User login
A Fixed Drug Eruption to Medroxyprogesterone Acetate Injectable Suspension
To the Editor:
A fixed drug eruption (FDE) is a well-documented form of cutaneous hypersensitivity that typically manifests as a sharply demarcated, dusky, round to oval, edematous, red-violaceous macule or patch on the skin and mucous membranes. The lesion often resolves with residual postinflammatory hyperpigmentation, most commonly as a reaction to ingested drugs or drug components.1 Lesions generally occur at the same anatomic site with repeated exposure to the offending drug. Typically, a single site is affected, but additional sites with more generalized involvement have been reported to occur with subsequent exposure to the offending medication. The diagnosis usually is clinical, but histopathologic findings can help confirm the diagnosis in unusual presentations. We present a novel case of a patient with an FDE from medroxyprogesterone acetate, a contraceptive injection that contains the hormone progestin.
A 35-year-old woman presented to the dermatology clinic for evaluation of a lesion on the left lower buttock of 1 year’s duration. She reported periodic swelling and associated pruritus of the lesion. She denied any growth in size, and no other similar lesions were present. The patient reported a medication history of medroxyprogesterone acetate for birth control, but she denied any other prescription or over-the-counter medication, oral supplements, or recreational drug use. Upon further inquiry, she reported that the recurrence of symptoms appeared to coincide with each administration of medroxyprogesterone acetate, which occurred approximately every 3 months. The eruption cleared between injections and recurred in the same location following subsequent injections. The lesion appeared approximately 2 weeks after the first injection (approximately 1 year prior to presentation to dermatology) and within 2 to 3 days after each subsequent injection. Physical examination revealed a 2×2-cm, circular, slightly violaceous patch on the left buttock (Figure 1). A biopsy was recommended to aid in diagnosis, and the patient was offered a topical steroid for symptomatic relief. A punch biopsy revealed subtle interface dermatitis with superficial perivascular lymphoid infiltrate and marked pigmentary incontinence consistent with an FDE (Figure 2).

An FDE was first reported in 1889 by Bourns,2 and over time more implicated agents and varying clinical presentations have been linked to the disease. The FDE can be accompanied by symptoms of pruritus or paresthesia. Most cases are devoid of systemic symptoms. An FDE can be located anywhere on the body, but it most frequently manifests on the lips, face, hands, feet, and genitalia. Although the eruption often heals with residual postinflammatory hyperpigmentation, a nonpigmenting FDE due to pseudoephedrine has been reported.3

Common culprits include antibiotics (eg, sulfonamides, trimethoprim, fluoroquinolones, tetracyclines), nonsteroidal anti-inflammatory medications (eg, naproxen sodium, ibuprofen, celecoxib), barbiturates, antimalarials, and anticonvulsants. Rare cases of FDE induced by foods and food additives also have been reported.4 Oral fluconazole, levocetirizine dihydrochloride, loperamide, and multivitamin-mineral preparations are other rare inducers of FDE.5-8 In 2004, Ritter and Meffert9 described an FDE to the green dye used in inactive oral contraceptive pills. A similar case was reported by Rea et al10 that described an FDE from the inactive sugar pills in ethinyl estradiol and levonorgestrel, which is another combined oral contraceptive.
The time between ingestion of the offending agent and the manifestation of the disease usually is 1 to 2 weeks; however, upon subsequent exposure, the disease has been reported to manifest within hours.1 CD8+ memory T cells have been shown to be major players in the development of FDE and can be found along the dermoepidermal junction as part of a delayed type IV hypersensitivity reaction.11 Histopathology reveals superficial and deep interstitial and perivascular infiltrates consisting of lymphocytes with admixed eosinophils and possibly neutrophils in the dermis. In the epidermis, necrotic keratinocytes can be present. In rare cases, FDE may have atypical features, such as in generalized bullous FDE and nonpigmenting FDE, the latter of which more commonly is associated with pseudoephedrine.1
The differential diagnosis for FDE includes erythema multiforme, Stevens-Johnson syndrome/toxic epidermal necrolysis, autoimmune progesterone dermatitis, and large plaque parapsoriasis. The number and morphology of lesions in erythema multiforme help differentiate it from FDE, as erythema multiforme presents with multiple targetoid lesions. The lesions of generalized bullous FDE can be similar to those of Stevens-Johnson syndrome/toxic epidermal necrolysis, and the pigmented patches of FDE can resemble large plaque parapsoriasis.
It is important to consider any medication ingested in the 1- to 2-week period before FDE onset, including over-the-counter medications, health food supplements, and prescription medications. Discontinuation of the implicated medication or any medication potentially cross-reacting with another medication is the most important step in management. Wound care may be needed for any bullous or eroded lesions. Lesions typically resolve within a few days to weeks of stopping the offending agent. Importantly, patients should be counseled on the secondary pigment alterations that may be persistent for several months. Other treatment for FDEs is aimed at symptomatic relief and may include topical corticosteroids and oral antihistamines.1
Medroxyprogesterone acetate is a highly effective contraceptive drug with low rates of failure.12 It is a weak androgenic progestin that is administered as a single 150-mg intramuscular injection every 3 months and inhibits gonadotropins. Common side effects include local injection-site reactions, unscheduled bleeding, amenorrhea, weight gain, headache, and mood changes. However, FDE has not been reported as an adverse effect to medroxyprogesterone acetate, both in official US Food and Drug Administration information and in the current literature.12
Autoimmune progesterone dermatitis (also known as progestin hypersensitivity) is a well-characterized cyclic hypersensitivity reaction to the hormone progesterone that occurs during the luteal phase of the menstrual cycle. It is known to have a variable clinical presentation including urticaria, erythema multiforme, eczema, and angioedema.13 Autoimmune progesterone dermatitis also has been reported to present as an FDE.14-16 The onset of the cutaneous manifestation often starts a few days before the onset of menses, with spontaneous resolution occurring after the onset of menstruation. The mechanism by which endogenous progesterone or other secretory products become antigenic is unknown. It has been suggested that there is an alteration in the properties of the hormone that would predispose it to be antigenic as it would not be considered self. In 2001, Warin17 proposed the following diagnostic criteria for autoimmune progesterone dermatitis: (1) skin lesions associated with menstrual cycle (premenstrual flare); (2) a positive response to the progesterone intradermal or intramuscular test; and (3) symptomatic improvement after inhibiting progesterone secretion by suppressing ovulation.17 The treatment includes antiallergy medications, progesterone desensitization, omalizumab injection, and leuprolide acetate injection.
Our case represents FDE from medroxyprogesterone acetate. Although we did not formally investigate the antigenicity of the exogenous progesterone, we postulate that the pathophysiology likely is similar to an FDE associated with endogenous progesterone. This reasoning is supported by the time course of the patient’s lesion as well as the worsening of symptoms in the days following the administration of the medication. Additionally, the patient had no history of skin lesions prior to the initiation of medroxyprogesterone acetate or similar lesions associated with her menstrual cycles.
A careful and detailed review of medication history is necessary to evaluate FDEs. Our case emphasizes that not only endogenous but also exogenous forms of progesterone may cause hypersensitivity, leading to an FDE. With more than 2 million prescriptions of medroxyprogesterone acetate written every year, dermatologists should be aware of the rare but potential risk for an FDE in patients using this medication.18
- Bolognia J, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby; 2008.
- Bourns DCG. Unusual effects of antipyrine. Br Med J. 1889;2:818-820.
- Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
- Sohn KH, Kim BK, Kim JY, et al. Fixed food eruption caused by Actinidia arguta (hardy kiwi): a case report and literature review. Allergy Asthma Immunol Res. 2017;9:182-184.
- Nakai N, Katoh N. Fixed drug eruption caused by fluconazole: a case report and mini-review of the literature. Allergol Int. 2013;6:139-141.
- An I, Demir V, Ibiloglu I, et al. Fixed drug eruption induced by levocetirizine. Indian Dermatol Online J. 2017;8:276-278.
- Matarredona J, Borrás Blasco J, Navarro-Ruiz A, et al. Fixed drug eruption associated to loperamide [in Spanish]. Med Clin (Barc). 2005;124:198-199.
- Gohel D. Fixed drug eruption due to multi-vitamin multi-mineral preparation. J Assoc Physicians India. 2000;48:268.
- Ritter SE, Meffert J. A refractory fixed drug reaction to a dye used in an oral contraceptive. Cutis. 2004;74:243-244.
- Rea S, McMeniman E, Darch K, et al. A fixed drug eruption to the sugar pills of a combined oral contraceptive. Poster presented at: The Australasian College of Dermatologists 51st Annual Scientific Meeting; May 22, 2018; Queensland, Australia.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflicted responses of intraepidermal T cells. Eur J Dermatol. 2007;17:201-208.
- Depo-Provera CI. Prescribing information. Pfizer; 2020. Accessed March 10, 2022. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=522
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report. Case Rep Obstet Gynecol. 2012;2012:757854.
- Mokhtari R, Sepaskhah M, Aslani FS, et al. Autoimmune progesterone dermatitis presenting as fixed drug eruption: a case report. Dermatol Online J. 2017;23:13030/qt685685p4.
- Asai J, Katoh N, Nakano M, et al. Case of autoimmune progesterone dermatitis presenting as fixed drug eruption. J Dermatol. 2009;36:643-645.
- Bhardwaj N, Jindal R, Chauhan P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019;12:E231873.
- Warin AP. Case 2. diagnosis: erythema multiforme as a presentation of autoimmune progesterone dermatitis. Clin Exp Dermatol. 2001;26:107-108.
- Medroxyprogesterone Drug Usage Statistics, United States, 2013-2019. ClinCalc website. Updated September 15, 2021. Accessed March 17, 2022. https://clincalc.com/DrugStats/Drugs/Medroxyprogesterone
To the Editor:
A fixed drug eruption (FDE) is a well-documented form of cutaneous hypersensitivity that typically manifests as a sharply demarcated, dusky, round to oval, edematous, red-violaceous macule or patch on the skin and mucous membranes. The lesion often resolves with residual postinflammatory hyperpigmentation, most commonly as a reaction to ingested drugs or drug components.1 Lesions generally occur at the same anatomic site with repeated exposure to the offending drug. Typically, a single site is affected, but additional sites with more generalized involvement have been reported to occur with subsequent exposure to the offending medication. The diagnosis usually is clinical, but histopathologic findings can help confirm the diagnosis in unusual presentations. We present a novel case of a patient with an FDE from medroxyprogesterone acetate, a contraceptive injection that contains the hormone progestin.
