User login
2019 at a glance: Hem-onc U.S. drug approvals
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.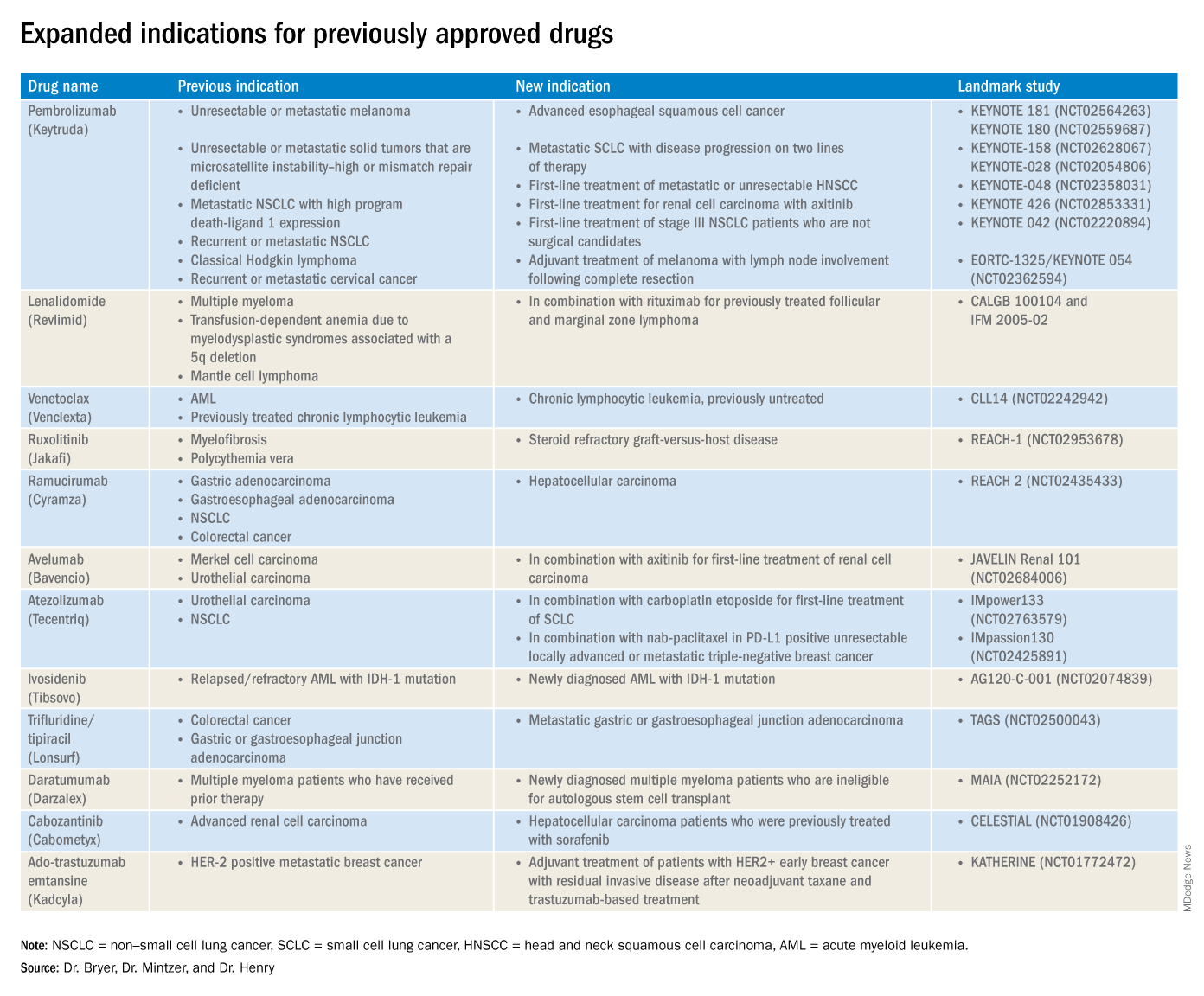
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
Dose-reduced NOACs may be safer than warfarin in some AFib patients
Background: Prior studies have suggested that NOACs have a favorable risk-benefit profile when compared with warfarin, but it is unclear if this advantage also is present for those high-risk patients for whom NOAC dose reduction is recommended.

Study design: A meta-analysis.
Setting: Three phase 3 randomized, control trials.
Synopsis: From the three randomized, control trials, the authors identified 7,351 of the 46,426 patients as being eligible for dose-reduced NOACs. Of these patients, 3,702 were randomized to take a NOAC and 3,649 were randomized to take warfarin. For the primary outcomes of stroke or systemic embolism, there was no significant difference between patients randomized to receive dose-reduced NOAC versus warfarin. For outcomes of major bleeding, hemorrhagic stroke, intracranial hemorrhage, and fatal bleeding, dose-reduced NOACs had a significantly lower risk, compared with warfarin.
Bottom line: In patients eligible for dose-reduced NOACs, the use of dose-reduced NOACs may have a better safety profile without significant difference in the rate of ischemic stroke or systemic embolism.
Citation: Wang KL et al. Efficacy and safety of reduced-dose non–vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: A meta-analysis of randomized controlled trials. Eur Heart J. 2018 Dec 22. doi: 10.1093/eurheartj/ehy802.
Dr. Biddick is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine Harvard Medical School.
Background: Prior studies have suggested that NOACs have a favorable risk-benefit profile when compared with warfarin, but it is unclear if this advantage also is present for those high-risk patients for whom NOAC dose reduction is recommended.
Study design: A meta-analysis.
Setting: Three phase 3 randomized, control trials.
Synopsis: From the three randomized, control trials, the authors identified 7,351 of the 46,426 patients as being eligible for dose-reduced NOACs. Of these patients, 3,702 were randomized to take a NOAC and 3,649 were randomized to take warfarin. For the primary outcomes of stroke or systemic embolism, there was no significant difference between patients randomized to receive dose-reduced NOAC versus warfarin. For outcomes of major bleeding, hemorrhagic stroke, intracranial hemorrhage, and fatal bleeding, dose-reduced NOACs had a significantly lower risk, compared with warfarin.
Bottom line: In patients eligible for dose-reduced NOACs, the use of dose-reduced NOACs may have a better safety profile without significant difference in the rate of ischemic stroke or systemic embolism.
Citation: Wang KL et al. Efficacy and safety of reduced-dose non–vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: A meta-analysis of randomized controlled trials. Eur Heart J. 2018 Dec 22. doi: 10.1093/eurheartj/ehy802.
Dr. Biddick is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine Harvard Medical School.
Background: Prior studies have suggested that NOACs have a favorable risk-benefit profile when compared with warfarin, but it is unclear if this advantage also is present for those high-risk patients for whom NOAC dose reduction is recommended.
Study design: A meta-analysis.
Setting: Three phase 3 randomized, control trials.
Synopsis: From the three randomized, control trials, the authors identified 7,351 of the 46,426 patients as being eligible for dose-reduced NOACs. Of these patients, 3,702 were randomized to take a NOAC and 3,649 were randomized to take warfarin. For the primary outcomes of stroke or systemic embolism, there was no significant difference between patients randomized to receive dose-reduced NOAC versus warfarin. For outcomes of major bleeding, hemorrhagic stroke, intracranial hemorrhage, and fatal bleeding, dose-reduced NOACs had a significantly lower risk, compared with warfarin.
Bottom line: In patients eligible for dose-reduced NOACs, the use of dose-reduced NOACs may have a better safety profile without significant difference in the rate of ischemic stroke or systemic embolism.
Citation: Wang KL et al. Efficacy and safety of reduced-dose non–vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: A meta-analysis of randomized controlled trials. Eur Heart J. 2018 Dec 22. doi: 10.1093/eurheartj/ehy802.
Dr. Biddick is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine Harvard Medical School.
Thromboembolic events more likely among CIDP patients with CVAD
AUSTIN, TEX. – Patients with chronic inflammatory demyelinating polyneuropathy (CIDP) who receive intravenous immunoglobulin (IVIg) appear to have an increased risk of thromboembolic events if it is administered with a central venous access device (CVAD) when compared against those without a CVAD, according to a recent study.
Although CVADs can reliably deliver IVIg, they also represent an established risk factor for thromboembolic events, Ami Patel, PhD, a senior epidemiologist at CSL Behring, and colleagues noted on their poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The results suggest a need for physicians to be vigilant about patients’ potential risk factors for thromboembolic events, Dr. Patel said in an interview. Further research is planned, however, because the current study did not control for other risk factors or explore other possible confounding, she said.
Dr. Patel and her associates analyzed U.S. claims data (IBM/Truven MarketScan) from 2006 to 2018 and included all patients with a CIDP diagnosis claim and a postdiagnosis code for IVIg. A code for CVAD up to 2 months before CIDP diagnosis without removal before IVIg treatment ended determined those with CVAD exposure, and thromboembolic events included any codes related to arterial, venous, or vascular prostheses.
The researchers then compared patients in a case-control fashion, matching each one with a CVAD to five patients of similar demographics without a CVAD. Characteristics used for matching included medical insurance type, prescription data availability, sex, age, geographic region, and years enrolled in the database.
