User login
Painful Growing Nodule on the Right Calf
The Diagnosis: Merkel Cell Carcinoma
Multiple diagnoses should be considered for a small, round, blue cell neoplasm of the skin, including both primary and metastatic entities. In our patient, histopathology revealed sheets and nests of infiltrative neoplastic cells with dispersed chromatin, minimal cytoplasm, and multiple mitoses (quiz image 1).1 The lesional cells were in the dermis and superficial subcutaneous tissue but did not appear to be arising from the epidermis. Lymphovascular invasion also was evident on additional sections. Metastatic disease was identified in 3 sentinel lymph nodes from the right inguinal and right iliac regions. These features were compatible with a diagnosis of Merkel cell carcinoma (MCC).
Merkel cell carcinoma is a rare malignant neuroendocrine cutaneous tumor with a worldwide incidence of 0.1 to 1.6 cases per 100,000 individuals annually.2 The typical patient is older than 75 years with fair skin and a history of extensive sun exposure. Immunocompromised individuals are predisposed and more susceptible to infection with the Merkel cell polyomavirus, which promotes oncogenesis in the majority of MCCs. Our patient’s history of combined variable immunodeficiency likely explains her presentation at a younger age.
The prognosis in patients with MCC is poor, with 5-year survival rates of 51% for local disease, 35% for nodal disease, and 14% for systemic metastases. Survival also is reduced in cases with head/ neck primary tumors and polyomavirus-negative tumors, as well as in immunocompromised patients.2 Treatment of resectable MCC consists of Mohs micrographic surgery or wide local excision depending on the patient’s cosmetic concerns. Radiation therapy is recommended for cases with increased risk for recurrence or positive surgical margins, as well as when additional resection is impossible. A study investigating immunotherapy with nivolumab demonstrated complete pathologic response and radiographic tumor regression in nearly half of patients when given 4 weeks prior to surgery.3
Immunohistochemistry is essential in discerning MCC from other small blue cell tumors. Most MCC cases show positive expression of neuroendocrine markers such as synaptophysin, chromogranin, and insulinomaassociated protein 1. Perinuclear dotlike staining with cytokeratin (CK) 20 (quiz image 2) commonly is seen, but up to 15% of cases may be CK20 negative. Many of these CK20-negative cases also express CK7. This tumor also may stain with paired box 5 (PAX-5), CD99, terminal deoxynucleotidyl transferase, Ber-EP4, and CD1171,4; melanoma stains (ie, human melanoma black [HMB] 45, SRYrelated HMB-box 10 [SOX-10], S-100, melanoma antigen recognized by T-cells 1 [MART-1]) should be negative. However, PAX-5 expression may be a potential pitfall given that B-cell lymphomas also would express that marker and could mimic MCC histologically. Therefore, other universal lymphoid markers such as CD45 should be ordered to rule out this entity. Even with one or a few aberrant stains, a diagnosis of MCC still can be rendered using the histomorphology and the overall staining profile.4 Of prognostic significance, p63 expression is associated with more aggressive tumors, while Bcl-2 expression is favorable, as it offers an additional targeted treatment option.5,6
Basal cell carcinoma (BCC) is linked to excessive sun exposure and is the most common skin cancer. Similar to MCC, it typically is mitotically active and hyperchromatic; however, lymphovascular invasion or metastasis almost never is observed in BCC, whereas approximately one-third of MCC cases have metastasized by the time of diagnosis. Additionally, BCC lacks the perinuclear dotlike staining seen with CK20.2,7 Features present in BCC that are unusual for MCC include peripheral nuclear palisading, mucin, and retraction artifact on paraffin-embedded sections (Figure 1).7

Leukemia cutis (or cutaneous infiltrates of leukemia) commonly displays a perivascular and periadnexal pattern in the dermis and subcutis. These infiltrates of neoplastic leukocytes can congregate into sheets, sometimes with an overlying Grenz zone, or form single-file infiltrates (Figure 2).1,4 The neoplastic cells can be monomorphic or atypical and commonly are susceptible to crush artifact.4 Although the immunohistochemical profile varies depending on the etiology of the underlying leukemia, broad hematologic markers such as CD43 and CD45 are helpful to discern these malignancies from MCC.4

Being neuroendocrine in origin, metastatic small cell carcinoma (Figure 3) strongly mimics MCC histologically and usually stains with synaptophysin, chromogranin, and insulinoma-associated protein 1. Both tumor cells typically exhibit nuclear molding and high mitotic rates. Although small cell carcinoma is more likely to stain with high-molecular-weight cytokeratins (ie, CK7), it is not uncommon for these tumors to express lowmolecular- weight cytokeratins such as CK20. Because most cases originate from the lungs, these lesions should be positive for thyroid transcription factor 1 and negative for PAX-5, whereas MCC would show the reverse for those stains.1 Ultimately, however, clinical correlation with imaging results is the single best methodology for differentiation.

Small cell melanoma, a variant of nevoid melanoma, can strongly resemble an MCC or a lymphoma. Usually located on the scalp or arising from a congenital nevus, small cell melanomas are aggressive and confer an unfavorable prognosis. Histologically, they consist of nests to sheets of atypical cells within the epidermis and dermis. These cells typically exhibit hyperchromatic nuclei, minimal cytoplasm, and frequent mitoses (Figure 4). Furthermore, the cells do not display maturation based on depth.8 These tumors usually are positive for HMB45, S-100, MART-1, SOX-10, and tyrosinase, all of which are extremely unlikely to stain an MCC.1

- Patterson JW, Hosler GA. Weedon’s Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016.
- Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48:411-421.
- Topalian SL, Bhatia S, Amin A, et al. Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol. 2020;38:2476-2488.
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Asioli S, Righi A, Volante M, et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640-647.
- Verhaegen ME, Mangelberger D, Weick JW, et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J Invest Dermatol. 2014;134:2241-2250.
- Le MD, O’Steen LH, Cassarino DS. A rare case of CK20/CK7 double negative Merkel cell carcinoma. Am J Dermatopathol. 2017;39:208-211.
- North JP, Bastian BC, Lazar AJ. Melanoma. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin With Clinical Correlations. 5th ed. Elsevier; 2020.
The Diagnosis: Merkel Cell Carcinoma
Multiple diagnoses should be considered for a small, round, blue cell neoplasm of the skin, including both primary and metastatic entities. In our patient, histopathology revealed sheets and nests of infiltrative neoplastic cells with dispersed chromatin, minimal cytoplasm, and multiple mitoses (quiz image 1).1 The lesional cells were in the dermis and superficial subcutaneous tissue but did not appear to be arising from the epidermis. Lymphovascular invasion also was evident on additional sections. Metastatic disease was identified in 3 sentinel lymph nodes from the right inguinal and right iliac regions. These features were compatible with a diagnosis of Merkel cell carcinoma (MCC).
Merkel cell carcinoma is a rare malignant neuroendocrine cutaneous tumor with a worldwide incidence of 0.1 to 1.6 cases per 100,000 individuals annually.2 The typical patient is older than 75 years with fair skin and a history of extensive sun exposure. Immunocompromised individuals are predisposed and more susceptible to infection with the Merkel cell polyomavirus, which promotes oncogenesis in the majority of MCCs. Our patient’s history of combined variable immunodeficiency likely explains her presentation at a younger age.
The prognosis in patients with MCC is poor, with 5-year survival rates of 51% for local disease, 35% for nodal disease, and 14% for systemic metastases. Survival also is reduced in cases with head/ neck primary tumors and polyomavirus-negative tumors, as well as in immunocompromised patients.2 Treatment of resectable MCC consists of Mohs micrographic surgery or wide local excision depending on the patient’s cosmetic concerns. Radiation therapy is recommended for cases with increased risk for recurrence or positive surgical margins, as well as when additional resection is impossible. A study investigating immunotherapy with nivolumab demonstrated complete pathologic response and radiographic tumor regression in nearly half of patients when given 4 weeks prior to surgery.3
Immunohistochemistry is essential in discerning MCC from other small blue cell tumors. Most MCC cases show positive expression of neuroendocrine markers such as synaptophysin, chromogranin, and insulinomaassociated protein 1. Perinuclear dotlike staining with cytokeratin (CK) 20 (quiz image 2) commonly is seen, but up to 15% of cases may be CK20 negative. Many of these CK20-negative cases also express CK7. This tumor also may stain with paired box 5 (PAX-5), CD99, terminal deoxynucleotidyl transferase, Ber-EP4, and CD1171,4; melanoma stains (ie, human melanoma black [HMB] 45, SRYrelated HMB-box 10 [SOX-10], S-100, melanoma antigen recognized by T-cells 1 [MART-1]) should be negative. However, PAX-5 expression may be a potential pitfall given that B-cell lymphomas also would express that marker and could mimic MCC histologically. Therefore, other universal lymphoid markers such as CD45 should be ordered to rule out this entity. Even with one or a few aberrant stains, a diagnosis of MCC still can be rendered using the histomorphology and the overall staining profile.4 Of prognostic significance, p63 expression is associated with more aggressive tumors, while Bcl-2 expression is favorable, as it offers an additional targeted treatment option.5,6
Basal cell carcinoma (BCC) is linked to excessive sun exposure and is the most common skin cancer. Similar to MCC, it typically is mitotically active and hyperchromatic; however, lymphovascular invasion or metastasis almost never is observed in BCC, whereas approximately one-third of MCC cases have metastasized by the time of diagnosis. Additionally, BCC lacks the perinuclear dotlike staining seen with CK20.2,7 Features present in BCC that are unusual for MCC include peripheral nuclear palisading, mucin, and retraction artifact on paraffin-embedded sections (Figure 1).7
Leukemia cutis (or cutaneous infiltrates of leukemia) commonly displays a perivascular and periadnexal pattern in the dermis and subcutis. These infiltrates of neoplastic leukocytes can congregate into sheets, sometimes with an overlying Grenz zone, or form single-file infiltrates (Figure 2).1,4 The neoplastic cells can be monomorphic or atypical and commonly are susceptible to crush artifact.4 Although the immunohistochemical profile varies depending on the etiology of the underlying leukemia, broad hematologic markers such as CD43 and CD45 are helpful to discern these malignancies from MCC.4
Being neuroendocrine in origin, metastatic small cell carcinoma (Figure 3) strongly mimics MCC histologically and usually stains with synaptophysin, chromogranin, and insulinoma-associated protein 1. Both tumor cells typically exhibit nuclear molding and high mitotic rates. Although small cell carcinoma is more likely to stain with high-molecular-weight cytokeratins (ie, CK7), it is not uncommon for these tumors to express lowmolecular- weight cytokeratins such as CK20. Because most cases originate from the lungs, these lesions should be positive for thyroid transcription factor 1 and negative for PAX-5, whereas MCC would show the reverse for those stains.1 Ultimately, however, clinical correlation with imaging results is the single best methodology for differentiation.
Small cell melanoma, a variant of nevoid melanoma, can strongly resemble an MCC or a lymphoma. Usually located on the scalp or arising from a congenital nevus, small cell melanomas are aggressive and confer an unfavorable prognosis. Histologically, they consist of nests to sheets of atypical cells within the epidermis and dermis. These cells typically exhibit hyperchromatic nuclei, minimal cytoplasm, and frequent mitoses (Figure 4). Furthermore, the cells do not display maturation based on depth.8 These tumors usually are positive for HMB45, S-100, MART-1, SOX-10, and tyrosinase, all of which are extremely unlikely to stain an MCC.1
The Diagnosis: Merkel Cell Carcinoma
Multiple diagnoses should be considered for a small, round, blue cell neoplasm of the skin, including both primary and metastatic entities. In our patient, histopathology revealed sheets and nests of infiltrative neoplastic cells with dispersed chromatin, minimal cytoplasm, and multiple mitoses (quiz image 1).1 The lesional cells were in the dermis and superficial subcutaneous tissue but did not appear to be arising from the epidermis. Lymphovascular invasion also was evident on additional sections. Metastatic disease was identified in 3 sentinel lymph nodes from the right inguinal and right iliac regions. These features were compatible with a diagnosis of Merkel cell carcinoma (MCC).
Merkel cell carcinoma is a rare malignant neuroendocrine cutaneous tumor with a worldwide incidence of 0.1 to 1.6 cases per 100,000 individuals annually.2 The typical patient is older than 75 years with fair skin and a history of extensive sun exposure. Immunocompromised individuals are predisposed and more susceptible to infection with the Merkel cell polyomavirus, which promotes oncogenesis in the majority of MCCs. Our patient’s history of combined variable immunodeficiency likely explains her presentation at a younger age.
The prognosis in patients with MCC is poor, with 5-year survival rates of 51% for local disease, 35% for nodal disease, and 14% for systemic metastases. Survival also is reduced in cases with head/ neck primary tumors and polyomavirus-negative tumors, as well as in immunocompromised patients.2 Treatment of resectable MCC consists of Mohs micrographic surgery or wide local excision depending on the patient’s cosmetic concerns. Radiation therapy is recommended for cases with increased risk for recurrence or positive surgical margins, as well as when additional resection is impossible. A study investigating immunotherapy with nivolumab demonstrated complete pathologic response and radiographic tumor regression in nearly half of patients when given 4 weeks prior to surgery.3
Immunohistochemistry is essential in discerning MCC from other small blue cell tumors. Most MCC cases show positive expression of neuroendocrine markers such as synaptophysin, chromogranin, and insulinomaassociated protein 1. Perinuclear dotlike staining with cytokeratin (CK) 20 (quiz image 2) commonly is seen, but up to 15% of cases may be CK20 negative. Many of these CK20-negative cases also express CK7. This tumor also may stain with paired box 5 (PAX-5), CD99, terminal deoxynucleotidyl transferase, Ber-EP4, and CD1171,4; melanoma stains (ie, human melanoma black [HMB] 45, SRYrelated HMB-box 10 [SOX-10], S-100, melanoma antigen recognized by T-cells 1 [MART-1]) should be negative. However, PAX-5 expression may be a potential pitfall given that B-cell lymphomas also would express that marker and could mimic MCC histologically. Therefore, other universal lymphoid markers such as CD45 should be ordered to rule out this entity. Even with one or a few aberrant stains, a diagnosis of MCC still can be rendered using the histomorphology and the overall staining profile.4 Of prognostic significance, p63 expression is associated with more aggressive tumors, while Bcl-2 expression is favorable, as it offers an additional targeted treatment option.5,6
Basal cell carcinoma (BCC) is linked to excessive sun exposure and is the most common skin cancer. Similar to MCC, it typically is mitotically active and hyperchromatic; however, lymphovascular invasion or metastasis almost never is observed in BCC, whereas approximately one-third of MCC cases have metastasized by the time of diagnosis. Additionally, BCC lacks the perinuclear dotlike staining seen with CK20.2,7 Features present in BCC that are unusual for MCC include peripheral nuclear palisading, mucin, and retraction artifact on paraffin-embedded sections (Figure 1).7
Leukemia cutis (or cutaneous infiltrates of leukemia) commonly displays a perivascular and periadnexal pattern in the dermis and subcutis. These infiltrates of neoplastic leukocytes can congregate into sheets, sometimes with an overlying Grenz zone, or form single-file infiltrates (Figure 2).1,4 The neoplastic cells can be monomorphic or atypical and commonly are susceptible to crush artifact.4 Although the immunohistochemical profile varies depending on the etiology of the underlying leukemia, broad hematologic markers such as CD43 and CD45 are helpful to discern these malignancies from MCC.4
Being neuroendocrine in origin, metastatic small cell carcinoma (Figure 3) strongly mimics MCC histologically and usually stains with synaptophysin, chromogranin, and insulinoma-associated protein 1. Both tumor cells typically exhibit nuclear molding and high mitotic rates. Although small cell carcinoma is more likely to stain with high-molecular-weight cytokeratins (ie, CK7), it is not uncommon for these tumors to express lowmolecular- weight cytokeratins such as CK20. Because most cases originate from the lungs, these lesions should be positive for thyroid transcription factor 1 and negative for PAX-5, whereas MCC would show the reverse for those stains.1 Ultimately, however, clinical correlation with imaging results is the single best methodology for differentiation.
Small cell melanoma, a variant of nevoid melanoma, can strongly resemble an MCC or a lymphoma. Usually located on the scalp or arising from a congenital nevus, small cell melanomas are aggressive and confer an unfavorable prognosis. Histologically, they consist of nests to sheets of atypical cells within the epidermis and dermis. These cells typically exhibit hyperchromatic nuclei, minimal cytoplasm, and frequent mitoses (Figure 4). Furthermore, the cells do not display maturation based on depth.8 These tumors usually are positive for HMB45, S-100, MART-1, SOX-10, and tyrosinase, all of which are extremely unlikely to stain an MCC.1
- Patterson JW, Hosler GA. Weedon’s Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016.
- Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48:411-421.
- Topalian SL, Bhatia S, Amin A, et al. Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol. 2020;38:2476-2488.
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Asioli S, Righi A, Volante M, et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640-647.
- Verhaegen ME, Mangelberger D, Weick JW, et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J Invest Dermatol. 2014;134:2241-2250.
- Le MD, O’Steen LH, Cassarino DS. A rare case of CK20/CK7 double negative Merkel cell carcinoma. Am J Dermatopathol. 2017;39:208-211.
- North JP, Bastian BC, Lazar AJ. Melanoma. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin With Clinical Correlations. 5th ed. Elsevier; 2020.
- Patterson JW, Hosler GA. Weedon’s Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016.
- Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48:411-421.
- Topalian SL, Bhatia S, Amin A, et al. Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol. 2020;38:2476-2488.
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Asioli S, Righi A, Volante M, et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640-647.
- Verhaegen ME, Mangelberger D, Weick JW, et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J Invest Dermatol. 2014;134:2241-2250.
- Le MD, O’Steen LH, Cassarino DS. A rare case of CK20/CK7 double negative Merkel cell carcinoma. Am J Dermatopathol. 2017;39:208-211.
- North JP, Bastian BC, Lazar AJ. Melanoma. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin With Clinical Correlations. 5th ed. Elsevier; 2020.
A 47-year-old woman with a history of combined variable immunodeficiency presented with a 2.6×2.4-cm nodule on the lateral aspect of the right calf that was first noticed 2 years prior as a smaller nodule. It increased in size and became painful to touch over the last 3 to 4 months. Following diagnostic biopsy, the nodule was removed by wide local excision and was tan-brown on gross dissection. The lesion showed dotlike perinuclear positivity with cytokeratin 20 immunostaining. Positron emission tomography–computed tomography showed no evidence of lung lesions. A complete blood cell count was within reference range.

