User login
Early ankylosing spondylitis treatment stops transition from inflammatory to bone-forming fatty lesions
Structural damage in axial and peripheral ankylosing spondylitis is the result of a combination of destruction and new bone formation, which can be halted if treated early and for long enough, according to Dirk Elewaut, MD, PhD.
“There is a longstanding paradox in the field of ankylosing spondylitis, as to whether there is a strict relationship between the inflammation that leads to the signs and symptoms in patients, and the structural progression,” said Dr. Elewaut, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “ ”

One key reason why the early TNF inhibition trials showed no effect of treatment on structural progression stemmed from inadequate study design. “The original studies were underpowered, and the patients used NSAIDs,” he said. “We also know that the anti-inflammatory effect [of TNF inhibition] is not 100%. So you have a clear drop in inflammation with biologics, but it’s not completely stopped. This led to a lot of speculation as to whether inflammation and ankyloses are coupled or not.”
More recently, researchers have honed in on the transition from acute inflammatory lesions visible on MRI by bone marrow edema to so-called fatty lesions, which are also visible on MRI. They have observed that fatty lesions are associated with new bone formation. “Lesions containing more fat are thought to be more difficult to modulate by biological therapy,” Dr. Elewaut said. “Once you have a fatty lesion, you have some kind of metabolic disease of the bone, and it’s a one-way road to the development of new syndesmophytes. So in other words, it is essential to assess the relationship between inflammation and new bone formation with both the STIR [short tau inversion recovery images] and T1-weighted MRI.”
A working hypothesis among AS researchers is that the effect of anti-TNF therapy on radiologic progression depends on the relative number of acute and mature inflammatory lesions that individual patients have. “Early diagnosis is a prerequisite for advances in disease modification,” he said. At least three “windows of opportunity” for disease modification exist, Dr. Elewaut continued. One is that the link between inflammation as measured by clinical parameters and new bone formation will be more evident in early AS. The second is that early treatment of patients with axial AS and spinal inflammation with anti-TNF agents will prevent new bone formation. The third is that reduction of new bone formation in established AS will be observed only with long-term anti-TNF therapy after mature lesions have resolved/repaired, and no new inflammatory lesions are being formed. One study found that patients with a delay of more than 10 years starting anti-TNF therapy were more likely to progress, compared with patients who received therapy earlier (odds ratio, 2.4; P = .03; see Arthritis Rheum. 2013;65:645-54). “The message here is quite clear,” Dr. Elewaut said. “The earlier we treat intensively, the better impact you have on structural outcomes.”
In a trial known as CRESPA (Clinical Remission in peripheral SPondyloArthritis), researchers including Dr. Elewaut evaluated the efficacy and safety of golimumab (Simponi) to induce clinical remission in patients with early, active peripheral AS (Ann Rheum Dis. 2017;76:1389-95). In all, 60 patients were randomized to golimumab or placebo for 24 weeks. At week 24, a significantly higher percentage of patients receiving golimumab achieved clinical remission, compared with placebo (75% vs. 20%, respectively; P less than .001). At week 12, similar results were observed (70% vs. 15%; P less than .001).
“These were very striking results in this early disease population,” Dr. Elewaut said. In a follow-up analysis, the researchers withdrew therapy in patients who reached clinical remission, to see what would happen. “At 1.5 years of follow-up, 57% of patients are in drug-free remission,” Dr. Elewaut said. “This is interesting and suggests that at least in a fraction of those patients, you can actually achieve a drug-free remission.” Predictors for relapse included pre-existing psoriasis and having more than five swollen joints.
Dr. Elewaut disclosed that he is a member of the speakers bureau for AbbVie, Novartis, Pfizer, and UCB. Global Academy for Medical Education and this news organization are owned by the same parent company.
Structural damage in axial and peripheral ankylosing spondylitis is the result of a combination of destruction and new bone formation, which can be halted if treated early and for long enough, according to Dirk Elewaut, MD, PhD.
“There is a longstanding paradox in the field of ankylosing spondylitis, as to whether there is a strict relationship between the inflammation that leads to the signs and symptoms in patients, and the structural progression,” said Dr. Elewaut, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “ ”
One key reason why the early TNF inhibition trials showed no effect of treatment on structural progression stemmed from inadequate study design. “The original studies were underpowered, and the patients used NSAIDs,” he said. “We also know that the anti-inflammatory effect [of TNF inhibition] is not 100%. So you have a clear drop in inflammation with biologics, but it’s not completely stopped. This led to a lot of speculation as to whether inflammation and ankyloses are coupled or not.”
More recently, researchers have honed in on the transition from acute inflammatory lesions visible on MRI by bone marrow edema to so-called fatty lesions, which are also visible on MRI. They have observed that fatty lesions are associated with new bone formation. “Lesions containing more fat are thought to be more difficult to modulate by biological therapy,” Dr. Elewaut said. “Once you have a fatty lesion, you have some kind of metabolic disease of the bone, and it’s a one-way road to the development of new syndesmophytes. So in other words, it is essential to assess the relationship between inflammation and new bone formation with both the STIR [short tau inversion recovery images] and T1-weighted MRI.”
A working hypothesis among AS researchers is that the effect of anti-TNF therapy on radiologic progression depends on the relative number of acute and mature inflammatory lesions that individual patients have. “Early diagnosis is a prerequisite for advances in disease modification,” he said. At least three “windows of opportunity” for disease modification exist, Dr. Elewaut continued. One is that the link between inflammation as measured by clinical parameters and new bone formation will be more evident in early AS. The second is that early treatment of patients with axial AS and spinal inflammation with anti-TNF agents will prevent new bone formation. The third is that reduction of new bone formation in established AS will be observed only with long-term anti-TNF therapy after mature lesions have resolved/repaired, and no new inflammatory lesions are being formed. One study found that patients with a delay of more than 10 years starting anti-TNF therapy were more likely to progress, compared with patients who received therapy earlier (odds ratio, 2.4; P = .03; see Arthritis Rheum. 2013;65:645-54). “The message here is quite clear,” Dr. Elewaut said. “The earlier we treat intensively, the better impact you have on structural outcomes.”
In a trial known as CRESPA (Clinical Remission in peripheral SPondyloArthritis), researchers including Dr. Elewaut evaluated the efficacy and safety of golimumab (Simponi) to induce clinical remission in patients with early, active peripheral AS (Ann Rheum Dis. 2017;76:1389-95). In all, 60 patients were randomized to golimumab or placebo for 24 weeks. At week 24, a significantly higher percentage of patients receiving golimumab achieved clinical remission, compared with placebo (75% vs. 20%, respectively; P less than .001). At week 12, similar results were observed (70% vs. 15%; P less than .001).
“These were very striking results in this early disease population,” Dr. Elewaut said. In a follow-up analysis, the researchers withdrew therapy in patients who reached clinical remission, to see what would happen. “At 1.5 years of follow-up, 57% of patients are in drug-free remission,” Dr. Elewaut said. “This is interesting and suggests that at least in a fraction of those patients, you can actually achieve a drug-free remission.” Predictors for relapse included pre-existing psoriasis and having more than five swollen joints.
Dr. Elewaut disclosed that he is a member of the speakers bureau for AbbVie, Novartis, Pfizer, and UCB. Global Academy for Medical Education and this news organization are owned by the same parent company.
Structural damage in axial and peripheral ankylosing spondylitis is the result of a combination of destruction and new bone formation, which can be halted if treated early and for long enough, according to Dirk Elewaut, MD, PhD.
“There is a longstanding paradox in the field of ankylosing spondylitis, as to whether there is a strict relationship between the inflammation that leads to the signs and symptoms in patients, and the structural progression,” said Dr. Elewaut, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “ ”
One key reason why the early TNF inhibition trials showed no effect of treatment on structural progression stemmed from inadequate study design. “The original studies were underpowered, and the patients used NSAIDs,” he said. “We also know that the anti-inflammatory effect [of TNF inhibition] is not 100%. So you have a clear drop in inflammation with biologics, but it’s not completely stopped. This led to a lot of speculation as to whether inflammation and ankyloses are coupled or not.”
