User login
Poor-prognosis cancers linked to highest suicide risk in first year
Suicide risk significantly increases within the first year of a cancer diagnosis, with risk varying by type of cancer, according to investigators who conducted a retrospective analysis representing nearly 4.7 million patients.
Risk of suicide in that first year after diagnosis was especially high in pancreatic and lung cancers, while by contrast, breast and prostate cancer did not increase suicide risk, reported the researchers, led by Hesham Hamoda, MD, MPH, of Boston Children’s Hospital/Harvard Medical School, and Ahmad Alfaar, MBBCh, MSc, of Charité–Universitätsmedizin Berlin.
That variation in suicide risk by cancer type suggests that prognosis and 5-year relative survival play a role in increasing suicide rates, according to Dr. Hamoda, Dr. Alfaar, and their coauthors.
“After the diagnosis, it is important that health care providers be vigilant in screening for suicide and ensuring that patients have access to social and emotional support,” they wrote in a report published in Cancer. Their analysis was based on 4,671,989 patients with a diagnosis of cancer in the Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2014. Out of 1,005,825 of those patients who died within the first year of diagnosis, the cause of death was suicide for 1,585, or 0.16%.
Overall, the risk of suicide increased significantly among cancer patients versus the general population, with an observed-to-expected (O/E) ratio of 2.51 per 10,000 person-years, the investigators found. The risk was highest in the first 6 months, with an O/E mortality of 3.13 versus 1.8 in the latter 6 months.
The highest ratios were seen for pancreatic cancer, with an O/E ratio of 8.01, and lung cancer, with a ratio of 6.05, the researchers found in further analysis.
Significant increases in suicide risk were also seen for colorectal cancer (2.08) and melanoma (1.45), though rates were not significantly different versus the general population for breast (1.23) and prostate (0.99), according to the reported data.
Suicide risk was relatively high for any cancer with distant metastases (5.63), though still significantly higher at 1.65 in persons with localized/regional disease, the data show.
The increased suicide risk persisted more than 1 year after the cancer diagnosis, though not to the degree observed within that first year, they added.
Most patients with suicide as a cause of death were white (90.2%) and male (87%). Nearly 60% were between the ages of 65 and 84 at the time of suicide.
Social support plays an integral role in suicide prevention among cancer patients, the researchers noted.
Previous studies suggest that support programs may decrease suicide risk by making patients better aware of their prognosis, receptive to decreased social stigma, or less likely to have stress related to cost of care, they said.
“Discussing the quality of life after diagnosis, the effectiveness of therapy, and the prognosis of the disease and maintaining a trusting relationship with health care professionals all decrease the likelihood of suicide immediately after a diagnosis of cancer,” they said.
Dr. Hamoda, Dr. Alfaar, and their coauthors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service (Dr. Alfaar).
SOURCE: Saad AM, et al. Cancer 2019 Jan 7. doi: 10.1002/cncr.31876.
Suicide risk significantly increases within the first year of a cancer diagnosis, with risk varying by type of cancer, according to investigators who conducted a retrospective analysis representing nearly 4.7 million patients.
Risk of suicide in that first year after diagnosis was especially high in pancreatic and lung cancers, while by contrast, breast and prostate cancer did not increase suicide risk, reported the researchers, led by Hesham Hamoda, MD, MPH, of Boston Children’s Hospital/Harvard Medical School, and Ahmad Alfaar, MBBCh, MSc, of Charité–Universitätsmedizin Berlin.
That variation in suicide risk by cancer type suggests that prognosis and 5-year relative survival play a role in increasing suicide rates, according to Dr. Hamoda, Dr. Alfaar, and their coauthors.
“After the diagnosis, it is important that health care providers be vigilant in screening for suicide and ensuring that patients have access to social and emotional support,” they wrote in a report published in Cancer. Their analysis was based on 4,671,989 patients with a diagnosis of cancer in the Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2014. Out of 1,005,825 of those patients who died within the first year of diagnosis, the cause of death was suicide for 1,585, or 0.16%.
Overall, the risk of suicide increased significantly among cancer patients versus the general population, with an observed-to-expected (O/E) ratio of 2.51 per 10,000 person-years, the investigators found. The risk was highest in the first 6 months, with an O/E mortality of 3.13 versus 1.8 in the latter 6 months.
The highest ratios were seen for pancreatic cancer, with an O/E ratio of 8.01, and lung cancer, with a ratio of 6.05, the researchers found in further analysis.
Significant increases in suicide risk were also seen for colorectal cancer (2.08) and melanoma (1.45), though rates were not significantly different versus the general population for breast (1.23) and prostate (0.99), according to the reported data.
Suicide risk was relatively high for any cancer with distant metastases (5.63), though still significantly higher at 1.65 in persons with localized/regional disease, the data show.
The increased suicide risk persisted more than 1 year after the cancer diagnosis, though not to the degree observed within that first year, they added.
Most patients with suicide as a cause of death were white (90.2%) and male (87%). Nearly 60% were between the ages of 65 and 84 at the time of suicide.
Social support plays an integral role in suicide prevention among cancer patients, the researchers noted.
Previous studies suggest that support programs may decrease suicide risk by making patients better aware of their prognosis, receptive to decreased social stigma, or less likely to have stress related to cost of care, they said.
“Discussing the quality of life after diagnosis, the effectiveness of therapy, and the prognosis of the disease and maintaining a trusting relationship with health care professionals all decrease the likelihood of suicide immediately after a diagnosis of cancer,” they said.
Dr. Hamoda, Dr. Alfaar, and their coauthors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service (Dr. Alfaar).
SOURCE: Saad AM, et al. Cancer 2019 Jan 7. doi: 10.1002/cncr.31876.
Suicide risk significantly increases within the first year of a cancer diagnosis, with risk varying by type of cancer, according to investigators who conducted a retrospective analysis representing nearly 4.7 million patients.
Risk of suicide in that first year after diagnosis was especially high in pancreatic and lung cancers, while by contrast, breast and prostate cancer did not increase suicide risk, reported the researchers, led by Hesham Hamoda, MD, MPH, of Boston Children’s Hospital/Harvard Medical School, and Ahmad Alfaar, MBBCh, MSc, of Charité–Universitätsmedizin Berlin.
That variation in suicide risk by cancer type suggests that prognosis and 5-year relative survival play a role in increasing suicide rates, according to Dr. Hamoda, Dr. Alfaar, and their coauthors.
“After the diagnosis, it is important that health care providers be vigilant in screening for suicide and ensuring that patients have access to social and emotional support,” they wrote in a report published in Cancer. Their analysis was based on 4,671,989 patients with a diagnosis of cancer in the Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2014. Out of 1,005,825 of those patients who died within the first year of diagnosis, the cause of death was suicide for 1,585, or 0.16%.
Overall, the risk of suicide increased significantly among cancer patients versus the general population, with an observed-to-expected (O/E) ratio of 2.51 per 10,000 person-years, the investigators found. The risk was highest in the first 6 months, with an O/E mortality of 3.13 versus 1.8 in the latter 6 months.
The highest ratios were seen for pancreatic cancer, with an O/E ratio of 8.01, and lung cancer, with a ratio of 6.05, the researchers found in further analysis.
Significant increases in suicide risk were also seen for colorectal cancer (2.08) and melanoma (1.45), though rates were not significantly different versus the general population for breast (1.23) and prostate (0.99), according to the reported data.
Suicide risk was relatively high for any cancer with distant metastases (5.63), though still significantly higher at 1.65 in persons with localized/regional disease, the data show.
The increased suicide risk persisted more than 1 year after the cancer diagnosis, though not to the degree observed within that first year, they added.
Most patients with suicide as a cause of death were white (90.2%) and male (87%). Nearly 60% were between the ages of 65 and 84 at the time of suicide.
Social support plays an integral role in suicide prevention among cancer patients, the researchers noted.
Previous studies suggest that support programs may decrease suicide risk by making patients better aware of their prognosis, receptive to decreased social stigma, or less likely to have stress related to cost of care, they said.
“Discussing the quality of life after diagnosis, the effectiveness of therapy, and the prognosis of the disease and maintaining a trusting relationship with health care professionals all decrease the likelihood of suicide immediately after a diagnosis of cancer,” they said.
Dr. Hamoda, Dr. Alfaar, and their coauthors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service (Dr. Alfaar).
SOURCE: Saad AM, et al. Cancer 2019 Jan 7. doi: 10.1002/cncr.31876.
FROM CANCER
Key clinical point: A cancer diagnosis significantly increases risk of suicide in comparison to the general population, particularly for poorer-prognosis cancers.
Major finding: The observed-to-expected mortality ratio was substantially higher for pancreatic cancer (8.01), and lung cancer (6.05), but not significantly increased for breast (1.23) and prostate (0.99).
Study details: A retrospective population-based study of 4,671,989 cancer patients.
Disclosures: The authors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service.
Source: Saad AM et al. Cancer. 2019 Jan 7. doi: 10.1002/cncr.31876.
No link between sex and survival on checkpoint inhibitors in latest meta-analysis
Men and women with cancer may derive a similar survival benefit from immune checkpoint inhibitor therapy, results of a recent meta-analysis suggest.
Both men and women had an overall survival benefit from immunotherapy versus standard of care therapy, with no significant difference between the sexes, according to authors of this meta-analysis, which included 23 randomized clinical trials comprising nearly 14,000 patients.
The findings, reported in JAMA Oncology, contrast with those of another recent analysis, which suggested that men had a greater advantage of receiving immunotherapy versus standard of care than women did.
“We found no evidence that sex should be considered when deciding whether to offer immunotherapy to patients with advanced cancers,” said Christopher J.D. Wallis, MD, PhD, of the University of Toronto, and his coauthors said in their report.
The present meta-analysis was based on a “more contemporary and comprehensive” literature search strategy than the earlier one, according to Dr. Wallis and his coinvestigators.
Specifically, they considered immunotherapy agents not included in the previous analysis, added seven new studies published since the previous analysis, and excluded three trials that compared immunotherapy agents, rather than comparing immunotherapy with standard of care, they explained in their report.
