User login
U.S. C. difficile mortality jumped sevenfold 1999-2008
VIENNA – U.S. deaths from Clostridium difficile infection jumped roughly sevenfold from 1999 to 2008, but in the ensuing years (2009-2012), remained stable at about the level first reached in 2008, according to analysis of cause-of-death data from the National Center for Health Statistics.
The increase occurred uniformly across all age groups older than 40 years; among patients 40 years or younger mortality from C. difficile infection remained low, at a rate at or below one death per one million people, Andrew Noymer, Ph.D., reported in a poster at the International Meeting on Emerging Diseases and Surveillance.
Among those patients aged 80 years or older, the mortality rate in recent years with C. difficile infection listed as the primary cause exceeded one death per 5,000 people, reported Dr. Noymer, a researcher in population health and disease prevention at the University of California, Irvine.
The mortality data provided no direct insight into the factors contributing to the rise in C. difficile deaths during 1999-2008, but previous reports documented an increased prevalence starting in 2000 of a “hypervirulent” strain of C. difficile circulating in North America and elsewhere (Critical Care 2008;12:203-10).
C. difficile infection as the underlying cause of death rose from less than one age-adjusted occurrence for every 200,000 people in 1999 to about 2.4 age-adjusted deaths per 100,000 in 2008, and then remained at about that level through 2012. Rates were similar in women and men.
When analyzed as any mention of C. difficile infection with another factor cited as the primary cause of death occurrences also rose roughly sevenfold from 1999 to 2008, from about one death for every 200,000 people to about 3.5 deaths per 100,000.

When C. difficile has been a contributing factor, the wide spectrum of causes of death that it can accompany “reads like a who’s who of conditions requiring inpatient clinical care,” Dr. Noymer said. “Given the nosocomial nature of C. difficile this is not surprising.”
The top three lethal conditions with C. difficile complication were atherosclerotic heart disease, chronic obstructive pulmonary disease, and septicemia.
Dr. Noymer had no disclosures.
VIENNA – U.S. deaths from Clostridium difficile infection jumped roughly sevenfold from 1999 to 2008, but in the ensuing years (2009-2012), remained stable at about the level first reached in 2008, according to analysis of cause-of-death data from the National Center for Health Statistics.
The increase occurred uniformly across all age groups older than 40 years; among patients 40 years or younger mortality from C. difficile infection remained low, at a rate at or below one death per one million people, Andrew Noymer, Ph.D., reported in a poster at the International Meeting on Emerging Diseases and Surveillance.
Among those patients aged 80 years or older, the mortality rate in recent years with C. difficile infection listed as the primary cause exceeded one death per 5,000 people, reported Dr. Noymer, a researcher in population health and disease prevention at the University of California, Irvine.
The mortality data provided no direct insight into the factors contributing to the rise in C. difficile deaths during 1999-2008, but previous reports documented an increased prevalence starting in 2000 of a “hypervirulent” strain of C. difficile circulating in North America and elsewhere (Critical Care 2008;12:203-10).
C. difficile infection as the underlying cause of death rose from less than one age-adjusted occurrence for every 200,000 people in 1999 to about 2.4 age-adjusted deaths per 100,000 in 2008, and then remained at about that level through 2012. Rates were similar in women and men.
When analyzed as any mention of C. difficile infection with another factor cited as the primary cause of death occurrences also rose roughly sevenfold from 1999 to 2008, from about one death for every 200,000 people to about 3.5 deaths per 100,000.
When C. difficile has been a contributing factor, the wide spectrum of causes of death that it can accompany “reads like a who’s who of conditions requiring inpatient clinical care,” Dr. Noymer said. “Given the nosocomial nature of C. difficile this is not surprising.”
The top three lethal conditions with C. difficile complication were atherosclerotic heart disease, chronic obstructive pulmonary disease, and septicemia.
Dr. Noymer had no disclosures.
VIENNA – U.S. deaths from Clostridium difficile infection jumped roughly sevenfold from 1999 to 2008, but in the ensuing years (2009-2012), remained stable at about the level first reached in 2008, according to analysis of cause-of-death data from the National Center for Health Statistics.
The increase occurred uniformly across all age groups older than 40 years; among patients 40 years or younger mortality from C. difficile infection remained low, at a rate at or below one death per one million people, Andrew Noymer, Ph.D., reported in a poster at the International Meeting on Emerging Diseases and Surveillance.
Among those patients aged 80 years or older, the mortality rate in recent years with C. difficile infection listed as the primary cause exceeded one death per 5,000 people, reported Dr. Noymer, a researcher in population health and disease prevention at the University of California, Irvine.
The mortality data provided no direct insight into the factors contributing to the rise in C. difficile deaths during 1999-2008, but previous reports documented an increased prevalence starting in 2000 of a “hypervirulent” strain of C. difficile circulating in North America and elsewhere (Critical Care 2008;12:203-10).
C. difficile infection as the underlying cause of death rose from less than one age-adjusted occurrence for every 200,000 people in 1999 to about 2.4 age-adjusted deaths per 100,000 in 2008, and then remained at about that level through 2012. Rates were similar in women and men.
When analyzed as any mention of C. difficile infection with another factor cited as the primary cause of death occurrences also rose roughly sevenfold from 1999 to 2008, from about one death for every 200,000 people to about 3.5 deaths per 100,000.
When C. difficile has been a contributing factor, the wide spectrum of causes of death that it can accompany “reads like a who’s who of conditions requiring inpatient clinical care,” Dr. Noymer said. “Given the nosocomial nature of C. difficile this is not surprising.”
The top three lethal conditions with C. difficile complication were atherosclerotic heart disease, chronic obstructive pulmonary disease, and septicemia.
Dr. Noymer had no disclosures.
AT IMED 2014
Key clinical point: During 1999-2008, U.S. deaths attributable to Clostridium difficile infection rose about sevenfold, and then plateaued at that high level through 2012.
Major finding: Age-adjusted C. difficile mortality was less than 1/200,000 in 1999 and about 2.4/100,000 in 2008.
Data source: Review of national mortality data collected by the U.S. National Center for Health Statistics.
Disclosures: Dr. Noymer had no disclosures.
Dabigatran raises major bleeding risk
Dabigatran significantly raises the risk of major bleeding and gastrointestinal bleeding across all subgroups of patients with atrial fibrillation, and particularly in African Americans and patients with chronic kidney disease, according to a report published online Nov. 3 in JAMA Internal Medicine.
Physicians should only prescribe dabigatran with caution, and should fully explain to patients who do take the drug how to identify abnormal bleeding so that it can be detected and controlled as early as possible, said Inmaculada Hernandez, Pharm.D., of the department of health policy and management, University of Pittsburgh, and her associates.
The FDA approved dabigatran in 2010 via an accelerated pathway after only 6 months of review, based largely on findings from a single clinical study, the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial, which did not adjust for patient characteristics (N. Engl. J. Med. 2009;361:1139-51). That study reported lower bleeding risks with dabigatran than with warfarin. Several months later, the agency’s Adverse Event Reporting System received “a large number” of reports of severe bleeding associated with dabigatran; and the relative bleeding risk associated with the two drugs is still unclear.
Dr. Hernandez and her colleagues examined the issue using data from a nationally representative random sample of 9,404 Medicare beneficiaries newly diagnosed as having nonvalvular atrial fibrillation during a 1-year period and treated in real-world practice. A total of 1,302 patients were given dabigatran and 8,102 were given warfarin to prevent stroke and systemic embolism. They were followed for a median of about 200 days, until discontinuing or switching their anticoagulant, dying, or reaching the study’s cutoff date. Nine categories of bleeding were assessed, and the data were adjusted to account for numerous demographic and clinical characteristics known to affect bleeding risk.
Compared with warfarin, dabigatran was associated with a significantly higher risk of major bleeding (9.0% vs 5.9%), with a hazard ratio of 1.58. Dabigatran also was associated with a significantly higher risk of GI bleeding (HR, 1.85), hematuria (HR, 1.41), vaginal bleeding (HR, 2.27), hemarthrosis (HR, 2.78), and hemoptysis (HR, 1.49). In contrast, dabigatran was associated with a slightly lower (0.6%) rate of intracranial bleeding, and also with lower rates of epistaxis and nonspecified bleeding, the investigators reported (JAMA Intern. Med. 2014 Nov. 3 [doi: 10.1001/jamainternmed.2014.5398]).
These differences were consistent across numerous subgroups of patients assessed, and were especially strong among African Americans and patients with chronic kidney disease.
This study was supported by the Commonwealth Foundation and the U.S. Agency for Healthcare Research and Quality. Dr. Hernandez and her associates reported having no financial conflicts of interest.
The bleeding risk for dabigatran appears to be higher than that for warfarin and significantly greater than it initially seemed at the time of FDA approval.
Hernandez et al. noted that the study on which the FDA based its approval failed to adjust for important differences in patient characteristics, which likely biased the results. They remind us that postmarketing data are crucial for us to advise our patients accurately.
Dr. Rita F. Redberg is the editor of JAMA Internal Medicine and director of women’s cardiovascular services at the Philip R. Lee Institute for Health Policy Studies at the University of California, San Francisco, Medical Center. She reported no financial conflicts of interest. Dr. Redberg made these remarks in an Editor’s Note accompanying Dr. Hernandez’s report (JAMA Intern. Med. 2014 Nov. 3).
The bleeding risk for dabigatran appears to be higher than that for warfarin and significantly greater than it initially seemed at the time of FDA approval.
Hernandez et al. noted that the study on which the FDA based its approval failed to adjust for important differences in patient characteristics, which likely biased the results. They remind us that postmarketing data are crucial for us to advise our patients accurately.
Dr. Rita F. Redberg is the editor of JAMA Internal Medicine and director of women’s cardiovascular services at the Philip R. Lee Institute for Health Policy Studies at the University of California, San Francisco, Medical Center. She reported no financial conflicts of interest. Dr. Redberg made these remarks in an Editor’s Note accompanying Dr. Hernandez’s report (JAMA Intern. Med. 2014 Nov. 3).
The bleeding risk for dabigatran appears to be higher than that for warfarin and significantly greater than it initially seemed at the time of FDA approval.
Hernandez et al. noted that the study on which the FDA based its approval failed to adjust for important differences in patient characteristics, which likely biased the results. They remind us that postmarketing data are crucial for us to advise our patients accurately.
Dr. Rita F. Redberg is the editor of JAMA Internal Medicine and director of women’s cardiovascular services at the Philip R. Lee Institute for Health Policy Studies at the University of California, San Francisco, Medical Center. She reported no financial conflicts of interest. Dr. Redberg made these remarks in an Editor’s Note accompanying Dr. Hernandez’s report (JAMA Intern. Med. 2014 Nov. 3).
Dabigatran significantly raises the risk of major bleeding and gastrointestinal bleeding across all subgroups of patients with atrial fibrillation, and particularly in African Americans and patients with chronic kidney disease, according to a report published online Nov. 3 in JAMA Internal Medicine.
Physicians should only prescribe dabigatran with caution, and should fully explain to patients who do take the drug how to identify abnormal bleeding so that it can be detected and controlled as early as possible, said Inmaculada Hernandez, Pharm.D., of the department of health policy and management, University of Pittsburgh, and her associates.
The FDA approved dabigatran in 2010 via an accelerated pathway after only 6 months of review, based largely on findings from a single clinical study, the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial, which did not adjust for patient characteristics (N. Engl. J. Med. 2009;361:1139-51). That study reported lower bleeding risks with dabigatran than with warfarin. Several months later, the agency’s Adverse Event Reporting System received “a large number” of reports of severe bleeding associated with dabigatran; and the relative bleeding risk associated with the two drugs is still unclear.
Dr. Hernandez and her colleagues examined the issue using data from a nationally representative random sample of 9,404 Medicare beneficiaries newly diagnosed as having nonvalvular atrial fibrillation during a 1-year period and treated in real-world practice. A total of 1,302 patients were given dabigatran and 8,102 were given warfarin to prevent stroke and systemic embolism. They were followed for a median of about 200 days, until discontinuing or switching their anticoagulant, dying, or reaching the study’s cutoff date. Nine categories of bleeding were assessed, and the data were adjusted to account for numerous demographic and clinical characteristics known to affect bleeding risk.
Compared with warfarin, dabigatran was associated with a significantly higher risk of major bleeding (9.0% vs 5.9%), with a hazard ratio of 1.58. Dabigatran also was associated with a significantly higher risk of GI bleeding (HR, 1.85), hematuria (HR, 1.41), vaginal bleeding (HR, 2.27), hemarthrosis (HR, 2.78), and hemoptysis (HR, 1.49). In contrast, dabigatran was associated with a slightly lower (0.6%) rate of intracranial bleeding, and also with lower rates of epistaxis and nonspecified bleeding, the investigators reported (JAMA Intern. Med. 2014 Nov. 3 [doi: 10.1001/jamainternmed.2014.5398]).
These differences were consistent across numerous subgroups of patients assessed, and were especially strong among African Americans and patients with chronic kidney disease.
This study was supported by the Commonwealth Foundation and the U.S. Agency for Healthcare Research and Quality. Dr. Hernandez and her associates reported having no financial conflicts of interest.
Dabigatran significantly raises the risk of major bleeding and gastrointestinal bleeding across all subgroups of patients with atrial fibrillation, and particularly in African Americans and patients with chronic kidney disease, according to a report published online Nov. 3 in JAMA Internal Medicine.
Physicians should only prescribe dabigatran with caution, and should fully explain to patients who do take the drug how to identify abnormal bleeding so that it can be detected and controlled as early as possible, said Inmaculada Hernandez, Pharm.D., of the department of health policy and management, University of Pittsburgh, and her associates.