Among 7,447 patients with at least one IVIg claim, 11.8% (n = 882) had CVAD exposure and 88.2% (n = 6,565) did not. Of those without a CVAD, 3,642 patients were matched to patients with CVAD. A quarter (25.4%) of patients with a CVAD had a thromboembolic event, compared with 11.2% of matched patients without CVADs (P less than .0001).
In the year leading up to IVIg therapy, 16.9% of those with a CVAD and 10.9% of matched patients without one had a previous thromboembolic event (P less than .0001). Patients with a CVAD also had significantly higher rates of hypertension (51.9% vs. 45.0% with placebo; P less than .001) and anticoagulation therapy (7.0% vs. 5.2% with placebo; P less than .05). Differences between the groups were not significant for diabetes (26.9% vs. 24.2%) and hyperlipidemia (19.1% vs. 17.8%).
Occlusion and stenosis of the carotid artery was the most common arterial thromboembolic outcome, occurring in 5.3% of those with a CVAD and in 2.8% of those without a CVAD. The most common venous thromboembolic event was acute venous embolism and thrombosis of lower-extremity deep vessels, which occurred in 7% of those with a CVAD and in 1.8% of those without.
The researchers also compared inpatient admissions and emergency department visits among those with and without a CVAD; both rates were higher in patients with a CVAD. Visits to the emergency department occurred at a rate of 0.14 events per month for those with a CVAD (2.01 distinct months with a claim) and 0.09 events per month for those without a CVAD (0.65 distinct months with a claim). Patients with a CVAD had 1.44 months with an inpatient admissions claim, in comparison with 0.41 months among matched patients without a CVAD. Inpatient admission frequency per month was 0.14 for those with a CVAD and 0.08 for those without.
The research was funded by CSL Behring. Dr. Patel and two of the other five authors are employees of CSL Behring.
SOURCE: Patel A et al. AANEM 2019, Abstract 94.
AUSTIN, TEX. – Patients with chronic inflammatory demyelinating polyneuropathy (CIDP) who receive intravenous immunoglobulin (IVIg) appear to have an increased risk of thromboembolic events if it is administered with a central venous access device (CVAD) when compared against those without a CVAD, according to a recent study.
Although CVADs can reliably deliver IVIg, they also represent an established risk factor for thromboembolic events, Ami Patel, PhD, a senior epidemiologist at CSL Behring, and colleagues noted on their poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The results suggest a need for physicians to be vigilant about patients’ potential risk factors for thromboembolic events, Dr. Patel said in an interview. Further research is planned, however, because the current study did not control for other risk factors or explore other possible confounding, she said.
Dr. Patel and her associates analyzed U.S. claims data (IBM/Truven MarketScan) from 2006 to 2018 and included all patients with a CIDP diagnosis claim and a postdiagnosis code for IVIg. A code for CVAD up to 2 months before CIDP diagnosis without removal before IVIg treatment ended determined those with CVAD exposure, and thromboembolic events included any codes related to arterial, venous, or vascular prostheses.
The researchers then compared patients in a case-control fashion, matching each one with a CVAD to five patients of similar demographics without a CVAD. Characteristics used for matching included medical insurance type, prescription data availability, sex, age, geographic region, and years enrolled in the database.
Among 7,447 patients with at least one IVIg claim, 11.8% (n = 882) had CVAD exposure and 88.2% (n = 6,565) did not. Of those without a CVAD, 3,642 patients were matched to patients with CVAD. A quarter (25.4%) of patients with a CVAD had a thromboembolic event, compared with 11.2% of matched patients without CVADs (P less than .0001).
In the year leading up to IVIg therapy, 16.9% of those with a CVAD and 10.9% of matched patients without one had a previous thromboembolic event (P less than .0001). Patients with a CVAD also had significantly higher rates of hypertension (51.9% vs. 45.0% with placebo; P less than .001) and anticoagulation therapy (7.0% vs. 5.2% with placebo; P less than .05). Differences between the groups were not significant for diabetes (26.9% vs. 24.2%) and hyperlipidemia (19.1% vs. 17.8%).
Occlusion and stenosis of the carotid artery was the most common arterial thromboembolic outcome, occurring in 5.3% of those with a CVAD and in 2.8% of those without a CVAD. The most common venous thromboembolic event was acute venous embolism and thrombosis of lower-extremity deep vessels, which occurred in 7% of those with a CVAD and in 1.8% of those without.
The researchers also compared inpatient admissions and emergency department visits among those with and without a CVAD; both rates were higher in patients with a CVAD. Visits to the emergency department occurred at a rate of 0.14 events per month for those with a CVAD (2.01 distinct months with a claim) and 0.09 events per month for those without a CVAD (0.65 distinct months with a claim). Patients with a CVAD had 1.44 months with an inpatient admissions claim, in comparison with 0.41 months among matched patients without a CVAD. Inpatient admission frequency per month was 0.14 for those with a CVAD and 0.08 for those without.
The research was funded by CSL Behring. Dr. Patel and two of the other five authors are employees of CSL Behring.
SOURCE: Patel A et al. AANEM 2019, Abstract 94.
AUSTIN, TEX. – Patients with chronic inflammatory demyelinating polyneuropathy (CIDP) who receive intravenous immunoglobulin (IVIg) appear to have an increased risk of thromboembolic events if it is administered with a central venous access device (CVAD) when compared against those without a CVAD, according to a recent study.
Although CVADs can reliably deliver IVIg, they also represent an established risk factor for thromboembolic events, Ami Patel, PhD, a senior epidemiologist at CSL Behring, and colleagues noted on their poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The results suggest a need for physicians to be vigilant about patients’ potential risk factors for thromboembolic events, Dr. Patel said in an interview. Further research is planned, however, because the current study did not control for other risk factors or explore other possible confounding, she said.
Dr. Patel and her associates analyzed U.S. claims data (IBM/Truven MarketScan) from 2006 to 2018 and included all patients with a CIDP diagnosis claim and a postdiagnosis code for IVIg. A code for CVAD up to 2 months before CIDP diagnosis without removal before IVIg treatment ended determined those with CVAD exposure, and thromboembolic events included any codes related to arterial, venous, or vascular prostheses.
The researchers then compared patients in a case-control fashion, matching each one with a CVAD to five patients of similar demographics without a CVAD. Characteristics used for matching included medical insurance type, prescription data availability, sex, age, geographic region, and years enrolled in the database.
Among 7,447 patients with at least one IVIg claim, 11.8% (n = 882) had CVAD exposure and 88.2% (n = 6,565) did not. Of those without a CVAD, 3,642 patients were matched to patients with CVAD. A quarter (25.4%) of patients with a CVAD had a thromboembolic event, compared with 11.2% of matched patients without CVADs (P less than .0001).
In the year leading up to IVIg therapy, 16.9% of those with a CVAD and 10.9% of matched patients without one had a previous thromboembolic event (P less than .0001). Patients with a CVAD also had significantly higher rates of hypertension (51.9% vs. 45.0% with placebo; P less than .001) and anticoagulation therapy (7.0% vs. 5.2% with placebo; P less than .05). Differences between the groups were not significant for diabetes (26.9% vs. 24.2%) and hyperlipidemia (19.1% vs. 17.8%).
Occlusion and stenosis of the carotid artery was the most common arterial thromboembolic outcome, occurring in 5.3% of those with a CVAD and in 2.8% of those without a CVAD. The most common venous thromboembolic event was acute venous embolism and thrombosis of lower-extremity deep vessels, which occurred in 7% of those with a CVAD and in 1.8% of those without.
The researchers also compared inpatient admissions and emergency department visits among those with and without a CVAD; both rates were higher in patients with a CVAD. Visits to the emergency department occurred at a rate of 0.14 events per month for those with a CVAD (2.01 distinct months with a claim) and 0.09 events per month for those without a CVAD (0.65 distinct months with a claim). Patients with a CVAD had 1.44 months with an inpatient admissions claim, in comparison with 0.41 months among matched patients without a CVAD. Inpatient admission frequency per month was 0.14 for those with a CVAD and 0.08 for those without.
The research was funded by CSL Behring. Dr. Patel and two of the other five authors are employees of CSL Behring.
SOURCE: Patel A et al. AANEM 2019, Abstract 94.
REPORTING FROM AANEM 2019
Emicizumab effective in children with hemophilia A and inhibitors
Emicizumab (Hemlibra) is well tolerated and has substantial, clinically meaningful efficacy in pediatric patients with hemophilia A and factor VIII inhibitors, according to an analysis of data from the HAVEN 2 trial in this bispecific humanized monoclonal antibody.
Among those receiving once-weekly emicizumab prophylaxis, 77% had no treated bleeding events and 100% of evaluable target joints resolved in the HAVEN 2 study, which investigators say is the largest prospective study so far of bleed prevention in pediatric patients with hemophilia A and inhibitors.
Moreover, emicizumab resulted in a 99% reduction in bleeding rate versus previous bypassing agent prophylaxis, subsequent, according to an intraindividual comparison described in the report.
Based on these results, emicizumab stands to become the “next-generation standard of care” for pediatric patients with hemophilia A and factor VIII inhibitors, reported Guy Young, MD, of Children’s Hospital Los Angeles and University of Southern California Keck School of Medicine, and his coinvestigators.
