User login
FDA approves rivaroxaban for VTE prevention in hospitalized, acutely ill patients
The Food and Drug Administration has approved rivaroxaban (Xarelto) for the prevention of venous thromboembolism (VTE) in hospitalized, acutely ill patients at risk for thromboembolic complications who do not have a high bleeding risk, according to a release from Janssen.

FDA approval for the new indication is based on results from the phase 3 MAGELLAN and MARINER trials, which included more than 20,000 hospitalized, acutely ill patients. In MAGELLAN, rivaroxaban demonstrated noninferiority to enoxaparin, a low-molecular-weight heparin, in short-term usage, and it was superior over the long term, compared with short-term enoxaparin followed by placebo.
While VTE and VTE-related deaths were not reduced in MARINER, compared with placebo, patients who received rivaroxaban did see a significantly reduction in symptomatic VTE with a favorable safety profile.
According to the indication, rivaroxaban can be administered to patients during hospitalization and can be continued after discharge for 31-39 days. The safety profile in MAGELLAN and MARINER was consistent with that already seen, with the most common adverse event being bleeding.
The new indication is the eighth for rivaroxaban, the most of any direct oral anticoagulant; six of these are specifically for the treatment, prevention, and reduction in the risk of VTE recurrence.
“With this new approval, Xarelto as an oral-only option now has the potential to change how acutely ill medical patients are managed for the prevention of blood clots, both in the hospital and for an extended period after discharge,” said Alex C. Spyropoulos, MD, of Northwell Health at Lenox Hill Hospital, New York, and a member of the steering committee of the MAGELLAN trial.
Find the full press release on the Janssen website.
The Food and Drug Administration has approved rivaroxaban (Xarelto) for the prevention of venous thromboembolism (VTE) in hospitalized, acutely ill patients at risk for thromboembolic complications who do not have a high bleeding risk, according to a release from Janssen.
FDA approval for the new indication is based on results from the phase 3 MAGELLAN and MARINER trials, which included more than 20,000 hospitalized, acutely ill patients. In MAGELLAN, rivaroxaban demonstrated noninferiority to enoxaparin, a low-molecular-weight heparin, in short-term usage, and it was superior over the long term, compared with short-term enoxaparin followed by placebo.
While VTE and VTE-related deaths were not reduced in MARINER, compared with placebo, patients who received rivaroxaban did see a significantly reduction in symptomatic VTE with a favorable safety profile.
According to the indication, rivaroxaban can be administered to patients during hospitalization and can be continued after discharge for 31-39 days. The safety profile in MAGELLAN and MARINER was consistent with that already seen, with the most common adverse event being bleeding.
The new indication is the eighth for rivaroxaban, the most of any direct oral anticoagulant; six of these are specifically for the treatment, prevention, and reduction in the risk of VTE recurrence.
“With this new approval, Xarelto as an oral-only option now has the potential to change how acutely ill medical patients are managed for the prevention of blood clots, both in the hospital and for an extended period after discharge,” said Alex C. Spyropoulos, MD, of Northwell Health at Lenox Hill Hospital, New York, and a member of the steering committee of the MAGELLAN trial.
Find the full press release on the Janssen website.
The Food and Drug Administration has approved rivaroxaban (Xarelto) for the prevention of venous thromboembolism (VTE) in hospitalized, acutely ill patients at risk for thromboembolic complications who do not have a high bleeding risk, according to a release from Janssen.
FDA approval for the new indication is based on results from the phase 3 MAGELLAN and MARINER trials, which included more than 20,000 hospitalized, acutely ill patients. In MAGELLAN, rivaroxaban demonstrated noninferiority to enoxaparin, a low-molecular-weight heparin, in short-term usage, and it was superior over the long term, compared with short-term enoxaparin followed by placebo.
While VTE and VTE-related deaths were not reduced in MARINER, compared with placebo, patients who received rivaroxaban did see a significantly reduction in symptomatic VTE with a favorable safety profile.
According to the indication, rivaroxaban can be administered to patients during hospitalization and can be continued after discharge for 31-39 days. The safety profile in MAGELLAN and MARINER was consistent with that already seen, with the most common adverse event being bleeding.
The new indication is the eighth for rivaroxaban, the most of any direct oral anticoagulant; six of these are specifically for the treatment, prevention, and reduction in the risk of VTE recurrence.
“With this new approval, Xarelto as an oral-only option now has the potential to change how acutely ill medical patients are managed for the prevention of blood clots, both in the hospital and for an extended period after discharge,” said Alex C. Spyropoulos, MD, of Northwell Health at Lenox Hill Hospital, New York, and a member of the steering committee of the MAGELLAN trial.
Find the full press release on the Janssen website.
Rivaroxaban trends toward higher thrombotic risk than vitamin K antagonists in APS
suggests a recent trial conducted in Spain.
Stroke was also more common among those taking rivaroxaban, while major bleeding was slightly less common, reported lead author Josep Ordi-Ros, MD, PhD, of Vall d’Hebrón University Hospital Research Institute in Barcelona, and colleagues in Annals of Internal Medicine.
“Two randomized, controlled trials comparing rivaroxaban with warfarin suggested that rivaroxaban may be efficacious in patients with previous venous thromboembolism who are receiving standard-intensity anticoagulation but showed an increased thrombotic risk in those with triple-positive antiphospholipid antibodies,” the investigators wrote. However, they also noted that these findings required a cautious interpretation because of study limitations, such as premature termination caused by an excess of study events and the use of a laboratory surrogate marker as a primary outcome.
To learn more, the investigators performed an open-label, phase 3 trial involving 190 patients with thrombotic APS. Patients were randomized in a 1:1 ratio to receive either rivaroxaban (20 mg per day, or 15 mg per day for patients with a creatinine clearance of 30-49 mL/min per 1.73 m2) or an adjusted dosage of vitamin K antagonists (target international normalized ratio of 2.0-3.0, or 3.1-4.0 for those with a history of recurrent thrombosis).
Patients underwent evaluations every month for the first 3 months and then every 3 months thereafter, each of which involved a variety of laboratory diagnostics such as checks for antinuclear antibodies and lupus anticoagulant, among others. Statistical analyses aimed to determine if rivaroxaban was noninferior to therapy with vitamin K antagonists based on parameters drawn from previous meta-analyses, as no studies had compared the two types of treatment when the present study was designed.
After 3 years of follow-up, almost twice as many patients in the rivaroxaban group had experienced recurrent thrombosis (11.6% vs. 6.3%), although this finding lacked statistical significance for both noninferiority of rivaroxaban (P = .29) and superiority of vitamin K antagonists (P = .20). Still, supporting a similar trend toward differences in efficacy, stroke was more common in the rivaroxaban group, in which nine events occurred, compared with none in the vitamin K antagonist group. In contrast, major bleeding was slightly less common with rivaroxaban than vitamin K antagonists (6.3% vs. 7.4%).
“In conclusion, rivaroxaban did not demonstrate noninferiority to dose-adjusted vitamin K antagonists for secondary thromboprophylaxis in patients with thrombotic APS,” the investigators wrote. “Instead, our results indicate a recurrent thrombotic rate that is nearly double, albeit without statistical significance.”
The study was funded by Bayer Hispania. One coauthor reported additional relationships with Pfizer, Lilly, Janssen, and others.
SOURCE: Ordi-Ros J et al. Ann Intern Med. 2019 Oct 15. doi: 10.7326/M19-0291.
The recent trial by Ordi-Ros et al. revealed similar findings to a previous trial, TRAPS, by Pengo et al., which compared rivaroxaban with warfarin among patients with thrombotic antiphospholipid syndrome and triple positivity for antiphospholipid antibodies. Despite the caveat that TRAPS was prematurely terminated, in both studies, a higher proportion of patients in the rivaroxaban group than the vitamin K antagonist group had thrombotic events, most of which were arterial, whether considering MI or stroke. Furthermore, both studies did not show noninferiority of rivaroxaban versus dose-adjusted vitamin K antagonists.
The reasons for this failure of noninferiority remain unclear.
Denis Wahl, MD, PhD, and Virginie Dufrost, MD, are with the University of Lorraine, Nancy, France, and the Centre Hospitalier Universitaire de Nancy. No conflicts of interest were reported. His remarks are adapted from an accompanying editorial (Ann Intern Med. 2019 Oct 15. doi: 10.7326/M19-2815).
The recent trial by Ordi-Ros et al. revealed similar findings to a previous trial, TRAPS, by Pengo et al., which compared rivaroxaban with warfarin among patients with thrombotic antiphospholipid syndrome and triple positivity for antiphospholipid antibodies. Despite the caveat that TRAPS was prematurely terminated, in both studies, a higher proportion of patients in the rivaroxaban group than the vitamin K antagonist group had thrombotic events, most of which were arterial, whether considering MI or stroke. Furthermore, both studies did not show noninferiority of rivaroxaban versus dose-adjusted vitamin K antagonists.
The reasons for this failure of noninferiority remain unclear.
Denis Wahl, MD, PhD, and Virginie Dufrost, MD, are with the University of Lorraine, Nancy, France, and the Centre Hospitalier Universitaire de Nancy. No conflicts of interest were reported. His remarks are adapted from an accompanying editorial (Ann Intern Med. 2019 Oct 15. doi: 10.7326/M19-2815).
The recent trial by Ordi-Ros et al. revealed similar findings to a previous trial, TRAPS, by Pengo et al., which compared rivaroxaban with warfarin among patients with thrombotic antiphospholipid syndrome and triple positivity for antiphospholipid antibodies. Despite the caveat that TRAPS was prematurely terminated, in both studies, a higher proportion of patients in the rivaroxaban group than the vitamin K antagonist group had thrombotic events, most of which were arterial, whether considering MI or stroke. Furthermore, both studies did not show noninferiority of rivaroxaban versus dose-adjusted vitamin K antagonists.
The reasons for this failure of noninferiority remain unclear.
Denis Wahl, MD, PhD, and Virginie Dufrost, MD, are with the University of Lorraine, Nancy, France, and the Centre Hospitalier Universitaire de Nancy. No conflicts of interest were reported. His remarks are adapted from an accompanying editorial (Ann Intern Med. 2019 Oct 15. doi: 10.7326/M19-2815).
suggests a recent trial conducted in Spain.
Stroke was also more common among those taking rivaroxaban, while major bleeding was slightly less common, reported lead author Josep Ordi-Ros, MD, PhD, of Vall d’Hebrón University Hospital Research Institute in Barcelona, and colleagues in Annals of Internal Medicine.
“Two randomized, controlled trials comparing rivaroxaban with warfarin suggested that rivaroxaban may be efficacious in patients with previous venous thromboembolism who are receiving standard-intensity anticoagulation but showed an increased thrombotic risk in those with triple-positive antiphospholipid antibodies,” the investigators wrote. However, they also noted that these findings required a cautious interpretation because of study limitations, such as premature termination caused by an excess of study events and the use of a laboratory surrogate marker as a primary outcome.
To learn more, the investigators performed an open-label, phase 3 trial involving 190 patients with thrombotic APS. Patients were randomized in a 1:1 ratio to receive either rivaroxaban (20 mg per day, or 15 mg per day for patients with a creatinine clearance of 30-49 mL/min per 1.73 m2) or an adjusted dosage of vitamin K antagonists (target international normalized ratio of 2.0-3.0, or 3.1-4.0 for those with a history of recurrent thrombosis).
Patients underwent evaluations every month for the first 3 months and then every 3 months thereafter, each of which involved a variety of laboratory diagnostics such as checks for antinuclear antibodies and lupus anticoagulant, among others. Statistical analyses aimed to determine if rivaroxaban was noninferior to therapy with vitamin K antagonists based on parameters drawn from previous meta-analyses, as no studies had compared the two types of treatment when the present study was designed.
After 3 years of follow-up, almost twice as many patients in the rivaroxaban group had experienced recurrent thrombosis (11.6% vs. 6.3%), although this finding lacked statistical significance for both noninferiority of rivaroxaban (P = .29) and superiority of vitamin K antagonists (P = .20). Still, supporting a similar trend toward differences in efficacy, stroke was more common in the rivaroxaban group, in which nine events occurred, compared with none in the vitamin K antagonist group. In contrast, major bleeding was slightly less common with rivaroxaban than vitamin K antagonists (6.3% vs. 7.4%).
“In conclusion, rivaroxaban did not demonstrate noninferiority to dose-adjusted vitamin K antagonists for secondary thromboprophylaxis in patients with thrombotic APS,” the investigators wrote. “Instead, our results indicate a recurrent thrombotic rate that is nearly double, albeit without statistical significance.”
The study was funded by Bayer Hispania. One coauthor reported additional relationships with Pfizer, Lilly, Janssen, and others.
SOURCE: Ordi-Ros J et al. Ann Intern Med. 2019 Oct 15. doi: 10.7326/M19-0291.
suggests a recent trial conducted in Spain.
Stroke was also more common among those taking rivaroxaban, while major bleeding was slightly less common, reported lead author Josep Ordi-Ros, MD, PhD, of Vall d’Hebrón University Hospital Research Institute in Barcelona, and colleagues in Annals of Internal Medicine.
“Two randomized, controlled trials comparing rivaroxaban with warfarin suggested that rivaroxaban may be efficacious in patients with previous venous thromboembolism who are receiving standard-intensity anticoagulation but showed an increased thrombotic risk in those with triple-positive antiphospholipid antibodies,” the investigators wrote. However, they also noted that these findings required a cautious interpretation because of study limitations, such as premature termination caused by an excess of study events and the use of a laboratory surrogate marker as a primary outcome.
To learn more, the investigators performed an open-label, phase 3 trial involving 190 patients with thrombotic APS. Patients were randomized in a 1:1 ratio to receive either rivaroxaban (20 mg per day, or 15 mg per day for patients with a creatinine clearance of 30-49 mL/min per 1.73 m2) or an adjusted dosage of vitamin K antagonists (target international normalized ratio of 2.0-3.0, or 3.1-4.0 for those with a history of recurrent thrombosis).
Patients underwent evaluations every month for the first 3 months and then every 3 months thereafter, each of which involved a variety of laboratory diagnostics such as checks for antinuclear antibodies and lupus anticoagulant, among others. Statistical analyses aimed to determine if rivaroxaban was noninferior to therapy with vitamin K antagonists based on parameters drawn from previous meta-analyses, as no studies had compared the two types of treatment when the present study was designed.
After 3 years of follow-up, almost twice as many patients in the rivaroxaban group had experienced recurrent thrombosis (11.6% vs. 6.3%), although this finding lacked statistical significance for both noninferiority of rivaroxaban (P = .29) and superiority of vitamin K antagonists (P = .20). Still, supporting a similar trend toward differences in efficacy, stroke was more common in the rivaroxaban group, in which nine events occurred, compared with none in the vitamin K antagonist group. In contrast, major bleeding was slightly less common with rivaroxaban than vitamin K antagonists (6.3% vs. 7.4%).
“In conclusion, rivaroxaban did not demonstrate noninferiority to dose-adjusted vitamin K antagonists for secondary thromboprophylaxis in patients with thrombotic APS,” the investigators wrote. “Instead, our results indicate a recurrent thrombotic rate that is nearly double, albeit without statistical significance.”
The study was funded by Bayer Hispania. One coauthor reported additional relationships with Pfizer, Lilly, Janssen, and others.
SOURCE: Ordi-Ros J et al. Ann Intern Med. 2019 Oct 15. doi: 10.7326/M19-0291.
FROM ANNALS OF INTERNAL MEDICINE
Readmission burden high for those with sickle cell disease
, according to the Agency for Healthcare Research and Quality.
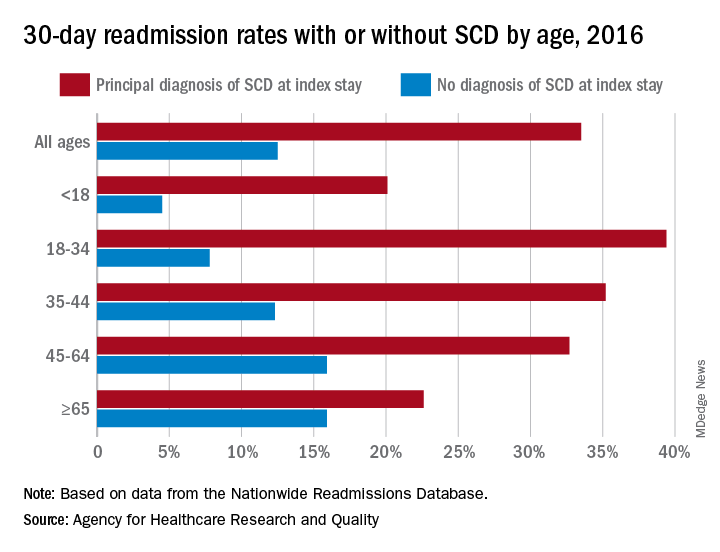
The 30-day all-cause readmission rate for index stays with a principal diagnosis of SCD was 33.5% in 2016, compared with 12.5% for non-SCD hospital stays. Patients with a secondary diagnosis of SCD had readmission rates of 32.9% with a pain crisis and 21.0% without one, and the overall readmission rate for an index stay with any SCD diagnosis was 31.1%, Kathryn R. Fingar, PhD, MPH, of IBM Watson Health, Sacramento, Calif., and associates wrote in an AHRQ statistical brief.
When age is factored in, the readmission gap between a principal SCD diagnosis and non-SCD becomes even greater – and smaller. The difference was greatest for patients aged 18-34 years – 39.4% with a principal diagnosis of SCD versus 7.8% without any SCD – and then narrowed as patients got older. For those aged 65 years and older, the rates were 22.6% with a principal diagnosis of SCD and 15.9% without, the investigators reported.
The approximately 100,000 Americans with SCD accounted for 134,000 admissions in 2016, and more than three-quarters of those stays involved a pain crisis. A principal diagnosis of SCD was recorded for almost 96,000 of those visits, and nearly all (96%) of those stays involved a pain crisis. For those with a secondary diagnosis of SCD, the most common reasons for hospitalization were diseases of the respiratory system (14.3% of those stays) and infectious and parasitic diseases (13.2%).
Patients with SCD were more likely than non-SCD patients to be admitted from the ED (79.6% vs. 51.3%), and they were more likely to discharged against medical advice (4.1% vs. 1.2%). Among those who left the hospital against medical advice, patients with SCD were much more likely to be readmitted than those without SCD (46.6% vs. 26.5%), based on data from the AHRQ’s Nationwide Readmissions Database.
Improved treatment of complications has “reduced mortality rates so that nearly 95% of individuals born with SCD in the United States reach 18 years of age [but] limited knowledge of SCD treatment guidelines among healthcare professionals continues to pose a barrier to effective patient-provider relationships, and this barrier contributes to lower quality of life,” Dr. Fingar and associates wrote.
, according to the Agency for Healthcare Research and Quality.
The 30-day all-cause readmission rate for index stays with a principal diagnosis of SCD was 33.5% in 2016, compared with 12.5% for non-SCD hospital stays. Patients with a secondary diagnosis of SCD had readmission rates of 32.9% with a pain crisis and 21.0% without one, and the overall readmission rate for an index stay with any SCD diagnosis was 31.1%, Kathryn R. Fingar, PhD, MPH, of IBM Watson Health, Sacramento, Calif., and associates wrote in an AHRQ statistical brief.
When age is factored in, the readmission gap between a principal SCD diagnosis and non-SCD becomes even greater – and smaller. The difference was greatest for patients aged 18-34 years – 39.4% with a principal diagnosis of SCD versus 7.8% without any SCD – and then narrowed as patients got older. For those aged 65 years and older, the rates were 22.6% with a principal diagnosis of SCD and 15.9% without, the investigators reported.
The approximately 100,000 Americans with SCD accounted for 134,000 admissions in 2016, and more than three-quarters of those stays involved a pain crisis. A principal diagnosis of SCD was recorded for almost 96,000 of those visits, and nearly all (96%) of those stays involved a pain crisis. For those with a secondary diagnosis of SCD, the most common reasons for hospitalization were diseases of the respiratory system (14.3% of those stays) and infectious and parasitic diseases (13.2%).
Patients with SCD were more likely than non-SCD patients to be admitted from the ED (79.6% vs. 51.3%), and they were more likely to discharged against medical advice (4.1% vs. 1.2%). Among those who left the hospital against medical advice, patients with SCD were much more likely to be readmitted than those without SCD (46.6% vs. 26.5%), based on data from the AHRQ’s Nationwide Readmissions Database.
Improved treatment of complications has “reduced mortality rates so that nearly 95% of individuals born with SCD in the United States reach 18 years of age [but] limited knowledge of SCD treatment guidelines among healthcare professionals continues to pose a barrier to effective patient-provider relationships, and this barrier contributes to lower quality of life,” Dr. Fingar and associates wrote.
, according to the Agency for Healthcare Research and Quality.
The 30-day all-cause readmission rate for index stays with a principal diagnosis of SCD was 33.5% in 2016, compared with 12.5% for non-SCD hospital stays. Patients with a secondary diagnosis of SCD had readmission rates of 32.9% with a pain crisis and 21.0% without one, and the overall readmission rate for an index stay with any SCD diagnosis was 31.1%, Kathryn R. Fingar, PhD, MPH, of IBM Watson Health, Sacramento, Calif., and associates wrote in an AHRQ statistical brief.
When age is factored in, the readmission gap between a principal SCD diagnosis and non-SCD becomes even greater – and smaller. The difference was greatest for patients aged 18-34 years – 39.4% with a principal diagnosis of SCD versus 7.8% without any SCD – and then narrowed as patients got older. For those aged 65 years and older, the rates were 22.6% with a principal diagnosis of SCD and 15.9% without, the investigators reported.
The approximately 100,000 Americans with SCD accounted for 134,000 admissions in 2016, and more than three-quarters of those stays involved a pain crisis. A principal diagnosis of SCD was recorded for almost 96,000 of those visits, and nearly all (96%) of those stays involved a pain crisis. For those with a secondary diagnosis of SCD, the most common reasons for hospitalization were diseases of the respiratory system (14.3% of those stays) and infectious and parasitic diseases (13.2%).
Patients with SCD were more likely than non-SCD patients to be admitted from the ED (79.6% vs. 51.3%), and they were more likely to discharged against medical advice (4.1% vs. 1.2%). Among those who left the hospital against medical advice, patients with SCD were much more likely to be readmitted than those without SCD (46.6% vs. 26.5%), based on data from the AHRQ’s Nationwide Readmissions Database.
Improved treatment of complications has “reduced mortality rates so that nearly 95% of individuals born with SCD in the United States reach 18 years of age [but] limited knowledge of SCD treatment guidelines among healthcare professionals continues to pose a barrier to effective patient-provider relationships, and this barrier contributes to lower quality of life,” Dr. Fingar and associates wrote.
Monthly and twice monthly emicizumab dosing safe for children with severe hemophilia A
Administration of twice-monthly or monthly emicizumab appears safe and effective for children with severe hemophilia A without inhibitors, according to a small cohort study.

After 24 weeks of treatment, only one moderate-intensity injection site reaction was reported, but no thrombotic microangiopathy or thromboembolic complications were observed.
The researchers evaluated the efficacy, safety, and pharmacokinetics of emicizumab in Japanese pediatric patients aged less than 12 years with severe hemophilia A without factor VIII inhibitors, wrote Midori Shima, MD, PhD, of Nara Medical University, Kashihara, Japan, and colleagues. The results were published in Haemophilia.
The open-label, nonrandomized study included 13 children who initially received weekly loading doses (3 mg/kg) of subcutaneous emicizumab for 4 weeks. Subsequently, patients received maintenance doses of 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks until week 24.
At baseline, the median age of patients in the 2- and 4-week dosing cohorts were 6.6 and 4.1 years, respectively. All participants had received factor VIII prophylaxis prior to starting emicizumab, with the exception of one patient.
Among six patients in the twice-monthly dosing cohort, two had no treated bleeding episodes, with an annualized bleeding rate for treated bleeding episodes of 1.3 (95% confidence interval, 0.6-2.9).
Among seven patients in the monthly dosing cohort, five had no treated bleeding episodes, with an annualized bleeding rate for treated bleeding episodes of 0.7 (95% CI, 0.2-2.6).
Caregivers completed a preference survey after the first 16 weeks of treatment, and “all reported a preference for emicizumab prophylaxis over the patient’s previous haemophilia treatment.” They cited the lower frequency of treatment and easier route of administration for favoring emicizumab.
With respect to pharmacokinetics, mean steady-state trough levels were within acceptable limits based on previous studies. No patients tested positive for anti-emicizumab antibodies.
The small sample size and nonrandomized design were key limitations of the study.
The results “confirm the appropriateness” of applying the every 2-week and every 4-week regimens of emicizumab in pediatric patients with hemophilia A without inhibitors, the researchers wrote.
The authors reported having financial affiliations with Chugai Pharmaceutical Co., which funded the study, and other companies.
SOURCE: Shima M et al. Haemophilia. 2019 Sep 12. doi: 10.1111/hae.13848.
Administration of twice-monthly or monthly emicizumab appears safe and effective for children with severe hemophilia A without inhibitors, according to a small cohort study.
After 24 weeks of treatment, only one moderate-intensity injection site reaction was reported, but no thrombotic microangiopathy or thromboembolic complications were observed.
The researchers evaluated the efficacy, safety, and pharmacokinetics of emicizumab in Japanese pediatric patients aged less than 12 years with severe hemophilia A without factor VIII inhibitors, wrote Midori Shima, MD, PhD, of Nara Medical University, Kashihara, Japan, and colleagues. The results were published in Haemophilia.
The open-label, nonrandomized study included 13 children who initially received weekly loading doses (3 mg/kg) of subcutaneous emicizumab for 4 weeks. Subsequently, patients received maintenance doses of 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks until week 24.
At baseline, the median age of patients in the 2- and 4-week dosing cohorts were 6.6 and 4.1 years, respectively. All participants had received factor VIII prophylaxis prior to starting emicizumab, with the exception of one patient.
Among six patients in the twice-monthly dosing cohort, two had no treated bleeding episodes, with an annualized bleeding rate for treated bleeding episodes of 1.3 (95% confidence interval, 0.6-2.9).
Among seven patients in the monthly dosing cohort, five had no treated bleeding episodes, with an annualized bleeding rate for treated bleeding episodes of 0.7 (95% CI, 0.2-2.6).
Caregivers completed a preference survey after the first 16 weeks of treatment, and “all reported a preference for emicizumab prophylaxis over the patient’s previous haemophilia treatment.” They cited the lower frequency of treatment and easier route of administration for favoring emicizumab.
With respect to pharmacokinetics, mean steady-state trough levels were within acceptable limits based on previous studies. No patients tested positive for anti-emicizumab antibodies.
The small sample size and nonrandomized design were key limitations of the study.
The results “confirm the appropriateness” of applying the every 2-week and every 4-week regimens of emicizumab in pediatric patients with hemophilia A without inhibitors, the researchers wrote.
The authors reported having financial affiliations with Chugai Pharmaceutical Co., which funded the study, and other companies.
SOURCE: Shima M et al. Haemophilia. 2019 Sep 12. doi: 10.1111/hae.13848.
Administration of twice-monthly or monthly emicizumab appears safe and effective for children with severe hemophilia A without inhibitors, according to a small cohort study.
After 24 weeks of treatment, only one moderate-intensity injection site reaction was reported, but no thrombotic microangiopathy or thromboembolic complications were observed.
The researchers evaluated the efficacy, safety, and pharmacokinetics of emicizumab in Japanese pediatric patients aged less than 12 years with severe hemophilia A without factor VIII inhibitors, wrote Midori Shima, MD, PhD, of Nara Medical University, Kashihara, Japan, and colleagues. The results were published in Haemophilia.
The open-label, nonrandomized study included 13 children who initially received weekly loading doses (3 mg/kg) of subcutaneous emicizumab for 4 weeks. Subsequently, patients received maintenance doses of 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks until week 24.
At baseline, the median age of patients in the 2- and 4-week dosing cohorts were 6.6 and 4.1 years, respectively. All participants had received factor VIII prophylaxis prior to starting emicizumab, with the exception of one patient.
Among six patients in the twice-monthly dosing cohort, two had no treated bleeding episodes, with an annualized bleeding rate for treated bleeding episodes of 1.3 (95% confidence interval, 0.6-2.9).
Among seven patients in the monthly dosing cohort, five had no treated bleeding episodes, with an annualized bleeding rate for treated bleeding episodes of 0.7 (95% CI, 0.2-2.6).
Caregivers completed a preference survey after the first 16 weeks of treatment, and “all reported a preference for emicizumab prophylaxis over the patient’s previous haemophilia treatment.” They cited the lower frequency of treatment and easier route of administration for favoring emicizumab.
With respect to pharmacokinetics, mean steady-state trough levels were within acceptable limits based on previous studies. No patients tested positive for anti-emicizumab antibodies.
The small sample size and nonrandomized design were key limitations of the study.
The results “confirm the appropriateness” of applying the every 2-week and every 4-week regimens of emicizumab in pediatric patients with hemophilia A without inhibitors, the researchers wrote.
The authors reported having financial affiliations with Chugai Pharmaceutical Co., which funded the study, and other companies.
SOURCE: Shima M et al. Haemophilia. 2019 Sep 12. doi: 10.1111/hae.13848.
FROM HAEMOPHILIA
New consensus recommendations on bleeding in acquired hemophilia
New consensus statements, released by a group of 36 experts, provide specific recommendations related to monitoring bleeding and assessing efficacy of treatment in patients with acquired hemophilia.
A global survey was developed by a nine-member steering committee with expertise in the hemostatic management of patients with acquired hemophilia. The Delphi methodology was used to obtain consensus on a list of statements on the location-specific treatment of bleeding in acquired hemophilia.
“The initial survey was circulated via email for refinement and was formally corroborated at a face-to-face meeting,” wrote Andreas Tiede, MD, PhD, of Hannover (Germany) Medical School and fellow experts. The report is published in Haemophilia.
The key areas outlined include the initial management of bleeding, and management of location-specific bleeding, including urological, gastrointestinal, muscle, and pharyngeal bleeds, as well as intracranial and postpartum hemorrhage.
If an expert hematologist is not available, and the bleeding event is life‐threatening, the emergency physician should initiate treatment in accordance with local or national recommendations, according to the initial management guidelines.
With respect to urological bleeds, the best interval for evaluating successful achievement of hemostasis is every 6-12 hours. The experts also reported that, if first-line hemostatic therapy is not effective, more intensive treatment should be considered every 6-12 hours.
In the management of intracranial hemorrhage, the frequency of clinical evaluation is subject to the particular scenario, and it can vary from every 2 hours (for clinical assessment) to every 24 hours (for imaging studies), they wrote.
If initial hemostatic treatment is not effective, more intensive therapy should be considered every 6 hours, they recommended.
“The statement addressing optimal frequency for assessing hemostasis in intracranial bleeds was the subject of much deliberation among the steering committee regarding timing of assessment,” the experts acknowledged.
The geographic diversity and global representation of expert participants were major strengths of these recommendations. However, these statements did not consider socioeconomic parameters or geopolitical differences that could affect patient care. As a result, they may not be applicable to all patient populations.
The manuscript was funded by Novo Nordisk AG. The authors reported having financial affiliations with Novo Nordisk and several other companies.