The FDA approved dabigatran in 2010 via an accelerated pathway after only 6 months of review, based largely on findings from a single clinical study, the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial, which did not adjust for patient characteristics (N. Engl. J. Med. 2009;361:1139-51). That study reported lower bleeding risks with dabigatran than with warfarin. Several months later, the agency’s Adverse Event Reporting System received “a large number” of reports of severe bleeding associated with dabigatran; and the relative bleeding risk associated with the two drugs is still unclear.
Dr. Hernandez and her colleagues examined the issue using data from a nationally representative random sample of 9,404 Medicare beneficiaries newly diagnosed as having nonvalvular atrial fibrillation during a 1-year period and treated in real-world practice. A total of 1,302 patients were given dabigatran and 8,102 were given warfarin to prevent stroke and systemic embolism. They were followed for a median of about 200 days, until discontinuing or switching their anticoagulant, dying, or reaching the study’s cutoff date. Nine categories of bleeding were assessed, and the data were adjusted to account for numerous demographic and clinical characteristics known to affect bleeding risk.
Compared with warfarin, dabigatran was associated with a significantly higher risk of major bleeding (9.0% vs 5.9%), with a hazard ratio of 1.58. Dabigatran also was associated with a significantly higher risk of GI bleeding (HR, 1.85), hematuria (HR, 1.41), vaginal bleeding (HR, 2.27), hemarthrosis (HR, 2.78), and hemoptysis (HR, 1.49). In contrast, dabigatran was associated with a slightly lower (0.6%) rate of intracranial bleeding, and also with lower rates of epistaxis and nonspecified bleeding, the investigators reported (JAMA Intern. Med. 2014 Nov. 3 [doi: 10.1001/jamainternmed.2014.5398]).
These differences were consistent across numerous subgroups of patients assessed, and were especially strong among African Americans and patients with chronic kidney disease.
This study was supported by the Commonwealth Foundation and the U.S. Agency for Healthcare Research and Quality. Dr. Hernandez and her associates reported having no financial conflicts of interest.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Dabigatran raises the risk of major bleeding, contrary to initial reports that fast-tracked FDA approval.
Major finding: Compared with warfarin, dabigatran was associated with a significantly higher risk of major bleeding (9.0% vs. 5.9%), with a hazard ratio of 1.58.
Data source: A retrospective cohort study of bleeding risks in 1,302 dabigatran users and 8,102 warfarin users who had newly diagnosed nonvalvular atrial fibrillation.
Disclosures: This study was supported by the Commonwealth Foundation and the U.S. Agency for Healthcare Research and Quality. Dr. Hernandez and her associates reported having no financial conflicts of interest.
Vedolizumab rated first line for ulcerative colitis
VIENNA – Vedolizumab, a second-line biologic for treating patients who have ulcerative colitis or Crohn’s disease and don’t respond to an anti-tumor necrosis factor drug, is gaining traction with experts who have used the drug investigationally for several years and consider vedolizumab to be their first-line biologic for ulcerative colitis. But they agree that for Crohn’s disease the available data support using vedolizumab as a second-line agent.
“I think vedolizumab [Entyvio] is a first-line biologic for ulcerative colitis. Full stop,” said Dr. Brian G. Feagan during a talk at the United European Gastroenterology Global Congress. He cited his 12-year experience using vedolizumab, which has compiled a strong record of efficacy as well as safety for ulcerative colitis.

“It’s the promise of a reduced risk of systemic adverse events, especially secondary infections,” by using vedolizumab instead of a drug that blocks tumor necrosis factor (TNF), said Dr. Feagan in an interview.
“For Crohn’s disease, vedolizumab seems to have a slower onset of action, so for sicker Crohn’s patients you may want to choose a TNF blocker,” said Dr. Feagan, a gastroenterologist and professor of medicine at the University of Western Ontario in London, Ont. “For Crohn’s disease you could use vedolizumab first in some patients and an anti-TNF first line in other patients.”
Another gastroenterologist experienced in using vedolizumab to treat inflammatory bowel diseases largely agreed. “Vedolizumab is a first-line biologic for ulcerative colitis, For Crohn’s disease, the anti-TNF drugs are still the way to go,” said Dr. Paul Rutgeerts, a gastroenterologist and professor of medicine at Catholic University in Leuven, Belgium.

Dr. Feagan and Dr. Rutgeerts as well as others cite a fundamental difference in the way vedolizumab mitigates autoimmunity compared with TNF blocking drugs as the likely explanation for why vedolizumab seems to work much better for ulcerative colitis than it does for Crohn’s disease.
As spelled out last year in an editorial by Dr. Fabio Cominelli (N. Engl. J. Med. 2013;369:775-6), vedolizumab specifically blocks the integrin that directs leukocytes to the gut musoca, and this limited, gut-specific action may explain why the drug has such a favorable adverse effect profile as well as why it is less effective at inducing remissions in Crohn’s disease, which can hit any site along the entire gastrointestinal tract and cause transmural fistulas and multi-organ involvement. In contrast, ulcerative colitis is limited to the superficial mucosa of the large bowel.
“With ulcerative colitis it is only the mucosa. Crohn’s disease is more of a transmural disorder that involves the entire wall of the gut so you have more need for systemic action, which is better delivered by anti-TNFs” than by vedolizumab, Dr. Rutgeerts said in an interview.
Perhaps vedolizumab’s only major drawback as first-line biologic treatment for ulcerative colitis and even for some Crohn’s disease patients is its high cost, especially at a time when the price for the anti-TNF drug infliximab has begun to fall following introduction of biosimilar infliximab in Europe and its expected appearance soon in the United States.
“The cost [of vedolizumab] is very high,” especially when administered every 4 weeks, which seems to be the dosage many patients need to maintain long-term responses and remissions, Dr. Rutgeerts said.
“Now we have biosimilar infliximab, which is cheaper than vedolizumab” and hence more attractive, at least for cost, noted Dr. C. Janneke van der Woude, head of the inflammatory bowel diseases unit at Erasmus University in Rotterdam, The Netherlands. She also noted that many patients prefer subcutaneous drug treatment with the anti-TNF agent adalimumab over intravenous treatments, which is the route for vedolizumab.
Two-year data show durable efficacy, safety
Vedolizumab demonstrated its ability to safely maintain remission in responsive patients with inflammatory bowel disease in results from long-term treatment and follow-up of patients enrolled in the pivotal phase III trials that supplied the data that led to vedolizumab’s marketing approval earlier this year in both the United States and in Europe.
Dr. Feagan presented outcome results after 80 and 104 weeks of vedolizumab treatment of 278 patients with ulcerative colitis who had completed a full year of treatment during the GEMINI 1 trial [Phase 3, Randomized, Placebo-Controlled, Blinded, Multicenter Study of the Induction and Maintenance of Clinical Response and Remission by Vedolizumab in Patients with Moderate to Severe Ulcerative Colitis] (N. Engl. J. Med. 2013;369:699-710). He reported that the percentage of patients in clinical remission grew from 66% after 52 weeks on treatment (the time of entry into the long-term phase of the study), to 77% after 80 weeks, which then dropped to 73% after 104 weeks. Patients with a clinical response increased from 78% after 52 weeks to 88% after 80 weeks, and then dropped to 83% after 104 weeks.
During weeks 53-104 on treatment the rates of adverse events, serious adverse events, serious infections, adverse events resulting in treatment discontinuation, enteric infections, and malignancies were all low and similar to the event rates seen among the patients randomized to placebo in the GEMINI 1 study. The same pattern of low event rates similar to the placebo patients also occurred for episodes of nasopharyngitis, upper respiratory tract infections, Clostridium difficile infections, and tuberculosis cases.
The results also showed better rates of long-term remission and response in the patients who had not previously failed treatment with an anti-TNF drug compared with those who had. Asked what might explain this, Dr. Feagan replied “we know that patients who fail prior drugs are always harder to treat.”
A very similar pattern occurred among the patients with Crohn’s disease who remained on vedolizumab for an additional 52 weeks following completion of a first year of treatment in the GEMINI 2 trial [the Randomized, Placebo-Controlled, Blinded, Multicenter Study of the Induction and Maintenance of Clinical Response and Remission by Vedolizumab in Patients with Moderate to Severe Crohn’s Disease], the pivotal, phase III study that led to the Crohn’s disease indication for vedolizumab (N. Engl. J. Med. 2013;369:711-21). Among 295 GEMINI 2 completer patients who remained on vedolizumab the rate of complete remissions was 57% at entry into the long-term study after 52 weeks on treatment, 64% after 80 weeks on treatment, and 61% after 104 weeks.The overall response rate was 81% at 52 weeks, 78% after 80 weeks, and 74% at 104 week, Dr. Rutgeerts reported. However, he noted that the study design allowed patients who experienced a flare to receive conventional rescue medications and still be counted a responder or in remission, which increased the number of patients in both of these two groups. “Patients did not need to be a responder or in remission at every time point,” he noted.
Once again, the rates of adverse events, serious adverse events, adverse events resulting in drug discontinuation, infections, serious infections, nasopharyngitis, and upper respiratory infection were all similar among patients on long-term vedolizumab compared with control patients with Crohn’s disease, Dr. Rutgeerts said.
The results suggest that with vedolizumab treatment of inflammatory bowel disease “once you achieve an effect it is long-lasting,” Dr. Rutgeerts said in an interview. But he cautioned that the long-lasting efficacy was achieved with treatment every 4 weeks. While this approach was safe, it would also be expensive in routine practice, he noted. “The safety looks good, but the cost would be very high.”
“A key concept of vedolizumab is that it builds efficacy over time,” commented Dr. Silvio Danese during a talk at the meeting. “Vedolizumab is not the fastest runner, but [treating inflammatory bowel disease] is a marathon, and the important thing is getting to the finish,” said Dr. Danese, head of the inflammatory bowel disease unit at the Humanitas Clinic and Research Center in Milan.
Dr. Danese also said that vedolizumab has a safety advantage over anti-TNF drugs when treating ulcerative colitis, although vedolizumab’s efficacy for treating steroid-refractory, fulminant ulcertative colitis remains untested. And he agreed that vedolizumab’s role as a go-to agent for inducing remission in active Crohn’s disease seems questionable.
Two other reports at the meeting further fleshed out vedolizumab’s performance in the GEMINI trials.
One analysis focused on 34 ulcerative colitis patients and 57 Crohn’s disease patients who responded to vedolizumab induction treatment but then lost their response when they subsequently received vedolizumab once every 8 weeks. These patients then switched to a regimen in which they received the drug every 4 weeks, and this resulted in significant reductions in disease activity among some of the ulcerative colitis and Crohn’s disease patients, reported Dr. Séverine Vermeire, a gastroenterologist and professor of medicine at Catholic University in Leuven.
She estimated that roughly a third of patients in both disease categories who lost response when put on the drug once every 8 weeks regained their complete response when their dosing frequency increased to once every 4 weeks.
The findings “suggest that dosing every 4 weeks may be beneficial for certain patients, with no apparent change in safety. These data provide insight into the potential value of dosing every 4 weeks, and in routine practice we will have patients who will lose response when they receive the drug every 8 weeks,” Dr. Vermeire said. “We should try to identify these patients early,” she said, and “we need to investigate why some patients lose their vedolizumab response.” The labeling for vedolizumab calls for treatment once every 8 weeks for maintenance.
Another analysis focused on the total of 1,443 inflammatory bowel disease patients who received vedolizumab for both induction and maintenance in the two GEMINI studies. This safety analysis examined the incidence of total infections and serious infections among patients who started on vedolizumab while on treatment with a corticosteroid, an immunomodulating drug, both of these agents, or neither of these agents.
The analysis showed similar rates of both total infections and serious infections in all four subgroups, suggesting no interaction between vedsolizumab and other immuno-active drugs in terms of vulnerability to infection, said Dr. Jean-Frédréic Colombel, professor of medicine and director of the inflammatory bowel disease center at Mount Sinai Medical Center in New York.
The low rate of serious infections, which occurred at 0.04-0.06 infections/person-year of follow-up was “reassuring,” said Dr. Colombel, but these infections were also so rare that it limited interpretation of the findings. He also noted that the analysis only focused on concomitant drugs at the time patients began vedolizumab treatment and did not take into account subsequent medication changes.
The GEMINI 1 and 2 trials were sponsored by Takeda, the company that markets vedolizumab (Entyvio). Dr. Feagan has been a consultant to Takeda, AbbVie, Merck, and UCB. Dr. Rutgeerts has been a consultant to Takeda, AbbVie, Janssen, Merch, and UCB. Dr. van der Woude has been an advisor to Dr. Falk, Abbvie, Janssen, Johnson&Johnson, and Cosmo. Dr. Danese has been a consultant to Takeda and to several other companies. Dr. Vermeire has been a consultant to Takeda and to several other companies. Dr. Colombel has been a consultant to Takeda and to several other companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AGA Resources
• IBD Clinical Service Line: www.gastro.org/practice/clinical-service-line/ibd-
clinical-service-line
• Clinical Decision Support Tool: Use of Biologic Drugs for Inflammatory Crohn’s Disease: http://campaigns.gastro.org/algorithms/crohns
VIENNA – Vedolizumab, a second-line biologic for treating patients who have ulcerative colitis or Crohn’s disease and don’t respond to an anti-tumor necrosis factor drug, is gaining traction with experts who have used the drug investigationally for several years and consider vedolizumab to be their first-line biologic for ulcerative colitis. But they agree that for Crohn’s disease the available data support using vedolizumab as a second-line agent.
“I think vedolizumab [Entyvio] is a first-line biologic for ulcerative colitis. Full stop,” said Dr. Brian G. Feagan during a talk at the United European Gastroenterology Global Congress. He cited his 12-year experience using vedolizumab, which has compiled a strong record of efficacy as well as safety for ulcerative colitis.