A 35-year-old woman presented to the dermatology clinic for evaluation of a lesion on the left lower buttock of 1 year’s duration. She reported periodic swelling and associated pruritus of the lesion. She denied any growth in size, and no other similar lesions were present. The patient reported a medication history of medroxyprogesterone acetate for birth control, but she denied any other prescription or over-the-counter medication, oral supplements, or recreational drug use. Upon further inquiry, she reported that the recurrence of symptoms appeared to coincide with each administration of medroxyprogesterone acetate, which occurred approximately every 3 months. The eruption cleared between injections and recurred in the same location following subsequent injections. The lesion appeared approximately 2 weeks after the first injection (approximately 1 year prior to presentation to dermatology) and within 2 to 3 days after each subsequent injection. Physical examination revealed a 2×2-cm, circular, slightly violaceous patch on the left buttock (Figure 1). A biopsy was recommended to aid in diagnosis, and the patient was offered a topical steroid for symptomatic relief. A punch biopsy revealed subtle interface dermatitis with superficial perivascular lymphoid infiltrate and marked pigmentary incontinence consistent with an FDE (Figure 2).
An FDE was first reported in 1889 by Bourns,2 and over time more implicated agents and varying clinical presentations have been linked to the disease. The FDE can be accompanied by symptoms of pruritus or paresthesia. Most cases are devoid of systemic symptoms. An FDE can be located anywhere on the body, but it most frequently manifests on the lips, face, hands, feet, and genitalia. Although the eruption often heals with residual postinflammatory hyperpigmentation, a nonpigmenting FDE due to pseudoephedrine has been reported.3
Common culprits include antibiotics (eg, sulfonamides, trimethoprim, fluoroquinolones, tetracyclines), nonsteroidal anti-inflammatory medications (eg, naproxen sodium, ibuprofen, celecoxib), barbiturates, antimalarials, and anticonvulsants. Rare cases of FDE induced by foods and food additives also have been reported.4 Oral fluconazole, levocetirizine dihydrochloride, loperamide, and multivitamin-mineral preparations are other rare inducers of FDE.5-8 In 2004, Ritter and Meffert9 described an FDE to the green dye used in inactive oral contraceptive pills. A similar case was reported by Rea et al10 that described an FDE from the inactive sugar pills in ethinyl estradiol and levonorgestrel, which is another combined oral contraceptive.
The time between ingestion of the offending agent and the manifestation of the disease usually is 1 to 2 weeks; however, upon subsequent exposure, the disease has been reported to manifest within hours.1 CD8+ memory T cells have been shown to be major players in the development of FDE and can be found along the dermoepidermal junction as part of a delayed type IV hypersensitivity reaction.11 Histopathology reveals superficial and deep interstitial and perivascular infiltrates consisting of lymphocytes with admixed eosinophils and possibly neutrophils in the dermis. In the epidermis, necrotic keratinocytes can be present. In rare cases, FDE may have atypical features, such as in generalized bullous FDE and nonpigmenting FDE, the latter of which more commonly is associated with pseudoephedrine.1
The differential diagnosis for FDE includes erythema multiforme, Stevens-Johnson syndrome/toxic epidermal necrolysis, autoimmune progesterone dermatitis, and large plaque parapsoriasis. The number and morphology of lesions in erythema multiforme help differentiate it from FDE, as erythema multiforme presents with multiple targetoid lesions. The lesions of generalized bullous FDE can be similar to those of Stevens-Johnson syndrome/toxic epidermal necrolysis, and the pigmented patches of FDE can resemble large plaque parapsoriasis.
It is important to consider any medication ingested in the 1- to 2-week period before FDE onset, including over-the-counter medications, health food supplements, and prescription medications. Discontinuation of the implicated medication or any medication potentially cross-reacting with another medication is the most important step in management. Wound care may be needed for any bullous or eroded lesions. Lesions typically resolve within a few days to weeks of stopping the offending agent. Importantly, patients should be counseled on the secondary pigment alterations that may be persistent for several months. Other treatment for FDEs is aimed at symptomatic relief and may include topical corticosteroids and oral antihistamines.1
Medroxyprogesterone acetate is a highly effective contraceptive drug with low rates of failure.12 It is a weak androgenic progestin that is administered as a single 150-mg intramuscular injection every 3 months and inhibits gonadotropins. Common side effects include local injection-site reactions, unscheduled bleeding, amenorrhea, weight gain, headache, and mood changes. However, FDE has not been reported as an adverse effect to medroxyprogesterone acetate, both in official US Food and Drug Administration information and in the current literature.12
Autoimmune progesterone dermatitis (also known as progestin hypersensitivity) is a well-characterized cyclic hypersensitivity reaction to the hormone progesterone that occurs during the luteal phase of the menstrual cycle. It is known to have a variable clinical presentation including urticaria, erythema multiforme, eczema, and angioedema.13 Autoimmune progesterone dermatitis also has been reported to present as an FDE.14-16 The onset of the cutaneous manifestation often starts a few days before the onset of menses, with spontaneous resolution occurring after the onset of menstruation. The mechanism by which endogenous progesterone or other secretory products become antigenic is unknown. It has been suggested that there is an alteration in the properties of the hormone that would predispose it to be antigenic as it would not be considered self. In 2001, Warin17 proposed the following diagnostic criteria for autoimmune progesterone dermatitis: (1) skin lesions associated with menstrual cycle (premenstrual flare); (2) a positive response to the progesterone intradermal or intramuscular test; and (3) symptomatic improvement after inhibiting progesterone secretion by suppressing ovulation.17 The treatment includes antiallergy medications, progesterone desensitization, omalizumab injection, and leuprolide acetate injection.
Our case represents FDE from medroxyprogesterone acetate. Although we did not formally investigate the antigenicity of the exogenous progesterone, we postulate that the pathophysiology likely is similar to an FDE associated with endogenous progesterone. This reasoning is supported by the time course of the patient’s lesion as well as the worsening of symptoms in the days following the administration of the medication. Additionally, the patient had no history of skin lesions prior to the initiation of medroxyprogesterone acetate or similar lesions associated with her menstrual cycles.
A careful and detailed review of medication history is necessary to evaluate FDEs. Our case emphasizes that not only endogenous but also exogenous forms of progesterone may cause hypersensitivity, leading to an FDE. With more than 2 million prescriptions of medroxyprogesterone acetate written every year, dermatologists should be aware of the rare but potential risk for an FDE in patients using this medication.18
To the Editor:
A fixed drug eruption (FDE) is a well-documented form of cutaneous hypersensitivity that typically manifests as a sharply demarcated, dusky, round to oval, edematous, red-violaceous macule or patch on the skin and mucous membranes. The lesion often resolves with residual postinflammatory hyperpigmentation, most commonly as a reaction to ingested drugs or drug components.1 Lesions generally occur at the same anatomic site with repeated exposure to the offending drug. Typically, a single site is affected, but additional sites with more generalized involvement have been reported to occur with subsequent exposure to the offending medication. The diagnosis usually is clinical, but histopathologic findings can help confirm the diagnosis in unusual presentations. We present a novel case of a patient with an FDE from medroxyprogesterone acetate, a contraceptive injection that contains the hormone progestin.
A 35-year-old woman presented to the dermatology clinic for evaluation of a lesion on the left lower buttock of 1 year’s duration. She reported periodic swelling and associated pruritus of the lesion. She denied any growth in size, and no other similar lesions were present. The patient reported a medication history of medroxyprogesterone acetate for birth control, but she denied any other prescription or over-the-counter medication, oral supplements, or recreational drug use. Upon further inquiry, she reported that the recurrence of symptoms appeared to coincide with each administration of medroxyprogesterone acetate, which occurred approximately every 3 months. The eruption cleared between injections and recurred in the same location following subsequent injections. The lesion appeared approximately 2 weeks after the first injection (approximately 1 year prior to presentation to dermatology) and within 2 to 3 days after each subsequent injection. Physical examination revealed a 2×2-cm, circular, slightly violaceous patch on the left buttock (Figure 1). A biopsy was recommended to aid in diagnosis, and the patient was offered a topical steroid for symptomatic relief. A punch biopsy revealed subtle interface dermatitis with superficial perivascular lymphoid infiltrate and marked pigmentary incontinence consistent with an FDE (Figure 2).
An FDE was first reported in 1889 by Bourns,2 and over time more implicated agents and varying clinical presentations have been linked to the disease. The FDE can be accompanied by symptoms of pruritus or paresthesia. Most cases are devoid of systemic symptoms. An FDE can be located anywhere on the body, but it most frequently manifests on the lips, face, hands, feet, and genitalia. Although the eruption often heals with residual postinflammatory hyperpigmentation, a nonpigmenting FDE due to pseudoephedrine has been reported.3
Common culprits include antibiotics (eg, sulfonamides, trimethoprim, fluoroquinolones, tetracyclines), nonsteroidal anti-inflammatory medications (eg, naproxen sodium, ibuprofen, celecoxib), barbiturates, antimalarials, and anticonvulsants. Rare cases of FDE induced by foods and food additives also have been reported.4 Oral fluconazole, levocetirizine dihydrochloride, loperamide, and multivitamin-mineral preparations are other rare inducers of FDE.5-8 In 2004, Ritter and Meffert9 described an FDE to the green dye used in inactive oral contraceptive pills. A similar case was reported by Rea et al10 that described an FDE from the inactive sugar pills in ethinyl estradiol and levonorgestrel, which is another combined oral contraceptive.
