User login
‘Pot’ is still hot for Dravet, Lennox-Gastaut
BANGKOK – Interim results of long-term, open-label extension trials of add-on prescription cannabidiol in patients with Dravet syndrome or Lennox-Gastaut syndrome show sustained, clinically meaningful seizure reductions with no new safety concerns, Anup D. Patel, MD, reported at the International Epilepsy Congress.

“Overall, this is a very promising and sustainable result that we were happy to see,” said Dr. Patel, chief of child neurology at Nationwide Children’s Hospital in Columbus, Ohio.
Epidiolex is the brand name for the plant-derived, highly purified cannabidiol (CBD) in an oil-based oral solution at 100 mg/mL. Dr. Patel has been involved in the medication’s development program since the earliest open-label compassionate use study, which was followed by rigorous phase 3, double-blind, placebo-controlled randomized trials, eventually leading to Food and Drug Administration marketing approval for the treatment of Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age or older.
“On June 25th, 2018, history was made: for the first time in United States history, a plant-based derivative of marijuana was approved for use as a medication, and it was also the first FDA-approved treatment for Dravet syndrome,” Dr. Patel noted at the congress sponsored by the International League Against Epilepsy.
A total of 96% of the 289 children with Dravet syndrome who completed the 14-week, double-blind, controlled randomized trials enrolled in the open-label, long-term extension study, during which they were on a median of three concurrent antiepileptic drugs along with a mean modal dose of CBD at 22 mg/kg/day. Although the target maintenance dose of CBD was 20 mg/kg/day, as advised in the product labeling, physicians could reduce or increase the dose up to 30 mg/kg/day.
“In the initial compassionate-use study, our site could go up to 50 mg/kg/day,” according to Dr. Patel. “We have plenty of data showing efficacy and continued safety beyond the FDA-recommended dose.”
In the open-label extension study, the median reduction from baseline in monthly seizure frequency assessed in 12-week intervals up to a maximum of week 72 was 44%-57% for convulsive seizures and 49%-67% for total seizures. More than 80% of patients and/or caregivers reported improvement in the patient’s overall condition as assessed on the Subject/Caregiver Global Impression of Change scale.
The pattern of adverse events associated with CBD has been consistent across all of the studies. The most common side effects are diarrhea in about one-third of patients, sleepiness in one-quarter, and decreased appetite in about one-quarter. Seven percent of patients discontinued the long-term extension trial because of adverse events.
Seventy percent of patients remained in the long-term extension study at 1 year.
Twenty-six patients developed liver transaminase levels greater than three times the upper limit of normal, and of note, 23 of the 26 were on concomitant valproic acid. None met criteria for severe drug-induced liver injury, and all recovered either spontaneously or after a reduction in the dose of CBD or valproic acid. But this association between CBD, valproic acid, and increased risk of mild liver injury has been a consistent finding across the clinical trials program.
“This is a very important clinical pearl to take away,” commented Dr. Patel, who is also a pediatric neurologist at Ohio State University.
The interim results of the long-term, open-label extension study of add-on CBD in patients with Lennox-Gastaut syndrome are similar to the Dravet syndrome study. Overall, 99% of the 368 patients with Lennox-Gastaut syndrome who completed the 14-week, double-blind, randomized trials signed up for the open-label extension. During a median follow-up of 61 weeks, the median percent reduction in seizure frequency as assessed in serial 12-week windows was 48%-70% for drop seizures and 48%-63% for total seizures. Twenty-four percent of patients withdrew from the study. Eighty-eight percent of patients or caregivers reported an improvement in overall condition when assessed at weeks 24 and 48. Forty-seven patients developed elevated transaminase levels – typically within the first 2 months on CBD – and 35 of them were on concomitant valproic acid.
More on drug-drug interactions
Elsewhere at IEC 2019, Gilmour Morrison of GW Pharmaceuticals, the Cambridge, England, company that markets Epidiolex, presented the findings of a series of drug-drug interaction studies involving coadministration of their CBD with clobazam (Sympazan and Onfi), valproate, stiripentol (Diacomit), or midazolam (Versed) in adult epilepsy patients and healthy volunteers. The researchers reported a bidirectional drug-drug interaction between Epidiolex and clobazam resulting in increased levels of the active metabolites of both drugs. The mechanism is believed to involve inhibition of cytochrome P450 2C19. However, there were no interactions with midazolam or valproate, and the slight bump in stiripentol levels when given with CBD didn’t reach the level of a clinically meaningful drug-drug interaction, according to the investigators.
On the horizon, Canadian researchers are investigating the possibility that since both the tetrahydrocannabinol (THC) and CBD components of marijuana have been shown to have anticonvulsant effects, adding a bit of THC to CBD will result in even better seizure control than with pure CBD in patients with Dravet syndrome. Investigators at Toronto’s Hospital for Sick Children have conducted a prospective, open-label study of a product containing CBD and THC in a 50:1 ratio as add-on therapy in 20 children with Dravet syndrome. The dose was 2-16 mg/kg/day of CBD and 0.04-0.32 mg/kg/day of THC. The cannabis plant extract used in the study was produced by Tilray, a Canadian pharmaceutical company.
Nineteen of the 20 patients completed the 20-week study. The sole noncompleter died of SUDEP (sudden unexpected death in epilepsy) deemed treatment unrelated. Patients experienced a median 71% reduction in motor seizures, compared with baseline. Sixty-three percent of patients had at least a 50% reduction in seizure frequency. Elevated liver transaminases occurred in patients on concomitant valproic acid, as did platelet abnormalities, which have not been seen in the Epidiolex studies, noted Dr. Patel, who was not involved in the Canadian study (Ann Clin Transl Neurol. 2018 Aug 1;5[9]:1077-88).
Dr. Patel reported serving as a consultant to Greenwich Biosciences, a U.S. offshoot of GW Pharmaceuticals. He receives research grants from that company as well as from the National Institutes of Health and the Pediatric Epilepsy Research Foundation.
BANGKOK – Interim results of long-term, open-label extension trials of add-on prescription cannabidiol in patients with Dravet syndrome or Lennox-Gastaut syndrome show sustained, clinically meaningful seizure reductions with no new safety concerns, Anup D. Patel, MD, reported at the International Epilepsy Congress.
“Overall, this is a very promising and sustainable result that we were happy to see,” said Dr. Patel, chief of child neurology at Nationwide Children’s Hospital in Columbus, Ohio.
Epidiolex is the brand name for the plant-derived, highly purified cannabidiol (CBD) in an oil-based oral solution at 100 mg/mL. Dr. Patel has been involved in the medication’s development program since the earliest open-label compassionate use study, which was followed by rigorous phase 3, double-blind, placebo-controlled randomized trials, eventually leading to Food and Drug Administration marketing approval for the treatment of Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age or older.
“On June 25th, 2018, history was made: for the first time in United States history, a plant-based derivative of marijuana was approved for use as a medication, and it was also the first FDA-approved treatment for Dravet syndrome,” Dr. Patel noted at the congress sponsored by the International League Against Epilepsy.
A total of 96% of the 289 children with Dravet syndrome who completed the 14-week, double-blind, controlled randomized trials enrolled in the open-label, long-term extension study, during which they were on a median of three concurrent antiepileptic drugs along with a mean modal dose of CBD at 22 mg/kg/day. Although the target maintenance dose of CBD was 20 mg/kg/day, as advised in the product labeling, physicians could reduce or increase the dose up to 30 mg/kg/day.
“In the initial compassionate-use study, our site could go up to 50 mg/kg/day,” according to Dr. Patel. “We have plenty of data showing efficacy and continued safety beyond the FDA-recommended dose.”
In the open-label extension study, the median reduction from baseline in monthly seizure frequency assessed in 12-week intervals up to a maximum of week 72 was 44%-57% for convulsive seizures and 49%-67% for total seizures. More than 80% of patients and/or caregivers reported improvement in the patient’s overall condition as assessed on the Subject/Caregiver Global Impression of Change scale.
The pattern of adverse events associated with CBD has been consistent across all of the studies. The most common side effects are diarrhea in about one-third of patients, sleepiness in one-quarter, and decreased appetite in about one-quarter. Seven percent of patients discontinued the long-term extension trial because of adverse events.
Seventy percent of patients remained in the long-term extension study at 1 year.
Twenty-six patients developed liver transaminase levels greater than three times the upper limit of normal, and of note, 23 of the 26 were on concomitant valproic acid. None met criteria for severe drug-induced liver injury, and all recovered either spontaneously or after a reduction in the dose of CBD or valproic acid. But this association between CBD, valproic acid, and increased risk of mild liver injury has been a consistent finding across the clinical trials program.
“This is a very important clinical pearl to take away,” commented Dr. Patel, who is also a pediatric neurologist at Ohio State University.
The interim results of the long-term, open-label extension study of add-on CBD in patients with Lennox-Gastaut syndrome are similar to the Dravet syndrome study. Overall, 99% of the 368 patients with Lennox-Gastaut syndrome who completed the 14-week, double-blind, randomized trials signed up for the open-label extension. During a median follow-up of 61 weeks, the median percent reduction in seizure frequency as assessed in serial 12-week windows was 48%-70% for drop seizures and 48%-63% for total seizures. Twenty-four percent of patients withdrew from the study. Eighty-eight percent of patients or caregivers reported an improvement in overall condition when assessed at weeks 24 and 48. Forty-seven patients developed elevated transaminase levels – typically within the first 2 months on CBD – and 35 of them were on concomitant valproic acid.
More on drug-drug interactions
Elsewhere at IEC 2019, Gilmour Morrison of GW Pharmaceuticals, the Cambridge, England, company that markets Epidiolex, presented the findings of a series of drug-drug interaction studies involving coadministration of their CBD with clobazam (Sympazan and Onfi), valproate, stiripentol (Diacomit), or midazolam (Versed) in adult epilepsy patients and healthy volunteers. The researchers reported a bidirectional drug-drug interaction between Epidiolex and clobazam resulting in increased levels of the active metabolites of both drugs. The mechanism is believed to involve inhibition of cytochrome P450 2C19. However, there were no interactions with midazolam or valproate, and the slight bump in stiripentol levels when given with CBD didn’t reach the level of a clinically meaningful drug-drug interaction, according to the investigators.
