User login
JAK-1 inhibitor upadacitinib advances to phase III for refractory Crohn’s
CHICAGO – An investigational inhibitor of the Janus-1 kinase receptor induced clinical and endoscopic remission at several doses in patients with long-standing, treatment-resistant Crohn’s disease.
The two highest doses of upadacitinib (ABT-494; AbbVie) also allowed about 30% of patients to rapidly withdraw systemic steroids and stay in remission during the 16-week dose-finding induction trial, William J. Sandborn, MD, said at the annual Digestive Disease Week®.
“This rapid steroid tapering was a unique feature of the trial,” said Dr. Sandborn of the University of California, San Diego. “Usually during induction trials, steroids are held fixed at 20 mg-30 mg throughout the trial and then withdrawn to maintenance levels.”

The preplanned analysis used a unique composite outcome of 7-day stool frequency and abdominal pain. At the time of trial design, the scale had only been validated in patients with mild-moderate Crohn’s, so the investigators used the validated stool frequency cutpoint of 1.5 per day as a measure of clinical remission.
That was not an appropriate target for this unique study group, Dr. Sandborn said.
“CELEST was the most refractory patient population ever recruited into a Crohn’s disease clinical trial. If we could do this over now, we would use a cutpoint of less than 3 instead of 1.5 or less. This is a really tough clinical endpoint” that probably isn’t a realistic clinical goal for patients in this category. “There’s no way we would do it this way today. A number of studies since then now suggest that the right cutpoint for remission in these patients would be about 3 per day.”
CELEST enrolled 220 patients who were randomized to five treatment arms comprising 30-35 patients each: placebo; twice daily upadacitinib at 3 mg, 6 mg, 12 mg, and 24 mg (24 mg BID); and 24 mg once daily (24 mg OD). The study lasted 16 weeks and was followed by 36 weeks of blinded extension treatment. Dr. Sandborn reported the 16-week induction phase data.
The patients had moderate to severe Crohn’s disease, with a mean baseline Crohn’s Disease Activity Index (CDAI) score of about 300 and a Simple Endoscopic Score-Crohns disease (SES-CD) of about 15. About 95% had already failed at least one anti–tumor necrosis factor (TNF) drug. Half had failed at least two.
The coprimary endpoints were the proportion of patients who achieved clinical remission (stool frequency of 1.5 or less per day and abdominal pain of 1 or lower) at week 16 and endoscopic remission at weeks 12 or 16. Secondary endpoints included CDAI response, clinical response (at least a 30% reduction from baseline in stool frequency or abdominal pain), and endoscopic response.
In the primary analysis, the rate of endoscopic remission was significant (P less than .05) in both the 24-mg BID and the 24-mg OD groups. However, clinical remission with the original stool frequency cutpoint of 1.5/day or less wasn’t significantly different from placebo in any group. Dr. Sandborn did point out a 27% rate of clinical remission in those taking 12 mg, which had a P value of less than 0.1, relative to placebo.
Among the secondary endpoints, remission as measured by the CDAI score (less than 150) occurred in 39% of those taking 12 mg – the only significant response in that category.
The rate of endoscopic response (at least a 50% improvement in endoscopic findings) was 21% in the 6-mg group and 25% in the 24-mg OD group (P less than 0.05) and in about 30% of the 12-mg and 24-mg BID group (P less than 0.01).
When the clinical remission analysis employed the revised stool frequency cutpoint of less than 2.8/day, clinical remission rates improved somewhat. Almost 40% of those taking 24-mg BID achieved clinical remission (P less than 0.01), and 30% of those taking 6 mg achieved clinical remission, but the significance was marginal (P less than 0.1).
Steroid-free remission rates were significantly better than placebo in the 18-mg group (39%) and the 15-mg group (33%), both with a P value less than 0.05.
Dr. Sandborn also showed dramatic changes in C-reactive protein and fecal calprotectin. These dropped precipitously in all active groups by week 2, in a dose-respondent manner, and stayed well-suppressed in the two highest-dose groups. In the placebo groups, C-reactive protein rose over the 16 weeks, and fecal calprotectin remained unchanged from baseline.
The drug was reasonably well-tolerated and safe. About 80% of each dosing group reported at least one adverse event. The 12-mg dose appeared particularly troublesome, with 25% stopping because of an adverse event. By comparison, the discontinuation rate was 8% in the 24-mg BID group and 14% in the 24-mg OD group.
Serious adverse events were consistent with what is known about the JAK1-inhibitor safety profile, Dr. Sandborn said. There were nine serious infections, including Escherichia coli bacteremia, subcutaneous abscess, and sepsis (3-mg group); anorectal abscess, urinary tract infection, and sepsis (12-mg group); sepsis (24 mg BID); and peritonitis and sepsis (24 mg QD). There was one nonmelanoma skin cancer, which Dr. Sandborn said was probably pre-existing but not recognized at baseline. Three cases of herpes zoster occurred, all in the 24-mg BID group.
One patient experienced a gastrointestinal perforation, which sometimes occurs in Crohn’s disease. Two patients experienced a myocardial infarction, a number “too small to understand fully,” Dr. Sandborn said.
The drug will move forward into phase III trials, but the final dose hasn’t been decided on.
Dr. Sandborn has received consulting fees from AbbVie, which is developing the drug and sponsored CELEST.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – An investigational inhibitor of the Janus-1 kinase receptor induced clinical and endoscopic remission at several doses in patients with long-standing, treatment-resistant Crohn’s disease.
The two highest doses of upadacitinib (ABT-494; AbbVie) also allowed about 30% of patients to rapidly withdraw systemic steroids and stay in remission during the 16-week dose-finding induction trial, William J. Sandborn, MD, said at the annual Digestive Disease Week®.
“This rapid steroid tapering was a unique feature of the trial,” said Dr. Sandborn of the University of California, San Diego. “Usually during induction trials, steroids are held fixed at 20 mg-30 mg throughout the trial and then withdrawn to maintenance levels.”
The preplanned analysis used a unique composite outcome of 7-day stool frequency and abdominal pain. At the time of trial design, the scale had only been validated in patients with mild-moderate Crohn’s, so the investigators used the validated stool frequency cutpoint of 1.5 per day as a measure of clinical remission.
That was not an appropriate target for this unique study group, Dr. Sandborn said.
“CELEST was the most refractory patient population ever recruited into a Crohn’s disease clinical trial. If we could do this over now, we would use a cutpoint of less than 3 instead of 1.5 or less. This is a really tough clinical endpoint” that probably isn’t a realistic clinical goal for patients in this category. “There’s no way we would do it this way today. A number of studies since then now suggest that the right cutpoint for remission in these patients would be about 3 per day.”
CELEST enrolled 220 patients who were randomized to five treatment arms comprising 30-35 patients each: placebo; twice daily upadacitinib at 3 mg, 6 mg, 12 mg, and 24 mg (24 mg BID); and 24 mg once daily (24 mg OD). The study lasted 16 weeks and was followed by 36 weeks of blinded extension treatment. Dr. Sandborn reported the 16-week induction phase data.
The patients had moderate to severe Crohn’s disease, with a mean baseline Crohn’s Disease Activity Index (CDAI) score of about 300 and a Simple Endoscopic Score-Crohns disease (SES-CD) of about 15. About 95% had already failed at least one anti–tumor necrosis factor (TNF) drug. Half had failed at least two.
The coprimary endpoints were the proportion of patients who achieved clinical remission (stool frequency of 1.5 or less per day and abdominal pain of 1 or lower) at week 16 and endoscopic remission at weeks 12 or 16. Secondary endpoints included CDAI response, clinical response (at least a 30% reduction from baseline in stool frequency or abdominal pain), and endoscopic response.
In the primary analysis, the rate of endoscopic remission was significant (P less than .05) in both the 24-mg BID and the 24-mg OD groups. However, clinical remission with the original stool frequency cutpoint of 1.5/day or less wasn’t significantly different from placebo in any group. Dr. Sandborn did point out a 27% rate of clinical remission in those taking 12 mg, which had a P value of less than 0.1, relative to placebo.
Among the secondary endpoints, remission as measured by the CDAI score (less than 150) occurred in 39% of those taking 12 mg – the only significant response in that category.
The rate of endoscopic response (at least a 50% improvement in endoscopic findings) was 21% in the 6-mg group and 25% in the 24-mg OD group (P less than 0.05) and in about 30% of the 12-mg and 24-mg BID group (P less than 0.01).
When the clinical remission analysis employed the revised stool frequency cutpoint of less than 2.8/day, clinical remission rates improved somewhat. Almost 40% of those taking 24-mg BID achieved clinical remission (P less than 0.01), and 30% of those taking 6 mg achieved clinical remission, but the significance was marginal (P less than 0.1).
Steroid-free remission rates were significantly better than placebo in the 18-mg group (39%) and the 15-mg group (33%), both with a P value less than 0.05.
Dr. Sandborn also showed dramatic changes in C-reactive protein and fecal calprotectin. These dropped precipitously in all active groups by week 2, in a dose-respondent manner, and stayed well-suppressed in the two highest-dose groups. In the placebo groups, C-reactive protein rose over the 16 weeks, and fecal calprotectin remained unchanged from baseline.
The drug was reasonably well-tolerated and safe. About 80% of each dosing group reported at least one adverse event. The 12-mg dose appeared particularly troublesome, with 25% stopping because of an adverse event. By comparison, the discontinuation rate was 8% in the 24-mg BID group and 14% in the 24-mg OD group.
Serious adverse events were consistent with what is known about the JAK1-inhibitor safety profile, Dr. Sandborn said. There were nine serious infections, including Escherichia coli bacteremia, subcutaneous abscess, and sepsis (3-mg group); anorectal abscess, urinary tract infection, and sepsis (12-mg group); sepsis (24 mg BID); and peritonitis and sepsis (24 mg QD). There was one nonmelanoma skin cancer, which Dr. Sandborn said was probably pre-existing but not recognized at baseline. Three cases of herpes zoster occurred, all in the 24-mg BID group.
One patient experienced a gastrointestinal perforation, which sometimes occurs in Crohn’s disease. Two patients experienced a myocardial infarction, a number “too small to understand fully,” Dr. Sandborn said.
The drug will move forward into phase III trials, but the final dose hasn’t been decided on.
Dr. Sandborn has received consulting fees from AbbVie, which is developing the drug and sponsored CELEST.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – An investigational inhibitor of the Janus-1 kinase receptor induced clinical and endoscopic remission at several doses in patients with long-standing, treatment-resistant Crohn’s disease.
The two highest doses of upadacitinib (ABT-494; AbbVie) also allowed about 30% of patients to rapidly withdraw systemic steroids and stay in remission during the 16-week dose-finding induction trial, William J. Sandborn, MD, said at the annual Digestive Disease Week®.
“This rapid steroid tapering was a unique feature of the trial,” said Dr. Sandborn of the University of California, San Diego. “Usually during induction trials, steroids are held fixed at 20 mg-30 mg throughout the trial and then withdrawn to maintenance levels.”
The preplanned analysis used a unique composite outcome of 7-day stool frequency and abdominal pain. At the time of trial design, the scale had only been validated in patients with mild-moderate Crohn’s, so the investigators used the validated stool frequency cutpoint of 1.5 per day as a measure of clinical remission.
That was not an appropriate target for this unique study group, Dr. Sandborn said.
“CELEST was the most refractory patient population ever recruited into a Crohn’s disease clinical trial. If we could do this over now, we would use a cutpoint of less than 3 instead of 1.5 or less. This is a really tough clinical endpoint” that probably isn’t a realistic clinical goal for patients in this category. “There’s no way we would do it this way today. A number of studies since then now suggest that the right cutpoint for remission in these patients would be about 3 per day.”
CELEST enrolled 220 patients who were randomized to five treatment arms comprising 30-35 patients each: placebo; twice daily upadacitinib at 3 mg, 6 mg, 12 mg, and 24 mg (24 mg BID); and 24 mg once daily (24 mg OD). The study lasted 16 weeks and was followed by 36 weeks of blinded extension treatment. Dr. Sandborn reported the 16-week induction phase data.
The patients had moderate to severe Crohn’s disease, with a mean baseline Crohn’s Disease Activity Index (CDAI) score of about 300 and a Simple Endoscopic Score-Crohns disease (SES-CD) of about 15. About 95% had already failed at least one anti–tumor necrosis factor (TNF) drug. Half had failed at least two.
The coprimary endpoints were the proportion of patients who achieved clinical remission (stool frequency of 1.5 or less per day and abdominal pain of 1 or lower) at week 16 and endoscopic remission at weeks 12 or 16. Secondary endpoints included CDAI response, clinical response (at least a 30% reduction from baseline in stool frequency or abdominal pain), and endoscopic response.
In the primary analysis, the rate of endoscopic remission was significant (P less than .05) in both the 24-mg BID and the 24-mg OD groups. However, clinical remission with the original stool frequency cutpoint of 1.5/day or less wasn’t significantly different from placebo in any group. Dr. Sandborn did point out a 27% rate of clinical remission in those taking 12 mg, which had a P value of less than 0.1, relative to placebo.
Among the secondary endpoints, remission as measured by the CDAI score (less than 150) occurred in 39% of those taking 12 mg – the only significant response in that category.
