User login
Intermittent pain and stiffness
The history and findings in this case are consistent with a diagnosis of psoriatic spondylitis.
Psoriatic spondylitis is a form of psoriatic arthritis (PsA) that affects the spine and the joints in the pelvis (axial involvement). PsA is a chronic, heterogeneous condition that affects approximately 25%-30% of patients with psoriasis, particularly those with severe psoriasis or nail or scalp involvement. It is characterized by musculoskeletal inflammation (arthritis, enthesitis, spondylitis, and dactylitis). PsA is a spondyloarthritis that can be found either in the peripheral or axial skeleton. If not treated, it may result in permanent joint damage and loss of function.
Patients with PsA may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Common symptoms of axial involvement in PsA include morning back/neck stiffness that lasts longer than 30 minutes, neck or back pain that improves with activity and worsens after prolonged inactivity, and diminished mobility. PsA affects men and women equally, and typically develops when patients are between 30 and 50 years of age. As with psoriasis, PsA is associated with numerous comorbidities, such as cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
The diagnosis of psoriatic spondylitis is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosus). Useful imaging tools for evaluating patients with PsA include plain radiography, CT, ultrasound, and MRI. Although MRI and ultrasound may be more sensitive than plain radiography for detecting early joint inflammation and damage and axial changes, including sacroiliitis, they are not mandatory for a diagnosis of PsA to be made.
International guidelines have been developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA), the European Alliance of Associations for Rheumatology (EULAR), and the Assessment of Spondyloarthritis International Society to guide the treatment of axial PsA. The goals of treatment include minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; improving functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Treatment options for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or have erosive disease or other indications of high disease activity, guidelines recommend initiation of a tumor necrosis factor (TNF) inhibitor (eg, adalimumab, etanercept, infliximab, golimumab, certolizumab pegol). Disease-modifying antirheumatic drugs (eg, methotrexate) are not routinely prescribed for patients with axial disease because they have not been shown to be effective. In patients with significant skin involvement, treatment with interleukin-17A inhibitors may be preferred to TNF inhibitors.
If patients have an inadequate response to a first trial of a TNF inhibitor, guidelines recommend trying a second TNF inhibitor before switching to a different class of biologic. For patients who do not respond to TNF inhibitors, a Janus kinase inhibitor (tofacitinib) may be considered. Additionally, nonpharmacologic therapies (eg, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are consistent with a diagnosis of psoriatic spondylitis.
Psoriatic spondylitis is a form of psoriatic arthritis (PsA) that affects the spine and the joints in the pelvis (axial involvement). PsA is a chronic, heterogeneous condition that affects approximately 25%-30% of patients with psoriasis, particularly those with severe psoriasis or nail or scalp involvement. It is characterized by musculoskeletal inflammation (arthritis, enthesitis, spondylitis, and dactylitis). PsA is a spondyloarthritis that can be found either in the peripheral or axial skeleton. If not treated, it may result in permanent joint damage and loss of function.
Patients with PsA may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Common symptoms of axial involvement in PsA include morning back/neck stiffness that lasts longer than 30 minutes, neck or back pain that improves with activity and worsens after prolonged inactivity, and diminished mobility. PsA affects men and women equally, and typically develops when patients are between 30 and 50 years of age. As with psoriasis, PsA is associated with numerous comorbidities, such as cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
The diagnosis of psoriatic spondylitis is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosus). Useful imaging tools for evaluating patients with PsA include plain radiography, CT, ultrasound, and MRI. Although MRI and ultrasound may be more sensitive than plain radiography for detecting early joint inflammation and damage and axial changes, including sacroiliitis, they are not mandatory for a diagnosis of PsA to be made.
International guidelines have been developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA), the European Alliance of Associations for Rheumatology (EULAR), and the Assessment of Spondyloarthritis International Society to guide the treatment of axial PsA. The goals of treatment include minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; improving functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Treatment options for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or have erosive disease or other indications of high disease activity, guidelines recommend initiation of a tumor necrosis factor (TNF) inhibitor (eg, adalimumab, etanercept, infliximab, golimumab, certolizumab pegol). Disease-modifying antirheumatic drugs (eg, methotrexate) are not routinely prescribed for patients with axial disease because they have not been shown to be effective. In patients with significant skin involvement, treatment with interleukin-17A inhibitors may be preferred to TNF inhibitors.
If patients have an inadequate response to a first trial of a TNF inhibitor, guidelines recommend trying a second TNF inhibitor before switching to a different class of biologic. For patients who do not respond to TNF inhibitors, a Janus kinase inhibitor (tofacitinib) may be considered. Additionally, nonpharmacologic therapies (eg, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are consistent with a diagnosis of psoriatic spondylitis.
Psoriatic spondylitis is a form of psoriatic arthritis (PsA) that affects the spine and the joints in the pelvis (axial involvement). PsA is a chronic, heterogeneous condition that affects approximately 25%-30% of patients with psoriasis, particularly those with severe psoriasis or nail or scalp involvement. It is characterized by musculoskeletal inflammation (arthritis, enthesitis, spondylitis, and dactylitis). PsA is a spondyloarthritis that can be found either in the peripheral or axial skeleton. If not treated, it may result in permanent joint damage and loss of function.
Patients with PsA may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Common symptoms of axial involvement in PsA include morning back/neck stiffness that lasts longer than 30 minutes, neck or back pain that improves with activity and worsens after prolonged inactivity, and diminished mobility. PsA affects men and women equally, and typically develops when patients are between 30 and 50 years of age. As with psoriasis, PsA is associated with numerous comorbidities, such as cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
The diagnosis of psoriatic spondylitis is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosus). Useful imaging tools for evaluating patients with PsA include plain radiography, CT, ultrasound, and MRI. Although MRI and ultrasound may be more sensitive than plain radiography for detecting early joint inflammation and damage and axial changes, including sacroiliitis, they are not mandatory for a diagnosis of PsA to be made.
International guidelines have been developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA), the European Alliance of Associations for Rheumatology (EULAR), and the Assessment of Spondyloarthritis International Society to guide the treatment of axial PsA. The goals of treatment include minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; improving functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Treatment options for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or have erosive disease or other indications of high disease activity, guidelines recommend initiation of a tumor necrosis factor (TNF) inhibitor (eg, adalimumab, etanercept, infliximab, golimumab, certolizumab pegol). Disease-modifying antirheumatic drugs (eg, methotrexate) are not routinely prescribed for patients with axial disease because they have not been shown to be effective. In patients with significant skin involvement, treatment with interleukin-17A inhibitors may be preferred to TNF inhibitors.
If patients have an inadequate response to a first trial of a TNF inhibitor, guidelines recommend trying a second TNF inhibitor before switching to a different class of biologic. For patients who do not respond to TNF inhibitors, a Janus kinase inhibitor (tofacitinib) may be considered. Additionally, nonpharmacologic therapies (eg, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
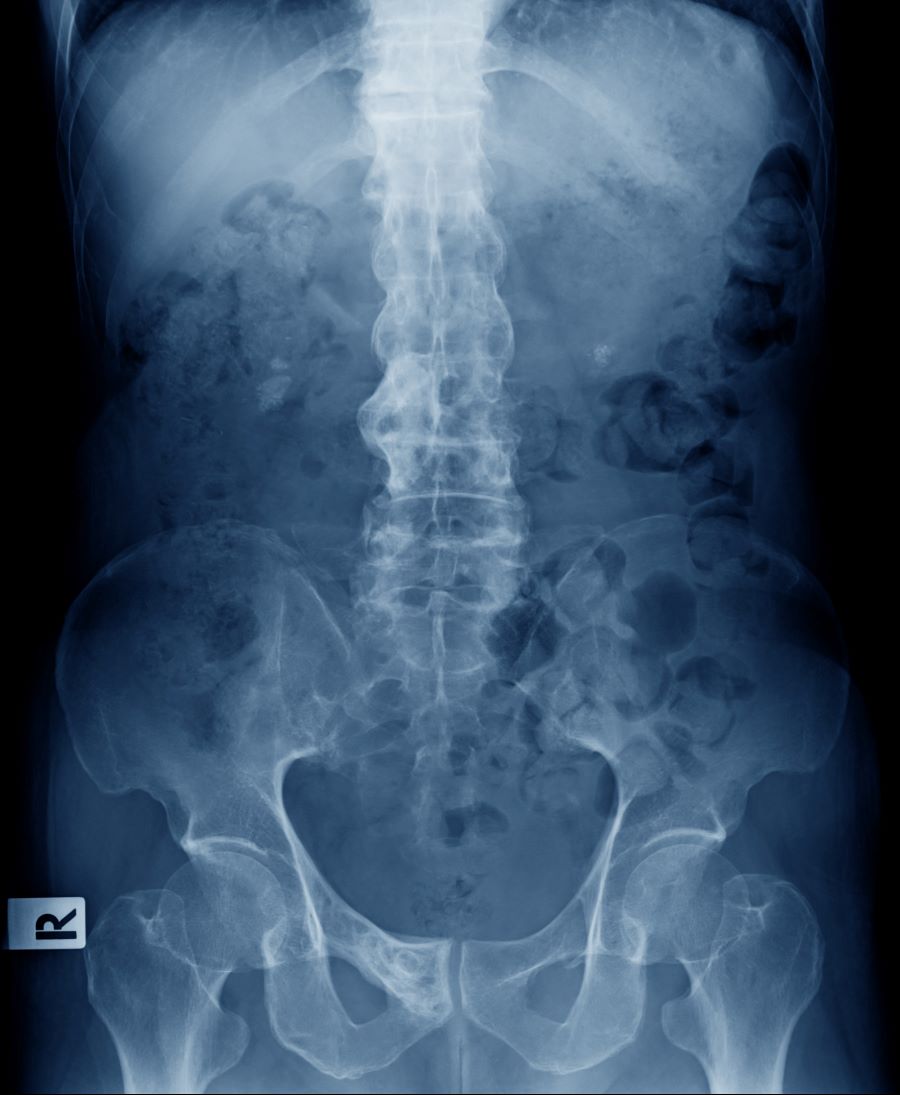
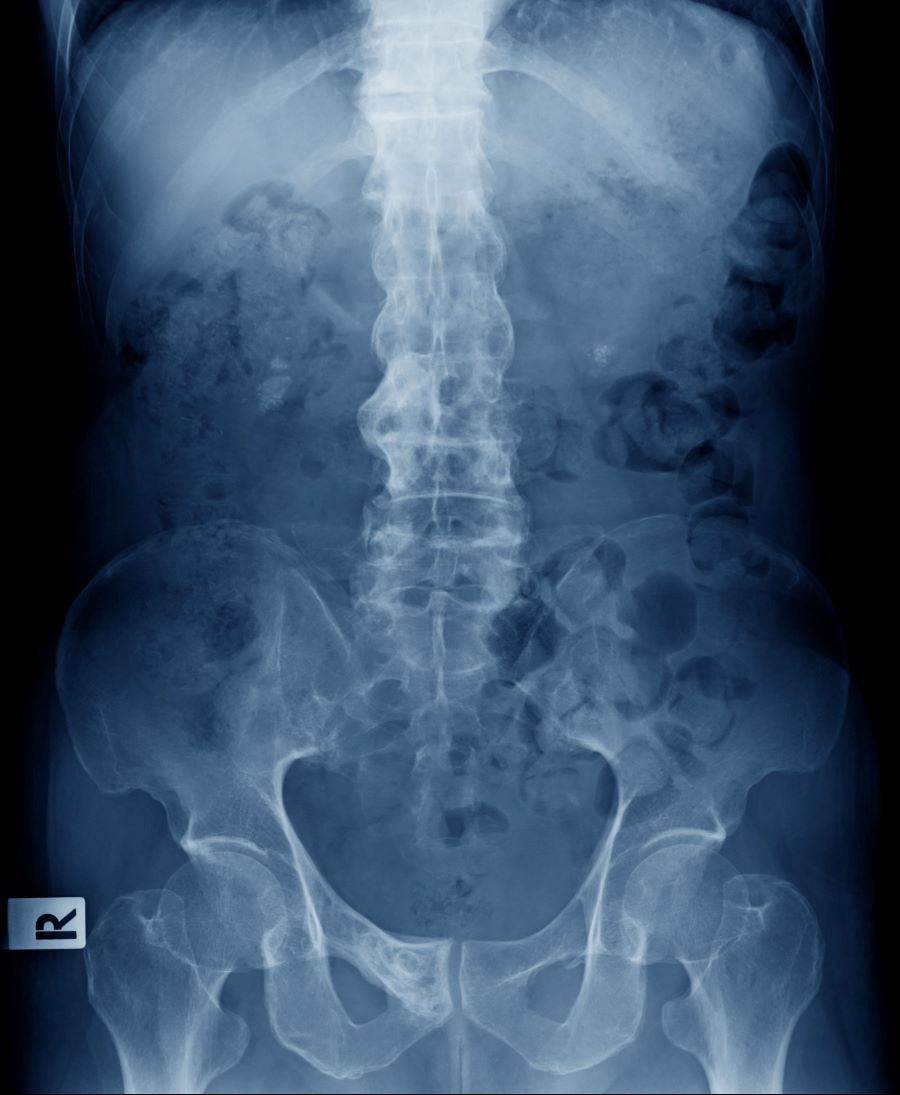
A 41-year-old man with a 5-year history of moderate to severe scalp psoriasis presents with complaints of intermittent pain and stiffness in his left hip and lower back of approximately 6 months' duration. The patient states that his back pain has been severe enough to wake him up on several occasions. Treatment with over-the-counter ibuprofen is moderately effective at relieving his pain. He also reports morning back stiffness that improves with motion, usually within an hour of awakening. The patient reports no fever, pain, swelling, or worsening of his scalp psoriasis. He is not aware of any injury or other triggering factor for his back pain. He takes an over-the-counter multivitamin daily and treats his scalp psoriasis with fluocinolone acetonide 0.01% oil. The patient is 5 ft 9 in and weighs 176 lb (BMI 26).
Physical examination reveals tenderness in the lumbar spine and associated decreased range of motion, as well as psoriatic plaques on the scalp. Vital signs are within normal ranges. Pertinent laboratory findings include erythrocyte sedimentation rate of 19 mm/h and C-reactive protein of 10 mg/L. Rheumatoid factor, antinuclear antibody, and anti-cyclic citrullinated peptide antibody were negative. Radiographic findings include sacroiliitis and bulky nonmarginal syndesmophytes.
Long-term Remission of Pyoderma Gangrenosum, Acne, and Hidradenitis Suppurativa Syndrome
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.

Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.

After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.

The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.

Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.

PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.
Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.
After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.
The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.
Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.
PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.
Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.
After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.
The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.
Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.
PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
Practice Points
- Despite phenotypic similarities among pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (PASH) syndrome; pyogenic arthritis, PG, and acne syndrome; and pyogenic arthritis–PASH syndrome, there are genotypic differences that contribute to unique inflammatory cytokine patterns and the need for distinct pharmacologic considerations within each entity.
- When formulating therapeutic regimens for patients with PASH syndrome, it is essential for dermatologists to consider the likelihood of hyperactivity in multiple pathways of the innate immune system and utilize a combination of multimodal antiinflammatory therapies.

Crusted scalp rash

Dermoscopy showed not only the erythema, inflammation, and crusting visible during the initial examination, but it also revealed that each lesion had a hair growing through it. This pointed to a diagnosis of superficial folliculitis of the scalp.
The physician ruled out tinea capitis, acne keloidalis nuchae, and scarring alopecia based on the dermoscopic exam. There were no broken hairs that one would expect with tinea capitis. Also, there was no polytrichia (multiple hairs pushed into a single follicular opening due to scarring of the skin) that would be expected with acne keloidalis nuchae and scarring alopecias.
There are multiple types of scalp folliculitis. This patient had superficial folliculitis, in which pustules develop at the ostium of the hair follicles. Deep folliculitis is more severe and includes furuncles and carbuncles.1
Folliculitis is usually caused by a bacterial infection and, less commonly, fungal infection. In addition to superficial and deep folliculitis, inflammation with scarring of the follicles occurs with folliculitis decalvans, which is one of the scarring alopecias.1
Mild cases of superficial bacterial folliculitis are treated with topical antibiotics (eg, topical clindamycin 1% applied twice daily). Depending on the severity, oral antibiotics including doxycycline 100 mg twice daily for 7 days or trimethoprim sulfamethoxazole 160 mg/800 mg (double strength) twice daily for 7 days may be used. There is also a chronic nonscarring form of scalp folliculitis that often responds initially to antibiotics but then recurs. This has been treated with longer courses of oral antibiotics and, if the lesions don’t respond or continue to recur, with low-dose isotretinoin.2
Due to the amount of scalp involvement, crusting, and inflammation seen on this patient’s scalp, he was treated with trimethoprim sulfamethoxazole 160 mg/800 mg twice daily for 7 days. After 1 week, he reported that he was doing much better and that the lesions had nearly resolved. He was told to return for reevaluation if the lesions did not completely resolve.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Lugović-Mihić L, Barisić F, Bulat V, et al. Differential diagnosis of the scalp hair folliculitis. Acta Clin Croat. 2011;50:395-402.
2. Romero-Maté A, Arias-Palomo D, Hernández-Núñez A, et al. Chronic nonscarring scalp folliculitis: retrospective case series study of 34 cases. J Am Acad Dermatol. 2019;81:1023-1024. doi: 10.1016/j.jaad.2019.02.065
Dermoscopy showed not only the erythema, inflammation, and crusting visible during the initial examination, but it also revealed that each lesion had a hair growing through it. This pointed to a diagnosis of superficial folliculitis of the scalp.
The physician ruled out tinea capitis, acne keloidalis nuchae, and scarring alopecia based on the dermoscopic exam. There were no broken hairs that one would expect with tinea capitis. Also, there was no polytrichia (multiple hairs pushed into a single follicular opening due to scarring of the skin) that would be expected with acne keloidalis nuchae and scarring alopecias.
There are multiple types of scalp folliculitis. This patient had superficial folliculitis, in which pustules develop at the ostium of the hair follicles. Deep folliculitis is more severe and includes furuncles and carbuncles.1
Folliculitis is usually caused by a bacterial infection and, less commonly, fungal infection. In addition to superficial and deep folliculitis, inflammation with scarring of the follicles occurs with folliculitis decalvans, which is one of the scarring alopecias.1
Mild cases of superficial bacterial folliculitis are treated with topical antibiotics (eg, topical clindamycin 1% applied twice daily). Depending on the severity, oral antibiotics including doxycycline 100 mg twice daily for 7 days or trimethoprim sulfamethoxazole 160 mg/800 mg (double strength) twice daily for 7 days may be used. There is also a chronic nonscarring form of scalp folliculitis that often responds initially to antibiotics but then recurs. This has been treated with longer courses of oral antibiotics and, if the lesions don’t respond or continue to recur, with low-dose isotretinoin.2
Due to the amount of scalp involvement, crusting, and inflammation seen on this patient’s scalp, he was treated with trimethoprim sulfamethoxazole 160 mg/800 mg twice daily for 7 days. After 1 week, he reported that he was doing much better and that the lesions had nearly resolved. He was told to return for reevaluation if the lesions did not completely resolve.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
Dermoscopy showed not only the erythema, inflammation, and crusting visible during the initial examination, but it also revealed that each lesion had a hair growing through it. This pointed to a diagnosis of superficial folliculitis of the scalp.
The physician ruled out tinea capitis, acne keloidalis nuchae, and scarring alopecia based on the dermoscopic exam. There were no broken hairs that one would expect with tinea capitis. Also, there was no polytrichia (multiple hairs pushed into a single follicular opening due to scarring of the skin) that would be expected with acne keloidalis nuchae and scarring alopecias.
There are multiple types of scalp folliculitis. This patient had superficial folliculitis, in which pustules develop at the ostium of the hair follicles. Deep folliculitis is more severe and includes furuncles and carbuncles.1
Folliculitis is usually caused by a bacterial infection and, less commonly, fungal infection. In addition to superficial and deep folliculitis, inflammation with scarring of the follicles occurs with folliculitis decalvans, which is one of the scarring alopecias.1
Mild cases of superficial bacterial folliculitis are treated with topical antibiotics (eg, topical clindamycin 1% applied twice daily). Depending on the severity, oral antibiotics including doxycycline 100 mg twice daily for 7 days or trimethoprim sulfamethoxazole 160 mg/800 mg (double strength) twice daily for 7 days may be used. There is also a chronic nonscarring form of scalp folliculitis that often responds initially to antibiotics but then recurs. This has been treated with longer courses of oral antibiotics and, if the lesions don’t respond or continue to recur, with low-dose isotretinoin.2
Due to the amount of scalp involvement, crusting, and inflammation seen on this patient’s scalp, he was treated with trimethoprim sulfamethoxazole 160 mg/800 mg twice daily for 7 days. After 1 week, he reported that he was doing much better and that the lesions had nearly resolved. He was told to return for reevaluation if the lesions did not completely resolve.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Lugović-Mihić L, Barisić F, Bulat V, et al. Differential diagnosis of the scalp hair folliculitis. Acta Clin Croat. 2011;50:395-402.
2. Romero-Maté A, Arias-Palomo D, Hernández-Núñez A, et al. Chronic nonscarring scalp folliculitis: retrospective case series study of 34 cases. J Am Acad Dermatol. 2019;81:1023-1024. doi: 10.1016/j.jaad.2019.02.065
1. Lugović-Mihić L, Barisić F, Bulat V, et al. Differential diagnosis of the scalp hair folliculitis. Acta Clin Croat. 2011;50:395-402.
2. Romero-Maté A, Arias-Palomo D, Hernández-Núñez A, et al. Chronic nonscarring scalp folliculitis: retrospective case series study of 34 cases. J Am Acad Dermatol. 2019;81:1023-1024. doi: 10.1016/j.jaad.2019.02.065