On the horizon, Canadian researchers are investigating the possibility that since both the tetrahydrocannabinol (THC) and CBD components of marijuana have been shown to have anticonvulsant effects, adding a bit of THC to CBD will result in even better seizure control than with pure CBD in patients with Dravet syndrome. Investigators at Toronto’s Hospital for Sick Children have conducted a prospective, open-label study of a product containing CBD and THC in a 50:1 ratio as add-on therapy in 20 children with Dravet syndrome. The dose was 2-16 mg/kg/day of CBD and 0.04-0.32 mg/kg/day of THC. The cannabis plant extract used in the study was produced by Tilray, a Canadian pharmaceutical company.
Nineteen of the 20 patients completed the 20-week study. The sole noncompleter died of SUDEP (sudden unexpected death in epilepsy) deemed treatment unrelated. Patients experienced a median 71% reduction in motor seizures, compared with baseline. Sixty-three percent of patients had at least a 50% reduction in seizure frequency. Elevated liver transaminases occurred in patients on concomitant valproic acid, as did platelet abnormalities, which have not been seen in the Epidiolex studies, noted Dr. Patel, who was not involved in the Canadian study (Ann Clin Transl Neurol. 2018 Aug 1;5[9]:1077-88).
Dr. Patel reported serving as a consultant to Greenwich Biosciences, a U.S. offshoot of GW Pharmaceuticals. He receives research grants from that company as well as from the National Institutes of Health and the Pediatric Epilepsy Research Foundation.
BANGKOK – Interim results of long-term, open-label extension trials of add-on prescription cannabidiol in patients with Dravet syndrome or Lennox-Gastaut syndrome show sustained, clinically meaningful seizure reductions with no new safety concerns, Anup D. Patel, MD, reported at the International Epilepsy Congress.
“Overall, this is a very promising and sustainable result that we were happy to see,” said Dr. Patel, chief of child neurology at Nationwide Children’s Hospital in Columbus, Ohio.
Epidiolex is the brand name for the plant-derived, highly purified cannabidiol (CBD) in an oil-based oral solution at 100 mg/mL. Dr. Patel has been involved in the medication’s development program since the earliest open-label compassionate use study, which was followed by rigorous phase 3, double-blind, placebo-controlled randomized trials, eventually leading to Food and Drug Administration marketing approval for the treatment of Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age or older.
“On June 25th, 2018, history was made: for the first time in United States history, a plant-based derivative of marijuana was approved for use as a medication, and it was also the first FDA-approved treatment for Dravet syndrome,” Dr. Patel noted at the congress sponsored by the International League Against Epilepsy.
A total of 96% of the 289 children with Dravet syndrome who completed the 14-week, double-blind, controlled randomized trials enrolled in the open-label, long-term extension study, during which they were on a median of three concurrent antiepileptic drugs along with a mean modal dose of CBD at 22 mg/kg/day. Although the target maintenance dose of CBD was 20 mg/kg/day, as advised in the product labeling, physicians could reduce or increase the dose up to 30 mg/kg/day.
“In the initial compassionate-use study, our site could go up to 50 mg/kg/day,” according to Dr. Patel. “We have plenty of data showing efficacy and continued safety beyond the FDA-recommended dose.”
In the open-label extension study, the median reduction from baseline in monthly seizure frequency assessed in 12-week intervals up to a maximum of week 72 was 44%-57% for convulsive seizures and 49%-67% for total seizures. More than 80% of patients and/or caregivers reported improvement in the patient’s overall condition as assessed on the Subject/Caregiver Global Impression of Change scale.
The pattern of adverse events associated with CBD has been consistent across all of the studies. The most common side effects are diarrhea in about one-third of patients, sleepiness in one-quarter, and decreased appetite in about one-quarter. Seven percent of patients discontinued the long-term extension trial because of adverse events.
Seventy percent of patients remained in the long-term extension study at 1 year.
Twenty-six patients developed liver transaminase levels greater than three times the upper limit of normal, and of note, 23 of the 26 were on concomitant valproic acid. None met criteria for severe drug-induced liver injury, and all recovered either spontaneously or after a reduction in the dose of CBD or valproic acid. But this association between CBD, valproic acid, and increased risk of mild liver injury has been a consistent finding across the clinical trials program.
“This is a very important clinical pearl to take away,” commented Dr. Patel, who is also a pediatric neurologist at Ohio State University.
The interim results of the long-term, open-label extension study of add-on CBD in patients with Lennox-Gastaut syndrome are similar to the Dravet syndrome study. Overall, 99% of the 368 patients with Lennox-Gastaut syndrome who completed the 14-week, double-blind, randomized trials signed up for the open-label extension. During a median follow-up of 61 weeks, the median percent reduction in seizure frequency as assessed in serial 12-week windows was 48%-70% for drop seizures and 48%-63% for total seizures. Twenty-four percent of patients withdrew from the study. Eighty-eight percent of patients or caregivers reported an improvement in overall condition when assessed at weeks 24 and 48. Forty-seven patients developed elevated transaminase levels – typically within the first 2 months on CBD – and 35 of them were on concomitant valproic acid.
More on drug-drug interactions
Elsewhere at IEC 2019, Gilmour Morrison of GW Pharmaceuticals, the Cambridge, England, company that markets Epidiolex, presented the findings of a series of drug-drug interaction studies involving coadministration of their CBD with clobazam (Sympazan and Onfi), valproate, stiripentol (Diacomit), or midazolam (Versed) in adult epilepsy patients and healthy volunteers. The researchers reported a bidirectional drug-drug interaction between Epidiolex and clobazam resulting in increased levels of the active metabolites of both drugs. The mechanism is believed to involve inhibition of cytochrome P450 2C19. However, there were no interactions with midazolam or valproate, and the slight bump in stiripentol levels when given with CBD didn’t reach the level of a clinically meaningful drug-drug interaction, according to the investigators.
On the horizon, Canadian researchers are investigating the possibility that since both the tetrahydrocannabinol (THC) and CBD components of marijuana have been shown to have anticonvulsant effects, adding a bit of THC to CBD will result in even better seizure control than with pure CBD in patients with Dravet syndrome. Investigators at Toronto’s Hospital for Sick Children have conducted a prospective, open-label study of a product containing CBD and THC in a 50:1 ratio as add-on therapy in 20 children with Dravet syndrome. The dose was 2-16 mg/kg/day of CBD and 0.04-0.32 mg/kg/day of THC. The cannabis plant extract used in the study was produced by Tilray, a Canadian pharmaceutical company.
Nineteen of the 20 patients completed the 20-week study. The sole noncompleter died of SUDEP (sudden unexpected death in epilepsy) deemed treatment unrelated. Patients experienced a median 71% reduction in motor seizures, compared with baseline. Sixty-three percent of patients had at least a 50% reduction in seizure frequency. Elevated liver transaminases occurred in patients on concomitant valproic acid, as did platelet abnormalities, which have not been seen in the Epidiolex studies, noted Dr. Patel, who was not involved in the Canadian study (Ann Clin Transl Neurol. 2018 Aug 1;5[9]:1077-88).
Dr. Patel reported serving as a consultant to Greenwich Biosciences, a U.S. offshoot of GW Pharmaceuticals. He receives research grants from that company as well as from the National Institutes of Health and the Pediatric Epilepsy Research Foundation.
REPORTING FROM IEC 2019
ASCO VTE guideline update: DOACs now an option for prevention, treatment
The direct oral anticoagulants (DOACs) apixaban and rivaroxaban are now among the options for thromboprophylaxis in high-risk cancer outpatients with low risk for bleeding and drug interactions, according to a practice guideline update from the American Society of Clinical Oncology.
Rivaroxaban also has been added as an option for initial anticoagulation for venous thromboembolism (VTE), and both rivaroxaban and edoxaban are now options for long-term anticoagulation, Nigel S. Key, MB ChB, and colleagues wrote in the updated guideline on the prophylaxis and treatment of VTE – including deep vein thrombosis (DVT) and pulmonary embolism (PE) – in cancer patients (J Clin Oncol. 2019 Aug 5. doi: 10.1200/JCO.19.19.01461).
The addition of DOACs as options for VTE prophylaxis and treatment represents the most notable change to the guideline.
“Oral anticoagulants that target thrombin (direct thrombin inhibitor, dabigatran) or activated factor X (antifactor Xa inhibitors, rivaroxaban, apixaban, and edoxaban) are now approved for treatment of DVT or PE as well as for DVT prophylaxis following orthopedic surgery and for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation,” the guideline panel wrote.
A systematic review of PubMed and the Cochrane Library for randomized controlled trials (RCTs) and meta-analyses of RCTs published from Aug. 1, 2014, through Dec. 4, 2018, identified 35 publications on VTE prophylaxis and treatment, including 2 RCTs of DOACs for prophylaxis and 2 others of DOAC treatment, as well as 8 publications on VTE risk assessment. A multidisciplinary expert panel appointed by ASCO and cochaired by Dr. Key of the University of North Carolina, Chapel Hill, used this evidence to develop the updated guideline.
The work was guided by “the ‘signals’ approach that is designed to identify only new, potentially practice-changing data – signals – that might translate into revised practice recommendations,” the authors explained.
DOAC-related updates
VTE prophylaxis. Based in part on findings from the recently published AVERT trial of apixaban in patients initiating a new course of chemotherapy and from the CASSINI trial of rivaroxaban in patients with solid tumors or lymphoma starting systemic antineoplastic therapy, the panel added both agents as thromboprophylactic options that can be offered to high-risk cancer outpatients with no significant risk factors for bleeding or drug interactions (N Engl J Med. 2019;380:711-19; N Engl J Med. 2019;380:720-8).
Low-molecular-weight heparin (LMWH) also remains an option in such patients; consideration of therapy should involve discussion with the patient about relative benefits and harms, drug costs, and “the uncertainty surrounding duration of prophylaxis in this setting,” they wrote.
Anticoagulation for VTE. Options for initial anticoagulation include LMWH, unfractionated heparin (UFH), fondaparinux, and now rivaroxaban, with the latter added based on findings from two RCTs – the SELECT-D trial and the Hokusai VTE-Cancer study – and multiple meta-analyses (J Clin Oncol. 2018;36:2017-23; N Engl J Med. 2018;378:615-24).