SOURCE: Tiede A et al. Haemophilia. 2019 Sep 13. doi: 10.1111/hae.13844.
New consensus statements, released by a group of 36 experts, provide specific recommendations related to monitoring bleeding and assessing efficacy of treatment in patients with acquired hemophilia.
A global survey was developed by a nine-member steering committee with expertise in the hemostatic management of patients with acquired hemophilia. The Delphi methodology was used to obtain consensus on a list of statements on the location-specific treatment of bleeding in acquired hemophilia.
“The initial survey was circulated via email for refinement and was formally corroborated at a face-to-face meeting,” wrote Andreas Tiede, MD, PhD, of Hannover (Germany) Medical School and fellow experts. The report is published in Haemophilia.
The key areas outlined include the initial management of bleeding, and management of location-specific bleeding, including urological, gastrointestinal, muscle, and pharyngeal bleeds, as well as intracranial and postpartum hemorrhage.
If an expert hematologist is not available, and the bleeding event is life‐threatening, the emergency physician should initiate treatment in accordance with local or national recommendations, according to the initial management guidelines.
With respect to urological bleeds, the best interval for evaluating successful achievement of hemostasis is every 6-12 hours. The experts also reported that, if first-line hemostatic therapy is not effective, more intensive treatment should be considered every 6-12 hours.
In the management of intracranial hemorrhage, the frequency of clinical evaluation is subject to the particular scenario, and it can vary from every 2 hours (for clinical assessment) to every 24 hours (for imaging studies), they wrote.
If initial hemostatic treatment is not effective, more intensive therapy should be considered every 6 hours, they recommended.
“The statement addressing optimal frequency for assessing hemostasis in intracranial bleeds was the subject of much deliberation among the steering committee regarding timing of assessment,” the experts acknowledged.
The geographic diversity and global representation of expert participants were major strengths of these recommendations. However, these statements did not consider socioeconomic parameters or geopolitical differences that could affect patient care. As a result, they may not be applicable to all patient populations.
The manuscript was funded by Novo Nordisk AG. The authors reported having financial affiliations with Novo Nordisk and several other companies.
SOURCE: Tiede A et al. Haemophilia. 2019 Sep 13. doi: 10.1111/hae.13844.
New consensus statements, released by a group of 36 experts, provide specific recommendations related to monitoring bleeding and assessing efficacy of treatment in patients with acquired hemophilia.
A global survey was developed by a nine-member steering committee with expertise in the hemostatic management of patients with acquired hemophilia. The Delphi methodology was used to obtain consensus on a list of statements on the location-specific treatment of bleeding in acquired hemophilia.
“The initial survey was circulated via email for refinement and was formally corroborated at a face-to-face meeting,” wrote Andreas Tiede, MD, PhD, of Hannover (Germany) Medical School and fellow experts. The report is published in Haemophilia.
The key areas outlined include the initial management of bleeding, and management of location-specific bleeding, including urological, gastrointestinal, muscle, and pharyngeal bleeds, as well as intracranial and postpartum hemorrhage.
If an expert hematologist is not available, and the bleeding event is life‐threatening, the emergency physician should initiate treatment in accordance with local or national recommendations, according to the initial management guidelines.
With respect to urological bleeds, the best interval for evaluating successful achievement of hemostasis is every 6-12 hours. The experts also reported that, if first-line hemostatic therapy is not effective, more intensive treatment should be considered every 6-12 hours.
In the management of intracranial hemorrhage, the frequency of clinical evaluation is subject to the particular scenario, and it can vary from every 2 hours (for clinical assessment) to every 24 hours (for imaging studies), they wrote.
If initial hemostatic treatment is not effective, more intensive therapy should be considered every 6 hours, they recommended.
“The statement addressing optimal frequency for assessing hemostasis in intracranial bleeds was the subject of much deliberation among the steering committee regarding timing of assessment,” the experts acknowledged.
The geographic diversity and global representation of expert participants were major strengths of these recommendations. However, these statements did not consider socioeconomic parameters or geopolitical differences that could affect patient care. As a result, they may not be applicable to all patient populations.
The manuscript was funded by Novo Nordisk AG. The authors reported having financial affiliations with Novo Nordisk and several other companies.
SOURCE: Tiede A et al. Haemophilia. 2019 Sep 13. doi: 10.1111/hae.13844.
FROM HAEMOPHILIA
Hemophilia prevalence is nearly three times higher than previously reported
The number of people with hemophilia worldwide is higher than previously estimated, and patients still face a shortened life expectancy, according to an international meta-analysis of registry data.

Approximately 1.125 million people have hemophilia worldwide, compared with the previous estimate of 400,000, reported lead author Alfonso Iorio, MD, PhD, of McMaster University, Hamilton, Ont., and colleagues.
The previous estimate, from the early 2000s, was based on prevalence in the United States and the global population at the time, the investigators explained. Their report is in Annals of Internal Medicine.
They noted a lack of clarity in prior estimates concerning type and severity of hemophilia, and aimed to correct this knowledge gap with the present meta-analysis.
Prevalence was estimated using data from registries in Australia, Canada, Italy, France, the United Kingdom, and New Zealand, which are all high-income countries. Prevalence at birth was estimated using the Canadian, French, and British registries, as these are the most established databases, according to the investigators. The World Federation of Hemophilia Annual Global survey was used to estimate the total global number of patients with hemophilia, while national statistics databases were used to determine the number of males and live male births.
Of the 1.125 million cases of hemophilia worldwide, the investigators estimated that 418,000 are likely severe. Proportionally, 17.1 out of 100,000 males have hemophilia A, with 6.0 out of 100,000 males exhibiting severe hemophilia A. Hemophilia B is less common, occurring in 3.8 out of 100,000 males, with a 1.1 out of 100,000 classified as severe.
Turning to prevalence at birth, the investigators estimated that there are 24.6 cases of hemophilia A per 100,000 male births and 5.0 cases of hemophilia B per 100,000 male births.
The associated life expectancy disadvantage in high-income countries is highest for severe hemophilia A (37%), followed by all severities of hemophilia A (30%), severe hemophilia B (27%), and all severities of hemophilia B (24%).
“Having 1,125,000 persons with hemophilia worldwide, of whom about 418,000 have severe and mostly undiagnosed disease, constitutes a formidable challenge and burden for researchers and health care systems, especially because only 196,706 patients have been identified and reported globally,” the investigators wrote. “More efficient diagnostic approaches are needed in less wealthy countries to take advantage of current and future treatment modalities, including gene therapy. Increased demand for care should drive new policy planning and spur renewed effort toward the development and manufacture of new drugs.”
The updated prevalence figures will serve as a valuable roadmap for the future, according to J. Michael Soucie, PhD, of the Centers for Disease Control and Prevention, Atlanta.
“Although the magnitude of the global gaps in care for persons with hemophilia is daunting, country specific data generated by application of the prevalence estimates reported by Iorio and colleagues are an important step toward prioritizing efforts to address these gaps,” Dr. Soucie wrote in an accompanying editorial. “Having more accurate prevalence data might also allow identification of ways in which regional efforts to improve care access could generate considerable benefits for patients and cost savings for countries. Armed with these data for action, we can hope to make substantial progress toward the goal of improving the lives of persons with hemophilia wherever they live.”
The study received no financial support. The investigators reported relationships with Pfizer, Roche, Novo Nordisk, and others. Dr. Soucie reported having no conflicts of interest.
SOURCE: Iorio A et al. Ann Intern Med. 2019 Sept 10. doi: 10.7326/M19-1208.
The number of people with hemophilia worldwide is higher than previously estimated, and patients still face a shortened life expectancy, according to an international meta-analysis of registry data.
Approximately 1.125 million people have hemophilia worldwide, compared with the previous estimate of 400,000, reported lead author Alfonso Iorio, MD, PhD, of McMaster University, Hamilton, Ont., and colleagues.
The previous estimate, from the early 2000s, was based on prevalence in the United States and the global population at the time, the investigators explained. Their report is in Annals of Internal Medicine.
They noted a lack of clarity in prior estimates concerning type and severity of hemophilia, and aimed to correct this knowledge gap with the present meta-analysis.
Prevalence was estimated using data from registries in Australia, Canada, Italy, France, the United Kingdom, and New Zealand, which are all high-income countries. Prevalence at birth was estimated using the Canadian, French, and British registries, as these are the most established databases, according to the investigators. The World Federation of Hemophilia Annual Global survey was used to estimate the total global number of patients with hemophilia, while national statistics databases were used to determine the number of males and live male births.
Of the 1.125 million cases of hemophilia worldwide, the investigators estimated that 418,000 are likely severe. Proportionally, 17.1 out of 100,000 males have hemophilia A, with 6.0 out of 100,000 males exhibiting severe hemophilia A. Hemophilia B is less common, occurring in 3.8 out of 100,000 males, with a 1.1 out of 100,000 classified as severe.
Turning to prevalence at birth, the investigators estimated that there are 24.6 cases of hemophilia A per 100,000 male births and 5.0 cases of hemophilia B per 100,000 male births.
The associated life expectancy disadvantage in high-income countries is highest for severe hemophilia A (37%), followed by all severities of hemophilia A (30%), severe hemophilia B (27%), and all severities of hemophilia B (24%).
“Having 1,125,000 persons with hemophilia worldwide, of whom about 418,000 have severe and mostly undiagnosed disease, constitutes a formidable challenge and burden for researchers and health care systems, especially because only 196,706 patients have been identified and reported globally,” the investigators wrote. “More efficient diagnostic approaches are needed in less wealthy countries to take advantage of current and future treatment modalities, including gene therapy. Increased demand for care should drive new policy planning and spur renewed effort toward the development and manufacture of new drugs.”
The updated prevalence figures will serve as a valuable roadmap for the future, according to J. Michael Soucie, PhD, of the Centers for Disease Control and Prevention, Atlanta.
“Although the magnitude of the global gaps in care for persons with hemophilia is daunting, country specific data generated by application of the prevalence estimates reported by Iorio and colleagues are an important step toward prioritizing efforts to address these gaps,” Dr. Soucie wrote in an accompanying editorial. “Having more accurate prevalence data might also allow identification of ways in which regional efforts to improve care access could generate considerable benefits for patients and cost savings for countries. Armed with these data for action, we can hope to make substantial progress toward the goal of improving the lives of persons with hemophilia wherever they live.”
The study received no financial support. The investigators reported relationships with Pfizer, Roche, Novo Nordisk, and others. Dr. Soucie reported having no conflicts of interest.
SOURCE: Iorio A et al. Ann Intern Med. 2019 Sept 10. doi: 10.7326/M19-1208.
The number of people with hemophilia worldwide is higher than previously estimated, and patients still face a shortened life expectancy, according to an international meta-analysis of registry data.
Approximately 1.125 million people have hemophilia worldwide, compared with the previous estimate of 400,000, reported lead author Alfonso Iorio, MD, PhD, of McMaster University, Hamilton, Ont., and colleagues.
The previous estimate, from the early 2000s, was based on prevalence in the United States and the global population at the time, the investigators explained. Their report is in Annals of Internal Medicine.
They noted a lack of clarity in prior estimates concerning type and severity of hemophilia, and aimed to correct this knowledge gap with the present meta-analysis.
Prevalence was estimated using data from registries in Australia, Canada, Italy, France, the United Kingdom, and New Zealand, which are all high-income countries. Prevalence at birth was estimated using the Canadian, French, and British registries, as these are the most established databases, according to the investigators. The World Federation of Hemophilia Annual Global survey was used to estimate the total global number of patients with hemophilia, while national statistics databases were used to determine the number of males and live male births.
Of the 1.125 million cases of hemophilia worldwide, the investigators estimated that 418,000 are likely severe. Proportionally, 17.1 out of 100,000 males have hemophilia A, with 6.0 out of 100,000 males exhibiting severe hemophilia A. Hemophilia B is less common, occurring in 3.8 out of 100,000 males, with a 1.1 out of 100,000 classified as severe.
Turning to prevalence at birth, the investigators estimated that there are 24.6 cases of hemophilia A per 100,000 male births and 5.0 cases of hemophilia B per 100,000 male births.
The associated life expectancy disadvantage in high-income countries is highest for severe hemophilia A (37%), followed by all severities of hemophilia A (30%), severe hemophilia B (27%), and all severities of hemophilia B (24%).
“Having 1,125,000 persons with hemophilia worldwide, of whom about 418,000 have severe and mostly undiagnosed disease, constitutes a formidable challenge and burden for researchers and health care systems, especially because only 196,706 patients have been identified and reported globally,” the investigators wrote. “More efficient diagnostic approaches are needed in less wealthy countries to take advantage of current and future treatment modalities, including gene therapy. Increased demand for care should drive new policy planning and spur renewed effort toward the development and manufacture of new drugs.”
The updated prevalence figures will serve as a valuable roadmap for the future, according to J. Michael Soucie, PhD, of the Centers for Disease Control and Prevention, Atlanta.
“Although the magnitude of the global gaps in care for persons with hemophilia is daunting, country specific data generated by application of the prevalence estimates reported by Iorio and colleagues are an important step toward prioritizing efforts to address these gaps,” Dr. Soucie wrote in an accompanying editorial. “Having more accurate prevalence data might also allow identification of ways in which regional efforts to improve care access could generate considerable benefits for patients and cost savings for countries. Armed with these data for action, we can hope to make substantial progress toward the goal of improving the lives of persons with hemophilia wherever they live.”
The study received no financial support. The investigators reported relationships with Pfizer, Roche, Novo Nordisk, and others. Dr. Soucie reported having no conflicts of interest.
SOURCE: Iorio A et al. Ann Intern Med. 2019 Sept 10. doi: 10.7326/M19-1208.
FROM ANNALS OF INTERNAL MEDICINE
Best treatment approach for early stage follicular lymphoma is unclear
Randomized trials are needed to determine the optimal treatment approach for early stage follicular lymphoma (FL), according to researchers.
A retrospective study showed similar outcomes among patients who received radiotherapy, immunochemotherapy, combined modality treatment (CMT), and watchful waiting (WW).
There were some differences in progression-free survival (PFS) according to treatment approach. However, there were no significant differences in overall survival (OS) between any of the active treatments or between patients who received active treatment and those managed with WW.
Joshua W. D. Tobin, MD, of Princess Alexandra Hospital in Brisbane, Queensland, Australia, and colleagues conducted this research and reported the results in Blood Advances.
The researchers analyzed 365 patients with newly diagnosed, stage I/II FL. The patients had a median age of 63 years and more than half were men. They were diagnosed between 2005 and 2017, and the median follow-up was 45 months.
Most patients (n = 280) received active treatment, but 85 were managed with WW. The WW patients were older and had more extranodal involvement.
Types of active treatment included radiotherapy alone (n = 171), immunochemotherapy alone (n = 63), and CMT (n = 46). Compared with the other groups, patients who received radiotherapy alone had less bulk, fewer nodal sites, and fewer B symptoms, and were more likely to have stage I disease. Patients who received CMT had fewer B symptoms and lower FLIPI scores compared with patients who received immunochemotherapy.
The immunochemotherapy regimens used were largely rituximab based. In all, 106 patients received rituximab (alone or in combination) for induction, and 49 received maintenance rituximab (37 in the immunochemotherapy group and 12 in the CMT group).
Results
Response rates were similar among the active treatment groups. The overall response rate was 95% in the radiotherapy group, 96% in the immunochemotherapy group, and 95% in the CMT group (P = .87).
There was a significant difference in PFS between the radiotherapy, immunochemotherapy, and CMT groups (P = .023), but there was no difference in OS between these groups (P = .38).
There was no significant difference in PFS between the immunochemotherapy and CMT groups (hazard ratio [HR], 1.78; P = .24), so the researchers combined these groups into a single group called “systemic therapy.” The patients treated with systemic therapy had PFS (HR, 1.32; P = .96) and OS (HR, 0.46; P = .21) similar to that of patients treated with radiotherapy alone.
Maintenance rituximab was associated with prolonged PFS among patients treated with systemic therapy (HR, 0.24; P = .017). However, there was no significant difference in OS between patients who received maintenance and those who did not (HR, 0.89; P = .90).
Relapse was less common among patients who received maintenance, and there were no cases of transformation in that group. Relapse occurred in 24.6% of the radiotherapy group, 18.3% of the systemic therapy group, and 4.1% of the group that received systemic therapy plus maintenance (P = .006). Transformation was less likely in the systemic therapy group (1.8%) than in the radiotherapy (6.4%) and WW (9.4%) groups (HR, 0.20; P = .034).
Overall, the active treatment group had better PFS than the WW group (HR, 0.52; P = .002), but there was no significant difference in OS between the groups (HR, 0.94; P = .90).
“Based on our comparable OS between WW and actively treated patients, WW could be considered as an initial management strategy in early stage FL,” Dr. Tobin and colleagues wrote. “However, long-term follow-up is required to determine if a survival benefit exists favoring active treatment.”
The researchers reported relationships with many pharmaceutical companies.
SOURCE: Tobin JWD et al. Blood Adv. 2019 Oct 8;3(19):2804-11.
Randomized trials are needed to determine the optimal treatment approach for early stage follicular lymphoma (FL), according to researchers.
A retrospective study showed similar outcomes among patients who received radiotherapy, immunochemotherapy, combined modality treatment (CMT), and watchful waiting (WW).
There were some differences in progression-free survival (PFS) according to treatment approach. However, there were no significant differences in overall survival (OS) between any of the active treatments or between patients who received active treatment and those managed with WW.
Joshua W. D. Tobin, MD, of Princess Alexandra Hospital in Brisbane, Queensland, Australia, and colleagues conducted this research and reported the results in Blood Advances.
The researchers analyzed 365 patients with newly diagnosed, stage I/II FL. The patients had a median age of 63 years and more than half were men. They were diagnosed between 2005 and 2017, and the median follow-up was 45 months.
Most patients (n = 280) received active treatment, but 85 were managed with WW. The WW patients were older and had more extranodal involvement.
Types of active treatment included radiotherapy alone (n = 171), immunochemotherapy alone (n = 63), and CMT (n = 46). Compared with the other groups, patients who received radiotherapy alone had less bulk, fewer nodal sites, and fewer B symptoms, and were more likely to have stage I disease. Patients who received CMT had fewer B symptoms and lower FLIPI scores compared with patients who received immunochemotherapy.
The immunochemotherapy regimens used were largely rituximab based. In all, 106 patients received rituximab (alone or in combination) for induction, and 49 received maintenance rituximab (37 in the immunochemotherapy group and 12 in the CMT group).
Results
Response rates were similar among the active treatment groups. The overall response rate was 95% in the radiotherapy group, 96% in the immunochemotherapy group, and 95% in the CMT group (P = .87).
There was a significant difference in PFS between the radiotherapy, immunochemotherapy, and CMT groups (P = .023), but there was no difference in OS between these groups (P = .38).
There was no significant difference in PFS between the immunochemotherapy and CMT groups (hazard ratio [HR], 1.78; P = .24), so the researchers combined these groups into a single group called “systemic therapy.” The patients treated with systemic therapy had PFS (HR, 1.32; P = .96) and OS (HR, 0.46; P = .21) similar to that of patients treated with radiotherapy alone.
Maintenance rituximab was associated with prolonged PFS among patients treated with systemic therapy (HR, 0.24; P = .017). However, there was no significant difference in OS between patients who received maintenance and those who did not (HR, 0.89; P = .90).
Relapse was less common among patients who received maintenance, and there were no cases of transformation in that group. Relapse occurred in 24.6% of the radiotherapy group, 18.3% of the systemic therapy group, and 4.1% of the group that received systemic therapy plus maintenance (P = .006). Transformation was less likely in the systemic therapy group (1.8%) than in the radiotherapy (6.4%) and WW (9.4%) groups (HR, 0.20; P = .034).
Overall, the active treatment group had better PFS than the WW group (HR, 0.52; P = .002), but there was no significant difference in OS between the groups (HR, 0.94; P = .90).
“Based on our comparable OS between WW and actively treated patients, WW could be considered as an initial management strategy in early stage FL,” Dr. Tobin and colleagues wrote. “However, long-term follow-up is required to determine if a survival benefit exists favoring active treatment.”
The researchers reported relationships with many pharmaceutical companies.
SOURCE: Tobin JWD et al. Blood Adv. 2019 Oct 8;3(19):2804-11.
Randomized trials are needed to determine the optimal treatment approach for early stage follicular lymphoma (FL), according to researchers.
A retrospective study showed similar outcomes among patients who received radiotherapy, immunochemotherapy, combined modality treatment (CMT), and watchful waiting (WW).
There were some differences in progression-free survival (PFS) according to treatment approach. However, there were no significant differences in overall survival (OS) between any of the active treatments or between patients who received active treatment and those managed with WW.
Joshua W. D. Tobin, MD, of Princess Alexandra Hospital in Brisbane, Queensland, Australia, and colleagues conducted this research and reported the results in Blood Advances.
The researchers analyzed 365 patients with newly diagnosed, stage I/II FL. The patients had a median age of 63 years and more than half were men. They were diagnosed between 2005 and 2017, and the median follow-up was 45 months.
Most patients (n = 280) received active treatment, but 85 were managed with WW. The WW patients were older and had more extranodal involvement.
Types of active treatment included radiotherapy alone (n = 171), immunochemotherapy alone (n = 63), and CMT (n = 46). Compared with the other groups, patients who received radiotherapy alone had less bulk, fewer nodal sites, and fewer B symptoms, and were more likely to have stage I disease. Patients who received CMT had fewer B symptoms and lower FLIPI scores compared with patients who received immunochemotherapy.
The immunochemotherapy regimens used were largely rituximab based. In all, 106 patients received rituximab (alone or in combination) for induction, and 49 received maintenance rituximab (37 in the immunochemotherapy group and 12 in the CMT group).
Results
Response rates were similar among the active treatment groups. The overall response rate was 95% in the radiotherapy group, 96% in the immunochemotherapy group, and 95% in the CMT group (P = .87).
There was a significant difference in PFS between the radiotherapy, immunochemotherapy, and CMT groups (P = .023), but there was no difference in OS between these groups (P = .38).
There was no significant difference in PFS between the immunochemotherapy and CMT groups (hazard ratio [HR], 1.78; P = .24), so the researchers combined these groups into a single group called “systemic therapy.” The patients treated with systemic therapy had PFS (HR, 1.32; P = .96) and OS (HR, 0.46; P = .21) similar to that of patients treated with radiotherapy alone.
Maintenance rituximab was associated with prolonged PFS among patients treated with systemic therapy (HR, 0.24; P = .017). However, there was no significant difference in OS between patients who received maintenance and those who did not (HR, 0.89; P = .90).
Relapse was less common among patients who received maintenance, and there were no cases of transformation in that group. Relapse occurred in 24.6% of the radiotherapy group, 18.3% of the systemic therapy group, and 4.1% of the group that received systemic therapy plus maintenance (P = .006). Transformation was less likely in the systemic therapy group (1.8%) than in the radiotherapy (6.4%) and WW (9.4%) groups (HR, 0.20; P = .034).
Overall, the active treatment group had better PFS than the WW group (HR, 0.52; P = .002), but there was no significant difference in OS between the groups (HR, 0.94; P = .90).
“Based on our comparable OS between WW and actively treated patients, WW could be considered as an initial management strategy in early stage FL,” Dr. Tobin and colleagues wrote. “However, long-term follow-up is required to determine if a survival benefit exists favoring active treatment.”
The researchers reported relationships with many pharmaceutical companies.
SOURCE: Tobin JWD et al. Blood Adv. 2019 Oct 8;3(19):2804-11.
FROM BLOOD ADVANCES
What is the optimal duration of maintenance in myeloma?
SAN FRANCISCO – Should patients with multiple myeloma receive maintenance therapy until progression?

Yvonne A. Efebera, MD, of The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital in Columbus, and Nina Shah, MD, of the University of California San Francisco Health, faced off on this question at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.

Dr. Shah said maintenance therapy improves survival in myeloma patients, so it follows that treating them until progression would confer a survival advantage. While Dr. Efebera agreed that maintenance can improve survival, she said the optimal duration of that treatment is unknown.
Treat until progression
Dr. Shah cited studies suggesting that maintenance improves progression-free survival (PFS) and may prolong overall survival (OS) in multiple myeloma.
A meta-analysis of data from the IFM 2005-02, CALGB 100104, and GIMEMA RV-MM-PI-209 trials showed that lenalidomide maintenance prolonged PFS and OS. The median PFS was 52.8 months in patients who received maintenance and 23.5 months in those who received placebo or observation (hazard ratio [HR], 0.48). At a median follow-up of 79.5 months, the median OS was not reached for the maintenance group and was 86.0 months for the no-maintenance group (HR, 0.75; P = .001; J Clin Oncol. 2017 Oct 10;35[29]:3279-89).
In the Myeloma XI trial, maintenance improved PFS, but not OS, in both transplant-eligible and ineligible patients. Overall, the median PFS was 39 months in the lenalidomide maintenance arm and 20 months in the observation arm (P less than .0001). Among transplant-eligible patients, the median PFS was 57 months and 30 months, respectively (P less than .0001). Among transplant-ineligible patients, the median PFS was 26 months and 11 months, respectively (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:57-73).
These data suggest maintenance can improve survival, “but the question is, how long should we have therapy,” Dr. Shah said. “No one has looked at this in a prospective manner, so we really have to look at our retrospective data.”
One study suggested a longer duration of lenalidomide maintenance improves PFS. The HR for progression or death was 0.39 for patients who received maintenance for 12-24 months, compared with those who received maintenance for less than 12 months. The HR was 0.13 for patients who received maintenance for more than 24 months, compared with less than 12 months (Leuk Lymphoma. 2019 Feb;60[2]:511-4).
Dr. Shah also cited a pooled analysis of three phase 3 trials suggesting that continuous therapy is superior to fixed-duration therapy in patients with newly diagnosed myeloma. The median PFS was 32 months with continuous therapy and 16 months with fixed-duration therapy (P less than .001). The 4-year OS was 69% and 60%, respectively (P = .003; J Clin Oncol. 2015 Oct 20;33[30]:3459-66).
These data suggest that “continuous therapy, more therapy, has a survival advantage,” Dr. Shah said.
Don’t treat until progression
Dr. Efebera also discussed data from studies showing that lenalidomide maintenance can prolong survival in multiple myeloma. However, she said, it’s unclear how long maintenance should last.
Different durations of maintenance have proved effective in different trials. In the CALGB 100104 trial, the median duration of maintenance was 31 months (Lancet Haematol. 2017 Sep;4[9]:e431-e442). In the meta-analysis of the CALGB, IFM, and GIMEMA trials, the median duration was 22 months. And in Myeloma XI, the median duration was 18 months.
As there is no randomized trial comparing different durations of maintenance, Dr. Efebera proposed that researchers conduct one. She said this “perfect study” would involve induction with an immunomodulatory agent, a proteasome inhibitor, dexamethasone, and perhaps an anti-CD38 therapy. Transplant-eligible patients would receive four cycles of induction before transplant. Transplant-ineligible patients would receive eight cycles of induction. Then, all patients would be randomized to lenalidomide maintenance for 3 years, 5 years, or 7-10 years.
Until a trial like this reveals the optimal duration of maintenance, we cannot conclude that treating patients until progression is better, Dr. Efebera said.
She added that maintenance has been shown to have detrimental effects, and these should be taken into consideration. For instance, neutropenia, other hematologic adverse events, and second primary malignancies have been shown to be more common among patients who receive lenalidomide maintenance (N Engl J Med. 2012; 366:1782-91).
The cost of maintenance is another factor to consider. Researchers analyzed data from the CALGB 100104 and IFM 2005-02 trials to compare the cost of lenalidomide maintenance with no maintenance. In the CALGB 100104 trial, patients who received lenalidomide maintenance had 5.72 quality-adjusted life years (QALYs), and those who received no maintenance had 4.61 QALYs. The incremental cost-utility ratio (ICUR) was more than 277,000 euros per QALY.
In the IFM2005-02 trial, patients in the lenalidomide group had 5.13 QALYs, and those who didn’t receive maintenance had 4.98 QALYs. The ICUR was more than 1.5 million euros per QALY. The researchers said the high ICURs and budgetary impact add “uncertainty about the maximum prudent duration of the treatment” (Bone Marrow Transplant. 2019 May 31. doi: 10.1038/s41409-019-0574-5).
Dr. Efebera reported relationships with Akcea Therapeutics, Janssen, and Takeda. Dr. Shah reported having no relevant financial relationships.
SAN FRANCISCO – Should patients with multiple myeloma receive maintenance therapy until progression?
Yvonne A. Efebera, MD, of The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital in Columbus, and Nina Shah, MD, of the University of California San Francisco Health, faced off on this question at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Shah said maintenance therapy improves survival in myeloma patients, so it follows that treating them until progression would confer a survival advantage. While Dr. Efebera agreed that maintenance can improve survival, she said the optimal duration of that treatment is unknown.
Treat until progression
Dr. Shah cited studies suggesting that maintenance improves progression-free survival (PFS) and may prolong overall survival (OS) in multiple myeloma.
A meta-analysis of data from the IFM 2005-02, CALGB 100104, and GIMEMA RV-MM-PI-209 trials showed that lenalidomide maintenance prolonged PFS and OS. The median PFS was 52.8 months in patients who received maintenance and 23.5 months in those who received placebo or observation (hazard ratio [HR], 0.48). At a median follow-up of 79.5 months, the median OS was not reached for the maintenance group and was 86.0 months for the no-maintenance group (HR, 0.75; P = .001; J Clin Oncol. 2017 Oct 10;35[29]:3279-89).
In the Myeloma XI trial, maintenance improved PFS, but not OS, in both transplant-eligible and ineligible patients. Overall, the median PFS was 39 months in the lenalidomide maintenance arm and 20 months in the observation arm (P less than .0001). Among transplant-eligible patients, the median PFS was 57 months and 30 months, respectively (P less than .0001). Among transplant-ineligible patients, the median PFS was 26 months and 11 months, respectively (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:57-73).
These data suggest maintenance can improve survival, “but the question is, how long should we have therapy,” Dr. Shah said. “No one has looked at this in a prospective manner, so we really have to look at our retrospective data.”
One study suggested a longer duration of lenalidomide maintenance improves PFS. The HR for progression or death was 0.39 for patients who received maintenance for 12-24 months, compared with those who received maintenance for less than 12 months. The HR was 0.13 for patients who received maintenance for more than 24 months, compared with less than 12 months (Leuk Lymphoma. 2019 Feb;60[2]:511-4).
Dr. Shah also cited a pooled analysis of three phase 3 trials suggesting that continuous therapy is superior to fixed-duration therapy in patients with newly diagnosed myeloma. The median PFS was 32 months with continuous therapy and 16 months with fixed-duration therapy (P less than .001). The 4-year OS was 69% and 60%, respectively (P = .003; J Clin Oncol. 2015 Oct 20;33[30]:3459-66).
These data suggest that “continuous therapy, more therapy, has a survival advantage,” Dr. Shah said.
Don’t treat until progression
Dr. Efebera also discussed data from studies showing that lenalidomide maintenance can prolong survival in multiple myeloma. However, she said, it’s unclear how long maintenance should last.