The rate of endoscopic response (at least a 50% improvement in endoscopic findings) was 21% in the 6-mg group and 25% in the 24-mg OD group (P less than 0.05) and in about 30% of the 12-mg and 24-mg BID group (P less than 0.01).
When the clinical remission analysis employed the revised stool frequency cutpoint of less than 2.8/day, clinical remission rates improved somewhat. Almost 40% of those taking 24-mg BID achieved clinical remission (P less than 0.01), and 30% of those taking 6 mg achieved clinical remission, but the significance was marginal (P less than 0.1).
Steroid-free remission rates were significantly better than placebo in the 18-mg group (39%) and the 15-mg group (33%), both with a P value less than 0.05.
Dr. Sandborn also showed dramatic changes in C-reactive protein and fecal calprotectin. These dropped precipitously in all active groups by week 2, in a dose-respondent manner, and stayed well-suppressed in the two highest-dose groups. In the placebo groups, C-reactive protein rose over the 16 weeks, and fecal calprotectin remained unchanged from baseline.
The drug was reasonably well-tolerated and safe. About 80% of each dosing group reported at least one adverse event. The 12-mg dose appeared particularly troublesome, with 25% stopping because of an adverse event. By comparison, the discontinuation rate was 8% in the 24-mg BID group and 14% in the 24-mg OD group.
Serious adverse events were consistent with what is known about the JAK1-inhibitor safety profile, Dr. Sandborn said. There were nine serious infections, including Escherichia coli bacteremia, subcutaneous abscess, and sepsis (3-mg group); anorectal abscess, urinary tract infection, and sepsis (12-mg group); sepsis (24 mg BID); and peritonitis and sepsis (24 mg QD). There was one nonmelanoma skin cancer, which Dr. Sandborn said was probably pre-existing but not recognized at baseline. Three cases of herpes zoster occurred, all in the 24-mg BID group.
One patient experienced a gastrointestinal perforation, which sometimes occurs in Crohn’s disease. Two patients experienced a myocardial infarction, a number “too small to understand fully,” Dr. Sandborn said.
The drug will move forward into phase III trials, but the final dose hasn’t been decided on.
Dr. Sandborn has received consulting fees from AbbVie, which is developing the drug and sponsored CELEST.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT DDW
Key clinical point:
Major finding: Several doses achieved statistically significant effects in endoscopic and clinical response. About 30% of patients taking a higher dose achieved steroid-free remission.
Data source: The phase II dose-ranging study comprised 220 patients.
Disclosures: AbbVie is developing the drug and sponsored the study. Dr. Sandborn is a consultant for the company.

Ribaxamase prevented C. difficile infections by protecting microbiome
VIENNA – An investigational beta-lactamase reduced Clostridium difficile infections by 71% in patients receiving extended antibiotic therapy for respiratory infections but not by killing the opportunistic bacteria.
Rather, ribaxamase prevented C. difficile infections (CDI) by breaking down excess therapeutic antibiotics in the gut before they could injure an otherwise healthy microbiome, John Kokai-Kun, PhD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.

Ribaxamase is an oral enzyme that breaks the lactam ring in penicillins and cephalosporins. It’s formulated to release at a pH of 5.5 or higher, an environment that begins to develop in the upper small intestine near the bile duct – the same place that excess antibiotics are excreted.
“The drug is intended to be administered during, and for a short time after, intravenous administration of specific beta-lactam–containing antibiotics,” Dr. Kokai-Kun said. Ribaxamase doesn’t work on carbapenem-type antibiotics, he noted, and Synthetic Biologics is working on an effective enzyme for those as well.
In early human studies, ribaxamase was well tolerated and didn’t interfere with the pharmacokinetics of therapeutic antibiotics (Antimicrob Agents Chemother. 2017 Mar;61[3]:e02197-16). It’s also effective in patients who are taking a proton pump inhibitor, he said.
Dr. Kokai-Kun reported the results of a phase IIb study of 412 patients who received IV ceftriaxone for lower respiratory infections. They were assigned 1:1 to either 150 mg ribaxamase daily or placebo throughout the IV treatment and for 3 days after.
The primary endpoint was prevention of C. difficile infection. The secondary endpoint was prevention of non–C. difficile antibiotic-associated diarrhea. An exploratory endpoint examined the drug’s ability to protect the microbiome. Patients were monitored for 6 weeks after treatment stopped.
The cohort was a mean 70 years old. One-third of patients also received a macrolide during their hospitalization, and one-third were taking proton pump inhibitors. The respiratory infection cure rate was about 99% in both groups at both 72 hours and 4 weeks.
Eight patients in the placebo group (3.8%) and two in the active group (less than 1%) developed C. difficile infection. That translated to a statistically significant 71% risk reduction, with a P value of .027, Dr. Kokai-Kun said. Ribaxamase did not hit its secondary endpoint of preventing all-cause diarrhea or antibiotic-associated diarrhea that was not caused by C. difficile infection.
Although not a primary finding, ribaxamase also inhibited colonization by vancomycin-resistant enterococci, which occurred in about 70 (40%) patients in the placebo group and 40 (20%) in the ribaxamase group at both 72 hours and 4 weeks.
All patients contributed stool samples at baseline and after treatment for microbiome analysis. That portion of the study is still ongoing, Dr. Kokai-Kun said.
Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
msullivan@frontlinemedcom.com
On Twitter @alz_gal

VIENNA – An investigational beta-lactamase reduced Clostridium difficile infections by 71% in patients receiving extended antibiotic therapy for respiratory infections but not by killing the opportunistic bacteria.
Rather, ribaxamase prevented C. difficile infections (CDI) by breaking down excess therapeutic antibiotics in the gut before they could injure an otherwise healthy microbiome, John Kokai-Kun, PhD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Ribaxamase is an oral enzyme that breaks the lactam ring in penicillins and cephalosporins. It’s formulated to release at a pH of 5.5 or higher, an environment that begins to develop in the upper small intestine near the bile duct – the same place that excess antibiotics are excreted.
“The drug is intended to be administered during, and for a short time after, intravenous administration of specific beta-lactam–containing antibiotics,” Dr. Kokai-Kun said. Ribaxamase doesn’t work on carbapenem-type antibiotics, he noted, and Synthetic Biologics is working on an effective enzyme for those as well.
In early human studies, ribaxamase was well tolerated and didn’t interfere with the pharmacokinetics of therapeutic antibiotics (Antimicrob Agents Chemother. 2017 Mar;61[3]:e02197-16). It’s also effective in patients who are taking a proton pump inhibitor, he said.
Dr. Kokai-Kun reported the results of a phase IIb study of 412 patients who received IV ceftriaxone for lower respiratory infections. They were assigned 1:1 to either 150 mg ribaxamase daily or placebo throughout the IV treatment and for 3 days after.
The primary endpoint was prevention of C. difficile infection. The secondary endpoint was prevention of non–C. difficile antibiotic-associated diarrhea. An exploratory endpoint examined the drug’s ability to protect the microbiome. Patients were monitored for 6 weeks after treatment stopped.
The cohort was a mean 70 years old. One-third of patients also received a macrolide during their hospitalization, and one-third were taking proton pump inhibitors. The respiratory infection cure rate was about 99% in both groups at both 72 hours and 4 weeks.
Eight patients in the placebo group (3.8%) and two in the active group (less than 1%) developed C. difficile infection. That translated to a statistically significant 71% risk reduction, with a P value of .027, Dr. Kokai-Kun said. Ribaxamase did not hit its secondary endpoint of preventing all-cause diarrhea or antibiotic-associated diarrhea that was not caused by C. difficile infection.
Although not a primary finding, ribaxamase also inhibited colonization by vancomycin-resistant enterococci, which occurred in about 70 (40%) patients in the placebo group and 40 (20%) in the ribaxamase group at both 72 hours and 4 weeks.
All patients contributed stool samples at baseline and after treatment for microbiome analysis. That portion of the study is still ongoing, Dr. Kokai-Kun said.
Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
VIENNA – An investigational beta-lactamase reduced Clostridium difficile infections by 71% in patients receiving extended antibiotic therapy for respiratory infections but not by killing the opportunistic bacteria.
Rather, ribaxamase prevented C. difficile infections (CDI) by breaking down excess therapeutic antibiotics in the gut before they could injure an otherwise healthy microbiome, John Kokai-Kun, PhD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Ribaxamase is an oral enzyme that breaks the lactam ring in penicillins and cephalosporins. It’s formulated to release at a pH of 5.5 or higher, an environment that begins to develop in the upper small intestine near the bile duct – the same place that excess antibiotics are excreted.
“The drug is intended to be administered during, and for a short time after, intravenous administration of specific beta-lactam–containing antibiotics,” Dr. Kokai-Kun said. Ribaxamase doesn’t work on carbapenem-type antibiotics, he noted, and Synthetic Biologics is working on an effective enzyme for those as well.
In early human studies, ribaxamase was well tolerated and didn’t interfere with the pharmacokinetics of therapeutic antibiotics (Antimicrob Agents Chemother. 2017 Mar;61[3]:e02197-16). It’s also effective in patients who are taking a proton pump inhibitor, he said.
Dr. Kokai-Kun reported the results of a phase IIb study of 412 patients who received IV ceftriaxone for lower respiratory infections. They were assigned 1:1 to either 150 mg ribaxamase daily or placebo throughout the IV treatment and for 3 days after.
The primary endpoint was prevention of C. difficile infection. The secondary endpoint was prevention of non–C. difficile antibiotic-associated diarrhea. An exploratory endpoint examined the drug’s ability to protect the microbiome. Patients were monitored for 6 weeks after treatment stopped.
The cohort was a mean 70 years old. One-third of patients also received a macrolide during their hospitalization, and one-third were taking proton pump inhibitors. The respiratory infection cure rate was about 99% in both groups at both 72 hours and 4 weeks.
Eight patients in the placebo group (3.8%) and two in the active group (less than 1%) developed C. difficile infection. That translated to a statistically significant 71% risk reduction, with a P value of .027, Dr. Kokai-Kun said. Ribaxamase did not hit its secondary endpoint of preventing all-cause diarrhea or antibiotic-associated diarrhea that was not caused by C. difficile infection.
Although not a primary finding, ribaxamase also inhibited colonization by vancomycin-resistant enterococci, which occurred in about 70 (40%) patients in the placebo group and 40 (20%) in the ribaxamase group at both 72 hours and 4 weeks.
All patients contributed stool samples at baseline and after treatment for microbiome analysis. That portion of the study is still ongoing, Dr. Kokai-Kun said.
Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT ECCMID 2017
Key clinical point:
Major finding: Ribaxamase reduced C. difficile infections by 71%, relative to a placebo.
Data source: The study randomized 412 patients to either placebo or ribaxamase in addition to their therapeutic antibiotics.
Disclosures: Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
Antacid use in infants linked to increased fracture risk
SAN FRANCISCO – Children were more likely to experience a fracture if they were prescribed antacids before age 1 year, according to a study of military families.
The large study revealed that use of proton pump inhibitors (PPIs) before age 1 year was linked to a 22% increased risk of fracture, compared with those not prescribed antacids. Similarly, children prescribed both PPIs and H2 blockers before age 1 year were 31% more likely to have a fracture compared to those not taking the drugs.
“A lot of data are coming out that proton pump inhibitors are not quite as benign as we used to think, and we are seeing that fracture risk is increased with use,” U.S. Air Force Capt. Laura Malchodi, MD, a pediatrics resident at Walter Reed National Military Medical Center in Bethesda, Md., told colleagues at the Pediatric Academic Societies meeting.
Antacid use has been increasing among both adults and children, but the biggest rise has been in children under age 1 year, she said. Previous research into adult use of antacids has revealed an increased incidence of fractures, so Dr. Malchodi investigated the incidence of fractures in children under age 1 year among those who had taken PPIs, H2 blockers, neither, or both.
“What this means for doctors is that when you do start to think of using proton pump inhibitors or any antacid therapy in children, we should really think of limiting it to one type if possible – H2 blockers are now preferable – and for the shortest amount of time as possible,” Dr. Malchodi said of her findings.
The retrospective study’s cohort comprised 874,447 children born between 2001 and 2013 who had been in the U.S. Military Health System for at least 2 years. Children who took antacids after age 1, spent more than a week in a neonatal intensive care unit, or had nonaccidental trauma (abuse) or osteogenesis imperfecta were excluded.
Ninety percent of the cohort had not received prescriptions for any antacids (789,631 children) in their first year of life, and 1.2% had received prescriptions for both PPIs and H2 blockers before age 1 year. Of the remaining children, 7.7% had received prescriptions for H2 blockers, and 0.8% for PPIs.
The children who had and had not been prescribed antacids were similar in median years enrolled in the system, but nearly twice as many who received antacid prescription had been preterm (6.4% vs. 3.5%, P less than .05). Similarly, 3.7% of those prescribed antacids had a low birth weight, compared with 2.2% of those not prescribed antacids (P less than .05). The median age of fracture also differed for the two groups: 3.9 years for those prescribed antacids and 4.5 years for those not (P less than .05).
In using medical records during their analysis, the researchers excluded follow-up visits for the same fracture within the previous 6 months. Before adjustment for covariates, boys had a slightly increased risk of fracture (hazard ratio [HR], 1.08), and those with a previous fracture had an 85% increased risk (HR, 1.85). Compared with children not prescribed antacids, those prescribed PPIs had a 23% increased risk of fracture (HR, 1.23), and those prescribed H2 blockers had a 13% increased risk (HR, 1.13). Those prescribed combination antacid therapy had a 32% increased risk of fractures (HR, 1.32).
