User login
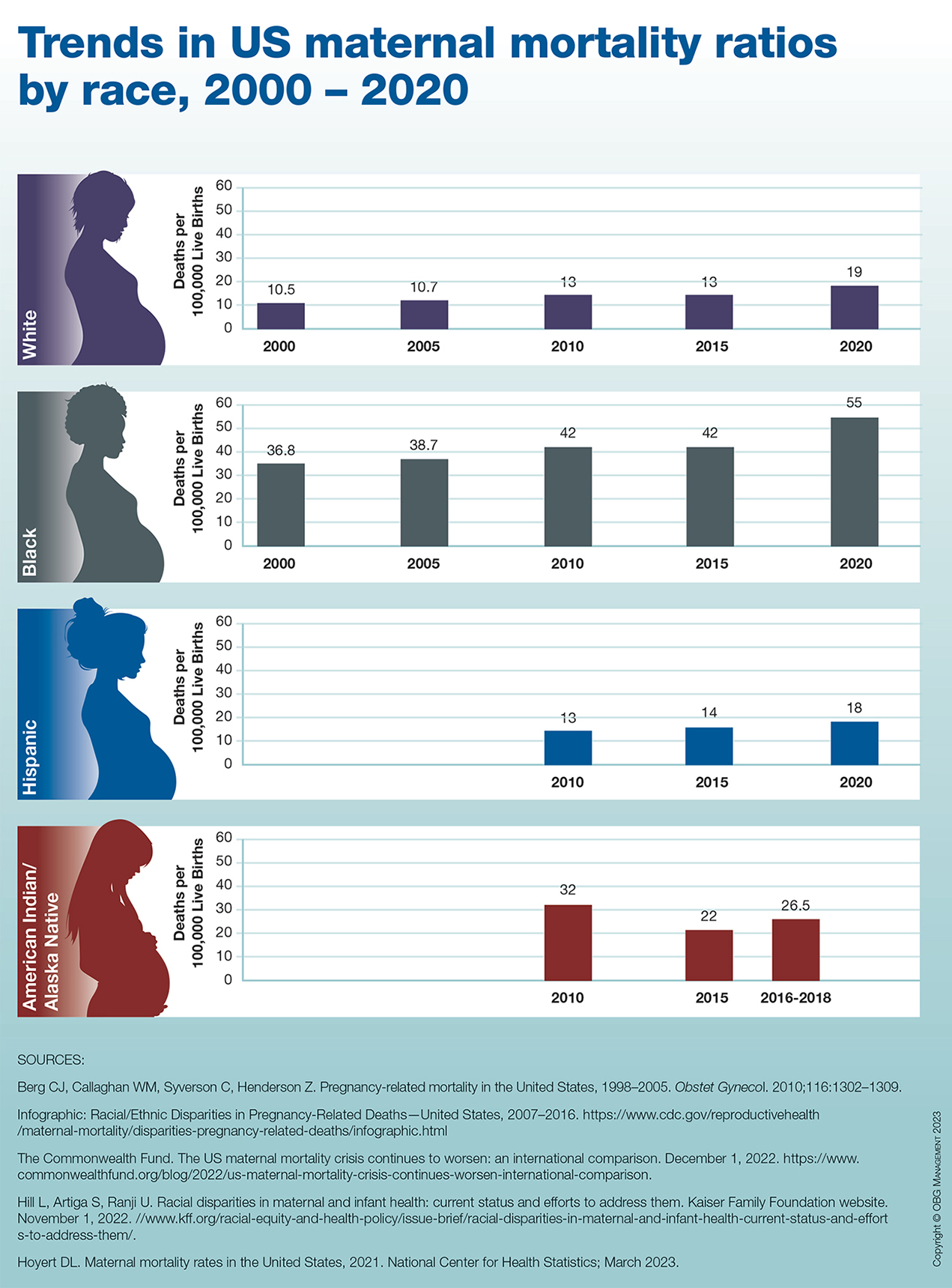
Trends in US maternal mortality ratios by race, 2000 – 2020
Therapeutic hypothermia to treat neonatal encephalopathy improves childhood outcomes
Therapeutic hypothermia (TH) for moderate and severe neonatal encephalopathy has been shown to reduce the risk of newborn death, major neurodevelopmental disability, developmental delay, and cerebral palsy.1 It is estimated that 8 newborns with moderate or severe neonatal encephalopathy need to be treated with TH to prevent 1 case of cerebral palsy.1 The key elements of TH include:
- initiate hypothermia within 6 hoursof birth
- cool the newborn to a core temperature of 33.5˚ C to 34.5˚ C (92.3˚ F to 94.1˚ F) for 72 hours
- obtain brain ultrasonography to assess for intracranial hemorrhage
- obtain sequential MRI studies to assess brain structure and function
- initiate EEG monitoring for seizure activity.
During hypothermia the newborn is sedated, and oral feedings are reduced. During TH, important physiological goals are to maintain normal oxygenation, blood pressure, fluid balance, and glucose levels.1,2
TH: The basics
Most of the major published randomized clinical trials used the following inclusion criteria to initiate TH2:
- gestational age at birth of ≥ 35 weeks
- neonate is within 6 hours of birth
- an Apgar score ≤ 5 at 10 minutes of life or prolonged resuscitation at birth or umbilical artery cord pH < 7.1 or neonatal blood gas within 60 minutes of life < 7.1
- moderate to severe encephalopathy or the presence of seizures
- absence of recognizable congenital abnormalities at birth.
However, in some institutions, expert neonatologists have developed more liberal criteria for the initiation of TH, to be considered on a case-by-case basis. These more inclusive criteria, which will result in more newborns being treated with TH, include3:
- gestational age at birth of ≥ 34 weeks
- neonate is within 12 hours of birth
- a sentinel event at birth or Apgar score ≤ 5 at 10 minutes of life or prolonged resuscitation or umbilical artery cord pH < 7.1 or neonatal blood gas within 60 minutes of life < 7.1 or postnatal cardiopulmonary failure
- moderate to severe encephalopathy or concern for the presence of seizures.
Birth at a gestational age ≤ 34 weeks is a contraindication to TH. Relative contraindications to initiation of TH include: birth weight < 1,750 g, severe congenital anomaly, major genetic disorders, known severe metabolic disorders, major intracranial hemorrhage, severe septicemia, and uncorrectable coagulopathy.3 Adverse outcomes of TH include thrombocytopenia, cardiac arrythmia, and fat necrosis.4
Diagnosing neonatal encephalopathy
Neonatal encephalopathy is a clinical diagnosis, defined as abnormal neurologic function in the first few days of life in an infant born at ≥ 35 weeks’ gestation. It is divided into 3 categories: mild (Stage 1), moderate (Stage 2), and severe (Stage 3).5,6 Institutions vary in the criteria used to differentiate mild from moderate neonatal encephalopathy, the two most frequent forms of encephalopathy. Newborns with mild encephalopathy are not routinely treated with TH because TH has not been shown to be helpful in this setting. Institutions with liberal criteria for diagnosing moderate encephalopathy will initiate TH in more cases. Involvement of a pediatric neurologist in the diagnosis of moderate encephalopathy may help confirm the diagnosis made by the primary neonatologist and provide an independent, second opinion about whether the newborn should be diagnosed with mild or moderate encephalopathy, a clinically important distinction. Physical examination and EEG findings associated with cases of mild, moderate, and severe encephalopathy are presented in TABLE 1.7

Continue: Obstetric factors that may be associated with neonatal encephalopathy...
Obstetric factors that may be associated with neonatal encephalopathy
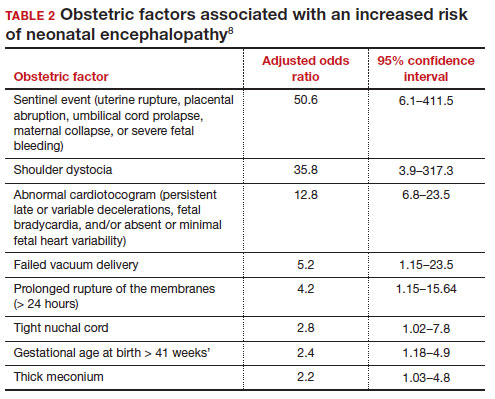
In a retrospective case-control study that included 405 newborns at ≥ 35 weeks’ gestational age with neonatal encephalopathy thought to be due to hypoxia, 8 obstetric factors were identified as being associated with an increased risk of neonatal encephalopathy, including (TABLE 2)8:
1. an obstetric sentinel event (uterine rupture, placental abruption, umbilical cord prolapse, maternal collapse, or severe fetal bleeding)
2. shoulder dystocia
3. abnormal cardiotocogram (persistent late or variable decelerations, fetal bradycardia, and/or absent or minimal fetal heart variability)
4. failed vacuum delivery
5. prolonged rupture of the membranes (> 24 hours)
6. tight nuchal cord
7. gestational age at birth > 41 weeks
8. thick meconium.
Similar findings have been reported by other investigators analyzing the obstetric risk factors for neonatal encephalopathy.7,9
Genetic causes of neonatal seizures and neonatal encephalopathy
Many neonatologists practice with the belief that for a newborn with encephalopathy in the setting of a sentinel labor event, a low Apgar score at 5 minutes, an umbilical cord artery pH < 7.00, and/or an elevated lactate level, the diagnosis of hypoxic ischemic encephalopathy is warranted. However, there are many causes of neonatal encephalopathy not related to intrapartum events. For example, neonatal encephalopathy and seizures may be caused by infectious, vascular, metabolic, medications, or congenital problems.10
There are genetic disorders that can be associated with both neonatal seizures and encephalopathy, suggesting that in some cases the primary cause of the encephalopathy is a genetic problem, not management of labor. Mutations in the potassium channel and sodium channel genes are well recognized causes of neonatal seizures.11,12 Cerebral palsy, a childhood outcome that may follow neonatal encephalopathy, also has numerous etiologies, including genetic causes. Among 1,345 children with cerebral palsy referred for exome sequencing, investigators reported that a genetic abnormality was identified in 33% of the cases.13 Mutations in 86 genes were identified in multiple children. Similar results have been reported in other cohorts.14-16 Maintaining an open mind about the causes of a case of neonatal encephalopathy and not jumping to a conclusion before completing an evaluation is an optimal approach.
Parent’s evolving emotional and intellectual reaction to the initiation of TH
Initiation of TH for a newborn with encephalopathy catalyzes parents to wonder, “How did my baby develop an encephalopathy?”, “Did my obstetrician’s management of labor and delivery contribute to the outcome?” and “What is the prognosis for my baby?” These are difficult questions with high emotional valence for both patients and clinicians. Obstetricians and neonatologists should collaborate to provide consistent responses to these questions.
The presence of a low umbilical cord artery pH and high lactate in combination with a low Apgar score at 5 minutes may lead the neonatologist to diagnose hypoxic-ischemic encephalopathy in the medical record. The diagnosis of brain hypoxia and ischemia in a newborn may be interpreted by parents as meaning that labor events caused or contributed to the encephalopathy. During the 72 hours of TH, the newborn is sedated and separated from the parents, causing additional emotional stress and uncertainty. When a baby is transferred from a community hospital to a neonatal intensive care unit (NICU) at a tertiary center, the parents may be geographically separated from their baby during a critical period of time, adding to their anxiety. At some point during the care process most newborns treated with TH will have an EEG, brain ultrasound, and brain magnetic resonance imaging (MRI). These data will be discussed with the parent(s) and may cause confusion and additional stress.
The optimal approach to communicating with parents whose newborn is treated with TH continues to evolve. Best practices may include17-20:
- in-person, regular multidisciplinary family meetings with the parents, including neonatologists, obstetricians, social service specialists and mental health experts when possible
- providing emotional support to parents, recognizing the psychological trauma of the clinical events
- encouraging parents to have physical contact with the newborn during TH
- elevating the role of the parents in the care process by having them participate in care events such as diapering the newborn
- ensuring that clinicians do not blame other clinicians for the clinical outcome
- communicating the results and interpretation of advanced physiological monitoring and imaging studies, with an emphasis on clarity, recognizing the limitations of the studies
- providing educational materials for parents about TH, early intervention programs, and support resources.
Coordinated and consistent communication with the parents is often difficult to facilitate due to many factors, including the unique perspectives and vocabularies of clinicians from different specialties and the difficulty of coordinating communications with all those involved over multiple shifts and sites of care. In terms of vocabulary, neonatologists are comfortable with making a diagnosis of hypoxic-ischemic encephalopathy in a newborn, but obstetricians would prefer that neonatologists use the more generic diagnosis of encephalopathy, holding judgment on the cause until additional data are available. In terms of coordinating communication over multiple shifts and sites of care, interactions between an obstetrician and their patient typically occurs in the postpartum unit, while interactions between neonatologists and parents occur in the NICU.
Parents of a baby with neonatal encephalopathy undergoing TH may have numerous traumatic experiences during the care process. For weeks or months after birth, they may recall or dream about the absence of sounds from their newborn at birth, the resuscitation events including chest compressions and intubation, the shivering of the baby during TH, and the jarring pivot from the expectation of holding and bonding with a healthy newborn to the reality of a sick newborn requiring intensive care. Obstetricians are also traumatized by these events and support from peers and mental health experts may help them recognize, explore, and adapt to the trauma. Neonatologists believe that TH can help improve the childhood outcomes of newborns with encephalopathy, a goal endorsed by all clinicians and family members. ●
- Jacobs SE, Berg M, Hunt R, et al. Cooling for newborns with hypoxic ischemic encephalopathy. Cochrane Database Syst Rev. 2013;CD003311.
- Committee on Fetus and Newborn; Papile E, Baley JE, Benitz W, et al. Hypothermia and neonatal encephalopathy. Pediatrics. 2014;133:1146-1150.
- Academic Medical Center Patient Safety Organization. Therapeutic hypothermia in neonates. Recommendations of the neonatal encephalopathy task force. 2016. https://www.rmf.harvard. edu/-/media/Files/_Global/KC/PDFs/Guide lines/crico_neonates.pdf. Accessed May 25, 2023.
- Zhang W, Ma J, Danzeng Q, et al. Safety of moderate hypothermia for perinatal hypoxic-ischemic encephalopathy: a meta-analysis. Pediatr Neurol. 2017;74:51-61.
- Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress: a clinical and electroencephalographic study. Arch Neurol. 1976;33:696-705.
- Thompson CM, Puterman AS, Linley LL, et al. The value of a scoring system for hypoxic ischemic encephalopathy in predicting neurodevelopmental outcome. Acta Pediatr. 1997;86:757-761.
- Lundgren C, Brudin L, Wanby AS, et al. Ante- and intrapartum risk factors for neonatal hypoxic ischemic encephalopathy. J Matern Fetal Neonatal Med. 2018;31:1595-1601.
- Martinez-Biarge M, Diez-Sebastian J, Wusthoff CJ, et al. Antepartum and intrapartum factors preceding neonatal hypoxic-ischemic encephalopathy. Pediatrics. 2013;132:e952-e959.
- Lorain P, Bower A, Gottardi E, et al. Risk factors for hypoxic-ischemic encephalopathy in cases of severe acidosis: a case-control study. Acta Obstet Gynecol Scand. 2022;101:471-478.
- Russ JB, Simmons R, Glass HC. Neonatal encephalopathy: beyond hypoxic-ischemic encephalopathy. Neo Reviews. 2021;22:e148-e162.
- Allen NM, Mannion M, Conroy J, et al. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. 2014;55:e99-e105.
- Zibro J, Shellhaas RA. Neonatal seizures: diagnosis, etiologies and management. Semin Neurol. 2020;40:246-256.
- Moreno-De-Luca A, Millan F, Peacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325:467-475.
- Srivastava S, Lewis SA, Cohen JS, et al. Molecular diagnostic yield of exome sequencing and chromosomal microarray in cerebral palsy. A systematic review and meta-analysis. JAMA Neurology. 2022;79:1287-1295.
- Gonzalez-Mantilla PJ, Hu Y, Myers SM, et al. Diagnostic yield of exome sequencing in cerebral palsy and implications for genetic testing guidelines. A systematic review and meta-analysis. JAMA Pediatr. Epub March 6, 2023.
- van Eyk C, MacLennon SC, MacLennan AH. All patients with cerebral palsy diagnosis merit genomic sequencing. JAMA Pediatr. Epub March 6, 2023.
- Craig AK, James C, Bainter J, et al. Parental perceptions of neonatal therapeutic hypothermia; emotional and healing experiences. J Matern Fetal Neonatal Med. 2020;33:2889-2896. doi: 10.1080/14767058.2018.1563592.
- Sagaser A, Pilon B, Goeller A, et al. Parent experience of hypoxic-ischemic encephalopathy and hypothermia: a call for trauma informed care. Am J Perinatol. Epub March 4, 2022.
- Cascio A, Ferrand A, Racine E, et al. Discussing brain magnetic resonance imaging results for neonates with hypoxic-ischemic encephalopathy treated with hypothermia: a challenge for clinicians and parents. E Neurological Sci. 2022;29:100424.
- Thyagarajan B, Baral V, Gunda R, et al. Parental perceptions of hypothermia treatment for neonatal hypoxic-ischaemic encephalopathy. J Matern Fetal Neonatal Med. 2018;31:2527-2533.

Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
The author reports no conflict of interest related to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
The author reports no conflict of interest related to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
The author reports no conflict of interest related to this article.
Therapeutic hypothermia (TH) for moderate and severe neonatal encephalopathy has been shown to reduce the risk of newborn death, major neurodevelopmental disability, developmental delay, and cerebral palsy.1 It is estimated that 8 newborns with moderate or severe neonatal encephalopathy need to be treated with TH to prevent 1 case of cerebral palsy.1 The key elements of TH include:
- initiate hypothermia within 6 hoursof birth
- cool the newborn to a core temperature of 33.5˚ C to 34.5˚ C (92.3˚ F to 94.1˚ F) for 72 hours
- obtain brain ultrasonography to assess for intracranial hemorrhage
- obtain sequential MRI studies to assess brain structure and function
- initiate EEG monitoring for seizure activity.
During hypothermia the newborn is sedated, and oral feedings are reduced. During TH, important physiological goals are to maintain normal oxygenation, blood pressure, fluid balance, and glucose levels.1,2
TH: The basics
Most of the major published randomized clinical trials used the following inclusion criteria to initiate TH2:
- gestational age at birth of ≥ 35 weeks
- neonate is within 6 hours of birth
- an Apgar score ≤ 5 at 10 minutes of life or prolonged resuscitation at birth or umbilical artery cord pH < 7.1 or neonatal blood gas within 60 minutes of life < 7.1
- moderate to severe encephalopathy or the presence of seizures
- absence of recognizable congenital abnormalities at birth.
However, in some institutions, expert neonatologists have developed more liberal criteria for the initiation of TH, to be considered on a case-by-case basis. These more inclusive criteria, which will result in more newborns being treated with TH, include3:
- gestational age at birth of ≥ 34 weeks
- neonate is within 12 hours of birth
- a sentinel event at birth or Apgar score ≤ 5 at 10 minutes of life or prolonged resuscitation or umbilical artery cord pH < 7.1 or neonatal blood gas within 60 minutes of life < 7.1 or postnatal cardiopulmonary failure
- moderate to severe encephalopathy or concern for the presence of seizures.
Birth at a gestational age ≤ 34 weeks is a contraindication to TH. Relative contraindications to initiation of TH include: birth weight < 1,750 g, severe congenital anomaly, major genetic disorders, known severe metabolic disorders, major intracranial hemorrhage, severe septicemia, and uncorrectable coagulopathy.3 Adverse outcomes of TH include thrombocytopenia, cardiac arrythmia, and fat necrosis.4
Diagnosing neonatal encephalopathy
Neonatal encephalopathy is a clinical diagnosis, defined as abnormal neurologic function in the first few days of life in an infant born at ≥ 35 weeks’ gestation. It is divided into 3 categories: mild (Stage 1), moderate (Stage 2), and severe (Stage 3).5,6 Institutions vary in the criteria used to differentiate mild from moderate neonatal encephalopathy, the two most frequent forms of encephalopathy. Newborns with mild encephalopathy are not routinely treated with TH because TH has not been shown to be helpful in this setting. Institutions with liberal criteria for diagnosing moderate encephalopathy will initiate TH in more cases. Involvement of a pediatric neurologist in the diagnosis of moderate encephalopathy may help confirm the diagnosis made by the primary neonatologist and provide an independent, second opinion about whether the newborn should be diagnosed with mild or moderate encephalopathy, a clinically important distinction. Physical examination and EEG findings associated with cases of mild, moderate, and severe encephalopathy are presented in TABLE 1.7
Continue: Obstetric factors that may be associated with neonatal encephalopathy...
Obstetric factors that may be associated with neonatal encephalopathy
In a retrospective case-control study that included 405 newborns at ≥ 35 weeks’ gestational age with neonatal encephalopathy thought to be due to hypoxia, 8 obstetric factors were identified as being associated with an increased risk of neonatal encephalopathy, including (TABLE 2)8:
1. an obstetric sentinel event (uterine rupture, placental abruption, umbilical cord prolapse, maternal collapse, or severe fetal bleeding)
2. shoulder dystocia
3. abnormal cardiotocogram (persistent late or variable decelerations, fetal bradycardia, and/or absent or minimal fetal heart variability)
4. failed vacuum delivery
5. prolonged rupture of the membranes (> 24 hours)
6. tight nuchal cord
7. gestational age at birth > 41 weeks
8. thick meconium.
Similar findings have been reported by other investigators analyzing the obstetric risk factors for neonatal encephalopathy.7,9
Genetic causes of neonatal seizures and neonatal encephalopathy
Many neonatologists practice with the belief that for a newborn with encephalopathy in the setting of a sentinel labor event, a low Apgar score at 5 minutes, an umbilical cord artery pH < 7.00, and/or an elevated lactate level, the diagnosis of hypoxic ischemic encephalopathy is warranted. However, there are many causes of neonatal encephalopathy not related to intrapartum events. For example, neonatal encephalopathy and seizures may be caused by infectious, vascular, metabolic, medications, or congenital problems.10
There are genetic disorders that can be associated with both neonatal seizures and encephalopathy, suggesting that in some cases the primary cause of the encephalopathy is a genetic problem, not management of labor. Mutations in the potassium channel and sodium channel genes are well recognized causes of neonatal seizures.11,12 Cerebral palsy, a childhood outcome that may follow neonatal encephalopathy, also has numerous etiologies, including genetic causes. Among 1,345 children with cerebral palsy referred for exome sequencing, investigators reported that a genetic abnormality was identified in 33% of the cases.13 Mutations in 86 genes were identified in multiple children. Similar results have been reported in other cohorts.14-16 Maintaining an open mind about the causes of a case of neonatal encephalopathy and not jumping to a conclusion before completing an evaluation is an optimal approach.
Parent’s evolving emotional and intellectual reaction to the initiation of TH
Initiation of TH for a newborn with encephalopathy catalyzes parents to wonder, “How did my baby develop an encephalopathy?”, “Did my obstetrician’s management of labor and delivery contribute to the outcome?” and “What is the prognosis for my baby?” These are difficult questions with high emotional valence for both patients and clinicians. Obstetricians and neonatologists should collaborate to provide consistent responses to these questions.
The presence of a low umbilical cord artery pH and high lactate in combination with a low Apgar score at 5 minutes may lead the neonatologist to diagnose hypoxic-ischemic encephalopathy in the medical record. The diagnosis of brain hypoxia and ischemia in a newborn may be interpreted by parents as meaning that labor events caused or contributed to the encephalopathy. During the 72 hours of TH, the newborn is sedated and separated from the parents, causing additional emotional stress and uncertainty. When a baby is transferred from a community hospital to a neonatal intensive care unit (NICU) at a tertiary center, the parents may be geographically separated from their baby during a critical period of time, adding to their anxiety. At some point during the care process most newborns treated with TH will have an EEG, brain ultrasound, and brain magnetic resonance imaging (MRI). These data will be discussed with the parent(s) and may cause confusion and additional stress.
The optimal approach to communicating with parents whose newborn is treated with TH continues to evolve. Best practices may include17-20:
- in-person, regular multidisciplinary family meetings with the parents, including neonatologists, obstetricians, social service specialists and mental health experts when possible
- providing emotional support to parents, recognizing the psychological trauma of the clinical events
- encouraging parents to have physical contact with the newborn during TH
- elevating the role of the parents in the care process by having them participate in care events such as diapering the newborn
- ensuring that clinicians do not blame other clinicians for the clinical outcome
- communicating the results and interpretation of advanced physiological monitoring and imaging studies, with an emphasis on clarity, recognizing the limitations of the studies
- providing educational materials for parents about TH, early intervention programs, and support resources.
Coordinated and consistent communication with the parents is often difficult to facilitate due to many factors, including the unique perspectives and vocabularies of clinicians from different specialties and the difficulty of coordinating communications with all those involved over multiple shifts and sites of care. In terms of vocabulary, neonatologists are comfortable with making a diagnosis of hypoxic-ischemic encephalopathy in a newborn, but obstetricians would prefer that neonatologists use the more generic diagnosis of encephalopathy, holding judgment on the cause until additional data are available. In terms of coordinating communication over multiple shifts and sites of care, interactions between an obstetrician and their patient typically occurs in the postpartum unit, while interactions between neonatologists and parents occur in the NICU.
Parents of a baby with neonatal encephalopathy undergoing TH may have numerous traumatic experiences during the care process. For weeks or months after birth, they may recall or dream about the absence of sounds from their newborn at birth, the resuscitation events including chest compressions and intubation, the shivering of the baby during TH, and the jarring pivot from the expectation of holding and bonding with a healthy newborn to the reality of a sick newborn requiring intensive care. Obstetricians are also traumatized by these events and support from peers and mental health experts may help them recognize, explore, and adapt to the trauma. Neonatologists believe that TH can help improve the childhood outcomes of newborns with encephalopathy, a goal endorsed by all clinicians and family members. ●
Therapeutic hypothermia (TH) for moderate and severe neonatal encephalopathy has been shown to reduce the risk of newborn death, major neurodevelopmental disability, developmental delay, and cerebral palsy.1 It is estimated that 8 newborns with moderate or severe neonatal encephalopathy need to be treated with TH to prevent 1 case of cerebral palsy.1 The key elements of TH include:
- initiate hypothermia within 6 hoursof birth
- cool the newborn to a core temperature of 33.5˚ C to 34.5˚ C (92.3˚ F to 94.1˚ F) for 72 hours
- obtain brain ultrasonography to assess for intracranial hemorrhage
- obtain sequential MRI studies to assess brain structure and function
- initiate EEG monitoring for seizure activity.
During hypothermia the newborn is sedated, and oral feedings are reduced. During TH, important physiological goals are to maintain normal oxygenation, blood pressure, fluid balance, and glucose levels.1,2
TH: The basics
Most of the major published randomized clinical trials used the following inclusion criteria to initiate TH2:
- gestational age at birth of ≥ 35 weeks
- neonate is within 6 hours of birth
- an Apgar score ≤ 5 at 10 minutes of life or prolonged resuscitation at birth or umbilical artery cord pH < 7.1 or neonatal blood gas within 60 minutes of life < 7.1
- moderate to severe encephalopathy or the presence of seizures
- absence of recognizable congenital abnormalities at birth.
However, in some institutions, expert neonatologists have developed more liberal criteria for the initiation of TH, to be considered on a case-by-case basis. These more inclusive criteria, which will result in more newborns being treated with TH, include3:
- gestational age at birth of ≥ 34 weeks
- neonate is within 12 hours of birth
- a sentinel event at birth or Apgar score ≤ 5 at 10 minutes of life or prolonged resuscitation or umbilical artery cord pH < 7.1 or neonatal blood gas within 60 minutes of life < 7.1 or postnatal cardiopulmonary failure
- moderate to severe encephalopathy or concern for the presence of seizures.
Birth at a gestational age ≤ 34 weeks is a contraindication to TH. Relative contraindications to initiation of TH include: birth weight < 1,750 g, severe congenital anomaly, major genetic disorders, known severe metabolic disorders, major intracranial hemorrhage, severe septicemia, and uncorrectable coagulopathy.3 Adverse outcomes of TH include thrombocytopenia, cardiac arrythmia, and fat necrosis.4
Diagnosing neonatal encephalopathy
Neonatal encephalopathy is a clinical diagnosis, defined as abnormal neurologic function in the first few days of life in an infant born at ≥ 35 weeks’ gestation. It is divided into 3 categories: mild (Stage 1), moderate (Stage 2), and severe (Stage 3).5,6 Institutions vary in the criteria used to differentiate mild from moderate neonatal encephalopathy, the two most frequent forms of encephalopathy. Newborns with mild encephalopathy are not routinely treated with TH because TH has not been shown to be helpful in this setting. Institutions with liberal criteria for diagnosing moderate encephalopathy will initiate TH in more cases. Involvement of a pediatric neurologist in the diagnosis of moderate encephalopathy may help confirm the diagnosis made by the primary neonatologist and provide an independent, second opinion about whether the newborn should be diagnosed with mild or moderate encephalopathy, a clinically important distinction. Physical examination and EEG findings associated with cases of mild, moderate, and severe encephalopathy are presented in TABLE 1.7
Continue: Obstetric factors that may be associated with neonatal encephalopathy...
Obstetric factors that may be associated with neonatal encephalopathy
In a retrospective case-control study that included 405 newborns at ≥ 35 weeks’ gestational age with neonatal encephalopathy thought to be due to hypoxia, 8 obstetric factors were identified as being associated with an increased risk of neonatal encephalopathy, including (TABLE 2)8:
1. an obstetric sentinel event (uterine rupture, placental abruption, umbilical cord prolapse, maternal collapse, or severe fetal bleeding)
2. shoulder dystocia
3. abnormal cardiotocogram (persistent late or variable decelerations, fetal bradycardia, and/or absent or minimal fetal heart variability)
4. failed vacuum delivery
5. prolonged rupture of the membranes (> 24 hours)
6. tight nuchal cord
7. gestational age at birth > 41 weeks
8. thick meconium.
Similar findings have been reported by other investigators analyzing the obstetric risk factors for neonatal encephalopathy.7,9
Genetic causes of neonatal seizures and neonatal encephalopathy
Many neonatologists practice with the belief that for a newborn with encephalopathy in the setting of a sentinel labor event, a low Apgar score at 5 minutes, an umbilical cord artery pH < 7.00, and/or an elevated lactate level, the diagnosis of hypoxic ischemic encephalopathy is warranted. However, there are many causes of neonatal encephalopathy not related to intrapartum events. For example, neonatal encephalopathy and seizures may be caused by infectious, vascular, metabolic, medications, or congenital problems.10
There are genetic disorders that can be associated with both neonatal seizures and encephalopathy, suggesting that in some cases the primary cause of the encephalopathy is a genetic problem, not management of labor. Mutations in the potassium channel and sodium channel genes are well recognized causes of neonatal seizures.11,12 Cerebral palsy, a childhood outcome that may follow neonatal encephalopathy, also has numerous etiologies, including genetic causes. Among 1,345 children with cerebral palsy referred for exome sequencing, investigators reported that a genetic abnormality was identified in 33% of the cases.13 Mutations in 86 genes were identified in multiple children. Similar results have been reported in other cohorts.14-16 Maintaining an open mind about the causes of a case of neonatal encephalopathy and not jumping to a conclusion before completing an evaluation is an optimal approach.
Parent’s evolving emotional and intellectual reaction to the initiation of TH
Initiation of TH for a newborn with encephalopathy catalyzes parents to wonder, “How did my baby develop an encephalopathy?”, “Did my obstetrician’s management of labor and delivery contribute to the outcome?” and “What is the prognosis for my baby?” These are difficult questions with high emotional valence for both patients and clinicians. Obstetricians and neonatologists should collaborate to provide consistent responses to these questions.
The presence of a low umbilical cord artery pH and high lactate in combination with a low Apgar score at 5 minutes may lead the neonatologist to diagnose hypoxic-ischemic encephalopathy in the medical record. The diagnosis of brain hypoxia and ischemia in a newborn may be interpreted by parents as meaning that labor events caused or contributed to the encephalopathy. During the 72 hours of TH, the newborn is sedated and separated from the parents, causing additional emotional stress and uncertainty. When a baby is transferred from a community hospital to a neonatal intensive care unit (NICU) at a tertiary center, the parents may be geographically separated from their baby during a critical period of time, adding to their anxiety. At some point during the care process most newborns treated with TH will have an EEG, brain ultrasound, and brain magnetic resonance imaging (MRI). These data will be discussed with the parent(s) and may cause confusion and additional stress.
The optimal approach to communicating with parents whose newborn is treated with TH continues to evolve. Best practices may include17-20:
- in-person, regular multidisciplinary family meetings with the parents, including neonatologists, obstetricians, social service specialists and mental health experts when possible
- providing emotional support to parents, recognizing the psychological trauma of the clinical events
- encouraging parents to have physical contact with the newborn during TH
- elevating the role of the parents in the care process by having them participate in care events such as diapering the newborn
- ensuring that clinicians do not blame other clinicians for the clinical outcome
- communicating the results and interpretation of advanced physiological monitoring and imaging studies, with an emphasis on clarity, recognizing the limitations of the studies
- providing educational materials for parents about TH, early intervention programs, and support resources.
Coordinated and consistent communication with the parents is often difficult to facilitate due to many factors, including the unique perspectives and vocabularies of clinicians from different specialties and the difficulty of coordinating communications with all those involved over multiple shifts and sites of care. In terms of vocabulary, neonatologists are comfortable with making a diagnosis of hypoxic-ischemic encephalopathy in a newborn, but obstetricians would prefer that neonatologists use the more generic diagnosis of encephalopathy, holding judgment on the cause until additional data are available. In terms of coordinating communication over multiple shifts and sites of care, interactions between an obstetrician and their patient typically occurs in the postpartum unit, while interactions between neonatologists and parents occur in the NICU.
Parents of a baby with neonatal encephalopathy undergoing TH may have numerous traumatic experiences during the care process. For weeks or months after birth, they may recall or dream about the absence of sounds from their newborn at birth, the resuscitation events including chest compressions and intubation, the shivering of the baby during TH, and the jarring pivot from the expectation of holding and bonding with a healthy newborn to the reality of a sick newborn requiring intensive care. Obstetricians are also traumatized by these events and support from peers and mental health experts may help them recognize, explore, and adapt to the trauma. Neonatologists believe that TH can help improve the childhood outcomes of newborns with encephalopathy, a goal endorsed by all clinicians and family members. ●
- Jacobs SE, Berg M, Hunt R, et al. Cooling for newborns with hypoxic ischemic encephalopathy. Cochrane Database Syst Rev. 2013;CD003311.
- Committee on Fetus and Newborn; Papile E, Baley JE, Benitz W, et al. Hypothermia and neonatal encephalopathy. Pediatrics. 2014;133:1146-1150.
- Academic Medical Center Patient Safety Organization. Therapeutic hypothermia in neonates. Recommendations of the neonatal encephalopathy task force. 2016. https://www.rmf.harvard. edu/-/media/Files/_Global/KC/PDFs/Guide lines/crico_neonates.pdf. Accessed May 25, 2023.
- Zhang W, Ma J, Danzeng Q, et al. Safety of moderate hypothermia for perinatal hypoxic-ischemic encephalopathy: a meta-analysis. Pediatr Neurol. 2017;74:51-61.
- Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress: a clinical and electroencephalographic study. Arch Neurol. 1976;33:696-705.
- Thompson CM, Puterman AS, Linley LL, et al. The value of a scoring system for hypoxic ischemic encephalopathy in predicting neurodevelopmental outcome. Acta Pediatr. 1997;86:757-761.
- Lundgren C, Brudin L, Wanby AS, et al. Ante- and intrapartum risk factors for neonatal hypoxic ischemic encephalopathy. J Matern Fetal Neonatal Med. 2018;31:1595-1601.
- Martinez-Biarge M, Diez-Sebastian J, Wusthoff CJ, et al. Antepartum and intrapartum factors preceding neonatal hypoxic-ischemic encephalopathy. Pediatrics. 2013;132:e952-e959.
- Lorain P, Bower A, Gottardi E, et al. Risk factors for hypoxic-ischemic encephalopathy in cases of severe acidosis: a case-control study. Acta Obstet Gynecol Scand. 2022;101:471-478.
- Russ JB, Simmons R, Glass HC. Neonatal encephalopathy: beyond hypoxic-ischemic encephalopathy. Neo Reviews. 2021;22:e148-e162.
- Allen NM, Mannion M, Conroy J, et al. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. 2014;55:e99-e105.
- Zibro J, Shellhaas RA. Neonatal seizures: diagnosis, etiologies and management. Semin Neurol. 2020;40:246-256.
- Moreno-De-Luca A, Millan F, Peacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325:467-475.
- Srivastava S, Lewis SA, Cohen JS, et al. Molecular diagnostic yield of exome sequencing and chromosomal microarray in cerebral palsy. A systematic review and meta-analysis. JAMA Neurology. 2022;79:1287-1295.
- Gonzalez-Mantilla PJ, Hu Y, Myers SM, et al. Diagnostic yield of exome sequencing in cerebral palsy and implications for genetic testing guidelines. A systematic review and meta-analysis. JAMA Pediatr. Epub March 6, 2023.
- van Eyk C, MacLennon SC, MacLennan AH. All patients with cerebral palsy diagnosis merit genomic sequencing. JAMA Pediatr. Epub March 6, 2023.
- Craig AK, James C, Bainter J, et al. Parental perceptions of neonatal therapeutic hypothermia; emotional and healing experiences. J Matern Fetal Neonatal Med. 2020;33:2889-2896. doi: 10.1080/14767058.2018.1563592.
- Sagaser A, Pilon B, Goeller A, et al. Parent experience of hypoxic-ischemic encephalopathy and hypothermia: a call for trauma informed care. Am J Perinatol. Epub March 4, 2022.
- Cascio A, Ferrand A, Racine E, et al. Discussing brain magnetic resonance imaging results for neonates with hypoxic-ischemic encephalopathy treated with hypothermia: a challenge for clinicians and parents. E Neurological Sci. 2022;29:100424.
- Thyagarajan B, Baral V, Gunda R, et al. Parental perceptions of hypothermia treatment for neonatal hypoxic-ischaemic encephalopathy. J Matern Fetal Neonatal Med. 2018;31:2527-2533.
- Jacobs SE, Berg M, Hunt R, et al. Cooling for newborns with hypoxic ischemic encephalopathy. Cochrane Database Syst Rev. 2013;CD003311.
- Committee on Fetus and Newborn; Papile E, Baley JE, Benitz W, et al. Hypothermia and neonatal encephalopathy. Pediatrics. 2014;133:1146-1150.
- Academic Medical Center Patient Safety Organization. Therapeutic hypothermia in neonates. Recommendations of the neonatal encephalopathy task force. 2016. https://www.rmf.harvard. edu/-/media/Files/_Global/KC/PDFs/Guide lines/crico_neonates.pdf. Accessed May 25, 2023.
- Zhang W, Ma J, Danzeng Q, et al. Safety of moderate hypothermia for perinatal hypoxic-ischemic encephalopathy: a meta-analysis. Pediatr Neurol. 2017;74:51-61.
- Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress: a clinical and electroencephalographic study. Arch Neurol. 1976;33:696-705.
- Thompson CM, Puterman AS, Linley LL, et al. The value of a scoring system for hypoxic ischemic encephalopathy in predicting neurodevelopmental outcome. Acta Pediatr. 1997;86:757-761.
- Lundgren C, Brudin L, Wanby AS, et al. Ante- and intrapartum risk factors for neonatal hypoxic ischemic encephalopathy. J Matern Fetal Neonatal Med. 2018;31:1595-1601.
- Martinez-Biarge M, Diez-Sebastian J, Wusthoff CJ, et al. Antepartum and intrapartum factors preceding neonatal hypoxic-ischemic encephalopathy. Pediatrics. 2013;132:e952-e959.
- Lorain P, Bower A, Gottardi E, et al. Risk factors for hypoxic-ischemic encephalopathy in cases of severe acidosis: a case-control study. Acta Obstet Gynecol Scand. 2022;101:471-478.
- Russ JB, Simmons R, Glass HC. Neonatal encephalopathy: beyond hypoxic-ischemic encephalopathy. Neo Reviews. 2021;22:e148-e162.
- Allen NM, Mannion M, Conroy J, et al. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. 2014;55:e99-e105.
- Zibro J, Shellhaas RA. Neonatal seizures: diagnosis, etiologies and management. Semin Neurol. 2020;40:246-256.
- Moreno-De-Luca A, Millan F, Peacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325:467-475.
- Srivastava S, Lewis SA, Cohen JS, et al. Molecular diagnostic yield of exome sequencing and chromosomal microarray in cerebral palsy. A systematic review and meta-analysis. JAMA Neurology. 2022;79:1287-1295.
- Gonzalez-Mantilla PJ, Hu Y, Myers SM, et al. Diagnostic yield of exome sequencing in cerebral palsy and implications for genetic testing guidelines. A systematic review and meta-analysis. JAMA Pediatr. Epub March 6, 2023.
- van Eyk C, MacLennon SC, MacLennan AH. All patients with cerebral palsy diagnosis merit genomic sequencing. JAMA Pediatr. Epub March 6, 2023.
- Craig AK, James C, Bainter J, et al. Parental perceptions of neonatal therapeutic hypothermia; emotional and healing experiences. J Matern Fetal Neonatal Med. 2020;33:2889-2896. doi: 10.1080/14767058.2018.1563592.
- Sagaser A, Pilon B, Goeller A, et al. Parent experience of hypoxic-ischemic encephalopathy and hypothermia: a call for trauma informed care. Am J Perinatol. Epub March 4, 2022.
- Cascio A, Ferrand A, Racine E, et al. Discussing brain magnetic resonance imaging results for neonates with hypoxic-ischemic encephalopathy treated with hypothermia: a challenge for clinicians and parents. E Neurological Sci. 2022;29:100424.
- Thyagarajan B, Baral V, Gunda R, et al. Parental perceptions of hypothermia treatment for neonatal hypoxic-ischaemic encephalopathy. J Matern Fetal Neonatal Med. 2018;31:2527-2533.
Can cffDNA technology be used to determine the underlying cause of pregnancy loss to better inform future pregnancy planning?
Hartwig TJ, Ambye L, Gruhn JR, et al. Cell-free fetal DNA for genetic evaluation in Copenhagen Pregnancy Loss Study (COPL): a prospective cohort study. Lancet. 2023;401:762-771. https://doi.org/10.1016/S0140-6736(22)02610-1.
Expert Commentary
A devastating outcome for women, pregnancy loss is directly proportional to maternal age, estimated to occur in approximately 15% of clinically recognized pregnancies and 30% of preclinical pregnancies.1 Approximately 80% of pregnancy losses occur in the first trimester.2 The frequency of clinically recognized early pregnancy loss for women aged 20–30 years is 9% to 17%, and these rates increase sharply, from 20% at age 35 years to 40% at age 40 years, and 80% at age 45 years. Recurrent pregnancy loss (RPL), defined as the spontaneous loss of 2 or more clinically recognized pregnancies, affects less than 5% of women.3 Genetic testing using chromosomal microarray analysis (CMA) has identified aneuploidy in about 55% of cases of miscarriage.4
Following ASRM guidelines for the evaluation of RPL, which consists of analyzing parental chromosomal abnormalities, congenital and acquired uterine anomalies, endocrine imbalances, and autoimmune factors (including antiphospholipid syndrome), no explainable cause is determined in 50% of cases.3 Recently, it has been shown that more than 90% of patients with RPL will have a probable or definitive cause identified when CMA testing on miscarriage tissue with the ASRM evaluation guidelines.5
Details of the study
In this prospective cohort study from Denmark, the authors analyzed maternal serum for cell-free fetal DNA (cffDNA) to determine the ploidy status of the pregnancy loss. One thousand women older than age 18 were included (those who demonstrated an ultrasound-confirmed intrauterine pregnancy loss prior to 22 weeks’ gestation). Maternal blood was obtained while pregnancy tissue was in situ or within 24 hours of passage of products of conception (POC), then analyzed by genome-wide sequencing of cffDNA.
For the first 333 recruited women (validation phase), direct sequencing of the POC was performed for sensitivity and specificity. Following the elimination of inconclusive samples, 302 of the 333 cases demonstrated a sensitivity of 85% and specificity of 93%. In the subsequent evaluation of 667 women, researchers analyzed maternal serum from the gestational age of fetuses ranging from 35 days to 149 days.
Results. In total, nearly 90% of cases yielded conclusive results, with 50% euploid, 46% aneuploid, and 4% multiple aneuploidies. Earlier gestational ages (less than 7 weeks) had a no-call rate (ie, inconclusive) of approximately 50% (only based on 16 patients), with results typically obtained in maternal serum following passage of POC; in pregnancies at gestational ages past 7 weeks, the no-call rate was about 10%. In general, the longer the time after the pregnancy tissue passed, the higher likelihood of a no-call result.
Applying the technology of single-nucleotide polymorphism (SNP)-based CMA can improve identification of fetal and/or maternal sources as causes of pregnancy loss with accuracy, but it does require collection of POC. Of note, samples were deficient in this study, the authors cite, in one-third of the cases. Given this limitation of collection, the authors argue for use of the noninvasive method of cffDNA, obtained from maternal serum.
Study strengths and weaknesses
Several weaknesses of this study are highlighted. Of the validation cohort, one-third of pregnancy tissue could not be analyzed due to insufficient collection. Only 73% of cases allowed for DNA isolation from fetal tissue or chorionic villi; in 27% of cases samples were labeled “unknown tissue.” In those cases classified as unknown, 70% were further determined to be maternal. When all female and monosomy cases were excluded in an effort to assuredly reduce the risk of contamination with maternal DNA, sensitivity of the cffDNA testing process declined to 78%. Another limitation was the required short window for maternal blood sampling (within 24 hours) and its impact on the no-call rate.
The authors note an association with later-life morbidity in patients with a history of pregnancy loss and RPL (including cardiovascular disease, type 2 diabetes, and mental health disorders), thereby arguing for cffDNA-based testing versus no causal testing; however, no treatment has been proven to be effective at reducing pregnancy loss. ●
The best management course for unexplained RPL is uncertain. Despite its use for a euploid miscarriage or parental chromosomal structural rearrangement, in vitro fertilization with preimplantation genetic testing remains an unproven modality.6,7 Given that approximately 70% of human conceptions never achieve viability, and 50% fail spontaneously before being detected,8 the authors’ findings demonstrate peripheral maternal blood can provide a reasonably high sensitivity and specificity for fetal ploidy status when compared with direct sequencing of pregnancy tissue. As fetal aneuploidy offers a higher percentage of subsequent successful pregnancy outcomes, cffDNA may offer reassurance, or direct further testing, following a pregnancy loss. As an application of their results, evaluation may be deferred for an aneuploid miscarriage.
—MARK P. TROLICE, MD, MBA
- Brown S. Miscarriage and its associations. Semin Reprod Med. 2008;26:391-400. doi: 10.1055/s-0028-1087105.
- Wang X, Chen C , Wang L, et al. Conception, early pregnancy loss, and time to clinical pregnancy: a population-based prospective study. Fertil Steril. 2003;79:577-584.
- Evaluation and treatment of recurrent pregnancy loss: a committee opinion. Practice Committee of the American Society for Reproductive Medicine. Fertil Steril. 2012;98: 1103-1111.
- Papas RS, Kutteh WH. Genetic testing for aneuploidy in patients who have had multiple miscarriages: a review of current literature. Appl Clin Genet. 2021;14:321-329. https://doi.org/10.2147/tacg.s320778.
- Popescu F, Jaslow FC, Kutteh WH. Recurrent pregnancy loss evaluation combined with 24-chromosome microarray of miscarriage tissue provides a probable or definite cause of pregnancy loss in over 90% of patients. Hum Reprod. 2018;33:579-587. https://doi.org/10.1093/humrep/dey021.
- Dahdouh EM, Balayla J, Garcia-Velasco JA, et al. PGT-A for recurrent pregnancy loss: evidence is growing but the issue is not resolved. Hum Reprod. 2021;36:2805-2806. https://doi.org/10.1093/humrep/deab194.
- Iews M, Tan J, Taskin O, et al. Does preimplantation genetic diagnosis improve reproductive outcome in couples with recurrent pregnancy loss owing to structural chromosomal rearrangement? A systematic review. Reproductive Bio Medicine Online. 2018;36:677-685. https://doi.org/10.1016 /j.rbmo.2018.03.005.
- Papas RS, Kutteh WH. Genetic testing for aneuploidy in patients who have had multiple miscarriages: a review of current literature. Appl Clin Genet. 2021;14:321-329. https://doi.org/10.2147/TACG.S320778.
Hartwig TJ, Ambye L, Gruhn JR, et al. Cell-free fetal DNA for genetic evaluation in Copenhagen Pregnancy Loss Study (COPL): a prospective cohort study. Lancet. 2023;401:762-771. https://doi.org/10.1016/S0140-6736(22)02610-1.
Expert Commentary
A devastating outcome for women, pregnancy loss is directly proportional to maternal age, estimated to occur in approximately 15% of clinically recognized pregnancies and 30% of preclinical pregnancies.1 Approximately 80% of pregnancy losses occur in the first trimester.2 The frequency of clinically recognized early pregnancy loss for women aged 20–30 years is 9% to 17%, and these rates increase sharply, from 20% at age 35 years to 40% at age 40 years, and 80% at age 45 years. Recurrent pregnancy loss (RPL), defined as the spontaneous loss of 2 or more clinically recognized pregnancies, affects less than 5% of women.3 Genetic testing using chromosomal microarray analysis (CMA) has identified aneuploidy in about 55% of cases of miscarriage.4
Following ASRM guidelines for the evaluation of RPL, which consists of analyzing parental chromosomal abnormalities, congenital and acquired uterine anomalies, endocrine imbalances, and autoimmune factors (including antiphospholipid syndrome), no explainable cause is determined in 50% of cases.3 Recently, it has been shown that more than 90% of patients with RPL will have a probable or definitive cause identified when CMA testing on miscarriage tissue with the ASRM evaluation guidelines.5
Details of the study
In this prospective cohort study from Denmark, the authors analyzed maternal serum for cell-free fetal DNA (cffDNA) to determine the ploidy status of the pregnancy loss. One thousand women older than age 18 were included (those who demonstrated an ultrasound-confirmed intrauterine pregnancy loss prior to 22 weeks’ gestation). Maternal blood was obtained while pregnancy tissue was in situ or within 24 hours of passage of products of conception (POC), then analyzed by genome-wide sequencing of cffDNA.
For the first 333 recruited women (validation phase), direct sequencing of the POC was performed for sensitivity and specificity. Following the elimination of inconclusive samples, 302 of the 333 cases demonstrated a sensitivity of 85% and specificity of 93%. In the subsequent evaluation of 667 women, researchers analyzed maternal serum from the gestational age of fetuses ranging from 35 days to 149 days.
Results. In total, nearly 90% of cases yielded conclusive results, with 50% euploid, 46% aneuploid, and 4% multiple aneuploidies. Earlier gestational ages (less than 7 weeks) had a no-call rate (ie, inconclusive) of approximately 50% (only based on 16 patients), with results typically obtained in maternal serum following passage of POC; in pregnancies at gestational ages past 7 weeks, the no-call rate was about 10%. In general, the longer the time after the pregnancy tissue passed, the higher likelihood of a no-call result.
Applying the technology of single-nucleotide polymorphism (SNP)-based CMA can improve identification of fetal and/or maternal sources as causes of pregnancy loss with accuracy, but it does require collection of POC. Of note, samples were deficient in this study, the authors cite, in one-third of the cases. Given this limitation of collection, the authors argue for use of the noninvasive method of cffDNA, obtained from maternal serum.
Study strengths and weaknesses
Several weaknesses of this study are highlighted. Of the validation cohort, one-third of pregnancy tissue could not be analyzed due to insufficient collection. Only 73% of cases allowed for DNA isolation from fetal tissue or chorionic villi; in 27% of cases samples were labeled “unknown tissue.” In those cases classified as unknown, 70% were further determined to be maternal. When all female and monosomy cases were excluded in an effort to assuredly reduce the risk of contamination with maternal DNA, sensitivity of the cffDNA testing process declined to 78%. Another limitation was the required short window for maternal blood sampling (within 24 hours) and its impact on the no-call rate.
The authors note an association with later-life morbidity in patients with a history of pregnancy loss and RPL (including cardiovascular disease, type 2 diabetes, and mental health disorders), thereby arguing for cffDNA-based testing versus no causal testing; however, no treatment has been proven to be effective at reducing pregnancy loss. ●
The best management course for unexplained RPL is uncertain. Despite its use for a euploid miscarriage or parental chromosomal structural rearrangement, in vitro fertilization with preimplantation genetic testing remains an unproven modality.6,7 Given that approximately 70% of human conceptions never achieve viability, and 50% fail spontaneously before being detected,8 the authors’ findings demonstrate peripheral maternal blood can provide a reasonably high sensitivity and specificity for fetal ploidy status when compared with direct sequencing of pregnancy tissue. As fetal aneuploidy offers a higher percentage of subsequent successful pregnancy outcomes, cffDNA may offer reassurance, or direct further testing, following a pregnancy loss. As an application of their results, evaluation may be deferred for an aneuploid miscarriage.
—MARK P. TROLICE, MD, MBA
Hartwig TJ, Ambye L, Gruhn JR, et al. Cell-free fetal DNA for genetic evaluation in Copenhagen Pregnancy Loss Study (COPL): a prospective cohort study. Lancet. 2023;401:762-771. https://doi.org/10.1016/S0140-6736(22)02610-1.
Expert Commentary
A devastating outcome for women, pregnancy loss is directly proportional to maternal age, estimated to occur in approximately 15% of clinically recognized pregnancies and 30% of preclinical pregnancies.1 Approximately 80% of pregnancy losses occur in the first trimester.2 The frequency of clinically recognized early pregnancy loss for women aged 20–30 years is 9% to 17%, and these rates increase sharply, from 20% at age 35 years to 40% at age 40 years, and 80% at age 45 years. Recurrent pregnancy loss (RPL), defined as the spontaneous loss of 2 or more clinically recognized pregnancies, affects less than 5% of women.3 Genetic testing using chromosomal microarray analysis (CMA) has identified aneuploidy in about 55% of cases of miscarriage.4
Following ASRM guidelines for the evaluation of RPL, which consists of analyzing parental chromosomal abnormalities, congenital and acquired uterine anomalies, endocrine imbalances, and autoimmune factors (including antiphospholipid syndrome), no explainable cause is determined in 50% of cases.3 Recently, it has been shown that more than 90% of patients with RPL will have a probable or definitive cause identified when CMA testing on miscarriage tissue with the ASRM evaluation guidelines.5
Details of the study
In this prospective cohort study from Denmark, the authors analyzed maternal serum for cell-free fetal DNA (cffDNA) to determine the ploidy status of the pregnancy loss. One thousand women older than age 18 were included (those who demonstrated an ultrasound-confirmed intrauterine pregnancy loss prior to 22 weeks’ gestation). Maternal blood was obtained while pregnancy tissue was in situ or within 24 hours of passage of products of conception (POC), then analyzed by genome-wide sequencing of cffDNA.
For the first 333 recruited women (validation phase), direct sequencing of the POC was performed for sensitivity and specificity. Following the elimination of inconclusive samples, 302 of the 333 cases demonstrated a sensitivity of 85% and specificity of 93%. In the subsequent evaluation of 667 women, researchers analyzed maternal serum from the gestational age of fetuses ranging from 35 days to 149 days.
Results. In total, nearly 90% of cases yielded conclusive results, with 50% euploid, 46% aneuploid, and 4% multiple aneuploidies. Earlier gestational ages (less than 7 weeks) had a no-call rate (ie, inconclusive) of approximately 50% (only based on 16 patients), with results typically obtained in maternal serum following passage of POC; in pregnancies at gestational ages past 7 weeks, the no-call rate was about 10%. In general, the longer the time after the pregnancy tissue passed, the higher likelihood of a no-call result.
Applying the technology of single-nucleotide polymorphism (SNP)-based CMA can improve identification of fetal and/or maternal sources as causes of pregnancy loss with accuracy, but it does require collection of POC. Of note, samples were deficient in this study, the authors cite, in one-third of the cases. Given this limitation of collection, the authors argue for use of the noninvasive method of cffDNA, obtained from maternal serum.
Study strengths and weaknesses
Several weaknesses of this study are highlighted. Of the validation cohort, one-third of pregnancy tissue could not be analyzed due to insufficient collection. Only 73% of cases allowed for DNA isolation from fetal tissue or chorionic villi; in 27% of cases samples were labeled “unknown tissue.” In those cases classified as unknown, 70% were further determined to be maternal. When all female and monosomy cases were excluded in an effort to assuredly reduce the risk of contamination with maternal DNA, sensitivity of the cffDNA testing process declined to 78%. Another limitation was the required short window for maternal blood sampling (within 24 hours) and its impact on the no-call rate.
The authors note an association with later-life morbidity in patients with a history of pregnancy loss and RPL (including cardiovascular disease, type 2 diabetes, and mental health disorders), thereby arguing for cffDNA-based testing versus no causal testing; however, no treatment has been proven to be effective at reducing pregnancy loss. ●
The best management course for unexplained RPL is uncertain. Despite its use for a euploid miscarriage or parental chromosomal structural rearrangement, in vitro fertilization with preimplantation genetic testing remains an unproven modality.6,7 Given that approximately 70% of human conceptions never achieve viability, and 50% fail spontaneously before being detected,8 the authors’ findings demonstrate peripheral maternal blood can provide a reasonably high sensitivity and specificity for fetal ploidy status when compared with direct sequencing of pregnancy tissue. As fetal aneuploidy offers a higher percentage of subsequent successful pregnancy outcomes, cffDNA may offer reassurance, or direct further testing, following a pregnancy loss. As an application of their results, evaluation may be deferred for an aneuploid miscarriage.
—MARK P. TROLICE, MD, MBA
- Brown S. Miscarriage and its associations. Semin Reprod Med. 2008;26:391-400. doi: 10.1055/s-0028-1087105.
- Wang X, Chen C , Wang L, et al. Conception, early pregnancy loss, and time to clinical pregnancy: a population-based prospective study. Fertil Steril. 2003;79:577-584.
- Evaluation and treatment of recurrent pregnancy loss: a committee opinion. Practice Committee of the American Society for Reproductive Medicine. Fertil Steril. 2012;98: 1103-1111.
- Papas RS, Kutteh WH. Genetic testing for aneuploidy in patients who have had multiple miscarriages: a review of current literature. Appl Clin Genet. 2021;14:321-329. https://doi.org/10.2147/tacg.s320778.
- Popescu F, Jaslow FC, Kutteh WH. Recurrent pregnancy loss evaluation combined with 24-chromosome microarray of miscarriage tissue provides a probable or definite cause of pregnancy loss in over 90% of patients. Hum Reprod. 2018;33:579-587. https://doi.org/10.1093/humrep/dey021.
- Dahdouh EM, Balayla J, Garcia-Velasco JA, et al. PGT-A for recurrent pregnancy loss: evidence is growing but the issue is not resolved. Hum Reprod. 2021;36:2805-2806. https://doi.org/10.1093/humrep/deab194.
- Iews M, Tan J, Taskin O, et al. Does preimplantation genetic diagnosis improve reproductive outcome in couples with recurrent pregnancy loss owing to structural chromosomal rearrangement? A systematic review. Reproductive Bio Medicine Online. 2018;36:677-685. https://doi.org/10.1016 /j.rbmo.2018.03.005.
- Papas RS, Kutteh WH. Genetic testing for aneuploidy in patients who have had multiple miscarriages: a review of current literature. Appl Clin Genet. 2021;14:321-329. https://doi.org/10.2147/TACG.S320778.
- Brown S. Miscarriage and its associations. Semin Reprod Med. 2008;26:391-400. doi: 10.1055/s-0028-1087105.
- Wang X, Chen C , Wang L, et al. Conception, early pregnancy loss, and time to clinical pregnancy: a population-based prospective study. Fertil Steril. 2003;79:577-584.
- Evaluation and treatment of recurrent pregnancy loss: a committee opinion. Practice Committee of the American Society for Reproductive Medicine. Fertil Steril. 2012;98: 1103-1111.
- Papas RS, Kutteh WH. Genetic testing for aneuploidy in patients who have had multiple miscarriages: a review of current literature. Appl Clin Genet. 2021;14:321-329. https://doi.org/10.2147/tacg.s320778.
- Popescu F, Jaslow FC, Kutteh WH. Recurrent pregnancy loss evaluation combined with 24-chromosome microarray of miscarriage tissue provides a probable or definite cause of pregnancy loss in over 90% of patients. Hum Reprod. 2018;33:579-587. https://doi.org/10.1093/humrep/dey021.
- Dahdouh EM, Balayla J, Garcia-Velasco JA, et al. PGT-A for recurrent pregnancy loss: evidence is growing but the issue is not resolved. Hum Reprod. 2021;36:2805-2806. https://doi.org/10.1093/humrep/deab194.
- Iews M, Tan J, Taskin O, et al. Does preimplantation genetic diagnosis improve reproductive outcome in couples with recurrent pregnancy loss owing to structural chromosomal rearrangement? A systematic review. Reproductive Bio Medicine Online. 2018;36:677-685. https://doi.org/10.1016 /j.rbmo.2018.03.005.
- Papas RS, Kutteh WH. Genetic testing for aneuploidy in patients who have had multiple miscarriages: a review of current literature. Appl Clin Genet. 2021;14:321-329. https://doi.org/10.2147/TACG.S320778.

