User login
Promising add-on therapy for neonatal seizures found active in safety study
WASHINGTON – presented at the annual meeting of the American Epilepsy Society.
“This is an early-phase trial, but it did associate bumetanide with an additional reduction in seizure burden relative to phenobarbital alone,” reported Janet S. Soul, MD, director of the fetal-neonatal neurology program at Boston Children’s Hospital. She added, “The adverse events observed were not substantially different in the group that received the experimental agent.”

Of the 111 neonates with documented seizures enrolled at four participating hospitals, 43 proceeded to randomization if their seizures proved to be refractory to standard doses of phenobarbital. After randomization, the next dose of phenobarbital was administered either with placebo or with 0.1, 0.2, or 0.3 mg/kg of bumetanide. Seizure burden was evaluated at 0-2, 2-4, and 0-4 hours after study-drug administration and compared with the burden during the 2 hours before treatment.
All three doses were active, reducing the seizure burden by a median of 41%-75% in a dose-dependent manner. Whether assessed in the first 2 hours or the first 4 hours, the efficacy of bumetanide was significantly greater in those with the greatest, relative to the least, baseline seizure burden (P = .01 for hours 0-2; P = .04 for hours 0-4). The median seizure burden during the baseline period was higher in the 27 children randomized to bumetanide (114 minutes) relative to those randomized to placebo (33 minutes), although researchers attributed this to random effects in a small study.
The evidence of antiseizure activity from bumetanide as an add-on to phenobarbital is consistent with its mechanism of action, which is blockading the chloride transporter NKCC1. In the immature neurons of neonates, NKCC1 is highly expressed, and there is basic scientific evidence that this impairs the efficacy of gamma-aminobutyric acid–receptor agonists like phenobarbital, according to Dr. Soul. The hypothesis driving the study of bumetanide is that blockading NKCC1 would improve the efficacy of phenobarbital while adding its own antiseizure effects, which together could potentially provide synergistic benefit.
The efficacy and the safety of this study are somewhat discordant with a previously published study evaluating bumetanide in 14 neonates with hypoxic-ischemic encephalopathy (HIE) seizures (Lancet Neurol 2015;14:469-77). Even though there were seizure reductions in five children in this other series, which did not include a control arm, there were three cases of hearing loss considered potentially related to bumetanide. The authors of that study concluded that efficacy was not shown.
There were also three cases of hearing loss in the randomized trial presented by Dr. Soul, but one occurred in the placebo group. Although the potential for ototoxicity “still needs to be addressed” in the next set of studies, Dr. Soul noted that hearing loss in children with epilepsy is common and has numerous potential etiologies. Based on these data, she concluded, “All serious adverse events were related to severe HIE with multiorgan dysfunction and/or withdrawal of care for poor prognosis.”
Among nonserious adverse events, diuresis was the only one found significantly more common in the bumetanide group (P = .02).
Phenobarbital has been a standard in the treatment of neonatal seizures for several decades despite the substantial proportion of children who do not achieve an adequate response, according to Dr. Soul. She noted that bumetanide is one of several agents being evaluated as an adjunctive agent. For example, a phase 2 crossover trial with levetiracetam is now underway. She suggested that there is reason for optimism about gaining new treatments for neonates in an area in which she believes there are unmet needs.
“I think we may see a phase 2 trial with bumetanide within a year or 2,” Dr. Soul said. If bumetanide moves forward, she expects its role to be primarily for the treatment of acute seizures caused by HIE, stroke, or hemorrhage. She is less optimistic about its benefit for seizures caused by other etiologies, such as brain malformations.
SOURCE: Soul J Abstract 2.426
WASHINGTON – presented at the annual meeting of the American Epilepsy Society.
“This is an early-phase trial, but it did associate bumetanide with an additional reduction in seizure burden relative to phenobarbital alone,” reported Janet S. Soul, MD, director of the fetal-neonatal neurology program at Boston Children’s Hospital. She added, “The adverse events observed were not substantially different in the group that received the experimental agent.”
Of the 111 neonates with documented seizures enrolled at four participating hospitals, 43 proceeded to randomization if their seizures proved to be refractory to standard doses of phenobarbital. After randomization, the next dose of phenobarbital was administered either with placebo or with 0.1, 0.2, or 0.3 mg/kg of bumetanide. Seizure burden was evaluated at 0-2, 2-4, and 0-4 hours after study-drug administration and compared with the burden during the 2 hours before treatment.
All three doses were active, reducing the seizure burden by a median of 41%-75% in a dose-dependent manner. Whether assessed in the first 2 hours or the first 4 hours, the efficacy of bumetanide was significantly greater in those with the greatest, relative to the least, baseline seizure burden (P = .01 for hours 0-2; P = .04 for hours 0-4). The median seizure burden during the baseline period was higher in the 27 children randomized to bumetanide (114 minutes) relative to those randomized to placebo (33 minutes), although researchers attributed this to random effects in a small study.
The evidence of antiseizure activity from bumetanide as an add-on to phenobarbital is consistent with its mechanism of action, which is blockading the chloride transporter NKCC1. In the immature neurons of neonates, NKCC1 is highly expressed, and there is basic scientific evidence that this impairs the efficacy of gamma-aminobutyric acid–receptor agonists like phenobarbital, according to Dr. Soul. The hypothesis driving the study of bumetanide is that blockading NKCC1 would improve the efficacy of phenobarbital while adding its own antiseizure effects, which together could potentially provide synergistic benefit.
The efficacy and the safety of this study are somewhat discordant with a previously published study evaluating bumetanide in 14 neonates with hypoxic-ischemic encephalopathy (HIE) seizures (Lancet Neurol 2015;14:469-77). Even though there were seizure reductions in five children in this other series, which did not include a control arm, there were three cases of hearing loss considered potentially related to bumetanide. The authors of that study concluded that efficacy was not shown.
There were also three cases of hearing loss in the randomized trial presented by Dr. Soul, but one occurred in the placebo group. Although the potential for ototoxicity “still needs to be addressed” in the next set of studies, Dr. Soul noted that hearing loss in children with epilepsy is common and has numerous potential etiologies. Based on these data, she concluded, “All serious adverse events were related to severe HIE with multiorgan dysfunction and/or withdrawal of care for poor prognosis.”
Among nonserious adverse events, diuresis was the only one found significantly more common in the bumetanide group (P = .02).
Phenobarbital has been a standard in the treatment of neonatal seizures for several decades despite the substantial proportion of children who do not achieve an adequate response, according to Dr. Soul. She noted that bumetanide is one of several agents being evaluated as an adjunctive agent. For example, a phase 2 crossover trial with levetiracetam is now underway. She suggested that there is reason for optimism about gaining new treatments for neonates in an area in which she believes there are unmet needs.
“I think we may see a phase 2 trial with bumetanide within a year or 2,” Dr. Soul said. If bumetanide moves forward, she expects its role to be primarily for the treatment of acute seizures caused by HIE, stroke, or hemorrhage. She is less optimistic about its benefit for seizures caused by other etiologies, such as brain malformations.
SOURCE: Soul J Abstract 2.426
WASHINGTON – presented at the annual meeting of the American Epilepsy Society.
“This is an early-phase trial, but it did associate bumetanide with an additional reduction in seizure burden relative to phenobarbital alone,” reported Janet S. Soul, MD, director of the fetal-neonatal neurology program at Boston Children’s Hospital. She added, “The adverse events observed were not substantially different in the group that received the experimental agent.”
Of the 111 neonates with documented seizures enrolled at four participating hospitals, 43 proceeded to randomization if their seizures proved to be refractory to standard doses of phenobarbital. After randomization, the next dose of phenobarbital was administered either with placebo or with 0.1, 0.2, or 0.3 mg/kg of bumetanide. Seizure burden was evaluated at 0-2, 2-4, and 0-4 hours after study-drug administration and compared with the burden during the 2 hours before treatment.
All three doses were active, reducing the seizure burden by a median of 41%-75% in a dose-dependent manner. Whether assessed in the first 2 hours or the first 4 hours, the efficacy of bumetanide was significantly greater in those with the greatest, relative to the least, baseline seizure burden (P = .01 for hours 0-2; P = .04 for hours 0-4). The median seizure burden during the baseline period was higher in the 27 children randomized to bumetanide (114 minutes) relative to those randomized to placebo (33 minutes), although researchers attributed this to random effects in a small study.
The evidence of antiseizure activity from bumetanide as an add-on to phenobarbital is consistent with its mechanism of action, which is blockading the chloride transporter NKCC1. In the immature neurons of neonates, NKCC1 is highly expressed, and there is basic scientific evidence that this impairs the efficacy of gamma-aminobutyric acid–receptor agonists like phenobarbital, according to Dr. Soul. The hypothesis driving the study of bumetanide is that blockading NKCC1 would improve the efficacy of phenobarbital while adding its own antiseizure effects, which together could potentially provide synergistic benefit.
The efficacy and the safety of this study are somewhat discordant with a previously published study evaluating bumetanide in 14 neonates with hypoxic-ischemic encephalopathy (HIE) seizures (Lancet Neurol 2015;14:469-77). Even though there were seizure reductions in five children in this other series, which did not include a control arm, there were three cases of hearing loss considered potentially related to bumetanide. The authors of that study concluded that efficacy was not shown.
There were also three cases of hearing loss in the randomized trial presented by Dr. Soul, but one occurred in the placebo group. Although the potential for ototoxicity “still needs to be addressed” in the next set of studies, Dr. Soul noted that hearing loss in children with epilepsy is common and has numerous potential etiologies. Based on these data, she concluded, “All serious adverse events were related to severe HIE with multiorgan dysfunction and/or withdrawal of care for poor prognosis.”
Among nonserious adverse events, diuresis was the only one found significantly more common in the bumetanide group (P = .02).
Phenobarbital has been a standard in the treatment of neonatal seizures for several decades despite the substantial proportion of children who do not achieve an adequate response, according to Dr. Soul. She noted that bumetanide is one of several agents being evaluated as an adjunctive agent. For example, a phase 2 crossover trial with levetiracetam is now underway. She suggested that there is reason for optimism about gaining new treatments for neonates in an area in which she believes there are unmet needs.
“I think we may see a phase 2 trial with bumetanide within a year or 2,” Dr. Soul said. If bumetanide moves forward, she expects its role to be primarily for the treatment of acute seizures caused by HIE, stroke, or hemorrhage. She is less optimistic about its benefit for seizures caused by other etiologies, such as brain malformations.
SOURCE: Soul J Abstract 2.426
REPORTING FROM AES 2017
Key clinical point: Bumetanide is associated with antiseizure activity as add-on therapy to phenobarbital for neonatal seizures.
Major finding: Relative to pretreatment, there was greater reduction in seizure burden (P = .01) at 4 hours in those with the highest seizure burden.
Data source: Randomized, double-blind phase 1/2 trial.
Disclosures: Dr. Soul reports no potential conflicts of interest related to this topic.
Source: Soul J et al. Abstract 2.426
ACTH and other standard treatments prove best for infantile spasms
WASHINGTON – Standard infantile spasm therapies such as adrenocorticotropic hormone appear to be significantly more effective than nonstandard therapies, according to a prospective study presented at the annual meeting of the American Epilepsy Society.
If infants currently treated with nonstandard therapies switched to adrenocorticotropic hormone (ACTH), there would be an increase of “one additional responder for every four infants with infantile spasms,” according to Renee Shellhaas, MD, a pediatric neurologist at the University of Michigan, Ann Arbor.
Dr. Shellhaas and her colleagues conducted a prospective study of 352 infants recorded to have spasms in the National Infantile Spasms Consortium from 2012 to 2016 and compared successful responses with the use of ACTH and other standard therapies against those with nonstandard therapies. They defined a successful response as a patient who did not take any other medication for infantile spasms for 60 days and had no infantile spasms for 30 days after finishing 30 days of treatment. Infants were split into four treatment arms: ACTH (n = 150), vigabatrin (68), oral steroids (90), and nonstandard therapies (44). Nonstandard therapies included topiramate, levetiracetam, clobazam, zonisamide, ketogenic diet, oxcarbazepine, and phenobarbital.
The proportion of male infants across all arms was 50%-64%, with an average age of 6.2 months in the ACTH group, 5.5 months in the vigabatrin group, 6.7 months in the oral steroids group, and 5.5 months in the nonstandard group. A majority of infants across all arms had hypsarrhythmia on EEG, ranging from 57% to 84%.
Dr. Shellhaas and her colleagues sought to answer the question, “What would happen if this infant had been treated with ACTH instead of the given medication?” They controlled these comparisons for selection bias by weighting them for various factors that may have increased the odds of using the comparison treatment. They also controlled for potential medical center effects, but did not adjust for dosing regimen.
If the infants who had received nonstandard therapies had instead received ACTH, their response rate would have improved from 5% to 32%, according to this analysis (P less than .01).
In comparisons against other standard treatments, response rates would not have been significantly better if patients had instead received ACTH: 29% for vigabatrin vs. an estimated 37% for ACTH and 46% for oral steroids vs. an estimated 44% for ACTH.
If there was one thing to take away from this, it is that nonstandard therapies do not work nearly as well as ACTH or other standard treatments,” Dr. Shellhaas said. “It is crucial to treat these infants with treatments that are effective.”
Dr. Shellhaas and her associates uncovered certain clinical factors associated with treatment selections. Among infants with unknown infantile spasm etiology, 30% were given nonstandard treatment, whereas 47% received ACTH. Infants who were not already on antiepileptic drugs more often received nonstandard therapies than ACTH (45% vs. 17%).
However, ACTH was still more likely to be given over nonstandard therapies to infants who had hypsarrhythmia (84% vs. 57%) or a normal head circumference (77% vs. 57%).
Dr. Shellhaas reported no relevant financial disclosures. The Pediatric Epilepsy Research Foundation funded the study.
ezimmerman@frontlinemedcom.com
SOURCE: AES 2017 abstract 1.303
WASHINGTON – Standard infantile spasm therapies such as adrenocorticotropic hormone appear to be significantly more effective than nonstandard therapies, according to a prospective study presented at the annual meeting of the American Epilepsy Society.
If infants currently treated with nonstandard therapies switched to adrenocorticotropic hormone (ACTH), there would be an increase of “one additional responder for every four infants with infantile spasms,” according to Renee Shellhaas, MD, a pediatric neurologist at the University of Michigan, Ann Arbor.
Dr. Shellhaas and her colleagues conducted a prospective study of 352 infants recorded to have spasms in the National Infantile Spasms Consortium from 2012 to 2016 and compared successful responses with the use of ACTH and other standard therapies against those with nonstandard therapies. They defined a successful response as a patient who did not take any other medication for infantile spasms for 60 days and had no infantile spasms for 30 days after finishing 30 days of treatment. Infants were split into four treatment arms: ACTH (n = 150), vigabatrin (68), oral steroids (90), and nonstandard therapies (44). Nonstandard therapies included topiramate, levetiracetam, clobazam, zonisamide, ketogenic diet, oxcarbazepine, and phenobarbital.
The proportion of male infants across all arms was 50%-64%, with an average age of 6.2 months in the ACTH group, 5.5 months in the vigabatrin group, 6.7 months in the oral steroids group, and 5.5 months in the nonstandard group. A majority of infants across all arms had hypsarrhythmia on EEG, ranging from 57% to 84%.
Dr. Shellhaas and her colleagues sought to answer the question, “What would happen if this infant had been treated with ACTH instead of the given medication?” They controlled these comparisons for selection bias by weighting them for various factors that may have increased the odds of using the comparison treatment. They also controlled for potential medical center effects, but did not adjust for dosing regimen.
If the infants who had received nonstandard therapies had instead received ACTH, their response rate would have improved from 5% to 32%, according to this analysis (P less than .01).
In comparisons against other standard treatments, response rates would not have been significantly better if patients had instead received ACTH: 29% for vigabatrin vs. an estimated 37% for ACTH and 46% for oral steroids vs. an estimated 44% for ACTH.
If there was one thing to take away from this, it is that nonstandard therapies do not work nearly as well as ACTH or other standard treatments,” Dr. Shellhaas said. “It is crucial to treat these infants with treatments that are effective.”
Dr. Shellhaas and her associates uncovered certain clinical factors associated with treatment selections. Among infants with unknown infantile spasm etiology, 30% were given nonstandard treatment, whereas 47% received ACTH. Infants who were not already on antiepileptic drugs more often received nonstandard therapies than ACTH (45% vs. 17%).
However, ACTH was still more likely to be given over nonstandard therapies to infants who had hypsarrhythmia (84% vs. 57%) or a normal head circumference (77% vs. 57%).
Dr. Shellhaas reported no relevant financial disclosures. The Pediatric Epilepsy Research Foundation funded the study.
ezimmerman@frontlinemedcom.com
SOURCE: AES 2017 abstract 1.303
WASHINGTON – Standard infantile spasm therapies such as adrenocorticotropic hormone appear to be significantly more effective than nonstandard therapies, according to a prospective study presented at the annual meeting of the American Epilepsy Society.
If infants currently treated with nonstandard therapies switched to adrenocorticotropic hormone (ACTH), there would be an increase of “one additional responder for every four infants with infantile spasms,” according to Renee Shellhaas, MD, a pediatric neurologist at the University of Michigan, Ann Arbor.