More recently, researchers have honed in on the transition from acute inflammatory lesions visible on MRI by bone marrow edema to so-called fatty lesions, which are also visible on MRI. They have observed that fatty lesions are associated with new bone formation. “Lesions containing more fat are thought to be more difficult to modulate by biological therapy,” Dr. Elewaut said. “Once you have a fatty lesion, you have some kind of metabolic disease of the bone, and it’s a one-way road to the development of new syndesmophytes. So in other words, it is essential to assess the relationship between inflammation and new bone formation with both the STIR [short tau inversion recovery images] and T1-weighted MRI.”
A working hypothesis among AS researchers is that the effect of anti-TNF therapy on radiologic progression depends on the relative number of acute and mature inflammatory lesions that individual patients have. “Early diagnosis is a prerequisite for advances in disease modification,” he said. At least three “windows of opportunity” for disease modification exist, Dr. Elewaut continued. One is that the link between inflammation as measured by clinical parameters and new bone formation will be more evident in early AS. The second is that early treatment of patients with axial AS and spinal inflammation with anti-TNF agents will prevent new bone formation. The third is that reduction of new bone formation in established AS will be observed only with long-term anti-TNF therapy after mature lesions have resolved/repaired, and no new inflammatory lesions are being formed. One study found that patients with a delay of more than 10 years starting anti-TNF therapy were more likely to progress, compared with patients who received therapy earlier (odds ratio, 2.4; P = .03; see Arthritis Rheum. 2013;65:645-54). “The message here is quite clear,” Dr. Elewaut said. “The earlier we treat intensively, the better impact you have on structural outcomes.”
In a trial known as CRESPA (Clinical Remission in peripheral SPondyloArthritis), researchers including Dr. Elewaut evaluated the efficacy and safety of golimumab (Simponi) to induce clinical remission in patients with early, active peripheral AS (Ann Rheum Dis. 2017;76:1389-95). In all, 60 patients were randomized to golimumab or placebo for 24 weeks. At week 24, a significantly higher percentage of patients receiving golimumab achieved clinical remission, compared with placebo (75% vs. 20%, respectively; P less than .001). At week 12, similar results were observed (70% vs. 15%; P less than .001).
“These were very striking results in this early disease population,” Dr. Elewaut said. In a follow-up analysis, the researchers withdrew therapy in patients who reached clinical remission, to see what would happen. “At 1.5 years of follow-up, 57% of patients are in drug-free remission,” Dr. Elewaut said. “This is interesting and suggests that at least in a fraction of those patients, you can actually achieve a drug-free remission.” Predictors for relapse included pre-existing psoriasis and having more than five swollen joints.
Dr. Elewaut disclosed that he is a member of the speakers bureau for AbbVie, Novartis, Pfizer, and UCB. Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM ANNUAL PERSPECTIVES IN RHEUMATIC DISEASES
Ultrasound’s value for arthralgia may be to rule out IA
Ultrasound evaluations to look for subclinical inflammation in joints of patients with arthralgia appear best at ruling out inflammatory arthritis (IA) 1 year in the future rather than ruling it in, according to findings from a multicenter cohort study published online in Arthritis Research and Therapy.
The imaging modality’s ability to identify those who will not go on to develop IA complemented the serologic and clinical factors that help to discriminate the individuals with arthralgia who are most at risk of the condition.
The ultimate goal of using imaging such as ultrasound in patients with arthralgia is to identify those who would benefit from starting treatment with disease-modifying antirheumatic drugs as early as possible to potentially improve outcomes, but it also could help to discriminate between the anti-citrullinated protein antibody (ACPA)-positive and seronegative individuals without clinical signs of inflammation at baseline who may progress from arthralgia to IA.
“Although ACPA positivity is a very good predictor for those patients who will develop IA within 1 year, it is still difficult to identify the exact individuals who will develop IA, because any ACPA-positive individual has an a priori chance of 50% of developing IA. In seronegative patients, the prediction of IA is even more difficult, because only 5% develop IA within the subsequent year. Imaging techniques have been shown to be able to detect synovitis before its clinical appearance and could be of help in identifying those at risk of IA,” the investigators wrote (Arthritis Res Ther. 2017;19:202. doi: 10.1186/s13075-017-1405-y).
Dr. van der Ven and her associates found that 31 (16%) of 196 patients who had arthralgia for less than 1 year in the hands, feet, or shoulders went on to develop IA after 1 year of follow-up. In this group of 196 patients at baseline, 72 (37%) had synovitis on ultrasound – defined as a greyscale grade of 2 or 3 and/or the presence of power Doppler signal (grade 1, 2, or 3) – including 32 with a positive power Doppler. A total of 18 patients were lost to follow-up during the first 6 months and another 19 were lost during months 6-12.
Rheumatologists who were unaware of ultrasound findings had to confirm soft-tissue swelling as arthritis at 1 year to classify it as incident IA. The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Overall, at 1 year, 15 of the 31 patients with IA had started therapy with a disease-modifying antirheumatic drug and 22 did not have a definite diagnosis; 12 had monoarthritis and 10 had polyarthritis. The remaining nine patients included four with rheumatoid arthritis, four with psoriatic arthritis, and one with spondyloarthritis.
At baseline, individuals with IA were more often older (mean age 50 vs. 44 years; P = .005), had synovitis on ultrasound (59% vs. 32%; P = .007), and had a positive power Doppler signal (31% vs. 12%; P = .012). A multivariate analysis revealed that IA at 1 year of follow-up could be independently predicted according to age (odds ratio, 1.06), morning stiffness lasting more than 30 minutes (OR, 2.80), ACPA positivity (OR, 2.35), and synovitis on ultrasound (OR, 2.65).
The investigators noted that the study’s limitations relate to requirements for patients to have at least two painful joints in hands, feet, or shoulders at baseline and two criteria related to inflammation. The possible inflammation-related criteria required for entry included morning stiffness for more than 1 hour, inability to clench a fist in the morning, pain when shaking someone’s hand, pins and needles in the fingers, difficulties wearing rings or shoes, family history of rheumatoid arthritis, and/or unexplained fatigue for less than 1 year.
Rheumatologists who enrolled patients into the cohort also may have “recruited clinically suspected patients with possibly more severe symptoms,” the investigators noted. Another potential source of bias related to the group of 38 patients who chose not to participate: It’s possible these patients had less severe symptoms than those who participated in the study.
The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
Ultrasound evaluations to look for subclinical inflammation in joints of patients with arthralgia appear best at ruling out inflammatory arthritis (IA) 1 year in the future rather than ruling it in, according to findings from a multicenter cohort study published online in Arthritis Research and Therapy.
The imaging modality’s ability to identify those who will not go on to develop IA complemented the serologic and clinical factors that help to discriminate the individuals with arthralgia who are most at risk of the condition.
The ultimate goal of using imaging such as ultrasound in patients with arthralgia is to identify those who would benefit from starting treatment with disease-modifying antirheumatic drugs as early as possible to potentially improve outcomes, but it also could help to discriminate between the anti-citrullinated protein antibody (ACPA)-positive and seronegative individuals without clinical signs of inflammation at baseline who may progress from arthralgia to IA.
“Although ACPA positivity is a very good predictor for those patients who will develop IA within 1 year, it is still difficult to identify the exact individuals who will develop IA, because any ACPA-positive individual has an a priori chance of 50% of developing IA. In seronegative patients, the prediction of IA is even more difficult, because only 5% develop IA within the subsequent year. Imaging techniques have been shown to be able to detect synovitis before its clinical appearance and could be of help in identifying those at risk of IA,” the investigators wrote (Arthritis Res Ther. 2017;19:202. doi: 10.1186/s13075-017-1405-y).
Dr. van der Ven and her associates found that 31 (16%) of 196 patients who had arthralgia for less than 1 year in the hands, feet, or shoulders went on to develop IA after 1 year of follow-up. In this group of 196 patients at baseline, 72 (37%) had synovitis on ultrasound – defined as a greyscale grade of 2 or 3 and/or the presence of power Doppler signal (grade 1, 2, or 3) – including 32 with a positive power Doppler. A total of 18 patients were lost to follow-up during the first 6 months and another 19 were lost during months 6-12.