Acute diffuse rash on trunk

This patient’s diffusely erythematous and scaly rash, in association with recent antibiotic use, was a classic presentation of a drug eruption. Drug eruptions are adverse cutaneous reactions to various medications; they frequently involve antibiotics and anti-epileptics. They can manifest in a multitude of ways with different morphologies. Medication history and timing to onset of symptoms are paramount in making the diagnosis.
Classic reactions include those that are morbilliform (erythematous macules and papules), lichenoid (violaceous and hyperpigmented papules), exfoliative/erythrodermic, and/or urticarial.1 Petechiae and palpable purpura may also manifest.1 Severe reactions, while less common, must always be considered, given their significant morbidity and mortality. These include2:
- Stevens-Johnson syndrome/toxic epidermal necrolysis with diffuse erythema and areas of denuded, necrotic epidermis,
- Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and
- Acute, generalized, exanthematous pustulosis (AGEP) consisting of confluent, nonfollicular pustules.
A general principle in the management of drug eruptions is the discontinuation of the offending drug (if known) as soon as possible. If the agent is not known, it is important to discontinue all drugs that are not deemed as essential, particularly medications that are often associated with reactions, such as antibiotics and anti-epileptics. Additionally, evaluation of the oral mucosa, eyes, and genitourinary tract is helpful to diagnose Stevens-Johnson syndrome, if indicated by symptoms or history.
Wound care with cleansing and covering of denuded skin with emollients and wet dressings should be performed. Infections are common complications in these patients due to the increased inflammation, fissuring, and excoriations that accompany the rash, with sepsis from staphylococcal bacteria being the most concerning complication of infection. Additionally, the compromised skin barrier may lead to heat loss and hypothermia, a compensatory hypermetabolism with hyperthermia, and electrolyte imbalances from insensible water losses.2
Most mild eruptions can be treated with topical corticosteroids and antihistamines. However, in severe eruptions, systemic corticosteroids, or referral for immunosuppressive and anticytokine therapies, also should be considered.1
This patient was treated with both a short course of systemic corticosteroids (prednisone 40 mg/d for 5 days, then tapered over 15 days) and topical steroids (triamcinolone 0.1% ointment bid) for symptomatic care. He also was started on an antihistamine (cetirizine 10 mg bid) for itching. Doxycycline and Augmentin were added to his allergy list. At a 1-week follow up, the patient had near resolution of his rash.
Images courtesy of Jose L. Cortez, MD. Text courtesy of Jose L. Cortez, MD, Department of Dermatology, University of New Mexico School of Medicine, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781-1790.
2. Zhang J, Lei Z, Xu C, et al. Current perspectives on severe drug eruption. Clin Rev Allergy Immunol. 2021;61:282-298. doi: 10.1007/s12016-021-08859-0
This patient’s diffusely erythematous and scaly rash, in association with recent antibiotic use, was a classic presentation of a drug eruption. Drug eruptions are adverse cutaneous reactions to various medications; they frequently involve antibiotics and anti-epileptics. They can manifest in a multitude of ways with different morphologies. Medication history and timing to onset of symptoms are paramount in making the diagnosis.
Classic reactions include those that are morbilliform (erythematous macules and papules), lichenoid (violaceous and hyperpigmented papules), exfoliative/erythrodermic, and/or urticarial.1 Petechiae and palpable purpura may also manifest.1 Severe reactions, while less common, must always be considered, given their significant morbidity and mortality. These include2:
- Stevens-Johnson syndrome/toxic epidermal necrolysis with diffuse erythema and areas of denuded, necrotic epidermis,
- Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and
- Acute, generalized, exanthematous pustulosis (AGEP) consisting of confluent, nonfollicular pustules.
A general principle in the management of drug eruptions is the discontinuation of the offending drug (if known) as soon as possible. If the agent is not known, it is important to discontinue all drugs that are not deemed as essential, particularly medications that are often associated with reactions, such as antibiotics and anti-epileptics. Additionally, evaluation of the oral mucosa, eyes, and genitourinary tract is helpful to diagnose Stevens-Johnson syndrome, if indicated by symptoms or history.
Wound care with cleansing and covering of denuded skin with emollients and wet dressings should be performed. Infections are common complications in these patients due to the increased inflammation, fissuring, and excoriations that accompany the rash, with sepsis from staphylococcal bacteria being the most concerning complication of infection. Additionally, the compromised skin barrier may lead to heat loss and hypothermia, a compensatory hypermetabolism with hyperthermia, and electrolyte imbalances from insensible water losses.2
Most mild eruptions can be treated with topical corticosteroids and antihistamines. However, in severe eruptions, systemic corticosteroids, or referral for immunosuppressive and anticytokine therapies, also should be considered.1
This patient was treated with both a short course of systemic corticosteroids (prednisone 40 mg/d for 5 days, then tapered over 15 days) and topical steroids (triamcinolone 0.1% ointment bid) for symptomatic care. He also was started on an antihistamine (cetirizine 10 mg bid) for itching. Doxycycline and Augmentin were added to his allergy list. At a 1-week follow up, the patient had near resolution of his rash.
Images courtesy of Jose L. Cortez, MD. Text courtesy of Jose L. Cortez, MD, Department of Dermatology, University of New Mexico School of Medicine, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
This patient’s diffusely erythematous and scaly rash, in association with recent antibiotic use, was a classic presentation of a drug eruption. Drug eruptions are adverse cutaneous reactions to various medications; they frequently involve antibiotics and anti-epileptics. They can manifest in a multitude of ways with different morphologies. Medication history and timing to onset of symptoms are paramount in making the diagnosis.
Classic reactions include those that are morbilliform (erythematous macules and papules), lichenoid (violaceous and hyperpigmented papules), exfoliative/erythrodermic, and/or urticarial.1 Petechiae and palpable purpura may also manifest.1 Severe reactions, while less common, must always be considered, given their significant morbidity and mortality. These include2:
- Stevens-Johnson syndrome/toxic epidermal necrolysis with diffuse erythema and areas of denuded, necrotic epidermis,
- Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and
- Acute, generalized, exanthematous pustulosis (AGEP) consisting of confluent, nonfollicular pustules.
A general principle in the management of drug eruptions is the discontinuation of the offending drug (if known) as soon as possible. If the agent is not known, it is important to discontinue all drugs that are not deemed as essential, particularly medications that are often associated with reactions, such as antibiotics and anti-epileptics. Additionally, evaluation of the oral mucosa, eyes, and genitourinary tract is helpful to diagnose Stevens-Johnson syndrome, if indicated by symptoms or history.
Wound care with cleansing and covering of denuded skin with emollients and wet dressings should be performed. Infections are common complications in these patients due to the increased inflammation, fissuring, and excoriations that accompany the rash, with sepsis from staphylococcal bacteria being the most concerning complication of infection. Additionally, the compromised skin barrier may lead to heat loss and hypothermia, a compensatory hypermetabolism with hyperthermia, and electrolyte imbalances from insensible water losses.2
Most mild eruptions can be treated with topical corticosteroids and antihistamines. However, in severe eruptions, systemic corticosteroids, or referral for immunosuppressive and anticytokine therapies, also should be considered.1
This patient was treated with both a short course of systemic corticosteroids (prednisone 40 mg/d for 5 days, then tapered over 15 days) and topical steroids (triamcinolone 0.1% ointment bid) for symptomatic care. He also was started on an antihistamine (cetirizine 10 mg bid) for itching. Doxycycline and Augmentin were added to his allergy list. At a 1-week follow up, the patient had near resolution of his rash.
Images courtesy of Jose L. Cortez, MD. Text courtesy of Jose L. Cortez, MD, Department of Dermatology, University of New Mexico School of Medicine, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781-1790.
2. Zhang J, Lei Z, Xu C, et al. Current perspectives on severe drug eruption. Clin Rev Allergy Immunol. 2021;61:282-298. doi: 10.1007/s12016-021-08859-0
1. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781-1790.
2. Zhang J, Lei Z, Xu C, et al. Current perspectives on severe drug eruption. Clin Rev Allergy Immunol. 2021;61:282-298. doi: 10.1007/s12016-021-08859-0