“Despite a rapidly evolving treatment landscape in hemophilia, children have been largely excluded from recent trials of novel agents,” they said in the report, which appears in the journal Blood.
Before emicizumab, current treatment options for pediatric patients with factor VIII inhibitors were limited to immune tolerance induction, or use of bypassing agents with efficacy that “can be suboptimal and unpredictable,” said Dr. Young and colleagues.
“More effective prophylactic options with reduced treatment burden are needed,” they said.
Emicizumab works in hemophilia A by bridging activated factor IX and factor X, restoring the function of missing factor VIIIa, according to the report.
A total of 88 male pediatric patients with congenital hemophilia A were enrolled in HAVEN 2, an ongoing phase 3 multicenter study that is nonrandomized and open label. The median age of patients was 7 years (range, 1-15 years). Most participants (97%) had severe hemophilia A, 72% had previously undergone immune tolerance induction, and 75% were receiving treatment with prophylactic bypassing agents.
Most participants received 4 once-weekly loading doses of 3 mg/kg body weight of emicizumab subcutaneously, followed by a maintenance regimen of 1.5 mg/kg weekly.
The annualized bleed rate was 0.3, with 77% of participants having zero bleeding events, for the 65 participants in the trial who were under 12 and received emicizumab 1.5 mg/kg weekly, according to the report.
In the subset of those patients with target joints, the mean annualized bleed rate was 3.3 for the first 24 weeks, and 0 for the next 24 weeks; these findings suggest that bleed rates decrease over time with continuing emicizumab treatment “even in patients with a more severe phenotype,” the investigators wrote.
For 15 patients younger than 12 years of age receiving bypassing agent prophylaxis, emicizumab 1.5 mg/kg once weekly resulted in an annualized bleed rate of 0.3, compared to 21.1 for the prior bypassing agent, which translates into a 99% reduction in bleeding risk, according to investigators.
Annualized bleed rates for patients receiving emicizumab 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks were 0.2 and 2.2, respectively; hover, the numerically higher annualized bleed rate in the 6 mg/kg every-4-weeks group was based on 2 out of the 10 patients treated with that maintenance regimen, including one who had 6 target joint bleeds in the 24 weeks before enrolling in the study, and another who developed antidrug antibodies within the first 8 weeks of emicizumab treatment.
Out of 23 patients who had target joints and received emicizumab prophylaxis for at least 52 weeks, 100% (45 of 45) target joints resolved, according to the report.
“Notably, this is the first report of a treatment resolving target joints in an inhibitor population, which until now has only been reported when using factor VIII products in patients without inhibitors,” Dr. Young and colleagues said.
The most common of the 721 adverse events reported in HAVEN 2 were nasopharyngitis and injection-site reactions. Of 21 serious adverse events, only 1 (antidrug antibodies with neutralizing potential) was thought by investigators to be related to emicizumab.
The Food and Drug Administration (FDA) initially approved emicizumab in November 2017 on the basis of the HAVEN 2 pediatric trial, and on HAVEN 1, a randomized, multicenter, open-label, phase 3 trial including 109 adult and adolescent males with hemophilia A and FVIII inhibitors. The indication for emicizumab was expanded to include patients without inhibitors in October 2018, on the basis of the HAVEN 3 and HAVEN 4 randomized phase 3 trials.
Dr. Young reported disclosures related to Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Shire, Spark, and uniQure.
SOURCE: Young G, et al. Blood. 2019 Oct 10. doi: 10.1182/blood.2019001869.
Emicizumab (Hemlibra) is well tolerated and has substantial, clinically meaningful efficacy in pediatric patients with hemophilia A and factor VIII inhibitors, according to an analysis of data from the HAVEN 2 trial in this bispecific humanized monoclonal antibody.
Among those receiving once-weekly emicizumab prophylaxis, 77% had no treated bleeding events and 100% of evaluable target joints resolved in the HAVEN 2 study, which investigators say is the largest prospective study so far of bleed prevention in pediatric patients with hemophilia A and inhibitors.
Moreover, emicizumab resulted in a 99% reduction in bleeding rate versus previous bypassing agent prophylaxis, subsequent, according to an intraindividual comparison described in the report.
Based on these results, emicizumab stands to become the “next-generation standard of care” for pediatric patients with hemophilia A and factor VIII inhibitors, reported Guy Young, MD, of Children’s Hospital Los Angeles and University of Southern California Keck School of Medicine, and his coinvestigators.
“Despite a rapidly evolving treatment landscape in hemophilia, children have been largely excluded from recent trials of novel agents,” they said in the report, which appears in the journal Blood.
Before emicizumab, current treatment options for pediatric patients with factor VIII inhibitors were limited to immune tolerance induction, or use of bypassing agents with efficacy that “can be suboptimal and unpredictable,” said Dr. Young and colleagues.
“More effective prophylactic options with reduced treatment burden are needed,” they said.
Emicizumab works in hemophilia A by bridging activated factor IX and factor X, restoring the function of missing factor VIIIa, according to the report.
A total of 88 male pediatric patients with congenital hemophilia A were enrolled in HAVEN 2, an ongoing phase 3 multicenter study that is nonrandomized and open label. The median age of patients was 7 years (range, 1-15 years). Most participants (97%) had severe hemophilia A, 72% had previously undergone immune tolerance induction, and 75% were receiving treatment with prophylactic bypassing agents.
Most participants received 4 once-weekly loading doses of 3 mg/kg body weight of emicizumab subcutaneously, followed by a maintenance regimen of 1.5 mg/kg weekly.
The annualized bleed rate was 0.3, with 77% of participants having zero bleeding events, for the 65 participants in the trial who were under 12 and received emicizumab 1.5 mg/kg weekly, according to the report.
In the subset of those patients with target joints, the mean annualized bleed rate was 3.3 for the first 24 weeks, and 0 for the next 24 weeks; these findings suggest that bleed rates decrease over time with continuing emicizumab treatment “even in patients with a more severe phenotype,” the investigators wrote.
For 15 patients younger than 12 years of age receiving bypassing agent prophylaxis, emicizumab 1.5 mg/kg once weekly resulted in an annualized bleed rate of 0.3, compared to 21.1 for the prior bypassing agent, which translates into a 99% reduction in bleeding risk, according to investigators.
Annualized bleed rates for patients receiving emicizumab 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks were 0.2 and 2.2, respectively; hover, the numerically higher annualized bleed rate in the 6 mg/kg every-4-weeks group was based on 2 out of the 10 patients treated with that maintenance regimen, including one who had 6 target joint bleeds in the 24 weeks before enrolling in the study, and another who developed antidrug antibodies within the first 8 weeks of emicizumab treatment.
Out of 23 patients who had target joints and received emicizumab prophylaxis for at least 52 weeks, 100% (45 of 45) target joints resolved, according to the report.
“Notably, this is the first report of a treatment resolving target joints in an inhibitor population, which until now has only been reported when using factor VIII products in patients without inhibitors,” Dr. Young and colleagues said.
The most common of the 721 adverse events reported in HAVEN 2 were nasopharyngitis and injection-site reactions. Of 21 serious adverse events, only 1 (antidrug antibodies with neutralizing potential) was thought by investigators to be related to emicizumab.
The Food and Drug Administration (FDA) initially approved emicizumab in November 2017 on the basis of the HAVEN 2 pediatric trial, and on HAVEN 1, a randomized, multicenter, open-label, phase 3 trial including 109 adult and adolescent males with hemophilia A and FVIII inhibitors. The indication for emicizumab was expanded to include patients without inhibitors in October 2018, on the basis of the HAVEN 3 and HAVEN 4 randomized phase 3 trials.
Dr. Young reported disclosures related to Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Shire, Spark, and uniQure.
SOURCE: Young G, et al. Blood. 2019 Oct 10. doi: 10.1182/blood.2019001869.
Emicizumab (Hemlibra) is well tolerated and has substantial, clinically meaningful efficacy in pediatric patients with hemophilia A and factor VIII inhibitors, according to an analysis of data from the HAVEN 2 trial in this bispecific humanized monoclonal antibody.
Among those receiving once-weekly emicizumab prophylaxis, 77% had no treated bleeding events and 100% of evaluable target joints resolved in the HAVEN 2 study, which investigators say is the largest prospective study so far of bleed prevention in pediatric patients with hemophilia A and inhibitors.
Moreover, emicizumab resulted in a 99% reduction in bleeding rate versus previous bypassing agent prophylaxis, subsequent, according to an intraindividual comparison described in the report.
Based on these results, emicizumab stands to become the “next-generation standard of care” for pediatric patients with hemophilia A and factor VIII inhibitors, reported Guy Young, MD, of Children’s Hospital Los Angeles and University of Southern California Keck School of Medicine, and his coinvestigators.
“Despite a rapidly evolving treatment landscape in hemophilia, children have been largely excluded from recent trials of novel agents,” they said in the report, which appears in the journal Blood.
Before emicizumab, current treatment options for pediatric patients with factor VIII inhibitors were limited to immune tolerance induction, or use of bypassing agents with efficacy that “can be suboptimal and unpredictable,” said Dr. Young and colleagues.
