overall survival in the intent-to-treat population was 14.2 months with atezolizumab versus 13.5 months without it (HR, 0.88). Patients with high PD-L1 expression (14% and 13% of patients in the groups, respectively), experienced dramatic, clinically important improvement in overall survival with atezolizumab plus chemotherapy, compared with chemotherapy alone (median of 23.4 vs. 10.2 months; HR, 0.48).
In IMpower 131, PD-L1-high expression was defined as TC3 or IC3 – expression on greater than 50% of tumor cells or greater than 10% of immune cells. Patients were also categorized as PD-L1 positive (TC 1/2/3 or IC 1/2/3 – expression of PD-L1 on 1% or greater of tumor cells or immune cells) or PD-LI negative (TC 0 or IC 0 – expression on less than 1% of cells). The PD-L1-positive and negative subsets did not demonstrate improved overall survival with atezolizumab.
How these results influence clinical practice
As noted above, in NSCLC patients (regardless of histology), we need biomarkers that predict benefit from ICIs alone and additive benefit when ICIs are combined with other, potentially toxic therapies. In the subset analysis of IMpower 131, despite clinically relevant differences in overall survival for the “PD-L1-high” patients, the PD-L1-positive patients did not benefit, so PD-L1 tumor proportion score remains an imperfect biomarker.
To put this report in its proper context, it will be important to analyze the details of the final manuscript of IMpower 131, particularly the comparison of arms A plus B versus C and the proportion of arm C patients who ultimately received an ICI in the second- or later-line setting. In the meantime, clinicians will select their ICI and chemotherapy regimen of choice, utilizing PD-L1 expression as an “eyebrow raiser,” but not an exclusionary criteria – as they did prior to WCLC 2019.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I will treat my next patient,” I highlight three studies presented at the World Conference on Lung Cancer (WCLC 2019) regarding the use of tissue biomarkers to predict benefit when immune checkpoint inhibitors (ICIs) are combined with chemotherapy in the treatment of stage IV non–small cell lung cancer (NSCLC) patients.
TMB in nonsquamous NSCLC
Marina C. Garassino, MD, and colleagues examined tumor mutation burden (TMB) to predict benefit from pembrolizumab in the KEYNOTE-189 study. KEYNOTE-189 was a double-blind comparison of first-line chemotherapy plus either pembrolizumab or placebo in 616 patients with stage IV nonsquamous NSCLC who were randomized 2:1 to the treatment arms.
Overall, adding pembrolizumab to pemetrexed and platinum significantly improved overall survival (hazard ratio, 0.49), progression-free survival (HR, 0.52), and overall response rate (47.6% vs. 18.9%). Benefit was observed in all analyzed subgroups, including patients with programmed death-ligand 1 (PD-L1) of less than 1%, 1%-49%, and 50% or greater.
In 293 patients – less than 50% of the total participants in the trial – with evaluable TMB data, TMB as a continuous variable showed no significant association with overall survival, progression-free survival, or overall response rate. There was no cut point at which TMB predicted outcome from treatment.
Similarly, Corey Langer, MD, and colleagues presented an exploratory analysis of the randomized, phase 2 KEYNOTE-021 trial (open label, pembrolizumab plus chemotherapy in 70 stage IV nonsquamous NSCLC patients). There was no association between tissue TMB and overall survival, progression-free survival, or overall response rate. In patients with tissue TMB greater than 175 mutations/exome and less than 175 mutations/exome, the overall response rate was 71% and 61%, respectively.
Both presenters recommended that tissue TMB not yet be used in therapeutic decision making.
How these results influence clinical practice
TMB has been associated with response to ICIs, but there is little information regarding whether TMB predicts for response to chemotherapy, either given alone or with ICIs. Logistical issues have limited the clinical utility of TMB. There are a variety of methodologies to measure TMB and no consensus on the ideal cut point for defining benefit from ICI therapy.