Their resulting meta-analysis included a total of 9,322 men and 4,399 women, most of whom were in their 70s. Overall, they found that immune checkpoint inhibitor therapy offered a statistically significant overall survival advantage versus standard systemic therapy, with a hazard ratio of 0.75 (95% confidence interval, 0.70-0.81; P less than .001).
That overall survival advantage was found for both men, with a hazard ratio of 0.75 (95% CI, 0.69-0.81; P less than .001) and women, at 0.77 (95% CI, 0.67-0.88; P = .002), investigators further reported. There was no statistically significant difference in overall survival advantage between men and women, both overall (P = 0.60) and in subgroup analyses that accounted for tumor type, line of treatment, and prevalence of women in the study.
The previous meta-analysis, published in the Lancet, found an overall survival hazard ratio of 0.72 for men receiving checkpoint inhibitors and 0.86 for women receiving checkpoint inhibitors (P = .0019), prompting those investigators to conclude that the magnitude of benefit was sex-dependent and that different immunotherapeutic approaches may be needed for men versus women.
“The present meta-analysis provides a more specific assessment of the research question while including a greater number of immunotherapy agents and an updated search,” Dr. Wallis and his coauthors said in a discussion of their more recent findings.
Dr. Wallis reported no disclosures related to the study. Study coauthors provided disclosures related to Merck, AstraZeneca, Bristol-Myers Squibb, Illumina, Tempus, Novartis, Eli Lilly, Fate, Incyte, MedImmune, Pfizer, Roche/Genentech, Xcovery, Fate Therapeutics, Genocea, and Iovance.
SOURCE: Wallis CJD et al. JAMA Oncol. 2019 Jan 3. doi:10.1001/jamaoncol.2018.5904.
Men and women with cancer may derive a similar survival benefit from immune checkpoint inhibitor therapy, results of a recent meta-analysis suggest.
Both men and women had an overall survival benefit from immunotherapy versus standard of care therapy, with no significant difference between the sexes, according to authors of this meta-analysis, which included 23 randomized clinical trials comprising nearly 14,000 patients.
The findings, reported in JAMA Oncology, contrast with those of another recent analysis, which suggested that men had a greater advantage of receiving immunotherapy versus standard of care than women did.
“We found no evidence that sex should be considered when deciding whether to offer immunotherapy to patients with advanced cancers,” said Christopher J.D. Wallis, MD, PhD, of the University of Toronto, and his coauthors said in their report.
The present meta-analysis was based on a “more contemporary and comprehensive” literature search strategy than the earlier one, according to Dr. Wallis and his coinvestigators.
Specifically, they considered immunotherapy agents not included in the previous analysis, added seven new studies published since the previous analysis, and excluded three trials that compared immunotherapy agents, rather than comparing immunotherapy with standard of care, they explained in their report.
Their resulting meta-analysis included a total of 9,322 men and 4,399 women, most of whom were in their 70s. Overall, they found that immune checkpoint inhibitor therapy offered a statistically significant overall survival advantage versus standard systemic therapy, with a hazard ratio of 0.75 (95% confidence interval, 0.70-0.81; P less than .001).
That overall survival advantage was found for both men, with a hazard ratio of 0.75 (95% CI, 0.69-0.81; P less than .001) and women, at 0.77 (95% CI, 0.67-0.88; P = .002), investigators further reported. There was no statistically significant difference in overall survival advantage between men and women, both overall (P = 0.60) and in subgroup analyses that accounted for tumor type, line of treatment, and prevalence of women in the study.
The previous meta-analysis, published in the Lancet, found an overall survival hazard ratio of 0.72 for men receiving checkpoint inhibitors and 0.86 for women receiving checkpoint inhibitors (P = .0019), prompting those investigators to conclude that the magnitude of benefit was sex-dependent and that different immunotherapeutic approaches may be needed for men versus women.
“The present meta-analysis provides a more specific assessment of the research question while including a greater number of immunotherapy agents and an updated search,” Dr. Wallis and his coauthors said in a discussion of their more recent findings.
Dr. Wallis reported no disclosures related to the study. Study coauthors provided disclosures related to Merck, AstraZeneca, Bristol-Myers Squibb, Illumina, Tempus, Novartis, Eli Lilly, Fate, Incyte, MedImmune, Pfizer, Roche/Genentech, Xcovery, Fate Therapeutics, Genocea, and Iovance.
SOURCE: Wallis CJD et al. JAMA Oncol. 2019 Jan 3. doi:10.1001/jamaoncol.2018.5904.
Men and women with cancer may derive a similar survival benefit from immune checkpoint inhibitor therapy, results of a recent meta-analysis suggest.
Both men and women had an overall survival benefit from immunotherapy versus standard of care therapy, with no significant difference between the sexes, according to authors of this meta-analysis, which included 23 randomized clinical trials comprising nearly 14,000 patients.
The findings, reported in JAMA Oncology, contrast with those of another recent analysis, which suggested that men had a greater advantage of receiving immunotherapy versus standard of care than women did.
“We found no evidence that sex should be considered when deciding whether to offer immunotherapy to patients with advanced cancers,” said Christopher J.D. Wallis, MD, PhD, of the University of Toronto, and his coauthors said in their report.
The present meta-analysis was based on a “more contemporary and comprehensive” literature search strategy than the earlier one, according to Dr. Wallis and his coinvestigators.
Specifically, they considered immunotherapy agents not included in the previous analysis, added seven new studies published since the previous analysis, and excluded three trials that compared immunotherapy agents, rather than comparing immunotherapy with standard of care, they explained in their report.
Their resulting meta-analysis included a total of 9,322 men and 4,399 women, most of whom were in their 70s. Overall, they found that immune checkpoint inhibitor therapy offered a statistically significant overall survival advantage versus standard systemic therapy, with a hazard ratio of 0.75 (95% confidence interval, 0.70-0.81; P less than .001).
That overall survival advantage was found for both men, with a hazard ratio of 0.75 (95% CI, 0.69-0.81; P less than .001) and women, at 0.77 (95% CI, 0.67-0.88; P = .002), investigators further reported. There was no statistically significant difference in overall survival advantage between men and women, both overall (P = 0.60) and in subgroup analyses that accounted for tumor type, line of treatment, and prevalence of women in the study.
The previous meta-analysis, published in the Lancet, found an overall survival hazard ratio of 0.72 for men receiving checkpoint inhibitors and 0.86 for women receiving checkpoint inhibitors (P = .0019), prompting those investigators to conclude that the magnitude of benefit was sex-dependent and that different immunotherapeutic approaches may be needed for men versus women.
“The present meta-analysis provides a more specific assessment of the research question while including a greater number of immunotherapy agents and an updated search,” Dr. Wallis and his coauthors said in a discussion of their more recent findings.
Dr. Wallis reported no disclosures related to the study. Study coauthors provided disclosures related to Merck, AstraZeneca, Bristol-Myers Squibb, Illumina, Tempus, Novartis, Eli Lilly, Fate, Incyte, MedImmune, Pfizer, Roche/Genentech, Xcovery, Fate Therapeutics, Genocea, and Iovance.
SOURCE: Wallis CJD et al. JAMA Oncol. 2019 Jan 3. doi:10.1001/jamaoncol.2018.5904.
FROM JAMA ONCOLOGY
Key clinical point: In contrast to an earlier meta-analysis, a more recent meta-analysis suggests no difference in overall survival benefit between men and women with cancer receiving immune checkpoint inhibitor therapy.
Major finding: An overall survival advantage was found for both men and women receiving checkpoint inhibitors versus standard therapy, with hazard ratios of 0.75 and 0.77, respectively (P = .60).
Study details: A systematic review and meta-analysis including nearly 14,000 patients in 23 randomized, clinical trials.
Disclosures: Study authors provided disclosures related to Merck, AstraZeneca, Bristol-Myers Squibb, Illumina, Tempus, Novartis, Eli Lilly, Fate, Incyte, MedImmune, Pfizer, Roche/Genentech, Xcovery, Fate Therapeutics, Genocea, and Iovance.
Source: Wallis CJD et al. JAMA Oncol. 2019 Jan 3. doi:10.1001/jamaoncol.2018.5904.
Frontline veliparib/cisplatin/etoposide shows efficacy in advanced SCLC
For patients with extensive-stage small-cell lung cancer (ES-SCLC), the addition of the poly (ADP ribose) polymerase (PARP) inhibitor veliparib to frontline chemotherapy with cisplatin and etoposide resulted in a slight but significant improvement in progression-free survival but not overall survival, compared with cisplatin/etoposide alone, investigators reported in the Journal of Clinical Oncology.
Among 128 patients with newly diagnosed ES-SCLC, the median progression-free survival (PFS; the primary endpoint) for those randomized to veliparib/cisplatin/etoposide was 6.1 months, compared with 5.5 months for patients randomized to cisplatin/etoposide alone.
This translated into an unstratified hazard ratio for PFS with veliparib of 0.75 (one-sided P = .06) and a stratified HR of 0.63 (one-sided P = .01), reported Taofeek K. Owonikoko, MD, PhD, and his colleagues at Emory University in Atlanta.
“Although the initial result of our study is promising, additional confirmation in a larger definitive study is warranted, given the mixed results reported by other studies of PARP inhibitors in this patient population,” they wrote.
Median overall survival (OS) with cisplatin/etoposide in ES-SCLC is approximately 9-11 months, and fewer than 5% of patients survive out to 5 years. To see whether the addition of a PARP inhibitor to the standard of care could improve outcomes, the investigators first demonstrated in a phase 1 trial that the combination of veliparib with a platinum doublet of cisplatin and etoposide was safe (Lung Cancer. 2015 Jul;89[1]:66-70), and in the current study, they evaluated efficacy.
A total of 128 eligible patients (median age, 66 years; 52% men) with ES-SCLC were included in the analysis. Extensive-stage disease was defined as the presence of extrathoracic metastatic disease, malignant pleural effusion, and bilateral or contralateral supraclavicular adenopathy.
The patients were stratified by sex and serum lactate dehydrogenase (LDH) levels and then randomized to receive a maximum of four 3-week cycles of of cisplatin 75 mg/m2 intravenously on day 1 and etoposide 100 mg/m2 on days 1 through 3, plus either oral veliparib 100 mg twice daily on days 1 through 7 or placebo.