.")
Pedunculated Verrucous Tumor on the Buttock
The Diagnosis: Giant Acrochordon
Based on the clinical and histologic findings, our patient was diagnosed with a giant acrochordon. Acrochordons (also known as fibroepithelial polyps or skin tags) are among the most commonly identified skin lesions and are believed to affect up to 46% of the general population.1,2 These benign growths typically appear after middle age in men and women alike and are believed to be of ectodermal and mesenchymal origin.3 The most common locations include the axillae, neck, and inguinal folds. They generally are small, measuring only a few millimeters, and frequently present as multiple lesions that are called giant acrochordons when their size exceeds 5 cm in length.2 Acrochordons are benign lesions with only rare reports of the presence of basal or squamous cell carcinoma within the lesion on pathology.4 In addition to being cosmetically unsightly, patients with acrochordons often report pruritus. These lesions are easily removed in an outpatient setting via snip excision, cryosurgery, or electrodesiccation. Once removed, recurrence is unlikely. Despite the prevalence of fibroepithelial polyps worldwide, reports of giant acrochordons are limited. The histopathology of giant acrochordons is similar to smaller acrochordons, with features including epidermal acanthosis and a central core of fibrovascular tissue without adnexal structures (Figure).4
.")
The differential diagnosis of giant acrochordon includes neurofibroma, nodular melanoma, squamous cell carcinoma, and giant condylomata acuminata (Buschke-Löwenstein tumor).1 It is important to consider the clinical presentation and histopathologic findings to differentiate giant acrochordons from these other entities.
Neurofibromas typically present as multiple flesh-colored to brown nodules that invaginate into the skin when minimal external pressure is applied.5 Histopathology demonstrates a discrete, nonencapsulated, dermal collection of small nerve fibers and loosely arranged spindle cells. In contrast, giant acrochordons typically present as large, fleshcolored, pedunculated, verrucous tumors with a central stalk. Histopathology reveals epidermal acanthosis and a central core of fibrovascular tissue without adnexal structures.
Nodular melanomas usually are blue to black and grow rapidly over the course of several months.6 They have signs of hemorrhagic crust, and histopathology reveals atypical melanocytes, frequent mitoses, pleomorphic tumor cells, and irregular clumping of chromatin within the nuclei. Giant acrochordons are flesh colored, benign, and do not have these malignant features.
Squamous cell carcinoma often presents as an erythematous scaly patch or red plaque on sun-exposed areas of the skin.1 Histopathology of squamous cell carcinoma shows atypical keratinocytes with an invasive growth pattern; giant acrochordon does not show keratinocytic atypia or invasive epidermal growth.
Giant condylomata acuminata (Buschke-Löwenstein tumor) is a locally destructive verrucous plaque that typically appears on the penis but can occur elsewhere in the anogenital region.7 Histopathologic features include epidermal hyperplasia, papillomatosis, and koilocytes. In contrast, giant acrochordons typically are located on the buttocks and do not present with these epidermal changes.
Based on the clinical and histologic findings, our patient was diagnosed with a giant acrochordon, a rare variant of the common skin lesion. Excisional removal was critical for both diagnostic and treatment purposes. By considering the clinical presentation and histopathologic features of other conditions in the differential, giant acrochordons can be distinguished from other similar entities. Diagnosis and prompt surgical removal are important for management of these neoplasms and prevention of misdiagnosis.
- Alkhalili E, Prapasiri S, Russell J. Giant acrochordon of the axilla. BMJ Case Rep. 2015:bcr2015210623. doi:10.1136/bcr-2015-210623
- Banik R, Lubach D. Skin tags: localization and frequencies according to sex and age. Dermatologica. 1987;174:180-183. doi:10.1159/000249169
- Can B, Yildrim Ozluk A. Giant fibroepithelial polyps: why do they grow excessively? Med Bull Sisli Etfal Hastan Tip Bul. 2020;54:257-260. doi:10.14744/SEMB.2018.33603
- Ghosh SK, Bandyopadhyay D, Chatterjee G, et al. Giant skin tags on unusual locations. J Eur Acad Dermatol Venereol. 2009;23:233. doi:10.1111/j.1468-3083.2008.02816.x
- Messersmith L, Krauland K. Neurofibroma. StatPearls [Internet]. StatPearls Publishing; 2023.
- Saaiq M, Ashraf B, Siddiqui S. Nodular melanoma. Iran J Med Sci. 2016;41:164-165.
- Spinu D, Ra˘dulescu A, Bratu O, et al. Giant condyloma acuminatum. Buschke-Lowenstein disease: a literature review. Chirurgia (Bucur). 2014;109:445-450.
The Diagnosis: Giant Acrochordon
Based on the clinical and histologic findings, our patient was diagnosed with a giant acrochordon. Acrochordons (also known as fibroepithelial polyps or skin tags) are among the most commonly identified skin lesions and are believed to affect up to 46% of the general population.1,2 These benign growths typically appear after middle age in men and women alike and are believed to be of ectodermal and mesenchymal origin.3 The most common locations include the axillae, neck, and inguinal folds. They generally are small, measuring only a few millimeters, and frequently present as multiple lesions that are called giant acrochordons when their size exceeds 5 cm in length.2 Acrochordons are benign lesions with only rare reports of the presence of basal or squamous cell carcinoma within the lesion on pathology.4 In addition to being cosmetically unsightly, patients with acrochordons often report pruritus. These lesions are easily removed in an outpatient setting via snip excision, cryosurgery, or electrodesiccation. Once removed, recurrence is unlikely. Despite the prevalence of fibroepithelial polyps worldwide, reports of giant acrochordons are limited. The histopathology of giant acrochordons is similar to smaller acrochordons, with features including epidermal acanthosis and a central core of fibrovascular tissue without adnexal structures (Figure).4
The differential diagnosis of giant acrochordon includes neurofibroma, nodular melanoma, squamous cell carcinoma, and giant condylomata acuminata (Buschke-Löwenstein tumor).1 It is important to consider the clinical presentation and histopathologic findings to differentiate giant acrochordons from these other entities.
Neurofibromas typically present as multiple flesh-colored to brown nodules that invaginate into the skin when minimal external pressure is applied.5 Histopathology demonstrates a discrete, nonencapsulated, dermal collection of small nerve fibers and loosely arranged spindle cells. In contrast, giant acrochordons typically present as large, fleshcolored, pedunculated, verrucous tumors with a central stalk. Histopathology reveals epidermal acanthosis and a central core of fibrovascular tissue without adnexal structures.
Nodular melanomas usually are blue to black and grow rapidly over the course of several months.6 They have signs of hemorrhagic crust, and histopathology reveals atypical melanocytes, frequent mitoses, pleomorphic tumor cells, and irregular clumping of chromatin within the nuclei. Giant acrochordons are flesh colored, benign, and do not have these malignant features.
Squamous cell carcinoma often presents as an erythematous scaly patch or red plaque on sun-exposed areas of the skin.1 Histopathology of squamous cell carcinoma shows atypical keratinocytes with an invasive growth pattern; giant acrochordon does not show keratinocytic atypia or invasive epidermal growth.
Giant condylomata acuminata (Buschke-Löwenstein tumor) is a locally destructive verrucous plaque that typically appears on the penis but can occur elsewhere in the anogenital region.7 Histopathologic features include epidermal hyperplasia, papillomatosis, and koilocytes. In contrast, giant acrochordons typically are located on the buttocks and do not present with these epidermal changes.
Based on the clinical and histologic findings, our patient was diagnosed with a giant acrochordon, a rare variant of the common skin lesion. Excisional removal was critical for both diagnostic and treatment purposes. By considering the clinical presentation and histopathologic features of other conditions in the differential, giant acrochordons can be distinguished from other similar entities. Diagnosis and prompt surgical removal are important for management of these neoplasms and prevention of misdiagnosis.
The Diagnosis: Giant Acrochordon
Based on the clinical and histologic findings, our patient was diagnosed with a giant acrochordon. Acrochordons (also known as fibroepithelial polyps or skin tags) are among the most commonly identified skin lesions and are believed to affect up to 46% of the general population.1,2 These benign growths typically appear after middle age in men and women alike and are believed to be of ectodermal and mesenchymal origin.3 The most common locations include the axillae, neck, and inguinal folds. They generally are small, measuring only a few millimeters, and frequently present as multiple lesions that are called giant acrochordons when their size exceeds 5 cm in length.2 Acrochordons are benign lesions with only rare reports of the presence of basal or squamous cell carcinoma within the lesion on pathology.4 In addition to being cosmetically unsightly, patients with acrochordons often report pruritus. These lesions are easily removed in an outpatient setting via snip excision, cryosurgery, or electrodesiccation. Once removed, recurrence is unlikely. Despite the prevalence of fibroepithelial polyps worldwide, reports of giant acrochordons are limited. The histopathology of giant acrochordons is similar to smaller acrochordons, with features including epidermal acanthosis and a central core of fibrovascular tissue without adnexal structures (Figure).4
The differential diagnosis of giant acrochordon includes neurofibroma, nodular melanoma, squamous cell carcinoma, and giant condylomata acuminata (Buschke-Löwenstein tumor).1 It is important to consider the clinical presentation and histopathologic findings to differentiate giant acrochordons from these other entities.
Neurofibromas typically present as multiple flesh-colored to brown nodules that invaginate into the skin when minimal external pressure is applied.5 Histopathology demonstrates a discrete, nonencapsulated, dermal collection of small nerve fibers and loosely arranged spindle cells. In contrast, giant acrochordons typically present as large, fleshcolored, pedunculated, verrucous tumors with a central stalk. Histopathology reveals epidermal acanthosis and a central core of fibrovascular tissue without adnexal structures.
Nodular melanomas usually are blue to black and grow rapidly over the course of several months.6 They have signs of hemorrhagic crust, and histopathology reveals atypical melanocytes, frequent mitoses, pleomorphic tumor cells, and irregular clumping of chromatin within the nuclei. Giant acrochordons are flesh colored, benign, and do not have these malignant features.
Squamous cell carcinoma often presents as an erythematous scaly patch or red plaque on sun-exposed areas of the skin.1 Histopathology of squamous cell carcinoma shows atypical keratinocytes with an invasive growth pattern; giant acrochordon does not show keratinocytic atypia or invasive epidermal growth.
Giant condylomata acuminata (Buschke-Löwenstein tumor) is a locally destructive verrucous plaque that typically appears on the penis but can occur elsewhere in the anogenital region.7 Histopathologic features include epidermal hyperplasia, papillomatosis, and koilocytes. In contrast, giant acrochordons typically are located on the buttocks and do not present with these epidermal changes.
Based on the clinical and histologic findings, our patient was diagnosed with a giant acrochordon, a rare variant of the common skin lesion. Excisional removal was critical for both diagnostic and treatment purposes. By considering the clinical presentation and histopathologic features of other conditions in the differential, giant acrochordons can be distinguished from other similar entities. Diagnosis and prompt surgical removal are important for management of these neoplasms and prevention of misdiagnosis.
- Alkhalili E, Prapasiri S, Russell J. Giant acrochordon of the axilla. BMJ Case Rep. 2015:bcr2015210623. doi:10.1136/bcr-2015-210623
- Banik R, Lubach D. Skin tags: localization and frequencies according to sex and age. Dermatologica. 1987;174:180-183. doi:10.1159/000249169
- Can B, Yildrim Ozluk A. Giant fibroepithelial polyps: why do they grow excessively? Med Bull Sisli Etfal Hastan Tip Bul. 2020;54:257-260. doi:10.14744/SEMB.2018.33603
- Ghosh SK, Bandyopadhyay D, Chatterjee G, et al. Giant skin tags on unusual locations. J Eur Acad Dermatol Venereol. 2009;23:233. doi:10.1111/j.1468-3083.2008.02816.x
- Messersmith L, Krauland K. Neurofibroma. StatPearls [Internet]. StatPearls Publishing; 2023.
- Saaiq M, Ashraf B, Siddiqui S. Nodular melanoma. Iran J Med Sci. 2016;41:164-165.
- Spinu D, Ra˘dulescu A, Bratu O, et al. Giant condyloma acuminatum. Buschke-Lowenstein disease: a literature review. Chirurgia (Bucur). 2014;109:445-450.
- Alkhalili E, Prapasiri S, Russell J. Giant acrochordon of the axilla. BMJ Case Rep. 2015:bcr2015210623. doi:10.1136/bcr-2015-210623
- Banik R, Lubach D. Skin tags: localization and frequencies according to sex and age. Dermatologica. 1987;174:180-183. doi:10.1159/000249169
- Can B, Yildrim Ozluk A. Giant fibroepithelial polyps: why do they grow excessively? Med Bull Sisli Etfal Hastan Tip Bul. 2020;54:257-260. doi:10.14744/SEMB.2018.33603
- Ghosh SK, Bandyopadhyay D, Chatterjee G, et al. Giant skin tags on unusual locations. J Eur Acad Dermatol Venereol. 2009;23:233. doi:10.1111/j.1468-3083.2008.02816.x
- Messersmith L, Krauland K. Neurofibroma. StatPearls [Internet]. StatPearls Publishing; 2023.
- Saaiq M, Ashraf B, Siddiqui S. Nodular melanoma. Iran J Med Sci. 2016;41:164-165.
- Spinu D, Ra˘dulescu A, Bratu O, et al. Giant condyloma acuminatum. Buschke-Lowenstein disease: a literature review. Chirurgia (Bucur). 2014;109:445-450.
A 40-year-old man presented to our dermatology clinic with a growth on the left buttock of more than 22 years’ duration that progressively increased in size. He was otherwise in good health and reported no ongoing medical problems. Physical examination revealed a 19×12-cm, flesh-colored, pedunculated, verrucous tumor with a central stalk. The patient underwent an excisional removal, and the specimen was sent for histopathologic evaluation.


Hyperpigmented Flexural Plaques, Hypohidrosis, and Hypotrichosis
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
. B, Grocott-Gomori methenamine-silver staining showed numerous fungal elements")
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
A 61-year-old woman with a history of hypohidrosis and deafness presented with a pruritic rash on the neck and antecubital fossae of several years’ duration. Prior treatment with topical corticosteroids failed to resolve the rash. Physical examination revealed thick, velvety, hyperpigmented plaques on the inframammary folds, axillae, groin, posterior neck, and antecubital fossae with lichenification of the latter 2 areas. Many pedunculated papules were seen on the face, chest, shoulders, and trunk, as well as diffuse hair thinning, particularly of the frontal and vertex scalp. Eyebrows, eyelashes, and axillary hair were absent. Two 5-mm punch biopsies of the antecubital fossa and inframammary fold were obtained for histopathologic analysis.