Rheumatologists who were unaware of ultrasound findings had to confirm soft-tissue swelling as arthritis at 1 year to classify it as incident IA. The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Overall, at 1 year, 15 of the 31 patients with IA had started therapy with a disease-modifying antirheumatic drug and 22 did not have a definite diagnosis; 12 had monoarthritis and 10 had polyarthritis. The remaining nine patients included four with rheumatoid arthritis, four with psoriatic arthritis, and one with spondyloarthritis.
At baseline, individuals with IA were more often older (mean age 50 vs. 44 years; P = .005), had synovitis on ultrasound (59% vs. 32%; P = .007), and had a positive power Doppler signal (31% vs. 12%; P = .012). A multivariate analysis revealed that IA at 1 year of follow-up could be independently predicted according to age (odds ratio, 1.06), morning stiffness lasting more than 30 minutes (OR, 2.80), ACPA positivity (OR, 2.35), and synovitis on ultrasound (OR, 2.65).
The investigators noted that the study’s limitations relate to requirements for patients to have at least two painful joints in hands, feet, or shoulders at baseline and two criteria related to inflammation. The possible inflammation-related criteria required for entry included morning stiffness for more than 1 hour, inability to clench a fist in the morning, pain when shaking someone’s hand, pins and needles in the fingers, difficulties wearing rings or shoes, family history of rheumatoid arthritis, and/or unexplained fatigue for less than 1 year.
Rheumatologists who enrolled patients into the cohort also may have “recruited clinically suspected patients with possibly more severe symptoms,” the investigators noted. Another potential source of bias related to the group of 38 patients who chose not to participate: It’s possible these patients had less severe symptoms than those who participated in the study.
The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
Ultrasound evaluations to look for subclinical inflammation in joints of patients with arthralgia appear best at ruling out inflammatory arthritis (IA) 1 year in the future rather than ruling it in, according to findings from a multicenter cohort study published online in Arthritis Research and Therapy.
The imaging modality’s ability to identify those who will not go on to develop IA complemented the serologic and clinical factors that help to discriminate the individuals with arthralgia who are most at risk of the condition.
The ultimate goal of using imaging such as ultrasound in patients with arthralgia is to identify those who would benefit from starting treatment with disease-modifying antirheumatic drugs as early as possible to potentially improve outcomes, but it also could help to discriminate between the anti-citrullinated protein antibody (ACPA)-positive and seronegative individuals without clinical signs of inflammation at baseline who may progress from arthralgia to IA.
“Although ACPA positivity is a very good predictor for those patients who will develop IA within 1 year, it is still difficult to identify the exact individuals who will develop IA, because any ACPA-positive individual has an a priori chance of 50% of developing IA. In seronegative patients, the prediction of IA is even more difficult, because only 5% develop IA within the subsequent year. Imaging techniques have been shown to be able to detect synovitis before its clinical appearance and could be of help in identifying those at risk of IA,” the investigators wrote (Arthritis Res Ther. 2017;19:202. doi: 10.1186/s13075-017-1405-y).
Dr. van der Ven and her associates found that 31 (16%) of 196 patients who had arthralgia for less than 1 year in the hands, feet, or shoulders went on to develop IA after 1 year of follow-up. In this group of 196 patients at baseline, 72 (37%) had synovitis on ultrasound – defined as a greyscale grade of 2 or 3 and/or the presence of power Doppler signal (grade 1, 2, or 3) – including 32 with a positive power Doppler. A total of 18 patients were lost to follow-up during the first 6 months and another 19 were lost during months 6-12.
Rheumatologists who were unaware of ultrasound findings had to confirm soft-tissue swelling as arthritis at 1 year to classify it as incident IA. The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Overall, at 1 year, 15 of the 31 patients with IA had started therapy with a disease-modifying antirheumatic drug and 22 did not have a definite diagnosis; 12 had monoarthritis and 10 had polyarthritis. The remaining nine patients included four with rheumatoid arthritis, four with psoriatic arthritis, and one with spondyloarthritis.
At baseline, individuals with IA were more often older (mean age 50 vs. 44 years; P = .005), had synovitis on ultrasound (59% vs. 32%; P = .007), and had a positive power Doppler signal (31% vs. 12%; P = .012). A multivariate analysis revealed that IA at 1 year of follow-up could be independently predicted according to age (odds ratio, 1.06), morning stiffness lasting more than 30 minutes (OR, 2.80), ACPA positivity (OR, 2.35), and synovitis on ultrasound (OR, 2.65).
The investigators noted that the study’s limitations relate to requirements for patients to have at least two painful joints in hands, feet, or shoulders at baseline and two criteria related to inflammation. The possible inflammation-related criteria required for entry included morning stiffness for more than 1 hour, inability to clench a fist in the morning, pain when shaking someone’s hand, pins and needles in the fingers, difficulties wearing rings or shoes, family history of rheumatoid arthritis, and/or unexplained fatigue for less than 1 year.
Rheumatologists who enrolled patients into the cohort also may have “recruited clinically suspected patients with possibly more severe symptoms,” the investigators noted. Another potential source of bias related to the group of 38 patients who chose not to participate: It’s possible these patients had less severe symptoms than those who participated in the study.
The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
FROM ARTHRITIS RESEARCH AND THERAPY
Key clinical point:
Major finding: The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Data source: A multicenter cohort study of 196 patients with arthralgia in at least two joints for less than 1 year.
Disclosures: The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
Adjuvant-boosted shingles vaccine earns FDA panel’s unanimous nod
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
New recommendations tout biosimilars for rheumatologic diseases
The available evidence is sufficient to support switching appropriate patients with rheumatologic diseases from a bio-originator agent to an approved biosimilar agent, according to new consensus-based recommendations from an international multidisciplinary task force.
“Treatment with biological agents has dramatically improved the outcome for patients with inflammatory diseases. However, the high cost of these medications has limited access for many patients,” Jonathan Kay, MD, of UMass Memorial Medical Center and the University of Massachusetts, Worcester, and his colleagues wrote on behalf of the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases. Biosimilars of agents no longer protected by patent allow for increased availability at lower costs, they noted. In the European Union, the United States, Japan, and other countries, biosimilars of adalimumab, etanercept, infliximab, and rituximab have been approved, and those for which the bio-originator is no longer protected by patent have been marketed.

The task force, convened in 2016 to address the matter at an international level, included 25 experts from Europe, Japan, and the United States, including 17 rheumatologists, a rheumatologist/regulator, a dermatologist, a gastroenterologist, 2 pharmacologists, 2 patients, and a research fellow. The task force identified five overarching principles and made eight specific recommendations, based on expert opinion and an extensive literature review that yielded 29 relevant full-text papers and 20 relevant abstracts from the 2015 and 2016 American College of Rheumatology and European League Against Rheumatism annual meetings (Ann Rheum Dis. 2017 Sep 2. doi: 10.1136/annrheumdis-2017-211937).
“This statement was intended both to guide clinicians and to serve as a framework for future educational efforts,” they wrote.
The experts based all five overarching principles for the use of biosimilars on level 5, grade D evidence, indicating that they were derived mainly from expert opinion. They determined that:
- Treatment of rheumatic diseases is based on a shared decision-making process between patients and their rheumatologists.
- The contextual aspects of the health care system should be taken into consideration when treatment decisions are made.
- A biosimilar, as approved by authorities in a highly regulated area, is neither better nor worse in efficacy and is not inferior in safety to its bio-originator.
- Patients and health care providers should be informed about the nature of biosimilars, their approval process, and their safety and efficacy.
- Harmonized methods should be established to obtain reliable pharmacovigilance data, including traceability, about both biosimilars and bio-originators.
These principles represent the key issues regarding biosimilars as identified by the task force. As for the specific recommendations, the task force agreed that:
1. The availability of biosimilars must significantly lower the cost of treating an individual patient and increase access to optimal therapy for all patients with rheumatic diseases (level 5, grade D evidence).