The time between ingestion of the offending agent and the manifestation of the disease usually is 1 to 2 weeks; however, upon subsequent exposure, the disease has been reported to manifest within hours.1 CD8+ memory T cells have been shown to be major players in the development of FDE and can be found along the dermoepidermal junction as part of a delayed type IV hypersensitivity reaction.11 Histopathology reveals superficial and deep interstitial and perivascular infiltrates consisting of lymphocytes with admixed eosinophils and possibly neutrophils in the dermis. In the epidermis, necrotic keratinocytes can be present. In rare cases, FDE may have atypical features, such as in generalized bullous FDE and nonpigmenting FDE, the latter of which more commonly is associated with pseudoephedrine.1
The differential diagnosis for FDE includes erythema multiforme, Stevens-Johnson syndrome/toxic epidermal necrolysis, autoimmune progesterone dermatitis, and large plaque parapsoriasis. The number and morphology of lesions in erythema multiforme help differentiate it from FDE, as erythema multiforme presents with multiple targetoid lesions. The lesions of generalized bullous FDE can be similar to those of Stevens-Johnson syndrome/toxic epidermal necrolysis, and the pigmented patches of FDE can resemble large plaque parapsoriasis.
It is important to consider any medication ingested in the 1- to 2-week period before FDE onset, including over-the-counter medications, health food supplements, and prescription medications. Discontinuation of the implicated medication or any medication potentially cross-reacting with another medication is the most important step in management. Wound care may be needed for any bullous or eroded lesions. Lesions typically resolve within a few days to weeks of stopping the offending agent. Importantly, patients should be counseled on the secondary pigment alterations that may be persistent for several months. Other treatment for FDEs is aimed at symptomatic relief and may include topical corticosteroids and oral antihistamines.1
Medroxyprogesterone acetate is a highly effective contraceptive drug with low rates of failure.12 It is a weak androgenic progestin that is administered as a single 150-mg intramuscular injection every 3 months and inhibits gonadotropins. Common side effects include local injection-site reactions, unscheduled bleeding, amenorrhea, weight gain, headache, and mood changes. However, FDE has not been reported as an adverse effect to medroxyprogesterone acetate, both in official US Food and Drug Administration information and in the current literature.12
Autoimmune progesterone dermatitis (also known as progestin hypersensitivity) is a well-characterized cyclic hypersensitivity reaction to the hormone progesterone that occurs during the luteal phase of the menstrual cycle. It is known to have a variable clinical presentation including urticaria, erythema multiforme, eczema, and angioedema.13 Autoimmune progesterone dermatitis also has been reported to present as an FDE.14-16 The onset of the cutaneous manifestation often starts a few days before the onset of menses, with spontaneous resolution occurring after the onset of menstruation. The mechanism by which endogenous progesterone or other secretory products become antigenic is unknown. It has been suggested that there is an alteration in the properties of the hormone that would predispose it to be antigenic as it would not be considered self. In 2001, Warin17 proposed the following diagnostic criteria for autoimmune progesterone dermatitis: (1) skin lesions associated with menstrual cycle (premenstrual flare); (2) a positive response to the progesterone intradermal or intramuscular test; and (3) symptomatic improvement after inhibiting progesterone secretion by suppressing ovulation.17 The treatment includes antiallergy medications, progesterone desensitization, omalizumab injection, and leuprolide acetate injection.
Our case represents FDE from medroxyprogesterone acetate. Although we did not formally investigate the antigenicity of the exogenous progesterone, we postulate that the pathophysiology likely is similar to an FDE associated with endogenous progesterone. This reasoning is supported by the time course of the patient’s lesion as well as the worsening of symptoms in the days following the administration of the medication. Additionally, the patient had no history of skin lesions prior to the initiation of medroxyprogesterone acetate or similar lesions associated with her menstrual cycles.
A careful and detailed review of medication history is necessary to evaluate FDEs. Our case emphasizes that not only endogenous but also exogenous forms of progesterone may cause hypersensitivity, leading to an FDE. With more than 2 million prescriptions of medroxyprogesterone acetate written every year, dermatologists should be aware of the rare but potential risk for an FDE in patients using this medication.18
- Bolognia J, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby; 2008.
- Bourns DCG. Unusual effects of antipyrine. Br Med J. 1889;2:818-820.
- Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
- Sohn KH, Kim BK, Kim JY, et al. Fixed food eruption caused by Actinidia arguta (hardy kiwi): a case report and literature review. Allergy Asthma Immunol Res. 2017;9:182-184.
- Nakai N, Katoh N. Fixed drug eruption caused by fluconazole: a case report and mini-review of the literature. Allergol Int. 2013;6:139-141.
- An I, Demir V, Ibiloglu I, et al. Fixed drug eruption induced by levocetirizine. Indian Dermatol Online J. 2017;8:276-278.
- Matarredona J, Borrás Blasco J, Navarro-Ruiz A, et al. Fixed drug eruption associated to loperamide [in Spanish]. Med Clin (Barc). 2005;124:198-199.
- Gohel D. Fixed drug eruption due to multi-vitamin multi-mineral preparation. J Assoc Physicians India. 2000;48:268.
- Ritter SE, Meffert J. A refractory fixed drug reaction to a dye used in an oral contraceptive. Cutis. 2004;74:243-244.
- Rea S, McMeniman E, Darch K, et al. A fixed drug eruption to the sugar pills of a combined oral contraceptive. Poster presented at: The Australasian College of Dermatologists 51st Annual Scientific Meeting; May 22, 2018; Queensland, Australia.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflicted responses of intraepidermal T cells. Eur J Dermatol. 2007;17:201-208.
- Depo-Provera CI. Prescribing information. Pfizer; 2020. Accessed March 10, 2022. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=522
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report. Case Rep Obstet Gynecol. 2012;2012:757854.
- Mokhtari R, Sepaskhah M, Aslani FS, et al. Autoimmune progesterone dermatitis presenting as fixed drug eruption: a case report. Dermatol Online J. 2017;23:13030/qt685685p4.
- Asai J, Katoh N, Nakano M, et al. Case of autoimmune progesterone dermatitis presenting as fixed drug eruption. J Dermatol. 2009;36:643-645.
- Bhardwaj N, Jindal R, Chauhan P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019;12:E231873.
- Warin AP. Case 2. diagnosis: erythema multiforme as a presentation of autoimmune progesterone dermatitis. Clin Exp Dermatol. 2001;26:107-108.
- Medroxyprogesterone Drug Usage Statistics, United States, 2013-2019. ClinCalc website. Updated September 15, 2021. Accessed March 17, 2022. https://clincalc.com/DrugStats/Drugs/Medroxyprogesterone
- Bolognia J, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby; 2008.
- Bourns DCG. Unusual effects of antipyrine. Br Med J. 1889;2:818-820.
- Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
- Sohn KH, Kim BK, Kim JY, et al. Fixed food eruption caused by Actinidia arguta (hardy kiwi): a case report and literature review. Allergy Asthma Immunol Res. 2017;9:182-184.
- Nakai N, Katoh N. Fixed drug eruption caused by fluconazole: a case report and mini-review of the literature. Allergol Int. 2013;6:139-141.
- An I, Demir V, Ibiloglu I, et al. Fixed drug eruption induced by levocetirizine. Indian Dermatol Online J. 2017;8:276-278.
- Matarredona J, Borrás Blasco J, Navarro-Ruiz A, et al. Fixed drug eruption associated to loperamide [in Spanish]. Med Clin (Barc). 2005;124:198-199.
- Gohel D. Fixed drug eruption due to multi-vitamin multi-mineral preparation. J Assoc Physicians India. 2000;48:268.
- Ritter SE, Meffert J. A refractory fixed drug reaction to a dye used in an oral contraceptive. Cutis. 2004;74:243-244.
- Rea S, McMeniman E, Darch K, et al. A fixed drug eruption to the sugar pills of a combined oral contraceptive. Poster presented at: The Australasian College of Dermatologists 51st Annual Scientific Meeting; May 22, 2018; Queensland, Australia.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflicted responses of intraepidermal T cells. Eur J Dermatol. 2007;17:201-208.
- Depo-Provera CI. Prescribing information. Pfizer; 2020. Accessed March 10, 2022. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=522
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report. Case Rep Obstet Gynecol. 2012;2012:757854.
- Mokhtari R, Sepaskhah M, Aslani FS, et al. Autoimmune progesterone dermatitis presenting as fixed drug eruption: a case report. Dermatol Online J. 2017;23:13030/qt685685p4.
- Asai J, Katoh N, Nakano M, et al. Case of autoimmune progesterone dermatitis presenting as fixed drug eruption. J Dermatol. 2009;36:643-645.
- Bhardwaj N, Jindal R, Chauhan P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019;12:E231873.
- Warin AP. Case 2. diagnosis: erythema multiforme as a presentation of autoimmune progesterone dermatitis. Clin Exp Dermatol. 2001;26:107-108.
- Medroxyprogesterone Drug Usage Statistics, United States, 2013-2019. ClinCalc website. Updated September 15, 2021. Accessed March 17, 2022. https://clincalc.com/DrugStats/Drugs/Medroxyprogesterone
Practice Points
- Exogenous progesterone from the administration of the contraceptive injectable medroxyprogesterone acetate has the potential to cause a cutaneous hypersensitivity reaction in the form of a fixed drug eruption (FDE).
- Dermatologists should perform a careful and detailed review of medication history to evaluate drug eruptions.

Q&A With JAAD Editor Dirk M. Elston, MD
who has authored more than 600 peer-reviewed publications and 92 textbook chapters.
After earning his undergraduate degree from Pennsylvania State University and his medical degree from Jefferson Medical College in Philadelphia, Dr. Elston completed an internship and a dermatology residency at Walter Reed Army Medical Center in Washington, as well as a dermatopathology fellowship at the Cleveland Clinic. He currently is professor and chair of the department of dermatology and dermatologic surgery at the Medical University of South Carolina in Charleston.

Dr. Elston is one of five authors of “Andrews’ Diseases of the Skin),” coauthor with Tammie Ferringer, MD, of the “Dermatopathology” textbook, and editor in chief of the Requisites in Dermatology series of textbooks. In 2018, he succeeded Bruce H. Thiers, MD, as editor of the Journal of the American Academy of Dermatology and in 2021, received the AAD’s Gold Medal Award, which is the academy’s highest honor.
In an interview, Dr. Elston reflected on his mentors, shared how he manages his many responsibilities as a clinician, teacher, and editor, and talked about the promising future of dermatology.
Who inspired you most to pursue a career in medicine? My grandmother, Annie Elston, was a physician and dedicated her life to helping others. She was a front-line medic during World War I, helped to run a neonatal syphilis ward after the war, and practiced pediatrics in New York City until her death. She was a great role model.