Different durations of maintenance have proved effective in different trials. In the CALGB 100104 trial, the median duration of maintenance was 31 months (Lancet Haematol. 2017 Sep;4[9]:e431-e442). In the meta-analysis of the CALGB, IFM, and GIMEMA trials, the median duration was 22 months. And in Myeloma XI, the median duration was 18 months.
As there is no randomized trial comparing different durations of maintenance, Dr. Efebera proposed that researchers conduct one. She said this “perfect study” would involve induction with an immunomodulatory agent, a proteasome inhibitor, dexamethasone, and perhaps an anti-CD38 therapy. Transplant-eligible patients would receive four cycles of induction before transplant. Transplant-ineligible patients would receive eight cycles of induction. Then, all patients would be randomized to lenalidomide maintenance for 3 years, 5 years, or 7-10 years.
Until a trial like this reveals the optimal duration of maintenance, we cannot conclude that treating patients until progression is better, Dr. Efebera said.
She added that maintenance has been shown to have detrimental effects, and these should be taken into consideration. For instance, neutropenia, other hematologic adverse events, and second primary malignancies have been shown to be more common among patients who receive lenalidomide maintenance (N Engl J Med. 2012; 366:1782-91).
The cost of maintenance is another factor to consider. Researchers analyzed data from the CALGB 100104 and IFM 2005-02 trials to compare the cost of lenalidomide maintenance with no maintenance. In the CALGB 100104 trial, patients who received lenalidomide maintenance had 5.72 quality-adjusted life years (QALYs), and those who received no maintenance had 4.61 QALYs. The incremental cost-utility ratio (ICUR) was more than 277,000 euros per QALY.
In the IFM2005-02 trial, patients in the lenalidomide group had 5.13 QALYs, and those who didn’t receive maintenance had 4.98 QALYs. The ICUR was more than 1.5 million euros per QALY. The researchers said the high ICURs and budgetary impact add “uncertainty about the maximum prudent duration of the treatment” (Bone Marrow Transplant. 2019 May 31. doi: 10.1038/s41409-019-0574-5).
Dr. Efebera reported relationships with Akcea Therapeutics, Janssen, and Takeda. Dr. Shah reported having no relevant financial relationships.
SAN FRANCISCO – Should patients with multiple myeloma receive maintenance therapy until progression?
Yvonne A. Efebera, MD, of The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital in Columbus, and Nina Shah, MD, of the University of California San Francisco Health, faced off on this question at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Shah said maintenance therapy improves survival in myeloma patients, so it follows that treating them until progression would confer a survival advantage. While Dr. Efebera agreed that maintenance can improve survival, she said the optimal duration of that treatment is unknown.
Treat until progression
Dr. Shah cited studies suggesting that maintenance improves progression-free survival (PFS) and may prolong overall survival (OS) in multiple myeloma.
A meta-analysis of data from the IFM 2005-02, CALGB 100104, and GIMEMA RV-MM-PI-209 trials showed that lenalidomide maintenance prolonged PFS and OS. The median PFS was 52.8 months in patients who received maintenance and 23.5 months in those who received placebo or observation (hazard ratio [HR], 0.48). At a median follow-up of 79.5 months, the median OS was not reached for the maintenance group and was 86.0 months for the no-maintenance group (HR, 0.75; P = .001; J Clin Oncol. 2017 Oct 10;35[29]:3279-89).
In the Myeloma XI trial, maintenance improved PFS, but not OS, in both transplant-eligible and ineligible patients. Overall, the median PFS was 39 months in the lenalidomide maintenance arm and 20 months in the observation arm (P less than .0001). Among transplant-eligible patients, the median PFS was 57 months and 30 months, respectively (P less than .0001). Among transplant-ineligible patients, the median PFS was 26 months and 11 months, respectively (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:57-73).
These data suggest maintenance can improve survival, “but the question is, how long should we have therapy,” Dr. Shah said. “No one has looked at this in a prospective manner, so we really have to look at our retrospective data.”
One study suggested a longer duration of lenalidomide maintenance improves PFS. The HR for progression or death was 0.39 for patients who received maintenance for 12-24 months, compared with those who received maintenance for less than 12 months. The HR was 0.13 for patients who received maintenance for more than 24 months, compared with less than 12 months (Leuk Lymphoma. 2019 Feb;60[2]:511-4).
Dr. Shah also cited a pooled analysis of three phase 3 trials suggesting that continuous therapy is superior to fixed-duration therapy in patients with newly diagnosed myeloma. The median PFS was 32 months with continuous therapy and 16 months with fixed-duration therapy (P less than .001). The 4-year OS was 69% and 60%, respectively (P = .003; J Clin Oncol. 2015 Oct 20;33[30]:3459-66).
These data suggest that “continuous therapy, more therapy, has a survival advantage,” Dr. Shah said.
Don’t treat until progression
Dr. Efebera also discussed data from studies showing that lenalidomide maintenance can prolong survival in multiple myeloma. However, she said, it’s unclear how long maintenance should last.
Different durations of maintenance have proved effective in different trials. In the CALGB 100104 trial, the median duration of maintenance was 31 months (Lancet Haematol. 2017 Sep;4[9]:e431-e442). In the meta-analysis of the CALGB, IFM, and GIMEMA trials, the median duration was 22 months. And in Myeloma XI, the median duration was 18 months.
As there is no randomized trial comparing different durations of maintenance, Dr. Efebera proposed that researchers conduct one. She said this “perfect study” would involve induction with an immunomodulatory agent, a proteasome inhibitor, dexamethasone, and perhaps an anti-CD38 therapy. Transplant-eligible patients would receive four cycles of induction before transplant. Transplant-ineligible patients would receive eight cycles of induction. Then, all patients would be randomized to lenalidomide maintenance for 3 years, 5 years, or 7-10 years.
Until a trial like this reveals the optimal duration of maintenance, we cannot conclude that treating patients until progression is better, Dr. Efebera said.
She added that maintenance has been shown to have detrimental effects, and these should be taken into consideration. For instance, neutropenia, other hematologic adverse events, and second primary malignancies have been shown to be more common among patients who receive lenalidomide maintenance (N Engl J Med. 2012; 366:1782-91).
The cost of maintenance is another factor to consider. Researchers analyzed data from the CALGB 100104 and IFM 2005-02 trials to compare the cost of lenalidomide maintenance with no maintenance. In the CALGB 100104 trial, patients who received lenalidomide maintenance had 5.72 quality-adjusted life years (QALYs), and those who received no maintenance had 4.61 QALYs. The incremental cost-utility ratio (ICUR) was more than 277,000 euros per QALY.
In the IFM2005-02 trial, patients in the lenalidomide group had 5.13 QALYs, and those who didn’t receive maintenance had 4.98 QALYs. The ICUR was more than 1.5 million euros per QALY. The researchers said the high ICURs and budgetary impact add “uncertainty about the maximum prudent duration of the treatment” (Bone Marrow Transplant. 2019 May 31. doi: 10.1038/s41409-019-0574-5).
Dr. Efebera reported relationships with Akcea Therapeutics, Janssen, and Takeda. Dr. Shah reported having no relevant financial relationships.
EXPERT ANALYSIS FROM NCCN HEMATOLOGIC MALIGNANCIES
Primary care for the declining cancer survivor
As a family physician (FP), you are well positioned to optimize the quality of life of advanced cancer patients as they decline and approach death. You can help them understand their evolving prognosis so that treatment goals can be adjusted, and you can ensure that hospice is implemented early to improve the end-of-life experience. This practical review will help you to provide the best care possible for these patients.
Family physicians can fill a care gap
The term cancer survivor describes a patient who has completed initial cancer treatment. Within this population, many have declining health and ultimately succumb to their disease. There were 16.9 million cancer survivors in the United States as of January 1, 2019,1 with 53% likely to experience significant symptoms and disability.2 More than 600,000 American cancer survivors will die in 2019.3
In 2011, the Commission on Cancer mandated available outpatient palliative care services at certified cancer centers.4 Unfortunately, current palliative care resources fall far short of expected needs. A 2010 estimate of required hospice and palliative care physicians demonstrated a staffing gap of more than 50% among those providing outpatient services.5 The shortage continues,6 and many cancer patients will look to their FP for supportive care.
FPs, in addition to easing symptoms and adverse effects of medication, can educate patients and families about their disease and prognosis. By providing longitudinal care, FPs can identify critical health declines that oncologists, patients, and families often overlook. FPs can also readily appreciate decline, guide patients toward their care goals, and facilitate comfort care—including at the end of life.
Early outpatient palliative care improves quality of life and patient satisfaction. It also may improve survival time and ward off depression.7,8 Some patients and providers resist palliative care due to a misconception that it requires abandoning treatment.9 Actually, palliative care can be given in concert with all active treatments. Many experts recommend a name change from “palliative care” to “supportive care” to dispel this misconception.10
Estimate prognosis using the “surprise question”
Several algorithms are available—using between 2 and 13 patient parameters—to estimate advanced cancer survival. Most of these algorithms are designed to identify the last months or weeks of life, but their utility to predict death within these periods is limited.11
The “surprise question” may be the most valuable prognostic test for primary care. In this test, the physician asks him- or herself: Would I be surprised if this patient died in 1 year? Researchers found that when primary care physicians answered No, their patient was 4 times more likely to die within the year than when they answered Yes.12 This test has a positive predictive value of 20% and a negative predictive value of 95%, making it valuable in distinguishing patients with longer life expectancy.12 Although it overidentifies at-risk patients, the "surprise question" is a simple and sensitive tool for defining prognosis.
Continue to: Priorities for patients likely to live more than a year
Priorities for patients likely to live more than a year
For patients who likely have more than a year to live, the focus is on symptom management and preparation for future decline. Initiate and facilitate discussions about end-of-life topics. Cancer survivors are often open to discussions on these topics, which include advanced directives, home health aides, and hospice.13 Patients can set specific goals for their remaining time, such as engaging in travel, personal projects, or special events. Cancer patients have better end-of-life experiences and families have improved mental health after these discussions.14 Although cancer patients are more likely than other terminal patients to have end-of-life discussions, fewer than 40% ever do.15
Address distressing symptoms with a focus on maintaining function. More than 50% of advanced cancer patients experience fatigue, weakness, pain, weight loss, and anorexia,16 and up to 60% experience psychological distress.17 Deprescribing most preventive medications is recommended with transition to symptomatic treatment.18
Priorities for patients with less than a year to live
For patients who may have less than a year to live, focus shifts to their wishes for the time remaining and priorities for the dying process. Most patients start out with prognostic views more optimistic than those of their physicians, but this gap narrows after end-of-life discussions.19,20 Patients with incurable cancer are less likely to choose aggressive therapy if they believe their 6-month survival probability is less than 90%.21 Honest conversations, with best- and worst-case scenarios, are important to patients and families, and should occur while the patient is well enough to participate and set goals.22
In the last months of life, opioids become the primary treatment for pain and air hunger. As function declines, concerns about such adverse effects as falls and confusion decrease. Opioids have been shown to be most effective over the course of 4 weeks, and avoiding their use in earlier stages may increase their efficacy at the end of life.23
Hospice benefit—more comfort, with limitations
Hospice care consists of services administered by nonprofit and for-profit entities covered by Medicare, Medicaid, and many private insurers.24 Hospice strives to allow patients to approach death in comfort, meeting their goal of a “good death.” A recent literature review identified 4 aspects of a good death that terminally ill patients and their families considered most important: control of the dying process, relief of pain, spirituality, and emotional well-being (TABLE 1).25

Continue to: Hospice use is increasing...
Hospice use is increasing, yet many enroll too late to fully benefit. While cancer patients alone are not currently tracked, the use of hospice by Medicare beneficiaries increased from 44% in 2012 to 48% in 2019.24 In 2017, the median hospice stay was 19 days.24 Unfortunately, though, just 28% of hospice-eligible patients enrolled in hospice in their last week of life.24 Without hospice, patients often receive excessive care near death. More than 6% receive aggressive chemotherapy in their last 2 weeks of life, and nearly 10% receive a life-prolonging procedure in their last month.26
Hospice care replaces standard hospital care, although patients can elect to be followed by their primary care physician.9 Most hospice services are provided as needed or continuously at the patient’s home, including assisted living facilities. And it is also offered as part of hospital care. Hospice services are interdisciplinary, provided by physicians, nurses, social workers, chaplains, and health aides. Hospices have on-call staff to assess and treat complications, avoiding emergency hospital visits.9 And hospice includes up to 5 days respite care for family caregivers, although with a 5% copay.9 Most hospice entities run inpatient facilities for care that cannot be effectively provided at home.
Hospice care has limitations—many set by insurance. Medicare, for example, stipulates that a primary care or hospice physician must certify the patient has a reasonable prognosis of 6 months or less and is expected to have a declining course.27 Patients who survive longer than 6 months are recertified by the same criteria every 60 days.27
Hospice patients forgo treatments aimed at curing their terminal diagnosis.28 Some hospice entities allow noncurative therapies while others do not. Hospice covers prescription medications for symptom control only, although patients can receive care unrelated to the terminal diagnosis under regular benefits.28 Hospice care practices differ from standard care in ways that may surprise patients and families (TABLE 227,28). Patients can disenroll and re-enroll in hospice as they wish.28

Symptom control in advanced cancer
General symptoms
Pain affects 64% of patients with advanced cancer.29 Evidence shows that cancer pain is often undertreated, with a recent systematic review reporting undertreated pain in 32% of patients.30 State and national chronic opioid guidelines do not restrict use for cancer pain.31 Opioids are effective in 75% of cancer patients over 1 month, but there is no evidence of benefit after this period.23 In fact, increasing evidence demonstrates that pain is likely negatively responsive to opioids over longer periods.32 Opioid adverse effects can worsen other cancer symptoms, including depression, anxiety, fatigue, constipation, hypogonadism, and cognitive dysfunction.32 Delaying opioid therapy to end of life can limit adverse effects and may preserve pain-control efficacy for the dying process.
Continue to: Most cancer pain...
Most cancer pain is partially neuropathic, so anticonvulsant and antidepressant medications can help.33 Gabapentin, pregabalin, and duloxetine are recommended based on evidence not restricted to cancer.34 Cannabinoids have been evaluated in 2 trials of cancer pain with 440 patients and showed a borderline significant reduction of pain.35
Palliative radiation therapy can sometimes reduce pain. Bone metastases pain has been studied the most, and the literature suggests that palliative radiation provides improvement for 60% of patients and complete relief to 25% of patients.36 Palliative thoracic radiotherapy for primary or metastatic lung masses reduces pain by more than 70% while improving dyspnea, hemoptysis, and cough in a majority of patients.36
Other uses of palliative radiation have varied evidence. Palliative chemotherapy has less evidence of benefit. In a recent multicenter cohort trial, chemotherapy in end-stage cancer reduced quality of life in patients with good functional status, without affecting quality of life when function was limited.37 Palliative chemotherapy may be beneficial if combined with corticosteroids or radiation therapy.38
Treatment in the last weeks of life centers on opioids; dose increases do not shorten survival.39 Cancer patients are 4 times as likely as noncancer patients to have severe or excruciating pain during the last 3 days of life.40 Narcotics can be titrated aggressively near end of life with less concern for hypotension, respiratory depression, or level of consciousness. Palliative sedation remains an option for uncontrolled pain.41
Anorexia is only a problem if quality of life is affected. Cachexia is caused by increases in cytokines more than reduced calorie intake.42 Reversible causes of reduced eating may be found, including candidiasis, dental problems, depression, or constipation. Megestrol acetate improves weight (number needed to treat = 12), although it significantly increases mortality (number needed to harm = 23), making its use controversial.43 Limited study of cannabinoids has not shown effectiveness in treating anorexia.35
Continue to: Constipation...
Constipation in advanced cancer is often related to opioid therapy, although bowel obstruction must be considered. Opioid-induced constipation affects 40% to 90% of patients on long-term treatment,44 and 5 days of opioid treatment nearly doubles gastrointestinal transit time.45 Opioid-induced constipation can be treated by adding a stimulating laxative followed by a peripheral acting μ-opioid receptor antagonist, such as subcutaneous methylnaltrexone or oral naloxegol.46 These medications are contraindicated if ileus or bowel obstruction is suspected.46
Nausea and vomiting are common in advanced cancer and have numerous causes. Approximately half of reversible causes are medication adverse effects from either chemotherapy or pain medication.47 Opioid rotation may improve symptoms.47 A suspected bowel obstruction should be evaluated by specialists; surgery, palliative chemotherapy, radiation therapy, or stenting may be required. Oncologists can best manage adverse effects of chemotherapy. For nausea and vomiting unrelated to chemotherapy, consider treating constipation and pain. Medication can also be helpful; a systemic review suggests metoclopramide works best, with some evidence supporting other dopaminergic agonists, including haloperidol.47
Fatigue. Both methylphenidate and modafinil have been studied to treat cancer-related fatigue.48 A majority of patients treated with methylphenidate reported less cancer-related fatigue at 4 weeks and wished to continue treatment.49 Modafinil demonstrated minimal improvement in fatigue.50 Sleep disorders, often due to anxiety or sleep apnea, may be a correctable cause.
Later symptoms
Delirium occurs in up to 90% of cancer patients near the end of life, and can signal death.51 Up to half of the delirium seen in palliative care is reversible.51 Reversible causes include uncontrolled pain, medication adverse effects, and urinary and fecal retention (TABLE 348,51). Addressing these factors reduces delirium, based on studies in postoperative patients.52 Consider opioid rotation if neurotoxicity is suspected.51

Delirium can be accompanied by agitation or decreased responsiveness.53 Agitated delirium commonly presents with moaning, facial grimacing, and purposeless repetitive movements, such as plucking bedsheets or removing clothes.51 Delirious patients without agitation have reported, following recovery, distress similar to that experienced by agitated patients.54 Caregivers are most likely to recognize delirium and often become upset. Educating family members about the frequency of delirium can lessen this distress.54
Continue to: Delirium can be treated with...
Delirium can be treated with antipsychotics; haloperidol has been most frequently studied.54 Antipsychotics are effective at reducing agitation but not at restoring cognition.55 Case reports suggest that use of atypical antipsychotics can be beneficial if adverse effects limit haloperidol dosing.56 Agitated delirium is the most frequent indication for palliative sedation.57
Dyspnea. In the last weeks, days, or hours of life, dyspnea is common and often distressing. Dyspnea appears to be multifactorial, worsened by poor control of secretions, airway hyperactivity, and lung pathologies.58 Intravenous hydration may unintentionally exacerbate dyspnea. Hospice providers generally discourage intravenous hydration because relative dehydration reduces terminal respiratory secretions (“death rattle”) and increases patient comfort.59
Some simple nonpharmacologic interventions have benefit. Oxygen is commonly employed, although multiple studies show no benefit over room air.59 Directing a handheld fan at the face does reduce dyspnea, likely by activation of the maxillary branch of the trigeminal nerve.60
Opioids effectively treat dyspnea near the end of life with oral and parenteral dosing, but the evidence does not support nebulized opioids.61 Opioid doses required to treat dyspnea are less than those for pain and do not cause significant respiratory depression.62 If a patient taking opioids experiences dyspnea, a 25% dose increase is recommended.63
Anticholinergic medications can improve excessive airway secretions associated with dyspnea. Glycopyrrolate causes less delirium because it does not cross the blood-brain barrier, while scopolamine patches have reduced anticholinergic adverse effects, but effects are delayed until 12 hours after patch placement.64 Atropine eye drops given sublingually were effective in a small study.65
Continue to: Palliative sedation
Palliative sedation
Palliative sedation can manage intractable symptoms near the end of life. A recent systematic review suggests that palliative sedation does not shorten life.57 Sedation is most often initiated by gradual increases in medication doses.57 Midazolam is most often employed, but antipsychotics are also used.57
CORRESPONDENCE
CDR Michael J. Arnold, MD, Uniformed Services University of the Health Sciences, 4501 Jones Bridge Road, Bethesda, MD 20814; michael.arnold@usuhs.edu.
ACKNOWLEDGEMENT
Kristian Sanchack, MD, and James Higgins, DO, assisted in the preparation of this manuscript.
1. American Cancer Society. Cancer Treatment & Survivorship Facts & Figures 2019-2021. www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/cancer-treatment-and-survivorship-facts-and-figures/cancer-treatment-and-survivorship-facts-and-figures-2019-2021.pdf. Accessed September 4, 2019.
2. Stein KD, Syrjala KL, Andrykowski MA. Physical and psychological long-term and late effects of cancer. Cancer. 2008;112(11 suppl):2577-2592.
3. National Comprehensive Cancer Network. NCCN Guidelines Version 2. 2019. Palliative Care. www.nccn.org/professionals/physician_gls/pdf/palliative.pdf. (Must register an account for access.) Accessed September 4, 2019.
4. American Cancer Society. New CoC accreditation standards gain strong support. www.facs.org/media/press-releases/2011/coc-standards0811. Accessed September 11, 2019.
5. Lupu D; American Academy of Hospice and Palliative Medicine Workforce Task Force. Estimate of current hospice and palliative medicine physician workforce shortage. J Pain Symptom Manage. 2010;40:899-911.
6. Lupu D, Quigley L, Mehfoud N, et al. The growing demand for hospice and palliative medicine physicians: will the supply keep up? J Pain Symptom Manage. 2018;55:1216-1223.
7. Rabow MW, Dahlin C, Calton B, et al. New frontiers in outpatient palliative care for patients with cancer. Cancer Control. 2015;22:465-474.
8. Haun MW, Estel S, Rücker G, et al. Early palliative care for adults with advanced cancer. Cochrane Database of Syst Rev. 2017:CD01129.
9. Buss MK, Rock LK, McCarthy EP. Understanding palliative care and hospice: a review for primary care providers. Mayo Clin Proc. 2017;92:280-286.
10. Hui D. Definition of supportive care: does the semantic matter? Curr Opin Oncol. 2014;26:372-379.
11. Simmons CPL, McMillan DC, McWilliams K, et al. Prognostic tools in patients with advanced cancer: a systematic review. J Pain Symptom Manage. 2017;53:962-970.
12. Lakin JR, Robinson MG, Bernacki RE, et al. Estimating 1-year mortality for high-risk primary care patients using the “surprise” question. JAMA Int Med. 2016;176:1863-1865.
13. Walczak A, Henselmans I, Tattersall MH, et al. A qualitative analysis of responses to a question prompt list and prognosis and end-of-life care discussion prompts delivered in a communication support program. Psychoonchology. 2015;24:287-293.
14. Yamaguchi T, Maeda I, Hatano Y, et al. Effects of end-of-life discussions on the mental health of bereaved family members and quality of patient death and care. J Pain Symptom Manage. 2017;54:17-26.
15. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, caregiver bereavement adjustment. JAMA. 2008;300:1665-1673.
16. Teunissen SC, Wesker W, Kruitwagen C, et al. Symptom prevalence in patients with incurable cancer: a systematic review. J Pain Symptom Manage. 2007;34:94-104.
17. Gao W, Bennett MI, Stark D, et al. Psychological distress in cancer from survivorship to end of life: prevalence, associated factors and clinical implications. Eur J Cancer. 2010;46:2036-2044.
18. Scott IA, Gray LC, Martin JH, et al. Deciding when to stop: towards evidence-based deprescribing of drugs in older populations. Evid Based Med. 2013;18:121-124.
19. Gramling R, Fiscella K, Xing G, et al. Determinants of patient-oncologist prognostic discordance in advanced cancer. JAMA Oncol. 2016;2:1421-1426.
20. Epstein AS, Prigerson HG, O’Reilly EM, et al. Discussions of life expectancy and changes in illness understanding in patients with advanced cancer. J Clin Oncol. 2016;34:2398-2403.
21. Weeks JC, Cook EF, O’Day SJ, et al. Relationship between cancer patients’ predictions of prognosis and their treatment preferences. JAMA. 1998;279:1709-1714.
22. Myers J. Improving the quality of end-of-life discussions. Curr Opin Support Palliat Care. 2015;9:72-76.
23. Corli O, Floriani I, Roberto A, et al. Are strong opioids equally effective and safe in the treatment of chronic cancer pain? A multicenter randomized phase IV ‘real life’ trial on the variability of response to opioids. Ann Oncolog. 2016;27:1107-1115.
24. National Hospice and Palliative Care Organization. NHPCO Facts and Figures. 2018. www.nhpco.org/wp-content/uploads/2019/07/2018_NHPCO_Facts_Figures.pdf. Accessed September 24, 2019.
25. Meier EA, Gallegos JV, Thomas LP, et al. Defining a good death (successful dying): literature review and a call for research and public dialogue. Am J Geriatr Psychiatry. 2016;24:261-271.
26. Morden NE, Chang CH, Jacobson JO, et al. End-of-life care for Medicare beneficiaries with cancer is highly intensive overall and varies widely. Health Aff (Millwood). 2012;31:786-796.
27. Centers for Medicare & Medicaid Services. Medicare Hospice Benefit Facts. www.cgsmedicare.com/hhh/education/materials/pdf/Medicare_Hospice_Benefit_Facts.pdf. Accessed September 11, 2019.
28. Centers for Medicare & Medicaid Services. Medicare Hospice Benefits. www.medicare.gov/pubs/pdf/02154-medicare-hospice-benefits.pdf. Accessed September 11, 2019.
29. van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, et al. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol. 2007;18:1437-1449.
30. Greco MT, Roberto A, Corli O, et al. Quality of cancer pain management: an update of a systematic review of undertreatment of patients with cancer. J Clin Oncol. 2014;32:4149-4154.
31. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm Rep. 2016;65:1-49.
32. Davis MP, Mehta Z. Opioids and chronic pain: where is the balance? Curr Oncol Rep. 2016;18:71.
33. Leppert W, Zajaczkowska R, Wordliczek J, et al. Pathophysiology and clinical characteristics of pain in most common locations in cancer patients. J Physiol Pharmacol. 2016;67:787-799.
34. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14:162-173.
35. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313:2456-2473.
36. Jones JA, Lutz ST, Chow E. et al. Palliative radiotherapy at the end of life: a critical review. CA Cancer J Clin. 2014;64:296-310.
37. Prigerson HG, Bao Y, Shah MA, et al. Chemotherapy use, performance status, and quality of life at the end of life. JAMA Oncol. 2015;1:778-784.
38. Kongsgaard U, Kaasa S, Dale O, et al. Palliative treatment of cancer-related pain. 2005. www.ncbi.nlm.nih.gov/books/NBK464794/. Accessed September 24, 2019.
39. Sathornviriyapong A, Nagaviroj K, Anothaisintawee T. The association between different opioid doses and the survival of advanced cancer patients receiving palliative care. BMC Palliat Care. 2016;15:95.
40. Steindal SA, Bredal IS. Sørbye LW, et al. Pain control at the end of life: a comparative study of hospitalized cancer and noncancer patients. Scand J Caring Sci. 2011;25:771-779.
41. Maltoni M, Setola E. Palliative sedation in patients with cancer. Cancer Control. 2015;22:433-441.
42. Cooper C, Burden ST, Cheng H, et al. Understanding and managing cancer-related weight loss and anorexia: insights from a systematic review of qualitative research. J Cachexia Sarcopenia Muscle. 2015;6:99-111.
43. Ruiz Garcia V, LÓpez-Briz E, Carbonell Sanchis R, et al. Megesterol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database Syst Rev. 2013;28:CD004310.
44. Chey WD, Webster L, Sostek M, et al. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387-2396.
45. Poulsen JL, Nilsson M, Brock C, et al. The impact of opioid treatment on regional gastrointestinal transit. J Neurogastroenterol Motil. 2016;22:282-291.
46. Pergolizzi JV, Raffa RB, Pappagallo M, et al. Peripherally acting μ-opioid receptor antagonists as treatment options for constipation in noncancer pain patients on chronic opioid therapy. Patient Prefer Adherence. 2017;11:107-119.
47. Walsh D, Davis M, Ripamonti C, et al. 2016 updated MASCC/ESMO consensus recommendations: management of nausea and vomiting in advanced cancer. Support Care Cancer. 2017;25:333-340.
48. Mücke M, Mochamat, Cuhls H, et al. Pharmacological treatments for fatigue associated with palliative care. Cochrane Database Syst Rev. 2015(5):CD006788.
49. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J. 2014;20:8-14.
50. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer. 2014;22:1233-1242.
51. Hosker CM, Bennett MI. Delirium and agitation at the end of life. BMJ. 2016;353:i3085.
52. Mercantonio ER, Flacker JM, Wright RJ, et al. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc. 2001;49:516-522.
53. Casarett DJ, Inouye SK. Diagnosis and management of delirium near the end of life. Ann Int Med. 2001;135:32-40.
54. Breitbart W, Alici Y. Agitation and delirium at the end of life: “We couldn’t manage him." JAMA. 2008;300:2898-2910.
55. Candy B, Jackson KC, Jones L, et al. Drug therapy for delirium in terminally ill patients. Cochrane Database Syst Rev. 2012;11:CD004770.
56. Bascom PB, Bordley JL, Lawton AJ. High-dose neuroleptics and neuroleptic rotation for agitated delirium near the end of life. Am J Hosp Palliat Med. 2014;31:808-811.
57. Maltoni M, Scarpi E, Rosati M, et al. Palliative sedation in end-of-life care and survival: a systematic review. J Clin Oncol. 2012;30:1378-1383.
58. Albert RH. End-of-life care: managing common symptoms. Am Fam Physician. 2017;95:356-361.
59. Arenella C. Artificial nutrition and hydration at the end of life: beneficial or harmful? https://americanhospice.org/caregiving/artificial-nutrition-and-hydration-at-the-end-of-life-beneficial-or-harmful/ Accessed September 11, 2019.
60. Booth S, Moffat C, Burkin J, et al. Nonpharmacological interventions for breathlessness. Curr Opinion Support Pall Care. 2011;5:77-86.
61. Barnes H, McDonald J, Smallwood N, et al. Opioids for the palliation of refractory breathlessness in adults with advanced disease and terminal illness. Cochrane Database Syst Rev. 2016(3)CD011008.
62. Lim RB. End-of-life care in patients with advanced lung cancer. Ther Adv Resp Dis. 2016;10:455-467.
63. Kreher M. Symptom control at the end of life. Med Clin North Am. 2016;100:1111-1122.
64. Baralatei FT, Ackerman RJ. Care of patients at the end of life: management of nonpain symptoms. FP Essent. 2016;447:18-24.
65. Protus BM, Grauer PA, Kimbrel JM. Evaluation of atropine 1% ophthalmic solution administered sublingual for the management of terminal respiratory secretions. Am J Hosp Palliat Med. 2013;30:388-392.
As a family physician (FP), you are well positioned to optimize the quality of life of advanced cancer patients as they decline and approach death. You can help them understand their evolving prognosis so that treatment goals can be adjusted, and you can ensure that hospice is implemented early to improve the end-of-life experience. This practical review will help you to provide the best care possible for these patients.
Family physicians can fill a care gap
The term cancer survivor describes a patient who has completed initial cancer treatment. Within this population, many have declining health and ultimately succumb to their disease. There were 16.9 million cancer survivors in the United States as of January 1, 2019,1 with 53% likely to experience significant symptoms and disability.2 More than 600,000 American cancer survivors will die in 2019.3
In 2011, the Commission on Cancer mandated available outpatient palliative care services at certified cancer centers.4 Unfortunately, current palliative care resources fall far short of expected needs. A 2010 estimate of required hospice and palliative care physicians demonstrated a staffing gap of more than 50% among those providing outpatient services.5 The shortage continues,6 and many cancer patients will look to their FP for supportive care.