While TMB remains a marker of interest, the two presentations at WCLC 2019 demonstrate that additional research is needed to define whether TMB needs to be combined with other markers in an algorithm or matrix to guide decision making or whether we should focus entirely on identifying better biomarkers of immunogenicity.
PD-L1 expression and overall survival
Federico Cappuzzo, MD, and colleagues reported a subset analysis of IMpower131, a randomized, phase 3 trial of chemotherapy plus or minus atezolizumab as first-line therapy in 1,021 patients with stage IV squamous NSCLC. Patients were randomized to arm A (atezolizumab plus carboplatin plus paclitaxel), arm B (atezolizumab plus carboplatin plus nab-paclitaxel) or arm C (carboplatin plus nab-paclitaxel). Investigator-assessed progression-free survival, reported at the 2018 annual meeting of the American Society of Clinical Oncology, showed a small (about 21 days), but statistically significant, improvement in median progression-free survival in arm B versus arm C. The progression-free survival benefit was seen in all PD-L1-positive subgroups. At WCLC 2019, he reported the final overall survival results of arms B versus C.
Median overall survival in the intent-to-treat population was 14.2 months with atezolizumab versus 13.5 months without it (HR, 0.88). Patients with high PD-L1 expression (14% and 13% of patients in the groups, respectively), experienced dramatic, clinically important improvement in overall survival with atezolizumab plus chemotherapy, compared with chemotherapy alone (median of 23.4 vs. 10.2 months; HR, 0.48).
In IMpower 131, PD-L1-high expression was defined as TC3 or IC3 – expression on greater than 50% of tumor cells or greater than 10% of immune cells. Patients were also categorized as PD-L1 positive (TC 1/2/3 or IC 1/2/3 – expression of PD-L1 on 1% or greater of tumor cells or immune cells) or PD-LI negative (TC 0 or IC 0 – expression on less than 1% of cells). The PD-L1-positive and negative subsets did not demonstrate improved overall survival with atezolizumab.
How these results influence clinical practice
As noted above, in NSCLC patients (regardless of histology), we need biomarkers that predict benefit from ICIs alone and additive benefit when ICIs are combined with other, potentially toxic therapies. In the subset analysis of IMpower 131, despite clinically relevant differences in overall survival for the “PD-L1-high” patients, the PD-L1-positive patients did not benefit, so PD-L1 tumor proportion score remains an imperfect biomarker.
To put this report in its proper context, it will be important to analyze the details of the final manuscript of IMpower 131, particularly the comparison of arms A plus B versus C and the proportion of arm C patients who ultimately received an ICI in the second- or later-line setting. In the meantime, clinicians will select their ICI and chemotherapy regimen of choice, utilizing PD-L1 expression as an “eyebrow raiser,” but not an exclusionary criteria – as they did prior to WCLC 2019.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
Private equity and independent gastroenterology practices – what do I need to know?
A few years ago, private equity (PE) firms began to focus on independent gastroenterology practices as a target for investment. The first PE investment transaction closed in March of 2016, and now an additional three such partnerships have occurred. Investment firms believe gastroenterology is ripe for investment and subsequent consolidation for the following reasons:
- Gastroenterology is a highly fragmented specialty with many small and mid-sized groups that could be rolled up into larger practice entities that create favorable scalability.
- There are multiple revenue streams through ancillary services that can be packaged into a comprehensive, high-quality gastroenterology practice that has high value for patients and that are delivered outside of a hospital environment.
- There is a growing need for gastroenterology care with increasing demand for chronic GI disease management (fatty liver disease, inflammatory bowel disease, and obesity management, for example) and increasing demand for colon cancer screening.
- Most independent gastroenterologists have natural entrepreneurial spirit.
- The current financial environment is favorable for investment and other sectors of the health care market are rapidly consolidating.
A PE transaction is not appropriate for every practice nor every physician. Further, not every physician group will be desirable for a PE firm. Nonetheless, the current business climate in the GI sector is generally favorable for accepting the PE capital model.