“It’s the promise of a reduced risk of systemic adverse events, especially secondary infections,” by using vedolizumab instead of a drug that blocks tumor necrosis factor (TNF), said Dr. Feagan in an interview.
“For Crohn’s disease, vedolizumab seems to have a slower onset of action, so for sicker Crohn’s patients you may want to choose a TNF blocker,” said Dr. Feagan, a gastroenterologist and professor of medicine at the University of Western Ontario in London, Ont. “For Crohn’s disease you could use vedolizumab first in some patients and an anti-TNF first line in other patients.”
Another gastroenterologist experienced in using vedolizumab to treat inflammatory bowel diseases largely agreed. “Vedolizumab is a first-line biologic for ulcerative colitis, For Crohn’s disease, the anti-TNF drugs are still the way to go,” said Dr. Paul Rutgeerts, a gastroenterologist and professor of medicine at Catholic University in Leuven, Belgium.
Dr. Feagan and Dr. Rutgeerts as well as others cite a fundamental difference in the way vedolizumab mitigates autoimmunity compared with TNF blocking drugs as the likely explanation for why vedolizumab seems to work much better for ulcerative colitis than it does for Crohn’s disease.
As spelled out last year in an editorial by Dr. Fabio Cominelli (N. Engl. J. Med. 2013;369:775-6), vedolizumab specifically blocks the integrin that directs leukocytes to the gut musoca, and this limited, gut-specific action may explain why the drug has such a favorable adverse effect profile as well as why it is less effective at inducing remissions in Crohn’s disease, which can hit any site along the entire gastrointestinal tract and cause transmural fistulas and multi-organ involvement. In contrast, ulcerative colitis is limited to the superficial mucosa of the large bowel.
“With ulcerative colitis it is only the mucosa. Crohn’s disease is more of a transmural disorder that involves the entire wall of the gut so you have more need for systemic action, which is better delivered by anti-TNFs” than by vedolizumab, Dr. Rutgeerts said in an interview.
Perhaps vedolizumab’s only major drawback as first-line biologic treatment for ulcerative colitis and even for some Crohn’s disease patients is its high cost, especially at a time when the price for the anti-TNF drug infliximab has begun to fall following introduction of biosimilar infliximab in Europe and its expected appearance soon in the United States.
“The cost [of vedolizumab] is very high,” especially when administered every 4 weeks, which seems to be the dosage many patients need to maintain long-term responses and remissions, Dr. Rutgeerts said.
“Now we have biosimilar infliximab, which is cheaper than vedolizumab” and hence more attractive, at least for cost, noted Dr. C. Janneke van der Woude, head of the inflammatory bowel diseases unit at Erasmus University in Rotterdam, The Netherlands. She also noted that many patients prefer subcutaneous drug treatment with the anti-TNF agent adalimumab over intravenous treatments, which is the route for vedolizumab.
Two-year data show durable efficacy, safety
Vedolizumab demonstrated its ability to safely maintain remission in responsive patients with inflammatory bowel disease in results from long-term treatment and follow-up of patients enrolled in the pivotal phase III trials that supplied the data that led to vedolizumab’s marketing approval earlier this year in both the United States and in Europe.
Dr. Feagan presented outcome results after 80 and 104 weeks of vedolizumab treatment of 278 patients with ulcerative colitis who had completed a full year of treatment during the GEMINI 1 trial [Phase 3, Randomized, Placebo-Controlled, Blinded, Multicenter Study of the Induction and Maintenance of Clinical Response and Remission by Vedolizumab in Patients with Moderate to Severe Ulcerative Colitis] (N. Engl. J. Med. 2013;369:699-710). He reported that the percentage of patients in clinical remission grew from 66% after 52 weeks on treatment (the time of entry into the long-term phase of the study), to 77% after 80 weeks, which then dropped to 73% after 104 weeks. Patients with a clinical response increased from 78% after 52 weeks to 88% after 80 weeks, and then dropped to 83% after 104 weeks.
During weeks 53-104 on treatment the rates of adverse events, serious adverse events, serious infections, adverse events resulting in treatment discontinuation, enteric infections, and malignancies were all low and similar to the event rates seen among the patients randomized to placebo in the GEMINI 1 study. The same pattern of low event rates similar to the placebo patients also occurred for episodes of nasopharyngitis, upper respiratory tract infections, Clostridium difficile infections, and tuberculosis cases.
The results also showed better rates of long-term remission and response in the patients who had not previously failed treatment with an anti-TNF drug compared with those who had. Asked what might explain this, Dr. Feagan replied “we know that patients who fail prior drugs are always harder to treat.”
A very similar pattern occurred among the patients with Crohn’s disease who remained on vedolizumab for an additional 52 weeks following completion of a first year of treatment in the GEMINI 2 trial [the Randomized, Placebo-Controlled, Blinded, Multicenter Study of the Induction and Maintenance of Clinical Response and Remission by Vedolizumab in Patients with Moderate to Severe Crohn’s Disease], the pivotal, phase III study that led to the Crohn’s disease indication for vedolizumab (N. Engl. J. Med. 2013;369:711-21). Among 295 GEMINI 2 completer patients who remained on vedolizumab the rate of complete remissions was 57% at entry into the long-term study after 52 weeks on treatment, 64% after 80 weeks on treatment, and 61% after 104 weeks.The overall response rate was 81% at 52 weeks, 78% after 80 weeks, and 74% at 104 week, Dr. Rutgeerts reported. However, he noted that the study design allowed patients who experienced a flare to receive conventional rescue medications and still be counted a responder or in remission, which increased the number of patients in both of these two groups. “Patients did not need to be a responder or in remission at every time point,” he noted.
Once again, the rates of adverse events, serious adverse events, adverse events resulting in drug discontinuation, infections, serious infections, nasopharyngitis, and upper respiratory infection were all similar among patients on long-term vedolizumab compared with control patients with Crohn’s disease, Dr. Rutgeerts said.
The results suggest that with vedolizumab treatment of inflammatory bowel disease “once you achieve an effect it is long-lasting,” Dr. Rutgeerts said in an interview. But he cautioned that the long-lasting efficacy was achieved with treatment every 4 weeks. While this approach was safe, it would also be expensive in routine practice, he noted. “The safety looks good, but the cost would be very high.”
“A key concept of vedolizumab is that it builds efficacy over time,” commented Dr. Silvio Danese during a talk at the meeting. “Vedolizumab is not the fastest runner, but [treating inflammatory bowel disease] is a marathon, and the important thing is getting to the finish,” said Dr. Danese, head of the inflammatory bowel disease unit at the Humanitas Clinic and Research Center in Milan.
Dr. Danese also said that vedolizumab has a safety advantage over anti-TNF drugs when treating ulcerative colitis, although vedolizumab’s efficacy for treating steroid-refractory, fulminant ulcertative colitis remains untested. And he agreed that vedolizumab’s role as a go-to agent for inducing remission in active Crohn’s disease seems questionable.
Two other reports at the meeting further fleshed out vedolizumab’s performance in the GEMINI trials.
One analysis focused on 34 ulcerative colitis patients and 57 Crohn’s disease patients who responded to vedolizumab induction treatment but then lost their response when they subsequently received vedolizumab once every 8 weeks. These patients then switched to a regimen in which they received the drug every 4 weeks, and this resulted in significant reductions in disease activity among some of the ulcerative colitis and Crohn’s disease patients, reported Dr. Séverine Vermeire, a gastroenterologist and professor of medicine at Catholic University in Leuven.
She estimated that roughly a third of patients in both disease categories who lost response when put on the drug once every 8 weeks regained their complete response when their dosing frequency increased to once every 4 weeks.
The findings “suggest that dosing every 4 weeks may be beneficial for certain patients, with no apparent change in safety. These data provide insight into the potential value of dosing every 4 weeks, and in routine practice we will have patients who will lose response when they receive the drug every 8 weeks,” Dr. Vermeire said. “We should try to identify these patients early,” she said, and “we need to investigate why some patients lose their vedolizumab response.” The labeling for vedolizumab calls for treatment once every 8 weeks for maintenance.
Another analysis focused on the total of 1,443 inflammatory bowel disease patients who received vedolizumab for both induction and maintenance in the two GEMINI studies. This safety analysis examined the incidence of total infections and serious infections among patients who started on vedolizumab while on treatment with a corticosteroid, an immunomodulating drug, both of these agents, or neither of these agents.
The analysis showed similar rates of both total infections and serious infections in all four subgroups, suggesting no interaction between vedsolizumab and other immuno-active drugs in terms of vulnerability to infection, said Dr. Jean-Frédréic Colombel, professor of medicine and director of the inflammatory bowel disease center at Mount Sinai Medical Center in New York.
The low rate of serious infections, which occurred at 0.04-0.06 infections/person-year of follow-up was “reassuring,” said Dr. Colombel, but these infections were also so rare that it limited interpretation of the findings. He also noted that the analysis only focused on concomitant drugs at the time patients began vedolizumab treatment and did not take into account subsequent medication changes.
The GEMINI 1 and 2 trials were sponsored by Takeda, the company that markets vedolizumab (Entyvio). Dr. Feagan has been a consultant to Takeda, AbbVie, Merck, and UCB. Dr. Rutgeerts has been a consultant to Takeda, AbbVie, Janssen, Merch, and UCB. Dr. van der Woude has been an advisor to Dr. Falk, Abbvie, Janssen, Johnson&Johnson, and Cosmo. Dr. Danese has been a consultant to Takeda and to several other companies. Dr. Vermeire has been a consultant to Takeda and to several other companies. Dr. Colombel has been a consultant to Takeda and to several other companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AGA Resources
• IBD Clinical Service Line: www.gastro.org/practice/clinical-service-line/ibd-
clinical-service-line
• Clinical Decision Support Tool: Use of Biologic Drugs for Inflammatory Crohn’s Disease: http://campaigns.gastro.org/algorithms/crohns
VIENNA – Vedolizumab, a second-line biologic for treating patients who have ulcerative colitis or Crohn’s disease and don’t respond to an anti-tumor necrosis factor drug, is gaining traction with experts who have used the drug investigationally for several years and consider vedolizumab to be their first-line biologic for ulcerative colitis. But they agree that for Crohn’s disease the available data support using vedolizumab as a second-line agent.
“I think vedolizumab [Entyvio] is a first-line biologic for ulcerative colitis. Full stop,” said Dr. Brian G. Feagan during a talk at the United European Gastroenterology Global Congress. He cited his 12-year experience using vedolizumab, which has compiled a strong record of efficacy as well as safety for ulcerative colitis.
“It’s the promise of a reduced risk of systemic adverse events, especially secondary infections,” by using vedolizumab instead of a drug that blocks tumor necrosis factor (TNF), said Dr. Feagan in an interview.
“For Crohn’s disease, vedolizumab seems to have a slower onset of action, so for sicker Crohn’s patients you may want to choose a TNF blocker,” said Dr. Feagan, a gastroenterologist and professor of medicine at the University of Western Ontario in London, Ont. “For Crohn’s disease you could use vedolizumab first in some patients and an anti-TNF first line in other patients.”
Another gastroenterologist experienced in using vedolizumab to treat inflammatory bowel diseases largely agreed. “Vedolizumab is a first-line biologic for ulcerative colitis, For Crohn’s disease, the anti-TNF drugs are still the way to go,” said Dr. Paul Rutgeerts, a gastroenterologist and professor of medicine at Catholic University in Leuven, Belgium.
Dr. Feagan and Dr. Rutgeerts as well as others cite a fundamental difference in the way vedolizumab mitigates autoimmunity compared with TNF blocking drugs as the likely explanation for why vedolizumab seems to work much better for ulcerative colitis than it does for Crohn’s disease.
As spelled out last year in an editorial by Dr. Fabio Cominelli (N. Engl. J. Med. 2013;369:775-6), vedolizumab specifically blocks the integrin that directs leukocytes to the gut musoca, and this limited, gut-specific action may explain why the drug has such a favorable adverse effect profile as well as why it is less effective at inducing remissions in Crohn’s disease, which can hit any site along the entire gastrointestinal tract and cause transmural fistulas and multi-organ involvement. In contrast, ulcerative colitis is limited to the superficial mucosa of the large bowel.
“With ulcerative colitis it is only the mucosa. Crohn’s disease is more of a transmural disorder that involves the entire wall of the gut so you have more need for systemic action, which is better delivered by anti-TNFs” than by vedolizumab, Dr. Rutgeerts said in an interview.
Perhaps vedolizumab’s only major drawback as first-line biologic treatment for ulcerative colitis and even for some Crohn’s disease patients is its high cost, especially at a time when the price for the anti-TNF drug infliximab has begun to fall following introduction of biosimilar infliximab in Europe and its expected appearance soon in the United States.
“The cost [of vedolizumab] is very high,” especially when administered every 4 weeks, which seems to be the dosage many patients need to maintain long-term responses and remissions, Dr. Rutgeerts said.
“Now we have biosimilar infliximab, which is cheaper than vedolizumab” and hence more attractive, at least for cost, noted Dr. C. Janneke van der Woude, head of the inflammatory bowel diseases unit at Erasmus University in Rotterdam, The Netherlands. She also noted that many patients prefer subcutaneous drug treatment with the anti-TNF agent adalimumab over intravenous treatments, which is the route for vedolizumab.
Two-year data show durable efficacy, safety
Vedolizumab demonstrated its ability to safely maintain remission in responsive patients with inflammatory bowel disease in results from long-term treatment and follow-up of patients enrolled in the pivotal phase III trials that supplied the data that led to vedolizumab’s marketing approval earlier this year in both the United States and in Europe.