Why Is There a Lack of Representation of Skin of Color in the COVID-19 Literature?
Throughout the COVID-19 pandemic, there has been a striking paucity of representations of patients with skin of color (SOC) in the dermatology literature. Was COVID-19 underdiagnosed in this patient population due to a lack of patient-centered resources and inadequate dermatology training; reduced access to care, resulting from social determinants of health and reduced skin-color concordance; or the absence of population-based prevalence studies?
Tan et al1 reviewed 51 articles describing skin findings secondary to COVID-19. Patients were stratified by country of origin, which yielded an increased prevalence of cutaneous manifestations among Americans and Europeans compared to Asians, but patients were not stratified by race.1 However, in one case series of 318 predominantly American patients, 89% were White and 0.7% were Black.2 This systematic review by Tan et al1 suggested that skin manifestations of COVID-19 were present in patients with SOC but less frequently than in White patients. However, case series are not a strong proxy for population-level prevalence.
More broadly, patients with SOC are underrepresented in Google image search results, as the medical resource websites (eg, DermNet [https://dermnetnz.org], MedicalNewsToday [www.medicalnewstoday.com], and Healthline [www.healthline.com]) are lacking these images.3 As a result, it is difficult for patients with SOC to recognize diseases presenting in darker skin types. This same tendency may exist for COVID-19 skin manifestations. A systematic review found that articles describing cutaneous manifestations of COVID-19 almost exclusively presented images of lighter skin and completely omitted darker skin.4 If images of patients with SOC are absent from online resources, it is increasingly unlikely for these patients to recognize if their skin lesions are associated with COVID-19, which may result in a decrease in the number of patients with SOC presenting with skin lesions secondary to COVID-19, thereby influencing the representation of patients with SOC in case studies.
The lack of representation of SOC in online resources mirrors the paucity of images in dermatology textbooks. According to a search of 7170 images in major dermatology textbooks, most images depicted light or white skin (80.6%), followed by medium or brown skin in 15.5% of images and dark or black skin in only 3.9%.5 Physicians rely on online and print resources for making diagnoses; inadequate resources highlight a component of a larger issue: inadequate training of dermatologists in SOC. In a survey of American dermatologists and dermatology residents (N=262), 47% thought that their medical education had not adequately trained them on skin conditions in Black patients.6
A lack of adequate training for dermatologists may decrease the rate of correct diagnosis of skin lesions secondary to COVID-19 in patients with SOC. A lack of trust in the health care system and social determinants of health may hinder patients with SOC from seeking medical help. Dermatology is the second least diverse of medical specialties; only 3% of dermatologists are Black.7 This is impactful: First, because minority physicians are increasingly likely to provide care for patients of the same race or background, and second, because race-concordant physician visits are associated with greater patient-reported positive affect.7 A lack of availability of race-concordant physicians or physicians with perceived cultural competence may deter patients with SOC from seeking help, which may be further prevalent in dermatologic practice.
Barriers at all levels of social determinants of health hinder access to health care. Patients with SOC experience greater housing insecurity, increased reliance on public transportation, more issues with health literacy, and limited English-language fluency.8 Combined, these factors equate to decreased access to health care resources and subsequently a lack of inclusion in case studies.
COVID-19 infection disproportionately affects patients with SOC,8 but there is a clear lack of representation of SOC in the COVID-19 dermatology literature. It is imperative to investigate factors that may contribute to this inequity. Recognizing skin manifestations can play a role in diagnosing COVID-19; increased awareness of its presentation in darker skin types may help bridge existing racial inequities. It is vital that physicians receive adequate resources and training to be able to recognize cutaneous manifestations of COVID-19 in all skin types. Finally, it is important to recognize that the lack of representation of SOC in the COVID-19 literature represents a larger trend that exists in dermatologic research that warrants further investigation and advocacy for inclusivity.
- Tan SW, Tam YC, Oh CC. Skin manifestations of COVID-19: a worldwide review. JAAD Int. 2021;2:119-133. doi:10.1016/j.jdin.2020.12.003
- Freeman EE, McMahon DE, Lipoff JB, et al; American Academy of Dermatology Ad Hoc Task Force on COVID-19. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dematol. 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Fathy R, Lipoff JB. Lack of skin of color in Google image searches may reflect under-representation in all educational resources. J Am Acad Dermatol. 2022;86:E113-E114. doi:10.1016/j.jaad.2021.04.097
- Lester JC, Jia JL, Zhang L, et al. Absence of images of skin of colour in publications of COVID-19 skin manifestations. Br J Dermatol. 2020;183:593-595. doi:10.1111/bjd.19258
- Kamath P, Sundaram N, Morillo-Hernandez C, et al. Visual racism in internet searches and dermatology textbooks. J Am Acad Dermatol. 2021;85:1348-1349. doi:10.1016/j.jaad.2020.10.072
- Buster KJ, Stevens EI, Elmets CA. Dermatologic health disparities. Dermatol Clin. 2012;30:53-59,viii. doi:10.1016/j.det.2011.08.002
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Tai DBG, Shah A, Doubeni CA, et al. The disproportionate impact of COVID-19 on racial and ethnic minorities in the United States. Clin Infect Dis. 2021;72:703-706. doi:10.1093/cid/ciaa815
Throughout the COVID-19 pandemic, there has been a striking paucity of representations of patients with skin of color (SOC) in the dermatology literature. Was COVID-19 underdiagnosed in this patient population due to a lack of patient-centered resources and inadequate dermatology training; reduced access to care, resulting from social determinants of health and reduced skin-color concordance; or the absence of population-based prevalence studies?
Tan et al1 reviewed 51 articles describing skin findings secondary to COVID-19. Patients were stratified by country of origin, which yielded an increased prevalence of cutaneous manifestations among Americans and Europeans compared to Asians, but patients were not stratified by race.1 However, in one case series of 318 predominantly American patients, 89% were White and 0.7% were Black.2 This systematic review by Tan et al1 suggested that skin manifestations of COVID-19 were present in patients with SOC but less frequently than in White patients. However, case series are not a strong proxy for population-level prevalence.
More broadly, patients with SOC are underrepresented in Google image search results, as the medical resource websites (eg, DermNet [https://dermnetnz.org], MedicalNewsToday [www.medicalnewstoday.com], and Healthline [www.healthline.com]) are lacking these images.3 As a result, it is difficult for patients with SOC to recognize diseases presenting in darker skin types. This same tendency may exist for COVID-19 skin manifestations. A systematic review found that articles describing cutaneous manifestations of COVID-19 almost exclusively presented images of lighter skin and completely omitted darker skin.4 If images of patients with SOC are absent from online resources, it is increasingly unlikely for these patients to recognize if their skin lesions are associated with COVID-19, which may result in a decrease in the number of patients with SOC presenting with skin lesions secondary to COVID-19, thereby influencing the representation of patients with SOC in case studies.
The lack of representation of SOC in online resources mirrors the paucity of images in dermatology textbooks. According to a search of 7170 images in major dermatology textbooks, most images depicted light or white skin (80.6%), followed by medium or brown skin in 15.5% of images and dark or black skin in only 3.9%.5 Physicians rely on online and print resources for making diagnoses; inadequate resources highlight a component of a larger issue: inadequate training of dermatologists in SOC. In a survey of American dermatologists and dermatology residents (N=262), 47% thought that their medical education had not adequately trained them on skin conditions in Black patients.6
A lack of adequate training for dermatologists may decrease the rate of correct diagnosis of skin lesions secondary to COVID-19 in patients with SOC. A lack of trust in the health care system and social determinants of health may hinder patients with SOC from seeking medical help. Dermatology is the second least diverse of medical specialties; only 3% of dermatologists are Black.7 This is impactful: First, because minority physicians are increasingly likely to provide care for patients of the same race or background, and second, because race-concordant physician visits are associated with greater patient-reported positive affect.7 A lack of availability of race-concordant physicians or physicians with perceived cultural competence may deter patients with SOC from seeking help, which may be further prevalent in dermatologic practice.
Barriers at all levels of social determinants of health hinder access to health care. Patients with SOC experience greater housing insecurity, increased reliance on public transportation, more issues with health literacy, and limited English-language fluency.8 Combined, these factors equate to decreased access to health care resources and subsequently a lack of inclusion in case studies.
COVID-19 infection disproportionately affects patients with SOC,8 but there is a clear lack of representation of SOC in the COVID-19 dermatology literature. It is imperative to investigate factors that may contribute to this inequity. Recognizing skin manifestations can play a role in diagnosing COVID-19; increased awareness of its presentation in darker skin types may help bridge existing racial inequities. It is vital that physicians receive adequate resources and training to be able to recognize cutaneous manifestations of COVID-19 in all skin types. Finally, it is important to recognize that the lack of representation of SOC in the COVID-19 literature represents a larger trend that exists in dermatologic research that warrants further investigation and advocacy for inclusivity.
Throughout the COVID-19 pandemic, there has been a striking paucity of representations of patients with skin of color (SOC) in the dermatology literature. Was COVID-19 underdiagnosed in this patient population due to a lack of patient-centered resources and inadequate dermatology training; reduced access to care, resulting from social determinants of health and reduced skin-color concordance; or the absence of population-based prevalence studies?
Tan et al1 reviewed 51 articles describing skin findings secondary to COVID-19. Patients were stratified by country of origin, which yielded an increased prevalence of cutaneous manifestations among Americans and Europeans compared to Asians, but patients were not stratified by race.1 However, in one case series of 318 predominantly American patients, 89% were White and 0.7% were Black.2 This systematic review by Tan et al1 suggested that skin manifestations of COVID-19 were present in patients with SOC but less frequently than in White patients. However, case series are not a strong proxy for population-level prevalence.
More broadly, patients with SOC are underrepresented in Google image search results, as the medical resource websites (eg, DermNet [https://dermnetnz.org], MedicalNewsToday [www.medicalnewstoday.com], and Healthline [www.healthline.com]) are lacking these images.3 As a result, it is difficult for patients with SOC to recognize diseases presenting in darker skin types. This same tendency may exist for COVID-19 skin manifestations. A systematic review found that articles describing cutaneous manifestations of COVID-19 almost exclusively presented images of lighter skin and completely omitted darker skin.4 If images of patients with SOC are absent from online resources, it is increasingly unlikely for these patients to recognize if their skin lesions are associated with COVID-19, which may result in a decrease in the number of patients with SOC presenting with skin lesions secondary to COVID-19, thereby influencing the representation of patients with SOC in case studies.
The lack of representation of SOC in online resources mirrors the paucity of images in dermatology textbooks. According to a search of 7170 images in major dermatology textbooks, most images depicted light or white skin (80.6%), followed by medium or brown skin in 15.5% of images and dark or black skin in only 3.9%.5 Physicians rely on online and print resources for making diagnoses; inadequate resources highlight a component of a larger issue: inadequate training of dermatologists in SOC. In a survey of American dermatologists and dermatology residents (N=262), 47% thought that their medical education had not adequately trained them on skin conditions in Black patients.6
A lack of adequate training for dermatologists may decrease the rate of correct diagnosis of skin lesions secondary to COVID-19 in patients with SOC. A lack of trust in the health care system and social determinants of health may hinder patients with SOC from seeking medical help. Dermatology is the second least diverse of medical specialties; only 3% of dermatologists are Black.7 This is impactful: First, because minority physicians are increasingly likely to provide care for patients of the same race or background, and second, because race-concordant physician visits are associated with greater patient-reported positive affect.7 A lack of availability of race-concordant physicians or physicians with perceived cultural competence may deter patients with SOC from seeking help, which may be further prevalent in dermatologic practice.
Barriers at all levels of social determinants of health hinder access to health care. Patients with SOC experience greater housing insecurity, increased reliance on public transportation, more issues with health literacy, and limited English-language fluency.8 Combined, these factors equate to decreased access to health care resources and subsequently a lack of inclusion in case studies.
COVID-19 infection disproportionately affects patients with SOC,8 but there is a clear lack of representation of SOC in the COVID-19 dermatology literature. It is imperative to investigate factors that may contribute to this inequity. Recognizing skin manifestations can play a role in diagnosing COVID-19; increased awareness of its presentation in darker skin types may help bridge existing racial inequities. It is vital that physicians receive adequate resources and training to be able to recognize cutaneous manifestations of COVID-19 in all skin types. Finally, it is important to recognize that the lack of representation of SOC in the COVID-19 literature represents a larger trend that exists in dermatologic research that warrants further investigation and advocacy for inclusivity.
- Tan SW, Tam YC, Oh CC. Skin manifestations of COVID-19: a worldwide review. JAAD Int. 2021;2:119-133. doi:10.1016/j.jdin.2020.12.003
- Freeman EE, McMahon DE, Lipoff JB, et al; American Academy of Dermatology Ad Hoc Task Force on COVID-19. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dematol. 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Fathy R, Lipoff JB. Lack of skin of color in Google image searches may reflect under-representation in all educational resources. J Am Acad Dermatol. 2022;86:E113-E114. doi:10.1016/j.jaad.2021.04.097
- Lester JC, Jia JL, Zhang L, et al. Absence of images of skin of colour in publications of COVID-19 skin manifestations. Br J Dermatol. 2020;183:593-595. doi:10.1111/bjd.19258
- Kamath P, Sundaram N, Morillo-Hernandez C, et al. Visual racism in internet searches and dermatology textbooks. J Am Acad Dermatol. 2021;85:1348-1349. doi:10.1016/j.jaad.2020.10.072
- Buster KJ, Stevens EI, Elmets CA. Dermatologic health disparities. Dermatol Clin. 2012;30:53-59,viii. doi:10.1016/j.det.2011.08.002
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Tai DBG, Shah A, Doubeni CA, et al. The disproportionate impact of COVID-19 on racial and ethnic minorities in the United States. Clin Infect Dis. 2021;72:703-706. doi:10.1093/cid/ciaa815
- Tan SW, Tam YC, Oh CC. Skin manifestations of COVID-19: a worldwide review. JAAD Int. 2021;2:119-133. doi:10.1016/j.jdin.2020.12.003
- Freeman EE, McMahon DE, Lipoff JB, et al; American Academy of Dermatology Ad Hoc Task Force on COVID-19. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dematol. 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Fathy R, Lipoff JB. Lack of skin of color in Google image searches may reflect under-representation in all educational resources. J Am Acad Dermatol. 2022;86:E113-E114. doi:10.1016/j.jaad.2021.04.097
- Lester JC, Jia JL, Zhang L, et al. Absence of images of skin of colour in publications of COVID-19 skin manifestations. Br J Dermatol. 2020;183:593-595. doi:10.1111/bjd.19258
- Kamath P, Sundaram N, Morillo-Hernandez C, et al. Visual racism in internet searches and dermatology textbooks. J Am Acad Dermatol. 2021;85:1348-1349. doi:10.1016/j.jaad.2020.10.072
- Buster KJ, Stevens EI, Elmets CA. Dermatologic health disparities. Dermatol Clin. 2012;30:53-59,viii. doi:10.1016/j.det.2011.08.002
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Tai DBG, Shah A, Doubeni CA, et al. The disproportionate impact of COVID-19 on racial and ethnic minorities in the United States. Clin Infect Dis. 2021;72:703-706. doi:10.1093/cid/ciaa815
Systemic Lupus Erythematosus
THE COMPARISON
A A 23-year-old White woman with malar erythema from acute cutaneous lupus erythematosus. The erythema also can be seen on the nose and eyelids but spares the nasolabial folds.
B A Black woman with malar erythema and hyperpigmentation from acute cutaneous lupus erythematosus. The nasolabial folds are spared.
C A 19-year-old Latina woman with malar erythema from acute cutaneous lupus erythematosus. The erythema also can be seen on the nose, chin, and eyelids but spares the nasolabial folds. Cutaneous erosions are present on the right cheek as part of the lupus flare. Systemic lupus erythematosus (SLE) is a chronic autoimmune condition that affects the kidneys, lungs, brain, and heart, though it is not limited to these organs. Dermatologists and primary care physicians play a critical role in the early identification of SLE, particularly in those with skin of color, as the standardized mortality rate is 2.6-fold higher in patients with SLE compared to the general population.1 The clinical manifestations of SLE vary.

Epidemiology
A meta-analysis of data from the Centers for Disease Control and Prevention National Lupus Registry network including 5417 patients revealed a prevalence of 72.8 cases per 100,000 person-years.2 The prevalence was higher in females than males and highest among females identifying as Black. White and Asian/Pacific Islander females had the lowest prevalence. The American Indian (indigenous)/Alaska Native–identifying population had the highest race-specific SLE estimates among both females and males compared to other racial/ethnic groups.2
Key clinical features in people with darker skin tones
The diagnosis of SLE is based on clinical and immunologic criteria from the European League Against Rheumatism/American College of Rheumatology.3,4 An antinuclear antibody titer of 1:80 or higher at least once is required for the diagnosis of SLE, as long as there is not another more likely diagnosis. If it is present, 22 additive weighted classification criteria are considered; each criterion is assigned points, ranging from 2 to 10. Patients with at least 1 clinical criterion and 10 or more points are classified as having SLE. If more than 1 of the criteria are met in a domain, then the one with the highest numerical value is counted.3,4 Aringer et al3,4 outline the criteria and numerical points to make the diagnosis of SLE. The mucocutaneous component of the SLE diagnostic criteria3,4 includes nonscarring alopecia, oral ulcers, subacute cutaneous or discoid lupus erythematosus,5 and acute cutaneous lupus erythematosus, with acute cutaneous lupus erythematosus being the highest-weighted criterion in that domain. The other clinical domains are constitutional, hematologic, neuropsychiatric, serosal, musculoskeletal, renal, antiphosopholipid antibodies, complement proteins, and SLE-specific antibodies.3,4
The malar (“butterfly”) rash of SLE characteristically includes erythema that spares the nasolabial folds but affects the nasal bridge and cheeks.6 The rash occasionally may be pruritic and painful, lasting days to weeks. Photosensitivity occurs, resulting in rashes or even an overall worsening of SLE symptoms. In those with darker skin tones, erythema may appear violaceous or may not be as readily appreciated.6
Worth noting
• Patients with skin of color are at an increased risk for postinflammatory hypopigmentation and hyperpigmentation (pigment alteration), hypertrophic scars, and keloids.7,8
• The mortality rate for those with SLE is high despite early recognition and treatment when compared to the general population.1,9
Health disparity highlight
Those at greatest risk for death from SLE in the United States are those of African descent, Hispanic individuals, men, and those with low socioeconomic status,9 which likely is primarily driven by social determinants of health instead of genetic patterns. Income level, educational attainment, insurance status, and environmental factors10 have far-reaching effects, negatively impacting quality of life and even mortality.
- Lee YH, Choi SJ, Ji JD, et al. Overall and cause-specific mortality in systemic lupus erythematosus: an updated meta-analysis. Lupus. 2016;25:727-734.
- Izmirly PM, Parton H, Wang L, et al. Prevalence of systemic lupus erythematosus in the United States: estimates from a meta-analysis of the Centers for Disease Control and Prevention National Lupus Registries [published online April 23, 2021]. Arthritis Rheumatol. 2021;73:991-996. doi:10.1002/art.41632
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71:1400-1412. doi:10.1002/art.40930
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78:1151-1159.
- Heath CR, Usatine RP. Discoid lupus. Cutis. 2022;109:172-173.
- Firestein GS, Budd RC, Harris ED Jr, et al, eds. Kelley’s Textbook of Rheumatology. 8th ed. Saunders Elsevier; 2008.
- Nozile W, Adgerson CH, Cohen GF. Cutaneous lupus erythematosus in skin of color. J Drugs Dermatol. 2015;14:343-349.
- Cardinali F, Kovacs D, Picardo M. Mechanisms underlying postinflammatory hyperpigmentation: lessons for solar. Ann Dermatol Venereol. 2012;139(suppl 4):S148-S152.
- Ocampo-Piraquive V, Nieto-Aristizábal I, Cañas CA, et al. Mortality in systemic lupus erythematosus: causes, predictors and interventions. Expert Rev Clin Immunol. 2018;14:1043-1053. doi:10.1080/17446 66X.2018.1538789
- Carter EE, Barr SG, Clarke AE. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. 2016;12:605-620. doi:10.1038/nrrheum.2016.137
THE COMPARISON
A A 23-year-old White woman with malar erythema from acute cutaneous lupus erythematosus. The erythema also can be seen on the nose and eyelids but spares the nasolabial folds.
B A Black woman with malar erythema and hyperpigmentation from acute cutaneous lupus erythematosus. The nasolabial folds are spared.
C A 19-year-old Latina woman with malar erythema from acute cutaneous lupus erythematosus. The erythema also can be seen on the nose, chin, and eyelids but spares the nasolabial folds. Cutaneous erosions are present on the right cheek as part of the lupus flare. Systemic lupus erythematosus (SLE) is a chronic autoimmune condition that affects the kidneys, lungs, brain, and heart, though it is not limited to these organs. Dermatologists and primary care physicians play a critical role in the early identification of SLE, particularly in those with skin of color, as the standardized mortality rate is 2.6-fold higher in patients with SLE compared to the general population.1 The clinical manifestations of SLE vary.
Epidemiology
A meta-analysis of data from the Centers for Disease Control and Prevention National Lupus Registry network including 5417 patients revealed a prevalence of 72.8 cases per 100,000 person-years.2 The prevalence was higher in females than males and highest among females identifying as Black. White and Asian/Pacific Islander females had the lowest prevalence. The American Indian (indigenous)/Alaska Native–identifying population had the highest race-specific SLE estimates among both females and males compared to other racial/ethnic groups.2
Key clinical features in people with darker skin tones
The diagnosis of SLE is based on clinical and immunologic criteria from the European League Against Rheumatism/American College of Rheumatology.3,4 An antinuclear antibody titer of 1:80 or higher at least once is required for the diagnosis of SLE, as long as there is not another more likely diagnosis. If it is present, 22 additive weighted classification criteria are considered; each criterion is assigned points, ranging from 2 to 10. Patients with at least 1 clinical criterion and 10 or more points are classified as having SLE. If more than 1 of the criteria are met in a domain, then the one with the highest numerical value is counted.3,4 Aringer et al3,4 outline the criteria and numerical points to make the diagnosis of SLE. The mucocutaneous component of the SLE diagnostic criteria3,4 includes nonscarring alopecia, oral ulcers, subacute cutaneous or discoid lupus erythematosus,5 and acute cutaneous lupus erythematosus, with acute cutaneous lupus erythematosus being the highest-weighted criterion in that domain. The other clinical domains are constitutional, hematologic, neuropsychiatric, serosal, musculoskeletal, renal, antiphosopholipid antibodies, complement proteins, and SLE-specific antibodies.3,4
The malar (“butterfly”) rash of SLE characteristically includes erythema that spares the nasolabial folds but affects the nasal bridge and cheeks.6 The rash occasionally may be pruritic and painful, lasting days to weeks. Photosensitivity occurs, resulting in rashes or even an overall worsening of SLE symptoms. In those with darker skin tones, erythema may appear violaceous or may not be as readily appreciated.6
Worth noting
• Patients with skin of color are at an increased risk for postinflammatory hypopigmentation and hyperpigmentation (pigment alteration), hypertrophic scars, and keloids.7,8
• The mortality rate for those with SLE is high despite early recognition and treatment when compared to the general population.1,9
Health disparity highlight
Those at greatest risk for death from SLE in the United States are those of African descent, Hispanic individuals, men, and those with low socioeconomic status,9 which likely is primarily driven by social determinants of health instead of genetic patterns. Income level, educational attainment, insurance status, and environmental factors10 have far-reaching effects, negatively impacting quality of life and even mortality.
THE COMPARISON
A A 23-year-old White woman with malar erythema from acute cutaneous lupus erythematosus. The erythema also can be seen on the nose and eyelids but spares the nasolabial folds.
B A Black woman with malar erythema and hyperpigmentation from acute cutaneous lupus erythematosus. The nasolabial folds are spared.
C A 19-year-old Latina woman with malar erythema from acute cutaneous lupus erythematosus. The erythema also can be seen on the nose, chin, and eyelids but spares the nasolabial folds. Cutaneous erosions are present on the right cheek as part of the lupus flare. Systemic lupus erythematosus (SLE) is a chronic autoimmune condition that affects the kidneys, lungs, brain, and heart, though it is not limited to these organs. Dermatologists and primary care physicians play a critical role in the early identification of SLE, particularly in those with skin of color, as the standardized mortality rate is 2.6-fold higher in patients with SLE compared to the general population.1 The clinical manifestations of SLE vary.
Epidemiology
A meta-analysis of data from the Centers for Disease Control and Prevention National Lupus Registry network including 5417 patients revealed a prevalence of 72.8 cases per 100,000 person-years.2 The prevalence was higher in females than males and highest among females identifying as Black. White and Asian/Pacific Islander females had the lowest prevalence. The American Indian (indigenous)/Alaska Native–identifying population had the highest race-specific SLE estimates among both females and males compared to other racial/ethnic groups.2
Key clinical features in people with darker skin tones
The diagnosis of SLE is based on clinical and immunologic criteria from the European League Against Rheumatism/American College of Rheumatology.3,4 An antinuclear antibody titer of 1:80 or higher at least once is required for the diagnosis of SLE, as long as there is not another more likely diagnosis. If it is present, 22 additive weighted classification criteria are considered; each criterion is assigned points, ranging from 2 to 10. Patients with at least 1 clinical criterion and 10 or more points are classified as having SLE. If more than 1 of the criteria are met in a domain, then the one with the highest numerical value is counted.3,4 Aringer et al3,4 outline the criteria and numerical points to make the diagnosis of SLE. The mucocutaneous component of the SLE diagnostic criteria3,4 includes nonscarring alopecia, oral ulcers, subacute cutaneous or discoid lupus erythematosus,5 and acute cutaneous lupus erythematosus, with acute cutaneous lupus erythematosus being the highest-weighted criterion in that domain. The other clinical domains are constitutional, hematologic, neuropsychiatric, serosal, musculoskeletal, renal, antiphosopholipid antibodies, complement proteins, and SLE-specific antibodies.3,4
The malar (“butterfly”) rash of SLE characteristically includes erythema that spares the nasolabial folds but affects the nasal bridge and cheeks.6 The rash occasionally may be pruritic and painful, lasting days to weeks. Photosensitivity occurs, resulting in rashes or even an overall worsening of SLE symptoms. In those with darker skin tones, erythema may appear violaceous or may not be as readily appreciated.6
Worth noting
• Patients with skin of color are at an increased risk for postinflammatory hypopigmentation and hyperpigmentation (pigment alteration), hypertrophic scars, and keloids.7,8
• The mortality rate for those with SLE is high despite early recognition and treatment when compared to the general population.1,9
Health disparity highlight
Those at greatest risk for death from SLE in the United States are those of African descent, Hispanic individuals, men, and those with low socioeconomic status,9 which likely is primarily driven by social determinants of health instead of genetic patterns. Income level, educational attainment, insurance status, and environmental factors10 have far-reaching effects, negatively impacting quality of life and even mortality.
- Lee YH, Choi SJ, Ji JD, et al. Overall and cause-specific mortality in systemic lupus erythematosus: an updated meta-analysis. Lupus. 2016;25:727-734.
- Izmirly PM, Parton H, Wang L, et al. Prevalence of systemic lupus erythematosus in the United States: estimates from a meta-analysis of the Centers for Disease Control and Prevention National Lupus Registries [published online April 23, 2021]. Arthritis Rheumatol. 2021;73:991-996. doi:10.1002/art.41632
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71:1400-1412. doi:10.1002/art.40930
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78:1151-1159.
- Heath CR, Usatine RP. Discoid lupus. Cutis. 2022;109:172-173.
- Firestein GS, Budd RC, Harris ED Jr, et al, eds. Kelley’s Textbook of Rheumatology. 8th ed. Saunders Elsevier; 2008.
- Nozile W, Adgerson CH, Cohen GF. Cutaneous lupus erythematosus in skin of color. J Drugs Dermatol. 2015;14:343-349.
- Cardinali F, Kovacs D, Picardo M. Mechanisms underlying postinflammatory hyperpigmentation: lessons for solar. Ann Dermatol Venereol. 2012;139(suppl 4):S148-S152.
- Ocampo-Piraquive V, Nieto-Aristizábal I, Cañas CA, et al. Mortality in systemic lupus erythematosus: causes, predictors and interventions. Expert Rev Clin Immunol. 2018;14:1043-1053. doi:10.1080/17446 66X.2018.1538789
- Carter EE, Barr SG, Clarke AE. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. 2016;12:605-620. doi:10.1038/nrrheum.2016.137
- Lee YH, Choi SJ, Ji JD, et al. Overall and cause-specific mortality in systemic lupus erythematosus: an updated meta-analysis. Lupus. 2016;25:727-734.
- Izmirly PM, Parton H, Wang L, et al. Prevalence of systemic lupus erythematosus in the United States: estimates from a meta-analysis of the Centers for Disease Control and Prevention National Lupus Registries [published online April 23, 2021]. Arthritis Rheumatol. 2021;73:991-996. doi:10.1002/art.41632
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71:1400-1412. doi:10.1002/art.40930
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78:1151-1159.
- Heath CR, Usatine RP. Discoid lupus. Cutis. 2022;109:172-173.
- Firestein GS, Budd RC, Harris ED Jr, et al, eds. Kelley’s Textbook of Rheumatology. 8th ed. Saunders Elsevier; 2008.
- Nozile W, Adgerson CH, Cohen GF. Cutaneous lupus erythematosus in skin of color. J Drugs Dermatol. 2015;14:343-349.
- Cardinali F, Kovacs D, Picardo M. Mechanisms underlying postinflammatory hyperpigmentation: lessons for solar. Ann Dermatol Venereol. 2012;139(suppl 4):S148-S152.
- Ocampo-Piraquive V, Nieto-Aristizábal I, Cañas CA, et al. Mortality in systemic lupus erythematosus: causes, predictors and interventions. Expert Rev Clin Immunol. 2018;14:1043-1053. doi:10.1080/17446 66X.2018.1538789
- Carter EE, Barr SG, Clarke AE. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. 2016;12:605-620. doi:10.1038/nrrheum.2016.137

Cross-sectional Analysis of Matched Dermatology Residency Applicants Without US Home Programs
To the Editor:
Dermatology is one of the most competitive residencies for matching, with a 57.5% match rate in 2022.1 Our prior study of research-mentor relationships among matched dermatology applicants corroborated the importance of home programs (HPs) and program connections.2 Therefore, our current objective was to compare profiles of matched dermatology applicants without HPs vs those with HPs.
We searched websites of 139 dermatology programs nationwide and found 1736 matched applicants from 2016 to 2020; of them, 323 did not have HPs. We determined program rank by research output using Doximity Residency Navigator (https://www.doximity.com/residency/). Advanced degrees (ADs) of applicants were identified using program websites and LinkedIn. A PubMed search was conducted for number of articles published by each applicant before September 15 of their match year. For applicants without HPs, we identified the senior author on each publication. The senior author publishing with an applicant most often was considered the research mentor. Two-tailed independent t tests and χ2 tests were used to determine statistical significance (P<.05).
On average, matched applicants without HPs matched in lower-ranked (74.4) and smaller (12.4) programs compared with matched applicants with HPs (45.3 [P<.0001] and 15.1 [P<.0001], respectively)(eTable). The mean number of publications was similar between matched applicants with HPs and without HPs (5.64 and 4.80, respectively; P=.0525) as well as the percentage with ADs (14.7% and 11.5%, respectively; P=.0953). Overall, 14.8% of matched applicants without HPs matched at their mentors’ institutions.