The primary endpoint of PFS was as noted before. Median overall survival was 10.3 months in the veliparib arm versus 8.9 months in the cisplatin/etoposide alone arm, a difference that was not statistically significant. The respective overall response rates were 71.9% vs. 65.6%, but this difference was also not significant.
Looking at the treatment effect by strata, the investigators found that men with high serum LDH levels had a significant PFS benefit with veliparib (HR, 0.34; one-sided P less than .001), but no significant benefit was seen in men with normal LDH or in women regardless of LDH status.
Grade 3 or greater hematologic toxicities that occurred more frequently in the veliparib arm included CD4 lymphopenia in 8% vs. 0% and neutropenia in 49% vs. 32%.
The investigators noted that the discrepancy between the magnitude of the risk reduction as measured by the hazard ratio and the actual, modest difference in median PFS between the study arms may be explained by the fact that men with elevated LDH represented the largest patient strata.
“Therefore, we hypothesize that this cohort probably contained a sufficient proportion of patients with SCLC who harbored some biologic vulnerability to this therapeutic strategy,” they wrote.
They acknowledged that although toxicities were higher with veliparib combination, the ability to deliver chemotherapy was equal between the arms.
Further exploration of the combination of veliparib/cisplatin/etoposide may be justified if results of another ongoing phase 2 study (NCT02289690) of a carboplatin-based chemotherapy doublet plus veliparib has similar efficacy results, Dr. Owonikoko and his associates concluded.
The study was coordinated by the Eastern Cooperative Oncology Group–American College of Radiology Imaging Network Cancer Research Group and supported by the National Cancer Institute. Dr. Owonikoko disclosed a consulting or advisory role with AbbVie, developer of veliparib, and other companies, as well as institutional research funding from AbbVie and others.
SOURCE: Owonikoko TK et al. J Clin Oncol. 2018 Dec 5. doi: 10.1200/JCO.18.00264.
For patients with extensive-stage small-cell lung cancer (ES-SCLC), the addition of the poly (ADP ribose) polymerase (PARP) inhibitor veliparib to frontline chemotherapy with cisplatin and etoposide resulted in a slight but significant improvement in progression-free survival but not overall survival, compared with cisplatin/etoposide alone, investigators reported in the Journal of Clinical Oncology.
Among 128 patients with newly diagnosed ES-SCLC, the median progression-free survival (PFS; the primary endpoint) for those randomized to veliparib/cisplatin/etoposide was 6.1 months, compared with 5.5 months for patients randomized to cisplatin/etoposide alone.
This translated into an unstratified hazard ratio for PFS with veliparib of 0.75 (one-sided P = .06) and a stratified HR of 0.63 (one-sided P = .01), reported Taofeek K. Owonikoko, MD, PhD, and his colleagues at Emory University in Atlanta.
“Although the initial result of our study is promising, additional confirmation in a larger definitive study is warranted, given the mixed results reported by other studies of PARP inhibitors in this patient population,” they wrote.
Median overall survival (OS) with cisplatin/etoposide in ES-SCLC is approximately 9-11 months, and fewer than 5% of patients survive out to 5 years. To see whether the addition of a PARP inhibitor to the standard of care could improve outcomes, the investigators first demonstrated in a phase 1 trial that the combination of veliparib with a platinum doublet of cisplatin and etoposide was safe (Lung Cancer. 2015 Jul;89[1]:66-70), and in the current study, they evaluated efficacy.
A total of 128 eligible patients (median age, 66 years; 52% men) with ES-SCLC were included in the analysis. Extensive-stage disease was defined as the presence of extrathoracic metastatic disease, malignant pleural effusion, and bilateral or contralateral supraclavicular adenopathy.
The patients were stratified by sex and serum lactate dehydrogenase (LDH) levels and then randomized to receive a maximum of four 3-week cycles of of cisplatin 75 mg/m2 intravenously on day 1 and etoposide 100 mg/m2 on days 1 through 3, plus either oral veliparib 100 mg twice daily on days 1 through 7 or placebo.
The primary endpoint of PFS was as noted before. Median overall survival was 10.3 months in the veliparib arm versus 8.9 months in the cisplatin/etoposide alone arm, a difference that was not statistically significant. The respective overall response rates were 71.9% vs. 65.6%, but this difference was also not significant.
Looking at the treatment effect by strata, the investigators found that men with high serum LDH levels had a significant PFS benefit with veliparib (HR, 0.34; one-sided P less than .001), but no significant benefit was seen in men with normal LDH or in women regardless of LDH status.
Grade 3 or greater hematologic toxicities that occurred more frequently in the veliparib arm included CD4 lymphopenia in 8% vs. 0% and neutropenia in 49% vs. 32%.
The investigators noted that the discrepancy between the magnitude of the risk reduction as measured by the hazard ratio and the actual, modest difference in median PFS between the study arms may be explained by the fact that men with elevated LDH represented the largest patient strata.
“Therefore, we hypothesize that this cohort probably contained a sufficient proportion of patients with SCLC who harbored some biologic vulnerability to this therapeutic strategy,” they wrote.
They acknowledged that although toxicities were higher with veliparib combination, the ability to deliver chemotherapy was equal between the arms.
Further exploration of the combination of veliparib/cisplatin/etoposide may be justified if results of another ongoing phase 2 study (NCT02289690) of a carboplatin-based chemotherapy doublet plus veliparib has similar efficacy results, Dr. Owonikoko and his associates concluded.
The study was coordinated by the Eastern Cooperative Oncology Group–American College of Radiology Imaging Network Cancer Research Group and supported by the National Cancer Institute. Dr. Owonikoko disclosed a consulting or advisory role with AbbVie, developer of veliparib, and other companies, as well as institutional research funding from AbbVie and others.
SOURCE: Owonikoko TK et al. J Clin Oncol. 2018 Dec 5. doi: 10.1200/JCO.18.00264.
For patients with extensive-stage small-cell lung cancer (ES-SCLC), the addition of the poly (ADP ribose) polymerase (PARP) inhibitor veliparib to frontline chemotherapy with cisplatin and etoposide resulted in a slight but significant improvement in progression-free survival but not overall survival, compared with cisplatin/etoposide alone, investigators reported in the Journal of Clinical Oncology.
Among 128 patients with newly diagnosed ES-SCLC, the median progression-free survival (PFS; the primary endpoint) for those randomized to veliparib/cisplatin/etoposide was 6.1 months, compared with 5.5 months for patients randomized to cisplatin/etoposide alone.
This translated into an unstratified hazard ratio for PFS with veliparib of 0.75 (one-sided P = .06) and a stratified HR of 0.63 (one-sided P = .01), reported Taofeek K. Owonikoko, MD, PhD, and his colleagues at Emory University in Atlanta.
“Although the initial result of our study is promising, additional confirmation in a larger definitive study is warranted, given the mixed results reported by other studies of PARP inhibitors in this patient population,” they wrote.
Median overall survival (OS) with cisplatin/etoposide in ES-SCLC is approximately 9-11 months, and fewer than 5% of patients survive out to 5 years. To see whether the addition of a PARP inhibitor to the standard of care could improve outcomes, the investigators first demonstrated in a phase 1 trial that the combination of veliparib with a platinum doublet of cisplatin and etoposide was safe (Lung Cancer. 2015 Jul;89[1]:66-70), and in the current study, they evaluated efficacy.
A total of 128 eligible patients (median age, 66 years; 52% men) with ES-SCLC were included in the analysis. Extensive-stage disease was defined as the presence of extrathoracic metastatic disease, malignant pleural effusion, and bilateral or contralateral supraclavicular adenopathy.
The patients were stratified by sex and serum lactate dehydrogenase (LDH) levels and then randomized to receive a maximum of four 3-week cycles of of cisplatin 75 mg/m2 intravenously on day 1 and etoposide 100 mg/m2 on days 1 through 3, plus either oral veliparib 100 mg twice daily on days 1 through 7 or placebo.
The primary endpoint of PFS was as noted before. Median overall survival was 10.3 months in the veliparib arm versus 8.9 months in the cisplatin/etoposide alone arm, a difference that was not statistically significant. The respective overall response rates were 71.9% vs. 65.6%, but this difference was also not significant.
Looking at the treatment effect by strata, the investigators found that men with high serum LDH levels had a significant PFS benefit with veliparib (HR, 0.34; one-sided P less than .001), but no significant benefit was seen in men with normal LDH or in women regardless of LDH status.
Grade 3 or greater hematologic toxicities that occurred more frequently in the veliparib arm included CD4 lymphopenia in 8% vs. 0% and neutropenia in 49% vs. 32%.
The investigators noted that the discrepancy between the magnitude of the risk reduction as measured by the hazard ratio and the actual, modest difference in median PFS between the study arms may be explained by the fact that men with elevated LDH represented the largest patient strata.
“Therefore, we hypothesize that this cohort probably contained a sufficient proportion of patients with SCLC who harbored some biologic vulnerability to this therapeutic strategy,” they wrote.
They acknowledged that although toxicities were higher with veliparib combination, the ability to deliver chemotherapy was equal between the arms.
Further exploration of the combination of veliparib/cisplatin/etoposide may be justified if results of another ongoing phase 2 study (NCT02289690) of a carboplatin-based chemotherapy doublet plus veliparib has similar efficacy results, Dr. Owonikoko and his associates concluded.
The study was coordinated by the Eastern Cooperative Oncology Group–American College of Radiology Imaging Network Cancer Research Group and supported by the National Cancer Institute. Dr. Owonikoko disclosed a consulting or advisory role with AbbVie, developer of veliparib, and other companies, as well as institutional research funding from AbbVie and others.
SOURCE: Owonikoko TK et al. J Clin Oncol. 2018 Dec 5. doi: 10.1200/JCO.18.00264.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Adding a PARP inhibitor to standard chemotherapy may improve progression-free survival in patients with extensive-stage small-cell lung cancer (ES-SCLC).
Major finding: The stratified hazard ratio for PFS with veliparib/cisplatin/etoposide was 0.63 (one-sided P = .01).
Study details: Randomized phase 2 trial in 128 patients with newly diagnosed ES-SCLC.