“More effective prophylactic options with reduced treatment burden are needed,” they said.
Emicizumab works in hemophilia A by bridging activated factor IX and factor X, restoring the function of missing factor VIIIa, according to the report.
A total of 88 male pediatric patients with congenital hemophilia A were enrolled in HAVEN 2, an ongoing phase 3 multicenter study that is nonrandomized and open label. The median age of patients was 7 years (range, 1-15 years). Most participants (97%) had severe hemophilia A, 72% had previously undergone immune tolerance induction, and 75% were receiving treatment with prophylactic bypassing agents.
Most participants received 4 once-weekly loading doses of 3 mg/kg body weight of emicizumab subcutaneously, followed by a maintenance regimen of 1.5 mg/kg weekly.
The annualized bleed rate was 0.3, with 77% of participants having zero bleeding events, for the 65 participants in the trial who were under 12 and received emicizumab 1.5 mg/kg weekly, according to the report.
In the subset of those patients with target joints, the mean annualized bleed rate was 3.3 for the first 24 weeks, and 0 for the next 24 weeks; these findings suggest that bleed rates decrease over time with continuing emicizumab treatment “even in patients with a more severe phenotype,” the investigators wrote.
For 15 patients younger than 12 years of age receiving bypassing agent prophylaxis, emicizumab 1.5 mg/kg once weekly resulted in an annualized bleed rate of 0.3, compared to 21.1 for the prior bypassing agent, which translates into a 99% reduction in bleeding risk, according to investigators.
Annualized bleed rates for patients receiving emicizumab 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks were 0.2 and 2.2, respectively; hover, the numerically higher annualized bleed rate in the 6 mg/kg every-4-weeks group was based on 2 out of the 10 patients treated with that maintenance regimen, including one who had 6 target joint bleeds in the 24 weeks before enrolling in the study, and another who developed antidrug antibodies within the first 8 weeks of emicizumab treatment.
Out of 23 patients who had target joints and received emicizumab prophylaxis for at least 52 weeks, 100% (45 of 45) target joints resolved, according to the report.
“Notably, this is the first report of a treatment resolving target joints in an inhibitor population, which until now has only been reported when using factor VIII products in patients without inhibitors,” Dr. Young and colleagues said.
The most common of the 721 adverse events reported in HAVEN 2 were nasopharyngitis and injection-site reactions. Of 21 serious adverse events, only 1 (antidrug antibodies with neutralizing potential) was thought by investigators to be related to emicizumab.
The Food and Drug Administration (FDA) initially approved emicizumab in November 2017 on the basis of the HAVEN 2 pediatric trial, and on HAVEN 1, a randomized, multicenter, open-label, phase 3 trial including 109 adult and adolescent males with hemophilia A and FVIII inhibitors. The indication for emicizumab was expanded to include patients without inhibitors in October 2018, on the basis of the HAVEN 3 and HAVEN 4 randomized phase 3 trials.
Dr. Young reported disclosures related to Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Shire, Spark, and uniQure.
SOURCE: Young G, et al. Blood. 2019 Oct 10. doi: 10.1182/blood.2019001869.
FROM Blood
NIH seeks gene-based cures for HIV, sickle cell disease
The National Institutes of Health and the Bill & Melinda Gates Foundation have announced that they plan to invest $100 million each over the next 4 years to develop affordable, gene-based cures for sickle cell disease (SCD) and HIV.
The initiative follows an announcement from President Trump that set a goal of ending the HIV epidemic in the United States in the next 10 years, seeking to reduce the number of diagnoses by 90% by 2030. The Trump administration has also identified SCD as an “intractable health challenge with the potential for dramatic advances in the coming years,” the NIH said in a statement.
Gene-based therapy has become a reality in recent years thanks to dramatic advances, but the cost is prohibitive in many parts of the world. “The collaboration between the NIH and the Gates Foundation sets out a bold goal of advancing safe, effective, and durable gene-based cures to clinical trials in the United States and relevant countries in sub-Saharan Africa within the next 7-10 years. The ultimate goal is to scale and implement these treatments globally in areas hardest hit by these diseases,” the NIH said.
Both diseases are a significant burden on low- and middle-income countries, as 95% of the 38 million people living with HIV globally are in the developing world, with 67% living in sub-Saharan Africa; about half of the HIV-infected population receives no treatment for the disease. An estimated 15 million children will be born with SCD over the next 30 years, with three-quarters of those births occurring in sub-Saharan Africa. About 50%-90% of children born with SCD will die before age 5 years.
The collaboration will focus on coordination in two areas: identifying potential candidate cures for SCD and HIV for preclinical and clinical evaluation, and defining long-term opportunities to work together and with African partners on advancing promising candidates to late-phase clinical trials, with funding to be determined as candidates progress.
“In recent years, gene-based treatments have been groundbreaking for rare genetic disorders and infectious diseases. While these treatments are exciting, people in low- and middle-income countries do not have access to these breakthroughs. By working with the NIH and scientists across Africa, we aim to ensure these approaches will improve the lives of those most in need and bring the incredible promise of gene-based treatments to the world of public health,” said Trevor Mundel, MD, PhD, president of the global health program at the Bill & Melinda Gates Foundation.
The National Institutes of Health and the Bill & Melinda Gates Foundation have announced that they plan to invest $100 million each over the next 4 years to develop affordable, gene-based cures for sickle cell disease (SCD) and HIV.
The initiative follows an announcement from President Trump that set a goal of ending the HIV epidemic in the United States in the next 10 years, seeking to reduce the number of diagnoses by 90% by 2030. The Trump administration has also identified SCD as an “intractable health challenge with the potential for dramatic advances in the coming years,” the NIH said in a statement.
Gene-based therapy has become a reality in recent years thanks to dramatic advances, but the cost is prohibitive in many parts of the world. “The collaboration between the NIH and the Gates Foundation sets out a bold goal of advancing safe, effective, and durable gene-based cures to clinical trials in the United States and relevant countries in sub-Saharan Africa within the next 7-10 years. The ultimate goal is to scale and implement these treatments globally in areas hardest hit by these diseases,” the NIH said.
Both diseases are a significant burden on low- and middle-income countries, as 95% of the 38 million people living with HIV globally are in the developing world, with 67% living in sub-Saharan Africa; about half of the HIV-infected population receives no treatment for the disease. An estimated 15 million children will be born with SCD over the next 30 years, with three-quarters of those births occurring in sub-Saharan Africa. About 50%-90% of children born with SCD will die before age 5 years.
The collaboration will focus on coordination in two areas: identifying potential candidate cures for SCD and HIV for preclinical and clinical evaluation, and defining long-term opportunities to work together and with African partners on advancing promising candidates to late-phase clinical trials, with funding to be determined as candidates progress.
“In recent years, gene-based treatments have been groundbreaking for rare genetic disorders and infectious diseases. While these treatments are exciting, people in low- and middle-income countries do not have access to these breakthroughs. By working with the NIH and scientists across Africa, we aim to ensure these approaches will improve the lives of those most in need and bring the incredible promise of gene-based treatments to the world of public health,” said Trevor Mundel, MD, PhD, president of the global health program at the Bill & Melinda Gates Foundation.
The National Institutes of Health and the Bill & Melinda Gates Foundation have announced that they plan to invest $100 million each over the next 4 years to develop affordable, gene-based cures for sickle cell disease (SCD) and HIV.
The initiative follows an announcement from President Trump that set a goal of ending the HIV epidemic in the United States in the next 10 years, seeking to reduce the number of diagnoses by 90% by 2030. The Trump administration has also identified SCD as an “intractable health challenge with the potential for dramatic advances in the coming years,” the NIH said in a statement.
Gene-based therapy has become a reality in recent years thanks to dramatic advances, but the cost is prohibitive in many parts of the world. “The collaboration between the NIH and the Gates Foundation sets out a bold goal of advancing safe, effective, and durable gene-based cures to clinical trials in the United States and relevant countries in sub-Saharan Africa within the next 7-10 years. The ultimate goal is to scale and implement these treatments globally in areas hardest hit by these diseases,” the NIH said.
Both diseases are a significant burden on low- and middle-income countries, as 95% of the 38 million people living with HIV globally are in the developing world, with 67% living in sub-Saharan Africa; about half of the HIV-infected population receives no treatment for the disease. An estimated 15 million children will be born with SCD over the next 30 years, with three-quarters of those births occurring in sub-Saharan Africa. About 50%-90% of children born with SCD will die before age 5 years.
The collaboration will focus on coordination in two areas: identifying potential candidate cures for SCD and HIV for preclinical and clinical evaluation, and defining long-term opportunities to work together and with African partners on advancing promising candidates to late-phase clinical trials, with funding to be determined as candidates progress.
“In recent years, gene-based treatments have been groundbreaking for rare genetic disorders and infectious diseases. While these treatments are exciting, people in low- and middle-income countries do not have access to these breakthroughs. By working with the NIH and scientists across Africa, we aim to ensure these approaches will improve the lives of those most in need and bring the incredible promise of gene-based treatments to the world of public health,” said Trevor Mundel, MD, PhD, president of the global health program at the Bill & Melinda Gates Foundation.