Dr. Feagan presented outcome results after 80 and 104 weeks of vedolizumab treatment of 278 patients with ulcerative colitis who had completed a full year of treatment during the GEMINI 1 trial [Phase 3, Randomized, Placebo-Controlled, Blinded, Multicenter Study of the Induction and Maintenance of Clinical Response and Remission by Vedolizumab in Patients with Moderate to Severe Ulcerative Colitis] (N. Engl. J. Med. 2013;369:699-710). He reported that the percentage of patients in clinical remission grew from 66% after 52 weeks on treatment (the time of entry into the long-term phase of the study), to 77% after 80 weeks, which then dropped to 73% after 104 weeks. Patients with a clinical response increased from 78% after 52 weeks to 88% after 80 weeks, and then dropped to 83% after 104 weeks.
During weeks 53-104 on treatment the rates of adverse events, serious adverse events, serious infections, adverse events resulting in treatment discontinuation, enteric infections, and malignancies were all low and similar to the event rates seen among the patients randomized to placebo in the GEMINI 1 study. The same pattern of low event rates similar to the placebo patients also occurred for episodes of nasopharyngitis, upper respiratory tract infections, Clostridium difficile infections, and tuberculosis cases.
The results also showed better rates of long-term remission and response in the patients who had not previously failed treatment with an anti-TNF drug compared with those who had. Asked what might explain this, Dr. Feagan replied “we know that patients who fail prior drugs are always harder to treat.”
A very similar pattern occurred among the patients with Crohn’s disease who remained on vedolizumab for an additional 52 weeks following completion of a first year of treatment in the GEMINI 2 trial [the Randomized, Placebo-Controlled, Blinded, Multicenter Study of the Induction and Maintenance of Clinical Response and Remission by Vedolizumab in Patients with Moderate to Severe Crohn’s Disease], the pivotal, phase III study that led to the Crohn’s disease indication for vedolizumab (N. Engl. J. Med. 2013;369:711-21). Among 295 GEMINI 2 completer patients who remained on vedolizumab the rate of complete remissions was 57% at entry into the long-term study after 52 weeks on treatment, 64% after 80 weeks on treatment, and 61% after 104 weeks.The overall response rate was 81% at 52 weeks, 78% after 80 weeks, and 74% at 104 week, Dr. Rutgeerts reported. However, he noted that the study design allowed patients who experienced a flare to receive conventional rescue medications and still be counted a responder or in remission, which increased the number of patients in both of these two groups. “Patients did not need to be a responder or in remission at every time point,” he noted.
Once again, the rates of adverse events, serious adverse events, adverse events resulting in drug discontinuation, infections, serious infections, nasopharyngitis, and upper respiratory infection were all similar among patients on long-term vedolizumab compared with control patients with Crohn’s disease, Dr. Rutgeerts said.
The results suggest that with vedolizumab treatment of inflammatory bowel disease “once you achieve an effect it is long-lasting,” Dr. Rutgeerts said in an interview. But he cautioned that the long-lasting efficacy was achieved with treatment every 4 weeks. While this approach was safe, it would also be expensive in routine practice, he noted. “The safety looks good, but the cost would be very high.”
“A key concept of vedolizumab is that it builds efficacy over time,” commented Dr. Silvio Danese during a talk at the meeting. “Vedolizumab is not the fastest runner, but [treating inflammatory bowel disease] is a marathon, and the important thing is getting to the finish,” said Dr. Danese, head of the inflammatory bowel disease unit at the Humanitas Clinic and Research Center in Milan.
Dr. Danese also said that vedolizumab has a safety advantage over anti-TNF drugs when treating ulcerative colitis, although vedolizumab’s efficacy for treating steroid-refractory, fulminant ulcertative colitis remains untested. And he agreed that vedolizumab’s role as a go-to agent for inducing remission in active Crohn’s disease seems questionable.
Two other reports at the meeting further fleshed out vedolizumab’s performance in the GEMINI trials.
One analysis focused on 34 ulcerative colitis patients and 57 Crohn’s disease patients who responded to vedolizumab induction treatment but then lost their response when they subsequently received vedolizumab once every 8 weeks. These patients then switched to a regimen in which they received the drug every 4 weeks, and this resulted in significant reductions in disease activity among some of the ulcerative colitis and Crohn’s disease patients, reported Dr. Séverine Vermeire, a gastroenterologist and professor of medicine at Catholic University in Leuven.
She estimated that roughly a third of patients in both disease categories who lost response when put on the drug once every 8 weeks regained their complete response when their dosing frequency increased to once every 4 weeks.
The findings “suggest that dosing every 4 weeks may be beneficial for certain patients, with no apparent change in safety. These data provide insight into the potential value of dosing every 4 weeks, and in routine practice we will have patients who will lose response when they receive the drug every 8 weeks,” Dr. Vermeire said. “We should try to identify these patients early,” she said, and “we need to investigate why some patients lose their vedolizumab response.” The labeling for vedolizumab calls for treatment once every 8 weeks for maintenance.
Another analysis focused on the total of 1,443 inflammatory bowel disease patients who received vedolizumab for both induction and maintenance in the two GEMINI studies. This safety analysis examined the incidence of total infections and serious infections among patients who started on vedolizumab while on treatment with a corticosteroid, an immunomodulating drug, both of these agents, or neither of these agents.
The analysis showed similar rates of both total infections and serious infections in all four subgroups, suggesting no interaction between vedsolizumab and other immuno-active drugs in terms of vulnerability to infection, said Dr. Jean-Frédréic Colombel, professor of medicine and director of the inflammatory bowel disease center at Mount Sinai Medical Center in New York.
The low rate of serious infections, which occurred at 0.04-0.06 infections/person-year of follow-up was “reassuring,” said Dr. Colombel, but these infections were also so rare that it limited interpretation of the findings. He also noted that the analysis only focused on concomitant drugs at the time patients began vedolizumab treatment and did not take into account subsequent medication changes.
The GEMINI 1 and 2 trials were sponsored by Takeda, the company that markets vedolizumab (Entyvio). Dr. Feagan has been a consultant to Takeda, AbbVie, Merck, and UCB. Dr. Rutgeerts has been a consultant to Takeda, AbbVie, Janssen, Merch, and UCB. Dr. van der Woude has been an advisor to Dr. Falk, Abbvie, Janssen, Johnson&Johnson, and Cosmo. Dr. Danese has been a consultant to Takeda and to several other companies. Dr. Vermeire has been a consultant to Takeda and to several other companies. Dr. Colombel has been a consultant to Takeda and to several other companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AGA Resources
• IBD Clinical Service Line: www.gastro.org/practice/clinical-service-line/ibd-
clinical-service-line
• Clinical Decision Support Tool: Use of Biologic Drugs for Inflammatory Crohn’s Disease: http://campaigns.gastro.org/algorithms/crohns
EXPERT ANALYSIS FROM UEG WEEK VIENNA 2014

Oral oligonucleotide shows efficacy, safety in Crohn’s
VIENNA – Fourteen days of daily treatment with mongersen, an oral, antisense oligonucleotide, safely produced remissions in two-thirds of patients with Crohn’s disease in a phase II study with a total of 163 patients.
The responses were also durable, with patients in remission continuing to show low disease activity at 10 weeks after they received their last dose of mongersen, Dr. Giovanni Monteleone reported at he United European Gastroenterology Global Congress.
The durability of patient response was not a surprise as similar findings occurred during phase I testing, he said. Mongensen “is not an anti-inflammatory drug. It removes a brake on the normal immunosupressant mechanism in the gut, and that is why the effect is long lasting. It allows TGF [transforming growth factor]-beta to work again [as an endogenous immunosuppressant] and promote healing,” said Dr. Monteleone, a professor of gastroenterology at the University of Rome Tor Vergata.

Dr. Silvio Danese, director of the IBD center at the Humanitis Research Hospital in Milan, said he was impressed by the frequent, durable, and apparently safe remissions achieved by mongensen treatment. “If the phase III studies show efficacy, mongersen will probably be a game changer” for treating inflammatory bowel disease, he commented.
Dr. Monteleone traced development on mongensen to work he published 13 years ago, which showed that oligonucleotide blockade of the production of the Smad7 intracellular protein prevented the inhibitory effect of Smad7 on TGF-beta and thus allowed TGF-beta to inhibit cytokine production and T lymphocyte signaling (J. Clin. Invest. 2001;108:601-9). His work also showed that patients with inflammatory bowel disease have elevated Smad7 levels in their intestinal mucosa, suggesting the potential for therapeutically cutting Smad7 levels in the gut of patients with inflammatory bowel disease.
The study he reported at the meeting included 163 patients with active Crohn’s disease at about a dozen hospitals in Italy. Patients had a Crohn’s disease activity index (CDAI) score of 221-400, were steroid dependent, steroid resistant, or both, and had documented lesions in their terminal ileum, right colon, or both. Patients randomized to active treatment received a daily pill for 14 days with one of three dosages of mongersen formulated into a tablet with a pH dependent coating designed to release the drug in the terminal ileum and right colon and avoid systemic absorption. The researchers then followed patients for an additional 70 days during which they received usual care but no additional treatment with mongersen or placebo.
The study’s primary endpoint was the frequency of remission, designed as a CDAI score of less than 150 on day 15 (the day after their last investigational dose) that was then maintained through day 28 of the study. This endpoint occurred in 65% of the 43 patients who received the highest mongersen dosage, 160 mg/day, in 55% of patients who received 40 mg/day of mongersen, in 12% of patients who received 10 mg/day of mongersen, and in 10% of placebo patients. The differences were statistically significant for the two highest drug dosages.
At 84 days into the study, 10 weeks after receiving their final dosage of mongersen, the rate of patients who were given the highest dosage and continued to have a CDAI of less than 150 stood at 67%, Dr. Monteleone reported. An additional analysis focused on the 61% of patients who entered the study with a C-reactive protein level greater than 3 mg/L. Within this subgroup, 68% of patients who received the highest mongersen dosage achieved remission compared with 12% of patients who received placebo, a statistically significant difference.
The safety analysis showed that mongersen was safe and well tolerated, with low rates of total and serious adverse events that were similar across all four treatment arms. Patient follow-up also showed no clinically meaningful changes in laboratory values, vital signs, physical findings, or strictures.
Celgene has stated that it will proceed into phase III testing of mongersen.
The study was sponsored by Nogra, and then subsequently by Celgene which acquired a license for morgensen from Nogra. Dr. Monteleone has received research support from Nogra and from several other companies and has been a speaker on behalf of AbbVie and Zambon. Dr. Danese has been a consultant to several drug companies.
On Twitter @mitchelzoler
VIENNA – Fourteen days of daily treatment with mongersen, an oral, antisense oligonucleotide, safely produced remissions in two-thirds of patients with Crohn’s disease in a phase II study with a total of 163 patients.
The responses were also durable, with patients in remission continuing to show low disease activity at 10 weeks after they received their last dose of mongersen, Dr. Giovanni Monteleone reported at he United European Gastroenterology Global Congress.
The durability of patient response was not a surprise as similar findings occurred during phase I testing, he said. Mongensen “is not an anti-inflammatory drug. It removes a brake on the normal immunosupressant mechanism in the gut, and that is why the effect is long lasting. It allows TGF [transforming growth factor]-beta to work again [as an endogenous immunosuppressant] and promote healing,” said Dr. Monteleone, a professor of gastroenterology at the University of Rome Tor Vergata.
Dr. Silvio Danese, director of the IBD center at the Humanitis Research Hospital in Milan, said he was impressed by the frequent, durable, and apparently safe remissions achieved by mongensen treatment. “If the phase III studies show efficacy, mongersen will probably be a game changer” for treating inflammatory bowel disease, he commented.
Dr. Monteleone traced development on mongensen to work he published 13 years ago, which showed that oligonucleotide blockade of the production of the Smad7 intracellular protein prevented the inhibitory effect of Smad7 on TGF-beta and thus allowed TGF-beta to inhibit cytokine production and T lymphocyte signaling (J. Clin. Invest. 2001;108:601-9). His work also showed that patients with inflammatory bowel disease have elevated Smad7 levels in their intestinal mucosa, suggesting the potential for therapeutically cutting Smad7 levels in the gut of patients with inflammatory bowel disease.
The study he reported at the meeting included 163 patients with active Crohn’s disease at about a dozen hospitals in Italy. Patients had a Crohn’s disease activity index (CDAI) score of 221-400, were steroid dependent, steroid resistant, or both, and had documented lesions in their terminal ileum, right colon, or both. Patients randomized to active treatment received a daily pill for 14 days with one of three dosages of mongersen formulated into a tablet with a pH dependent coating designed to release the drug in the terminal ileum and right colon and avoid systemic absorption. The researchers then followed patients for an additional 70 days during which they received usual care but no additional treatment with mongersen or placebo.
The study’s primary endpoint was the frequency of remission, designed as a CDAI score of less than 150 on day 15 (the day after their last investigational dose) that was then maintained through day 28 of the study. This endpoint occurred in 65% of the 43 patients who received the highest mongersen dosage, 160 mg/day, in 55% of patients who received 40 mg/day of mongersen, in 12% of patients who received 10 mg/day of mongersen, and in 10% of placebo patients. The differences were statistically significant for the two highest drug dosages.
At 84 days into the study, 10 weeks after receiving their final dosage of mongersen, the rate of patients who were given the highest dosage and continued to have a CDAI of less than 150 stood at 67%, Dr. Monteleone reported. An additional analysis focused on the 61% of patients who entered the study with a C-reactive protein level greater than 3 mg/L. Within this subgroup, 68% of patients who received the highest mongersen dosage achieved remission compared with 12% of patients who received placebo, a statistically significant difference.
The safety analysis showed that mongersen was safe and well tolerated, with low rates of total and serious adverse events that were similar across all four treatment arms. Patient follow-up also showed no clinically meaningful changes in laboratory values, vital signs, physical findings, or strictures.