Data were obtained for matched international applicants as a subset of non-HP applicants. Despite attending medical schools without associated HPs in the United States, international applicants matched at similarly ranked (44.3) and sized (15.0) programs, on average, compared with HP applicants. The mean number of publications was higher for international applicants (11.4) vs domestic applicants (5.33). International applicants more often had ADs (23.8%) and 60.1% of them held doctor of philosophy degrees. Overall, 40.5% of international applicants matched at their mentors’ institutions.
Our study suggests that matched dermatology applicants with and without HPs had similar achievements, on average, for the number of publications and percentage with ADs. However, non-HP applicants matched at lower-ranked programs than HP applicants. Therefore, applicants without HPs should strongly consider cultivating program connections, especially if they desire to match at higher-ranked dermatology programs. To illustrate, the rate of matching at research mentors’ institutions was approximately 3-times higher for international applicants than non-HP applicants overall. Despite the disadvantages of applying as international applicants, they were able to match at substantially higher-ranked dermatology programs than non-HP applicants. International applicants may have a longer time investment—the number of years from obtaining their medical degree or US medical license to matching—giving them time to produce quality research and develop meaningful relationships at an institution. Additionally, our prior study of the top 25 dermatology residencies showed that 26.2% of successful applicants matched at their research mentors’ institutions, with almost half of this subset matching at their HPs, where their mentors also practiced.2 Because of the potential benefits of having program connections, applicants without HPs should seek dermatology research mentors, especially via highly beneficial in-person networking opportunities (eg, away rotations, conferences) that had previously been limited during the COVID-19 pandemic.3 Formal mentorship programs giving priority to students without HPs recently have been developed, which only begins to address the inequities in the dermatology residency application process.4
Study limitations include lack of resident information on 15 program websites, missed publications due to applicant name changes, not accounting for abstracts and posters, and inability to collect data on unmatched applicants.
We hope that our study alleviates some concerns that applicants without HPs may have regarding applying for dermatology residency and encourages those with a genuine interest in dermatology to pursue the specialty, provided they find a strong research mentor. Residency programs should be cognizant of the unique challenges that non-HP applicants face for matching.
- National Resident Matching Program. Results and Data: 2022 Main Residency Match. National Resident Matching Program; May 2022. Accessed May 30, 2023. https://www.nrmp.org/wp-content/uploads/2022/11 /2022-Main-Match-Results-and-Data-Final-Revised.pdf
- Yeh C, Desai AD, Wilson BN, et al. Cross-sectional analysis of scholarly work and mentor relationships in matched dermatology residency applicants. J Am Acad Dermatol. 2022;86:1437-1439.
- Association of American Medical Colleges. Specialty recommendations on away rotations for 2021-22 academic year. Accessed May 24, 2023. https://students-residents.aamc.org/researching-residency-programs -and-building-application-strategy/specialty-response-covid-19
- derminterest Instagram page. DIGA is excited for the second year of our mentor-mentee program! Mentors are dermatology residents. Please keep in mind due to the current circumstances, dermatology residency 2021-2022 applicants without home programs will be prioritized as mentees. Please refrain from signing up if you were paired with a faculty mentor for the APD-DIGA Mentorship Program in May 2021. Contact @suryasweetie123 only if you have specific questions, otherwise all information is on our website and the link is here. Link is below and in our bio! #DIGA #derm #mentee #residencyapplication. Accessed May 24, 2023. https://www.instagram.com/p/CSrq0exMchY/
To the Editor:
Dermatology is one of the most competitive residencies for matching, with a 57.5% match rate in 2022.1 Our prior study of research-mentor relationships among matched dermatology applicants corroborated the importance of home programs (HPs) and program connections.2 Therefore, our current objective was to compare profiles of matched dermatology applicants without HPs vs those with HPs.
We searched websites of 139 dermatology programs nationwide and found 1736 matched applicants from 2016 to 2020; of them, 323 did not have HPs. We determined program rank by research output using Doximity Residency Navigator (https://www.doximity.com/residency/). Advanced degrees (ADs) of applicants were identified using program websites and LinkedIn. A PubMed search was conducted for number of articles published by each applicant before September 15 of their match year. For applicants without HPs, we identified the senior author on each publication. The senior author publishing with an applicant most often was considered the research mentor. Two-tailed independent t tests and χ2 tests were used to determine statistical significance (P<.05).
On average, matched applicants without HPs matched in lower-ranked (74.4) and smaller (12.4) programs compared with matched applicants with HPs (45.3 [P<.0001] and 15.1 [P<.0001], respectively)(eTable). The mean number of publications was similar between matched applicants with HPs and without HPs (5.64 and 4.80, respectively; P=.0525) as well as the percentage with ADs (14.7% and 11.5%, respectively; P=.0953). Overall, 14.8% of matched applicants without HPs matched at their mentors’ institutions.
Data were obtained for matched international applicants as a subset of non-HP applicants. Despite attending medical schools without associated HPs in the United States, international applicants matched at similarly ranked (44.3) and sized (15.0) programs, on average, compared with HP applicants. The mean number of publications was higher for international applicants (11.4) vs domestic applicants (5.33). International applicants more often had ADs (23.8%) and 60.1% of them held doctor of philosophy degrees. Overall, 40.5% of international applicants matched at their mentors’ institutions.
Our study suggests that matched dermatology applicants with and without HPs had similar achievements, on average, for the number of publications and percentage with ADs. However, non-HP applicants matched at lower-ranked programs than HP applicants. Therefore, applicants without HPs should strongly consider cultivating program connections, especially if they desire to match at higher-ranked dermatology programs. To illustrate, the rate of matching at research mentors’ institutions was approximately 3-times higher for international applicants than non-HP applicants overall. Despite the disadvantages of applying as international applicants, they were able to match at substantially higher-ranked dermatology programs than non-HP applicants. International applicants may have a longer time investment—the number of years from obtaining their medical degree or US medical license to matching—giving them time to produce quality research and develop meaningful relationships at an institution. Additionally, our prior study of the top 25 dermatology residencies showed that 26.2% of successful applicants matched at their research mentors’ institutions, with almost half of this subset matching at their HPs, where their mentors also practiced.2 Because of the potential benefits of having program connections, applicants without HPs should seek dermatology research mentors, especially via highly beneficial in-person networking opportunities (eg, away rotations, conferences) that had previously been limited during the COVID-19 pandemic.3 Formal mentorship programs giving priority to students without HPs recently have been developed, which only begins to address the inequities in the dermatology residency application process.4
Study limitations include lack of resident information on 15 program websites, missed publications due to applicant name changes, not accounting for abstracts and posters, and inability to collect data on unmatched applicants.
We hope that our study alleviates some concerns that applicants without HPs may have regarding applying for dermatology residency and encourages those with a genuine interest in dermatology to pursue the specialty, provided they find a strong research mentor. Residency programs should be cognizant of the unique challenges that non-HP applicants face for matching.
To the Editor:
Dermatology is one of the most competitive residencies for matching, with a 57.5% match rate in 2022.1 Our prior study of research-mentor relationships among matched dermatology applicants corroborated the importance of home programs (HPs) and program connections.2 Therefore, our current objective was to compare profiles of matched dermatology applicants without HPs vs those with HPs.
We searched websites of 139 dermatology programs nationwide and found 1736 matched applicants from 2016 to 2020; of them, 323 did not have HPs. We determined program rank by research output using Doximity Residency Navigator (https://www.doximity.com/residency/). Advanced degrees (ADs) of applicants were identified using program websites and LinkedIn. A PubMed search was conducted for number of articles published by each applicant before September 15 of their match year. For applicants without HPs, we identified the senior author on each publication. The senior author publishing with an applicant most often was considered the research mentor. Two-tailed independent t tests and χ2 tests were used to determine statistical significance (P<.05).
On average, matched applicants without HPs matched in lower-ranked (74.4) and smaller (12.4) programs compared with matched applicants with HPs (45.3 [P<.0001] and 15.1 [P<.0001], respectively)(eTable). The mean number of publications was similar between matched applicants with HPs and without HPs (5.64 and 4.80, respectively; P=.0525) as well as the percentage with ADs (14.7% and 11.5%, respectively; P=.0953). Overall, 14.8% of matched applicants without HPs matched at their mentors’ institutions.
Data were obtained for matched international applicants as a subset of non-HP applicants. Despite attending medical schools without associated HPs in the United States, international applicants matched at similarly ranked (44.3) and sized (15.0) programs, on average, compared with HP applicants. The mean number of publications was higher for international applicants (11.4) vs domestic applicants (5.33). International applicants more often had ADs (23.8%) and 60.1% of them held doctor of philosophy degrees. Overall, 40.5% of international applicants matched at their mentors’ institutions.
Our study suggests that matched dermatology applicants with and without HPs had similar achievements, on average, for the number of publications and percentage with ADs. However, non-HP applicants matched at lower-ranked programs than HP applicants. Therefore, applicants without HPs should strongly consider cultivating program connections, especially if they desire to match at higher-ranked dermatology programs. To illustrate, the rate of matching at research mentors’ institutions was approximately 3-times higher for international applicants than non-HP applicants overall. Despite the disadvantages of applying as international applicants, they were able to match at substantially higher-ranked dermatology programs than non-HP applicants. International applicants may have a longer time investment—the number of years from obtaining their medical degree or US medical license to matching—giving them time to produce quality research and develop meaningful relationships at an institution. Additionally, our prior study of the top 25 dermatology residencies showed that 26.2% of successful applicants matched at their research mentors’ institutions, with almost half of this subset matching at their HPs, where their mentors also practiced.2 Because of the potential benefits of having program connections, applicants without HPs should seek dermatology research mentors, especially via highly beneficial in-person networking opportunities (eg, away rotations, conferences) that had previously been limited during the COVID-19 pandemic.3 Formal mentorship programs giving priority to students without HPs recently have been developed, which only begins to address the inequities in the dermatology residency application process.4
Study limitations include lack of resident information on 15 program websites, missed publications due to applicant name changes, not accounting for abstracts and posters, and inability to collect data on unmatched applicants.
We hope that our study alleviates some concerns that applicants without HPs may have regarding applying for dermatology residency and encourages those with a genuine interest in dermatology to pursue the specialty, provided they find a strong research mentor. Residency programs should be cognizant of the unique challenges that non-HP applicants face for matching.
- National Resident Matching Program. Results and Data: 2022 Main Residency Match. National Resident Matching Program; May 2022. Accessed May 30, 2023. https://www.nrmp.org/wp-content/uploads/2022/11 /2022-Main-Match-Results-and-Data-Final-Revised.pdf
- Yeh C, Desai AD, Wilson BN, et al. Cross-sectional analysis of scholarly work and mentor relationships in matched dermatology residency applicants. J Am Acad Dermatol. 2022;86:1437-1439.
- Association of American Medical Colleges. Specialty recommendations on away rotations for 2021-22 academic year. Accessed May 24, 2023. https://students-residents.aamc.org/researching-residency-programs -and-building-application-strategy/specialty-response-covid-19
- derminterest Instagram page. DIGA is excited for the second year of our mentor-mentee program! Mentors are dermatology residents. Please keep in mind due to the current circumstances, dermatology residency 2021-2022 applicants without home programs will be prioritized as mentees. Please refrain from signing up if you were paired with a faculty mentor for the APD-DIGA Mentorship Program in May 2021. Contact @suryasweetie123 only if you have specific questions, otherwise all information is on our website and the link is here. Link is below and in our bio! #DIGA #derm #mentee #residencyapplication. Accessed May 24, 2023. https://www.instagram.com/p/CSrq0exMchY/
- National Resident Matching Program. Results and Data: 2022 Main Residency Match. National Resident Matching Program; May 2022. Accessed May 30, 2023. https://www.nrmp.org/wp-content/uploads/2022/11 /2022-Main-Match-Results-and-Data-Final-Revised.pdf
- Yeh C, Desai AD, Wilson BN, et al. Cross-sectional analysis of scholarly work and mentor relationships in matched dermatology residency applicants. J Am Acad Dermatol. 2022;86:1437-1439.
- Association of American Medical Colleges. Specialty recommendations on away rotations for 2021-22 academic year. Accessed May 24, 2023. https://students-residents.aamc.org/researching-residency-programs -and-building-application-strategy/specialty-response-covid-19
- derminterest Instagram page. DIGA is excited for the second year of our mentor-mentee program! Mentors are dermatology residents. Please keep in mind due to the current circumstances, dermatology residency 2021-2022 applicants without home programs will be prioritized as mentees. Please refrain from signing up if you were paired with a faculty mentor for the APD-DIGA Mentorship Program in May 2021. Contact @suryasweetie123 only if you have specific questions, otherwise all information is on our website and the link is here. Link is below and in our bio! #DIGA #derm #mentee #residencyapplication. Accessed May 24, 2023. https://www.instagram.com/p/CSrq0exMchY/
Practice Points
- Our study suggests that matched dermatology applicants with and without home programs (HPs) had similar achievements, on average, for number of publications and holding advanced degrees.
- Because of the potential benefits of having program connections for matching in dermatology, applicants without HPs should seek dermatology research mentors.
What’s Eating You? Triatoma and Arilus cristatus Bugs
Classification
Triatomine bugs (Triatoma) and the wheel bug (Arilus cristatus) are part of the family Reduviidae (order Hemiptera, a name that describes the sucking proboscis on the front of the insect’s head).1,2 Both arthropods are found in multiple countries and are especially common in warmer areas, including in the United States, where they can be seen from Texas to California.3,4 Because blood-feeding triatomines need a blood meal to survive while laying eggs and then throughout their 5 developmental nymph stages to undergo molting, they feed on mammals, such as opossums, raccoons, pack rats, and armadillos, whereas wheel bugs mainly prey on soft-bodied insects.1,4-6
Triatoma bugs seek cutaneous blood vessels using thermosensors in their antennae to locate blood flow under the skin for feeding. After inserting the proboscis, they release nitric oxide and an anticoagulant that allows for continuous blood flow while feeding.7 It has been reported that triatomine bugs are not able to bite through clothing, instead seeking exposed skin, particularly near mucous membranes, such as the hands, arms, feet, head, and trunk. The name kissing bug for triatomines was coined because bites near the mouth are common.6 The bite typically is painless and occurs mainly at night when the insect is most active. After obtaining a blood meal, triatomine bugs seek shelter and hide in mud and daub structures, cracks, crevices, and furniture.1,8
Unlike Triatoma species, A cristatus does not require a blood meal for development and survival, leading it to prey on soft-bodied insects. Piercing prey with the proboscis, wheel bugs inject a toxin to digest the contents and suck the digested contents through this apparatus.4 Because the wheel bug does not require a blood meal, it typically bites a human only for defense if it feels threatened. Unlike the painless bite of a triatomine bug, the bite of A cristatus is extremely painful; it has been described as the worst arthropod bite—worse than a hornet’s sting. The pain from the bite is caused by the toxin being injected into the skin; possible retention of the proboscis makes the pain worse.4,9 In addition, when A cristatus is disturbed, it exudes pungent material from a pair of bright orange subrectal glands while stridulating to repulse predators.9
Although Triatoma species and A cristatus have separate roles in nature and vastly different impacts on health, they often are mistaken for the same arthropod when seen in nature. Features that members of Reduviidae share include large bodies (relative to their overall length); long thin legs; a narrow head; wings; and a long sucking proboscis on the front of the head.10
(Triatoma species).")
Characteristics that differentiate Triatoma and A cristatus species include size, color, and distinctive markings. Most triatomine bugs are 12- to 36-mm long; are dark brown or black; and have what are called tiger-stripe orange markings on the peripheral two-thirds of the body (Figure 1).11 In contrast, wheel bugs commonly are bigger—measuring longer than 1.25 inches—and gray, with a cogwheel-like structure on the thorax (Figure 2).10
.")
Dermatologic Presentation and Clinical Symptoms
The area of involved skin on patients presenting with Triatoma or A cristatus bites may resemble other insect bites. Many bites from Triatoma bugs and A cristatus initially present as an erythematous, raised, pruritic papule with a central punctum that is visible because of the involvement of the proboscis. However, other presentations of bites from both arthropods have been reported4,6,7: grouped vesicles on an erythematous base; indurated, giant, urticarial-type wheels measuring 10 to 15 mm in diameter; and hemorrhagic bullous nodules (Figure 3). Associated lymphangitis or lymphadenitis is typical of the latter 2 variations.6 These variations in presentation can be mistaken for other causes of similarly presenting lesions, such as shingles or spider bites, leading to delayed or missed diagnosis.