Long-term anticoagulation can involve treatment with LMWH, edoxaban, or rivaroxaban for at least 6 months, all of which have improved efficacy versus vitamin K agonists (VKAs), the panel noted. However, VKAs may be used if LMWH and DOACs are not accessible.
Importantly, the literature indicates an increased risk of major bleeding with DOACs, particularly in patients with gastrointestinal malignancies and potentially in those with genitourinary malignancies. “Caution with DOACs is also warranted in other settings with high risk for mucosal bleeding,” the panel wrote.
Additional updates
CNS metastases. The anticoagulation recommendations were also updated to include patients with metastatic central nervous system malignancies (those with primary CNS malignancies were included previously). Both those with primary and metastatic CNS malignancy should be offered anticoagulation for established VTE as described for patients with other types of cancer. However, the panel stressed that “uncertainties remain about choice of agents and selection of patients most likely to benefit.”
“Patients with intracranial tumors are at increased risk for thrombotic complications and intracranial hemorrhage (ICH), but the presence of a stable or active primary intracranial malignancy or brain metastases is not an absolute contraindication to anticoagulation,” they wrote.
Limited evidence suggests that therapeutic anticoagulation does not increase ICH risk in patients with brain metastases, but it may increase risk in those with primary brain tumors, the panel added.
Additionally, preliminary data from a retrospective cohort of patients with metastatic brain disease and venous thrombosis suggest that DOACs may be associated with a lower risk of ICH than is LMWH in this population.
Long-term postoperative LMWH. Extended prophylaxis with LMWH for up to 4 weeks is recommended after major open or laparoscopic abdominal or pelvic surgery in cancer patients with high-risk features, such as restricted mobility, obesity, history of VTE, or with additional risk factors. Lower-risk surgical settings require case-by-case decision making about appropriate thromboprophylaxis duration, according to the update.
A 2014 RCT looking at thromboprophylaxis duration in 225 patients undergoing laparoscopic surgery for colorectal cancer prompted the addition of laparoscopic surgery to this recommendation. In that study, VTE occurred by 4 weeks in nearly 10% of patients receiving 1 week of prophylaxis and in no patients in the 4-week arm. Major bleeding occurred in one versus zero patients in the thromboprophylaxis arms, respectively (Ann Surg. April 2014;259[4]:665-9).
Reaffirmed recommendations
Based on the latest available data, the panel reaffirmed that most hospitalized patients with cancer and an acute medical condition require thromboprophylaxis for the duration of their hospitalization and that thromboprophylaxis should not be routinely recommended for all outpatients with cancer.
The panel also reaffirmed the need for thromboprophylaxis starting preoperatively and continuing for at least 7-10 days in patients undergoing major cancer surgery, the need for periodic assessment of VTE risk in cancer patients, and the importance of patient education about the signs and symptoms of VTE.
Perspective and future directions
In an interview, David H. Henry, MD, said he was pleased to see ASCO incorporate the latest DOAC data into the VTE guideline.

The AVERT and CASSINI studies, in particular, highlight the value of using the Khorana Risk Score, which considers cancer type, blood counts, and body mass index to predict the risk of thrombosis in cancer patients and to guide decisions regarding prophylaxis, said Dr. Henry, vice chair of the department of medicine and clinical professor of medicine at Penn Medicine’s Abramson Cancer Center, Philadelphia.
The DOACs also represent “a nice new development in the treatment setting,” he said, adding that it’s been long known – since the 2003 CLOT trial – that cancer patients with VTE had much lower recurrence rates with LMWH versus warfarin (Coumadin).
“Now fast forward to the modern era ... and DOACs now appear to be a good idea,” he said.
Dr. Henry also addressed the recommendation for expanded postoperative LMWH use.
“That I found interesting; I’m not sure what took them so long,” he said, explaining that National Comprehensive Cancer Network and European Society of Medical Oncology recommendations have long stated that, for patients with abdominal cancers who undergo abdominopelvic surgery, DVT prophylaxis should continue for 4 weeks.
Dr. Henry said that a survey at his center showed that those recommendations were “very poorly followed,” with surgeons giving 4 weeks of prophylaxis in just 5% of cases.
“The good news from our survey was that not many people had a VTE, despite not many people following the recommendations, but I must say I think our surgeons are catching on,” he said.
Overall, the updated guideline highlights the importance of considering the “cancer variable” when it comes to VTE prevention and treatment.
“We’ve known forever that when we diagnose a DVT or PE in the outpatient setting – and this is independent of cancer – that you should treat it. Add the cancer variable and we now know that we should worry and try to prevent the VTE in certain high-risk patients, and there are some drugs to do it with,” he said, adding that “you should worry about the person you’ve just provoked [with surgery] as well.”
An important question not addressed in the guideline update is the indefinite use of DOACs in cancer patients with ongoing risk, he said.
“When we see DVT or PE, we usually treat for 3 months – that’s the industry standard – and at the end of 3 months ... you do a time out and you say to yourself, ‘Was this person provoked?’ ” he said.
For example, if they took a long flight or if pregnancy was a factor, treatment can usually be safely stopped. However, in a cancer patient who still has cancer, the provocation continues, and the patient may require indefinite treatment.
Questions that remain involve defining “indefinite” and include whether (and which of) these drugs can be used indefinitely in such patients, Dr. Henry said.
Dr. Key reported receiving honoraria from Novo Nordisk, research funding to his institution from Baxter Biosciences, Grifols, and Pfizer, and serving as a consultant or advisor for Genentech, Roche, Uniqure, Seattle Genetics, and Shire Human Genetic Therapies. Numerous disclosures were also reported by other expert panel members.
The direct oral anticoagulants (DOACs) apixaban and rivaroxaban are now among the options for thromboprophylaxis in high-risk cancer outpatients with low risk for bleeding and drug interactions, according to a practice guideline update from the American Society of Clinical Oncology.
Rivaroxaban also has been added as an option for initial anticoagulation for venous thromboembolism (VTE), and both rivaroxaban and edoxaban are now options for long-term anticoagulation, Nigel S. Key, MB ChB, and colleagues wrote in the updated guideline on the prophylaxis and treatment of VTE – including deep vein thrombosis (DVT) and pulmonary embolism (PE) – in cancer patients (J Clin Oncol. 2019 Aug 5. doi: 10.1200/JCO.19.19.01461).
The addition of DOACs as options for VTE prophylaxis and treatment represents the most notable change to the guideline.
“Oral anticoagulants that target thrombin (direct thrombin inhibitor, dabigatran) or activated factor X (antifactor Xa inhibitors, rivaroxaban, apixaban, and edoxaban) are now approved for treatment of DVT or PE as well as for DVT prophylaxis following orthopedic surgery and for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation,” the guideline panel wrote.
A systematic review of PubMed and the Cochrane Library for randomized controlled trials (RCTs) and meta-analyses of RCTs published from Aug. 1, 2014, through Dec. 4, 2018, identified 35 publications on VTE prophylaxis and treatment, including 2 RCTs of DOACs for prophylaxis and 2 others of DOAC treatment, as well as 8 publications on VTE risk assessment. A multidisciplinary expert panel appointed by ASCO and cochaired by Dr. Key of the University of North Carolina, Chapel Hill, used this evidence to develop the updated guideline.
The work was guided by “the ‘signals’ approach that is designed to identify only new, potentially practice-changing data – signals – that might translate into revised practice recommendations,” the authors explained.
DOAC-related updates
VTE prophylaxis. Based in part on findings from the recently published AVERT trial of apixaban in patients initiating a new course of chemotherapy and from the CASSINI trial of rivaroxaban in patients with solid tumors or lymphoma starting systemic antineoplastic therapy, the panel added both agents as thromboprophylactic options that can be offered to high-risk cancer outpatients with no significant risk factors for bleeding or drug interactions (N Engl J Med. 2019;380:711-19; N Engl J Med. 2019;380:720-8).
Low-molecular-weight heparin (LMWH) also remains an option in such patients; consideration of therapy should involve discussion with the patient about relative benefits and harms, drug costs, and “the uncertainty surrounding duration of prophylaxis in this setting,” they wrote.
Anticoagulation for VTE. Options for initial anticoagulation include LMWH, unfractionated heparin (UFH), fondaparinux, and now rivaroxaban, with the latter added based on findings from two RCTs – the SELECT-D trial and the Hokusai VTE-Cancer study – and multiple meta-analyses (J Clin Oncol. 2018;36:2017-23; N Engl J Med. 2018;378:615-24).
Long-term anticoagulation can involve treatment with LMWH, edoxaban, or rivaroxaban for at least 6 months, all of which have improved efficacy versus vitamin K agonists (VKAs), the panel noted. However, VKAs may be used if LMWH and DOACs are not accessible.
Importantly, the literature indicates an increased risk of major bleeding with DOACs, particularly in patients with gastrointestinal malignancies and potentially in those with genitourinary malignancies. “Caution with DOACs is also warranted in other settings with high risk for mucosal bleeding,” the panel wrote.
Additional updates
CNS metastases. The anticoagulation recommendations were also updated to include patients with metastatic central nervous system malignancies (those with primary CNS malignancies were included previously). Both those with primary and metastatic CNS malignancy should be offered anticoagulation for established VTE as described for patients with other types of cancer. However, the panel stressed that “uncertainties remain about choice of agents and selection of patients most likely to benefit.”
“Patients with intracranial tumors are at increased risk for thrombotic complications and intracranial hemorrhage (ICH), but the presence of a stable or active primary intracranial malignancy or brain metastases is not an absolute contraindication to anticoagulation,” they wrote.
Limited evidence suggests that therapeutic anticoagulation does not increase ICH risk in patients with brain metastases, but it may increase risk in those with primary brain tumors, the panel added.
Additionally, preliminary data from a retrospective cohort of patients with metastatic brain disease and venous thrombosis suggest that DOACs may be associated with a lower risk of ICH than is LMWH in this population.
Long-term postoperative LMWH. Extended prophylaxis with LMWH for up to 4 weeks is recommended after major open or laparoscopic abdominal or pelvic surgery in cancer patients with high-risk features, such as restricted mobility, obesity, history of VTE, or with additional risk factors. Lower-risk surgical settings require case-by-case decision making about appropriate thromboprophylaxis duration, according to the update.