Adjustment for preterm birth, low birth weight, sex, and a previous fracture barely reduced those risks: 22% increased risk for PPI use, 4% increased risk for H2 blocker use, and 31% increased risk for using both. The vast majority of children who took antacids had been prescribed them in their first 6 months, so the researchers calculated adjusted risk by age of exposure. For H2 blockers, no statistically significant increased risk of fracture existed in those taking them before or after 6 months old.
Those taking PPIs, however, had a 25% increased risk of fracture if they took them before 6 months old, compared with a 20% increased risk if prescribed PPIs between 6 and 12 months. Likewise, children taking both PPIs and H2 blockers before 6 months old had a 32% increased risk of fracture, compared with a 23% increased risk between 6 and 12 months old.
Analysis of the duration of children’s use of antacids revealed a dose-response relationship, with an increasing risk alongside increasing days taking the medication. For example, those on PPIs for a month or less had a 19% increased risk of fracture, compared with children not prescribed antacids, but that rose to a 23% increased risk for those taking PPIs from 60 to 150 days and to a 42% increased risk for taking them longer than 150 days.
Similarly, the risk of fracture after having taken H2 blockers for up to a month was 14%, which increased to 22% for medication durations over 120 days. Children on combination therapy took the medication for much longer than did children prescribed either antacid. The risk of fracture was 17% greater for those taking them for up to 4 months, but that increased to a 50% greater risk for children taking both antacids for longer than 338 days.
“A couple of decades ago, we thought these medications were super safe, that there could be no problem with them,” Dr. Malchodi said, suggesting that their availability over the counter for adults may contribute to that perception. “With this growing evidence, there’s at least a lot more caution about using them,” she said.
Because the study relied on prescriptions for antacids, the researchers could not take into account which children actually took the antacids. Another limitation was their inability to consider other potential confounders, such as socioeconomic status or comorbidities that may later increase the risk of fracture. Further, exclusion of 6 months of follow-up after one fracture may have missed new fractures in that time period. Using a military cohort, on the other hand, meant having a geographically and socioeconomically diverse population with less risk of care bias because all the children had universal health care coverage.
No external funding was used. Dr. Malchodi reported having no disclosures.
*The View on the News was added on 6/13/17.
Acid suppression is frequently prescribed in infants for the treatment of symptoms such as fussiness, arching, and poor feeding, despite randomized controlled trials showing no benefit for these symptoms over placebo. These medications are often prescribed because physicians think they are useful; families are frustrated, exhausted, and worried about the infant’s symptoms; and these medications are considered safe and well tolerated. Recent adult studies have raised the possibility that these medications may not be as safe as once thought, with case-controlled studies linking them to increased risk of infectious, renal, cardiac, neurologic, and orthopedic complications. While there are pediatric studies supporting an increased infectious risk from both PPI and H2 antagonist use, there are no pediatric studies that address other complications. In this study by Dr. Malchodi et al., acid suppression use in infants under the age of 1 year was associated with an increased risk of fractures over the duration of enrollment in the U.S. Military Health System. They also found a dose-dependent effect, which further strengthens the conclusions that acid suppression may predispose patients to fractures. This research is a critical first step in elucidating the relationship of acid suppression and fracture risk in infants.
As with all database studies, there are some limitations to this study. First, patients taking acid suppression often have more comorbidities than do patients who are not taking the medications; because these patients are sicker, they may have more risk factors including compromised nutritional status and malabsorption predisposing them to fractures. The authors controlled for some of these comorbidities, but future studies should address additional ones. Second, as with all case-control studies, proving causality, not just association, is difficult so any future prospective acid suppression trials should include an assessment of bone health. Third, because the dosing per kilogram is not included, it is difficult to determine if there is a safe level of acid suppression for those children who need it. Fourth, because this is a database review, it is not clear if patients actually took the prescribed medication.
Because of the safety concerns regarding acid suppression as well as the lack of benefit in reducing symptoms in infants, nonpharmacologic therapies should be considered as first-line therapy for the treatment of bothersome symptoms. In the fussy, arching, or irritable child, changing the frequency or volume of feeds, thickening feeds, or changing to partially hydrolyzed formulas or eliminating dairy from the maternal diet (for breastfed infants) should be considered before starting acid suppression therapy. Other diagnoses besides gastroesophageal reflux disease, such as colic and cow’s milk protein allergy, need to be considered as well to ensure that the therapy matches the diagnosis. For those patients in whom acid suppression is required, using the lowest dose possible for the shortest amount of time is critical. Finally, for patients on multiple medications that may impact fracture risk (such as acid suppression, steroids), extra vigilance is needed to stop unnecessary medications as soon as possible.
Acid suppression is frequently prescribed in infants for the treatment of symptoms such as fussiness, arching, and poor feeding, despite randomized controlled trials showing no benefit for these symptoms over placebo. These medications are often prescribed because physicians think they are useful; families are frustrated, exhausted, and worried about the infant’s symptoms; and these medications are considered safe and well tolerated. Recent adult studies have raised the possibility that these medications may not be as safe as once thought, with case-controlled studies linking them to increased risk of infectious, renal, cardiac, neurologic, and orthopedic complications. While there are pediatric studies supporting an increased infectious risk from both PPI and H2 antagonist use, there are no pediatric studies that address other complications. In this study by Dr. Malchodi et al., acid suppression use in infants under the age of 1 year was associated with an increased risk of fractures over the duration of enrollment in the U.S. Military Health System. They also found a dose-dependent effect, which further strengthens the conclusions that acid suppression may predispose patients to fractures. This research is a critical first step in elucidating the relationship of acid suppression and fracture risk in infants.
As with all database studies, there are some limitations to this study. First, patients taking acid suppression often have more comorbidities than do patients who are not taking the medications; because these patients are sicker, they may have more risk factors including compromised nutritional status and malabsorption predisposing them to fractures. The authors controlled for some of these comorbidities, but future studies should address additional ones. Second, as with all case-control studies, proving causality, not just association, is difficult so any future prospective acid suppression trials should include an assessment of bone health. Third, because the dosing per kilogram is not included, it is difficult to determine if there is a safe level of acid suppression for those children who need it. Fourth, because this is a database review, it is not clear if patients actually took the prescribed medication.
Because of the safety concerns regarding acid suppression as well as the lack of benefit in reducing symptoms in infants, nonpharmacologic therapies should be considered as first-line therapy for the treatment of bothersome symptoms. In the fussy, arching, or irritable child, changing the frequency or volume of feeds, thickening feeds, or changing to partially hydrolyzed formulas or eliminating dairy from the maternal diet (for breastfed infants) should be considered before starting acid suppression therapy. Other diagnoses besides gastroesophageal reflux disease, such as colic and cow’s milk protein allergy, need to be considered as well to ensure that the therapy matches the diagnosis. For those patients in whom acid suppression is required, using the lowest dose possible for the shortest amount of time is critical. Finally, for patients on multiple medications that may impact fracture risk (such as acid suppression, steroids), extra vigilance is needed to stop unnecessary medications as soon as possible.
Acid suppression is frequently prescribed in infants for the treatment of symptoms such as fussiness, arching, and poor feeding, despite randomized controlled trials showing no benefit for these symptoms over placebo. These medications are often prescribed because physicians think they are useful; families are frustrated, exhausted, and worried about the infant’s symptoms; and these medications are considered safe and well tolerated. Recent adult studies have raised the possibility that these medications may not be as safe as once thought, with case-controlled studies linking them to increased risk of infectious, renal, cardiac, neurologic, and orthopedic complications. While there are pediatric studies supporting an increased infectious risk from both PPI and H2 antagonist use, there are no pediatric studies that address other complications. In this study by Dr. Malchodi et al., acid suppression use in infants under the age of 1 year was associated with an increased risk of fractures over the duration of enrollment in the U.S. Military Health System. They also found a dose-dependent effect, which further strengthens the conclusions that acid suppression may predispose patients to fractures. This research is a critical first step in elucidating the relationship of acid suppression and fracture risk in infants.
As with all database studies, there are some limitations to this study. First, patients taking acid suppression often have more comorbidities than do patients who are not taking the medications; because these patients are sicker, they may have more risk factors including compromised nutritional status and malabsorption predisposing them to fractures. The authors controlled for some of these comorbidities, but future studies should address additional ones. Second, as with all case-control studies, proving causality, not just association, is difficult so any future prospective acid suppression trials should include an assessment of bone health. Third, because the dosing per kilogram is not included, it is difficult to determine if there is a safe level of acid suppression for those children who need it. Fourth, because this is a database review, it is not clear if patients actually took the prescribed medication.
Because of the safety concerns regarding acid suppression as well as the lack of benefit in reducing symptoms in infants, nonpharmacologic therapies should be considered as first-line therapy for the treatment of bothersome symptoms. In the fussy, arching, or irritable child, changing the frequency or volume of feeds, thickening feeds, or changing to partially hydrolyzed formulas or eliminating dairy from the maternal diet (for breastfed infants) should be considered before starting acid suppression therapy. Other diagnoses besides gastroesophageal reflux disease, such as colic and cow’s milk protein allergy, need to be considered as well to ensure that the therapy matches the diagnosis. For those patients in whom acid suppression is required, using the lowest dose possible for the shortest amount of time is critical. Finally, for patients on multiple medications that may impact fracture risk (such as acid suppression, steroids), extra vigilance is needed to stop unnecessary medications as soon as possible.
SAN FRANCISCO – Children were more likely to experience a fracture if they were prescribed antacids before age 1 year, according to a study of military families.
The large study revealed that use of proton pump inhibitors (PPIs) before age 1 year was linked to a 22% increased risk of fracture, compared with those not prescribed antacids. Similarly, children prescribed both PPIs and H2 blockers before age 1 year were 31% more likely to have a fracture compared to those not taking the drugs.
“A lot of data are coming out that proton pump inhibitors are not quite as benign as we used to think, and we are seeing that fracture risk is increased with use,” U.S. Air Force Capt. Laura Malchodi, MD, a pediatrics resident at Walter Reed National Military Medical Center in Bethesda, Md., told colleagues at the Pediatric Academic Societies meeting.
Antacid use has been increasing among both adults and children, but the biggest rise has been in children under age 1 year, she said. Previous research into adult use of antacids has revealed an increased incidence of fractures, so Dr. Malchodi investigated the incidence of fractures in children under age 1 year among those who had taken PPIs, H2 blockers, neither, or both.
“What this means for doctors is that when you do start to think of using proton pump inhibitors or any antacid therapy in children, we should really think of limiting it to one type if possible – H2 blockers are now preferable – and for the shortest amount of time as possible,” Dr. Malchodi said of her findings.
The retrospective study’s cohort comprised 874,447 children born between 2001 and 2013 who had been in the U.S. Military Health System for at least 2 years. Children who took antacids after age 1, spent more than a week in a neonatal intensive care unit, or had nonaccidental trauma (abuse) or osteogenesis imperfecta were excluded.
Ninety percent of the cohort had not received prescriptions for any antacids (789,631 children) in their first year of life, and 1.2% had received prescriptions for both PPIs and H2 blockers before age 1 year. Of the remaining children, 7.7% had received prescriptions for H2 blockers, and 0.8% for PPIs.
The children who had and had not been prescribed antacids were similar in median years enrolled in the system, but nearly twice as many who received antacid prescription had been preterm (6.4% vs. 3.5%, P less than .05). Similarly, 3.7% of those prescribed antacids had a low birth weight, compared with 2.2% of those not prescribed antacids (P less than .05). The median age of fracture also differed for the two groups: 3.9 years for those prescribed antacids and 4.5 years for those not (P less than .05).
In using medical records during their analysis, the researchers excluded follow-up visits for the same fracture within the previous 6 months. Before adjustment for covariates, boys had a slightly increased risk of fracture (hazard ratio [HR], 1.08), and those with a previous fracture had an 85% increased risk (HR, 1.85). Compared with children not prescribed antacids, those prescribed PPIs had a 23% increased risk of fracture (HR, 1.23), and those prescribed H2 blockers had a 13% increased risk (HR, 1.13). Those prescribed combination antacid therapy had a 32% increased risk of fractures (HR, 1.32).
Adjustment for preterm birth, low birth weight, sex, and a previous fracture barely reduced those risks: 22% increased risk for PPI use, 4% increased risk for H2 blocker use, and 31% increased risk for using both. The vast majority of children who took antacids had been prescribed them in their first 6 months, so the researchers calculated adjusted risk by age of exposure. For H2 blockers, no statistically significant increased risk of fracture existed in those taking them before or after 6 months old.
Those taking PPIs, however, had a 25% increased risk of fracture if they took them before 6 months old, compared with a 20% increased risk if prescribed PPIs between 6 and 12 months. Likewise, children taking both PPIs and H2 blockers before 6 months old had a 32% increased risk of fracture, compared with a 23% increased risk between 6 and 12 months old.
Analysis of the duration of children’s use of antacids revealed a dose-response relationship, with an increasing risk alongside increasing days taking the medication. For example, those on PPIs for a month or less had a 19% increased risk of fracture, compared with children not prescribed antacids, but that rose to a 23% increased risk for those taking PPIs from 60 to 150 days and to a 42% increased risk for taking them longer than 150 days.