Celgene has stated that it will proceed into phase III testing of mongersen.
The study was sponsored by Nogra, and then subsequently by Celgene which acquired a license for morgensen from Nogra. Dr. Monteleone has received research support from Nogra and from several other companies and has been a speaker on behalf of AbbVie and Zambon. Dr. Danese has been a consultant to several drug companies.
On Twitter @mitchelzoler
VIENNA – Fourteen days of daily treatment with mongersen, an oral, antisense oligonucleotide, safely produced remissions in two-thirds of patients with Crohn’s disease in a phase II study with a total of 163 patients.
The responses were also durable, with patients in remission continuing to show low disease activity at 10 weeks after they received their last dose of mongersen, Dr. Giovanni Monteleone reported at he United European Gastroenterology Global Congress.
The durability of patient response was not a surprise as similar findings occurred during phase I testing, he said. Mongensen “is not an anti-inflammatory drug. It removes a brake on the normal immunosupressant mechanism in the gut, and that is why the effect is long lasting. It allows TGF [transforming growth factor]-beta to work again [as an endogenous immunosuppressant] and promote healing,” said Dr. Monteleone, a professor of gastroenterology at the University of Rome Tor Vergata.
Dr. Silvio Danese, director of the IBD center at the Humanitis Research Hospital in Milan, said he was impressed by the frequent, durable, and apparently safe remissions achieved by mongensen treatment. “If the phase III studies show efficacy, mongersen will probably be a game changer” for treating inflammatory bowel disease, he commented.
Dr. Monteleone traced development on mongensen to work he published 13 years ago, which showed that oligonucleotide blockade of the production of the Smad7 intracellular protein prevented the inhibitory effect of Smad7 on TGF-beta and thus allowed TGF-beta to inhibit cytokine production and T lymphocyte signaling (J. Clin. Invest. 2001;108:601-9). His work also showed that patients with inflammatory bowel disease have elevated Smad7 levels in their intestinal mucosa, suggesting the potential for therapeutically cutting Smad7 levels in the gut of patients with inflammatory bowel disease.
The study he reported at the meeting included 163 patients with active Crohn’s disease at about a dozen hospitals in Italy. Patients had a Crohn’s disease activity index (CDAI) score of 221-400, were steroid dependent, steroid resistant, or both, and had documented lesions in their terminal ileum, right colon, or both. Patients randomized to active treatment received a daily pill for 14 days with one of three dosages of mongersen formulated into a tablet with a pH dependent coating designed to release the drug in the terminal ileum and right colon and avoid systemic absorption. The researchers then followed patients for an additional 70 days during which they received usual care but no additional treatment with mongersen or placebo.
The study’s primary endpoint was the frequency of remission, designed as a CDAI score of less than 150 on day 15 (the day after their last investigational dose) that was then maintained through day 28 of the study. This endpoint occurred in 65% of the 43 patients who received the highest mongersen dosage, 160 mg/day, in 55% of patients who received 40 mg/day of mongersen, in 12% of patients who received 10 mg/day of mongersen, and in 10% of placebo patients. The differences were statistically significant for the two highest drug dosages.
At 84 days into the study, 10 weeks after receiving their final dosage of mongersen, the rate of patients who were given the highest dosage and continued to have a CDAI of less than 150 stood at 67%, Dr. Monteleone reported. An additional analysis focused on the 61% of patients who entered the study with a C-reactive protein level greater than 3 mg/L. Within this subgroup, 68% of patients who received the highest mongersen dosage achieved remission compared with 12% of patients who received placebo, a statistically significant difference.
The safety analysis showed that mongersen was safe and well tolerated, with low rates of total and serious adverse events that were similar across all four treatment arms. Patient follow-up also showed no clinically meaningful changes in laboratory values, vital signs, physical findings, or strictures.
Celgene has stated that it will proceed into phase III testing of mongersen.
The study was sponsored by Nogra, and then subsequently by Celgene which acquired a license for morgensen from Nogra. Dr. Monteleone has received research support from Nogra and from several other companies and has been a speaker on behalf of AbbVie and Zambon. Dr. Danese has been a consultant to several drug companies.
On Twitter @mitchelzoler
AT UEG WEEK VIENNA 2014
Key clinical point: Mongersen may prove to be an effective oral therapy that can be used on a short-term basis to produce durable remissions in Crohn’s.
Major finding: Two weeks of oral treatment with mongersen produced remissions in 65% of Crohn’s disease patients while the remission rate was 10% in placebo-treated controls.
Data source: A multicenter, placebo-controlled phase II study that included 163 patients.
Disclosures: The study was sponsored by Nogra, and then subsequently by Celgene which acquired a license for morgensen from Nogra. Dr. Monteleone has received research support from Nogra and from several other companies and has been a speaker on behalf of AbbVie and Zambon. Dr. Danese has been a consultant to several drug companies.

Higher dose of mesalamine can decrease risk of relapse for patients with quiescent ulcerative colitis
Increasing dosages of mesalamine by 2.4 g/d for patients with quiescent ulcerative colitis can dramatically reduce concentrations of fecal calprotectin and ultimately decrease the likelihood of relapse, according to the results of a new Dose Escalation and Remission (DEAR) study published in the November issue of Clinical Gastroenterology and Hepatology (2014;12:1887-93.e3).
In an open-label, randomized controlled trial, 119 patients with quiescent ulcerative colitis (UC) in remission were screened for Simple Clinical Colitis Activity Index scores, fecal calprotectin (FC) of 50 mcg/gram, and intake of no more than 3 g of mesalamine per day between October 2008 and March 2012. These subjects were divided into four groups based on FC levels, with Group 1 comprising 58 representing a baseline of FC < 50 mcg/gram, and groups 2-4 having 61 individuals with elevated baseline FC levels. Group 1 patients were retained for an observational follow-up study but were not included in the randomized controlled trial.
Participants were either told to maintain their current medication regimen or were prescribed a multimatrix mesalamine of either 2.4 or 4.8 g/d for 6 weeks, depending on their initial FC levels, after which point FC levels were taken. Participants in Group 2 with FC < 50 mcg/g were kept in an observational follow-up study group, while the rest of the participants were randomly assigned to continue their 2.4 g/d regimen, or increase their intake to 4.8 g/d.
Individuals in Group 3 with FC levels greater than 50 mcg/g were randomly assigned to either the 2.4- or 4.8-g/d regimen for 6 weeks; after that point, those whose FC levels had decreased below 50 continued with their regimen while those whose FC levels remained high were upped to 4.8 g/d. Subjects in Group 4 who were not already on a multimatrix mesalamine regimen and whose FC levels were above 50 mcg/g were randomized to continue their medication or add 2.4 g/d of mesalamine; after 6 weeks, if FC levels remained above 50, the dosage was increased to 4.8 g/d.
The primary outcome of continued remission with FC no greater than 50 mcg/g was observed in 3.8% of controls and 26.9% of participants in the dose escalation group (P = .0496). Others within the same group experienced secondary outcomes of FC levels below 100 mcg/gram (P = .04) and below 200 mcg/g (P = .005). Furthermore, among patients who were in remission during the randomization phase of the study, relapse occurred far sooner in those whose FC was above 200 mcg/g than those whose FC levels were below that mark.
“Our study adds to the evidence supporting FC concentration as a valid biomarker for UC,” said lead authors Dr. Mark T. Osterman and Dr. Faten N. Aberra of the University of Pennsylvania’s department of medicine, Philadelphia. “Perhaps more importantly, our results offer a novel way to use FC testing to positively impact outcomes in UC. We demonstrated efficacy of intervention to reduce FC (a surrogate for mucosal inflammation) before symptoms develop in patients at potentially increased risk of relapse.”
Dr. Osterman is a consultant for AbbVie, Elan, Janssen, and UCB and receives research funding from UCB. Dr. Aberra is a consultant for Janssen and research investigator for Amgen, Janssen, and UCB. Other coauthors disclosed ties to numerous pharmaceutical companies.
Increasing dosages of mesalamine by 2.4 g/d for patients with quiescent ulcerative colitis can dramatically reduce concentrations of fecal calprotectin and ultimately decrease the likelihood of relapse, according to the results of a new Dose Escalation and Remission (DEAR) study published in the November issue of Clinical Gastroenterology and Hepatology (2014;12:1887-93.e3).
In an open-label, randomized controlled trial, 119 patients with quiescent ulcerative colitis (UC) in remission were screened for Simple Clinical Colitis Activity Index scores, fecal calprotectin (FC) of 50 mcg/gram, and intake of no more than 3 g of mesalamine per day between October 2008 and March 2012. These subjects were divided into four groups based on FC levels, with Group 1 comprising 58 representing a baseline of FC < 50 mcg/gram, and groups 2-4 having 61 individuals with elevated baseline FC levels. Group 1 patients were retained for an observational follow-up study but were not included in the randomized controlled trial.
Participants were either told to maintain their current medication regimen or were prescribed a multimatrix mesalamine of either 2.4 or 4.8 g/d for 6 weeks, depending on their initial FC levels, after which point FC levels were taken. Participants in Group 2 with FC < 50 mcg/g were kept in an observational follow-up study group, while the rest of the participants were randomly assigned to continue their 2.4 g/d regimen, or increase their intake to 4.8 g/d.
Individuals in Group 3 with FC levels greater than 50 mcg/g were randomly assigned to either the 2.4- or 4.8-g/d regimen for 6 weeks; after that point, those whose FC levels had decreased below 50 continued with their regimen while those whose FC levels remained high were upped to 4.8 g/d. Subjects in Group 4 who were not already on a multimatrix mesalamine regimen and whose FC levels were above 50 mcg/g were randomized to continue their medication or add 2.4 g/d of mesalamine; after 6 weeks, if FC levels remained above 50, the dosage was increased to 4.8 g/d.
The primary outcome of continued remission with FC no greater than 50 mcg/g was observed in 3.8% of controls and 26.9% of participants in the dose escalation group (P = .0496). Others within the same group experienced secondary outcomes of FC levels below 100 mcg/gram (P = .04) and below 200 mcg/g (P = .005). Furthermore, among patients who were in remission during the randomization phase of the study, relapse occurred far sooner in those whose FC was above 200 mcg/g than those whose FC levels were below that mark.
“Our study adds to the evidence supporting FC concentration as a valid biomarker for UC,” said lead authors Dr. Mark T. Osterman and Dr. Faten N. Aberra of the University of Pennsylvania’s department of medicine, Philadelphia. “Perhaps more importantly, our results offer a novel way to use FC testing to positively impact outcomes in UC. We demonstrated efficacy of intervention to reduce FC (a surrogate for mucosal inflammation) before symptoms develop in patients at potentially increased risk of relapse.”
Dr. Osterman is a consultant for AbbVie, Elan, Janssen, and UCB and receives research funding from UCB. Dr. Aberra is a consultant for Janssen and research investigator for Amgen, Janssen, and UCB. Other coauthors disclosed ties to numerous pharmaceutical companies.
Increasing dosages of mesalamine by 2.4 g/d for patients with quiescent ulcerative colitis can dramatically reduce concentrations of fecal calprotectin and ultimately decrease the likelihood of relapse, according to the results of a new Dose Escalation and Remission (DEAR) study published in the November issue of Clinical Gastroenterology and Hepatology (2014;12:1887-93.e3).
In an open-label, randomized controlled trial, 119 patients with quiescent ulcerative colitis (UC) in remission were screened for Simple Clinical Colitis Activity Index scores, fecal calprotectin (FC) of 50 mcg/gram, and intake of no more than 3 g of mesalamine per day between October 2008 and March 2012. These subjects were divided into four groups based on FC levels, with Group 1 comprising 58 representing a baseline of FC < 50 mcg/gram, and groups 2-4 having 61 individuals with elevated baseline FC levels. Group 1 patients were retained for an observational follow-up study but were not included in the randomized controlled trial.
Participants were either told to maintain their current medication regimen or were prescribed a multimatrix mesalamine of either 2.4 or 4.8 g/d for 6 weeks, depending on their initial FC levels, after which point FC levels were taken. Participants in Group 2 with FC < 50 mcg/g were kept in an observational follow-up study group, while the rest of the participants were randomly assigned to continue their 2.4 g/d regimen, or increase their intake to 4.8 g/d.
Individuals in Group 3 with FC levels greater than 50 mcg/g were randomly assigned to either the 2.4- or 4.8-g/d regimen for 6 weeks; after that point, those whose FC levels had decreased below 50 continued with their regimen while those whose FC levels remained high were upped to 4.8 g/d. Subjects in Group 4 who were not already on a multimatrix mesalamine regimen and whose FC levels were above 50 mcg/g were randomized to continue their medication or add 2.4 g/d of mesalamine; after 6 weeks, if FC levels remained above 50, the dosage was increased to 4.8 g/d.
The primary outcome of continued remission with FC no greater than 50 mcg/g was observed in 3.8% of controls and 26.9% of participants in the dose escalation group (P = .0496). Others within the same group experienced secondary outcomes of FC levels below 100 mcg/gram (P = .04) and below 200 mcg/g (P = .005). Furthermore, among patients who were in remission during the randomization phase of the study, relapse occurred far sooner in those whose FC was above 200 mcg/g than those whose FC levels were below that mark.
“Our study adds to the evidence supporting FC concentration as a valid biomarker for UC,” said lead authors Dr. Mark T. Osterman and Dr. Faten N. Aberra of the University of Pennsylvania’s department of medicine, Philadelphia. “Perhaps more importantly, our results offer a novel way to use FC testing to positively impact outcomes in UC. We demonstrated efficacy of intervention to reduce FC (a surrogate for mucosal inflammation) before symptoms develop in patients at potentially increased risk of relapse.”