The following are 10 common questions dealing with a PE transaction:
1. What does a PE deal mean for the independent gastroenterologist? A PE transaction and the resulting formation of a managed services organization (MSO) will be a liquidity event for all current owners in the acquired practice. Financial benefits are typically substantial, especially when considering the funds can then be invested by the individual physician and often the money paid can be taxed as capital gains rather than ordinary income. In exchange for the pay-out, the physician group relinquishes managerial control of nonclinical decisions through a managed services agreement (MSA) with the MSO. The MSO is typically formed by the partnership between the practice and the PE firm and provides all nonclinical services to the physician group.
2. What autonomy will be left after signing a PE deal/MSA? Autonomy after the deal closes is determined largely by terms written into the contract prior to the closing and will differ among the various PE firms. There will be conditions important to the MSO and some important to the practice that can be codified in the contract. These conditions are spelled out in an employment agreement with the continuing physician group. Both the PE group and physicians will want to ensure that practice culture is not negatively impacted through an acquisition. Physicians must feel that they retain complete autonomy when it comes to clinical decisions, and the PE group must avoid interfering in the patient-doctor relationship. The PE group wants to improve nonclinical management of the practice, without interfering with the actual care of a patient. Physicians may influence nonclinical managerial decisions, but providers must understand that all nonclinical managerial decisions ultimately will be made by the MSO and PE firm.
3. What makes a good PE partnership? The asset that a PE firm is purchasing and hoping to grow is the revenue from a medical practice that they hope to improve by increasing profitability (through enhanced efficiency), expanding ancillary services and through multiple additional acquisitions to gain scale and size. Ensuring both sides are respected and aligned in decisions helps move the organization forward. A good partnership will build and bridge three types of capital – financial, experiential, and educational. Various factors must be considered; however, most important is mutual respect and admiration between the MSO and the physicians. Managerial styles will vary, but, a shared vision of the future will lead to success.
4. What changes are ahead with a PE deal? A PE firm and the MSO that it controls will put its management team in place to optimize revenue and contain expenses. The PE firm will look to combine practices where synergies exist and growth potential is strategically beneficial. For example, one practice might bring a pathology lab, the other geographic coverage, and the third an infusion center. Larger scale will usually improve negotiating influence with payers and hospitals as well as buying power for operational necessities. The MSO will roll out best practice protocols throughout the group, both back-office as well as patient-facing services. Finally, all PE groups will transition accounting to accrual from cash based as well as work with outside auditors and consultants due to the MSO’s bank covenants.
5. What is a platform company? A variation on the PE-based MSO is the formation of a “platform company”. This structure typically comes from a more sophisticated, mature practice that already has substantial business structures and managerial team members in place. This type of company can provide services not just to the founding practice, but to others that are “added on” as the organization grows. The investment hold period by the PE firm is typically 4-6 years, and thus, adding expertise to existing processes is usually more efficient and effective than starting from the ground up. Platform companies are typically paid a higher multiple than a company or practice that is “added on” to an existing platform, especially since these owners are taking the greatest risk by being the initial investor.
6. Explain the idiom “second bite of the apple”? A portion of each owner’s proceeds from the initial sale (“first bite of the apple”) of the practice is typically converted into stock of the MSO in a tax favorable method. The PE firm will maintain the largest shareholder position in the MSO (often majority), while the physicians and management team will be minority shareholders in the MSO. The proportion of proceeds rolled into stock depends on negotiations and ranges anywhere from 20% to 50% of the proceeds. The “second bite” is when the PE firm sells the stock of the MSO to the next investor. At the time of that transaction, all shareholders have a liquidity event and often another portion of the proceeds are rolled for the “next bite of the apple”. Specific terms of the shares are defined during negotiations, specifically the vesting terms, voting rights associated, and the value of each share.