Disclosures: The study was coordinated by the Eastern Cooperative Oncology Group–American College of Radiology Imaging Network Cancer Research Group and supported by the National Cancer Institute. Dr. Owonikoko disclosed a consulting or advisory role with AbbVie, the developer of veliparib, and other companies, as well as institutional research funding from AbbVie and others.
Source: Owonikoko TK et al. J Clin Oncol. 2018 Dec 5. doi: 10.1200/JCO.18.00264.
ICYMI: Durvalumab boosts overall survival in stage III NSCLC
Patients with unresectable stage III non–small cell lung cancer who received durvalumab had their overall and progression-free survival boosted by about 12 months, compared with patients who received a placebo, according to results of the multicenter, randomized, double-blind, placebo-controlled, phase 3 PACIFIC trial published in the New England Journal of Medicine (2018 Sep 25. doi: 10.1056/NEJMoa1809697).
We covered this story at the World Conference on Lung Cancer before it was published in the journal. Find our coverage at the link below.
Patients with unresectable stage III non–small cell lung cancer who received durvalumab had their overall and progression-free survival boosted by about 12 months, compared with patients who received a placebo, according to results of the multicenter, randomized, double-blind, placebo-controlled, phase 3 PACIFIC trial published in the New England Journal of Medicine (2018 Sep 25. doi: 10.1056/NEJMoa1809697).
We covered this story at the World Conference on Lung Cancer before it was published in the journal. Find our coverage at the link below.
Patients with unresectable stage III non–small cell lung cancer who received durvalumab had their overall and progression-free survival boosted by about 12 months, compared with patients who received a placebo, according to results of the multicenter, randomized, double-blind, placebo-controlled, phase 3 PACIFIC trial published in the New England Journal of Medicine (2018 Sep 25. doi: 10.1056/NEJMoa1809697).
We covered this story at the World Conference on Lung Cancer before it was published in the journal. Find our coverage at the link below.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Perioperative M&M similar for lobar, sublobar surgeries in early lung cancer
Though lobectomy is the long-held standard of care for people with early stage non–small cell lung cancer, a noninferiority study shows little difference in perioperative morbidity and mortality outcomes when sublobar resections are performed instead.
The study, published online in The Lancet Respiratory Medicine, compared results from 697 functionally and physically fit patients with stage I cancer randomized over a 10-year period to lobar resection (n = 357) or sublobar resection (n = 340). Patients were analyzed for morbidity and mortality outcomes at 30 and 90 days post surgery. Nasser K. Altorki, MD, of Weill Cornell Medicine–New York Presbyterian Hospital, led the study as a post hoc, exploratory analysis of CALGB/Alliance 140503, a multinational phase 3 trial whose primary outcome – still pending – is disease-free survival associated with the two different surgeries.
Dr. Altorki and his colleagues found 30- and 90-day survival to be comparable between surgery types. At 30 days, six patients in the study had died; four in the lobar resection group and two in the sublobar group (1.1% and 0.6%). At 90 days, 10 patients had died, or 1.4% of the cohort; 6 following lobar resection and 4 following sublobar resection. The between-group difference at 30 days was 0.5% (95% confidence interval, –1.1 to 2.3) and at 90 days remained 0.5% (95% CI, –1.5 to 2.6).
Similar rates of serious (grade 3 or worse) adverse advents were seen between surgery groups at 15% and 14%, respectively, and no differences were seen for cardiac or pulmonary complications. In the study, the type of sublobar approach was left to the surgeon’s discretion, and a majority of the sublobar procedures (59%) were found to comprise wedge resections, with the rest segmentectomies. Dr. Altorki and colleagues noted the high rate of wedge resections as striking, because “conventional wisdom … holds that an anatomical segmentectomy, involving individual ligation of segmental vessels and bronchi and wider parenchymal resection, is oncologically superior to nonanatomical wedge resections.” In their analysis the researchers conceded that a three-arm trial allocating patients to lobectomy, segmentectomy, or wedge resection “would have answered more precisely the posited research question,” but said that the sample size needed would have been too large.
The study was funded by the National Cancer Institute. Dr. Altorki reported a research grant from AstraZeneca unrelated to the study; two more coauthors disclosed funding from pharmaceutical or device manufacturers, and an additional 17 coauthors listed no competing interests.
SOURCE: Altorki NK et al. Lancet Respir Med. 2018 Nov 12. doi: 10.1016/S2213-2600(18)30411-9 .
Though lobectomy is the long-held standard of care for people with early stage non–small cell lung cancer, a noninferiority study shows little difference in perioperative morbidity and mortality outcomes when sublobar resections are performed instead.
The study, published online in The Lancet Respiratory Medicine, compared results from 697 functionally and physically fit patients with stage I cancer randomized over a 10-year period to lobar resection (n = 357) or sublobar resection (n = 340). Patients were analyzed for morbidity and mortality outcomes at 30 and 90 days post surgery. Nasser K. Altorki, MD, of Weill Cornell Medicine–New York Presbyterian Hospital, led the study as a post hoc, exploratory analysis of CALGB/Alliance 140503, a multinational phase 3 trial whose primary outcome – still pending – is disease-free survival associated with the two different surgeries.
Dr. Altorki and his colleagues found 30- and 90-day survival to be comparable between surgery types. At 30 days, six patients in the study had died; four in the lobar resection group and two in the sublobar group (1.1% and 0.6%). At 90 days, 10 patients had died, or 1.4% of the cohort; 6 following lobar resection and 4 following sublobar resection. The between-group difference at 30 days was 0.5% (95% confidence interval, –1.1 to 2.3) and at 90 days remained 0.5% (95% CI, –1.5 to 2.6).
Similar rates of serious (grade 3 or worse) adverse advents were seen between surgery groups at 15% and 14%, respectively, and no differences were seen for cardiac or pulmonary complications. In the study, the type of sublobar approach was left to the surgeon’s discretion, and a majority of the sublobar procedures (59%) were found to comprise wedge resections, with the rest segmentectomies. Dr. Altorki and colleagues noted the high rate of wedge resections as striking, because “conventional wisdom … holds that an anatomical segmentectomy, involving individual ligation of segmental vessels and bronchi and wider parenchymal resection, is oncologically superior to nonanatomical wedge resections.” In their analysis the researchers conceded that a three-arm trial allocating patients to lobectomy, segmentectomy, or wedge resection “would have answered more precisely the posited research question,” but said that the sample size needed would have been too large.
The study was funded by the National Cancer Institute. Dr. Altorki reported a research grant from AstraZeneca unrelated to the study; two more coauthors disclosed funding from pharmaceutical or device manufacturers, and an additional 17 coauthors listed no competing interests.
SOURCE: Altorki NK et al. Lancet Respir Med. 2018 Nov 12. doi: 10.1016/S2213-2600(18)30411-9 .
Though lobectomy is the long-held standard of care for people with early stage non–small cell lung cancer, a noninferiority study shows little difference in perioperative morbidity and mortality outcomes when sublobar resections are performed instead.
The study, published online in The Lancet Respiratory Medicine, compared results from 697 functionally and physically fit patients with stage I cancer randomized over a 10-year period to lobar resection (n = 357) or sublobar resection (n = 340). Patients were analyzed for morbidity and mortality outcomes at 30 and 90 days post surgery. Nasser K. Altorki, MD, of Weill Cornell Medicine–New York Presbyterian Hospital, led the study as a post hoc, exploratory analysis of CALGB/Alliance 140503, a multinational phase 3 trial whose primary outcome – still pending – is disease-free survival associated with the two different surgeries.
Dr. Altorki and his colleagues found 30- and 90-day survival to be comparable between surgery types. At 30 days, six patients in the study had died; four in the lobar resection group and two in the sublobar group (1.1% and 0.6%). At 90 days, 10 patients had died, or 1.4% of the cohort; 6 following lobar resection and 4 following sublobar resection. The between-group difference at 30 days was 0.5% (95% confidence interval, –1.1 to 2.3) and at 90 days remained 0.5% (95% CI, –1.5 to 2.6).
Similar rates of serious (grade 3 or worse) adverse advents were seen between surgery groups at 15% and 14%, respectively, and no differences were seen for cardiac or pulmonary complications. In the study, the type of sublobar approach was left to the surgeon’s discretion, and a majority of the sublobar procedures (59%) were found to comprise wedge resections, with the rest segmentectomies. Dr. Altorki and colleagues noted the high rate of wedge resections as striking, because “conventional wisdom … holds that an anatomical segmentectomy, involving individual ligation of segmental vessels and bronchi and wider parenchymal resection, is oncologically superior to nonanatomical wedge resections.” In their analysis the researchers conceded that a three-arm trial allocating patients to lobectomy, segmentectomy, or wedge resection “would have answered more precisely the posited research question,” but said that the sample size needed would have been too large.
The study was funded by the National Cancer Institute. Dr. Altorki reported a research grant from AstraZeneca unrelated to the study; two more coauthors disclosed funding from pharmaceutical or device manufacturers, and an additional 17 coauthors listed no competing interests.
SOURCE: Altorki NK et al. Lancet Respir Med. 2018 Nov 12. doi: 10.1016/S2213-2600(18)30411-9 .
FROM THE LANCET RESPIRATORY MEDICINE
Key clinical point: Patients with 30 and 90 days post surgery.
Major finding: Mortality at 30 days and 90 days was 0.5% for both trial groups and serious adverse advents were similar between groups.
Study details: A post hoc analysis from a multinational trial randomizing about 700 stage I NSCLC patients to lobar or sublobar surgery
Disclosures: National Cancer Institute sponsored the study; three authors including the lead author reported financial ties to manufacturers.
Source: Altorki et al. Lancet Respir Med. 2018 Nov 12. doi: 10.1016/S2213-2600(18)30411-9.
Atezolizumab combination regimen approved for advanced non-squamous NSCLC
The Food and Drug Administration has approved atezolizumab (Tecentriq) in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations.

Approval was based on greater overall survival (OS) among patients receiving the four drug combination, compared with patients who did not receive the checkpoint inhibitor but received the other three drugs in the randomized IMpower150 trial.