Dr. Osterman is a consultant for AbbVie, Elan, Janssen, and UCB and receives research funding from UCB. Dr. Aberra is a consultant for Janssen and research investigator for Amgen, Janssen, and UCB. Other coauthors disclosed ties to numerous pharmaceutical companies.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Increasing mesalamine dosage by 2.4 g/d significantly decreases risk of relapse for patients with UC.
Major finding: 26.9% of subjects with dose increase experienced drop in fecal calprotectin level below 50 mcg/g.
Data source: Dose Escalation and Remission (DEAR) study; open-label, randomized controlled trial.
Disclosures: Dr. Osterman is a consultant for AbbVie, Elan, Janssen, and UCB and receives research funding from UCB. Dr. Aberra is a consultant for Janssen and research investigator for Amgen, Janssen, and UCB. Other coauthors disclosed ties to numerous pharmaceutical companies.
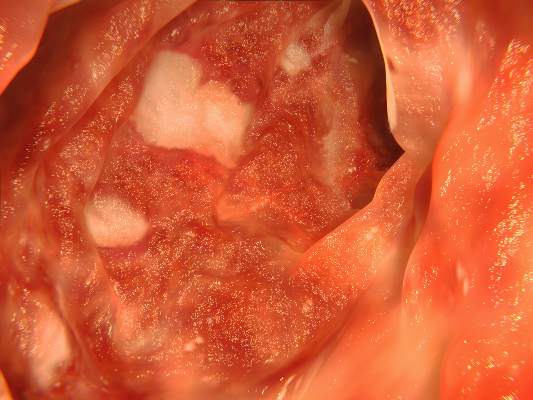
Vedolizumab infection concerns diminish with growing data
PHILADELPHIA – In long-term follow-up for the treatment of inflammatory bowel disease, the novel monoclonal antibody vedolizumab continues to remain free of serious infections or other signs of major complications from altered immune function, according to pooled data presented at the annual meeting of the American College of Gastroenterology.
Over the course of follow-up, which now extends to 104 weeks in these trials, called GEMINI 1 and GEMINI 2, overall infection rates have been about the same in the vedolizumab and placebo arms even when corticosteroids or immunosuppressive agents are used concurrently, according to Dr. Edward V. Loftus Jr., professor of medicine at the Mayo Clinic, Rochester, Minn.

There is particular focus on the safety of vedolizumab, because of the theoretical risks of biologics overall and vedolizumab specifically. Vedolizumab has been shown to provide sustained remissions relative to placebo in both Crohn’s disease and ulcerative colitis in the previously presented and updated GEMINI trials, but this agent, like other biologics, alters mediators of immune function.
The follow-up summarized at the ACG meeting, which evaluated risk of adverse events of vedolizumab alone or in combination with other inflammatory bowel disease (IBD) agents that affect immune function, suggests that the risk of adverse events is low, but the conclusion is not definitive.
“There was not a signal of more side effects with combination therapy, which is reassuring. However, the study is limited by the number of adverse events available to analyze,” said Dr. David T. Rubin, section chief of gastroenterology, hepatology, and nutrition at the University of Chicago. The likelihood of a high risk of adverse events is diminishing as follow-up and patient sample size increase, but Dr. Rubin suggested that clinicians should monitor any IBD patient taking an immunosuppressive agent for infectious complications.
“At the University of Chicago IBD Center, we have now treated more than 100 patients and are studying this therapy to better understand its performance in postmarket,” Dr. Rubin said in an interview. “So far, we have made similar observations to the clinical trials and to those made by Dr. Loftus here at the ACG.”
Unlike tumor necrosis factor (TNF) inhibitors, which were the first biologics to be licensed for use in IBD, vedolizumab inhibits the migration of leukocytes in the GI tract by preventing the alpha4beta7 subunit from binding to the adhesion molecule MAdCAM-1. It has been unclear whether or to what degree vedolizumab shares the known risk, albeit small, of inducing opportunistic infections observed with TNF inhibitors.
The most recent data continue to suggest that there is no greater risk of infection or other complications of immune function for vedolizumab than other biologics, and the risk may be lower. In the pooled GEMINI data with 1,434 patients followed during induction and maintenance phases with data in some groups out to 104 weeks, the overall percentages of infection and the individual infections rates have been “similar,” Dr. Loftus reported.
“The exception has been nasopharyngitis, which was less common in the placebo group,” said Dr. Loftus, providing relative rates of approximately 13% and 9% for vedolizumab and placebo, respectively. He noted, however, that the length of follow-up has been longer in the vedolizumab relative to the placebo groups. Rates of tuberculosis, Clostridium difficile colitis, and other serious infections have not differed significantly.
In addition, there has been no association between vedolizumab and progressive multifocal leukoencephalopathy (PML) so far. Concern about a risk for PML was raised by the relationship of vedolizumab to natalizumab, which blocks the same adhesion compounds and has been associated with PML. The absence of PML in follow-up so far is consistent with evidence that the activity of vedolizumab, unlike the activity of natalizumab, is largely or entirely confined to the GI tract.
No multicenter, randomized trials have yet to be conducted comparing either the efficacy or safety of vedolizumab, which was approved in May 2014 for the treatment of moderate to severe Crohn’s disease and ulcerative colitis, to other biologics used in IBD.
PHILADELPHIA – In long-term follow-up for the treatment of inflammatory bowel disease, the novel monoclonal antibody vedolizumab continues to remain free of serious infections or other signs of major complications from altered immune function, according to pooled data presented at the annual meeting of the American College of Gastroenterology.
Over the course of follow-up, which now extends to 104 weeks in these trials, called GEMINI 1 and GEMINI 2, overall infection rates have been about the same in the vedolizumab and placebo arms even when corticosteroids or immunosuppressive agents are used concurrently, according to Dr. Edward V. Loftus Jr., professor of medicine at the Mayo Clinic, Rochester, Minn.
There is particular focus on the safety of vedolizumab, because of the theoretical risks of biologics overall and vedolizumab specifically. Vedolizumab has been shown to provide sustained remissions relative to placebo in both Crohn’s disease and ulcerative colitis in the previously presented and updated GEMINI trials, but this agent, like other biologics, alters mediators of immune function.
The follow-up summarized at the ACG meeting, which evaluated risk of adverse events of vedolizumab alone or in combination with other inflammatory bowel disease (IBD) agents that affect immune function, suggests that the risk of adverse events is low, but the conclusion is not definitive.
“There was not a signal of more side effects with combination therapy, which is reassuring. However, the study is limited by the number of adverse events available to analyze,” said Dr. David T. Rubin, section chief of gastroenterology, hepatology, and nutrition at the University of Chicago. The likelihood of a high risk of adverse events is diminishing as follow-up and patient sample size increase, but Dr. Rubin suggested that clinicians should monitor any IBD patient taking an immunosuppressive agent for infectious complications.
“At the University of Chicago IBD Center, we have now treated more than 100 patients and are studying this therapy to better understand its performance in postmarket,” Dr. Rubin said in an interview. “So far, we have made similar observations to the clinical trials and to those made by Dr. Loftus here at the ACG.”
Unlike tumor necrosis factor (TNF) inhibitors, which were the first biologics to be licensed for use in IBD, vedolizumab inhibits the migration of leukocytes in the GI tract by preventing the alpha4beta7 subunit from binding to the adhesion molecule MAdCAM-1. It has been unclear whether or to what degree vedolizumab shares the known risk, albeit small, of inducing opportunistic infections observed with TNF inhibitors.
The most recent data continue to suggest that there is no greater risk of infection or other complications of immune function for vedolizumab than other biologics, and the risk may be lower. In the pooled GEMINI data with 1,434 patients followed during induction and maintenance phases with data in some groups out to 104 weeks, the overall percentages of infection and the individual infections rates have been “similar,” Dr. Loftus reported.
“The exception has been nasopharyngitis, which was less common in the placebo group,” said Dr. Loftus, providing relative rates of approximately 13% and 9% for vedolizumab and placebo, respectively. He noted, however, that the length of follow-up has been longer in the vedolizumab relative to the placebo groups. Rates of tuberculosis, Clostridium difficile colitis, and other serious infections have not differed significantly.
In addition, there has been no association between vedolizumab and progressive multifocal leukoencephalopathy (PML) so far. Concern about a risk for PML was raised by the relationship of vedolizumab to natalizumab, which blocks the same adhesion compounds and has been associated with PML. The absence of PML in follow-up so far is consistent with evidence that the activity of vedolizumab, unlike the activity of natalizumab, is largely or entirely confined to the GI tract.
No multicenter, randomized trials have yet to be conducted comparing either the efficacy or safety of vedolizumab, which was approved in May 2014 for the treatment of moderate to severe Crohn’s disease and ulcerative colitis, to other biologics used in IBD.
PHILADELPHIA – In long-term follow-up for the treatment of inflammatory bowel disease, the novel monoclonal antibody vedolizumab continues to remain free of serious infections or other signs of major complications from altered immune function, according to pooled data presented at the annual meeting of the American College of Gastroenterology.
Over the course of follow-up, which now extends to 104 weeks in these trials, called GEMINI 1 and GEMINI 2, overall infection rates have been about the same in the vedolizumab and placebo arms even when corticosteroids or immunosuppressive agents are used concurrently, according to Dr. Edward V. Loftus Jr., professor of medicine at the Mayo Clinic, Rochester, Minn.
There is particular focus on the safety of vedolizumab, because of the theoretical risks of biologics overall and vedolizumab specifically. Vedolizumab has been shown to provide sustained remissions relative to placebo in both Crohn’s disease and ulcerative colitis in the previously presented and updated GEMINI trials, but this agent, like other biologics, alters mediators of immune function.
The follow-up summarized at the ACG meeting, which evaluated risk of adverse events of vedolizumab alone or in combination with other inflammatory bowel disease (IBD) agents that affect immune function, suggests that the risk of adverse events is low, but the conclusion is not definitive.
“There was not a signal of more side effects with combination therapy, which is reassuring. However, the study is limited by the number of adverse events available to analyze,” said Dr. David T. Rubin, section chief of gastroenterology, hepatology, and nutrition at the University of Chicago. The likelihood of a high risk of adverse events is diminishing as follow-up and patient sample size increase, but Dr. Rubin suggested that clinicians should monitor any IBD patient taking an immunosuppressive agent for infectious complications.
“At the University of Chicago IBD Center, we have now treated more than 100 patients and are studying this therapy to better understand its performance in postmarket,” Dr. Rubin said in an interview. “So far, we have made similar observations to the clinical trials and to those made by Dr. Loftus here at the ACG.”
Unlike tumor necrosis factor (TNF) inhibitors, which were the first biologics to be licensed for use in IBD, vedolizumab inhibits the migration of leukocytes in the GI tract by preventing the alpha4beta7 subunit from binding to the adhesion molecule MAdCAM-1. It has been unclear whether or to what degree vedolizumab shares the known risk, albeit small, of inducing opportunistic infections observed with TNF inhibitors.
The most recent data continue to suggest that there is no greater risk of infection or other complications of immune function for vedolizumab than other biologics, and the risk may be lower. In the pooled GEMINI data with 1,434 patients followed during induction and maintenance phases with data in some groups out to 104 weeks, the overall percentages of infection and the individual infections rates have been “similar,” Dr. Loftus reported.
“The exception has been nasopharyngitis, which was less common in the placebo group,” said Dr. Loftus, providing relative rates of approximately 13% and 9% for vedolizumab and placebo, respectively. He noted, however, that the length of follow-up has been longer in the vedolizumab relative to the placebo groups. Rates of tuberculosis, Clostridium difficile colitis, and other serious infections have not differed significantly.
In addition, there has been no association between vedolizumab and progressive multifocal leukoencephalopathy (PML) so far. Concern about a risk for PML was raised by the relationship of vedolizumab to natalizumab, which blocks the same adhesion compounds and has been associated with PML. The absence of PML in follow-up so far is consistent with evidence that the activity of vedolizumab, unlike the activity of natalizumab, is largely or entirely confined to the GI tract.
No multicenter, randomized trials have yet to be conducted comparing either the efficacy or safety of vedolizumab, which was approved in May 2014 for the treatment of moderate to severe Crohn’s disease and ulcerative colitis, to other biologics used in IBD.
AT ACG 2014
Key clinical point: Vedolizumab, a novel biologic for the treatment of inflammatory bowel disease, has yet to be associated with serious risks of infection or other signs of impaired immune function.
Major finding: In pooled analysis from phase III studies of vedolizumab with up to 2 years of follow-up, rates of infection have been similar with only nasopharyngitis occurring somewhat more frequently.
Data source: Pooled analysis of initial and follow-up data from phase III studies.
Disclosures: Dr. Edward V. Loftus Jr. reports research support and consultancy relationships with more than 10 pharmaceutical companies, including Takeda, the sponsor of the phase III studies he evaluated.

Psoriasiform lesions linked to anti-TNF treatment
VIENNA – Patients treated with an anti–tumor necrosis factor drug had a 5% annual rate of developing one or more psoriasiform skin lesions in a review of more than 400 patients who received these drugs to treat inflammatory bowel disease at a single center in Rome.
The cohort review confirmed prior reports that smoking is a risk factor for the appearance of psoriasislike skin lesions on patients being treated with an anti–tumor necrosis factor (TNF) drug, such as infliximab (Remicade) or adalimumab (Humira). The Rome experience also showed that in 28 of the 42 patients who developed a psoriasiform lesion, the eruption responded to topical treatment without need to stop or change the anti-TNF regimen. Ten of the 42 patients ultimately had to stop their anti-TNF regimen, Dr. Daniela Pugliese said at the United European Gastroenterology Global Congress.

“There have been several case reports of this, but this is the largest cohort review yet reported by one center,” said Dr. Pugliese, a gastroenterologist in the inflammatory bowel disease (IBD) unit of Complesso Integrato Columbus, Catholic University in Rome.