7. Why would one practice receive a higher multiple compared to another practice? Each practice will have a different intrinsic value to the MSO and PE firm. The range of multiples on the purchased earnings before interest, depreciation, taxes, and amortization (EBIDTA) will depend on the timing of the transaction in the lifecycle of the investment as well as market forces. The number, age, and productivity of a practice’s providers, the ability to add certain ancillary services (i.e., revenue sources), the quality of contracts and associated payer mix, and the location of a practice are often the critical elements which the PE firm evaluates in the determination of a group’s value. The investment strategy will not be successful if exorbitant multiples are used for every practice. Strategically, a group with multiple providers in a desirable location with limited ancillary services early in the lifecycle will likely receive a higher multiple than a smaller group.
8. What outside professional assistance is needed to consummate a PE deal? Some groups may depend on an investment banker or health care mergers and acquisitions consultant to assist in the process or even seek out a partnership. Larger, more complicated groups with various existing relationships and competing forces often require such professional assistance. However, other smaller groups being approached by the MSO/PE firm as a “bolt-on” acquisition might not require a professional banker as the terms of joining may be more uniform to create a cohesive group of providers upon closing. All transactions, however, will require experienced health care transaction attorneys to ensure compliance with the myriad regulations. Some may engage a tax law attorney or accountant to ensure terms of the transaction are favorable. The PE firm will almost certainly require a quality of earnings evaluation by an outside, third-party financial auditor. One can probably assume close to 5% of proceeds may go to various professionals assisting in the process of the deal.
9. What are the common governance structures in PE transactions for physician provider service organizations? Like most businesses, a group of individuals typically form a board of directors which work in a decision making capacity and provide advice to the management team of the MSO. The board of directors usually includes successful leaders from other industries or business which bring specific talents, connections, and experiences, as well as individuals from the PE group and management team. Often, the platform practice will have a representative physician sit on the MSO board to ensure the medical provider perspective is prominent. The board of directors typically approves acquisitions and entry into new MSAs with additional practices, sets quarterly or yearly strategic goals, approves the budget and management team compensation structure and ultimately works on an exit strategy for the PE firm. Finally, pros and cons exist to having a physician as the CEO of the MSO; regardless, the CEO must be a strong leader with a vision and solid ability to communicate, as the PE sponsor and board of directors will have certain expectations, just as the independent gastroenterologist becoming a part of a new entity will have significant insecurities and hesitancies which must be appreciated and reassured.
10. In 3-5 years, what opportunities will a gastroenterologist leaving fellowship face as far as the GI landscape? Beyond the typical hospital-based employment opportunities or academic positions, consolidation of groups from PE acquisitions will likely have led to regional and maybe even national companies competing amongst themselves for talent. Over the coming years, there may be a total of 6-8 entities consisting of 15% of all gastroenterologists. Likely, one or two of the currently backed PE companies will have a new investor (i.e., initial exit completed/“second bite”). Each group will try to provide a differing value-based proposition beyond just the location a provider will be practicing. Fellows entering a practice already owned by a PE firm (or if a sale is pending) must clearly understand the legal, financial, and governance implications of these structures. This type of business structure is much different than one would encounter when hired by a physician-owned practice. It is not yet clear how a PE exit (4-6 years after acquisition) will play out for physicians not part of the original practice.
Dr. Sonenshine is a member of Atlanta Gastroenterology Associates.
A few years ago, private equity (PE) firms began to focus on independent gastroenterology practices as a target for investment. The first PE investment transaction closed in March of 2016, and now an additional three such partnerships have occurred. Investment firms believe gastroenterology is ripe for investment and subsequent consolidation for the following reasons:
- Gastroenterology is a highly fragmented specialty with many small and mid-sized groups that could be rolled up into larger practice entities that create favorable scalability.
- There are multiple revenue streams through ancillary services that can be packaged into a comprehensive, high-quality gastroenterology practice that has high value for patients and that are delivered outside of a hospital environment.
- There is a growing need for gastroenterology care with increasing demand for chronic GI disease management (fatty liver disease, inflammatory bowel disease, and obesity management, for example) and increasing demand for colon cancer screening.