Persistent scaling rash

The clinical pattern of a scaly herald patch predating the eruption of multiple scaly macules is the hallmark of pityriasis rosea (PR). This patient’s severe itching is also classic for PR.
PR’s etiology is believed to be a reactivation of infection from human herpes viruses 6 and 7.1 Prodromal viral symptoms of malaise, sore throat, myalgias, and fever are common.2 Along with the prodromal symptoms, there is often a several-centimeter herald patch that occurs on the trunk. It is often confused with eczema or tinea due to its erythema and scale. (Secondary syphilis is also in the differential.) Sometimes PR can be differentiated by the scale pattern being a collarette instead of diffuse. The diagnosis becomes clearer 1 to 2 weeks later with the onset of multiple small scaly macules across the trunk following the Langer’s skin lines. The course is self-limited but takes several weeks to months to resolve.
If severe, PR may be treated with acyclovir 800 mg orally 5 times daily for 5 days; this is the same regimen for treating varicella zoster (shingles).1,2 Estimated recurrence rates are 4% to 24%.1,3
At age 49 years, this woman was older than the average patient with PR, as the usual age range is 10 to 35 years.1 Her physician advised her that the outbreak might recur. She was also given a prescription for oral hydroxyzine 25 mg to be taken at bedtime if the itching was interfering with her sleep. Her physician told her to return for reevaluation if the rash did not resolve in 3 months. She did not return for reevaluation.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Drago F, Ciccarese G, Parodi A. Commentary on: "pityriasis rosea recurrence is much higher than previously known: a prospective study." Acta Derm Venereol. 2019;99:1053-1054. doi: 10.2340/00015555-3265
2. Villalon-Gomez JM. Pityriasis rosea: diagnosis and treatment. Am Fam Physician. 2018;97:38-44.
3. Yüksel M. Pityriasis rosea recurrence is much higher than previously known: a prospective study. Acta Derm Venereol. 2019;99:664-667. doi: 10.2340/00015555-3169
The clinical pattern of a scaly herald patch predating the eruption of multiple scaly macules is the hallmark of pityriasis rosea (PR). This patient’s severe itching is also classic for PR.
PR’s etiology is believed to be a reactivation of infection from human herpes viruses 6 and 7.1 Prodromal viral symptoms of malaise, sore throat, myalgias, and fever are common.2 Along with the prodromal symptoms, there is often a several-centimeter herald patch that occurs on the trunk. It is often confused with eczema or tinea due to its erythema and scale. (Secondary syphilis is also in the differential.) Sometimes PR can be differentiated by the scale pattern being a collarette instead of diffuse. The diagnosis becomes clearer 1 to 2 weeks later with the onset of multiple small scaly macules across the trunk following the Langer’s skin lines. The course is self-limited but takes several weeks to months to resolve.
If severe, PR may be treated with acyclovir 800 mg orally 5 times daily for 5 days; this is the same regimen for treating varicella zoster (shingles).1,2 Estimated recurrence rates are 4% to 24%.1,3
At age 49 years, this woman was older than the average patient with PR, as the usual age range is 10 to 35 years.1 Her physician advised her that the outbreak might recur. She was also given a prescription for oral hydroxyzine 25 mg to be taken at bedtime if the itching was interfering with her sleep. Her physician told her to return for reevaluation if the rash did not resolve in 3 months. She did not return for reevaluation.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
The clinical pattern of a scaly herald patch predating the eruption of multiple scaly macules is the hallmark of pityriasis rosea (PR). This patient’s severe itching is also classic for PR.
PR’s etiology is believed to be a reactivation of infection from human herpes viruses 6 and 7.1 Prodromal viral symptoms of malaise, sore throat, myalgias, and fever are common.2 Along with the prodromal symptoms, there is often a several-centimeter herald patch that occurs on the trunk. It is often confused with eczema or tinea due to its erythema and scale. (Secondary syphilis is also in the differential.) Sometimes PR can be differentiated by the scale pattern being a collarette instead of diffuse. The diagnosis becomes clearer 1 to 2 weeks later with the onset of multiple small scaly macules across the trunk following the Langer’s skin lines. The course is self-limited but takes several weeks to months to resolve.
If severe, PR may be treated with acyclovir 800 mg orally 5 times daily for 5 days; this is the same regimen for treating varicella zoster (shingles).1,2 Estimated recurrence rates are 4% to 24%.1,3
At age 49 years, this woman was older than the average patient with PR, as the usual age range is 10 to 35 years.1 Her physician advised her that the outbreak might recur. She was also given a prescription for oral hydroxyzine 25 mg to be taken at bedtime if the itching was interfering with her sleep. Her physician told her to return for reevaluation if the rash did not resolve in 3 months. She did not return for reevaluation.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Drago F, Ciccarese G, Parodi A. Commentary on: "pityriasis rosea recurrence is much higher than previously known: a prospective study." Acta Derm Venereol. 2019;99:1053-1054. doi: 10.2340/00015555-3265
2. Villalon-Gomez JM. Pityriasis rosea: diagnosis and treatment. Am Fam Physician. 2018;97:38-44.
3. Yüksel M. Pityriasis rosea recurrence is much higher than previously known: a prospective study. Acta Derm Venereol. 2019;99:664-667. doi: 10.2340/00015555-3169
1. Drago F, Ciccarese G, Parodi A. Commentary on: "pityriasis rosea recurrence is much higher than previously known: a prospective study." Acta Derm Venereol. 2019;99:1053-1054. doi: 10.2340/00015555-3265
2. Villalon-Gomez JM. Pityriasis rosea: diagnosis and treatment. Am Fam Physician. 2018;97:38-44.
3. Yüksel M. Pityriasis rosea recurrence is much higher than previously known: a prospective study. Acta Derm Venereol. 2019;99:664-667. doi: 10.2340/00015555-3169