Dr. Shellhaas and her colleagues conducted a prospective study of 352 infants recorded to have spasms in the National Infantile Spasms Consortium from 2012 to 2016 and compared successful responses with the use of ACTH and other standard therapies against those with nonstandard therapies. They defined a successful response as a patient who did not take any other medication for infantile spasms for 60 days and had no infantile spasms for 30 days after finishing 30 days of treatment. Infants were split into four treatment arms: ACTH (n = 150), vigabatrin (68), oral steroids (90), and nonstandard therapies (44). Nonstandard therapies included topiramate, levetiracetam, clobazam, zonisamide, ketogenic diet, oxcarbazepine, and phenobarbital.
The proportion of male infants across all arms was 50%-64%, with an average age of 6.2 months in the ACTH group, 5.5 months in the vigabatrin group, 6.7 months in the oral steroids group, and 5.5 months in the nonstandard group. A majority of infants across all arms had hypsarrhythmia on EEG, ranging from 57% to 84%.
Dr. Shellhaas and her colleagues sought to answer the question, “What would happen if this infant had been treated with ACTH instead of the given medication?” They controlled these comparisons for selection bias by weighting them for various factors that may have increased the odds of using the comparison treatment. They also controlled for potential medical center effects, but did not adjust for dosing regimen.
If the infants who had received nonstandard therapies had instead received ACTH, their response rate would have improved from 5% to 32%, according to this analysis (P less than .01).
In comparisons against other standard treatments, response rates would not have been significantly better if patients had instead received ACTH: 29% for vigabatrin vs. an estimated 37% for ACTH and 46% for oral steroids vs. an estimated 44% for ACTH.
If there was one thing to take away from this, it is that nonstandard therapies do not work nearly as well as ACTH or other standard treatments,” Dr. Shellhaas said. “It is crucial to treat these infants with treatments that are effective.”
Dr. Shellhaas and her associates uncovered certain clinical factors associated with treatment selections. Among infants with unknown infantile spasm etiology, 30% were given nonstandard treatment, whereas 47% received ACTH. Infants who were not already on antiepileptic drugs more often received nonstandard therapies than ACTH (45% vs. 17%).
However, ACTH was still more likely to be given over nonstandard therapies to infants who had hypsarrhythmia (84% vs. 57%) or a normal head circumference (77% vs. 57%).
Dr. Shellhaas reported no relevant financial disclosures. The Pediatric Epilepsy Research Foundation funded the study.
ezimmerman@frontlinemedcom.com
SOURCE: AES 2017 abstract 1.303
REPORTING FROM AES 2017
Key clinical point:
Major finding: If the infants who had received nonstandard therapies had instead received ACTH, their response rate would have improved from 5% to 32% (P less than .01).
Data source: Prospective study of 352 infants gathered from the National Infantile Spasms Consortium database from 2012-2016.
Disclosures: The presenter reported no relevant financial disclosures. The Pediatric Epilepsy Research Foundation funded the study.
Source: R. Shellhaas, et al. AES 2017 abstract 1.303
Rheumatology 911: Inside the rheumatologic emergency
SAN DIEGO – At first glance, rheumatology may seem like the perfect specialty for physicians who don’t want to be bothered by medical emergencies. But the reality can be more complicated.
As Bharat Kumar, MD, explained to an audience at the annual meeting of the American College of Rheumatology, rheumatologists will at times encounter patients in urgent need of their care due to dire medical conditions. In these situations, he said, there may be no time for careful and cautious diagnostics.
“You have to have an awareness of how you think about things,” advised Dr. Kumar, a rheumatologist/immunologist and clinical assistant professor of internal medicine at the University of Iowa, Iowa City. “During emergencies, you have to rely more on intuition to quickly get at answers.”

Q: When do rheumatologists have to deal with medical emergencies?
A: Rheumatology is considered mostly an outpatient specialty. Most of the time, rheumatologists don’t receive off-hour emergency calls.
But there are conditions in which rheumatologists have to be at the front lines in diagnosing and managing medical emergencies. These range from issues like septic arthritis to scleroderma renal crisis and vasculitis affecting vital organs such as the heart, lungs, and kidneys. These are more common at academic settings, but even rheumatologists in private practice should be aware of these conditions.
Q: How often do rheumatologists come across true emergencies in normal practice?
A: It depends on where the rheumatologist is practicing. In our academic setting, we have to see patients in the hospital several times per week.
Rarer are the emergencies that show up to clinic and require evaluation in the emergency department or hospitalization. Over the past year, that has happened perhaps three times to me.
This is likely much less in the private setting, where patients tend to be less sick and less complicated. But that is no guarantee that an emergency won’t crop up.
Q: What is the scariest emergency situation that you’ve come across?
A: It occurred when I entered a room to see a patient of mine with adult-onset Still’s disease.
She was huddled, shivering, barely answering questions. Her eyes were glazed. Her blood pressure was below 90/60 mm Hg, and her pulse was 130 beats per minute. I was petrified that she was in the midst of a cytokine storm secondary to either hemophagocytic lymphohistiocytosis (HLH) or sepsis. Given the high mortality of both, we immediately called our colleagues in the emergency department and sent her for hospitalization. It turned out that she did have HLH, and we had to pursue intensive immunosuppression to abate that cytokine storm.
It was particularly scary because there is no good way to differentiate between the two conditions, apart from going with clinical intuition.
Treating a patient who is potentially septic with immunosuppression is extremely dangerous, and ultimately, we would not have known if our intuition was correct until the infection presented itself.
Fortunately, we were correct. She recovered after 1 week of hospitalization, and we have been following her since then. But it still gives me goosebumps to think, “What if we were wrong?”
Q: Do emergencies in rheumatology tend to appear suddenly or are they more likely to occur because of a long-standing and perhaps untreated condition?
A: While it is true that uncontrolled disease activity can predispose patients to emergencies, other emergencies can occur sporadically and out of the blue.
Many times, an emergency is the first manifestation of disease. The literature is littered with cases of renal crisis being the first manifestation of systemic sclerosis. And internists are often baffled by sudden kidney failure due to previously undiagnosed lupus.
In addition, all rheumatologists have great reverence for septic arthritis and know that it can mimic gout very closely. If a swollen joint is mistaken for gout instead of septic arthritis, this can lead to worsening infection and ultimately, loss of joint function.
Q: What are some potentially dire conditions that may test the diagnostic powers of rheumatologists?
A: Rheumatologists are becoming more aware of HLH. Because it may look clinically indistinguishable from severe infection but needs to be treated with immunosuppression instead of antimicrobial therapy, rheumatologists have to keep it in mind and revisit the diagnosis often in case patients are not improving on the prescribed therapy.
Pulmonary vasculitis is another concerning condition because an otherwise negligible cough can turn into massive pulmonary hemorrhage very quickly.
Q: Do you have tips about dealing with ER doctors, primary doctors and others who may be involved with an emergency?
A: Rheumatologists think differently from other specialists. We are cognitive specialists and think more in the long term. Emergency medicine doctors are more concerned about the short term and how to deal with more immediate issues.
Signposting concerns is essential to optimizing communication. Education of other physicians is also important because more frequently than not, patients with rheumatologic diseases present very differently.
Lastly, there’s a very fine line between advocating for patients and overstepping your bounds as a consultant rheumatologist. Maintaining close collaboration and establishing clear and open lines of communication can prevent this.
Dr. Kumar has no relevant disclosures.
SAN DIEGO – At first glance, rheumatology may seem like the perfect specialty for physicians who don’t want to be bothered by medical emergencies. But the reality can be more complicated.
As Bharat Kumar, MD, explained to an audience at the annual meeting of the American College of Rheumatology, rheumatologists will at times encounter patients in urgent need of their care due to dire medical conditions. In these situations, he said, there may be no time for careful and cautious diagnostics.
“You have to have an awareness of how you think about things,” advised Dr. Kumar, a rheumatologist/immunologist and clinical assistant professor of internal medicine at the University of Iowa, Iowa City. “During emergencies, you have to rely more on intuition to quickly get at answers.”
Q: When do rheumatologists have to deal with medical emergencies?
A: Rheumatology is considered mostly an outpatient specialty. Most of the time, rheumatologists don’t receive off-hour emergency calls.
But there are conditions in which rheumatologists have to be at the front lines in diagnosing and managing medical emergencies. These range from issues like septic arthritis to scleroderma renal crisis and vasculitis affecting vital organs such as the heart, lungs, and kidneys. These are more common at academic settings, but even rheumatologists in private practice should be aware of these conditions.
Q: How often do rheumatologists come across true emergencies in normal practice?
A: It depends on where the rheumatologist is practicing. In our academic setting, we have to see patients in the hospital several times per week.
Rarer are the emergencies that show up to clinic and require evaluation in the emergency department or hospitalization. Over the past year, that has happened perhaps three times to me.
This is likely much less in the private setting, where patients tend to be less sick and less complicated. But that is no guarantee that an emergency won’t crop up.
Q: What is the scariest emergency situation that you’ve come across?
A: It occurred when I entered a room to see a patient of mine with adult-onset Still’s disease.
She was huddled, shivering, barely answering questions. Her eyes were glazed. Her blood pressure was below 90/60 mm Hg, and her pulse was 130 beats per minute. I was petrified that she was in the midst of a cytokine storm secondary to either hemophagocytic lymphohistiocytosis (HLH) or sepsis. Given the high mortality of both, we immediately called our colleagues in the emergency department and sent her for hospitalization. It turned out that she did have HLH, and we had to pursue intensive immunosuppression to abate that cytokine storm.
It was particularly scary because there is no good way to differentiate between the two conditions, apart from going with clinical intuition.
Treating a patient who is potentially septic with immunosuppression is extremely dangerous, and ultimately, we would not have known if our intuition was correct until the infection presented itself.
Fortunately, we were correct. She recovered after 1 week of hospitalization, and we have been following her since then. But it still gives me goosebumps to think, “What if we were wrong?”
Q: Do emergencies in rheumatology tend to appear suddenly or are they more likely to occur because of a long-standing and perhaps untreated condition?
A: While it is true that uncontrolled disease activity can predispose patients to emergencies, other emergencies can occur sporadically and out of the blue.
Many times, an emergency is the first manifestation of disease. The literature is littered with cases of renal crisis being the first manifestation of systemic sclerosis. And internists are often baffled by sudden kidney failure due to previously undiagnosed lupus.
In addition, all rheumatologists have great reverence for septic arthritis and know that it can mimic gout very closely. If a swollen joint is mistaken for gout instead of septic arthritis, this can lead to worsening infection and ultimately, loss of joint function.
Q: What are some potentially dire conditions that may test the diagnostic powers of rheumatologists?
A: Rheumatologists are becoming more aware of HLH. Because it may look clinically indistinguishable from severe infection but needs to be treated with immunosuppression instead of antimicrobial therapy, rheumatologists have to keep it in mind and revisit the diagnosis often in case patients are not improving on the prescribed therapy.
Pulmonary vasculitis is another concerning condition because an otherwise negligible cough can turn into massive pulmonary hemorrhage very quickly.
Q: Do you have tips about dealing with ER doctors, primary doctors and others who may be involved with an emergency?
A: Rheumatologists think differently from other specialists. We are cognitive specialists and think more in the long term. Emergency medicine doctors are more concerned about the short term and how to deal with more immediate issues.
Signposting concerns is essential to optimizing communication. Education of other physicians is also important because more frequently than not, patients with rheumatologic diseases present very differently.
Lastly, there’s a very fine line between advocating for patients and overstepping your bounds as a consultant rheumatologist. Maintaining close collaboration and establishing clear and open lines of communication can prevent this.
Dr. Kumar has no relevant disclosures.
SAN DIEGO – At first glance, rheumatology may seem like the perfect specialty for physicians who don’t want to be bothered by medical emergencies. But the reality can be more complicated.
As Bharat Kumar, MD, explained to an audience at the annual meeting of the American College of Rheumatology, rheumatologists will at times encounter patients in urgent need of their care due to dire medical conditions. In these situations, he said, there may be no time for careful and cautious diagnostics.
“You have to have an awareness of how you think about things,” advised Dr. Kumar, a rheumatologist/immunologist and clinical assistant professor of internal medicine at the University of Iowa, Iowa City. “During emergencies, you have to rely more on intuition to quickly get at answers.”
Q: When do rheumatologists have to deal with medical emergencies?
A: Rheumatology is considered mostly an outpatient specialty. Most of the time, rheumatologists don’t receive off-hour emergency calls.
But there are conditions in which rheumatologists have to be at the front lines in diagnosing and managing medical emergencies. These range from issues like septic arthritis to scleroderma renal crisis and vasculitis affecting vital organs such as the heart, lungs, and kidneys. These are more common at academic settings, but even rheumatologists in private practice should be aware of these conditions.
Q: How often do rheumatologists come across true emergencies in normal practice?
A: It depends on where the rheumatologist is practicing. In our academic setting, we have to see patients in the hospital several times per week.
Rarer are the emergencies that show up to clinic and require evaluation in the emergency department or hospitalization. Over the past year, that has happened perhaps three times to me.
This is likely much less in the private setting, where patients tend to be less sick and less complicated. But that is no guarantee that an emergency won’t crop up.
Q: What is the scariest emergency situation that you’ve come across?
A: It occurred when I entered a room to see a patient of mine with adult-onset Still’s disease.
She was huddled, shivering, barely answering questions. Her eyes were glazed. Her blood pressure was below 90/60 mm Hg, and her pulse was 130 beats per minute. I was petrified that she was in the midst of a cytokine storm secondary to either hemophagocytic lymphohistiocytosis (HLH) or sepsis. Given the high mortality of both, we immediately called our colleagues in the emergency department and sent her for hospitalization. It turned out that she did have HLH, and we had to pursue intensive immunosuppression to abate that cytokine storm.
It was particularly scary because there is no good way to differentiate between the two conditions, apart from going with clinical intuition.
Treating a patient who is potentially septic with immunosuppression is extremely dangerous, and ultimately, we would not have known if our intuition was correct until the infection presented itself.
Fortunately, we were correct. She recovered after 1 week of hospitalization, and we have been following her since then. But it still gives me goosebumps to think, “What if we were wrong?”
Q: Do emergencies in rheumatology tend to appear suddenly or are they more likely to occur because of a long-standing and perhaps untreated condition?
A: While it is true that uncontrolled disease activity can predispose patients to emergencies, other emergencies can occur sporadically and out of the blue.
Many times, an emergency is the first manifestation of disease. The literature is littered with cases of renal crisis being the first manifestation of systemic sclerosis. And internists are often baffled by sudden kidney failure due to previously undiagnosed lupus.
In addition, all rheumatologists have great reverence for septic arthritis and know that it can mimic gout very closely. If a swollen joint is mistaken for gout instead of septic arthritis, this can lead to worsening infection and ultimately, loss of joint function.
Q: What are some potentially dire conditions that may test the diagnostic powers of rheumatologists?
A: Rheumatologists are becoming more aware of HLH. Because it may look clinically indistinguishable from severe infection but needs to be treated with immunosuppression instead of antimicrobial therapy, rheumatologists have to keep it in mind and revisit the diagnosis often in case patients are not improving on the prescribed therapy.
Pulmonary vasculitis is another concerning condition because an otherwise negligible cough can turn into massive pulmonary hemorrhage very quickly.
Q: Do you have tips about dealing with ER doctors, primary doctors and others who may be involved with an emergency?
A: Rheumatologists think differently from other specialists. We are cognitive specialists and think more in the long term. Emergency medicine doctors are more concerned about the short term and how to deal with more immediate issues.
Signposting concerns is essential to optimizing communication. Education of other physicians is also important because more frequently than not, patients with rheumatologic diseases present very differently.
Lastly, there’s a very fine line between advocating for patients and overstepping your bounds as a consultant rheumatologist. Maintaining close collaboration and establishing clear and open lines of communication can prevent this.
Dr. Kumar has no relevant disclosures.
EXPERT ANALYSIS FROM ACR 2017
Fenfluramine trials in Dravet syndrome yield highly positive results
WASHINGTON – The oral experimental agent fenfluramine, also known as ZX008, has been associated with a high degree of efficacy and good tolerability for the adjunctive treatment of Dravet syndrome, according to combined results of the first patients enrolled in two phase III trials.
“For me, a highlight of this study is the finding that 45% of patients on the higher dose achieved at least a 75% reduction from baseline in monthly convulsive seizures. This is a life-changing improvement,” reported Joseph Sullivan, MD, director of the pediatric epilepsy center at the University of California, San Francisco. He presented the results at the annual meeting of the American Epilepsy Society.
“If fenfluramine is approved as an adjunctive agent, it is likely to be introduced as the second or third medication in an effort to gain adequate symptom control,” Dr. Sullivan speculated.
Three phase 3 trials with fenfluramine are underway. The data presented at the American Epilepsy Society meeting were based on the first 119 patients who had participated in either of the two identical trials conducted in Europe and North America. The data from these two trials has now been combined, and the outcomes in the remaining patients in these two trials will be presented at a later time along with results from a third phase 3 study.
Patients between the ages of 2 and 18 years with a clinical diagnosis of Dravet syndrome were eligible for the European and North American trials if they were not controlled on current therapy, which could include multiple agents. However, patients had to be on stable therapies prior to enrollment for at least 4 weeks. Once enrolled, they were observed for 6 weeks prior to randomization.
After randomization to placebo, 0.2 mg/kg fenfluramine, or 0.8 mg/kg fenfluramine, patients completed a 2-week titration before they reached their maintenance dose. They were then evaluated over an additional 12-week treatment period. There were three withdrawals over the course of treatment in the placebo group, none in the lower-dose fenfluramine group, and six in the higher-dose fenfluramine group.