Aberrant Expression of CD56 in Metastatic Malignant Melanoma
To the Editor:
Many types of neoplasms can show aberrant immunoreactivity or unexpected expression of markers.1 Malignant melanoma is a tumor that can show not only aberrant immunohistochemical staining patterns but also notable histologic diversity,1,2 which often makes the diagnosis of melanoma challenging and ultimately can lead to diagnostic uncertainty.2
The incidence of malignant melanoma continues to grow.3 Maintaining a high degree of suspicion for this disease, recognizing its heterogeneity and divergent differentiation, and knowing potential aberrant immunohistochemical staining patterns are imperative for accurate diagnosis.
A 36-year-old man presented to a primary care physician with right-sided chest pain, upper and lower back aches, bilateral hip pain, neck pain, headache, night sweats, chills, and nausea. After infectious causes were ruled out, he was placed on a steroid taper without improvement. He presented to the emergency department a few days later with muscle spasms and was found to also have diffuse abdominal tenderness and guarding. The patient’s medical history was noncontributory; he was a lifelong nonsmoker. Laboratory studies revealed elevated levels of alanine aminotransferase and C-reactive protein. Computed tomography of the chest and abdomen revealed innumerable liver and lung lesions that were suspicious for metastatic malignancy. A liver biopsy revealed nests and sheets of metastatic tumor with pleomorphic nuclei, inconspicuous nucleoli, and areas of intranuclear clearing (Figures 1 and 2). Immunohistochemical staining was performed to further characterize the tumor. Neoplastic cells were positive for MART-1 (also known as Melan-A and melanoma-associated antigen recognized by T cells)(Figure 3), SOX10, S-100, HMB-45, and vimentin. Nonspecific staining with CD56 (Figure 4), a neuroendocrine marker, also was noted; however, the neoplasm was negative for synaptophysin, another neuroendocrine marker. Other markers for which staining was negative included pan-keratin, CD138 (syndecan-1), desmin, placental alkaline phosphatase (PLAP), inhibin, OCT-4, cytokeratin 7, and cytokeratin 20. This staining pattern was compatible with metastatic melanoma with aberrant CD56 expression.
.")
BRAF V600E immunohistochemical staining also was performed and showed strong and diffuse positivity within neoplastic cells. A subsequent positron emission tomography scan revealed widespread metastatic disease involving the lungs, liver, spleen, and bones. The patient did not have a history of an excised skin lesion; no primary cutaneous or mucosal lesions were identified.
.")
The patient was started on targeted therapy with trametinib, a mitogen-activated extracellular signal-related kinase kinase (MEK) inhibitor, and dabrafenib, a BRAF inhibitor. The disease continued to progress; he developed extensive leptomeningeal metastatic disease for which palliative radiation therapy was administered. The patient died 4 months after the initial diagnosis.
 immunostain (original magnification ×100).")
More than 90% of melanoma cases are of cutaneous origin; however, 4% to 8% of cases present as a metastatic lesion in the absence of an identified primary lesion,4 similar to our patient. The diagnosis of melanoma often is challenging; the tumor can show notable histologic diversity and has the potential to express aberrant immunophenotypes.1,2 The histologic diversity of melanoma includes a variety of architectural patterns (eg, nests, trabeculae, fascicular, pseudoglandular, pseudopapillary, or pseudorosette patterns), cytomorphologic features, and stromal changes. Cytomorphologic features of melanoma can be large pleomorphic cells; small cells; spindle cells; clear cells; signet-ring cells; and rhabdoid, plasmacytoid, and balloon cells.5
.")
Melanoma can mimic carcinoma, sarcoma, lymphoma, benign stromal tumors, plasmacytoma, and germ-cell tumors.5 Nuclei can binucleated, multinucleated, or lobated and may contain inclusions or grooves. Stroma may become myxoid or desmoplastic in appearance or rarely show granulomatous inflammation or osteoclastic giant cells.5 These variations render the diagnosis of melanoma challenging and ultimately can lead to diagnostic uncertainty.
Melanomas typically express MART-1, HMB-45, S-100, tyrosinase, NK1C3, vimentin, and neuron-specific enolase. However, melanoma is among the many neoplasms that sometimes exhibit aberrant immunoreactivity and differentiation toward nonmelanocytic elements.6 The most commonly expressed immunophenotypic aberration is cytokeratin, especially the low-molecular-weight keratin marker CAM5.2.5 CAM5.2 positivity also is seen more often in metastatic melanoma. Melanomas rarely express other intermediate filaments, including desmin, neurofilament protein, and glial fibrillary acidic protein; expression of smooth-muscle actin is rare.5
Only a few cases of melanoma showing expression of neuroendocrine markers have been reported. However, one study reported synaptophysin positivity in 29% (10/34) of cases of primary and metastatic melanoma, making the stain a relatively common finding.1
In contrast, expression of CD56 (also known as neural-cell adhesion molecule 1) in melanoma has been reported only rarely. CD56 is a nonspecific neuroendocrine marker that normally is expressed on neurons, glial tissue, skeletal muscle, and natural killer cells. Riddle and Bui7 reported a case of metastatic malignant melanoma with focal CD56 positivity and no expression of other neuroendocrine markers, similar to our patient. Suzuki and colleagues4 also reported a case of melanoma metastatic to bone marrow that showed CD56 expression in true nonhematologic tumor cells and negative immunoreactivity with synaptophysin and chromogranin A.
It is important to document cases of melanoma that express neuroendocrine markers to prevent an incorrect diagnosis of a neuroendocrine tumor.1 In some cases, distinguishing amelanotic melanoma from poorly differentiated squamous cell carcinoma, neuroendocrine tumor, and lymphoma can be difficult.5
The term neuroendocrine differentiation is reserved for cases of melanoma that show areas of ultrastructural change consistent with a neuroendocrine tumor.2 Neuroendocrine differentiation in melanoma is not common; its prognostic significance is unknown.8 We do not consider our case to be true neuroendocrine differentiation, as the tumor lacked the morphologic changes of a neuroendocrine tumor. Furthermore, CD56 is a nonspecific neuroendocrine marker, and the tumor was negative for synaptophysin.
Melanoma has the potential to show notable histologic diversity as well as aberrant immunohistochemical staining patterns.1,2 Our patient had metastatic melanoma with aberrant neuroendocrine expression of CD56, which could have been a potential diagnostic pitfall. Because expression of CD56 in melanoma is rare, it is imperative to recognize this potential aberrant staining pattern to ensure the accurate diagnosis of melanoma and appropriate provision of care.
1. Romano RC, Carter JM, Folpe AL. Aberrant intermediate filament and synaptophysin expression is a frequent event in malignant melanoma: an immunohistochemical study of 73 cases. Mod Pathol. 2015;28:1033-1042. doi:10.1038/modpathol.2015.62
2. Eyden B, Pandit D, Banerjee SS. Malignant melanoma with neuroendocrine differentiation: clinical, histological, immunohistochemical and ultrastructural features of three cases. Histopathology. 2005;47:402-409. doi:10.1111/j.1365-2559.2005.02240.x
3. Katerji H, Childs JM, Bratton LE, et al. Primary esophageal melanoma with aberrant CD56 expression: a potential diagnostic pitfall. Case Rep Pathol. 2017;2017:9052637. doi:10.1155/2017/9052637
4. Suzuki T, Kusumoto S, Iida S, et al. Amelanotic malignant melanoma of unknown primary origin metastasizing to the bone marrow: a case report and review of the literature. Intern Med. 2014;53:325-328. doi:10.2169/internalmedicine.53.1412
5. Banerjee SS, Harris M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology. 2000;36:387-402. doi:10.1046/j.1365-2559.2000.00894.x
6. Banerjee SS, Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology. 2008;52:119-129. doi:10.1111/j.1365-2559.2007.02823.x
7. Riddle ND, Bui MM. When melanoma is negative for S100: diagnostic pitfalls. Arch Pathol Lab Med. 2012;136:237-239. doi:10.5858/arpa.2011-0405-LE
8. Ilardi G, Caroppo D, Varricchio S, et al. Anal melanoma with neuroendocrine differentiation: report of a case. Int J Surg Pathol. 2015;23:329-332. doi:10.1177/1066896915573568
To the Editor:
Many types of neoplasms can show aberrant immunoreactivity or unexpected expression of markers.1 Malignant melanoma is a tumor that can show not only aberrant immunohistochemical staining patterns but also notable histologic diversity,1,2 which often makes the diagnosis of melanoma challenging and ultimately can lead to diagnostic uncertainty.2
The incidence of malignant melanoma continues to grow.3 Maintaining a high degree of suspicion for this disease, recognizing its heterogeneity and divergent differentiation, and knowing potential aberrant immunohistochemical staining patterns are imperative for accurate diagnosis.
A 36-year-old man presented to a primary care physician with right-sided chest pain, upper and lower back aches, bilateral hip pain, neck pain, headache, night sweats, chills, and nausea. After infectious causes were ruled out, he was placed on a steroid taper without improvement. He presented to the emergency department a few days later with muscle spasms and was found to also have diffuse abdominal tenderness and guarding. The patient’s medical history was noncontributory; he was a lifelong nonsmoker. Laboratory studies revealed elevated levels of alanine aminotransferase and C-reactive protein. Computed tomography of the chest and abdomen revealed innumerable liver and lung lesions that were suspicious for metastatic malignancy. A liver biopsy revealed nests and sheets of metastatic tumor with pleomorphic nuclei, inconspicuous nucleoli, and areas of intranuclear clearing (Figures 1 and 2). Immunohistochemical staining was performed to further characterize the tumor. Neoplastic cells were positive for MART-1 (also known as Melan-A and melanoma-associated antigen recognized by T cells)(Figure 3), SOX10, S-100, HMB-45, and vimentin. Nonspecific staining with CD56 (Figure 4), a neuroendocrine marker, also was noted; however, the neoplasm was negative for synaptophysin, another neuroendocrine marker. Other markers for which staining was negative included pan-keratin, CD138 (syndecan-1), desmin, placental alkaline phosphatase (PLAP), inhibin, OCT-4, cytokeratin 7, and cytokeratin 20. This staining pattern was compatible with metastatic melanoma with aberrant CD56 expression.
BRAF V600E immunohistochemical staining also was performed and showed strong and diffuse positivity within neoplastic cells. A subsequent positron emission tomography scan revealed widespread metastatic disease involving the lungs, liver, spleen, and bones. The patient did not have a history of an excised skin lesion; no primary cutaneous or mucosal lesions were identified.
The patient was started on targeted therapy with trametinib, a mitogen-activated extracellular signal-related kinase kinase (MEK) inhibitor, and dabrafenib, a BRAF inhibitor. The disease continued to progress; he developed extensive leptomeningeal metastatic disease for which palliative radiation therapy was administered. The patient died 4 months after the initial diagnosis.
More than 90% of melanoma cases are of cutaneous origin; however, 4% to 8% of cases present as a metastatic lesion in the absence of an identified primary lesion,4 similar to our patient. The diagnosis of melanoma often is challenging; the tumor can show notable histologic diversity and has the potential to express aberrant immunophenotypes.1,2 The histologic diversity of melanoma includes a variety of architectural patterns (eg, nests, trabeculae, fascicular, pseudoglandular, pseudopapillary, or pseudorosette patterns), cytomorphologic features, and stromal changes. Cytomorphologic features of melanoma can be large pleomorphic cells; small cells; spindle cells; clear cells; signet-ring cells; and rhabdoid, plasmacytoid, and balloon cells.5
Melanoma can mimic carcinoma, sarcoma, lymphoma, benign stromal tumors, plasmacytoma, and germ-cell tumors.5 Nuclei can binucleated, multinucleated, or lobated and may contain inclusions or grooves. Stroma may become myxoid or desmoplastic in appearance or rarely show granulomatous inflammation or osteoclastic giant cells.5 These variations render the diagnosis of melanoma challenging and ultimately can lead to diagnostic uncertainty.
Melanomas typically express MART-1, HMB-45, S-100, tyrosinase, NK1C3, vimentin, and neuron-specific enolase. However, melanoma is among the many neoplasms that sometimes exhibit aberrant immunoreactivity and differentiation toward nonmelanocytic elements.6 The most commonly expressed immunophenotypic aberration is cytokeratin, especially the low-molecular-weight keratin marker CAM5.2.5 CAM5.2 positivity also is seen more often in metastatic melanoma. Melanomas rarely express other intermediate filaments, including desmin, neurofilament protein, and glial fibrillary acidic protein; expression of smooth-muscle actin is rare.5
Only a few cases of melanoma showing expression of neuroendocrine markers have been reported. However, one study reported synaptophysin positivity in 29% (10/34) of cases of primary and metastatic melanoma, making the stain a relatively common finding.1
In contrast, expression of CD56 (also known as neural-cell adhesion molecule 1) in melanoma has been reported only rarely. CD56 is a nonspecific neuroendocrine marker that normally is expressed on neurons, glial tissue, skeletal muscle, and natural killer cells. Riddle and Bui7 reported a case of metastatic malignant melanoma with focal CD56 positivity and no expression of other neuroendocrine markers, similar to our patient. Suzuki and colleagues4 also reported a case of melanoma metastatic to bone marrow that showed CD56 expression in true nonhematologic tumor cells and negative immunoreactivity with synaptophysin and chromogranin A.
It is important to document cases of melanoma that express neuroendocrine markers to prevent an incorrect diagnosis of a neuroendocrine tumor.1 In some cases, distinguishing amelanotic melanoma from poorly differentiated squamous cell carcinoma, neuroendocrine tumor, and lymphoma can be difficult.5
The term neuroendocrine differentiation is reserved for cases of melanoma that show areas of ultrastructural change consistent with a neuroendocrine tumor.2 Neuroendocrine differentiation in melanoma is not common; its prognostic significance is unknown.8 We do not consider our case to be true neuroendocrine differentiation, as the tumor lacked the morphologic changes of a neuroendocrine tumor. Furthermore, CD56 is a nonspecific neuroendocrine marker, and the tumor was negative for synaptophysin.
Melanoma has the potential to show notable histologic diversity as well as aberrant immunohistochemical staining patterns.1,2 Our patient had metastatic melanoma with aberrant neuroendocrine expression of CD56, which could have been a potential diagnostic pitfall. Because expression of CD56 in melanoma is rare, it is imperative to recognize this potential aberrant staining pattern to ensure the accurate diagnosis of melanoma and appropriate provision of care.
To the Editor:
Many types of neoplasms can show aberrant immunoreactivity or unexpected expression of markers.1 Malignant melanoma is a tumor that can show not only aberrant immunohistochemical staining patterns but also notable histologic diversity,1,2 which often makes the diagnosis of melanoma challenging and ultimately can lead to diagnostic uncertainty.2
The incidence of malignant melanoma continues to grow.3 Maintaining a high degree of suspicion for this disease, recognizing its heterogeneity and divergent differentiation, and knowing potential aberrant immunohistochemical staining patterns are imperative for accurate diagnosis.
A 36-year-old man presented to a primary care physician with right-sided chest pain, upper and lower back aches, bilateral hip pain, neck pain, headache, night sweats, chills, and nausea. After infectious causes were ruled out, he was placed on a steroid taper without improvement. He presented to the emergency department a few days later with muscle spasms and was found to also have diffuse abdominal tenderness and guarding. The patient’s medical history was noncontributory; he was a lifelong nonsmoker. Laboratory studies revealed elevated levels of alanine aminotransferase and C-reactive protein. Computed tomography of the chest and abdomen revealed innumerable liver and lung lesions that were suspicious for metastatic malignancy. A liver biopsy revealed nests and sheets of metastatic tumor with pleomorphic nuclei, inconspicuous nucleoli, and areas of intranuclear clearing (Figures 1 and 2). Immunohistochemical staining was performed to further characterize the tumor. Neoplastic cells were positive for MART-1 (also known as Melan-A and melanoma-associated antigen recognized by T cells)(Figure 3), SOX10, S-100, HMB-45, and vimentin. Nonspecific staining with CD56 (Figure 4), a neuroendocrine marker, also was noted; however, the neoplasm was negative for synaptophysin, another neuroendocrine marker. Other markers for which staining was negative included pan-keratin, CD138 (syndecan-1), desmin, placental alkaline phosphatase (PLAP), inhibin, OCT-4, cytokeratin 7, and cytokeratin 20. This staining pattern was compatible with metastatic melanoma with aberrant CD56 expression.
BRAF V600E immunohistochemical staining also was performed and showed strong and diffuse positivity within neoplastic cells. A subsequent positron emission tomography scan revealed widespread metastatic disease involving the lungs, liver, spleen, and bones. The patient did not have a history of an excised skin lesion; no primary cutaneous or mucosal lesions were identified.
The patient was started on targeted therapy with trametinib, a mitogen-activated extracellular signal-related kinase kinase (MEK) inhibitor, and dabrafenib, a BRAF inhibitor. The disease continued to progress; he developed extensive leptomeningeal metastatic disease for which palliative radiation therapy was administered. The patient died 4 months after the initial diagnosis.
More than 90% of melanoma cases are of cutaneous origin; however, 4% to 8% of cases present as a metastatic lesion in the absence of an identified primary lesion,4 similar to our patient. The diagnosis of melanoma often is challenging; the tumor can show notable histologic diversity and has the potential to express aberrant immunophenotypes.1,2 The histologic diversity of melanoma includes a variety of architectural patterns (eg, nests, trabeculae, fascicular, pseudoglandular, pseudopapillary, or pseudorosette patterns), cytomorphologic features, and stromal changes. Cytomorphologic features of melanoma can be large pleomorphic cells; small cells; spindle cells; clear cells; signet-ring cells; and rhabdoid, plasmacytoid, and balloon cells.5
Melanoma can mimic carcinoma, sarcoma, lymphoma, benign stromal tumors, plasmacytoma, and germ-cell tumors.5 Nuclei can binucleated, multinucleated, or lobated and may contain inclusions or grooves. Stroma may become myxoid or desmoplastic in appearance or rarely show granulomatous inflammation or osteoclastic giant cells.5 These variations render the diagnosis of melanoma challenging and ultimately can lead to diagnostic uncertainty.
Melanomas typically express MART-1, HMB-45, S-100, tyrosinase, NK1C3, vimentin, and neuron-specific enolase. However, melanoma is among the many neoplasms that sometimes exhibit aberrant immunoreactivity and differentiation toward nonmelanocytic elements.6 The most commonly expressed immunophenotypic aberration is cytokeratin, especially the low-molecular-weight keratin marker CAM5.2.5 CAM5.2 positivity also is seen more often in metastatic melanoma. Melanomas rarely express other intermediate filaments, including desmin, neurofilament protein, and glial fibrillary acidic protein; expression of smooth-muscle actin is rare.5
Only a few cases of melanoma showing expression of neuroendocrine markers have been reported. However, one study reported synaptophysin positivity in 29% (10/34) of cases of primary and metastatic melanoma, making the stain a relatively common finding.1
In contrast, expression of CD56 (also known as neural-cell adhesion molecule 1) in melanoma has been reported only rarely. CD56 is a nonspecific neuroendocrine marker that normally is expressed on neurons, glial tissue, skeletal muscle, and natural killer cells. Riddle and Bui7 reported a case of metastatic malignant melanoma with focal CD56 positivity and no expression of other neuroendocrine markers, similar to our patient. Suzuki and colleagues4 also reported a case of melanoma metastatic to bone marrow that showed CD56 expression in true nonhematologic tumor cells and negative immunoreactivity with synaptophysin and chromogranin A.
It is important to document cases of melanoma that express neuroendocrine markers to prevent an incorrect diagnosis of a neuroendocrine tumor.1 In some cases, distinguishing amelanotic melanoma from poorly differentiated squamous cell carcinoma, neuroendocrine tumor, and lymphoma can be difficult.5
The term neuroendocrine differentiation is reserved for cases of melanoma that show areas of ultrastructural change consistent with a neuroendocrine tumor.2 Neuroendocrine differentiation in melanoma is not common; its prognostic significance is unknown.8 We do not consider our case to be true neuroendocrine differentiation, as the tumor lacked the morphologic changes of a neuroendocrine tumor. Furthermore, CD56 is a nonspecific neuroendocrine marker, and the tumor was negative for synaptophysin.
Melanoma has the potential to show notable histologic diversity as well as aberrant immunohistochemical staining patterns.1,2 Our patient had metastatic melanoma with aberrant neuroendocrine expression of CD56, which could have been a potential diagnostic pitfall. Because expression of CD56 in melanoma is rare, it is imperative to recognize this potential aberrant staining pattern to ensure the accurate diagnosis of melanoma and appropriate provision of care.
1. Romano RC, Carter JM, Folpe AL. Aberrant intermediate filament and synaptophysin expression is a frequent event in malignant melanoma: an immunohistochemical study of 73 cases. Mod Pathol. 2015;28:1033-1042. doi:10.1038/modpathol.2015.62
2. Eyden B, Pandit D, Banerjee SS. Malignant melanoma with neuroendocrine differentiation: clinical, histological, immunohistochemical and ultrastructural features of three cases. Histopathology. 2005;47:402-409. doi:10.1111/j.1365-2559.2005.02240.x
3. Katerji H, Childs JM, Bratton LE, et al. Primary esophageal melanoma with aberrant CD56 expression: a potential diagnostic pitfall. Case Rep Pathol. 2017;2017:9052637. doi:10.1155/2017/9052637
4. Suzuki T, Kusumoto S, Iida S, et al. Amelanotic malignant melanoma of unknown primary origin metastasizing to the bone marrow: a case report and review of the literature. Intern Med. 2014;53:325-328. doi:10.2169/internalmedicine.53.1412
5. Banerjee SS, Harris M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology. 2000;36:387-402. doi:10.1046/j.1365-2559.2000.00894.x
6. Banerjee SS, Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology. 2008;52:119-129. doi:10.1111/j.1365-2559.2007.02823.x
7. Riddle ND, Bui MM. When melanoma is negative for S100: diagnostic pitfalls. Arch Pathol Lab Med. 2012;136:237-239. doi:10.5858/arpa.2011-0405-LE
8. Ilardi G, Caroppo D, Varricchio S, et al. Anal melanoma with neuroendocrine differentiation: report of a case. Int J Surg Pathol. 2015;23:329-332. doi:10.1177/1066896915573568
1. Romano RC, Carter JM, Folpe AL. Aberrant intermediate filament and synaptophysin expression is a frequent event in malignant melanoma: an immunohistochemical study of 73 cases. Mod Pathol. 2015;28:1033-1042. doi:10.1038/modpathol.2015.62
2. Eyden B, Pandit D, Banerjee SS. Malignant melanoma with neuroendocrine differentiation: clinical, histological, immunohistochemical and ultrastructural features of three cases. Histopathology. 2005;47:402-409. doi:10.1111/j.1365-2559.2005.02240.x
3. Katerji H, Childs JM, Bratton LE, et al. Primary esophageal melanoma with aberrant CD56 expression: a potential diagnostic pitfall. Case Rep Pathol. 2017;2017:9052637. doi:10.1155/2017/9052637
4. Suzuki T, Kusumoto S, Iida S, et al. Amelanotic malignant melanoma of unknown primary origin metastasizing to the bone marrow: a case report and review of the literature. Intern Med. 2014;53:325-328. doi:10.2169/internalmedicine.53.1412
5. Banerjee SS, Harris M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology. 2000;36:387-402. doi:10.1046/j.1365-2559.2000.00894.x
6. Banerjee SS, Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology. 2008;52:119-129. doi:10.1111/j.1365-2559.2007.02823.x
7. Riddle ND, Bui MM. When melanoma is negative for S100: diagnostic pitfalls. Arch Pathol Lab Med. 2012;136:237-239. doi:10.5858/arpa.2011-0405-LE
8. Ilardi G, Caroppo D, Varricchio S, et al. Anal melanoma with neuroendocrine differentiation: report of a case. Int J Surg Pathol. 2015;23:329-332. doi:10.1177/1066896915573568
Practice Points
- The diagnosis of melanoma often is challenging as tumors can show notable histologic diversity and have the potential to express aberrant immunophenotypes including CD56 expression.
- Because expression of CD56 in melanoma is rare, it is important to be aware of this potential aberrant staining pattern.
- Recognizing this heterogeneity and divergent differentiation as well as knowing potential aberrant immunohistochemical staining patterns are imperative for accurate and timely diagnosis.
.")
Specialty-trained pathologists more likely to make higher-grade diagnoses for melanocytic lesions
, results from an exploratory study showed.
The findings “could in part play a role in the rising incidence of early-stage melanoma with low risk of progression or patient morbidity, thereby contributing to increasing rates of overdiagnosis,” researchers led by co–senior authors Joann G. Elmore, MD, MPH, of the University of California, Los Angeles, and Raymond L. Barnhill, MD, MBA, of the Institut Curie, Paris, wrote in their study, published online in JAMA Dermatology.
To investigate the characteristics associated with rendering higher-grade diagnoses, including invasive melanoma, the researchers drew from two national data sets: the Melanoma Pathology (M-Path) study, conducted from July 2013 to May 2016, and the Reducing Errors in Melanocytic Interpretations (REMI) study, conducted from August 2018 to March 2021. In both studies, pathologists who interpreted melanocytic lesions in their clinical practices interpreted study cases in glass slide format. For the current study, researchers used logistic regression to examine the association of pathologist characteristics with diagnosis of a study case as higher grade (including severely dysplastic and melanoma in situ) vs. lower grade (including mild to moderately dysplastic nevi) and diagnosis of invasive melanoma vs. any less severe diagnosis.
A total of 338 pathologists were included in the analysis. Of these, 113 were general pathologists and 225 were dermatopathologists (those who were board certified and/or fellowship trained in dermatopathology).
The researchers found that, compared with general pathologists, dermatopathologists were 2.63 times more likely to render higher-grade diagnoses and 1.95 times more likely to diagnose invasive melanoma (P < .001 for both associations). Diagnoses of stage pT1a melanomas with no mitotic activity completely accounted for the difference between dermatopathologists and general pathologists in diagnosing invasive melanoma.
For the analysis limited to the 225 dermatopathologists, those with a higher practice caseload of melanocytic lesions were more likely to assign higher-grade diagnoses (odds ratio for trend, 1.27; P = .02), while those affiliated with an academic center had lower odds of diagnosing invasive melanoma (OR, 0.61; P = .049).
The researchers acknowledged limitations of their analysis, including the lack of data on patient outcomes, “so we could not make conclusions about the clinical outcome of any particular diagnosis by a study participant,” they wrote. “While our analyses revealed pathologist characteristics associated with assigning more vs. less severe diagnoses of melanocytic lesions, we could not conclude that any particular diagnosis by a study participant was overcalling or undercalling. However, the epidemiologic evidence that melanoma is overdiagnosed suggests that overcalling by some pathologists may be contributing to increasing rates of low-risk melanoma diagnoses.”
In an accompanying editorial, authors Klaus J. Busam, MD, of the department of pathology and laboratory medicine at Memorial Sloan Kettering Cancer Center, New York, Pedram Gerami, MD, of the department of dermatology at Northwestern University, Chicago, and Richard A. Scolyer, MD, of the Melanoma Institute, Wollstonecraft, Australia, wrote that the study findings “raise the question of whether subspecialization in dermatopathology may be a factor contributing to the epidemiologic phenomenon of overdiagnosis – that is, the discordance in the rise of melanoma incidence and relatively constant annual mortality rates over many decades. The findings also invite a discussion about strategies to minimize harm from overdiagnosis for both patients and the health care system.”
To minimize misdiagnoses, they continued, efforts to facilitate diagnostic accuracy should be encouraged. “Excisional (rather than partial) biopsies and provision of relevant clinical information would facilitate rendering of the correct histopathologic diagnosis,” they wrote. “When the diagnosis is uncertain, this is best acknowledged. If felt necessary, a reexcision of a lesion with an uncertain diagnosis can be recommended without upgrading the diagnosis.”
In addition, “improvements in prognosis are needed beyond American Joint Committee on Cancer staging,” they noted. “This will likely require a multimodal approach with novel methods, including artificial intelligence and biomarkers that help distinguish low-risk melanomas, for which a conservative approach may be appropriate, from those that require surgical intervention.”
The study was supported by the National Center for Advancing Translational Sciences and by the National Institutes of Health. One author disclosed receiving grants from the National Cancer Institute during the conduct of the study, and another disclosed serving as editor in chief of Primary Care topics at UpToDate; other authors had no disclosures. Dr. Busam reported receiving nonfinancial support from the American Society of Dermatopathology. Dr. Gerami reported receiving consulting fees from Castle Biosciences. Dr. Scolyer reported receiving an investigator grant from the National Health and Medical Research Council of Australia during the conduct of the study and personal fees from several pharmaceutical companies outside the submitted work.
, results from an exploratory study showed.
The findings “could in part play a role in the rising incidence of early-stage melanoma with low risk of progression or patient morbidity, thereby contributing to increasing rates of overdiagnosis,” researchers led by co–senior authors Joann G. Elmore, MD, MPH, of the University of California, Los Angeles, and Raymond L. Barnhill, MD, MBA, of the Institut Curie, Paris, wrote in their study, published online in JAMA Dermatology.
To investigate the characteristics associated with rendering higher-grade diagnoses, including invasive melanoma, the researchers drew from two national data sets: the Melanoma Pathology (M-Path) study, conducted from July 2013 to May 2016, and the Reducing Errors in Melanocytic Interpretations (REMI) study, conducted from August 2018 to March 2021. In both studies, pathologists who interpreted melanocytic lesions in their clinical practices interpreted study cases in glass slide format. For the current study, researchers used logistic regression to examine the association of pathologist characteristics with diagnosis of a study case as higher grade (including severely dysplastic and melanoma in situ) vs. lower grade (including mild to moderately dysplastic nevi) and diagnosis of invasive melanoma vs. any less severe diagnosis.
A total of 338 pathologists were included in the analysis. Of these, 113 were general pathologists and 225 were dermatopathologists (those who were board certified and/or fellowship trained in dermatopathology).
The researchers found that, compared with general pathologists, dermatopathologists were 2.63 times more likely to render higher-grade diagnoses and 1.95 times more likely to diagnose invasive melanoma (P < .001 for both associations). Diagnoses of stage pT1a melanomas with no mitotic activity completely accounted for the difference between dermatopathologists and general pathologists in diagnosing invasive melanoma.
For the analysis limited to the 225 dermatopathologists, those with a higher practice caseload of melanocytic lesions were more likely to assign higher-grade diagnoses (odds ratio for trend, 1.27; P = .02), while those affiliated with an academic center had lower odds of diagnosing invasive melanoma (OR, 0.61; P = .049).
The researchers acknowledged limitations of their analysis, including the lack of data on patient outcomes, “so we could not make conclusions about the clinical outcome of any particular diagnosis by a study participant,” they wrote. “While our analyses revealed pathologist characteristics associated with assigning more vs. less severe diagnoses of melanocytic lesions, we could not conclude that any particular diagnosis by a study participant was overcalling or undercalling. However, the epidemiologic evidence that melanoma is overdiagnosed suggests that overcalling by some pathologists may be contributing to increasing rates of low-risk melanoma diagnoses.”
In an accompanying editorial, authors Klaus J. Busam, MD, of the department of pathology and laboratory medicine at Memorial Sloan Kettering Cancer Center, New York, Pedram Gerami, MD, of the department of dermatology at Northwestern University, Chicago, and Richard A. Scolyer, MD, of the Melanoma Institute, Wollstonecraft, Australia, wrote that the study findings “raise the question of whether subspecialization in dermatopathology may be a factor contributing to the epidemiologic phenomenon of overdiagnosis – that is, the discordance in the rise of melanoma incidence and relatively constant annual mortality rates over many decades. The findings also invite a discussion about strategies to minimize harm from overdiagnosis for both patients and the health care system.”
To minimize misdiagnoses, they continued, efforts to facilitate diagnostic accuracy should be encouraged. “Excisional (rather than partial) biopsies and provision of relevant clinical information would facilitate rendering of the correct histopathologic diagnosis,” they wrote. “When the diagnosis is uncertain, this is best acknowledged. If felt necessary, a reexcision of a lesion with an uncertain diagnosis can be recommended without upgrading the diagnosis.”
In addition, “improvements in prognosis are needed beyond American Joint Committee on Cancer staging,” they noted. “This will likely require a multimodal approach with novel methods, including artificial intelligence and biomarkers that help distinguish low-risk melanomas, for which a conservative approach may be appropriate, from those that require surgical intervention.”
The study was supported by the National Center for Advancing Translational Sciences and by the National Institutes of Health. One author disclosed receiving grants from the National Cancer Institute during the conduct of the study, and another disclosed serving as editor in chief of Primary Care topics at UpToDate; other authors had no disclosures. Dr. Busam reported receiving nonfinancial support from the American Society of Dermatopathology. Dr. Gerami reported receiving consulting fees from Castle Biosciences. Dr. Scolyer reported receiving an investigator grant from the National Health and Medical Research Council of Australia during the conduct of the study and personal fees from several pharmaceutical companies outside the submitted work.
, results from an exploratory study showed.
The findings “could in part play a role in the rising incidence of early-stage melanoma with low risk of progression or patient morbidity, thereby contributing to increasing rates of overdiagnosis,” researchers led by co–senior authors Joann G. Elmore, MD, MPH, of the University of California, Los Angeles, and Raymond L. Barnhill, MD, MBA, of the Institut Curie, Paris, wrote in their study, published online in JAMA Dermatology.
To investigate the characteristics associated with rendering higher-grade diagnoses, including invasive melanoma, the researchers drew from two national data sets: the Melanoma Pathology (M-Path) study, conducted from July 2013 to May 2016, and the Reducing Errors in Melanocytic Interpretations (REMI) study, conducted from August 2018 to March 2021. In both studies, pathologists who interpreted melanocytic lesions in their clinical practices interpreted study cases in glass slide format. For the current study, researchers used logistic regression to examine the association of pathologist characteristics with diagnosis of a study case as higher grade (including severely dysplastic and melanoma in situ) vs. lower grade (including mild to moderately dysplastic nevi) and diagnosis of invasive melanoma vs. any less severe diagnosis.
A total of 338 pathologists were included in the analysis. Of these, 113 were general pathologists and 225 were dermatopathologists (those who were board certified and/or fellowship trained in dermatopathology).
The researchers found that, compared with general pathologists, dermatopathologists were 2.63 times more likely to render higher-grade diagnoses and 1.95 times more likely to diagnose invasive melanoma (P < .001 for both associations). Diagnoses of stage pT1a melanomas with no mitotic activity completely accounted for the difference between dermatopathologists and general pathologists in diagnosing invasive melanoma.
For the analysis limited to the 225 dermatopathologists, those with a higher practice caseload of melanocytic lesions were more likely to assign higher-grade diagnoses (odds ratio for trend, 1.27; P = .02), while those affiliated with an academic center had lower odds of diagnosing invasive melanoma (OR, 0.61; P = .049).
The researchers acknowledged limitations of their analysis, including the lack of data on patient outcomes, “so we could not make conclusions about the clinical outcome of any particular diagnosis by a study participant,” they wrote. “While our analyses revealed pathologist characteristics associated with assigning more vs. less severe diagnoses of melanocytic lesions, we could not conclude that any particular diagnosis by a study participant was overcalling or undercalling. However, the epidemiologic evidence that melanoma is overdiagnosed suggests that overcalling by some pathologists may be contributing to increasing rates of low-risk melanoma diagnoses.”
In an accompanying editorial, authors Klaus J. Busam, MD, of the department of pathology and laboratory medicine at Memorial Sloan Kettering Cancer Center, New York, Pedram Gerami, MD, of the department of dermatology at Northwestern University, Chicago, and Richard A. Scolyer, MD, of the Melanoma Institute, Wollstonecraft, Australia, wrote that the study findings “raise the question of whether subspecialization in dermatopathology may be a factor contributing to the epidemiologic phenomenon of overdiagnosis – that is, the discordance in the rise of melanoma incidence and relatively constant annual mortality rates over many decades. The findings also invite a discussion about strategies to minimize harm from overdiagnosis for both patients and the health care system.”
To minimize misdiagnoses, they continued, efforts to facilitate diagnostic accuracy should be encouraged. “Excisional (rather than partial) biopsies and provision of relevant clinical information would facilitate rendering of the correct histopathologic diagnosis,” they wrote. “When the diagnosis is uncertain, this is best acknowledged. If felt necessary, a reexcision of a lesion with an uncertain diagnosis can be recommended without upgrading the diagnosis.”
In addition, “improvements in prognosis are needed beyond American Joint Committee on Cancer staging,” they noted. “This will likely require a multimodal approach with novel methods, including artificial intelligence and biomarkers that help distinguish low-risk melanomas, for which a conservative approach may be appropriate, from those that require surgical intervention.”
The study was supported by the National Center for Advancing Translational Sciences and by the National Institutes of Health. One author disclosed receiving grants from the National Cancer Institute during the conduct of the study, and another disclosed serving as editor in chief of Primary Care topics at UpToDate; other authors had no disclosures. Dr. Busam reported receiving nonfinancial support from the American Society of Dermatopathology. Dr. Gerami reported receiving consulting fees from Castle Biosciences. Dr. Scolyer reported receiving an investigator grant from the National Health and Medical Research Council of Australia during the conduct of the study and personal fees from several pharmaceutical companies outside the submitted work.
FROM JAMA DERMATOLOGY
An 88-year-old Black woman presented with 3 months duration of asymptomatic, violaceous patches on the left breast
Angiosarcomas are uncommon, high-grade malignant tumors of endothelial cell origin that can arise via the lymphatics or vasculature. They typically occur spontaneously; however, there have been cases reported of benign vascular transformation. These tumors are more commonly found in elderly men on the head and neck in sun-damaged skin. . This is a late complication, typically occurring about 5-10 years after radiation. Stewart-Treves syndrome, chronic lymphedema occurring after breast cancer treatment with axillary node dissection, increases the risk of angiosarcoma. As a vascular tumor, angiosarcoma spreads hematogenously and carries a poor prognosis if not caught early. Differential diagnoses include other vascular tumors such as retiform hemangioendothelioma. In this specific patient, the differential diagnosis includes Paget’s disease, chronic radiation skin changes, and eczema.