Applications of ChatGPT and Large Language Models in Medicine and Health Care: Benefits and Pitfalls
The development of [artificial intelligence] is as fundamental as the creation of the microprocessor, the personal computer, the Internet, and the mobile phone. It will change the way people work, learn, travel, get health care, and communicate with each other.
Bill Gates 1
As the world emerges from the pandemic and the health care system faces new challenges, technology has become an increasingly important tool for health care professionals (HCPs). One such technology is the large language model (LLM), which has the potential to revolutionize the health care industry. ChatGPT, a popular LLM developed by OpenAI, has gained particular attention in the medical community for its ability to pass the United States Medical Licensing Exam.2 This article will explore the benefits and potential pitfalls of using LLMs like ChatGPT in medicine and health care.
Benefits
HCP burnout is a serious issue that can lead to lower productivity, increased medical errors, and decreased patient satisfaction.3 LLMs can alleviate some administrative burdens on HCPs, allowing them to focus on patient care. By assisting with billing, coding, insurance claims, and organizing schedules, LLMs like ChatGPT can free up time for HCPs to focus on what they do best: providing quality patient care.4 ChatGPT also can assist with diagnoses by providing accurate and reliable information based on a vast amount of clinical data. By learning the relationships between different medical conditions, symptoms, and treatment options, ChatGPT can provide an appropriate differential diagnosis (Figure 1). 
Imaging medical specialists like radiologists, pathologists, dermatologists, and others can benefit from combining computer vision diagnostics with ChatGPT report creation abilities to streamline the diagnostic workflow and improve diagnostic accuracy (Figure 2). 
Although using ChatGPT and other LLMs in mental health care has potential benefits, it is essential to note that they are not a substitute for human interaction and personalized care. While ChatGPT can remember information from previous conversations, it cannot provide the same level of personalized, high-quality care that a professional therapist or HCP can. However, by augmenting the work of HCPs, ChatGPT and other LLMs have the potential to make mental health care more accessible and efficient. In addition to providing effective screening in underserved areas, ChatGPT technology may improve the competence of physician assistants and nurse practitioners in delivering mental health care. With the increased incidence of mental health problems in veterans, the pertinence of a ChatGPT-like feature will only increase with time.9
ChatGPT can also be integrated into health care organizations’ websites and mobile apps, providing patients instant access to medical information, self-care advice, symptom checkers, scheduling appointments, and arranging transportation. These features can reduce the burden on health care staff and help patients stay informed and motivated to take an active role in their health. Additionally, health care organizations can use ChatGPT to engage patients by providing reminders for medication renewals and assistance with self-care.4,6,10,11
The potential of artificial intelligence (AI) in the field of medical education and research is immense. According to a study by Gilson and colleagues, ChatGPT has shown promising results as a medical education tool.12 ChatGPT can simulate clinical scenarios, provide real-time feedback, and improve diagnostic skills. It also offers new interactive and personalized learning opportunities for medical students and HCPs.13 ChatGPT can help researchers by streamlining the process of data analysis. It can also administer surveys or questionnaires, facilitate data collection on preferences and experiences, and help in writing scientific publications.14 Nevertheless, to fully unlock the potential of these AI models, additional models that perform checks for factual accuracy, plagiarism, and copyright infringement must be developed.15,16
AI Bill of Rights
In order to protect the American public, the White House Office of Science and Technology Policy (OSTP) has released a blueprint for an AI Bill of Rights that emphasizes 5 principles to protect the public from the harmful effects of AI models, including safe and effective systems; algorithmic discrimination protection; data privacy; notice and explanation; and human alternatives, considerations, and fallback (Figure 3).17 
One of the biggest challenges with LLMs like ChatGPT is the prevalence of inaccurate information or so-called hallucinations.16 These inaccuracies stem from the inability of LLMs to distinguish between real and fake information. To prevent hallucinations, researchers have proposed several methods, including training models on more diverse data, using adversarial training methods, and human-in-the-loop approaches.21 In addition, medicine-specific models like GatorTron, medPaLM, and Almanac were developed, increasing the accuracy of factual results.22-24 Unfortunately, only the GatorTron model is available to the public through the NVIDIA developers’ program.25
Despite these shortcomings, the future of LLMs in health care is promising. Although these models will not replace HCPs, they can help reduce the unnecessary burden on them, prevent burnout, and enable HCPs and patients spend more time together. Establishing an official hospital AI oversight governing body that would promote best practices could ensure the trustworthy implementation of these new technologies.26
Conclusions
The use of ChatGPT and other LLMs in health care has the potential to revolutionize the industry. By assisting HCPs with administrative tasks, improving the accuracy and reliability of diagnoses, and engaging patients, ChatGPT can help health care organizations provide better care to their patients. While LLMs are not a substitute for human interaction and personalized care, they can augment the work of HCPs, making health care more accessible and efficient. As the health care industry continues to evolve, it will be exciting to see how ChatGPT and other LLMs are used to improve patient outcomes and quality of care. In addition, AI technologies like ChatGPT offer enormous potential in medical education and research. To ensure that the benefits outweigh the risks, developing trustworthy AI health care products and establishing oversight governing bodies to ensure their implementation is essential. By doing so, we can help HCPs focus on what matters most, providing high-quality care to patients.
Acknowledgments
This material is the result of work supported by resources and the use of facilities at the James A. Haley Veterans’ Hospital.
1. Bill Gates. The age of AI has begun. March 21, 2023. Accessed May 10, 2023. https://www.gatesnotes.com/the-age-of-ai-has-begun
2. Kung TH, Cheatham M, Medenilla A, et al. Performance of ChatGPT on USMLE: Potential for AI-assisted medical education using large language models. PLOS Digit Health. 2023;2(2):e0000198. Published 2023 Feb 9. doi:10.1371/journal.pdig.0000198
3. Shanafelt TD, West CP, Sinsky C, et al. Changes in burnout and satisfaction with work-life integration in physicians and the general US working population between 2011 and 2020. Mayo Clin Proc. 2022;97(3):491-506. doi:10.1016/j.mayocp.2021.11.021
4. Goodman RS, Patrinely JR Jr, Osterman T, Wheless L, Johnson DB. On the cusp: considering the impact of artificial intelligence language models in healthcare. Med. 2023;4(3):139-140. doi:10.1016/j.medj.2023.02.008
5. Will ChatGPT transform healthcare? Nat Med. 2023;29(3):505-506. doi:10.1038/s41591-023-02289-5
6. Hopkins AM, Logan JM, Kichenadasse G, Sorich MJ. Artificial intelligence chatbots will revolutionize how cancer patients access information: ChatGPT represents a paradigm-shift. JNCI Cancer Spectr. 2023;7(2):pkad010. doi:10.1093/jncics/pkad010
7. Babar Z, van Laarhoven T, Zanzotto FM, Marchiori E. Evaluating diagnostic content of AI-generated radiology reports of chest X-rays. Artif Intell Med. 2021;116:102075. doi:10.1016/j.artmed.2021.102075
8. Lecler A, Duron L, Soyer P. Revolutionizing radiology with GPT-based models: current applications, future possibilities and limitations of ChatGPT. Diagn Interv Imaging. 2023;S2211-5684(23)00027-X. doi:10.1016/j.diii.2023.02.003
9. Germain JM. Is ChatGPT smart enough to practice mental health therapy? March 23, 2023. Accessed May 11, 2023. https://www.technewsworld.com/story/is-chatgpt-smart-enough-to-practice-mental-health-therapy-178064.html
10. Cascella M, Montomoli J, Bellini V, Bignami E. Evaluating the feasibility of ChatGPT in healthcare: an analysis of multiple clinical and research scenarios. J Med Syst. 2023;47(1):33. Published 2023 Mar 4. doi:10.1007/s10916-023-01925-4
11. Jungwirth D, Haluza D. Artificial intelligence and public health: an exploratory study. Int J Environ Res Public Health. 2023;20(5):4541. Published 2023 Mar 3. doi:10.3390/ijerph20054541
12. Gilson A, Safranek CW, Huang T, et al. How does ChatGPT perform on the United States Medical Licensing Examination? The implications of large language models for medical education and knowledge assessment. JMIR Med Educ. 2023;9:e45312. Published 2023 Feb 8. doi:10.2196/45312
13. Eysenbach G. The role of ChatGPT, generative language models, and artificial intelligence in medical education: a conversation with ChatGPT and a call for papers. JMIR Med Educ. 2023;9:e46885. Published 2023 Mar 6. doi:10.2196/46885
14. Macdonald C, Adeloye D, Sheikh A, Rudan I. Can ChatGPT draft a research article? An example of population-level vaccine effectiveness analysis. J Glob Health. 2023;13:01003. Published 2023 Feb 17. doi:10.7189/jogh.13.01003
15. Masters K. Ethical use of artificial intelligence in health professions education: AMEE Guide No.158. Med Teach. 2023;1-11. doi:10.1080/0142159X.2023.2186203
16. Smith CS. Hallucinations could blunt ChatGPT’s success. IEEE Spectrum. March 13, 2023. Accessed May 11, 2023. https://spectrum.ieee.org/ai-hallucination
17. Executive Office of the President, Office of Science and Technology Policy. Blueprint for an AI Bill of Rights. Accessed May 11, 2023. https://www.whitehouse.gov/ostp/ai-bill-of-rights
18. Executive office of the President. Executive Order 13960: promoting the use of trustworthy artificial intelligence in the federal government. Fed Regist. 2020;89(236):78939-78943.
19. US Department of Commerce, National institute of Standards and Technology. Artificial Intelligence Risk Management Framework (AI RMF 1.0). Published January 2023. doi:10.6028/NIST.AI.100-1
20. Microsoft. Azure Cognitive Search—Cloud Search Service. Accessed May 11, 2023. https://azure.microsoft.com/en-us/products/search
21. Aiyappa R, An J, Kwak H, Ahn YY. Can we trust the evaluation on ChatGPT? March 22, 2023. Accessed May 11, 2023. https://arxiv.org/abs/2303.12767v1
22. Yang X, Chen A, Pournejatian N, et al. GatorTron: a large clinical language model to unlock patient information from unstructured electronic health records. Updated December 16, 2022. Accessed May 11, 2023. https://arxiv.org/abs/2203.03540v3
23. Singhal K, Azizi S, Tu T, et al. Large language models encode clinical knowledge. December 26, 2022. Accessed May 11, 2023. https://arxiv.org/abs/2212.13138v1
24. Zakka C, Chaurasia A, Shad R, Hiesinger W. Almanac: knowledge-grounded language models for clinical medicine. March 1, 2023. Accessed May 11, 2023. https://arxiv.org/abs/2303.01229v1
25. NVIDIA. GatorTron-OG. Accessed May 11, 2023. https://catalog.ngc.nvidia.com/orgs/nvidia/teams/clara/models/gatortron_og
26. Borkowski AA, Jakey CE, Thomas LB, Viswanadhan N, Mastorides SM. Establishing a hospital artificial intelligence committee to improve patient care. Fed Pract. 2022;39(8):334-336. doi:10.12788/fp.0299
The development of [artificial intelligence] is as fundamental as the creation of the microprocessor, the personal computer, the Internet, and the mobile phone. It will change the way people work, learn, travel, get health care, and communicate with each other.
Bill Gates 1
As the world emerges from the pandemic and the health care system faces new challenges, technology has become an increasingly important tool for health care professionals (HCPs). One such technology is the large language model (LLM), which has the potential to revolutionize the health care industry. ChatGPT, a popular LLM developed by OpenAI, has gained particular attention in the medical community for its ability to pass the United States Medical Licensing Exam.2 This article will explore the benefits and potential pitfalls of using LLMs like ChatGPT in medicine and health care.
Benefits
HCP burnout is a serious issue that can lead to lower productivity, increased medical errors, and decreased patient satisfaction.3 LLMs can alleviate some administrative burdens on HCPs, allowing them to focus on patient care. By assisting with billing, coding, insurance claims, and organizing schedules, LLMs like ChatGPT can free up time for HCPs to focus on what they do best: providing quality patient care.4 ChatGPT also can assist with diagnoses by providing accurate and reliable information based on a vast amount of clinical data. By learning the relationships between different medical conditions, symptoms, and treatment options, ChatGPT can provide an appropriate differential diagnosis (Figure 1).
Imaging medical specialists like radiologists, pathologists, dermatologists, and others can benefit from combining computer vision diagnostics with ChatGPT report creation abilities to streamline the diagnostic workflow and improve diagnostic accuracy (Figure 2).
Although using ChatGPT and other LLMs in mental health care has potential benefits, it is essential to note that they are not a substitute for human interaction and personalized care. While ChatGPT can remember information from previous conversations, it cannot provide the same level of personalized, high-quality care that a professional therapist or HCP can. However, by augmenting the work of HCPs, ChatGPT and other LLMs have the potential to make mental health care more accessible and efficient. In addition to providing effective screening in underserved areas, ChatGPT technology may improve the competence of physician assistants and nurse practitioners in delivering mental health care. With the increased incidence of mental health problems in veterans, the pertinence of a ChatGPT-like feature will only increase with time.9
ChatGPT can also be integrated into health care organizations’ websites and mobile apps, providing patients instant access to medical information, self-care advice, symptom checkers, scheduling appointments, and arranging transportation. These features can reduce the burden on health care staff and help patients stay informed and motivated to take an active role in their health. Additionally, health care organizations can use ChatGPT to engage patients by providing reminders for medication renewals and assistance with self-care.4,6,10,11
The potential of artificial intelligence (AI) in the field of medical education and research is immense. According to a study by Gilson and colleagues, ChatGPT has shown promising results as a medical education tool.12 ChatGPT can simulate clinical scenarios, provide real-time feedback, and improve diagnostic skills. It also offers new interactive and personalized learning opportunities for medical students and HCPs.13 ChatGPT can help researchers by streamlining the process of data analysis. It can also administer surveys or questionnaires, facilitate data collection on preferences and experiences, and help in writing scientific publications.14 Nevertheless, to fully unlock the potential of these AI models, additional models that perform checks for factual accuracy, plagiarism, and copyright infringement must be developed.15,16
AI Bill of Rights
In order to protect the American public, the White House Office of Science and Technology Policy (OSTP) has released a blueprint for an AI Bill of Rights that emphasizes 5 principles to protect the public from the harmful effects of AI models, including safe and effective systems; algorithmic discrimination protection; data privacy; notice and explanation; and human alternatives, considerations, and fallback (Figure 3).17
One of the biggest challenges with LLMs like ChatGPT is the prevalence of inaccurate information or so-called hallucinations.16 These inaccuracies stem from the inability of LLMs to distinguish between real and fake information. To prevent hallucinations, researchers have proposed several methods, including training models on more diverse data, using adversarial training methods, and human-in-the-loop approaches.21 In addition, medicine-specific models like GatorTron, medPaLM, and Almanac were developed, increasing the accuracy of factual results.22-24 Unfortunately, only the GatorTron model is available to the public through the NVIDIA developers’ program.25
Despite these shortcomings, the future of LLMs in health care is promising. Although these models will not replace HCPs, they can help reduce the unnecessary burden on them, prevent burnout, and enable HCPs and patients spend more time together. Establishing an official hospital AI oversight governing body that would promote best practices could ensure the trustworthy implementation of these new technologies.26
Conclusions
The use of ChatGPT and other LLMs in health care has the potential to revolutionize the industry. By assisting HCPs with administrative tasks, improving the accuracy and reliability of diagnoses, and engaging patients, ChatGPT can help health care organizations provide better care to their patients. While LLMs are not a substitute for human interaction and personalized care, they can augment the work of HCPs, making health care more accessible and efficient. As the health care industry continues to evolve, it will be exciting to see how ChatGPT and other LLMs are used to improve patient outcomes and quality of care. In addition, AI technologies like ChatGPT offer enormous potential in medical education and research. To ensure that the benefits outweigh the risks, developing trustworthy AI health care products and establishing oversight governing bodies to ensure their implementation is essential. By doing so, we can help HCPs focus on what matters most, providing high-quality care to patients.
Acknowledgments
This material is the result of work supported by resources and the use of facilities at the James A. Haley Veterans’ Hospital.
The development of [artificial intelligence] is as fundamental as the creation of the microprocessor, the personal computer, the Internet, and the mobile phone. It will change the way people work, learn, travel, get health care, and communicate with each other.
Bill Gates 1
As the world emerges from the pandemic and the health care system faces new challenges, technology has become an increasingly important tool for health care professionals (HCPs). One such technology is the large language model (LLM), which has the potential to revolutionize the health care industry. ChatGPT, a popular LLM developed by OpenAI, has gained particular attention in the medical community for its ability to pass the United States Medical Licensing Exam.2 This article will explore the benefits and potential pitfalls of using LLMs like ChatGPT in medicine and health care.
Benefits
HCP burnout is a serious issue that can lead to lower productivity, increased medical errors, and decreased patient satisfaction.3 LLMs can alleviate some administrative burdens on HCPs, allowing them to focus on patient care. By assisting with billing, coding, insurance claims, and organizing schedules, LLMs like ChatGPT can free up time for HCPs to focus on what they do best: providing quality patient care.4 ChatGPT also can assist with diagnoses by providing accurate and reliable information based on a vast amount of clinical data. By learning the relationships between different medical conditions, symptoms, and treatment options, ChatGPT can provide an appropriate differential diagnosis (Figure 1).
Imaging medical specialists like radiologists, pathologists, dermatologists, and others can benefit from combining computer vision diagnostics with ChatGPT report creation abilities to streamline the diagnostic workflow and improve diagnostic accuracy (Figure 2).
Although using ChatGPT and other LLMs in mental health care has potential benefits, it is essential to note that they are not a substitute for human interaction and personalized care. While ChatGPT can remember information from previous conversations, it cannot provide the same level of personalized, high-quality care that a professional therapist or HCP can. However, by augmenting the work of HCPs, ChatGPT and other LLMs have the potential to make mental health care more accessible and efficient. In addition to providing effective screening in underserved areas, ChatGPT technology may improve the competence of physician assistants and nurse practitioners in delivering mental health care. With the increased incidence of mental health problems in veterans, the pertinence of a ChatGPT-like feature will only increase with time.9
ChatGPT can also be integrated into health care organizations’ websites and mobile apps, providing patients instant access to medical information, self-care advice, symptom checkers, scheduling appointments, and arranging transportation. These features can reduce the burden on health care staff and help patients stay informed and motivated to take an active role in their health. Additionally, health care organizations can use ChatGPT to engage patients by providing reminders for medication renewals and assistance with self-care.4,6,10,11
The potential of artificial intelligence (AI) in the field of medical education and research is immense. According to a study by Gilson and colleagues, ChatGPT has shown promising results as a medical education tool.12 ChatGPT can simulate clinical scenarios, provide real-time feedback, and improve diagnostic skills. It also offers new interactive and personalized learning opportunities for medical students and HCPs.13 ChatGPT can help researchers by streamlining the process of data analysis. It can also administer surveys or questionnaires, facilitate data collection on preferences and experiences, and help in writing scientific publications.14 Nevertheless, to fully unlock the potential of these AI models, additional models that perform checks for factual accuracy, plagiarism, and copyright infringement must be developed.15,16
AI Bill of Rights
In order to protect the American public, the White House Office of Science and Technology Policy (OSTP) has released a blueprint for an AI Bill of Rights that emphasizes 5 principles to protect the public from the harmful effects of AI models, including safe and effective systems; algorithmic discrimination protection; data privacy; notice and explanation; and human alternatives, considerations, and fallback (Figure 3).17
One of the biggest challenges with LLMs like ChatGPT is the prevalence of inaccurate information or so-called hallucinations.16 These inaccuracies stem from the inability of LLMs to distinguish between real and fake information. To prevent hallucinations, researchers have proposed several methods, including training models on more diverse data, using adversarial training methods, and human-in-the-loop approaches.21 In addition, medicine-specific models like GatorTron, medPaLM, and Almanac were developed, increasing the accuracy of factual results.22-24 Unfortunately, only the GatorTron model is available to the public through the NVIDIA developers’ program.25
Despite these shortcomings, the future of LLMs in health care is promising. Although these models will not replace HCPs, they can help reduce the unnecessary burden on them, prevent burnout, and enable HCPs and patients spend more time together. Establishing an official hospital AI oversight governing body that would promote best practices could ensure the trustworthy implementation of these new technologies.26
Conclusions
The use of ChatGPT and other LLMs in health care has the potential to revolutionize the industry. By assisting HCPs with administrative tasks, improving the accuracy and reliability of diagnoses, and engaging patients, ChatGPT can help health care organizations provide better care to their patients. While LLMs are not a substitute for human interaction and personalized care, they can augment the work of HCPs, making health care more accessible and efficient. As the health care industry continues to evolve, it will be exciting to see how ChatGPT and other LLMs are used to improve patient outcomes and quality of care. In addition, AI technologies like ChatGPT offer enormous potential in medical education and research. To ensure that the benefits outweigh the risks, developing trustworthy AI health care products and establishing oversight governing bodies to ensure their implementation is essential. By doing so, we can help HCPs focus on what matters most, providing high-quality care to patients.
Acknowledgments
This material is the result of work supported by resources and the use of facilities at the James A. Haley Veterans’ Hospital.
1. Bill Gates. The age of AI has begun. March 21, 2023. Accessed May 10, 2023. https://www.gatesnotes.com/the-age-of-ai-has-begun
2. Kung TH, Cheatham M, Medenilla A, et al. Performance of ChatGPT on USMLE: Potential for AI-assisted medical education using large language models. PLOS Digit Health. 2023;2(2):e0000198. Published 2023 Feb 9. doi:10.1371/journal.pdig.0000198
3. Shanafelt TD, West CP, Sinsky C, et al. Changes in burnout and satisfaction with work-life integration in physicians and the general US working population between 2011 and 2020. Mayo Clin Proc. 2022;97(3):491-506. doi:10.1016/j.mayocp.2021.11.021
4. Goodman RS, Patrinely JR Jr, Osterman T, Wheless L, Johnson DB. On the cusp: considering the impact of artificial intelligence language models in healthcare. Med. 2023;4(3):139-140. doi:10.1016/j.medj.2023.02.008
5. Will ChatGPT transform healthcare? Nat Med. 2023;29(3):505-506. doi:10.1038/s41591-023-02289-5
6. Hopkins AM, Logan JM, Kichenadasse G, Sorich MJ. Artificial intelligence chatbots will revolutionize how cancer patients access information: ChatGPT represents a paradigm-shift. JNCI Cancer Spectr. 2023;7(2):pkad010. doi:10.1093/jncics/pkad010
7. Babar Z, van Laarhoven T, Zanzotto FM, Marchiori E. Evaluating diagnostic content of AI-generated radiology reports of chest X-rays. Artif Intell Med. 2021;116:102075. doi:10.1016/j.artmed.2021.102075
8. Lecler A, Duron L, Soyer P. Revolutionizing radiology with GPT-based models: current applications, future possibilities and limitations of ChatGPT. Diagn Interv Imaging. 2023;S2211-5684(23)00027-X. doi:10.1016/j.diii.2023.02.003
9. Germain JM. Is ChatGPT smart enough to practice mental health therapy? March 23, 2023. Accessed May 11, 2023. https://www.technewsworld.com/story/is-chatgpt-smart-enough-to-practice-mental-health-therapy-178064.html
10. Cascella M, Montomoli J, Bellini V, Bignami E. Evaluating the feasibility of ChatGPT in healthcare: an analysis of multiple clinical and research scenarios. J Med Syst. 2023;47(1):33. Published 2023 Mar 4. doi:10.1007/s10916-023-01925-4
11. Jungwirth D, Haluza D. Artificial intelligence and public health: an exploratory study. Int J Environ Res Public Health. 2023;20(5):4541. Published 2023 Mar 3. doi:10.3390/ijerph20054541
12. Gilson A, Safranek CW, Huang T, et al. How does ChatGPT perform on the United States Medical Licensing Examination? The implications of large language models for medical education and knowledge assessment. JMIR Med Educ. 2023;9:e45312. Published 2023 Feb 8. doi:10.2196/45312
13. Eysenbach G. The role of ChatGPT, generative language models, and artificial intelligence in medical education: a conversation with ChatGPT and a call for papers. JMIR Med Educ. 2023;9:e46885. Published 2023 Mar 6. doi:10.2196/46885
14. Macdonald C, Adeloye D, Sheikh A, Rudan I. Can ChatGPT draft a research article? An example of population-level vaccine effectiveness analysis. J Glob Health. 2023;13:01003. Published 2023 Feb 17. doi:10.7189/jogh.13.01003