The primary endpoint was change in mean monthly convulsive seizure frequency from the observation period. When compared with placebo, these reductions were 63.9% (P less than .001) in the 0.8-mg/kg group and 33.7% (P = .019) in the 0.2-mg/kg group. When expressed as the median percent reduction in convulsive seizures from the observation period per 28 days, the reductions were 72.4% for the 0.8-mg/kg dose (P less than .001 vs. placebo), 37.6% for the 0.2-mg/kg group (P = .185 vs. placebo), and 17.4% for placebo.
Other efficacy measures supported the relative advantage of fenfluramine. For example, 70% and 41% of the patients in the 0.8-mg/kg and 0.2-mg/kg groups, respectively, versus 8% of placebo patients, had at least a 50% reduction in seizure frequency. Median seizure-free intervals for the three groups were 20.5, 14, and 9 days, respectively. Seizure activity was reduced to one or no seizures over the treatment period in 25% of the 0.8-mg/kg group, 12.8% of the 0.2-mg/kg group, and 0% of the placebo group.
The most common adverse events on the 0.8-mg/kg dose of fenfluramine, compared with placebo, were decreased appetite (37.5% vs. 5%) and lethargy (17.5% vs. 5%). The proportion of patients with weight loss was also greater on 0.8 mg/kg (5%) and 0.2 mg/kg (12.8%) versus placebo (0%). Diarrhea was more common in the 0.2-mg/kg group (30.8%) than in the 0.8-mg/kg group (17.5%) or in the placebo group (7.5%).
Although monitored closely, cardiotoxicity was not observed in this study. Concern about potential cardiotoxic effects was generated by the increased risk of valvular disease observed in patients taking fenfluramine with phentermine (fen-phen) for weight loss in the 1990s. This combination was withdrawn from the market in 1997.
“The potential for cardiotoxicity will continue to be monitored closely, but these initial results were reassuring,” reported Dr. Sullivan, who noted that a history of cardiovascular or cerebrovascular disease were exclusion criteria from this study.
Application for regulatory approval is not anticipated until all the phase 3 trial data are available, but Dr. Sullivan said that the results so far suggest that fenfluramine as an adjunctive agent “may represent a significant advance over existing treatment options for Dravet syndrome.”
The studies are funded by Zogenix. Dr. Sullivan reported financial relationships with Epygenix and Zogenix.
WASHINGTON – The oral experimental agent fenfluramine, also known as ZX008, has been associated with a high degree of efficacy and good tolerability for the adjunctive treatment of Dravet syndrome, according to combined results of the first patients enrolled in two phase III trials.
“For me, a highlight of this study is the finding that 45% of patients on the higher dose achieved at least a 75% reduction from baseline in monthly convulsive seizures. This is a life-changing improvement,” reported Joseph Sullivan, MD, director of the pediatric epilepsy center at the University of California, San Francisco. He presented the results at the annual meeting of the American Epilepsy Society.
“If fenfluramine is approved as an adjunctive agent, it is likely to be introduced as the second or third medication in an effort to gain adequate symptom control,” Dr. Sullivan speculated.
Three phase 3 trials with fenfluramine are underway. The data presented at the American Epilepsy Society meeting were based on the first 119 patients who had participated in either of the two identical trials conducted in Europe and North America. The data from these two trials has now been combined, and the outcomes in the remaining patients in these two trials will be presented at a later time along with results from a third phase 3 study.
Patients between the ages of 2 and 18 years with a clinical diagnosis of Dravet syndrome were eligible for the European and North American trials if they were not controlled on current therapy, which could include multiple agents. However, patients had to be on stable therapies prior to enrollment for at least 4 weeks. Once enrolled, they were observed for 6 weeks prior to randomization.
After randomization to placebo, 0.2 mg/kg fenfluramine, or 0.8 mg/kg fenfluramine, patients completed a 2-week titration before they reached their maintenance dose. They were then evaluated over an additional 12-week treatment period. There were three withdrawals over the course of treatment in the placebo group, none in the lower-dose fenfluramine group, and six in the higher-dose fenfluramine group.
The primary endpoint was change in mean monthly convulsive seizure frequency from the observation period. When compared with placebo, these reductions were 63.9% (P less than .001) in the 0.8-mg/kg group and 33.7% (P = .019) in the 0.2-mg/kg group. When expressed as the median percent reduction in convulsive seizures from the observation period per 28 days, the reductions were 72.4% for the 0.8-mg/kg dose (P less than .001 vs. placebo), 37.6% for the 0.2-mg/kg group (P = .185 vs. placebo), and 17.4% for placebo.
Other efficacy measures supported the relative advantage of fenfluramine. For example, 70% and 41% of the patients in the 0.8-mg/kg and 0.2-mg/kg groups, respectively, versus 8% of placebo patients, had at least a 50% reduction in seizure frequency. Median seizure-free intervals for the three groups were 20.5, 14, and 9 days, respectively. Seizure activity was reduced to one or no seizures over the treatment period in 25% of the 0.8-mg/kg group, 12.8% of the 0.2-mg/kg group, and 0% of the placebo group.
The most common adverse events on the 0.8-mg/kg dose of fenfluramine, compared with placebo, were decreased appetite (37.5% vs. 5%) and lethargy (17.5% vs. 5%). The proportion of patients with weight loss was also greater on 0.8 mg/kg (5%) and 0.2 mg/kg (12.8%) versus placebo (0%). Diarrhea was more common in the 0.2-mg/kg group (30.8%) than in the 0.8-mg/kg group (17.5%) or in the placebo group (7.5%).
Although monitored closely, cardiotoxicity was not observed in this study. Concern about potential cardiotoxic effects was generated by the increased risk of valvular disease observed in patients taking fenfluramine with phentermine (fen-phen) for weight loss in the 1990s. This combination was withdrawn from the market in 1997.
“The potential for cardiotoxicity will continue to be monitored closely, but these initial results were reassuring,” reported Dr. Sullivan, who noted that a history of cardiovascular or cerebrovascular disease were exclusion criteria from this study.
Application for regulatory approval is not anticipated until all the phase 3 trial data are available, but Dr. Sullivan said that the results so far suggest that fenfluramine as an adjunctive agent “may represent a significant advance over existing treatment options for Dravet syndrome.”
The studies are funded by Zogenix. Dr. Sullivan reported financial relationships with Epygenix and Zogenix.
WASHINGTON – The oral experimental agent fenfluramine, also known as ZX008, has been associated with a high degree of efficacy and good tolerability for the adjunctive treatment of Dravet syndrome, according to combined results of the first patients enrolled in two phase III trials.
“For me, a highlight of this study is the finding that 45% of patients on the higher dose achieved at least a 75% reduction from baseline in monthly convulsive seizures. This is a life-changing improvement,” reported Joseph Sullivan, MD, director of the pediatric epilepsy center at the University of California, San Francisco. He presented the results at the annual meeting of the American Epilepsy Society.
“If fenfluramine is approved as an adjunctive agent, it is likely to be introduced as the second or third medication in an effort to gain adequate symptom control,” Dr. Sullivan speculated.
Three phase 3 trials with fenfluramine are underway. The data presented at the American Epilepsy Society meeting were based on the first 119 patients who had participated in either of the two identical trials conducted in Europe and North America. The data from these two trials has now been combined, and the outcomes in the remaining patients in these two trials will be presented at a later time along with results from a third phase 3 study.
Patients between the ages of 2 and 18 years with a clinical diagnosis of Dravet syndrome were eligible for the European and North American trials if they were not controlled on current therapy, which could include multiple agents. However, patients had to be on stable therapies prior to enrollment for at least 4 weeks. Once enrolled, they were observed for 6 weeks prior to randomization.
After randomization to placebo, 0.2 mg/kg fenfluramine, or 0.8 mg/kg fenfluramine, patients completed a 2-week titration before they reached their maintenance dose. They were then evaluated over an additional 12-week treatment period. There were three withdrawals over the course of treatment in the placebo group, none in the lower-dose fenfluramine group, and six in the higher-dose fenfluramine group.
The primary endpoint was change in mean monthly convulsive seizure frequency from the observation period. When compared with placebo, these reductions were 63.9% (P less than .001) in the 0.8-mg/kg group and 33.7% (P = .019) in the 0.2-mg/kg group. When expressed as the median percent reduction in convulsive seizures from the observation period per 28 days, the reductions were 72.4% for the 0.8-mg/kg dose (P less than .001 vs. placebo), 37.6% for the 0.2-mg/kg group (P = .185 vs. placebo), and 17.4% for placebo.
Other efficacy measures supported the relative advantage of fenfluramine. For example, 70% and 41% of the patients in the 0.8-mg/kg and 0.2-mg/kg groups, respectively, versus 8% of placebo patients, had at least a 50% reduction in seizure frequency. Median seizure-free intervals for the three groups were 20.5, 14, and 9 days, respectively. Seizure activity was reduced to one or no seizures over the treatment period in 25% of the 0.8-mg/kg group, 12.8% of the 0.2-mg/kg group, and 0% of the placebo group.
The most common adverse events on the 0.8-mg/kg dose of fenfluramine, compared with placebo, were decreased appetite (37.5% vs. 5%) and lethargy (17.5% vs. 5%). The proportion of patients with weight loss was also greater on 0.8 mg/kg (5%) and 0.2 mg/kg (12.8%) versus placebo (0%). Diarrhea was more common in the 0.2-mg/kg group (30.8%) than in the 0.8-mg/kg group (17.5%) or in the placebo group (7.5%).
Although monitored closely, cardiotoxicity was not observed in this study. Concern about potential cardiotoxic effects was generated by the increased risk of valvular disease observed in patients taking fenfluramine with phentermine (fen-phen) for weight loss in the 1990s. This combination was withdrawn from the market in 1997.
“The potential for cardiotoxicity will continue to be monitored closely, but these initial results were reassuring,” reported Dr. Sullivan, who noted that a history of cardiovascular or cerebrovascular disease were exclusion criteria from this study.
Application for regulatory approval is not anticipated until all the phase 3 trial data are available, but Dr. Sullivan said that the results so far suggest that fenfluramine as an adjunctive agent “may represent a significant advance over existing treatment options for Dravet syndrome.”
The studies are funded by Zogenix. Dr. Sullivan reported financial relationships with Epygenix and Zogenix.
REPORTING FROM AES 2017
Key clinical point:
Major finding: For the highest dose, the mean reduction in convulsive seizure frequency was 63.9% (P less than .001) versus placebo over a 14-week treatment period.
Data source: An analysis of the first 119 patients with Dravet syndrome enrolled in two ongoing randomized, double-blind, multicenter, phase 3 trials.
Disclosures: The studies are funded by Zogenix. The presenter reported financial relationships with Epygenix and Zogenix.
Source: L Lagae et al. AES 2017 Abstract 2.434
Adolescents with chronic health conditions often undervaccinated
said Annika M. Hofstetter, MD, PhD, of Columbia University, New York, and her associates.
The National Health Interview Survey on Disability in 1994-1995 estimated that chronic conditions of any type affected 15%-18% of U.S. children and adolescents. The Advisory Committee on Immunization Practices recommends that all adolescents, whether or not they have chronic medical condition, be vaccinated with human papillomavirus (HPV), Tdap, meningococcal, and flu vaccines.
Fewer adolescents with CMCs had received one more doses of HPV (81%), than did those without CMCs (85%; P less than .01). Fewer adolescents with epilepsy (63%), mental retardation (58%), cerebral palsy (54%), and autism spectrum disorder (46%) had started HPV vaccination, compared with those without each of these conditions (84%; all comparisons, P less than .001). No differences were seen for asthma or congenital heart disease, the investigators said.
More adolescents with CMCs had gotten their flu shot than did those without CMCs during the 2011-2012 season (67% vs. 50%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). More adolescents with asthma got their flu shot than did those without asthma during the 2011-2012 season (69% vs. 51%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). No differences were seen for the other common CMCs.
Nonetheless, the mean number of missed opportunities was significantly higher among unvaccinated adolescents with CMCs, compared with those without CMCs, for the first HPV vaccination, meningococcal vaccination, and influenza vaccination in both seasons measured (P less than .001 for all).
“Missed opportunities for the third HPV vaccine dose or Tdap did not differ by CMC status,” Dr. Hofstetter and her associates said.
Read more in the American Journal of Preventive Medicine (2017 Nov;53[5]:680-8).
said Annika M. Hofstetter, MD, PhD, of Columbia University, New York, and her associates.
The National Health Interview Survey on Disability in 1994-1995 estimated that chronic conditions of any type affected 15%-18% of U.S. children and adolescents. The Advisory Committee on Immunization Practices recommends that all adolescents, whether or not they have chronic medical condition, be vaccinated with human papillomavirus (HPV), Tdap, meningococcal, and flu vaccines.
Fewer adolescents with CMCs had received one more doses of HPV (81%), than did those without CMCs (85%; P less than .01). Fewer adolescents with epilepsy (63%), mental retardation (58%), cerebral palsy (54%), and autism spectrum disorder (46%) had started HPV vaccination, compared with those without each of these conditions (84%; all comparisons, P less than .001). No differences were seen for asthma or congenital heart disease, the investigators said.
More adolescents with CMCs had gotten their flu shot than did those without CMCs during the 2011-2012 season (67% vs. 50%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). More adolescents with asthma got their flu shot than did those without asthma during the 2011-2012 season (69% vs. 51%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). No differences were seen for the other common CMCs.
Nonetheless, the mean number of missed opportunities was significantly higher among unvaccinated adolescents with CMCs, compared with those without CMCs, for the first HPV vaccination, meningococcal vaccination, and influenza vaccination in both seasons measured (P less than .001 for all).
“Missed opportunities for the third HPV vaccine dose or Tdap did not differ by CMC status,” Dr. Hofstetter and her associates said.
Read more in the American Journal of Preventive Medicine (2017 Nov;53[5]:680-8).
said Annika M. Hofstetter, MD, PhD, of Columbia University, New York, and her associates.
The National Health Interview Survey on Disability in 1994-1995 estimated that chronic conditions of any type affected 15%-18% of U.S. children and adolescents. The Advisory Committee on Immunization Practices recommends that all adolescents, whether or not they have chronic medical condition, be vaccinated with human papillomavirus (HPV), Tdap, meningococcal, and flu vaccines.
Fewer adolescents with CMCs had received one more doses of HPV (81%), than did those without CMCs (85%; P less than .01). Fewer adolescents with epilepsy (63%), mental retardation (58%), cerebral palsy (54%), and autism spectrum disorder (46%) had started HPV vaccination, compared with those without each of these conditions (84%; all comparisons, P less than .001). No differences were seen for asthma or congenital heart disease, the investigators said.
More adolescents with CMCs had gotten their flu shot than did those without CMCs during the 2011-2012 season (67% vs. 50%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). More adolescents with asthma got their flu shot than did those without asthma during the 2011-2012 season (69% vs. 51%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). No differences were seen for the other common CMCs.
Nonetheless, the mean number of missed opportunities was significantly higher among unvaccinated adolescents with CMCs, compared with those without CMCs, for the first HPV vaccination, meningococcal vaccination, and influenza vaccination in both seasons measured (P less than .001 for all).
“Missed opportunities for the third HPV vaccine dose or Tdap did not differ by CMC status,” Dr. Hofstetter and her associates said.
Read more in the American Journal of Preventive Medicine (2017 Nov;53[5]:680-8).
FROM THE AMERICAN JOURNAL OF PREVENTIVE MEDICINE
System helps predict RFS, OS in BCP-ALL

Researchers say they have developed a more accurate risk scoring system for children with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) who are typically thought to have standard- or medium-risk disease.
The scoring system includes 3 factors associated with higher-risk BCP-ALL—the presence of high-risk ALL gene microdeletions, having minimal residual disease (MRD) greater than 5 x 10-5 at day 33, and being high-risk according to National Cancer Institute (NCI) classification.
The researchers found that children with 2 or more of these characteristics were most likely to relapse or die within 7 years of treatment initiation.
On the other hand, children without any of the 3 characteristics had high rates of relapse-free survival (RFS) and overall survival (OS).
Rosemary Sutton, PhD, of Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues devised this risk scoring system and described it in the British Journal of Haematology.
The researchers created their system with the help of data from 475 patients (ages 1 to 18) who had BCP-ALL and were considered non-high-risk. The patients were enrolled on the ANZCHOG ALL8 trial.
Dr Sutton and her colleagues noted that children with standard- or medium-risk BCP-ALL typically receive less intensive treatment than children with high-risk BCP-ALL. However, some of the standard- and medium-risk patients do relapse.
“For the standard- to medium-risk group, we needed more information to get a better handle on the biology of the child’s cancer to better determine their risk,” Dr Sutton said. “So we supplemented MRD results with 2 other pieces of patient information—the presence or absence of specific gene microdeletions and a score called the NCI risk, based on age and white blood cell count.”
“We tested for microdeletions in 9 genes involved in leukemia and found that 2 of the genes—IKZF1 and P2RY8-CRLF2—were important predictors of relapse.”
The researchers combined patients with IKZF1 intragenic deletions, P2RY8-CRLF2 gene fusion, or both into a “high-risk deletion group.”
And the team based the scoring system on 3 factors—the high-risk deletion group, MRD >5 x 10-5 at day 33, and high risk according to NCI risk classification. Patients received 1 point for each of these factors.
The RFS rate was 93% for patients with a score of 0, 78% for those with a score of 1, and 49% for patients with a score of 2 or 3. The OS rate was 99%, 91%, and 71%, respectively.