Did you enter medical school knowing that you wanted to become a dermatologist? If not, what was the turning point for you? I didn’t really know much about dermatology when I entered medical school. I fell in love with the specialty during a rotation.
What was the most memorable experience from your dermatology residency at Walter Reed Army Medical Center? There were so many interesting patients, including many tropical diseases.
Why did you choose to pursue a fellowship in dermatopathology? What was it about this subspeciality that piqued your interest? Great teachers, including Tim Berger, MD, George Lupton, MD, and Dean Pearson, MD. They inspired me to seek a dermpath fellowship and I was lucky enough to train with Wilma Bergfeld, MD.
In your opinion, what’s been the most important advance in dermatopathology to date?
Immunohistochemistry changed the specialty. Now molecular diagnostics is a second wave of major advancement.
How do you stay passionate about both dermatology and dermatopathology? The patients, residents, and fellows keep it interesting. It’s a two-way street. I learn as much as I teach.
You’ve had a remarkable run at the Journal of the American Academy of Dermatology, starting as deputy editor in 2008 before becoming editor in 2018. What’s been most rewarding about this role for you? It is a labor of love and such a privilege to see everyone’s best work.
During the peak of the COVID-19 pandemic, what were your most significant challenges from both a clinical and a personal standpoint? Fear of the unknown is always a challenge with a new epidemic and worse with a pandemic. The patients still needed to be seen but it was a challenge with some buildings closed and some personnel afraid to come to work.
Is there anything you would tell your younger self in terms of career advice? Enjoy every step of the journey.
Considering your various work responsibilities as a clinician, teacher, and editor, what’s your strategy for achieving a work-life balance? A good friend of mine is fond of saying that balance is an illusion. There is only resilience. I believe the truth lies somewhere in between. Make time for family, and decide what has to get done today and what can wait until tomorrow.
What development in dermatology are you most excited about in the next 5 years? We are in a golden age of therapeutic innovations that are life changing and lifesaving for our patients. I never would have believed I would see complete cures of patients with widely metastatic melanoma. From psoriasis to eczema to malignancy, our therapeutic armamentarium is dramatically better each year. It makes the practice of medicine exciting.
who has authored more than 600 peer-reviewed publications and 92 textbook chapters.
After earning his undergraduate degree from Pennsylvania State University and his medical degree from Jefferson Medical College in Philadelphia, Dr. Elston completed an internship and a dermatology residency at Walter Reed Army Medical Center in Washington, as well as a dermatopathology fellowship at the Cleveland Clinic. He currently is professor and chair of the department of dermatology and dermatologic surgery at the Medical University of South Carolina in Charleston.
Dr. Elston is one of five authors of “Andrews’ Diseases of the Skin),” coauthor with Tammie Ferringer, MD, of the “Dermatopathology” textbook, and editor in chief of the Requisites in Dermatology series of textbooks. In 2018, he succeeded Bruce H. Thiers, MD, as editor of the Journal of the American Academy of Dermatology and in 2021, received the AAD’s Gold Medal Award, which is the academy’s highest honor.
In an interview, Dr. Elston reflected on his mentors, shared how he manages his many responsibilities as a clinician, teacher, and editor, and talked about the promising future of dermatology.
Who inspired you most to pursue a career in medicine? My grandmother, Annie Elston, was a physician and dedicated her life to helping others. She was a front-line medic during World War I, helped to run a neonatal syphilis ward after the war, and practiced pediatrics in New York City until her death. She was a great role model.
Did you enter medical school knowing that you wanted to become a dermatologist? If not, what was the turning point for you? I didn’t really know much about dermatology when I entered medical school. I fell in love with the specialty during a rotation.
What was the most memorable experience from your dermatology residency at Walter Reed Army Medical Center? There were so many interesting patients, including many tropical diseases.
Why did you choose to pursue a fellowship in dermatopathology? What was it about this subspeciality that piqued your interest? Great teachers, including Tim Berger, MD, George Lupton, MD, and Dean Pearson, MD. They inspired me to seek a dermpath fellowship and I was lucky enough to train with Wilma Bergfeld, MD.
In your opinion, what’s been the most important advance in dermatopathology to date?
Immunohistochemistry changed the specialty. Now molecular diagnostics is a second wave of major advancement.
How do you stay passionate about both dermatology and dermatopathology? The patients, residents, and fellows keep it interesting. It’s a two-way street. I learn as much as I teach.
You’ve had a remarkable run at the Journal of the American Academy of Dermatology, starting as deputy editor in 2008 before becoming editor in 2018. What’s been most rewarding about this role for you? It is a labor of love and such a privilege to see everyone’s best work.
During the peak of the COVID-19 pandemic, what were your most significant challenges from both a clinical and a personal standpoint? Fear of the unknown is always a challenge with a new epidemic and worse with a pandemic. The patients still needed to be seen but it was a challenge with some buildings closed and some personnel afraid to come to work.
Is there anything you would tell your younger self in terms of career advice? Enjoy every step of the journey.
Considering your various work responsibilities as a clinician, teacher, and editor, what’s your strategy for achieving a work-life balance? A good friend of mine is fond of saying that balance is an illusion. There is only resilience. I believe the truth lies somewhere in between. Make time for family, and decide what has to get done today and what can wait until tomorrow.
What development in dermatology are you most excited about in the next 5 years? We are in a golden age of therapeutic innovations that are life changing and lifesaving for our patients. I never would have believed I would see complete cures of patients with widely metastatic melanoma. From psoriasis to eczema to malignancy, our therapeutic armamentarium is dramatically better each year. It makes the practice of medicine exciting.
who has authored more than 600 peer-reviewed publications and 92 textbook chapters.
After earning his undergraduate degree from Pennsylvania State University and his medical degree from Jefferson Medical College in Philadelphia, Dr. Elston completed an internship and a dermatology residency at Walter Reed Army Medical Center in Washington, as well as a dermatopathology fellowship at the Cleveland Clinic. He currently is professor and chair of the department of dermatology and dermatologic surgery at the Medical University of South Carolina in Charleston.
Dr. Elston is one of five authors of “Andrews’ Diseases of the Skin),” coauthor with Tammie Ferringer, MD, of the “Dermatopathology” textbook, and editor in chief of the Requisites in Dermatology series of textbooks. In 2018, he succeeded Bruce H. Thiers, MD, as editor of the Journal of the American Academy of Dermatology and in 2021, received the AAD’s Gold Medal Award, which is the academy’s highest honor.
In an interview, Dr. Elston reflected on his mentors, shared how he manages his many responsibilities as a clinician, teacher, and editor, and talked about the promising future of dermatology.
Who inspired you most to pursue a career in medicine? My grandmother, Annie Elston, was a physician and dedicated her life to helping others. She was a front-line medic during World War I, helped to run a neonatal syphilis ward after the war, and practiced pediatrics in New York City until her death. She was a great role model.
Did you enter medical school knowing that you wanted to become a dermatologist? If not, what was the turning point for you? I didn’t really know much about dermatology when I entered medical school. I fell in love with the specialty during a rotation.
What was the most memorable experience from your dermatology residency at Walter Reed Army Medical Center? There were so many interesting patients, including many tropical diseases.
Why did you choose to pursue a fellowship in dermatopathology? What was it about this subspeciality that piqued your interest? Great teachers, including Tim Berger, MD, George Lupton, MD, and Dean Pearson, MD. They inspired me to seek a dermpath fellowship and I was lucky enough to train with Wilma Bergfeld, MD.
In your opinion, what’s been the most important advance in dermatopathology to date?
Immunohistochemistry changed the specialty. Now molecular diagnostics is a second wave of major advancement.
How do you stay passionate about both dermatology and dermatopathology? The patients, residents, and fellows keep it interesting. It’s a two-way street. I learn as much as I teach.
You’ve had a remarkable run at the Journal of the American Academy of Dermatology, starting as deputy editor in 2008 before becoming editor in 2018. What’s been most rewarding about this role for you? It is a labor of love and such a privilege to see everyone’s best work.
During the peak of the COVID-19 pandemic, what were your most significant challenges from both a clinical and a personal standpoint? Fear of the unknown is always a challenge with a new epidemic and worse with a pandemic. The patients still needed to be seen but it was a challenge with some buildings closed and some personnel afraid to come to work.
Is there anything you would tell your younger self in terms of career advice? Enjoy every step of the journey.
Considering your various work responsibilities as a clinician, teacher, and editor, what’s your strategy for achieving a work-life balance? A good friend of mine is fond of saying that balance is an illusion. There is only resilience. I believe the truth lies somewhere in between. Make time for family, and decide what has to get done today and what can wait until tomorrow.
What development in dermatology are you most excited about in the next 5 years? We are in a golden age of therapeutic innovations that are life changing and lifesaving for our patients. I never would have believed I would see complete cures of patients with widely metastatic melanoma. From psoriasis to eczema to malignancy, our therapeutic armamentarium is dramatically better each year. It makes the practice of medicine exciting.
Painful Ulcerating Lesions on the Breast
The Diagnosis: Cystic Neutrophilic Granulomatous Mastitis
The histopathologic findings in our patient were characteristic of cystic neutrophilic granulomatous mastitis (CNGM), a rare granulomatous mastitis associated with Corynebacterium and suppurative lipogranulomas. Although not seen in our patient, the lipid vacuoles may contain gram-positive bacilli.1 The surrounding mixed inflammatory infiltrate contains Langerhans giant cells, lymphocytes, and neutrophils. Cystic neutrophilic granulomatous mastitis is seen in parous women of reproductive age. Physical examination demonstrates a palpable painful mass on the breast. Wound cultures frequently are negative, likely due to difficulty culturing Corynebacterium and prophylactic antibiotic treatment. Given the association with Corynebacterium species, early diagnosis of CNGM is essential in offering patients the most appropriate treatment. Prolonged antibiotic therapy specifically directed to corynebacteria is required, sometimes even beyond resolution of clinical symptoms. The diagnosis of CNGM often is missed or delayed due to its rarity and many potential mimickers. Clinically, CNGM may be virtually impossible to discern from invasive carcinoma.1
Our patient was treated with vancomycin and cefepime with incision and drainage as an inpatient. Upon discharge, she was started on prednisone 1 mg/kg daily tapered by 10 mg every 5 days over 1 month and doxycycline 100 mg twice daily. She was then transitioned to topical hydrocortisone and bacitracin; she reported decreased swelling and pain. No new lesions formed after the initiation of therapy; however, most lesions remained open. Cystic neutrophilic granulomatous mastitis remains a challenging entity to treat, with a variable response rate reported in the literature for antibiotics such as doxycycline and systemic and topical steroids as well as immunosuppressants including methotrexate.2,3
Cystic neutrophilic granulomatous mastitis can be distinguished from hidradenitis suppurativa clinically because ulcerating lesions can involve the superior portions of the breast in CNGM, whereas hidradenitis suppurativa typically is restricted to the lower intertriginous parts of the breast. Other mimics of CNGM can be distinguished with biopsy. Histology of pyoderma gangrenosum lacks prominent granuloma formation. Although sarcoidosis and mycobacterial infection show prominent granulomas, neither show the characteristic lipogranulomas seen in CNGM. Additionally, the granulomas of sarcoidosis are much larger and deeper than CNGM. Mycobacterial granulomas also typically reveal bacilli with acid-fast bacilli staining or via wound culture.