FPs, in addition to easing symptoms and adverse effects of medication, can educate patients and families about their disease and prognosis. By providing longitudinal care, FPs can identify critical health declines that oncologists, patients, and families often overlook. FPs can also readily appreciate decline, guide patients toward their care goals, and facilitate comfort care—including at the end of life.
Early outpatient palliative care improves quality of life and patient satisfaction. It also may improve survival time and ward off depression.7,8 Some patients and providers resist palliative care due to a misconception that it requires abandoning treatment.9 Actually, palliative care can be given in concert with all active treatments. Many experts recommend a name change from “palliative care” to “supportive care” to dispel this misconception.10
Estimate prognosis using the “surprise question”
Several algorithms are available—using between 2 and 13 patient parameters—to estimate advanced cancer survival. Most of these algorithms are designed to identify the last months or weeks of life, but their utility to predict death within these periods is limited.11
The “surprise question” may be the most valuable prognostic test for primary care. In this test, the physician asks him- or herself: Would I be surprised if this patient died in 1 year? Researchers found that when primary care physicians answered No, their patient was 4 times more likely to die within the year than when they answered Yes.12 This test has a positive predictive value of 20% and a negative predictive value of 95%, making it valuable in distinguishing patients with longer life expectancy.12 Although it overidentifies at-risk patients, the "surprise question" is a simple and sensitive tool for defining prognosis.
Continue to: Priorities for patients likely to live more than a year
Priorities for patients likely to live more than a year
For patients who likely have more than a year to live, the focus is on symptom management and preparation for future decline. Initiate and facilitate discussions about end-of-life topics. Cancer survivors are often open to discussions on these topics, which include advanced directives, home health aides, and hospice.13 Patients can set specific goals for their remaining time, such as engaging in travel, personal projects, or special events. Cancer patients have better end-of-life experiences and families have improved mental health after these discussions.14 Although cancer patients are more likely than other terminal patients to have end-of-life discussions, fewer than 40% ever do.15
Address distressing symptoms with a focus on maintaining function. More than 50% of advanced cancer patients experience fatigue, weakness, pain, weight loss, and anorexia,16 and up to 60% experience psychological distress.17 Deprescribing most preventive medications is recommended with transition to symptomatic treatment.18
Priorities for patients with less than a year to live
For patients who may have less than a year to live, focus shifts to their wishes for the time remaining and priorities for the dying process. Most patients start out with prognostic views more optimistic than those of their physicians, but this gap narrows after end-of-life discussions.19,20 Patients with incurable cancer are less likely to choose aggressive therapy if they believe their 6-month survival probability is less than 90%.21 Honest conversations, with best- and worst-case scenarios, are important to patients and families, and should occur while the patient is well enough to participate and set goals.22
In the last months of life, opioids become the primary treatment for pain and air hunger. As function declines, concerns about such adverse effects as falls and confusion decrease. Opioids have been shown to be most effective over the course of 4 weeks, and avoiding their use in earlier stages may increase their efficacy at the end of life.23
Hospice benefit—more comfort, with limitations
Hospice care consists of services administered by nonprofit and for-profit entities covered by Medicare, Medicaid, and many private insurers.24 Hospice strives to allow patients to approach death in comfort, meeting their goal of a “good death.” A recent literature review identified 4 aspects of a good death that terminally ill patients and their families considered most important: control of the dying process, relief of pain, spirituality, and emotional well-being (TABLE 1).25
Continue to: Hospice use is increasing...
Hospice use is increasing, yet many enroll too late to fully benefit. While cancer patients alone are not currently tracked, the use of hospice by Medicare beneficiaries increased from 44% in 2012 to 48% in 2019.24 In 2017, the median hospice stay was 19 days.24 Unfortunately, though, just 28% of hospice-eligible patients enrolled in hospice in their last week of life.24 Without hospice, patients often receive excessive care near death. More than 6% receive aggressive chemotherapy in their last 2 weeks of life, and nearly 10% receive a life-prolonging procedure in their last month.26
Hospice care replaces standard hospital care, although patients can elect to be followed by their primary care physician.9 Most hospice services are provided as needed or continuously at the patient’s home, including assisted living facilities. And it is also offered as part of hospital care. Hospice services are interdisciplinary, provided by physicians, nurses, social workers, chaplains, and health aides. Hospices have on-call staff to assess and treat complications, avoiding emergency hospital visits.9 And hospice includes up to 5 days respite care for family caregivers, although with a 5% copay.9 Most hospice entities run inpatient facilities for care that cannot be effectively provided at home.
Hospice care has limitations—many set by insurance. Medicare, for example, stipulates that a primary care or hospice physician must certify the patient has a reasonable prognosis of 6 months or less and is expected to have a declining course.27 Patients who survive longer than 6 months are recertified by the same criteria every 60 days.27
Hospice patients forgo treatments aimed at curing their terminal diagnosis.28 Some hospice entities allow noncurative therapies while others do not. Hospice covers prescription medications for symptom control only, although patients can receive care unrelated to the terminal diagnosis under regular benefits.28 Hospice care practices differ from standard care in ways that may surprise patients and families (TABLE 227,28). Patients can disenroll and re-enroll in hospice as they wish.28
Symptom control in advanced cancer
General symptoms
Pain affects 64% of patients with advanced cancer.29 Evidence shows that cancer pain is often undertreated, with a recent systematic review reporting undertreated pain in 32% of patients.30 State and national chronic opioid guidelines do not restrict use for cancer pain.31 Opioids are effective in 75% of cancer patients over 1 month, but there is no evidence of benefit after this period.23 In fact, increasing evidence demonstrates that pain is likely negatively responsive to opioids over longer periods.32 Opioid adverse effects can worsen other cancer symptoms, including depression, anxiety, fatigue, constipation, hypogonadism, and cognitive dysfunction.32 Delaying opioid therapy to end of life can limit adverse effects and may preserve pain-control efficacy for the dying process.
Continue to: Most cancer pain...
Most cancer pain is partially neuropathic, so anticonvulsant and antidepressant medications can help.33 Gabapentin, pregabalin, and duloxetine are recommended based on evidence not restricted to cancer.34 Cannabinoids have been evaluated in 2 trials of cancer pain with 440 patients and showed a borderline significant reduction of pain.35
Palliative radiation therapy can sometimes reduce pain. Bone metastases pain has been studied the most, and the literature suggests that palliative radiation provides improvement for 60% of patients and complete relief to 25% of patients.36 Palliative thoracic radiotherapy for primary or metastatic lung masses reduces pain by more than 70% while improving dyspnea, hemoptysis, and cough in a majority of patients.36
Other uses of palliative radiation have varied evidence. Palliative chemotherapy has less evidence of benefit. In a recent multicenter cohort trial, chemotherapy in end-stage cancer reduced quality of life in patients with good functional status, without affecting quality of life when function was limited.37 Palliative chemotherapy may be beneficial if combined with corticosteroids or radiation therapy.38
Treatment in the last weeks of life centers on opioids; dose increases do not shorten survival.39 Cancer patients are 4 times as likely as noncancer patients to have severe or excruciating pain during the last 3 days of life.40 Narcotics can be titrated aggressively near end of life with less concern for hypotension, respiratory depression, or level of consciousness. Palliative sedation remains an option for uncontrolled pain.41
Anorexia is only a problem if quality of life is affected. Cachexia is caused by increases in cytokines more than reduced calorie intake.42 Reversible causes of reduced eating may be found, including candidiasis, dental problems, depression, or constipation. Megestrol acetate improves weight (number needed to treat = 12), although it significantly increases mortality (number needed to harm = 23), making its use controversial.43 Limited study of cannabinoids has not shown effectiveness in treating anorexia.35
Continue to: Constipation...
Constipation in advanced cancer is often related to opioid therapy, although bowel obstruction must be considered. Opioid-induced constipation affects 40% to 90% of patients on long-term treatment,44 and 5 days of opioid treatment nearly doubles gastrointestinal transit time.45 Opioid-induced constipation can be treated by adding a stimulating laxative followed by a peripheral acting μ-opioid receptor antagonist, such as subcutaneous methylnaltrexone or oral naloxegol.46 These medications are contraindicated if ileus or bowel obstruction is suspected.46
Nausea and vomiting are common in advanced cancer and have numerous causes. Approximately half of reversible causes are medication adverse effects from either chemotherapy or pain medication.47 Opioid rotation may improve symptoms.47 A suspected bowel obstruction should be evaluated by specialists; surgery, palliative chemotherapy, radiation therapy, or stenting may be required. Oncologists can best manage adverse effects of chemotherapy. For nausea and vomiting unrelated to chemotherapy, consider treating constipation and pain. Medication can also be helpful; a systemic review suggests metoclopramide works best, with some evidence supporting other dopaminergic agonists, including haloperidol.47
Fatigue. Both methylphenidate and modafinil have been studied to treat cancer-related fatigue.48 A majority of patients treated with methylphenidate reported less cancer-related fatigue at 4 weeks and wished to continue treatment.49 Modafinil demonstrated minimal improvement in fatigue.50 Sleep disorders, often due to anxiety or sleep apnea, may be a correctable cause.
Later symptoms
Delirium occurs in up to 90% of cancer patients near the end of life, and can signal death.51 Up to half of the delirium seen in palliative care is reversible.51 Reversible causes include uncontrolled pain, medication adverse effects, and urinary and fecal retention (TABLE 348,51). Addressing these factors reduces delirium, based on studies in postoperative patients.52 Consider opioid rotation if neurotoxicity is suspected.51
Delirium can be accompanied by agitation or decreased responsiveness.53 Agitated delirium commonly presents with moaning, facial grimacing, and purposeless repetitive movements, such as plucking bedsheets or removing clothes.51 Delirious patients without agitation have reported, following recovery, distress similar to that experienced by agitated patients.54 Caregivers are most likely to recognize delirium and often become upset. Educating family members about the frequency of delirium can lessen this distress.54
Continue to: Delirium can be treated with...
Delirium can be treated with antipsychotics; haloperidol has been most frequently studied.54 Antipsychotics are effective at reducing agitation but not at restoring cognition.55 Case reports suggest that use of atypical antipsychotics can be beneficial if adverse effects limit haloperidol dosing.56 Agitated delirium is the most frequent indication for palliative sedation.57
Dyspnea. In the last weeks, days, or hours of life, dyspnea is common and often distressing. Dyspnea appears to be multifactorial, worsened by poor control of secretions, airway hyperactivity, and lung pathologies.58 Intravenous hydration may unintentionally exacerbate dyspnea. Hospice providers generally discourage intravenous hydration because relative dehydration reduces terminal respiratory secretions (“death rattle”) and increases patient comfort.59
Some simple nonpharmacologic interventions have benefit. Oxygen is commonly employed, although multiple studies show no benefit over room air.59 Directing a handheld fan at the face does reduce dyspnea, likely by activation of the maxillary branch of the trigeminal nerve.60
Opioids effectively treat dyspnea near the end of life with oral and parenteral dosing, but the evidence does not support nebulized opioids.61 Opioid doses required to treat dyspnea are less than those for pain and do not cause significant respiratory depression.62 If a patient taking opioids experiences dyspnea, a 25% dose increase is recommended.63
Anticholinergic medications can improve excessive airway secretions associated with dyspnea. Glycopyrrolate causes less delirium because it does not cross the blood-brain barrier, while scopolamine patches have reduced anticholinergic adverse effects, but effects are delayed until 12 hours after patch placement.64 Atropine eye drops given sublingually were effective in a small study.65
Continue to: Palliative sedation
Palliative sedation
Palliative sedation can manage intractable symptoms near the end of life. A recent systematic review suggests that palliative sedation does not shorten life.57 Sedation is most often initiated by gradual increases in medication doses.57 Midazolam is most often employed, but antipsychotics are also used.57
CORRESPONDENCE
CDR Michael J. Arnold, MD, Uniformed Services University of the Health Sciences, 4501 Jones Bridge Road, Bethesda, MD 20814; michael.arnold@usuhs.edu.
ACKNOWLEDGEMENT
Kristian Sanchack, MD, and James Higgins, DO, assisted in the preparation of this manuscript.
As a family physician (FP), you are well positioned to optimize the quality of life of advanced cancer patients as they decline and approach death. You can help them understand their evolving prognosis so that treatment goals can be adjusted, and you can ensure that hospice is implemented early to improve the end-of-life experience. This practical review will help you to provide the best care possible for these patients.
Family physicians can fill a care gap
The term cancer survivor describes a patient who has completed initial cancer treatment. Within this population, many have declining health and ultimately succumb to their disease. There were 16.9 million cancer survivors in the United States as of January 1, 2019,1 with 53% likely to experience significant symptoms and disability.2 More than 600,000 American cancer survivors will die in 2019.3
In 2011, the Commission on Cancer mandated available outpatient palliative care services at certified cancer centers.4 Unfortunately, current palliative care resources fall far short of expected needs. A 2010 estimate of required hospice and palliative care physicians demonstrated a staffing gap of more than 50% among those providing outpatient services.5 The shortage continues,6 and many cancer patients will look to their FP for supportive care.
FPs, in addition to easing symptoms and adverse effects of medication, can educate patients and families about their disease and prognosis. By providing longitudinal care, FPs can identify critical health declines that oncologists, patients, and families often overlook. FPs can also readily appreciate decline, guide patients toward their care goals, and facilitate comfort care—including at the end of life.
Early outpatient palliative care improves quality of life and patient satisfaction. It also may improve survival time and ward off depression.7,8 Some patients and providers resist palliative care due to a misconception that it requires abandoning treatment.9 Actually, palliative care can be given in concert with all active treatments. Many experts recommend a name change from “palliative care” to “supportive care” to dispel this misconception.10
Estimate prognosis using the “surprise question”
Several algorithms are available—using between 2 and 13 patient parameters—to estimate advanced cancer survival. Most of these algorithms are designed to identify the last months or weeks of life, but their utility to predict death within these periods is limited.11
The “surprise question” may be the most valuable prognostic test for primary care. In this test, the physician asks him- or herself: Would I be surprised if this patient died in 1 year? Researchers found that when primary care physicians answered No, their patient was 4 times more likely to die within the year than when they answered Yes.12 This test has a positive predictive value of 20% and a negative predictive value of 95%, making it valuable in distinguishing patients with longer life expectancy.12 Although it overidentifies at-risk patients, the "surprise question" is a simple and sensitive tool for defining prognosis.
Continue to: Priorities for patients likely to live more than a year
Priorities for patients likely to live more than a year
For patients who likely have more than a year to live, the focus is on symptom management and preparation for future decline. Initiate and facilitate discussions about end-of-life topics. Cancer survivors are often open to discussions on these topics, which include advanced directives, home health aides, and hospice.13 Patients can set specific goals for their remaining time, such as engaging in travel, personal projects, or special events. Cancer patients have better end-of-life experiences and families have improved mental health after these discussions.14 Although cancer patients are more likely than other terminal patients to have end-of-life discussions, fewer than 40% ever do.15
Address distressing symptoms with a focus on maintaining function. More than 50% of advanced cancer patients experience fatigue, weakness, pain, weight loss, and anorexia,16 and up to 60% experience psychological distress.17 Deprescribing most preventive medications is recommended with transition to symptomatic treatment.18
Priorities for patients with less than a year to live
For patients who may have less than a year to live, focus shifts to their wishes for the time remaining and priorities for the dying process. Most patients start out with prognostic views more optimistic than those of their physicians, but this gap narrows after end-of-life discussions.19,20 Patients with incurable cancer are less likely to choose aggressive therapy if they believe their 6-month survival probability is less than 90%.21 Honest conversations, with best- and worst-case scenarios, are important to patients and families, and should occur while the patient is well enough to participate and set goals.22
In the last months of life, opioids become the primary treatment for pain and air hunger. As function declines, concerns about such adverse effects as falls and confusion decrease. Opioids have been shown to be most effective over the course of 4 weeks, and avoiding their use in earlier stages may increase their efficacy at the end of life.23
Hospice benefit—more comfort, with limitations
Hospice care consists of services administered by nonprofit and for-profit entities covered by Medicare, Medicaid, and many private insurers.24 Hospice strives to allow patients to approach death in comfort, meeting their goal of a “good death.” A recent literature review identified 4 aspects of a good death that terminally ill patients and their families considered most important: control of the dying process, relief of pain, spirituality, and emotional well-being (TABLE 1).25
Continue to: Hospice use is increasing...
Hospice use is increasing, yet many enroll too late to fully benefit. While cancer patients alone are not currently tracked, the use of hospice by Medicare beneficiaries increased from 44% in 2012 to 48% in 2019.24 In 2017, the median hospice stay was 19 days.24 Unfortunately, though, just 28% of hospice-eligible patients enrolled in hospice in their last week of life.24 Without hospice, patients often receive excessive care near death. More than 6% receive aggressive chemotherapy in their last 2 weeks of life, and nearly 10% receive a life-prolonging procedure in their last month.26
Hospice care replaces standard hospital care, although patients can elect to be followed by their primary care physician.9 Most hospice services are provided as needed or continuously at the patient’s home, including assisted living facilities. And it is also offered as part of hospital care. Hospice services are interdisciplinary, provided by physicians, nurses, social workers, chaplains, and health aides. Hospices have on-call staff to assess and treat complications, avoiding emergency hospital visits.9 And hospice includes up to 5 days respite care for family caregivers, although with a 5% copay.9 Most hospice entities run inpatient facilities for care that cannot be effectively provided at home.
Hospice care has limitations—many set by insurance. Medicare, for example, stipulates that a primary care or hospice physician must certify the patient has a reasonable prognosis of 6 months or less and is expected to have a declining course.27 Patients who survive longer than 6 months are recertified by the same criteria every 60 days.27
Hospice patients forgo treatments aimed at curing their terminal diagnosis.28 Some hospice entities allow noncurative therapies while others do not. Hospice covers prescription medications for symptom control only, although patients can receive care unrelated to the terminal diagnosis under regular benefits.28 Hospice care practices differ from standard care in ways that may surprise patients and families (TABLE 227,28). Patients can disenroll and re-enroll in hospice as they wish.28
Symptom control in advanced cancer
General symptoms
Pain affects 64% of patients with advanced cancer.29 Evidence shows that cancer pain is often undertreated, with a recent systematic review reporting undertreated pain in 32% of patients.30 State and national chronic opioid guidelines do not restrict use for cancer pain.31 Opioids are effective in 75% of cancer patients over 1 month, but there is no evidence of benefit after this period.23 In fact, increasing evidence demonstrates that pain is likely negatively responsive to opioids over longer periods.32 Opioid adverse effects can worsen other cancer symptoms, including depression, anxiety, fatigue, constipation, hypogonadism, and cognitive dysfunction.32 Delaying opioid therapy to end of life can limit adverse effects and may preserve pain-control efficacy for the dying process.
Continue to: Most cancer pain...
Most cancer pain is partially neuropathic, so anticonvulsant and antidepressant medications can help.33 Gabapentin, pregabalin, and duloxetine are recommended based on evidence not restricted to cancer.34 Cannabinoids have been evaluated in 2 trials of cancer pain with 440 patients and showed a borderline significant reduction of pain.35
Palliative radiation therapy can sometimes reduce pain. Bone metastases pain has been studied the most, and the literature suggests that palliative radiation provides improvement for 60% of patients and complete relief to 25% of patients.36 Palliative thoracic radiotherapy for primary or metastatic lung masses reduces pain by more than 70% while improving dyspnea, hemoptysis, and cough in a majority of patients.36
Other uses of palliative radiation have varied evidence. Palliative chemotherapy has less evidence of benefit. In a recent multicenter cohort trial, chemotherapy in end-stage cancer reduced quality of life in patients with good functional status, without affecting quality of life when function was limited.37 Palliative chemotherapy may be beneficial if combined with corticosteroids or radiation therapy.38
Treatment in the last weeks of life centers on opioids; dose increases do not shorten survival.39 Cancer patients are 4 times as likely as noncancer patients to have severe or excruciating pain during the last 3 days of life.40 Narcotics can be titrated aggressively near end of life with less concern for hypotension, respiratory depression, or level of consciousness. Palliative sedation remains an option for uncontrolled pain.41
Anorexia is only a problem if quality of life is affected. Cachexia is caused by increases in cytokines more than reduced calorie intake.42 Reversible causes of reduced eating may be found, including candidiasis, dental problems, depression, or constipation. Megestrol acetate improves weight (number needed to treat = 12), although it significantly increases mortality (number needed to harm = 23), making its use controversial.43 Limited study of cannabinoids has not shown effectiveness in treating anorexia.35
Continue to: Constipation...
Constipation in advanced cancer is often related to opioid therapy, although bowel obstruction must be considered. Opioid-induced constipation affects 40% to 90% of patients on long-term treatment,44 and 5 days of opioid treatment nearly doubles gastrointestinal transit time.45 Opioid-induced constipation can be treated by adding a stimulating laxative followed by a peripheral acting μ-opioid receptor antagonist, such as subcutaneous methylnaltrexone or oral naloxegol.46 These medications are contraindicated if ileus or bowel obstruction is suspected.46
Nausea and vomiting are common in advanced cancer and have numerous causes. Approximately half of reversible causes are medication adverse effects from either chemotherapy or pain medication.47 Opioid rotation may improve symptoms.47 A suspected bowel obstruction should be evaluated by specialists; surgery, palliative chemotherapy, radiation therapy, or stenting may be required. Oncologists can best manage adverse effects of chemotherapy. For nausea and vomiting unrelated to chemotherapy, consider treating constipation and pain. Medication can also be helpful; a systemic review suggests metoclopramide works best, with some evidence supporting other dopaminergic agonists, including haloperidol.47
Fatigue. Both methylphenidate and modafinil have been studied to treat cancer-related fatigue.48 A majority of patients treated with methylphenidate reported less cancer-related fatigue at 4 weeks and wished to continue treatment.49 Modafinil demonstrated minimal improvement in fatigue.50 Sleep disorders, often due to anxiety or sleep apnea, may be a correctable cause.
Later symptoms
Delirium occurs in up to 90% of cancer patients near the end of life, and can signal death.51 Up to half of the delirium seen in palliative care is reversible.51 Reversible causes include uncontrolled pain, medication adverse effects, and urinary and fecal retention (TABLE 348,51). Addressing these factors reduces delirium, based on studies in postoperative patients.52 Consider opioid rotation if neurotoxicity is suspected.51
Delirium can be accompanied by agitation or decreased responsiveness.53 Agitated delirium commonly presents with moaning, facial grimacing, and purposeless repetitive movements, such as plucking bedsheets or removing clothes.51 Delirious patients without agitation have reported, following recovery, distress similar to that experienced by agitated patients.54 Caregivers are most likely to recognize delirium and often become upset. Educating family members about the frequency of delirium can lessen this distress.54
Continue to: Delirium can be treated with...
Delirium can be treated with antipsychotics; haloperidol has been most frequently studied.54 Antipsychotics are effective at reducing agitation but not at restoring cognition.55 Case reports suggest that use of atypical antipsychotics can be beneficial if adverse effects limit haloperidol dosing.56 Agitated delirium is the most frequent indication for palliative sedation.57
Dyspnea. In the last weeks, days, or hours of life, dyspnea is common and often distressing. Dyspnea appears to be multifactorial, worsened by poor control of secretions, airway hyperactivity, and lung pathologies.58 Intravenous hydration may unintentionally exacerbate dyspnea. Hospice providers generally discourage intravenous hydration because relative dehydration reduces terminal respiratory secretions (“death rattle”) and increases patient comfort.59
Some simple nonpharmacologic interventions have benefit. Oxygen is commonly employed, although multiple studies show no benefit over room air.59 Directing a handheld fan at the face does reduce dyspnea, likely by activation of the maxillary branch of the trigeminal nerve.60
Opioids effectively treat dyspnea near the end of life with oral and parenteral dosing, but the evidence does not support nebulized opioids.61 Opioid doses required to treat dyspnea are less than those for pain and do not cause significant respiratory depression.62 If a patient taking opioids experiences dyspnea, a 25% dose increase is recommended.63
Anticholinergic medications can improve excessive airway secretions associated with dyspnea. Glycopyrrolate causes less delirium because it does not cross the blood-brain barrier, while scopolamine patches have reduced anticholinergic adverse effects, but effects are delayed until 12 hours after patch placement.64 Atropine eye drops given sublingually were effective in a small study.65
Continue to: Palliative sedation
Palliative sedation
Palliative sedation can manage intractable symptoms near the end of life. A recent systematic review suggests that palliative sedation does not shorten life.57 Sedation is most often initiated by gradual increases in medication doses.57 Midazolam is most often employed, but antipsychotics are also used.57
CORRESPONDENCE
CDR Michael J. Arnold, MD, Uniformed Services University of the Health Sciences, 4501 Jones Bridge Road, Bethesda, MD 20814; michael.arnold@usuhs.edu.
ACKNOWLEDGEMENT
Kristian Sanchack, MD, and James Higgins, DO, assisted in the preparation of this manuscript.
1. American Cancer Society. Cancer Treatment & Survivorship Facts & Figures 2019-2021. www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/cancer-treatment-and-survivorship-facts-and-figures/cancer-treatment-and-survivorship-facts-and-figures-2019-2021.pdf. Accessed September 4, 2019.
2. Stein KD, Syrjala KL, Andrykowski MA. Physical and psychological long-term and late effects of cancer. Cancer. 2008;112(11 suppl):2577-2592.
3. National Comprehensive Cancer Network. NCCN Guidelines Version 2. 2019. Palliative Care. www.nccn.org/professionals/physician_gls/pdf/palliative.pdf. (Must register an account for access.) Accessed September 4, 2019.
4. American Cancer Society. New CoC accreditation standards gain strong support. www.facs.org/media/press-releases/2011/coc-standards0811. Accessed September 11, 2019.
5. Lupu D; American Academy of Hospice and Palliative Medicine Workforce Task Force. Estimate of current hospice and palliative medicine physician workforce shortage. J Pain Symptom Manage. 2010;40:899-911.
6. Lupu D, Quigley L, Mehfoud N, et al. The growing demand for hospice and palliative medicine physicians: will the supply keep up? J Pain Symptom Manage. 2018;55:1216-1223.
7. Rabow MW, Dahlin C, Calton B, et al. New frontiers in outpatient palliative care for patients with cancer. Cancer Control. 2015;22:465-474.
8. Haun MW, Estel S, Rücker G, et al. Early palliative care for adults with advanced cancer. Cochrane Database of Syst Rev. 2017:CD01129.
9. Buss MK, Rock LK, McCarthy EP. Understanding palliative care and hospice: a review for primary care providers. Mayo Clin Proc. 2017;92:280-286.
10. Hui D. Definition of supportive care: does the semantic matter? Curr Opin Oncol. 2014;26:372-379.
11. Simmons CPL, McMillan DC, McWilliams K, et al. Prognostic tools in patients with advanced cancer: a systematic review. J Pain Symptom Manage. 2017;53:962-970.
12. Lakin JR, Robinson MG, Bernacki RE, et al. Estimating 1-year mortality for high-risk primary care patients using the “surprise” question. JAMA Int Med. 2016;176:1863-1865.
13. Walczak A, Henselmans I, Tattersall MH, et al. A qualitative analysis of responses to a question prompt list and prognosis and end-of-life care discussion prompts delivered in a communication support program. Psychoonchology. 2015;24:287-293.
14. Yamaguchi T, Maeda I, Hatano Y, et al. Effects of end-of-life discussions on the mental health of bereaved family members and quality of patient death and care. J Pain Symptom Manage. 2017;54:17-26.
15. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, caregiver bereavement adjustment. JAMA. 2008;300:1665-1673.
16. Teunissen SC, Wesker W, Kruitwagen C, et al. Symptom prevalence in patients with incurable cancer: a systematic review. J Pain Symptom Manage. 2007;34:94-104.
17. Gao W, Bennett MI, Stark D, et al. Psychological distress in cancer from survivorship to end of life: prevalence, associated factors and clinical implications. Eur J Cancer. 2010;46:2036-2044.
18. Scott IA, Gray LC, Martin JH, et al. Deciding when to stop: towards evidence-based deprescribing of drugs in older populations. Evid Based Med. 2013;18:121-124.
19. Gramling R, Fiscella K, Xing G, et al. Determinants of patient-oncologist prognostic discordance in advanced cancer. JAMA Oncol. 2016;2:1421-1426.
20. Epstein AS, Prigerson HG, O’Reilly EM, et al. Discussions of life expectancy and changes in illness understanding in patients with advanced cancer. J Clin Oncol. 2016;34:2398-2403.
21. Weeks JC, Cook EF, O’Day SJ, et al. Relationship between cancer patients’ predictions of prognosis and their treatment preferences. JAMA. 1998;279:1709-1714.
22. Myers J. Improving the quality of end-of-life discussions. Curr Opin Support Palliat Care. 2015;9:72-76.
23. Corli O, Floriani I, Roberto A, et al. Are strong opioids equally effective and safe in the treatment of chronic cancer pain? A multicenter randomized phase IV ‘real life’ trial on the variability of response to opioids. Ann Oncolog. 2016;27:1107-1115.
24. National Hospice and Palliative Care Organization. NHPCO Facts and Figures. 2018. www.nhpco.org/wp-content/uploads/2019/07/2018_NHPCO_Facts_Figures.pdf. Accessed September 24, 2019.
25. Meier EA, Gallegos JV, Thomas LP, et al. Defining a good death (successful dying): literature review and a call for research and public dialogue. Am J Geriatr Psychiatry. 2016;24:261-271.
26. Morden NE, Chang CH, Jacobson JO, et al. End-of-life care for Medicare beneficiaries with cancer is highly intensive overall and varies widely. Health Aff (Millwood). 2012;31:786-796.
27. Centers for Medicare & Medicaid Services. Medicare Hospice Benefit Facts. www.cgsmedicare.com/hhh/education/materials/pdf/Medicare_Hospice_Benefit_Facts.pdf. Accessed September 11, 2019.
28. Centers for Medicare & Medicaid Services. Medicare Hospice Benefits. www.medicare.gov/pubs/pdf/02154-medicare-hospice-benefits.pdf. Accessed September 11, 2019.
29. van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, et al. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol. 2007;18:1437-1449.
30. Greco MT, Roberto A, Corli O, et al. Quality of cancer pain management: an update of a systematic review of undertreatment of patients with cancer. J Clin Oncol. 2014;32:4149-4154.
31. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm Rep. 2016;65:1-49.
32. Davis MP, Mehta Z. Opioids and chronic pain: where is the balance? Curr Oncol Rep. 2016;18:71.
33. Leppert W, Zajaczkowska R, Wordliczek J, et al. Pathophysiology and clinical characteristics of pain in most common locations in cancer patients. J Physiol Pharmacol. 2016;67:787-799.
34. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14:162-173.
35. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313:2456-2473.
36. Jones JA, Lutz ST, Chow E. et al. Palliative radiotherapy at the end of life: a critical review. CA Cancer J Clin. 2014;64:296-310.
37. Prigerson HG, Bao Y, Shah MA, et al. Chemotherapy use, performance status, and quality of life at the end of life. JAMA Oncol. 2015;1:778-784.