Fibrinogen concentrate effective, safe for postop bleeding
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.

Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.
Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.
Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
REPORTING FROM AABB 2019
Hematopoietic cell transplant offers realistic cure in secondary AML
yielding significantly better survival outcomes, according to findings from an observational study.
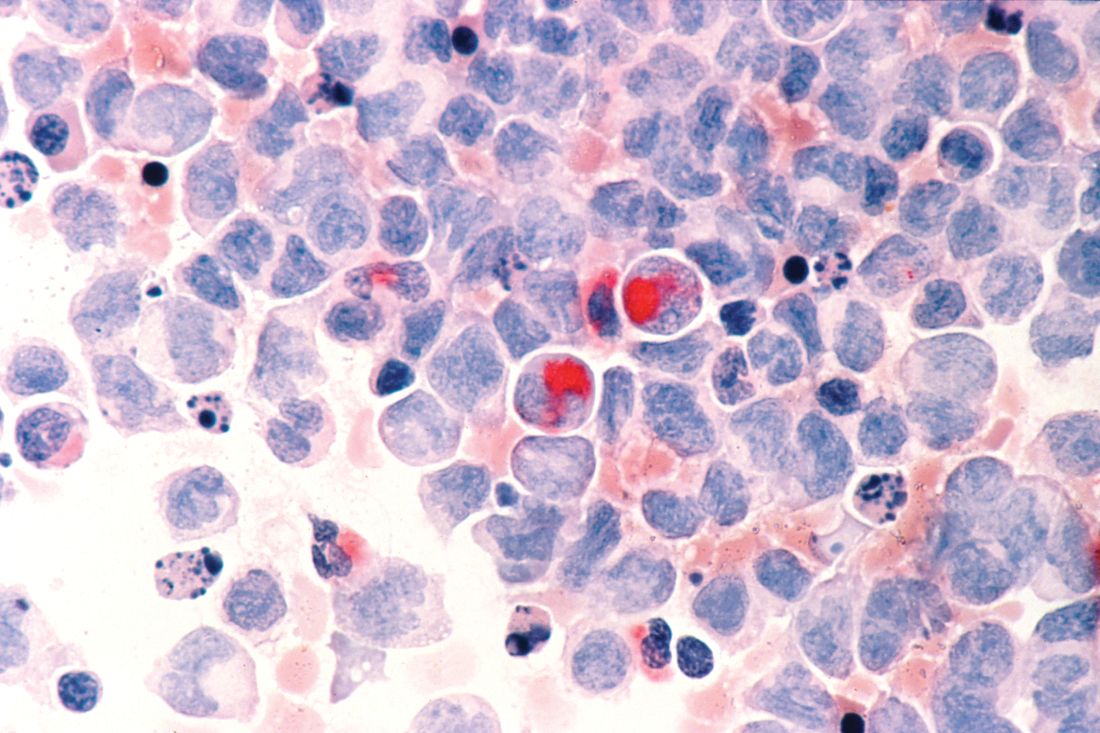
Although secondary AML has been identified as an independent predictor of poor prognosis, it is not included in current risk classifications that provide the basis of deciding when to perform HCT.
Christer Nilsson, MD, of Karolinska Institute, Stockholm, and colleagues, used two nationwide Swedish registries – the Swedish AML Registry and the Swedish Cancer Registry – to characterize how often HCT is performed in these patients and to evaluate its impact in a real-world setting. The registries include all patients with AML diagnosed between 1997 and 2013.
Their findings are in Biology of Blood and Marrow Transplantation.
The analysis included 3,337 adult patients with AML who were intensively treated and did not have acute promyelocytic leukemia. More than three-quarters of the patients had de novo AML and the remainder had secondary AML that was either therapy related or developed after an antecedent myeloid disease. In total, 100 patients with secondary AML underwent HCT while in first complete remission.
In terms of crude survival at 5 years after diagnosis, patients with secondary AML who did not undergo HCT did very poorly. The survival rate was 0% in those with AML preceded by myeloproliferative neoplasm (MPN-AML), 2% in patients with AML preceded by myelodysplastic syndrome (MDS-AML), and 4% in patients with therapy-related AML (t-AML). In contrast, the 5-year overall survival in patients who underwent HCT at any time point or disease stage was 32% for patients with MPN-AML, 18% for patients with MDS-AML, and 25% for patients t-AML.
These crude survival figures suggest that “HCT is the sole realistic curable treatment option for [secondary] AML,” the researchers wrote.
The researchers also performed a propensity score matching analysis of HCT versus chemotherapy consolidation in patients with secondary AML who had been in first complete remission for more than 90 days. The model matched 45 patients who underwent HCT with 66 patients treated with chemotherapy consolidation. The projected 5-year overall survival was 48% in the HCT group, compared with 20% in the consolidation group (P = .01). Similarly, 5-year relapse-free survival was also higher in the HCT group, compared with the consolidation group (43% vs. 21%, P = .02).
“Ideally, the role of transplantation in [secondary] AML should be evaluated in a prospective randomized trial, minimizing the risk of any bias,” the researchers wrote. “However, such a trial is lacking and most likely will never be performed.”
The researchers concluded that HCT should be considered for all patients with secondary AML who are eligible and fit for transplantation.
The study was supported by the Swedish Cancer Foundation, Swedish Research Council, Stockholm County Council, Gothenberg Medical Society, and Assar Gabrielsson Foundation. The researchers reported having no conflicts of interest.
SOURCE: Nilson C et al. Biol Blood Marrow Tranplant. 2019;25:1770-8.
yielding significantly better survival outcomes, according to findings from an observational study.
Although secondary AML has been identified as an independent predictor of poor prognosis, it is not included in current risk classifications that provide the basis of deciding when to perform HCT.
Christer Nilsson, MD, of Karolinska Institute, Stockholm, and colleagues, used two nationwide Swedish registries – the Swedish AML Registry and the Swedish Cancer Registry – to characterize how often HCT is performed in these patients and to evaluate its impact in a real-world setting. The registries include all patients with AML diagnosed between 1997 and 2013.
Their findings are in Biology of Blood and Marrow Transplantation.
The analysis included 3,337 adult patients with AML who were intensively treated and did not have acute promyelocytic leukemia. More than three-quarters of the patients had de novo AML and the remainder had secondary AML that was either therapy related or developed after an antecedent myeloid disease. In total, 100 patients with secondary AML underwent HCT while in first complete remission.
In terms of crude survival at 5 years after diagnosis, patients with secondary AML who did not undergo HCT did very poorly. The survival rate was 0% in those with AML preceded by myeloproliferative neoplasm (MPN-AML), 2% in patients with AML preceded by myelodysplastic syndrome (MDS-AML), and 4% in patients with therapy-related AML (t-AML). In contrast, the 5-year overall survival in patients who underwent HCT at any time point or disease stage was 32% for patients with MPN-AML, 18% for patients with MDS-AML, and 25% for patients t-AML.
These crude survival figures suggest that “HCT is the sole realistic curable treatment option for [secondary] AML,” the researchers wrote.
The researchers also performed a propensity score matching analysis of HCT versus chemotherapy consolidation in patients with secondary AML who had been in first complete remission for more than 90 days. The model matched 45 patients who underwent HCT with 66 patients treated with chemotherapy consolidation. The projected 5-year overall survival was 48% in the HCT group, compared with 20% in the consolidation group (P = .01). Similarly, 5-year relapse-free survival was also higher in the HCT group, compared with the consolidation group (43% vs. 21%, P = .02).
“Ideally, the role of transplantation in [secondary] AML should be evaluated in a prospective randomized trial, minimizing the risk of any bias,” the researchers wrote. “However, such a trial is lacking and most likely will never be performed.”
The researchers concluded that HCT should be considered for all patients with secondary AML who are eligible and fit for transplantation.
The study was supported by the Swedish Cancer Foundation, Swedish Research Council, Stockholm County Council, Gothenberg Medical Society, and Assar Gabrielsson Foundation. The researchers reported having no conflicts of interest.
SOURCE: Nilson C et al. Biol Blood Marrow Tranplant. 2019;25:1770-8.
yielding significantly better survival outcomes, according to findings from an observational study.
Although secondary AML has been identified as an independent predictor of poor prognosis, it is not included in current risk classifications that provide the basis of deciding when to perform HCT.
Christer Nilsson, MD, of Karolinska Institute, Stockholm, and colleagues, used two nationwide Swedish registries – the Swedish AML Registry and the Swedish Cancer Registry – to characterize how often HCT is performed in these patients and to evaluate its impact in a real-world setting. The registries include all patients with AML diagnosed between 1997 and 2013.
Their findings are in Biology of Blood and Marrow Transplantation.
The analysis included 3,337 adult patients with AML who were intensively treated and did not have acute promyelocytic leukemia. More than three-quarters of the patients had de novo AML and the remainder had secondary AML that was either therapy related or developed after an antecedent myeloid disease. In total, 100 patients with secondary AML underwent HCT while in first complete remission.