The researchers said their scoring system provided greater discrimination than MRD-based risk stratification into a standard-risk group—which had an RFS of 89% and an OS of 96%—and a medium-risk group—which had an RFS of 79% and an OS of 91%.
Study author Toby Trahair, MBBS, PhD, of Sydney Children’s Hospital in Randwick, New South Wales, said this scoring system could make a big difference to the success of BCP-ALL treatment.
“We are always trying to improve how we diagnose and treat children with this most common childhood cancer,” Dr Trahair said. “This risk score will mean doctors can fine tune a child’s risk category and so fine tune their treatment. It will mean more kids can conquer this horrible disease, which, only 50 years ago, had survival rates of close to 0.” ![]()
Researchers say they have developed a more accurate risk scoring system for children with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) who are typically thought to have standard- or medium-risk disease.
The scoring system includes 3 factors associated with higher-risk BCP-ALL—the presence of high-risk ALL gene microdeletions, having minimal residual disease (MRD) greater than 5 x 10-5 at day 33, and being high-risk according to National Cancer Institute (NCI) classification.
The researchers found that children with 2 or more of these characteristics were most likely to relapse or die within 7 years of treatment initiation.
On the other hand, children without any of the 3 characteristics had high rates of relapse-free survival (RFS) and overall survival (OS).
Rosemary Sutton, PhD, of Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues devised this risk scoring system and described it in the British Journal of Haematology.
The researchers created their system with the help of data from 475 patients (ages 1 to 18) who had BCP-ALL and were considered non-high-risk. The patients were enrolled on the ANZCHOG ALL8 trial.
Dr Sutton and her colleagues noted that children with standard- or medium-risk BCP-ALL typically receive less intensive treatment than children with high-risk BCP-ALL. However, some of the standard- and medium-risk patients do relapse.
“For the standard- to medium-risk group, we needed more information to get a better handle on the biology of the child’s cancer to better determine their risk,” Dr Sutton said. “So we supplemented MRD results with 2 other pieces of patient information—the presence or absence of specific gene microdeletions and a score called the NCI risk, based on age and white blood cell count.”
“We tested for microdeletions in 9 genes involved in leukemia and found that 2 of the genes—IKZF1 and P2RY8-CRLF2—were important predictors of relapse.”
The researchers combined patients with IKZF1 intragenic deletions, P2RY8-CRLF2 gene fusion, or both into a “high-risk deletion group.”
And the team based the scoring system on 3 factors—the high-risk deletion group, MRD >5 x 10-5 at day 33, and high risk according to NCI risk classification. Patients received 1 point for each of these factors.
The RFS rate was 93% for patients with a score of 0, 78% for those with a score of 1, and 49% for patients with a score of 2 or 3. The OS rate was 99%, 91%, and 71%, respectively.
The researchers said their scoring system provided greater discrimination than MRD-based risk stratification into a standard-risk group—which had an RFS of 89% and an OS of 96%—and a medium-risk group—which had an RFS of 79% and an OS of 91%.
Study author Toby Trahair, MBBS, PhD, of Sydney Children’s Hospital in Randwick, New South Wales, said this scoring system could make a big difference to the success of BCP-ALL treatment.
“We are always trying to improve how we diagnose and treat children with this most common childhood cancer,” Dr Trahair said. “This risk score will mean doctors can fine tune a child’s risk category and so fine tune their treatment. It will mean more kids can conquer this horrible disease, which, only 50 years ago, had survival rates of close to 0.” ![]()
Researchers say they have developed a more accurate risk scoring system for children with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) who are typically thought to have standard- or medium-risk disease.
The scoring system includes 3 factors associated with higher-risk BCP-ALL—the presence of high-risk ALL gene microdeletions, having minimal residual disease (MRD) greater than 5 x 10-5 at day 33, and being high-risk according to National Cancer Institute (NCI) classification.
The researchers found that children with 2 or more of these characteristics were most likely to relapse or die within 7 years of treatment initiation.
On the other hand, children without any of the 3 characteristics had high rates of relapse-free survival (RFS) and overall survival (OS).
Rosemary Sutton, PhD, of Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues devised this risk scoring system and described it in the British Journal of Haematology.
The researchers created their system with the help of data from 475 patients (ages 1 to 18) who had BCP-ALL and were considered non-high-risk. The patients were enrolled on the ANZCHOG ALL8 trial.
Dr Sutton and her colleagues noted that children with standard- or medium-risk BCP-ALL typically receive less intensive treatment than children with high-risk BCP-ALL. However, some of the standard- and medium-risk patients do relapse.
“For the standard- to medium-risk group, we needed more information to get a better handle on the biology of the child’s cancer to better determine their risk,” Dr Sutton said. “So we supplemented MRD results with 2 other pieces of patient information—the presence or absence of specific gene microdeletions and a score called the NCI risk, based on age and white blood cell count.”
“We tested for microdeletions in 9 genes involved in leukemia and found that 2 of the genes—IKZF1 and P2RY8-CRLF2—were important predictors of relapse.”
The researchers combined patients with IKZF1 intragenic deletions, P2RY8-CRLF2 gene fusion, or both into a “high-risk deletion group.”
And the team based the scoring system on 3 factors—the high-risk deletion group, MRD >5 x 10-5 at day 33, and high risk according to NCI risk classification. Patients received 1 point for each of these factors.
The RFS rate was 93% for patients with a score of 0, 78% for those with a score of 1, and 49% for patients with a score of 2 or 3. The OS rate was 99%, 91%, and 71%, respectively.
The researchers said their scoring system provided greater discrimination than MRD-based risk stratification into a standard-risk group—which had an RFS of 89% and an OS of 96%—and a medium-risk group—which had an RFS of 79% and an OS of 91%.
Study author Toby Trahair, MBBS, PhD, of Sydney Children’s Hospital in Randwick, New South Wales, said this scoring system could make a big difference to the success of BCP-ALL treatment.
“We are always trying to improve how we diagnose and treat children with this most common childhood cancer,” Dr Trahair said. “This risk score will mean doctors can fine tune a child’s risk category and so fine tune their treatment. It will mean more kids can conquer this horrible disease, which, only 50 years ago, had survival rates of close to 0.” ![]()
Hippocampal features may predispose children with febrile status epilepticus to poorer memory
WASHINGTON – Children with febrile status epilepticus (FSE) may be at risk for memory impairment when abnormal hippocampal development or acute injury is also present, according to research presented at the annual meeting of the American Epilepsy Society.
This analysis of patients enrolled in the FEBSTAT study presents some of the first prospective data available regarding significant risk factors for cognitive dysfunction in children with FSE.
“Overall, children with FSE have generally intact memory function and generally intact IQ,” said Erica Weiss, PhD, a neurology instructor at the Albert Einstein College of Medicine, New York. “However, children with acute T2 [hyperintensities on MRI] and kids who have hippocampal malrotation [HIMAL] tend to have weaker memory scores.”
The investigators conducted a prospective study of 113 children with FSE using data gathered from five medical centers across the United States between June 2003 and March 2010.
Children included in the study were followed with serial MRIs and electroencephalograms for more than 5 years after their having FSE; during this time, their verbal, visual, and screening memory abilities were tested using the Wide Range Assessment of Memory and Learning, Second Edition, (WRAML2) test.
Patients had an average age of 15.5 months at time of FSE. Of the children in the study, 46% were female, and 46% were non-white.
Overall, mean scores at baseline on the WRAML2 were significantly lower for children with acute hippocampal injury shown on T2 hyperintensities or HIMAL than they were for children with a normal MRI scan. On individual memory functions of the WRAML2, mean scores at baseline for children with acute T2 hyperintensities were lower than they were for those with a normal MRI on the verbal index (79 vs. 102.3), visual index (81 vs. 93.7), and screening memory index (76 vs. 97.7). Children with HIMAL at baseline also had lower scores on those indexes (94.9 for verbal memory, 82.5 for visual memory, and 97 for screening memory) than did children with a normal MRI.
The differences were statistically significant for lower verbal memory and screening memory scores in patients with acute T2 hyperintensities and for lower visual memory scores in patients with HIMAL. The differences trended toward statistical significance for lower visual memory scores in children with acute T2 hyperintensities and for lower verbal memory scores in children with focal FSE seizures.
The researchers found no significant differences in memory task performances when stratifying for age at time of FSE, duration of FSE, or patients’ sex, according to Dr. Weiss.
With this initial connection uncovered, Dr. Weiss and her colleagues are looking to dive deeper into different aspects of hippocampal properties and FSE.
“We’re looking into the relationship between hippocampus size and memory performances, as well as continue to track these studies,” Dr. Weiss said in an interview. “Another factor to consider when you talk about memory is attention, and we have looked into it a bit, but we need more information.”
This study was funded by a grant from the National Institute of Neurological Disorders and Stroke. The presenters reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
On Twitter @eaztweets
WASHINGTON – Children with febrile status epilepticus (FSE) may be at risk for memory impairment when abnormal hippocampal development or acute injury is also present, according to research presented at the annual meeting of the American Epilepsy Society.
This analysis of patients enrolled in the FEBSTAT study presents some of the first prospective data available regarding significant risk factors for cognitive dysfunction in children with FSE.
“Overall, children with FSE have generally intact memory function and generally intact IQ,” said Erica Weiss, PhD, a neurology instructor at the Albert Einstein College of Medicine, New York. “However, children with acute T2 [hyperintensities on MRI] and kids who have hippocampal malrotation [HIMAL] tend to have weaker memory scores.”
The investigators conducted a prospective study of 113 children with FSE using data gathered from five medical centers across the United States between June 2003 and March 2010.
Children included in the study were followed with serial MRIs and electroencephalograms for more than 5 years after their having FSE; during this time, their verbal, visual, and screening memory abilities were tested using the Wide Range Assessment of Memory and Learning, Second Edition, (WRAML2) test.
Patients had an average age of 15.5 months at time of FSE. Of the children in the study, 46% were female, and 46% were non-white.
Overall, mean scores at baseline on the WRAML2 were significantly lower for children with acute hippocampal injury shown on T2 hyperintensities or HIMAL than they were for children with a normal MRI scan. On individual memory functions of the WRAML2, mean scores at baseline for children with acute T2 hyperintensities were lower than they were for those with a normal MRI on the verbal index (79 vs. 102.3), visual index (81 vs. 93.7), and screening memory index (76 vs. 97.7). Children with HIMAL at baseline also had lower scores on those indexes (94.9 for verbal memory, 82.5 for visual memory, and 97 for screening memory) than did children with a normal MRI.
The differences were statistically significant for lower verbal memory and screening memory scores in patients with acute T2 hyperintensities and for lower visual memory scores in patients with HIMAL. The differences trended toward statistical significance for lower visual memory scores in children with acute T2 hyperintensities and for lower verbal memory scores in children with focal FSE seizures.
The researchers found no significant differences in memory task performances when stratifying for age at time of FSE, duration of FSE, or patients’ sex, according to Dr. Weiss.
With this initial connection uncovered, Dr. Weiss and her colleagues are looking to dive deeper into different aspects of hippocampal properties and FSE.
“We’re looking into the relationship between hippocampus size and memory performances, as well as continue to track these studies,” Dr. Weiss said in an interview. “Another factor to consider when you talk about memory is attention, and we have looked into it a bit, but we need more information.”
This study was funded by a grant from the National Institute of Neurological Disorders and Stroke. The presenters reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
On Twitter @eaztweets
WASHINGTON – Children with febrile status epilepticus (FSE) may be at risk for memory impairment when abnormal hippocampal development or acute injury is also present, according to research presented at the annual meeting of the American Epilepsy Society.
This analysis of patients enrolled in the FEBSTAT study presents some of the first prospective data available regarding significant risk factors for cognitive dysfunction in children with FSE.
“Overall, children with FSE have generally intact memory function and generally intact IQ,” said Erica Weiss, PhD, a neurology instructor at the Albert Einstein College of Medicine, New York. “However, children with acute T2 [hyperintensities on MRI] and kids who have hippocampal malrotation [HIMAL] tend to have weaker memory scores.”
The investigators conducted a prospective study of 113 children with FSE using data gathered from five medical centers across the United States between June 2003 and March 2010.
Children included in the study were followed with serial MRIs and electroencephalograms for more than 5 years after their having FSE; during this time, their verbal, visual, and screening memory abilities were tested using the Wide Range Assessment of Memory and Learning, Second Edition, (WRAML2) test.
Patients had an average age of 15.5 months at time of FSE. Of the children in the study, 46% were female, and 46% were non-white.
Overall, mean scores at baseline on the WRAML2 were significantly lower for children with acute hippocampal injury shown on T2 hyperintensities or HIMAL than they were for children with a normal MRI scan. On individual memory functions of the WRAML2, mean scores at baseline for children with acute T2 hyperintensities were lower than they were for those with a normal MRI on the verbal index (79 vs. 102.3), visual index (81 vs. 93.7), and screening memory index (76 vs. 97.7). Children with HIMAL at baseline also had lower scores on those indexes (94.9 for verbal memory, 82.5 for visual memory, and 97 for screening memory) than did children with a normal MRI.
The differences were statistically significant for lower verbal memory and screening memory scores in patients with acute T2 hyperintensities and for lower visual memory scores in patients with HIMAL. The differences trended toward statistical significance for lower visual memory scores in children with acute T2 hyperintensities and for lower verbal memory scores in children with focal FSE seizures.
The researchers found no significant differences in memory task performances when stratifying for age at time of FSE, duration of FSE, or patients’ sex, according to Dr. Weiss.
With this initial connection uncovered, Dr. Weiss and her colleagues are looking to dive deeper into different aspects of hippocampal properties and FSE.
“We’re looking into the relationship between hippocampus size and memory performances, as well as continue to track these studies,” Dr. Weiss said in an interview. “Another factor to consider when you talk about memory is attention, and we have looked into it a bit, but we need more information.”
This study was funded by a grant from the National Institute of Neurological Disorders and Stroke. The presenters reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
On Twitter @eaztweets
AT AES 2017
Key clinical point:
Major finding: On individual memory functions of the Wide Range Assessment of Memory and Learning, Second Edition, test, mean scores at baseline for children with acute T2 hyperintensities or hippocampal malrotation were lower than they were for those with a normal MRI on the verbal index (79 and 94.9, respectively, vs. 102.3), the visual index (81 and 82.5 vs. 93.7), and the screening memory (76 and 97 vs. 97.7) index.
Data source: Prospective study of 113 children, the data for which was gathered from five medical centers across the United States between 2003 and 2010.
Disclosures: The study was funded by a grant from the National Institute of Neurological Disorders and Stroke. The presenters reported no relevant financial disclosures.
Genital Ulcers and Swelling in an Adolescent Girl
The Diagnosis: Epstein-Barr Virus
Physical examination revealed bilateral 1-cm ulcerated lesions on the labia minora with vulvar edema (Figure). She had a palpable liver edge but no splenomegaly, oral ulcers or lesions, conjunctivitis or scleral icterus, or cervical or inguinal lymphadenopathy. A detailed genitourinary examination was performed under anesthesia, but the hymen was not commented on. Inflammatory markers were elevated with a C-reactive protein level of 16.4 mg/L (reference range, 0.08-3.1 mg/mL), erythrocyte sedimentation rate of39 mm/h (reference range, 0-20 mm/h), white blood cell count of 7.1×109/L (reference range, 4.5-11.0×109/L) with 57% neutrophils and 30% lymphocytes, an alanine aminotransferase level of 41 U/L (reference range, 10-40 U/L), and an aspartate aminotransferase level of 126 U/L (reference range, 10-30 U/L).
Bacterial and fungal cultures of vulvar tissue were negative as well as blood and urine cultures. Serological tests for herpes simplex virus (HSV), syphilis, and cytomegalovirus were negative, and urine testing for gonorrhea and chlamydia were negative. Serologies for Epstein-Barr virus (EBV) all were strongly positive with an EBV viral capsid antigen (VCA) IgM greater than 160 U/mL, early antigen IgG of 68 U/mL, and EBV VCA IgG of 456 U/mL. Two years after the initial presentation, repeat EBV serologies were obtained, showing a strongly positive EBV VCA IgG (>8.0 antibody index; reference range, 0-0.8), and a negative EBV VCA IgM.
Infectious etiologies of genital ulcers in a sexually active female include HSV, syphilis, lymphogranuloma venereum, and chancroid. Herpes simplex virus often is the assumed etiology of genital ulcers, especially in sexually active patients, and misdiagnosis in the setting of negative HSV testing may be high. Less common infectious causes such as mumps and cytomegalovirus also have been reported.1,2 Lichen planus and lichen sclerosus are noninfectious inflammatory causes, both of which may involve and be limited to the genitals. Autoimmune disorders include Crohn disease and Behçet disease, and vulvar ulcers with an eschar, consistent with aphthous major or complex apotheosis, has been used to describe patients with severe recurrent oral and genital ulcerations without other systemic manifestations of Behçet disease.3
Genital ulcers are an uncommon manifestation of EBV infection. The formation of genital ulcers in EBV infection has been hypothesized to be due to immune complex formation during the acute phase that becomes activated in the vasculature, leading to microthrombosis and eventually necrosis of the tissue.4 The mode of transmission for EBV-related acute genital ulcers has been postulated to be hematogenous spread in lymphocytes or EBV shedding in the urine with subsequent transfer to the genital mucosa.5
Epstein-Barr virus-related acute genital ulcers are self-limiting. The average healing time for the ulcers is 14 to 18 days.6,7 Antivirals are ineffective in treating this condition; however, supportive treatment with systemic glucocorticoids for associated swelling and pain medications could be considered. Our patient was treated symptomatically. Two weeks after debridement, granulation tissue was noted at the site and her pain and discomfort had resolved. This case illustrates an uncommon manifestation of EBV in a sexually inactive adolescent and is a reminder for the dermatologist of the diverse spectrum of illness caused by this common virus.