A 2014 RCT looking at thromboprophylaxis duration in 225 patients undergoing laparoscopic surgery for colorectal cancer prompted the addition of laparoscopic surgery to this recommendation. In that study, VTE occurred by 4 weeks in nearly 10% of patients receiving 1 week of prophylaxis and in no patients in the 4-week arm. Major bleeding occurred in one versus zero patients in the thromboprophylaxis arms, respectively (Ann Surg. April 2014;259[4]:665-9).
Reaffirmed recommendations
Based on the latest available data, the panel reaffirmed that most hospitalized patients with cancer and an acute medical condition require thromboprophylaxis for the duration of their hospitalization and that thromboprophylaxis should not be routinely recommended for all outpatients with cancer.
The panel also reaffirmed the need for thromboprophylaxis starting preoperatively and continuing for at least 7-10 days in patients undergoing major cancer surgery, the need for periodic assessment of VTE risk in cancer patients, and the importance of patient education about the signs and symptoms of VTE.
Perspective and future directions
In an interview, David H. Henry, MD, said he was pleased to see ASCO incorporate the latest DOAC data into the VTE guideline.
The AVERT and CASSINI studies, in particular, highlight the value of using the Khorana Risk Score, which considers cancer type, blood counts, and body mass index to predict the risk of thrombosis in cancer patients and to guide decisions regarding prophylaxis, said Dr. Henry, vice chair of the department of medicine and clinical professor of medicine at Penn Medicine’s Abramson Cancer Center, Philadelphia.
The DOACs also represent “a nice new development in the treatment setting,” he said, adding that it’s been long known – since the 2003 CLOT trial – that cancer patients with VTE had much lower recurrence rates with LMWH versus warfarin (Coumadin).
“Now fast forward to the modern era ... and DOACs now appear to be a good idea,” he said.
Dr. Henry also addressed the recommendation for expanded postoperative LMWH use.
“That I found interesting; I’m not sure what took them so long,” he said, explaining that National Comprehensive Cancer Network and European Society of Medical Oncology recommendations have long stated that, for patients with abdominal cancers who undergo abdominopelvic surgery, DVT prophylaxis should continue for 4 weeks.
Dr. Henry said that a survey at his center showed that those recommendations were “very poorly followed,” with surgeons giving 4 weeks of prophylaxis in just 5% of cases.
“The good news from our survey was that not many people had a VTE, despite not many people following the recommendations, but I must say I think our surgeons are catching on,” he said.
Overall, the updated guideline highlights the importance of considering the “cancer variable” when it comes to VTE prevention and treatment.
“We’ve known forever that when we diagnose a DVT or PE in the outpatient setting – and this is independent of cancer – that you should treat it. Add the cancer variable and we now know that we should worry and try to prevent the VTE in certain high-risk patients, and there are some drugs to do it with,” he said, adding that “you should worry about the person you’ve just provoked [with surgery] as well.”
An important question not addressed in the guideline update is the indefinite use of DOACs in cancer patients with ongoing risk, he said.
“When we see DVT or PE, we usually treat for 3 months – that’s the industry standard – and at the end of 3 months ... you do a time out and you say to yourself, ‘Was this person provoked?’ ” he said.
For example, if they took a long flight or if pregnancy was a factor, treatment can usually be safely stopped. However, in a cancer patient who still has cancer, the provocation continues, and the patient may require indefinite treatment.
Questions that remain involve defining “indefinite” and include whether (and which of) these drugs can be used indefinitely in such patients, Dr. Henry said.
Dr. Key reported receiving honoraria from Novo Nordisk, research funding to his institution from Baxter Biosciences, Grifols, and Pfizer, and serving as a consultant or advisor for Genentech, Roche, Uniqure, Seattle Genetics, and Shire Human Genetic Therapies. Numerous disclosures were also reported by other expert panel members.
The direct oral anticoagulants (DOACs) apixaban and rivaroxaban are now among the options for thromboprophylaxis in high-risk cancer outpatients with low risk for bleeding and drug interactions, according to a practice guideline update from the American Society of Clinical Oncology.
Rivaroxaban also has been added as an option for initial anticoagulation for venous thromboembolism (VTE), and both rivaroxaban and edoxaban are now options for long-term anticoagulation, Nigel S. Key, MB ChB, and colleagues wrote in the updated guideline on the prophylaxis and treatment of VTE – including deep vein thrombosis (DVT) and pulmonary embolism (PE) – in cancer patients (J Clin Oncol. 2019 Aug 5. doi: 10.1200/JCO.19.19.01461).
The addition of DOACs as options for VTE prophylaxis and treatment represents the most notable change to the guideline.
“Oral anticoagulants that target thrombin (direct thrombin inhibitor, dabigatran) or activated factor X (antifactor Xa inhibitors, rivaroxaban, apixaban, and edoxaban) are now approved for treatment of DVT or PE as well as for DVT prophylaxis following orthopedic surgery and for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation,” the guideline panel wrote.
A systematic review of PubMed and the Cochrane Library for randomized controlled trials (RCTs) and meta-analyses of RCTs published from Aug. 1, 2014, through Dec. 4, 2018, identified 35 publications on VTE prophylaxis and treatment, including 2 RCTs of DOACs for prophylaxis and 2 others of DOAC treatment, as well as 8 publications on VTE risk assessment. A multidisciplinary expert panel appointed by ASCO and cochaired by Dr. Key of the University of North Carolina, Chapel Hill, used this evidence to develop the updated guideline.
The work was guided by “the ‘signals’ approach that is designed to identify only new, potentially practice-changing data – signals – that might translate into revised practice recommendations,” the authors explained.
DOAC-related updates
VTE prophylaxis. Based in part on findings from the recently published AVERT trial of apixaban in patients initiating a new course of chemotherapy and from the CASSINI trial of rivaroxaban in patients with solid tumors or lymphoma starting systemic antineoplastic therapy, the panel added both agents as thromboprophylactic options that can be offered to high-risk cancer outpatients with no significant risk factors for bleeding or drug interactions (N Engl J Med. 2019;380:711-19; N Engl J Med. 2019;380:720-8).
Low-molecular-weight heparin (LMWH) also remains an option in such patients; consideration of therapy should involve discussion with the patient about relative benefits and harms, drug costs, and “the uncertainty surrounding duration of prophylaxis in this setting,” they wrote.
Anticoagulation for VTE. Options for initial anticoagulation include LMWH, unfractionated heparin (UFH), fondaparinux, and now rivaroxaban, with the latter added based on findings from two RCTs – the SELECT-D trial and the Hokusai VTE-Cancer study – and multiple meta-analyses (J Clin Oncol. 2018;36:2017-23; N Engl J Med. 2018;378:615-24).
Long-term anticoagulation can involve treatment with LMWH, edoxaban, or rivaroxaban for at least 6 months, all of which have improved efficacy versus vitamin K agonists (VKAs), the panel noted. However, VKAs may be used if LMWH and DOACs are not accessible.
Importantly, the literature indicates an increased risk of major bleeding with DOACs, particularly in patients with gastrointestinal malignancies and potentially in those with genitourinary malignancies. “Caution with DOACs is also warranted in other settings with high risk for mucosal bleeding,” the panel wrote.
Additional updates
CNS metastases. The anticoagulation recommendations were also updated to include patients with metastatic central nervous system malignancies (those with primary CNS malignancies were included previously). Both those with primary and metastatic CNS malignancy should be offered anticoagulation for established VTE as described for patients with other types of cancer. However, the panel stressed that “uncertainties remain about choice of agents and selection of patients most likely to benefit.”
“Patients with intracranial tumors are at increased risk for thrombotic complications and intracranial hemorrhage (ICH), but the presence of a stable or active primary intracranial malignancy or brain metastases is not an absolute contraindication to anticoagulation,” they wrote.
Limited evidence suggests that therapeutic anticoagulation does not increase ICH risk in patients with brain metastases, but it may increase risk in those with primary brain tumors, the panel added.
Additionally, preliminary data from a retrospective cohort of patients with metastatic brain disease and venous thrombosis suggest that DOACs may be associated with a lower risk of ICH than is LMWH in this population.
Long-term postoperative LMWH. Extended prophylaxis with LMWH for up to 4 weeks is recommended after major open or laparoscopic abdominal or pelvic surgery in cancer patients with high-risk features, such as restricted mobility, obesity, history of VTE, or with additional risk factors. Lower-risk surgical settings require case-by-case decision making about appropriate thromboprophylaxis duration, according to the update.
A 2014 RCT looking at thromboprophylaxis duration in 225 patients undergoing laparoscopic surgery for colorectal cancer prompted the addition of laparoscopic surgery to this recommendation. In that study, VTE occurred by 4 weeks in nearly 10% of patients receiving 1 week of prophylaxis and in no patients in the 4-week arm. Major bleeding occurred in one versus zero patients in the thromboprophylaxis arms, respectively (Ann Surg. April 2014;259[4]:665-9).
Reaffirmed recommendations
Based on the latest available data, the panel reaffirmed that most hospitalized patients with cancer and an acute medical condition require thromboprophylaxis for the duration of their hospitalization and that thromboprophylaxis should not be routinely recommended for all outpatients with cancer.
The panel also reaffirmed the need for thromboprophylaxis starting preoperatively and continuing for at least 7-10 days in patients undergoing major cancer surgery, the need for periodic assessment of VTE risk in cancer patients, and the importance of patient education about the signs and symptoms of VTE.
Perspective and future directions
In an interview, David H. Henry, MD, said he was pleased to see ASCO incorporate the latest DOAC data into the VTE guideline.
The AVERT and CASSINI studies, in particular, highlight the value of using the Khorana Risk Score, which considers cancer type, blood counts, and body mass index to predict the risk of thrombosis in cancer patients and to guide decisions regarding prophylaxis, said Dr. Henry, vice chair of the department of medicine and clinical professor of medicine at Penn Medicine’s Abramson Cancer Center, Philadelphia.
The DOACs also represent “a nice new development in the treatment setting,” he said, adding that it’s been long known – since the 2003 CLOT trial – that cancer patients with VTE had much lower recurrence rates with LMWH versus warfarin (Coumadin).
“Now fast forward to the modern era ... and DOACs now appear to be a good idea,” he said.
Dr. Henry also addressed the recommendation for expanded postoperative LMWH use.