15. Masters K. Ethical use of artificial intelligence in health professions education: AMEE Guide No.158. Med Teach. 2023;1-11. doi:10.1080/0142159X.2023.2186203
16. Smith CS. Hallucinations could blunt ChatGPT’s success. IEEE Spectrum. March 13, 2023. Accessed May 11, 2023. https://spectrum.ieee.org/ai-hallucination
17. Executive Office of the President, Office of Science and Technology Policy. Blueprint for an AI Bill of Rights. Accessed May 11, 2023. https://www.whitehouse.gov/ostp/ai-bill-of-rights
18. Executive office of the President. Executive Order 13960: promoting the use of trustworthy artificial intelligence in the federal government. Fed Regist. 2020;89(236):78939-78943.
19. US Department of Commerce, National institute of Standards and Technology. Artificial Intelligence Risk Management Framework (AI RMF 1.0). Published January 2023. doi:10.6028/NIST.AI.100-1
20. Microsoft. Azure Cognitive Search—Cloud Search Service. Accessed May 11, 2023. https://azure.microsoft.com/en-us/products/search
21. Aiyappa R, An J, Kwak H, Ahn YY. Can we trust the evaluation on ChatGPT? March 22, 2023. Accessed May 11, 2023. https://arxiv.org/abs/2303.12767v1
22. Yang X, Chen A, Pournejatian N, et al. GatorTron: a large clinical language model to unlock patient information from unstructured electronic health records. Updated December 16, 2022. Accessed May 11, 2023. https://arxiv.org/abs/2203.03540v3
23. Singhal K, Azizi S, Tu T, et al. Large language models encode clinical knowledge. December 26, 2022. Accessed May 11, 2023. https://arxiv.org/abs/2212.13138v1
24. Zakka C, Chaurasia A, Shad R, Hiesinger W. Almanac: knowledge-grounded language models for clinical medicine. March 1, 2023. Accessed May 11, 2023. https://arxiv.org/abs/2303.01229v1
25. NVIDIA. GatorTron-OG. Accessed May 11, 2023. https://catalog.ngc.nvidia.com/orgs/nvidia/teams/clara/models/gatortron_og
26. Borkowski AA, Jakey CE, Thomas LB, Viswanadhan N, Mastorides SM. Establishing a hospital artificial intelligence committee to improve patient care. Fed Pract. 2022;39(8):334-336. doi:10.12788/fp.0299
1. Bill Gates. The age of AI has begun. March 21, 2023. Accessed May 10, 2023. https://www.gatesnotes.com/the-age-of-ai-has-begun
2. Kung TH, Cheatham M, Medenilla A, et al. Performance of ChatGPT on USMLE: Potential for AI-assisted medical education using large language models. PLOS Digit Health. 2023;2(2):e0000198. Published 2023 Feb 9. doi:10.1371/journal.pdig.0000198
3. Shanafelt TD, West CP, Sinsky C, et al. Changes in burnout and satisfaction with work-life integration in physicians and the general US working population between 2011 and 2020. Mayo Clin Proc. 2022;97(3):491-506. doi:10.1016/j.mayocp.2021.11.021
4. Goodman RS, Patrinely JR Jr, Osterman T, Wheless L, Johnson DB. On the cusp: considering the impact of artificial intelligence language models in healthcare. Med. 2023;4(3):139-140. doi:10.1016/j.medj.2023.02.008
5. Will ChatGPT transform healthcare? Nat Med. 2023;29(3):505-506. doi:10.1038/s41591-023-02289-5
6. Hopkins AM, Logan JM, Kichenadasse G, Sorich MJ. Artificial intelligence chatbots will revolutionize how cancer patients access information: ChatGPT represents a paradigm-shift. JNCI Cancer Spectr. 2023;7(2):pkad010. doi:10.1093/jncics/pkad010
7. Babar Z, van Laarhoven T, Zanzotto FM, Marchiori E. Evaluating diagnostic content of AI-generated radiology reports of chest X-rays. Artif Intell Med. 2021;116:102075. doi:10.1016/j.artmed.2021.102075
8. Lecler A, Duron L, Soyer P. Revolutionizing radiology with GPT-based models: current applications, future possibilities and limitations of ChatGPT. Diagn Interv Imaging. 2023;S2211-5684(23)00027-X. doi:10.1016/j.diii.2023.02.003
9. Germain JM. Is ChatGPT smart enough to practice mental health therapy? March 23, 2023. Accessed May 11, 2023. https://www.technewsworld.com/story/is-chatgpt-smart-enough-to-practice-mental-health-therapy-178064.html
10. Cascella M, Montomoli J, Bellini V, Bignami E. Evaluating the feasibility of ChatGPT in healthcare: an analysis of multiple clinical and research scenarios. J Med Syst. 2023;47(1):33. Published 2023 Mar 4. doi:10.1007/s10916-023-01925-4
11. Jungwirth D, Haluza D. Artificial intelligence and public health: an exploratory study. Int J Environ Res Public Health. 2023;20(5):4541. Published 2023 Mar 3. doi:10.3390/ijerph20054541
12. Gilson A, Safranek CW, Huang T, et al. How does ChatGPT perform on the United States Medical Licensing Examination? The implications of large language models for medical education and knowledge assessment. JMIR Med Educ. 2023;9:e45312. Published 2023 Feb 8. doi:10.2196/45312
13. Eysenbach G. The role of ChatGPT, generative language models, and artificial intelligence in medical education: a conversation with ChatGPT and a call for papers. JMIR Med Educ. 2023;9:e46885. Published 2023 Mar 6. doi:10.2196/46885
14. Macdonald C, Adeloye D, Sheikh A, Rudan I. Can ChatGPT draft a research article? An example of population-level vaccine effectiveness analysis. J Glob Health. 2023;13:01003. Published 2023 Feb 17. doi:10.7189/jogh.13.01003
15. Masters K. Ethical use of artificial intelligence in health professions education: AMEE Guide No.158. Med Teach. 2023;1-11. doi:10.1080/0142159X.2023.2186203
16. Smith CS. Hallucinations could blunt ChatGPT’s success. IEEE Spectrum. March 13, 2023. Accessed May 11, 2023. https://spectrum.ieee.org/ai-hallucination
17. Executive Office of the President, Office of Science and Technology Policy. Blueprint for an AI Bill of Rights. Accessed May 11, 2023. https://www.whitehouse.gov/ostp/ai-bill-of-rights
18. Executive office of the President. Executive Order 13960: promoting the use of trustworthy artificial intelligence in the federal government. Fed Regist. 2020;89(236):78939-78943.
19. US Department of Commerce, National institute of Standards and Technology. Artificial Intelligence Risk Management Framework (AI RMF 1.0). Published January 2023. doi:10.6028/NIST.AI.100-1
20. Microsoft. Azure Cognitive Search—Cloud Search Service. Accessed May 11, 2023. https://azure.microsoft.com/en-us/products/search
21. Aiyappa R, An J, Kwak H, Ahn YY. Can we trust the evaluation on ChatGPT? March 22, 2023. Accessed May 11, 2023. https://arxiv.org/abs/2303.12767v1
22. Yang X, Chen A, Pournejatian N, et al. GatorTron: a large clinical language model to unlock patient information from unstructured electronic health records. Updated December 16, 2022. Accessed May 11, 2023. https://arxiv.org/abs/2203.03540v3
23. Singhal K, Azizi S, Tu T, et al. Large language models encode clinical knowledge. December 26, 2022. Accessed May 11, 2023. https://arxiv.org/abs/2212.13138v1
24. Zakka C, Chaurasia A, Shad R, Hiesinger W. Almanac: knowledge-grounded language models for clinical medicine. March 1, 2023. Accessed May 11, 2023. https://arxiv.org/abs/2303.01229v1
25. NVIDIA. GatorTron-OG. Accessed May 11, 2023. https://catalog.ngc.nvidia.com/orgs/nvidia/teams/clara/models/gatortron_og
26. Borkowski AA, Jakey CE, Thomas LB, Viswanadhan N, Mastorides SM. Establishing a hospital artificial intelligence committee to improve patient care. Fed Pract. 2022;39(8):334-336. doi:10.12788/fp.0299
55-year-old woman • unilateral nasal drainage • salty taste • nasal redness • recent COVID-19 nasal swabs • Dx?
THE CASE
A 55-year-old woman was evaluated in a family medicine clinic for clear, right-side nasal drainage. She stated that the drainage began 5 months earlier after 2 hospitalizations for severe anxiety leading to emesis and hypokalemia. She reported 3 different COVID-19 nasal swab tests performed on the right nare. Chart review showed 2 negative COVID-19 tests, 6 days apart. Since the hospitalizations, the patient had been given antihistamines for rhinorrhea at an urgent care visit. Despite this treatment, the patient reported a constant drip from the right nare with a salty taste. She also reported experiencing occasional headaches but denied nausea/vomiting.
The patient’s history included uncontrolled hypertension, treatment-resistant anxiety and depression, obstructive sleep apnea, chronic sinus disease (observed on computed tomography [CT] scans), and type 2 diabetes. She was on amlodipine 10 mg/d for hypertension and was not taking any medication for diabetes.
On examination, the patient’s vital signs were within normal limits except for an elevated blood pressure of 158/88 mm Hg. The patient had persistent clear rhinorrhea fluid draining from the right nostril that was exacerbated when she looked down. Right nasal erythema was present.
THE DIAGNOSIS
The patient’s negative COVID-19 tests, lack of improvement on antihistamines, and description of the nasal fluid as salty tasting prompted us to suspect a cerebrospinal fluid (CSF) leak. The clinical work-up included a halo (“double-ring”) sign test, a β-2 transferrin test, and a sinus x-ray.
The halo sign test was negative for CSF fluid. Sinus/skull x-ray did not show a cribriform or other fracture. However, a sample of the nasal fluid collected in a sterile container was positive for β-2 transferrin, the gold-standard laboratory test to confirm a CSF leak.
The patient was sent for a maxillofacial CT scan without contrast. Results showed a 3-mm defect over the right ethmoid roof associated with a 10 × 16–mm low-attenuation structure in the right ethmoid labyrinth, suspicious for encephalocele. This defect, in the setting of the patient’s history of chronic sinus disease, furthered our suspicion of a CSF leak secondary to COVID-19 testing. Radiology confirmed the diagnosis.
DISCUSSION
CSF rhinorrhea is CSF leakage through the nasal cavity due to abnormal communication between the arachnoid membrane and nasal mucosa.1 The most commonly reported risk factors for this include female sex, middle age (fourth to fifth decade), obesity (body mass index > 40), intracranial hypertension, and obstructive sleep apnea.1,2
Continue to: Clear, unilateral rhinorrhea...
Clear, unilateral rhinorrhea drainage that increases at times of relatively increased intracranial pressure and has a metallic or salty taste is suspicious for CSF rhinorrhea.3 It can occur following skull‐base trauma (eg, cribriform plate, temporal bone), endoscopic sinus surgery, or neurosurgical procedures, or have a spontaneous etiology.3,4
Modalities to confirm CSF rhinorrhea include radionuclide cisternography and testing of fluid for the halo sign, glucose, and the CSF-specific proteins β‐2 transferrin and β-trace protein.3,4 High‐resolution CT is the imaging method most commonly used for localizing a CSF leak.4
Treatment is provided in the hospital
Patients with CSF rhinorrhea typically require inpatient management with bed rest, head-of-bed elevation, and frequent neurologic evaluation, as persistent CSF rhinorrhea increases the risk for meningitis, thus necessitating surgical intervention.3,5 Some cases resolve with bed rest alone. Endonasal endoscopic repair of CSF leaks has become the standard of care because of its high success rate and lower morbidity profile.4
The preferred treatment method for encephalocele is surgical removal after diagnosis is confirmed with CT or magnetic resonance imaging.6
Our patient underwent surgery to remove the encephalocele. The surgeons reported no evidence of fracture.
The final cause of her CSF leak is still uncertain. The surgeons felt confident it was due to ethmoidal encephalocele, a form of neural tube defect in which brain tissue herniates through structural weaknesses of the skull.6-8 While more common in infants, encephalocele can manifest in adulthood due to traumatic or iatrogenic causes.7,8
There is a previous report of encephalocele with CSF leak after COVID-19 testing.9 This case report suggests the possibility of a nasal swab causing trauma to a patient’s pre‐existing encephalocele—a probability in our patient’s case. It is unlikely, however, that the nasal swab itself violated the bony skull base.
THE TAKEAWAY
This case exemplifies how unexplained local symptoms, a high index of suspicion, and adequate work-up can lead to a rare diagnosis. Diagnostic strategies employed for cases of CSF rhinorrhea vary widely due to limited evidence-based guidance.4 Unilateral rhinorrhea with clear fluid that increases at times of increased intracranial pressure, such as bending over, should prompt suspicion for CSF rhinorrhea. With millions of people getting nasal swabs daily during the COVID-19 pandemic, it is even more important to keep CSF leak in our differential diagnosis.
CORRESPONDENCE
Eliana Lizeth Garcia, MD, BS, BA, University of New Mexico Health Sciences Center, 1209 University Boulevard NE, Albuquerque, NM 87131-5001; elianalg@hs.uci.edu
1. Keshri A, Jain R, Manogaran RS, et al. Management of spontaneous CSF rhinorrhea: an institutional experience. J Neurol Surg B Skull Base. 2019;80:493-499. doi: 10.1055/s-0038-1676334
2. Lobo BC, Baumanis MM, Nelson RF. Surgical repair of spontaneous cerebrospinal fluid (CSF) leaks: a systematic review. Laryngoscope Investig Otolaryngol. 2017;2:215-224. doi: 10.1002/lio2.75
3. Van Zele T, Dewaele F. Traumatic CSF leaks of the anterior skull base. B-ENT. 2016;suppl 26:19-27.
4. Oakley GM, Alt JA, Schlosser RJ, et al. Diagnosis of cerebrospinal fluid rhinorrhea: an evidence-based review with recommendations. Int Forum Allergy Rhinol. 2016;6:8-16. doi: 10.1002/alr.21637
5. Friedman JA, Ebersold MJ, Quast LM. Post-traumatic cerebrospinal fluid leakage. World J Surg. 2001;25:1062-1066. doi: 10.1007/s00268-001-0059-7
6. Tirumandas M, Sharma A, Gbenimacho I, et al. Nasal encephaloceles: a review of etiology, pathophysiology, clinical presentations, diagnosis, treatment, and complications. Childs Nerv Syst. 2013;29:739-744. doi: 10.1007/s00381-012-1998-z
7. Junaid M, Sobani ZU, Shamim AA, et al. Nasal encephaloceles presenting at later ages: experience of otorhinolaryngology department at a tertiary care center in Karachi, Pakistan. J Pak Med Assoc. 2012;62:74-76.
8. Dhirawani RB, Gupta R, Pathak S, et al. Frontoethmoidal encephalocele: case report and review on management. Ann Maxillofac Surg. 2014;4:195-197. doi: 10.4103/2231-0746.147140
9. Paquin R, Ryan L, Vale FL, et al. CSF leak after COVID-19 nasopharyngeal swab: a case report. Laryngoscope. 2021;131:1927-1929. doi: 10.1002/lary.29462
THE CASE
A 55-year-old woman was evaluated in a family medicine clinic for clear, right-side nasal drainage. She stated that the drainage began 5 months earlier after 2 hospitalizations for severe anxiety leading to emesis and hypokalemia. She reported 3 different COVID-19 nasal swab tests performed on the right nare. Chart review showed 2 negative COVID-19 tests, 6 days apart. Since the hospitalizations, the patient had been given antihistamines for rhinorrhea at an urgent care visit. Despite this treatment, the patient reported a constant drip from the right nare with a salty taste. She also reported experiencing occasional headaches but denied nausea/vomiting.
The patient’s history included uncontrolled hypertension, treatment-resistant anxiety and depression, obstructive sleep apnea, chronic sinus disease (observed on computed tomography [CT] scans), and type 2 diabetes. She was on amlodipine 10 mg/d for hypertension and was not taking any medication for diabetes.
On examination, the patient’s vital signs were within normal limits except for an elevated blood pressure of 158/88 mm Hg. The patient had persistent clear rhinorrhea fluid draining from the right nostril that was exacerbated when she looked down. Right nasal erythema was present.
THE DIAGNOSIS
The patient’s negative COVID-19 tests, lack of improvement on antihistamines, and description of the nasal fluid as salty tasting prompted us to suspect a cerebrospinal fluid (CSF) leak. The clinical work-up included a halo (“double-ring”) sign test, a β-2 transferrin test, and a sinus x-ray.
The halo sign test was negative for CSF fluid. Sinus/skull x-ray did not show a cribriform or other fracture. However, a sample of the nasal fluid collected in a sterile container was positive for β-2 transferrin, the gold-standard laboratory test to confirm a CSF leak.
The patient was sent for a maxillofacial CT scan without contrast. Results showed a 3-mm defect over the right ethmoid roof associated with a 10 × 16–mm low-attenuation structure in the right ethmoid labyrinth, suspicious for encephalocele. This defect, in the setting of the patient’s history of chronic sinus disease, furthered our suspicion of a CSF leak secondary to COVID-19 testing. Radiology confirmed the diagnosis.
DISCUSSION
CSF rhinorrhea is CSF leakage through the nasal cavity due to abnormal communication between the arachnoid membrane and nasal mucosa.1 The most commonly reported risk factors for this include female sex, middle age (fourth to fifth decade), obesity (body mass index > 40), intracranial hypertension, and obstructive sleep apnea.1,2
Continue to: Clear, unilateral rhinorrhea...
Clear, unilateral rhinorrhea drainage that increases at times of relatively increased intracranial pressure and has a metallic or salty taste is suspicious for CSF rhinorrhea.3 It can occur following skull‐base trauma (eg, cribriform plate, temporal bone), endoscopic sinus surgery, or neurosurgical procedures, or have a spontaneous etiology.3,4
Modalities to confirm CSF rhinorrhea include radionuclide cisternography and testing of fluid for the halo sign, glucose, and the CSF-specific proteins β‐2 transferrin and β-trace protein.3,4 High‐resolution CT is the imaging method most commonly used for localizing a CSF leak.4
Treatment is provided in the hospital
Patients with CSF rhinorrhea typically require inpatient management with bed rest, head-of-bed elevation, and frequent neurologic evaluation, as persistent CSF rhinorrhea increases the risk for meningitis, thus necessitating surgical intervention.3,5 Some cases resolve with bed rest alone. Endonasal endoscopic repair of CSF leaks has become the standard of care because of its high success rate and lower morbidity profile.4
The preferred treatment method for encephalocele is surgical removal after diagnosis is confirmed with CT or magnetic resonance imaging.6
Our patient underwent surgery to remove the encephalocele. The surgeons reported no evidence of fracture.
The final cause of her CSF leak is still uncertain. The surgeons felt confident it was due to ethmoidal encephalocele, a form of neural tube defect in which brain tissue herniates through structural weaknesses of the skull.6-8 While more common in infants, encephalocele can manifest in adulthood due to traumatic or iatrogenic causes.7,8
There is a previous report of encephalocele with CSF leak after COVID-19 testing.9 This case report suggests the possibility of a nasal swab causing trauma to a patient’s pre‐existing encephalocele—a probability in our patient’s case. It is unlikely, however, that the nasal swab itself violated the bony skull base.
THE TAKEAWAY
This case exemplifies how unexplained local symptoms, a high index of suspicion, and adequate work-up can lead to a rare diagnosis. Diagnostic strategies employed for cases of CSF rhinorrhea vary widely due to limited evidence-based guidance.4 Unilateral rhinorrhea with clear fluid that increases at times of increased intracranial pressure, such as bending over, should prompt suspicion for CSF rhinorrhea. With millions of people getting nasal swabs daily during the COVID-19 pandemic, it is even more important to keep CSF leak in our differential diagnosis.
CORRESPONDENCE
Eliana Lizeth Garcia, MD, BS, BA, University of New Mexico Health Sciences Center, 1209 University Boulevard NE, Albuquerque, NM 87131-5001; elianalg@hs.uci.edu
THE CASE
A 55-year-old woman was evaluated in a family medicine clinic for clear, right-side nasal drainage. She stated that the drainage began 5 months earlier after 2 hospitalizations for severe anxiety leading to emesis and hypokalemia. She reported 3 different COVID-19 nasal swab tests performed on the right nare. Chart review showed 2 negative COVID-19 tests, 6 days apart. Since the hospitalizations, the patient had been given antihistamines for rhinorrhea at an urgent care visit. Despite this treatment, the patient reported a constant drip from the right nare with a salty taste. She also reported experiencing occasional headaches but denied nausea/vomiting.
The patient’s history included uncontrolled hypertension, treatment-resistant anxiety and depression, obstructive sleep apnea, chronic sinus disease (observed on computed tomography [CT] scans), and type 2 diabetes. She was on amlodipine 10 mg/d for hypertension and was not taking any medication for diabetes.
On examination, the patient’s vital signs were within normal limits except for an elevated blood pressure of 158/88 mm Hg. The patient had persistent clear rhinorrhea fluid draining from the right nostril that was exacerbated when she looked down. Right nasal erythema was present.
THE DIAGNOSIS
The patient’s negative COVID-19 tests, lack of improvement on antihistamines, and description of the nasal fluid as salty tasting prompted us to suspect a cerebrospinal fluid (CSF) leak. The clinical work-up included a halo (“double-ring”) sign test, a β-2 transferrin test, and a sinus x-ray.
The halo sign test was negative for CSF fluid. Sinus/skull x-ray did not show a cribriform or other fracture. However, a sample of the nasal fluid collected in a sterile container was positive for β-2 transferrin, the gold-standard laboratory test to confirm a CSF leak.
The patient was sent for a maxillofacial CT scan without contrast. Results showed a 3-mm defect over the right ethmoid roof associated with a 10 × 16–mm low-attenuation structure in the right ethmoid labyrinth, suspicious for encephalocele. This defect, in the setting of the patient’s history of chronic sinus disease, furthered our suspicion of a CSF leak secondary to COVID-19 testing. Radiology confirmed the diagnosis.
DISCUSSION
CSF rhinorrhea is CSF leakage through the nasal cavity due to abnormal communication between the arachnoid membrane and nasal mucosa.1 The most commonly reported risk factors for this include female sex, middle age (fourth to fifth decade), obesity (body mass index > 40), intracranial hypertension, and obstructive sleep apnea.1,2
Continue to: Clear, unilateral rhinorrhea...
Clear, unilateral rhinorrhea drainage that increases at times of relatively increased intracranial pressure and has a metallic or salty taste is suspicious for CSF rhinorrhea.3 It can occur following skull‐base trauma (eg, cribriform plate, temporal bone), endoscopic sinus surgery, or neurosurgical procedures, or have a spontaneous etiology.3,4
Modalities to confirm CSF rhinorrhea include radionuclide cisternography and testing of fluid for the halo sign, glucose, and the CSF-specific proteins β‐2 transferrin and β-trace protein.3,4 High‐resolution CT is the imaging method most commonly used for localizing a CSF leak.4
Treatment is provided in the hospital
Patients with CSF rhinorrhea typically require inpatient management with bed rest, head-of-bed elevation, and frequent neurologic evaluation, as persistent CSF rhinorrhea increases the risk for meningitis, thus necessitating surgical intervention.3,5 Some cases resolve with bed rest alone. Endonasal endoscopic repair of CSF leaks has become the standard of care because of its high success rate and lower morbidity profile.4
The preferred treatment method for encephalocele is surgical removal after diagnosis is confirmed with CT or magnetic resonance imaging.6
Our patient underwent surgery to remove the encephalocele. The surgeons reported no evidence of fracture.
The final cause of her CSF leak is still uncertain. The surgeons felt confident it was due to ethmoidal encephalocele, a form of neural tube defect in which brain tissue herniates through structural weaknesses of the skull.6-8 While more common in infants, encephalocele can manifest in adulthood due to traumatic or iatrogenic causes.7,8
There is a previous report of encephalocele with CSF leak after COVID-19 testing.9 This case report suggests the possibility of a nasal swab causing trauma to a patient’s pre‐existing encephalocele—a probability in our patient’s case. It is unlikely, however, that the nasal swab itself violated the bony skull base.
THE TAKEAWAY
This case exemplifies how unexplained local symptoms, a high index of suspicion, and adequate work-up can lead to a rare diagnosis. Diagnostic strategies employed for cases of CSF rhinorrhea vary widely due to limited evidence-based guidance.4 Unilateral rhinorrhea with clear fluid that increases at times of increased intracranial pressure, such as bending over, should prompt suspicion for CSF rhinorrhea. With millions of people getting nasal swabs daily during the COVID-19 pandemic, it is even more important to keep CSF leak in our differential diagnosis.
CORRESPONDENCE
Eliana Lizeth Garcia, MD, BS, BA, University of New Mexico Health Sciences Center, 1209 University Boulevard NE, Albuquerque, NM 87131-5001; elianalg@hs.uci.edu
1. Keshri A, Jain R, Manogaran RS, et al. Management of spontaneous CSF rhinorrhea: an institutional experience. J Neurol Surg B Skull Base. 2019;80:493-499. doi: 10.1055/s-0038-1676334
2. Lobo BC, Baumanis MM, Nelson RF. Surgical repair of spontaneous cerebrospinal fluid (CSF) leaks: a systematic review. Laryngoscope Investig Otolaryngol. 2017;2:215-224. doi: 10.1002/lio2.75
3. Van Zele T, Dewaele F. Traumatic CSF leaks of the anterior skull base. B-ENT. 2016;suppl 26:19-27.
4. Oakley GM, Alt JA, Schlosser RJ, et al. Diagnosis of cerebrospinal fluid rhinorrhea: an evidence-based review with recommendations. Int Forum Allergy Rhinol. 2016;6:8-16. doi: 10.1002/alr.21637
5. Friedman JA, Ebersold MJ, Quast LM. Post-traumatic cerebrospinal fluid leakage. World J Surg. 2001;25:1062-1066. doi: 10.1007/s00268-001-0059-7
6. Tirumandas M, Sharma A, Gbenimacho I, et al. Nasal encephaloceles: a review of etiology, pathophysiology, clinical presentations, diagnosis, treatment, and complications. Childs Nerv Syst. 2013;29:739-744. doi: 10.1007/s00381-012-1998-z
7. Junaid M, Sobani ZU, Shamim AA, et al. Nasal encephaloceles presenting at later ages: experience of otorhinolaryngology department at a tertiary care center in Karachi, Pakistan. J Pak Med Assoc. 2012;62:74-76.
8. Dhirawani RB, Gupta R, Pathak S, et al. Frontoethmoidal encephalocele: case report and review on management. Ann Maxillofac Surg. 2014;4:195-197. doi: 10.4103/2231-0746.147140
9. Paquin R, Ryan L, Vale FL, et al. CSF leak after COVID-19 nasopharyngeal swab: a case report. Laryngoscope. 2021;131:1927-1929. doi: 10.1002/lary.29462
1. Keshri A, Jain R, Manogaran RS, et al. Management of spontaneous CSF rhinorrhea: an institutional experience. J Neurol Surg B Skull Base. 2019;80:493-499. doi: 10.1055/s-0038-1676334
2. Lobo BC, Baumanis MM, Nelson RF. Surgical repair of spontaneous cerebrospinal fluid (CSF) leaks: a systematic review. Laryngoscope Investig Otolaryngol. 2017;2:215-224. doi: 10.1002/lio2.75
3. Van Zele T, Dewaele F. Traumatic CSF leaks of the anterior skull base. B-ENT. 2016;suppl 26:19-27.
4. Oakley GM, Alt JA, Schlosser RJ, et al. Diagnosis of cerebrospinal fluid rhinorrhea: an evidence-based review with recommendations. Int Forum Allergy Rhinol. 2016;6:8-16. doi: 10.1002/alr.21637
5. Friedman JA, Ebersold MJ, Quast LM. Post-traumatic cerebrospinal fluid leakage. World J Surg. 2001;25:1062-1066. doi: 10.1007/s00268-001-0059-7
6. Tirumandas M, Sharma A, Gbenimacho I, et al. Nasal encephaloceles: a review of etiology, pathophysiology, clinical presentations, diagnosis, treatment, and complications. Childs Nerv Syst. 2013;29:739-744. doi: 10.1007/s00381-012-1998-z
7. Junaid M, Sobani ZU, Shamim AA, et al. Nasal encephaloceles presenting at later ages: experience of otorhinolaryngology department at a tertiary care center in Karachi, Pakistan. J Pak Med Assoc. 2012;62:74-76.
8. Dhirawani RB, Gupta R, Pathak S, et al. Frontoethmoidal encephalocele: case report and review on management. Ann Maxillofac Surg. 2014;4:195-197. doi: 10.4103/2231-0746.147140
9. Paquin R, Ryan L, Vale FL, et al. CSF leak after COVID-19 nasopharyngeal swab: a case report. Laryngoscope. 2021;131:1927-1929. doi: 10.1002/lary.29462
► Unilateral nasal drainage
► Salty taste
► Nasal redness
► Recent COVID-19 nasal swabs