Patients may present with a single bite or multiple bites due to the feeding pattern of Triatoma bugs; if the host moves or disrupts its feeding, the arthropod takes multiple bites to finish feeding.8 In comparison, 4 common variations of wheel bug bites have been reported: (1) a painful bite without complications; (2) a cutaneous horn and papilloma at the site of toxin injection; (3) a necrotic ulcer around the central punctum caused by injected toxin; and (4) an abscess under the central punctum due to secondary infection.4
Anaphylaxis—Although the bites of Triatoma and A cristatus present differently, both can cause anaphylaxis. Triatoma is implicated more often than A cristatus as the cause of anaphylaxis.12 In fact, Triatoma bites are among the more common causes of anaphylaxis from bug bites, with multiple cases of these reactions reported in the literature.12,13
Symptoms of Triatoma anaphylaxis include acute-onset urticarial rash, flushing, dyspnea, wheezing, nausea, vomiting, and localized edema.2 The cause of anaphylaxis is proteins in Triatoma saliva, including 20-kDa procalin, which incites the systemic reaction. Other potential causes of anaphylaxis include serine protease, which has similarities to salivary protein and desmoglein in humans.11
The degree of reaction to a bite depends on the patient's sensitization to antigenic proteins in each insect’s saliva.4,6 Patients who have a bite from a triatomine bug are at risk for subsequent bites, as household infestation is likely due to the pliability of the insect, allowing it to hide in small spaces unnoticed.8 In the case of a bite from Triatoma or A cristatus, sensitization may lead to severe and worsening reactions with subsequent bites, which ultimately can result in life-threatening anaphylaxis.1,6
Treatment and Prevention
Treatment of Triatoma and A cristatus bites depends on the severity of the patient’s reaction to the bite. A local reaction to a bite from either insect can be treated supportively with local corticosteroids and antihistamines.3 If the patient is sensitized to proteins associated with a bite, standard anaphylaxis treatment such as epinephrine and intravenous antihistamines may be indicated.14 Secondary infection can be treated with antibiotics; a formed abscess might need to be drained or debrided.15
There’s No Place Like Home—Because Triatoma bugs have a pliable exoskeleton and can squeeze into small spaces, they commonly infest dwellings where they find multiple attractants: light, heat, carbon dioxide, and lactic acid.8 The more household occupants (including pets), the higher the levels of carbon dioxide and lactic acid, thus the greater the attraction. Infestation of a home can lead to the spread of diseases harbored by Triatoma, including Chagas disease, which is caused by the parasite Trypanosoma cruzi.5
Preventive measures can be taken to reduce the risk and extent of home infestation by Triatoma bugs, including insecticides, a solid foundation, window screens, air conditioning, sealing of cracks and crevices, outdoor light management, and removal of clutter throughout the house.8 Because Triatoma bugs cannot bite through clothing, protective clothing and bug repellent on exposed skin can be employed. Another degree of protection is offered by pest management, especially control of rodents by removing food, water, and nests in areas where triatomine bugs feed off of that population.8,14
Unlike triatomine bugs, wheel bugs tend not to invade houses; therefore, these preventive measures are unnecessary. If a wheel bug is identified, do not engage the arthropod due to the defensive nature of its attack.4,9 Such deliberate avoidance should ensure protection from the wheel bug’s painful bite.
Medical Complications
Although triatomine bugs and wheel bugs are in the same taxonomic family, subsequent infection is unique to Triatoma bugs because they need a blood meal to survive. Because Triatoma bugs feed on mammals, they present an increased opportunity for transmitting the causative agents of infection from hosts on which they have fed.12 The principal parasite transmitted by triatomines is T cruzi, which causes Chagas disease and lives in the gastrointestinal (GI) tract of the insect.5 When a triatomine bug seeks out a mucosal surface to bite, including the mouth, it defecates and urinates during or shortly after feeding, leading to contamination of the initial wound or mucosal surfaces. In addition, Triatoma bugs are vectors for transmission of Serratia marcescans, Bartonella henselae, and Mycobacterium leprae.16
Chagas Disease—This infection has 3 stages: acute, intermediate, and chronic.5 The acute stage can present with symptoms of conjunctivitis, fever, lymphadenopathy, hepatosplenomegaly, and anemia. The intermediate stage typically is asymptomatic. The chronic stage usually involves the heart and GI tract and causes cardiac aneurysms, cardiomegaly, megacolon, and megaesophagus. Initial symptoms can be a localized nodule (chagoma) at the inoculation site, fever, fatigue, lymphadenopathy, and hepatosplenomegaly.2 Unilateral palpebral edema with associated lymphadenopathy (Romaña sign) also can be seen—not to be confused with bilateral swelling in an acute reaction to an insect bite. Romaña sign is pathognomonic of T cruzi infection; bilateral palpebral swelling is typical of an allergic reaction.12
Identification of a triatomine bite is the first step in diagnosing Chagas disease, which can be life-threatening. Among chronic carriers of Chagas disease, 30% develop GI and cardiac symptoms, of which 20% to 30% develop cardiomyopathy, with serious symptoms that present 10 to 20 years after the asymptomatic intermediate phase.2
Chagas disease is endemic to Central and South America but is also seen in North America; 28,000 new cases are reported annually in South America and North America combined. Human migration from endemic areas and from rural to urban areas has promoted the spread of Chagas disease.2 However, patients in the United States have a relatively low risk for Chagas disease, largely because of the quality of housing construction and use of insecticides.
Treatment options for Chagas disease include nifurtimox and benznidazole. Without treatment, the host immune response typically controls acute replication of the parasite but will lead to a chronic state, ultimately involving the heart and GI tract.5
- Vetter R. Kissing bugs (Triatoma) and the skin. Dermatol Online J. 2001;7:6.
- Zemore ZM, Wills BK. Kissing bug bite. StatPearls [Internet]. StatPearlsPublishing; 2023.
- Edwards L, Lynch PJ. Anaphylactic reaction to kissing bug bites. Ariz Med. 1984;41:159-161.
- Smith FD, Miller NG, Carnazzo SJ, et al. Insect bite by Arilus cristatus, a North American reduviid. AMA Arch Derm. 1958;77:324-330. doi:10.1001/archderm.1958.01560030070011
- Nguyen T, Waseem M. Chagas disease. StatPearls [Internet]. StatPearls Publishing; 2022.
- Shields TL, Walsh EN. Kissing bug bite. AMA Arch Derm. 1956;74:14-21. doi:10.1001/archderm.1956.01550070016004
- Beatty NL, Klotz SA. The midnight bite! a kissing bug nightmare. Am J Med. 2018;131:E43-E44. doi:10.1016/j.amjmed.2017.10.013
- Klotz SA, Smith SL, Schmidt JO. Kissing bug intrusions into homes in the Southwest United States. Insects. 2021;12:654. doi:10.3390/insects12070654
- Aldrich JR, Chauhan KR, Zhang A, et al. Exocrine secretions of wheel bugs (Heteroptera: Reduviidae: Arilus spp.): clarification and chemistry. Z Naturforsch C J Biosci. 2013;68:522-526.
- Boggs J. They’re wheel bugs, NOT kissing bugs. Buckeye Yard and Garden onLine [Internet]. September 17, 2020. Accessed May 25, 2023. https://bygl.osu.edu/node/1688
- Weber RW. Allergen of the month—assassin bug. Ann Allergy Asthma Immunol. 2015;115:A9.
- Klotz JH, Dorn PL, Logan JL, et al. “Kissing bugs”: potential disease vectors and cause of anaphylaxis. Clin Infect Dis 2010;50:1629-1634. doi:10.1086/652769
- Anderson C, Belnap C. The kiss of death: a rare case of anaphylaxis to the bite of the “red margined kissing bug”. Hawaii J Med Public Health. 2015;74(9 suppl 2):33-35.
- Moffitt JE, Venarske D, Goddard J, et al. Allergic reactions to Triatoma bites. Ann Allergy Asthma Immunol. 2003;91:122-128. doi:10.1016/s1081-1206(10)62165-5
- Burnett JW, Calton GJ, Morgan RJ. Triatoma: the “kissing bug”. Cutis. 1987;39:399.
- Vieira CB, Praça YR, Bentes K, et al. Triatomines: Trypanosomatids, bacteria, and viruses potential vectors? Front Cell Infect Microbiol. 2018;8:405. doi:10.3389/fcimb.2018.00405
Classification
Triatomine bugs (Triatoma) and the wheel bug (Arilus cristatus) are part of the family Reduviidae (order Hemiptera, a name that describes the sucking proboscis on the front of the insect’s head).1,2 Both arthropods are found in multiple countries and are especially common in warmer areas, including in the United States, where they can be seen from Texas to California.3,4 Because blood-feeding triatomines need a blood meal to survive while laying eggs and then throughout their 5 developmental nymph stages to undergo molting, they feed on mammals, such as opossums, raccoons, pack rats, and armadillos, whereas wheel bugs mainly prey on soft-bodied insects.1,4-6
Triatoma bugs seek cutaneous blood vessels using thermosensors in their antennae to locate blood flow under the skin for feeding. After inserting the proboscis, they release nitric oxide and an anticoagulant that allows for continuous blood flow while feeding.7 It has been reported that triatomine bugs are not able to bite through clothing, instead seeking exposed skin, particularly near mucous membranes, such as the hands, arms, feet, head, and trunk. The name kissing bug for triatomines was coined because bites near the mouth are common.6 The bite typically is painless and occurs mainly at night when the insect is most active. After obtaining a blood meal, triatomine bugs seek shelter and hide in mud and daub structures, cracks, crevices, and furniture.1,8
Unlike Triatoma species, A cristatus does not require a blood meal for development and survival, leading it to prey on soft-bodied insects. Piercing prey with the proboscis, wheel bugs inject a toxin to digest the contents and suck the digested contents through this apparatus.4 Because the wheel bug does not require a blood meal, it typically bites a human only for defense if it feels threatened. Unlike the painless bite of a triatomine bug, the bite of A cristatus is extremely painful; it has been described as the worst arthropod bite—worse than a hornet’s sting. The pain from the bite is caused by the toxin being injected into the skin; possible retention of the proboscis makes the pain worse.4,9 In addition, when A cristatus is disturbed, it exudes pungent material from a pair of bright orange subrectal glands while stridulating to repulse predators.9
Although Triatoma species and A cristatus have separate roles in nature and vastly different impacts on health, they often are mistaken for the same arthropod when seen in nature. Features that members of Reduviidae share include large bodies (relative to their overall length); long thin legs; a narrow head; wings; and a long sucking proboscis on the front of the head.10
Characteristics that differentiate Triatoma and A cristatus species include size, color, and distinctive markings. Most triatomine bugs are 12- to 36-mm long; are dark brown or black; and have what are called tiger-stripe orange markings on the peripheral two-thirds of the body (Figure 1).11 In contrast, wheel bugs commonly are bigger—measuring longer than 1.25 inches—and gray, with a cogwheel-like structure on the thorax (Figure 2).10
Dermatologic Presentation and Clinical Symptoms
The area of involved skin on patients presenting with Triatoma or A cristatus bites may resemble other insect bites. Many bites from Triatoma bugs and A cristatus initially present as an erythematous, raised, pruritic papule with a central punctum that is visible because of the involvement of the proboscis. However, other presentations of bites from both arthropods have been reported4,6,7: grouped vesicles on an erythematous base; indurated, giant, urticarial-type wheels measuring 10 to 15 mm in diameter; and hemorrhagic bullous nodules (Figure 3). Associated lymphangitis or lymphadenitis is typical of the latter 2 variations.6 These variations in presentation can be mistaken for other causes of similarly presenting lesions, such as shingles or spider bites, leading to delayed or missed diagnosis.
Patients may present with a single bite or multiple bites due to the feeding pattern of Triatoma bugs; if the host moves or disrupts its feeding, the arthropod takes multiple bites to finish feeding.8 In comparison, 4 common variations of wheel bug bites have been reported: (1) a painful bite without complications; (2) a cutaneous horn and papilloma at the site of toxin injection; (3) a necrotic ulcer around the central punctum caused by injected toxin; and (4) an abscess under the central punctum due to secondary infection.4
Anaphylaxis—Although the bites of Triatoma and A cristatus present differently, both can cause anaphylaxis. Triatoma is implicated more often than A cristatus as the cause of anaphylaxis.12 In fact, Triatoma bites are among the more common causes of anaphylaxis from bug bites, with multiple cases of these reactions reported in the literature.12,13
Symptoms of Triatoma anaphylaxis include acute-onset urticarial rash, flushing, dyspnea, wheezing, nausea, vomiting, and localized edema.2 The cause of anaphylaxis is proteins in Triatoma saliva, including 20-kDa procalin, which incites the systemic reaction. Other potential causes of anaphylaxis include serine protease, which has similarities to salivary protein and desmoglein in humans.11
The degree of reaction to a bite depends on the patient's sensitization to antigenic proteins in each insect’s saliva.4,6 Patients who have a bite from a triatomine bug are at risk for subsequent bites, as household infestation is likely due to the pliability of the insect, allowing it to hide in small spaces unnoticed.8 In the case of a bite from Triatoma or A cristatus, sensitization may lead to severe and worsening reactions with subsequent bites, which ultimately can result in life-threatening anaphylaxis.1,6
Treatment and Prevention
Treatment of Triatoma and A cristatus bites depends on the severity of the patient’s reaction to the bite. A local reaction to a bite from either insect can be treated supportively with local corticosteroids and antihistamines.3 If the patient is sensitized to proteins associated with a bite, standard anaphylaxis treatment such as epinephrine and intravenous antihistamines may be indicated.14 Secondary infection can be treated with antibiotics; a formed abscess might need to be drained or debrided.15
There’s No Place Like Home—Because Triatoma bugs have a pliable exoskeleton and can squeeze into small spaces, they commonly infest dwellings where they find multiple attractants: light, heat, carbon dioxide, and lactic acid.8 The more household occupants (including pets), the higher the levels of carbon dioxide and lactic acid, thus the greater the attraction. Infestation of a home can lead to the spread of diseases harbored by Triatoma, including Chagas disease, which is caused by the parasite Trypanosoma cruzi.5
Preventive measures can be taken to reduce the risk and extent of home infestation by Triatoma bugs, including insecticides, a solid foundation, window screens, air conditioning, sealing of cracks and crevices, outdoor light management, and removal of clutter throughout the house.8 Because Triatoma bugs cannot bite through clothing, protective clothing and bug repellent on exposed skin can be employed. Another degree of protection is offered by pest management, especially control of rodents by removing food, water, and nests in areas where triatomine bugs feed off of that population.8,14
Unlike triatomine bugs, wheel bugs tend not to invade houses; therefore, these preventive measures are unnecessary. If a wheel bug is identified, do not engage the arthropod due to the defensive nature of its attack.4,9 Such deliberate avoidance should ensure protection from the wheel bug’s painful bite.
Medical Complications
Although triatomine bugs and wheel bugs are in the same taxonomic family, subsequent infection is unique to Triatoma bugs because they need a blood meal to survive. Because Triatoma bugs feed on mammals, they present an increased opportunity for transmitting the causative agents of infection from hosts on which they have fed.12 The principal parasite transmitted by triatomines is T cruzi, which causes Chagas disease and lives in the gastrointestinal (GI) tract of the insect.5 When a triatomine bug seeks out a mucosal surface to bite, including the mouth, it defecates and urinates during or shortly after feeding, leading to contamination of the initial wound or mucosal surfaces. In addition, Triatoma bugs are vectors for transmission of Serratia marcescans, Bartonella henselae, and Mycobacterium leprae.16
Chagas Disease—This infection has 3 stages: acute, intermediate, and chronic.5 The acute stage can present with symptoms of conjunctivitis, fever, lymphadenopathy, hepatosplenomegaly, and anemia. The intermediate stage typically is asymptomatic. The chronic stage usually involves the heart and GI tract and causes cardiac aneurysms, cardiomegaly, megacolon, and megaesophagus. Initial symptoms can be a localized nodule (chagoma) at the inoculation site, fever, fatigue, lymphadenopathy, and hepatosplenomegaly.2 Unilateral palpebral edema with associated lymphadenopathy (Romaña sign) also can be seen—not to be confused with bilateral swelling in an acute reaction to an insect bite. Romaña sign is pathognomonic of T cruzi infection; bilateral palpebral swelling is typical of an allergic reaction.12
Identification of a triatomine bite is the first step in diagnosing Chagas disease, which can be life-threatening. Among chronic carriers of Chagas disease, 30% develop GI and cardiac symptoms, of which 20% to 30% develop cardiomyopathy, with serious symptoms that present 10 to 20 years after the asymptomatic intermediate phase.2
Chagas disease is endemic to Central and South America but is also seen in North America; 28,000 new cases are reported annually in South America and North America combined. Human migration from endemic areas and from rural to urban areas has promoted the spread of Chagas disease.2 However, patients in the United States have a relatively low risk for Chagas disease, largely because of the quality of housing construction and use of insecticides.
Treatment options for Chagas disease include nifurtimox and benznidazole. Without treatment, the host immune response typically controls acute replication of the parasite but will lead to a chronic state, ultimately involving the heart and GI tract.5
Classification
Triatomine bugs (Triatoma) and the wheel bug (Arilus cristatus) are part of the family Reduviidae (order Hemiptera, a name that describes the sucking proboscis on the front of the insect’s head).1,2 Both arthropods are found in multiple countries and are especially common in warmer areas, including in the United States, where they can be seen from Texas to California.3,4 Because blood-feeding triatomines need a blood meal to survive while laying eggs and then throughout their 5 developmental nymph stages to undergo molting, they feed on mammals, such as opossums, raccoons, pack rats, and armadillos, whereas wheel bugs mainly prey on soft-bodied insects.1,4-6
Triatoma bugs seek cutaneous blood vessels using thermosensors in their antennae to locate blood flow under the skin for feeding. After inserting the proboscis, they release nitric oxide and an anticoagulant that allows for continuous blood flow while feeding.7 It has been reported that triatomine bugs are not able to bite through clothing, instead seeking exposed skin, particularly near mucous membranes, such as the hands, arms, feet, head, and trunk. The name kissing bug for triatomines was coined because bites near the mouth are common.6 The bite typically is painless and occurs mainly at night when the insect is most active. After obtaining a blood meal, triatomine bugs seek shelter and hide in mud and daub structures, cracks, crevices, and furniture.1,8
Unlike Triatoma species, A cristatus does not require a blood meal for development and survival, leading it to prey on soft-bodied insects. Piercing prey with the proboscis, wheel bugs inject a toxin to digest the contents and suck the digested contents through this apparatus.4 Because the wheel bug does not require a blood meal, it typically bites a human only for defense if it feels threatened. Unlike the painless bite of a triatomine bug, the bite of A cristatus is extremely painful; it has been described as the worst arthropod bite—worse than a hornet’s sting. The pain from the bite is caused by the toxin being injected into the skin; possible retention of the proboscis makes the pain worse.4,9 In addition, when A cristatus is disturbed, it exudes pungent material from a pair of bright orange subrectal glands while stridulating to repulse predators.9
Although Triatoma species and A cristatus have separate roles in nature and vastly different impacts on health, they often are mistaken for the same arthropod when seen in nature. Features that members of Reduviidae share include large bodies (relative to their overall length); long thin legs; a narrow head; wings; and a long sucking proboscis on the front of the head.10
Characteristics that differentiate Triatoma and A cristatus species include size, color, and distinctive markings. Most triatomine bugs are 12- to 36-mm long; are dark brown or black; and have what are called tiger-stripe orange markings on the peripheral two-thirds of the body (Figure 1).11 In contrast, wheel bugs commonly are bigger—measuring longer than 1.25 inches—and gray, with a cogwheel-like structure on the thorax (Figure 2).10
Dermatologic Presentation and Clinical Symptoms
The area of involved skin on patients presenting with Triatoma or A cristatus bites may resemble other insect bites. Many bites from Triatoma bugs and A cristatus initially present as an erythematous, raised, pruritic papule with a central punctum that is visible because of the involvement of the proboscis. However, other presentations of bites from both arthropods have been reported4,6,7: grouped vesicles on an erythematous base; indurated, giant, urticarial-type wheels measuring 10 to 15 mm in diameter; and hemorrhagic bullous nodules (Figure 3). Associated lymphangitis or lymphadenitis is typical of the latter 2 variations.6 These variations in presentation can be mistaken for other causes of similarly presenting lesions, such as shingles or spider bites, leading to delayed or missed diagnosis.
Patients may present with a single bite or multiple bites due to the feeding pattern of Triatoma bugs; if the host moves or disrupts its feeding, the arthropod takes multiple bites to finish feeding.8 In comparison, 4 common variations of wheel bug bites have been reported: (1) a painful bite without complications; (2) a cutaneous horn and papilloma at the site of toxin injection; (3) a necrotic ulcer around the central punctum caused by injected toxin; and (4) an abscess under the central punctum due to secondary infection.4
Anaphylaxis—Although the bites of Triatoma and A cristatus present differently, both can cause anaphylaxis. Triatoma is implicated more often than A cristatus as the cause of anaphylaxis.12 In fact, Triatoma bites are among the more common causes of anaphylaxis from bug bites, with multiple cases of these reactions reported in the literature.12,13
Symptoms of Triatoma anaphylaxis include acute-onset urticarial rash, flushing, dyspnea, wheezing, nausea, vomiting, and localized edema.2 The cause of anaphylaxis is proteins in Triatoma saliva, including 20-kDa procalin, which incites the systemic reaction. Other potential causes of anaphylaxis include serine protease, which has similarities to salivary protein and desmoglein in humans.11
The degree of reaction to a bite depends on the patient's sensitization to antigenic proteins in each insect’s saliva.4,6 Patients who have a bite from a triatomine bug are at risk for subsequent bites, as household infestation is likely due to the pliability of the insect, allowing it to hide in small spaces unnoticed.8 In the case of a bite from Triatoma or A cristatus, sensitization may lead to severe and worsening reactions with subsequent bites, which ultimately can result in life-threatening anaphylaxis.1,6
Treatment and Prevention
Treatment of Triatoma and A cristatus bites depends on the severity of the patient’s reaction to the bite. A local reaction to a bite from either insect can be treated supportively with local corticosteroids and antihistamines.3 If the patient is sensitized to proteins associated with a bite, standard anaphylaxis treatment such as epinephrine and intravenous antihistamines may be indicated.14 Secondary infection can be treated with antibiotics; a formed abscess might need to be drained or debrided.15
There’s No Place Like Home—Because Triatoma bugs have a pliable exoskeleton and can squeeze into small spaces, they commonly infest dwellings where they find multiple attractants: light, heat, carbon dioxide, and lactic acid.8 The more household occupants (including pets), the higher the levels of carbon dioxide and lactic acid, thus the greater the attraction. Infestation of a home can lead to the spread of diseases harbored by Triatoma, including Chagas disease, which is caused by the parasite Trypanosoma cruzi.5
Preventive measures can be taken to reduce the risk and extent of home infestation by Triatoma bugs, including insecticides, a solid foundation, window screens, air conditioning, sealing of cracks and crevices, outdoor light management, and removal of clutter throughout the house.8 Because Triatoma bugs cannot bite through clothing, protective clothing and bug repellent on exposed skin can be employed. Another degree of protection is offered by pest management, especially control of rodents by removing food, water, and nests in areas where triatomine bugs feed off of that population.8,14
Unlike triatomine bugs, wheel bugs tend not to invade houses; therefore, these preventive measures are unnecessary. If a wheel bug is identified, do not engage the arthropod due to the defensive nature of its attack.4,9 Such deliberate avoidance should ensure protection from the wheel bug’s painful bite.
Medical Complications
Although triatomine bugs and wheel bugs are in the same taxonomic family, subsequent infection is unique to Triatoma bugs because they need a blood meal to survive. Because Triatoma bugs feed on mammals, they present an increased opportunity for transmitting the causative agents of infection from hosts on which they have fed.12 The principal parasite transmitted by triatomines is T cruzi, which causes Chagas disease and lives in the gastrointestinal (GI) tract of the insect.5 When a triatomine bug seeks out a mucosal surface to bite, including the mouth, it defecates and urinates during or shortly after feeding, leading to contamination of the initial wound or mucosal surfaces. In addition, Triatoma bugs are vectors for transmission of Serratia marcescans, Bartonella henselae, and Mycobacterium leprae.16
Chagas Disease—This infection has 3 stages: acute, intermediate, and chronic.5 The acute stage can present with symptoms of conjunctivitis, fever, lymphadenopathy, hepatosplenomegaly, and anemia. The intermediate stage typically is asymptomatic. The chronic stage usually involves the heart and GI tract and causes cardiac aneurysms, cardiomegaly, megacolon, and megaesophagus. Initial symptoms can be a localized nodule (chagoma) at the inoculation site, fever, fatigue, lymphadenopathy, and hepatosplenomegaly.2 Unilateral palpebral edema with associated lymphadenopathy (Romaña sign) also can be seen—not to be confused with bilateral swelling in an acute reaction to an insect bite. Romaña sign is pathognomonic of T cruzi infection; bilateral palpebral swelling is typical of an allergic reaction.12
Identification of a triatomine bite is the first step in diagnosing Chagas disease, which can be life-threatening. Among chronic carriers of Chagas disease, 30% develop GI and cardiac symptoms, of which 20% to 30% develop cardiomyopathy, with serious symptoms that present 10 to 20 years after the asymptomatic intermediate phase.2
Chagas disease is endemic to Central and South America but is also seen in North America; 28,000 new cases are reported annually in South America and North America combined. Human migration from endemic areas and from rural to urban areas has promoted the spread of Chagas disease.2 However, patients in the United States have a relatively low risk for Chagas disease, largely because of the quality of housing construction and use of insecticides.
Treatment options for Chagas disease include nifurtimox and benznidazole. Without treatment, the host immune response typically controls acute replication of the parasite but will lead to a chronic state, ultimately involving the heart and GI tract.5
- Vetter R. Kissing bugs (Triatoma) and the skin. Dermatol Online J. 2001;7:6.
- Zemore ZM, Wills BK. Kissing bug bite. StatPearls [Internet]. StatPearlsPublishing; 2023.
- Edwards L, Lynch PJ. Anaphylactic reaction to kissing bug bites. Ariz Med. 1984;41:159-161.
- Smith FD, Miller NG, Carnazzo SJ, et al. Insect bite by Arilus cristatus, a North American reduviid. AMA Arch Derm. 1958;77:324-330. doi:10.1001/archderm.1958.01560030070011
- Nguyen T, Waseem M. Chagas disease. StatPearls [Internet]. StatPearls Publishing; 2022.
- Shields TL, Walsh EN. Kissing bug bite. AMA Arch Derm. 1956;74:14-21. doi:10.1001/archderm.1956.01550070016004
- Beatty NL, Klotz SA. The midnight bite! a kissing bug nightmare. Am J Med. 2018;131:E43-E44. doi:10.1016/j.amjmed.2017.10.013
- Klotz SA, Smith SL, Schmidt JO. Kissing bug intrusions into homes in the Southwest United States. Insects. 2021;12:654. doi:10.3390/insects12070654
- Aldrich JR, Chauhan KR, Zhang A, et al. Exocrine secretions of wheel bugs (Heteroptera: Reduviidae: Arilus spp.): clarification and chemistry. Z Naturforsch C J Biosci. 2013;68:522-526.
- Boggs J. They’re wheel bugs, NOT kissing bugs. Buckeye Yard and Garden onLine [Internet]. September 17, 2020. Accessed May 25, 2023. https://bygl.osu.edu/node/1688
- Weber RW. Allergen of the month—assassin bug. Ann Allergy Asthma Immunol. 2015;115:A9.
- Klotz JH, Dorn PL, Logan JL, et al. “Kissing bugs”: potential disease vectors and cause of anaphylaxis. Clin Infect Dis 2010;50:1629-1634. doi:10.1086/652769
- Anderson C, Belnap C. The kiss of death: a rare case of anaphylaxis to the bite of the “red margined kissing bug”. Hawaii J Med Public Health. 2015;74(9 suppl 2):33-35.
- Moffitt JE, Venarske D, Goddard J, et al. Allergic reactions to Triatoma bites. Ann Allergy Asthma Immunol. 2003;91:122-128. doi:10.1016/s1081-1206(10)62165-5
- Burnett JW, Calton GJ, Morgan RJ. Triatoma: the “kissing bug”. Cutis. 1987;39:399.
- Vieira CB, Praça YR, Bentes K, et al. Triatomines: Trypanosomatids, bacteria, and viruses potential vectors? Front Cell Infect Microbiol. 2018;8:405. doi:10.3389/fcimb.2018.00405
- Vetter R. Kissing bugs (Triatoma) and the skin. Dermatol Online J. 2001;7:6.
- Zemore ZM, Wills BK. Kissing bug bite. StatPearls [Internet]. StatPearlsPublishing; 2023.
- Edwards L, Lynch PJ. Anaphylactic reaction to kissing bug bites. Ariz Med. 1984;41:159-161.
- Smith FD, Miller NG, Carnazzo SJ, et al. Insect bite by Arilus cristatus, a North American reduviid. AMA Arch Derm. 1958;77:324-330. doi:10.1001/archderm.1958.01560030070011
- Nguyen T, Waseem M. Chagas disease. StatPearls [Internet]. StatPearls Publishing; 2022.
- Shields TL, Walsh EN. Kissing bug bite. AMA Arch Derm. 1956;74:14-21. doi:10.1001/archderm.1956.01550070016004
- Beatty NL, Klotz SA. The midnight bite! a kissing bug nightmare. Am J Med. 2018;131:E43-E44. doi:10.1016/j.amjmed.2017.10.013
- Klotz SA, Smith SL, Schmidt JO. Kissing bug intrusions into homes in the Southwest United States. Insects. 2021;12:654. doi:10.3390/insects12070654
- Aldrich JR, Chauhan KR, Zhang A, et al. Exocrine secretions of wheel bugs (Heteroptera: Reduviidae: Arilus spp.): clarification and chemistry. Z Naturforsch C J Biosci. 2013;68:522-526.
- Boggs J. They’re wheel bugs, NOT kissing bugs. Buckeye Yard and Garden onLine [Internet]. September 17, 2020. Accessed May 25, 2023. https://bygl.osu.edu/node/1688
- Weber RW. Allergen of the month—assassin bug. Ann Allergy Asthma Immunol. 2015;115:A9.
- Klotz JH, Dorn PL, Logan JL, et al. “Kissing bugs”: potential disease vectors and cause of anaphylaxis. Clin Infect Dis 2010;50:1629-1634. doi:10.1086/652769
- Anderson C, Belnap C. The kiss of death: a rare case of anaphylaxis to the bite of the “red margined kissing bug”. Hawaii J Med Public Health. 2015;74(9 suppl 2):33-35.
- Moffitt JE, Venarske D, Goddard J, et al. Allergic reactions to Triatoma bites. Ann Allergy Asthma Immunol. 2003;91:122-128. doi:10.1016/s1081-1206(10)62165-5
- Burnett JW, Calton GJ, Morgan RJ. Triatoma: the “kissing bug”. Cutis. 1987;39:399.
- Vieira CB, Praça YR, Bentes K, et al. Triatomines: Trypanosomatids, bacteria, and viruses potential vectors? Front Cell Infect Microbiol. 2018;8:405. doi:10.3389/fcimb.2018.00405
Practice Points
- Triatomine bugs (Triatoma) and the wheel bug (Arilus cristatus) are found throughout North America with a concentration in southern regions.
- Bites of triatomine bugs can cause anaphylaxis; prevention of bites to diminish household infestation is important because sensitization can result in increased severity of anaphylaxis upon subsequent exposure.
- Chagas disease—caused by transmission of the parasite Trypanosoma cruzi—can be a complication of a Triatoma bite in endemic areas; treatments include nifurtimox and benznidazole.
- Left undiagnosed and untreated, Chagas disease can have long-lasting implications for cardiac and gastrointestinal pathology.