The review included 402 patients treated with an anti-TNF drug at the unit during 2008-2013, with a median follow-up of 17 months. During follow-up, 42 patients developed a psoriasiform lesion with a biopsy-proven diagnosis, a rate of 5 cases/100 person-years on anti-TNF treatment. The IBD patients averaged 40 years old, and 60% had Crohn’s disease and 40% had ulcerative colitis. About 60% received infliximab treatment, and 40% adalimumab. Most lesions appeared in predilection sites, as well as on palmoplantar surfaces or on the scalp; nearly half the patients had lesions in two or more locations.
A multivariate regression analysis that assessed several demographic and clinical features of all 402 patients identified two parameters that significantly linked with lesion development. Smoking linked with a greater than twofold increased risk for having a psoriasiform lesion (78 of the patients, 19%, smoked), and concurrent treatment with a thiopurine such as azathioprine, linked with a 67% reduced rate of lesion development (85 patients, 21%, were on concurrent thiopurine treatment). All IBD patients seen at the Rome unit who smoked were counseled regarding smoking cessation, Dr. Pugliese said in an interview.
Among the patients who did not respond to topical treatment, four received some benefit from starting treatment with ustekinumab (Stelara), which especially benefited patients who were otherwise difficult to treat, Dr. Pugliese said. Other patients benefited from starting treatment with cyclosporine, methotrexate, or a transient treatment with an oral steroid. Two patients who developed lesions on infliximab switched to adalimumab, and two other patients who had lesions on adalimumab switched to infliximab.

Dr. Pugliese and her associates did not have a good explanation of why patients on anti-TNF drugs develop these lesions, which Dr. Pugliese called “paradoxical.” One possible etiology is that inhibition of TNF-alpha results in uncontrolled production of interferon-alpha by plasmacytoid dendritic cells and this then triggers the psoriasiform eruptions.
A key element in managing these lesions may be early detection and topical treatment while they remain small, commented Dr. C. Janneke van der Woude, head of the IBD unit at Erasmus University Medical Center in Rotterdam, the Netherlands. “Every time we see an IBD patient who is on an anti-TNF drug, we ask whether they have had any itching, allergic reaction, arthritis, eye problem, or headache,” to facilitate early detection of an adverse effect from treatment, she said in an interview.
Dr. Pugliese had no disclosures. Dr. van der Woude said that she has been an adviser to Dr Falk, AbbVie, Janssen, Johnson & Johnson, and Cosmo.
On Twitter@mitchelzoler
VIENNA – Patients treated with an anti–tumor necrosis factor drug had a 5% annual rate of developing one or more psoriasiform skin lesions in a review of more than 400 patients who received these drugs to treat inflammatory bowel disease at a single center in Rome.
The cohort review confirmed prior reports that smoking is a risk factor for the appearance of psoriasislike skin lesions on patients being treated with an anti–tumor necrosis factor (TNF) drug, such as infliximab (Remicade) or adalimumab (Humira). The Rome experience also showed that in 28 of the 42 patients who developed a psoriasiform lesion, the eruption responded to topical treatment without need to stop or change the anti-TNF regimen. Ten of the 42 patients ultimately had to stop their anti-TNF regimen, Dr. Daniela Pugliese said at the United European Gastroenterology Global Congress.
“There have been several case reports of this, but this is the largest cohort review yet reported by one center,” said Dr. Pugliese, a gastroenterologist in the inflammatory bowel disease (IBD) unit of Complesso Integrato Columbus, Catholic University in Rome.
The review included 402 patients treated with an anti-TNF drug at the unit during 2008-2013, with a median follow-up of 17 months. During follow-up, 42 patients developed a psoriasiform lesion with a biopsy-proven diagnosis, a rate of 5 cases/100 person-years on anti-TNF treatment. The IBD patients averaged 40 years old, and 60% had Crohn’s disease and 40% had ulcerative colitis. About 60% received infliximab treatment, and 40% adalimumab. Most lesions appeared in predilection sites, as well as on palmoplantar surfaces or on the scalp; nearly half the patients had lesions in two or more locations.
A multivariate regression analysis that assessed several demographic and clinical features of all 402 patients identified two parameters that significantly linked with lesion development. Smoking linked with a greater than twofold increased risk for having a psoriasiform lesion (78 of the patients, 19%, smoked), and concurrent treatment with a thiopurine such as azathioprine, linked with a 67% reduced rate of lesion development (85 patients, 21%, were on concurrent thiopurine treatment). All IBD patients seen at the Rome unit who smoked were counseled regarding smoking cessation, Dr. Pugliese said in an interview.
Among the patients who did not respond to topical treatment, four received some benefit from starting treatment with ustekinumab (Stelara), which especially benefited patients who were otherwise difficult to treat, Dr. Pugliese said. Other patients benefited from starting treatment with cyclosporine, methotrexate, or a transient treatment with an oral steroid. Two patients who developed lesions on infliximab switched to adalimumab, and two other patients who had lesions on adalimumab switched to infliximab.
Dr. Pugliese and her associates did not have a good explanation of why patients on anti-TNF drugs develop these lesions, which Dr. Pugliese called “paradoxical.” One possible etiology is that inhibition of TNF-alpha results in uncontrolled production of interferon-alpha by plasmacytoid dendritic cells and this then triggers the psoriasiform eruptions.
A key element in managing these lesions may be early detection and topical treatment while they remain small, commented Dr. C. Janneke van der Woude, head of the IBD unit at Erasmus University Medical Center in Rotterdam, the Netherlands. “Every time we see an IBD patient who is on an anti-TNF drug, we ask whether they have had any itching, allergic reaction, arthritis, eye problem, or headache,” to facilitate early detection of an adverse effect from treatment, she said in an interview.
Dr. Pugliese had no disclosures. Dr. van der Woude said that she has been an adviser to Dr Falk, AbbVie, Janssen, Johnson & Johnson, and Cosmo.
On Twitter@mitchelzoler
VIENNA – Patients treated with an anti–tumor necrosis factor drug had a 5% annual rate of developing one or more psoriasiform skin lesions in a review of more than 400 patients who received these drugs to treat inflammatory bowel disease at a single center in Rome.
The cohort review confirmed prior reports that smoking is a risk factor for the appearance of psoriasislike skin lesions on patients being treated with an anti–tumor necrosis factor (TNF) drug, such as infliximab (Remicade) or adalimumab (Humira). The Rome experience also showed that in 28 of the 42 patients who developed a psoriasiform lesion, the eruption responded to topical treatment without need to stop or change the anti-TNF regimen. Ten of the 42 patients ultimately had to stop their anti-TNF regimen, Dr. Daniela Pugliese said at the United European Gastroenterology Global Congress.
“There have been several case reports of this, but this is the largest cohort review yet reported by one center,” said Dr. Pugliese, a gastroenterologist in the inflammatory bowel disease (IBD) unit of Complesso Integrato Columbus, Catholic University in Rome.
The review included 402 patients treated with an anti-TNF drug at the unit during 2008-2013, with a median follow-up of 17 months. During follow-up, 42 patients developed a psoriasiform lesion with a biopsy-proven diagnosis, a rate of 5 cases/100 person-years on anti-TNF treatment. The IBD patients averaged 40 years old, and 60% had Crohn’s disease and 40% had ulcerative colitis. About 60% received infliximab treatment, and 40% adalimumab. Most lesions appeared in predilection sites, as well as on palmoplantar surfaces or on the scalp; nearly half the patients had lesions in two or more locations.
A multivariate regression analysis that assessed several demographic and clinical features of all 402 patients identified two parameters that significantly linked with lesion development. Smoking linked with a greater than twofold increased risk for having a psoriasiform lesion (78 of the patients, 19%, smoked), and concurrent treatment with a thiopurine such as azathioprine, linked with a 67% reduced rate of lesion development (85 patients, 21%, were on concurrent thiopurine treatment). All IBD patients seen at the Rome unit who smoked were counseled regarding smoking cessation, Dr. Pugliese said in an interview.
Among the patients who did not respond to topical treatment, four received some benefit from starting treatment with ustekinumab (Stelara), which especially benefited patients who were otherwise difficult to treat, Dr. Pugliese said. Other patients benefited from starting treatment with cyclosporine, methotrexate, or a transient treatment with an oral steroid. Two patients who developed lesions on infliximab switched to adalimumab, and two other patients who had lesions on adalimumab switched to infliximab.
Dr. Pugliese and her associates did not have a good explanation of why patients on anti-TNF drugs develop these lesions, which Dr. Pugliese called “paradoxical.” One possible etiology is that inhibition of TNF-alpha results in uncontrolled production of interferon-alpha by plasmacytoid dendritic cells and this then triggers the psoriasiform eruptions.
A key element in managing these lesions may be early detection and topical treatment while they remain small, commented Dr. C. Janneke van der Woude, head of the IBD unit at Erasmus University Medical Center in Rotterdam, the Netherlands. “Every time we see an IBD patient who is on an anti-TNF drug, we ask whether they have had any itching, allergic reaction, arthritis, eye problem, or headache,” to facilitate early detection of an adverse effect from treatment, she said in an interview.
Dr. Pugliese had no disclosures. Dr. van der Woude said that she has been an adviser to Dr Falk, AbbVie, Janssen, Johnson & Johnson, and Cosmo.
On Twitter@mitchelzoler
AT UEG WEEK VIENNA 2014
Key clinical point: Inflammatory bowel disease patients on anti–tumor necrosis factor treatment often develop psoriasiform skin lesions.
Major finding: Patients receiving an anti-TNF drug developed psoriasiform lesions at an annual rate of 5%.
Data source: Retrospective review of 402 patients with inflammatory bowel disease treated with an anti-TNF drug at one center in Rome.
Disclosures: Dr. Pugliese had no disclosures. Dr. van der Woude said that she has been an adviser to Dr Falk, AbbVie, Janssen, Johnson & Johnson, and Cosmo.

Uncomplicated diverticulitis patients safely avoid routine antibiotics
VIENNA – Patients with first-episode, uncomplicated diverticulitis can safely be managed by observation alone without routine antibiotic treatment, based on results from a prospective, controlled, multicenter Dutch trial with 528 patients.
Coupled with similar results from a 2012 Swedish randomized trial, the new findings show that an observational approach without up-front antibiotic treatment is safe and effective for routine practice for patients with a modified Hinchey stage 1 classification, the most common diverticulitis presentation, Dr. Lidewine Daniels said at the United European Gastroenterology Global Congress.

The 2012 trial, done in Sweden and Iceland, randomized 623 patients with acute, uncomplicated diverticulitis, and found no statistically significant difference in the rate of recovery without complications during 12 months of follow-up, regardless of whether or not patients received routine antibiotic treatment at the time of initial diagnosis (Br. J. Surg. 2012;99:532-9).
This approach “can now be adopted into guidelines,” said Dr. Daniels, a researcher in the surgery department at the Academic Medical Center in Amsterdam. This would shift current practice recommendations, such as those published earlier this year by the American College of Colon and Rectal Surgeons (Dis. Colon Rectum 2014;57:284-94), which have generally called for routine antibiotic management of these types of diverticulitis patients, Dr. Daniels said.
The DIABOLO (Multicenter Randomized Clinical Trial Investigating the Cost-effectiveness of Treatment Strategies With or Without Antibiotics for Uncomplicated Acute Diverticulitis) trial enrolled patients with CT-proven, left-sided, first-episode, uncomplicated diverticulitis at 22 Dutch centers. All patients were classified as having stage 1 disease by the modified Hinchey criteria (Int. J. Colorectal Dis. 2012;27:207-14), with about 92% of patients classified as Hinchey 1a.
The 266 patients randomized to initial antibiotic treatment began at least 2 days on intravenous treatment with amoxicillin and clavulanic acid, followed by a switch to oral delivery of the same combination when appropriate for a total of 10 days on the antibiotic.
Patients remained hospitalized until they switched from intravenous to oral treatment. The 262 patients randomized to no initial antibiotic treatment were hospitalized and observed, and received supportive treatment until they were judged ready for discharge by being able to eat a normal diet, had a temperature of less than 38 °C, and had a self-rated pain score of less than 4 on a visual analog scale of 0-10 without use of pain medication; in addition, the patient’s agreement was needed for discharge.
During 6 months of follow-up, the median time to complete recovery – defined as the criteria for hospital discharge plus a return to normal activity – occurred after a median of 12 days among patients in the antibiotic group and 14 days among those in the observation group, a difference that was not statistically significant for the study’s primary endpoint.
The results also showed no statistically significant differences between the two study arms for almost all the other outcomes assessed, including need for readmission, complications, need for surgery, morbidities, serious morbidities, and deaths. The only significant differences in outcomes were a decreased rate of hospitalization at the time of initial treatment among patients in the observation-only arm, and fewer hospitalized days among the observation-only patients.
“Observational treatment is without short- or long-term repercussions,” Dr. Daniels concluded.
Dr. Daniels had no disclosures.
On Twitter @mitchelzoler
VIENNA – Patients with first-episode, uncomplicated diverticulitis can safely be managed by observation alone without routine antibiotic treatment, based on results from a prospective, controlled, multicenter Dutch trial with 528 patients.
Coupled with similar results from a 2012 Swedish randomized trial, the new findings show that an observational approach without up-front antibiotic treatment is safe and effective for routine practice for patients with a modified Hinchey stage 1 classification, the most common diverticulitis presentation, Dr. Lidewine Daniels said at the United European Gastroenterology Global Congress.
The 2012 trial, done in Sweden and Iceland, randomized 623 patients with acute, uncomplicated diverticulitis, and found no statistically significant difference in the rate of recovery without complications during 12 months of follow-up, regardless of whether or not patients received routine antibiotic treatment at the time of initial diagnosis (Br. J. Surg. 2012;99:532-9).