Is there benefit to adding ezetimibe to a statin for the secondary prevention of CVD?
Evidence summary
Adding ezetimibe reduces nonfatal events but does not improve mortality
A 2018 Cochrane meta-analysis included 10 RCTs (N = 21,919 patients) that evaluated the efficacy and safety of ezetimibe plus a statin (dual therapy) vs a statin alone or plus placebo (monotherapy) for the secondary prevention of CVD. Mean age of patients ranged from 55 to 84 years. Almost all of the patients (> 99%) included in the analyses had existing ASCVD. The dose of ezetimibe was 10 mg; statins used included atorvastatin 10 to 80 mg, pitavastatin 2 to 4 mg, rosuvastatin 10 mg, and simvastatin 20 to 80 mg.1
The primary outcomes were MACE and all-cause mortality. MACE is defined as a composite of CVD, nonfatal myocardial infarction (MI), nonfatal stroke, hospitalization for unstable angina, or coronary revascularization procedures. The TABLE1 provides a detailed breakdown of each of the outcomes.

The dual-therapy group compared to the monotherapy group had a lower risk for MACE (26.6% vs 28.3%; 1.7% absolute risk reduction; 6% relative risk reduction; NNT = 59) and little or no difference in the reduction of all-cause mortality. For secondary outcomes, the dual-therapy group had a lower risk for nonfatal MI, nonfatal stroke, and coronary revascularization. There was no difference in cardiovascular mortality or adverse events between the 2 groups. The quality of evidence was high for all-cause mortality and moderate for cardiovascular mortality, MACE, MI, and stroke.1
The 2015 IMPROVE-IT study, the largest included in the Cochrane review, was a double-blind RCT (N = 18,144) conducted at 1147 sites in 39 countries comparing simvastatin 40 mg/d plus ezetimibe 10 mg/d (dual therapy) vs simvastatin 40 mg/d plus placebo (monotherapy). Patients were at least 50 years old (average age, 64 years) and had been hospitalized for acute coronary syndrome (ACS) within the previous 10 days; 76% were male and 84% were White. The average low-density lipoprotein (LDL) concentration at baseline was 94 mg/dL in both groups.2
The primary endpoint was a composite of cardiovascular death, a major coronary event (nonfatal MI, unstable angina requiring hospitalization, coronary revascularization at least 30 days after randomization), or nonfatal stroke, with a median follow-up of 6 years. The simvastatin plus ezetimibe group compared to the simvastatin-only group had a lower risk for the primary end point (HR = 0.94; 95% CI, 0.89-0.99; NNT = 50), but no differences in cardiovascular or all-cause mortality. Since the study only recruited patients with recent ACS, results are only applicable to that specific population.2
The 2022 RACING study was a multicenter, open-label, randomized, noninferiority trial that evaluated the combination of ezetimibe 10 mg and a moderate-intensity statin (rosuvastatin 10 mg) compared to a high-intensity statin alone (rosuvastatin 20 mg) in adults (N = 3780) with ASCVD. Included patients were ages 19 to 80 years (mean, 64 years) and had a baseline LDL concentration of 80 mg/dL (standard deviation, 64-100 mg/dL) with known ASCVD (defined by prior MI, ACS, history of coronary or other arterial revascularization, ischemic stroke, or peripheral artery disease); 75% were male.3
The primary outcome was a composite of cardiovascular death, major cardiovascular events, or nonfatal stroke. At 3 years, an intention-to-treat analysis found no significant difference between the combination and monotherapy groups (9% vs 9.9%; absolute difference, –0.78%; 95% CI, –2.39% to 0.83%). Dose reduction or discontinuation of the study drug(s) due to intolerance was lower in the combination group than in the monotherapy group (4.8% vs 8.2%; P < 0.0001). The study may be limited by the fact that it was nonblinded and all participants were South Korean, which limits generalizability.3
Recommendations from others
A 2022 evidence-based clinical practice guideline published in BMJ recommends adding ezetimibe to a statin to decrease all-cause mortality, cardiovascular mortality, nonfatal stroke, and nonfatal MI in patients with known CVD, regardless of their LDL concentration (weak recommendation based on a systematic review and network meta-analysis).4
In 2019, the American Heart Association and the American College of Cardiology recommended ezetimibe for patients with clinical ASCVD who are on maximally tolerated statin therapy and have an LDL concentration of 70 mg/dL or higher (Class 2b recommendation [meaning it can be considered] based on a meta-analysis of moderate-quality RCTs).5
Editor’s takeaway
The data on this important and well-studied question have inched closer to firm and clear answers. First, adding ezetimibe to a lower-intensity statin when a higher-intensity statin is not tolerated is an effective treatment. Second, adding ezetimibe to a statin improves nonfatal ASCVD outcomes but not fatal ones. What has not yet been made clear, because a noninferiority trial does not answer this question, is whether the highest intensity statin plus ezetimibe is superior to that high-intensity statin alone, regardless of LDL concentration.
1. Zhan S, Tang M, Liu F, et al. Ezetimibe for the prevention of cardiovascular disease and all‐cause mortality events. Cochrane Database System Rev. 2018;11:CD012502. doi: 10.1002/14651858.CD012502.pub2
2. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387-2397. doi: 10.1056/NEJMoa1410489 pmid:26039521
3. Kim BK, Hong SJ, Lee YJ, et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet. 2022;400:380-390. doi: 10.1016/S0140-6736(22)00916-3
4. Hao Q, Aertgeerts B, Guyatt G, et al. PCSK9 inhibitors and ezetimibe for the reduction of cardiovascular events: a clinical practice guideline with risk-stratified recommendations. BMJ. 2022;377:e069066. doi: 10.1136/bmj-2021-069066
5. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73:e285-e350. doi: 10.1016/j.jacc.2018.11.003
Evidence summary
Adding ezetimibe reduces nonfatal events but does not improve mortality
A 2018 Cochrane meta-analysis included 10 RCTs (N = 21,919 patients) that evaluated the efficacy and safety of ezetimibe plus a statin (dual therapy) vs a statin alone or plus placebo (monotherapy) for the secondary prevention of CVD. Mean age of patients ranged from 55 to 84 years. Almost all of the patients (> 99%) included in the analyses had existing ASCVD. The dose of ezetimibe was 10 mg; statins used included atorvastatin 10 to 80 mg, pitavastatin 2 to 4 mg, rosuvastatin 10 mg, and simvastatin 20 to 80 mg.1
The primary outcomes were MACE and all-cause mortality. MACE is defined as a composite of CVD, nonfatal myocardial infarction (MI), nonfatal stroke, hospitalization for unstable angina, or coronary revascularization procedures. The TABLE1 provides a detailed breakdown of each of the outcomes.
The dual-therapy group compared to the monotherapy group had a lower risk for MACE (26.6% vs 28.3%; 1.7% absolute risk reduction; 6% relative risk reduction; NNT = 59) and little or no difference in the reduction of all-cause mortality. For secondary outcomes, the dual-therapy group had a lower risk for nonfatal MI, nonfatal stroke, and coronary revascularization. There was no difference in cardiovascular mortality or adverse events between the 2 groups. The quality of evidence was high for all-cause mortality and moderate for cardiovascular mortality, MACE, MI, and stroke.1
The 2015 IMPROVE-IT study, the largest included in the Cochrane review, was a double-blind RCT (N = 18,144) conducted at 1147 sites in 39 countries comparing simvastatin 40 mg/d plus ezetimibe 10 mg/d (dual therapy) vs simvastatin 40 mg/d plus placebo (monotherapy). Patients were at least 50 years old (average age, 64 years) and had been hospitalized for acute coronary syndrome (ACS) within the previous 10 days; 76% were male and 84% were White. The average low-density lipoprotein (LDL) concentration at baseline was 94 mg/dL in both groups.2
The primary endpoint was a composite of cardiovascular death, a major coronary event (nonfatal MI, unstable angina requiring hospitalization, coronary revascularization at least 30 days after randomization), or nonfatal stroke, with a median follow-up of 6 years. The simvastatin plus ezetimibe group compared to the simvastatin-only group had a lower risk for the primary end point (HR = 0.94; 95% CI, 0.89-0.99; NNT = 50), but no differences in cardiovascular or all-cause mortality. Since the study only recruited patients with recent ACS, results are only applicable to that specific population.2
The 2022 RACING study was a multicenter, open-label, randomized, noninferiority trial that evaluated the combination of ezetimibe 10 mg and a moderate-intensity statin (rosuvastatin 10 mg) compared to a high-intensity statin alone (rosuvastatin 20 mg) in adults (N = 3780) with ASCVD. Included patients were ages 19 to 80 years (mean, 64 years) and had a baseline LDL concentration of 80 mg/dL (standard deviation, 64-100 mg/dL) with known ASCVD (defined by prior MI, ACS, history of coronary or other arterial revascularization, ischemic stroke, or peripheral artery disease); 75% were male.3
The primary outcome was a composite of cardiovascular death, major cardiovascular events, or nonfatal stroke. At 3 years, an intention-to-treat analysis found no significant difference between the combination and monotherapy groups (9% vs 9.9%; absolute difference, –0.78%; 95% CI, –2.39% to 0.83%). Dose reduction or discontinuation of the study drug(s) due to intolerance was lower in the combination group than in the monotherapy group (4.8% vs 8.2%; P < 0.0001). The study may be limited by the fact that it was nonblinded and all participants were South Korean, which limits generalizability.3
Recommendations from others
A 2022 evidence-based clinical practice guideline published in BMJ recommends adding ezetimibe to a statin to decrease all-cause mortality, cardiovascular mortality, nonfatal stroke, and nonfatal MI in patients with known CVD, regardless of their LDL concentration (weak recommendation based on a systematic review and network meta-analysis).4
In 2019, the American Heart Association and the American College of Cardiology recommended ezetimibe for patients with clinical ASCVD who are on maximally tolerated statin therapy and have an LDL concentration of 70 mg/dL or higher (Class 2b recommendation [meaning it can be considered] based on a meta-analysis of moderate-quality RCTs).5
Editor’s takeaway
The data on this important and well-studied question have inched closer to firm and clear answers. First, adding ezetimibe to a lower-intensity statin when a higher-intensity statin is not tolerated is an effective treatment. Second, adding ezetimibe to a statin improves nonfatal ASCVD outcomes but not fatal ones. What has not yet been made clear, because a noninferiority trial does not answer this question, is whether the highest intensity statin plus ezetimibe is superior to that high-intensity statin alone, regardless of LDL concentration.
Evidence summary
Adding ezetimibe reduces nonfatal events but does not improve mortality
A 2018 Cochrane meta-analysis included 10 RCTs (N = 21,919 patients) that evaluated the efficacy and safety of ezetimibe plus a statin (dual therapy) vs a statin alone or plus placebo (monotherapy) for the secondary prevention of CVD. Mean age of patients ranged from 55 to 84 years. Almost all of the patients (> 99%) included in the analyses had existing ASCVD. The dose of ezetimibe was 10 mg; statins used included atorvastatin 10 to 80 mg, pitavastatin 2 to 4 mg, rosuvastatin 10 mg, and simvastatin 20 to 80 mg.1
The primary outcomes were MACE and all-cause mortality. MACE is defined as a composite of CVD, nonfatal myocardial infarction (MI), nonfatal stroke, hospitalization for unstable angina, or coronary revascularization procedures. The TABLE1 provides a detailed breakdown of each of the outcomes.
The dual-therapy group compared to the monotherapy group had a lower risk for MACE (26.6% vs 28.3%; 1.7% absolute risk reduction; 6% relative risk reduction; NNT = 59) and little or no difference in the reduction of all-cause mortality. For secondary outcomes, the dual-therapy group had a lower risk for nonfatal MI, nonfatal stroke, and coronary revascularization. There was no difference in cardiovascular mortality or adverse events between the 2 groups. The quality of evidence was high for all-cause mortality and moderate for cardiovascular mortality, MACE, MI, and stroke.1
The 2015 IMPROVE-IT study, the largest included in the Cochrane review, was a double-blind RCT (N = 18,144) conducted at 1147 sites in 39 countries comparing simvastatin 40 mg/d plus ezetimibe 10 mg/d (dual therapy) vs simvastatin 40 mg/d plus placebo (monotherapy). Patients were at least 50 years old (average age, 64 years) and had been hospitalized for acute coronary syndrome (ACS) within the previous 10 days; 76% were male and 84% were White. The average low-density lipoprotein (LDL) concentration at baseline was 94 mg/dL in both groups.2
The primary endpoint was a composite of cardiovascular death, a major coronary event (nonfatal MI, unstable angina requiring hospitalization, coronary revascularization at least 30 days after randomization), or nonfatal stroke, with a median follow-up of 6 years. The simvastatin plus ezetimibe group compared to the simvastatin-only group had a lower risk for the primary end point (HR = 0.94; 95% CI, 0.89-0.99; NNT = 50), but no differences in cardiovascular or all-cause mortality. Since the study only recruited patients with recent ACS, results are only applicable to that specific population.2
The 2022 RACING study was a multicenter, open-label, randomized, noninferiority trial that evaluated the combination of ezetimibe 10 mg and a moderate-intensity statin (rosuvastatin 10 mg) compared to a high-intensity statin alone (rosuvastatin 20 mg) in adults (N = 3780) with ASCVD. Included patients were ages 19 to 80 years (mean, 64 years) and had a baseline LDL concentration of 80 mg/dL (standard deviation, 64-100 mg/dL) with known ASCVD (defined by prior MI, ACS, history of coronary or other arterial revascularization, ischemic stroke, or peripheral artery disease); 75% were male.3
The primary outcome was a composite of cardiovascular death, major cardiovascular events, or nonfatal stroke. At 3 years, an intention-to-treat analysis found no significant difference between the combination and monotherapy groups (9% vs 9.9%; absolute difference, –0.78%; 95% CI, –2.39% to 0.83%). Dose reduction or discontinuation of the study drug(s) due to intolerance was lower in the combination group than in the monotherapy group (4.8% vs 8.2%; P < 0.0001). The study may be limited by the fact that it was nonblinded and all participants were South Korean, which limits generalizability.3
Recommendations from others
A 2022 evidence-based clinical practice guideline published in BMJ recommends adding ezetimibe to a statin to decrease all-cause mortality, cardiovascular mortality, nonfatal stroke, and nonfatal MI in patients with known CVD, regardless of their LDL concentration (weak recommendation based on a systematic review and network meta-analysis).4
In 2019, the American Heart Association and the American College of Cardiology recommended ezetimibe for patients with clinical ASCVD who are on maximally tolerated statin therapy and have an LDL concentration of 70 mg/dL or higher (Class 2b recommendation [meaning it can be considered] based on a meta-analysis of moderate-quality RCTs).5
Editor’s takeaway
The data on this important and well-studied question have inched closer to firm and clear answers. First, adding ezetimibe to a lower-intensity statin when a higher-intensity statin is not tolerated is an effective treatment. Second, adding ezetimibe to a statin improves nonfatal ASCVD outcomes but not fatal ones. What has not yet been made clear, because a noninferiority trial does not answer this question, is whether the highest intensity statin plus ezetimibe is superior to that high-intensity statin alone, regardless of LDL concentration.
1. Zhan S, Tang M, Liu F, et al. Ezetimibe for the prevention of cardiovascular disease and all‐cause mortality events. Cochrane Database System Rev. 2018;11:CD012502. doi: 10.1002/14651858.CD012502.pub2
2. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387-2397. doi: 10.1056/NEJMoa1410489 pmid:26039521
3. Kim BK, Hong SJ, Lee YJ, et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet. 2022;400:380-390. doi: 10.1016/S0140-6736(22)00916-3
4. Hao Q, Aertgeerts B, Guyatt G, et al. PCSK9 inhibitors and ezetimibe for the reduction of cardiovascular events: a clinical practice guideline with risk-stratified recommendations. BMJ. 2022;377:e069066. doi: 10.1136/bmj-2021-069066
5. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73:e285-e350. doi: 10.1016/j.jacc.2018.11.003
1. Zhan S, Tang M, Liu F, et al. Ezetimibe for the prevention of cardiovascular disease and all‐cause mortality events. Cochrane Database System Rev. 2018;11:CD012502. doi: 10.1002/14651858.CD012502.pub2
2. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387-2397. doi: 10.1056/NEJMoa1410489 pmid:26039521
3. Kim BK, Hong SJ, Lee YJ, et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet. 2022;400:380-390. doi: 10.1016/S0140-6736(22)00916-3
4. Hao Q, Aertgeerts B, Guyatt G, et al. PCSK9 inhibitors and ezetimibe for the reduction of cardiovascular events: a clinical practice guideline with risk-stratified recommendations. BMJ. 2022;377:e069066. doi: 10.1136/bmj-2021-069066
5. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73:e285-e350. doi: 10.1016/j.jacc.2018.11.003
EVIDENCE-BASED REVIEW:
YES. In patients with known cardio- vascular disease (CVD), ezetimibe with a statin decreases
In adults with atherosclerotic CVD (ASCVD), the combination of ezetimibe and a moderate-intensity statin (rosuvastatin 10 mg) was noninferior at decreasing cardiovascular death, major cardiovascular events, and nonfatal stroke, but was more tolerable, compared to a high-intensity statin (rosuvastatin 20 mg) alone (SOR, B; 1 RCT).