Coding the “Spot Check”: Part 2
When the Current Procedural Terminology (CPT) evaluation and management (E/M) reporting rules changed dramatically in January 2021, “bullet counting” became unnecessary and the coding level became based on either the new medical decision making (MDM) table or time spent on all activities relating to the care of the patient on the day of the encounter. 1
To make your documentation more likely to pass audits, explicitly link parts of your documentation to CPT MDM descriptors. Part 1 of this series discussed how to approach the “spot check,” a commonly encountered chief concern (CC) within dermatology, with 2 scenarios presented.2 The American Medical Association3 and American Academy of Dermatology4 have provided education that focuses on how to report a given vignette, but specific examples of documentation with commentary are uncommon. In part 2, we describe how to best code an encounter that includes a “spot check” with other concerns.
Scenario 3: By the Way, Doc
A 34-year-old presents with a new spot on the left cheek that seems to be growing and changing shape rapidly. You examine the patient and discuss treatment options. The documentation reads as follows:
- CC: New spot on left cheek that seems to be growing and changing shape rapidly.
- History: No family history of skin cancer; concerned about scarring, no blood thinner.
- Examination: Irregular tan to brown to black 8-mm macule. No lymphadenopathy.
- Impression: Rule out melanoma (undiagnosed new problem with uncertain prognosis).
- Plan: Discuss risks, benefits, and alternatives, including biopsy (decision regarding minor surgery with identified patient or procedure risk factors) vs a noninvasive gene expression profiling (GEP) melanoma rule-out test. (Based on the decision you and the patient make, you also would document which option was chosen, so a biopsy would include your standard documentation, and if the GEP is chosen, you would simply state that this was chosen and performed.)
As you turn to leave the room, the patient says:“By the way, Doc, can you do anything about these
How would it be best to approach this scenario? It depends on which treatment option the patient chooses.
If you performed a noninvasive GEP melanoma rule-out test, the CPT reporting does not change with the addition of the new problem, and only the codes 99204 (new patient office or other outpatient visit) or 99214 (established patient office or other outpatient visit) would be reported. This would be because, with the original documentation, the number and complexity of problems would be an “undiagnosed new problem with uncertain prognosis,” which would be moderate complexity (column 1, level 4). There are no data that are reviewed or analyzed, which would be straightforward (column 2, level 2). For risk, the discussion of the biopsy as a diagnostic choice should include possible scarring, bleeding, pain, and infection, which would be best described as a decision regarding minor surgery with identified patient or procedure risk factors, given the identified patient concerns, making this of moderate complexity (column 3, level 4).1
Importantly, even if the procedure is not chosen as the final treatment plan, the discussion regarding the surgery, including the risks, benefits, and alternatives, can still count toward this category in the MDM table. Therefore, in this scenario, documentation would best fit with CPT code 99204 for a new patient or 99214 for an established patient. The addition of the psoriasis diagnosis would not change the level of service but also should include documentation of the psoriasis as medically necessary.
However, if you perform the biopsy, then the documentation above would only allow reporting the biopsy, as the decision to perform a 0- or 10-day global procedure is “bundled” with the procedure if performed on the same date of service. Therefore, with the addition of the psoriasis diagnosis, you would now use a separate E/M code to report the psoriasis. You must append a modifier −25 to the E/M code to certify that you are dealing with a separate and discrete problem with no overlap in physician work.
Clearly you also have an E/M to report. But what level? Is this chronic? Yes, as CPT clearly defines chronic as “[a] problem with an expected duration of at least one year or until the death of the patient.”1,5
But is this stable progressive or showing side effects of treatment? “‘Stable’ for the purposes of categorizing MDM is defined by the specific treatment goals for an individual patient. A patient who is not at his or her treatment goal is not stable, even if the condition has not changed and there is no short-term threat to life or function,” according to the CPT descriptors. Therefore, in this scenario, the documentation would best fit a chronic illness with exacerbation, progression, or side effects of treatment (column 1, level 4), which is of moderate complexity.1
But what about column 3, where we look at risks of testing and treatment? This would depend on the type of treatment given. If an over-the-counter product such as a tar gel is recommended, this is a low risk (column 3, level 3), which would mean this lower value determines the E/M code to be 99213 or 99203 depending on whether this is an established or new patient, respectively. If we treat with a prescription medication such as a topical corticosteroid, we are providing prescription drug management (column 3, level 4), which is moderate risk, and we would use codes 99204 or 99214, assuming we document appropriately. Again, including the CPT terminology of “not at treatment goal” in your impression and “prescription drug management” in your plan tells an auditor what you are thinking and doing.1,5
The Takeaway—Clearly if a GEP is performed, there is a single CPT code used—99204 or 99214. If the biopsy is performed, there would be a biopsy code and an E/M code with a modifier −25 attached to the latter. For the documentation below, a 99204 or 99214 would be the chosen E/M code:
- CC: (1) New spot on left cheek that seems to be growing and changing shape rapidly; (2) Silvery spots on elbows, knees, and buttocks for which patient desires treatment.
- History: No family history of skin cancer; concerned about scarring, no blood thinner. Mom has psoriasis. Tried petroleum jelly on scaly areas but no better.
- Examination: Irregular tan to brown to black 8-mm macule. No lymphadenopathy. Silver scaly erythematous plaques on elbows, knees, sacrum.
- Impression: (1) Rule out melanoma (undiagnosed new problem with uncertain prognosis); (2) Psoriasis (chronic disease not at treatment goal).
- Plan: (1) Discuss risks, benefits, and alternatives, including biopsy (decision regarding minor surgery with identified patient or procedure risk factors) vs a noninvasive GEP melanoma rule-out test. Patient wants biopsy. Consent, biopsy via shave technique. Lidocaine hydrochloride 1% with epinephrine 1 cc, prepare and drape, aluminum chloride for hemostasis, ointment and bandage applied, care instructions provided; (2) Discuss options. Calcipotriene cream daily; triamcinolone ointment 0.1% twice a day (prescription drug management). Review bathing, avoiding trauma to site, no picking.
Scenario 4: Here for a Total-Body Screening Examination
Medicare does not cover skin cancer screenings as a primary CC. Being worried or knowing someone with melanoma are not CCs that are covered. However, “spot of concern,” “changing mole,” or ”new growth” would be. Conversely, if the patient has a history of skin cancer, actinic keratoses, or other premalignant lesions, and/or is immunosuppressed or has a high-risk genetic syndrome, the visit may be covered if these factors are documented in the note.6
For the diagnosis, the International Classification of Diseases, Tenth Revision, code Z12.83—“encounter for screening for malignant neoplasm of skin”—is not an appropriate primary billing code. However, D48.5—“neoplasm of behavior of skin”—can be, unless there is a specific diagnosis you are able to make (eg, melanocytic nevus, seborrheic keratosis).6
Let’s look at documentation examples:
- CC: 1-year follow-up on basal cell carcinoma (BCC) excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose and prior BCC treatment site with no sign of recurrence.
- Plan: Reassure. Annual surveillance in 1 year.
Using what we have previously discussed, this would likely be considered CPT code 99212 (established patient office visit). However, it is important to ensure all concerns and treatment interventions are fully documented. Consider this fuller documentation with bolded additions:
- CC: 1-year follow-up on BCC excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion. Also unhappy with generally looking older.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Diffuse changes of chronic sun damage. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose and prior BCC treatment site with no sign of recurrence and heliodermatosis/chronic sun damage not at treat-ment goal.
- Plan: Reassure. Annual surveillance in 1 year. Over-the-counter broad-spectrum sun protection factor 30+ sunscreen daily.
This is better but still possibly confusing to an auditor. Consider instead with bolded additions to the changes to the impression:
- CC: 1-year follow-up on BCC excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion. Also unhappy with generally looking older.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Diffuse changes of chronic sun damage. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose (D22.39)7 and prior BCC treatment site with no sign of recurrence (Z85.828: “personal history of other malignant neoplasm of skin) and heliodermatosis/chronic sun damage not at treatment goal (L57.8: “other skin changes due to chronic exposure to nonionizing radiation”).
- Plan: Reassure. Annual surveillance 1 year. Over-the-counter broad-spectrum sun protection factor 30+ sunscreen daily.
We now have chronic heliodermatitis not at treatment goal, which is moderate (column 1, level 4), and the over-the-counter broad-spectrum sun protection factor 30+ sunscreen (column 1, low) would be best coded as CPT code 99213.
Final Thoughts
“Spot check” encounters are common dermatologic visits, both on their own and in combination with other concerns. With the updated E/M guidelines, it is crucial to clarify and streamline your documentation. In particular, utilize language clearly defining the number and complexity of problems, data to be reviewed and/or analyzed, and appropriate risk stratification to ensure appropriate reimbursement and minimize your difficulties with audits.
- American Medical Association. CPT evaluation and management (E/M) code and guideline changes; 2023. Accessed May 15, 2023. https://www.ama-assn.org/system/files/2023-e-m-descriptors-guidelines.pdf
- Flamm A, Siegel DM. Coding the “spot check”: part 1. Cutis. 2023;111:224-226. doi:10.12788/cutis.0762
- American Medical Association. Evaluation and management (E/M) coding. Accessed May 15, 2023. https://www.ama-assn.org/topics/evaluation-and-management-em-coding
- American Academy of Dermatology Association. Coding resource center. Accessed May 15, 2023. https://www.aad.org/member/practice/coding
- American Medical Association. CPT Professional Edition 2023. American Medical Association; 2022.
- Elizey Coding Solutions, Inc. Dermatology preventive/screening exam visit caution. Updated September 18, 2016. Accessed May 2, 2023. https://www.ellzeycodingsolutions.com/kb_results.asp?ID=9
- 2023 ICD-10-CM diagnosis code D22.39: melanocytic nevi of other parts of the face. Accessed May 2, 2023. https://www.icd10data.com/ICD10CM/Codes/C00-D49/D10-D36/D22-/D22.39
When the Current Procedural Terminology (CPT) evaluation and management (E/M) reporting rules changed dramatically in January 2021, “bullet counting” became unnecessary and the coding level became based on either the new medical decision making (MDM) table or time spent on all activities relating to the care of the patient on the day of the encounter. 1
To make your documentation more likely to pass audits, explicitly link parts of your documentation to CPT MDM descriptors. Part 1 of this series discussed how to approach the “spot check,” a commonly encountered chief concern (CC) within dermatology, with 2 scenarios presented.2 The American Medical Association3 and American Academy of Dermatology4 have provided education that focuses on how to report a given vignette, but specific examples of documentation with commentary are uncommon. In part 2, we describe how to best code an encounter that includes a “spot check” with other concerns.
Scenario 3: By the Way, Doc
A 34-year-old presents with a new spot on the left cheek that seems to be growing and changing shape rapidly. You examine the patient and discuss treatment options. The documentation reads as follows:
- CC: New spot on left cheek that seems to be growing and changing shape rapidly.
- History: No family history of skin cancer; concerned about scarring, no blood thinner.
- Examination: Irregular tan to brown to black 8-mm macule. No lymphadenopathy.
- Impression: Rule out melanoma (undiagnosed new problem with uncertain prognosis).
- Plan: Discuss risks, benefits, and alternatives, including biopsy (decision regarding minor surgery with identified patient or procedure risk factors) vs a noninvasive gene expression profiling (GEP) melanoma rule-out test. (Based on the decision you and the patient make, you also would document which option was chosen, so a biopsy would include your standard documentation, and if the GEP is chosen, you would simply state that this was chosen and performed.)
As you turn to leave the room, the patient says:“By the way, Doc, can you do anything about these
How would it be best to approach this scenario? It depends on which treatment option the patient chooses.
If you performed a noninvasive GEP melanoma rule-out test, the CPT reporting does not change with the addition of the new problem, and only the codes 99204 (new patient office or other outpatient visit) or 99214 (established patient office or other outpatient visit) would be reported. This would be because, with the original documentation, the number and complexity of problems would be an “undiagnosed new problem with uncertain prognosis,” which would be moderate complexity (column 1, level 4). There are no data that are reviewed or analyzed, which would be straightforward (column 2, level 2). For risk, the discussion of the biopsy as a diagnostic choice should include possible scarring, bleeding, pain, and infection, which would be best described as a decision regarding minor surgery with identified patient or procedure risk factors, given the identified patient concerns, making this of moderate complexity (column 3, level 4).1
Importantly, even if the procedure is not chosen as the final treatment plan, the discussion regarding the surgery, including the risks, benefits, and alternatives, can still count toward this category in the MDM table. Therefore, in this scenario, documentation would best fit with CPT code 99204 for a new patient or 99214 for an established patient. The addition of the psoriasis diagnosis would not change the level of service but also should include documentation of the psoriasis as medically necessary.
However, if you perform the biopsy, then the documentation above would only allow reporting the biopsy, as the decision to perform a 0- or 10-day global procedure is “bundled” with the procedure if performed on the same date of service. Therefore, with the addition of the psoriasis diagnosis, you would now use a separate E/M code to report the psoriasis. You must append a modifier −25 to the E/M code to certify that you are dealing with a separate and discrete problem with no overlap in physician work.
Clearly you also have an E/M to report. But what level? Is this chronic? Yes, as CPT clearly defines chronic as “[a] problem with an expected duration of at least one year or until the death of the patient.”1,5
But is this stable progressive or showing side effects of treatment? “‘Stable’ for the purposes of categorizing MDM is defined by the specific treatment goals for an individual patient. A patient who is not at his or her treatment goal is not stable, even if the condition has not changed and there is no short-term threat to life or function,” according to the CPT descriptors. Therefore, in this scenario, the documentation would best fit a chronic illness with exacerbation, progression, or side effects of treatment (column 1, level 4), which is of moderate complexity.1
But what about column 3, where we look at risks of testing and treatment? This would depend on the type of treatment given. If an over-the-counter product such as a tar gel is recommended, this is a low risk (column 3, level 3), which would mean this lower value determines the E/M code to be 99213 or 99203 depending on whether this is an established or new patient, respectively. If we treat with a prescription medication such as a topical corticosteroid, we are providing prescription drug management (column 3, level 4), which is moderate risk, and we would use codes 99204 or 99214, assuming we document appropriately. Again, including the CPT terminology of “not at treatment goal” in your impression and “prescription drug management” in your plan tells an auditor what you are thinking and doing.1,5
The Takeaway—Clearly if a GEP is performed, there is a single CPT code used—99204 or 99214. If the biopsy is performed, there would be a biopsy code and an E/M code with a modifier −25 attached to the latter. For the documentation below, a 99204 or 99214 would be the chosen E/M code:
- CC: (1) New spot on left cheek that seems to be growing and changing shape rapidly; (2) Silvery spots on elbows, knees, and buttocks for which patient desires treatment.
- History: No family history of skin cancer; concerned about scarring, no blood thinner. Mom has psoriasis. Tried petroleum jelly on scaly areas but no better.
- Examination: Irregular tan to brown to black 8-mm macule. No lymphadenopathy. Silver scaly erythematous plaques on elbows, knees, sacrum.
- Impression: (1) Rule out melanoma (undiagnosed new problem with uncertain prognosis); (2) Psoriasis (chronic disease not at treatment goal).
- Plan: (1) Discuss risks, benefits, and alternatives, including biopsy (decision regarding minor surgery with identified patient or procedure risk factors) vs a noninvasive GEP melanoma rule-out test. Patient wants biopsy. Consent, biopsy via shave technique. Lidocaine hydrochloride 1% with epinephrine 1 cc, prepare and drape, aluminum chloride for hemostasis, ointment and bandage applied, care instructions provided; (2) Discuss options. Calcipotriene cream daily; triamcinolone ointment 0.1% twice a day (prescription drug management). Review bathing, avoiding trauma to site, no picking.
Scenario 4: Here for a Total-Body Screening Examination
Medicare does not cover skin cancer screenings as a primary CC. Being worried or knowing someone with melanoma are not CCs that are covered. However, “spot of concern,” “changing mole,” or ”new growth” would be. Conversely, if the patient has a history of skin cancer, actinic keratoses, or other premalignant lesions, and/or is immunosuppressed or has a high-risk genetic syndrome, the visit may be covered if these factors are documented in the note.6
For the diagnosis, the International Classification of Diseases, Tenth Revision, code Z12.83—“encounter for screening for malignant neoplasm of skin”—is not an appropriate primary billing code. However, D48.5—“neoplasm of behavior of skin”—can be, unless there is a specific diagnosis you are able to make (eg, melanocytic nevus, seborrheic keratosis).6
Let’s look at documentation examples:
- CC: 1-year follow-up on basal cell carcinoma (BCC) excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose and prior BCC treatment site with no sign of recurrence.
- Plan: Reassure. Annual surveillance in 1 year.
Using what we have previously discussed, this would likely be considered CPT code 99212 (established patient office visit). However, it is important to ensure all concerns and treatment interventions are fully documented. Consider this fuller documentation with bolded additions:
- CC: 1-year follow-up on BCC excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion. Also unhappy with generally looking older.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Diffuse changes of chronic sun damage. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose and prior BCC treatment site with no sign of recurrence and heliodermatosis/chronic sun damage not at treat-ment goal.
- Plan: Reassure. Annual surveillance in 1 year. Over-the-counter broad-spectrum sun protection factor 30+ sunscreen daily.
This is better but still possibly confusing to an auditor. Consider instead with bolded additions to the changes to the impression:
- CC: 1-year follow-up on BCC excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion. Also unhappy with generally looking older.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Diffuse changes of chronic sun damage. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose (D22.39)7 and prior BCC treatment site with no sign of recurrence (Z85.828: “personal history of other malignant neoplasm of skin) and heliodermatosis/chronic sun damage not at treatment goal (L57.8: “other skin changes due to chronic exposure to nonionizing radiation”).
- Plan: Reassure. Annual surveillance 1 year. Over-the-counter broad-spectrum sun protection factor 30+ sunscreen daily.
We now have chronic heliodermatitis not at treatment goal, which is moderate (column 1, level 4), and the over-the-counter broad-spectrum sun protection factor 30+ sunscreen (column 1, low) would be best coded as CPT code 99213.
Final Thoughts
“Spot check” encounters are common dermatologic visits, both on their own and in combination with other concerns. With the updated E/M guidelines, it is crucial to clarify and streamline your documentation. In particular, utilize language clearly defining the number and complexity of problems, data to be reviewed and/or analyzed, and appropriate risk stratification to ensure appropriate reimbursement and minimize your difficulties with audits.
When the Current Procedural Terminology (CPT) evaluation and management (E/M) reporting rules changed dramatically in January 2021, “bullet counting” became unnecessary and the coding level became based on either the new medical decision making (MDM) table or time spent on all activities relating to the care of the patient on the day of the encounter. 1
To make your documentation more likely to pass audits, explicitly link parts of your documentation to CPT MDM descriptors. Part 1 of this series discussed how to approach the “spot check,” a commonly encountered chief concern (CC) within dermatology, with 2 scenarios presented.2 The American Medical Association3 and American Academy of Dermatology4 have provided education that focuses on how to report a given vignette, but specific examples of documentation with commentary are uncommon. In part 2, we describe how to best code an encounter that includes a “spot check” with other concerns.
Scenario 3: By the Way, Doc
A 34-year-old presents with a new spot on the left cheek that seems to be growing and changing shape rapidly. You examine the patient and discuss treatment options. The documentation reads as follows:
- CC: New spot on left cheek that seems to be growing and changing shape rapidly.
- History: No family history of skin cancer; concerned about scarring, no blood thinner.
- Examination: Irregular tan to brown to black 8-mm macule. No lymphadenopathy.
- Impression: Rule out melanoma (undiagnosed new problem with uncertain prognosis).
- Plan: Discuss risks, benefits, and alternatives, including biopsy (decision regarding minor surgery with identified patient or procedure risk factors) vs a noninvasive gene expression profiling (GEP) melanoma rule-out test. (Based on the decision you and the patient make, you also would document which option was chosen, so a biopsy would include your standard documentation, and if the GEP is chosen, you would simply state that this was chosen and performed.)
As you turn to leave the room, the patient says:“By the way, Doc, can you do anything about these
How would it be best to approach this scenario? It depends on which treatment option the patient chooses.
If you performed a noninvasive GEP melanoma rule-out test, the CPT reporting does not change with the addition of the new problem, and only the codes 99204 (new patient office or other outpatient visit) or 99214 (established patient office or other outpatient visit) would be reported. This would be because, with the original documentation, the number and complexity of problems would be an “undiagnosed new problem with uncertain prognosis,” which would be moderate complexity (column 1, level 4). There are no data that are reviewed or analyzed, which would be straightforward (column 2, level 2). For risk, the discussion of the biopsy as a diagnostic choice should include possible scarring, bleeding, pain, and infection, which would be best described as a decision regarding minor surgery with identified patient or procedure risk factors, given the identified patient concerns, making this of moderate complexity (column 3, level 4).1
Importantly, even if the procedure is not chosen as the final treatment plan, the discussion regarding the surgery, including the risks, benefits, and alternatives, can still count toward this category in the MDM table. Therefore, in this scenario, documentation would best fit with CPT code 99204 for a new patient or 99214 for an established patient. The addition of the psoriasis diagnosis would not change the level of service but also should include documentation of the psoriasis as medically necessary.
However, if you perform the biopsy, then the documentation above would only allow reporting the biopsy, as the decision to perform a 0- or 10-day global procedure is “bundled” with the procedure if performed on the same date of service. Therefore, with the addition of the psoriasis diagnosis, you would now use a separate E/M code to report the psoriasis. You must append a modifier −25 to the E/M code to certify that you are dealing with a separate and discrete problem with no overlap in physician work.
Clearly you also have an E/M to report. But what level? Is this chronic? Yes, as CPT clearly defines chronic as “[a] problem with an expected duration of at least one year or until the death of the patient.”1,5
But is this stable progressive or showing side effects of treatment? “‘Stable’ for the purposes of categorizing MDM is defined by the specific treatment goals for an individual patient. A patient who is not at his or her treatment goal is not stable, even if the condition has not changed and there is no short-term threat to life or function,” according to the CPT descriptors. Therefore, in this scenario, the documentation would best fit a chronic illness with exacerbation, progression, or side effects of treatment (column 1, level 4), which is of moderate complexity.1
But what about column 3, where we look at risks of testing and treatment? This would depend on the type of treatment given. If an over-the-counter product such as a tar gel is recommended, this is a low risk (column 3, level 3), which would mean this lower value determines the E/M code to be 99213 or 99203 depending on whether this is an established or new patient, respectively. If we treat with a prescription medication such as a topical corticosteroid, we are providing prescription drug management (column 3, level 4), which is moderate risk, and we would use codes 99204 or 99214, assuming we document appropriately. Again, including the CPT terminology of “not at treatment goal” in your impression and “prescription drug management” in your plan tells an auditor what you are thinking and doing.1,5
The Takeaway—Clearly if a GEP is performed, there is a single CPT code used—99204 or 99214. If the biopsy is performed, there would be a biopsy code and an E/M code with a modifier −25 attached to the latter. For the documentation below, a 99204 or 99214 would be the chosen E/M code:
- CC: (1) New spot on left cheek that seems to be growing and changing shape rapidly; (2) Silvery spots on elbows, knees, and buttocks for which patient desires treatment.
- History: No family history of skin cancer; concerned about scarring, no blood thinner. Mom has psoriasis. Tried petroleum jelly on scaly areas but no better.
- Examination: Irregular tan to brown to black 8-mm macule. No lymphadenopathy. Silver scaly erythematous plaques on elbows, knees, sacrum.
- Impression: (1) Rule out melanoma (undiagnosed new problem with uncertain prognosis); (2) Psoriasis (chronic disease not at treatment goal).
- Plan: (1) Discuss risks, benefits, and alternatives, including biopsy (decision regarding minor surgery with identified patient or procedure risk factors) vs a noninvasive GEP melanoma rule-out test. Patient wants biopsy. Consent, biopsy via shave technique. Lidocaine hydrochloride 1% with epinephrine 1 cc, prepare and drape, aluminum chloride for hemostasis, ointment and bandage applied, care instructions provided; (2) Discuss options. Calcipotriene cream daily; triamcinolone ointment 0.1% twice a day (prescription drug management). Review bathing, avoiding trauma to site, no picking.
Scenario 4: Here for a Total-Body Screening Examination
Medicare does not cover skin cancer screenings as a primary CC. Being worried or knowing someone with melanoma are not CCs that are covered. However, “spot of concern,” “changing mole,” or ”new growth” would be. Conversely, if the patient has a history of skin cancer, actinic keratoses, or other premalignant lesions, and/or is immunosuppressed or has a high-risk genetic syndrome, the visit may be covered if these factors are documented in the note.6
For the diagnosis, the International Classification of Diseases, Tenth Revision, code Z12.83—“encounter for screening for malignant neoplasm of skin”—is not an appropriate primary billing code. However, D48.5—“neoplasm of behavior of skin”—can be, unless there is a specific diagnosis you are able to make (eg, melanocytic nevus, seborrheic keratosis).6
Let’s look at documentation examples:
- CC: 1-year follow-up on basal cell carcinoma (BCC) excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose and prior BCC treatment site with no sign of recurrence.
- Plan: Reassure. Annual surveillance in 1 year.
Using what we have previously discussed, this would likely be considered CPT code 99212 (established patient office visit). However, it is important to ensure all concerns and treatment interventions are fully documented. Consider this fuller documentation with bolded additions:
- CC: 1-year follow-up on BCC excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion. Also unhappy with generally looking older.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Diffuse changes of chronic sun damage. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose and prior BCC treatment site with no sign of recurrence and heliodermatosis/chronic sun damage not at treat-ment goal.
- Plan: Reassure. Annual surveillance in 1 year. Over-the-counter broad-spectrum sun protection factor 30+ sunscreen daily.
This is better but still possibly confusing to an auditor. Consider instead with bolded additions to the changes to the impression:
- CC: 1-year follow-up on BCC excision and concern about a new spot on the nose.
- History: Notice new spot on the nose; due for annual follow-up and came early for nose lesion. Also unhappy with generally looking older.
- Examination: Left ala with flesh-colored papule dermoscopically banal. Prior left back BCC excision site soft and supple. Diffuse changes of chronic sun damage. Total-body examination performed, except perianal and external genitalia, and is unremarkable.
- Impression: Fibrous papule of nose (D22.39)7 and prior BCC treatment site with no sign of recurrence (Z85.828: “personal history of other malignant neoplasm of skin) and heliodermatosis/chronic sun damage not at treatment goal (L57.8: “other skin changes due to chronic exposure to nonionizing radiation”).
- Plan: Reassure. Annual surveillance 1 year. Over-the-counter broad-spectrum sun protection factor 30+ sunscreen daily.
We now have chronic heliodermatitis not at treatment goal, which is moderate (column 1, level 4), and the over-the-counter broad-spectrum sun protection factor 30+ sunscreen (column 1, low) would be best coded as CPT code 99213.
Final Thoughts
“Spot check” encounters are common dermatologic visits, both on their own and in combination with other concerns. With the updated E/M guidelines, it is crucial to clarify and streamline your documentation. In particular, utilize language clearly defining the number and complexity of problems, data to be reviewed and/or analyzed, and appropriate risk stratification to ensure appropriate reimbursement and minimize your difficulties with audits.
- American Medical Association. CPT evaluation and management (E/M) code and guideline changes; 2023. Accessed May 15, 2023. https://www.ama-assn.org/system/files/2023-e-m-descriptors-guidelines.pdf
- Flamm A, Siegel DM. Coding the “spot check”: part 1. Cutis. 2023;111:224-226. doi:10.12788/cutis.0762
- American Medical Association. Evaluation and management (E/M) coding. Accessed May 15, 2023. https://www.ama-assn.org/topics/evaluation-and-management-em-coding
- American Academy of Dermatology Association. Coding resource center. Accessed May 15, 2023. https://www.aad.org/member/practice/coding
- American Medical Association. CPT Professional Edition 2023. American Medical Association; 2022.
- Elizey Coding Solutions, Inc. Dermatology preventive/screening exam visit caution. Updated September 18, 2016. Accessed May 2, 2023. https://www.ellzeycodingsolutions.com/kb_results.asp?ID=9
- 2023 ICD-10-CM diagnosis code D22.39: melanocytic nevi of other parts of the face. Accessed May 2, 2023. https://www.icd10data.com/ICD10CM/Codes/C00-D49/D10-D36/D22-/D22.39
- American Medical Association. CPT evaluation and management (E/M) code and guideline changes; 2023. Accessed May 15, 2023. https://www.ama-assn.org/system/files/2023-e-m-descriptors-guidelines.pdf
- Flamm A, Siegel DM. Coding the “spot check”: part 1. Cutis. 2023;111:224-226. doi:10.12788/cutis.0762
- American Medical Association. Evaluation and management (E/M) coding. Accessed May 15, 2023. https://www.ama-assn.org/topics/evaluation-and-management-em-coding
- American Academy of Dermatology Association. Coding resource center. Accessed May 15, 2023. https://www.aad.org/member/practice/coding
- American Medical Association. CPT Professional Edition 2023. American Medical Association; 2022.
- Elizey Coding Solutions, Inc. Dermatology preventive/screening exam visit caution. Updated September 18, 2016. Accessed May 2, 2023. https://www.ellzeycodingsolutions.com/kb_results.asp?ID=9
- 2023 ICD-10-CM diagnosis code D22.39: melanocytic nevi of other parts of the face. Accessed May 2, 2023. https://www.icd10data.com/ICD10CM/Codes/C00-D49/D10-D36/D22-/D22.39
Practice Points
- Clear documentation that reflects your thought process is an important component of effective coding and billing.
- Include Current Procedural Terminology–defined language within documentation to help ensure appropriate reimbursement and decrease the risk of audits.