Similarly, the risk of fracture after having taken H2 blockers for up to a month was 14%, which increased to 22% for medication durations over 120 days. Children on combination therapy took the medication for much longer than did children prescribed either antacid. The risk of fracture was 17% greater for those taking them for up to 4 months, but that increased to a 50% greater risk for children taking both antacids for longer than 338 days.
“A couple of decades ago, we thought these medications were super safe, that there could be no problem with them,” Dr. Malchodi said, suggesting that their availability over the counter for adults may contribute to that perception. “With this growing evidence, there’s at least a lot more caution about using them,” she said.
Because the study relied on prescriptions for antacids, the researchers could not take into account which children actually took the antacids. Another limitation was their inability to consider other potential confounders, such as socioeconomic status or comorbidities that may later increase the risk of fracture. Further, exclusion of 6 months of follow-up after one fracture may have missed new fractures in that time period. Using a military cohort, on the other hand, meant having a geographically and socioeconomically diverse population with less risk of care bias because all the children had universal health care coverage.
No external funding was used. Dr. Malchodi reported having no disclosures.
*The View on the News was added on 6/13/17.
SAN FRANCISCO – Children were more likely to experience a fracture if they were prescribed antacids before age 1 year, according to a study of military families.
The large study revealed that use of proton pump inhibitors (PPIs) before age 1 year was linked to a 22% increased risk of fracture, compared with those not prescribed antacids. Similarly, children prescribed both PPIs and H2 blockers before age 1 year were 31% more likely to have a fracture compared to those not taking the drugs.
“A lot of data are coming out that proton pump inhibitors are not quite as benign as we used to think, and we are seeing that fracture risk is increased with use,” U.S. Air Force Capt. Laura Malchodi, MD, a pediatrics resident at Walter Reed National Military Medical Center in Bethesda, Md., told colleagues at the Pediatric Academic Societies meeting.
Antacid use has been increasing among both adults and children, but the biggest rise has been in children under age 1 year, she said. Previous research into adult use of antacids has revealed an increased incidence of fractures, so Dr. Malchodi investigated the incidence of fractures in children under age 1 year among those who had taken PPIs, H2 blockers, neither, or both.
“What this means for doctors is that when you do start to think of using proton pump inhibitors or any antacid therapy in children, we should really think of limiting it to one type if possible – H2 blockers are now preferable – and for the shortest amount of time as possible,” Dr. Malchodi said of her findings.
The retrospective study’s cohort comprised 874,447 children born between 2001 and 2013 who had been in the U.S. Military Health System for at least 2 years. Children who took antacids after age 1, spent more than a week in a neonatal intensive care unit, or had nonaccidental trauma (abuse) or osteogenesis imperfecta were excluded.
Ninety percent of the cohort had not received prescriptions for any antacids (789,631 children) in their first year of life, and 1.2% had received prescriptions for both PPIs and H2 blockers before age 1 year. Of the remaining children, 7.7% had received prescriptions for H2 blockers, and 0.8% for PPIs.
The children who had and had not been prescribed antacids were similar in median years enrolled in the system, but nearly twice as many who received antacid prescription had been preterm (6.4% vs. 3.5%, P less than .05). Similarly, 3.7% of those prescribed antacids had a low birth weight, compared with 2.2% of those not prescribed antacids (P less than .05). The median age of fracture also differed for the two groups: 3.9 years for those prescribed antacids and 4.5 years for those not (P less than .05).
In using medical records during their analysis, the researchers excluded follow-up visits for the same fracture within the previous 6 months. Before adjustment for covariates, boys had a slightly increased risk of fracture (hazard ratio [HR], 1.08), and those with a previous fracture had an 85% increased risk (HR, 1.85). Compared with children not prescribed antacids, those prescribed PPIs had a 23% increased risk of fracture (HR, 1.23), and those prescribed H2 blockers had a 13% increased risk (HR, 1.13). Those prescribed combination antacid therapy had a 32% increased risk of fractures (HR, 1.32).
Adjustment for preterm birth, low birth weight, sex, and a previous fracture barely reduced those risks: 22% increased risk for PPI use, 4% increased risk for H2 blocker use, and 31% increased risk for using both. The vast majority of children who took antacids had been prescribed them in their first 6 months, so the researchers calculated adjusted risk by age of exposure. For H2 blockers, no statistically significant increased risk of fracture existed in those taking them before or after 6 months old.
Those taking PPIs, however, had a 25% increased risk of fracture if they took them before 6 months old, compared with a 20% increased risk if prescribed PPIs between 6 and 12 months. Likewise, children taking both PPIs and H2 blockers before 6 months old had a 32% increased risk of fracture, compared with a 23% increased risk between 6 and 12 months old.
Analysis of the duration of children’s use of antacids revealed a dose-response relationship, with an increasing risk alongside increasing days taking the medication. For example, those on PPIs for a month or less had a 19% increased risk of fracture, compared with children not prescribed antacids, but that rose to a 23% increased risk for those taking PPIs from 60 to 150 days and to a 42% increased risk for taking them longer than 150 days.
Similarly, the risk of fracture after having taken H2 blockers for up to a month was 14%, which increased to 22% for medication durations over 120 days. Children on combination therapy took the medication for much longer than did children prescribed either antacid. The risk of fracture was 17% greater for those taking them for up to 4 months, but that increased to a 50% greater risk for children taking both antacids for longer than 338 days.
“A couple of decades ago, we thought these medications were super safe, that there could be no problem with them,” Dr. Malchodi said, suggesting that their availability over the counter for adults may contribute to that perception. “With this growing evidence, there’s at least a lot more caution about using them,” she said.
Because the study relied on prescriptions for antacids, the researchers could not take into account which children actually took the antacids. Another limitation was their inability to consider other potential confounders, such as socioeconomic status or comorbidities that may later increase the risk of fracture. Further, exclusion of 6 months of follow-up after one fracture may have missed new fractures in that time period. Using a military cohort, on the other hand, meant having a geographically and socioeconomically diverse population with less risk of care bias because all the children had universal health care coverage.
No external funding was used. Dr. Malchodi reported having no disclosures.
*The View on the News was added on 6/13/17.
AT PAS 17
Key clinical point:
Major finding: Risk of fracture increased 22% among children who took proton pump inhibitors in their first year of life and increased 31% among children taking both PPIs and H2 blockers.
Data source: A retrospective cohort study of 874,447 children born between 2001 and 2013 and who were in the U.S. Military Health System for at least 2 years.
Disclosures: No external funding was used. Dr. Malchodi reported having no relevant financial disclosures.
Adalimumab is good first-line anti-TNF therapy for pediatric Crohn’s disease
Adalimumab (ADA) as a first-line anti–tumor necrosis factor therapy induced and maintained clinical remission in children with Crohn’s disease, said Víctor Manuel Navas-López, MD, PhD, of the Hospital Materno Infantil, Málaga, Spain, and his associates.
Infliximab is the usual first-line anti–tumor necrosis factor treatment given to children with Crohn’s disease, with ADA used in patients who don’t respond or who develop tolerance to infliximab.
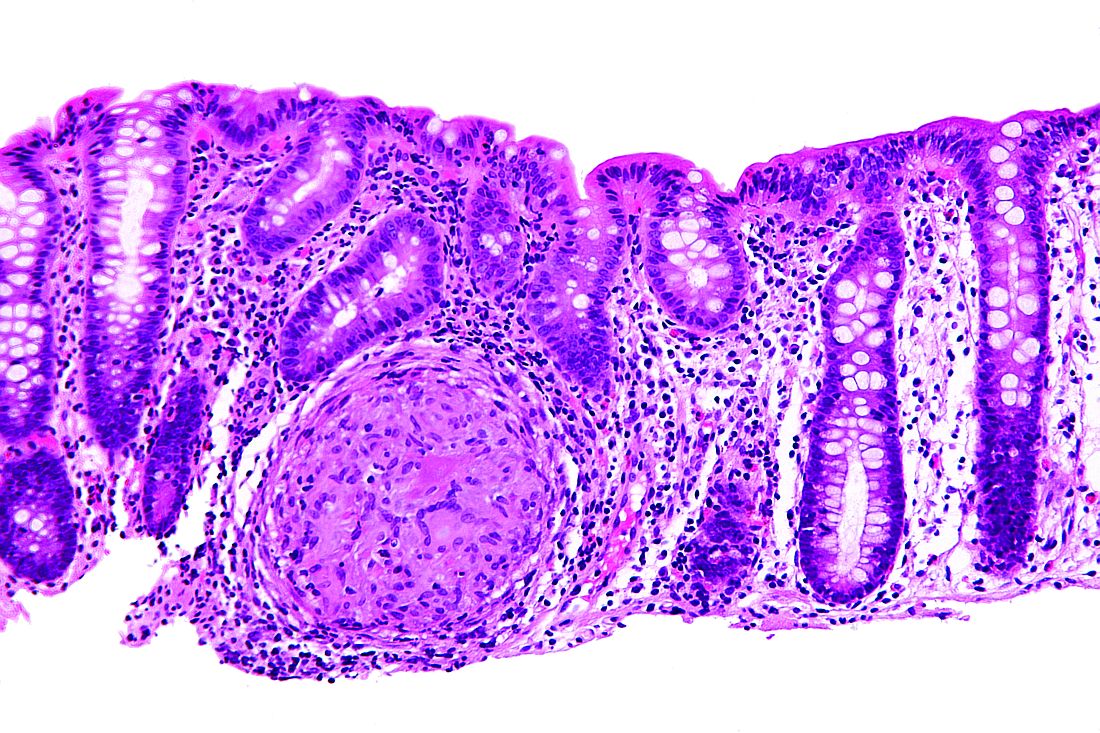
Dose escalation was necessary for 26% of the 62 patients. Thirty-nine percent of patients had growth retardation.
“ADA treatment significantly improved z-score growth rate in children with Crohn’s disease, especially in those with severe growth failure at baseline,” the researchers said. Only 13% of patients reported adverse events, none of them severe.
Read more in the Anales de Pediatría (2017 Apr 14. doi: 10.1016/j.anpedi.2017.01.013).
Adalimumab (ADA) as a first-line anti–tumor necrosis factor therapy induced and maintained clinical remission in children with Crohn’s disease, said Víctor Manuel Navas-López, MD, PhD, of the Hospital Materno Infantil, Málaga, Spain, and his associates.
Infliximab is the usual first-line anti–tumor necrosis factor treatment given to children with Crohn’s disease, with ADA used in patients who don’t respond or who develop tolerance to infliximab.
Dose escalation was necessary for 26% of the 62 patients. Thirty-nine percent of patients had growth retardation.
“ADA treatment significantly improved z-score growth rate in children with Crohn’s disease, especially in those with severe growth failure at baseline,” the researchers said. Only 13% of patients reported adverse events, none of them severe.
Read more in the Anales de Pediatría (2017 Apr 14. doi: 10.1016/j.anpedi.2017.01.013).
Adalimumab (ADA) as a first-line anti–tumor necrosis factor therapy induced and maintained clinical remission in children with Crohn’s disease, said Víctor Manuel Navas-López, MD, PhD, of the Hospital Materno Infantil, Málaga, Spain, and his associates.
Infliximab is the usual first-line anti–tumor necrosis factor treatment given to children with Crohn’s disease, with ADA used in patients who don’t respond or who develop tolerance to infliximab.
Dose escalation was necessary for 26% of the 62 patients. Thirty-nine percent of patients had growth retardation.
“ADA treatment significantly improved z-score growth rate in children with Crohn’s disease, especially in those with severe growth failure at baseline,” the researchers said. Only 13% of patients reported adverse events, none of them severe.
Read more in the Anales de Pediatría (2017 Apr 14. doi: 10.1016/j.anpedi.2017.01.013).
FROM ANALES DE PEDIATRIA
Anti-TNF drugs reduce mortality in Crohn’s disease
CHICAGO – As compared with prolonged use of corticosteroids, the use of anti–tumor necrosis factor (TNF) drugs was associated with reduced mortality in patients with Crohn’s disease, according to new findings presented here at Digestive Disease Week®.
The reduced mortality seen in this population may be secondary to the lower rates of major adverse cardiovascular events and hip fracture that are associated with anti-TNF use as compared to corticosteroid use. However, the same reduction in mortality risk was not observed in patients with ulcerative colitis using anti-TNF drugs.
Anti-TNF therapy has become a cornerstone in the management of inflammatory bowel disease (IBD), and Dr. Lewis noted that these agents have been shown to be useful for induction, maintenance, and remission, and to reduce surgical and hospitalization rates.
“However, fear of adverse events and cost has deterred greater use of these agents,” he told attendees.
In their study, Dr. Lewis and his colleagues compared the mortality risk with prolonged corticosteroids use versus anti-TNF drugs in patients with IBD.
They conducted a retrospective cohort study using data from 2006-2013 of a population of Medicaid and Medicare beneficiaries in the United States. The cohort included individuals who had received treatment with corticosteroids within the prior year and subsequently had been treated with either additional corticosteroid therapy for a total of greater than 3,000 mg of prednisone or equivalent within 12 months or newly initiated anti-TNF therapy.