- Wu JM, Turashvili G. Cystic neutrophilic granulomatous mastitis: an update. J Clin Pathol. 2020;73:445-453. doi:10.1136/jclinpath-2019-206180
- Steuer AB, Stern MJ, Cobos G, et al. Clinical characteristics and medical management of idiopathic granulomatous mastitis. JAMA Dermatol. 2020;156:460-464. doi:10.1001/jamadermatol.2019.4516
- Dobinson HC, Anderson TP, Chambers ST, et al. Antimicrobial treatment options for granulomatous mastitis caused by Corynebacterium species [published online July 1, 2015]. J Clin Microbiol. 2015;53:2895-2899. doi:10.1128/JCM.00760-15
The Diagnosis: Cystic Neutrophilic Granulomatous Mastitis
The histopathologic findings in our patient were characteristic of cystic neutrophilic granulomatous mastitis (CNGM), a rare granulomatous mastitis associated with Corynebacterium and suppurative lipogranulomas. Although not seen in our patient, the lipid vacuoles may contain gram-positive bacilli.1 The surrounding mixed inflammatory infiltrate contains Langerhans giant cells, lymphocytes, and neutrophils. Cystic neutrophilic granulomatous mastitis is seen in parous women of reproductive age. Physical examination demonstrates a palpable painful mass on the breast. Wound cultures frequently are negative, likely due to difficulty culturing Corynebacterium and prophylactic antibiotic treatment. Given the association with Corynebacterium species, early diagnosis of CNGM is essential in offering patients the most appropriate treatment. Prolonged antibiotic therapy specifically directed to corynebacteria is required, sometimes even beyond resolution of clinical symptoms. The diagnosis of CNGM often is missed or delayed due to its rarity and many potential mimickers. Clinically, CNGM may be virtually impossible to discern from invasive carcinoma.1
Our patient was treated with vancomycin and cefepime with incision and drainage as an inpatient. Upon discharge, she was started on prednisone 1 mg/kg daily tapered by 10 mg every 5 days over 1 month and doxycycline 100 mg twice daily. She was then transitioned to topical hydrocortisone and bacitracin; she reported decreased swelling and pain. No new lesions formed after the initiation of therapy; however, most lesions remained open. Cystic neutrophilic granulomatous mastitis remains a challenging entity to treat, with a variable response rate reported in the literature for antibiotics such as doxycycline and systemic and topical steroids as well as immunosuppressants including methotrexate.2,3
Cystic neutrophilic granulomatous mastitis can be distinguished from hidradenitis suppurativa clinically because ulcerating lesions can involve the superior portions of the breast in CNGM, whereas hidradenitis suppurativa typically is restricted to the lower intertriginous parts of the breast. Other mimics of CNGM can be distinguished with biopsy. Histology of pyoderma gangrenosum lacks prominent granuloma formation. Although sarcoidosis and mycobacterial infection show prominent granulomas, neither show the characteristic lipogranulomas seen in CNGM. Additionally, the granulomas of sarcoidosis are much larger and deeper than CNGM. Mycobacterial granulomas also typically reveal bacilli with acid-fast bacilli staining or via wound culture.
The Diagnosis: Cystic Neutrophilic Granulomatous Mastitis
The histopathologic findings in our patient were characteristic of cystic neutrophilic granulomatous mastitis (CNGM), a rare granulomatous mastitis associated with Corynebacterium and suppurative lipogranulomas. Although not seen in our patient, the lipid vacuoles may contain gram-positive bacilli.1 The surrounding mixed inflammatory infiltrate contains Langerhans giant cells, lymphocytes, and neutrophils. Cystic neutrophilic granulomatous mastitis is seen in parous women of reproductive age. Physical examination demonstrates a palpable painful mass on the breast. Wound cultures frequently are negative, likely due to difficulty culturing Corynebacterium and prophylactic antibiotic treatment. Given the association with Corynebacterium species, early diagnosis of CNGM is essential in offering patients the most appropriate treatment. Prolonged antibiotic therapy specifically directed to corynebacteria is required, sometimes even beyond resolution of clinical symptoms. The diagnosis of CNGM often is missed or delayed due to its rarity and many potential mimickers. Clinically, CNGM may be virtually impossible to discern from invasive carcinoma.1
Our patient was treated with vancomycin and cefepime with incision and drainage as an inpatient. Upon discharge, she was started on prednisone 1 mg/kg daily tapered by 10 mg every 5 days over 1 month and doxycycline 100 mg twice daily. She was then transitioned to topical hydrocortisone and bacitracin; she reported decreased swelling and pain. No new lesions formed after the initiation of therapy; however, most lesions remained open. Cystic neutrophilic granulomatous mastitis remains a challenging entity to treat, with a variable response rate reported in the literature for antibiotics such as doxycycline and systemic and topical steroids as well as immunosuppressants including methotrexate.2,3
Cystic neutrophilic granulomatous mastitis can be distinguished from hidradenitis suppurativa clinically because ulcerating lesions can involve the superior portions of the breast in CNGM, whereas hidradenitis suppurativa typically is restricted to the lower intertriginous parts of the breast. Other mimics of CNGM can be distinguished with biopsy. Histology of pyoderma gangrenosum lacks prominent granuloma formation. Although sarcoidosis and mycobacterial infection show prominent granulomas, neither show the characteristic lipogranulomas seen in CNGM. Additionally, the granulomas of sarcoidosis are much larger and deeper than CNGM. Mycobacterial granulomas also typically reveal bacilli with acid-fast bacilli staining or via wound culture.
- Wu JM, Turashvili G. Cystic neutrophilic granulomatous mastitis: an update. J Clin Pathol. 2020;73:445-453. doi:10.1136/jclinpath-2019-206180
- Steuer AB, Stern MJ, Cobos G, et al. Clinical characteristics and medical management of idiopathic granulomatous mastitis. JAMA Dermatol. 2020;156:460-464. doi:10.1001/jamadermatol.2019.4516
- Dobinson HC, Anderson TP, Chambers ST, et al. Antimicrobial treatment options for granulomatous mastitis caused by Corynebacterium species [published online July 1, 2015]. J Clin Microbiol. 2015;53:2895-2899. doi:10.1128/JCM.00760-15
- Wu JM, Turashvili G. Cystic neutrophilic granulomatous mastitis: an update. J Clin Pathol. 2020;73:445-453. doi:10.1136/jclinpath-2019-206180
- Steuer AB, Stern MJ, Cobos G, et al. Clinical characteristics and medical management of idiopathic granulomatous mastitis. JAMA Dermatol. 2020;156:460-464. doi:10.1001/jamadermatol.2019.4516
- Dobinson HC, Anderson TP, Chambers ST, et al. Antimicrobial treatment options for granulomatous mastitis caused by Corynebacterium species [published online July 1, 2015]. J Clin Microbiol. 2015;53:2895-2899. doi:10.1128/JCM.00760-15
A 36-year-old puerperal woman presented with painful, unilateral, ulcerating breast lesions (top) of 3 months’ duration that developed during pregnancy and drained pus with blood. No improvement was seen with antibiotics or incision and drainage. Biopsy of a lesion showed stellate granulomas with cystic spaces and suppurative lipogranulomas where central lipid vacuoles were rimmed by neutrophils and an outer cuff of epithelioid histiocytes (bottom). Acid-fast bacilli, Grocott-Gomori methenamine-silver, Gram, and Steiner staining did not reveal any microorganisms. Additionally, wound cultures were negative.


Isolated Nodule and Generalized Lymphadenopathy
The Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm
A diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN) was rendered. Subsequent needle core biopsy of a left axillary lymph node as well as bone marrow aspiration and biopsy revealed a similar diffuse blastoid infiltrate with an identical immunophenotype to that in the skin biopsy from the pretibial mass and peripheral blood.
Previously known as blastic natural killer cell leukemia/lymphoma or agranular CD4+/CD56+ hematodermic neoplasm/tumor, BPDCN is a rare, clinically aggressive hematologic malignancy derived from the precursors of plasmacytoid dendritic cells. It often is diagnostically challenging, particularly when presenting at noncutaneous sites and in unusual (young) patient populations.1 It was included with other myeloid neoplasms in the 2008 World Health Organization classification; however, in the 2017 classification it was categorized as a separate entity. Blastic plasmacytoid dendritic cell neoplasm typically presents in the skin of elderly patients (age range at diagnosis, 61–67 years) with or without bone marrow involvement and systemic dissemination.1,2 The skin is the most common clinical site of disease in typical cases of BPDCN and often precedes bone marrow involvement. Thus, skin biopsy often is the key to making the diagnosis. Diagnosis of BPDCN may be delayed because of diagnostic pitfalls. Patients usually present with asymptomatic solitary or multiple lesions.3-5 Blastic plasmacytoid dendritic cell neoplasm can present as an isolated purplish nodule or bruiselike papule or more commonly as disseminated purplish nodules, papules, and macules. Isolated nodules are found on the head and lower limbs and can be more than 10 cm in diameter. Peripheral blood and bone marrow may be minimally involved at presentation but invariably become involved with the progression of disease. Cytopenia can occur at diagnosis and in a minority of severe cases indicates bone marrow failure.2-6
Skin involvement of BPDCN is thought to be secondary to the expression of skin migration molecules, such as cutaneous lymphocyte-associated antigen, one of the E-selectin ligands, which binds to E-selectin on high endothelial venules. In addition, the local dermal microenvironment of chemokines binding CXCR3, CXCR4, CCR6, or CCR7 present on neoplastic cells possibly leads to skin involvement. The full mechanism underlying the cutaneous tropism is still to be elucidated.4-7 Infiltration of the oral mucosa is seen in some patients, but it may be underreported. Mucosal disease typically appears similarly to cutaneous disease.