“That I found interesting; I’m not sure what took them so long,” he said, explaining that National Comprehensive Cancer Network and European Society of Medical Oncology recommendations have long stated that, for patients with abdominal cancers who undergo abdominopelvic surgery, DVT prophylaxis should continue for 4 weeks.
Dr. Henry said that a survey at his center showed that those recommendations were “very poorly followed,” with surgeons giving 4 weeks of prophylaxis in just 5% of cases.
“The good news from our survey was that not many people had a VTE, despite not many people following the recommendations, but I must say I think our surgeons are catching on,” he said.
Overall, the updated guideline highlights the importance of considering the “cancer variable” when it comes to VTE prevention and treatment.
“We’ve known forever that when we diagnose a DVT or PE in the outpatient setting – and this is independent of cancer – that you should treat it. Add the cancer variable and we now know that we should worry and try to prevent the VTE in certain high-risk patients, and there are some drugs to do it with,” he said, adding that “you should worry about the person you’ve just provoked [with surgery] as well.”
An important question not addressed in the guideline update is the indefinite use of DOACs in cancer patients with ongoing risk, he said.
“When we see DVT or PE, we usually treat for 3 months – that’s the industry standard – and at the end of 3 months ... you do a time out and you say to yourself, ‘Was this person provoked?’ ” he said.
For example, if they took a long flight or if pregnancy was a factor, treatment can usually be safely stopped. However, in a cancer patient who still has cancer, the provocation continues, and the patient may require indefinite treatment.
Questions that remain involve defining “indefinite” and include whether (and which of) these drugs can be used indefinitely in such patients, Dr. Henry said.
Dr. Key reported receiving honoraria from Novo Nordisk, research funding to his institution from Baxter Biosciences, Grifols, and Pfizer, and serving as a consultant or advisor for Genentech, Roche, Uniqure, Seattle Genetics, and Shire Human Genetic Therapies. Numerous disclosures were also reported by other expert panel members.
Pediatric, adolescent migraine treatment and prevention guidelines are updated
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
FROM NEUROLOGY
FDA approves drug combo to treat highly resistant TB
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.

The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
NEWS FROM THE FDA
Treatment of episodic cluster headache deviates from recommendations
PHILADELPHIA – , according to an analysis presented at the annual meeting of the American Headache Society.

Although consensus treatment guidelines do not exist for episodic cluster headache, treatment of this disorder did not follow many established recommendations that call for the use of preventive medications (e.g., MacGregor et al., 2010; Sarchielli et al., 2012; and May et al., 2006). Additional preventive medication options may be needed.
Patients with episodic cluster headache have several unilateral headache attacks per day. Little information is available to guide the selection of treatments for this population, and little is known about how available treatments are used in routine practice.
Analyzing cross-sectional survey data
To address this paucity of evidence, Jeffrey Scott Andrews, PharmD, a senior research scientist at Eli Lilly in Indianapolis, and colleagues examined data from the Adelphi 2017 Cluster Headache Disease Specific Programme, a large, international, cross-sectional survey. Physicians and patients in Germany, the United Kingdom, and the United States responded to the survey. Eligible physicians consulted with at least four patients with cluster headache per month, and eligible patients had a diagnosis of episodic cluster headache that was consistent with ICHD-3 beta criteria. Additional data were collected from all participants through questionnaires.
The analysis included 309 patients in Germany, 328 in the United Kingdom, and 375 in the United States. The average age of the patients was 40 years, and most of the patients were male. Less than 70% of patients reported working full time, which may indicate “the impact of this condition on work status,” said Dr. Andrews. Patients’ average number of attacks per day within an active period was 2.4. The two most commonly reported comorbidities were anxiety and depression. About 40% of cases of depression were reported to have occurred after the receipt of a diagnosis of cluster headache.
Use of inhaled oxygen was low
Most patients received acute treatments. The proportion of patients who received acute therapy only was 53% in Germany, 48% in the United Kingdom, and 43% in the United States. Approximately 34% of patients in Germany received a combination of acute and preventive therapy, compared with 37% in the United Kingdom and 42% in the United States. The proportion of patients who received preventive therapy only was 10% in Germany, 8% in the United Kingdom, and 12% in the United States.
The most commonly prescribed acute treatment, regardless of formulation, was sumatriptan. About 60% of patients received this medication. Less than one-third of patients used inhaled oxygen. Oxygen was prescribed more often in Germany (45%) and the United Kingdom (33%), compared with the United States (19%). U.S. patients face well-known obstacles in getting access to, and reimbursement for, oxygen, said Dr. Andrews. “That’s an area that deserves increased attention.” Zolmitriptan was the third most commonly prescribed acute medication.
Among prescriptions for sumatriptan, oral and injectable formulations were approximately equally common. Recommendations, however, indicate formulations with potentially fast onset of action. “The average duration of one of these attacks is between 15 and 180 minutes, so that certainly suggests that a formulation that gives you a faster onset of action might improve outcomes,” said Dr. Andrews. The use of injectable sumatriptan was lowest in the United States and highest in the United Kingdom.
“The most common decision regarding preventive treatment was [to give] no preventive treatment,” said Dr. Andrews. Verapamil was the most commonly prescribed preventive therapy (34% in Germany, 29% in the United States, and 25% in the United Kingdom), followed by topiramate, lithium, and valproate.
Nonadherence and noncompliance was common
Fewer U.K. patients (32%) reported taking their preventive therapy as advised, compared with German patients (60%) and U.S. patients (80%). Common reasons for noncompliance, regardless of location, were forgetfulness, the belief that a dose was not needed, and side effects. Most patients in the United Kingdom (60%) and the United States (54%) reported the need to take an extra dose of their acute medication to relieve pain symptoms, compared with 30% in Germany. Furthermore, 13% of U.S. patients indicated that they took extra doses all the time or nearly all the time, compared with 2% in Germany and 7% in the United Kingdom. Among patients who had discontinued a preventive treatment in the past, the most common reasons for discontinuation were lack of efficacy and problems with tolerability.
One limitation of the study was that the survey was not designed to represent the general cluster headache or treating physician populations fully. The data may reflect selection bias in favor of physicians who treat high volumes of patients and in favor of patients who frequently seek health care. In addition, the data were based on self-reports.
“Increased awareness and educational efforts that aim at promoting the need and benefit of the preventive treatment for these patients is warranted,” Dr. Andrews concluded.
Dr. Andrews is an employee of Eli Lilly, which funded the study.
SOURCE: Nichols R et al. AHS 2019. Abstract OR04.
PHILADELPHIA – , according to an analysis presented at the annual meeting of the American Headache Society.
Although consensus treatment guidelines do not exist for episodic cluster headache, treatment of this disorder did not follow many established recommendations that call for the use of preventive medications (e.g., MacGregor et al., 2010; Sarchielli et al., 2012; and May et al., 2006). Additional preventive medication options may be needed.
Patients with episodic cluster headache have several unilateral headache attacks per day. Little information is available to guide the selection of treatments for this population, and little is known about how available treatments are used in routine practice.
Analyzing cross-sectional survey data
To address this paucity of evidence, Jeffrey Scott Andrews, PharmD, a senior research scientist at Eli Lilly in Indianapolis, and colleagues examined data from the Adelphi 2017 Cluster Headache Disease Specific Programme, a large, international, cross-sectional survey. Physicians and patients in Germany, the United Kingdom, and the United States responded to the survey. Eligible physicians consulted with at least four patients with cluster headache per month, and eligible patients had a diagnosis of episodic cluster headache that was consistent with ICHD-3 beta criteria. Additional data were collected from all participants through questionnaires.
The analysis included 309 patients in Germany, 328 in the United Kingdom, and 375 in the United States. The average age of the patients was 40 years, and most of the patients were male. Less than 70% of patients reported working full time, which may indicate “the impact of this condition on work status,” said Dr. Andrews. Patients’ average number of attacks per day within an active period was 2.4. The two most commonly reported comorbidities were anxiety and depression. About 40% of cases of depression were reported to have occurred after the receipt of a diagnosis of cluster headache.
Use of inhaled oxygen was low
Most patients received acute treatments. The proportion of patients who received acute therapy only was 53% in Germany, 48% in the United Kingdom, and 43% in the United States. Approximately 34% of patients in Germany received a combination of acute and preventive therapy, compared with 37% in the United Kingdom and 42% in the United States. The proportion of patients who received preventive therapy only was 10% in Germany, 8% in the United Kingdom, and 12% in the United States.
The most commonly prescribed acute treatment, regardless of formulation, was sumatriptan. About 60% of patients received this medication. Less than one-third of patients used inhaled oxygen. Oxygen was prescribed more often in Germany (45%) and the United Kingdom (33%), compared with the United States (19%). U.S. patients face well-known obstacles in getting access to, and reimbursement for, oxygen, said Dr. Andrews. “That’s an area that deserves increased attention.” Zolmitriptan was the third most commonly prescribed acute medication.
Among prescriptions for sumatriptan, oral and injectable formulations were approximately equally common. Recommendations, however, indicate formulations with potentially fast onset of action. “The average duration of one of these attacks is between 15 and 180 minutes, so that certainly suggests that a formulation that gives you a faster onset of action might improve outcomes,” said Dr. Andrews. The use of injectable sumatriptan was lowest in the United States and highest in the United Kingdom.
“The most common decision regarding preventive treatment was [to give] no preventive treatment,” said Dr. Andrews. Verapamil was the most commonly prescribed preventive therapy (34% in Germany, 29% in the United States, and 25% in the United Kingdom), followed by topiramate, lithium, and valproate.
Nonadherence and noncompliance was common
Fewer U.K. patients (32%) reported taking their preventive therapy as advised, compared with German patients (60%) and U.S. patients (80%). Common reasons for noncompliance, regardless of location, were forgetfulness, the belief that a dose was not needed, and side effects. Most patients in the United Kingdom (60%) and the United States (54%) reported the need to take an extra dose of their acute medication to relieve pain symptoms, compared with 30% in Germany. Furthermore, 13% of U.S. patients indicated that they took extra doses all the time or nearly all the time, compared with 2% in Germany and 7% in the United Kingdom. Among patients who had discontinued a preventive treatment in the past, the most common reasons for discontinuation were lack of efficacy and problems with tolerability.
One limitation of the study was that the survey was not designed to represent the general cluster headache or treating physician populations fully. The data may reflect selection bias in favor of physicians who treat high volumes of patients and in favor of patients who frequently seek health care. In addition, the data were based on self-reports.