Feeling disconnected? Focus on what you can do
This is the exciting time of year when we graduate new classes of medical students and residents. Med school graduation brings mixed emotions; the new doctors and I both know residency will bring growth and challenges. Residency graduation is a wistful passage as well. It is so rewarding to welcome the newly board-certified family physicians to family medicine, but we miss them even as we orient a new class.
Every year, a few months (or even a few years) after graduation, I hear from a former resident, sometimes several. They ask to talk and, although it can be hard for them to explain exactly the ennui and disillusionment they’re feeling, their concerns boil down to: Is this all there is?
They are not burnt out, exactly, but they were hoping for more from their careers in family medicine.1 They find their hopes and expectations are not fulfilled by seeing patients in the office 8 hours per day, 4.5 days per week. Even those who report rewarding relationships with patients express less overall enthusiasm for jobs they were excited to start just months or years earlier.
Some of the difficulties I hear the graduates report are expected growing pains. It is a transition to go from supervised practice with attending backup to a setting where you are on your own, typically with a 4-fold increase in volume compared with residency. But the monotony is real for family physicians in full-time outpatient practice.
Research suggests an expanded scope of practice—including hospital medicine, obstetrics, and procedures—is associated with physician well-being.2,3 A broad scope of practice can bring stress, but it also brings meaning, and that meaning is protective to our well-being. However, a robust scope of practice is not always supported by medical groups or hospital systems, who prefer a more compartmentalized, widgetized physician.4 It would be easier for their algorithms if family physicians picked a lane and stayed in it. Alas, the broader our scope of practice, the healthier our population, the more equitable our care,5,6 and the happier our physicians.
The disconnect and hopelessness experienced by family physicians is more concerning. Many of my graduates report feeling disconnected from their patients, because they begin to feel disillusioned by the demands and requests that practice and patients place on them. The paperwork, “permission slips,” and requests for tests and studies not only feel overwhelming and exhausting but also create distance between physicians and patients.7 We want to help our patients, so we do the forms and order the tests. As the quantity of forms, slips, and requests adds up, we begin to feel resentful at what the forms take away: time with our patients, perhaps, or time with our families. We get angry at the forms and the “asks,” and then begin to get angry at the patients simply for having needs. Administrative burden is a hassle, but it is also insidiously destructive.8
Family physicians confront hopelessness when, day after day, we diagnose problems that no physician is likely to fix in a single office visit: chronic stress, family dysfunction, violence, unemployment, poverty, racism, loneliness, and the hopelessness of the patients themselves. This is not to say that we ignore these concerns or their impact on health. It is because we see and feel them, and deeply understand their consequences for our patients, that we grow frustrated with the lack of solutions.9,10
Thankfully, we have strong teams working at the policy level to improve the primary care and public health infrastructure so that we can maintain some hope that it will be better in the future. Sometimes when I counsel a former resident, they decide to join those teams so that they can work on the solutions. Others decide to expand their scope of practice. Others seek out virtual scribes to streamline charting and regain time. Some build better boundaries with their EHR inboxes.
The key is figuring out what we can do and making peace with our limits. When disillusionment hits, what we can do includes seeking connection and social contact and remembering that we are not trapped in our situation, even if we are practicing in a less-than-functional health care system. There are many ways to “be” a family physician—if what you’re doing isn’t working for you, look for opportunities (big or small) that make it better. We can all reach out to coaches, therapists, colleagues, and friends for support to remain steadfast in our purpose as family physicians. This support and the power of change means that from residency to the latter parts of our careers, we will continue to bring the tremendous good of family medicine to the communities we serve.
1. Coutinho AJ, Cochrane A, Stelter K, et al. Comparison of intended scope of practice for family medicine residents with reported scope of practice among practicing family physicians. JAMA. 2015;314:2364-2372. doi: 10.1001/jama.2015.13734
2. Weidner AKH, Phillips RL, Fang B, et al. Burnout and scope of practice in new family physicians. Ann Fam Med. 2018;16:200-205. doi: 10.1370/afm.2221
3. Zomahoun HT, Samson I, Sawadogo J, et al. Effects of the scope of practice on family physicians: a systematic review. BMC Family Practice. 2021;22. doi: 10.1186/s12875-020-01328-1
4. Killeen D, Jetty A, Peterson LE, et al. The association of practice type and the scope of care of family physicians. J Am Board Fam Med. 2023;36:79-87. doi: 10.3122/jabfm.2022.220172R1
5. Starfield B, Shi L, Macinko J. Contribution of primary care to health systems and health. Milbank Q. 2005;83:457-502. doi: 10.1111/j.1468-0009.2005.00409.x
6. Ferrer RL. Pursuing equity: contact with primary care and specialist clinicians by demographics, insurance, and health status. Ann Fam Med. 2007;5:492-502. doi: 10.1370/afm.746
7. Rao SK, Kimball AB, Lehrhoff SR, et al. The impact of administrative burden on academic physicians: results of a hospital-wide physician survey. Acad Med. 2017;92:237-243. doi: 10.1097/ACM.0000000000001461
8. McMahon LF, Rize K, Irby-Johnson N, et al. Designed to fail? The future of primary care. J Gen Intern Med. 2021;36:515-517. doi: 10.1007/s11606-020-06077-6
9. Welles CC, Tong A, Brereton E, et al. Sources of clinician burnout in providing care for underserved patients in a safety-net healthcare system. J Gen Intern Med. 2023;38:1468-1475. doi: 10.1007/s11606-022-07896-5
10. Kung A, Cheung T, Knox M, et al. Capacity to address social needs affects primary care clinician burnout. Ann Fam Med. 2019;17:487-494. doi: 10.1370/afm.2470
This is the exciting time of year when we graduate new classes of medical students and residents. Med school graduation brings mixed emotions; the new doctors and I both know residency will bring growth and challenges. Residency graduation is a wistful passage as well. It is so rewarding to welcome the newly board-certified family physicians to family medicine, but we miss them even as we orient a new class.
Every year, a few months (or even a few years) after graduation, I hear from a former resident, sometimes several. They ask to talk and, although it can be hard for them to explain exactly the ennui and disillusionment they’re feeling, their concerns boil down to: Is this all there is?
They are not burnt out, exactly, but they were hoping for more from their careers in family medicine.1 They find their hopes and expectations are not fulfilled by seeing patients in the office 8 hours per day, 4.5 days per week. Even those who report rewarding relationships with patients express less overall enthusiasm for jobs they were excited to start just months or years earlier.
Some of the difficulties I hear the graduates report are expected growing pains. It is a transition to go from supervised practice with attending backup to a setting where you are on your own, typically with a 4-fold increase in volume compared with residency. But the monotony is real for family physicians in full-time outpatient practice.
Research suggests an expanded scope of practice—including hospital medicine, obstetrics, and procedures—is associated with physician well-being.2,3 A broad scope of practice can bring stress, but it also brings meaning, and that meaning is protective to our well-being. However, a robust scope of practice is not always supported by medical groups or hospital systems, who prefer a more compartmentalized, widgetized physician.4 It would be easier for their algorithms if family physicians picked a lane and stayed in it. Alas, the broader our scope of practice, the healthier our population, the more equitable our care,5,6 and the happier our physicians.
The disconnect and hopelessness experienced by family physicians is more concerning. Many of my graduates report feeling disconnected from their patients, because they begin to feel disillusioned by the demands and requests that practice and patients place on them. The paperwork, “permission slips,” and requests for tests and studies not only feel overwhelming and exhausting but also create distance between physicians and patients.7 We want to help our patients, so we do the forms and order the tests. As the quantity of forms, slips, and requests adds up, we begin to feel resentful at what the forms take away: time with our patients, perhaps, or time with our families. We get angry at the forms and the “asks,” and then begin to get angry at the patients simply for having needs. Administrative burden is a hassle, but it is also insidiously destructive.8
Family physicians confront hopelessness when, day after day, we diagnose problems that no physician is likely to fix in a single office visit: chronic stress, family dysfunction, violence, unemployment, poverty, racism, loneliness, and the hopelessness of the patients themselves. This is not to say that we ignore these concerns or their impact on health. It is because we see and feel them, and deeply understand their consequences for our patients, that we grow frustrated with the lack of solutions.9,10
Thankfully, we have strong teams working at the policy level to improve the primary care and public health infrastructure so that we can maintain some hope that it will be better in the future. Sometimes when I counsel a former resident, they decide to join those teams so that they can work on the solutions. Others decide to expand their scope of practice. Others seek out virtual scribes to streamline charting and regain time. Some build better boundaries with their EHR inboxes.
The key is figuring out what we can do and making peace with our limits. When disillusionment hits, what we can do includes seeking connection and social contact and remembering that we are not trapped in our situation, even if we are practicing in a less-than-functional health care system. There are many ways to “be” a family physician—if what you’re doing isn’t working for you, look for opportunities (big or small) that make it better. We can all reach out to coaches, therapists, colleagues, and friends for support to remain steadfast in our purpose as family physicians. This support and the power of change means that from residency to the latter parts of our careers, we will continue to bring the tremendous good of family medicine to the communities we serve.
This is the exciting time of year when we graduate new classes of medical students and residents. Med school graduation brings mixed emotions; the new doctors and I both know residency will bring growth and challenges. Residency graduation is a wistful passage as well. It is so rewarding to welcome the newly board-certified family physicians to family medicine, but we miss them even as we orient a new class.
Every year, a few months (or even a few years) after graduation, I hear from a former resident, sometimes several. They ask to talk and, although it can be hard for them to explain exactly the ennui and disillusionment they’re feeling, their concerns boil down to: Is this all there is?
They are not burnt out, exactly, but they were hoping for more from their careers in family medicine.1 They find their hopes and expectations are not fulfilled by seeing patients in the office 8 hours per day, 4.5 days per week. Even those who report rewarding relationships with patients express less overall enthusiasm for jobs they were excited to start just months or years earlier.
Some of the difficulties I hear the graduates report are expected growing pains. It is a transition to go from supervised practice with attending backup to a setting where you are on your own, typically with a 4-fold increase in volume compared with residency. But the monotony is real for family physicians in full-time outpatient practice.
Research suggests an expanded scope of practice—including hospital medicine, obstetrics, and procedures—is associated with physician well-being.2,3 A broad scope of practice can bring stress, but it also brings meaning, and that meaning is protective to our well-being. However, a robust scope of practice is not always supported by medical groups or hospital systems, who prefer a more compartmentalized, widgetized physician.4 It would be easier for their algorithms if family physicians picked a lane and stayed in it. Alas, the broader our scope of practice, the healthier our population, the more equitable our care,5,6 and the happier our physicians.
The disconnect and hopelessness experienced by family physicians is more concerning. Many of my graduates report feeling disconnected from their patients, because they begin to feel disillusioned by the demands and requests that practice and patients place on them. The paperwork, “permission slips,” and requests for tests and studies not only feel overwhelming and exhausting but also create distance between physicians and patients.7 We want to help our patients, so we do the forms and order the tests. As the quantity of forms, slips, and requests adds up, we begin to feel resentful at what the forms take away: time with our patients, perhaps, or time with our families. We get angry at the forms and the “asks,” and then begin to get angry at the patients simply for having needs. Administrative burden is a hassle, but it is also insidiously destructive.8
Family physicians confront hopelessness when, day after day, we diagnose problems that no physician is likely to fix in a single office visit: chronic stress, family dysfunction, violence, unemployment, poverty, racism, loneliness, and the hopelessness of the patients themselves. This is not to say that we ignore these concerns or their impact on health. It is because we see and feel them, and deeply understand their consequences for our patients, that we grow frustrated with the lack of solutions.9,10
Thankfully, we have strong teams working at the policy level to improve the primary care and public health infrastructure so that we can maintain some hope that it will be better in the future. Sometimes when I counsel a former resident, they decide to join those teams so that they can work on the solutions. Others decide to expand their scope of practice. Others seek out virtual scribes to streamline charting and regain time. Some build better boundaries with their EHR inboxes.
The key is figuring out what we can do and making peace with our limits. When disillusionment hits, what we can do includes seeking connection and social contact and remembering that we are not trapped in our situation, even if we are practicing in a less-than-functional health care system. There are many ways to “be” a family physician—if what you’re doing isn’t working for you, look for opportunities (big or small) that make it better. We can all reach out to coaches, therapists, colleagues, and friends for support to remain steadfast in our purpose as family physicians. This support and the power of change means that from residency to the latter parts of our careers, we will continue to bring the tremendous good of family medicine to the communities we serve.
1. Coutinho AJ, Cochrane A, Stelter K, et al. Comparison of intended scope of practice for family medicine residents with reported scope of practice among practicing family physicians. JAMA. 2015;314:2364-2372. doi: 10.1001/jama.2015.13734
2. Weidner AKH, Phillips RL, Fang B, et al. Burnout and scope of practice in new family physicians. Ann Fam Med. 2018;16:200-205. doi: 10.1370/afm.2221
3. Zomahoun HT, Samson I, Sawadogo J, et al. Effects of the scope of practice on family physicians: a systematic review. BMC Family Practice. 2021;22. doi: 10.1186/s12875-020-01328-1
4. Killeen D, Jetty A, Peterson LE, et al. The association of practice type and the scope of care of family physicians. J Am Board Fam Med. 2023;36:79-87. doi: 10.3122/jabfm.2022.220172R1
5. Starfield B, Shi L, Macinko J. Contribution of primary care to health systems and health. Milbank Q. 2005;83:457-502. doi: 10.1111/j.1468-0009.2005.00409.x
6. Ferrer RL. Pursuing equity: contact with primary care and specialist clinicians by demographics, insurance, and health status. Ann Fam Med. 2007;5:492-502. doi: 10.1370/afm.746
7. Rao SK, Kimball AB, Lehrhoff SR, et al. The impact of administrative burden on academic physicians: results of a hospital-wide physician survey. Acad Med. 2017;92:237-243. doi: 10.1097/ACM.0000000000001461
8. McMahon LF, Rize K, Irby-Johnson N, et al. Designed to fail? The future of primary care. J Gen Intern Med. 2021;36:515-517. doi: 10.1007/s11606-020-06077-6
9. Welles CC, Tong A, Brereton E, et al. Sources of clinician burnout in providing care for underserved patients in a safety-net healthcare system. J Gen Intern Med. 2023;38:1468-1475. doi: 10.1007/s11606-022-07896-5
10. Kung A, Cheung T, Knox M, et al. Capacity to address social needs affects primary care clinician burnout. Ann Fam Med. 2019;17:487-494. doi: 10.1370/afm.2470
1. Coutinho AJ, Cochrane A, Stelter K, et al. Comparison of intended scope of practice for family medicine residents with reported scope of practice among practicing family physicians. JAMA. 2015;314:2364-2372. doi: 10.1001/jama.2015.13734
2. Weidner AKH, Phillips RL, Fang B, et al. Burnout and scope of practice in new family physicians. Ann Fam Med. 2018;16:200-205. doi: 10.1370/afm.2221
3. Zomahoun HT, Samson I, Sawadogo J, et al. Effects of the scope of practice on family physicians: a systematic review. BMC Family Practice. 2021;22. doi: 10.1186/s12875-020-01328-1
4. Killeen D, Jetty A, Peterson LE, et al. The association of practice type and the scope of care of family physicians. J Am Board Fam Med. 2023;36:79-87. doi: 10.3122/jabfm.2022.220172R1
5. Starfield B, Shi L, Macinko J. Contribution of primary care to health systems and health. Milbank Q. 2005;83:457-502. doi: 10.1111/j.1468-0009.2005.00409.x
6. Ferrer RL. Pursuing equity: contact with primary care and specialist clinicians by demographics, insurance, and health status. Ann Fam Med. 2007;5:492-502. doi: 10.1370/afm.746
7. Rao SK, Kimball AB, Lehrhoff SR, et al. The impact of administrative burden on academic physicians: results of a hospital-wide physician survey. Acad Med. 2017;92:237-243. doi: 10.1097/ACM.0000000000001461
8. McMahon LF, Rize K, Irby-Johnson N, et al. Designed to fail? The future of primary care. J Gen Intern Med. 2021;36:515-517. doi: 10.1007/s11606-020-06077-6
9. Welles CC, Tong A, Brereton E, et al. Sources of clinician burnout in providing care for underserved patients in a safety-net healthcare system. J Gen Intern Med. 2023;38:1468-1475. doi: 10.1007/s11606-022-07896-5
10. Kung A, Cheung T, Knox M, et al. Capacity to address social needs affects primary care clinician burnout. Ann Fam Med. 2019;17:487-494. doi: 10.1370/afm.2470
Pedunculated gluteal mass
A 30-YEAR-OLD MAN presented for evaluation of a solitary, flesh-colored, pedunculated mass on his right inner gluteal area (FIGURE) that had gradually enlarged over the previous 18 months. The lesion had manifested 4 years prior as a small papule that was stable for many years. It began to grow steadily after the patient compressed the papule forcefully. Activities of daily living, such as sitting, were now uncomfortable, so he sought treatment. He denied pain, pruritis, and bleeding and reported no history of trauma or surgery in the area of the mass.