Histopathologically, angiosarcomas exhibit abnormal, pleomorphic, malignant endothelial cells. As the tumor progresses, the cell architecture becomes more distorted and cells form layers with papillary projections into the vascular lumen. Malignant cells may stain positive for CD31, CD34, the oncogene ERG and the proto-oncogene FLI-1. Histology in this patient revealed radiation changes in the dermis, as well as few vascular channels lined by large endothelial cells with marked nuclear atypia, in the form of large nucleoli and variably coarse chromatin. The cells were positive for MYC.
Treatment of angiosarcoma involves a multidisciplinary approach. Resection with wide margins is generally the treatment of choice. However, recurrence is relatively common, which may be a result of microsatellite deposits of the tumor. Perioperative radiation is recommended, and adjuvant chemotherapy often is recommended for metastatic disease. Specifically, paclitaxel has been found to promote survival in some cases of cutaneous angiosarcoma. Metastatic disease may be treated with cytotoxic drugs such as anthracyclines and taxanes. Additionally, targeted therapy including anti-VEGF drugs and tyrosine kinase inhibitors have been tested.
The case and photo were submitted by Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Dr. Bilu Martin. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
Cohen-Hallaleh RB et al. Clin Sarcoma Res. 2017 Aug 7:7:15.
Cozzi S et al. Rep Pract Oncol Radiother. 2021 Sep 30;26(5):827-32.
Spiker AM, Mangla A, Ramsey ML. Angiosarcoma. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing; 2023 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK441983/
Angiosarcomas are uncommon, high-grade malignant tumors of endothelial cell origin that can arise via the lymphatics or vasculature. They typically occur spontaneously; however, there have been cases reported of benign vascular transformation. These tumors are more commonly found in elderly men on the head and neck in sun-damaged skin. . This is a late complication, typically occurring about 5-10 years after radiation. Stewart-Treves syndrome, chronic lymphedema occurring after breast cancer treatment with axillary node dissection, increases the risk of angiosarcoma. As a vascular tumor, angiosarcoma spreads hematogenously and carries a poor prognosis if not caught early. Differential diagnoses include other vascular tumors such as retiform hemangioendothelioma. In this specific patient, the differential diagnosis includes Paget’s disease, chronic radiation skin changes, and eczema.
Histopathologically, angiosarcomas exhibit abnormal, pleomorphic, malignant endothelial cells. As the tumor progresses, the cell architecture becomes more distorted and cells form layers with papillary projections into the vascular lumen. Malignant cells may stain positive for CD31, CD34, the oncogene ERG and the proto-oncogene FLI-1. Histology in this patient revealed radiation changes in the dermis, as well as few vascular channels lined by large endothelial cells with marked nuclear atypia, in the form of large nucleoli and variably coarse chromatin. The cells were positive for MYC.
Treatment of angiosarcoma involves a multidisciplinary approach. Resection with wide margins is generally the treatment of choice. However, recurrence is relatively common, which may be a result of microsatellite deposits of the tumor. Perioperative radiation is recommended, and adjuvant chemotherapy often is recommended for metastatic disease. Specifically, paclitaxel has been found to promote survival in some cases of cutaneous angiosarcoma. Metastatic disease may be treated with cytotoxic drugs such as anthracyclines and taxanes. Additionally, targeted therapy including anti-VEGF drugs and tyrosine kinase inhibitors have been tested.
The case and photo were submitted by Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Dr. Bilu Martin. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
Cohen-Hallaleh RB et al. Clin Sarcoma Res. 2017 Aug 7:7:15.
Cozzi S et al. Rep Pract Oncol Radiother. 2021 Sep 30;26(5):827-32.
Spiker AM, Mangla A, Ramsey ML. Angiosarcoma. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing; 2023 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK441983/
Angiosarcomas are uncommon, high-grade malignant tumors of endothelial cell origin that can arise via the lymphatics or vasculature. They typically occur spontaneously; however, there have been cases reported of benign vascular transformation. These tumors are more commonly found in elderly men on the head and neck in sun-damaged skin. . This is a late complication, typically occurring about 5-10 years after radiation. Stewart-Treves syndrome, chronic lymphedema occurring after breast cancer treatment with axillary node dissection, increases the risk of angiosarcoma. As a vascular tumor, angiosarcoma spreads hematogenously and carries a poor prognosis if not caught early. Differential diagnoses include other vascular tumors such as retiform hemangioendothelioma. In this specific patient, the differential diagnosis includes Paget’s disease, chronic radiation skin changes, and eczema.
Histopathologically, angiosarcomas exhibit abnormal, pleomorphic, malignant endothelial cells. As the tumor progresses, the cell architecture becomes more distorted and cells form layers with papillary projections into the vascular lumen. Malignant cells may stain positive for CD31, CD34, the oncogene ERG and the proto-oncogene FLI-1. Histology in this patient revealed radiation changes in the dermis, as well as few vascular channels lined by large endothelial cells with marked nuclear atypia, in the form of large nucleoli and variably coarse chromatin. The cells were positive for MYC.
Treatment of angiosarcoma involves a multidisciplinary approach. Resection with wide margins is generally the treatment of choice. However, recurrence is relatively common, which may be a result of microsatellite deposits of the tumor. Perioperative radiation is recommended, and adjuvant chemotherapy often is recommended for metastatic disease. Specifically, paclitaxel has been found to promote survival in some cases of cutaneous angiosarcoma. Metastatic disease may be treated with cytotoxic drugs such as anthracyclines and taxanes. Additionally, targeted therapy including anti-VEGF drugs and tyrosine kinase inhibitors have been tested.
The case and photo were submitted by Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Dr. Bilu Martin. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
Cohen-Hallaleh RB et al. Clin Sarcoma Res. 2017 Aug 7:7:15.
Cozzi S et al. Rep Pract Oncol Radiother. 2021 Sep 30;26(5):827-32.
Spiker AM, Mangla A, Ramsey ML. Angiosarcoma. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing; 2023 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK441983/
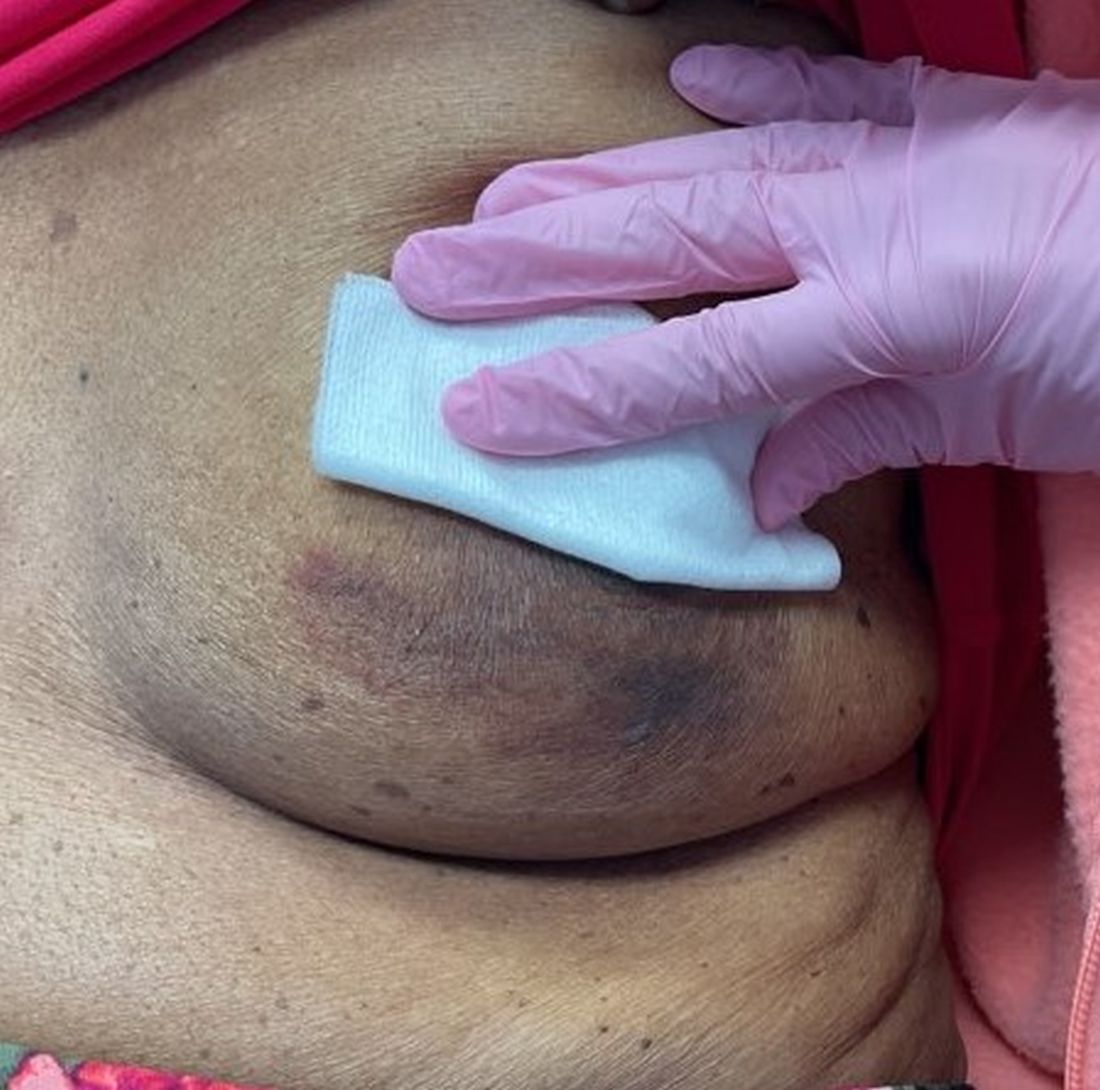
Keratotic Nodules in a Patient With End-Stage Renal Disease
The Diagnosis: Reactive Perforating Collagenosis
Reactive perforating collagenosis (RPC) is the most common type of primary perforating dermatosis and is characterized by the transepithelial elimination of collagen from the dermis. Although familial RPC usually presents in infancy or early childhood, the acquired form has a strong association with type 2 diabetes mellitus and chronic renal disease. Up to 10% of hemodialysis patients develop RPC.1 Patients with RPC develop red-brown, umbilicated, papulonodular lesions, often with a central keratotic crust and erythematous halo. The lesions are variable in shape and size (typically up to 10 mm in diameter) and commonly are located on the trunk or extensor aspects of the limbs. Pruritus is the primary concern, and the Koebner phenomenon commonly is seen.2
Although the histopathology can vary depending on the stage of the lesion, an invaginating epidermal process with prominent epidermal hyperplasia surrounding a central plug of keratin, basophilic inflammatory debris, and degenerated collagen are findings indicative of RPC. At the base of the invagination, the altered collagen perforates through the epidermis by the process of transepidermal elimination.3 Trichrome stains can highlight the collagen, while Verhoeff–van Gieson staining is negative (no elastic fiber elimination). Anecdotal reports have described a variety of successful therapies including retinoids, allopurinol, doxycycline, dupilumab, and phototherapy, with phototherapy being especially effective in patients with coexistent renal disease.4-8 Our patient was started on dupilumab 300 mg every other week and triamcinolone cream 0.1% twice daily (Monday through Friday) for itchy areas. The efficacy of the treatment was to be assessed at the next visit.
Elastosis perforans serpiginosa (EPS) is a rare skin disease that presents as small papules arranged in serpiginous or annular patterns on the neck, face, arms, or other flexural areas in early adulthood. It more commonly is seen in males and can be associated with other inherited disorders such as Down syndrome, Ehlers-Danlos syndrome, and Marfan syndrome. In rare instances, EPS has been linked to D-penicillamine.9 Elastosis perforans serpiginosa is characterized by focal dermal elastosis and transepithelial elimination of abnormal elastic fibers instead of collagen. The formation of narrow channels extending upward from the dermis in straight or corkscrew patterns commonly is seen (Figure 1). The dermis also may contain a chronic inflammatory infiltrate consisting of lymphocytes, macrophages, or multinucleated giant cells.10 Verhoeff– van Gieson stain highlights the altered elastic fibers in the papillary dermis.