- Most independent gastroenterologists have natural entrepreneurial spirit.
- The current financial environment is favorable for investment and other sectors of the health care market are rapidly consolidating.
A PE transaction is not appropriate for every practice nor every physician. Further, not every physician group will be desirable for a PE firm. Nonetheless, the current business climate in the GI sector is generally favorable for accepting the PE capital model.
The following are 10 common questions dealing with a PE transaction:
1. What does a PE deal mean for the independent gastroenterologist? A PE transaction and the resulting formation of a managed services organization (MSO) will be a liquidity event for all current owners in the acquired practice. Financial benefits are typically substantial, especially when considering the funds can then be invested by the individual physician and often the money paid can be taxed as capital gains rather than ordinary income. In exchange for the pay-out, the physician group relinquishes managerial control of nonclinical decisions through a managed services agreement (MSA) with the MSO. The MSO is typically formed by the partnership between the practice and the PE firm and provides all nonclinical services to the physician group.
2. What autonomy will be left after signing a PE deal/MSA? Autonomy after the deal closes is determined largely by terms written into the contract prior to the closing and will differ among the various PE firms. There will be conditions important to the MSO and some important to the practice that can be codified in the contract. These conditions are spelled out in an employment agreement with the continuing physician group. Both the PE group and physicians will want to ensure that practice culture is not negatively impacted through an acquisition. Physicians must feel that they retain complete autonomy when it comes to clinical decisions, and the PE group must avoid interfering in the patient-doctor relationship. The PE group wants to improve nonclinical management of the practice, without interfering with the actual care of a patient. Physicians may influence nonclinical managerial decisions, but providers must understand that all nonclinical managerial decisions ultimately will be made by the MSO and PE firm.
3. What makes a good PE partnership? The asset that a PE firm is purchasing and hoping to grow is the revenue from a medical practice that they hope to improve by increasing profitability (through enhanced efficiency), expanding ancillary services and through multiple additional acquisitions to gain scale and size. Ensuring both sides are respected and aligned in decisions helps move the organization forward. A good partnership will build and bridge three types of capital – financial, experiential, and educational. Various factors must be considered; however, most important is mutual respect and admiration between the MSO and the physicians. Managerial styles will vary, but, a shared vision of the future will lead to success.
4. What changes are ahead with a PE deal? A PE firm and the MSO that it controls will put its management team in place to optimize revenue and contain expenses. The PE firm will look to combine practices where synergies exist and growth potential is strategically beneficial. For example, one practice might bring a pathology lab, the other geographic coverage, and the third an infusion center. Larger scale will usually improve negotiating influence with payers and hospitals as well as buying power for operational necessities. The MSO will roll out best practice protocols throughout the group, both back-office as well as patient-facing services. Finally, all PE groups will transition accounting to accrual from cash based as well as work with outside auditors and consultants due to the MSO’s bank covenants.
5. What is a platform company? A variation on the PE-based MSO is the formation of a “platform company”. This structure typically comes from a more sophisticated, mature practice that already has substantial business structures and managerial team members in place. This type of company can provide services not just to the founding practice, but to others that are “added on” as the organization grows. The investment hold period by the PE firm is typically 4-6 years, and thus, adding expertise to existing processes is usually more efficient and effective than starting from the ground up. Platform companies are typically paid a higher multiple than a company or practice that is “added on” to an existing platform, especially since these owners are taking the greatest risk by being the initial investor.
6. Explain the idiom “second bite of the apple”? A portion of each owner’s proceeds from the initial sale (“first bite of the apple”) of the practice is typically converted into stock of the MSO in a tax favorable method. The PE firm will maintain the largest shareholder position in the MSO (often majority), while the physicians and management team will be minority shareholders in the MSO. The proportion of proceeds rolled into stock depends on negotiations and ranges anywhere from 20% to 50% of the proceeds. The “second bite” is when the PE firm sells the stock of the MSO to the next investor. At the time of that transaction, all shareholders have a liquidity event and often another portion of the proceeds are rolled for the “next bite of the apple”. Specific terms of the shares are defined during negotiations, specifically the vesting terms, voting rights associated, and the value of each share.