On physical examination, the mass measured 3.5 × 4.5 cm with a 1.2-cm base. It was smooth, soft, nontender, and compressible—but nonfluctuant. There were no signs of ulceration or bleeding. No regional lymphadenopathy was noted. An excisional biopsy was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Fibrolipoma
The biopsy confirmed a diagnosis of fibrolipoma—a rare variant of lipoma composed of a mixture of adipocytes and thick bands of fibrous connective tissues.1 Etiology for fibrolipomas is unknown. Blunt trauma rupture of the fibrous septa that prevent fat migration may result in a proliferation of adipose tissue and thereby enlargement of fibrolipomas and other lipoma variants.2 In this case, the patient’s compression of the original papule likely served as the trauma that led to its enlargement. Malignant change has not been reported with fibrolipomas.
What you’ll see—and on whom. Fibrolipomas typically are flesh-colored, pedunculated, compressible, and relatively asymptomatic.3 They have been reported on the face, neck, back, and pubic areas, among other locations. Size is variable; they can be as small as 1 cm in diameter and as large as 10 cm in diameter.4 However, fibrolipomas can grow to be “giant” if they exceed 10 cm (or 1000 g).2
Men and women are affected equally by fibrolipomas. Prevalence does not differ by race or ethnicity.
The differential include sother lipomas and skin tags
The differential for a mass such as this one includes lipomas, acrochordons (also known as skin tags), and fibrokeratomas.
Lipomas are the most common benign soft-tissue tumors and are composed of adipocytes.5 The fibrolipoma is just one variant of lipoma; others include the myxolipoma, myolipoma, spindle cell lipoma, angiolipoma, osteolipoma, and chondrolipoma.2 Lipomas typically are subcutaneous and located over the scalp, neck, and upper trunk area but can occur anywhere on the body. They are mobile and typically well circumscribed. Lipomas have a broad base with well-demarcated swelling; fibrolipomas are usually pedunculated.
Continue to: Acrochordons ("skin tags")
Acrochordons (“skin tags”) usually contain a peduncle but may be sessile. They range from 1 mm to 1 cm in diameter and typically are located in skin folds, especially in the neck, axillae, and inguinal areas.6 Obesity, older
Fibrokeratomas typically are benign, solitary, fibrous tissue tumors that are found on fingers and seldom are pedunculated. They are flesh-colored and conical or nodular, with a hyperkeratotic collar. Fibrokeratomas are smaller and thicker than fibromas, as well as firm in consistency. They are acquired tumors that have been shown to be related to repetitive trauma.6
Treatment involves surgical excision
The preferred treatment for fibrolipoma is complete surgical excision, although cryotherapy is another option for lesions < 1 cm.4 Without surgical excision, the mass will continue to grow, albeit slowly.
This patient’s mass was excised successfully in its entirety; there were no complications. Follow-up is usually unnecessary.
1. Kim YT, Kim WS, Park YL, et al. A case of fibrolipoma. Korean J Dermatol. 2003;41:939-941.
2. Mazzocchi M, Onesti MG, Pasquini P, et al. Giant fibrolipoma in the leg—a case report. Anticancer Res. 2006;26:3649-3654.
3. Shin SJ. Subcutaneous fibrolipoma on the back. J Craniofac Surg. 2013;24:1051-1053. doi: 10.1097/SCS.0b013e3182802517
4. Suleiman J, Suleman M, Amsi P, et al. Giant pedunculated lipofibroma of the thigh. J Surg Case Rep. 2023;2023(3):rjad153. doi: 10.1093/jscr/rjad153
5. Dai X-M, Li Y-S, Liu H, et al. Giant pedunculated fibrolipoma arising from right facial and cervical region. J Oral and Maxillofac Surg. 2009;67:1323-1326. doi: 10.1016/j.joms.2008.12.037
6. Lee JA, Khodaee M. Enlarging, pedunculated skin lesion. Am Fam Physician. 2012;85:1191-1192.
7. Banik R, Lubach D. Skin tags: localization and frequencies according to sex and age. Dermatologica. 1987;174:180-183. doi: 10.1159/000249169
A 30-YEAR-OLD MAN presented for evaluation of a solitary, flesh-colored, pedunculated mass on his right inner gluteal area (FIGURE) that had gradually enlarged over the previous 18 months. The lesion had manifested 4 years prior as a small papule that was stable for many years. It began to grow steadily after the patient compressed the papule forcefully. Activities of daily living, such as sitting, were now uncomfortable, so he sought treatment. He denied pain, pruritis, and bleeding and reported no history of trauma or surgery in the area of the mass.
On physical examination, the mass measured 3.5 × 4.5 cm with a 1.2-cm base. It was smooth, soft, nontender, and compressible—but nonfluctuant. There were no signs of ulceration or bleeding. No regional lymphadenopathy was noted. An excisional biopsy was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Fibrolipoma
The biopsy confirmed a diagnosis of fibrolipoma—a rare variant of lipoma composed of a mixture of adipocytes and thick bands of fibrous connective tissues.1 Etiology for fibrolipomas is unknown. Blunt trauma rupture of the fibrous septa that prevent fat migration may result in a proliferation of adipose tissue and thereby enlargement of fibrolipomas and other lipoma variants.2 In this case, the patient’s compression of the original papule likely served as the trauma that led to its enlargement. Malignant change has not been reported with fibrolipomas.
What you’ll see—and on whom. Fibrolipomas typically are flesh-colored, pedunculated, compressible, and relatively asymptomatic.3 They have been reported on the face, neck, back, and pubic areas, among other locations. Size is variable; they can be as small as 1 cm in diameter and as large as 10 cm in diameter.4 However, fibrolipomas can grow to be “giant” if they exceed 10 cm (or 1000 g).2
Men and women are affected equally by fibrolipomas. Prevalence does not differ by race or ethnicity.
The differential include sother lipomas and skin tags
The differential for a mass such as this one includes lipomas, acrochordons (also known as skin tags), and fibrokeratomas.
Lipomas are the most common benign soft-tissue tumors and are composed of adipocytes.5 The fibrolipoma is just one variant of lipoma; others include the myxolipoma, myolipoma, spindle cell lipoma, angiolipoma, osteolipoma, and chondrolipoma.2 Lipomas typically are subcutaneous and located over the scalp, neck, and upper trunk area but can occur anywhere on the body. They are mobile and typically well circumscribed. Lipomas have a broad base with well-demarcated swelling; fibrolipomas are usually pedunculated.
Continue to: Acrochordons ("skin tags")
Acrochordons (“skin tags”) usually contain a peduncle but may be sessile. They range from 1 mm to 1 cm in diameter and typically are located in skin folds, especially in the neck, axillae, and inguinal areas.6 Obesity, older
Fibrokeratomas typically are benign, solitary, fibrous tissue tumors that are found on fingers and seldom are pedunculated. They are flesh-colored and conical or nodular, with a hyperkeratotic collar. Fibrokeratomas are smaller and thicker than fibromas, as well as firm in consistency. They are acquired tumors that have been shown to be related to repetitive trauma.6
Treatment involves surgical excision
The preferred treatment for fibrolipoma is complete surgical excision, although cryotherapy is another option for lesions < 1 cm.4 Without surgical excision, the mass will continue to grow, albeit slowly.
This patient’s mass was excised successfully in its entirety; there were no complications. Follow-up is usually unnecessary.
A 30-YEAR-OLD MAN presented for evaluation of a solitary, flesh-colored, pedunculated mass on his right inner gluteal area (FIGURE) that had gradually enlarged over the previous 18 months. The lesion had manifested 4 years prior as a small papule that was stable for many years. It began to grow steadily after the patient compressed the papule forcefully. Activities of daily living, such as sitting, were now uncomfortable, so he sought treatment. He denied pain, pruritis, and bleeding and reported no history of trauma or surgery in the area of the mass.
On physical examination, the mass measured 3.5 × 4.5 cm with a 1.2-cm base. It was smooth, soft, nontender, and compressible—but nonfluctuant. There were no signs of ulceration or bleeding. No regional lymphadenopathy was noted. An excisional biopsy was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Fibrolipoma
The biopsy confirmed a diagnosis of fibrolipoma—a rare variant of lipoma composed of a mixture of adipocytes and thick bands of fibrous connective tissues.1 Etiology for fibrolipomas is unknown. Blunt trauma rupture of the fibrous septa that prevent fat migration may result in a proliferation of adipose tissue and thereby enlargement of fibrolipomas and other lipoma variants.2 In this case, the patient’s compression of the original papule likely served as the trauma that led to its enlargement. Malignant change has not been reported with fibrolipomas.
What you’ll see—and on whom. Fibrolipomas typically are flesh-colored, pedunculated, compressible, and relatively asymptomatic.3 They have been reported on the face, neck, back, and pubic areas, among other locations. Size is variable; they can be as small as 1 cm in diameter and as large as 10 cm in diameter.4 However, fibrolipomas can grow to be “giant” if they exceed 10 cm (or 1000 g).2
Men and women are affected equally by fibrolipomas. Prevalence does not differ by race or ethnicity.
The differential include sother lipomas and skin tags
The differential for a mass such as this one includes lipomas, acrochordons (also known as skin tags), and fibrokeratomas.
Lipomas are the most common benign soft-tissue tumors and are composed of adipocytes.5 The fibrolipoma is just one variant of lipoma; others include the myxolipoma, myolipoma, spindle cell lipoma, angiolipoma, osteolipoma, and chondrolipoma.2 Lipomas typically are subcutaneous and located over the scalp, neck, and upper trunk area but can occur anywhere on the body. They are mobile and typically well circumscribed. Lipomas have a broad base with well-demarcated swelling; fibrolipomas are usually pedunculated.
Continue to: Acrochordons ("skin tags")
Acrochordons (“skin tags”) usually contain a peduncle but may be sessile. They range from 1 mm to 1 cm in diameter and typically are located in skin folds, especially in the neck, axillae, and inguinal areas.6 Obesity, older
Fibrokeratomas typically are benign, solitary, fibrous tissue tumors that are found on fingers and seldom are pedunculated. They are flesh-colored and conical or nodular, with a hyperkeratotic collar. Fibrokeratomas are smaller and thicker than fibromas, as well as firm in consistency. They are acquired tumors that have been shown to be related to repetitive trauma.6
Treatment involves surgical excision
The preferred treatment for fibrolipoma is complete surgical excision, although cryotherapy is another option for lesions < 1 cm.4 Without surgical excision, the mass will continue to grow, albeit slowly.
This patient’s mass was excised successfully in its entirety; there were no complications. Follow-up is usually unnecessary.
1. Kim YT, Kim WS, Park YL, et al. A case of fibrolipoma. Korean J Dermatol. 2003;41:939-941.
2. Mazzocchi M, Onesti MG, Pasquini P, et al. Giant fibrolipoma in the leg—a case report. Anticancer Res. 2006;26:3649-3654.
3. Shin SJ. Subcutaneous fibrolipoma on the back. J Craniofac Surg. 2013;24:1051-1053. doi: 10.1097/SCS.0b013e3182802517
4. Suleiman J, Suleman M, Amsi P, et al. Giant pedunculated lipofibroma of the thigh. J Surg Case Rep. 2023;2023(3):rjad153. doi: 10.1093/jscr/rjad153
5. Dai X-M, Li Y-S, Liu H, et al. Giant pedunculated fibrolipoma arising from right facial and cervical region. J Oral and Maxillofac Surg. 2009;67:1323-1326. doi: 10.1016/j.joms.2008.12.037
6. Lee JA, Khodaee M. Enlarging, pedunculated skin lesion. Am Fam Physician. 2012;85:1191-1192.
7. Banik R, Lubach D. Skin tags: localization and frequencies according to sex and age. Dermatologica. 1987;174:180-183. doi: 10.1159/000249169
1. Kim YT, Kim WS, Park YL, et al. A case of fibrolipoma. Korean J Dermatol. 2003;41:939-941.
2. Mazzocchi M, Onesti MG, Pasquini P, et al. Giant fibrolipoma in the leg—a case report. Anticancer Res. 2006;26:3649-3654.
3. Shin SJ. Subcutaneous fibrolipoma on the back. J Craniofac Surg. 2013;24:1051-1053. doi: 10.1097/SCS.0b013e3182802517
4. Suleiman J, Suleman M, Amsi P, et al. Giant pedunculated lipofibroma of the thigh. J Surg Case Rep. 2023;2023(3):rjad153. doi: 10.1093/jscr/rjad153
5. Dai X-M, Li Y-S, Liu H, et al. Giant pedunculated fibrolipoma arising from right facial and cervical region. J Oral and Maxillofac Surg. 2009;67:1323-1326. doi: 10.1016/j.joms.2008.12.037
6. Lee JA, Khodaee M. Enlarging, pedunculated skin lesion. Am Fam Physician. 2012;85:1191-1192.
7. Banik R, Lubach D. Skin tags: localization and frequencies according to sex and age. Dermatologica. 1987;174:180-183. doi: 10.1159/000249169

64-year-old woman • hot flashes, facial flushing, excessive sweating, and palpitations • daily headaches • history of hypertension • Dx?
THE CASE
A 64-year-old woman sought care after having hot flashes, facial flushing, excessive sweating, palpitations, and daily headaches for 1 month. She had a history of hypertension that was well controlled with hydrochlorothiazide 25 mg/d but over the previous month, it had become more difficult to control. Her blood pressure remained elevated to 150/100 mm Hg despite the addition of lisinopril 40 mg/d and amlodipine 10 mg/d, indicating resistant hypertension. She had no family history of hypertension, diabetes, or obesity or any other pertinent medical or surgical history. Physical examination was negative for weight gain, stretch marks, or muscle weakness.

Laboratory tests revealed a normal serum aldosterone-renin ratio, renal function, and thyroid function; however, she had elevated levels of normetanephrine (2429 pg/mL; normal range, 0-145 pg/mL) and metanephrine (143 pg/mL; normal range, 0-62 pg/mL). Computed tomography (CT) revealed an 8.6-cm complex, hemorrhagic, necrotic left adrenal mass with attenuation of 33.1 Hounsfield units (HU) (FIGURE 1). Magnetic resonance imaging (MRI) demonstrated a T2 hyperintense left adrenal mass. An evaluation for Cushing syndrome was negative, and positron emission tomography (PET)/CT with gallium-68 dotatate was ordered. It showed intense radiotracer uptake in the left adrenal gland, with a maximum standardized uptake value of 70.1 (FIGURE 2).