Prurigo nodularis involves chronic, intensely pruritic, lichenified, excoriated nodules that often present as grouped symmetric lesions predominantly on the extensor aspects of the distal extremities and occasionally the trunk. Histologically, prurigo nodularis appears similar to lichen simplex chronicus but in a nodular form with pronounced hyperkeratosis and acanthosis, sometimes to the degree of pseudoepitheliomatous hyperplasia (Figure 2).11 Its features may resemble chronic eczema with mild spongiosis and focal parakeratosis. In the dermis, there is vascular hyperplasia surrounded by perivascular inflammatory infiltrates. Immunohistochemical staining for calcitonin gene-related peptide and substance P may show a large increase of immunoreactive nerves in the lesional skin of nodular prurigo patients compared to the lichenified skin of eczema patients.12 However, neural hyperplasia is not a diagnostic prerequisite in prurigo nodularis.13 Rarely, hyperplasic nerve trunks associated with Schwann cell proliferation may give rise to small neuromata that can be detected on electron microscopy.14 Screening for underlying systemic disease is recommended to rule out cancer, liver disease, chronic kidney disease, thyroid disorders, or HIV.

Ecthyma can affect children, adults, and especially immunocompromised patients at sites of trauma that allow entry of Streptococcus pyogenes or Staphylococcus aureus. Histologically, there is ulceration of the epidermis with a thick overlying inflammatory crust (Figure 3). The heavy infiltrate of neutrophils in the reticular dermis forms the base of the ulcer, and gram-positive cocci may be detected within the inflammatory crust. Ecthyma lesions may resemble the excoriations and shallow ulcers that are seen in a variety of other pruritic conditions.15

Pityriasis lichenoides et varioliformis acuta is a T-cell–mediated disease that is characterized by crops of lesions in varying sizes and stages including vesicular, hemorrhagic, ulcerated, and necrotic. It often results in varioliform scarring. Histologic findings can include parakeratosis, lichenoid inflammation, extravasation of red blood cells, vasculitis, and apoptotic keratinocytes (Figure 4).16

- Hong SB, Park JH, Ihm CG, et al. Acquired perforating dermatosis in patients with chronic renal failure and diabetes mellitus. J Korean Med Sci. 2004;19:283-288. doi:10.3346/jkms.2004.19.2.283
- Mullins TB, Sickinger M, Zito PM. Reactive perforating collagenosis. StatPearls [Internet]. StatPearls Publishing; 2022.
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritus! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Cullen SI. Successful treatment of reactive perforating collagenosis with tretinoin. Cutis. 1979;23:187-193.
- Tilz H, Becker JC, Legat F, et al. Allopurinol in the treatment of acquired reactive perforating collagenosis. An Bras Dermatol. 2013;88:94-97. doi:10.1590/s0365-05962013000100012
- Brinkmeier T, Schaller J, Herbst RA, et al. Successful treatment of acquired reactive perforating collagenosis with doxycycline. Acta Derm Venereol. 2002;82:393-395. doi:10.1080/000155502320624249
- Gil-Lianes J, Riquelme-McLoughlin C, Mascaró JM Jr. Reactive perforating collagenosis successfully treated with dupilumab. Australas J Dermatol. 2022;63:398-400. doi:10.1111/ajd.13874
- Gambichler T, Altmeyer P, Kreuter A. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol. 2005;52:363-364. doi:10.1016/j.jaad.2004.08.018
- Na SY, Choi M, Kim MJ, et al. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson’s disease. Ann Dermatol. 2010;22:468-471. doi:10.5021/ad.2010.22.4.468
- Lee SH, Choi Y, Kim SC. Elastosis perforans serpiginosa. Ann Dermatol. 2014;26:103-106. doi:10.5021/ad.2014.26.1.103
- Weigelt N, Metze D, Ständer S. Prurigo nodularis: systematic analysis of 58 histological criteria in 136 patients. J Cutan Pathol. 2010;37:578-586. doi:10.1111/j.1600-0560.2009.01484.x
- Abadía Molina F, Burrows NP, Jones RR, et al. Increased sensory neuropeptides in nodular prurigo: a quantitative immunohistochemical analysis. Br J Dermatol. 1992;127:344-351. doi:10.1111/j.1365-2133.1992.tb00452.x
- Lindley RP, Payne CM. Neural hyperplasia is not a diagnostic prerequisite in nodular prurigo. a controlled morphometric microscopic study of 26 biopsy specimens. J Cutan Pathol. 1989;16:14-18. doi:10.1111/j.1600-0560.1989.tb00003.x
- Feuerman EJ, Sandbank M. Prurigo nodularis. histological and electron microscopical study. Arch Dermatol. 1975;111:1472-1477. doi:10.1001/archderm.111.11.1472
- Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone; 2010. 16. Clarey DD, Lauer SR, Trowbridge RM. Clinical, dermatoscopic, and histological findings in a diagnosis of pityriasis lichenoides [published online June 20, 2020]. Cureus. 2020;12:E8725. doi:10.7759 /cureus.8725
The Diagnosis: Reactive Perforating Collagenosis
Reactive perforating collagenosis (RPC) is the most common type of primary perforating dermatosis and is characterized by the transepithelial elimination of collagen from the dermis. Although familial RPC usually presents in infancy or early childhood, the acquired form has a strong association with type 2 diabetes mellitus and chronic renal disease. Up to 10% of hemodialysis patients develop RPC.1 Patients with RPC develop red-brown, umbilicated, papulonodular lesions, often with a central keratotic crust and erythematous halo. The lesions are variable in shape and size (typically up to 10 mm in diameter) and commonly are located on the trunk or extensor aspects of the limbs. Pruritus is the primary concern, and the Koebner phenomenon commonly is seen.2
Although the histopathology can vary depending on the stage of the lesion, an invaginating epidermal process with prominent epidermal hyperplasia surrounding a central plug of keratin, basophilic inflammatory debris, and degenerated collagen are findings indicative of RPC. At the base of the invagination, the altered collagen perforates through the epidermis by the process of transepidermal elimination.3 Trichrome stains can highlight the collagen, while Verhoeff–van Gieson staining is negative (no elastic fiber elimination). Anecdotal reports have described a variety of successful therapies including retinoids, allopurinol, doxycycline, dupilumab, and phototherapy, with phototherapy being especially effective in patients with coexistent renal disease.4-8 Our patient was started on dupilumab 300 mg every other week and triamcinolone cream 0.1% twice daily (Monday through Friday) for itchy areas. The efficacy of the treatment was to be assessed at the next visit.
Elastosis perforans serpiginosa (EPS) is a rare skin disease that presents as small papules arranged in serpiginous or annular patterns on the neck, face, arms, or other flexural areas in early adulthood. It more commonly is seen in males and can be associated with other inherited disorders such as Down syndrome, Ehlers-Danlos syndrome, and Marfan syndrome. In rare instances, EPS has been linked to D-penicillamine.9 Elastosis perforans serpiginosa is characterized by focal dermal elastosis and transepithelial elimination of abnormal elastic fibers instead of collagen. The formation of narrow channels extending upward from the dermis in straight or corkscrew patterns commonly is seen (Figure 1). The dermis also may contain a chronic inflammatory infiltrate consisting of lymphocytes, macrophages, or multinucleated giant cells.10 Verhoeff– van Gieson stain highlights the altered elastic fibers in the papillary dermis.
Prurigo nodularis involves chronic, intensely pruritic, lichenified, excoriated nodules that often present as grouped symmetric lesions predominantly on the extensor aspects of the distal extremities and occasionally the trunk. Histologically, prurigo nodularis appears similar to lichen simplex chronicus but in a nodular form with pronounced hyperkeratosis and acanthosis, sometimes to the degree of pseudoepitheliomatous hyperplasia (Figure 2).11 Its features may resemble chronic eczema with mild spongiosis and focal parakeratosis. In the dermis, there is vascular hyperplasia surrounded by perivascular inflammatory infiltrates. Immunohistochemical staining for calcitonin gene-related peptide and substance P may show a large increase of immunoreactive nerves in the lesional skin of nodular prurigo patients compared to the lichenified skin of eczema patients.12 However, neural hyperplasia is not a diagnostic prerequisite in prurigo nodularis.13 Rarely, hyperplasic nerve trunks associated with Schwann cell proliferation may give rise to small neuromata that can be detected on electron microscopy.14 Screening for underlying systemic disease is recommended to rule out cancer, liver disease, chronic kidney disease, thyroid disorders, or HIV.
Ecthyma can affect children, adults, and especially immunocompromised patients at sites of trauma that allow entry of Streptococcus pyogenes or Staphylococcus aureus. Histologically, there is ulceration of the epidermis with a thick overlying inflammatory crust (Figure 3). The heavy infiltrate of neutrophils in the reticular dermis forms the base of the ulcer, and gram-positive cocci may be detected within the inflammatory crust. Ecthyma lesions may resemble the excoriations and shallow ulcers that are seen in a variety of other pruritic conditions.15
Pityriasis lichenoides et varioliformis acuta is a T-cell–mediated disease that is characterized by crops of lesions in varying sizes and stages including vesicular, hemorrhagic, ulcerated, and necrotic. It often results in varioliform scarring. Histologic findings can include parakeratosis, lichenoid inflammation, extravasation of red blood cells, vasculitis, and apoptotic keratinocytes (Figure 4).16
The Diagnosis: Reactive Perforating Collagenosis
Reactive perforating collagenosis (RPC) is the most common type of primary perforating dermatosis and is characterized by the transepithelial elimination of collagen from the dermis. Although familial RPC usually presents in infancy or early childhood, the acquired form has a strong association with type 2 diabetes mellitus and chronic renal disease. Up to 10% of hemodialysis patients develop RPC.1 Patients with RPC develop red-brown, umbilicated, papulonodular lesions, often with a central keratotic crust and erythematous halo. The lesions are variable in shape and size (typically up to 10 mm in diameter) and commonly are located on the trunk or extensor aspects of the limbs. Pruritus is the primary concern, and the Koebner phenomenon commonly is seen.2
Although the histopathology can vary depending on the stage of the lesion, an invaginating epidermal process with prominent epidermal hyperplasia surrounding a central plug of keratin, basophilic inflammatory debris, and degenerated collagen are findings indicative of RPC. At the base of the invagination, the altered collagen perforates through the epidermis by the process of transepidermal elimination.3 Trichrome stains can highlight the collagen, while Verhoeff–van Gieson staining is negative (no elastic fiber elimination). Anecdotal reports have described a variety of successful therapies including retinoids, allopurinol, doxycycline, dupilumab, and phototherapy, with phototherapy being especially effective in patients with coexistent renal disease.4-8 Our patient was started on dupilumab 300 mg every other week and triamcinolone cream 0.1% twice daily (Monday through Friday) for itchy areas. The efficacy of the treatment was to be assessed at the next visit.
Elastosis perforans serpiginosa (EPS) is a rare skin disease that presents as small papules arranged in serpiginous or annular patterns on the neck, face, arms, or other flexural areas in early adulthood. It more commonly is seen in males and can be associated with other inherited disorders such as Down syndrome, Ehlers-Danlos syndrome, and Marfan syndrome. In rare instances, EPS has been linked to D-penicillamine.9 Elastosis perforans serpiginosa is characterized by focal dermal elastosis and transepithelial elimination of abnormal elastic fibers instead of collagen. The formation of narrow channels extending upward from the dermis in straight or corkscrew patterns commonly is seen (Figure 1). The dermis also may contain a chronic inflammatory infiltrate consisting of lymphocytes, macrophages, or multinucleated giant cells.10 Verhoeff– van Gieson stain highlights the altered elastic fibers in the papillary dermis.
Prurigo nodularis involves chronic, intensely pruritic, lichenified, excoriated nodules that often present as grouped symmetric lesions predominantly on the extensor aspects of the distal extremities and occasionally the trunk. Histologically, prurigo nodularis appears similar to lichen simplex chronicus but in a nodular form with pronounced hyperkeratosis and acanthosis, sometimes to the degree of pseudoepitheliomatous hyperplasia (Figure 2).11 Its features may resemble chronic eczema with mild spongiosis and focal parakeratosis. In the dermis, there is vascular hyperplasia surrounded by perivascular inflammatory infiltrates. Immunohistochemical staining for calcitonin gene-related peptide and substance P may show a large increase of immunoreactive nerves in the lesional skin of nodular prurigo patients compared to the lichenified skin of eczema patients.12 However, neural hyperplasia is not a diagnostic prerequisite in prurigo nodularis.13 Rarely, hyperplasic nerve trunks associated with Schwann cell proliferation may give rise to small neuromata that can be detected on electron microscopy.14 Screening for underlying systemic disease is recommended to rule out cancer, liver disease, chronic kidney disease, thyroid disorders, or HIV.
Ecthyma can affect children, adults, and especially immunocompromised patients at sites of trauma that allow entry of Streptococcus pyogenes or Staphylococcus aureus. Histologically, there is ulceration of the epidermis with a thick overlying inflammatory crust (Figure 3). The heavy infiltrate of neutrophils in the reticular dermis forms the base of the ulcer, and gram-positive cocci may be detected within the inflammatory crust. Ecthyma lesions may resemble the excoriations and shallow ulcers that are seen in a variety of other pruritic conditions.15
Pityriasis lichenoides et varioliformis acuta is a T-cell–mediated disease that is characterized by crops of lesions in varying sizes and stages including vesicular, hemorrhagic, ulcerated, and necrotic. It often results in varioliform scarring. Histologic findings can include parakeratosis, lichenoid inflammation, extravasation of red blood cells, vasculitis, and apoptotic keratinocytes (Figure 4).16
- Hong SB, Park JH, Ihm CG, et al. Acquired perforating dermatosis in patients with chronic renal failure and diabetes mellitus. J Korean Med Sci. 2004;19:283-288. doi:10.3346/jkms.2004.19.2.283
- Mullins TB, Sickinger M, Zito PM. Reactive perforating collagenosis. StatPearls [Internet]. StatPearls Publishing; 2022.
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritus! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Cullen SI. Successful treatment of reactive perforating collagenosis with tretinoin. Cutis. 1979;23:187-193.
- Tilz H, Becker JC, Legat F, et al. Allopurinol in the treatment of acquired reactive perforating collagenosis. An Bras Dermatol. 2013;88:94-97. doi:10.1590/s0365-05962013000100012
- Brinkmeier T, Schaller J, Herbst RA, et al. Successful treatment of acquired reactive perforating collagenosis with doxycycline. Acta Derm Venereol. 2002;82:393-395. doi:10.1080/000155502320624249
- Gil-Lianes J, Riquelme-McLoughlin C, Mascaró JM Jr. Reactive perforating collagenosis successfully treated with dupilumab. Australas J Dermatol. 2022;63:398-400. doi:10.1111/ajd.13874
- Gambichler T, Altmeyer P, Kreuter A. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol. 2005;52:363-364. doi:10.1016/j.jaad.2004.08.018
- Na SY, Choi M, Kim MJ, et al. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson’s disease. Ann Dermatol. 2010;22:468-471. doi:10.5021/ad.2010.22.4.468
- Lee SH, Choi Y, Kim SC. Elastosis perforans serpiginosa. Ann Dermatol. 2014;26:103-106. doi:10.5021/ad.2014.26.1.103
- Weigelt N, Metze D, Ständer S. Prurigo nodularis: systematic analysis of 58 histological criteria in 136 patients. J Cutan Pathol. 2010;37:578-586. doi:10.1111/j.1600-0560.2009.01484.x
- Abadía Molina F, Burrows NP, Jones RR, et al. Increased sensory neuropeptides in nodular prurigo: a quantitative immunohistochemical analysis. Br J Dermatol. 1992;127:344-351. doi:10.1111/j.1365-2133.1992.tb00452.x
- Lindley RP, Payne CM. Neural hyperplasia is not a diagnostic prerequisite in nodular prurigo. a controlled morphometric microscopic study of 26 biopsy specimens. J Cutan Pathol. 1989;16:14-18. doi:10.1111/j.1600-0560.1989.tb00003.x
- Feuerman EJ, Sandbank M. Prurigo nodularis. histological and electron microscopical study. Arch Dermatol. 1975;111:1472-1477. doi:10.1001/archderm.111.11.1472
- Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone; 2010. 16. Clarey DD, Lauer SR, Trowbridge RM. Clinical, dermatoscopic, and histological findings in a diagnosis of pityriasis lichenoides [published online June 20, 2020]. Cureus. 2020;12:E8725. doi:10.7759 /cureus.8725
- Hong SB, Park JH, Ihm CG, et al. Acquired perforating dermatosis in patients with chronic renal failure and diabetes mellitus. J Korean Med Sci. 2004;19:283-288. doi:10.3346/jkms.2004.19.2.283
- Mullins TB, Sickinger M, Zito PM. Reactive perforating collagenosis. StatPearls [Internet]. StatPearls Publishing; 2022.
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritus! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Cullen SI. Successful treatment of reactive perforating collagenosis with tretinoin. Cutis. 1979;23:187-193.
- Tilz H, Becker JC, Legat F, et al. Allopurinol in the treatment of acquired reactive perforating collagenosis. An Bras Dermatol. 2013;88:94-97. doi:10.1590/s0365-05962013000100012
- Brinkmeier T, Schaller J, Herbst RA, et al. Successful treatment of acquired reactive perforating collagenosis with doxycycline. Acta Derm Venereol. 2002;82:393-395. doi:10.1080/000155502320624249
- Gil-Lianes J, Riquelme-McLoughlin C, Mascaró JM Jr. Reactive perforating collagenosis successfully treated with dupilumab. Australas J Dermatol. 2022;63:398-400. doi:10.1111/ajd.13874
- Gambichler T, Altmeyer P, Kreuter A. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol. 2005;52:363-364. doi:10.1016/j.jaad.2004.08.018
- Na SY, Choi M, Kim MJ, et al. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson’s disease. Ann Dermatol. 2010;22:468-471. doi:10.5021/ad.2010.22.4.468
- Lee SH, Choi Y, Kim SC. Elastosis perforans serpiginosa. Ann Dermatol. 2014;26:103-106. doi:10.5021/ad.2014.26.1.103
- Weigelt N, Metze D, Ständer S. Prurigo nodularis: systematic analysis of 58 histological criteria in 136 patients. J Cutan Pathol. 2010;37:578-586. doi:10.1111/j.1600-0560.2009.01484.x
- Abadía Molina F, Burrows NP, Jones RR, et al. Increased sensory neuropeptides in nodular prurigo: a quantitative immunohistochemical analysis. Br J Dermatol. 1992;127:344-351. doi:10.1111/j.1365-2133.1992.tb00452.x
- Lindley RP, Payne CM. Neural hyperplasia is not a diagnostic prerequisite in nodular prurigo. a controlled morphometric microscopic study of 26 biopsy specimens. J Cutan Pathol. 1989;16:14-18. doi:10.1111/j.1600-0560.1989.tb00003.x
- Feuerman EJ, Sandbank M. Prurigo nodularis. histological and electron microscopical study. Arch Dermatol. 1975;111:1472-1477. doi:10.1001/archderm.111.11.1472
- Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone; 2010. 16. Clarey DD, Lauer SR, Trowbridge RM. Clinical, dermatoscopic, and histological findings in a diagnosis of pityriasis lichenoides [published online June 20, 2020]. Cureus. 2020;12:E8725. doi:10.7759 /cureus.8725
A 42-year-old man with end-stage renal disease on hemodialysis presented with generalized body itching and nodules on the scalp and back of 1 year’s duration. Physical examination revealed diffuse, hyperpigmented, pruritic, keratotic nodules and macules on the scalp and back (top). A punch biopsy was performed (bottom).


Plaquelike Syringoma Mimicking Microcystic Adnexal Carcinoma: A Potential Histologic Pitfall
To the Editor:
Plaquelike or plaque-type syringoma is a lesser-known variant of syringoma that can appear histologically indistinguishable from the superficial portion of microcystic adnexal carcinoma (MAC). The plaquelike variant of syringoma holds a benign clinical course, and no treatment is necessary. Microcystic adnexal carcinoma is distinguished from plaquelike syringoma by an aggressive growth pattern with a high risk for local invasion and recurrence if inadequately treated. Thus, treatment with Mohs micrographic surgery (MMS) has been recommended as the mainstay for MAC. If superficial biopsy specimens reveal suspicion for MAC and patients are referred for MMS, careful consideration should be made to differentiate MAC and plaquelike syringoma early to prevent unnecessary morbidity.
.")
A 78-year-old woman was referred for MMS for a left forehead lesion that was diagnosed via shave biopsy as a desmoplastic and cystic adnexal neoplasm with suspicion for desmoplastic trichoepithelioma or MAC (Figure 1). Upon presentation for MMS, a well-healed, 1.0×0.9-cm scar at the biopsy site on the left forehead was observed (Figure 2A). One stage was obtained by standard MMS technique and sent for intraoperative processing (Figure 2B). Frozen section examination of the first stage demonstrated peripheral margin involvement with syringomatous change confined to the superficial and mid dermis (Figure 3). Before proceeding further, these findings were reviewed with an in-house dermatopathologist, and it was determined that no infiltrative tumor, perineural involvement, or other features to indicate malignancy were noted. A decision was made to refrain from obtaining any additional layers and to send excised Burow triangles for permanent section analysis. A primary linear closure was performed without complication, and the patient was discharged from the ambulatory surgery suite. Histopathologic examination of the Burow triangles later confirmed findings consistent with plaquelike syringoma with no evidence of malignancy (Figure 4).