“Increased awareness and educational efforts that aim at promoting the need and benefit of the preventive treatment for these patients is warranted,” Dr. Andrews concluded.
Dr. Andrews is an employee of Eli Lilly, which funded the study.
SOURCE: Nichols R et al. AHS 2019. Abstract OR04.
PHILADELPHIA – , according to an analysis presented at the annual meeting of the American Headache Society.
Although consensus treatment guidelines do not exist for episodic cluster headache, treatment of this disorder did not follow many established recommendations that call for the use of preventive medications (e.g., MacGregor et al., 2010; Sarchielli et al., 2012; and May et al., 2006). Additional preventive medication options may be needed.
Patients with episodic cluster headache have several unilateral headache attacks per day. Little information is available to guide the selection of treatments for this population, and little is known about how available treatments are used in routine practice.
Analyzing cross-sectional survey data
To address this paucity of evidence, Jeffrey Scott Andrews, PharmD, a senior research scientist at Eli Lilly in Indianapolis, and colleagues examined data from the Adelphi 2017 Cluster Headache Disease Specific Programme, a large, international, cross-sectional survey. Physicians and patients in Germany, the United Kingdom, and the United States responded to the survey. Eligible physicians consulted with at least four patients with cluster headache per month, and eligible patients had a diagnosis of episodic cluster headache that was consistent with ICHD-3 beta criteria. Additional data were collected from all participants through questionnaires.
The analysis included 309 patients in Germany, 328 in the United Kingdom, and 375 in the United States. The average age of the patients was 40 years, and most of the patients were male. Less than 70% of patients reported working full time, which may indicate “the impact of this condition on work status,” said Dr. Andrews. Patients’ average number of attacks per day within an active period was 2.4. The two most commonly reported comorbidities were anxiety and depression. About 40% of cases of depression were reported to have occurred after the receipt of a diagnosis of cluster headache.
Use of inhaled oxygen was low
Most patients received acute treatments. The proportion of patients who received acute therapy only was 53% in Germany, 48% in the United Kingdom, and 43% in the United States. Approximately 34% of patients in Germany received a combination of acute and preventive therapy, compared with 37% in the United Kingdom and 42% in the United States. The proportion of patients who received preventive therapy only was 10% in Germany, 8% in the United Kingdom, and 12% in the United States.
The most commonly prescribed acute treatment, regardless of formulation, was sumatriptan. About 60% of patients received this medication. Less than one-third of patients used inhaled oxygen. Oxygen was prescribed more often in Germany (45%) and the United Kingdom (33%), compared with the United States (19%). U.S. patients face well-known obstacles in getting access to, and reimbursement for, oxygen, said Dr. Andrews. “That’s an area that deserves increased attention.” Zolmitriptan was the third most commonly prescribed acute medication.
Among prescriptions for sumatriptan, oral and injectable formulations were approximately equally common. Recommendations, however, indicate formulations with potentially fast onset of action. “The average duration of one of these attacks is between 15 and 180 minutes, so that certainly suggests that a formulation that gives you a faster onset of action might improve outcomes,” said Dr. Andrews. The use of injectable sumatriptan was lowest in the United States and highest in the United Kingdom.
“The most common decision regarding preventive treatment was [to give] no preventive treatment,” said Dr. Andrews. Verapamil was the most commonly prescribed preventive therapy (34% in Germany, 29% in the United States, and 25% in the United Kingdom), followed by topiramate, lithium, and valproate.
Nonadherence and noncompliance was common
Fewer U.K. patients (32%) reported taking their preventive therapy as advised, compared with German patients (60%) and U.S. patients (80%). Common reasons for noncompliance, regardless of location, were forgetfulness, the belief that a dose was not needed, and side effects. Most patients in the United Kingdom (60%) and the United States (54%) reported the need to take an extra dose of their acute medication to relieve pain symptoms, compared with 30% in Germany. Furthermore, 13% of U.S. patients indicated that they took extra doses all the time or nearly all the time, compared with 2% in Germany and 7% in the United Kingdom. Among patients who had discontinued a preventive treatment in the past, the most common reasons for discontinuation were lack of efficacy and problems with tolerability.
One limitation of the study was that the survey was not designed to represent the general cluster headache or treating physician populations fully. The data may reflect selection bias in favor of physicians who treat high volumes of patients and in favor of patients who frequently seek health care. In addition, the data were based on self-reports.
“Increased awareness and educational efforts that aim at promoting the need and benefit of the preventive treatment for these patients is warranted,” Dr. Andrews concluded.
Dr. Andrews is an employee of Eli Lilly, which funded the study.
SOURCE: Nichols R et al. AHS 2019. Abstract OR04.
REPORTING FROM AHS 2019
Translating the 2019 AAD-NPF Guidelines of Care for the Management of Psoriasis With Biologics to Clinical Practice
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
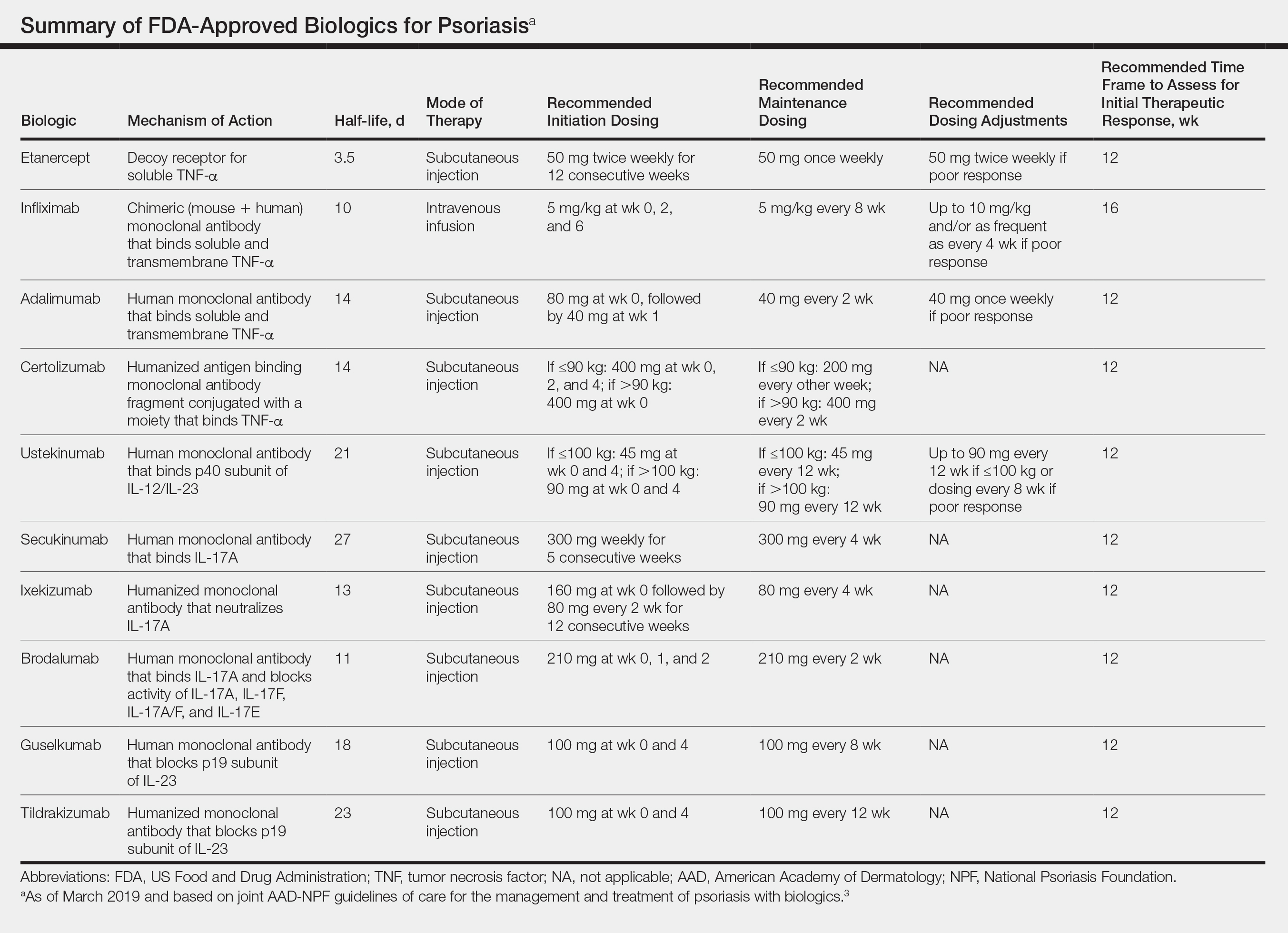
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
Practice Points
- There are currently 11 biologics approved for psoriasis, but there is no first-line or optimalbiologic. The choice must be made using clinical judgment based on a variety of medical and social factors.
- Frequent assessment for efficacy of and adverse events due to biologic therapy is warranted, as lack of response, loss of response, or severe side effects may warrant addition of concurrent therapies or switching to a different biologic.
- There are important considerations to make when immunizing and planning for surgery in patients on biologics.
Oral semaglutide monotherapy delivers HbA1c improvements in type 2 diabetes
Oral semaglutide monotherapy was superior to placebo for improving glycated hemoglobin (HbA1c) levels at all doses tested in adults with type 2 diabetes who had been previously insufficiently managed with diet and exercise, according to findings from a global, randomized trial.
The drug also showed dose-dependent weight loss, with a statistically significant effect on body weight, compared with placebo, at higher doses.
To date, the glucagon-like peptide–1 receptor agonist has been available as weekly subcutaneous shots for patients with type 2 diabetes, and in that form they have been shown to be effective in reducing HbA1c, inducing weight loss, and lowering the risk of cardiovascular events in patients with cardiovascular disease or those who are at high risk for it, wrote Vanita R. Aroda, MD, of Brigham and Women’s Hospital, Boston, and colleagues. The report is in Diabetes Care.
The novel oral semaglutide tablet is designed to enhance medication absorption, and the pharmacokinetics and dosage were established in phase 2 studies, they noted.