38. Kongsgaard U, Kaasa S, Dale O, et al. Palliative treatment of cancer-related pain. 2005. www.ncbi.nlm.nih.gov/books/NBK464794/. Accessed September 24, 2019.
39. Sathornviriyapong A, Nagaviroj K, Anothaisintawee T. The association between different opioid doses and the survival of advanced cancer patients receiving palliative care. BMC Palliat Care. 2016;15:95.
40. Steindal SA, Bredal IS. Sørbye LW, et al. Pain control at the end of life: a comparative study of hospitalized cancer and noncancer patients. Scand J Caring Sci. 2011;25:771-779.
41. Maltoni M, Setola E. Palliative sedation in patients with cancer. Cancer Control. 2015;22:433-441.
42. Cooper C, Burden ST, Cheng H, et al. Understanding and managing cancer-related weight loss and anorexia: insights from a systematic review of qualitative research. J Cachexia Sarcopenia Muscle. 2015;6:99-111.
43. Ruiz Garcia V, LÓpez-Briz E, Carbonell Sanchis R, et al. Megesterol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database Syst Rev. 2013;28:CD004310.
44. Chey WD, Webster L, Sostek M, et al. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387-2396.
45. Poulsen JL, Nilsson M, Brock C, et al. The impact of opioid treatment on regional gastrointestinal transit. J Neurogastroenterol Motil. 2016;22:282-291.
46. Pergolizzi JV, Raffa RB, Pappagallo M, et al. Peripherally acting μ-opioid receptor antagonists as treatment options for constipation in noncancer pain patients on chronic opioid therapy. Patient Prefer Adherence. 2017;11:107-119.
47. Walsh D, Davis M, Ripamonti C, et al. 2016 updated MASCC/ESMO consensus recommendations: management of nausea and vomiting in advanced cancer. Support Care Cancer. 2017;25:333-340.
48. Mücke M, Mochamat, Cuhls H, et al. Pharmacological treatments for fatigue associated with palliative care. Cochrane Database Syst Rev. 2015(5):CD006788.
49. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J. 2014;20:8-14.
50. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer. 2014;22:1233-1242.
51. Hosker CM, Bennett MI. Delirium and agitation at the end of life. BMJ. 2016;353:i3085.
52. Mercantonio ER, Flacker JM, Wright RJ, et al. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc. 2001;49:516-522.
53. Casarett DJ, Inouye SK. Diagnosis and management of delirium near the end of life. Ann Int Med. 2001;135:32-40.
54. Breitbart W, Alici Y. Agitation and delirium at the end of life: “We couldn’t manage him." JAMA. 2008;300:2898-2910.
55. Candy B, Jackson KC, Jones L, et al. Drug therapy for delirium in terminally ill patients. Cochrane Database Syst Rev. 2012;11:CD004770.
56. Bascom PB, Bordley JL, Lawton AJ. High-dose neuroleptics and neuroleptic rotation for agitated delirium near the end of life. Am J Hosp Palliat Med. 2014;31:808-811.
57. Maltoni M, Scarpi E, Rosati M, et al. Palliative sedation in end-of-life care and survival: a systematic review. J Clin Oncol. 2012;30:1378-1383.
58. Albert RH. End-of-life care: managing common symptoms. Am Fam Physician. 2017;95:356-361.
59. Arenella C. Artificial nutrition and hydration at the end of life: beneficial or harmful? https://americanhospice.org/caregiving/artificial-nutrition-and-hydration-at-the-end-of-life-beneficial-or-harmful/ Accessed September 11, 2019.
60. Booth S, Moffat C, Burkin J, et al. Nonpharmacological interventions for breathlessness. Curr Opinion Support Pall Care. 2011;5:77-86.
61. Barnes H, McDonald J, Smallwood N, et al. Opioids for the palliation of refractory breathlessness in adults with advanced disease and terminal illness. Cochrane Database Syst Rev. 2016(3)CD011008.
62. Lim RB. End-of-life care in patients with advanced lung cancer. Ther Adv Resp Dis. 2016;10:455-467.
63. Kreher M. Symptom control at the end of life. Med Clin North Am. 2016;100:1111-1122.
64. Baralatei FT, Ackerman RJ. Care of patients at the end of life: management of nonpain symptoms. FP Essent. 2016;447:18-24.
65. Protus BM, Grauer PA, Kimbrel JM. Evaluation of atropine 1% ophthalmic solution administered sublingual for the management of terminal respiratory secretions. Am J Hosp Palliat Med. 2013;30:388-392.
1. American Cancer Society. Cancer Treatment & Survivorship Facts & Figures 2019-2021. www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/cancer-treatment-and-survivorship-facts-and-figures/cancer-treatment-and-survivorship-facts-and-figures-2019-2021.pdf. Accessed September 4, 2019.
2. Stein KD, Syrjala KL, Andrykowski MA. Physical and psychological long-term and late effects of cancer. Cancer. 2008;112(11 suppl):2577-2592.
3. National Comprehensive Cancer Network. NCCN Guidelines Version 2. 2019. Palliative Care. www.nccn.org/professionals/physician_gls/pdf/palliative.pdf. (Must register an account for access.) Accessed September 4, 2019.
4. American Cancer Society. New CoC accreditation standards gain strong support. www.facs.org/media/press-releases/2011/coc-standards0811. Accessed September 11, 2019.
5. Lupu D; American Academy of Hospice and Palliative Medicine Workforce Task Force. Estimate of current hospice and palliative medicine physician workforce shortage. J Pain Symptom Manage. 2010;40:899-911.
6. Lupu D, Quigley L, Mehfoud N, et al. The growing demand for hospice and palliative medicine physicians: will the supply keep up? J Pain Symptom Manage. 2018;55:1216-1223.
7. Rabow MW, Dahlin C, Calton B, et al. New frontiers in outpatient palliative care for patients with cancer. Cancer Control. 2015;22:465-474.
8. Haun MW, Estel S, Rücker G, et al. Early palliative care for adults with advanced cancer. Cochrane Database of Syst Rev. 2017:CD01129.
9. Buss MK, Rock LK, McCarthy EP. Understanding palliative care and hospice: a review for primary care providers. Mayo Clin Proc. 2017;92:280-286.
10. Hui D. Definition of supportive care: does the semantic matter? Curr Opin Oncol. 2014;26:372-379.
11. Simmons CPL, McMillan DC, McWilliams K, et al. Prognostic tools in patients with advanced cancer: a systematic review. J Pain Symptom Manage. 2017;53:962-970.
12. Lakin JR, Robinson MG, Bernacki RE, et al. Estimating 1-year mortality for high-risk primary care patients using the “surprise” question. JAMA Int Med. 2016;176:1863-1865.
13. Walczak A, Henselmans I, Tattersall MH, et al. A qualitative analysis of responses to a question prompt list and prognosis and end-of-life care discussion prompts delivered in a communication support program. Psychoonchology. 2015;24:287-293.
14. Yamaguchi T, Maeda I, Hatano Y, et al. Effects of end-of-life discussions on the mental health of bereaved family members and quality of patient death and care. J Pain Symptom Manage. 2017;54:17-26.
15. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, caregiver bereavement adjustment. JAMA. 2008;300:1665-1673.
16. Teunissen SC, Wesker W, Kruitwagen C, et al. Symptom prevalence in patients with incurable cancer: a systematic review. J Pain Symptom Manage. 2007;34:94-104.
17. Gao W, Bennett MI, Stark D, et al. Psychological distress in cancer from survivorship to end of life: prevalence, associated factors and clinical implications. Eur J Cancer. 2010;46:2036-2044.
18. Scott IA, Gray LC, Martin JH, et al. Deciding when to stop: towards evidence-based deprescribing of drugs in older populations. Evid Based Med. 2013;18:121-124.
19. Gramling R, Fiscella K, Xing G, et al. Determinants of patient-oncologist prognostic discordance in advanced cancer. JAMA Oncol. 2016;2:1421-1426.
20. Epstein AS, Prigerson HG, O’Reilly EM, et al. Discussions of life expectancy and changes in illness understanding in patients with advanced cancer. J Clin Oncol. 2016;34:2398-2403.
21. Weeks JC, Cook EF, O’Day SJ, et al. Relationship between cancer patients’ predictions of prognosis and their treatment preferences. JAMA. 1998;279:1709-1714.
22. Myers J. Improving the quality of end-of-life discussions. Curr Opin Support Palliat Care. 2015;9:72-76.
23. Corli O, Floriani I, Roberto A, et al. Are strong opioids equally effective and safe in the treatment of chronic cancer pain? A multicenter randomized phase IV ‘real life’ trial on the variability of response to opioids. Ann Oncolog. 2016;27:1107-1115.
24. National Hospice and Palliative Care Organization. NHPCO Facts and Figures. 2018. www.nhpco.org/wp-content/uploads/2019/07/2018_NHPCO_Facts_Figures.pdf. Accessed September 24, 2019.
25. Meier EA, Gallegos JV, Thomas LP, et al. Defining a good death (successful dying): literature review and a call for research and public dialogue. Am J Geriatr Psychiatry. 2016;24:261-271.
26. Morden NE, Chang CH, Jacobson JO, et al. End-of-life care for Medicare beneficiaries with cancer is highly intensive overall and varies widely. Health Aff (Millwood). 2012;31:786-796.
27. Centers for Medicare & Medicaid Services. Medicare Hospice Benefit Facts. www.cgsmedicare.com/hhh/education/materials/pdf/Medicare_Hospice_Benefit_Facts.pdf. Accessed September 11, 2019.
28. Centers for Medicare & Medicaid Services. Medicare Hospice Benefits. www.medicare.gov/pubs/pdf/02154-medicare-hospice-benefits.pdf. Accessed September 11, 2019.
29. van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, et al. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol. 2007;18:1437-1449.
30. Greco MT, Roberto A, Corli O, et al. Quality of cancer pain management: an update of a systematic review of undertreatment of patients with cancer. J Clin Oncol. 2014;32:4149-4154.
31. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm Rep. 2016;65:1-49.
32. Davis MP, Mehta Z. Opioids and chronic pain: where is the balance? Curr Oncol Rep. 2016;18:71.
33. Leppert W, Zajaczkowska R, Wordliczek J, et al. Pathophysiology and clinical characteristics of pain in most common locations in cancer patients. J Physiol Pharmacol. 2016;67:787-799.
34. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14:162-173.
35. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313:2456-2473.
36. Jones JA, Lutz ST, Chow E. et al. Palliative radiotherapy at the end of life: a critical review. CA Cancer J Clin. 2014;64:296-310.
37. Prigerson HG, Bao Y, Shah MA, et al. Chemotherapy use, performance status, and quality of life at the end of life. JAMA Oncol. 2015;1:778-784.
38. Kongsgaard U, Kaasa S, Dale O, et al. Palliative treatment of cancer-related pain. 2005. www.ncbi.nlm.nih.gov/books/NBK464794/. Accessed September 24, 2019.
39. Sathornviriyapong A, Nagaviroj K, Anothaisintawee T. The association between different opioid doses and the survival of advanced cancer patients receiving palliative care. BMC Palliat Care. 2016;15:95.
40. Steindal SA, Bredal IS. Sørbye LW, et al. Pain control at the end of life: a comparative study of hospitalized cancer and noncancer patients. Scand J Caring Sci. 2011;25:771-779.
41. Maltoni M, Setola E. Palliative sedation in patients with cancer. Cancer Control. 2015;22:433-441.
42. Cooper C, Burden ST, Cheng H, et al. Understanding and managing cancer-related weight loss and anorexia: insights from a systematic review of qualitative research. J Cachexia Sarcopenia Muscle. 2015;6:99-111.
43. Ruiz Garcia V, LÓpez-Briz E, Carbonell Sanchis R, et al. Megesterol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database Syst Rev. 2013;28:CD004310.
44. Chey WD, Webster L, Sostek M, et al. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387-2396.
45. Poulsen JL, Nilsson M, Brock C, et al. The impact of opioid treatment on regional gastrointestinal transit. J Neurogastroenterol Motil. 2016;22:282-291.
46. Pergolizzi JV, Raffa RB, Pappagallo M, et al. Peripherally acting μ-opioid receptor antagonists as treatment options for constipation in noncancer pain patients on chronic opioid therapy. Patient Prefer Adherence. 2017;11:107-119.
47. Walsh D, Davis M, Ripamonti C, et al. 2016 updated MASCC/ESMO consensus recommendations: management of nausea and vomiting in advanced cancer. Support Care Cancer. 2017;25:333-340.
48. Mücke M, Mochamat, Cuhls H, et al. Pharmacological treatments for fatigue associated with palliative care. Cochrane Database Syst Rev. 2015(5):CD006788.
49. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J. 2014;20:8-14.
50. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer. 2014;22:1233-1242.
51. Hosker CM, Bennett MI. Delirium and agitation at the end of life. BMJ. 2016;353:i3085.
52. Mercantonio ER, Flacker JM, Wright RJ, et al. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc. 2001;49:516-522.
53. Casarett DJ, Inouye SK. Diagnosis and management of delirium near the end of life. Ann Int Med. 2001;135:32-40.
54. Breitbart W, Alici Y. Agitation and delirium at the end of life: “We couldn’t manage him." JAMA. 2008;300:2898-2910.
55. Candy B, Jackson KC, Jones L, et al. Drug therapy for delirium in terminally ill patients. Cochrane Database Syst Rev. 2012;11:CD004770.
56. Bascom PB, Bordley JL, Lawton AJ. High-dose neuroleptics and neuroleptic rotation for agitated delirium near the end of life. Am J Hosp Palliat Med. 2014;31:808-811.
57. Maltoni M, Scarpi E, Rosati M, et al. Palliative sedation in end-of-life care and survival: a systematic review. J Clin Oncol. 2012;30:1378-1383.
58. Albert RH. End-of-life care: managing common symptoms. Am Fam Physician. 2017;95:356-361.
59. Arenella C. Artificial nutrition and hydration at the end of life: beneficial or harmful? https://americanhospice.org/caregiving/artificial-nutrition-and-hydration-at-the-end-of-life-beneficial-or-harmful/ Accessed September 11, 2019.
60. Booth S, Moffat C, Burkin J, et al. Nonpharmacological interventions for breathlessness. Curr Opinion Support Pall Care. 2011;5:77-86.
61. Barnes H, McDonald J, Smallwood N, et al. Opioids for the palliation of refractory breathlessness in adults with advanced disease and terminal illness. Cochrane Database Syst Rev. 2016(3)CD011008.
62. Lim RB. End-of-life care in patients with advanced lung cancer. Ther Adv Resp Dis. 2016;10:455-467.
63. Kreher M. Symptom control at the end of life. Med Clin North Am. 2016;100:1111-1122.
64. Baralatei FT, Ackerman RJ. Care of patients at the end of life: management of nonpain symptoms. FP Essent. 2016;447:18-24.
65. Protus BM, Grauer PA, Kimbrel JM. Evaluation of atropine 1% ophthalmic solution administered sublingual for the management of terminal respiratory secretions. Am J Hosp Palliat Med. 2013;30:388-392.
PRACTICE RECOMMENDATIONS
› Implement palliative/ supportive care shortly after the diagnosis of an incurable cancer. A
› Candidly communicate prognoses to patients and help them adjust their goals of care. B
› Recommend hospice care for patients who likely have less than 6 months to live, especially with treatmentrelated complications or significant caregiver stress. B
› Delay opioid therapy— if possible—to better control symptoms near the end of life. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Aplastic Anemia: Diagnosis and Treatment
From the Oregon Health and Science University, Portland, OR.
Abstract
- Objective: To describe the current approach to diagnosis and treatment of aplastic anemia.
- Methods: Review of the literature.
- Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), and the treatment and prognosis vary dramatically between these 2 etiologies. Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to life-threatening neutropenic infections or bleeding. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia.
- Conclusion: Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marro
w transplant program to evaluate for early transplantation is the new standard of care for aplastic anemia.
Keywords: inherited marrow failure syndrome; Fanconi anemia; immunosuppression; transplant; stem cell.
Aplastic anemia is a clinical and pathological entity of bone marrow failure that causes progressive loss of hematopoietic progenitor stem cells (HPSC), resulting in pancytopenia.1 Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to having life-threatening neutropenic infections or bleeding. Aplastic anemia results from either inherited or acquired causes, and the pathophysiology and treatment approach vary significantly between these 2 causes. Therefore, recognition of inherited marrow failure diseases, such as Fanconi anemia and telomere biology disorders, is critical to establishing the management plan.
Epidemiology
Aplastic anemia is a rare disorder, with an incidence of approximately 1.5 to 7 cases per million individuals per year.2,3 A recent Scandinavian study reported that the incidence of aplastic anemia among the Swedish population is 2.3 cases per million individuals per year, with a median age at diagnosis of 60 years and a slight female predominance (52% versus 48%, respectively).2 This data is congruent with prior observations made in Barcelona, where the incidence was 2.34 cases per million individuals per year, albeit with a slightly higher incidence in males compared to females (2.54 versus 2.16, respectively).4 The incidence of aplastic anemia varies globally, with a disproportionate increase in incidence seen among Asian populations, with rates as high as 8.8 per million individuals per year.3-5 This variation in incidence in Asia versus other countries has not been well explained. There appears to be a bimodal distribution, with incidence peaks seen in young adults and in older adults.2,3,6
Pathophysiology
Acquired Aplastic Anemia
The leading hypothesis as to the cause of most cases of acquired aplastic anemia is that a dysregulated immune system destroys HPSCs. Inciting etiologies implicated in the development of acquired aplastic anemia include pregnancy, infection, medications, and exposure to certain chemicals, such as benzene.1,7 The historical understanding of acquired aplastic anemia implicates cytotoxic T-lymphocyte–mediated destruction of CD34+ hematopoietic stem cells.1,8,9 This hypothesis served as the basis for treatment of acquired aplastic anemia with immunosuppressive therapy, predominantly anti-thymocyte globulin (ATG) combined with cyclosporine A.1,8 More recent work has focused on cytokine interactions, particularly the suppressive role of interferon (IFN)-γ on hematopoietic stem cells independent of T-lymphocyte–mediated destruction, which has been demonstrated in a murine model.8 The interaction of IFN-γ with the hematopoietic stem cell pool is dynamic. IFN-γ levels are elevated during an acute inflammatory response, such as a viral infection, providing further basis for the immune-mediated nature of the acquired disease.10 Specifically, in vitro studies suggest the effects of IFN-γ on HPSC may be secondary to interruption of thrombopoietin and its respective signaling pathways, which play a key role in hematopoietic stem cell renewal.11 Eltrombopag, a thrombopoietin receptor antagonist, has shown promise in the treatment of refractory aplastic anemia, with studies indicating that its effectiveness is independent of IFN-γ levels.11,12
Inherited Aplastic Anemia
The inherited marrow failure syndromes (IMFSs) are a group of disorders characterized by cellular maintenance and repair defects, leading to cytopenias, increased cancer risk, structural defects, and risk of end organ damage, such as liver cirrhosis and pulmonary fibrosis.13-15 The most common diseases include Fanconi anemia, dyskeratosis congenita/telomere biology disorders, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome, but with the advent of whole exome sequencing, new syndromes continue to be discovered. While classically these disorders present in children, adult presentations are now commonplace. Broadly, the pathophysiology of inherited aplastic anemia relates to the defective HPSCs and an accelerated decline of the hematopoietic stem cell compartment.
The most common IMFSs, Fanconi anemia and telomere biology disorders, are associated with numerous mutations in DNA damage repair pathways and telomere maintenance pathways. TERT, DKC, and TERC mutations are most commonly associated with dyskeratosis congenita, but may also be found infrequently in patients with aplastic anemia presenting at an older age in the absence of the classic phenotypical features.1,16,17 The recognition of an underlying genetic disorder or telomere biology disorder leading to constitutional aplastic anemia is significant, as these conditions are associated not only with marrow failure, but also with endocrinopathies, organ fibrosis, and and hematopoietic and solid organ malignancies.13-15 In particular, TERT and TERC gene mutations have been associated with dyskeratosis congenita as well as pulmonary fibrosis and cirrhosis.18,19 The implications of early diagnosis of an IMFS lie in the approach to treatment and prognosis.
Clonal Disorders and Secondary Malignancies
Myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) are 2 clonal disorders that may arise from a background of aplastic anemia.9,20,21 Hypoplastic MDS can be difficult to differentiate from aplastic anemia at diagnosis based on morphology alone, although recent work has demonstrated that molecular testing for somatic mutations in ASXL1, DNMT3A, and BCOR can aid in differentiating a subset of aplastic anemia patients who are more likely to progress to MDS.21 Clonal populations of cells harboring 6p uniparental disomy are seen in more than 10% of patients with aplastic anemia on cytogenetic analysis, which can help differentiate the diseases.9 Yoshizato and colleagues found lower rates of ASXL1 and DNMT3A mutations in patients with aplastic anemia as compared with patients with MDS or AML. In this study, patients with aplastic anemia had higher rates of mutations in PIGA (reflecting the increased paroxysmal nocturnal hemoglobinuria [PNH] clonality seen in aplastic anemia) and BCOR.9 Mutations were also found in genes commonly mutated in MDS and AML, including TET2, RUNX1, TP53, and JAK2, albeit at lower frequencies.9 These mutations as a whole have not predicted response to therapy or prognosis. However, when performing survival analysis in patients with specific mutations, those commonly encountered in MDS/AML (ASXL1, DNMT3A, TP53, RUNX1, CSMD1) are associated with faster progression to overt MDS/AML and decreased overall survival (OS),20,21 suggesting these mutations may represent early clonality that can lead to clonal evolution and the development of secondary malignancies. Conversely, mutations in BCOR and BCORL appear to identify patients who may have a favorable outcome in response to immunosuppressive therapy and, similar to patients with PIGA mutations, improved OS.9
Paroxysmal Nocturnal Hemoglobinuria
In addition to having an increased risk of myelodysplasia and malignancy due to the development of a dominant pre-malignant clone, patients with aplastic anemia often harbor progenitor cell clones associated with PNH.1,17 PNH clones have been identified in more than 50% of patients with aplastic anemia.22,23 PNH represents a clonal disorder of hematopoiesis in which cells harbor X-linked somatic mutations in the PIGA gene; this gene encodes a protein responsible for the synthesis of glycosylphosphatidylinositol anchors on the cell surface.22,24 The lack of these cell surface proteins, specifically CD55 (also known as decay accelerating factor) and CD59 (also known as membrane inhibitor of reactive lysis), predisposes red cells to increased complement-mediated lysis.25 The exact mechanism for the development of these clones in patients with aplastic anemia is not fully understood. Current theories hypothesize that the clones are protected from the immune-mediated destruction of normal hematopoietic stem cells due to the absence of the cell surface proteins.1,20 The role of these clones over time in patients with aplastic anemia is less clear, though recent work demonstrated that despite differences in clonality over the disease course, aplastic anemia patients with small PNH clones are less likely to develop overt hemolysis and larger PNH clones compared to patients harboring larger (≥ 50%) PNH clones at diagnosis.23,26,27 Additionally, PNH clones in patients with aplastic anemia infrequently become clinically significant.27 It should be noted that these conditions exist along a continuum; that is, patients with aplastic anemia may develop PNH clones, while conversely patients with PNH may develop aplastic anemia.20 Patients with PNH clones should be followed via peripheral blood flow cytometry and complete blood count to track clonal stability and identify clinically significant PNH among aplastic anemia patients.28
Clinical Presentation
Patients with aplastic anemia typically are diagnosed either due to asymptomatic cytopenias found on peripheral blood sampling, symptomatic anemia, bleeding secondary to thrombocytopenia, or wound healing and infectious complications related to neutropenia.29 A thorough history to understand the timing of symptoms, recent infectious symptoms/exposure, habits, and chemical or toxin exposures (including medications, travel, and supplements) helps guide diagnostic testing. Family history is also critical, with attention given to premature graying; pulmonary, renal, and liver disease; and blood disorders.
Patients with an IMFS (eg, Fanconi anemia or dyskeratosis congenita) may have associated phenotypical findings such as urogenital abnormalities or short stature; in addition, those with dyskeratosis congenita may present with the classic triad of oral leukoplakia, lacy skin pigmentation, and dystrophic nails.7 However, classic phenotypical findings may be lacking in up to 30% to 40% of patients with an IMFS.7 As described previously, while congenital malformations are common in Fanconi anemia and dyskeratosis congenita, a third of patients may have no or only subtle phenotypical abnormalities, including alterations in skin or hair pigmentation, skeletal and growth abnormalities, and endocrine disorders.30 The International Fanconi Anemia Registry identified central nervous system, genitourinary, skin and musculoskeletal, ophthalmic, and gastrointestinal system malformations among children with Fanconi anemia.31,32 Patients with dyskeratosis congenita may present with pulmonary fibrosis, hepatic cirrhosis, or premature graying, as highlighted in a recent study by DiNardo and colleagues.33 Therefore, physicians must have a heightened index of suspicion in patients with subtle phenotypical findings and associated cytopenias.
Diagnosis
The diagnosis of aplastic anemia should be suspected in any patient presenting with pancytopenia. Aplastic anemia is a diagnosis of exclusion.34 Other conditions associated with peripheral blood pancytopenia should be considered, including infections (HIV, hepatitis, parvovirus B19, cytomegalovirus, Epstein-Barr virus, varicella-zoster virus), nutritional deficiencies (vitamin B12, folate, copper, zinc), autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, hemophagocytic lymphohistiocytosis), hypersplenism, marrow-occupying diseases (eg, leukemia, lymphoma, MDS), solid malignancies, and fibrosis (Table).7

Diagnostic Evaluation
The workup for aplastic anemia should include a thorough history and physical exam to search simultaneously for alternative diagnoses and clues pointing to potential etiologic agents.7 Diagnostic tests to be performed include a complete blood count with differential, reticulocyte count, immature platelet fraction, flow cytometry (to rule out lymphoproliferative disorders and atypical myeloid cells and to evaluate for PNH), and bone marrow biopsy with subsequent cytogenetic, immunohistochemical, and molecular testing.35 Typical findings in aplastic anemia include peripheral blood pancytopenia without dysplastic features and bone marrow biopsy demonstrating a hypocellular marrow.7 A relative lymphocytosis in the peripheral blood is common.7 In patients with a significant PNH clone, a macrocytosis along with elevated lactate dehydrogenase and elevated reticulocyte and granulocyte counts may be present.36
The diagnosis (based on the Camitta criteria37 and modified Camitta criteria38 for severe aplastic anemia) requires 2 of the following findings on peripheral blood samples:
- Absolute neutrophil count (ANC) < 500 cells/µL
- Platelet count < 20,000 cells/µL
- Reticulocyte count < 1% corrected or < 20,000 cells/µL.35
In addition to peripheral blood findings, bone marrow biopsy is essential for the diagnosis, and should demonstrate a markedly hypocellular marrow (cellularity < 25%), occasionally with an increase in T lymphocytes.7,39 Because marrow cellularity varies with age and can be challenging to assess, additional biopsies may be needed to confirm the diagnosis.29 A 1- to 2-cm core biopsy is necessary to confirm hypocellularity, as small areas of residual hematopoiesis may be present and obscure the diagnosis.35
Excluding Hypocellular MDS and IMFS
Excluding hypocellular MDS is challenging, especially in the older adult presenting with aplastic anemia, as patients with aplastic anemia may have some degree of erythroid dysplasia on bone marrow morphology.36 The presence of a PNH clone on flow cytometry can aid in diagnosing aplastic anemia and excluding MDS,34 although PNH clones can be present in refractory anemia MDS. Patients with aplastic anemia have a lower ratio of CD34+ cells compared to those with hypoplastic MDS, with 1 study demonstrating a mean CD34+ percentage of < 0.5% in aplastic anemia versus 3.7% in hypoplastic MDS.40 Cytogenetic and molecular testing can also aid in making this distinction by identifying mutations commonly implicated in MDS.7 The presence of monosomy 7 (-7) in aplastic anemia patients is associated with a poor overall prognosis.34,41
Peripheral blood screening using chromosome breakage analysis (done using either mitomycin C or diepoxybutane as in vitro DNA-crosslinking agents)42 and telomere length testing (of peripheral blood leukocytes) is necessary to exclude the main IMFSs, Fanconi anemia and telomere biology disorders, respectively. Ruling out these conditions is imperative, as the approach to treatment varies significantly between IMFS and aplastic anemia. Patients with shortened telomeres should undergo genetic screening for mutations in the telomere maintenance genes to evaluate the underlying defect leading to shortened telomeres. Patients with increased peripheral blood breakage should have genetic testing to detect mutations associated with Fanconi anemia.
Classification
Once the diagnosis of aplastic anemia has been made, the patient should be classified according to the severity of their disease. Disease severity is determined based on peripheral blood ANC: non-severe aplastic anemia (NSAA), ANC > 500 polymorphonuclear neutrophils (PMNs)/µL; severe aplastic anemia (SAA), 200–500 PMNs/µL; and very severe aplastic anemia (VSAA), 0–200 PMNs/µL.4,34 Disease classification is important, as VSAA is associated with a decreased OS compared to SAA.2 Disease classification may affect treatment decisions, as patients with NSAA may be observed for a short period of time, while, conversely, patients with SAA have a worse prognosis with delays in therapy.43-45
Treatment of Inherited Aplastic Anemia
First-line treatment options for patients with IMFS are androgen therapy and hematopoietic stem cell transplant (HSCT). When evaluating patients for HSCT, it is critical to identify the presence of an IMFS, as the risk and mortality associated with the conditioning regimen, stem cell source, graft-versus-host disease (GVHD), and secondary malignancies differ between patients with IMFS and those with acquired marrow failure syndromes or hematologic malignancies.
Potential sibling donors need to be screened for donor candidacy as well as for the inherited defect. Among patients with Fanconi anemia or a telomere biology disorder, the stem cell source must be considered, with multiple studies in IMFSs and SAA showing superior outcomes with a bone marrow product compared to peripheral blood stem cells.46-48 In IMFS patients, the donor cell type may affect the choice of conditioning regimen.5,6 Reduced-intensity conditioning in lieu of myeloablative conditioning without total body irradiation has proved feasible in patients with Fanconi anemia, and is associated with a reduced risk of secondary malignancies.49,50 Incorporation of fludarabine in the conditioning regimen of patients without a matched sibling donor is associated with superior engraftment and survival46,49,51 compared to cyclophosphamide conditioning, which was historically used in matched related donors.50,52 Adding fludarabine appears to be especially beneficial in older patients, in whom its use is associated with lower rates of graft failure, likely due to increased immunosuppression at the time of engraftment.51,53 Fludarabine has also been incorporated into conditioning regimens for patients with a telomere biology disorder, but outcomes data are limited.5
For patients presenting with AML or a high-risk MDS who are subsequently diagnosed with an IMFS, treatment can be more complex, as these patients are at high risk for toxicity from standard chemotherapy. Limited data suggest that induction therapy and transplantation are feasible in this group of patients, and this approach is associated with increased OS, despite lower OS rates than those of IMFS patients who present prior to the development of MDS or AML.54,55 Further work is needed to determine the optimal induction regimen that balances the risks of treatment-related mortality and complications associated with conditioning regimens, risk of relapse, and risk of secondary malignancies, especially in the cohort of patients diagnosed at an older age.