Syringomas present as small flesh-colored papules in the periorbital areas. These benign neoplasms previously have been classified into 4 major clinical variants: localized, generalized, Down syndrome associated, and familial.1 The lesser-known plaquelike variant of syringoma was first described by Kikuchi et al2 in 1979. Aside from our report, a PubMed search of articles indexed for MEDLINE using the terms plaquelike or plaque-type syringoma yielded 16 cases in the literature.2-14 Of these, 6 were referred to or encountered in the MMS setting.8,9,11,12,14 Plaquelike syringoma can be solitary or multiple in presentation.6 It most commonly involves the head and neck but also can present on the trunk, arms, legs, and groin areas. The clinical size of plaquelike syringoma is variable, with the largest reported cases extending several centimeters in diameter.2,6 Similar to reported associations with conventional syringoma, the plaquelike subtype of syringoma has been reported in association with Down syndrome.13
.")
Histopathologically, plaquelike syringoma shares features with MAC as well as desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma. Plaquelike syringoma demonstrates broad proliferations of small tubules morphologically reminiscent of tadpoles confined within the dermis. Ducts typically are lined with 2 or 3 layers of small cuboidal cells. Microcystic adnexal carcinoma typically features asymmetric ductal structures lined with single cells extending from the dermis into the subcutis and even underlying muscle, cartilage, or bone.8 There are no reliable immunohistochemical stains to differentiate between these 2 entities; thus, the primary distinction lies in the depth of involvement. Desmoplastic trichoepithelioma is composed of narrow cords and nests of basaloid cells of follicular origin commonly admixed with small cornifying cysts appearing in the dermis.8 Colonizing Merkel cells positive for cytokeratin 20 often are present in desmoplastic trichoepithelioma and not in syringoma or MAC.15 Desmoplastic basal cell carcinoma demonstrates narrow strands of basaloid cells of follicular origin appearing in the dermis. Desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma are each fundamentally differentiated from plaquelike syringoma in that proliferations of cords and nests are not of eccrine or apocrine origin.
.")
Several cases of plaquelike syringoma have been challenging to distinguish from MAC in performing MMS.8,9,11 Underlying extension of this syringoma variant can be far-reaching, extending to several centimeters in size and involving multiple cosmetic subunits.6,11,14 Inadvertent overtreatment with multiple MMS stages can be avoided with careful recognition of the differentiating histopathologic features. Syringomatous lesions commonly are encountered in MMS and may even be present at the edge of other tumor types. Plaquelike syringoma has been reported as a coexistent entity with nodular basal cell carcinoma.12 Boos et al16 similarly reported the presence of deceptive ductal proliferations along the immediate peripheral margin of MAC, which prompted multiple re-excisions. Pursuit of permanent section analysis in these cases revealed the appearance of small syringomas, and a diagnosis of benign subclinical syringomatous proliferations was made, averting further intervention.16
Our case sheds light on the threat of commission bias in dermatologic surgery, which is the tendency for action rather than inaction.17 In this context, it is important to avoid the perspective that harm to the patient can only be prevented by active intervention. Cognitive bias has been increasingly recognized as a source of medical error, and methods to mitigate bias in medical practice have been well described.17 Microcystic adnexal carcinoma and plaquelike syringoma can be hard to differentiate especially initially, as demonstrated in our case, which particularly illustrates the importance of slowing down a surgical case at the appropriate time, considering and revisiting alternative diagnoses, implementing checklists, and seeking histopathologic collaboration with colleagues when necessary. Our attempted implementation of these principles, especially early collaboration with colleagues, led to intraoperative recognition of plaquelike syringoma within the first stage of MMS.
We seek to raise the index of suspicion for plaquelike syringoma among dermatologists and dermatologic surgeons, especially when syringomatous structures are limited to the superficial dermis. We encourage familiarity with the plaquelike syringoma entity as well as careful consideration of further investigation via scouting biopsies or permanent section analysis when other characteristic features of MAC are unclear or lacking. Adequate sampling as well as collaboration with a dermatopathologist in cases of suspected syringoma can help to reduce the susceptibility to commission bias and prevent histopathologic pitfalls and unwarranted surgical morbidity.
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Dekio S, Jidoi J. Submammary syringoma—report of a case. J Dermatol. 1988;15:351-352.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Nguyen DB, Patterson JW, Wilson BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49:337-339.
- Rongioletti F, Semino MT, Rebora A. Unilateral multiple plaque-like syringomas. Br J Dermatol. 1996;135:623-625.
- Chi HI. A case of unusual syringoma: unilateral linear distribution and plaque formation. J Dermatol. 1996;23:505-506.
- Suwatee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:E206-E207.
- Schleich C, Ferringer T, Petrick M. Plaque type syringoma mimicking a microcystic adnexal carcinoma. J Am Acad Dermatol. 2016;74(suppl 1):AB287.
- Yang Y, Srivastava D. Plaque-type syringoma coexisting with basal cell carcinoma. Dermatol Surg. 2018;44:1464-1466.
- Motegi SI, Sekiguchi A, Fujiwara C, et al. Milia-like idiopathic calcinosis cutis and plaque-type syringoma in a girl with Down syndrome. J Dermatol. 2019;46:E136-E137.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Boos MD, Elenitsas R, Seykora J, et al. Benign subclinical syringomatous proliferations adjacent to a microcystic adnexal carcinoma: a tumor mimic with significant patient implications. Am J Dermatopathol. 2014;36:174-178.
- O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48:225-232.
To the Editor:
Plaquelike or plaque-type syringoma is a lesser-known variant of syringoma that can appear histologically indistinguishable from the superficial portion of microcystic adnexal carcinoma (MAC). The plaquelike variant of syringoma holds a benign clinical course, and no treatment is necessary. Microcystic adnexal carcinoma is distinguished from plaquelike syringoma by an aggressive growth pattern with a high risk for local invasion and recurrence if inadequately treated. Thus, treatment with Mohs micrographic surgery (MMS) has been recommended as the mainstay for MAC. If superficial biopsy specimens reveal suspicion for MAC and patients are referred for MMS, careful consideration should be made to differentiate MAC and plaquelike syringoma early to prevent unnecessary morbidity.
A 78-year-old woman was referred for MMS for a left forehead lesion that was diagnosed via shave biopsy as a desmoplastic and cystic adnexal neoplasm with suspicion for desmoplastic trichoepithelioma or MAC (Figure 1). Upon presentation for MMS, a well-healed, 1.0×0.9-cm scar at the biopsy site on the left forehead was observed (Figure 2A). One stage was obtained by standard MMS technique and sent for intraoperative processing (Figure 2B). Frozen section examination of the first stage demonstrated peripheral margin involvement with syringomatous change confined to the superficial and mid dermis (Figure 3). Before proceeding further, these findings were reviewed with an in-house dermatopathologist, and it was determined that no infiltrative tumor, perineural involvement, or other features to indicate malignancy were noted. A decision was made to refrain from obtaining any additional layers and to send excised Burow triangles for permanent section analysis. A primary linear closure was performed without complication, and the patient was discharged from the ambulatory surgery suite. Histopathologic examination of the Burow triangles later confirmed findings consistent with plaquelike syringoma with no evidence of malignancy (Figure 4).
Syringomas present as small flesh-colored papules in the periorbital areas. These benign neoplasms previously have been classified into 4 major clinical variants: localized, generalized, Down syndrome associated, and familial.1 The lesser-known plaquelike variant of syringoma was first described by Kikuchi et al2 in 1979. Aside from our report, a PubMed search of articles indexed for MEDLINE using the terms plaquelike or plaque-type syringoma yielded 16 cases in the literature.2-14 Of these, 6 were referred to or encountered in the MMS setting.8,9,11,12,14 Plaquelike syringoma can be solitary or multiple in presentation.6 It most commonly involves the head and neck but also can present on the trunk, arms, legs, and groin areas. The clinical size of plaquelike syringoma is variable, with the largest reported cases extending several centimeters in diameter.2,6 Similar to reported associations with conventional syringoma, the plaquelike subtype of syringoma has been reported in association with Down syndrome.13
Histopathologically, plaquelike syringoma shares features with MAC as well as desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma. Plaquelike syringoma demonstrates broad proliferations of small tubules morphologically reminiscent of tadpoles confined within the dermis. Ducts typically are lined with 2 or 3 layers of small cuboidal cells. Microcystic adnexal carcinoma typically features asymmetric ductal structures lined with single cells extending from the dermis into the subcutis and even underlying muscle, cartilage, or bone.8 There are no reliable immunohistochemical stains to differentiate between these 2 entities; thus, the primary distinction lies in the depth of involvement. Desmoplastic trichoepithelioma is composed of narrow cords and nests of basaloid cells of follicular origin commonly admixed with small cornifying cysts appearing in the dermis.8 Colonizing Merkel cells positive for cytokeratin 20 often are present in desmoplastic trichoepithelioma and not in syringoma or MAC.15 Desmoplastic basal cell carcinoma demonstrates narrow strands of basaloid cells of follicular origin appearing in the dermis. Desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma are each fundamentally differentiated from plaquelike syringoma in that proliferations of cords and nests are not of eccrine or apocrine origin.
Several cases of plaquelike syringoma have been challenging to distinguish from MAC in performing MMS.8,9,11 Underlying extension of this syringoma variant can be far-reaching, extending to several centimeters in size and involving multiple cosmetic subunits.6,11,14 Inadvertent overtreatment with multiple MMS stages can be avoided with careful recognition of the differentiating histopathologic features. Syringomatous lesions commonly are encountered in MMS and may even be present at the edge of other tumor types. Plaquelike syringoma has been reported as a coexistent entity with nodular basal cell carcinoma.12 Boos et al16 similarly reported the presence of deceptive ductal proliferations along the immediate peripheral margin of MAC, which prompted multiple re-excisions. Pursuit of permanent section analysis in these cases revealed the appearance of small syringomas, and a diagnosis of benign subclinical syringomatous proliferations was made, averting further intervention.16
Our case sheds light on the threat of commission bias in dermatologic surgery, which is the tendency for action rather than inaction.17 In this context, it is important to avoid the perspective that harm to the patient can only be prevented by active intervention. Cognitive bias has been increasingly recognized as a source of medical error, and methods to mitigate bias in medical practice have been well described.17 Microcystic adnexal carcinoma and plaquelike syringoma can be hard to differentiate especially initially, as demonstrated in our case, which particularly illustrates the importance of slowing down a surgical case at the appropriate time, considering and revisiting alternative diagnoses, implementing checklists, and seeking histopathologic collaboration with colleagues when necessary. Our attempted implementation of these principles, especially early collaboration with colleagues, led to intraoperative recognition of plaquelike syringoma within the first stage of MMS.
We seek to raise the index of suspicion for plaquelike syringoma among dermatologists and dermatologic surgeons, especially when syringomatous structures are limited to the superficial dermis. We encourage familiarity with the plaquelike syringoma entity as well as careful consideration of further investigation via scouting biopsies or permanent section analysis when other characteristic features of MAC are unclear or lacking. Adequate sampling as well as collaboration with a dermatopathologist in cases of suspected syringoma can help to reduce the susceptibility to commission bias and prevent histopathologic pitfalls and unwarranted surgical morbidity.
To the Editor:
Plaquelike or plaque-type syringoma is a lesser-known variant of syringoma that can appear histologically indistinguishable from the superficial portion of microcystic adnexal carcinoma (MAC). The plaquelike variant of syringoma holds a benign clinical course, and no treatment is necessary. Microcystic adnexal carcinoma is distinguished from plaquelike syringoma by an aggressive growth pattern with a high risk for local invasion and recurrence if inadequately treated. Thus, treatment with Mohs micrographic surgery (MMS) has been recommended as the mainstay for MAC. If superficial biopsy specimens reveal suspicion for MAC and patients are referred for MMS, careful consideration should be made to differentiate MAC and plaquelike syringoma early to prevent unnecessary morbidity.
A 78-year-old woman was referred for MMS for a left forehead lesion that was diagnosed via shave biopsy as a desmoplastic and cystic adnexal neoplasm with suspicion for desmoplastic trichoepithelioma or MAC (Figure 1). Upon presentation for MMS, a well-healed, 1.0×0.9-cm scar at the biopsy site on the left forehead was observed (Figure 2A). One stage was obtained by standard MMS technique and sent for intraoperative processing (Figure 2B). Frozen section examination of the first stage demonstrated peripheral margin involvement with syringomatous change confined to the superficial and mid dermis (Figure 3). Before proceeding further, these findings were reviewed with an in-house dermatopathologist, and it was determined that no infiltrative tumor, perineural involvement, or other features to indicate malignancy were noted. A decision was made to refrain from obtaining any additional layers and to send excised Burow triangles for permanent section analysis. A primary linear closure was performed without complication, and the patient was discharged from the ambulatory surgery suite. Histopathologic examination of the Burow triangles later confirmed findings consistent with plaquelike syringoma with no evidence of malignancy (Figure 4).
Syringomas present as small flesh-colored papules in the periorbital areas. These benign neoplasms previously have been classified into 4 major clinical variants: localized, generalized, Down syndrome associated, and familial.1 The lesser-known plaquelike variant of syringoma was first described by Kikuchi et al2 in 1979. Aside from our report, a PubMed search of articles indexed for MEDLINE using the terms plaquelike or plaque-type syringoma yielded 16 cases in the literature.2-14 Of these, 6 were referred to or encountered in the MMS setting.8,9,11,12,14 Plaquelike syringoma can be solitary or multiple in presentation.6 It most commonly involves the head and neck but also can present on the trunk, arms, legs, and groin areas. The clinical size of plaquelike syringoma is variable, with the largest reported cases extending several centimeters in diameter.2,6 Similar to reported associations with conventional syringoma, the plaquelike subtype of syringoma has been reported in association with Down syndrome.13
Histopathologically, plaquelike syringoma shares features with MAC as well as desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma. Plaquelike syringoma demonstrates broad proliferations of small tubules morphologically reminiscent of tadpoles confined within the dermis. Ducts typically are lined with 2 or 3 layers of small cuboidal cells. Microcystic adnexal carcinoma typically features asymmetric ductal structures lined with single cells extending from the dermis into the subcutis and even underlying muscle, cartilage, or bone.8 There are no reliable immunohistochemical stains to differentiate between these 2 entities; thus, the primary distinction lies in the depth of involvement. Desmoplastic trichoepithelioma is composed of narrow cords and nests of basaloid cells of follicular origin commonly admixed with small cornifying cysts appearing in the dermis.8 Colonizing Merkel cells positive for cytokeratin 20 often are present in desmoplastic trichoepithelioma and not in syringoma or MAC.15 Desmoplastic basal cell carcinoma demonstrates narrow strands of basaloid cells of follicular origin appearing in the dermis. Desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma are each fundamentally differentiated from plaquelike syringoma in that proliferations of cords and nests are not of eccrine or apocrine origin.
Several cases of plaquelike syringoma have been challenging to distinguish from MAC in performing MMS.8,9,11 Underlying extension of this syringoma variant can be far-reaching, extending to several centimeters in size and involving multiple cosmetic subunits.6,11,14 Inadvertent overtreatment with multiple MMS stages can be avoided with careful recognition of the differentiating histopathologic features. Syringomatous lesions commonly are encountered in MMS and may even be present at the edge of other tumor types. Plaquelike syringoma has been reported as a coexistent entity with nodular basal cell carcinoma.12 Boos et al16 similarly reported the presence of deceptive ductal proliferations along the immediate peripheral margin of MAC, which prompted multiple re-excisions. Pursuit of permanent section analysis in these cases revealed the appearance of small syringomas, and a diagnosis of benign subclinical syringomatous proliferations was made, averting further intervention.16
Our case sheds light on the threat of commission bias in dermatologic surgery, which is the tendency for action rather than inaction.17 In this context, it is important to avoid the perspective that harm to the patient can only be prevented by active intervention. Cognitive bias has been increasingly recognized as a source of medical error, and methods to mitigate bias in medical practice have been well described.17 Microcystic adnexal carcinoma and plaquelike syringoma can be hard to differentiate especially initially, as demonstrated in our case, which particularly illustrates the importance of slowing down a surgical case at the appropriate time, considering and revisiting alternative diagnoses, implementing checklists, and seeking histopathologic collaboration with colleagues when necessary. Our attempted implementation of these principles, especially early collaboration with colleagues, led to intraoperative recognition of plaquelike syringoma within the first stage of MMS.
We seek to raise the index of suspicion for plaquelike syringoma among dermatologists and dermatologic surgeons, especially when syringomatous structures are limited to the superficial dermis. We encourage familiarity with the plaquelike syringoma entity as well as careful consideration of further investigation via scouting biopsies or permanent section analysis when other characteristic features of MAC are unclear or lacking. Adequate sampling as well as collaboration with a dermatopathologist in cases of suspected syringoma can help to reduce the susceptibility to commission bias and prevent histopathologic pitfalls and unwarranted surgical morbidity.
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Dekio S, Jidoi J. Submammary syringoma—report of a case. J Dermatol. 1988;15:351-352.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Nguyen DB, Patterson JW, Wilson BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49:337-339.
- Rongioletti F, Semino MT, Rebora A. Unilateral multiple plaque-like syringomas. Br J Dermatol. 1996;135:623-625.
- Chi HI. A case of unusual syringoma: unilateral linear distribution and plaque formation. J Dermatol. 1996;23:505-506.
- Suwatee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:E206-E207.
- Schleich C, Ferringer T, Petrick M. Plaque type syringoma mimicking a microcystic adnexal carcinoma. J Am Acad Dermatol. 2016;74(suppl 1):AB287.
- Yang Y, Srivastava D. Plaque-type syringoma coexisting with basal cell carcinoma. Dermatol Surg. 2018;44:1464-1466.
- Motegi SI, Sekiguchi A, Fujiwara C, et al. Milia-like idiopathic calcinosis cutis and plaque-type syringoma in a girl with Down syndrome. J Dermatol. 2019;46:E136-E137.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Boos MD, Elenitsas R, Seykora J, et al. Benign subclinical syringomatous proliferations adjacent to a microcystic adnexal carcinoma: a tumor mimic with significant patient implications. Am J Dermatopathol. 2014;36:174-178.
- O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48:225-232.
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Dekio S, Jidoi J. Submammary syringoma—report of a case. J Dermatol. 1988;15:351-352.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Nguyen DB, Patterson JW, Wilson BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49:337-339.
- Rongioletti F, Semino MT, Rebora A. Unilateral multiple plaque-like syringomas. Br J Dermatol. 1996;135:623-625.
- Chi HI. A case of unusual syringoma: unilateral linear distribution and plaque formation. J Dermatol. 1996;23:505-506.
- Suwatee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:E206-E207.
- Schleich C, Ferringer T, Petrick M. Plaque type syringoma mimicking a microcystic adnexal carcinoma. J Am Acad Dermatol. 2016;74(suppl 1):AB287.
- Yang Y, Srivastava D. Plaque-type syringoma coexisting with basal cell carcinoma. Dermatol Surg. 2018;44:1464-1466.
- Motegi SI, Sekiguchi A, Fujiwara C, et al. Milia-like idiopathic calcinosis cutis and plaque-type syringoma in a girl with Down syndrome. J Dermatol. 2019;46:E136-E137.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Boos MD, Elenitsas R, Seykora J, et al. Benign subclinical syringomatous proliferations adjacent to a microcystic adnexal carcinoma: a tumor mimic with significant patient implications. Am J Dermatopathol. 2014;36:174-178.
- O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48:225-232.
Practice Points
- Dermatologists should familiarize themselves with the plaquelike subtype of syringoma, which can histologically mimic the superficial portion of microcystic adnexal carcinoma (MAC).
- Careful recognition of plaquelike syringoma in the Mohs micrographic surgery setting may prevent unnecessary surgical morbidity.
- Further diagnostic investigation is warranted for superficial biopsies suggestive of MAC or when other characteristic features are lacking.

Blue Nodules on the Forearms in an Active-Duty Military Servicemember
The Diagnosis: Glomangiomyoma
A punch biopsy of the right forearm revealed a collection of vascular and smooth muscle components with small and spindled bland cells containing minimal eosinophilic cytoplasm (Figure 1), confirming the diagnosis of glomangiomyoma. Immunohistochemical stains also supported the diagnosis and were positive for smooth muscle actin, desmin, and CD34 (Figure 2). Magnetic resonance imaging from a prior attempt at treatment with sclerotherapy demonstrated scattered vascular malformations with no notable internal derangement. There was no improvement with sclerotherapy. Given the number and vascular nature of the lesions, a trial of pulsed dye laser (PDL) therapy was administered and tolerated by the patient. He subsequently moved to a new military duty station. On follow-up, he reported no noticeable clinical improvement in the lesions after PDL and opted not to continue with laser treatment.