In terms of crude survival at 5 years after diagnosis, patients with secondary AML who did not undergo HCT did very poorly. The survival rate was 0% in those with AML preceded by myeloproliferative neoplasm (MPN-AML), 2% in patients with AML preceded by myelodysplastic syndrome (MDS-AML), and 4% in patients with therapy-related AML (t-AML). In contrast, the 5-year overall survival in patients who underwent HCT at any time point or disease stage was 32% for patients with MPN-AML, 18% for patients with MDS-AML, and 25% for patients t-AML.
These crude survival figures suggest that “HCT is the sole realistic curable treatment option for [secondary] AML,” the researchers wrote.
The researchers also performed a propensity score matching analysis of HCT versus chemotherapy consolidation in patients with secondary AML who had been in first complete remission for more than 90 days. The model matched 45 patients who underwent HCT with 66 patients treated with chemotherapy consolidation. The projected 5-year overall survival was 48% in the HCT group, compared with 20% in the consolidation group (P = .01). Similarly, 5-year relapse-free survival was also higher in the HCT group, compared with the consolidation group (43% vs. 21%, P = .02).
“Ideally, the role of transplantation in [secondary] AML should be evaluated in a prospective randomized trial, minimizing the risk of any bias,” the researchers wrote. “However, such a trial is lacking and most likely will never be performed.”
The researchers concluded that HCT should be considered for all patients with secondary AML who are eligible and fit for transplantation.
The study was supported by the Swedish Cancer Foundation, Swedish Research Council, Stockholm County Council, Gothenberg Medical Society, and Assar Gabrielsson Foundation. The researchers reported having no conflicts of interest.
SOURCE: Nilson C et al. Biol Blood Marrow Tranplant. 2019;25:1770-8.
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
Updated international consensus recommendations on management of acute upper GI bleeding
Guidelines on the management of acute upper gastrointestinal bleeding have been updated, including recommendations on managing patients on antiplatelet or anticoagulant therapy and on use of endoscopy and new therapeutic approaches.
Writing in Annals of Internal Medicine, an international group of experts published an update to the 2010 International Consensus Recommendations on the Management of Patients With Nonvariceal Upper Gastrointestinal Bleeding, with a focus on resuscitation and risk assessment; pre-endoscopic, endoscopic, and pharmacologic management; and secondary prophylaxis.
Alan N. Barkun, MDCM, MSc, from McGill University, Montreal, and coauthors first recommended that fluid resuscitation should be initiated in patients with acute upper gastrointestinal bleeding and hemodynamic instability to avoid hemorrhagic shock and restore end-organ perfusion and tissue oxygenation while the bleeding is brought under control.
They acknowledged the uncertainty around whether colloid or crystalloid fluid should be used, but suggested routine use of colloids was not justified because they were more expensive and did not appear to increase survival.
On the question of whether the resuscitation should be aggressive or restrictive in its timing and rate, the group said there was not enough evidence to support a recommendation on this. “The important issue in patients with hemorrhagic shock due to trauma or UGIB [upper gastrointestinal bleeding] is to stop the bleeding while minimizing hemodynamic compromise,” they wrote.
They also advised blood transfusions in patients with a hemoglobin level below 80 g/L who did not have underlying cardiovascular disease, but suggested a higher hemoglobin threshold for those with underlying cardiovascular disease.
The second recommendation was that patients with a Glasgow Blatchford score of 1 or less were at very low risk for rebleeding and mortality, and these patients may therefore not need hospitalization or inpatient endoscopy. They advised against using the AIMS65 prognostic score for this purpose because it was designed to identify patients at high risk of death, not those at low risk for safe discharge.
In regard to endoscopic management, they advocated that all patients with acute upper gastrointestinal bleeding – whether low or high risk – undergo endoscopy within 24 hours of presentation. This was even more urgent in patients being treated with anticoagulants. “Because of the recognized benefits of early endoscopy, coagulopathy should be treated as necessary but endoscopy should not be delayed,” they wrote.
Patients with acutely bleeding ulcers with high-risk stigmata should undergo endoscopic therapy preferably with thermocoagulation or sclerosant injection, or with hemoclips depending on the bleeding location and patient characteristics.
The group also included two conditional recommendations, based on very-low-quality evidence, that patients with actively bleeding ulcers receive TC-325 hemostatic powder as temporizing therapy to stop the bleeding if conventional endoscopic therapies aren’t available or fail. However, they stressed that TC-325 should not be used as a single therapeutic strategy.
Because of a lack of efficacy data and low availability of expertise in the technology, the authors said they could not make a recommendation for or against using a Doppler endoscopic probe (DEP) to assess the need for further endoscopic therapy.
“The group generally agreed that although making a recommendation for or against using DEP to manage UGIB is premature, DEP has the potential to alter the usual approach to visually assessing bleeding lesion risk when evaluating the need for, and adequacy of, endoscopic hemostasis.”
The guidelines also addressed the issue of pharmacologic management of acute upper gastrointestinal bleeding. They strongly recommended that patients with bleeding ulcers and high-risk stigmata who have undergone successful endoscopic therapy should then receive an intravenous loading dose of proton pump inhibitor (PPI) therapy, followed by continuous intravenous infusion.
“Cost-effectiveness studies have suggested that high-dose intravenous PPIs after successful endoscopic hemostasis improve outcomes at a modest cost increase relative to non–high-dose intravenous or oral PPI strategies,” they wrote.
A second conditional recommendation, based on very-low-quality evidence, was that patients with a bleeding ulcer who were at high risk for rebleeding be also treated twice-daily with oral PPIs for 2 weeks, then once-daily. They also recommended patients on cardiovascular prophylaxis with single or dual antiplatelet therapy or anticoagulant therapy be given PPIs.
“The consensus group concluded that, for high-risk patients with an ongoing need for anticoagulants, the evidence suggests that the benefits of secondary prophylaxis outweigh the risks.”
The group was supported by a grant from CIHR Institute of Nutrition, Metabolism and Diabetes and from the Saudi Gastroenterology Association. Nine authors declared grants, personal fees, honoraria and other funding from the pharmaceutical and medical device sector outside the submitted work. No other conflicts of interest were declared.
SOURCE: Barkun A et al. Ann Intern Med 2019, October 22. doi: 10.7326/M19-1795.
These updated consensus guidelines provide a rigorous review of evidence on managing nonvariceal upper gastrointestinal bleeding. The recommendations for patients on anticoagulant or antiplatelet therapy will be particularly helpful because of increasing use of these medications. The advice on proton pump inhibitor therapy in patients on these drugs who have had previous ulcer bleeding can help allay concerns about possible integrations between PPIs and clopidogrel.
While the guidelines recommend using the Glasgow Blatchford scale to guide hospitalization decisions, prognostic scores are not commonly used in the emergency department, and many patients present with a Glasgow Blatchford score greater than 1, so this tool may have little impact on hospitalization rates. More studies are needed to compare clinical judgment with these prognostic scores.
Angel Lanas, MD, is from the University Clinic Hospital at the University of Zaragoza (Spain). These comments are adapted from an accompanying editorial (Ann Intern Med 2019, October 22. doi: 10.7326/M19-2789). Dr. Lanas declared unrelated personal fees from the pharmaceutical sector.
These updated consensus guidelines provide a rigorous review of evidence on managing nonvariceal upper gastrointestinal bleeding. The recommendations for patients on anticoagulant or antiplatelet therapy will be particularly helpful because of increasing use of these medications. The advice on proton pump inhibitor therapy in patients on these drugs who have had previous ulcer bleeding can help allay concerns about possible integrations between PPIs and clopidogrel.
While the guidelines recommend using the Glasgow Blatchford scale to guide hospitalization decisions, prognostic scores are not commonly used in the emergency department, and many patients present with a Glasgow Blatchford score greater than 1, so this tool may have little impact on hospitalization rates. More studies are needed to compare clinical judgment with these prognostic scores.
Angel Lanas, MD, is from the University Clinic Hospital at the University of Zaragoza (Spain). These comments are adapted from an accompanying editorial (Ann Intern Med 2019, October 22. doi: 10.7326/M19-2789). Dr. Lanas declared unrelated personal fees from the pharmaceutical sector.
These updated consensus guidelines provide a rigorous review of evidence on managing nonvariceal upper gastrointestinal bleeding. The recommendations for patients on anticoagulant or antiplatelet therapy will be particularly helpful because of increasing use of these medications. The advice on proton pump inhibitor therapy in patients on these drugs who have had previous ulcer bleeding can help allay concerns about possible integrations between PPIs and clopidogrel.
While the guidelines recommend using the Glasgow Blatchford scale to guide hospitalization decisions, prognostic scores are not commonly used in the emergency department, and many patients present with a Glasgow Blatchford score greater than 1, so this tool may have little impact on hospitalization rates. More studies are needed to compare clinical judgment with these prognostic scores.
Angel Lanas, MD, is from the University Clinic Hospital at the University of Zaragoza (Spain). These comments are adapted from an accompanying editorial (Ann Intern Med 2019, October 22. doi: 10.7326/M19-2789). Dr. Lanas declared unrelated personal fees from the pharmaceutical sector.
Guidelines on the management of acute upper gastrointestinal bleeding have been updated, including recommendations on managing patients on antiplatelet or anticoagulant therapy and on use of endoscopy and new therapeutic approaches.