Treatment of Acquired Aplastic Anemia
Supportive Care
While the workup and treatment plan are being established, attention should be directed at supportive care for prevention of complications. The most common complications leading to death in patients with significant pancytopenia and neutropenia are opportunistic infections and hemorrhagic complications.2
Transfusion support is critical to avoid symptomatic anemia and hemorrhagic complications related to thrombocytopenia, which typically occur with platelet counts lower than 10,000 cells/µL. However, transfusion carries the risk of alloimmunization (which may persist for years following transfusion) and transfusion-related graft versus host disease (trGVHD), and thus use of transfusion should be minimized when possible.56,57 All blood products given to patients with aplastic anemia should be irradiated and leukoreduced to reduce the risk of both alloimmunization and trGVHD. Guidelines from the British Society for Haematology recommend routine screening for Rh and Kell antibodies to reduce the risk of alloimmunization.58 Infectious complications remain a common cause of morbidity and mortality in patients with aplastic anemia who have prolonged neutropenia (defined as an ANC < 500 cells/µL).59-62 Therefore, patients should receive broad-spectrum antibiotics with antipseudomonal coverage. In a study evaluating the role of granulocyte-colony stimulating factor (G-CSF) in patients with SAA receiving immunosuppressive therapy, 55% of all patient deaths were secondary to infection.63 There was no OS benefit seen in patients who received G-CSF, though a significantly lower rate of infection was observed in the G-CSF arm compared to those not receiving G-CSF (56% versus 81%, P = 0.006). This difference was largely driven by a decrease in infectious episodes in patients with VSAA treated with G-CSF as compared to those who did not receive this therapy (22% versus 48%, P = 0.014).63
Angio-invasive pulmonary aspergillosis and Zygomycetes (eg, Rhizopus, Mucor species) remain major causes of mortality related to opportunistic mycotic infections in patients with aplastic anemia.18 The infectious risk is directly related to the duration and severity of neutropenia, with one study demonstrating a significant increase in risk in AML patients with neutropenia lasting longer than 3 weeks.64 Invasive fungal infections carry a high mortality in patients with severe neutropenia, though due to earlier recognition and empiric antifungal therapy with extended-spectrum azoles, overall mortality secondary to invasive fungal infections is declining.62,65
While neutropenia related to cytotoxic chemotherapy is commonly associated with gram-negative bacteria due to disruption of mucosal barriers, patients with aplastic anemia have an increased incidence of gram-positive bacteremia with staphylococcal species compared to other neutropenic populations.61,62 This appears to be changing with time. Valdez et al demonstrated a decrease in prevalence of coagulase-negative staphylococcal infections, increased prevalence of gram-positive bacilli bacteremia, and no change in prevalence of gram-negative bacteremia in patients with aplastic anemia treated between 1989 and 2008.65 Gram-negative bacteremia caused by Stenotrophomonas maltophila, Escherichia coli, Klebsiella pneumoniae, Citrobacter, and Proteus has also been reported.62 Despite a lack of clinical trials investigating the role of antifungal and antibacterial prophylaxis for patients with aplastic anemia, most centers initiate antifungal prophylaxis in patients with SAA or VSAA with an anti-mold agent such as voriconazole or posaconazole (which has the additional benefit compared to voriconazole of covering Mucor species).60,66 This is especially true for patients who have received ATG or undergone HSCT. For antimicrobial prophylaxis, a fluoroquinolone antibiotic with a spectrum of activity against Pseudomonas should be considered for patients with an ANC < 500 cells/µL.60 Acyclovir or valacyclovir prophylaxis is recommended for varicella-zoster virus and herpes simplex virus. Cytomegalovirus reactivation is minimal in patients with aplastic anemia, unless multiple courses of ATG are used.
Iron overload is another complication the provider must be aware of in the setting of increased transfusions in aplastic anemia patients. Lee and colleagues showed that iron chelation therapy using deferasirox is effective at reducing serum ferritin levels in patients with aplastic anemia (median ferritin level of 3254 ng/mL prior to therapy, 1854 ng/mL following), and is associated with no serious adverse events (most common adverse events included nausea, diarrhea, vomiting, and rash).67 Approximately 25% of patients in this trial had an increase in creatinine, with patients taking concomitant cyclosporine affected to a greater degree than those on chelation therapy alone. For patients following HSCT or with improved hematopoiesis following immunosuppressive therapy, phlebotomy can be used to treat iron overload in lieu of chelation therapy.58
Approach to Therapy
The main treatment options for SAA and VSAA include allogeneic bone marrow transplant and immunosuppression. The deciding factors as to which treatment is best initially depends on the availability of HLA-matched related donors and age (Figure 1 and Figure 2). Survival is decreased in patients with SAA or VSAA who delay initiation of therapy, and therefore prompt referral for HLA typing and evaluation for bone marrow transplant is a very important first step in managing aplastic anemia.

Matched Sibling Donor Transplant. Current standards of care recommend HLA-matched sibling donor transplant for patients with SAA or VSAA who are younger than 50 years, with the caveat that integration of fludarabine and reduced cyclophosphamide dosing along with ATG shows the best overall outcomes. Locasciulli and colleagues examined outcomes in patients given either immunosuppressive therapy or sibling HSCT between 1991-1996 and 1997-2002, respectively, and found that sibling HSCT was associated with a superior 10-year OS compared to immunosuppressive therapy (73% versus 68%).43 Interestingly in this study, there was no OS improvement seen with immunosuppressive therapy alone (69% versus 73%) between the 2 time periods, despite increased OS in both sibling HSCT (74% and 80%) and MUD HSCT (38% and 65%).43 Though total body irradiation has been used in the past, it is typically not included in current conditioning regimens for matched related donor transplants.68

Current conditioning regimens typically use a combination of cyclophosphamide and ATG,69,70 with or without fludarabine. Fludarabine-based conditioning regimens have shown promise in patients undergoing sibling HSCT. Maury and colleagues evaluated the role of fludarabine in addition to low-dose cyclophosphamide and ATG compared to cyclophosphamide alone or in combination with ATG in patients over age 30 undergoing sibling HSCT.53 There was a nonsignificant improvement in 5-year OS in the fludarabine arm compared to controls (77% ± 8% versus 60% ± 3%, P = 0.14) in the pooled analysis, but when adjusted for age the fludarabine arm had a significantly lower relative risk (RR) of death (0.44; P = 0.04) compared to the control arm. Shin et al reported outcomes with fludarabine/cyclophosphamide/ATG, with excellent overall outcomes and no difference in patients older or younger than 40 years.71
Kim et al evaluated their experience with patients older than 40 years receiving matched related donors, finding comparable outcomes in those ages 41 to 50 years compared to younger patients. Outcomes declined in those over the age of 50 years.72 Long-term data for matched related donor transplant for aplastic anemia show excellent long-term outcomes, with minimal chronic GVHD and good performance status.73 Hence, these factors support the role of matched related donor transplant as the initial treatment in SAA and VSAA.
Regarding the role of transplant for patients who lack a matched related donor, a growing body of literature demonstrating identical outcomes between matched related and MUD transplants for pediatric patients74,75 supports recent recommendations for upfront unrelated donor transplantation for aplastic anemia.76,77
Immunosuppressive Therapy. For patients without an HLA-matched sibling donor or those who are older than 50 years of age, immunosuppressive therapy is the first-line therapy. ATG and cyclosporine A are the treatments of choice.78 The potential effectiveness of immunosuppressive therapy in treating aplastic anemia was initially observed in patients in whom autologous transplant failed but who still experienced hematopoietic reconstitution despite the failed graft; this observation led to the hypothesis that the conditioning regimen may have an effect on hematopoiesis.59,78,79
Immunosuppressive therapy with ATG has been used for the treatment of aplastic anemia since the 1980s.80 Historically, rabbit ATG had been used, but a 2011 study of horse ATG demonstrated superior hematological response at 6 months compared to rabbit ATG (68% versus 37%).59 Superior survival was also seen with horse ATG compared to rabbit ATG (3-year OS, 96% versus 76%). Due to these results, horse ATG is preferred over rabbit ATG. ATG should be used in combination with cyclosporine A to optimize outcomes.
Early studies also demonstrated the efficacy of cyclosporine A in the treatment of aplastic anemia, with response rates equivalent to that of ATG monotherapy.81 Recent publications still note the efficacy of cyclosporine A in the treatment of aplastic anemia. Its role as an affordable option for single-agent therapy in developing countries is intriguing.81 The combination of ATG and cyclosporine A was proven superior to either agent alone in a study by Frickhofen et al.79 In this study, patients were randomly assigned to a control arm that received ATG plus methylprednisolone or to an arm that received ATG plus cyclosporine A and methylprednisolone. At 6 months, 70% of patients in the cyclosporine A arm had a complete remission (CR) or partial remission compared to 46% in the control arm.82 Further work confirmed the long-term efficacy of this regimen, reporting a 7-year OS of 55%.83 Among a pediatric population, immunosuppressive therapy was associated with an 83% 10-year OS.84
It is recommended that patients remain on cyclosporine therapy for a minimum of 6 months, after which a gradual taper may be considered, although there is variation among practitioners, with some continuing immunosuppressive therapy for a minimum of 12 months due to a proportion of patients being cyclosporine dependent.34,84 A study found that within a population of patients who responded to immunosuppressive therapy, 18% became cyclosporine dependent.84 The median duration of cyclosporine A treatment at full dose was 12 months, with tapering completed over a median of 19 months after patients had been in a stable CR for a minimum of 3 months. Relapse occurred more often when patients were tapered quickly (decrease ≥ 0.8 mg/kg/month) compared to slowly (0.4-0.7 mg/kg/month) or very slowly (< 0.3 mg/kg/month).
Townsley and colleagues recently investigated incorporating the use of the thrombopoietin receptor agonist eltrombopag with immunosuppressive therapy as first-line therapy in aplastic anemia.85 When given at a dose of 150 mg daily in patients ages 12 years and older or 75 mg daily in patients younger than 12 years, in conjunction with cyclosporine A and ATG, patients demonstrated markedly improved hematological response compared to historical treatment with standard immunosuppressive therapy alone.45 In the patient cohort administered eltrombopag starting on day 1 and continuing for 6 months, the complete response rate was 58%. Eltrombopag led to improvement in all cell lines among all treatment subgroups, and OS (censored for patients who proceeded to transplant) was 99% at 2 years.12 Overall, toxicities associated with this therapy were low, with liver enzyme elevations most commonly observed.85 Recently, a phase 2 trial of immunosuppressive therapy with or without eltrombopag was reported. Of the 38 patients enrolled, overall response, complete response, and time to response were not statistically different.86 With this recent finding, the role of eltrombopag in addition to immunosuppressive therapy is not clearly defined, and further studies are warranted.
OS for patients who do not respond to immunosuppressive therapy is approximately 57% at 5 years, largely due to improved supportive measures among this patient population.48,65 Therefore, it is important to recognize those patients who have a low chance of response so that second-line therapy can be pursued to improve outcomes.
Matched Unrelated Donor Transplant. For patients with refractory disease following immunosuppressive therapy who lack a matched sibling donor, MUD HSCT is considered standard therapy given the marked improvement in overall outcomes with modulating conditioning regimens and high-resolution HLA typing. A European Society for Blood and Marrow Transplantation (EBMT) analysis comparing matched sibling HSCT to MUD HSCT noted significantly higher rates of acute grade II-IV and grade III-IV GVHD (grade II-IV 13% versus 25%, grade III-IV 5% versus 10%) among patients undergoing MUD transplant.47 Chronic GVHD rates were 14% in the sibling group, as compared to 26% in the MUD group. Factors associated with improved survival in this analysis include transplant under age 20 years (84% versus 72%), transplant within 6 months of diagnosis (85% versus 72%), the use of ATG in the conditioning regimen (81% versus 73%), and cytomegalovirus-negative donor and recipient as compared to other combinations (82% versus 76%).87 Interestingly, this study demonstrated that OS was not significantly increased when using a sibling HSCT compared to a MUD HSCT, likely as a result of improved understanding of conditioning regimens, GVHD prophylaxis, and supportive care.
Additional studies of MUD HSCT have shown outcomes similar to those seen in sibling HSCT.34,48 A French study found a significant increase in survival in patients undergoing MUD HSCT compared to historical cohorts (2000-2005: OS 52%; 2006-2012: OS 74%).75 The majority of patients underwent conditioning with cyclophosphamide or a combination of busulfan and cyclophosphamide, with or without fludarabine; 81% of patients underwent in vivo T-cell depletion, and a bone marrow donor source was utilized. OS was significantly lower in patients over age 30 years undergoing MUD HSCT (57%) compared to those under age 30 years (70%). Improved OS was also seen when patients underwent transplant within 1 year of diagnosis and when a 10/10 matched donor (compared to a 9/10 mismatched donor) was utilized.48
A 2015 study investigated the role of MUD HSCT as frontline therapy instead of immunosuppressive therapy in patients without a matched sibling donor.75 The 2-year OS was 96% in the MUD HSCT cohort compared to 91%, 94%, and 74% in historical cohorts of sibling HSCT, frontline immunosuppressive therapy, and second-line MUD HSCT following failed immunosuppressive therapy, respectively. Additionally, event-free survival in the MUD HSCT cohort (defined by the authors as death, lack of response, relapse, occurrence of clonal evolution/clinical PNH, malignancies developing over follow‐up, and transplant for patients receiving immunosuppressive therapy frontline) was similar compared to sibling HSCT and superior to frontline immunosuppressive therapy and second-line MUD HSCT. Furthermore, Samarasinghe et al highlighted the importance of in vivo T-cell depletion with either ATG or alemtuzumab (anti-CD52 monoclonal antibody) in the prevention of acute and chronic GVHD in both sibling HSCT and MUD HSCT.88
With continued improvement of less toxic and more immunomodulating conditioning regimens,utilization of bone marrow as a donor cell source, in vivo T-cell depletion, and use of GVHD and antimicrobial prophylaxis, more clinical evidence supports elevating MUD HSCT in the treatment plan for patients without a matched sibling donor.89 However, there is still a large population of patients without matched sibling or unrelated donor options. Given the need to expand the transplant pool and thus avoid clonal hematopoiesis, clinically significant PNH, and relapsed aplastic anemia, more work continues to recognize the expanding role of alternative donor transplants (cord blood and haploidentical) as another viable treatment strategy for aplastic anemia after immunosuppressive therapy failure.90
Summary
Aplastic anemia is a rare but potentially life-threatening disorder with pancytopenia and a marked reduction in the HSC compartment. It can be acquired or associated with an IMFS, and the treatment and prognosis vary dramatically between these 2 etiologies. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia. Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is the new standard of care.
Corresponding author: Gabrielle Meyers, MD, 3181 SW Sam Jackson Park Road, Mail Code UHN73C, Portland, OR 97239.
Financial disclosures: None.
1. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
2. Vaht K, Göransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica. 2017;102:1683-1690.
3. Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718-1721.
4. Montané E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93:518-523.
5. Ohta A, Nagai M, Nishina M, et al. Incidence of aplastic anemia in Japan: analysis of data from a nationwide registration system. Int J Epidemiol. 2015; 44(suppl_1):i178.
6. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
7. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013;139:9-29.
8. Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoiesis stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699-3708.
9. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35-47.
10. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479-2486.
11. Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870.
12. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11-19.
13. Townsley DM, Dumitriu B, Young NS, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922-1931.
14. Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43:598-608.
15. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775-2783.
16. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.
17. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446-4455.
18. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
19. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48:1721-1731.
20. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337-347.
21. Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124:2698-2704.
22. Mukhina GL, Buckley JT, Barber JP, et al. Multilineage glycosylphosphatidylinositol anchor‐deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476-482.
23. Pu JJ, Mukhina G, Wang H, et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87:37-45.
24. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-5340.
25. Devalet B, Mullier F, Chatelain B, et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190-198.
26. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314.
27. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95:1075-1080.
28. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
29. Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76-81.
30. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genetics. 1997;68:58-61.
31. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4-10.
32. Giampietro PF, Davis JG, Adler-Brecher B, et al. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116-1120.
33. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129:1428-1436.
35. DeZern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol. 2011;4:221-230.
36. Tichelli A, Gratwohl A, Nissen C, et al. Morphology in patients with severe aplastic anemia treated with antilymphocyte globulin. Blood. 1992;80:337-345.
37. Camitta BM, Storb R, Thomas ED. Aplastic anemia: pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306:645-652.
38. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
39. Brodsky RA, Chen AR, Dorr D, et al. High-dose cyclophosphamide for severe aplastic anemia: long-term follow-up. Blood. 2010;115:2136-2141.
40. Matsui WH, Brodsky RA, Smith BD, et al. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458-462.
41. Maciejewski JP, Risitano AM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
42. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1-17.
43. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:11-18.
44. Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90:858-864.
45. Gupta V, Eapen M, Brazauskas R, et al. Impact of age on outcomes after transplantation for acquired aplastic anemia using HLA-identical sibling donors. Haematologica. 2010;95:2119-2125.
46. Peffault de Latour R, Le Rademacher J, Antin JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279-4286.
47. Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118:2618-2621.
48. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the Severe Aplastic Anemia Working Party of EBMT. Haematologica. 2016;101:884-890.
49. Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50:1168-1172.
50. De Medeiros CR, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849-852.
51. Mohanan E, Panetta JC, Lakshmi KM, et al. Population pharmacokinetics of fludarabine in patients with aplastic anemia and Fanconi anemia undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:977-983.
52. Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856-2862.
53. Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94:1312-1315.
54. Talbot A, Peffault de Latour R, Raffoux E, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199-e200.
55. Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for Fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669-1676.
56. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
57. Laundy GJ, Bradley BA, Rees BM, et al. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44:814-825.
58. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
59. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430-438.
60. Höchsmann B, Moicean A, Risitano A, et al. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168-173.
61. Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269-276.
62. Torres HA, Bodey GP, Rolston KV, et al. Infections in patients with aplastic anemia: experience at a tertiary care cancer center. Cancer. 2003;98:86-93.
63. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434-4441.
64. Gerson SL, Talbot GH, Hurwitz S, et al. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345-351.
65. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726-735.
66. Robenshtok E, Gafter-Gvili A, Goldberg E, et al. Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol. 2007;25:5471-5489.
67. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood. 2010;116:2448-2454.
68. Deeg HJ, Amylon MD, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208-215.
69. Kahl C, Leisenring W, Joachim Deeg H, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long‐term follow‐up. Br J Haematol. 2005;130:747-751.
70. Socié G. Allogeneic BM transplantation for the treatment of aplastic anemia: current results and expanding donor possibilities. Hematology Am Soc Hematol Educ Program. 2013;2013:82-86.
71. Shin SH, Jeon YW, Yoon JH, et al. Comparable outcomes between younger (<40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016;51:1456-1463.
72. Kim H, Lee KH, Yoon SS, et al; Korean Society of Blood and Marrow Transplantation. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biol Blood Marrow Transplant. 2012;18:1500-1508.
73. Mortensen BK, Jacobsen N, Heilmann C, Sengelov H. Allogeneic hematopoietic cell transplantation for severe aplastic anemia: similar long-term overall survival after transplantation with related donors compared to unrelated donors. Bone Marrow Transplant. 2016;51:288-290.
74. Dufour C, Svahn J, Bacigalupo A. Front-line immunosuppressive treatment of acquired aplastic anemia. Bone Marrow Transplant. 2013;48:174-177.
75. Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on the behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of the EBMT. Br J Haematol. 2015;171:585-594.
76. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2:2020-2028.
77. Yoshida N, Kojima S. Updated guidelines for the treatment of acquired aplastic anemia in children. Curr Oncol Rep. 2018;20:67.
78. Mathe G, Amiel JL, Schwarzenberg L, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J. 1970;2:131-136.
79. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al; German Aplastic Anemia Study Group. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med. 1991;324:1297-1304.
80. Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J. 1981;282:860-863.
81. Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK, et al. Cyclosporine monotherapy for severe aplastic anemia: a developing country experience. Ann Saudi Med. 2005;25:375-379.
82. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185-1196.
83. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135.
84. Saracco P, Quarello P, Iori AP, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long‐term observation follow‐up. Br J Haematol. 2008;140:197-205.
85. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540-1550.
86. Assi R, Garcia-Manero G, Ravandi F, et al. Addition of eltrombopag to immunosuppressive therapy in patients with newly diagnosed aplastic anemia. Cancer. 2018;124:4192-4201.
87. Bacigalupo A, Socié G, Hamladji RM, et al. Current outcome of HLA identical sibling vs. unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100:696-702.
88. Samarasinghe S, Iacobelli S, Knol C, et al. Impact of different in vivo T cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anaemia: a study on behalf of the EBMT SAA Working Party. Am J Hematol. 2019; 94:80-86.
89. Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619-628.
90. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am. 2018;32:629-642.
From the Oregon Health and Science University, Portland, OR.
Abstract
- Objective: To describe the current approach to diagnosis and treatment of aplastic anemia.
- Methods: Review of the literature.
- Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), and the treatment and prognosis vary dramatically between these 2 etiologies. Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to life-threatening neutropenic infections or bleeding. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia.
- Conclusion: Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marro
w transplant program to evaluate for early transplantation is the new standard of care for aplastic anemia.
Keywords: inherited marrow failure syndrome; Fanconi anemia; immunosuppression; transplant; stem cell.
Aplastic anemia is a clinical and pathological entity of bone marrow failure that causes progressive loss of hematopoietic progenitor stem cells (HPSC), resulting in pancytopenia.1 Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to having life-threatening neutropenic infections or bleeding. Aplastic anemia results from either inherited or acquired causes, and the pathophysiology and treatment approach vary significantly between these 2 causes. Therefore, recognition of inherited marrow failure diseases, such as Fanconi anemia and telomere biology disorders, is critical to establishing the management plan.
Epidemiology
Aplastic anemia is a rare disorder, with an incidence of approximately 1.5 to 7 cases per million individuals per year.2,3 A recent Scandinavian study reported that the incidence of aplastic anemia among the Swedish population is 2.3 cases per million individuals per year, with a median age at diagnosis of 60 years and a slight female predominance (52% versus 48%, respectively).2 This data is congruent with prior observations made in Barcelona, where the incidence was 2.34 cases per million individuals per year, albeit with a slightly higher incidence in males compared to females (2.54 versus 2.16, respectively).4 The incidence of aplastic anemia varies globally, with a disproportionate increase in incidence seen among Asian populations, with rates as high as 8.8 per million individuals per year.3-5 This variation in incidence in Asia versus other countries has not been well explained. There appears to be a bimodal distribution, with incidence peaks seen in young adults and in older adults.2,3,6
Pathophysiology
Acquired Aplastic Anemia
The leading hypothesis as to the cause of most cases of acquired aplastic anemia is that a dysregulated immune system destroys HPSCs. Inciting etiologies implicated in the development of acquired aplastic anemia include pregnancy, infection, medications, and exposure to certain chemicals, such as benzene.1,7 The historical understanding of acquired aplastic anemia implicates cytotoxic T-lymphocyte–mediated destruction of CD34+ hematopoietic stem cells.1,8,9 This hypothesis served as the basis for treatment of acquired aplastic anemia with immunosuppressive therapy, predominantly anti-thymocyte globulin (ATG) combined with cyclosporine A.1,8 More recent work has focused on cytokine interactions, particularly the suppressive role of interferon (IFN)-γ on hematopoietic stem cells independent of T-lymphocyte–mediated destruction, which has been demonstrated in a murine model.8 The interaction of IFN-γ with the hematopoietic stem cell pool is dynamic. IFN-γ levels are elevated during an acute inflammatory response, such as a viral infection, providing further basis for the immune-mediated nature of the acquired disease.10 Specifically, in vitro studies suggest the effects of IFN-γ on HPSC may be secondary to interruption of thrombopoietin and its respective signaling pathways, which play a key role in hematopoietic stem cell renewal.11 Eltrombopag, a thrombopoietin receptor antagonist, has shown promise in the treatment of refractory aplastic anemia, with studies indicating that its effectiveness is independent of IFN-γ levels.11,12
Inherited Aplastic Anemia
The inherited marrow failure syndromes (IMFSs) are a group of disorders characterized by cellular maintenance and repair defects, leading to cytopenias, increased cancer risk, structural defects, and risk of end organ damage, such as liver cirrhosis and pulmonary fibrosis.13-15 The most common diseases include Fanconi anemia, dyskeratosis congenita/telomere biology disorders, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome, but with the advent of whole exome sequencing, new syndromes continue to be discovered. While classically these disorders present in children, adult presentations are now commonplace. Broadly, the pathophysiology of inherited aplastic anemia relates to the defective HPSCs and an accelerated decline of the hematopoietic stem cell compartment.
The most common IMFSs, Fanconi anemia and telomere biology disorders, are associated with numerous mutations in DNA damage repair pathways and telomere maintenance pathways. TERT, DKC, and TERC mutations are most commonly associated with dyskeratosis congenita, but may also be found infrequently in patients with aplastic anemia presenting at an older age in the absence of the classic phenotypical features.1,16,17 The recognition of an underlying genetic disorder or telomere biology disorder leading to constitutional aplastic anemia is significant, as these conditions are associated not only with marrow failure, but also with endocrinopathies, organ fibrosis, and and hematopoietic and solid organ malignancies.13-15 In particular, TERT and TERC gene mutations have been associated with dyskeratosis congenita as well as pulmonary fibrosis and cirrhosis.18,19 The implications of early diagnosis of an IMFS lie in the approach to treatment and prognosis.
Clonal Disorders and Secondary Malignancies
Myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) are 2 clonal disorders that may arise from a background of aplastic anemia.9,20,21 Hypoplastic MDS can be difficult to differentiate from aplastic anemia at diagnosis based on morphology alone, although recent work has demonstrated that molecular testing for somatic mutations in ASXL1, DNMT3A, and BCOR can aid in differentiating a subset of aplastic anemia patients who are more likely to progress to MDS.21 Clonal populations of cells harboring 6p uniparental disomy are seen in more than 10% of patients with aplastic anemia on cytogenetic analysis, which can help differentiate the diseases.9 Yoshizato and colleagues found lower rates of ASXL1 and DNMT3A mutations in patients with aplastic anemia as compared with patients with MDS or AML. In this study, patients with aplastic anemia had higher rates of mutations in PIGA (reflecting the increased paroxysmal nocturnal hemoglobinuria [PNH] clonality seen in aplastic anemia) and BCOR.9 Mutations were also found in genes commonly mutated in MDS and AML, including TET2, RUNX1, TP53, and JAK2, albeit at lower frequencies.9 These mutations as a whole have not predicted response to therapy or prognosis. However, when performing survival analysis in patients with specific mutations, those commonly encountered in MDS/AML (ASXL1, DNMT3A, TP53, RUNX1, CSMD1) are associated with faster progression to overt MDS/AML and decreased overall survival (OS),20,21 suggesting these mutations may represent early clonality that can lead to clonal evolution and the development of secondary malignancies. Conversely, mutations in BCOR and BCORL appear to identify patients who may have a favorable outcome in response to immunosuppressive therapy and, similar to patients with PIGA mutations, improved OS.9
Paroxysmal Nocturnal Hemoglobinuria
In addition to having an increased risk of myelodysplasia and malignancy due to the development of a dominant pre-malignant clone, patients with aplastic anemia often harbor progenitor cell clones associated with PNH.1,17 PNH clones have been identified in more than 50% of patients with aplastic anemia.22,23 PNH represents a clonal disorder of hematopoiesis in which cells harbor X-linked somatic mutations in the PIGA gene; this gene encodes a protein responsible for the synthesis of glycosylphosphatidylinositol anchors on the cell surface.22,24 The lack of these cell surface proteins, specifically CD55 (also known as decay accelerating factor) and CD59 (also known as membrane inhibitor of reactive lysis), predisposes red cells to increased complement-mediated lysis.25 The exact mechanism for the development of these clones in patients with aplastic anemia is not fully understood. Current theories hypothesize that the clones are protected from the immune-mediated destruction of normal hematopoietic stem cells due to the absence of the cell surface proteins.1,20 The role of these clones over time in patients with aplastic anemia is less clear, though recent work demonstrated that despite differences in clonality over the disease course, aplastic anemia patients with small PNH clones are less likely to develop overt hemolysis and larger PNH clones compared to patients harboring larger (≥ 50%) PNH clones at diagnosis.23,26,27 Additionally, PNH clones in patients with aplastic anemia infrequently become clinically significant.27 It should be noted that these conditions exist along a continuum; that is, patients with aplastic anemia may develop PNH clones, while conversely patients with PNH may develop aplastic anemia.20 Patients with PNH clones should be followed via peripheral blood flow cytometry and complete blood count to track clonal stability and identify clinically significant PNH among aplastic anemia patients.28
Clinical Presentation
Patients with aplastic anemia typically are diagnosed either due to asymptomatic cytopenias found on peripheral blood sampling, symptomatic anemia, bleeding secondary to thrombocytopenia, or wound healing and infectious complications related to neutropenia.29 A thorough history to understand the timing of symptoms, recent infectious symptoms/exposure, habits, and chemical or toxin exposures (including medications, travel, and supplements) helps guide diagnostic testing. Family history is also critical, with attention given to premature graying; pulmonary, renal, and liver disease; and blood disorders.
Patients with an IMFS (eg, Fanconi anemia or dyskeratosis congenita) may have associated phenotypical findings such as urogenital abnormalities or short stature; in addition, those with dyskeratosis congenita may present with the classic triad of oral leukoplakia, lacy skin pigmentation, and dystrophic nails.7 However, classic phenotypical findings may be lacking in up to 30% to 40% of patients with an IMFS.7 As described previously, while congenital malformations are common in Fanconi anemia and dyskeratosis congenita, a third of patients may have no or only subtle phenotypical abnormalities, including alterations in skin or hair pigmentation, skeletal and growth abnormalities, and endocrine disorders.30 The International Fanconi Anemia Registry identified central nervous system, genitourinary, skin and musculoskeletal, ophthalmic, and gastrointestinal system malformations among children with Fanconi anemia.31,32 Patients with dyskeratosis congenita may present with pulmonary fibrosis, hepatic cirrhosis, or premature graying, as highlighted in a recent study by DiNardo and colleagues.33 Therefore, physicians must have a heightened index of suspicion in patients with subtle phenotypical findings and associated cytopenias.