Glomangiomyoma is a rare and benign glomus tumor variant that demonstrates differentiation into the smooth muscle and potentially can result in substantial complications.1 Glomus tumors generally are benign neoplasms of the glomus apparatus, and glomus cells function as thermoregulators in the reticular dermis.2 Glomus tumors comprise less than 2% of soft tissue neoplasms and generally are solitary nodules; only 10% of glomus tumors occur with multiple lesions, and among them, glomangiomyoma is the rarest subtype, presenting in only 15% of cases.2,3 The 3 main subtypes of glomus tumors are solid, glomangioma, and glomangiomyoma.4 Clinically, the lesions may present as small blue nodules with associated pain and cold or pressure sensitivity. Although there appears to be variation of the nomenclature depending on the source in the literature, glomangiomas are characterized by their predominant vascular malformations on biopsy. Glomangiomyomas are a subset of glomus tumors with distinct smooth muscle differentiation.4 Given their pathologic presentation, our patient’s lesions were most consistent with the diagnosis of glomangiomyoma.

The small size of the lesions may result in difficulty establishing a clinical diagnosis, particularly if there is no hand involvement, where lesions most commonly occur.2 Therefore, histopathologic evaluation is essential and is the best initial step in evaluating glomangiomyomas.4 Biopsy is the most reliable means of confirming a diagnosis2,4,5; however, diagnostic imaging such as a computed tomography also should be performed if considering blue rubber bleb nevus syndrome due to the primary site of involvement. Surgical excision is the treatment of choice after confirming the diagnosis in most cases of symptomatic glomangiomyomas, particularly with painful lesions.6
Neurilemmomas (also known as schwannomas) are benign lesions that generally present as asymptomatic, soft, smooth nodules most often on the neck; however, they also may present on the flexor extremities or in internal organs. Although primarily asymptomatic, the tumors may be associated with pain and paresthesia as they enlarge and affect surrounding structures. Neurilemmomas may occur spontaneously or as part of a syndrome, such as neurofibromatosis type 2 or Carney complex.7
Hereditary hemorrhagic telangiectasia (formerly known as Osler-Weber-Rendu syndrome) is an autosomal-dominant disease that presents with arteriovenous malformations and telangiectases. Patients generally present in the third decade of life, with the main concern generally being epistaxis.8
Kaposi sarcoma is a viral infection secondary to human herpesvirus 8 that results in red-purple lesions commonly on mucocutaneous sites. Kaposi sarcoma can be AIDS associated and non-HIV associated. Although clinically indistinguishable, a few subtle histologic features can assist in differentiating the 2 etiologies. In addition to a potential history of immunodeficiency, evaluating for involvement of the lymphatic system, respiratory tract, or gastrointestinal tract can aid in differentiating this entity from glomus tumors.9
Leiomyomas are smooth muscle lesions divided into 3 subcategories: angioleiomyoma, piloleiomyoma, and genital leiomyoma. The clinical presentation and histopathology will vary depending on the subcategory. Although cutaneous leiomyomas are benign, further workup for piloleiomyoma may be required given the reported association with hereditary leiomyomatosis and renal cell cancer (Reed syndrome).10
Imaging can be helpful when the clinical diagnosis of a glomus tumor vs other painful neoplasms of the skin is unclear, such as in blue rubber bleb nevus syndrome, angioleiomyomas, neuromas, glomus tumors, leiomyomas, eccrine spiradenomas, congenital vascular malformations, schwannomas, or hemangiomas.4 Radiologic findings for glomus tumors may demonstrate cortical or cystic osseous defects. Magnetic resonance imaging and ultrasonography can help provide additional information on the lesion size and depth of involvement.1 Additionally, deeper glomangiomyomas have been associated with malignancy,2 potentially highlighting the benefit of early incorporation of imaging in the workup for this condition. Malignant transformation is rare and has been reported in less than 1% of cases.6
Treatment of glomus tumors predominantly is directed to the patient’s symptoms; asymptomatic lesions may be monitored.4 For symptomatic lesions, therapeutic options include wide local excision; sclerotherapy; and incorporation of various lasers, including Nd:YAG, CO2, and flashlamp tunable dye laser.4,5 One case report documented use of a PDL that successfully eliminated the pain associated with glomangiomyoma; however, the lesion in that report was not biopsy proven.11
Our case highlights the need to consider glomus tumors in patients presenting with multiple small nodules given the potential for misdiagnosis, impact on quality of life with associated psychological distress, and potential utility of incorporating PDL in treatment. Although our patient did not report clinical improvement in the appearance of the lesions with PDL therapy, additional treatment sessions may have helped,11 but he opted to discontinue. Follow-up for persistently symptomatic or changing lesions is necessary, given the minimal risk for malignant transformation.6
- Lee DY, Hwang SC, Jeong ST, et al. The value of diagnostic ultrasonography in the assessment of a glomus tumor of the subcutaneous layer of the forearm mimicking a hemangioma: a case report. J Med Case Rep. 2015;9:191. doi:10.1186/s13256-015-0672-y
- Li L, Bardsley V, Grainger A, et al. Extradigital glomangiomyoma of the forearm mimicking peripheral nerve sheath tumour and thrombosed varicose vein. BMJ Case Rep. 2021;14:E241221. doi: 10.1136 /bcr-2020-241221
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mohammadi O, Suarez M. Glomus cancer. StatPearls [Internet]. StatPearls Publishing; 2021.
- Maxey ML, Houghton CC, Mastriani KS, et al. Large prepatellar glomangioma: a case report [published online July 10, 2015]. Int J Surg Case Rep. 2015;14:80-84. doi:10.1016/j.ijscr.2015.07.002
- Brathwaite CD, Poppiti RJ Jr. Malignant glomus tumor. a case report of widespread metastases in a patient with multiple glomus body hamartomas. Am J Surg Pathol. 1996;20:233-238. doi:10.1097/00000478-199602000-00012
- Davis DD, Kane SM. Neurilemmoma. StatPearls [Internet]. StatPearls Publishing; 2022.
- Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary hemorrhagic telangiectasia. Otolaryngol Clin North Am. 2018;51:237-254. doi:10.1016/j.otc.2017.09.017
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. StatPearls [Internet]. StatPearls Publishing; 2022.
- Antony FC, Cliff S, Cowley N. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol. 2003;28:617-619. doi:10.1046/j.1365-2230.2003.01403.x
The Diagnosis: Glomangiomyoma
A punch biopsy of the right forearm revealed a collection of vascular and smooth muscle components with small and spindled bland cells containing minimal eosinophilic cytoplasm (Figure 1), confirming the diagnosis of glomangiomyoma. Immunohistochemical stains also supported the diagnosis and were positive for smooth muscle actin, desmin, and CD34 (Figure 2). Magnetic resonance imaging from a prior attempt at treatment with sclerotherapy demonstrated scattered vascular malformations with no notable internal derangement. There was no improvement with sclerotherapy. Given the number and vascular nature of the lesions, a trial of pulsed dye laser (PDL) therapy was administered and tolerated by the patient. He subsequently moved to a new military duty station. On follow-up, he reported no noticeable clinical improvement in the lesions after PDL and opted not to continue with laser treatment.
Glomangiomyoma is a rare and benign glomus tumor variant that demonstrates differentiation into the smooth muscle and potentially can result in substantial complications.1 Glomus tumors generally are benign neoplasms of the glomus apparatus, and glomus cells function as thermoregulators in the reticular dermis.2 Glomus tumors comprise less than 2% of soft tissue neoplasms and generally are solitary nodules; only 10% of glomus tumors occur with multiple lesions, and among them, glomangiomyoma is the rarest subtype, presenting in only 15% of cases.2,3 The 3 main subtypes of glomus tumors are solid, glomangioma, and glomangiomyoma.4 Clinically, the lesions may present as small blue nodules with associated pain and cold or pressure sensitivity. Although there appears to be variation of the nomenclature depending on the source in the literature, glomangiomas are characterized by their predominant vascular malformations on biopsy. Glomangiomyomas are a subset of glomus tumors with distinct smooth muscle differentiation.4 Given their pathologic presentation, our patient’s lesions were most consistent with the diagnosis of glomangiomyoma.
The small size of the lesions may result in difficulty establishing a clinical diagnosis, particularly if there is no hand involvement, where lesions most commonly occur.2 Therefore, histopathologic evaluation is essential and is the best initial step in evaluating glomangiomyomas.4 Biopsy is the most reliable means of confirming a diagnosis2,4,5; however, diagnostic imaging such as a computed tomography also should be performed if considering blue rubber bleb nevus syndrome due to the primary site of involvement. Surgical excision is the treatment of choice after confirming the diagnosis in most cases of symptomatic glomangiomyomas, particularly with painful lesions.6
Neurilemmomas (also known as schwannomas) are benign lesions that generally present as asymptomatic, soft, smooth nodules most often on the neck; however, they also may present on the flexor extremities or in internal organs. Although primarily asymptomatic, the tumors may be associated with pain and paresthesia as they enlarge and affect surrounding structures. Neurilemmomas may occur spontaneously or as part of a syndrome, such as neurofibromatosis type 2 or Carney complex.7
Hereditary hemorrhagic telangiectasia (formerly known as Osler-Weber-Rendu syndrome) is an autosomal-dominant disease that presents with arteriovenous malformations and telangiectases. Patients generally present in the third decade of life, with the main concern generally being epistaxis.8
Kaposi sarcoma is a viral infection secondary to human herpesvirus 8 that results in red-purple lesions commonly on mucocutaneous sites. Kaposi sarcoma can be AIDS associated and non-HIV associated. Although clinically indistinguishable, a few subtle histologic features can assist in differentiating the 2 etiologies. In addition to a potential history of immunodeficiency, evaluating for involvement of the lymphatic system, respiratory tract, or gastrointestinal tract can aid in differentiating this entity from glomus tumors.9
Leiomyomas are smooth muscle lesions divided into 3 subcategories: angioleiomyoma, piloleiomyoma, and genital leiomyoma. The clinical presentation and histopathology will vary depending on the subcategory. Although cutaneous leiomyomas are benign, further workup for piloleiomyoma may be required given the reported association with hereditary leiomyomatosis and renal cell cancer (Reed syndrome).10
Imaging can be helpful when the clinical diagnosis of a glomus tumor vs other painful neoplasms of the skin is unclear, such as in blue rubber bleb nevus syndrome, angioleiomyomas, neuromas, glomus tumors, leiomyomas, eccrine spiradenomas, congenital vascular malformations, schwannomas, or hemangiomas.4 Radiologic findings for glomus tumors may demonstrate cortical or cystic osseous defects. Magnetic resonance imaging and ultrasonography can help provide additional information on the lesion size and depth of involvement.1 Additionally, deeper glomangiomyomas have been associated with malignancy,2 potentially highlighting the benefit of early incorporation of imaging in the workup for this condition. Malignant transformation is rare and has been reported in less than 1% of cases.6
Treatment of glomus tumors predominantly is directed to the patient’s symptoms; asymptomatic lesions may be monitored.4 For symptomatic lesions, therapeutic options include wide local excision; sclerotherapy; and incorporation of various lasers, including Nd:YAG, CO2, and flashlamp tunable dye laser.4,5 One case report documented use of a PDL that successfully eliminated the pain associated with glomangiomyoma; however, the lesion in that report was not biopsy proven.11
Our case highlights the need to consider glomus tumors in patients presenting with multiple small nodules given the potential for misdiagnosis, impact on quality of life with associated psychological distress, and potential utility of incorporating PDL in treatment. Although our patient did not report clinical improvement in the appearance of the lesions with PDL therapy, additional treatment sessions may have helped,11 but he opted to discontinue. Follow-up for persistently symptomatic or changing lesions is necessary, given the minimal risk for malignant transformation.6
The Diagnosis: Glomangiomyoma
A punch biopsy of the right forearm revealed a collection of vascular and smooth muscle components with small and spindled bland cells containing minimal eosinophilic cytoplasm (Figure 1), confirming the diagnosis of glomangiomyoma. Immunohistochemical stains also supported the diagnosis and were positive for smooth muscle actin, desmin, and CD34 (Figure 2). Magnetic resonance imaging from a prior attempt at treatment with sclerotherapy demonstrated scattered vascular malformations with no notable internal derangement. There was no improvement with sclerotherapy. Given the number and vascular nature of the lesions, a trial of pulsed dye laser (PDL) therapy was administered and tolerated by the patient. He subsequently moved to a new military duty station. On follow-up, he reported no noticeable clinical improvement in the lesions after PDL and opted not to continue with laser treatment.
Glomangiomyoma is a rare and benign glomus tumor variant that demonstrates differentiation into the smooth muscle and potentially can result in substantial complications.1 Glomus tumors generally are benign neoplasms of the glomus apparatus, and glomus cells function as thermoregulators in the reticular dermis.2 Glomus tumors comprise less than 2% of soft tissue neoplasms and generally are solitary nodules; only 10% of glomus tumors occur with multiple lesions, and among them, glomangiomyoma is the rarest subtype, presenting in only 15% of cases.2,3 The 3 main subtypes of glomus tumors are solid, glomangioma, and glomangiomyoma.4 Clinically, the lesions may present as small blue nodules with associated pain and cold or pressure sensitivity. Although there appears to be variation of the nomenclature depending on the source in the literature, glomangiomas are characterized by their predominant vascular malformations on biopsy. Glomangiomyomas are a subset of glomus tumors with distinct smooth muscle differentiation.4 Given their pathologic presentation, our patient’s lesions were most consistent with the diagnosis of glomangiomyoma.
The small size of the lesions may result in difficulty establishing a clinical diagnosis, particularly if there is no hand involvement, where lesions most commonly occur.2 Therefore, histopathologic evaluation is essential and is the best initial step in evaluating glomangiomyomas.4 Biopsy is the most reliable means of confirming a diagnosis2,4,5; however, diagnostic imaging such as a computed tomography also should be performed if considering blue rubber bleb nevus syndrome due to the primary site of involvement. Surgical excision is the treatment of choice after confirming the diagnosis in most cases of symptomatic glomangiomyomas, particularly with painful lesions.6
Neurilemmomas (also known as schwannomas) are benign lesions that generally present as asymptomatic, soft, smooth nodules most often on the neck; however, they also may present on the flexor extremities or in internal organs. Although primarily asymptomatic, the tumors may be associated with pain and paresthesia as they enlarge and affect surrounding structures. Neurilemmomas may occur spontaneously or as part of a syndrome, such as neurofibromatosis type 2 or Carney complex.7
Hereditary hemorrhagic telangiectasia (formerly known as Osler-Weber-Rendu syndrome) is an autosomal-dominant disease that presents with arteriovenous malformations and telangiectases. Patients generally present in the third decade of life, with the main concern generally being epistaxis.8
Kaposi sarcoma is a viral infection secondary to human herpesvirus 8 that results in red-purple lesions commonly on mucocutaneous sites. Kaposi sarcoma can be AIDS associated and non-HIV associated. Although clinically indistinguishable, a few subtle histologic features can assist in differentiating the 2 etiologies. In addition to a potential history of immunodeficiency, evaluating for involvement of the lymphatic system, respiratory tract, or gastrointestinal tract can aid in differentiating this entity from glomus tumors.9
Leiomyomas are smooth muscle lesions divided into 3 subcategories: angioleiomyoma, piloleiomyoma, and genital leiomyoma. The clinical presentation and histopathology will vary depending on the subcategory. Although cutaneous leiomyomas are benign, further workup for piloleiomyoma may be required given the reported association with hereditary leiomyomatosis and renal cell cancer (Reed syndrome).10
Imaging can be helpful when the clinical diagnosis of a glomus tumor vs other painful neoplasms of the skin is unclear, such as in blue rubber bleb nevus syndrome, angioleiomyomas, neuromas, glomus tumors, leiomyomas, eccrine spiradenomas, congenital vascular malformations, schwannomas, or hemangiomas.4 Radiologic findings for glomus tumors may demonstrate cortical or cystic osseous defects. Magnetic resonance imaging and ultrasonography can help provide additional information on the lesion size and depth of involvement.1 Additionally, deeper glomangiomyomas have been associated with malignancy,2 potentially highlighting the benefit of early incorporation of imaging in the workup for this condition. Malignant transformation is rare and has been reported in less than 1% of cases.6
Treatment of glomus tumors predominantly is directed to the patient’s symptoms; asymptomatic lesions may be monitored.4 For symptomatic lesions, therapeutic options include wide local excision; sclerotherapy; and incorporation of various lasers, including Nd:YAG, CO2, and flashlamp tunable dye laser.4,5 One case report documented use of a PDL that successfully eliminated the pain associated with glomangiomyoma; however, the lesion in that report was not biopsy proven.11
Our case highlights the need to consider glomus tumors in patients presenting with multiple small nodules given the potential for misdiagnosis, impact on quality of life with associated psychological distress, and potential utility of incorporating PDL in treatment. Although our patient did not report clinical improvement in the appearance of the lesions with PDL therapy, additional treatment sessions may have helped,11 but he opted to discontinue. Follow-up for persistently symptomatic or changing lesions is necessary, given the minimal risk for malignant transformation.6
- Lee DY, Hwang SC, Jeong ST, et al. The value of diagnostic ultrasonography in the assessment of a glomus tumor of the subcutaneous layer of the forearm mimicking a hemangioma: a case report. J Med Case Rep. 2015;9:191. doi:10.1186/s13256-015-0672-y
- Li L, Bardsley V, Grainger A, et al. Extradigital glomangiomyoma of the forearm mimicking peripheral nerve sheath tumour and thrombosed varicose vein. BMJ Case Rep. 2021;14:E241221. doi: 10.1136 /bcr-2020-241221
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mohammadi O, Suarez M. Glomus cancer. StatPearls [Internet]. StatPearls Publishing; 2021.
- Maxey ML, Houghton CC, Mastriani KS, et al. Large prepatellar glomangioma: a case report [published online July 10, 2015]. Int J Surg Case Rep. 2015;14:80-84. doi:10.1016/j.ijscr.2015.07.002
- Brathwaite CD, Poppiti RJ Jr. Malignant glomus tumor. a case report of widespread metastases in a patient with multiple glomus body hamartomas. Am J Surg Pathol. 1996;20:233-238. doi:10.1097/00000478-199602000-00012
- Davis DD, Kane SM. Neurilemmoma. StatPearls [Internet]. StatPearls Publishing; 2022.
- Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary hemorrhagic telangiectasia. Otolaryngol Clin North Am. 2018;51:237-254. doi:10.1016/j.otc.2017.09.017
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. StatPearls [Internet]. StatPearls Publishing; 2022.
- Antony FC, Cliff S, Cowley N. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol. 2003;28:617-619. doi:10.1046/j.1365-2230.2003.01403.x
- Lee DY, Hwang SC, Jeong ST, et al. The value of diagnostic ultrasonography in the assessment of a glomus tumor of the subcutaneous layer of the forearm mimicking a hemangioma: a case report. J Med Case Rep. 2015;9:191. doi:10.1186/s13256-015-0672-y
- Li L, Bardsley V, Grainger A, et al. Extradigital glomangiomyoma of the forearm mimicking peripheral nerve sheath tumour and thrombosed varicose vein. BMJ Case Rep. 2021;14:E241221. doi: 10.1136 /bcr-2020-241221
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mohammadi O, Suarez M. Glomus cancer. StatPearls [Internet]. StatPearls Publishing; 2021.
- Maxey ML, Houghton CC, Mastriani KS, et al. Large prepatellar glomangioma: a case report [published online July 10, 2015]. Int J Surg Case Rep. 2015;14:80-84. doi:10.1016/j.ijscr.2015.07.002
- Brathwaite CD, Poppiti RJ Jr. Malignant glomus tumor. a case report of widespread metastases in a patient with multiple glomus body hamartomas. Am J Surg Pathol. 1996;20:233-238. doi:10.1097/00000478-199602000-00012
- Davis DD, Kane SM. Neurilemmoma. StatPearls [Internet]. StatPearls Publishing; 2022.
- Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary hemorrhagic telangiectasia. Otolaryngol Clin North Am. 2018;51:237-254. doi:10.1016/j.otc.2017.09.017
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. StatPearls [Internet]. StatPearls Publishing; 2022.
- Antony FC, Cliff S, Cowley N. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol. 2003;28:617-619. doi:10.1046/j.1365-2230.2003.01403.x
A 31-year-old active-duty military servicemember presented to the dermatology clinic for evaluation of 0.3- to 2-cm, tender, blue nodules on the wrists and forearms. The lesions first appeared on the right volar wrist secondary to a presumed injury sustained approximately 10 years prior to presentation and spread to the proximal forearm as well as the left wrist and forearm. He denied fevers, chills, chest pain, hematochezia, hematuria, or other skin findings. Physical examination revealed blue-violaceous, firm nodules on the right volar wrist and forearm that were tender to palpation. Blue-violaceous, papulonodular lesions on the left volar wrist and dorsal hand were not tender to palpation. A punch biopsy was performed.