For the trial, 1,202 patients with metastatic NSq NSCLC were randomized to three arms for first-line treatment:
• atezolizumab, carboplatin, paclitaxel, and bevacizumab (4-drug regimen);
• atezolizumab, carboplatin and paclitaxel (3-drug regimen); or
• carboplatin, paclitaxel, and bevacizumab (control arm).
Among patients with NSq NSCLC without an EGFR or ALK mutation (87%), the estimated median OS was 19.2 months for patients receiving the 4-drug regimen and 14.7 months for those in the control arm (hazard ratio [HR] 0.78; 95% CI: 0.64, 0.96; P = .016), the FDA said in a press statement announcing the approval.
The median progression-free survival was 8.5 months for patients receiving the 4-drug regimen and 7.0 months for those in the control arm (HR 0.71; 95% CI 0.59, 0.85; P = .0002). The overall response rates were 55% in the 4-drug arm and 42% in the control arm. There were no significant differences in OS or final progression-free survival between the 3-drug arm containing atezolizumab and the control arm.
The most common adverse reactions with atezolizumab were fatigue/asthenia, alopecia, nausea, diarrhea, constipation, decreased appetite, arthralgia, hypertension, and neuropathy. Treatment with atezolizumab was discontinued in 15% of patients due to adverse reactions, the most common reason being pneumonitis.
The recommended atezolizumab dose is 1,200 mg intravenously over 60 minutes every 3 weeks, the FDA said.
The Food and Drug Administration has approved atezolizumab (Tecentriq) in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations.
Approval was based on greater overall survival (OS) among patients receiving the four drug combination, compared with patients who did not receive the checkpoint inhibitor but received the other three drugs in the randomized IMpower150 trial.
For the trial, 1,202 patients with metastatic NSq NSCLC were randomized to three arms for first-line treatment:
• atezolizumab, carboplatin, paclitaxel, and bevacizumab (4-drug regimen);
• atezolizumab, carboplatin and paclitaxel (3-drug regimen); or
• carboplatin, paclitaxel, and bevacizumab (control arm).
Among patients with NSq NSCLC without an EGFR or ALK mutation (87%), the estimated median OS was 19.2 months for patients receiving the 4-drug regimen and 14.7 months for those in the control arm (hazard ratio [HR] 0.78; 95% CI: 0.64, 0.96; P = .016), the FDA said in a press statement announcing the approval.
The median progression-free survival was 8.5 months for patients receiving the 4-drug regimen and 7.0 months for those in the control arm (HR 0.71; 95% CI 0.59, 0.85; P = .0002). The overall response rates were 55% in the 4-drug arm and 42% in the control arm. There were no significant differences in OS or final progression-free survival between the 3-drug arm containing atezolizumab and the control arm.
The most common adverse reactions with atezolizumab were fatigue/asthenia, alopecia, nausea, diarrhea, constipation, decreased appetite, arthralgia, hypertension, and neuropathy. Treatment with atezolizumab was discontinued in 15% of patients due to adverse reactions, the most common reason being pneumonitis.
The recommended atezolizumab dose is 1,200 mg intravenously over 60 minutes every 3 weeks, the FDA said.
The Food and Drug Administration has approved atezolizumab (Tecentriq) in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations.
Approval was based on greater overall survival (OS) among patients receiving the four drug combination, compared with patients who did not receive the checkpoint inhibitor but received the other three drugs in the randomized IMpower150 trial.
For the trial, 1,202 patients with metastatic NSq NSCLC were randomized to three arms for first-line treatment:
• atezolizumab, carboplatin, paclitaxel, and bevacizumab (4-drug regimen);
• atezolizumab, carboplatin and paclitaxel (3-drug regimen); or
• carboplatin, paclitaxel, and bevacizumab (control arm).
Among patients with NSq NSCLC without an EGFR or ALK mutation (87%), the estimated median OS was 19.2 months for patients receiving the 4-drug regimen and 14.7 months for those in the control arm (hazard ratio [HR] 0.78; 95% CI: 0.64, 0.96; P = .016), the FDA said in a press statement announcing the approval.
The median progression-free survival was 8.5 months for patients receiving the 4-drug regimen and 7.0 months for those in the control arm (HR 0.71; 95% CI 0.59, 0.85; P = .0002). The overall response rates were 55% in the 4-drug arm and 42% in the control arm. There were no significant differences in OS or final progression-free survival between the 3-drug arm containing atezolizumab and the control arm.
The most common adverse reactions with atezolizumab were fatigue/asthenia, alopecia, nausea, diarrhea, constipation, decreased appetite, arthralgia, hypertension, and neuropathy. Treatment with atezolizumab was discontinued in 15% of patients due to adverse reactions, the most common reason being pneumonitis.
The recommended atezolizumab dose is 1,200 mg intravenously over 60 minutes every 3 weeks, the FDA said.
Trio of biosimilars have good showing
Biosimilars for three widely used oncology drugs showed efficacy and safety in lung cancer and breast cancer similar to those of the reference products, according to findings reported at the 2018 annual meeting of the American Society of Clinical Oncology in Chicago.
Oncology biosimilars for bevacizumab (Avastin), trastuzumab (Herceptin), and filgrastim (Neupogen and others) have yielded positive results in various patient populations and clinical settings, investigators reported at the annual ASCO meeting. The findings advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
“Biosimilars are here,” said Michael A Thompson, MD, PhD, of Aurora Health Care in Milwaukee, Wisconsin, “[although] issues remain, including clinical decision support and pathway adoption, naming differences across the world, competition and lower prices versus the illusion of a free market, and adoption to decrease costs and increase value to our patients.” Dr Thompson was commenting during an invited discussion at the meeting. He is the medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health (also see Commentary at end of article).
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous non–small-cell lung cancer (NSCLC). Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The overall response rate by week 19, confirmed by week 25 – the trial’s primary endpoint – was 45.3% with the biosimilar and 44.6% with bevacizumab, reported lead author Mark A Socinski, MD, executive medical director of the Florida Hospital Cancer Institute in Orlando. The confidence interval (CI) for the risk difference fell within the equivalence margins set by European Union regulators (-13% and +13% for the 95% CI). And the confidence interval for the risk ratio fell within the equivalence margins set by the US Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival (PFS) was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio [HR], 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, respectively, Dr Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the 2 agents. Patients also had similar rates of grade 3 or higher serious adverse events (AEs) and of fatal (grade 5) serious AEs with the biosimilar and bevacizumab (5.3% and 5.9%, respectively).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr Socinski summarized. “These results confirm the similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU.”
Funding Pfizer sponsored the REFLECTIONS trial. Disclosures Dr Socinski disclosed that his institution receives research funding from Pfizer. Source Socinski MA et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. ASCO 2018, Abstract 109. https://meetinglibrary.asco.org/record/161702/abstract. Clinical trial registry number NCT02364999 https://clinicaltrials.gov/ct2/show/NCT02364999
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer. The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various AEs were essentially the same.
The 48-week results showed a median PFS of 11.1 months with trastuzumab-dkst and 11.1 months with trastuzumab (HR, 0.95; P = .842), reported senior investigator Hope S Rugo, MD, a clinical professor of medicine and director of the Breast Oncology Clinical Trials Program at the University of California, San Francisco, Helen Diller Family Comprehensive Cancer Center. “The overall survival is immature but is impressive at over 80% at 52 weeks,” she noted.
Presence of overall response at 24 weeks correlated with duration of PFS at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr Rugo commented.
Common AEs through week 48 were much the same as those seen at week 24, with few additional [events] occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell to a very low number and was identical between the 2 arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of AEs of special interest – respiratory events, cardiac disorders, and infusion-related AEs – and of serious AEs were similar for the 2 agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.”
Dr Rugo emphasized that trastuzumab-dkst provides “an additional high-quality treatment option for patients with HER2+ breast cancers in any setting. This study shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Funding Mylan sponsored the HERITAGE trial. Disclosures Dr Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan. Source Manikhas A et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: Toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. ASCO 2018, Abstract 110. https://meetinglibrary.asco.org/record/161572/abstract. Clinical trial registry number NCT02472964 https://clinicaltrials.gov/ct2/show/NCT02472964
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of OB&GYN at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony-stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Data for the former came from PIONEER, a phase 3, randomized, controlled trial among patients with nonmetastatic breast cancer undergoing docetaxel, doxorubicin, and cyclophosphamide (TAC) chemotherapy in the neoadjuvant or adjuvant setting (Ann Oncol. 2015;26[9]:1948-53). Data for the latter came from MONITOR-GCSF, a postmarketing, open-label, observational cohort study among patients from 12 European countries receiving chemotherapy for various solid and hematologic malignancies (Support Care Cancer. 2016;24[2]:911-25).
Dr Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial/biosimilar population. Findings were similar for temperature exceeding 38.5°C in any cycle: 3.4% and 5.6%, respectively. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% and 74.3%) and higher rates of infection (15.5% and 7.9%) and hospitalization caused by febrile neutropenia (3.9% and 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 and 261 events, respectively) and skin/subcutaneous tissue disorders (5 and 258 events). “Seeing these data, you have to keep in mind that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr Harbeck commented. “Having seen the discrepancies in the data, I think it’s important to have randomized controlled trials to assess and monitor AEs for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Funding Sandoz sponsored the PIONEER and MONITOR-GCSF trials. Disclosures Dr Harbeck disclosed that she has a consulting or advisory role with Sandoz. Source Harbeck N et al. Comparison of efficacy and safety of biosimilar filgrastim in a RCT (PIONEER) and real-world practice (MONITOR-GCSF). ASCO 2018, Abstract 111. https://meetinglibrary.asco.org/record/161688/abstract. Clinical trial registry number NCT01519700 https://clinicaltrials.gov/ct2/show/NCT01519700
Biosimilars for three widely used oncology drugs showed efficacy and safety in lung cancer and breast cancer similar to those of the reference products, according to findings reported at the 2018 annual meeting of the American Society of Clinical Oncology in Chicago.
Oncology biosimilars for bevacizumab (Avastin), trastuzumab (Herceptin), and filgrastim (Neupogen and others) have yielded positive results in various patient populations and clinical settings, investigators reported at the annual ASCO meeting. The findings advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
“Biosimilars are here,” said Michael A Thompson, MD, PhD, of Aurora Health Care in Milwaukee, Wisconsin, “[although] issues remain, including clinical decision support and pathway adoption, naming differences across the world, competition and lower prices versus the illusion of a free market, and adoption to decrease costs and increase value to our patients.” Dr Thompson was commenting during an invited discussion at the meeting. He is the medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health (also see Commentary at end of article).