The primary outcome of the study was all-cause mortality and secondary outcomes included common causes of death.
Dr. Lewis explained that 57 potential confounding variables were thought to be associated with the choice between corticosteroid use or anti-TNF therapy. These variables, which included demographic characteristics, medications, diagnostic tests, and comorbidities, were measured.
Among Crohn’s disease patients, 7,694 who were prolonged corticosteroid users and 1,879 were new to anti-TNF therapy. Among patients with ulcerative colitis, 3,224 were long-term corticosteroid users and 459 were new anti-TNF users.
The researchers found that the weighted annual incidences of death per 1,000 Crohn’s disease treated patients were 21.4 for those using anti-TNF therapy and 30.1 for those with prolonged corticosteroid use. For those with ulcerative colitis, these figures were 23.0 and 30.9, respectively.
The risk of death was statistically significantly reduced in Crohn’s disease patients who used anti-TNF therapy (odds ratio, 0.78; 95% confidence interval, 0.65-0.93). However, the benefit was not as pronounced in ulcerative colitis.
“We did not see the same effect for ulcerative colitis but for mortality, it was in the same direction, with a hazard ratio of 0.87,” he said.
Among the Crohn’s disease patients, anti-TNF therapy was associated with lower rates of major adverse cardiovascular events (OR, 0.68; 95% CI, 0.55-0.85) and hip fracture (OR, 0.5; 95% CI, 0.34-0.83), which were statistically significant. The use of anti-TNF therapy also reduced the risk of stroke in Crohn’s disease patients (OR, 0.72; 95% CI, 0.51-1.03), but there was also an increase in the risk of cancer (OR, 0.27; 95% CI, 0.98-1.65). Both of these findings nearly reached statistical significance.
In the model that censored for any of the secondary outcomes, the lower mortality risk was attenuated and very close to a null result (OR, 0.97; 95% CI 0.63-1.47).
Dr. Lewis also pointed out that in some of their models, the magnitude of benefit with anti-TNF therapy appears to be greatest in the patients with the most comorbidities.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
Dr. Lewis has disclosed financial relationships with Takeda, Pfizer, Lilly, Gilead, Johnson and Johnson, Samsung Bioepis, AbbVie, and Dark Canyon Laboratories.
CHICAGO – As compared with prolonged use of corticosteroids, the use of anti–tumor necrosis factor (TNF) drugs was associated with reduced mortality in patients with Crohn’s disease, according to new findings presented here at Digestive Disease Week®.
The reduced mortality seen in this population may be secondary to the lower rates of major adverse cardiovascular events and hip fracture that are associated with anti-TNF use as compared to corticosteroid use. However, the same reduction in mortality risk was not observed in patients with ulcerative colitis using anti-TNF drugs.
Anti-TNF therapy has become a cornerstone in the management of inflammatory bowel disease (IBD), and Dr. Lewis noted that these agents have been shown to be useful for induction, maintenance, and remission, and to reduce surgical and hospitalization rates.
“However, fear of adverse events and cost has deterred greater use of these agents,” he told attendees.
In their study, Dr. Lewis and his colleagues compared the mortality risk with prolonged corticosteroids use versus anti-TNF drugs in patients with IBD.
They conducted a retrospective cohort study using data from 2006-2013 of a population of Medicaid and Medicare beneficiaries in the United States. The cohort included individuals who had received treatment with corticosteroids within the prior year and subsequently had been treated with either additional corticosteroid therapy for a total of greater than 3,000 mg of prednisone or equivalent within 12 months or newly initiated anti-TNF therapy.
The primary outcome of the study was all-cause mortality and secondary outcomes included common causes of death.
Dr. Lewis explained that 57 potential confounding variables were thought to be associated with the choice between corticosteroid use or anti-TNF therapy. These variables, which included demographic characteristics, medications, diagnostic tests, and comorbidities, were measured.
Among Crohn’s disease patients, 7,694 who were prolonged corticosteroid users and 1,879 were new to anti-TNF therapy. Among patients with ulcerative colitis, 3,224 were long-term corticosteroid users and 459 were new anti-TNF users.
The researchers found that the weighted annual incidences of death per 1,000 Crohn’s disease treated patients were 21.4 for those using anti-TNF therapy and 30.1 for those with prolonged corticosteroid use. For those with ulcerative colitis, these figures were 23.0 and 30.9, respectively.
The risk of death was statistically significantly reduced in Crohn’s disease patients who used anti-TNF therapy (odds ratio, 0.78; 95% confidence interval, 0.65-0.93). However, the benefit was not as pronounced in ulcerative colitis.
“We did not see the same effect for ulcerative colitis but for mortality, it was in the same direction, with a hazard ratio of 0.87,” he said.
Among the Crohn’s disease patients, anti-TNF therapy was associated with lower rates of major adverse cardiovascular events (OR, 0.68; 95% CI, 0.55-0.85) and hip fracture (OR, 0.5; 95% CI, 0.34-0.83), which were statistically significant. The use of anti-TNF therapy also reduced the risk of stroke in Crohn’s disease patients (OR, 0.72; 95% CI, 0.51-1.03), but there was also an increase in the risk of cancer (OR, 0.27; 95% CI, 0.98-1.65). Both of these findings nearly reached statistical significance.
In the model that censored for any of the secondary outcomes, the lower mortality risk was attenuated and very close to a null result (OR, 0.97; 95% CI 0.63-1.47).
Dr. Lewis also pointed out that in some of their models, the magnitude of benefit with anti-TNF therapy appears to be greatest in the patients with the most comorbidities.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
Dr. Lewis has disclosed financial relationships with Takeda, Pfizer, Lilly, Gilead, Johnson and Johnson, Samsung Bioepis, AbbVie, and Dark Canyon Laboratories.
CHICAGO – As compared with prolonged use of corticosteroids, the use of anti–tumor necrosis factor (TNF) drugs was associated with reduced mortality in patients with Crohn’s disease, according to new findings presented here at Digestive Disease Week®.
The reduced mortality seen in this population may be secondary to the lower rates of major adverse cardiovascular events and hip fracture that are associated with anti-TNF use as compared to corticosteroid use. However, the same reduction in mortality risk was not observed in patients with ulcerative colitis using anti-TNF drugs.
Anti-TNF therapy has become a cornerstone in the management of inflammatory bowel disease (IBD), and Dr. Lewis noted that these agents have been shown to be useful for induction, maintenance, and remission, and to reduce surgical and hospitalization rates.
“However, fear of adverse events and cost has deterred greater use of these agents,” he told attendees.
In their study, Dr. Lewis and his colleagues compared the mortality risk with prolonged corticosteroids use versus anti-TNF drugs in patients with IBD.
They conducted a retrospective cohort study using data from 2006-2013 of a population of Medicaid and Medicare beneficiaries in the United States. The cohort included individuals who had received treatment with corticosteroids within the prior year and subsequently had been treated with either additional corticosteroid therapy for a total of greater than 3,000 mg of prednisone or equivalent within 12 months or newly initiated anti-TNF therapy.
The primary outcome of the study was all-cause mortality and secondary outcomes included common causes of death.
Dr. Lewis explained that 57 potential confounding variables were thought to be associated with the choice between corticosteroid use or anti-TNF therapy. These variables, which included demographic characteristics, medications, diagnostic tests, and comorbidities, were measured.
Among Crohn’s disease patients, 7,694 who were prolonged corticosteroid users and 1,879 were new to anti-TNF therapy. Among patients with ulcerative colitis, 3,224 were long-term corticosteroid users and 459 were new anti-TNF users.
The researchers found that the weighted annual incidences of death per 1,000 Crohn’s disease treated patients were 21.4 for those using anti-TNF therapy and 30.1 for those with prolonged corticosteroid use. For those with ulcerative colitis, these figures were 23.0 and 30.9, respectively.
The risk of death was statistically significantly reduced in Crohn’s disease patients who used anti-TNF therapy (odds ratio, 0.78; 95% confidence interval, 0.65-0.93). However, the benefit was not as pronounced in ulcerative colitis.
“We did not see the same effect for ulcerative colitis but for mortality, it was in the same direction, with a hazard ratio of 0.87,” he said.
Among the Crohn’s disease patients, anti-TNF therapy was associated with lower rates of major adverse cardiovascular events (OR, 0.68; 95% CI, 0.55-0.85) and hip fracture (OR, 0.5; 95% CI, 0.34-0.83), which were statistically significant. The use of anti-TNF therapy also reduced the risk of stroke in Crohn’s disease patients (OR, 0.72; 95% CI, 0.51-1.03), but there was also an increase in the risk of cancer (OR, 0.27; 95% CI, 0.98-1.65). Both of these findings nearly reached statistical significance.
In the model that censored for any of the secondary outcomes, the lower mortality risk was attenuated and very close to a null result (OR, 0.97; 95% CI 0.63-1.47).
Dr. Lewis also pointed out that in some of their models, the magnitude of benefit with anti-TNF therapy appears to be greatest in the patients with the most comorbidities.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
Dr. Lewis has disclosed financial relationships with Takeda, Pfizer, Lilly, Gilead, Johnson and Johnson, Samsung Bioepis, AbbVie, and Dark Canyon Laboratories.
AT DDW
Key clinical point:
Major finding: Anti-TNF drugs were associated with reduced mortality in patients with Crohn’s disease (OR, 0.78) but to a much lesser extent in ulcerative colitis (OR, 0.87).
Data source: Database of Medicare and Medicaid recipients with 9,573 patients with Crohn’s disease and 3,683 with ulcerative colitis.
Disclosures: Dr. Lewis has disclosed financial relationships with Takeda, Pfizer, Lilly, Gilead, Johnson and Johnson, Samsung Bioepis, AbbVie, and Dark Canyon Laboratories.
VIDEO: Registry study will follow 4,000 fecal transplant patients for 10 years
CHICAGO – A 10-year registry study aims to gather clinical and patient-reported outcomes on 4,000 adult and pediatric patients who undergo fecal microbiota transplant in the United States, officials of the American Gastroenterological Association announced during Digestive Disease Week®.
The AGA Fecal Microbiota Transplantation National Registry will be the first study to assess both short- and long-term patient outcomes associated with fecal microbiota transplant (FMT) in both adults and children, Colleen Kelly, MD, said in an video interview. Most subjects will have received FMT for recurrent or refractory Clostridium difficile infections – the only indication for which Food and Drug Administration currently allows independent clinician action. But the investigational uses of FMT are expanding rapidly, and patients who undergo the procedure during any registered study will be eligible for enrollment, said Dr. Kelly, co-chair of the study’s steering committee.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The study’s primary objectives are short- and long-term safety outcomes, said Dr. Kelly of Brown University, Providence, R.I. While generally considered quite safe, short-term adverse events have been reported with FMT, and some of them have been serious – including one death from aspiration pneumonia in a patient who received donor stool via nasogastric tube (Clin Infect Dis. 2015 Mar;61[1]:136-7). Other adverse events are usually self-limited but can include low-grade fever, abdominal pain, distention, bloating, and diarrhea.
Researchers seek to illuminate many of the unknowns associated with this relatively new procedure. Scientists are only now beginning to unravel the myriad ways the human microbiome promotes both health and disease. Specific alterations, for example, have been associated with obesity and other conditions; there is concern that transplanting a new microbial population could induce a disease phenotype in a recipient who might not have otherwise been at risk.
With the planned cohort size and follow-up period, the study should be able to detect any unanticipated adverse events that occur in more than 1% of the population, Dr. Kelly said. It will include a comparator group of patients with recurrent or refractory C. difficile infection from a large insurance claims database to allow comparison between patients treated with FMT and those treated with antibiotics only.
The registry study also aims to discover which method or methods of transplant material delivery are best, she said. Right now, there are a number of methods (colonoscopy/sigmoidoscopy, enema, upper gastrointestinal endoscopy, nasogastric or nasoduodenal tube, and capsules), and no consensus on which is the best. As indications for FMT expand, there may be no single best method. The approach will probably be matched to the disorder being treated, and the study may help illuminate this as well.
For the first 2 years after a transplant, clinicians will follow patients and enter data into the registry. After that, an electronic patient-reported outcomes system will automatically contact the patient annually for follow-up information by email or text message. When patients enter their data, they can access educational material that will help keep them up-to-date on potential adverse events.
The study will also include a biobank of stool samples obtained during the procedures, hosted by the American Gut Project and the Microbiome Initiative at the University of California, San Diego. This arm of the project will analyze the microbiome of 3,000 stool samples from recipients, both before and after their transplant, as well as the corresponding donors whose material was used in the fecal transplant.
The registry study, a project of the AGA Center for Gut Microbiome Research and Education, is funded by a $3.3 million grant from the National Institute of Allergy and Infectious Diseases. It will be conducted in partnership with the Crohn’s and Colitis Foundation, Infectious Diseases Society, and the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition.
The registry study currently is accepting applications. Physicians who perform FMT for C. difficile infections, and centers that conduct FMT research for other potential indications, can fill out a short survey to indicate their interest.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE) and the Society for Surgery of the Alimentary Tract (SSAT).
This article was updated June 8, 2017.
msullivan@frontlinemedcom.com
On Twitter @Alz_gal
CHICAGO – A 10-year registry study aims to gather clinical and patient-reported outcomes on 4,000 adult and pediatric patients who undergo fecal microbiota transplant in the United States, officials of the American Gastroenterological Association announced during Digestive Disease Week®.