Iododerma Simulating Cryptococcal Infection
To the Editor:
A woman in her 40s presented with acute onset of rapidly spreading lesions on the face, trunk, and extremities. She reported high fever and endorsed malaise. She had a history of end-stage renal disease and was on renal dialysis. She recently underwent revision of an arteriovenous fistula.
Physical examination revealed diffuse, erythematous, firm papules and plaques with central hemorrhage and umbilication on the dorsal aspect of the nose, forehead, temples, and cheeks. There also were purpuric papules and plaques with a peripheral rim of vesiculation (Figure 1) on the medial and posterior thighs and buttocks. Histopathology of a biopsy specimen revealed an interstitial neutrophilic infiltrate in the superficial dermis and mid dermis with scattered, haloed, acellular structures simulating cryptococcal organisms (Figure 2). Periodic acid–Schiff (PAS), Grocott methenamine-silver, and mucicarmine staining was negative. Repeat biopsy showed similar findings. A (1-3)-β-

The findings compatible with a diagnosis of iododerma included umbilicated hemorrhagic papules and plaques, cryptococcal-like structures with negative staining on histopathology, and elevated iodine levels with a negative infectious workup. The patient was treated with topical corticosteroids. At 1-month follow-up, the lesions had resolved.
. Reference bar indicates 200 μm.")
Iododerma is a halogenoderma, a skin eruption that occurs after ingestion of or exposure to a halogen-containing substance (eg, iodine, bromine, fluorine) or medication (eg, lithium).1 Common sources of iodine include iodinated contrast media, potassium iodide ingestion, topical application of povidone–iodine, radioactive iodine administration, and the antiarrhythmic amiodarone. Excess exposure to iodine-containing compounds typically occurs in the setting of kidney disease or failure as well as due to reduced iodine clearance.1 Although the pathogenesis of iododerma is unknown, the most common hypothesis is that lesions are delayed hypersensitivity reactions secondary to formation of a protein-halogen complex.2
The presentation of iododerma is polymorphous and includes acneform, vegetative, or pustular eruptions; umbilicated papules and plaques can be present.2,3 Lesions can be either asymptomatic or painful and pruritic. Timing between iodine exposure and onset of lesions varies from hours to days to years.2,4
Systemic symptoms of iododerma can occur, including salivary gland swelling, hypotension and bradycardia, kidney injury, or thyroid and liver abnormalities. Histopathologic analysis demonstrates a dense neutrophilic dermatitis with negative staining for infectious causes.4,5 Cryptococcal-like structures have been described in iododerma3; neutrophilic dermatoses of various causes that mimic cryptococcal infection have been reported.6 Ultimately, iododerma remains a diagnosis of exclusion.
Withdrawal of an offending compound is remedial. Dialysis is beneficial in end-stage renal disease. Topical, intralesional, and systemic corticosteroids, as well as antibiotics, provide variable benefit.4,7 Lesions can take 4 to 6 weeks to clear after withdrawal of the offending agent. It is unclear whether recurrences happen; iodine-containing compounds need to be avoided after a patient has been affected.
Iododerma has a broad differential diagnosis due to the polymorphous presentation of the disorder, including acute febrile neutrophilic dermatosis (also known as Sweet syndrome), cutaneous cryptococcosis, and cutaneous histoplasmosis. Sweet syndrome presents as abrupt onset of edematous erythematous plaques with fever and leukocytosis. It is associated with infection, inflammatory disorders, medication, and malignancy.8 Histopathologic analysis reveals papillary dermal edema and a neutrophilic dermatosis. Cytoplasmic vacuolization resembling C neoformans has been reported.9 The diagnosis is less favored in the presence of renal disease, temporal association of the eruption with iodine exposure, and elevated blood and urine iodine levels, as in our patient.
Cutaneous cryptococcosis, an infection caused by C neoformans, typically occurs secondary to dissemination from the lungs; rarely, the disease is primary. Acneform plaques, vegetative plaques, and umbilicated lesions are seen.10 Histopathologic analysis shows characteristic yeast forms of cryptococcosis surrounded by gelatinous edema, which create a haloed effect, typically throughout the dermis. Capsules are positive for PAS or mucicarmine staining. Although C neoformans can closely mimic iododerma both clinically and histopathologically, negative infectious staining, localization of haloed structures to the upper dermis, a negative test for cryptococcal antigen, and elevated blood and urine iodine levels in this case all favored iododerma.
Cutaneous histoplasmosis is an infection caused by Histoplasma capsulatum, most commonly as secondary dissemination from pulmonary infection but rarely from direct inoculation of the skin.11 Presentation includes erythematous to hemorrhagic, umbilicated papules and plaques. Histopathologic findings are round to oval, narrow-based, budding yeasts that stain positive for PAS or mucicarmine. Although histoplasmosis can clinically mimic iododerma, the disease is distinguished histologically by the presence of fungal microorganisms that lack the gelatinous edema and haloed effect of iododerma.
We presented a unique case of iododerma simulating cryptococcal infection both clinically and histopathologically. Prompt recognition of histologic mimickers of true infectious microorganisms is essential to prevent unnecessary delay of withdrawal of the offending substance and to initiate appropriate therapy.
- Alagheband M, Engineer L. Lithium and halogenoderma. Arch Dermatol. 2000;136:126-127. doi:10.1001/archderm.136.1.126
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379. doi:10.1111/bjd.12852
- Runge M, Williams K, Scharnitz T, et al. Iodine toxicity after iodinated contrast: new observations in iododerma. JAAD Case Rep. 2020;6:319-322. doi:10.1016/j.jdcr.2020.02.006
- Chalela JG, Aguilar L. Iododerma from contrast material. N Engl J Med. 2016;374:2477. doi:10.1056/NEJMicm1512512
- Chang MW, Miner JE, Moiin A, et al. Iododerma after computed tomographic scan with intravenous radiopaque contrast media. J Am Acad Dermatol. 1997;36:1014-1016. doi:10.1016/s0190-9622(97)80291-5
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi:10.1111/cup.12019
- Pranteda G, Grimaldi M, Salzetta M, et al. Vegetating iododerma and pulmonary eosinophilic infiltration. a simple co-occurrence? Acta Derm Venereol. 2004;84:480-481.
- Nelson CA, Stephen S, Ashchyan HJ, et al. M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.064
- Wilson J, Gleghorn K, Kelly B. Cryptococcoid Sweet’s syndrome: two reports of Sweet’s syndrome mimicking cutaneous cryptococcosis. J Cutan Pathol. 2017;44:413-419. doi:10.1111/cup.12921
- Beatson M, Harwood M, Reese V, et al. Primary cutaneous cryptococcosis in an elderly pigeon breeder. JAAD Case Rep. 2019;5:433-435. doi:10.1016/j.jdcr.2019.03.006
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348. doi:10.1177/0145561318097010-1108
To the Editor:
A woman in her 40s presented with acute onset of rapidly spreading lesions on the face, trunk, and extremities. She reported high fever and endorsed malaise. She had a history of end-stage renal disease and was on renal dialysis. She recently underwent revision of an arteriovenous fistula.
Physical examination revealed diffuse, erythematous, firm papules and plaques with central hemorrhage and umbilication on the dorsal aspect of the nose, forehead, temples, and cheeks. There also were purpuric papules and plaques with a peripheral rim of vesiculation (Figure 1) on the medial and posterior thighs and buttocks. Histopathology of a biopsy specimen revealed an interstitial neutrophilic infiltrate in the superficial dermis and mid dermis with scattered, haloed, acellular structures simulating cryptococcal organisms (Figure 2). Periodic acid–Schiff (PAS), Grocott methenamine-silver, and mucicarmine staining was negative. Repeat biopsy showed similar findings. A (1-3)-β-
The findings compatible with a diagnosis of iododerma included umbilicated hemorrhagic papules and plaques, cryptococcal-like structures with negative staining on histopathology, and elevated iodine levels with a negative infectious workup. The patient was treated with topical corticosteroids. At 1-month follow-up, the lesions had resolved.
Iododerma is a halogenoderma, a skin eruption that occurs after ingestion of or exposure to a halogen-containing substance (eg, iodine, bromine, fluorine) or medication (eg, lithium).1 Common sources of iodine include iodinated contrast media, potassium iodide ingestion, topical application of povidone–iodine, radioactive iodine administration, and the antiarrhythmic amiodarone. Excess exposure to iodine-containing compounds typically occurs in the setting of kidney disease or failure as well as due to reduced iodine clearance.1 Although the pathogenesis of iododerma is unknown, the most common hypothesis is that lesions are delayed hypersensitivity reactions secondary to formation of a protein-halogen complex.2
The presentation of iododerma is polymorphous and includes acneform, vegetative, or pustular eruptions; umbilicated papules and plaques can be present.2,3 Lesions can be either asymptomatic or painful and pruritic. Timing between iodine exposure and onset of lesions varies from hours to days to years.2,4
Systemic symptoms of iododerma can occur, including salivary gland swelling, hypotension and bradycardia, kidney injury, or thyroid and liver abnormalities. Histopathologic analysis demonstrates a dense neutrophilic dermatitis with negative staining for infectious causes.4,5 Cryptococcal-like structures have been described in iododerma3; neutrophilic dermatoses of various causes that mimic cryptococcal infection have been reported.6 Ultimately, iododerma remains a diagnosis of exclusion.
Withdrawal of an offending compound is remedial. Dialysis is beneficial in end-stage renal disease. Topical, intralesional, and systemic corticosteroids, as well as antibiotics, provide variable benefit.4,7 Lesions can take 4 to 6 weeks to clear after withdrawal of the offending agent. It is unclear whether recurrences happen; iodine-containing compounds need to be avoided after a patient has been affected.
Iododerma has a broad differential diagnosis due to the polymorphous presentation of the disorder, including acute febrile neutrophilic dermatosis (also known as Sweet syndrome), cutaneous cryptococcosis, and cutaneous histoplasmosis. Sweet syndrome presents as abrupt onset of edematous erythematous plaques with fever and leukocytosis. It is associated with infection, inflammatory disorders, medication, and malignancy.8 Histopathologic analysis reveals papillary dermal edema and a neutrophilic dermatosis. Cytoplasmic vacuolization resembling C neoformans has been reported.9 The diagnosis is less favored in the presence of renal disease, temporal association of the eruption with iodine exposure, and elevated blood and urine iodine levels, as in our patient.
Cutaneous cryptococcosis, an infection caused by C neoformans, typically occurs secondary to dissemination from the lungs; rarely, the disease is primary. Acneform plaques, vegetative plaques, and umbilicated lesions are seen.10 Histopathologic analysis shows characteristic yeast forms of cryptococcosis surrounded by gelatinous edema, which create a haloed effect, typically throughout the dermis. Capsules are positive for PAS or mucicarmine staining. Although C neoformans can closely mimic iododerma both clinically and histopathologically, negative infectious staining, localization of haloed structures to the upper dermis, a negative test for cryptococcal antigen, and elevated blood and urine iodine levels in this case all favored iododerma.
Cutaneous histoplasmosis is an infection caused by Histoplasma capsulatum, most commonly as secondary dissemination from pulmonary infection but rarely from direct inoculation of the skin.11 Presentation includes erythematous to hemorrhagic, umbilicated papules and plaques. Histopathologic findings are round to oval, narrow-based, budding yeasts that stain positive for PAS or mucicarmine. Although histoplasmosis can clinically mimic iododerma, the disease is distinguished histologically by the presence of fungal microorganisms that lack the gelatinous edema and haloed effect of iododerma.
We presented a unique case of iododerma simulating cryptococcal infection both clinically and histopathologically. Prompt recognition of histologic mimickers of true infectious microorganisms is essential to prevent unnecessary delay of withdrawal of the offending substance and to initiate appropriate therapy.
To the Editor:
A woman in her 40s presented with acute onset of rapidly spreading lesions on the face, trunk, and extremities. She reported high fever and endorsed malaise. She had a history of end-stage renal disease and was on renal dialysis. She recently underwent revision of an arteriovenous fistula.
Physical examination revealed diffuse, erythematous, firm papules and plaques with central hemorrhage and umbilication on the dorsal aspect of the nose, forehead, temples, and cheeks. There also were purpuric papules and plaques with a peripheral rim of vesiculation (Figure 1) on the medial and posterior thighs and buttocks. Histopathology of a biopsy specimen revealed an interstitial neutrophilic infiltrate in the superficial dermis and mid dermis with scattered, haloed, acellular structures simulating cryptococcal organisms (Figure 2). Periodic acid–Schiff (PAS), Grocott methenamine-silver, and mucicarmine staining was negative. Repeat biopsy showed similar findings. A (1-3)-β-
The findings compatible with a diagnosis of iododerma included umbilicated hemorrhagic papules and plaques, cryptococcal-like structures with negative staining on histopathology, and elevated iodine levels with a negative infectious workup. The patient was treated with topical corticosteroids. At 1-month follow-up, the lesions had resolved.
Iododerma is a halogenoderma, a skin eruption that occurs after ingestion of or exposure to a halogen-containing substance (eg, iodine, bromine, fluorine) or medication (eg, lithium).1 Common sources of iodine include iodinated contrast media, potassium iodide ingestion, topical application of povidone–iodine, radioactive iodine administration, and the antiarrhythmic amiodarone. Excess exposure to iodine-containing compounds typically occurs in the setting of kidney disease or failure as well as due to reduced iodine clearance.1 Although the pathogenesis of iododerma is unknown, the most common hypothesis is that lesions are delayed hypersensitivity reactions secondary to formation of a protein-halogen complex.2
The presentation of iododerma is polymorphous and includes acneform, vegetative, or pustular eruptions; umbilicated papules and plaques can be present.2,3 Lesions can be either asymptomatic or painful and pruritic. Timing between iodine exposure and onset of lesions varies from hours to days to years.2,4
Systemic symptoms of iododerma can occur, including salivary gland swelling, hypotension and bradycardia, kidney injury, or thyroid and liver abnormalities. Histopathologic analysis demonstrates a dense neutrophilic dermatitis with negative staining for infectious causes.4,5 Cryptococcal-like structures have been described in iododerma3; neutrophilic dermatoses of various causes that mimic cryptococcal infection have been reported.6 Ultimately, iododerma remains a diagnosis of exclusion.
Withdrawal of an offending compound is remedial. Dialysis is beneficial in end-stage renal disease. Topical, intralesional, and systemic corticosteroids, as well as antibiotics, provide variable benefit.4,7 Lesions can take 4 to 6 weeks to clear after withdrawal of the offending agent. It is unclear whether recurrences happen; iodine-containing compounds need to be avoided after a patient has been affected.
Iododerma has a broad differential diagnosis due to the polymorphous presentation of the disorder, including acute febrile neutrophilic dermatosis (also known as Sweet syndrome), cutaneous cryptococcosis, and cutaneous histoplasmosis. Sweet syndrome presents as abrupt onset of edematous erythematous plaques with fever and leukocytosis. It is associated with infection, inflammatory disorders, medication, and malignancy.8 Histopathologic analysis reveals papillary dermal edema and a neutrophilic dermatosis. Cytoplasmic vacuolization resembling C neoformans has been reported.9 The diagnosis is less favored in the presence of renal disease, temporal association of the eruption with iodine exposure, and elevated blood and urine iodine levels, as in our patient.
Cutaneous cryptococcosis, an infection caused by C neoformans, typically occurs secondary to dissemination from the lungs; rarely, the disease is primary. Acneform plaques, vegetative plaques, and umbilicated lesions are seen.10 Histopathologic analysis shows characteristic yeast forms of cryptococcosis surrounded by gelatinous edema, which create a haloed effect, typically throughout the dermis. Capsules are positive for PAS or mucicarmine staining. Although C neoformans can closely mimic iododerma both clinically and histopathologically, negative infectious staining, localization of haloed structures to the upper dermis, a negative test for cryptococcal antigen, and elevated blood and urine iodine levels in this case all favored iododerma.
Cutaneous histoplasmosis is an infection caused by Histoplasma capsulatum, most commonly as secondary dissemination from pulmonary infection but rarely from direct inoculation of the skin.11 Presentation includes erythematous to hemorrhagic, umbilicated papules and plaques. Histopathologic findings are round to oval, narrow-based, budding yeasts that stain positive for PAS or mucicarmine. Although histoplasmosis can clinically mimic iododerma, the disease is distinguished histologically by the presence of fungal microorganisms that lack the gelatinous edema and haloed effect of iododerma.
We presented a unique case of iododerma simulating cryptococcal infection both clinically and histopathologically. Prompt recognition of histologic mimickers of true infectious microorganisms is essential to prevent unnecessary delay of withdrawal of the offending substance and to initiate appropriate therapy.
- Alagheband M, Engineer L. Lithium and halogenoderma. Arch Dermatol. 2000;136:126-127. doi:10.1001/archderm.136.1.126
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379. doi:10.1111/bjd.12852
- Runge M, Williams K, Scharnitz T, et al. Iodine toxicity after iodinated contrast: new observations in iododerma. JAAD Case Rep. 2020;6:319-322. doi:10.1016/j.jdcr.2020.02.006
- Chalela JG, Aguilar L. Iododerma from contrast material. N Engl J Med. 2016;374:2477. doi:10.1056/NEJMicm1512512
- Chang MW, Miner JE, Moiin A, et al. Iododerma after computed tomographic scan with intravenous radiopaque contrast media. J Am Acad Dermatol. 1997;36:1014-1016. doi:10.1016/s0190-9622(97)80291-5
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi:10.1111/cup.12019
- Pranteda G, Grimaldi M, Salzetta M, et al. Vegetating iododerma and pulmonary eosinophilic infiltration. a simple co-occurrence? Acta Derm Venereol. 2004;84:480-481.
- Nelson CA, Stephen S, Ashchyan HJ, et al. M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.064
- Wilson J, Gleghorn K, Kelly B. Cryptococcoid Sweet’s syndrome: two reports of Sweet’s syndrome mimicking cutaneous cryptococcosis. J Cutan Pathol. 2017;44:413-419. doi:10.1111/cup.12921
- Beatson M, Harwood M, Reese V, et al. Primary cutaneous cryptococcosis in an elderly pigeon breeder. JAAD Case Rep. 2019;5:433-435. doi:10.1016/j.jdcr.2019.03.006
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348. doi:10.1177/0145561318097010-1108
- Alagheband M, Engineer L. Lithium and halogenoderma. Arch Dermatol. 2000;136:126-127. doi:10.1001/archderm.136.1.126
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379. doi:10.1111/bjd.12852
- Runge M, Williams K, Scharnitz T, et al. Iodine toxicity after iodinated contrast: new observations in iododerma. JAAD Case Rep. 2020;6:319-322. doi:10.1016/j.jdcr.2020.02.006
- Chalela JG, Aguilar L. Iododerma from contrast material. N Engl J Med. 2016;374:2477. doi:10.1056/NEJMicm1512512
- Chang MW, Miner JE, Moiin A, et al. Iododerma after computed tomographic scan with intravenous radiopaque contrast media. J Am Acad Dermatol. 1997;36:1014-1016. doi:10.1016/s0190-9622(97)80291-5
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi:10.1111/cup.12019
- Pranteda G, Grimaldi M, Salzetta M, et al. Vegetating iododerma and pulmonary eosinophilic infiltration. a simple co-occurrence? Acta Derm Venereol. 2004;84:480-481.
- Nelson CA, Stephen S, Ashchyan HJ, et al. M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.064
- Wilson J, Gleghorn K, Kelly B. Cryptococcoid Sweet’s syndrome: two reports of Sweet’s syndrome mimicking cutaneous cryptococcosis. J Cutan Pathol. 2017;44:413-419. doi:10.1111/cup.12921
- Beatson M, Harwood M, Reese V, et al. Primary cutaneous cryptococcosis in an elderly pigeon breeder. JAAD Case Rep. 2019;5:433-435. doi:10.1016/j.jdcr.2019.03.006
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348. doi:10.1177/0145561318097010-1108
Practice Points
- Halogenodermas are rare cutaneous reactions to excess exposure to or ingestion of halogen-containing drugs or substances such as bromine, iodine (iododerma), fluorine, and rarely lithium.
- The clinical presentation of a halogenoderma varies; the most characteristic manifestation is a vegetative or exudative plaque with a peripheral rim of pustules.
- Histologically, lesions of a halogenoderma are characterized by pseudoepitheliomatous hyperplasia associated with numerous intraepidermal microabscesses overlying a dense mixed inflammatory infiltrate of neutrophils, plasma cells, eosinophils, histiocytes, and scattered multinucleated giant cells.
- Rarely, the dermal infiltrate of a halogenoderma contains abundant acellular bodies surrounded by capsulelike vacuolated spaces mimicking Cryptococcus neoformans.