This approach “can now be adopted into guidelines,” said Dr. Daniels, a researcher in the surgery department at the Academic Medical Center in Amsterdam. This would shift current practice recommendations, such as those published earlier this year by the American College of Colon and Rectal Surgeons (Dis. Colon Rectum 2014;57:284-94), which have generally called for routine antibiotic management of these types of diverticulitis patients, Dr. Daniels said.
The DIABOLO (Multicenter Randomized Clinical Trial Investigating the Cost-effectiveness of Treatment Strategies With or Without Antibiotics for Uncomplicated Acute Diverticulitis) trial enrolled patients with CT-proven, left-sided, first-episode, uncomplicated diverticulitis at 22 Dutch centers. All patients were classified as having stage 1 disease by the modified Hinchey criteria (Int. J. Colorectal Dis. 2012;27:207-14), with about 92% of patients classified as Hinchey 1a.
The 266 patients randomized to initial antibiotic treatment began at least 2 days on intravenous treatment with amoxicillin and clavulanic acid, followed by a switch to oral delivery of the same combination when appropriate for a total of 10 days on the antibiotic.
Patients remained hospitalized until they switched from intravenous to oral treatment. The 262 patients randomized to no initial antibiotic treatment were hospitalized and observed, and received supportive treatment until they were judged ready for discharge by being able to eat a normal diet, had a temperature of less than 38 °C, and had a self-rated pain score of less than 4 on a visual analog scale of 0-10 without use of pain medication; in addition, the patient’s agreement was needed for discharge.
During 6 months of follow-up, the median time to complete recovery – defined as the criteria for hospital discharge plus a return to normal activity – occurred after a median of 12 days among patients in the antibiotic group and 14 days among those in the observation group, a difference that was not statistically significant for the study’s primary endpoint.
The results also showed no statistically significant differences between the two study arms for almost all the other outcomes assessed, including need for readmission, complications, need for surgery, morbidities, serious morbidities, and deaths. The only significant differences in outcomes were a decreased rate of hospitalization at the time of initial treatment among patients in the observation-only arm, and fewer hospitalized days among the observation-only patients.
“Observational treatment is without short- or long-term repercussions,” Dr. Daniels concluded.
Dr. Daniels had no disclosures.
On Twitter @mitchelzoler
VIENNA – Patients with first-episode, uncomplicated diverticulitis can safely be managed by observation alone without routine antibiotic treatment, based on results from a prospective, controlled, multicenter Dutch trial with 528 patients.
Coupled with similar results from a 2012 Swedish randomized trial, the new findings show that an observational approach without up-front antibiotic treatment is safe and effective for routine practice for patients with a modified Hinchey stage 1 classification, the most common diverticulitis presentation, Dr. Lidewine Daniels said at the United European Gastroenterology Global Congress.
The 2012 trial, done in Sweden and Iceland, randomized 623 patients with acute, uncomplicated diverticulitis, and found no statistically significant difference in the rate of recovery without complications during 12 months of follow-up, regardless of whether or not patients received routine antibiotic treatment at the time of initial diagnosis (Br. J. Surg. 2012;99:532-9).
This approach “can now be adopted into guidelines,” said Dr. Daniels, a researcher in the surgery department at the Academic Medical Center in Amsterdam. This would shift current practice recommendations, such as those published earlier this year by the American College of Colon and Rectal Surgeons (Dis. Colon Rectum 2014;57:284-94), which have generally called for routine antibiotic management of these types of diverticulitis patients, Dr. Daniels said.
The DIABOLO (Multicenter Randomized Clinical Trial Investigating the Cost-effectiveness of Treatment Strategies With or Without Antibiotics for Uncomplicated Acute Diverticulitis) trial enrolled patients with CT-proven, left-sided, first-episode, uncomplicated diverticulitis at 22 Dutch centers. All patients were classified as having stage 1 disease by the modified Hinchey criteria (Int. J. Colorectal Dis. 2012;27:207-14), with about 92% of patients classified as Hinchey 1a.
The 266 patients randomized to initial antibiotic treatment began at least 2 days on intravenous treatment with amoxicillin and clavulanic acid, followed by a switch to oral delivery of the same combination when appropriate for a total of 10 days on the antibiotic.
Patients remained hospitalized until they switched from intravenous to oral treatment. The 262 patients randomized to no initial antibiotic treatment were hospitalized and observed, and received supportive treatment until they were judged ready for discharge by being able to eat a normal diet, had a temperature of less than 38 °C, and had a self-rated pain score of less than 4 on a visual analog scale of 0-10 without use of pain medication; in addition, the patient’s agreement was needed for discharge.
During 6 months of follow-up, the median time to complete recovery – defined as the criteria for hospital discharge plus a return to normal activity – occurred after a median of 12 days among patients in the antibiotic group and 14 days among those in the observation group, a difference that was not statistically significant for the study’s primary endpoint.
The results also showed no statistically significant differences between the two study arms for almost all the other outcomes assessed, including need for readmission, complications, need for surgery, morbidities, serious morbidities, and deaths. The only significant differences in outcomes were a decreased rate of hospitalization at the time of initial treatment among patients in the observation-only arm, and fewer hospitalized days among the observation-only patients.
“Observational treatment is without short- or long-term repercussions,” Dr. Daniels concluded.
Dr. Daniels had no disclosures.
On Twitter @mitchelzoler
AT UEG WEEK VIENNA 2014
Key clinical point: Observation-only worked as well as routine antibiotic treatment for patients with uncomplicated diverticulitis.
Major finding: Median time to full recovery was 12 days for antibiotic-treated patients and 14 days for observation-only patients.
Data source: The DIABOLO study, which randomized 528 patients with first-time, CT-proven, acute, uncomplicated diverticulitis at 22 Dutch centers.
Disclosures: Dr. Daniels had no disclosures.

FDA approves budesonide rectal foam for distal UC
A rectal foam formulation of 2% budesonide has been approved by the Food and Drug Administration for treating distal ulcerative colitis, the manufacturer, Salix Pharmaceuticals, has announced.
The approved indication is for the induction of remission in patients with mild to moderate distal ulcerative colitis extending up to 40 cm from the anal verge. The foam is administered rectally and “overcomes treatment limitations associated with currently approved therapies which are often ineffective due to insufficient distribution of active drug to the distal colon,” the company said in a statement announcing the final approval on Oct. 8.
The company will be marketing the product as Uceris.
The company also markets budesonide oral extended-release tablets under the same name.
A rectal foam formulation of 2% budesonide has been approved by the Food and Drug Administration for treating distal ulcerative colitis, the manufacturer, Salix Pharmaceuticals, has announced.
The approved indication is for the induction of remission in patients with mild to moderate distal ulcerative colitis extending up to 40 cm from the anal verge. The foam is administered rectally and “overcomes treatment limitations associated with currently approved therapies which are often ineffective due to insufficient distribution of active drug to the distal colon,” the company said in a statement announcing the final approval on Oct. 8.
The company will be marketing the product as Uceris.
The company also markets budesonide oral extended-release tablets under the same name.
A rectal foam formulation of 2% budesonide has been approved by the Food and Drug Administration for treating distal ulcerative colitis, the manufacturer, Salix Pharmaceuticals, has announced.
The approved indication is for the induction of remission in patients with mild to moderate distal ulcerative colitis extending up to 40 cm from the anal verge. The foam is administered rectally and “overcomes treatment limitations associated with currently approved therapies which are often ineffective due to insufficient distribution of active drug to the distal colon,” the company said in a statement announcing the final approval on Oct. 8.
The company will be marketing the product as Uceris.
The company also markets budesonide oral extended-release tablets under the same name.
Fidaxomicin appears safe, effective for CDAD in children
PHILADELPHIA – Fidaxomicin, an approved treatment for Clostridium difficile-associated diarrhea in adults, appears safe and effective in children, according to findings from an open-label study.
The overall clinical response rate was 92% in 38 children aged 11 months through 17 years who were treated with 32 mg/kg/day for 10 days. Although 74% of patients had at least one adverse event, most of the events were mild (45%) or moderate (21%) in severity and were the type of events that would be expected in children with moderate to severe underlying illness, Pam Sears, Ph.D. of Cubist Pharmaceuticals, San Diego, reported at an annual scientific meeting on infectious diseases.
The children included in this first pediatric study of fidaxomicin had diarrhea and tested positive for C. difficile toxin. Most had relatively complex medical histories; 24% had neoplasms and 79% had gastrointestinal disorders. They received either the commercial tablet formulation of fidaxomicin or an oral suspension.
Plasma concentrations were generally low, with a mean across the strata ranging from 8.87-16.6 ng/mL and metabolite OP-1118 levels that ranged from 27.5-130 ng/mL at 1-2 hours following the first dose. No age-related trends in concentrations were seen.
Fecal concentrations of fidaxomicin averaged 3,230 mcg/g with a trend toward higher concentrations in the youngest age group. This could be because samples from the youngest group were collected from diapers and could have become dehydrated, Dr. Sears explained the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
Of the children in the study with a clinical response, 29% experienced a relapse, but all but one of these children had a history of recurrent C. difficile-related diarrhea.
Fidaxomicin is a first-in class narrow spectrum macrocyclic antibiotic drug approved in the United States and several other countries for the treatment of C. difficile-associated diarrhea in adults.
These findings demonstrate that the pharmacokinetic profile in children is similar to that in adults – with low plasma levels and high fecal concentrations, and suggest that clinical response in children is also excellent, Dr. Sears said.
This study was funded by Cubist Pharmaceuticals. Dr. Sears is an employee of Cubist Pharmaceuticals.
PHILADELPHIA – Fidaxomicin, an approved treatment for Clostridium difficile-associated diarrhea in adults, appears safe and effective in children, according to findings from an open-label study.
The overall clinical response rate was 92% in 38 children aged 11 months through 17 years who were treated with 32 mg/kg/day for 10 days. Although 74% of patients had at least one adverse event, most of the events were mild (45%) or moderate (21%) in severity and were the type of events that would be expected in children with moderate to severe underlying illness, Pam Sears, Ph.D. of Cubist Pharmaceuticals, San Diego, reported at an annual scientific meeting on infectious diseases.
The children included in this first pediatric study of fidaxomicin had diarrhea and tested positive for C. difficile toxin. Most had relatively complex medical histories; 24% had neoplasms and 79% had gastrointestinal disorders. They received either the commercial tablet formulation of fidaxomicin or an oral suspension.
Plasma concentrations were generally low, with a mean across the strata ranging from 8.87-16.6 ng/mL and metabolite OP-1118 levels that ranged from 27.5-130 ng/mL at 1-2 hours following the first dose. No age-related trends in concentrations were seen.
Fecal concentrations of fidaxomicin averaged 3,230 mcg/g with a trend toward higher concentrations in the youngest age group. This could be because samples from the youngest group were collected from diapers and could have become dehydrated, Dr. Sears explained the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
Of the children in the study with a clinical response, 29% experienced a relapse, but all but one of these children had a history of recurrent C. difficile-related diarrhea.
Fidaxomicin is a first-in class narrow spectrum macrocyclic antibiotic drug approved in the United States and several other countries for the treatment of C. difficile-associated diarrhea in adults.
These findings demonstrate that the pharmacokinetic profile in children is similar to that in adults – with low plasma levels and high fecal concentrations, and suggest that clinical response in children is also excellent, Dr. Sears said.
This study was funded by Cubist Pharmaceuticals. Dr. Sears is an employee of Cubist Pharmaceuticals.
PHILADELPHIA – Fidaxomicin, an approved treatment for Clostridium difficile-associated diarrhea in adults, appears safe and effective in children, according to findings from an open-label study.
The overall clinical response rate was 92% in 38 children aged 11 months through 17 years who were treated with 32 mg/kg/day for 10 days. Although 74% of patients had at least one adverse event, most of the events were mild (45%) or moderate (21%) in severity and were the type of events that would be expected in children with moderate to severe underlying illness, Pam Sears, Ph.D. of Cubist Pharmaceuticals, San Diego, reported at an annual scientific meeting on infectious diseases.
The children included in this first pediatric study of fidaxomicin had diarrhea and tested positive for C. difficile toxin. Most had relatively complex medical histories; 24% had neoplasms and 79% had gastrointestinal disorders. They received either the commercial tablet formulation of fidaxomicin or an oral suspension.
Plasma concentrations were generally low, with a mean across the strata ranging from 8.87-16.6 ng/mL and metabolite OP-1118 levels that ranged from 27.5-130 ng/mL at 1-2 hours following the first dose. No age-related trends in concentrations were seen.
Fecal concentrations of fidaxomicin averaged 3,230 mcg/g with a trend toward higher concentrations in the youngest age group. This could be because samples from the youngest group were collected from diapers and could have become dehydrated, Dr. Sears explained the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
Of the children in the study with a clinical response, 29% experienced a relapse, but all but one of these children had a history of recurrent C. difficile-related diarrhea.
Fidaxomicin is a first-in class narrow spectrum macrocyclic antibiotic drug approved in the United States and several other countries for the treatment of C. difficile-associated diarrhea in adults.
These findings demonstrate that the pharmacokinetic profile in children is similar to that in adults – with low plasma levels and high fecal concentrations, and suggest that clinical response in children is also excellent, Dr. Sears said.
This study was funded by Cubist Pharmaceuticals. Dr. Sears is an employee of Cubist Pharmaceuticals.
Key clinical point: Fidaxomicin appear to have excellent clinical efficacy in children with C. difficile-associated diarrhea and a pharmacokinetic profile similar to that seen in adults.
Major finding: The overall clinical response rate was 92%.
Data source: An open-label study in 38 children.
Disclosures: This study was funded by Cubist Pharmaceuticals. Dr. Sears is an employee of Cubist Pharmaceuticals.