The cutaneous differential diagnosis for BPDCN depends on the clinical presentation, extent of disease spread, and thickness of infiltration. It includes common nonneoplastic diseases such as traumatic ecchymoses; purpuric disorders; extramedullary hematopoiesis; and soft-tissue neoplasms such as angiosarcoma, Kaposi sarcoma, neuroblastoma, and vascular metastases, as well as skin involvement by other hematologic neoplasms. An adequate incisional biopsy rather than a punch or shave biopsy is recommended for diagnosis. Dermatologists should alert the pathologist that BPDCN is in the clinical differential diagnosis when possible so that judicious use of appropriate immunophenotypic markers such as CD123, CD4, CD56, and T-cell leukemia/lymphoma protein 1 will avoid misdiagnosis of this aggressive condition, in addition to excluding acute myeloid leukemia, which also may express 3 of the above markers. However, most cases of acute myeloid leukemia lack terminal deoxynucleotidyl transferase (TdT) and express monocytic and other myeloid markers. Terminal deoxynucleotidyl transferase is positive in approximately one-third of cases of BPDCN, with expression in 10% to 80% of cells.1
It is important to include BPDCN in the differential diagnosis of immunophenotypically aberrant hematologic tumors. Diffuse large B-cell lymphoma, leg type, accounts for 4% of all primary cutaneous B-cell lymphomas.1 Compared with BPDCN, diffuse large B-cell lymphoma usually occurs in an older age group and is of B-cell lineage. Morphologically, these neoplasms are composed of a monotonous, diffuse, nonepidermotropic infiltrate of confluent sheets of centroblasts and immunoblasts (Figure 1). They may share immunohistochemical markers of CD79a, multiple myeloma 1, Bcl-2, and Bcl-6; however, they lack plasmacytoid dendritic cell (PDC)– associated antigens such as CD4, CD56, CD123, and T-cell leukemia/lymphoma protein 1.1

Adult T-cell leukemia/lymphoma is a neoplasm histologically composed of highly pleomorphic medium- to large-sized T cells with an irregular multilobated nuclear contour, so-called flower cells, in the peripheral blood. The nuclear chromatin is coarse and clumped with prominent nucleoli. Blastlike cells with dispersed chromatin are present in variable proportions. Most patients present with widespread lymph node and peripheral blood involvement. Skin is involved in more than half of patients with an epidermal as well as dermal pattern of infiltration (mainly perivascular)(Figure 2). Adult T-cell leukemia/lymphoma is endemic in several regions of the world, and the distribution is closely linked to the prevalence of human T-cell lymphotropic virus type 1 in the population. This neoplasm is of T-cell lineage and may share CD4 but not PDC-associated antigens with BPDCN.1

Cutaneous involvement by T-cell lymphoblastic leukemia/lymphoma (T-LBL) is a rare occurrence with a frequency of approximately 4.3%.8 T-cell lymphoblastic leukemia/lymphoma usually presents as multiple skin lesions throughout the body. Almost all cutaneous T-LBL cases are seen in association with bone marrow and/or mediastinal, lymph node, or extranodal involvement. Cutaneous T-LBLs present as a diffuse monomorphous infiltrate located in the entire dermis and subcutis without epidermotropism, composed of medium to large blasts with finely dispersed chromatin and relatively prominent nucleoli (Figure 3). Immunophenotyping studies show an immature T-cell immunophenotype, with expression of TdT (usually uniform), CD7, and cytoplasmic CD3 and an absence of PDC-associated antigens.8

Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a neoplasm primarily involving the skin. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection. Histologically, they show variable epidermotropism as well as dermal and subcutaneous involvement by medium to large cells with coarse clumped chromatin (Figure 4). Large blastic cells with vesicular nuclei and prominent nucleoli are infrequent. In contrast to BPCDN, the neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are positive for T-cell intracellular antigen-1 and granzyme B with loss of CD4.9

At the time of presentation, 27% to 87% of BPDCN patients will have bone marrow involvement, 22% to 28% will have blood involvement, and 6% to 41% will have lymph node involvement.1-4,6,7,10,11 The clinical course is aggressive, with a median survival of 10.0 to 19.8 months, irrespective of the initial pattern of disease.1 Most cases have shown initial response to multiagent chemotherapy, but relapses with subsequent resistance to drugs regularly have been observed. Age has an adverse impact of prognosis. Low TdT expression has been associated with shorter survival.1 Approximately 10% to 20% of cases of BPDCN are associated with or develop into chronic myelogenous leukemia, myelodysplastic syndrome, or acute myeloid leukemia.1,4 Pediatric patients have a greater 5-year overall survival rate than older patients, and overall survival worsens with increasing age. The extent of cutaneous involvement and presence of systemic involvement at initial presentation do not seem to be strong predictors of survival.1,2,5-7,10-12 In a retrospective analysis of 90 patients, Julia et al12 found that the type of skin disease did not predict survival. Specifically, the presence of nodular lesions and disseminated skin involvement were not adverse prognostic factors compared with macular lesions limited to 1 or 2 body areas.12
- Facchetti F, Petrella T, Pileri SA. Blastic plasmacytoid dendritic cells neoplasm. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. World Health Organization; 2017:174-177.
- Jegalian AG, Facchetti F, Jaffe ES. Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv Anat Pathol. 2009;16:392-404.
- Shi Y, Wang E. Blastic plasmacytoid dendritic cell neoplasm: a clinicopathologic review. Arch Pathol Lab Med. 2014;138:564-569.
- Khoury JD, Medeiros LJ, Manning JT, et al. CD56(+) TdT(+) blastic natural killer cell tumor of the skin: a primitive systemic malignancy related to myelomonocytic leukemia. Cancer. 2002;94:2401-2408.
- Kolerova A, Sergeeva I, Krinitsyna J, et al. Blastic plasmacytoid dendritic cell neoplasm: case report and literature overview. Indian J Dermatol. 2020;65:217-221.
- Hirner JP, O’Malley JT, LeBoeuf NR. Blastic plasmacytoid dendritic cell neoplasm: the dermatologist’s perspective. Hematol Oncol Clin North Am. 2020;34:501-509.
- Guiducii C, Tripodo C, Gong M, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207:2931-2942.
- Khurana S, Beltran M, Jiang L, et al. Primary cutaneous T-cell lymphoblastic lymphoma: case report and literature review. Case Rep Hematol. 2019;2019:3540487. doi:10.1155/2019/3540487
- Gladys TE, Helm MF, Anderson BE, et al. Rapid onset of widespread nodules and lymphadenopathy. Cutis. 2020;106:132, 153-155.
- Gregorio J, Meller S, Conrad C, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. 2010;207:2921-2930.
- Guru Murthy GS, Pemmaraju N, Attallah E. Epidemiology and survival of blastic plasmacytoid dendritic cell neoplasm. Leuk Res. 2018;73:21-23.
- Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2012;169:579-586.
The Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm
A diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN) was rendered. Subsequent needle core biopsy of a left axillary lymph node as well as bone marrow aspiration and biopsy revealed a similar diffuse blastoid infiltrate with an identical immunophenotype to that in the skin biopsy from the pretibial mass and peripheral blood.
Previously known as blastic natural killer cell leukemia/lymphoma or agranular CD4+/CD56+ hematodermic neoplasm/tumor, BPDCN is a rare, clinically aggressive hematologic malignancy derived from the precursors of plasmacytoid dendritic cells. It often is diagnostically challenging, particularly when presenting at noncutaneous sites and in unusual (young) patient populations.1 It was included with other myeloid neoplasms in the 2008 World Health Organization classification; however, in the 2017 classification it was categorized as a separate entity. Blastic plasmacytoid dendritic cell neoplasm typically presents in the skin of elderly patients (age range at diagnosis, 61–67 years) with or without bone marrow involvement and systemic dissemination.1,2 The skin is the most common clinical site of disease in typical cases of BPDCN and often precedes bone marrow involvement. Thus, skin biopsy often is the key to making the diagnosis. Diagnosis of BPDCN may be delayed because of diagnostic pitfalls. Patients usually present with asymptomatic solitary or multiple lesions.3-5 Blastic plasmacytoid dendritic cell neoplasm can present as an isolated purplish nodule or bruiselike papule or more commonly as disseminated purplish nodules, papules, and macules. Isolated nodules are found on the head and lower limbs and can be more than 10 cm in diameter. Peripheral blood and bone marrow may be minimally involved at presentation but invariably become involved with the progression of disease. Cytopenia can occur at diagnosis and in a minority of severe cases indicates bone marrow failure.2-6
Skin involvement of BPDCN is thought to be secondary to the expression of skin migration molecules, such as cutaneous lymphocyte-associated antigen, one of the E-selectin ligands, which binds to E-selectin on high endothelial venules. In addition, the local dermal microenvironment of chemokines binding CXCR3, CXCR4, CCR6, or CCR7 present on neoplastic cells possibly leads to skin involvement. The full mechanism underlying the cutaneous tropism is still to be elucidated.4-7 Infiltration of the oral mucosa is seen in some patients, but it may be underreported. Mucosal disease typically appears similarly to cutaneous disease.