2. Approved biosimilars can be used to treat appropriate patients in the same way as their bio-originators (level 1b, grade A evidence, indicating that the recommendation is based on an individual randomized, controlled trial and that the level 1 evidence is consistent).
3. Antidrug antibodies to biosimilars need not be measured in clinical practice as no significant differences have been detected between biosimilars and their bio-originators (level 2b, grade B evidence, indicating that the recommendation is based on an individual cohort study/low-quality randomized, controlled trial and consistent level 2 or 3 evidence).
4. Relevant preclinical and phase 1 data on a biosimilar should be available when phase 3 data are published (level 5, grade D evidence).
5. Confirmation of efficacy and safety in a single indication is sufficient for extrapolation to other diseases for which the bio-originator has been approved because biosimilars are equivalent in physiochemical, functional, and pharmacokinetic properties to the bio-originator (level 5, grade D evidence).
6. Available evidence suggests that a single switch from a bio-originator to one of its biosimilars is safe and effective; there is no reason to expect a different clinical outcome. However, patient perspectives must be considered (level 1b, grade A evidence).
7. Multiple switching between biosimilars and their bio-originators or other biosimilars should be assessed in registries (level 5, grade D evidence).
8. No switch to or among biosimilars should be initiated without the prior awareness of the patient and the treating health care provider (level 5, grade D evidence).
Differing opinions about the use of biosimilars as published by various subspecialty organizations highlight a lack of confidence among many clinicians with respect to appropriate use of the products, but that is changing amid a rapidly growing body of evidence, the task force said. The group achieved a high level of agreement about both the evaluation of biosimilars and their use to treat rheumatologic diseases, reaching 100% consensus for six of the recommendations and 91% and 96% for the other two.
“Data available as of December 2016 support the use of biosimilars by rheumatologists to encourage a fair and competitive market for biologics. Biosimilars now provide an opportunity to expand access to effective but expensive medications, increasing the number of available treatment choices and helping to control rapidly increasing drug expenditures,” they concluded.
The task force’s work was funded by an unrestricted educational grant from Amgen. Dr. Kay and his coauthors reported financial relationships with multiple pharmaceutical companies, many of which are developing biosimilars.
The available evidence is sufficient to support switching appropriate patients with rheumatologic diseases from a bio-originator agent to an approved biosimilar agent, according to new consensus-based recommendations from an international multidisciplinary task force.
“Treatment with biological agents has dramatically improved the outcome for patients with inflammatory diseases. However, the high cost of these medications has limited access for many patients,” Jonathan Kay, MD, of UMass Memorial Medical Center and the University of Massachusetts, Worcester, and his colleagues wrote on behalf of the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases. Biosimilars of agents no longer protected by patent allow for increased availability at lower costs, they noted. In the European Union, the United States, Japan, and other countries, biosimilars of adalimumab, etanercept, infliximab, and rituximab have been approved, and those for which the bio-originator is no longer protected by patent have been marketed.
The task force, convened in 2016 to address the matter at an international level, included 25 experts from Europe, Japan, and the United States, including 17 rheumatologists, a rheumatologist/regulator, a dermatologist, a gastroenterologist, 2 pharmacologists, 2 patients, and a research fellow. The task force identified five overarching principles and made eight specific recommendations, based on expert opinion and an extensive literature review that yielded 29 relevant full-text papers and 20 relevant abstracts from the 2015 and 2016 American College of Rheumatology and European League Against Rheumatism annual meetings (Ann Rheum Dis. 2017 Sep 2. doi: 10.1136/annrheumdis-2017-211937).
“This statement was intended both to guide clinicians and to serve as a framework for future educational efforts,” they wrote.
The experts based all five overarching principles for the use of biosimilars on level 5, grade D evidence, indicating that they were derived mainly from expert opinion. They determined that:
- Treatment of rheumatic diseases is based on a shared decision-making process between patients and their rheumatologists.
- The contextual aspects of the health care system should be taken into consideration when treatment decisions are made.
- A biosimilar, as approved by authorities in a highly regulated area, is neither better nor worse in efficacy and is not inferior in safety to its bio-originator.
- Patients and health care providers should be informed about the nature of biosimilars, their approval process, and their safety and efficacy.
- Harmonized methods should be established to obtain reliable pharmacovigilance data, including traceability, about both biosimilars and bio-originators.
These principles represent the key issues regarding biosimilars as identified by the task force. As for the specific recommendations, the task force agreed that:
1. The availability of biosimilars must significantly lower the cost of treating an individual patient and increase access to optimal therapy for all patients with rheumatic diseases (level 5, grade D evidence).
2. Approved biosimilars can be used to treat appropriate patients in the same way as their bio-originators (level 1b, grade A evidence, indicating that the recommendation is based on an individual randomized, controlled trial and that the level 1 evidence is consistent).
3. Antidrug antibodies to biosimilars need not be measured in clinical practice as no significant differences have been detected between biosimilars and their bio-originators (level 2b, grade B evidence, indicating that the recommendation is based on an individual cohort study/low-quality randomized, controlled trial and consistent level 2 or 3 evidence).
4. Relevant preclinical and phase 1 data on a biosimilar should be available when phase 3 data are published (level 5, grade D evidence).
5. Confirmation of efficacy and safety in a single indication is sufficient for extrapolation to other diseases for which the bio-originator has been approved because biosimilars are equivalent in physiochemical, functional, and pharmacokinetic properties to the bio-originator (level 5, grade D evidence).
6. Available evidence suggests that a single switch from a bio-originator to one of its biosimilars is safe and effective; there is no reason to expect a different clinical outcome. However, patient perspectives must be considered (level 1b, grade A evidence).
7. Multiple switching between biosimilars and their bio-originators or other biosimilars should be assessed in registries (level 5, grade D evidence).
8. No switch to or among biosimilars should be initiated without the prior awareness of the patient and the treating health care provider (level 5, grade D evidence).
Differing opinions about the use of biosimilars as published by various subspecialty organizations highlight a lack of confidence among many clinicians with respect to appropriate use of the products, but that is changing amid a rapidly growing body of evidence, the task force said. The group achieved a high level of agreement about both the evaluation of biosimilars and their use to treat rheumatologic diseases, reaching 100% consensus for six of the recommendations and 91% and 96% for the other two.
“Data available as of December 2016 support the use of biosimilars by rheumatologists to encourage a fair and competitive market for biologics. Biosimilars now provide an opportunity to expand access to effective but expensive medications, increasing the number of available treatment choices and helping to control rapidly increasing drug expenditures,” they concluded.
The task force’s work was funded by an unrestricted educational grant from Amgen. Dr. Kay and his coauthors reported financial relationships with multiple pharmaceutical companies, many of which are developing biosimilars.
The available evidence is sufficient to support switching appropriate patients with rheumatologic diseases from a bio-originator agent to an approved biosimilar agent, according to new consensus-based recommendations from an international multidisciplinary task force.
“Treatment with biological agents has dramatically improved the outcome for patients with inflammatory diseases. However, the high cost of these medications has limited access for many patients,” Jonathan Kay, MD, of UMass Memorial Medical Center and the University of Massachusetts, Worcester, and his colleagues wrote on behalf of the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases. Biosimilars of agents no longer protected by patent allow for increased availability at lower costs, they noted. In the European Union, the United States, Japan, and other countries, biosimilars of adalimumab, etanercept, infliximab, and rituximab have been approved, and those for which the bio-originator is no longer protected by patent have been marketed.
The task force, convened in 2016 to address the matter at an international level, included 25 experts from Europe, Japan, and the United States, including 17 rheumatologists, a rheumatologist/regulator, a dermatologist, a gastroenterologist, 2 pharmacologists, 2 patients, and a research fellow. The task force identified five overarching principles and made eight specific recommendations, based on expert opinion and an extensive literature review that yielded 29 relevant full-text papers and 20 relevant abstracts from the 2015 and 2016 American College of Rheumatology and European League Against Rheumatism annual meetings (Ann Rheum Dis. 2017 Sep 2. doi: 10.1136/annrheumdis-2017-211937).