The AGA Fecal Microbiota Transplantation National Registry will be the first study to assess both short- and long-term patient outcomes associated with fecal microbiota transplant (FMT) in both adults and children, Colleen Kelly, MD, said in an video interview. Most subjects will have received FMT for recurrent or refractory Clostridium difficile infections – the only indication for which Food and Drug Administration currently allows independent clinician action. But the investigational uses of FMT are expanding rapidly, and patients who undergo the procedure during any registered study will be eligible for enrollment, said Dr. Kelly, co-chair of the study’s steering committee.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The study’s primary objectives are short- and long-term safety outcomes, said Dr. Kelly of Brown University, Providence, R.I. While generally considered quite safe, short-term adverse events have been reported with FMT, and some of them have been serious – including one death from aspiration pneumonia in a patient who received donor stool via nasogastric tube (Clin Infect Dis. 2015 Mar;61[1]:136-7). Other adverse events are usually self-limited but can include low-grade fever, abdominal pain, distention, bloating, and diarrhea.
Researchers seek to illuminate many of the unknowns associated with this relatively new procedure. Scientists are only now beginning to unravel the myriad ways the human microbiome promotes both health and disease. Specific alterations, for example, have been associated with obesity and other conditions; there is concern that transplanting a new microbial population could induce a disease phenotype in a recipient who might not have otherwise been at risk.
With the planned cohort size and follow-up period, the study should be able to detect any unanticipated adverse events that occur in more than 1% of the population, Dr. Kelly said. It will include a comparator group of patients with recurrent or refractory C. difficile infection from a large insurance claims database to allow comparison between patients treated with FMT and those treated with antibiotics only.
The registry study also aims to discover which method or methods of transplant material delivery are best, she said. Right now, there are a number of methods (colonoscopy/sigmoidoscopy, enema, upper gastrointestinal endoscopy, nasogastric or nasoduodenal tube, and capsules), and no consensus on which is the best. As indications for FMT expand, there may be no single best method. The approach will probably be matched to the disorder being treated, and the study may help illuminate this as well.
For the first 2 years after a transplant, clinicians will follow patients and enter data into the registry. After that, an electronic patient-reported outcomes system will automatically contact the patient annually for follow-up information by email or text message. When patients enter their data, they can access educational material that will help keep them up-to-date on potential adverse events.
The study will also include a biobank of stool samples obtained during the procedures, hosted by the American Gut Project and the Microbiome Initiative at the University of California, San Diego. This arm of the project will analyze the microbiome of 3,000 stool samples from recipients, both before and after their transplant, as well as the corresponding donors whose material was used in the fecal transplant.
The registry study, a project of the AGA Center for Gut Microbiome Research and Education, is funded by a $3.3 million grant from the National Institute of Allergy and Infectious Diseases. It will be conducted in partnership with the Crohn’s and Colitis Foundation, Infectious Diseases Society, and the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition.
The registry study currently is accepting applications. Physicians who perform FMT for C. difficile infections, and centers that conduct FMT research for other potential indications, can fill out a short survey to indicate their interest.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE) and the Society for Surgery of the Alimentary Tract (SSAT).
This article was updated June 8, 2017.
msullivan@frontlinemedcom.com
On Twitter @Alz_gal
CHICAGO – A 10-year registry study aims to gather clinical and patient-reported outcomes on 4,000 adult and pediatric patients who undergo fecal microbiota transplant in the United States, officials of the American Gastroenterological Association announced during Digestive Disease Week®.
The AGA Fecal Microbiota Transplantation National Registry will be the first study to assess both short- and long-term patient outcomes associated with fecal microbiota transplant (FMT) in both adults and children, Colleen Kelly, MD, said in an video interview. Most subjects will have received FMT for recurrent or refractory Clostridium difficile infections – the only indication for which Food and Drug Administration currently allows independent clinician action. But the investigational uses of FMT are expanding rapidly, and patients who undergo the procedure during any registered study will be eligible for enrollment, said Dr. Kelly, co-chair of the study’s steering committee.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The study’s primary objectives are short- and long-term safety outcomes, said Dr. Kelly of Brown University, Providence, R.I. While generally considered quite safe, short-term adverse events have been reported with FMT, and some of them have been serious – including one death from aspiration pneumonia in a patient who received donor stool via nasogastric tube (Clin Infect Dis. 2015 Mar;61[1]:136-7). Other adverse events are usually self-limited but can include low-grade fever, abdominal pain, distention, bloating, and diarrhea.
Researchers seek to illuminate many of the unknowns associated with this relatively new procedure. Scientists are only now beginning to unravel the myriad ways the human microbiome promotes both health and disease. Specific alterations, for example, have been associated with obesity and other conditions; there is concern that transplanting a new microbial population could induce a disease phenotype in a recipient who might not have otherwise been at risk.
With the planned cohort size and follow-up period, the study should be able to detect any unanticipated adverse events that occur in more than 1% of the population, Dr. Kelly said. It will include a comparator group of patients with recurrent or refractory C. difficile infection from a large insurance claims database to allow comparison between patients treated with FMT and those treated with antibiotics only.
The registry study also aims to discover which method or methods of transplant material delivery are best, she said. Right now, there are a number of methods (colonoscopy/sigmoidoscopy, enema, upper gastrointestinal endoscopy, nasogastric or nasoduodenal tube, and capsules), and no consensus on which is the best. As indications for FMT expand, there may be no single best method. The approach will probably be matched to the disorder being treated, and the study may help illuminate this as well.
For the first 2 years after a transplant, clinicians will follow patients and enter data into the registry. After that, an electronic patient-reported outcomes system will automatically contact the patient annually for follow-up information by email or text message. When patients enter their data, they can access educational material that will help keep them up-to-date on potential adverse events.
The study will also include a biobank of stool samples obtained during the procedures, hosted by the American Gut Project and the Microbiome Initiative at the University of California, San Diego. This arm of the project will analyze the microbiome of 3,000 stool samples from recipients, both before and after their transplant, as well as the corresponding donors whose material was used in the fecal transplant.
The registry study, a project of the AGA Center for Gut Microbiome Research and Education, is funded by a $3.3 million grant from the National Institute of Allergy and Infectious Diseases. It will be conducted in partnership with the Crohn’s and Colitis Foundation, Infectious Diseases Society, and the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition.
The registry study currently is accepting applications. Physicians who perform FMT for C. difficile infections, and centers that conduct FMT research for other potential indications, can fill out a short survey to indicate their interest.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE) and the Society for Surgery of the Alimentary Tract (SSAT).
This article was updated June 8, 2017.
msullivan@frontlinemedcom.com
On Twitter @Alz_gal
AT DDW

Enzyme tablet eases pain of gluten consumption
CHICAGO – Gluten-sensitive patients were able to tolerate small amounts of gluten after consuming an enzyme supplement derived from Aspergillus niger as part of a meal in a randomized, placebo-controlled trial of 18 adults. The data were presented at the annual Digestive Disease Week.
The enzyme, A. niger-derived prolyl endoprotease (AN-PEP), has demonstrated the ability to degrade gluten into nonimmunogenic compounds in vivo in healthy subjects, according to Julia König, PhD, of the School of Medical Sciences, Örebro (Sweden) University and her colleagues. The researchers tested the enzyme at two separate doses in 18 adults with self-reported gluten sensitivity.
The participants’ gastric and duodenal content was sampled several times over 180 minutes and analyzed for gluten epitopes using an enzyme-linked immunosorbent assay test. Participants also completed questionnaires after each day of testing.
After taking the enzyme in conjunction with the gluten, stomach gluten content averaged 31 microg x min/mL in the high-dose and low-dose enzyme patients (P = 0.001 for both doses), compared with 281 microg x min/mL in the placebo patients.
By the time the gluten reached the duodenum, the average levels had dropped to 12 microg x min/mL in the high-dose patients (P = 0.019) and 8 microg x min/mL in the low-dose patients (P = 0.015), compared with an average of 65 microg x min/mL in the placebo patients.
Overall, the enzyme was well tolerated by the patients, the researchers said. However, Dr. König emphasized that the enzyme tablet is meant to help avoid symptoms when gluten-sensitive patients encounter small amounts of gluten, and these patients should still follow a gluten-free diet.
AN-PEP is available in the United States in supplement form under several names and is manufactured by the Dutch company DSM. The AN-PEP enzyme used in the study was provided by DSM, but the company provided no other support. Dr. König had no relevant financial conflicts to disclose.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
CHICAGO – Gluten-sensitive patients were able to tolerate small amounts of gluten after consuming an enzyme supplement derived from Aspergillus niger as part of a meal in a randomized, placebo-controlled trial of 18 adults. The data were presented at the annual Digestive Disease Week.
The enzyme, A. niger-derived prolyl endoprotease (AN-PEP), has demonstrated the ability to degrade gluten into nonimmunogenic compounds in vivo in healthy subjects, according to Julia König, PhD, of the School of Medical Sciences, Örebro (Sweden) University and her colleagues. The researchers tested the enzyme at two separate doses in 18 adults with self-reported gluten sensitivity.
The participants’ gastric and duodenal content was sampled several times over 180 minutes and analyzed for gluten epitopes using an enzyme-linked immunosorbent assay test. Participants also completed questionnaires after each day of testing.
After taking the enzyme in conjunction with the gluten, stomach gluten content averaged 31 microg x min/mL in the high-dose and low-dose enzyme patients (P = 0.001 for both doses), compared with 281 microg x min/mL in the placebo patients.
By the time the gluten reached the duodenum, the average levels had dropped to 12 microg x min/mL in the high-dose patients (P = 0.019) and 8 microg x min/mL in the low-dose patients (P = 0.015), compared with an average of 65 microg x min/mL in the placebo patients.
Overall, the enzyme was well tolerated by the patients, the researchers said. However, Dr. König emphasized that the enzyme tablet is meant to help avoid symptoms when gluten-sensitive patients encounter small amounts of gluten, and these patients should still follow a gluten-free diet.
AN-PEP is available in the United States in supplement form under several names and is manufactured by the Dutch company DSM. The AN-PEP enzyme used in the study was provided by DSM, but the company provided no other support. Dr. König had no relevant financial conflicts to disclose.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
CHICAGO – Gluten-sensitive patients were able to tolerate small amounts of gluten after consuming an enzyme supplement derived from Aspergillus niger as part of a meal in a randomized, placebo-controlled trial of 18 adults. The data were presented at the annual Digestive Disease Week.
The enzyme, A. niger-derived prolyl endoprotease (AN-PEP), has demonstrated the ability to degrade gluten into nonimmunogenic compounds in vivo in healthy subjects, according to Julia König, PhD, of the School of Medical Sciences, Örebro (Sweden) University and her colleagues. The researchers tested the enzyme at two separate doses in 18 adults with self-reported gluten sensitivity.
The participants’ gastric and duodenal content was sampled several times over 180 minutes and analyzed for gluten epitopes using an enzyme-linked immunosorbent assay test. Participants also completed questionnaires after each day of testing.
After taking the enzyme in conjunction with the gluten, stomach gluten content averaged 31 microg x min/mL in the high-dose and low-dose enzyme patients (P = 0.001 for both doses), compared with 281 microg x min/mL in the placebo patients.
By the time the gluten reached the duodenum, the average levels had dropped to 12 microg x min/mL in the high-dose patients (P = 0.019) and 8 microg x min/mL in the low-dose patients (P = 0.015), compared with an average of 65 microg x min/mL in the placebo patients.
Overall, the enzyme was well tolerated by the patients, the researchers said. However, Dr. König emphasized that the enzyme tablet is meant to help avoid symptoms when gluten-sensitive patients encounter small amounts of gluten, and these patients should still follow a gluten-free diet.
AN-PEP is available in the United States in supplement form under several names and is manufactured by the Dutch company DSM. The AN-PEP enzyme used in the study was provided by DSM, but the company provided no other support. Dr. König had no relevant financial conflicts to disclose.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
FROM DDW
Key clinical point: Consuming an enzyme tablet simultaneously with small amounts of gluten may reduce discomfort in gluten-sensitive individuals.
Major finding: On average, gluten levels in the stomach after enzyme consumption were 31 microg x min/mL in both high- and low-dose groups, vs. 281 microg x min/mL in a placebo group.
Data source: A randomized, placebo-controlled trial of 18 gluten-sensitive adults.
Disclosures: The enzyme used in the study, AN-PEP, was provided by the Dutch company DSM, but the company provided no other support. Dr. König had no relevant financial conflicts to disclose.
VIDEO: Bile acid malabsorption as a cause of chronic diarrhea
CHICAGO – Bile acid malabsorption increasingly is recognized as a cause of persistent, chronic diarrhea, but patients often receive suboptimal treatment because medical and public awareness is low, Julian Walters, MD, of Imperial College London, said at Digestive Disease Week®.
Members of two patient support groups in the United Kingdom were invited to complete an online survey to provide information on how this condition affects them. The first 100 responses were analyzed. The majority of respondents were female (91). More than 35 respondents were diagnosed after the age of 50 years, and 35 felt their condition had not been taken seriously by multiple practitioners prior to their eventual diagnosis, Dr. Walters reported.