7. Why would one practice receive a higher multiple compared to another practice? Each practice will have a different intrinsic value to the MSO and PE firm. The range of multiples on the purchased earnings before interest, depreciation, taxes, and amortization (EBIDTA) will depend on the timing of the transaction in the lifecycle of the investment as well as market forces. The number, age, and productivity of a practice’s providers, the ability to add certain ancillary services (i.e., revenue sources), the quality of contracts and associated payer mix, and the location of a practice are often the critical elements which the PE firm evaluates in the determination of a group’s value. The investment strategy will not be successful if exorbitant multiples are used for every practice. Strategically, a group with multiple providers in a desirable location with limited ancillary services early in the lifecycle will likely receive a higher multiple than a smaller group.
8. What outside professional assistance is needed to consummate a PE deal? Some groups may depend on an investment banker or health care mergers and acquisitions consultant to assist in the process or even seek out a partnership. Larger, more complicated groups with various existing relationships and competing forces often require such professional assistance. However, other smaller groups being approached by the MSO/PE firm as a “bolt-on” acquisition might not require a professional banker as the terms of joining may be more uniform to create a cohesive group of providers upon closing. All transactions, however, will require experienced health care transaction attorneys to ensure compliance with the myriad regulations. Some may engage a tax law attorney or accountant to ensure terms of the transaction are favorable. The PE firm will almost certainly require a quality of earnings evaluation by an outside, third-party financial auditor. One can probably assume close to 5% of proceeds may go to various professionals assisting in the process of the deal.
9. What are the common governance structures in PE transactions for physician provider service organizations? Like most businesses, a group of individuals typically form a board of directors which work in a decision making capacity and provide advice to the management team of the MSO. The board of directors usually includes successful leaders from other industries or business which bring specific talents, connections, and experiences, as well as individuals from the PE group and management team. Often, the platform practice will have a representative physician sit on the MSO board to ensure the medical provider perspective is prominent. The board of directors typically approves acquisitions and entry into new MSAs with additional practices, sets quarterly or yearly strategic goals, approves the budget and management team compensation structure and ultimately works on an exit strategy for the PE firm. Finally, pros and cons exist to having a physician as the CEO of the MSO; regardless, the CEO must be a strong leader with a vision and solid ability to communicate, as the PE sponsor and board of directors will have certain expectations, just as the independent gastroenterologist becoming a part of a new entity will have significant insecurities and hesitancies which must be appreciated and reassured.
10. In 3-5 years, what opportunities will a gastroenterologist leaving fellowship face as far as the GI landscape? Beyond the typical hospital-based employment opportunities or academic positions, consolidation of groups from PE acquisitions will likely have led to regional and maybe even national companies competing amongst themselves for talent. Over the coming years, there may be a total of 6-8 entities consisting of 15% of all gastroenterologists. Likely, one or two of the currently backed PE companies will have a new investor (i.e., initial exit completed/“second bite”). Each group will try to provide a differing value-based proposition beyond just the location a provider will be practicing. Fellows entering a practice already owned by a PE firm (or if a sale is pending) must clearly understand the legal, financial, and governance implications of these structures. This type of business structure is much different than one would encounter when hired by a physician-owned practice. It is not yet clear how a PE exit (4-6 years after acquisition) will play out for physicians not part of the original practice.
Dr. Sonenshine is a member of Atlanta Gastroenterology Associates.
A few years ago, private equity (PE) firms began to focus on independent gastroenterology practices as a target for investment. The first PE investment transaction closed in March of 2016, and now an additional three such partnerships have occurred. Investment firms believe gastroenterology is ripe for investment and subsequent consolidation for the following reasons:
- Gastroenterology is a highly fragmented specialty with many small and mid-sized groups that could be rolled up into larger practice entities that create favorable scalability.