The cutaneous differential diagnosis for BPDCN depends on the clinical presentation, extent of disease spread, and thickness of infiltration. It includes common nonneoplastic diseases such as traumatic ecchymoses; purpuric disorders; extramedullary hematopoiesis; and soft-tissue neoplasms such as angiosarcoma, Kaposi sarcoma, neuroblastoma, and vascular metastases, as well as skin involvement by other hematologic neoplasms. An adequate incisional biopsy rather than a punch or shave biopsy is recommended for diagnosis. Dermatologists should alert the pathologist that BPDCN is in the clinical differential diagnosis when possible so that judicious use of appropriate immunophenotypic markers such as CD123, CD4, CD56, and T-cell leukemia/lymphoma protein 1 will avoid misdiagnosis of this aggressive condition, in addition to excluding acute myeloid leukemia, which also may express 3 of the above markers. However, most cases of acute myeloid leukemia lack terminal deoxynucleotidyl transferase (TdT) and express monocytic and other myeloid markers. Terminal deoxynucleotidyl transferase is positive in approximately one-third of cases of BPDCN, with expression in 10% to 80% of cells.1
It is important to include BPDCN in the differential diagnosis of immunophenotypically aberrant hematologic tumors. Diffuse large B-cell lymphoma, leg type, accounts for 4% of all primary cutaneous B-cell lymphomas.1 Compared with BPDCN, diffuse large B-cell lymphoma usually occurs in an older age group and is of B-cell lineage. Morphologically, these neoplasms are composed of a monotonous, diffuse, nonepidermotropic infiltrate of confluent sheets of centroblasts and immunoblasts (Figure 1). They may share immunohistochemical markers of CD79a, multiple myeloma 1, Bcl-2, and Bcl-6; however, they lack plasmacytoid dendritic cell (PDC)– associated antigens such as CD4, CD56, CD123, and T-cell leukemia/lymphoma protein 1.1
Adult T-cell leukemia/lymphoma is a neoplasm histologically composed of highly pleomorphic medium- to large-sized T cells with an irregular multilobated nuclear contour, so-called flower cells, in the peripheral blood. The nuclear chromatin is coarse and clumped with prominent nucleoli. Blastlike cells with dispersed chromatin are present in variable proportions. Most patients present with widespread lymph node and peripheral blood involvement. Skin is involved in more than half of patients with an epidermal as well as dermal pattern of infiltration (mainly perivascular)(Figure 2). Adult T-cell leukemia/lymphoma is endemic in several regions of the world, and the distribution is closely linked to the prevalence of human T-cell lymphotropic virus type 1 in the population. This neoplasm is of T-cell lineage and may share CD4 but not PDC-associated antigens with BPDCN.1
Cutaneous involvement by T-cell lymphoblastic leukemia/lymphoma (T-LBL) is a rare occurrence with a frequency of approximately 4.3%.8 T-cell lymphoblastic leukemia/lymphoma usually presents as multiple skin lesions throughout the body. Almost all cutaneous T-LBL cases are seen in association with bone marrow and/or mediastinal, lymph node, or extranodal involvement. Cutaneous T-LBLs present as a diffuse monomorphous infiltrate located in the entire dermis and subcutis without epidermotropism, composed of medium to large blasts with finely dispersed chromatin and relatively prominent nucleoli (Figure 3). Immunophenotyping studies show an immature T-cell immunophenotype, with expression of TdT (usually uniform), CD7, and cytoplasmic CD3 and an absence of PDC-associated antigens.8
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a neoplasm primarily involving the skin. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection. Histologically, they show variable epidermotropism as well as dermal and subcutaneous involvement by medium to large cells with coarse clumped chromatin (Figure 4). Large blastic cells with vesicular nuclei and prominent nucleoli are infrequent. In contrast to BPCDN, the neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are positive for T-cell intracellular antigen-1 and granzyme B with loss of CD4.9
At the time of presentation, 27% to 87% of BPDCN patients will have bone marrow involvement, 22% to 28% will have blood involvement, and 6% to 41% will have lymph node involvement.1-4,6,7,10,11 The clinical course is aggressive, with a median survival of 10.0 to 19.8 months, irrespective of the initial pattern of disease.1 Most cases have shown initial response to multiagent chemotherapy, but relapses with subsequent resistance to drugs regularly have been observed. Age has an adverse impact of prognosis. Low TdT expression has been associated with shorter survival.1 Approximately 10% to 20% of cases of BPDCN are associated with or develop into chronic myelogenous leukemia, myelodysplastic syndrome, or acute myeloid leukemia.1,4 Pediatric patients have a greater 5-year overall survival rate than older patients, and overall survival worsens with increasing age. The extent of cutaneous involvement and presence of systemic involvement at initial presentation do not seem to be strong predictors of survival.1,2,5-7,10-12 In a retrospective analysis of 90 patients, Julia et al12 found that the type of skin disease did not predict survival. Specifically, the presence of nodular lesions and disseminated skin involvement were not adverse prognostic factors compared with macular lesions limited to 1 or 2 body areas.12
The Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm
A diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN) was rendered. Subsequent needle core biopsy of a left axillary lymph node as well as bone marrow aspiration and biopsy revealed a similar diffuse blastoid infiltrate with an identical immunophenotype to that in the skin biopsy from the pretibial mass and peripheral blood.
Previously known as blastic natural killer cell leukemia/lymphoma or agranular CD4+/CD56+ hematodermic neoplasm/tumor, BPDCN is a rare, clinically aggressive hematologic malignancy derived from the precursors of plasmacytoid dendritic cells. It often is diagnostically challenging, particularly when presenting at noncutaneous sites and in unusual (young) patient populations.1 It was included with other myeloid neoplasms in the 2008 World Health Organization classification; however, in the 2017 classification it was categorized as a separate entity. Blastic plasmacytoid dendritic cell neoplasm typically presents in the skin of elderly patients (age range at diagnosis, 61–67 years) with or without bone marrow involvement and systemic dissemination.1,2 The skin is the most common clinical site of disease in typical cases of BPDCN and often precedes bone marrow involvement. Thus, skin biopsy often is the key to making the diagnosis. Diagnosis of BPDCN may be delayed because of diagnostic pitfalls. Patients usually present with asymptomatic solitary or multiple lesions.3-5 Blastic plasmacytoid dendritic cell neoplasm can present as an isolated purplish nodule or bruiselike papule or more commonly as disseminated purplish nodules, papules, and macules. Isolated nodules are found on the head and lower limbs and can be more than 10 cm in diameter. Peripheral blood and bone marrow may be minimally involved at presentation but invariably become involved with the progression of disease. Cytopenia can occur at diagnosis and in a minority of severe cases indicates bone marrow failure.2-6
Skin involvement of BPDCN is thought to be secondary to the expression of skin migration molecules, such as cutaneous lymphocyte-associated antigen, one of the E-selectin ligands, which binds to E-selectin on high endothelial venules. In addition, the local dermal microenvironment of chemokines binding CXCR3, CXCR4, CCR6, or CCR7 present on neoplastic cells possibly leads to skin involvement. The full mechanism underlying the cutaneous tropism is still to be elucidated.4-7 Infiltration of the oral mucosa is seen in some patients, but it may be underreported. Mucosal disease typically appears similarly to cutaneous disease.
The cutaneous differential diagnosis for BPDCN depends on the clinical presentation, extent of disease spread, and thickness of infiltration. It includes common nonneoplastic diseases such as traumatic ecchymoses; purpuric disorders; extramedullary hematopoiesis; and soft-tissue neoplasms such as angiosarcoma, Kaposi sarcoma, neuroblastoma, and vascular metastases, as well as skin involvement by other hematologic neoplasms. An adequate incisional biopsy rather than a punch or shave biopsy is recommended for diagnosis. Dermatologists should alert the pathologist that BPDCN is in the clinical differential diagnosis when possible so that judicious use of appropriate immunophenotypic markers such as CD123, CD4, CD56, and T-cell leukemia/lymphoma protein 1 will avoid misdiagnosis of this aggressive condition, in addition to excluding acute myeloid leukemia, which also may express 3 of the above markers. However, most cases of acute myeloid leukemia lack terminal deoxynucleotidyl transferase (TdT) and express monocytic and other myeloid markers. Terminal deoxynucleotidyl transferase is positive in approximately one-third of cases of BPDCN, with expression in 10% to 80% of cells.1
It is important to include BPDCN in the differential diagnosis of immunophenotypically aberrant hematologic tumors. Diffuse large B-cell lymphoma, leg type, accounts for 4% of all primary cutaneous B-cell lymphomas.1 Compared with BPDCN, diffuse large B-cell lymphoma usually occurs in an older age group and is of B-cell lineage. Morphologically, these neoplasms are composed of a monotonous, diffuse, nonepidermotropic infiltrate of confluent sheets of centroblasts and immunoblasts (Figure 1). They may share immunohistochemical markers of CD79a, multiple myeloma 1, Bcl-2, and Bcl-6; however, they lack plasmacytoid dendritic cell (PDC)– associated antigens such as CD4, CD56, CD123, and T-cell leukemia/lymphoma protein 1.1
Adult T-cell leukemia/lymphoma is a neoplasm histologically composed of highly pleomorphic medium- to large-sized T cells with an irregular multilobated nuclear contour, so-called flower cells, in the peripheral blood. The nuclear chromatin is coarse and clumped with prominent nucleoli. Blastlike cells with dispersed chromatin are present in variable proportions. Most patients present with widespread lymph node and peripheral blood involvement. Skin is involved in more than half of patients with an epidermal as well as dermal pattern of infiltration (mainly perivascular)(Figure 2). Adult T-cell leukemia/lymphoma is endemic in several regions of the world, and the distribution is closely linked to the prevalence of human T-cell lymphotropic virus type 1 in the population. This neoplasm is of T-cell lineage and may share CD4 but not PDC-associated antigens with BPDCN.1
Cutaneous involvement by T-cell lymphoblastic leukemia/lymphoma (T-LBL) is a rare occurrence with a frequency of approximately 4.3%.8 T-cell lymphoblastic leukemia/lymphoma usually presents as multiple skin lesions throughout the body. Almost all cutaneous T-LBL cases are seen in association with bone marrow and/or mediastinal, lymph node, or extranodal involvement. Cutaneous T-LBLs present as a diffuse monomorphous infiltrate located in the entire dermis and subcutis without epidermotropism, composed of medium to large blasts with finely dispersed chromatin and relatively prominent nucleoli (Figure 3). Immunophenotyping studies show an immature T-cell immunophenotype, with expression of TdT (usually uniform), CD7, and cytoplasmic CD3 and an absence of PDC-associated antigens.8
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a neoplasm primarily involving the skin. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection. Histologically, they show variable epidermotropism as well as dermal and subcutaneous involvement by medium to large cells with coarse clumped chromatin (Figure 4). Large blastic cells with vesicular nuclei and prominent nucleoli are infrequent. In contrast to BPCDN, the neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are positive for T-cell intracellular antigen-1 and granzyme B with loss of CD4.9
At the time of presentation, 27% to 87% of BPDCN patients will have bone marrow involvement, 22% to 28% will have blood involvement, and 6% to 41% will have lymph node involvement.1-4,6,7,10,11 The clinical course is aggressive, with a median survival of 10.0 to 19.8 months, irrespective of the initial pattern of disease.1 Most cases have shown initial response to multiagent chemotherapy, but relapses with subsequent resistance to drugs regularly have been observed. Age has an adverse impact of prognosis. Low TdT expression has been associated with shorter survival.1 Approximately 10% to 20% of cases of BPDCN are associated with or develop into chronic myelogenous leukemia, myelodysplastic syndrome, or acute myeloid leukemia.1,4 Pediatric patients have a greater 5-year overall survival rate than older patients, and overall survival worsens with increasing age. The extent of cutaneous involvement and presence of systemic involvement at initial presentation do not seem to be strong predictors of survival.1,2,5-7,10-12 In a retrospective analysis of 90 patients, Julia et al12 found that the type of skin disease did not predict survival. Specifically, the presence of nodular lesions and disseminated skin involvement were not adverse prognostic factors compared with macular lesions limited to 1 or 2 body areas.12
- Facchetti F, Petrella T, Pileri SA. Blastic plasmacytoid dendritic cells neoplasm. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. World Health Organization; 2017:174-177.