Two-thirds of respondents had been diagnosed with irritable bowel syndrome; the majority (68) of these had more than 10 interactions with medical professionals before being diagnosed with bile acid malabsorption.
Once appropriately diagnosed, most respondents reported doing very well on drugs such as cholestyramine and colesevelam, Dr. Walters said. He stressed that mental health issues are an important part of this condition because of its pervasive effects on daily life.
He discusses the survey and bile acid malabsorption in this video interview.
Dr. Walters disclosed that he has been a consultant to or has received research funds from GE Healthcare, Intercept, Albireo, and Novartis.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
CHICAGO – Bile acid malabsorption increasingly is recognized as a cause of persistent, chronic diarrhea, but patients often receive suboptimal treatment because medical and public awareness is low, Julian Walters, MD, of Imperial College London, said at Digestive Disease Week®.
Members of two patient support groups in the United Kingdom were invited to complete an online survey to provide information on how this condition affects them. The first 100 responses were analyzed. The majority of respondents were female (91). More than 35 respondents were diagnosed after the age of 50 years, and 35 felt their condition had not been taken seriously by multiple practitioners prior to their eventual diagnosis, Dr. Walters reported.
Two-thirds of respondents had been diagnosed with irritable bowel syndrome; the majority (68) of these had more than 10 interactions with medical professionals before being diagnosed with bile acid malabsorption.
Once appropriately diagnosed, most respondents reported doing very well on drugs such as cholestyramine and colesevelam, Dr. Walters said. He stressed that mental health issues are an important part of this condition because of its pervasive effects on daily life.
He discusses the survey and bile acid malabsorption in this video interview.
Dr. Walters disclosed that he has been a consultant to or has received research funds from GE Healthcare, Intercept, Albireo, and Novartis.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
CHICAGO – Bile acid malabsorption increasingly is recognized as a cause of persistent, chronic diarrhea, but patients often receive suboptimal treatment because medical and public awareness is low, Julian Walters, MD, of Imperial College London, said at Digestive Disease Week®.
Members of two patient support groups in the United Kingdom were invited to complete an online survey to provide information on how this condition affects them. The first 100 responses were analyzed. The majority of respondents were female (91). More than 35 respondents were diagnosed after the age of 50 years, and 35 felt their condition had not been taken seriously by multiple practitioners prior to their eventual diagnosis, Dr. Walters reported.
Two-thirds of respondents had been diagnosed with irritable bowel syndrome; the majority (68) of these had more than 10 interactions with medical professionals before being diagnosed with bile acid malabsorption.
Once appropriately diagnosed, most respondents reported doing very well on drugs such as cholestyramine and colesevelam, Dr. Walters said. He stressed that mental health issues are an important part of this condition because of its pervasive effects on daily life.
He discusses the survey and bile acid malabsorption in this video interview.
Dr. Walters disclosed that he has been a consultant to or has received research funds from GE Healthcare, Intercept, Albireo, and Novartis.
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
AT DDW

VIDEO: Rifamycin matches ciprofloxacin’s efficacy in travelers’ diarrhea with less antibiotic resistance
CHICAGO – An investigational antibiotic was just as effective as ciprofloxacin at curing travelers’ diarrhea but was associated with a significantly lower rate of colonization with extended spectrum beta-lactam–resistant Escherichia coli, a phase III trial has determined.
“Rifamycin was noninferior to ciprofloxacin on every endpoint in this trial,” Robert Steffen, MD, said at the annual Digestive Disease Week. “However, there was no increase in extended spectrum beta-lactamase–producing Enterobacteriaceae (ESBL-E) associated with rifamycin, and significantly less new acquisition of these pathogens than in the ciprofloxacin group.”
Rifamycin is a poorly absorbed, broad-spectrum antibiotic in the same chemical family as rifaximin. It’s designed, both molecularly and in packaging, to become active only in the lower ileum and colon with limited systemic absorption. The drug is approved in Europe for infectious colitis, Clostridium difficile, diverticulitis, and also as supportive treatment of inflammatory bowel diseases and hepatic encephalopathy.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Subjects were randomized to 3 days of rifamycin 800 mg, or ciprofloxacin 1,000 mg. Follow-up visits occurred on days 2, 5, and 6, with a final follow-up by mail 4 weeks later. The primary endpoint was time to last unformed stool from the first dose of study medication. Secondary endpoints were clinical cure (24 hours with no clinical symptoms, fever, or watery stools, or 48 hours with no fever; and either no stools or only formed stools), need for rescue therapy, treatment failure, pathogen eradication in posttreatment stool, and the rate of ESBL-E colonization.
The time to last unformed stool was 43 hours in the rifamycin group and 37 hours in the ciprofloxacin group, which were not significantly different. The results were similar when broken down by infective organism, by gender, and by study location.
Rifamycin was also noninferior to ciprofloxacin in several secondary endpoints, including clinical cure (85% each), treatment failure (15% each), and need for rescue therapy (1% vs. 2.6%). The drugs were also virtually identical in the number of unformed stools per 24-hour interval, which fell precipitously from 5.5 on day 1, to 1 by day 5, and in complete resolution of gastrointestinal symptoms, which were about 75% resolved in each group by day 5.
Rifamycin was equally effective in eradicating all of the pathogens identified in the cohort. This included all pathogens in the E. coli group, all in the potentially invasive group (Shigella, Campylobacter, Salmonella, and Aeromonas), norovirus, giardia, and Cryptosporidium.
Treatment-emergent adverse events occurred in 12% of each group; none were serious. About 8% of each group experienced an adverse drug reaction.
Where the drugs did differ, and sharply so, was in antibiotic resistance, said Dr. Steffen, of the University of Zürich and the University of Texas School of Public Health, Houston. At baseline, about 16% of the group was infected with ESBL–E coli. At last follow-up, those species were present in 16% of the rifamycin group, but in 21% of the ciprofloxacin group. Similarly, there was less new ESBL–E. coli colonization in patients who had been negative at baseline (10% vs. 17%).
The findings are particularly important in light of the increasing worldwide emergence of antibiotic-resistant bacteria, Dr. Steffen said. In fact, new guidelines released April 29 by the International Society of Travel Medicine recommend that antibiotics be reserved for moderate to severe cases of traveler’s diarrhea and not be used at all in milder cases (J Travel Med. 2017 Apr 29;24[suppl. 1]:S57-S74).
“The widespread use of ciprofloxacin and other antibiotics for travelers’ diarrhea has contributed to the rise of these resistant bacteria,” Dr. Steffen said in an interview. “We need to rethink the way we use these drugs and to focus instead on drugs that are not systemically absorbed. If rifamycin is eventually approved for this indication, it would be a good alternative to systemic antibiotics, curing the acute illness, and not contributing as much to the emergence of these worrisome pathogens.”
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
In a video interview at the meeting, Dr. Steffen spoke about the trial and concerns about antibiotic resistance that are addressed in the new guidelines and by this new study.
Dr. Falk Pharma GmbH of Freiburg, Germany, is developing rifamycin and conducted the study. Dr. Steffen has received consulting and travel fees from the company.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – An investigational antibiotic was just as effective as ciprofloxacin at curing travelers’ diarrhea but was associated with a significantly lower rate of colonization with extended spectrum beta-lactam–resistant Escherichia coli, a phase III trial has determined.
“Rifamycin was noninferior to ciprofloxacin on every endpoint in this trial,” Robert Steffen, MD, said at the annual Digestive Disease Week. “However, there was no increase in extended spectrum beta-lactamase–producing Enterobacteriaceae (ESBL-E) associated with rifamycin, and significantly less new acquisition of these pathogens than in the ciprofloxacin group.”
Rifamycin is a poorly absorbed, broad-spectrum antibiotic in the same chemical family as rifaximin. It’s designed, both molecularly and in packaging, to become active only in the lower ileum and colon with limited systemic absorption. The drug is approved in Europe for infectious colitis, Clostridium difficile, diverticulitis, and also as supportive treatment of inflammatory bowel diseases and hepatic encephalopathy.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Subjects were randomized to 3 days of rifamycin 800 mg, or ciprofloxacin 1,000 mg. Follow-up visits occurred on days 2, 5, and 6, with a final follow-up by mail 4 weeks later. The primary endpoint was time to last unformed stool from the first dose of study medication. Secondary endpoints were clinical cure (24 hours with no clinical symptoms, fever, or watery stools, or 48 hours with no fever; and either no stools or only formed stools), need for rescue therapy, treatment failure, pathogen eradication in posttreatment stool, and the rate of ESBL-E colonization.
The time to last unformed stool was 43 hours in the rifamycin group and 37 hours in the ciprofloxacin group, which were not significantly different. The results were similar when broken down by infective organism, by gender, and by study location.
Rifamycin was also noninferior to ciprofloxacin in several secondary endpoints, including clinical cure (85% each), treatment failure (15% each), and need for rescue therapy (1% vs. 2.6%). The drugs were also virtually identical in the number of unformed stools per 24-hour interval, which fell precipitously from 5.5 on day 1, to 1 by day 5, and in complete resolution of gastrointestinal symptoms, which were about 75% resolved in each group by day 5.
Rifamycin was equally effective in eradicating all of the pathogens identified in the cohort. This included all pathogens in the E. coli group, all in the potentially invasive group (Shigella, Campylobacter, Salmonella, and Aeromonas), norovirus, giardia, and Cryptosporidium.
Treatment-emergent adverse events occurred in 12% of each group; none were serious. About 8% of each group experienced an adverse drug reaction.
Where the drugs did differ, and sharply so, was in antibiotic resistance, said Dr. Steffen, of the University of Zürich and the University of Texas School of Public Health, Houston. At baseline, about 16% of the group was infected with ESBL–E coli. At last follow-up, those species were present in 16% of the rifamycin group, but in 21% of the ciprofloxacin group. Similarly, there was less new ESBL–E. coli colonization in patients who had been negative at baseline (10% vs. 17%).
The findings are particularly important in light of the increasing worldwide emergence of antibiotic-resistant bacteria, Dr. Steffen said. In fact, new guidelines released April 29 by the International Society of Travel Medicine recommend that antibiotics be reserved for moderate to severe cases of traveler’s diarrhea and not be used at all in milder cases (J Travel Med. 2017 Apr 29;24[suppl. 1]:S57-S74).
“The widespread use of ciprofloxacin and other antibiotics for travelers’ diarrhea has contributed to the rise of these resistant bacteria,” Dr. Steffen said in an interview. “We need to rethink the way we use these drugs and to focus instead on drugs that are not systemically absorbed. If rifamycin is eventually approved for this indication, it would be a good alternative to systemic antibiotics, curing the acute illness, and not contributing as much to the emergence of these worrisome pathogens.”
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
In a video interview at the meeting, Dr. Steffen spoke about the trial and concerns about antibiotic resistance that are addressed in the new guidelines and by this new study.
Dr. Falk Pharma GmbH of Freiburg, Germany, is developing rifamycin and conducted the study. Dr. Steffen has received consulting and travel fees from the company.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – An investigational antibiotic was just as effective as ciprofloxacin at curing travelers’ diarrhea but was associated with a significantly lower rate of colonization with extended spectrum beta-lactam–resistant Escherichia coli, a phase III trial has determined.
“Rifamycin was noninferior to ciprofloxacin on every endpoint in this trial,” Robert Steffen, MD, said at the annual Digestive Disease Week. “However, there was no increase in extended spectrum beta-lactamase–producing Enterobacteriaceae (ESBL-E) associated with rifamycin, and significantly less new acquisition of these pathogens than in the ciprofloxacin group.”
Rifamycin is a poorly absorbed, broad-spectrum antibiotic in the same chemical family as rifaximin. It’s designed, both molecularly and in packaging, to become active only in the lower ileum and colon with limited systemic absorption. The drug is approved in Europe for infectious colitis, Clostridium difficile, diverticulitis, and also as supportive treatment of inflammatory bowel diseases and hepatic encephalopathy.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Subjects were randomized to 3 days of rifamycin 800 mg, or ciprofloxacin 1,000 mg. Follow-up visits occurred on days 2, 5, and 6, with a final follow-up by mail 4 weeks later. The primary endpoint was time to last unformed stool from the first dose of study medication. Secondary endpoints were clinical cure (24 hours with no clinical symptoms, fever, or watery stools, or 48 hours with no fever; and either no stools or only formed stools), need for rescue therapy, treatment failure, pathogen eradication in posttreatment stool, and the rate of ESBL-E colonization.
The time to last unformed stool was 43 hours in the rifamycin group and 37 hours in the ciprofloxacin group, which were not significantly different. The results were similar when broken down by infective organism, by gender, and by study location.
Rifamycin was also noninferior to ciprofloxacin in several secondary endpoints, including clinical cure (85% each), treatment failure (15% each), and need for rescue therapy (1% vs. 2.6%). The drugs were also virtually identical in the number of unformed stools per 24-hour interval, which fell precipitously from 5.5 on day 1, to 1 by day 5, and in complete resolution of gastrointestinal symptoms, which were about 75% resolved in each group by day 5.
Rifamycin was equally effective in eradicating all of the pathogens identified in the cohort. This included all pathogens in the E. coli group, all in the potentially invasive group (Shigella, Campylobacter, Salmonella, and Aeromonas), norovirus, giardia, and Cryptosporidium.
Treatment-emergent adverse events occurred in 12% of each group; none were serious. About 8% of each group experienced an adverse drug reaction.