- There are multiple revenue streams through ancillary services that can be packaged into a comprehensive, high-quality gastroenterology practice that has high value for patients and that are delivered outside of a hospital environment.
- There is a growing need for gastroenterology care with increasing demand for chronic GI disease management (fatty liver disease, inflammatory bowel disease, and obesity management, for example) and increasing demand for colon cancer screening.
- Most independent gastroenterologists have natural entrepreneurial spirit.
- The current financial environment is favorable for investment and other sectors of the health care market are rapidly consolidating.
A PE transaction is not appropriate for every practice nor every physician. Further, not every physician group will be desirable for a PE firm. Nonetheless, the current business climate in the GI sector is generally favorable for accepting the PE capital model.
The following are 10 common questions dealing with a PE transaction:
1. What does a PE deal mean for the independent gastroenterologist? A PE transaction and the resulting formation of a managed services organization (MSO) will be a liquidity event for all current owners in the acquired practice. Financial benefits are typically substantial, especially when considering the funds can then be invested by the individual physician and often the money paid can be taxed as capital gains rather than ordinary income. In exchange for the pay-out, the physician group relinquishes managerial control of nonclinical decisions through a managed services agreement (MSA) with the MSO. The MSO is typically formed by the partnership between the practice and the PE firm and provides all nonclinical services to the physician group.
2. What autonomy will be left after signing a PE deal/MSA? Autonomy after the deal closes is determined largely by terms written into the contract prior to the closing and will differ among the various PE firms. There will be conditions important to the MSO and some important to the practice that can be codified in the contract. These conditions are spelled out in an employment agreement with the continuing physician group. Both the PE group and physicians will want to ensure that practice culture is not negatively impacted through an acquisition. Physicians must feel that they retain complete autonomy when it comes to clinical decisions, and the PE group must avoid interfering in the patient-doctor relationship. The PE group wants to improve nonclinical management of the practice, without interfering with the actual care of a patient. Physicians may influence nonclinical managerial decisions, but providers must understand that all nonclinical managerial decisions ultimately will be made by the MSO and PE firm.
3. What makes a good PE partnership? The asset that a PE firm is purchasing and hoping to grow is the revenue from a medical practice that they hope to improve by increasing profitability (through enhanced efficiency), expanding ancillary services and through multiple additional acquisitions to gain scale and size. Ensuring both sides are respected and aligned in decisions helps move the organization forward. A good partnership will build and bridge three types of capital – financial, experiential, and educational. Various factors must be considered; however, most important is mutual respect and admiration between the MSO and the physicians. Managerial styles will vary, but, a shared vision of the future will lead to success.
4. What changes are ahead with a PE deal? A PE firm and the MSO that it controls will put its management team in place to optimize revenue and contain expenses. The PE firm will look to combine practices where synergies exist and growth potential is strategically beneficial. For example, one practice might bring a pathology lab, the other geographic coverage, and the third an infusion center. Larger scale will usually improve negotiating influence with payers and hospitals as well as buying power for operational necessities. The MSO will roll out best practice protocols throughout the group, both back-office as well as patient-facing services. Finally, all PE groups will transition accounting to accrual from cash based as well as work with outside auditors and consultants due to the MSO’s bank covenants.
5. What is a platform company? A variation on the PE-based MSO is the formation of a “platform company”. This structure typically comes from a more sophisticated, mature practice that already has substantial business structures and managerial team members in place. This type of company can provide services not just to the founding practice, but to others that are “added on” as the organization grows. The investment hold period by the PE firm is typically 4-6 years, and thus, adding expertise to existing processes is usually more efficient and effective than starting from the ground up. Platform companies are typically paid a higher multiple than a company or practice that is “added on” to an existing platform, especially since these owners are taking the greatest risk by being the initial investor.