“This statement was intended both to guide clinicians and to serve as a framework for future educational efforts,” they wrote.
The experts based all five overarching principles for the use of biosimilars on level 5, grade D evidence, indicating that they were derived mainly from expert opinion. They determined that:
- Treatment of rheumatic diseases is based on a shared decision-making process between patients and their rheumatologists.
- The contextual aspects of the health care system should be taken into consideration when treatment decisions are made.
- A biosimilar, as approved by authorities in a highly regulated area, is neither better nor worse in efficacy and is not inferior in safety to its bio-originator.
- Patients and health care providers should be informed about the nature of biosimilars, their approval process, and their safety and efficacy.
- Harmonized methods should be established to obtain reliable pharmacovigilance data, including traceability, about both biosimilars and bio-originators.
These principles represent the key issues regarding biosimilars as identified by the task force. As for the specific recommendations, the task force agreed that:
1. The availability of biosimilars must significantly lower the cost of treating an individual patient and increase access to optimal therapy for all patients with rheumatic diseases (level 5, grade D evidence).
2. Approved biosimilars can be used to treat appropriate patients in the same way as their bio-originators (level 1b, grade A evidence, indicating that the recommendation is based on an individual randomized, controlled trial and that the level 1 evidence is consistent).
3. Antidrug antibodies to biosimilars need not be measured in clinical practice as no significant differences have been detected between biosimilars and their bio-originators (level 2b, grade B evidence, indicating that the recommendation is based on an individual cohort study/low-quality randomized, controlled trial and consistent level 2 or 3 evidence).
4. Relevant preclinical and phase 1 data on a biosimilar should be available when phase 3 data are published (level 5, grade D evidence).
5. Confirmation of efficacy and safety in a single indication is sufficient for extrapolation to other diseases for which the bio-originator has been approved because biosimilars are equivalent in physiochemical, functional, and pharmacokinetic properties to the bio-originator (level 5, grade D evidence).
6. Available evidence suggests that a single switch from a bio-originator to one of its biosimilars is safe and effective; there is no reason to expect a different clinical outcome. However, patient perspectives must be considered (level 1b, grade A evidence).
7. Multiple switching between biosimilars and their bio-originators or other biosimilars should be assessed in registries (level 5, grade D evidence).
8. No switch to or among biosimilars should be initiated without the prior awareness of the patient and the treating health care provider (level 5, grade D evidence).
Differing opinions about the use of biosimilars as published by various subspecialty organizations highlight a lack of confidence among many clinicians with respect to appropriate use of the products, but that is changing amid a rapidly growing body of evidence, the task force said. The group achieved a high level of agreement about both the evaluation of biosimilars and their use to treat rheumatologic diseases, reaching 100% consensus for six of the recommendations and 91% and 96% for the other two.
“Data available as of December 2016 support the use of biosimilars by rheumatologists to encourage a fair and competitive market for biologics. Biosimilars now provide an opportunity to expand access to effective but expensive medications, increasing the number of available treatment choices and helping to control rapidly increasing drug expenditures,” they concluded.
The task force’s work was funded by an unrestricted educational grant from Amgen. Dr. Kay and his coauthors reported financial relationships with multiple pharmaceutical companies, many of which are developing biosimilars.
FROM ANNALS OF THE RHEUMATIC DISEASES
FDA approves second adalimumab biosimilar for multiple conditions
The Food and Drug Administration has approved Cyltezo (adalimumab-adbm) for multiple conditions.
Cyltezo is an injectable tumor necrosis factor blocker, and is a biosimilar to adalimumab (Humira). The drug is indicated to treat moderate to severe active rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate to severe active Crohn’s disease, moderate to severe active ulcerative colitis, moderately to severely active polyarticular juvenile idiopathic arthritis in patients 4 years of age and older, and moderate to severe plaque psoriasis.![]()
Find the Cyltezo labeling information here.
The Food and Drug Administration has approved Cyltezo (adalimumab-adbm) for multiple conditions.
Cyltezo is an injectable tumor necrosis factor blocker, and is a biosimilar to adalimumab (Humira). The drug is indicated to treat moderate to severe active rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate to severe active Crohn’s disease, moderate to severe active ulcerative colitis, moderately to severely active polyarticular juvenile idiopathic arthritis in patients 4 years of age and older, and moderate to severe plaque psoriasis.![]()
Find the Cyltezo labeling information here.
The Food and Drug Administration has approved Cyltezo (adalimumab-adbm) for multiple conditions.
Cyltezo is an injectable tumor necrosis factor blocker, and is a biosimilar to adalimumab (Humira). The drug is indicated to treat moderate to severe active rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate to severe active Crohn’s disease, moderate to severe active ulcerative colitis, moderately to severely active polyarticular juvenile idiopathic arthritis in patients 4 years of age and older, and moderate to severe plaque psoriasis.![]()
Find the Cyltezo labeling information here.
Axial SpA features don’t guarantee its diagnosis in chronic back pain
The manifestation of multiple features of spondyloarthritis (SpA) in patients with chronic back pain is not sufficient for a diagnosis of axial spondyloarthritis, according to a report from Zineb Ez-Zaitouni and associates.
In a group of 250 people with chronic back pain who were not diagnosed with axial SpA, the most common alternative diagnosis was nonspecific back pain, followed by mechanical back pain, degenerative disc disease, and myalgia/fibromyalgia. Sacroiliitis on either radiographs or MRI and HLA-B27 was uncommon, and HLA-B27 positivity was also infrequent.
A total of 18 patients within the study group had at least four features of SpA but did not have axial SpA. Within this group, the most common SpA features were inflammatory back pain, a positive family history of SpA, a good response to nonsteroidal anti-inflammatory drugs, elevated C-reactive protein or erythrocyte sedimentation rate, and enthesitis. No patients had positive imaging, and only four were positive for HLA-B27.
“These findings show that rheumatologists in clinical practice rightly dispute a diagnosis of axSpA even when there is a high number of SpA features, especially when imaging is normal and patients are negative for HLA-B27,” the investigators concluded.
Find the full report in Annals of the Rheumatic Diseases (doi: 10.1136/annrheumdis-2017-212175)
The manifestation of multiple features of spondyloarthritis (SpA) in patients with chronic back pain is not sufficient for a diagnosis of axial spondyloarthritis, according to a report from Zineb Ez-Zaitouni and associates.
In a group of 250 people with chronic back pain who were not diagnosed with axial SpA, the most common alternative diagnosis was nonspecific back pain, followed by mechanical back pain, degenerative disc disease, and myalgia/fibromyalgia. Sacroiliitis on either radiographs or MRI and HLA-B27 was uncommon, and HLA-B27 positivity was also infrequent.
A total of 18 patients within the study group had at least four features of SpA but did not have axial SpA. Within this group, the most common SpA features were inflammatory back pain, a positive family history of SpA, a good response to nonsteroidal anti-inflammatory drugs, elevated C-reactive protein or erythrocyte sedimentation rate, and enthesitis. No patients had positive imaging, and only four were positive for HLA-B27.
“These findings show that rheumatologists in clinical practice rightly dispute a diagnosis of axSpA even when there is a high number of SpA features, especially when imaging is normal and patients are negative for HLA-B27,” the investigators concluded.
Find the full report in Annals of the Rheumatic Diseases (doi: 10.1136/annrheumdis-2017-212175)
The manifestation of multiple features of spondyloarthritis (SpA) in patients with chronic back pain is not sufficient for a diagnosis of axial spondyloarthritis, according to a report from Zineb Ez-Zaitouni and associates.
In a group of 250 people with chronic back pain who were not diagnosed with axial SpA, the most common alternative diagnosis was nonspecific back pain, followed by mechanical back pain, degenerative disc disease, and myalgia/fibromyalgia. Sacroiliitis on either radiographs or MRI and HLA-B27 was uncommon, and HLA-B27 positivity was also infrequent.
A total of 18 patients within the study group had at least four features of SpA but did not have axial SpA. Within this group, the most common SpA features were inflammatory back pain, a positive family history of SpA, a good response to nonsteroidal anti-inflammatory drugs, elevated C-reactive protein or erythrocyte sedimentation rate, and enthesitis. No patients had positive imaging, and only four were positive for HLA-B27.