Where the drugs did differ, and sharply so, was in antibiotic resistance, said Dr. Steffen, of the University of Zürich and the University of Texas School of Public Health, Houston. At baseline, about 16% of the group was infected with ESBL–E coli. At last follow-up, those species were present in 16% of the rifamycin group, but in 21% of the ciprofloxacin group. Similarly, there was less new ESBL–E. coli colonization in patients who had been negative at baseline (10% vs. 17%).
The findings are particularly important in light of the increasing worldwide emergence of antibiotic-resistant bacteria, Dr. Steffen said. In fact, new guidelines released April 29 by the International Society of Travel Medicine recommend that antibiotics be reserved for moderate to severe cases of traveler’s diarrhea and not be used at all in milder cases (J Travel Med. 2017 Apr 29;24[suppl. 1]:S57-S74).
“The widespread use of ciprofloxacin and other antibiotics for travelers’ diarrhea has contributed to the rise of these resistant bacteria,” Dr. Steffen said in an interview. “We need to rethink the way we use these drugs and to focus instead on drugs that are not systemically absorbed. If rifamycin is eventually approved for this indication, it would be a good alternative to systemic antibiotics, curing the acute illness, and not contributing as much to the emergence of these worrisome pathogens.”
Digestive Disease Week® is jointly sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA) Institute, the American Society for Gastrointestinal Endoscopy (ASGE), and the Society for Surgery of the Alimentary Tract (SSAT).
In a video interview at the meeting, Dr. Steffen spoke about the trial and concerns about antibiotic resistance that are addressed in the new guidelines and by this new study.
Dr. Falk Pharma GmbH of Freiburg, Germany, is developing rifamycin and conducted the study. Dr. Steffen has received consulting and travel fees from the company.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT DDW
Key clinical point:
Major finding: Clinical cure occurred in 85% of each group, but new beta-lactam–resistant E. coli colonization occurred in 16% of the rifamycin group and 21% of the ciprofloxacin group.
Data source: The randomized study comprised 835 subjects.
Disclosures: Dr. Falk Pharma GmbH of Freiburg, Germany, is developing the drug and sponsored the study. Dr. Steffen has received consulting and travel fees from the company.

Twice-daily tofacitinib induces ulcerative colitis remission
A 10-mg dose of tofacitinib twice daily was significantly more effective than placebo for inducing remission in ulcerative colitis patients, based on data from a group of three randomized trials totaling approximately 1,500 adults. The findings were published online May 3 in the New England Journal of Medicine (2017;376:1723-36).
The series of OCTAVE trials (Oral Clinical Trials for Tofacitinib in Ulcerative Colitis) included adults with moderately to severely active ulcerative colitis (UC). Patients were randomized to 10 mg of tofacitinib, 5 mg tofacitinib, or placebo. The studies were conducted over a 4-year period, at 144 sites for OCTAVE 1, 169 sites for OCTAVE 2, and 297 sites for OCTAVE Sustain.
In both OCTAVE 1 and OCTAVE 2, the remission rates at 8 weeks were significantly higher in the 10-mg tofacitinib groups, compared with the placebo groups (18.5% vs. 8.2%, respectively; 16.6% vs. 3.6%, respectively). The rate of remission at 52 weeks was significantly higher in the 5-mg and 10-mg tofacitinib groups (34.3% and 40.6%, respectively) than in the placebo group (11.1%) in the OCTAVE Sustain trial.
“Pharmacokinetic results in the OCTAVE trials did not indicate a decrease in plasma tofacitinib concentrations during the course of treatment at any given dose in individual patients. These results are consistent with the previously established physicochemical characteristics and clearance mechanisms of tofacitinib,” noted William J. Sandborn, MD, of the University of California, San Diego, and his colleagues.
In the OCTAVE 1 trial, serious adverse events occurred in 4.2% and 8.0% of patients in the 10-mg and placebo groups, respectively. In the OCTAVE 2 trial, they occured in 3.4% and 4.1% of the 10-mg and placebo groups, respectively. The rate of serious adverse events in the OCTAVE Sustain trial was 5.1%, 5.6%, and 6.6% in the 10-mg, 5-mg, and placebo groups, respectively. Tofacitinib was associated with increased lipid levels, as well as higher rates of overall infection and herpes zoster infection, compared with placebo.
The study was supported by Pfizer. Lead author Dr. Sandborn and several coauthors disclosed financial relationships with multiple companies including Pfizer.
AGA Resource
AGA offers materials to help you manage patients with Crohn’s disease and ulcerative colitis, and integrate patient education into your practice, including multimedia publications that are accessible and informative. Learn more at http://www.gastro.org/ibd
“This report is the culmination of an international effort,” over a 4-year period, wrote Sonia Friedman, MD, of Harvard University in an accompanying editorial.
“This study has all the elements of a high-quality trial: a large, international cohort of patients and investigators; reasonable enrollment criteria that would apply to many of my own patients with moderate-to-severe ulcerative colitis; rigorous and unbiased scoring for response, remission, and mucosal healing; fair adjudication of adverse events; and meticulous reporting of all meaningful outcomes and laboratory test results,” she said. Tofacitinib has proven its efficacy – its exact role will be determined by additional research, she noted.
“Only a continued combination of human ingenuity, worldwide cooperation, and enthusiastic funding will allow investigators to further explore the mechanisms by which JAK inhibition ameliorates inflammation in patients with ulcerative colitis and ... identify the specific subsets of patients who will most likely benefit from this new therapy,” Dr. Friedman emphasized (N. Engl. J. Med. 2017;376:1792-3).
Dr. Friedman is affiliated with Harvard University, Boston, Mass., and Brigham and Women’s Hospital Center for Crohn’s and Colitis, Chestnut Hill, Mass. She disclosed receiving personal fees from Boston University.
“This report is the culmination of an international effort,” over a 4-year period, wrote Sonia Friedman, MD, of Harvard University in an accompanying editorial.
“This study has all the elements of a high-quality trial: a large, international cohort of patients and investigators; reasonable enrollment criteria that would apply to many of my own patients with moderate-to-severe ulcerative colitis; rigorous and unbiased scoring for response, remission, and mucosal healing; fair adjudication of adverse events; and meticulous reporting of all meaningful outcomes and laboratory test results,” she said. Tofacitinib has proven its efficacy – its exact role will be determined by additional research, she noted.
“Only a continued combination of human ingenuity, worldwide cooperation, and enthusiastic funding will allow investigators to further explore the mechanisms by which JAK inhibition ameliorates inflammation in patients with ulcerative colitis and ... identify the specific subsets of patients who will most likely benefit from this new therapy,” Dr. Friedman emphasized (N. Engl. J. Med. 2017;376:1792-3).
Dr. Friedman is affiliated with Harvard University, Boston, Mass., and Brigham and Women’s Hospital Center for Crohn’s and Colitis, Chestnut Hill, Mass. She disclosed receiving personal fees from Boston University.
“This report is the culmination of an international effort,” over a 4-year period, wrote Sonia Friedman, MD, of Harvard University in an accompanying editorial.
“This study has all the elements of a high-quality trial: a large, international cohort of patients and investigators; reasonable enrollment criteria that would apply to many of my own patients with moderate-to-severe ulcerative colitis; rigorous and unbiased scoring for response, remission, and mucosal healing; fair adjudication of adverse events; and meticulous reporting of all meaningful outcomes and laboratory test results,” she said. Tofacitinib has proven its efficacy – its exact role will be determined by additional research, she noted.
“Only a continued combination of human ingenuity, worldwide cooperation, and enthusiastic funding will allow investigators to further explore the mechanisms by which JAK inhibition ameliorates inflammation in patients with ulcerative colitis and ... identify the specific subsets of patients who will most likely benefit from this new therapy,” Dr. Friedman emphasized (N. Engl. J. Med. 2017;376:1792-3).
Dr. Friedman is affiliated with Harvard University, Boston, Mass., and Brigham and Women’s Hospital Center for Crohn’s and Colitis, Chestnut Hill, Mass. She disclosed receiving personal fees from Boston University.
A 10-mg dose of tofacitinib twice daily was significantly more effective than placebo for inducing remission in ulcerative colitis patients, based on data from a group of three randomized trials totaling approximately 1,500 adults. The findings were published online May 3 in the New England Journal of Medicine (2017;376:1723-36).
The series of OCTAVE trials (Oral Clinical Trials for Tofacitinib in Ulcerative Colitis) included adults with moderately to severely active ulcerative colitis (UC). Patients were randomized to 10 mg of tofacitinib, 5 mg tofacitinib, or placebo. The studies were conducted over a 4-year period, at 144 sites for OCTAVE 1, 169 sites for OCTAVE 2, and 297 sites for OCTAVE Sustain.
In both OCTAVE 1 and OCTAVE 2, the remission rates at 8 weeks were significantly higher in the 10-mg tofacitinib groups, compared with the placebo groups (18.5% vs. 8.2%, respectively; 16.6% vs. 3.6%, respectively). The rate of remission at 52 weeks was significantly higher in the 5-mg and 10-mg tofacitinib groups (34.3% and 40.6%, respectively) than in the placebo group (11.1%) in the OCTAVE Sustain trial.
“Pharmacokinetic results in the OCTAVE trials did not indicate a decrease in plasma tofacitinib concentrations during the course of treatment at any given dose in individual patients. These results are consistent with the previously established physicochemical characteristics and clearance mechanisms of tofacitinib,” noted William J. Sandborn, MD, of the University of California, San Diego, and his colleagues.
In the OCTAVE 1 trial, serious adverse events occurred in 4.2% and 8.0% of patients in the 10-mg and placebo groups, respectively. In the OCTAVE 2 trial, they occured in 3.4% and 4.1% of the 10-mg and placebo groups, respectively. The rate of serious adverse events in the OCTAVE Sustain trial was 5.1%, 5.6%, and 6.6% in the 10-mg, 5-mg, and placebo groups, respectively. Tofacitinib was associated with increased lipid levels, as well as higher rates of overall infection and herpes zoster infection, compared with placebo.
The study was supported by Pfizer. Lead author Dr. Sandborn and several coauthors disclosed financial relationships with multiple companies including Pfizer.
AGA Resource
AGA offers materials to help you manage patients with Crohn’s disease and ulcerative colitis, and integrate patient education into your practice, including multimedia publications that are accessible and informative. Learn more at http://www.gastro.org/ibd
A 10-mg dose of tofacitinib twice daily was significantly more effective than placebo for inducing remission in ulcerative colitis patients, based on data from a group of three randomized trials totaling approximately 1,500 adults. The findings were published online May 3 in the New England Journal of Medicine (2017;376:1723-36).
The series of OCTAVE trials (Oral Clinical Trials for Tofacitinib in Ulcerative Colitis) included adults with moderately to severely active ulcerative colitis (UC). Patients were randomized to 10 mg of tofacitinib, 5 mg tofacitinib, or placebo. The studies were conducted over a 4-year period, at 144 sites for OCTAVE 1, 169 sites for OCTAVE 2, and 297 sites for OCTAVE Sustain.
In both OCTAVE 1 and OCTAVE 2, the remission rates at 8 weeks were significantly higher in the 10-mg tofacitinib groups, compared with the placebo groups (18.5% vs. 8.2%, respectively; 16.6% vs. 3.6%, respectively). The rate of remission at 52 weeks was significantly higher in the 5-mg and 10-mg tofacitinib groups (34.3% and 40.6%, respectively) than in the placebo group (11.1%) in the OCTAVE Sustain trial.
“Pharmacokinetic results in the OCTAVE trials did not indicate a decrease in plasma tofacitinib concentrations during the course of treatment at any given dose in individual patients. These results are consistent with the previously established physicochemical characteristics and clearance mechanisms of tofacitinib,” noted William J. Sandborn, MD, of the University of California, San Diego, and his colleagues.
In the OCTAVE 1 trial, serious adverse events occurred in 4.2% and 8.0% of patients in the 10-mg and placebo groups, respectively. In the OCTAVE 2 trial, they occured in 3.4% and 4.1% of the 10-mg and placebo groups, respectively. The rate of serious adverse events in the OCTAVE Sustain trial was 5.1%, 5.6%, and 6.6% in the 10-mg, 5-mg, and placebo groups, respectively. Tofacitinib was associated with increased lipid levels, as well as higher rates of overall infection and herpes zoster infection, compared with placebo.
The study was supported by Pfizer. Lead author Dr. Sandborn and several coauthors disclosed financial relationships with multiple companies including Pfizer.
AGA Resource
AGA offers materials to help you manage patients with Crohn’s disease and ulcerative colitis, and integrate patient education into your practice, including multimedia publications that are accessible and informative. Learn more at http://www.gastro.org/ibd
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Tofacitinib, a JAK inhibitor, was a significantly more effective induction and maintenance therapy for patients with moderate to severe ulcerative colitis compared with placebo.
Major finding: Tofacitinib dosed at 10 mg twice daily yielded a remission rate of 41% at 52 weeks, compared with 11% in a placebo group.
Data source: The OCTAVE series of three randomized trials totaled approximately 1,500 adults with moderate to severely active ulcerative colitis.
Disclosures: The study was supported by Pfizer. Lead author Dr. Sandborn and several coauthors disclosed financial relationships with multiple companies, including Pfizer.