- Chanal J, Carlotti A, Laude H, et al. Lipschütz genital ulceration associated with mumps. Dermatology. 2010;221:292-295.
- Martin JM, Godoy R, Calduch L, et al. Lipschütz acute vulval ulcers associated with primary cytomegalovirus infection. Pediatr Dermatol. 2008;25:113-115.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Sárdy M, Wollenberg A, Niedermeier A, et al. Genital ulcers associated with Epstein-Barr virus infection (ulcus vulvae acutum). Acta Derm Venereol. 2011;91:55-59.
- Di Lernia V, Mansouri Y. Epstein-Barr virus and skin manifestations in childhood. Int J Dermatol. 2013;52:1177-1184.
- Halvorsen JA, Brevig T, Aas T, et al. Genital ulcers as initial manifestation of Epstein-Barr virus infection: two new cases and a review of the literature. Acta Derm Venereol. 2006;86:439-442.
- Jerdan K, Aronson I, Hernandez C, et al. Genital ulcers associated with Epstein-Barr virus. Cutis. 2013;91:273-276.
The Diagnosis: Epstein-Barr Virus
Physical examination revealed bilateral 1-cm ulcerated lesions on the labia minora with vulvar edema (Figure). She had a palpable liver edge but no splenomegaly, oral ulcers or lesions, conjunctivitis or scleral icterus, or cervical or inguinal lymphadenopathy. A detailed genitourinary examination was performed under anesthesia, but the hymen was not commented on. Inflammatory markers were elevated with a C-reactive protein level of 16.4 mg/L (reference range, 0.08-3.1 mg/mL), erythrocyte sedimentation rate of39 mm/h (reference range, 0-20 mm/h), white blood cell count of 7.1×109/L (reference range, 4.5-11.0×109/L) with 57% neutrophils and 30% lymphocytes, an alanine aminotransferase level of 41 U/L (reference range, 10-40 U/L), and an aspartate aminotransferase level of 126 U/L (reference range, 10-30 U/L).
Bacterial and fungal cultures of vulvar tissue were negative as well as blood and urine cultures. Serological tests for herpes simplex virus (HSV), syphilis, and cytomegalovirus were negative, and urine testing for gonorrhea and chlamydia were negative. Serologies for Epstein-Barr virus (EBV) all were strongly positive with an EBV viral capsid antigen (VCA) IgM greater than 160 U/mL, early antigen IgG of 68 U/mL, and EBV VCA IgG of 456 U/mL. Two years after the initial presentation, repeat EBV serologies were obtained, showing a strongly positive EBV VCA IgG (>8.0 antibody index; reference range, 0-0.8), and a negative EBV VCA IgM.
Infectious etiologies of genital ulcers in a sexually active female include HSV, syphilis, lymphogranuloma venereum, and chancroid. Herpes simplex virus often is the assumed etiology of genital ulcers, especially in sexually active patients, and misdiagnosis in the setting of negative HSV testing may be high. Less common infectious causes such as mumps and cytomegalovirus also have been reported.1,2 Lichen planus and lichen sclerosus are noninfectious inflammatory causes, both of which may involve and be limited to the genitals. Autoimmune disorders include Crohn disease and Behçet disease, and vulvar ulcers with an eschar, consistent with aphthous major or complex apotheosis, has been used to describe patients with severe recurrent oral and genital ulcerations without other systemic manifestations of Behçet disease.3
Genital ulcers are an uncommon manifestation of EBV infection. The formation of genital ulcers in EBV infection has been hypothesized to be due to immune complex formation during the acute phase that becomes activated in the vasculature, leading to microthrombosis and eventually necrosis of the tissue.4 The mode of transmission for EBV-related acute genital ulcers has been postulated to be hematogenous spread in lymphocytes or EBV shedding in the urine with subsequent transfer to the genital mucosa.5
Epstein-Barr virus-related acute genital ulcers are self-limiting. The average healing time for the ulcers is 14 to 18 days.6,7 Antivirals are ineffective in treating this condition; however, supportive treatment with systemic glucocorticoids for associated swelling and pain medications could be considered. Our patient was treated symptomatically. Two weeks after debridement, granulation tissue was noted at the site and her pain and discomfort had resolved. This case illustrates an uncommon manifestation of EBV in a sexually inactive adolescent and is a reminder for the dermatologist of the diverse spectrum of illness caused by this common virus.
The Diagnosis: Epstein-Barr Virus
Physical examination revealed bilateral 1-cm ulcerated lesions on the labia minora with vulvar edema (Figure). She had a palpable liver edge but no splenomegaly, oral ulcers or lesions, conjunctivitis or scleral icterus, or cervical or inguinal lymphadenopathy. A detailed genitourinary examination was performed under anesthesia, but the hymen was not commented on. Inflammatory markers were elevated with a C-reactive protein level of 16.4 mg/L (reference range, 0.08-3.1 mg/mL), erythrocyte sedimentation rate of39 mm/h (reference range, 0-20 mm/h), white blood cell count of 7.1×109/L (reference range, 4.5-11.0×109/L) with 57% neutrophils and 30% lymphocytes, an alanine aminotransferase level of 41 U/L (reference range, 10-40 U/L), and an aspartate aminotransferase level of 126 U/L (reference range, 10-30 U/L).
Bacterial and fungal cultures of vulvar tissue were negative as well as blood and urine cultures. Serological tests for herpes simplex virus (HSV), syphilis, and cytomegalovirus were negative, and urine testing for gonorrhea and chlamydia were negative. Serologies for Epstein-Barr virus (EBV) all were strongly positive with an EBV viral capsid antigen (VCA) IgM greater than 160 U/mL, early antigen IgG of 68 U/mL, and EBV VCA IgG of 456 U/mL. Two years after the initial presentation, repeat EBV serologies were obtained, showing a strongly positive EBV VCA IgG (>8.0 antibody index; reference range, 0-0.8), and a negative EBV VCA IgM.
Infectious etiologies of genital ulcers in a sexually active female include HSV, syphilis, lymphogranuloma venereum, and chancroid. Herpes simplex virus often is the assumed etiology of genital ulcers, especially in sexually active patients, and misdiagnosis in the setting of negative HSV testing may be high. Less common infectious causes such as mumps and cytomegalovirus also have been reported.1,2 Lichen planus and lichen sclerosus are noninfectious inflammatory causes, both of which may involve and be limited to the genitals. Autoimmune disorders include Crohn disease and Behçet disease, and vulvar ulcers with an eschar, consistent with aphthous major or complex apotheosis, has been used to describe patients with severe recurrent oral and genital ulcerations without other systemic manifestations of Behçet disease.3
Genital ulcers are an uncommon manifestation of EBV infection. The formation of genital ulcers in EBV infection has been hypothesized to be due to immune complex formation during the acute phase that becomes activated in the vasculature, leading to microthrombosis and eventually necrosis of the tissue.4 The mode of transmission for EBV-related acute genital ulcers has been postulated to be hematogenous spread in lymphocytes or EBV shedding in the urine with subsequent transfer to the genital mucosa.5
Epstein-Barr virus-related acute genital ulcers are self-limiting. The average healing time for the ulcers is 14 to 18 days.6,7 Antivirals are ineffective in treating this condition; however, supportive treatment with systemic glucocorticoids for associated swelling and pain medications could be considered. Our patient was treated symptomatically. Two weeks after debridement, granulation tissue was noted at the site and her pain and discomfort had resolved. This case illustrates an uncommon manifestation of EBV in a sexually inactive adolescent and is a reminder for the dermatologist of the diverse spectrum of illness caused by this common virus.
- Chanal J, Carlotti A, Laude H, et al. Lipschütz genital ulceration associated with mumps. Dermatology. 2010;221:292-295.
- Martin JM, Godoy R, Calduch L, et al. Lipschütz acute vulval ulcers associated with primary cytomegalovirus infection. Pediatr Dermatol. 2008;25:113-115.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Sárdy M, Wollenberg A, Niedermeier A, et al. Genital ulcers associated with Epstein-Barr virus infection (ulcus vulvae acutum). Acta Derm Venereol. 2011;91:55-59.
- Di Lernia V, Mansouri Y. Epstein-Barr virus and skin manifestations in childhood. Int J Dermatol. 2013;52:1177-1184.
- Halvorsen JA, Brevig T, Aas T, et al. Genital ulcers as initial manifestation of Epstein-Barr virus infection: two new cases and a review of the literature. Acta Derm Venereol. 2006;86:439-442.
- Jerdan K, Aronson I, Hernandez C, et al. Genital ulcers associated with Epstein-Barr virus. Cutis. 2013;91:273-276.
- Chanal J, Carlotti A, Laude H, et al. Lipschütz genital ulceration associated with mumps. Dermatology. 2010;221:292-295.
- Martin JM, Godoy R, Calduch L, et al. Lipschütz acute vulval ulcers associated with primary cytomegalovirus infection. Pediatr Dermatol. 2008;25:113-115.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Sárdy M, Wollenberg A, Niedermeier A, et al. Genital ulcers associated with Epstein-Barr virus infection (ulcus vulvae acutum). Acta Derm Venereol. 2011;91:55-59.
- Di Lernia V, Mansouri Y. Epstein-Barr virus and skin manifestations in childhood. Int J Dermatol. 2013;52:1177-1184.
- Halvorsen JA, Brevig T, Aas T, et al. Genital ulcers as initial manifestation of Epstein-Barr virus infection: two new cases and a review of the literature. Acta Derm Venereol. 2006;86:439-442.
- Jerdan K, Aronson I, Hernandez C, et al. Genital ulcers associated with Epstein-Barr virus. Cutis. 2013;91:273-276.
A 14-year-old previously healthy, postmenarcheal adolescent girl with a family history of thyroid disease and rheumatoid arthritis presented with vulvar pain and swelling. Vulvar pruritus was noted 6 days prior, which worsened and became associated with vulvar swelling, yellow vaginal discharge, difficulty walking, and a fever (temperature, 39.3.2 °C). Her condition did not improve after a course of cephalexin and trimethoprim-sulfamethoxazole. She denied being sexually active or exposing foreign objects or chemicals to the vaginal area.
Sjögren-Larsson Syndrome: Definitive Diagnosis on Magnetic Resonance Spectroscopy
Sjögren-Larsson syndrome (SLS) is a rare autosomal-recessive neurocutaneous disorder comprising a triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.1 The disorder was first described by Sjögren and Larsson2 in 1957. Early reports of SLS were mainly in white patients, with a particularly high prevalence of 8.3 cases per 100,000 individuals in the county of Västerbotten in Sweden.3 Reports of SLS in Asian and Indian populations are rare.4,5 We report a case of SLS in an Indian boy.
Case Report
A 12-year-old Indian boy born to nonconsanguineous parents after a full-term pregnancy with normal vaginal delivery presented with generalized dry scaly skin that had been present since 2 months of age. He had a history of delayed milestones (ie, facial recognition, sitting without support at 3 years of age), inability to walk, dysarthria, mental retardation). He had never attended school due to subnormal intellectual functioning. He had a single episode of a tonic-clonic seizure at 4 years of age but was not on any regular antiepileptic medication. There was a history of similar skin lesions in one male sibling of the patient and in 2 maternal uncles. None of them survived beyond early childhood, but detailed information regarding the cutaneous and neurologic manifestations in these family members was not available.
Cutaneous examination revealed lamellarlike ichthyosis on the dorsal aspects of the arms and legs (Figure 1A). Ichthyosis with lichenification was present on the neck, axillae, cubital and popliteal fossae, and abdomen (Figure 1B). The palms and soles showed keratoderma. Neurologic examination of the arms revealed mild rigidity and brisk reflexes. Examination of the legs showed marked rigidity, brisk knee jerks, ankle clonus, extensor plantar reflexes, flexion deformity with contractures, and scissor gait. A Goddard (Seguin) formboard test was performed and indicated a mental age of 4 years. The patient’s IQ was in the range of 25 to 30, indicating a severe degree of subnormality in intellectual functioning. The clinical presentation suggested a diagnosis of SLS.
A skin biopsy from the ichthyotic lesion showed hyperkeratosis, acanthosis, and papillomatosis with sparse superficial perivascular lymphocytic infiltrate, thus confirming the diagnosis of lamellar ichthyosis. Fundus examination was normal. Magnetic resonance imaging (MRI) of the brain revealed confluent symmetrical signal abnormalities along the body of the lateral ventricles, white matter in the perioccipital horn, and in deep white matter of centrum semiovale (Figure 2). Magnetic resonance spectroscopy revealed a narrow lipid peak at approximately 1.3 ppm in the region of signal abnormality (Figure 3). Thus, the diagnosis of SLS was confirmed. Measurement of fatty aldehyde dehydrogenase (FALDH) activity and genetic analysis were not performed due to unavailability.
The patient was treated with topical emollients for the ichthyosis. To reduce his dietary intake of long-chain fatty acids and increase the intake of omega-3 and omega-6 fatty acids, the patient’s parents were advised to use canola, mustard, and/or coconut oil for cooking for the patient, and skim milk was recommended instead of whole milk. Neurodevelopmental techniques in the form of stretching exercises were given to maintain his range of movements. Gutter splints were given to maintain the knees in extension for physiological standing and to prevent osteoporosis. Subsequently, the patient also underwent a multilevel soft-tissue release (hip and knee joints) to relieve the contractures. These measures resulted in considerable improvement and the patient was able to walk with support.
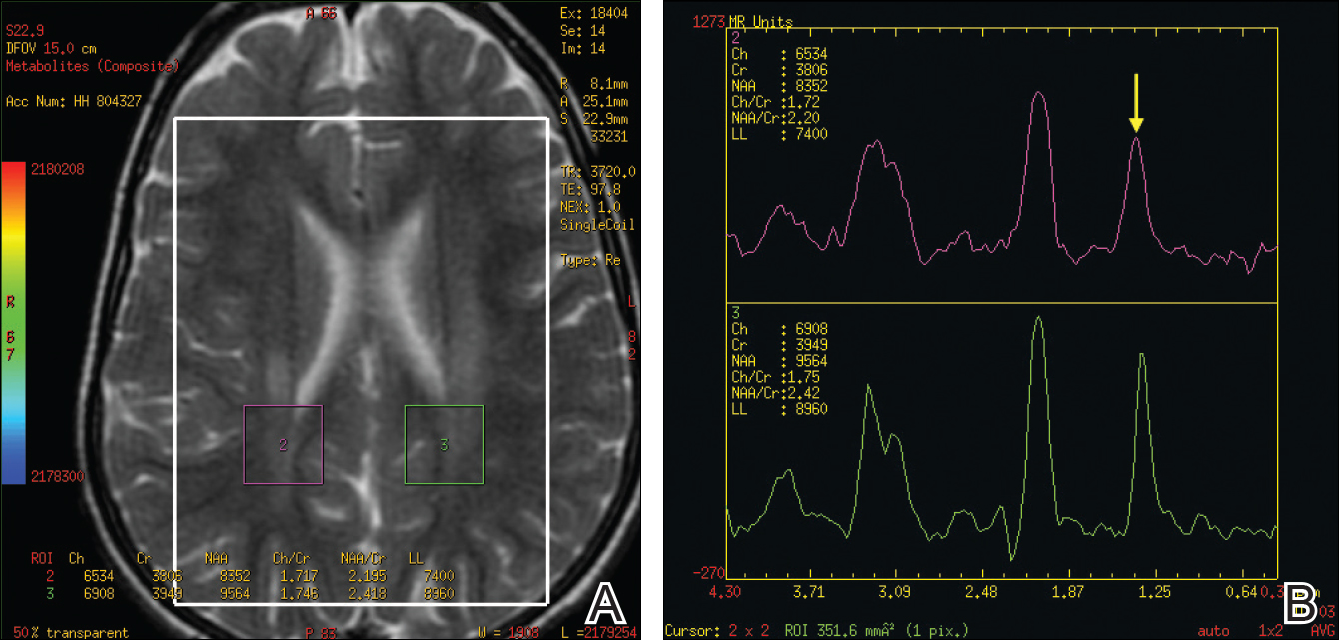
Comment
Presentation
The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations
Sjögren-Larsson syndrome is caused by mutation in the aldehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
Lipid Metabolism
Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.

Histopathology
The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings
Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment
Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.
- Judge MR, McLean WHI, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, eds. Rook’s Textbook of Dermatology. 7th ed. West Sussex, United Kingdom: Wiley & Sons; 2004:34.37-34.39.
- Sjögren T, Larsson T. Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1-113.
- Jagell S, Gustavson KH, Holmgren G. Sjögren-Larsson syndrome in Sweden. a clinical, genetic and epidemiological study. Clin Genet. 1981;19:233-256.
- Sood M, Trehan A, Dinakaran J, et al. Sjögren-Larsson syndrome. Indian J Pediatr. 2002;69:193-194.
- Uppal M, Srinivas CR, Thowfeeq KT. Sjögren-Larsson syndrome: report of two cases. Indian J Dermatol Venereol Leprol. 2004;70:110-111.
- Rizzo WB. Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1-9.
- Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124(pt 7):1426-1437.
- Willemsen MA, Cruysberg JR, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. Am J Ophthalmol. 2000;130:782-789.
- Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Arch Neurol. 2006;63:278-280.
- Rizzo WB, Craft DA, Somer T, et al. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410-419.
- Rizzo WB, S’Aulis D, Jennings MA, et al. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443-451.
- Rizzo WB. The role of fatty aldehyde dehydrogenase in epidermal structure and function. Dermatoendocrinol. 2011;2:91-99.
- Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18:338-341.
- Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Larsson’s Syndrome. Am J Neuroradiol. 1999;20:1671-1673.
- Willemsen MA, van der Graf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
- Kaminaga T, Mano T, Ono J, et al. Proton magnetic resonance spectroscopy of Sjögren-Larsson Syndrome. Magn Reson Med. 2001;45:1112-1115.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870-875.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711-717.
- Pirgon O, Aydin K, Atabek ME. Proton magnetic resonance spectroscopy findings and clinical effects of montelukast sodium in a case with Sjögren-Larsson syndrome. J Child Neurol. 2006;21:1092-1095.
- Gloerich J, Ijlst L, Wanders RJ, et al. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome Mol Genet Metab. 2006;89:111-115.
- Auada MP, Taube MB, Collares EF, et al. Sjögren-Larsson syndrome: biochemical defects and follow up in three cases. Eur J Dermatol. 2002;12:263-266.
- Taube B, Billeaud C, Labreze C, et al. Sjögren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology. 1999;198:340-345.
- Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opin Orphan Drugs. 2016;4:395-406.
- Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjögren-Larsson syndrome. Gene Ther. 2006;13:1021-1026.
Sjögren-Larsson syndrome (SLS) is a rare autosomal-recessive neurocutaneous disorder comprising a triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.1 The disorder was first described by Sjögren and Larsson2 in 1957. Early reports of SLS were mainly in white patients, with a particularly high prevalence of 8.3 cases per 100,000 individuals in the county of Västerbotten in Sweden.3 Reports of SLS in Asian and Indian populations are rare.4,5 We report a case of SLS in an Indian boy.
Case Report
A 12-year-old Indian boy born to nonconsanguineous parents after a full-term pregnancy with normal vaginal delivery presented with generalized dry scaly skin that had been present since 2 months of age. He had a history of delayed milestones (ie, facial recognition, sitting without support at 3 years of age), inability to walk, dysarthria, mental retardation). He had never attended school due to subnormal intellectual functioning. He had a single episode of a tonic-clonic seizure at 4 years of age but was not on any regular antiepileptic medication. There was a history of similar skin lesions in one male sibling of the patient and in 2 maternal uncles. None of them survived beyond early childhood, but detailed information regarding the cutaneous and neurologic manifestations in these family members was not available.
Cutaneous examination revealed lamellarlike ichthyosis on the dorsal aspects of the arms and legs (Figure 1A). Ichthyosis with lichenification was present on the neck, axillae, cubital and popliteal fossae, and abdomen (Figure 1B). The palms and soles showed keratoderma. Neurologic examination of the arms revealed mild rigidity and brisk reflexes. Examination of the legs showed marked rigidity, brisk knee jerks, ankle clonus, extensor plantar reflexes, flexion deformity with contractures, and scissor gait. A Goddard (Seguin) formboard test was performed and indicated a mental age of 4 years. The patient’s IQ was in the range of 25 to 30, indicating a severe degree of subnormality in intellectual functioning. The clinical presentation suggested a diagnosis of SLS.
A skin biopsy from the ichthyotic lesion showed hyperkeratosis, acanthosis, and papillomatosis with sparse superficial perivascular lymphocytic infiltrate, thus confirming the diagnosis of lamellar ichthyosis. Fundus examination was normal. Magnetic resonance imaging (MRI) of the brain revealed confluent symmetrical signal abnormalities along the body of the lateral ventricles, white matter in the perioccipital horn, and in deep white matter of centrum semiovale (Figure 2). Magnetic resonance spectroscopy revealed a narrow lipid peak at approximately 1.3 ppm in the region of signal abnormality (Figure 3). Thus, the diagnosis of SLS was confirmed. Measurement of fatty aldehyde dehydrogenase (FALDH) activity and genetic analysis were not performed due to unavailability.
The patient was treated with topical emollients for the ichthyosis. To reduce his dietary intake of long-chain fatty acids and increase the intake of omega-3 and omega-6 fatty acids, the patient’s parents were advised to use canola, mustard, and/or coconut oil for cooking for the patient, and skim milk was recommended instead of whole milk. Neurodevelopmental techniques in the form of stretching exercises were given to maintain his range of movements. Gutter splints were given to maintain the knees in extension for physiological standing and to prevent osteoporosis. Subsequently, the patient also underwent a multilevel soft-tissue release (hip and knee joints) to relieve the contractures. These measures resulted in considerable improvement and the patient was able to walk with support.
Comment
Presentation
The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations
Sjögren-Larsson syndrome is caused by mutation in the aldehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
Lipid Metabolism
Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.
Histopathology
The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings
Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment
Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.
Sjögren-Larsson syndrome (SLS) is a rare autosomal-recessive neurocutaneous disorder comprising a triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.1 The disorder was first described by Sjögren and Larsson2 in 1957. Early reports of SLS were mainly in white patients, with a particularly high prevalence of 8.3 cases per 100,000 individuals in the county of Västerbotten in Sweden.3 Reports of SLS in Asian and Indian populations are rare.4,5 We report a case of SLS in an Indian boy.
Case Report
A 12-year-old Indian boy born to nonconsanguineous parents after a full-term pregnancy with normal vaginal delivery presented with generalized dry scaly skin that had been present since 2 months of age. He had a history of delayed milestones (ie, facial recognition, sitting without support at 3 years of age), inability to walk, dysarthria, mental retardation). He had never attended school due to subnormal intellectual functioning. He had a single episode of a tonic-clonic seizure at 4 years of age but was not on any regular antiepileptic medication. There was a history of similar skin lesions in one male sibling of the patient and in 2 maternal uncles. None of them survived beyond early childhood, but detailed information regarding the cutaneous and neurologic manifestations in these family members was not available.
Cutaneous examination revealed lamellarlike ichthyosis on the dorsal aspects of the arms and legs (Figure 1A). Ichthyosis with lichenification was present on the neck, axillae, cubital and popliteal fossae, and abdomen (Figure 1B). The palms and soles showed keratoderma. Neurologic examination of the arms revealed mild rigidity and brisk reflexes. Examination of the legs showed marked rigidity, brisk knee jerks, ankle clonus, extensor plantar reflexes, flexion deformity with contractures, and scissor gait. A Goddard (Seguin) formboard test was performed and indicated a mental age of 4 years. The patient’s IQ was in the range of 25 to 30, indicating a severe degree of subnormality in intellectual functioning. The clinical presentation suggested a diagnosis of SLS.
A skin biopsy from the ichthyotic lesion showed hyperkeratosis, acanthosis, and papillomatosis with sparse superficial perivascular lymphocytic infiltrate, thus confirming the diagnosis of lamellar ichthyosis. Fundus examination was normal. Magnetic resonance imaging (MRI) of the brain revealed confluent symmetrical signal abnormalities along the body of the lateral ventricles, white matter in the perioccipital horn, and in deep white matter of centrum semiovale (Figure 2). Magnetic resonance spectroscopy revealed a narrow lipid peak at approximately 1.3 ppm in the region of signal abnormality (Figure 3). Thus, the diagnosis of SLS was confirmed. Measurement of fatty aldehyde dehydrogenase (FALDH) activity and genetic analysis were not performed due to unavailability.
The patient was treated with topical emollients for the ichthyosis. To reduce his dietary intake of long-chain fatty acids and increase the intake of omega-3 and omega-6 fatty acids, the patient’s parents were advised to use canola, mustard, and/or coconut oil for cooking for the patient, and skim milk was recommended instead of whole milk. Neurodevelopmental techniques in the form of stretching exercises were given to maintain his range of movements. Gutter splints were given to maintain the knees in extension for physiological standing and to prevent osteoporosis. Subsequently, the patient also underwent a multilevel soft-tissue release (hip and knee joints) to relieve the contractures. These measures resulted in considerable improvement and the patient was able to walk with support.
Comment
Presentation
The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations
Sjögren-Larsson syndrome is caused by mutation in the aldehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
Lipid Metabolism
Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.
Histopathology
The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings
Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment
Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.
- Judge MR, McLean WHI, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, eds. Rook’s Textbook of Dermatology. 7th ed. West Sussex, United Kingdom: Wiley & Sons; 2004:34.37-34.39.
- Sjögren T, Larsson T. Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1-113.
- Jagell S, Gustavson KH, Holmgren G. Sjögren-Larsson syndrome in Sweden. a clinical, genetic and epidemiological study. Clin Genet. 1981;19:233-256.
- Sood M, Trehan A, Dinakaran J, et al. Sjögren-Larsson syndrome. Indian J Pediatr. 2002;69:193-194.
- Uppal M, Srinivas CR, Thowfeeq KT. Sjögren-Larsson syndrome: report of two cases. Indian J Dermatol Venereol Leprol. 2004;70:110-111.
- Rizzo WB. Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1-9.
- Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124(pt 7):1426-1437.
- Willemsen MA, Cruysberg JR, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. Am J Ophthalmol. 2000;130:782-789.
- Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Arch Neurol. 2006;63:278-280.
- Rizzo WB, Craft DA, Somer T, et al. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410-419.
- Rizzo WB, S’Aulis D, Jennings MA, et al. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443-451.
- Rizzo WB. The role of fatty aldehyde dehydrogenase in epidermal structure and function. Dermatoendocrinol. 2011;2:91-99.
- Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18:338-341.
- Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Larsson’s Syndrome. Am J Neuroradiol. 1999;20:1671-1673.
- Willemsen MA, van der Graf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
- Kaminaga T, Mano T, Ono J, et al. Proton magnetic resonance spectroscopy of Sjögren-Larsson Syndrome. Magn Reson Med. 2001;45:1112-1115.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870-875.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711-717.
- Pirgon O, Aydin K, Atabek ME. Proton magnetic resonance spectroscopy findings and clinical effects of montelukast sodium in a case with Sjögren-Larsson syndrome. J Child Neurol. 2006;21:1092-1095.
- Gloerich J, Ijlst L, Wanders RJ, et al. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome Mol Genet Metab. 2006;89:111-115.
- Auada MP, Taube MB, Collares EF, et al. Sjögren-Larsson syndrome: biochemical defects and follow up in three cases. Eur J Dermatol. 2002;12:263-266.
- Taube B, Billeaud C, Labreze C, et al. Sjögren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology. 1999;198:340-345.
- Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opin Orphan Drugs. 2016;4:395-406.
- Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjögren-Larsson syndrome. Gene Ther. 2006;13:1021-1026.
- Judge MR, McLean WHI, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, eds. Rook’s Textbook of Dermatology. 7th ed. West Sussex, United Kingdom: Wiley & Sons; 2004:34.37-34.39.
- Sjögren T, Larsson T. Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1-113.
- Jagell S, Gustavson KH, Holmgren G. Sjögren-Larsson syndrome in Sweden. a clinical, genetic and epidemiological study. Clin Genet. 1981;19:233-256.
- Sood M, Trehan A, Dinakaran J, et al. Sjögren-Larsson syndrome. Indian J Pediatr. 2002;69:193-194.
- Uppal M, Srinivas CR, Thowfeeq KT. Sjögren-Larsson syndrome: report of two cases. Indian J Dermatol Venereol Leprol. 2004;70:110-111.
- Rizzo WB. Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1-9.
- Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124(pt 7):1426-1437.
- Willemsen MA, Cruysberg JR, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. Am J Ophthalmol. 2000;130:782-789.
- Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Arch Neurol. 2006;63:278-280.
- Rizzo WB, Craft DA, Somer T, et al. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410-419.
- Rizzo WB, S’Aulis D, Jennings MA, et al. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443-451.
- Rizzo WB. The role of fatty aldehyde dehydrogenase in epidermal structure and function. Dermatoendocrinol. 2011;2:91-99.
- Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18:338-341.
- Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Larsson’s Syndrome. Am J Neuroradiol. 1999;20:1671-1673.
- Willemsen MA, van der Graf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
- Kaminaga T, Mano T, Ono J, et al. Proton magnetic resonance spectroscopy of Sjögren-Larsson Syndrome. Magn Reson Med. 2001;45:1112-1115.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870-875.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711-717.
- Pirgon O, Aydin K, Atabek ME. Proton magnetic resonance spectroscopy findings and clinical effects of montelukast sodium in a case with Sjögren-Larsson syndrome. J Child Neurol. 2006;21:1092-1095.
- Gloerich J, Ijlst L, Wanders RJ, et al. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome Mol Genet Metab. 2006;89:111-115.
- Auada MP, Taube MB, Collares EF, et al. Sjögren-Larsson syndrome: biochemical defects and follow up in three cases. Eur J Dermatol. 2002;12:263-266.
- Taube B, Billeaud C, Labreze C, et al. Sjögren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology. 1999;198:340-345.
- Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opin Orphan Drugs. 2016;4:395-406.
- Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjögren-Larsson syndrome. Gene Ther. 2006;13:1021-1026.
Practice Points
- Sjögren-Larsson syndrome (SLS) is characterized by a clinical triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.
- A characteristic lipid peak at 1.3 ppm on magnetic resonance spectroscopy is diagnostic of SLS.
Pediatric Nevoid Basal Cell Carcinoma Syndrome
In 1960, Gorlin and Goltz1 first described nevoid basal cell carcinoma syndrome (NBCCS) as a distinct clinical entity with multiple basal cell carcinomas (BCCs), jaw cysts, and bifid ribs. This rare autosomal-dominant genodermatosis has a minimal prevalence of 1 case per 57,000 individuals2 and no sexual predilection.3 Nevoid basal cell carcinoma syndrome is caused by a mutation in the human homolog of a Drosophila gene, patched 1 (PTCH1), which is located on chromosome 9q22.3.4,5 The major clinical diagnostic criteria includes multiple BCCs, odontogenic keratocysts, palmar or plantar pits, ectopic calcification of the falx cerebri, and a family history of NBCCS.6 Basal cell carcinoma formation is affected by both skin pigmentation and sun exposure; 80% of white patients with NBCCS will develop at least 1 BCC compared to only 40% of black patients with NBCCS.7 Goldstein et al8 postulated that this disparity is associated with increased skin pigmentation providing UV radiation protection, thus decreasing the tumor burden. We report a case of an 11-year-old black boy with NBCCS to highlight the treatment considerations in pediatric cases of NBCCS.
Case Report
An 11-year-old boy with Fitzpatrick skin type V presented with a history of multiple facial lesions after undergoing excision of large keratocysts from the right maxilla, left maxilla, and right mandible. Physical examination revealed multiple light to dark brown facial papules (Figure 1), palmar and plantar pitting (Figure 2), and frontal bossing.
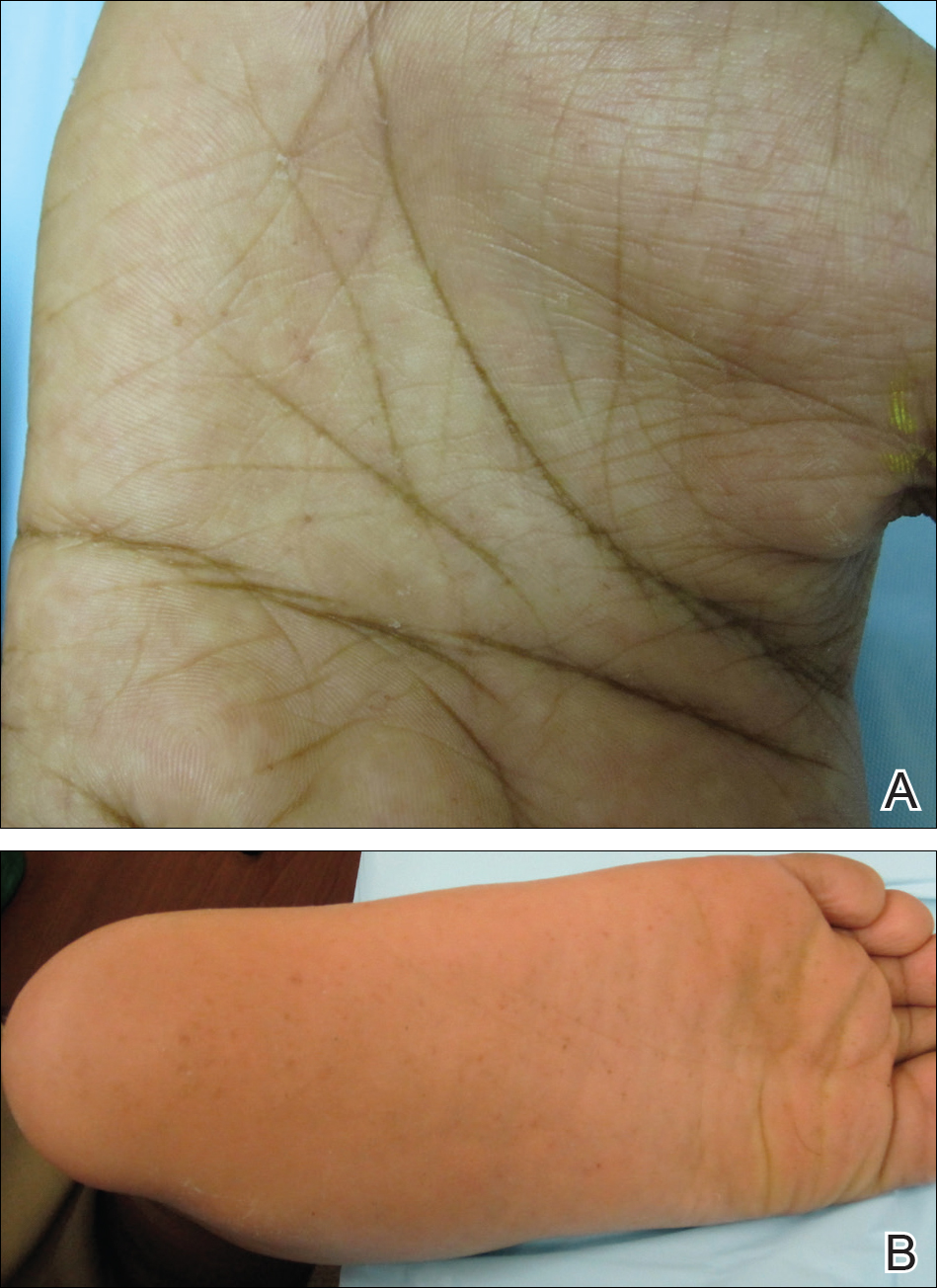
He was previously diagnosed with autism and his surgical history was notable only for excision of the keratocysts. The patient was not taking any medications and did not have any drug allergies. There was no maternal family history of skin cancer or related syndromes; his paternal family history was unknown. A shave biopsy was performed on a facial papule from the right nasolabial fold. Histopathologic evaluation revealed findings consistent with a pigmented nodular BCC (Figure 3). The patient was subsequently sent for magnetic resonance imaging of the brain, which demonstrated calcifications along the tentorium. Genetic consultation confirmed a heterozygous mutation of the PTCH1 gene.