Diagnosis
The diagnosis of aplastic anemia should be suspected in any patient presenting with pancytopenia. Aplastic anemia is a diagnosis of exclusion.34 Other conditions associated with peripheral blood pancytopenia should be considered, including infections (HIV, hepatitis, parvovirus B19, cytomegalovirus, Epstein-Barr virus, varicella-zoster virus), nutritional deficiencies (vitamin B12, folate, copper, zinc), autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, hemophagocytic lymphohistiocytosis), hypersplenism, marrow-occupying diseases (eg, leukemia, lymphoma, MDS), solid malignancies, and fibrosis (Table).7
Diagnostic Evaluation
The workup for aplastic anemia should include a thorough history and physical exam to search simultaneously for alternative diagnoses and clues pointing to potential etiologic agents.7 Diagnostic tests to be performed include a complete blood count with differential, reticulocyte count, immature platelet fraction, flow cytometry (to rule out lymphoproliferative disorders and atypical myeloid cells and to evaluate for PNH), and bone marrow biopsy with subsequent cytogenetic, immunohistochemical, and molecular testing.35 Typical findings in aplastic anemia include peripheral blood pancytopenia without dysplastic features and bone marrow biopsy demonstrating a hypocellular marrow.7 A relative lymphocytosis in the peripheral blood is common.7 In patients with a significant PNH clone, a macrocytosis along with elevated lactate dehydrogenase and elevated reticulocyte and granulocyte counts may be present.36
The diagnosis (based on the Camitta criteria37 and modified Camitta criteria38 for severe aplastic anemia) requires 2 of the following findings on peripheral blood samples:
- Absolute neutrophil count (ANC) < 500 cells/µL
- Platelet count < 20,000 cells/µL
- Reticulocyte count < 1% corrected or < 20,000 cells/µL.35
In addition to peripheral blood findings, bone marrow biopsy is essential for the diagnosis, and should demonstrate a markedly hypocellular marrow (cellularity < 25%), occasionally with an increase in T lymphocytes.7,39 Because marrow cellularity varies with age and can be challenging to assess, additional biopsies may be needed to confirm the diagnosis.29 A 1- to 2-cm core biopsy is necessary to confirm hypocellularity, as small areas of residual hematopoiesis may be present and obscure the diagnosis.35
Excluding Hypocellular MDS and IMFS
Excluding hypocellular MDS is challenging, especially in the older adult presenting with aplastic anemia, as patients with aplastic anemia may have some degree of erythroid dysplasia on bone marrow morphology.36 The presence of a PNH clone on flow cytometry can aid in diagnosing aplastic anemia and excluding MDS,34 although PNH clones can be present in refractory anemia MDS. Patients with aplastic anemia have a lower ratio of CD34+ cells compared to those with hypoplastic MDS, with 1 study demonstrating a mean CD34+ percentage of < 0.5% in aplastic anemia versus 3.7% in hypoplastic MDS.40 Cytogenetic and molecular testing can also aid in making this distinction by identifying mutations commonly implicated in MDS.7 The presence of monosomy 7 (-7) in aplastic anemia patients is associated with a poor overall prognosis.34,41
Peripheral blood screening using chromosome breakage analysis (done using either mitomycin C or diepoxybutane as in vitro DNA-crosslinking agents)42 and telomere length testing (of peripheral blood leukocytes) is necessary to exclude the main IMFSs, Fanconi anemia and telomere biology disorders, respectively. Ruling out these conditions is imperative, as the approach to treatment varies significantly between IMFS and aplastic anemia. Patients with shortened telomeres should undergo genetic screening for mutations in the telomere maintenance genes to evaluate the underlying defect leading to shortened telomeres. Patients with increased peripheral blood breakage should have genetic testing to detect mutations associated with Fanconi anemia.
Classification
Once the diagnosis of aplastic anemia has been made, the patient should be classified according to the severity of their disease. Disease severity is determined based on peripheral blood ANC: non-severe aplastic anemia (NSAA), ANC > 500 polymorphonuclear neutrophils (PMNs)/µL; severe aplastic anemia (SAA), 200–500 PMNs/µL; and very severe aplastic anemia (VSAA), 0–200 PMNs/µL.4,34 Disease classification is important, as VSAA is associated with a decreased OS compared to SAA.2 Disease classification may affect treatment decisions, as patients with NSAA may be observed for a short period of time, while, conversely, patients with SAA have a worse prognosis with delays in therapy.43-45
Treatment of Inherited Aplastic Anemia
First-line treatment options for patients with IMFS are androgen therapy and hematopoietic stem cell transplant (HSCT). When evaluating patients for HSCT, it is critical to identify the presence of an IMFS, as the risk and mortality associated with the conditioning regimen, stem cell source, graft-versus-host disease (GVHD), and secondary malignancies differ between patients with IMFS and those with acquired marrow failure syndromes or hematologic malignancies.
Potential sibling donors need to be screened for donor candidacy as well as for the inherited defect. Among patients with Fanconi anemia or a telomere biology disorder, the stem cell source must be considered, with multiple studies in IMFSs and SAA showing superior outcomes with a bone marrow product compared to peripheral blood stem cells.46-48 In IMFS patients, the donor cell type may affect the choice of conditioning regimen.5,6 Reduced-intensity conditioning in lieu of myeloablative conditioning without total body irradiation has proved feasible in patients with Fanconi anemia, and is associated with a reduced risk of secondary malignancies.49,50 Incorporation of fludarabine in the conditioning regimen of patients without a matched sibling donor is associated with superior engraftment and survival46,49,51 compared to cyclophosphamide conditioning, which was historically used in matched related donors.50,52 Adding fludarabine appears to be especially beneficial in older patients, in whom its use is associated with lower rates of graft failure, likely due to increased immunosuppression at the time of engraftment.51,53 Fludarabine has also been incorporated into conditioning regimens for patients with a telomere biology disorder, but outcomes data are limited.5
For patients presenting with AML or a high-risk MDS who are subsequently diagnosed with an IMFS, treatment can be more complex, as these patients are at high risk for toxicity from standard chemotherapy. Limited data suggest that induction therapy and transplantation are feasible in this group of patients, and this approach is associated with increased OS, despite lower OS rates than those of IMFS patients who present prior to the development of MDS or AML.54,55 Further work is needed to determine the optimal induction regimen that balances the risks of treatment-related mortality and complications associated with conditioning regimens, risk of relapse, and risk of secondary malignancies, especially in the cohort of patients diagnosed at an older age.
Treatment of Acquired Aplastic Anemia
Supportive Care
While the workup and treatment plan are being established, attention should be directed at supportive care for prevention of complications. The most common complications leading to death in patients with significant pancytopenia and neutropenia are opportunistic infections and hemorrhagic complications.2
Transfusion support is critical to avoid symptomatic anemia and hemorrhagic complications related to thrombocytopenia, which typically occur with platelet counts lower than 10,000 cells/µL. However, transfusion carries the risk of alloimmunization (which may persist for years following transfusion) and transfusion-related graft versus host disease (trGVHD), and thus use of transfusion should be minimized when possible.56,57 All blood products given to patients with aplastic anemia should be irradiated and leukoreduced to reduce the risk of both alloimmunization and trGVHD. Guidelines from the British Society for Haematology recommend routine screening for Rh and Kell antibodies to reduce the risk of alloimmunization.58 Infectious complications remain a common cause of morbidity and mortality in patients with aplastic anemia who have prolonged neutropenia (defined as an ANC < 500 cells/µL).59-62 Therefore, patients should receive broad-spectrum antibiotics with antipseudomonal coverage. In a study evaluating the role of granulocyte-colony stimulating factor (G-CSF) in patients with SAA receiving immunosuppressive therapy, 55% of all patient deaths were secondary to infection.63 There was no OS benefit seen in patients who received G-CSF, though a significantly lower rate of infection was observed in the G-CSF arm compared to those not receiving G-CSF (56% versus 81%, P = 0.006). This difference was largely driven by a decrease in infectious episodes in patients with VSAA treated with G-CSF as compared to those who did not receive this therapy (22% versus 48%, P = 0.014).63
Angio-invasive pulmonary aspergillosis and Zygomycetes (eg, Rhizopus, Mucor species) remain major causes of mortality related to opportunistic mycotic infections in patients with aplastic anemia.18 The infectious risk is directly related to the duration and severity of neutropenia, with one study demonstrating a significant increase in risk in AML patients with neutropenia lasting longer than 3 weeks.64 Invasive fungal infections carry a high mortality in patients with severe neutropenia, though due to earlier recognition and empiric antifungal therapy with extended-spectrum azoles, overall mortality secondary to invasive fungal infections is declining.62,65
While neutropenia related to cytotoxic chemotherapy is commonly associated with gram-negative bacteria due to disruption of mucosal barriers, patients with aplastic anemia have an increased incidence of gram-positive bacteremia with staphylococcal species compared to other neutropenic populations.61,62 This appears to be changing with time. Valdez et al demonstrated a decrease in prevalence of coagulase-negative staphylococcal infections, increased prevalence of gram-positive bacilli bacteremia, and no change in prevalence of gram-negative bacteremia in patients with aplastic anemia treated between 1989 and 2008.65 Gram-negative bacteremia caused by Stenotrophomonas maltophila, Escherichia coli, Klebsiella pneumoniae, Citrobacter, and Proteus has also been reported.62 Despite a lack of clinical trials investigating the role of antifungal and antibacterial prophylaxis for patients with aplastic anemia, most centers initiate antifungal prophylaxis in patients with SAA or VSAA with an anti-mold agent such as voriconazole or posaconazole (which has the additional benefit compared to voriconazole of covering Mucor species).60,66 This is especially true for patients who have received ATG or undergone HSCT. For antimicrobial prophylaxis, a fluoroquinolone antibiotic with a spectrum of activity against Pseudomonas should be considered for patients with an ANC < 500 cells/µL.60 Acyclovir or valacyclovir prophylaxis is recommended for varicella-zoster virus and herpes simplex virus. Cytomegalovirus reactivation is minimal in patients with aplastic anemia, unless multiple courses of ATG are used.
Iron overload is another complication the provider must be aware of in the setting of increased transfusions in aplastic anemia patients. Lee and colleagues showed that iron chelation therapy using deferasirox is effective at reducing serum ferritin levels in patients with aplastic anemia (median ferritin level of 3254 ng/mL prior to therapy, 1854 ng/mL following), and is associated with no serious adverse events (most common adverse events included nausea, diarrhea, vomiting, and rash).67 Approximately 25% of patients in this trial had an increase in creatinine, with patients taking concomitant cyclosporine affected to a greater degree than those on chelation therapy alone. For patients following HSCT or with improved hematopoiesis following immunosuppressive therapy, phlebotomy can be used to treat iron overload in lieu of chelation therapy.58
Approach to Therapy
The main treatment options for SAA and VSAA include allogeneic bone marrow transplant and immunosuppression. The deciding factors as to which treatment is best initially depends on the availability of HLA-matched related donors and age (Figure 1 and Figure 2). Survival is decreased in patients with SAA or VSAA who delay initiation of therapy, and therefore prompt referral for HLA typing and evaluation for bone marrow transplant is a very important first step in managing aplastic anemia.
Matched Sibling Donor Transplant. Current standards of care recommend HLA-matched sibling donor transplant for patients with SAA or VSAA who are younger than 50 years, with the caveat that integration of fludarabine and reduced cyclophosphamide dosing along with ATG shows the best overall outcomes. Locasciulli and colleagues examined outcomes in patients given either immunosuppressive therapy or sibling HSCT between 1991-1996 and 1997-2002, respectively, and found that sibling HSCT was associated with a superior 10-year OS compared to immunosuppressive therapy (73% versus 68%).43 Interestingly in this study, there was no OS improvement seen with immunosuppressive therapy alone (69% versus 73%) between the 2 time periods, despite increased OS in both sibling HSCT (74% and 80%) and MUD HSCT (38% and 65%).43 Though total body irradiation has been used in the past, it is typically not included in current conditioning regimens for matched related donor transplants.68
Current conditioning regimens typically use a combination of cyclophosphamide and ATG,69,70 with or without fludarabine. Fludarabine-based conditioning regimens have shown promise in patients undergoing sibling HSCT. Maury and colleagues evaluated the role of fludarabine in addition to low-dose cyclophosphamide and ATG compared to cyclophosphamide alone or in combination with ATG in patients over age 30 undergoing sibling HSCT.53 There was a nonsignificant improvement in 5-year OS in the fludarabine arm compared to controls (77% ± 8% versus 60% ± 3%, P = 0.14) in the pooled analysis, but when adjusted for age the fludarabine arm had a significantly lower relative risk (RR) of death (0.44; P = 0.04) compared to the control arm. Shin et al reported outcomes with fludarabine/cyclophosphamide/ATG, with excellent overall outcomes and no difference in patients older or younger than 40 years.71
Kim et al evaluated their experience with patients older than 40 years receiving matched related donors, finding comparable outcomes in those ages 41 to 50 years compared to younger patients. Outcomes declined in those over the age of 50 years.72 Long-term data for matched related donor transplant for aplastic anemia show excellent long-term outcomes, with minimal chronic GVHD and good performance status.73 Hence, these factors support the role of matched related donor transplant as the initial treatment in SAA and VSAA.
Regarding the role of transplant for patients who lack a matched related donor, a growing body of literature demonstrating identical outcomes between matched related and MUD transplants for pediatric patients74,75 supports recent recommendations for upfront unrelated donor transplantation for aplastic anemia.76,77
Immunosuppressive Therapy. For patients without an HLA-matched sibling donor or those who are older than 50 years of age, immunosuppressive therapy is the first-line therapy. ATG and cyclosporine A are the treatments of choice.78 The potential effectiveness of immunosuppressive therapy in treating aplastic anemia was initially observed in patients in whom autologous transplant failed but who still experienced hematopoietic reconstitution despite the failed graft; this observation led to the hypothesis that the conditioning regimen may have an effect on hematopoiesis.59,78,79
Immunosuppressive therapy with ATG has been used for the treatment of aplastic anemia since the 1980s.80 Historically, rabbit ATG had been used, but a 2011 study of horse ATG demonstrated superior hematological response at 6 months compared to rabbit ATG (68% versus 37%).59 Superior survival was also seen with horse ATG compared to rabbit ATG (3-year OS, 96% versus 76%). Due to these results, horse ATG is preferred over rabbit ATG. ATG should be used in combination with cyclosporine A to optimize outcomes.
Early studies also demonstrated the efficacy of cyclosporine A in the treatment of aplastic anemia, with response rates equivalent to that of ATG monotherapy.81 Recent publications still note the efficacy of cyclosporine A in the treatment of aplastic anemia. Its role as an affordable option for single-agent therapy in developing countries is intriguing.81 The combination of ATG and cyclosporine A was proven superior to either agent alone in a study by Frickhofen et al.79 In this study, patients were randomly assigned to a control arm that received ATG plus methylprednisolone or to an arm that received ATG plus cyclosporine A and methylprednisolone. At 6 months, 70% of patients in the cyclosporine A arm had a complete remission (CR) or partial remission compared to 46% in the control arm.82 Further work confirmed the long-term efficacy of this regimen, reporting a 7-year OS of 55%.83 Among a pediatric population, immunosuppressive therapy was associated with an 83% 10-year OS.84
It is recommended that patients remain on cyclosporine therapy for a minimum of 6 months, after which a gradual taper may be considered, although there is variation among practitioners, with some continuing immunosuppressive therapy for a minimum of 12 months due to a proportion of patients being cyclosporine dependent.34,84 A study found that within a population of patients who responded to immunosuppressive therapy, 18% became cyclosporine dependent.84 The median duration of cyclosporine A treatment at full dose was 12 months, with tapering completed over a median of 19 months after patients had been in a stable CR for a minimum of 3 months. Relapse occurred more often when patients were tapered quickly (decrease ≥ 0.8 mg/kg/month) compared to slowly (0.4-0.7 mg/kg/month) or very slowly (< 0.3 mg/kg/month).
Townsley and colleagues recently investigated incorporating the use of the thrombopoietin receptor agonist eltrombopag with immunosuppressive therapy as first-line therapy in aplastic anemia.85 When given at a dose of 150 mg daily in patients ages 12 years and older or 75 mg daily in patients younger than 12 years, in conjunction with cyclosporine A and ATG, patients demonstrated markedly improved hematological response compared to historical treatment with standard immunosuppressive therapy alone.45 In the patient cohort administered eltrombopag starting on day 1 and continuing for 6 months, the complete response rate was 58%. Eltrombopag led to improvement in all cell lines among all treatment subgroups, and OS (censored for patients who proceeded to transplant) was 99% at 2 years.12 Overall, toxicities associated with this therapy were low, with liver enzyme elevations most commonly observed.85 Recently, a phase 2 trial of immunosuppressive therapy with or without eltrombopag was reported. Of the 38 patients enrolled, overall response, complete response, and time to response were not statistically different.86 With this recent finding, the role of eltrombopag in addition to immunosuppressive therapy is not clearly defined, and further studies are warranted.
OS for patients who do not respond to immunosuppressive therapy is approximately 57% at 5 years, largely due to improved supportive measures among this patient population.48,65 Therefore, it is important to recognize those patients who have a low chance of response so that second-line therapy can be pursued to improve outcomes.
Matched Unrelated Donor Transplant. For patients with refractory disease following immunosuppressive therapy who lack a matched sibling donor, MUD HSCT is considered standard therapy given the marked improvement in overall outcomes with modulating conditioning regimens and high-resolution HLA typing. A European Society for Blood and Marrow Transplantation (EBMT) analysis comparing matched sibling HSCT to MUD HSCT noted significantly higher rates of acute grade II-IV and grade III-IV GVHD (grade II-IV 13% versus 25%, grade III-IV 5% versus 10%) among patients undergoing MUD transplant.47 Chronic GVHD rates were 14% in the sibling group, as compared to 26% in the MUD group. Factors associated with improved survival in this analysis include transplant under age 20 years (84% versus 72%), transplant within 6 months of diagnosis (85% versus 72%), the use of ATG in the conditioning regimen (81% versus 73%), and cytomegalovirus-negative donor and recipient as compared to other combinations (82% versus 76%).87 Interestingly, this study demonstrated that OS was not significantly increased when using a sibling HSCT compared to a MUD HSCT, likely as a result of improved understanding of conditioning regimens, GVHD prophylaxis, and supportive care.
Additional studies of MUD HSCT have shown outcomes similar to those seen in sibling HSCT.34,48 A French study found a significant increase in survival in patients undergoing MUD HSCT compared to historical cohorts (2000-2005: OS 52%; 2006-2012: OS 74%).75 The majority of patients underwent conditioning with cyclophosphamide or a combination of busulfan and cyclophosphamide, with or without fludarabine; 81% of patients underwent in vivo T-cell depletion, and a bone marrow donor source was utilized. OS was significantly lower in patients over age 30 years undergoing MUD HSCT (57%) compared to those under age 30 years (70%). Improved OS was also seen when patients underwent transplant within 1 year of diagnosis and when a 10/10 matched donor (compared to a 9/10 mismatched donor) was utilized.48
A 2015 study investigated the role of MUD HSCT as frontline therapy instead of immunosuppressive therapy in patients without a matched sibling donor.75 The 2-year OS was 96% in the MUD HSCT cohort compared to 91%, 94%, and 74% in historical cohorts of sibling HSCT, frontline immunosuppressive therapy, and second-line MUD HSCT following failed immunosuppressive therapy, respectively. Additionally, event-free survival in the MUD HSCT cohort (defined by the authors as death, lack of response, relapse, occurrence of clonal evolution/clinical PNH, malignancies developing over follow‐up, and transplant for patients receiving immunosuppressive therapy frontline) was similar compared to sibling HSCT and superior to frontline immunosuppressive therapy and second-line MUD HSCT. Furthermore, Samarasinghe et al highlighted the importance of in vivo T-cell depletion with either ATG or alemtuzumab (anti-CD52 monoclonal antibody) in the prevention of acute and chronic GVHD in both sibling HSCT and MUD HSCT.88
With continued improvement of less toxic and more immunomodulating conditioning regimens,utilization of bone marrow as a donor cell source, in vivo T-cell depletion, and use of GVHD and antimicrobial prophylaxis, more clinical evidence supports elevating MUD HSCT in the treatment plan for patients without a matched sibling donor.89 However, there is still a large population of patients without matched sibling or unrelated donor options. Given the need to expand the transplant pool and thus avoid clonal hematopoiesis, clinically significant PNH, and relapsed aplastic anemia, more work continues to recognize the expanding role of alternative donor transplants (cord blood and haploidentical) as another viable treatment strategy for aplastic anemia after immunosuppressive therapy failure.90
Summary
Aplastic anemia is a rare but potentially life-threatening disorder with pancytopenia and a marked reduction in the HSC compartment. It can be acquired or associated with an IMFS, and the treatment and prognosis vary dramatically between these 2 etiologies. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia. Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is the new standard of care.
Corresponding author: Gabrielle Meyers, MD, 3181 SW Sam Jackson Park Road, Mail Code UHN73C, Portland, OR 97239.
Financial disclosures: None.
From the Oregon Health and Science University, Portland, OR.
Abstract
- Objective: To describe the current approach to diagnosis and treatment of aplastic anemia.
- Methods: Review of the literature.
- Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), and the treatment and prognosis vary dramatically between these 2 etiologies. Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to life-threatening neutropenic infections or bleeding. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia.
- Conclusion: Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marro
w transplant program to evaluate for early transplantation is the new standard of care for aplastic anemia.
Keywords: inherited marrow failure syndrome; Fanconi anemia; immunosuppression; transplant; stem cell.
Aplastic anemia is a clinical and pathological entity of bone marrow failure that causes progressive loss of hematopoietic progenitor stem cells (HPSC), resulting in pancytopenia.1 Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to having life-threatening neutropenic infections or bleeding. Aplastic anemia results from either inherited or acquired causes, and the pathophysiology and treatment approach vary significantly between these 2 causes. Therefore, recognition of inherited marrow failure diseases, such as Fanconi anemia and telomere biology disorders, is critical to establishing the management plan.
Epidemiology
Aplastic anemia is a rare disorder, with an incidence of approximately 1.5 to 7 cases per million individuals per year.2,3 A recent Scandinavian study reported that the incidence of aplastic anemia among the Swedish population is 2.3 cases per million individuals per year, with a median age at diagnosis of 60 years and a slight female predominance (52% versus 48%, respectively).2 This data is congruent with prior observations made in Barcelona, where the incidence was 2.34 cases per million individuals per year, albeit with a slightly higher incidence in males compared to females (2.54 versus 2.16, respectively).4 The incidence of aplastic anemia varies globally, with a disproportionate increase in incidence seen among Asian populations, with rates as high as 8.8 per million individuals per year.3-5 This variation in incidence in Asia versus other countries has not been well explained. There appears to be a bimodal distribution, with incidence peaks seen in young adults and in older adults.2,3,6
Pathophysiology
Acquired Aplastic Anemia
The leading hypothesis as to the cause of most cases of acquired aplastic anemia is that a dysregulated immune system destroys HPSCs. Inciting etiologies implicated in the development of acquired aplastic anemia include pregnancy, infection, medications, and exposure to certain chemicals, such as benzene.1,7 The historical understanding of acquired aplastic anemia implicates cytotoxic T-lymphocyte–mediated destruction of CD34+ hematopoietic stem cells.1,8,9 This hypothesis served as the basis for treatment of acquired aplastic anemia with immunosuppressive therapy, predominantly anti-thymocyte globulin (ATG) combined with cyclosporine A.1,8 More recent work has focused on cytokine interactions, particularly the suppressive role of interferon (IFN)-γ on hematopoietic stem cells independent of T-lymphocyte–mediated destruction, which has been demonstrated in a murine model.8 The interaction of IFN-γ with the hematopoietic stem cell pool is dynamic. IFN-γ levels are elevated during an acute inflammatory response, such as a viral infection, providing further basis for the immune-mediated nature of the acquired disease.10 Specifically, in vitro studies suggest the effects of IFN-γ on HPSC may be secondary to interruption of thrombopoietin and its respective signaling pathways, which play a key role in hematopoietic stem cell renewal.11 Eltrombopag, a thrombopoietin receptor antagonist, has shown promise in the treatment of refractory aplastic anemia, with studies indicating that its effectiveness is independent of IFN-γ levels.11,12
Inherited Aplastic Anemia
The inherited marrow failure syndromes (IMFSs) are a group of disorders characterized by cellular maintenance and repair defects, leading to cytopenias, increased cancer risk, structural defects, and risk of end organ damage, such as liver cirrhosis and pulmonary fibrosis.13-15 The most common diseases include Fanconi anemia, dyskeratosis congenita/telomere biology disorders, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome, but with the advent of whole exome sequencing, new syndromes continue to be discovered. While classically these disorders present in children, adult presentations are now commonplace. Broadly, the pathophysiology of inherited aplastic anemia relates to the defective HPSCs and an accelerated decline of the hematopoietic stem cell compartment.
The most common IMFSs, Fanconi anemia and telomere biology disorders, are associated with numerous mutations in DNA damage repair pathways and telomere maintenance pathways. TERT, DKC, and TERC mutations are most commonly associated with dyskeratosis congenita, but may also be found infrequently in patients with aplastic anemia presenting at an older age in the absence of the classic phenotypical features.1,16,17 The recognition of an underlying genetic disorder or telomere biology disorder leading to constitutional aplastic anemia is significant, as these conditions are associated not only with marrow failure, but also with endocrinopathies, organ fibrosis, and and hematopoietic and solid organ malignancies.13-15 In particular, TERT and TERC gene mutations have been associated with dyskeratosis congenita as well as pulmonary fibrosis and cirrhosis.18,19 The implications of early diagnosis of an IMFS lie in the approach to treatment and prognosis.
Clonal Disorders and Secondary Malignancies
Myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) are 2 clonal disorders that may arise from a background of aplastic anemia.9,20,21 Hypoplastic MDS can be difficult to differentiate from aplastic anemia at diagnosis based on morphology alone, although recent work has demonstrated that molecular testing for somatic mutations in ASXL1, DNMT3A, and BCOR can aid in differentiating a subset of aplastic anemia patients who are more likely to progress to MDS.21 Clonal populations of cells harboring 6p uniparental disomy are seen in more than 10% of patients with aplastic anemia on cytogenetic analysis, which can help differentiate the diseases.9 Yoshizato and colleagues found lower rates of ASXL1 and DNMT3A mutations in patients with aplastic anemia as compared with patients with MDS or AML. In this study, patients with aplastic anemia had higher rates of mutations in PIGA (reflecting the increased paroxysmal nocturnal hemoglobinuria [PNH] clonality seen in aplastic anemia) and BCOR.9 Mutations were also found in genes commonly mutated in MDS and AML, including TET2, RUNX1, TP53, and JAK2, albeit at lower frequencies.9 These mutations as a whole have not predicted response to therapy or prognosis. However, when performing survival analysis in patients with specific mutations, those commonly encountered in MDS/AML (ASXL1, DNMT3A, TP53, RUNX1, CSMD1) are associated with faster progression to overt MDS/AML and decreased overall survival (OS),20,21 suggesting these mutations may represent early clonality that can lead to clonal evolution and the development of secondary malignancies. Conversely, mutations in BCOR and BCORL appear to identify patients who may have a favorable outcome in response to immunosuppressive therapy and, similar to patients with PIGA mutations, improved OS.9
Paroxysmal Nocturnal Hemoglobinuria
In addition to having an increased risk of myelodysplasia and malignancy due to the development of a dominant pre-malignant clone, patients with aplastic anemia often harbor progenitor cell clones associated with PNH.1,17 PNH clones have been identified in more than 50% of patients with aplastic anemia.22,23 PNH represents a clonal disorder of hematopoiesis in which cells harbor X-linked somatic mutations in the PIGA gene; this gene encodes a protein responsible for the synthesis of glycosylphosphatidylinositol anchors on the cell surface.22,24 The lack of these cell surface proteins, specifically CD55 (also known as decay accelerating factor) and CD59 (also known as membrane inhibitor of reactive lysis), predisposes red cells to increased complement-mediated lysis.25 The exact mechanism for the development of these clones in patients with aplastic anemia is not fully understood. Current theories hypothesize that the clones are protected from the immune-mediated destruction of normal hematopoietic stem cells due to the absence of the cell surface proteins.1,20 The role of these clones over time in patients with aplastic anemia is less clear, though recent work demonstrated that despite differences in clonality over the disease course, aplastic anemia patients with small PNH clones are less likely to develop overt hemolysis and larger PNH clones compared to patients harboring larger (≥ 50%) PNH clones at diagnosis.23,26,27 Additionally, PNH clones in patients with aplastic anemia infrequently become clinically significant.27 It should be noted that these conditions exist along a continuum; that is, patients with aplastic anemia may develop PNH clones, while conversely patients with PNH may develop aplastic anemia.20 Patients with PNH clones should be followed via peripheral blood flow cytometry and complete blood count to track clonal stability and identify clinically significant PNH among aplastic anemia patients.28
Clinical Presentation
Patients with aplastic anemia typically are diagnosed either due to asymptomatic cytopenias found on peripheral blood sampling, symptomatic anemia, bleeding secondary to thrombocytopenia, or wound healing and infectious complications related to neutropenia.29 A thorough history to understand the timing of symptoms, recent infectious symptoms/exposure, habits, and chemical or toxin exposures (including medications, travel, and supplements) helps guide diagnostic testing. Family history is also critical, with attention given to premature graying; pulmonary, renal, and liver disease; and blood disorders.
Patients with an IMFS (eg, Fanconi anemia or dyskeratosis congenita) may have associated phenotypical findings such as urogenital abnormalities or short stature; in addition, those with dyskeratosis congenita may present with the classic triad of oral leukoplakia, lacy skin pigmentation, and dystrophic nails.7 However, classic phenotypical findings may be lacking in up to 30% to 40% of patients with an IMFS.7 As described previously, while congenital malformations are common in Fanconi anemia and dyskeratosis congenita, a third of patients may have no or only subtle phenotypical abnormalities, including alterations in skin or hair pigmentation, skeletal and growth abnormalities, and endocrine disorders.30 The International Fanconi Anemia Registry identified central nervous system, genitourinary, skin and musculoskeletal, ophthalmic, and gastrointestinal system malformations among children with Fanconi anemia.31,32 Patients with dyskeratosis congenita may present with pulmonary fibrosis, hepatic cirrhosis, or premature graying, as highlighted in a recent study by DiNardo and colleagues.33 Therefore, physicians must have a heightened index of suspicion in patients with subtle phenotypical findings and associated cytopenias.