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous non–small-cell lung cancer (NSCLC). Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The overall response rate by week 19, confirmed by week 25 – the trial’s primary endpoint – was 45.3% with the biosimilar and 44.6% with bevacizumab, reported lead author Mark A Socinski, MD, executive medical director of the Florida Hospital Cancer Institute in Orlando. The confidence interval (CI) for the risk difference fell within the equivalence margins set by European Union regulators (-13% and +13% for the 95% CI). And the confidence interval for the risk ratio fell within the equivalence margins set by the US Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival (PFS) was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio [HR], 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, respectively, Dr Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the 2 agents. Patients also had similar rates of grade 3 or higher serious adverse events (AEs) and of fatal (grade 5) serious AEs with the biosimilar and bevacizumab (5.3% and 5.9%, respectively).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr Socinski summarized. “These results confirm the similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU.”
Funding Pfizer sponsored the REFLECTIONS trial. Disclosures Dr Socinski disclosed that his institution receives research funding from Pfizer. Source Socinski MA et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. ASCO 2018, Abstract 109. https://meetinglibrary.asco.org/record/161702/abstract. Clinical trial registry number NCT02364999 https://clinicaltrials.gov/ct2/show/NCT02364999
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer. The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various AEs were essentially the same.
The 48-week results showed a median PFS of 11.1 months with trastuzumab-dkst and 11.1 months with trastuzumab (HR, 0.95; P = .842), reported senior investigator Hope S Rugo, MD, a clinical professor of medicine and director of the Breast Oncology Clinical Trials Program at the University of California, San Francisco, Helen Diller Family Comprehensive Cancer Center. “The overall survival is immature but is impressive at over 80% at 52 weeks,” she noted.
Presence of overall response at 24 weeks correlated with duration of PFS at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr Rugo commented.
Common AEs through week 48 were much the same as those seen at week 24, with few additional [events] occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell to a very low number and was identical between the 2 arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of AEs of special interest – respiratory events, cardiac disorders, and infusion-related AEs – and of serious AEs were similar for the 2 agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.”
Dr Rugo emphasized that trastuzumab-dkst provides “an additional high-quality treatment option for patients with HER2+ breast cancers in any setting. This study shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Funding Mylan sponsored the HERITAGE trial. Disclosures Dr Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan. Source Manikhas A et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: Toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. ASCO 2018, Abstract 110. https://meetinglibrary.asco.org/record/161572/abstract. Clinical trial registry number NCT02472964 https://clinicaltrials.gov/ct2/show/NCT02472964
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of OB&GYN at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony-stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Data for the former came from PIONEER, a phase 3, randomized, controlled trial among patients with nonmetastatic breast cancer undergoing docetaxel, doxorubicin, and cyclophosphamide (TAC) chemotherapy in the neoadjuvant or adjuvant setting (Ann Oncol. 2015;26[9]:1948-53). Data for the latter came from MONITOR-GCSF, a postmarketing, open-label, observational cohort study among patients from 12 European countries receiving chemotherapy for various solid and hematologic malignancies (Support Care Cancer. 2016;24[2]:911-25).
Dr Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial/biosimilar population. Findings were similar for temperature exceeding 38.5°C in any cycle: 3.4% and 5.6%, respectively. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% and 74.3%) and higher rates of infection (15.5% and 7.9%) and hospitalization caused by febrile neutropenia (3.9% and 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 and 261 events, respectively) and skin/subcutaneous tissue disorders (5 and 258 events). “Seeing these data, you have to keep in mind that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr Harbeck commented. “Having seen the discrepancies in the data, I think it’s important to have randomized controlled trials to assess and monitor AEs for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Funding Sandoz sponsored the PIONEER and MONITOR-GCSF trials. Disclosures Dr Harbeck disclosed that she has a consulting or advisory role with Sandoz. Source Harbeck N et al. Comparison of efficacy and safety of biosimilar filgrastim in a RCT (PIONEER) and real-world practice (MONITOR-GCSF). ASCO 2018, Abstract 111. https://meetinglibrary.asco.org/record/161688/abstract. Clinical trial registry number NCT01519700 https://clinicaltrials.gov/ct2/show/NCT01519700
Biosimilars for three widely used oncology drugs showed efficacy and safety in lung cancer and breast cancer similar to those of the reference products, according to findings reported at the 2018 annual meeting of the American Society of Clinical Oncology in Chicago.
Oncology biosimilars for bevacizumab (Avastin), trastuzumab (Herceptin), and filgrastim (Neupogen and others) have yielded positive results in various patient populations and clinical settings, investigators reported at the annual ASCO meeting. The findings advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
“Biosimilars are here,” said Michael A Thompson, MD, PhD, of Aurora Health Care in Milwaukee, Wisconsin, “[although] issues remain, including clinical decision support and pathway adoption, naming differences across the world, competition and lower prices versus the illusion of a free market, and adoption to decrease costs and increase value to our patients.” Dr Thompson was commenting during an invited discussion at the meeting. He is the medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health (also see Commentary at end of article).
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous non–small-cell lung cancer (NSCLC). Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The overall response rate by week 19, confirmed by week 25 – the trial’s primary endpoint – was 45.3% with the biosimilar and 44.6% with bevacizumab, reported lead author Mark A Socinski, MD, executive medical director of the Florida Hospital Cancer Institute in Orlando. The confidence interval (CI) for the risk difference fell within the equivalence margins set by European Union regulators (-13% and +13% for the 95% CI). And the confidence interval for the risk ratio fell within the equivalence margins set by the US Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival (PFS) was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio [HR], 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, respectively, Dr Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the 2 agents. Patients also had similar rates of grade 3 or higher serious adverse events (AEs) and of fatal (grade 5) serious AEs with the biosimilar and bevacizumab (5.3% and 5.9%, respectively).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr Socinski summarized. “These results confirm the similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU.”
Funding Pfizer sponsored the REFLECTIONS trial. Disclosures Dr Socinski disclosed that his institution receives research funding from Pfizer. Source Socinski MA et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. ASCO 2018, Abstract 109. https://meetinglibrary.asco.org/record/161702/abstract. Clinical trial registry number NCT02364999 https://clinicaltrials.gov/ct2/show/NCT02364999
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer. The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various AEs were essentially the same.
The 48-week results showed a median PFS of 11.1 months with trastuzumab-dkst and 11.1 months with trastuzumab (HR, 0.95; P = .842), reported senior investigator Hope S Rugo, MD, a clinical professor of medicine and director of the Breast Oncology Clinical Trials Program at the University of California, San Francisco, Helen Diller Family Comprehensive Cancer Center. “The overall survival is immature but is impressive at over 80% at 52 weeks,” she noted.
Presence of overall response at 24 weeks correlated with duration of PFS at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr Rugo commented.
Common AEs through week 48 were much the same as those seen at week 24, with few additional [events] occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell to a very low number and was identical between the 2 arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of AEs of special interest – respiratory events, cardiac disorders, and infusion-related AEs – and of serious AEs were similar for the 2 agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.”
Dr Rugo emphasized that trastuzumab-dkst provides “an additional high-quality treatment option for patients with HER2+ breast cancers in any setting. This study shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Funding Mylan sponsored the HERITAGE trial. Disclosures Dr Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan. Source Manikhas A et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: Toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. ASCO 2018, Abstract 110. https://meetinglibrary.asco.org/record/161572/abstract. Clinical trial registry number NCT02472964 https://clinicaltrials.gov/ct2/show/NCT02472964
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of OB&GYN at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony-stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Data for the former came from PIONEER, a phase 3, randomized, controlled trial among patients with nonmetastatic breast cancer undergoing docetaxel, doxorubicin, and cyclophosphamide (TAC) chemotherapy in the neoadjuvant or adjuvant setting (Ann Oncol. 2015;26[9]:1948-53). Data for the latter came from MONITOR-GCSF, a postmarketing, open-label, observational cohort study among patients from 12 European countries receiving chemotherapy for various solid and hematologic malignancies (Support Care Cancer. 2016;24[2]:911-25).
Dr Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial/biosimilar population. Findings were similar for temperature exceeding 38.5°C in any cycle: 3.4% and 5.6%, respectively. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% and 74.3%) and higher rates of infection (15.5% and 7.9%) and hospitalization caused by febrile neutropenia (3.9% and 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 and 261 events, respectively) and skin/subcutaneous tissue disorders (5 and 258 events). “Seeing these data, you have to keep in mind that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr Harbeck commented. “Having seen the discrepancies in the data, I think it’s important to have randomized controlled trials to assess and monitor AEs for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Funding Sandoz sponsored the PIONEER and MONITOR-GCSF trials. Disclosures Dr Harbeck disclosed that she has a consulting or advisory role with Sandoz. Source Harbeck N et al. Comparison of efficacy and safety of biosimilar filgrastim in a RCT (PIONEER) and real-world practice (MONITOR-GCSF). ASCO 2018, Abstract 111. https://meetinglibrary.asco.org/record/161688/abstract. Clinical trial registry number NCT01519700 https://clinicaltrials.gov/ct2/show/NCT01519700
Key clinical points Biosimilars for bevacizumab, trastuzumab, and filgrastim showed similar efficacy and safety compared with their reference drugs.
Major findings Bevacizumab In patients with advanced nonsquamous NSCLC, the ORR was 45.3% with a candidate bevacizumab biosimilar and 44.6% with bevacizumab. Trastuzumab In patients with HER2+ advanced breast cancer, 48-week median PFS was 11.1 months for both trastuzumab-dkst and trastuzumab. Filgrastim The rate of chemotherapy-induced febrile neutropenia among breast cancer patients given a biosimilar for filgrastim was 5.1% in a trial population and 6.2% in a real-world population.