THE DIAGNOSIS
After appropriate preparation with alpha blockade (phenoxybenzamine 20 mg twice daily for 7 days) and fluid resuscitation (normal saline run over 12 hours preoperatively), the patient underwent successful open surgical resection of the adrenal mass, during which her blood pressure was controlled with a nitroprusside infusion and boluses of esmolol and labetalol. Pathology results showed cells in a nested pattern with round to oval nuclei in a vascular background. There was no necrosis, increased mitotic figures, capsular invasion, or increased cellularity. Chromogranin immunohistochemical staining was positive. Given her resistant hypertension, clinical symptoms, and pathology results, the patient was given a diagnosis of pheochromocytoma.
DISCUSSION
Resistant hypertension is defined as blood pressure that is elevated above goal despite the use of 3 maximally titrated antihypertensive agents from different classes or that is well controlled with at least 4 antihypertensive medications.1 The prevalence of resistant hypertension is 12% to 18% in adults being treated for hypertension.1 Patients with resistant hypertension have a higher risk for cardiovascular events and death, are more likely to have a secondary cause of hypertension, and may benefit from special diagnostic testing or treatment approaches to control their blood pressure.1
There are many causes of resistant hypertension; primary aldosteronism is the most common cause (prevalence as high as 20%).2 Given the increased risk for cardiovascular/cerebrovascular disease, all patients with resistant hypertension should be screened for this condition.2 Other causes of resistant hypertension include renal parenchymal disease, renal artery stenosis, coarctation of the aorta, thyroid dysfunction, Cushing syndrome, paraganglioma, and as seen in our case, pheochromocytoma. Although pheochromocytoma is a rare cause of resistant hypertension (0.01%-4%),1 it is associated with high rates of morbidity and mortality if left untreated and may be inherited, making it an essential diagnosis to consider in all patients with resistant hypertension.1,3
Common symptoms of pheochromocytoma are hypertension (paroxysmal or sustained), headaches, palpitations, pallor, and piloerection (or cold sweats).1 Patients with pheochromocytoma typically exhibit metanephrine levels that are more than 4 times the upper limit of normal.4 Therefore, measurement of plasma free metanephrines or urinary fractionated metanephrines is recommended.5 Elevated metanephrine levels also are caused by obesity, obstructive sleep apnea, and certain medications and should be ruled out.5
All pheochromocytomas are potentially malignant. Despite the existence of pathologic scoring systems6,7 and radiographic features that suggest malignancy,8,9 no single risk-stratification tool is recommended in the current literature.10 Ultimately, the only way to confirm malignancy is to see metastases where chromaffin tissue is not normally found on imaging.10
Continue to: Pathologic features to look for...
Pathologic features to look for include capsular/periadrenal adipose invasion, increased cellularity, necrosis, tumor cell spindling, increased/atypical mitotic figures, and nuclear pleomorphism. Radiographic features include larger size (≥ 4-6 cm),11 an irregular shape, necrosis, calcifications, attenuation of 10 HU or higher on noncontrast CT, absolute washout of 60% or lower, and relative washout of 40% or lower.8,12 On MRI, malignant lesions appear hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging.9 Fluorodeoxyglucose avidity on PET scan also is indicative of malignancy.8,9
Treatment for pheochromocytoma is surgical resection. An experienced surgical team and proper preoperative preparation are necessary because the induction of anesthesia, endotracheal intubation, and tumor manipulation can lead to a release of catecholamines, potentially resulting in an intraoperative hypertensive crisis, cardiac arrhythmias, and multiorgan failure.
Proper preoperative preparation includes taking an alpha-adrenergic blocker, such as phenoxybenzamine, prazosin, terazosin, or doxazosin, for at least 7 days to normalize the patient’s blood pressure. Patients should be counseled that they may experience nasal congestion, orthostasis, and fatigue while taking these medications. Volume expansion with intravenous fluids also should be performed and a high-salt diet considered. Beta-adrenergic blockade can be initiated once appropriate alpha-adrenergic blockade is achieved to control the patient’s heart rate; beta-blockers should never be started first because of the risk for severe hypertension. Careful hemodynamic monitoring is vital intraoperatively and postoperatively.5,13 Because metastatic lesions can occur decades after resection, long-term follow-up is critical.5,10
Following tumor resection, our patient’s blood pressure was supported with intravenous fluids and phenylephrine. She was able to discontinue all her antihypertensive medications postoperatively, and her plasma free and urinary fractionated metanephrine levels returned to within normal limits 8 weeks after surgery. Five years after surgery, she continues to have no signs of recurrence, as evidenced by annual negative plasma free metanephrines testing and abdominal/pelvic CT.
THE TAKEAWAY
This case highlights the importance of recognizing resistant hypertension and a potential secondary cause of this disease—pheochromocytoma. Although rare, pheochromocytomas confer increased risk for cardiovascular disease and death. Thus, swift recognition and proper preparation for surgical resection are necessary. Malignant lesions can be diagnosed only upon discovery of metastatic disease and can recur for decades after surgical resection, making diligent long-term follow-up imperative.
CORRESPONDENCE
Nicole O. Vietor, MD, Division of Endocrinology, Walter Reed National Military Medical Center, 8901 Wisconsin Avenue, Bethesda, MD 20889; nicole.o.vietor.mil@health.mil
1. Carey RM, Calhoun DA, Bakris GL, et al. Resistant hypertension: detection, evaluation, and management: a scientific statement from the American Heart Association. Hypertension. 2018;72:e53-e90. doi: 10.1161/HYP.0000000000000084
2. Young WF Jr. Diagnosis and treatment of primary aldosteronism: practical clinical perspectives. J Intern Med. 2019;285:126-148. doi: 10.1111/joim.12831
3. Young WF Jr, Calhoun DA, Lenders JWM, et al. Screening for endocrine hypertension: an Endocrine Society Scientific Statement. Endocr Rev. 2017;38:103-122. doi: 10.1210/er.2017-00054
4. Lenders JWM, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427-1434. doi: 10.1001/jama.287.11.1427
5. Lenders JW, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:1915-1942. doi: 10.1210/jc.2014-1498
6. Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21:405-414. doi: 10.1530/ERC-13-0494
7. Thompson LDR. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551-566. doi: 10.1097/00000478-200205000-00002
8. Vaidya A, Hamrahian A, Bancos I, et al. The evaluation of incidentally discovered adrenal masses. Endocr Pract. 2019;25:178-192. doi: 10.4158/DSCR-2018-0565
9. Young WF Jr. Conventional imaging in adrenocortical carcinoma: update and perspectives. Horm Cancer. 2011;2:341-347. doi: 10.1007/s12672-011-0089-z
10. Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019;381:552-565. doi: 10.1056/NEJMra1806651
11. Iñiguez-Ariza NM, Kohlenberg JD, Delivanis DA, et al. Clinical, biochemical, and radiological characteristics of a single-center retrospective cohort of 705 large adrenal tumors. Mayo Clin Proc Innov Qual Outcomes. 2017;2:30-39. doi: 10.1016/j.mayocpiqo.2017.11.002
12. Marty M, Gaye D, Perez P, et al. Diagnostic accuracy of computed tomography to identify adenomas among adrenal incidentalomas in an endocrinological population. Eur J Endocrinol. 2018;178:439-446. doi: 10.1530/EJE-17-1056
13. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069-4079. doi: 10.1210/jc.2007-1720
THE CASE
A 64-year-old woman sought care after having hot flashes, facial flushing, excessive sweating, palpitations, and daily headaches for 1 month. She had a history of hypertension that was well controlled with hydrochlorothiazide 25 mg/d but over the previous month, it had become more difficult to control. Her blood pressure remained elevated to 150/100 mm Hg despite the addition of lisinopril 40 mg/d and amlodipine 10 mg/d, indicating resistant hypertension. She had no family history of hypertension, diabetes, or obesity or any other pertinent medical or surgical history. Physical examination was negative for weight gain, stretch marks, or muscle weakness.
Laboratory tests revealed a normal serum aldosterone-renin ratio, renal function, and thyroid function; however, she had elevated levels of normetanephrine (2429 pg/mL; normal range, 0-145 pg/mL) and metanephrine (143 pg/mL; normal range, 0-62 pg/mL). Computed tomography (CT) revealed an 8.6-cm complex, hemorrhagic, necrotic left adrenal mass with attenuation of 33.1 Hounsfield units (HU) (FIGURE 1). Magnetic resonance imaging (MRI) demonstrated a T2 hyperintense left adrenal mass. An evaluation for Cushing syndrome was negative, and positron emission tomography (PET)/CT with gallium-68 dotatate was ordered. It showed intense radiotracer uptake in the left adrenal gland, with a maximum standardized uptake value of 70.1 (FIGURE 2).
THE DIAGNOSIS
After appropriate preparation with alpha blockade (phenoxybenzamine 20 mg twice daily for 7 days) and fluid resuscitation (normal saline run over 12 hours preoperatively), the patient underwent successful open surgical resection of the adrenal mass, during which her blood pressure was controlled with a nitroprusside infusion and boluses of esmolol and labetalol. Pathology results showed cells in a nested pattern with round to oval nuclei in a vascular background. There was no necrosis, increased mitotic figures, capsular invasion, or increased cellularity. Chromogranin immunohistochemical staining was positive. Given her resistant hypertension, clinical symptoms, and pathology results, the patient was given a diagnosis of pheochromocytoma.
DISCUSSION
Resistant hypertension is defined as blood pressure that is elevated above goal despite the use of 3 maximally titrated antihypertensive agents from different classes or that is well controlled with at least 4 antihypertensive medications.1 The prevalence of resistant hypertension is 12% to 18% in adults being treated for hypertension.1 Patients with resistant hypertension have a higher risk for cardiovascular events and death, are more likely to have a secondary cause of hypertension, and may benefit from special diagnostic testing or treatment approaches to control their blood pressure.1
There are many causes of resistant hypertension; primary aldosteronism is the most common cause (prevalence as high as 20%).2 Given the increased risk for cardiovascular/cerebrovascular disease, all patients with resistant hypertension should be screened for this condition.2 Other causes of resistant hypertension include renal parenchymal disease, renal artery stenosis, coarctation of the aorta, thyroid dysfunction, Cushing syndrome, paraganglioma, and as seen in our case, pheochromocytoma. Although pheochromocytoma is a rare cause of resistant hypertension (0.01%-4%),1 it is associated with high rates of morbidity and mortality if left untreated and may be inherited, making it an essential diagnosis to consider in all patients with resistant hypertension.1,3
Common symptoms of pheochromocytoma are hypertension (paroxysmal or sustained), headaches, palpitations, pallor, and piloerection (or cold sweats).1 Patients with pheochromocytoma typically exhibit metanephrine levels that are more than 4 times the upper limit of normal.4 Therefore, measurement of plasma free metanephrines or urinary fractionated metanephrines is recommended.5 Elevated metanephrine levels also are caused by obesity, obstructive sleep apnea, and certain medications and should be ruled out.5
All pheochromocytomas are potentially malignant. Despite the existence of pathologic scoring systems6,7 and radiographic features that suggest malignancy,8,9 no single risk-stratification tool is recommended in the current literature.10 Ultimately, the only way to confirm malignancy is to see metastases where chromaffin tissue is not normally found on imaging.10
Continue to: Pathologic features to look for...
Pathologic features to look for include capsular/periadrenal adipose invasion, increased cellularity, necrosis, tumor cell spindling, increased/atypical mitotic figures, and nuclear pleomorphism. Radiographic features include larger size (≥ 4-6 cm),11 an irregular shape, necrosis, calcifications, attenuation of 10 HU or higher on noncontrast CT, absolute washout of 60% or lower, and relative washout of 40% or lower.8,12 On MRI, malignant lesions appear hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging.9 Fluorodeoxyglucose avidity on PET scan also is indicative of malignancy.8,9
Treatment for pheochromocytoma is surgical resection. An experienced surgical team and proper preoperative preparation are necessary because the induction of anesthesia, endotracheal intubation, and tumor manipulation can lead to a release of catecholamines, potentially resulting in an intraoperative hypertensive crisis, cardiac arrhythmias, and multiorgan failure.
Proper preoperative preparation includes taking an alpha-adrenergic blocker, such as phenoxybenzamine, prazosin, terazosin, or doxazosin, for at least 7 days to normalize the patient’s blood pressure. Patients should be counseled that they may experience nasal congestion, orthostasis, and fatigue while taking these medications. Volume expansion with intravenous fluids also should be performed and a high-salt diet considered. Beta-adrenergic blockade can be initiated once appropriate alpha-adrenergic blockade is achieved to control the patient’s heart rate; beta-blockers should never be started first because of the risk for severe hypertension. Careful hemodynamic monitoring is vital intraoperatively and postoperatively.5,13 Because metastatic lesions can occur decades after resection, long-term follow-up is critical.5,10
Following tumor resection, our patient’s blood pressure was supported with intravenous fluids and phenylephrine. She was able to discontinue all her antihypertensive medications postoperatively, and her plasma free and urinary fractionated metanephrine levels returned to within normal limits 8 weeks after surgery. Five years after surgery, she continues to have no signs of recurrence, as evidenced by annual negative plasma free metanephrines testing and abdominal/pelvic CT.
THE TAKEAWAY
This case highlights the importance of recognizing resistant hypertension and a potential secondary cause of this disease—pheochromocytoma. Although rare, pheochromocytomas confer increased risk for cardiovascular disease and death. Thus, swift recognition and proper preparation for surgical resection are necessary. Malignant lesions can be diagnosed only upon discovery of metastatic disease and can recur for decades after surgical resection, making diligent long-term follow-up imperative.
CORRESPONDENCE
Nicole O. Vietor, MD, Division of Endocrinology, Walter Reed National Military Medical Center, 8901 Wisconsin Avenue, Bethesda, MD 20889; nicole.o.vietor.mil@health.mil
THE CASE
A 64-year-old woman sought care after having hot flashes, facial flushing, excessive sweating, palpitations, and daily headaches for 1 month. She had a history of hypertension that was well controlled with hydrochlorothiazide 25 mg/d but over the previous month, it had become more difficult to control. Her blood pressure remained elevated to 150/100 mm Hg despite the addition of lisinopril 40 mg/d and amlodipine 10 mg/d, indicating resistant hypertension. She had no family history of hypertension, diabetes, or obesity or any other pertinent medical or surgical history. Physical examination was negative for weight gain, stretch marks, or muscle weakness.
Laboratory tests revealed a normal serum aldosterone-renin ratio, renal function, and thyroid function; however, she had elevated levels of normetanephrine (2429 pg/mL; normal range, 0-145 pg/mL) and metanephrine (143 pg/mL; normal range, 0-62 pg/mL). Computed tomography (CT) revealed an 8.6-cm complex, hemorrhagic, necrotic left adrenal mass with attenuation of 33.1 Hounsfield units (HU) (FIGURE 1). Magnetic resonance imaging (MRI) demonstrated a T2 hyperintense left adrenal mass. An evaluation for Cushing syndrome was negative, and positron emission tomography (PET)/CT with gallium-68 dotatate was ordered. It showed intense radiotracer uptake in the left adrenal gland, with a maximum standardized uptake value of 70.1 (FIGURE 2).
THE DIAGNOSIS
After appropriate preparation with alpha blockade (phenoxybenzamine 20 mg twice daily for 7 days) and fluid resuscitation (normal saline run over 12 hours preoperatively), the patient underwent successful open surgical resection of the adrenal mass, during which her blood pressure was controlled with a nitroprusside infusion and boluses of esmolol and labetalol. Pathology results showed cells in a nested pattern with round to oval nuclei in a vascular background. There was no necrosis, increased mitotic figures, capsular invasion, or increased cellularity. Chromogranin immunohistochemical staining was positive. Given her resistant hypertension, clinical symptoms, and pathology results, the patient was given a diagnosis of pheochromocytoma.
DISCUSSION
Resistant hypertension is defined as blood pressure that is elevated above goal despite the use of 3 maximally titrated antihypertensive agents from different classes or that is well controlled with at least 4 antihypertensive medications.1 The prevalence of resistant hypertension is 12% to 18% in adults being treated for hypertension.1 Patients with resistant hypertension have a higher risk for cardiovascular events and death, are more likely to have a secondary cause of hypertension, and may benefit from special diagnostic testing or treatment approaches to control their blood pressure.1
There are many causes of resistant hypertension; primary aldosteronism is the most common cause (prevalence as high as 20%).2 Given the increased risk for cardiovascular/cerebrovascular disease, all patients with resistant hypertension should be screened for this condition.2 Other causes of resistant hypertension include renal parenchymal disease, renal artery stenosis, coarctation of the aorta, thyroid dysfunction, Cushing syndrome, paraganglioma, and as seen in our case, pheochromocytoma. Although pheochromocytoma is a rare cause of resistant hypertension (0.01%-4%),1 it is associated with high rates of morbidity and mortality if left untreated and may be inherited, making it an essential diagnosis to consider in all patients with resistant hypertension.1,3
Common symptoms of pheochromocytoma are hypertension (paroxysmal or sustained), headaches, palpitations, pallor, and piloerection (or cold sweats).1 Patients with pheochromocytoma typically exhibit metanephrine levels that are more than 4 times the upper limit of normal.4 Therefore, measurement of plasma free metanephrines or urinary fractionated metanephrines is recommended.5 Elevated metanephrine levels also are caused by obesity, obstructive sleep apnea, and certain medications and should be ruled out.5
All pheochromocytomas are potentially malignant. Despite the existence of pathologic scoring systems6,7 and radiographic features that suggest malignancy,8,9 no single risk-stratification tool is recommended in the current literature.10 Ultimately, the only way to confirm malignancy is to see metastases where chromaffin tissue is not normally found on imaging.10
Continue to: Pathologic features to look for...
Pathologic features to look for include capsular/periadrenal adipose invasion, increased cellularity, necrosis, tumor cell spindling, increased/atypical mitotic figures, and nuclear pleomorphism. Radiographic features include larger size (≥ 4-6 cm),11 an irregular shape, necrosis, calcifications, attenuation of 10 HU or higher on noncontrast CT, absolute washout of 60% or lower, and relative washout of 40% or lower.8,12 On MRI, malignant lesions appear hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging.9 Fluorodeoxyglucose avidity on PET scan also is indicative of malignancy.8,9
Treatment for pheochromocytoma is surgical resection. An experienced surgical team and proper preoperative preparation are necessary because the induction of anesthesia, endotracheal intubation, and tumor manipulation can lead to a release of catecholamines, potentially resulting in an intraoperative hypertensive crisis, cardiac arrhythmias, and multiorgan failure.
Proper preoperative preparation includes taking an alpha-adrenergic blocker, such as phenoxybenzamine, prazosin, terazosin, or doxazosin, for at least 7 days to normalize the patient’s blood pressure. Patients should be counseled that they may experience nasal congestion, orthostasis, and fatigue while taking these medications. Volume expansion with intravenous fluids also should be performed and a high-salt diet considered. Beta-adrenergic blockade can be initiated once appropriate alpha-adrenergic blockade is achieved to control the patient’s heart rate; beta-blockers should never be started first because of the risk for severe hypertension. Careful hemodynamic monitoring is vital intraoperatively and postoperatively.5,13 Because metastatic lesions can occur decades after resection, long-term follow-up is critical.5,10
Following tumor resection, our patient’s blood pressure was supported with intravenous fluids and phenylephrine. She was able to discontinue all her antihypertensive medications postoperatively, and her plasma free and urinary fractionated metanephrine levels returned to within normal limits 8 weeks after surgery. Five years after surgery, she continues to have no signs of recurrence, as evidenced by annual negative plasma free metanephrines testing and abdominal/pelvic CT.
THE TAKEAWAY
This case highlights the importance of recognizing resistant hypertension and a potential secondary cause of this disease—pheochromocytoma. Although rare, pheochromocytomas confer increased risk for cardiovascular disease and death. Thus, swift recognition and proper preparation for surgical resection are necessary. Malignant lesions can be diagnosed only upon discovery of metastatic disease and can recur for decades after surgical resection, making diligent long-term follow-up imperative.
CORRESPONDENCE
Nicole O. Vietor, MD, Division of Endocrinology, Walter Reed National Military Medical Center, 8901 Wisconsin Avenue, Bethesda, MD 20889; nicole.o.vietor.mil@health.mil
1. Carey RM, Calhoun DA, Bakris GL, et al. Resistant hypertension: detection, evaluation, and management: a scientific statement from the American Heart Association. Hypertension. 2018;72:e53-e90. doi: 10.1161/HYP.0000000000000084
2. Young WF Jr. Diagnosis and treatment of primary aldosteronism: practical clinical perspectives. J Intern Med. 2019;285:126-148. doi: 10.1111/joim.12831
3. Young WF Jr, Calhoun DA, Lenders JWM, et al. Screening for endocrine hypertension: an Endocrine Society Scientific Statement. Endocr Rev. 2017;38:103-122. doi: 10.1210/er.2017-00054
4. Lenders JWM, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427-1434. doi: 10.1001/jama.287.11.1427
5. Lenders JW, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:1915-1942. doi: 10.1210/jc.2014-1498
6. Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21:405-414. doi: 10.1530/ERC-13-0494
7. Thompson LDR. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551-566. doi: 10.1097/00000478-200205000-00002
8. Vaidya A, Hamrahian A, Bancos I, et al. The evaluation of incidentally discovered adrenal masses. Endocr Pract. 2019;25:178-192. doi: 10.4158/DSCR-2018-0565
9. Young WF Jr. Conventional imaging in adrenocortical carcinoma: update and perspectives. Horm Cancer. 2011;2:341-347. doi: 10.1007/s12672-011-0089-z
10. Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019;381:552-565. doi: 10.1056/NEJMra1806651
11. Iñiguez-Ariza NM, Kohlenberg JD, Delivanis DA, et al. Clinical, biochemical, and radiological characteristics of a single-center retrospective cohort of 705 large adrenal tumors. Mayo Clin Proc Innov Qual Outcomes. 2017;2:30-39. doi: 10.1016/j.mayocpiqo.2017.11.002
12. Marty M, Gaye D, Perez P, et al. Diagnostic accuracy of computed tomography to identify adenomas among adrenal incidentalomas in an endocrinological population. Eur J Endocrinol. 2018;178:439-446. doi: 10.1530/EJE-17-1056
13. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069-4079. doi: 10.1210/jc.2007-1720
1. Carey RM, Calhoun DA, Bakris GL, et al. Resistant hypertension: detection, evaluation, and management: a scientific statement from the American Heart Association. Hypertension. 2018;72:e53-e90. doi: 10.1161/HYP.0000000000000084
2. Young WF Jr. Diagnosis and treatment of primary aldosteronism: practical clinical perspectives. J Intern Med. 2019;285:126-148. doi: 10.1111/joim.12831
3. Young WF Jr, Calhoun DA, Lenders JWM, et al. Screening for endocrine hypertension: an Endocrine Society Scientific Statement. Endocr Rev. 2017;38:103-122. doi: 10.1210/er.2017-00054
4. Lenders JWM, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427-1434. doi: 10.1001/jama.287.11.1427
5. Lenders JW, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:1915-1942. doi: 10.1210/jc.2014-1498
6. Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21:405-414. doi: 10.1530/ERC-13-0494
7. Thompson LDR. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551-566. doi: 10.1097/00000478-200205000-00002
8. Vaidya A, Hamrahian A, Bancos I, et al. The evaluation of incidentally discovered adrenal masses. Endocr Pract. 2019;25:178-192. doi: 10.4158/DSCR-2018-0565
9. Young WF Jr. Conventional imaging in adrenocortical carcinoma: update and perspectives. Horm Cancer. 2011;2:341-347. doi: 10.1007/s12672-011-0089-z
10. Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019;381:552-565. doi: 10.1056/NEJMra1806651
11. Iñiguez-Ariza NM, Kohlenberg JD, Delivanis DA, et al. Clinical, biochemical, and radiological characteristics of a single-center retrospective cohort of 705 large adrenal tumors. Mayo Clin Proc Innov Qual Outcomes. 2017;2:30-39. doi: 10.1016/j.mayocpiqo.2017.11.002
12. Marty M, Gaye D, Perez P, et al. Diagnostic accuracy of computed tomography to identify adenomas among adrenal incidentalomas in an endocrinological population. Eur J Endocrinol. 2018;178:439-446. doi: 10.1530/EJE-17-1056
13. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069-4079. doi: 10.1210/jc.2007-1720
► Hot flashes, facial flushing, excessive sweating, and palpitations
► Daily headaches
► History of hypertension