Diagnosis
The diagnosis of aplastic anemia should be suspected in any patient presenting with pancytopenia. Aplastic anemia is a diagnosis of exclusion.34 Other conditions associated with peripheral blood pancytopenia should be considered, including infections (HIV, hepatitis, parvovirus B19, cytomegalovirus, Epstein-Barr virus, varicella-zoster virus), nutritional deficiencies (vitamin B12, folate, copper, zinc), autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, hemophagocytic lymphohistiocytosis), hypersplenism, marrow-occupying diseases (eg, leukemia, lymphoma, MDS), solid malignancies, and fibrosis (Table).7
Diagnostic Evaluation
The workup for aplastic anemia should include a thorough history and physical exam to search simultaneously for alternative diagnoses and clues pointing to potential etiologic agents.7 Diagnostic tests to be performed include a complete blood count with differential, reticulocyte count, immature platelet fraction, flow cytometry (to rule out lymphoproliferative disorders and atypical myeloid cells and to evaluate for PNH), and bone marrow biopsy with subsequent cytogenetic, immunohistochemical, and molecular testing.35 Typical findings in aplastic anemia include peripheral blood pancytopenia without dysplastic features and bone marrow biopsy demonstrating a hypocellular marrow.7 A relative lymphocytosis in the peripheral blood is common.7 In patients with a significant PNH clone, a macrocytosis along with elevated lactate dehydrogenase and elevated reticulocyte and granulocyte counts may be present.36
The diagnosis (based on the Camitta criteria37 and modified Camitta criteria38 for severe aplastic anemia) requires 2 of the following findings on peripheral blood samples:
- Absolute neutrophil count (ANC) < 500 cells/µL
- Platelet count < 20,000 cells/µL
- Reticulocyte count < 1% corrected or < 20,000 cells/µL.35
In addition to peripheral blood findings, bone marrow biopsy is essential for the diagnosis, and should demonstrate a markedly hypocellular marrow (cellularity < 25%), occasionally with an increase in T lymphocytes.7,39 Because marrow cellularity varies with age and can be challenging to assess, additional biopsies may be needed to confirm the diagnosis.29 A 1- to 2-cm core biopsy is necessary to confirm hypocellularity, as small areas of residual hematopoiesis may be present and obscure the diagnosis.35
Excluding Hypocellular MDS and IMFS
Excluding hypocellular MDS is challenging, especially in the older adult presenting with aplastic anemia, as patients with aplastic anemia may have some degree of erythroid dysplasia on bone marrow morphology.36 The presence of a PNH clone on flow cytometry can aid in diagnosing aplastic anemia and excluding MDS,34 although PNH clones can be present in refractory anemia MDS. Patients with aplastic anemia have a lower ratio of CD34+ cells compared to those with hypoplastic MDS, with 1 study demonstrating a mean CD34+ percentage of < 0.5% in aplastic anemia versus 3.7% in hypoplastic MDS.40 Cytogenetic and molecular testing can also aid in making this distinction by identifying mutations commonly implicated in MDS.7 The presence of monosomy 7 (-7) in aplastic anemia patients is associated with a poor overall prognosis.34,41
Peripheral blood screening using chromosome breakage analysis (done using either mitomycin C or diepoxybutane as in vitro DNA-crosslinking agents)42 and telomere length testing (of peripheral blood leukocytes) is necessary to exclude the main IMFSs, Fanconi anemia and telomere biology disorders, respectively. Ruling out these conditions is imperative, as the approach to treatment varies significantly between IMFS and aplastic anemia. Patients with shortened telomeres should undergo genetic screening for mutations in the telomere maintenance genes to evaluate the underlying defect leading to shortened telomeres. Patients with increased peripheral blood breakage should have genetic testing to detect mutations associated with Fanconi anemia.
Classification
Once the diagnosis of aplastic anemia has been made, the patient should be classified according to the severity of their disease. Disease severity is determined based on peripheral blood ANC: non-severe aplastic anemia (NSAA), ANC > 500 polymorphonuclear neutrophils (PMNs)/µL; severe aplastic anemia (SAA), 200–500 PMNs/µL; and very severe aplastic anemia (VSAA), 0–200 PMNs/µL.4,34 Disease classification is important, as VSAA is associated with a decreased OS compared to SAA.2 Disease classification may affect treatment decisions, as patients with NSAA may be observed for a short period of time, while, conversely, patients with SAA have a worse prognosis with delays in therapy.43-45
Treatment of Inherited Aplastic Anemia
First-line treatment options for patients with IMFS are androgen therapy and hematopoietic stem cell transplant (HSCT). When evaluating patients for HSCT, it is critical to identify the presence of an IMFS, as the risk and mortality associated with the conditioning regimen, stem cell source, graft-versus-host disease (GVHD), and secondary malignancies differ between patients with IMFS and those with acquired marrow failure syndromes or hematologic malignancies.
Potential sibling donors need to be screened for donor candidacy as well as for the inherited defect. Among patients with Fanconi anemia or a telomere biology disorder, the stem cell source must be considered, with multiple studies in IMFSs and SAA showing superior outcomes with a bone marrow product compared to peripheral blood stem cells.46-48 In IMFS patients, the donor cell type may affect the choice of conditioning regimen.5,6 Reduced-intensity conditioning in lieu of myeloablative conditioning without total body irradiation has proved feasible in patients with Fanconi anemia, and is associated with a reduced risk of secondary malignancies.49,50 Incorporation of fludarabine in the conditioning regimen of patients without a matched sibling donor is associated with superior engraftment and survival46,49,51 compared to cyclophosphamide conditioning, which was historically used in matched related donors.50,52 Adding fludarabine appears to be especially beneficial in older patients, in whom its use is associated with lower rates of graft failure, likely due to increased immunosuppression at the time of engraftment.51,53 Fludarabine has also been incorporated into conditioning regimens for patients with a telomere biology disorder, but outcomes data are limited.5
For patients presenting with AML or a high-risk MDS who are subsequently diagnosed with an IMFS, treatment can be more complex, as these patients are at high risk for toxicity from standard chemotherapy. Limited data suggest that induction therapy and transplantation are feasible in this group of patients, and this approach is associated with increased OS, despite lower OS rates than those of IMFS patients who present prior to the development of MDS or AML.54,55 Further work is needed to determine the optimal induction regimen that balances the risks of treatment-related mortality and complications associated with conditioning regimens, risk of relapse, and risk of secondary malignancies, especially in the cohort of patients diagnosed at an older age.
Treatment of Acquired Aplastic Anemia
Supportive Care
While the workup and treatment plan are being established, attention should be directed at supportive care for prevention of complications. The most common complications leading to death in patients with significant pancytopenia and neutropenia are opportunistic infections and hemorrhagic complications.2
Transfusion support is critical to avoid symptomatic anemia and hemorrhagic complications related to thrombocytopenia, which typically occur with platelet counts lower than 10,000 cells/µL. However, transfusion carries the risk of alloimmunization (which may persist for years following transfusion) and transfusion-related graft versus host disease (trGVHD), and thus use of transfusion should be minimized when possible.56,57 All blood products given to patients with aplastic anemia should be irradiated and leukoreduced to reduce the risk of both alloimmunization and trGVHD. Guidelines from the British Society for Haematology recommend routine screening for Rh and Kell antibodies to reduce the risk of alloimmunization.58 Infectious complications remain a common cause of morbidity and mortality in patients with aplastic anemia who have prolonged neutropenia (defined as an ANC < 500 cells/µL).59-62 Therefore, patients should receive broad-spectrum antibiotics with antipseudomonal coverage. In a study evaluating the role of granulocyte-colony stimulating factor (G-CSF) in patients with SAA receiving immunosuppressive therapy, 55% of all patient deaths were secondary to infection.63 There was no OS benefit seen in patients who received G-CSF, though a significantly lower rate of infection was observed in the G-CSF arm compared to those not receiving G-CSF (56% versus 81%, P = 0.006). This difference was largely driven by a decrease in infectious episodes in patients with VSAA treated with G-CSF as compared to those who did not receive this therapy (22% versus 48%, P = 0.014).63
Angio-invasive pulmonary aspergillosis and Zygomycetes (eg, Rhizopus, Mucor species) remain major causes of mortality related to opportunistic mycotic infections in patients with aplastic anemia.18 The infectious risk is directly related to the duration and severity of neutropenia, with one study demonstrating a significant increase in risk in AML patients with neutropenia lasting longer than 3 weeks.64 Invasive fungal infections carry a high mortality in patients with severe neutropenia, though due to earlier recognition and empiric antifungal therapy with extended-spectrum azoles, overall mortality secondary to invasive fungal infections is declining.62,65
While neutropenia related to cytotoxic chemotherapy is commonly associated with gram-negative bacteria due to disruption of mucosal barriers, patients with aplastic anemia have an increased incidence of gram-positive bacteremia with staphylococcal species compared to other neutropenic populations.61,62 This appears to be changing with time. Valdez et al demonstrated a decrease in prevalence of coagulase-negative staphylococcal infections, increased prevalence of gram-positive bacilli bacteremia, and no change in prevalence of gram-negative bacteremia in patients with aplastic anemia treated between 1989 and 2008.65 Gram-negative bacteremia caused by Stenotrophomonas maltophila, Escherichia coli, Klebsiella pneumoniae, Citrobacter, and Proteus has also been reported.62 Despite a lack of clinical trials investigating the role of antifungal and antibacterial prophylaxis for patients with aplastic anemia, most centers initiate antifungal prophylaxis in patients with SAA or VSAA with an anti-mold agent such as voriconazole or posaconazole (which has the additional benefit compared to voriconazole of covering Mucor species).60,66 This is especially true for patients who have received ATG or undergone HSCT. For antimicrobial prophylaxis, a fluoroquinolone antibiotic with a spectrum of activity against Pseudomonas should be considered for patients with an ANC < 500 cells/µL.60 Acyclovir or valacyclovir prophylaxis is recommended for varicella-zoster virus and herpes simplex virus. Cytomegalovirus reactivation is minimal in patients with aplastic anemia, unless multiple courses of ATG are used.
Iron overload is another complication the provider must be aware of in the setting of increased transfusions in aplastic anemia patients. Lee and colleagues showed that iron chelation therapy using deferasirox is effective at reducing serum ferritin levels in patients with aplastic anemia (median ferritin level of 3254 ng/mL prior to therapy, 1854 ng/mL following), and is associated with no serious adverse events (most common adverse events included nausea, diarrhea, vomiting, and rash).67 Approximately 25% of patients in this trial had an increase in creatinine, with patients taking concomitant cyclosporine affected to a greater degree than those on chelation therapy alone. For patients following HSCT or with improved hematopoiesis following immunosuppressive therapy, phlebotomy can be used to treat iron overload in lieu of chelation therapy.58
Approach to Therapy
The main treatment options for SAA and VSAA include allogeneic bone marrow transplant and immunosuppression. The deciding factors as to which treatment is best initially depends on the availability of HLA-matched related donors and age (Figure 1 and Figure 2). Survival is decreased in patients with SAA or VSAA who delay initiation of therapy, and therefore prompt referral for HLA typing and evaluation for bone marrow transplant is a very important first step in managing aplastic anemia.
Matched Sibling Donor Transplant. Current standards of care recommend HLA-matched sibling donor transplant for patients with SAA or VSAA who are younger than 50 years, with the caveat that integration of fludarabine and reduced cyclophosphamide dosing along with ATG shows the best overall outcomes. Locasciulli and colleagues examined outcomes in patients given either immunosuppressive therapy or sibling HSCT between 1991-1996 and 1997-2002, respectively, and found that sibling HSCT was associated with a superior 10-year OS compared to immunosuppressive therapy (73% versus 68%).43 Interestingly in this study, there was no OS improvement seen with immunosuppressive therapy alone (69% versus 73%) between the 2 time periods, despite increased OS in both sibling HSCT (74% and 80%) and MUD HSCT (38% and 65%).43 Though total body irradiation has been used in the past, it is typically not included in current conditioning regimens for matched related donor transplants.68
Current conditioning regimens typically use a combination of cyclophosphamide and ATG,69,70 with or without fludarabine. Fludarabine-based conditioning regimens have shown promise in patients undergoing sibling HSCT. Maury and colleagues evaluated the role of fludarabine in addition to low-dose cyclophosphamide and ATG compared to cyclophosphamide alone or in combination with ATG in patients over age 30 undergoing sibling HSCT.53 There was a nonsignificant improvement in 5-year OS in the fludarabine arm compared to controls (77% ± 8% versus 60% ± 3%, P = 0.14) in the pooled analysis, but when adjusted for age the fludarabine arm had a significantly lower relative risk (RR) of death (0.44; P = 0.04) compared to the control arm. Shin et al reported outcomes with fludarabine/cyclophosphamide/ATG, with excellent overall outcomes and no difference in patients older or younger than 40 years.71
Kim et al evaluated their experience with patients older than 40 years receiving matched related donors, finding comparable outcomes in those ages 41 to 50 years compared to younger patients. Outcomes declined in those over the age of 50 years.72 Long-term data for matched related donor transplant for aplastic anemia show excellent long-term outcomes, with minimal chronic GVHD and good performance status.73 Hence, these factors support the role of matched related donor transplant as the initial treatment in SAA and VSAA.
Regarding the role of transplant for patients who lack a matched related donor, a growing body of literature demonstrating identical outcomes between matched related and MUD transplants for pediatric patients74,75 supports recent recommendations for upfront unrelated donor transplantation for aplastic anemia.76,77
Immunosuppressive Therapy. For patients without an HLA-matched sibling donor or those who are older than 50 years of age, immunosuppressive therapy is the first-line therapy. ATG and cyclosporine A are the treatments of choice.78 The potential effectiveness of immunosuppressive therapy in treating aplastic anemia was initially observed in patients in whom autologous transplant failed but who still experienced hematopoietic reconstitution despite the failed graft; this observation led to the hypothesis that the conditioning regimen may have an effect on hematopoiesis.59,78,79
Immunosuppressive therapy with ATG has been used for the treatment of aplastic anemia since the 1980s.80 Historically, rabbit ATG had been used, but a 2011 study of horse ATG demonstrated superior hematological response at 6 months compared to rabbit ATG (68% versus 37%).59 Superior survival was also seen with horse ATG compared to rabbit ATG (3-year OS, 96% versus 76%). Due to these results, horse ATG is preferred over rabbit ATG. ATG should be used in combination with cyclosporine A to optimize outcomes.
Early studies also demonstrated the efficacy of cyclosporine A in the treatment of aplastic anemia, with response rates equivalent to that of ATG monotherapy.81 Recent publications still note the efficacy of cyclosporine A in the treatment of aplastic anemia. Its role as an affordable option for single-agent therapy in developing countries is intriguing.81 The combination of ATG and cyclosporine A was proven superior to either agent alone in a study by Frickhofen et al.79 In this study, patients were randomly assigned to a control arm that received ATG plus methylprednisolone or to an arm that received ATG plus cyclosporine A and methylprednisolone. At 6 months, 70% of patients in the cyclosporine A arm had a complete remission (CR) or partial remission compared to 46% in the control arm.82 Further work confirmed the long-term efficacy of this regimen, reporting a 7-year OS of 55%.83 Among a pediatric population, immunosuppressive therapy was associated with an 83% 10-year OS.84
It is recommended that patients remain on cyclosporine therapy for a minimum of 6 months, after which a gradual taper may be considered, although there is variation among practitioners, with some continuing immunosuppressive therapy for a minimum of 12 months due to a proportion of patients being cyclosporine dependent.34,84 A study found that within a population of patients who responded to immunosuppressive therapy, 18% became cyclosporine dependent.84 The median duration of cyclosporine A treatment at full dose was 12 months, with tapering completed over a median of 19 months after patients had been in a stable CR for a minimum of 3 months. Relapse occurred more often when patients were tapered quickly (decrease ≥ 0.8 mg/kg/month) compared to slowly (0.4-0.7 mg/kg/month) or very slowly (< 0.3 mg/kg/month).
Townsley and colleagues recently investigated incorporating the use of the thrombopoietin receptor agonist eltrombopag with immunosuppressive therapy as first-line therapy in aplastic anemia.85 When given at a dose of 150 mg daily in patients ages 12 years and older or 75 mg daily in patients younger than 12 years, in conjunction with cyclosporine A and ATG, patients demonstrated markedly improved hematological response compared to historical treatment with standard immunosuppressive therapy alone.45 In the patient cohort administered eltrombopag starting on day 1 and continuing for 6 months, the complete response rate was 58%. Eltrombopag led to improvement in all cell lines among all treatment subgroups, and OS (censored for patients who proceeded to transplant) was 99% at 2 years.12 Overall, toxicities associated with this therapy were low, with liver enzyme elevations most commonly observed.85 Recently, a phase 2 trial of immunosuppressive therapy with or without eltrombopag was reported. Of the 38 patients enrolled, overall response, complete response, and time to response were not statistically different.86 With this recent finding, the role of eltrombopag in addition to immunosuppressive therapy is not clearly defined, and further studies are warranted.
OS for patients who do not respond to immunosuppressive therapy is approximately 57% at 5 years, largely due to improved supportive measures among this patient population.48,65 Therefore, it is important to recognize those patients who have a low chance of response so that second-line therapy can be pursued to improve outcomes.
Matched Unrelated Donor Transplant. For patients with refractory disease following immunosuppressive therapy who lack a matched sibling donor, MUD HSCT is considered standard therapy given the marked improvement in overall outcomes with modulating conditioning regimens and high-resolution HLA typing. A European Society for Blood and Marrow Transplantation (EBMT) analysis comparing matched sibling HSCT to MUD HSCT noted significantly higher rates of acute grade II-IV and grade III-IV GVHD (grade II-IV 13% versus 25%, grade III-IV 5% versus 10%) among patients undergoing MUD transplant.47 Chronic GVHD rates were 14% in the sibling group, as compared to 26% in the MUD group. Factors associated with improved survival in this analysis include transplant under age 20 years (84% versus 72%), transplant within 6 months of diagnosis (85% versus 72%), the use of ATG in the conditioning regimen (81% versus 73%), and cytomegalovirus-negative donor and recipient as compared to other combinations (82% versus 76%).87 Interestingly, this study demonstrated that OS was not significantly increased when using a sibling HSCT compared to a MUD HSCT, likely as a result of improved understanding of conditioning regimens, GVHD prophylaxis, and supportive care.
Additional studies of MUD HSCT have shown outcomes similar to those seen in sibling HSCT.34,48 A French study found a significant increase in survival in patients undergoing MUD HSCT compared to historical cohorts (2000-2005: OS 52%; 2006-2012: OS 74%).75 The majority of patients underwent conditioning with cyclophosphamide or a combination of busulfan and cyclophosphamide, with or without fludarabine; 81% of patients underwent in vivo T-cell depletion, and a bone marrow donor source was utilized. OS was significantly lower in patients over age 30 years undergoing MUD HSCT (57%) compared to those under age 30 years (70%). Improved OS was also seen when patients underwent transplant within 1 year of diagnosis and when a 10/10 matched donor (compared to a 9/10 mismatched donor) was utilized.48
A 2015 study investigated the role of MUD HSCT as frontline therapy instead of immunosuppressive therapy in patients without a matched sibling donor.75 The 2-year OS was 96% in the MUD HSCT cohort compared to 91%, 94%, and 74% in historical cohorts of sibling HSCT, frontline immunosuppressive therapy, and second-line MUD HSCT following failed immunosuppressive therapy, respectively. Additionally, event-free survival in the MUD HSCT cohort (defined by the authors as death, lack of response, relapse, occurrence of clonal evolution/clinical PNH, malignancies developing over follow‐up, and transplant for patients receiving immunosuppressive therapy frontline) was similar compared to sibling HSCT and superior to frontline immunosuppressive therapy and second-line MUD HSCT. Furthermore, Samarasinghe et al highlighted the importance of in vivo T-cell depletion with either ATG or alemtuzumab (anti-CD52 monoclonal antibody) in the prevention of acute and chronic GVHD in both sibling HSCT and MUD HSCT.88
With continued improvement of less toxic and more immunomodulating conditioning regimens,utilization of bone marrow as a donor cell source, in vivo T-cell depletion, and use of GVHD and antimicrobial prophylaxis, more clinical evidence supports elevating MUD HSCT in the treatment plan for patients without a matched sibling donor.89 However, there is still a large population of patients without matched sibling or unrelated donor options. Given the need to expand the transplant pool and thus avoid clonal hematopoiesis, clinically significant PNH, and relapsed aplastic anemia, more work continues to recognize the expanding role of alternative donor transplants (cord blood and haploidentical) as another viable treatment strategy for aplastic anemia after immunosuppressive therapy failure.90
Summary
Aplastic anemia is a rare but potentially life-threatening disorder with pancytopenia and a marked reduction in the HSC compartment. It can be acquired or associated with an IMFS, and the treatment and prognosis vary dramatically between these 2 etiologies. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia. Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is the new standard of care.
Corresponding author: Gabrielle Meyers, MD, 3181 SW Sam Jackson Park Road, Mail Code UHN73C, Portland, OR 97239.
Financial disclosures: None.
1. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
2. Vaht K, Göransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica. 2017;102:1683-1690.
3. Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718-1721.
4. Montané E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93:518-523.
5. Ohta A, Nagai M, Nishina M, et al. Incidence of aplastic anemia in Japan: analysis of data from a nationwide registration system. Int J Epidemiol. 2015; 44(suppl_1):i178.
6. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
7. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013;139:9-29.
8. Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoiesis stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699-3708.
9. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35-47.
10. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479-2486.
11. Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870.
12. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11-19.
13. Townsley DM, Dumitriu B, Young NS, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922-1931.
14. Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43:598-608.
15. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775-2783.
16. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.
17. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446-4455.
18. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
19. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48:1721-1731.
20. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337-347.
21. Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124:2698-2704.
22. Mukhina GL, Buckley JT, Barber JP, et al. Multilineage glycosylphosphatidylinositol anchor‐deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476-482.
23. Pu JJ, Mukhina G, Wang H, et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87:37-45.
24. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-5340.
25. Devalet B, Mullier F, Chatelain B, et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190-198.
26. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314.
27. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95:1075-1080.
28. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
29. Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76-81.
30. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genetics. 1997;68:58-61.
31. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4-10.
32. Giampietro PF, Davis JG, Adler-Brecher B, et al. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116-1120.
33. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129:1428-1436.
35. DeZern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol. 2011;4:221-230.
36. Tichelli A, Gratwohl A, Nissen C, et al. Morphology in patients with severe aplastic anemia treated with antilymphocyte globulin. Blood. 1992;80:337-345.
37. Camitta BM, Storb R, Thomas ED. Aplastic anemia: pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306:645-652.
38. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
39. Brodsky RA, Chen AR, Dorr D, et al. High-dose cyclophosphamide for severe aplastic anemia: long-term follow-up. Blood. 2010;115:2136-2141.
40. Matsui WH, Brodsky RA, Smith BD, et al. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458-462.
41. Maciejewski JP, Risitano AM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
42. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1-17.
43. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:11-18.
44. Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90:858-864.
45. Gupta V, Eapen M, Brazauskas R, et al. Impact of age on outcomes after transplantation for acquired aplastic anemia using HLA-identical sibling donors. Haematologica. 2010;95:2119-2125.
46. Peffault de Latour R, Le Rademacher J, Antin JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279-4286.
47. Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118:2618-2621.
48. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the Severe Aplastic Anemia Working Party of EBMT. Haematologica. 2016;101:884-890.
49. Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50:1168-1172.
50. De Medeiros CR, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849-852.
51. Mohanan E, Panetta JC, Lakshmi KM, et al. Population pharmacokinetics of fludarabine in patients with aplastic anemia and Fanconi anemia undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:977-983.
52. Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856-2862.
53. Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94:1312-1315.
54. Talbot A, Peffault de Latour R, Raffoux E, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199-e200.
55. Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for Fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669-1676.
56. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
57. Laundy GJ, Bradley BA, Rees BM, et al. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44:814-825.
58. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
59. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430-438.
60. Höchsmann B, Moicean A, Risitano A, et al. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168-173.
61. Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269-276.
62. Torres HA, Bodey GP, Rolston KV, et al. Infections in patients with aplastic anemia: experience at a tertiary care cancer center. Cancer. 2003;98:86-93.
63. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434-4441.
64. Gerson SL, Talbot GH, Hurwitz S, et al. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345-351.
65. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726-735.
66. Robenshtok E, Gafter-Gvili A, Goldberg E, et al. Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol. 2007;25:5471-5489.
67. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood. 2010;116:2448-2454.
68. Deeg HJ, Amylon MD, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208-215.
69. Kahl C, Leisenring W, Joachim Deeg H, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long‐term follow‐up. Br J Haematol. 2005;130:747-751.
70. Socié G. Allogeneic BM transplantation for the treatment of aplastic anemia: current results and expanding donor possibilities. Hematology Am Soc Hematol Educ Program. 2013;2013:82-86.
71. Shin SH, Jeon YW, Yoon JH, et al. Comparable outcomes between younger (<40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016;51:1456-1463.
72. Kim H, Lee KH, Yoon SS, et al; Korean Society of Blood and Marrow Transplantation. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biol Blood Marrow Transplant. 2012;18:1500-1508.
73. Mortensen BK, Jacobsen N, Heilmann C, Sengelov H. Allogeneic hematopoietic cell transplantation for severe aplastic anemia: similar long-term overall survival after transplantation with related donors compared to unrelated donors. Bone Marrow Transplant. 2016;51:288-290.
74. Dufour C, Svahn J, Bacigalupo A. Front-line immunosuppressive treatment of acquired aplastic anemia. Bone Marrow Transplant. 2013;48:174-177.
75. Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on the behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of the EBMT. Br J Haematol. 2015;171:585-594.
76. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2:2020-2028.
77. Yoshida N, Kojima S. Updated guidelines for the treatment of acquired aplastic anemia in children. Curr Oncol Rep. 2018;20:67.
78. Mathe G, Amiel JL, Schwarzenberg L, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J. 1970;2:131-136.
79. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al; German Aplastic Anemia Study Group. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med. 1991;324:1297-1304.
80. Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J. 1981;282:860-863.
81. Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK, et al. Cyclosporine monotherapy for severe aplastic anemia: a developing country experience. Ann Saudi Med. 2005;25:375-379.
82. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185-1196.
83. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135.
84. Saracco P, Quarello P, Iori AP, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long‐term observation follow‐up. Br J Haematol. 2008;140:197-205.
85. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540-1550.
86. Assi R, Garcia-Manero G, Ravandi F, et al. Addition of eltrombopag to immunosuppressive therapy in patients with newly diagnosed aplastic anemia. Cancer. 2018;124:4192-4201.
87. Bacigalupo A, Socié G, Hamladji RM, et al. Current outcome of HLA identical sibling vs. unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100:696-702.
88. Samarasinghe S, Iacobelli S, Knol C, et al. Impact of different in vivo T cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anaemia: a study on behalf of the EBMT SAA Working Party. Am J Hematol. 2019; 94:80-86.
89. Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619-628.
90. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am. 2018;32:629-642.
1. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
2. Vaht K, Göransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica. 2017;102:1683-1690.
3. Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718-1721.
4. Montané E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93:518-523.
5. Ohta A, Nagai M, Nishina M, et al. Incidence of aplastic anemia in Japan: analysis of data from a nationwide registration system. Int J Epidemiol. 2015; 44(suppl_1):i178.
6. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
7. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013;139:9-29.
8. Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoiesis stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699-3708.
9. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35-47.
10. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479-2486.
11. Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870.
12. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11-19.
13. Townsley DM, Dumitriu B, Young NS, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922-1931.
14. Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43:598-608.
15. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775-2783.
16. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.
17. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446-4455.
18. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
19. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48:1721-1731.
20. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337-347.
21. Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124:2698-2704.
22. Mukhina GL, Buckley JT, Barber JP, et al. Multilineage glycosylphosphatidylinositol anchor‐deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476-482.
23. Pu JJ, Mukhina G, Wang H, et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87:37-45.
24. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-5340.
25. Devalet B, Mullier F, Chatelain B, et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190-198.
26. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314.
27. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95:1075-1080.
28. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
29. Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76-81.
30. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genetics. 1997;68:58-61.
31. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4-10.
32. Giampietro PF, Davis JG, Adler-Brecher B, et al. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116-1120.
33. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129:1428-1436.
35. DeZern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol. 2011;4:221-230.
36. Tichelli A, Gratwohl A, Nissen C, et al. Morphology in patients with severe aplastic anemia treated with antilymphocyte globulin. Blood. 1992;80:337-345.
37. Camitta BM, Storb R, Thomas ED. Aplastic anemia: pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306:645-652.
38. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
39. Brodsky RA, Chen AR, Dorr D, et al. High-dose cyclophosphamide for severe aplastic anemia: long-term follow-up. Blood. 2010;115:2136-2141.
40. Matsui WH, Brodsky RA, Smith BD, et al. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458-462.
41. Maciejewski JP, Risitano AM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
42. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1-17.
43. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:11-18.
44. Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90:858-864.
45. Gupta V, Eapen M, Brazauskas R, et al. Impact of age on outcomes after transplantation for acquired aplastic anemia using HLA-identical sibling donors. Haematologica. 2010;95:2119-2125.
46. Peffault de Latour R, Le Rademacher J, Antin JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279-4286.
47. Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118:2618-2621.
48. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the Severe Aplastic Anemia Working Party of EBMT. Haematologica. 2016;101:884-890.
49. Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50:1168-1172.
50. De Medeiros CR, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849-852.
51. Mohanan E, Panetta JC, Lakshmi KM, et al. Population pharmacokinetics of fludarabine in patients with aplastic anemia and Fanconi anemia undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:977-983.
52. Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856-2862.
53. Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94:1312-1315.
54. Talbot A, Peffault de Latour R, Raffoux E, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199-e200.
55. Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for Fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669-1676.
56. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
57. Laundy GJ, Bradley BA, Rees BM, et al. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44:814-825.
58. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
59. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430-438.
60. Höchsmann B, Moicean A, Risitano A, et al. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168-173.
61. Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269-276.
62. Torres HA, Bodey GP, Rolston KV, et al. Infections in patients with aplastic anemia: experience at a tertiary care cancer center. Cancer. 2003;98:86-93.
63. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434-4441.
64. Gerson SL, Talbot GH, Hurwitz S, et al. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345-351.
65. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726-735.
66. Robenshtok E, Gafter-Gvili A, Goldberg E, et al. Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol. 2007;25:5471-5489.
67. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood. 2010;116:2448-2454.
68. Deeg HJ, Amylon MD, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208-215.
69. Kahl C, Leisenring W, Joachim Deeg H, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long‐term follow‐up. Br J Haematol. 2005;130:747-751.
70. Socié G. Allogeneic BM transplantation for the treatment of aplastic anemia: current results and expanding donor possibilities. Hematology Am Soc Hematol Educ Program. 2013;2013:82-86.
71. Shin SH, Jeon YW, Yoon JH, et al. Comparable outcomes between younger (<40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016;51:1456-1463.
72. Kim H, Lee KH, Yoon SS, et al; Korean Society of Blood and Marrow Transplantation. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biol Blood Marrow Transplant. 2012;18:1500-1508.
73. Mortensen BK, Jacobsen N, Heilmann C, Sengelov H. Allogeneic hematopoietic cell transplantation for severe aplastic anemia: similar long-term overall survival after transplantation with related donors compared to unrelated donors. Bone Marrow Transplant. 2016;51:288-290.
74. Dufour C, Svahn J, Bacigalupo A. Front-line immunosuppressive treatment of acquired aplastic anemia. Bone Marrow Transplant. 2013;48:174-177.
75. Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on the behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of the EBMT. Br J Haematol. 2015;171:585-594.
76. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2:2020-2028.
77. Yoshida N, Kojima S. Updated guidelines for the treatment of acquired aplastic anemia in children. Curr Oncol Rep. 2018;20:67.
78. Mathe G, Amiel JL, Schwarzenberg L, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J. 1970;2:131-136.
79. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al; German Aplastic Anemia Study Group. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med. 1991;324:1297-1304.
80. Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J. 1981;282:860-863.
81. Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK, et al. Cyclosporine monotherapy for severe aplastic anemia: a developing country experience. Ann Saudi Med. 2005;25:375-379.
82. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185-1196.
83. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135.
84. Saracco P, Quarello P, Iori AP, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long‐term observation follow‐up. Br J Haematol. 2008;140:197-205.
85. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540-1550.
86. Assi R, Garcia-Manero G, Ravandi F, et al. Addition of eltrombopag to immunosuppressive therapy in patients with newly diagnosed aplastic anemia. Cancer. 2018;124:4192-4201.
87. Bacigalupo A, Socié G, Hamladji RM, et al. Current outcome of HLA identical sibling vs. unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100:696-702.
88. Samarasinghe S, Iacobelli S, Knol C, et al. Impact of different in vivo T cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anaemia: a study on behalf of the EBMT SAA Working Party. Am J Hematol. 2019; 94:80-86.
89. Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619-628.
90. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am. 2018;32:629-642.