In the phase 3 Peptide Innovation for Early Diabetes Treatment 1 (PIONEER 1) study, Dr. Aroda and colleagues randomized 703 adults with type 2 diabetes to receive either 3 mg, 7 mg, or 14 mg of oral semaglutide daily, or placebo. The average age of the patients was 55 years, about half were women, and the average baseline HbA1c was 8.0% (64 mmol/mol). The primary endpoint was change in HbA1c level from baseline to week 26, and the secondary endpoint was change in body weight over the same period.
After 26 weeks of once-daily treatment, patients in semaglutide group showed significant reductions in HbA1c from baseline with all three doses: –0.6% (3 mg), –0.9% (7 mg), and –1.1% (14 mg), with P less than .001 for all, based on an intention-to-treat analysis. Similar results occurred using an on-treatment analysis, with differences of –0.7%, –1.2%, and –1.4%, respectively, for the three doses.
In addition, patients in all dose groups achieved the secondary endpoint of reduction in body weight, compared with placebo, from baseline to 26 weeks based on both types of analyses. “Significantly more patients achieved body weight loss of at least 5% with oral semaglutide at 7 mg and 14 mg, compared with placebo,” Dr. Aroda and colleagues wrote (intention-to-treat: –0.1 for 3 mg daily [P = .87], –0.9 for 7 mg [P = .09], –2.3 for 14 mg [P less than .001]; and on-treatment: –0.2 for 3 mg [P = .71], –1.0 for 7 mg [P = .01], –2.6 for 14 mg [P less than .001]).
The overall incidence of adverse events and serious adverse events was similar in the treatment and placebo groups, with the most frequent being nausea and diarrhea. No deaths occurred among patients on the medication.
The findings were limited by several factors, including a patient population that had a relatively short duration of diabetes (mean, 3.5 years) and that the oral semaglutide was used as first-line monotherapy, without first using metformin, the researchers noted. However, oral semaglutide “achieved clinically meaningful and superior glucose lowering,” compared with placebo, at all three doses, they wrote.
“Ongoing additional studies in the PIONEER program will further define the effect when used in combination with other glucose-lowering therapies and in other populations of interest, such as those with high cardiovascular risk or renal impairment,” they emphasized
Novo Nordisk funded the study. The lead author disclosed relationships with Novo Nordisk, and several coauthors disclosed relationships with or employment by the company.
SOURCE: Aroda VR et al. Diabetes Care. 2019 Jul. doi: 10.2337/dc19-0749.
Oral semaglutide monotherapy was superior to placebo for improving glycated hemoglobin (HbA1c) levels at all doses tested in adults with type 2 diabetes who had been previously insufficiently managed with diet and exercise, according to findings from a global, randomized trial.
The drug also showed dose-dependent weight loss, with a statistically significant effect on body weight, compared with placebo, at higher doses.
To date, the glucagon-like peptide–1 receptor agonist has been available as weekly subcutaneous shots for patients with type 2 diabetes, and in that form they have been shown to be effective in reducing HbA1c, inducing weight loss, and lowering the risk of cardiovascular events in patients with cardiovascular disease or those who are at high risk for it, wrote Vanita R. Aroda, MD, of Brigham and Women’s Hospital, Boston, and colleagues. The report is in Diabetes Care.
The novel oral semaglutide tablet is designed to enhance medication absorption, and the pharmacokinetics and dosage were established in phase 2 studies, they noted.
In the phase 3 Peptide Innovation for Early Diabetes Treatment 1 (PIONEER 1) study, Dr. Aroda and colleagues randomized 703 adults with type 2 diabetes to receive either 3 mg, 7 mg, or 14 mg of oral semaglutide daily, or placebo. The average age of the patients was 55 years, about half were women, and the average baseline HbA1c was 8.0% (64 mmol/mol). The primary endpoint was change in HbA1c level from baseline to week 26, and the secondary endpoint was change in body weight over the same period.
After 26 weeks of once-daily treatment, patients in semaglutide group showed significant reductions in HbA1c from baseline with all three doses: –0.6% (3 mg), –0.9% (7 mg), and –1.1% (14 mg), with P less than .001 for all, based on an intention-to-treat analysis. Similar results occurred using an on-treatment analysis, with differences of –0.7%, –1.2%, and –1.4%, respectively, for the three doses.
In addition, patients in all dose groups achieved the secondary endpoint of reduction in body weight, compared with placebo, from baseline to 26 weeks based on both types of analyses. “Significantly more patients achieved body weight loss of at least 5% with oral semaglutide at 7 mg and 14 mg, compared with placebo,” Dr. Aroda and colleagues wrote (intention-to-treat: –0.1 for 3 mg daily [P = .87], –0.9 for 7 mg [P = .09], –2.3 for 14 mg [P less than .001]; and on-treatment: –0.2 for 3 mg [P = .71], –1.0 for 7 mg [P = .01], –2.6 for 14 mg [P less than .001]).
The overall incidence of adverse events and serious adverse events was similar in the treatment and placebo groups, with the most frequent being nausea and diarrhea. No deaths occurred among patients on the medication.
The findings were limited by several factors, including a patient population that had a relatively short duration of diabetes (mean, 3.5 years) and that the oral semaglutide was used as first-line monotherapy, without first using metformin, the researchers noted. However, oral semaglutide “achieved clinically meaningful and superior glucose lowering,” compared with placebo, at all three doses, they wrote.
“Ongoing additional studies in the PIONEER program will further define the effect when used in combination with other glucose-lowering therapies and in other populations of interest, such as those with high cardiovascular risk or renal impairment,” they emphasized
Novo Nordisk funded the study. The lead author disclosed relationships with Novo Nordisk, and several coauthors disclosed relationships with or employment by the company.
SOURCE: Aroda VR et al. Diabetes Care. 2019 Jul. doi: 10.2337/dc19-0749.
Oral semaglutide monotherapy was superior to placebo for improving glycated hemoglobin (HbA1c) levels at all doses tested in adults with type 2 diabetes who had been previously insufficiently managed with diet and exercise, according to findings from a global, randomized trial.
The drug also showed dose-dependent weight loss, with a statistically significant effect on body weight, compared with placebo, at higher doses.
To date, the glucagon-like peptide–1 receptor agonist has been available as weekly subcutaneous shots for patients with type 2 diabetes, and in that form they have been shown to be effective in reducing HbA1c, inducing weight loss, and lowering the risk of cardiovascular events in patients with cardiovascular disease or those who are at high risk for it, wrote Vanita R. Aroda, MD, of Brigham and Women’s Hospital, Boston, and colleagues. The report is in Diabetes Care.
The novel oral semaglutide tablet is designed to enhance medication absorption, and the pharmacokinetics and dosage were established in phase 2 studies, they noted.
In the phase 3 Peptide Innovation for Early Diabetes Treatment 1 (PIONEER 1) study, Dr. Aroda and colleagues randomized 703 adults with type 2 diabetes to receive either 3 mg, 7 mg, or 14 mg of oral semaglutide daily, or placebo. The average age of the patients was 55 years, about half were women, and the average baseline HbA1c was 8.0% (64 mmol/mol). The primary endpoint was change in HbA1c level from baseline to week 26, and the secondary endpoint was change in body weight over the same period.
After 26 weeks of once-daily treatment, patients in semaglutide group showed significant reductions in HbA1c from baseline with all three doses: –0.6% (3 mg), –0.9% (7 mg), and –1.1% (14 mg), with P less than .001 for all, based on an intention-to-treat analysis. Similar results occurred using an on-treatment analysis, with differences of –0.7%, –1.2%, and –1.4%, respectively, for the three doses.
In addition, patients in all dose groups achieved the secondary endpoint of reduction in body weight, compared with placebo, from baseline to 26 weeks based on both types of analyses. “Significantly more patients achieved body weight loss of at least 5% with oral semaglutide at 7 mg and 14 mg, compared with placebo,” Dr. Aroda and colleagues wrote (intention-to-treat: –0.1 for 3 mg daily [P = .87], –0.9 for 7 mg [P = .09], –2.3 for 14 mg [P less than .001]; and on-treatment: –0.2 for 3 mg [P = .71], –1.0 for 7 mg [P = .01], –2.6 for 14 mg [P less than .001]).
The overall incidence of adverse events and serious adverse events was similar in the treatment and placebo groups, with the most frequent being nausea and diarrhea. No deaths occurred among patients on the medication.
The findings were limited by several factors, including a patient population that had a relatively short duration of diabetes (mean, 3.5 years) and that the oral semaglutide was used as first-line monotherapy, without first using metformin, the researchers noted. However, oral semaglutide “achieved clinically meaningful and superior glucose lowering,” compared with placebo, at all three doses, they wrote.
“Ongoing additional studies in the PIONEER program will further define the effect when used in combination with other glucose-lowering therapies and in other populations of interest, such as those with high cardiovascular risk or renal impairment,” they emphasized
Novo Nordisk funded the study. The lead author disclosed relationships with Novo Nordisk, and several coauthors disclosed relationships with or employment by the company.
SOURCE: Aroda VR et al. Diabetes Care. 2019 Jul. doi: 10.2337/dc19-0749.
FROM DIABETES CARE
Aspirin interacts with epigenetics to influence breast cancer mortality
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
FROM CANCER
Quarterly intravenous eptinezumab prevents migraine
PHILADELPHIA – An intravenous formulation of a calcitonin gene–related peptide inhibitor monoclonal antibody showed efficacy for preventing chronic migraine headaches for 3 months in a dose-ranging, phase 3 trial with 1,072 patients.
In a separate study with 669 patients, a single IV dose of the antibody, eptinezumab, also significantly reduced the incidence of episodic migraine headaches during 3 months of follow-up, compared with placebo. And in both the chronic and episodic migraine studies a similar 3-month effect resulted from a second IV dose of the humanized antibody that binds the calcitonin gene–related peptide (CGRP) ligand, thereby blocking the pathway, Laszlo L. Mechtler, MD, and his associates reported in a poster at the annual meeting of the American Headache Society.
Eptinezumab follows the therapeutic approach already used by three Food and Drug Administration–approved monoclonal antibody drugs that cut migraine headache recurrences by blocking the CGRP pathway by binding either the peptide ligand or its receptor: erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). Eptinezumab differs from the three approved CGRP antibodies by using an IV route of administration – the other three are delivered by subcutaneous injection – and by a 3-month dosing interval. Both erenumab-aooe and galcanezumab-gnlm are labeled for monthly administration only, while fremanezumab-vfrm is labeled for both monthly and once every 3 months dosing schedules.