Writing in Annals of Internal Medicine, an international group of experts published an update to the 2010 International Consensus Recommendations on the Management of Patients With Nonvariceal Upper Gastrointestinal Bleeding, with a focus on resuscitation and risk assessment; pre-endoscopic, endoscopic, and pharmacologic management; and secondary prophylaxis.
Alan N. Barkun, MDCM, MSc, from McGill University, Montreal, and coauthors first recommended that fluid resuscitation should be initiated in patients with acute upper gastrointestinal bleeding and hemodynamic instability to avoid hemorrhagic shock and restore end-organ perfusion and tissue oxygenation while the bleeding is brought under control.
They acknowledged the uncertainty around whether colloid or crystalloid fluid should be used, but suggested routine use of colloids was not justified because they were more expensive and did not appear to increase survival.
On the question of whether the resuscitation should be aggressive or restrictive in its timing and rate, the group said there was not enough evidence to support a recommendation on this. “The important issue in patients with hemorrhagic shock due to trauma or UGIB [upper gastrointestinal bleeding] is to stop the bleeding while minimizing hemodynamic compromise,” they wrote.
They also advised blood transfusions in patients with a hemoglobin level below 80 g/L who did not have underlying cardiovascular disease, but suggested a higher hemoglobin threshold for those with underlying cardiovascular disease.
The second recommendation was that patients with a Glasgow Blatchford score of 1 or less were at very low risk for rebleeding and mortality, and these patients may therefore not need hospitalization or inpatient endoscopy. They advised against using the AIMS65 prognostic score for this purpose because it was designed to identify patients at high risk of death, not those at low risk for safe discharge.
In regard to endoscopic management, they advocated that all patients with acute upper gastrointestinal bleeding – whether low or high risk – undergo endoscopy within 24 hours of presentation. This was even more urgent in patients being treated with anticoagulants. “Because of the recognized benefits of early endoscopy, coagulopathy should be treated as necessary but endoscopy should not be delayed,” they wrote.
Patients with acutely bleeding ulcers with high-risk stigmata should undergo endoscopic therapy preferably with thermocoagulation or sclerosant injection, or with hemoclips depending on the bleeding location and patient characteristics.
The group also included two conditional recommendations, based on very-low-quality evidence, that patients with actively bleeding ulcers receive TC-325 hemostatic powder as temporizing therapy to stop the bleeding if conventional endoscopic therapies aren’t available or fail. However, they stressed that TC-325 should not be used as a single therapeutic strategy.
Because of a lack of efficacy data and low availability of expertise in the technology, the authors said they could not make a recommendation for or against using a Doppler endoscopic probe (DEP) to assess the need for further endoscopic therapy.
“The group generally agreed that although making a recommendation for or against using DEP to manage UGIB is premature, DEP has the potential to alter the usual approach to visually assessing bleeding lesion risk when evaluating the need for, and adequacy of, endoscopic hemostasis.”
The guidelines also addressed the issue of pharmacologic management of acute upper gastrointestinal bleeding. They strongly recommended that patients with bleeding ulcers and high-risk stigmata who have undergone successful endoscopic therapy should then receive an intravenous loading dose of proton pump inhibitor (PPI) therapy, followed by continuous intravenous infusion.
“Cost-effectiveness studies have suggested that high-dose intravenous PPIs after successful endoscopic hemostasis improve outcomes at a modest cost increase relative to non–high-dose intravenous or oral PPI strategies,” they wrote.
A second conditional recommendation, based on very-low-quality evidence, was that patients with a bleeding ulcer who were at high risk for rebleeding be also treated twice-daily with oral PPIs for 2 weeks, then once-daily. They also recommended patients on cardiovascular prophylaxis with single or dual antiplatelet therapy or anticoagulant therapy be given PPIs.
“The consensus group concluded that, for high-risk patients with an ongoing need for anticoagulants, the evidence suggests that the benefits of secondary prophylaxis outweigh the risks.”
The group was supported by a grant from CIHR Institute of Nutrition, Metabolism and Diabetes and from the Saudi Gastroenterology Association. Nine authors declared grants, personal fees, honoraria and other funding from the pharmaceutical and medical device sector outside the submitted work. No other conflicts of interest were declared.
SOURCE: Barkun A et al. Ann Intern Med 2019, October 22. doi: 10.7326/M19-1795.
Guidelines on the management of acute upper gastrointestinal bleeding have been updated, including recommendations on managing patients on antiplatelet or anticoagulant therapy and on use of endoscopy and new therapeutic approaches.
Writing in Annals of Internal Medicine, an international group of experts published an update to the 2010 International Consensus Recommendations on the Management of Patients With Nonvariceal Upper Gastrointestinal Bleeding, with a focus on resuscitation and risk assessment; pre-endoscopic, endoscopic, and pharmacologic management; and secondary prophylaxis.
Alan N. Barkun, MDCM, MSc, from McGill University, Montreal, and coauthors first recommended that fluid resuscitation should be initiated in patients with acute upper gastrointestinal bleeding and hemodynamic instability to avoid hemorrhagic shock and restore end-organ perfusion and tissue oxygenation while the bleeding is brought under control.
They acknowledged the uncertainty around whether colloid or crystalloid fluid should be used, but suggested routine use of colloids was not justified because they were more expensive and did not appear to increase survival.
On the question of whether the resuscitation should be aggressive or restrictive in its timing and rate, the group said there was not enough evidence to support a recommendation on this. “The important issue in patients with hemorrhagic shock due to trauma or UGIB [upper gastrointestinal bleeding] is to stop the bleeding while minimizing hemodynamic compromise,” they wrote.
They also advised blood transfusions in patients with a hemoglobin level below 80 g/L who did not have underlying cardiovascular disease, but suggested a higher hemoglobin threshold for those with underlying cardiovascular disease.
The second recommendation was that patients with a Glasgow Blatchford score of 1 or less were at very low risk for rebleeding and mortality, and these patients may therefore not need hospitalization or inpatient endoscopy. They advised against using the AIMS65 prognostic score for this purpose because it was designed to identify patients at high risk of death, not those at low risk for safe discharge.
In regard to endoscopic management, they advocated that all patients with acute upper gastrointestinal bleeding – whether low or high risk – undergo endoscopy within 24 hours of presentation. This was even more urgent in patients being treated with anticoagulants. “Because of the recognized benefits of early endoscopy, coagulopathy should be treated as necessary but endoscopy should not be delayed,” they wrote.
Patients with acutely bleeding ulcers with high-risk stigmata should undergo endoscopic therapy preferably with thermocoagulation or sclerosant injection, or with hemoclips depending on the bleeding location and patient characteristics.
The group also included two conditional recommendations, based on very-low-quality evidence, that patients with actively bleeding ulcers receive TC-325 hemostatic powder as temporizing therapy to stop the bleeding if conventional endoscopic therapies aren’t available or fail. However, they stressed that TC-325 should not be used as a single therapeutic strategy.
Because of a lack of efficacy data and low availability of expertise in the technology, the authors said they could not make a recommendation for or against using a Doppler endoscopic probe (DEP) to assess the need for further endoscopic therapy.
“The group generally agreed that although making a recommendation for or against using DEP to manage UGIB is premature, DEP has the potential to alter the usual approach to visually assessing bleeding lesion risk when evaluating the need for, and adequacy of, endoscopic hemostasis.”
The guidelines also addressed the issue of pharmacologic management of acute upper gastrointestinal bleeding. They strongly recommended that patients with bleeding ulcers and high-risk stigmata who have undergone successful endoscopic therapy should then receive an intravenous loading dose of proton pump inhibitor (PPI) therapy, followed by continuous intravenous infusion.
“Cost-effectiveness studies have suggested that high-dose intravenous PPIs after successful endoscopic hemostasis improve outcomes at a modest cost increase relative to non–high-dose intravenous or oral PPI strategies,” they wrote.
A second conditional recommendation, based on very-low-quality evidence, was that patients with a bleeding ulcer who were at high risk for rebleeding be also treated twice-daily with oral PPIs for 2 weeks, then once-daily. They also recommended patients on cardiovascular prophylaxis with single or dual antiplatelet therapy or anticoagulant therapy be given PPIs.
“The consensus group concluded that, for high-risk patients with an ongoing need for anticoagulants, the evidence suggests that the benefits of secondary prophylaxis outweigh the risks.”
The group was supported by a grant from CIHR Institute of Nutrition, Metabolism and Diabetes and from the Saudi Gastroenterology Association. Nine authors declared grants, personal fees, honoraria and other funding from the pharmaceutical and medical device sector outside the submitted work. No other conflicts of interest were declared.
SOURCE: Barkun A et al. Ann Intern Med 2019, October 22. doi: 10.7326/M19-1795.
FROM ANNALS OF INTERNAL MEDICINE

FDA gives nod to earlier use of Nplate in adults with ITP
Romiplostim (Nplate) earned a new indication from the Food and Drug Administration, allowing for earlier usage in adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.

The new indication is for newly diagnosed and persistent adult patients.
Romiplostim is already approved for the treatment of pediatric patients aged 1 year and older who have had ITP for at least 6 months and had an insufficient response to other treatments, as well as for adults patients with chronic ITP who had insufficient response to other therapies.