Study details Randomized, controlled trials of first-line therapy among 719 patients with advanced nonsquamous NSCLC (REFLECTIONS trial with bevacizumab) and among 458 patients with HER2+ advanced breast cancer (HERITAGE trial with trastuzumab). Comparison of outcomes in a randomized, controlled trial among 217 patients with nonmetastatic breast cancer (PIONEER trial with filgrastim) and a real-world cohort study of 466 patients with any-stage breast cancer (MONITOR-GCSF with filgrastim).
Disclosures and sources See article text.
Paradigm-changing osimertinib approval in front-line for advanced NSCLC
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.

Symptom burdens related to chemotherapy-induced anemia in stage IV cancer
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).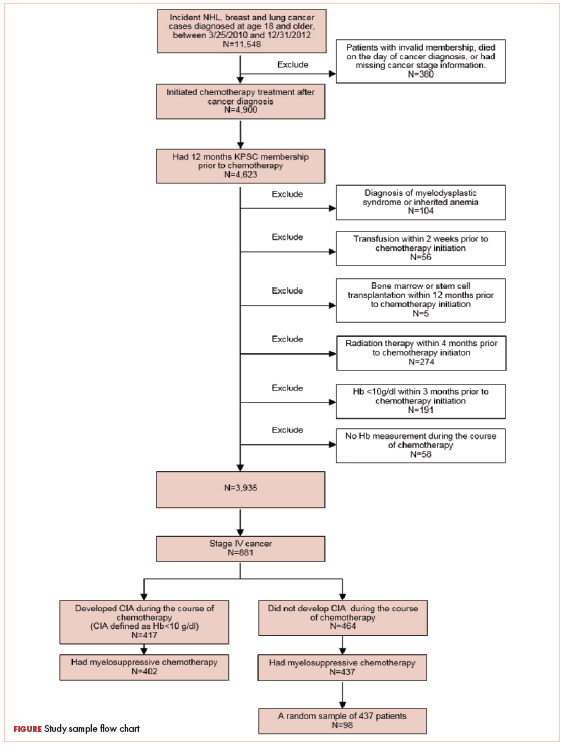
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).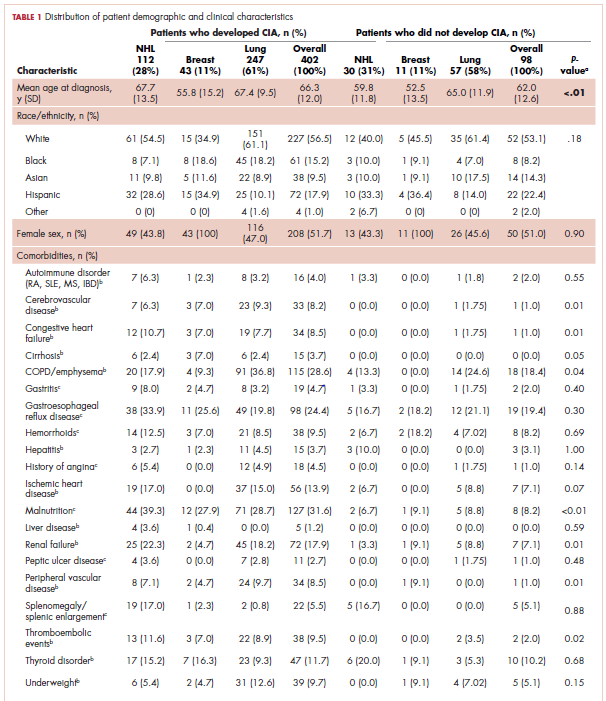
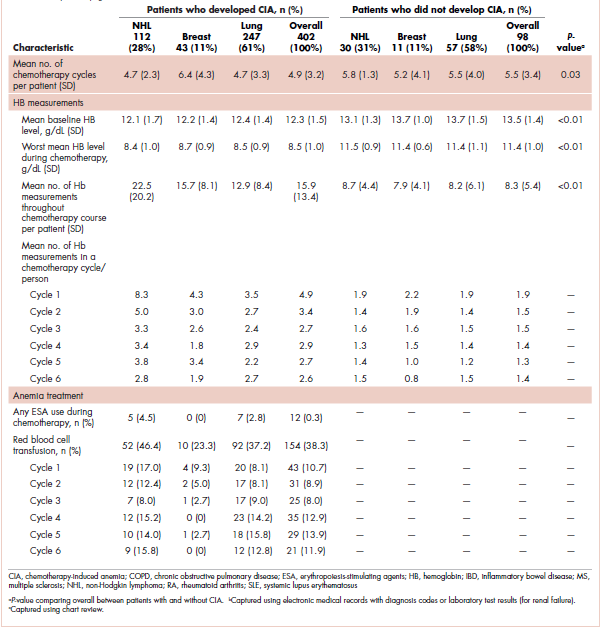
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2. 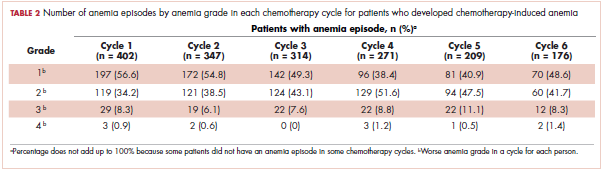
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA. 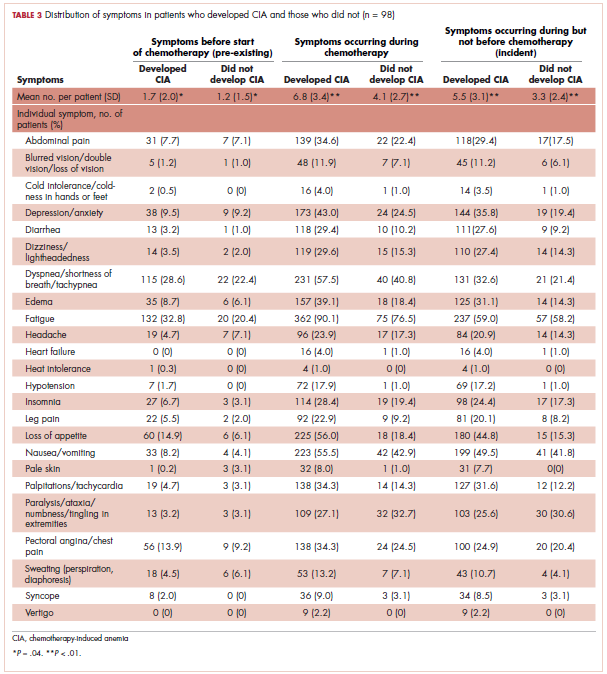
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).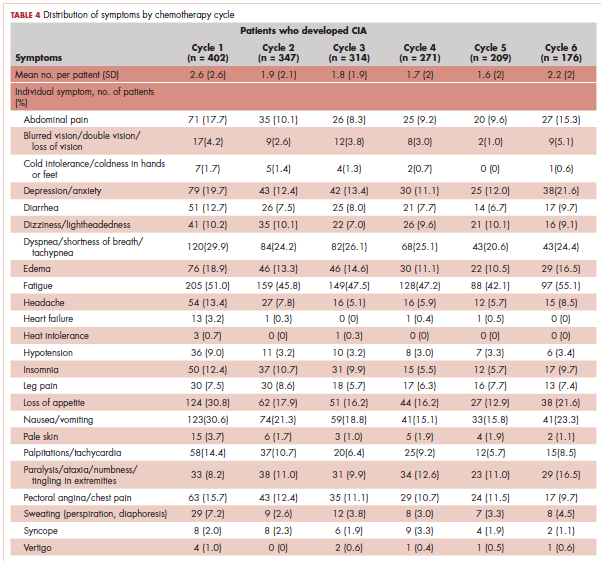
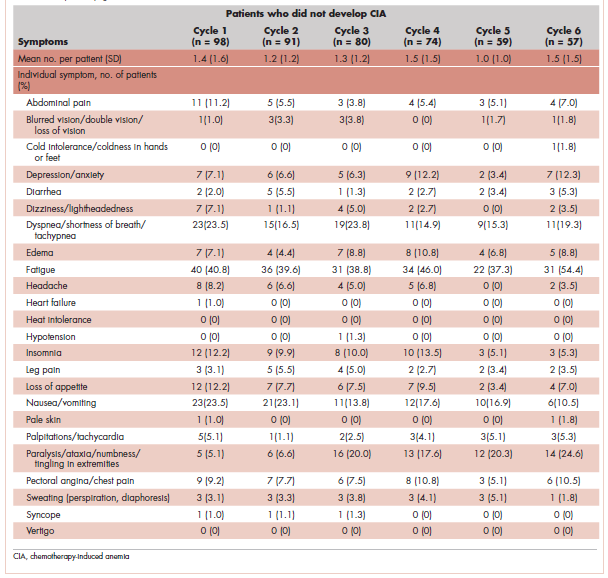
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).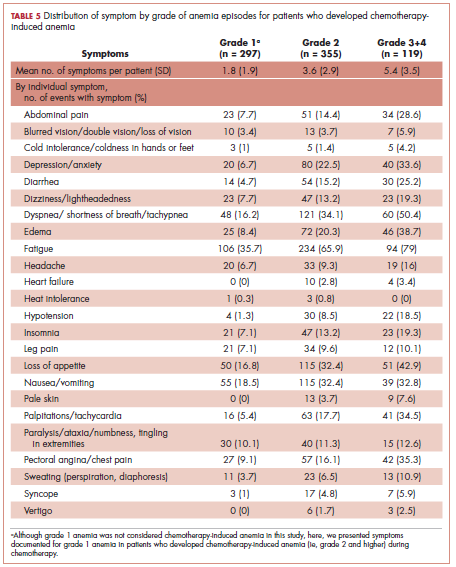
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2.
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA.
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2.
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA.
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
A novel tracer shows promise for detecting CD8 T-cells in advanced solid tumors
WASHINGTON – and reference tissue in an open-label, phase 1, first-in-human study.
The findings demonstrate the ability of the tracer–an anti-CD8 zirconium-labeled minibody–to noninvasively detect CD8 distribution in patients with metastatic solid tumors, potentially providing more information – and more quickly – than is possible with a single biopsy, Michael S. Gordon, MD, reported during a late-breaking abstract session at the annual meeting of the Society for Immunotherapy of Cancer.