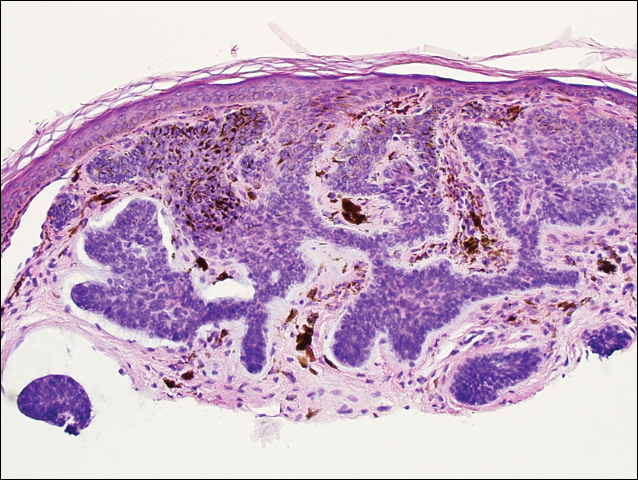
Over the next 12 months, the patient had multiple biopsy-proven pigmented BCCs. Initial management of these carcinomas located on cosmetically sensitive areas, including the upper eyelid and penis, were excised by a pediatric plastic surgeon. A truncal carcinoma was treated with electrodesiccation and curettage, which resulted in keloid formation. Early suspicious lesions were treated with imiquimod cream 5% 5 times weekly in combination with the prophylactic use of tretinoin cream 0.1%. Despite this treatment regimen, the patient continued to demonstrate multiple small clinical pigmented BCCs along the malar surfaces of the cheeks and dorsum of the nose. The patient’s mother deferred chemoprevention with an oral retinoid due to the extensive side-effect profile and long-term necessity of administration.
Management also encompassed BCC surveillance every 4 months; annual digital panorex of the jaw; routine dental screening; routine developmental screening; annual follow-up with a geneticist to ensure multidisciplinary care; and annual vision, hearing, and speech-screening examinations. Strict sun-protective measures were encouraged, including wearing a hat during physical education class.
Comment
Classification and Clinical Presentation
Nevoid basal cell carcinoma syndrome is a multisystem disorder that requires close monitoring under multidisciplinary care. Evans et al6 defined the diagnostic criteria of NBCCS to require the presence of 2 major criteria or 1 major and 2 minor criteria. The major criteria include multiple BCCs, an odontogenic keratocyst or polyostotic bone cyst, palmar or plantar pits, ectopic calcification of the falx cerebri, and family history of NBCCS. The minor criteria are defined as congenital skeletal anomalies; macrocephaly with frontal bossing; cardiac or ovarian fibromas; medulloblastoma; lymphomesenteric cysts; and congenital malformations such as cleft lip or palate, polydactyly, or eye anomalies.6 The mean age of initial BCC diagnosis is 21 years, with proliferation of cancers between puberty and 35 years of age.7,9 Our case is unique due to the patient’s young age at the time of diagnosis as well as his presentation with multiple BCCs with a darker skin type. Kimonis et al7 reported that approximately 20% of black patients develop their first BCC by the age of 21 years and 40% by 35 years. The presence of multiple BCCs is complicated by the limited treatment options in a pediatric patient. The patient’s inability to withstand multiple procedures contributed to our clinical decision to have multiple lesions removed under general anesthesia by a pediatric plastic surgeon.
Due to the patient’s young age of onset, we placed a great emphasis on close surveillance and management. A management protocol for pediatric patients with NBCCS was described by Bree and Shah; BCNS Colloquium Group10 (eTable). We closely followed this protocol for surveillance; however, we scheduled dermatologic examinations every 4 months due to his extensive history of BCCs.

Management
Our case presents a challenging therapeutic and management dilemma. The management of NBCCS utilizes a multitude of treatment modalities, but many of them posed cosmetic challenges in our patient such as postinflammatory hypopigmentation and the propensity for keloid formation. Although surgical excision or Mohs micrographic surgery is the standard of treatment of nodular BCCs, we were limited due to the patient’s inability to tolerate multiple surgical procedures without the use of general anesthesia.
Case reports have discussed the use of CO2 laser resurfacing for management of multiple facial BCCs in patients with NBCCS. Doctoroff et al11 treated a patient with 45 facial BCCs with full-face CO2 laser resurfacing, and in a 10-month follow-up period the patient developed 6 new BCCs on the face. Nouri et al12 described 3 cases of multiple BCCs on the face, trunk, and extremities treated with ultrapulse CO2 laser with postoperative Mohs sections verifying complete histologic clearance of tumors. All 3 patients had Fitzpatrick skin type IV; their ages were 2, 16, and 35 years. Local anesthesia was used in the 2-year-old patient and intravenous sedation in the 16-year-old patient.12 Although CO2 laser therapy may be a practical treatment option, it posed too many cosmetic concerns in our patient.
Photodynamic therapy (PDT) is an emerging treatment option for NBCCS patients. Itkin and Gilchrest13 treated 2 NBCCS patients with δ-aminolevulinic acid for 1 to 5 hours prior to treatment with blue light therapy. Complete clearance was documented in 89% (8/9) of superficial BCCs and 31% (5/16) of nodular BCCs on the face, indicating that blue light treatment may reduce the cutaneous tumor burden.13 Oseroff et al14 reported similar success in treating 3 children with NBCCS with 20% δ-aminolevulinic acid for 24 hours under occlusion followed by red light treatment. After 1 to 3 treatments, the children had 85% to 98% total clearance, demonstrating it as a viable treatment option in young patients that yields excellent cosmetic results and is well tolerated.14 Photodynamic therapy is reported to have a low risk of carcinogenicity15; however, there has been 1 reported case of melanoma developing at the site of multiple PDT treatments.16 Thus, the risk of carcinogenicity is increasingly bothersome in NBCCS patients due to their sensitivity to exposure. The limited number of studies using topical PDT on pediatric patients, the lack of treatment protocols for pediatric patients, and the need to use general anesthesia for pediatric patients all posed limitations to the use of PDT in our case.
Imiquimod cream 5% was shown in randomized, vehicle-controlled studies to be a safe and effective treatment of superficial BCCs when used 5 days weekly for 6 weeks.17 These studies excluded patients with NBCCS; however, other studies have been completed in patients with NBCCS. Kagy and Amonette18 successfully treated 3 nonfacial BCCs in a patient with NBCCS with imiquimod cream 5% daily for 18 weeks, with complete histologic resolution of the tumors. Micali et al19 also treated 4 patients with NBCCS using imiquimod cream 5% 3 to 5 times weekly for 8 to 14 weeks. Thirteen of 17 BCCs resolved, as confirmed with histologic evaluation.19 One case report revealed a child with NBCCS who was successfully managed with topical fluorouracil and topical tretinoin for more than 10 years.20 Our patient used imiquimod cream 5% 5 times weekly, which inhibited the growth of existing lesions but did not clear them entirely, as they were nodular in nature.
Chemoprevention with oral retinoids breaches a controversial treatment topic. In 1989, a case study of an NBCCS patient treated with surgical excision and oral etretinate for 12 months documented reduction of large tumors.21 A multicenter clinical trial reported that low-dose isotretinoin (10 mg daily) is ineffective in preventing the occurrence of new BCC formation in patients with a history of 2 or more sporadic BCCs.22 Chemoprevention with oral retinoids is well known for being effective for squamous cell carcinomas and actinic keratosis; however, the treatment is less effective for BCCs.22 Most importantly, the extensive side-effect profile and toxicity associated with long-term administration of oral retinoids prohibits many practitioners from routinely using them in pediatric NBCCS patients.
Nevoid basal cell carcinoma syndrome patients are exquisitely sensitive to ionizing radiation and the effects of UV exposure. Therefore, it is essential to emphasize the importance of sun-protective measures such as sun avoidance, broad-spectrum sunscreen use, and sun-protective clothing.
Conclusion
Nevoid basal cell carcinoma syndrome is a multisystem disorder with a notable predisposition for skin cancer. Our case demonstrates the treatment considerations in a pediatric patient with Fitzpatrick skin type V. Pediatric NBCCS patients develop BCCs at a young age and will continue to develop additional lesions throughout life; therefore, skin preservation is an important consideration when choosing the appropriate treatment regimen. Particularly in our patient, utilizing multiple strategic treatment modalities in combination with chemoprevention moving forward will be a continued management challenge. Strict adherence to a surveillance protocol is encouraged to closely monitor the systemic manifestations of the disorder.
- Gorlin RJ, Goltz R. Multiple nevoid basal cell epitheliomata, jaw cysts, bifid rib-a syndrome. N Engl J Med. 1960;262:908-911.
- Evans DGR, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
- Farndon PA, Del Mastro RG, Evans DG, et al. Location of gene for Gorlin Syndrome. Lancet. 1992;339:581-582.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10:757-761.
- Evans DGR, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460-464.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al. Clinical findings in two African-American families with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1994;50:272-281.
- Shanley S, Ratcliffe J, Hockey A, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282-290.
- Bree AF, Shah MR; BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155:2091-2097.
- Doctoroff A, Oberlender SA, Purcell SM. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg. 2003;29:1236-1240.
- Nouri K, Chang A, Trent JT, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg. 2002;28:287-290.
- Itkin A, Gilchrest BA. δ-Aminolevulinic acid and blue light photodynamic therapy for treatment of multiple basal cell carcinomas in two patients with nevoid basal cell carcinoma syndrome. Dermatol Surg. 2004;30:1054-1061.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141:60-67.
- Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146:552-567.
- Wolf P, Fink-Puches R, Reimann-Weber A, et al. Development of malignant melanoma after repeated topical photodynamic therapy with 5-aminolevulinic acid at the exposed site. Dermatology. 1997;194:53-54.
- Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-733.
- Kagy MK, Amonette R. The use of imiquimod 5% cream for the treatment of superficial basal cell carcinomas in a basal cell nevus syndrome patient. Dermatol Surg. 2000;26:577-579.
- Micali G, Lacarrubba F, Nasca MR, et al. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin Exp Dermatol. 2003;28:19-23.
- Strange PR, Lang PG. Long-term management of basal cell nevus syndrome with topical tretinoin and 5-fluorouracil. J Am Acad Dermatol. 1992;27:842-845.
- Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol. 1989;15:868-871.
- Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst. 1992;84:328-332.
In 1960, Gorlin and Goltz1 first described nevoid basal cell carcinoma syndrome (NBCCS) as a distinct clinical entity with multiple basal cell carcinomas (BCCs), jaw cysts, and bifid ribs. This rare autosomal-dominant genodermatosis has a minimal prevalence of 1 case per 57,000 individuals2 and no sexual predilection.3 Nevoid basal cell carcinoma syndrome is caused by a mutation in the human homolog of a Drosophila gene, patched 1 (PTCH1), which is located on chromosome 9q22.3.4,5 The major clinical diagnostic criteria includes multiple BCCs, odontogenic keratocysts, palmar or plantar pits, ectopic calcification of the falx cerebri, and a family history of NBCCS.6 Basal cell carcinoma formation is affected by both skin pigmentation and sun exposure; 80% of white patients with NBCCS will develop at least 1 BCC compared to only 40% of black patients with NBCCS.7 Goldstein et al8 postulated that this disparity is associated with increased skin pigmentation providing UV radiation protection, thus decreasing the tumor burden. We report a case of an 11-year-old black boy with NBCCS to highlight the treatment considerations in pediatric cases of NBCCS.
Case Report
An 11-year-old boy with Fitzpatrick skin type V presented with a history of multiple facial lesions after undergoing excision of large keratocysts from the right maxilla, left maxilla, and right mandible. Physical examination revealed multiple light to dark brown facial papules (Figure 1), palmar and plantar pitting (Figure 2), and frontal bossing.
He was previously diagnosed with autism and his surgical history was notable only for excision of the keratocysts. The patient was not taking any medications and did not have any drug allergies. There was no maternal family history of skin cancer or related syndromes; his paternal family history was unknown. A shave biopsy was performed on a facial papule from the right nasolabial fold. Histopathologic evaluation revealed findings consistent with a pigmented nodular BCC (Figure 3). The patient was subsequently sent for magnetic resonance imaging of the brain, which demonstrated calcifications along the tentorium. Genetic consultation confirmed a heterozygous mutation of the PTCH1 gene.
Over the next 12 months, the patient had multiple biopsy-proven pigmented BCCs. Initial management of these carcinomas located on cosmetically sensitive areas, including the upper eyelid and penis, were excised by a pediatric plastic surgeon. A truncal carcinoma was treated with electrodesiccation and curettage, which resulted in keloid formation. Early suspicious lesions were treated with imiquimod cream 5% 5 times weekly in combination with the prophylactic use of tretinoin cream 0.1%. Despite this treatment regimen, the patient continued to demonstrate multiple small clinical pigmented BCCs along the malar surfaces of the cheeks and dorsum of the nose. The patient’s mother deferred chemoprevention with an oral retinoid due to the extensive side-effect profile and long-term necessity of administration.
Management also encompassed BCC surveillance every 4 months; annual digital panorex of the jaw; routine dental screening; routine developmental screening; annual follow-up with a geneticist to ensure multidisciplinary care; and annual vision, hearing, and speech-screening examinations. Strict sun-protective measures were encouraged, including wearing a hat during physical education class.
Comment
Classification and Clinical Presentation
Nevoid basal cell carcinoma syndrome is a multisystem disorder that requires close monitoring under multidisciplinary care. Evans et al6 defined the diagnostic criteria of NBCCS to require the presence of 2 major criteria or 1 major and 2 minor criteria. The major criteria include multiple BCCs, an odontogenic keratocyst or polyostotic bone cyst, palmar or plantar pits, ectopic calcification of the falx cerebri, and family history of NBCCS. The minor criteria are defined as congenital skeletal anomalies; macrocephaly with frontal bossing; cardiac or ovarian fibromas; medulloblastoma; lymphomesenteric cysts; and congenital malformations such as cleft lip or palate, polydactyly, or eye anomalies.6 The mean age of initial BCC diagnosis is 21 years, with proliferation of cancers between puberty and 35 years of age.7,9 Our case is unique due to the patient’s young age at the time of diagnosis as well as his presentation with multiple BCCs with a darker skin type. Kimonis et al7 reported that approximately 20% of black patients develop their first BCC by the age of 21 years and 40% by 35 years. The presence of multiple BCCs is complicated by the limited treatment options in a pediatric patient. The patient’s inability to withstand multiple procedures contributed to our clinical decision to have multiple lesions removed under general anesthesia by a pediatric plastic surgeon.
Due to the patient’s young age of onset, we placed a great emphasis on close surveillance and management. A management protocol for pediatric patients with NBCCS was described by Bree and Shah; BCNS Colloquium Group10 (eTable). We closely followed this protocol for surveillance; however, we scheduled dermatologic examinations every 4 months due to his extensive history of BCCs.
Management
Our case presents a challenging therapeutic and management dilemma. The management of NBCCS utilizes a multitude of treatment modalities, but many of them posed cosmetic challenges in our patient such as postinflammatory hypopigmentation and the propensity for keloid formation. Although surgical excision or Mohs micrographic surgery is the standard of treatment of nodular BCCs, we were limited due to the patient’s inability to tolerate multiple surgical procedures without the use of general anesthesia.