Extensive Erosions and Ulcerations on the Trunk and Extremities in a Neonate
The Diagnosis: Dominant Dystrophic Epidermolysis Bullosa
Blisters in a neonate may be caused by infectious, traumatic, autoimmune, or congenital etiologies. Biopsy findings correlated with clinical findings usually can establish a prompt diagnosis when the clinical diagnosis is uncertain. Direct immunofluorescence (DIF) as well as indirect immunofluorescence studies are useful when autoimmune blistering disease or congenital or heritable disorders of skin fragility are in the differential diagnosis. Many genetic abnormalities of skin fragility are associated with marked morbidity and mortality, and prompt diagnosis is essential to provide proper care. Our patient’s parents had no history of skin disorders, and there was no known family history of blistering disease or traumatic birth. A heritable disorder of skin fragility was still a top consideration because of the extensive blistering in the absence of any other symptoms.
Although dystrophic epidermolysis bullosa (DEB) is an uncommon cause of skin fragility in neonates, our patient’s presentation was typical because of the extensive blistering and increased fragility of the skin at pressure points. Dystrophic epidermolysis bullosa has both dominant and recessive presentations that span a spectrum from mild and focal skin blistering to extensive blistering with esophageal involvement.1 Early diagnosis and treatment can mitigate potential failure to thrive or premature death. Inherited mutations in the type VII collagen gene, COL7A1, are causative.2 Dominant DEB may be associated with dental caries, swallowing problems secondary to esophageal scarring, and constipation, as well as dystrophic or absent nails. Immunomapping studies of the skin often reveal type VII collagen cytoplasmic granules in the epidermis and weaker reaction in the roof of the subepidermal separation (quiz image).3 Abnormalities in type VII collagen impact the production of anchoring fibrils. Blister cleavage occurs in the sublamina densa with type VII collagen staining evident on the blister roof (quiz image).4 Patients with severe generalized recessive DEB may have barely detectable type VII collagen. In our patient, the cytoplasmic staining and weak staining in the epidermal roof of the separation confirmed the clinical impression of dominant DEB.
Autoimmune blistering disease should be considered in the histologic differential diagnosis, but it usually is associated with obvious disease in the mother. Direct immunofluorescence of pemphigoid gestationis reveals linear deposition of C3 at the basement membrane zone, which also can be associated with IgG (Figure 1). Neonates receiving passive transfer of antibodies may develop annular erythema, vesicles, and even dyshidroticlike changes on the soles.5

Suction blisters are subepithelial.6,7 When they occur in the neonatal period, they often are localized and are thought to be the result of vigorous sucking in utero.6 They quickly resolve without treatment and do not reveal abnormalities on DIF. If immunomapping is done for type VII collagen, it will be located at the floor of the suction blister (Figure 2).

Bullous pemphigoid is associated with deposition of linear IgG along the dermoepidermal junction—IgG4 is most common—and/or C3 (Figure 3). Direct immunofluorescence on split-skin biopsy reveals IgG on the epidermal side of the blister in bullous pemphigoid in contrast to epidermolysis bullosa acquisita, where the immune deposits are found on the dermal side of the split.8,9 Linear IgA bullous disease is associated with IgA deposition (Figure 4).10,11 Secretory IgA derived from breast milk can be causative.11 Neonatal linear IgA bullous disease is a serious condition associated with marked mucosal involvement that can eventuate in respiratory compromise. Prompt recognition is important; breastfeeding must be stopped and supportive therapy must be provided.

Other types of vesicular or pustular eruptions in the newborn usually are easily diagnosed by their typical clinical presentation without biopsy. Erythema toxicum neonatorum usually presents within 1 to 2 days of birth. It is self-limited and often resembles acne, but it also occurs on the trunk and extremities. Transient neonatal pustular melanosis may be present at birth and predominantly is seen in newborns with skin of color. Lesions easily rupture and usually resolve within 1 to 2 days. Infectious causes of blistering often can be identified on clinical examination and confirmed by culture. Herpes simplex virus infection is associated with characteristic multinucleated giant cells as well as steel grey nuclei evident on routine histologic evaluation. Bullous impetigo reveals superficial acantholysis and will have negative findings on DIF.12

When a neonate presents with widespread blistering, both genetic disorders of skin fragility as well as passive transfer of antibodies from maternal autoimmune disease need to be considered. Direct immunofluorescence and indirect immunofluorescence immunomapping findings can be useful in clarifying the diagnosis when heritable disorders of skin fragility or autoimmune blistering diseases are a clinical consideration.
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-627. doi:10.1111/bjd.18921
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553-568. doi:10.1111/j.1600-0625.2008.00723.x
- Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol. 2016;136:E65-E71. doi:10.1016/j.jid.2016.05.093
- Rao R, Mellerio J, Bhogal BS, et al. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol. 2012;78:692-697.
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly followup of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168-1172. doi:10.1001/archderm.143.9.1168
- Afsar FS, Cun S, Seremet S. Neonatal sucking blister [published online November 15, 2019]. Dermatol Online J. 2019;25:13030 /qt33b1w59j.
- Yu WY, Wei ML. Suction blisters. JAMA Dermatol. 2019;155:237. doi:10.1001/jamadermatol.2018.3277
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Reis-Filho EG, Silva Tde A, Aguirre LH, et al. Bullous pemphigoid in a 3-month-old infant: case report and literature review of this dermatosis in childhood. An Bras Dermatol. 2013;88:961-965. doi:10.1590/abd1806-4841.20132378
- Hruza LL, Mallory SB, Fitzgibbons J, et al. Linear IgA bullous dermatosis in a neonate. Pediatr Dermatol. 1993;10:171-176. doi:10.1111/j.1525-1470
- Egami S, Suzuki C, Kurihara Y, et al. Neonatal linear IgA bullous dermatosis mediated by breast milk–borne maternal IgA. JAMA Dermatol. 2021;157:1107-1111. doi:10.1001/jamadermatol.2021.2392
- Ligtenberg KG, Hu JK, Panse G, et al. Bullous impetigo masquerading as pemphigus foliaceus in an adult patient. JAAD Case Rep. 2020; 6:428-430. doi:10.1016/j.jdcr.2020.02.040
The Diagnosis: Dominant Dystrophic Epidermolysis Bullosa
Blisters in a neonate may be caused by infectious, traumatic, autoimmune, or congenital etiologies. Biopsy findings correlated with clinical findings usually can establish a prompt diagnosis when the clinical diagnosis is uncertain. Direct immunofluorescence (DIF) as well as indirect immunofluorescence studies are useful when autoimmune blistering disease or congenital or heritable disorders of skin fragility are in the differential diagnosis. Many genetic abnormalities of skin fragility are associated with marked morbidity and mortality, and prompt diagnosis is essential to provide proper care. Our patient’s parents had no history of skin disorders, and there was no known family history of blistering disease or traumatic birth. A heritable disorder of skin fragility was still a top consideration because of the extensive blistering in the absence of any other symptoms.
Although dystrophic epidermolysis bullosa (DEB) is an uncommon cause of skin fragility in neonates, our patient’s presentation was typical because of the extensive blistering and increased fragility of the skin at pressure points. Dystrophic epidermolysis bullosa has both dominant and recessive presentations that span a spectrum from mild and focal skin blistering to extensive blistering with esophageal involvement.1 Early diagnosis and treatment can mitigate potential failure to thrive or premature death. Inherited mutations in the type VII collagen gene, COL7A1, are causative.2 Dominant DEB may be associated with dental caries, swallowing problems secondary to esophageal scarring, and constipation, as well as dystrophic or absent nails. Immunomapping studies of the skin often reveal type VII collagen cytoplasmic granules in the epidermis and weaker reaction in the roof of the subepidermal separation (quiz image).3 Abnormalities in type VII collagen impact the production of anchoring fibrils. Blister cleavage occurs in the sublamina densa with type VII collagen staining evident on the blister roof (quiz image).4 Patients with severe generalized recessive DEB may have barely detectable type VII collagen. In our patient, the cytoplasmic staining and weak staining in the epidermal roof of the separation confirmed the clinical impression of dominant DEB.
Autoimmune blistering disease should be considered in the histologic differential diagnosis, but it usually is associated with obvious disease in the mother. Direct immunofluorescence of pemphigoid gestationis reveals linear deposition of C3 at the basement membrane zone, which also can be associated with IgG (Figure 1). Neonates receiving passive transfer of antibodies may develop annular erythema, vesicles, and even dyshidroticlike changes on the soles.5
Suction blisters are subepithelial.6,7 When they occur in the neonatal period, they often are localized and are thought to be the result of vigorous sucking in utero.6 They quickly resolve without treatment and do not reveal abnormalities on DIF. If immunomapping is done for type VII collagen, it will be located at the floor of the suction blister (Figure 2).
Bullous pemphigoid is associated with deposition of linear IgG along the dermoepidermal junction—IgG4 is most common—and/or C3 (Figure 3). Direct immunofluorescence on split-skin biopsy reveals IgG on the epidermal side of the blister in bullous pemphigoid in contrast to epidermolysis bullosa acquisita, where the immune deposits are found on the dermal side of the split.8,9 Linear IgA bullous disease is associated with IgA deposition (Figure 4).10,11 Secretory IgA derived from breast milk can be causative.11 Neonatal linear IgA bullous disease is a serious condition associated with marked mucosal involvement that can eventuate in respiratory compromise. Prompt recognition is important; breastfeeding must be stopped and supportive therapy must be provided.
Other types of vesicular or pustular eruptions in the newborn usually are easily diagnosed by their typical clinical presentation without biopsy. Erythema toxicum neonatorum usually presents within 1 to 2 days of birth. It is self-limited and often resembles acne, but it also occurs on the trunk and extremities. Transient neonatal pustular melanosis may be present at birth and predominantly is seen in newborns with skin of color. Lesions easily rupture and usually resolve within 1 to 2 days. Infectious causes of blistering often can be identified on clinical examination and confirmed by culture. Herpes simplex virus infection is associated with characteristic multinucleated giant cells as well as steel grey nuclei evident on routine histologic evaluation. Bullous impetigo reveals superficial acantholysis and will have negative findings on DIF.12
When a neonate presents with widespread blistering, both genetic disorders of skin fragility as well as passive transfer of antibodies from maternal autoimmune disease need to be considered. Direct immunofluorescence and indirect immunofluorescence immunomapping findings can be useful in clarifying the diagnosis when heritable disorders of skin fragility or autoimmune blistering diseases are a clinical consideration.
The Diagnosis: Dominant Dystrophic Epidermolysis Bullosa
Blisters in a neonate may be caused by infectious, traumatic, autoimmune, or congenital etiologies. Biopsy findings correlated with clinical findings usually can establish a prompt diagnosis when the clinical diagnosis is uncertain. Direct immunofluorescence (DIF) as well as indirect immunofluorescence studies are useful when autoimmune blistering disease or congenital or heritable disorders of skin fragility are in the differential diagnosis. Many genetic abnormalities of skin fragility are associated with marked morbidity and mortality, and prompt diagnosis is essential to provide proper care. Our patient’s parents had no history of skin disorders, and there was no known family history of blistering disease or traumatic birth. A heritable disorder of skin fragility was still a top consideration because of the extensive blistering in the absence of any other symptoms.
Although dystrophic epidermolysis bullosa (DEB) is an uncommon cause of skin fragility in neonates, our patient’s presentation was typical because of the extensive blistering and increased fragility of the skin at pressure points. Dystrophic epidermolysis bullosa has both dominant and recessive presentations that span a spectrum from mild and focal skin blistering to extensive blistering with esophageal involvement.1 Early diagnosis and treatment can mitigate potential failure to thrive or premature death. Inherited mutations in the type VII collagen gene, COL7A1, are causative.2 Dominant DEB may be associated with dental caries, swallowing problems secondary to esophageal scarring, and constipation, as well as dystrophic or absent nails. Immunomapping studies of the skin often reveal type VII collagen cytoplasmic granules in the epidermis and weaker reaction in the roof of the subepidermal separation (quiz image).3 Abnormalities in type VII collagen impact the production of anchoring fibrils. Blister cleavage occurs in the sublamina densa with type VII collagen staining evident on the blister roof (quiz image).4 Patients with severe generalized recessive DEB may have barely detectable type VII collagen. In our patient, the cytoplasmic staining and weak staining in the epidermal roof of the separation confirmed the clinical impression of dominant DEB.
Autoimmune blistering disease should be considered in the histologic differential diagnosis, but it usually is associated with obvious disease in the mother. Direct immunofluorescence of pemphigoid gestationis reveals linear deposition of C3 at the basement membrane zone, which also can be associated with IgG (Figure 1). Neonates receiving passive transfer of antibodies may develop annular erythema, vesicles, and even dyshidroticlike changes on the soles.5
Suction blisters are subepithelial.6,7 When they occur in the neonatal period, they often are localized and are thought to be the result of vigorous sucking in utero.6 They quickly resolve without treatment and do not reveal abnormalities on DIF. If immunomapping is done for type VII collagen, it will be located at the floor of the suction blister (Figure 2).
Bullous pemphigoid is associated with deposition of linear IgG along the dermoepidermal junction—IgG4 is most common—and/or C3 (Figure 3). Direct immunofluorescence on split-skin biopsy reveals IgG on the epidermal side of the blister in bullous pemphigoid in contrast to epidermolysis bullosa acquisita, where the immune deposits are found on the dermal side of the split.8,9 Linear IgA bullous disease is associated with IgA deposition (Figure 4).10,11 Secretory IgA derived from breast milk can be causative.11 Neonatal linear IgA bullous disease is a serious condition associated with marked mucosal involvement that can eventuate in respiratory compromise. Prompt recognition is important; breastfeeding must be stopped and supportive therapy must be provided.
Other types of vesicular or pustular eruptions in the newborn usually are easily diagnosed by their typical clinical presentation without biopsy. Erythema toxicum neonatorum usually presents within 1 to 2 days of birth. It is self-limited and often resembles acne, but it also occurs on the trunk and extremities. Transient neonatal pustular melanosis may be present at birth and predominantly is seen in newborns with skin of color. Lesions easily rupture and usually resolve within 1 to 2 days. Infectious causes of blistering often can be identified on clinical examination and confirmed by culture. Herpes simplex virus infection is associated with characteristic multinucleated giant cells as well as steel grey nuclei evident on routine histologic evaluation. Bullous impetigo reveals superficial acantholysis and will have negative findings on DIF.12
When a neonate presents with widespread blistering, both genetic disorders of skin fragility as well as passive transfer of antibodies from maternal autoimmune disease need to be considered. Direct immunofluorescence and indirect immunofluorescence immunomapping findings can be useful in clarifying the diagnosis when heritable disorders of skin fragility or autoimmune blistering diseases are a clinical consideration.
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-627. doi:10.1111/bjd.18921
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553-568. doi:10.1111/j.1600-0625.2008.00723.x
- Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol. 2016;136:E65-E71. doi:10.1016/j.jid.2016.05.093
- Rao R, Mellerio J, Bhogal BS, et al. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol. 2012;78:692-697.
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly followup of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168-1172. doi:10.1001/archderm.143.9.1168
- Afsar FS, Cun S, Seremet S. Neonatal sucking blister [published online November 15, 2019]. Dermatol Online J. 2019;25:13030 /qt33b1w59j.
- Yu WY, Wei ML. Suction blisters. JAMA Dermatol. 2019;155:237. doi:10.1001/jamadermatol.2018.3277
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Reis-Filho EG, Silva Tde A, Aguirre LH, et al. Bullous pemphigoid in a 3-month-old infant: case report and literature review of this dermatosis in childhood. An Bras Dermatol. 2013;88:961-965. doi:10.1590/abd1806-4841.20132378
- Hruza LL, Mallory SB, Fitzgibbons J, et al. Linear IgA bullous dermatosis in a neonate. Pediatr Dermatol. 1993;10:171-176. doi:10.1111/j.1525-1470
- Egami S, Suzuki C, Kurihara Y, et al. Neonatal linear IgA bullous dermatosis mediated by breast milk–borne maternal IgA. JAMA Dermatol. 2021;157:1107-1111. doi:10.1001/jamadermatol.2021.2392
- Ligtenberg KG, Hu JK, Panse G, et al. Bullous impetigo masquerading as pemphigus foliaceus in an adult patient. JAAD Case Rep. 2020; 6:428-430. doi:10.1016/j.jdcr.2020.02.040
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-627. doi:10.1111/bjd.18921
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553-568. doi:10.1111/j.1600-0625.2008.00723.x
- Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol. 2016;136:E65-E71. doi:10.1016/j.jid.2016.05.093
- Rao R, Mellerio J, Bhogal BS, et al. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol. 2012;78:692-697.
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly followup of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168-1172. doi:10.1001/archderm.143.9.1168
- Afsar FS, Cun S, Seremet S. Neonatal sucking blister [published online November 15, 2019]. Dermatol Online J. 2019;25:13030 /qt33b1w59j.
- Yu WY, Wei ML. Suction blisters. JAMA Dermatol. 2019;155:237. doi:10.1001/jamadermatol.2018.3277
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Reis-Filho EG, Silva Tde A, Aguirre LH, et al. Bullous pemphigoid in a 3-month-old infant: case report and literature review of this dermatosis in childhood. An Bras Dermatol. 2013;88:961-965. doi:10.1590/abd1806-4841.20132378
- Hruza LL, Mallory SB, Fitzgibbons J, et al. Linear IgA bullous dermatosis in a neonate. Pediatr Dermatol. 1993;10:171-176. doi:10.1111/j.1525-1470
- Egami S, Suzuki C, Kurihara Y, et al. Neonatal linear IgA bullous dermatosis mediated by breast milk–borne maternal IgA. JAMA Dermatol. 2021;157:1107-1111. doi:10.1001/jamadermatol.2021.2392
- Ligtenberg KG, Hu JK, Panse G, et al. Bullous impetigo masquerading as pemphigus foliaceus in an adult patient. JAAD Case Rep. 2020; 6:428-430. doi:10.1016/j.jdcr.2020.02.040
A neonate was born with extensive erosions and ulcerations on the trunk and extremities. The eroded areas had a beefy red appearance. A biopsy taken from a small blister was stained for type VII collagen by indirect immunofluorescence.
.")
.")