During a dose escalation period (stage 1) of the study, six patients received 3 mCi of 89Zr-IAB22M2C once intravenously followed by serial PET scans over a period of 5-7 days. The patients received increasing protein doses of 0.2 through 10 mg to establish safety and determine a “recommended protein dose and scanning parameters for subsequent trials,” explained Dr. Gordon of HonorHealth Research Institute, Scottsdale, Ariz.
Stage 1 was followed by a dose expansion period (stage 2) in which an additional nine subjects were scanned to better delineate the recommended phase 2 study dose, he said.
All patients were monitored for drug-related adverse events and evaluated with blood chemistry, hematology, cytokine assay, and anti-drug antibodies. Biodistribution, radiodosimetry and semi-quantitative evaluation of CD8-tracer uptake were performed in all patients.
“We saw rapid clearance with excretion through the hepatobiliary mechanism, uptake in T-cell rich tissues, and no uptake in background normal tissues – so no uptake in muscle, heart, brain, or lungs,” he said, adding that “tumor uptake was variable and was clearly seen in 10 out of 15 patients.
“The protein dose that was considered to have favorable biodistribution was the range between 0.5 and 1.5, and based upon the analysis, the most favorable imaging time point ... was deemed to be 24 hours,” he said, noting that changes could be seen in as early as 6 hours.
The estimated mean effective radiation dose was 2.4 rem/mCi, “which is consistent with other zirconium-labeled antibody or minibody technologies,” Dr. Gordon said.
Study subjects ranged in age from 31 to 82 years and included nine men and six women with solid tumor malignancies who were eligible to receive checkpoint inhibitor therapy. Their primary cancer types were melanoma (eight patients), non–small-cell lung cancer (six patients), and hepatocellular carcinoma (one patient).
Two patients had received no prior treatment, three had discontinued prior checkpoint inhibitor therapy, and 10 were on immunotherapy.
No drug-related adverse events occurred during the course of the study, although one patient had a transient increase in anti-drug antibodies, Dr. Gordon said.
“Immunotherapy, and specifically checkpoint inhibitors (CPIs), have transformed the landscape of cancer care. Antitumor activity of CPIs is mediated by the CD8-positive T-cell cytotoxic effects, with preclinical and translational clinical studies demonstrating the importance of activated CD8-positive cells within the tumor microenvironment,” he explained, adding that currently available technology is limited in its ability to continually assess the presence of and change in the CD8 infiltrate; one biopsy may fail to capture the immunologic heterogeneity that exists among various tumors in an individual patient.
“As CPI therapy moves into front-line and earlier settings, the ability to have a noninvasive technology to assess whole body and intratumoral changes in CD8 trafficking or expansion in response to therapy is viewed as being crucial,” he said.
A phase 2 study to look closer at the potential for PET + 89Zr-IAB22M2C to fulfill that role will begin soon. The study will focus on correlating imaging with synchronous biopsies before and after primary immunotherapy to look for any predictive potential for this technology, he said.
This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
SOURCE: Gordon M et al., SITC 2018: Abstract LB49.
WASHINGTON – and reference tissue in an open-label, phase 1, first-in-human study.
The findings demonstrate the ability of the tracer–an anti-CD8 zirconium-labeled minibody–to noninvasively detect CD8 distribution in patients with metastatic solid tumors, potentially providing more information – and more quickly – than is possible with a single biopsy, Michael S. Gordon, MD, reported during a late-breaking abstract session at the annual meeting of the Society for Immunotherapy of Cancer.
During a dose escalation period (stage 1) of the study, six patients received 3 mCi of 89Zr-IAB22M2C once intravenously followed by serial PET scans over a period of 5-7 days. The patients received increasing protein doses of 0.2 through 10 mg to establish safety and determine a “recommended protein dose and scanning parameters for subsequent trials,” explained Dr. Gordon of HonorHealth Research Institute, Scottsdale, Ariz.
Stage 1 was followed by a dose expansion period (stage 2) in which an additional nine subjects were scanned to better delineate the recommended phase 2 study dose, he said.
All patients were monitored for drug-related adverse events and evaluated with blood chemistry, hematology, cytokine assay, and anti-drug antibodies. Biodistribution, radiodosimetry and semi-quantitative evaluation of CD8-tracer uptake were performed in all patients.
“We saw rapid clearance with excretion through the hepatobiliary mechanism, uptake in T-cell rich tissues, and no uptake in background normal tissues – so no uptake in muscle, heart, brain, or lungs,” he said, adding that “tumor uptake was variable and was clearly seen in 10 out of 15 patients.
“The protein dose that was considered to have favorable biodistribution was the range between 0.5 and 1.5, and based upon the analysis, the most favorable imaging time point ... was deemed to be 24 hours,” he said, noting that changes could be seen in as early as 6 hours.
The estimated mean effective radiation dose was 2.4 rem/mCi, “which is consistent with other zirconium-labeled antibody or minibody technologies,” Dr. Gordon said.
Study subjects ranged in age from 31 to 82 years and included nine men and six women with solid tumor malignancies who were eligible to receive checkpoint inhibitor therapy. Their primary cancer types were melanoma (eight patients), non–small-cell lung cancer (six patients), and hepatocellular carcinoma (one patient).
Two patients had received no prior treatment, three had discontinued prior checkpoint inhibitor therapy, and 10 were on immunotherapy.
No drug-related adverse events occurred during the course of the study, although one patient had a transient increase in anti-drug antibodies, Dr. Gordon said.
“Immunotherapy, and specifically checkpoint inhibitors (CPIs), have transformed the landscape of cancer care. Antitumor activity of CPIs is mediated by the CD8-positive T-cell cytotoxic effects, with preclinical and translational clinical studies demonstrating the importance of activated CD8-positive cells within the tumor microenvironment,” he explained, adding that currently available technology is limited in its ability to continually assess the presence of and change in the CD8 infiltrate; one biopsy may fail to capture the immunologic heterogeneity that exists among various tumors in an individual patient.
“As CPI therapy moves into front-line and earlier settings, the ability to have a noninvasive technology to assess whole body and intratumoral changes in CD8 trafficking or expansion in response to therapy is viewed as being crucial,” he said.
A phase 2 study to look closer at the potential for PET + 89Zr-IAB22M2C to fulfill that role will begin soon. The study will focus on correlating imaging with synchronous biopsies before and after primary immunotherapy to look for any predictive potential for this technology, he said.
This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
SOURCE: Gordon M et al., SITC 2018: Abstract LB49.
WASHINGTON – and reference tissue in an open-label, phase 1, first-in-human study.
The findings demonstrate the ability of the tracer–an anti-CD8 zirconium-labeled minibody–to noninvasively detect CD8 distribution in patients with metastatic solid tumors, potentially providing more information – and more quickly – than is possible with a single biopsy, Michael S. Gordon, MD, reported during a late-breaking abstract session at the annual meeting of the Society for Immunotherapy of Cancer.
During a dose escalation period (stage 1) of the study, six patients received 3 mCi of 89Zr-IAB22M2C once intravenously followed by serial PET scans over a period of 5-7 days. The patients received increasing protein doses of 0.2 through 10 mg to establish safety and determine a “recommended protein dose and scanning parameters for subsequent trials,” explained Dr. Gordon of HonorHealth Research Institute, Scottsdale, Ariz.
Stage 1 was followed by a dose expansion period (stage 2) in which an additional nine subjects were scanned to better delineate the recommended phase 2 study dose, he said.
All patients were monitored for drug-related adverse events and evaluated with blood chemistry, hematology, cytokine assay, and anti-drug antibodies. Biodistribution, radiodosimetry and semi-quantitative evaluation of CD8-tracer uptake were performed in all patients.
“We saw rapid clearance with excretion through the hepatobiliary mechanism, uptake in T-cell rich tissues, and no uptake in background normal tissues – so no uptake in muscle, heart, brain, or lungs,” he said, adding that “tumor uptake was variable and was clearly seen in 10 out of 15 patients.
“The protein dose that was considered to have favorable biodistribution was the range between 0.5 and 1.5, and based upon the analysis, the most favorable imaging time point ... was deemed to be 24 hours,” he said, noting that changes could be seen in as early as 6 hours.
The estimated mean effective radiation dose was 2.4 rem/mCi, “which is consistent with other zirconium-labeled antibody or minibody technologies,” Dr. Gordon said.
Study subjects ranged in age from 31 to 82 years and included nine men and six women with solid tumor malignancies who were eligible to receive checkpoint inhibitor therapy. Their primary cancer types were melanoma (eight patients), non–small-cell lung cancer (six patients), and hepatocellular carcinoma (one patient).
Two patients had received no prior treatment, three had discontinued prior checkpoint inhibitor therapy, and 10 were on immunotherapy.
No drug-related adverse events occurred during the course of the study, although one patient had a transient increase in anti-drug antibodies, Dr. Gordon said.
“Immunotherapy, and specifically checkpoint inhibitors (CPIs), have transformed the landscape of cancer care. Antitumor activity of CPIs is mediated by the CD8-positive T-cell cytotoxic effects, with preclinical and translational clinical studies demonstrating the importance of activated CD8-positive cells within the tumor microenvironment,” he explained, adding that currently available technology is limited in its ability to continually assess the presence of and change in the CD8 infiltrate; one biopsy may fail to capture the immunologic heterogeneity that exists among various tumors in an individual patient.
“As CPI therapy moves into front-line and earlier settings, the ability to have a noninvasive technology to assess whole body and intratumoral changes in CD8 trafficking or expansion in response to therapy is viewed as being crucial,” he said.
A phase 2 study to look closer at the potential for PET + 89Zr-IAB22M2C to fulfill that role will begin soon. The study will focus on correlating imaging with synchronous biopsies before and after primary immunotherapy to look for any predictive potential for this technology, he said.
This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
SOURCE: Gordon M et al., SITC 2018: Abstract LB49.
REPORTING FROM SITC 2018
Key clinical point: PET with CD8-tracer 89Zr-IAB22M2C is safe, provides detailed CD8 T-cell information.
Major finding: Tumor uptake of the CD8-tracer was seen in 10 of 15 subjects.
Study details: An open-label phase 1 study of 15 patients.
Disclosures: This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
Source: Gordon M et al. SITC 2018: Abstract LB49.