Case reports have discussed the use of CO2 laser resurfacing for management of multiple facial BCCs in patients with NBCCS. Doctoroff et al11 treated a patient with 45 facial BCCs with full-face CO2 laser resurfacing, and in a 10-month follow-up period the patient developed 6 new BCCs on the face. Nouri et al12 described 3 cases of multiple BCCs on the face, trunk, and extremities treated with ultrapulse CO2 laser with postoperative Mohs sections verifying complete histologic clearance of tumors. All 3 patients had Fitzpatrick skin type IV; their ages were 2, 16, and 35 years. Local anesthesia was used in the 2-year-old patient and intravenous sedation in the 16-year-old patient.12 Although CO2 laser therapy may be a practical treatment option, it posed too many cosmetic concerns in our patient.
Photodynamic therapy (PDT) is an emerging treatment option for NBCCS patients. Itkin and Gilchrest13 treated 2 NBCCS patients with δ-aminolevulinic acid for 1 to 5 hours prior to treatment with blue light therapy. Complete clearance was documented in 89% (8/9) of superficial BCCs and 31% (5/16) of nodular BCCs on the face, indicating that blue light treatment may reduce the cutaneous tumor burden.13 Oseroff et al14 reported similar success in treating 3 children with NBCCS with 20% δ-aminolevulinic acid for 24 hours under occlusion followed by red light treatment. After 1 to 3 treatments, the children had 85% to 98% total clearance, demonstrating it as a viable treatment option in young patients that yields excellent cosmetic results and is well tolerated.14 Photodynamic therapy is reported to have a low risk of carcinogenicity15; however, there has been 1 reported case of melanoma developing at the site of multiple PDT treatments.16 Thus, the risk of carcinogenicity is increasingly bothersome in NBCCS patients due to their sensitivity to exposure. The limited number of studies using topical PDT on pediatric patients, the lack of treatment protocols for pediatric patients, and the need to use general anesthesia for pediatric patients all posed limitations to the use of PDT in our case.
Imiquimod cream 5% was shown in randomized, vehicle-controlled studies to be a safe and effective treatment of superficial BCCs when used 5 days weekly for 6 weeks.17 These studies excluded patients with NBCCS; however, other studies have been completed in patients with NBCCS. Kagy and Amonette18 successfully treated 3 nonfacial BCCs in a patient with NBCCS with imiquimod cream 5% daily for 18 weeks, with complete histologic resolution of the tumors. Micali et al19 also treated 4 patients with NBCCS using imiquimod cream 5% 3 to 5 times weekly for 8 to 14 weeks. Thirteen of 17 BCCs resolved, as confirmed with histologic evaluation.19 One case report revealed a child with NBCCS who was successfully managed with topical fluorouracil and topical tretinoin for more than 10 years.20 Our patient used imiquimod cream 5% 5 times weekly, which inhibited the growth of existing lesions but did not clear them entirely, as they were nodular in nature.
Chemoprevention with oral retinoids breaches a controversial treatment topic. In 1989, a case study of an NBCCS patient treated with surgical excision and oral etretinate for 12 months documented reduction of large tumors.21 A multicenter clinical trial reported that low-dose isotretinoin (10 mg daily) is ineffective in preventing the occurrence of new BCC formation in patients with a history of 2 or more sporadic BCCs.22 Chemoprevention with oral retinoids is well known for being effective for squamous cell carcinomas and actinic keratosis; however, the treatment is less effective for BCCs.22 Most importantly, the extensive side-effect profile and toxicity associated with long-term administration of oral retinoids prohibits many practitioners from routinely using them in pediatric NBCCS patients.
Nevoid basal cell carcinoma syndrome patients are exquisitely sensitive to ionizing radiation and the effects of UV exposure. Therefore, it is essential to emphasize the importance of sun-protective measures such as sun avoidance, broad-spectrum sunscreen use, and sun-protective clothing.
Conclusion
Nevoid basal cell carcinoma syndrome is a multisystem disorder with a notable predisposition for skin cancer. Our case demonstrates the treatment considerations in a pediatric patient with Fitzpatrick skin type V. Pediatric NBCCS patients develop BCCs at a young age and will continue to develop additional lesions throughout life; therefore, skin preservation is an important consideration when choosing the appropriate treatment regimen. Particularly in our patient, utilizing multiple strategic treatment modalities in combination with chemoprevention moving forward will be a continued management challenge. Strict adherence to a surveillance protocol is encouraged to closely monitor the systemic manifestations of the disorder.
In 1960, Gorlin and Goltz1 first described nevoid basal cell carcinoma syndrome (NBCCS) as a distinct clinical entity with multiple basal cell carcinomas (BCCs), jaw cysts, and bifid ribs. This rare autosomal-dominant genodermatosis has a minimal prevalence of 1 case per 57,000 individuals2 and no sexual predilection.3 Nevoid basal cell carcinoma syndrome is caused by a mutation in the human homolog of a Drosophila gene, patched 1 (PTCH1), which is located on chromosome 9q22.3.4,5 The major clinical diagnostic criteria includes multiple BCCs, odontogenic keratocysts, palmar or plantar pits, ectopic calcification of the falx cerebri, and a family history of NBCCS.6 Basal cell carcinoma formation is affected by both skin pigmentation and sun exposure; 80% of white patients with NBCCS will develop at least 1 BCC compared to only 40% of black patients with NBCCS.7 Goldstein et al8 postulated that this disparity is associated with increased skin pigmentation providing UV radiation protection, thus decreasing the tumor burden. We report a case of an 11-year-old black boy with NBCCS to highlight the treatment considerations in pediatric cases of NBCCS.
Case Report
An 11-year-old boy with Fitzpatrick skin type V presented with a history of multiple facial lesions after undergoing excision of large keratocysts from the right maxilla, left maxilla, and right mandible. Physical examination revealed multiple light to dark brown facial papules (Figure 1), palmar and plantar pitting (Figure 2), and frontal bossing.
He was previously diagnosed with autism and his surgical history was notable only for excision of the keratocysts. The patient was not taking any medications and did not have any drug allergies. There was no maternal family history of skin cancer or related syndromes; his paternal family history was unknown. A shave biopsy was performed on a facial papule from the right nasolabial fold. Histopathologic evaluation revealed findings consistent with a pigmented nodular BCC (Figure 3). The patient was subsequently sent for magnetic resonance imaging of the brain, which demonstrated calcifications along the tentorium. Genetic consultation confirmed a heterozygous mutation of the PTCH1 gene.
Over the next 12 months, the patient had multiple biopsy-proven pigmented BCCs. Initial management of these carcinomas located on cosmetically sensitive areas, including the upper eyelid and penis, were excised by a pediatric plastic surgeon. A truncal carcinoma was treated with electrodesiccation and curettage, which resulted in keloid formation. Early suspicious lesions were treated with imiquimod cream 5% 5 times weekly in combination with the prophylactic use of tretinoin cream 0.1%. Despite this treatment regimen, the patient continued to demonstrate multiple small clinical pigmented BCCs along the malar surfaces of the cheeks and dorsum of the nose. The patient’s mother deferred chemoprevention with an oral retinoid due to the extensive side-effect profile and long-term necessity of administration.
Management also encompassed BCC surveillance every 4 months; annual digital panorex of the jaw; routine dental screening; routine developmental screening; annual follow-up with a geneticist to ensure multidisciplinary care; and annual vision, hearing, and speech-screening examinations. Strict sun-protective measures were encouraged, including wearing a hat during physical education class.
Comment
Classification and Clinical Presentation
Nevoid basal cell carcinoma syndrome is a multisystem disorder that requires close monitoring under multidisciplinary care. Evans et al6 defined the diagnostic criteria of NBCCS to require the presence of 2 major criteria or 1 major and 2 minor criteria. The major criteria include multiple BCCs, an odontogenic keratocyst or polyostotic bone cyst, palmar or plantar pits, ectopic calcification of the falx cerebri, and family history of NBCCS. The minor criteria are defined as congenital skeletal anomalies; macrocephaly with frontal bossing; cardiac or ovarian fibromas; medulloblastoma; lymphomesenteric cysts; and congenital malformations such as cleft lip or palate, polydactyly, or eye anomalies.6 The mean age of initial BCC diagnosis is 21 years, with proliferation of cancers between puberty and 35 years of age.7,9 Our case is unique due to the patient’s young age at the time of diagnosis as well as his presentation with multiple BCCs with a darker skin type. Kimonis et al7 reported that approximately 20% of black patients develop their first BCC by the age of 21 years and 40% by 35 years. The presence of multiple BCCs is complicated by the limited treatment options in a pediatric patient. The patient’s inability to withstand multiple procedures contributed to our clinical decision to have multiple lesions removed under general anesthesia by a pediatric plastic surgeon.
Due to the patient’s young age of onset, we placed a great emphasis on close surveillance and management. A management protocol for pediatric patients with NBCCS was described by Bree and Shah; BCNS Colloquium Group10 (eTable). We closely followed this protocol for surveillance; however, we scheduled dermatologic examinations every 4 months due to his extensive history of BCCs.
Management
Our case presents a challenging therapeutic and management dilemma. The management of NBCCS utilizes a multitude of treatment modalities, but many of them posed cosmetic challenges in our patient such as postinflammatory hypopigmentation and the propensity for keloid formation. Although surgical excision or Mohs micrographic surgery is the standard of treatment of nodular BCCs, we were limited due to the patient’s inability to tolerate multiple surgical procedures without the use of general anesthesia.
Case reports have discussed the use of CO2 laser resurfacing for management of multiple facial BCCs in patients with NBCCS. Doctoroff et al11 treated a patient with 45 facial BCCs with full-face CO2 laser resurfacing, and in a 10-month follow-up period the patient developed 6 new BCCs on the face. Nouri et al12 described 3 cases of multiple BCCs on the face, trunk, and extremities treated with ultrapulse CO2 laser with postoperative Mohs sections verifying complete histologic clearance of tumors. All 3 patients had Fitzpatrick skin type IV; their ages were 2, 16, and 35 years. Local anesthesia was used in the 2-year-old patient and intravenous sedation in the 16-year-old patient.12 Although CO2 laser therapy may be a practical treatment option, it posed too many cosmetic concerns in our patient.
Photodynamic therapy (PDT) is an emerging treatment option for NBCCS patients. Itkin and Gilchrest13 treated 2 NBCCS patients with δ-aminolevulinic acid for 1 to 5 hours prior to treatment with blue light therapy. Complete clearance was documented in 89% (8/9) of superficial BCCs and 31% (5/16) of nodular BCCs on the face, indicating that blue light treatment may reduce the cutaneous tumor burden.13 Oseroff et al14 reported similar success in treating 3 children with NBCCS with 20% δ-aminolevulinic acid for 24 hours under occlusion followed by red light treatment. After 1 to 3 treatments, the children had 85% to 98% total clearance, demonstrating it as a viable treatment option in young patients that yields excellent cosmetic results and is well tolerated.14 Photodynamic therapy is reported to have a low risk of carcinogenicity15; however, there has been 1 reported case of melanoma developing at the site of multiple PDT treatments.16 Thus, the risk of carcinogenicity is increasingly bothersome in NBCCS patients due to their sensitivity to exposure. The limited number of studies using topical PDT on pediatric patients, the lack of treatment protocols for pediatric patients, and the need to use general anesthesia for pediatric patients all posed limitations to the use of PDT in our case.
Imiquimod cream 5% was shown in randomized, vehicle-controlled studies to be a safe and effective treatment of superficial BCCs when used 5 days weekly for 6 weeks.17 These studies excluded patients with NBCCS; however, other studies have been completed in patients with NBCCS. Kagy and Amonette18 successfully treated 3 nonfacial BCCs in a patient with NBCCS with imiquimod cream 5% daily for 18 weeks, with complete histologic resolution of the tumors. Micali et al19 also treated 4 patients with NBCCS using imiquimod cream 5% 3 to 5 times weekly for 8 to 14 weeks. Thirteen of 17 BCCs resolved, as confirmed with histologic evaluation.19 One case report revealed a child with NBCCS who was successfully managed with topical fluorouracil and topical tretinoin for more than 10 years.20 Our patient used imiquimod cream 5% 5 times weekly, which inhibited the growth of existing lesions but did not clear them entirely, as they were nodular in nature.
Chemoprevention with oral retinoids breaches a controversial treatment topic. In 1989, a case study of an NBCCS patient treated with surgical excision and oral etretinate for 12 months documented reduction of large tumors.21 A multicenter clinical trial reported that low-dose isotretinoin (10 mg daily) is ineffective in preventing the occurrence of new BCC formation in patients with a history of 2 or more sporadic BCCs.22 Chemoprevention with oral retinoids is well known for being effective for squamous cell carcinomas and actinic keratosis; however, the treatment is less effective for BCCs.22 Most importantly, the extensive side-effect profile and toxicity associated with long-term administration of oral retinoids prohibits many practitioners from routinely using them in pediatric NBCCS patients.
Nevoid basal cell carcinoma syndrome patients are exquisitely sensitive to ionizing radiation and the effects of UV exposure. Therefore, it is essential to emphasize the importance of sun-protective measures such as sun avoidance, broad-spectrum sunscreen use, and sun-protective clothing.
Conclusion
Nevoid basal cell carcinoma syndrome is a multisystem disorder with a notable predisposition for skin cancer. Our case demonstrates the treatment considerations in a pediatric patient with Fitzpatrick skin type V. Pediatric NBCCS patients develop BCCs at a young age and will continue to develop additional lesions throughout life; therefore, skin preservation is an important consideration when choosing the appropriate treatment regimen. Particularly in our patient, utilizing multiple strategic treatment modalities in combination with chemoprevention moving forward will be a continued management challenge. Strict adherence to a surveillance protocol is encouraged to closely monitor the systemic manifestations of the disorder.
- Gorlin RJ, Goltz R. Multiple nevoid basal cell epitheliomata, jaw cysts, bifid rib-a syndrome. N Engl J Med. 1960;262:908-911.
- Evans DGR, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
- Farndon PA, Del Mastro RG, Evans DG, et al. Location of gene for Gorlin Syndrome. Lancet. 1992;339:581-582.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10:757-761.
- Evans DGR, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460-464.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al. Clinical findings in two African-American families with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1994;50:272-281.
- Shanley S, Ratcliffe J, Hockey A, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282-290.
- Bree AF, Shah MR; BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155:2091-2097.
- Doctoroff A, Oberlender SA, Purcell SM. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg. 2003;29:1236-1240.
- Nouri K, Chang A, Trent JT, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg. 2002;28:287-290.
- Itkin A, Gilchrest BA. δ-Aminolevulinic acid and blue light photodynamic therapy for treatment of multiple basal cell carcinomas in two patients with nevoid basal cell carcinoma syndrome. Dermatol Surg. 2004;30:1054-1061.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141:60-67.
- Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146:552-567.
- Wolf P, Fink-Puches R, Reimann-Weber A, et al. Development of malignant melanoma after repeated topical photodynamic therapy with 5-aminolevulinic acid at the exposed site. Dermatology. 1997;194:53-54.
- Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-733.
- Kagy MK, Amonette R. The use of imiquimod 5% cream for the treatment of superficial basal cell carcinomas in a basal cell nevus syndrome patient. Dermatol Surg. 2000;26:577-579.
- Micali G, Lacarrubba F, Nasca MR, et al. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin Exp Dermatol. 2003;28:19-23.
- Strange PR, Lang PG. Long-term management of basal cell nevus syndrome with topical tretinoin and 5-fluorouracil. J Am Acad Dermatol. 1992;27:842-845.
- Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol. 1989;15:868-871.
- Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst. 1992;84:328-332.
- Gorlin RJ, Goltz R. Multiple nevoid basal cell epitheliomata, jaw cysts, bifid rib-a syndrome. N Engl J Med. 1960;262:908-911.
- Evans DGR, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
- Farndon PA, Del Mastro RG, Evans DG, et al. Location of gene for Gorlin Syndrome. Lancet. 1992;339:581-582.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10:757-761.
- Evans DGR, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460-464.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al. Clinical findings in two African-American families with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1994;50:272-281.
- Shanley S, Ratcliffe J, Hockey A, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282-290.
- Bree AF, Shah MR; BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155:2091-2097.
- Doctoroff A, Oberlender SA, Purcell SM. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg. 2003;29:1236-1240.
- Nouri K, Chang A, Trent JT, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg. 2002;28:287-290.
- Itkin A, Gilchrest BA. δ-Aminolevulinic acid and blue light photodynamic therapy for treatment of multiple basal cell carcinomas in two patients with nevoid basal cell carcinoma syndrome. Dermatol Surg. 2004;30:1054-1061.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141:60-67.
- Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146:552-567.
- Wolf P, Fink-Puches R, Reimann-Weber A, et al. Development of malignant melanoma after repeated topical photodynamic therapy with 5-aminolevulinic acid at the exposed site. Dermatology. 1997;194:53-54.
- Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-733.
- Kagy MK, Amonette R. The use of imiquimod 5% cream for the treatment of superficial basal cell carcinomas in a basal cell nevus syndrome patient. Dermatol Surg. 2000;26:577-579.
- Micali G, Lacarrubba F, Nasca MR, et al. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin Exp Dermatol. 2003;28:19-23.
- Strange PR, Lang PG. Long-term management of basal cell nevus syndrome with topical tretinoin and 5-fluorouracil. J Am Acad Dermatol. 1992;27:842-845.
- Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol. 1989;15:868-871.
- Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst. 1992;84:328-332.
Practice Points
- Nevoid basal cell carcinoma syndrome (NBCCS) is a multisystem disorder that requires close monitoring under multidisciplinary care.
- The clinical manifestations of NBCCS include multiple basal cell carcinomas, odontogenic keratocysts, palmar or plantar pits, and calcification of the falx cerebri.