“These findings show that rheumatologists in clinical practice rightly dispute a diagnosis of axSpA even when there is a high number of SpA features, especially when imaging is normal and patients are negative for HLA-B27,” the investigators concluded.
Find the full report in Annals of the Rheumatic Diseases (doi: 10.1136/annrheumdis-2017-212175)
FROM ANNALS OF THE RHEUMATIC DISEASES
Radiographic progression in axial spondyloarthritis moves slowly in first 5 years
Sacroiliac joint radiographic progression during the first 5 years of the onset of axial spondyloarthritis occurs to an extent related to the degree of inflammation seen on MRI at baseline, according to new findings from 416 French patients in the DESIR cohort.
Maxime Dougados, MD, of Paris Descartes University, and his colleagues found that 15% of patients at baseline met modified New York (mNY) criteria – and therefore had radiographic axial spondyloarthritis (r-axSpA) – and this increased to 20% at 5 years. During the 5-year follow-up, the net percentage of patients who progressed was 5% (those who went from nonradiographic axial spondyloarthritis [nr-axSpA] to r-axSpA minus those who regressed from r-axSpA to nr-axSpA). Overall, 13% changed at least one grade on mNY criteria, and if an mNY criteria grade change from zero to one was not considered, only 10% experienced a change in at least one grade. These patients overall had a mean age of 34 years and had inflammatory back pain that had lasted at least 3 months but less than 3 years.

“The association between baseline MRI inflammation and 5-year SIJ damage was consistently found, regardless of the analytical method and the definition of SIJ progression,” the investigators wrote.
The estimated risk for progression by at least one mNY criteria grade varied from as high as 18% in HLA-B27–positive individuals with baseline SIJ inflammation on MRI and elevated C-reactive protein to just 1% in those who were negative for those three variables.
Read the full report online (Ann Rheum Dis. 2017 Jul 6. doi: 10.1136/annrheumdis-2017-211596).
Sacroiliac joint radiographic progression during the first 5 years of the onset of axial spondyloarthritis occurs to an extent related to the degree of inflammation seen on MRI at baseline, according to new findings from 416 French patients in the DESIR cohort.
Maxime Dougados, MD, of Paris Descartes University, and his colleagues found that 15% of patients at baseline met modified New York (mNY) criteria – and therefore had radiographic axial spondyloarthritis (r-axSpA) – and this increased to 20% at 5 years. During the 5-year follow-up, the net percentage of patients who progressed was 5% (those who went from nonradiographic axial spondyloarthritis [nr-axSpA] to r-axSpA minus those who regressed from r-axSpA to nr-axSpA). Overall, 13% changed at least one grade on mNY criteria, and if an mNY criteria grade change from zero to one was not considered, only 10% experienced a change in at least one grade. These patients overall had a mean age of 34 years and had inflammatory back pain that had lasted at least 3 months but less than 3 years.
“The association between baseline MRI inflammation and 5-year SIJ damage was consistently found, regardless of the analytical method and the definition of SIJ progression,” the investigators wrote.
The estimated risk for progression by at least one mNY criteria grade varied from as high as 18% in HLA-B27–positive individuals with baseline SIJ inflammation on MRI and elevated C-reactive protein to just 1% in those who were negative for those three variables.
Read the full report online (Ann Rheum Dis. 2017 Jul 6. doi: 10.1136/annrheumdis-2017-211596).
Sacroiliac joint radiographic progression during the first 5 years of the onset of axial spondyloarthritis occurs to an extent related to the degree of inflammation seen on MRI at baseline, according to new findings from 416 French patients in the DESIR cohort.
Maxime Dougados, MD, of Paris Descartes University, and his colleagues found that 15% of patients at baseline met modified New York (mNY) criteria – and therefore had radiographic axial spondyloarthritis (r-axSpA) – and this increased to 20% at 5 years. During the 5-year follow-up, the net percentage of patients who progressed was 5% (those who went from nonradiographic axial spondyloarthritis [nr-axSpA] to r-axSpA minus those who regressed from r-axSpA to nr-axSpA). Overall, 13% changed at least one grade on mNY criteria, and if an mNY criteria grade change from zero to one was not considered, only 10% experienced a change in at least one grade. These patients overall had a mean age of 34 years and had inflammatory back pain that had lasted at least 3 months but less than 3 years.
“The association between baseline MRI inflammation and 5-year SIJ damage was consistently found, regardless of the analytical method and the definition of SIJ progression,” the investigators wrote.
The estimated risk for progression by at least one mNY criteria grade varied from as high as 18% in HLA-B27–positive individuals with baseline SIJ inflammation on MRI and elevated C-reactive protein to just 1% in those who were negative for those three variables.
Read the full report online (Ann Rheum Dis. 2017 Jul 6. doi: 10.1136/annrheumdis-2017-211596).
FROM ANNALS OF THE RHEUMATIC DISEASES
First interchangeability study for an adalimumab biosimilar has begun
The VOLTAIRE-X study of a biosimilar candidate for adalimumab (Humira) for chronic plaque psoriasis has enrolled its first patient, announced Boehringer Ingelheim, the biosimilar’s developer, on July 27.
This is the first study in the United States to investigate whether a biosimilar candidate should be granted an interchangeability designation with adalimumab. The candidate, BI 695501, is up against adalimumab’s 40-mg injection.
In VOLTAIRE-X, some patients will alternate between adalimumab and BI 695501, and others will take adalimumab continuously. The study will compare the pharmacokinetics, clinical outcomes, safety, immunogenicity, and efficacy between the two groups of patients. The estimated enrollment of adult patients with moderate to severe chronic plaque psoriasis is 240, and the study is expected to conclude in July 2019.
A phase 3 study of BI 695501’s performance for rheumatoid arthritis patients, completed in 2016, demonstrated similar efficacy, safety, and immunogenicity.
The VOLTAIRE-X study of a biosimilar candidate for adalimumab (Humira) for chronic plaque psoriasis has enrolled its first patient, announced Boehringer Ingelheim, the biosimilar’s developer, on July 27.
This is the first study in the United States to investigate whether a biosimilar candidate should be granted an interchangeability designation with adalimumab. The candidate, BI 695501, is up against adalimumab’s 40-mg injection.
In VOLTAIRE-X, some patients will alternate between adalimumab and BI 695501, and others will take adalimumab continuously. The study will compare the pharmacokinetics, clinical outcomes, safety, immunogenicity, and efficacy between the two groups of patients. The estimated enrollment of adult patients with moderate to severe chronic plaque psoriasis is 240, and the study is expected to conclude in July 2019.
A phase 3 study of BI 695501’s performance for rheumatoid arthritis patients, completed in 2016, demonstrated similar efficacy, safety, and immunogenicity.
The VOLTAIRE-X study of a biosimilar candidate for adalimumab (Humira) for chronic plaque psoriasis has enrolled its first patient, announced Boehringer Ingelheim, the biosimilar’s developer, on July 27.
This is the first study in the United States to investigate whether a biosimilar candidate should be granted an interchangeability designation with adalimumab. The candidate, BI 695501, is up against adalimumab’s 40-mg injection.
In VOLTAIRE-X, some patients will alternate between adalimumab and BI 695501, and others will take adalimumab continuously. The study will compare the pharmacokinetics, clinical outcomes, safety, immunogenicity, and efficacy between the two groups of patients. The estimated enrollment of adult patients with moderate to severe chronic plaque psoriasis is 240, and the study is expected to conclude in July 2019.
A phase 3 study of BI 695501’s performance for rheumatoid arthritis patients, completed in 2016, demonstrated similar efficacy, safety, and immunogenicity.
CT scoring system may improve sacroiliitis treatment in IBD
A standardized scoring system to identify sacroiliitis could enable patients with inflammatory bowel disease to get earlier rheumatology referrals and improve treatment, according to an analysis of IBD patients with pre-existing abdominal CT scans.