6. Explain the idiom “second bite of the apple”? A portion of each owner’s proceeds from the initial sale (“first bite of the apple”) of the practice is typically converted into stock of the MSO in a tax favorable method. The PE firm will maintain the largest shareholder position in the MSO (often majority), while the physicians and management team will be minority shareholders in the MSO. The proportion of proceeds rolled into stock depends on negotiations and ranges anywhere from 20% to 50% of the proceeds. The “second bite” is when the PE firm sells the stock of the MSO to the next investor. At the time of that transaction, all shareholders have a liquidity event and often another portion of the proceeds are rolled for the “next bite of the apple”. Specific terms of the shares are defined during negotiations, specifically the vesting terms, voting rights associated, and the value of each share.
7. Why would one practice receive a higher multiple compared to another practice? Each practice will have a different intrinsic value to the MSO and PE firm. The range of multiples on the purchased earnings before interest, depreciation, taxes, and amortization (EBIDTA) will depend on the timing of the transaction in the lifecycle of the investment as well as market forces. The number, age, and productivity of a practice’s providers, the ability to add certain ancillary services (i.e., revenue sources), the quality of contracts and associated payer mix, and the location of a practice are often the critical elements which the PE firm evaluates in the determination of a group’s value. The investment strategy will not be successful if exorbitant multiples are used for every practice. Strategically, a group with multiple providers in a desirable location with limited ancillary services early in the lifecycle will likely receive a higher multiple than a smaller group.
8. What outside professional assistance is needed to consummate a PE deal? Some groups may depend on an investment banker or health care mergers and acquisitions consultant to assist in the process or even seek out a partnership. Larger, more complicated groups with various existing relationships and competing forces often require such professional assistance. However, other smaller groups being approached by the MSO/PE firm as a “bolt-on” acquisition might not require a professional banker as the terms of joining may be more uniform to create a cohesive group of providers upon closing. All transactions, however, will require experienced health care transaction attorneys to ensure compliance with the myriad regulations. Some may engage a tax law attorney or accountant to ensure terms of the transaction are favorable. The PE firm will almost certainly require a quality of earnings evaluation by an outside, third-party financial auditor. One can probably assume close to 5% of proceeds may go to various professionals assisting in the process of the deal.
9. What are the common governance structures in PE transactions for physician provider service organizations? Like most businesses, a group of individuals typically form a board of directors which work in a decision making capacity and provide advice to the management team of the MSO. The board of directors usually includes successful leaders from other industries or business which bring specific talents, connections, and experiences, as well as individuals from the PE group and management team. Often, the platform practice will have a representative physician sit on the MSO board to ensure the medical provider perspective is prominent. The board of directors typically approves acquisitions and entry into new MSAs with additional practices, sets quarterly or yearly strategic goals, approves the budget and management team compensation structure and ultimately works on an exit strategy for the PE firm. Finally, pros and cons exist to having a physician as the CEO of the MSO; regardless, the CEO must be a strong leader with a vision and solid ability to communicate, as the PE sponsor and board of directors will have certain expectations, just as the independent gastroenterologist becoming a part of a new entity will have significant insecurities and hesitancies which must be appreciated and reassured.
10. In 3-5 years, what opportunities will a gastroenterologist leaving fellowship face as far as the GI landscape? Beyond the typical hospital-based employment opportunities or academic positions, consolidation of groups from PE acquisitions will likely have led to regional and maybe even national companies competing amongst themselves for talent. Over the coming years, there may be a total of 6-8 entities consisting of 15% of all gastroenterologists. Likely, one or two of the currently backed PE companies will have a new investor (i.e., initial exit completed/“second bite”). Each group will try to provide a differing value-based proposition beyond just the location a provider will be practicing. Fellows entering a practice already owned by a PE firm (or if a sale is pending) must clearly understand the legal, financial, and governance implications of these structures. This type of business structure is much different than one would encounter when hired by a physician-owned practice. It is not yet clear how a PE exit (4-6 years after acquisition) will play out for physicians not part of the original practice.
Dr. Sonenshine is a member of Atlanta Gastroenterology Associates.