The PROMISE-1 (A Multicenter Assessment of ALD403 in Frequent Episodic Migraine) trial randomized 669 patients with episodic migraine (defined as 4-14 headache days/month with at least 4 classifiable as migraine headache days) at 87 centers mostly in the United States and with some in Georgia. The PROMISE-2 (Evaluation of ALD403 (Eptinezumab) in the Prevention of Chronic Migraine) trial randomized 1,072 patients with chronic migraine (defined as a history of 15-26 headache days/month and with at least 8 of the days involving a migraine headache) at any of 145 study sites, many in the United States, in several countries.
In PROMISE-1, patients could receive as many as four serial infusions every 3 months, and up to two serial infusions in PROMISE-2, but the primary endpoint in both studies was the change in monthly migraine count from baseline during the 3 months following the first dosage.
Among patients with chronic migraine in PROMISE-2, the average monthly migraine number fell by 8.2 migraine days/month, compared with an average 5.6 monthly migraine days drop from baseline among placebo patients, which was a statistically significant difference for the higher dosage of eptinezumab tested, 300 mg. A 100-mg dose linked with an average 7.7 migraine days/month reduction, also a statistically significant difference from the placebo patients, reported Dr. Mechtler, professor of neurology at the State University of New York at Buffalo and medical director of the Dent Neurologic Institute in Buffalo, and his associates.
Among patients with episodic migraine in PROMISE-1, the 300-mg dosage cut monthly migraines by an average 4.3 migraine headache days/month, compared with 3.2 in the placebo group, a statistically significant difference. Among patients who received the 100-mg dosage, the average cut was 3.9 migraine headache days/month, also a statistically significant difference from the placebo controls.
The researchers included no safety findings in their report, but in an interview Dr. Mechtler said that eptinezumab showed an excellent safety profile that was consistent with what’s been previously reported for the approved agents from this class. He cited the safety of the drugs in the class as a major feature of their clinical utility.
PROMISE-1 and PROMISE-2 were sponsored by Alder BioPharmaceuticals, the company developing eptinezumab. Dr. Mechtler has been a speaker on behalf of Allergan, Amgen/Novartis, Boston Biomedical, Promius, Avanir, and Teva, and he has received research funding from Allergan, Autonomic Technologies, Boston Biomedical, and Teva.
SOURCE: Mechtler LL et al. Headache. 2019 June;59[S1]:34, Abstract P12
PHILADELPHIA – An intravenous formulation of a calcitonin gene–related peptide inhibitor monoclonal antibody showed efficacy for preventing chronic migraine headaches for 3 months in a dose-ranging, phase 3 trial with 1,072 patients.
In a separate study with 669 patients, a single IV dose of the antibody, eptinezumab, also significantly reduced the incidence of episodic migraine headaches during 3 months of follow-up, compared with placebo. And in both the chronic and episodic migraine studies a similar 3-month effect resulted from a second IV dose of the humanized antibody that binds the calcitonin gene–related peptide (CGRP) ligand, thereby blocking the pathway, Laszlo L. Mechtler, MD, and his associates reported in a poster at the annual meeting of the American Headache Society.
Eptinezumab follows the therapeutic approach already used by three Food and Drug Administration–approved monoclonal antibody drugs that cut migraine headache recurrences by blocking the CGRP pathway by binding either the peptide ligand or its receptor: erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). Eptinezumab differs from the three approved CGRP antibodies by using an IV route of administration – the other three are delivered by subcutaneous injection – and by a 3-month dosing interval. Both erenumab-aooe and galcanezumab-gnlm are labeled for monthly administration only, while fremanezumab-vfrm is labeled for both monthly and once every 3 months dosing schedules.
The PROMISE-1 (A Multicenter Assessment of ALD403 in Frequent Episodic Migraine) trial randomized 669 patients with episodic migraine (defined as 4-14 headache days/month with at least 4 classifiable as migraine headache days) at 87 centers mostly in the United States and with some in Georgia. The PROMISE-2 (Evaluation of ALD403 (Eptinezumab) in the Prevention of Chronic Migraine) trial randomized 1,072 patients with chronic migraine (defined as a history of 15-26 headache days/month and with at least 8 of the days involving a migraine headache) at any of 145 study sites, many in the United States, in several countries.
In PROMISE-1, patients could receive as many as four serial infusions every 3 months, and up to two serial infusions in PROMISE-2, but the primary endpoint in both studies was the change in monthly migraine count from baseline during the 3 months following the first dosage.
Among patients with chronic migraine in PROMISE-2, the average monthly migraine number fell by 8.2 migraine days/month, compared with an average 5.6 monthly migraine days drop from baseline among placebo patients, which was a statistically significant difference for the higher dosage of eptinezumab tested, 300 mg. A 100-mg dose linked with an average 7.7 migraine days/month reduction, also a statistically significant difference from the placebo patients, reported Dr. Mechtler, professor of neurology at the State University of New York at Buffalo and medical director of the Dent Neurologic Institute in Buffalo, and his associates.
Among patients with episodic migraine in PROMISE-1, the 300-mg dosage cut monthly migraines by an average 4.3 migraine headache days/month, compared with 3.2 in the placebo group, a statistically significant difference. Among patients who received the 100-mg dosage, the average cut was 3.9 migraine headache days/month, also a statistically significant difference from the placebo controls.
The researchers included no safety findings in their report, but in an interview Dr. Mechtler said that eptinezumab showed an excellent safety profile that was consistent with what’s been previously reported for the approved agents from this class. He cited the safety of the drugs in the class as a major feature of their clinical utility.
PROMISE-1 and PROMISE-2 were sponsored by Alder BioPharmaceuticals, the company developing eptinezumab. Dr. Mechtler has been a speaker on behalf of Allergan, Amgen/Novartis, Boston Biomedical, Promius, Avanir, and Teva, and he has received research funding from Allergan, Autonomic Technologies, Boston Biomedical, and Teva.
SOURCE: Mechtler LL et al. Headache. 2019 June;59[S1]:34, Abstract P12
PHILADELPHIA – An intravenous formulation of a calcitonin gene–related peptide inhibitor monoclonal antibody showed efficacy for preventing chronic migraine headaches for 3 months in a dose-ranging, phase 3 trial with 1,072 patients.
In a separate study with 669 patients, a single IV dose of the antibody, eptinezumab, also significantly reduced the incidence of episodic migraine headaches during 3 months of follow-up, compared with placebo. And in both the chronic and episodic migraine studies a similar 3-month effect resulted from a second IV dose of the humanized antibody that binds the calcitonin gene–related peptide (CGRP) ligand, thereby blocking the pathway, Laszlo L. Mechtler, MD, and his associates reported in a poster at the annual meeting of the American Headache Society.
Eptinezumab follows the therapeutic approach already used by three Food and Drug Administration–approved monoclonal antibody drugs that cut migraine headache recurrences by blocking the CGRP pathway by binding either the peptide ligand or its receptor: erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). Eptinezumab differs from the three approved CGRP antibodies by using an IV route of administration – the other three are delivered by subcutaneous injection – and by a 3-month dosing interval. Both erenumab-aooe and galcanezumab-gnlm are labeled for monthly administration only, while fremanezumab-vfrm is labeled for both monthly and once every 3 months dosing schedules.
The PROMISE-1 (A Multicenter Assessment of ALD403 in Frequent Episodic Migraine) trial randomized 669 patients with episodic migraine (defined as 4-14 headache days/month with at least 4 classifiable as migraine headache days) at 87 centers mostly in the United States and with some in Georgia. The PROMISE-2 (Evaluation of ALD403 (Eptinezumab) in the Prevention of Chronic Migraine) trial randomized 1,072 patients with chronic migraine (defined as a history of 15-26 headache days/month and with at least 8 of the days involving a migraine headache) at any of 145 study sites, many in the United States, in several countries.
In PROMISE-1, patients could receive as many as four serial infusions every 3 months, and up to two serial infusions in PROMISE-2, but the primary endpoint in both studies was the change in monthly migraine count from baseline during the 3 months following the first dosage.
Among patients with chronic migraine in PROMISE-2, the average monthly migraine number fell by 8.2 migraine days/month, compared with an average 5.6 monthly migraine days drop from baseline among placebo patients, which was a statistically significant difference for the higher dosage of eptinezumab tested, 300 mg. A 100-mg dose linked with an average 7.7 migraine days/month reduction, also a statistically significant difference from the placebo patients, reported Dr. Mechtler, professor of neurology at the State University of New York at Buffalo and medical director of the Dent Neurologic Institute in Buffalo, and his associates.
Among patients with episodic migraine in PROMISE-1, the 300-mg dosage cut monthly migraines by an average 4.3 migraine headache days/month, compared with 3.2 in the placebo group, a statistically significant difference. Among patients who received the 100-mg dosage, the average cut was 3.9 migraine headache days/month, also a statistically significant difference from the placebo controls.
The researchers included no safety findings in their report, but in an interview Dr. Mechtler said that eptinezumab showed an excellent safety profile that was consistent with what’s been previously reported for the approved agents from this class. He cited the safety of the drugs in the class as a major feature of their clinical utility.
PROMISE-1 and PROMISE-2 were sponsored by Alder BioPharmaceuticals, the company developing eptinezumab. Dr. Mechtler has been a speaker on behalf of Allergan, Amgen/Novartis, Boston Biomedical, Promius, Avanir, and Teva, and he has received research funding from Allergan, Autonomic Technologies, Boston Biomedical, and Teva.
SOURCE: Mechtler LL et al. Headache. 2019 June;59[S1]:34, Abstract P12
REPORTING FROM AHS 2019

Hospital slashes S. aureus vancomycin resistance
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
REPORTING FROM ESPID 2019
Key clinical point: Staphylococcus aureus vancomycin MIC creep is reversible through dedicated antimicrobial stewardship.
Major finding: The prevalence of hGISA in MRSA and MSSA specimens improved from 25% during 2002-2009 to 6% during 2010-2017 at one German tertiary children’s hospital.
Study details: This was a retrospective single-center analysis of vancomycin resistance trends over time in 540 S. aureus specimens gathered in 2002-2017.
Disclosures: The presenter reported having no financial conflicts regarding this study, which was conducted free of commercial support.