Of the 316 patients recruited from an IBD clinic in Toronto, the validated CT scan scoring system identified 49 with sacroiliitis, of whom only 5 had been referred to an outpatient rheumatology clinic in the city. Rates of sacroiliitis were similar between the 233 patients with Crohn’s disease (15%) and the 83 with ulcerative colitis (16.9%). The scoring system indicated sacroiliitis in 6 (5.6%) of the 108 control subjects, who were recruited from a urology clinic and had no prior history of chronic back pain, reported Jonathan Chan, MD, of the University of Toronto and his associates (Arthritis Care Res. 2017 Jul 21. doi: 10.1002/acr.23323).“Previous studies using CT scan to detect sacroiliitis have relied upon an adaptation of the [modified New York] criteria or a radiologist’s gestalt. Such an adaptation may not be appropriate since changes suggestive of sacroiliitis can be found in the healthy population due to the increased sensitivity of CT scans,” the investigators said. They developed a CT-scan scoring system in which the sacroiliac joints are divided into left/right and iliac/sacral segments. The slice with the greatest number of erosions in each of the four segments contributes that value to the total erosion score, with a score of 3 or greater identifying the presence of sacroiliitis.
By demonstrating that sacroiliitis is three times more prevalent among IBD patients and can be reliably detected in CT scans performed in the clinical care of those patients, this study suggests that “more timely referral for rheumatology assessment” could avoid unnecessary treatment with biologics, Dr. Chan and his associates wrote.
The study was supported in part by a fellowship grant from the Assessment of Spondyloarthritis International Society and in part by Janssen. The investigators reported having no conflicts of interest for the study.
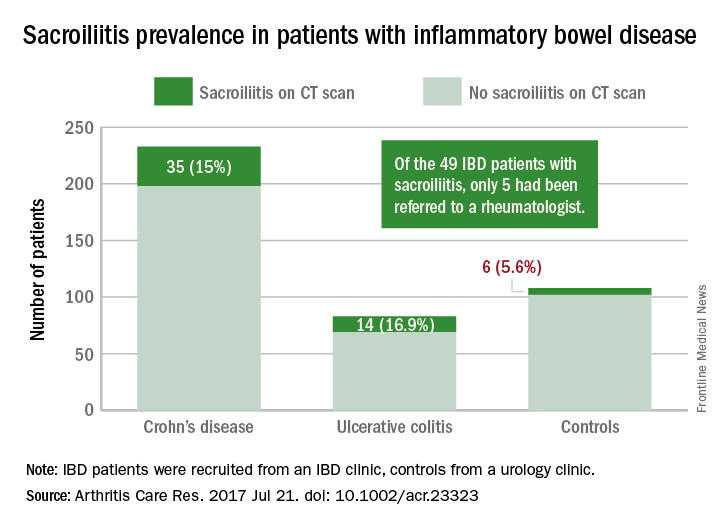
A standardized scoring system to identify sacroiliitis could enable patients with inflammatory bowel disease to get earlier rheumatology referrals and improve treatment, according to an analysis of IBD patients with pre-existing abdominal CT scans.
Of the 316 patients recruited from an IBD clinic in Toronto, the validated CT scan scoring system identified 49 with sacroiliitis, of whom only 5 had been referred to an outpatient rheumatology clinic in the city. Rates of sacroiliitis were similar between the 233 patients with Crohn’s disease (15%) and the 83 with ulcerative colitis (16.9%). The scoring system indicated sacroiliitis in 6 (5.6%) of the 108 control subjects, who were recruited from a urology clinic and had no prior history of chronic back pain, reported Jonathan Chan, MD, of the University of Toronto and his associates (Arthritis Care Res. 2017 Jul 21. doi: 10.1002/acr.23323).“Previous studies using CT scan to detect sacroiliitis have relied upon an adaptation of the [modified New York] criteria or a radiologist’s gestalt. Such an adaptation may not be appropriate since changes suggestive of sacroiliitis can be found in the healthy population due to the increased sensitivity of CT scans,” the investigators said. They developed a CT-scan scoring system in which the sacroiliac joints are divided into left/right and iliac/sacral segments. The slice with the greatest number of erosions in each of the four segments contributes that value to the total erosion score, with a score of 3 or greater identifying the presence of sacroiliitis.
By demonstrating that sacroiliitis is three times more prevalent among IBD patients and can be reliably detected in CT scans performed in the clinical care of those patients, this study suggests that “more timely referral for rheumatology assessment” could avoid unnecessary treatment with biologics, Dr. Chan and his associates wrote.
The study was supported in part by a fellowship grant from the Assessment of Spondyloarthritis International Society and in part by Janssen. The investigators reported having no conflicts of interest for the study.
A standardized scoring system to identify sacroiliitis could enable patients with inflammatory bowel disease to get earlier rheumatology referrals and improve treatment, according to an analysis of IBD patients with pre-existing abdominal CT scans.
Of the 316 patients recruited from an IBD clinic in Toronto, the validated CT scan scoring system identified 49 with sacroiliitis, of whom only 5 had been referred to an outpatient rheumatology clinic in the city. Rates of sacroiliitis were similar between the 233 patients with Crohn’s disease (15%) and the 83 with ulcerative colitis (16.9%). The scoring system indicated sacroiliitis in 6 (5.6%) of the 108 control subjects, who were recruited from a urology clinic and had no prior history of chronic back pain, reported Jonathan Chan, MD, of the University of Toronto and his associates (Arthritis Care Res. 2017 Jul 21. doi: 10.1002/acr.23323).“Previous studies using CT scan to detect sacroiliitis have relied upon an adaptation of the [modified New York] criteria or a radiologist’s gestalt. Such an adaptation may not be appropriate since changes suggestive of sacroiliitis can be found in the healthy population due to the increased sensitivity of CT scans,” the investigators said. They developed a CT-scan scoring system in which the sacroiliac joints are divided into left/right and iliac/sacral segments. The slice with the greatest number of erosions in each of the four segments contributes that value to the total erosion score, with a score of 3 or greater identifying the presence of sacroiliitis.
By demonstrating that sacroiliitis is three times more prevalent among IBD patients and can be reliably detected in CT scans performed in the clinical care of those patients, this study suggests that “more timely referral for rheumatology assessment” could avoid unnecessary treatment with biologics, Dr. Chan and his associates wrote.
The study was supported in part by a fellowship grant from the Assessment of Spondyloarthritis International Society and in part by Janssen. The investigators reported having no conflicts of interest for the study.
FROM ARTHRITIS CARE & RESEARCH
TNFi treatment halves ankylosing spondylitis progression
MADRID – At least 2 years of tumor necrosis factor–inhibitor treatment of patients with ankylosing spondylitis nearly halved the rate of spinal radiographic progression in a study involving 432 Swiss patients.
In addition, patients on a tumor necrosis factor inhibitor (TNFi) who achieved low disease activity, reflected in an Ankylosing Spondylitis (AS) Disease Activity Score of 1.3 or less, showed virtually no spinal radiographic progression during a 2-year follow-up, Adrian Ciurea, MD, reported at the European Congress of Rheumatology.
He cautioned, however, that the evidence only shows correlation and can’t prove a causal relationship between TNFi treatment and slowed spinal radiographic progression because of potential residual confounding.
Dr. Ciurea and his associates analyzed records for AS patients enrolled in the Swiss Clinical Quality Management in Rheumatic Diseases cohort who underwent at least two spinal radiographs separated by a 2-year gap. They assessed the radiographs using the modified Stoke AS Spinal Score (mSASSS), and they defined progression as a gain of at least two units on the mSASSS during a 2-year period between radiographs.
The 432 AS patients in the study averaged 40 years old, two-thirds were men, and they had AS symptoms for an average of nearly 14 years. Their average AS Disease Activity Score (ASDAS) at entry was 2.8.
A multivariate analysis that controlled for several variables, including sex, smoking history, baseline mSASSS, and exercise, identified three parameters that had significant correlations with radiographic progression: Men had more than double the rate of progression, compared with women; higher baseline mSASSS was linked with a higher rate of progression; and a greater-than-2-year history of treatment with a TNFi was linked with a 48% reduced rate of progression, reported Dr. Ciurea, a rheumatologist at the Zürich University Hospital.