The latest approval is based on positive results from a single-arm, phase 2 trial in adults with ITP who had an insufficient response to first-line treatment. The study met its primary endpoint – platelet response (50 x 109/L or greater). The median number of months with platelet response was 11 months during a 12-month treatment period. The median time to first platelet response was just over 2 weeks.
Adverse events of at least 5% incidence in patients taking romiplostim with an ITP duration up to 12 months included bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea, and oropharyngeal pain. There was a 2% incidence of thrombocytosis among adults with an ITP duration up to 12 months.
Romiplostim (Nplate) earned a new indication from the Food and Drug Administration, allowing for earlier usage in adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
The new indication is for newly diagnosed and persistent adult patients.
Romiplostim is already approved for the treatment of pediatric patients aged 1 year and older who have had ITP for at least 6 months and had an insufficient response to other treatments, as well as for adults patients with chronic ITP who had insufficient response to other therapies.
The latest approval is based on positive results from a single-arm, phase 2 trial in adults with ITP who had an insufficient response to first-line treatment. The study met its primary endpoint – platelet response (50 x 109/L or greater). The median number of months with platelet response was 11 months during a 12-month treatment period. The median time to first platelet response was just over 2 weeks.
Adverse events of at least 5% incidence in patients taking romiplostim with an ITP duration up to 12 months included bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea, and oropharyngeal pain. There was a 2% incidence of thrombocytosis among adults with an ITP duration up to 12 months.
Romiplostim (Nplate) earned a new indication from the Food and Drug Administration, allowing for earlier usage in adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
The new indication is for newly diagnosed and persistent adult patients.
Romiplostim is already approved for the treatment of pediatric patients aged 1 year and older who have had ITP for at least 6 months and had an insufficient response to other treatments, as well as for adults patients with chronic ITP who had insufficient response to other therapies.
The latest approval is based on positive results from a single-arm, phase 2 trial in adults with ITP who had an insufficient response to first-line treatment. The study met its primary endpoint – platelet response (50 x 109/L or greater). The median number of months with platelet response was 11 months during a 12-month treatment period. The median time to first platelet response was just over 2 weeks.
Adverse events of at least 5% incidence in patients taking romiplostim with an ITP duration up to 12 months included bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea, and oropharyngeal pain. There was a 2% incidence of thrombocytosis among adults with an ITP duration up to 12 months.
Global blood supply runs low
Nearly two-thirds of countries worldwide have an insufficient supply of blood for transfusion, according to findings from a recent modeling study including 195 countries and territories.
![]()
Low- and middle-income countries are most often affected, reported lead author Nicholas Roberts of the University of Washington in Seattle and colleagues. Among all countries in Oceania, south Asia, and eastern, central, and western sub-Saharan Africa, not one had enough blood to meet estimated needs, they noted.
Shortages are attributable to a variety of challenges, including resource constraints, insufficient component production, and a high prevalence of infectious diseases, according to investigators. And existing guidelines, which call for 10-20 donations per 1,000 people in the population, may need to be revised, investigators suggested.
“These estimates are important, as they can be used to guide further investments in blood transfusion services, for analysis of current transfusion practices, and to highlight need for alternatives to transfusions such as antifibrinolytics, blood saving devices, and implementation of blood management systems,” the investigators wrote in Lancet Haematology.
Blood availability was calculated using the 2016 WHO Global Status Report on Blood Safety and Availability, in which 92% of countries participated. Blood needs were calculated using multiple databases, including the (U.S.) National Inpatient Sample datasets from 2000-2014, the State Inpatient Databases from 2003-2007, and the Global Burden of Disease 2017 study.
The global blood need was almost 305 million blood product units, while the supply totaled approximately 272 million units. These shortages, however, were not distributed equally across the globe. Out of 195 countries, 119 (61%) had an insufficient supply of blood to meet anticipated demand. Within this group, the shortage equated to about 102 million blood product units.
Denmark had the greatest supply of blood products (red blood cell products, platelets, and plasma), at 14,704 units per 100,000 population, compared with South Sudan, the least prepared to meet transfusion needs, with just 46 blood product units available per 100,000 population. This pattern was echoed across the globe; high-income countries were usually better stocked than those with low or middle income.
To meet demands, some of the most affected countries would need to raise their collection goals from single-digit figures to as high as 40 donations per 1,000 population, the investigators advised.
“Many countries face critical undersupply of transfusions, which will become more pronounced as access to care improves,” the investigators wrote.
The study was funded by the National Institutes of Health. The investigators reported having no other financial disclosures.
SOURCE: Roberts N et al. Lancet Haematol. 2019 Oct 17. doi: 10.1016/ S2352-3026(19)30200-5.
Nearly two-thirds of countries worldwide have an insufficient supply of blood for transfusion, according to findings from a recent modeling study including 195 countries and territories.
![]()
Low- and middle-income countries are most often affected, reported lead author Nicholas Roberts of the University of Washington in Seattle and colleagues. Among all countries in Oceania, south Asia, and eastern, central, and western sub-Saharan Africa, not one had enough blood to meet estimated needs, they noted.
Shortages are attributable to a variety of challenges, including resource constraints, insufficient component production, and a high prevalence of infectious diseases, according to investigators. And existing guidelines, which call for 10-20 donations per 1,000 people in the population, may need to be revised, investigators suggested.
“These estimates are important, as they can be used to guide further investments in blood transfusion services, for analysis of current transfusion practices, and to highlight need for alternatives to transfusions such as antifibrinolytics, blood saving devices, and implementation of blood management systems,” the investigators wrote in Lancet Haematology.
Blood availability was calculated using the 2016 WHO Global Status Report on Blood Safety and Availability, in which 92% of countries participated. Blood needs were calculated using multiple databases, including the (U.S.) National Inpatient Sample datasets from 2000-2014, the State Inpatient Databases from 2003-2007, and the Global Burden of Disease 2017 study.
The global blood need was almost 305 million blood product units, while the supply totaled approximately 272 million units. These shortages, however, were not distributed equally across the globe. Out of 195 countries, 119 (61%) had an insufficient supply of blood to meet anticipated demand. Within this group, the shortage equated to about 102 million blood product units.
Denmark had the greatest supply of blood products (red blood cell products, platelets, and plasma), at 14,704 units per 100,000 population, compared with South Sudan, the least prepared to meet transfusion needs, with just 46 blood product units available per 100,000 population. This pattern was echoed across the globe; high-income countries were usually better stocked than those with low or middle income.
To meet demands, some of the most affected countries would need to raise their collection goals from single-digit figures to as high as 40 donations per 1,000 population, the investigators advised.
“Many countries face critical undersupply of transfusions, which will become more pronounced as access to care improves,” the investigators wrote.
The study was funded by the National Institutes of Health. The investigators reported having no other financial disclosures.
SOURCE: Roberts N et al. Lancet Haematol. 2019 Oct 17. doi: 10.1016/ S2352-3026(19)30200-5.
Nearly two-thirds of countries worldwide have an insufficient supply of blood for transfusion, according to findings from a recent modeling study including 195 countries and territories.
![]()
Low- and middle-income countries are most often affected, reported lead author Nicholas Roberts of the University of Washington in Seattle and colleagues. Among all countries in Oceania, south Asia, and eastern, central, and western sub-Saharan Africa, not one had enough blood to meet estimated needs, they noted.
Shortages are attributable to a variety of challenges, including resource constraints, insufficient component production, and a high prevalence of infectious diseases, according to investigators. And existing guidelines, which call for 10-20 donations per 1,000 people in the population, may need to be revised, investigators suggested.
“These estimates are important, as they can be used to guide further investments in blood transfusion services, for analysis of current transfusion practices, and to highlight need for alternatives to transfusions such as antifibrinolytics, blood saving devices, and implementation of blood management systems,” the investigators wrote in Lancet Haematology.
Blood availability was calculated using the 2016 WHO Global Status Report on Blood Safety and Availability, in which 92% of countries participated. Blood needs were calculated using multiple databases, including the (U.S.) National Inpatient Sample datasets from 2000-2014, the State Inpatient Databases from 2003-2007, and the Global Burden of Disease 2017 study.
The global blood need was almost 305 million blood product units, while the supply totaled approximately 272 million units. These shortages, however, were not distributed equally across the globe. Out of 195 countries, 119 (61%) had an insufficient supply of blood to meet anticipated demand. Within this group, the shortage equated to about 102 million blood product units.
Denmark had the greatest supply of blood products (red blood cell products, platelets, and plasma), at 14,704 units per 100,000 population, compared with South Sudan, the least prepared to meet transfusion needs, with just 46 blood product units available per 100,000 population. This pattern was echoed across the globe; high-income countries were usually better stocked than those with low or middle income.
To meet demands, some of the most affected countries would need to raise their collection goals from single-digit figures to as high as 40 donations per 1,000 population, the investigators advised.
“Many countries face critical undersupply of transfusions, which will become more pronounced as access to care improves,” the investigators wrote.
The study was funded by the National Institutes of Health. The investigators reported having no other financial disclosures.
SOURCE: Roberts N et al. Lancet Haematol. 2019 Oct 17. doi: 10.1016/ S2352-3026(19)30200-5.
FROM LANCET HAEMATOLOGY