The duration of treatment also mattered. Patients who received at least 4 years of TNFi treatment had a statistically significant 68% reduced rate of radiographic spinal progression. In contrast, patients who received a TNFi for fewer than 4 years but more than 2 years had a 42% lower rate of progression that was of borderline statistical significance. TNFi treatment that started during the 2 years immediately preceding the radiograph failed to show a significant link with reduced progression.
Further analysis also showed a tight correlation between patients’ disease activity while on TNFi treatment and radiographic progression. Patients who maintained an average ASDAS of 2.1 or less during the 2 years prior to radiographic assessment showed an average mSASSS gain of 0.31 units over that 2-year period, compared with an average 1.45-unit mSASSS gain among patients whose average ASDAS remained above 2.1, a statistically significant difference between these two groups. Patients with even more inactive disease on TNFi treatment – those who maintained an average ASDAS of 1.3 or less – had an average 0.01-unit rise in their mSASSS after 2 years of treatment, compared with an average 0.52-unit mSASSS rise after 2 years in patients with an ASDAS of more than 1.3 but less than 2.1, he said.
The cohort study received partial support from Merck Sharpe & Dohme. Dr. Ciurea has been a consultant to or speaker for Abbvie, Celgene, Eli Lilly, Janssen-Cilag, Merck Sharp & Dohme, Novartis, Pfizer, and UCB.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
MADRID – At least 2 years of tumor necrosis factor–inhibitor treatment of patients with ankylosing spondylitis nearly halved the rate of spinal radiographic progression in a study involving 432 Swiss patients.
In addition, patients on a tumor necrosis factor inhibitor (TNFi) who achieved low disease activity, reflected in an Ankylosing Spondylitis (AS) Disease Activity Score of 1.3 or less, showed virtually no spinal radiographic progression during a 2-year follow-up, Adrian Ciurea, MD, reported at the European Congress of Rheumatology.
He cautioned, however, that the evidence only shows correlation and can’t prove a causal relationship between TNFi treatment and slowed spinal radiographic progression because of potential residual confounding.
Dr. Ciurea and his associates analyzed records for AS patients enrolled in the Swiss Clinical Quality Management in Rheumatic Diseases cohort who underwent at least two spinal radiographs separated by a 2-year gap. They assessed the radiographs using the modified Stoke AS Spinal Score (mSASSS), and they defined progression as a gain of at least two units on the mSASSS during a 2-year period between radiographs.
The 432 AS patients in the study averaged 40 years old, two-thirds were men, and they had AS symptoms for an average of nearly 14 years. Their average AS Disease Activity Score (ASDAS) at entry was 2.8.
A multivariate analysis that controlled for several variables, including sex, smoking history, baseline mSASSS, and exercise, identified three parameters that had significant correlations with radiographic progression: Men had more than double the rate of progression, compared with women; higher baseline mSASSS was linked with a higher rate of progression; and a greater-than-2-year history of treatment with a TNFi was linked with a 48% reduced rate of progression, reported Dr. Ciurea, a rheumatologist at the Zürich University Hospital.
The duration of treatment also mattered. Patients who received at least 4 years of TNFi treatment had a statistically significant 68% reduced rate of radiographic spinal progression. In contrast, patients who received a TNFi for fewer than 4 years but more than 2 years had a 42% lower rate of progression that was of borderline statistical significance. TNFi treatment that started during the 2 years immediately preceding the radiograph failed to show a significant link with reduced progression.
Further analysis also showed a tight correlation between patients’ disease activity while on TNFi treatment and radiographic progression. Patients who maintained an average ASDAS of 2.1 or less during the 2 years prior to radiographic assessment showed an average mSASSS gain of 0.31 units over that 2-year period, compared with an average 1.45-unit mSASSS gain among patients whose average ASDAS remained above 2.1, a statistically significant difference between these two groups. Patients with even more inactive disease on TNFi treatment – those who maintained an average ASDAS of 1.3 or less – had an average 0.01-unit rise in their mSASSS after 2 years of treatment, compared with an average 0.52-unit mSASSS rise after 2 years in patients with an ASDAS of more than 1.3 but less than 2.1, he said.
The cohort study received partial support from Merck Sharpe & Dohme. Dr. Ciurea has been a consultant to or speaker for Abbvie, Celgene, Eli Lilly, Janssen-Cilag, Merck Sharp & Dohme, Novartis, Pfizer, and UCB.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
MADRID – At least 2 years of tumor necrosis factor–inhibitor treatment of patients with ankylosing spondylitis nearly halved the rate of spinal radiographic progression in a study involving 432 Swiss patients.
In addition, patients on a tumor necrosis factor inhibitor (TNFi) who achieved low disease activity, reflected in an Ankylosing Spondylitis (AS) Disease Activity Score of 1.3 or less, showed virtually no spinal radiographic progression during a 2-year follow-up, Adrian Ciurea, MD, reported at the European Congress of Rheumatology.
He cautioned, however, that the evidence only shows correlation and can’t prove a causal relationship between TNFi treatment and slowed spinal radiographic progression because of potential residual confounding.
Dr. Ciurea and his associates analyzed records for AS patients enrolled in the Swiss Clinical Quality Management in Rheumatic Diseases cohort who underwent at least two spinal radiographs separated by a 2-year gap. They assessed the radiographs using the modified Stoke AS Spinal Score (mSASSS), and they defined progression as a gain of at least two units on the mSASSS during a 2-year period between radiographs.
The 432 AS patients in the study averaged 40 years old, two-thirds were men, and they had AS symptoms for an average of nearly 14 years. Their average AS Disease Activity Score (ASDAS) at entry was 2.8.
A multivariate analysis that controlled for several variables, including sex, smoking history, baseline mSASSS, and exercise, identified three parameters that had significant correlations with radiographic progression: Men had more than double the rate of progression, compared with women; higher baseline mSASSS was linked with a higher rate of progression; and a greater-than-2-year history of treatment with a TNFi was linked with a 48% reduced rate of progression, reported Dr. Ciurea, a rheumatologist at the Zürich University Hospital.
The duration of treatment also mattered. Patients who received at least 4 years of TNFi treatment had a statistically significant 68% reduced rate of radiographic spinal progression. In contrast, patients who received a TNFi for fewer than 4 years but more than 2 years had a 42% lower rate of progression that was of borderline statistical significance. TNFi treatment that started during the 2 years immediately preceding the radiograph failed to show a significant link with reduced progression.
Further analysis also showed a tight correlation between patients’ disease activity while on TNFi treatment and radiographic progression. Patients who maintained an average ASDAS of 2.1 or less during the 2 years prior to radiographic assessment showed an average mSASSS gain of 0.31 units over that 2-year period, compared with an average 1.45-unit mSASSS gain among patients whose average ASDAS remained above 2.1, a statistically significant difference between these two groups. Patients with even more inactive disease on TNFi treatment – those who maintained an average ASDAS of 1.3 or less – had an average 0.01-unit rise in their mSASSS after 2 years of treatment, compared with an average 0.52-unit mSASSS rise after 2 years in patients with an ASDAS of more than 1.3 but less than 2.1, he said.
The cohort study received partial support from Merck Sharpe & Dohme. Dr. Ciurea has been a consultant to or speaker for Abbvie, Celgene, Eli Lilly, Janssen-Cilag, Merck Sharp & Dohme, Novartis, Pfizer, and UCB.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AT THE EULAR 2017 CONGRESS
Key clinical point:
Major finding: Prolonged TNFi treatment was linked with a 48% lower rate of spinal radiographic progression, compared with shorter treatment.
Data source: Review of 432 patients in the Swiss Clinical Quality Management in Rheumatic Diseases cohort.
Disclosures: The cohort study received partial support from Merck Sharpe & Dohme. Dr. Ciurea has been a consultant to or speaker for Abbvie, Celgene, Eli Lilly, Janssen-Cilag, Merck Sharp & Dohme, Novartis, Pfizer, and UCB.