- Jegalian AG, Facchetti F, Jaffe ES. Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv Anat Pathol. 2009;16:392-404.
- Shi Y, Wang E. Blastic plasmacytoid dendritic cell neoplasm: a clinicopathologic review. Arch Pathol Lab Med. 2014;138:564-569.
- Khoury JD, Medeiros LJ, Manning JT, et al. CD56(+) TdT(+) blastic natural killer cell tumor of the skin: a primitive systemic malignancy related to myelomonocytic leukemia. Cancer. 2002;94:2401-2408.
- Kolerova A, Sergeeva I, Krinitsyna J, et al. Blastic plasmacytoid dendritic cell neoplasm: case report and literature overview. Indian J Dermatol. 2020;65:217-221.
- Hirner JP, O’Malley JT, LeBoeuf NR. Blastic plasmacytoid dendritic cell neoplasm: the dermatologist’s perspective. Hematol Oncol Clin North Am. 2020;34:501-509.
- Guiducii C, Tripodo C, Gong M, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207:2931-2942.
- Khurana S, Beltran M, Jiang L, et al. Primary cutaneous T-cell lymphoblastic lymphoma: case report and literature review. Case Rep Hematol. 2019;2019:3540487. doi:10.1155/2019/3540487
- Gladys TE, Helm MF, Anderson BE, et al. Rapid onset of widespread nodules and lymphadenopathy. Cutis. 2020;106:132, 153-155.
- Gregorio J, Meller S, Conrad C, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. 2010;207:2921-2930.
- Guru Murthy GS, Pemmaraju N, Attallah E. Epidemiology and survival of blastic plasmacytoid dendritic cell neoplasm. Leuk Res. 2018;73:21-23.
- Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2012;169:579-586.
- Facchetti F, Petrella T, Pileri SA. Blastic plasmacytoid dendritic cells neoplasm. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. World Health Organization; 2017:174-177.
- Jegalian AG, Facchetti F, Jaffe ES. Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv Anat Pathol. 2009;16:392-404.
- Shi Y, Wang E. Blastic plasmacytoid dendritic cell neoplasm: a clinicopathologic review. Arch Pathol Lab Med. 2014;138:564-569.
- Khoury JD, Medeiros LJ, Manning JT, et al. CD56(+) TdT(+) blastic natural killer cell tumor of the skin: a primitive systemic malignancy related to myelomonocytic leukemia. Cancer. 2002;94:2401-2408.
- Kolerova A, Sergeeva I, Krinitsyna J, et al. Blastic plasmacytoid dendritic cell neoplasm: case report and literature overview. Indian J Dermatol. 2020;65:217-221.
- Hirner JP, O’Malley JT, LeBoeuf NR. Blastic plasmacytoid dendritic cell neoplasm: the dermatologist’s perspective. Hematol Oncol Clin North Am. 2020;34:501-509.
- Guiducii C, Tripodo C, Gong M, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207:2931-2942.
- Khurana S, Beltran M, Jiang L, et al. Primary cutaneous T-cell lymphoblastic lymphoma: case report and literature review. Case Rep Hematol. 2019;2019:3540487. doi:10.1155/2019/3540487
- Gladys TE, Helm MF, Anderson BE, et al. Rapid onset of widespread nodules and lymphadenopathy. Cutis. 2020;106:132, 153-155.
- Gregorio J, Meller S, Conrad C, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. 2010;207:2921-2930.
- Guru Murthy GS, Pemmaraju N, Attallah E. Epidemiology and survival of blastic plasmacytoid dendritic cell neoplasm. Leuk Res. 2018;73:21-23.
- Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2012;169:579-586.
A 23-year-old man presented with skin that bruised easily, pancytopenia, recent fatigue, fever, and loss of appetite, along with a nontender, brown-purple, left anterior pretibial mass of 2 years’ duration (top). Computed tomography showed diffuse lymphadenopathy involving the inguinal, mesenteric, retroperitoneal, mediastinal, and axillary regions. A biopsy of the mass showed a dense monomorphous infiltrate of medium-sized blastoid cells with small or inconspicuous nucleoli (bottom). The lesion diffusely involved the dermis and extended into the subcutaneous tissue but spared the epidermis. Flow cytometry immunophenotyping of peripheral blood neoplastic cells (bottom [inset]) showed high-level expression of CD123 together with expression of CD4, CD56, CD45RA, and CD43 but a lack of expression of any other myelomonocytic or lymphoid lineage–associated markers.



Dermatologic Management of Hidradenitis Suppurativa and Impact on Pregnancy and Breastfeeding
Hidradenitis suppurativa (HS) is a chronic inflammatory skin disease associated with hyperandrogenism and is caused by occlusion or rupture of follicular units and inflammation of the apocrine glands.1-3 The disease most commonly affects women (female to male ratio of 3:1) of childbearing age.1,2,4,5 Body areas affected include the axillae and groin, and less commonly the perineum; perianal region; and skin folds, such as gluteal, inframammary, and infraumbilical folds.1,2 Symptoms manifest as painful subcutaneous nodules with possible accompanying purulent drainage, sinus tracts, and/or dermal contractures. Although the pathophysiology is unclear, androgens affect the course of HS during pregnancy by stimulating the affected glands and altering cytokines.1,2,6
During pregnancy, maternal immune function switches from cell-mediated T helper cell (TH1) to humoral TH2 cytokine production. The activity of sebaceous and eccrine glands increases while the activity of apocrine glands decreases, thus changing the inflammatory course of HS during pregnancy.3 Approximately 20% of women with HS experience improvement of symptoms during pregnancy, while the remainder either experience no relief or deterioration of symptoms.1 Improvement in symptoms during pregnancy was found to occur more frequently in those who had worsening symptoms during menses owing to the possible hormonal effect estrogen has on inhibiting TH1 and TH17 proinflammatory cytokines, which promotes an immunosuppressive environment.4
Lactation and breastfeeding abilities may be hindered if a woman has HS affecting the apocrine glands of breast tissue and a symptom flare in the postpartum period. If HS causes notable inflammation in the nipple-areolar complex during pregnancy, the patient may experience difficulties with lactation and milk fistula formation, leading to inability to breastfeed.2 Another reason why mothers with HS may not be able to breastfeed is that the medications required to treat the disease are unsafe if passed to the infant via breast milk. In addition, the teratogenic effects of HS medications may necessitate therapy adjustments in pregnancy.1 Here, we provide a brief overview of the medical management considerations of HS in the setting of pregnancy and the impact on breastfeeding.
MEDICAL MANAGEMENT AND DRUG SAFETY
Dermatologists prescribe a myriad of topical and systemic medications to ameliorate symptoms of HS. Therapy regimens often are multimodal and include antibiotics, biologics, and immunosuppressants.1,3
Antibiotics
First-line antibiotics include clindamycin, metronidazole, tetracyclines, erythromycin, rifampin, dapsone, and fluoroquinolones. Topical clindamycin 1%, metronidazole 0.75%, and erythromycin 2% are used for open or active HS lesions and are all safe to use in pregnancy since there is minimal systemic absorption and minimal excretion into breast milk.1 Topical antimicrobial washes such as benzoyl peroxide and chlorhexidine often are used in combination with systemic medications to treat HS. These washes are safe during pregnancy and lactation, as they have minimal systemic absorption.7
Of these first-line antibiotics, only tetracyclines are contraindicated during pregnancy and lactation, as they are deemed to be in category D by the US Food and Drug Administration (FDA).1 Aside from tetracyclines, these antibiotics do not cause birth defects and are safe for nursing infants.1,8 Systemic clindamycin is safe during pregnancy and breastfeeding. Systemic metronidazole also is safe for use in pregnant patients but needs to be discontinued 12 to 24 hours prior to breastfeeding, which often prohibits appropriate dosing.1
Systemic Erythromycin—There are several forms of systemic erythromycin, including erythromycin base, erythromycin estolate, erythromycin ethylsuccinate (EES), and erythromycin stearate. Erythromycin estolate is contraindicated in pregnancy because it is associated with reversible maternal hepatoxicity and jaundice.9-11 Erythromycin ethylsuccinate is the preferred form for pregnant patients. Providers should exercise caution when prescribing EES to lactating mothers, as small amounts are still secreted through breast milk.11 Some studies have shown an increased risk for development of infantile hypertrophic pyloric stenosis with systemic erythromycin use, especially if a neonate is exposed in the first 14 days of life. Thus, we recommend withholding EES for 2 weeks after delivery if the patient is breastfeeding. A follow-up study did not find any association between erythromycin and infantile hypertrophic pyloric stenosis; however, the American Academy of Pediatrics still recommends short-term use only of erythromycin if it is to be used in the systemic form.8
Rifampin—Rifampin is excreted into breast milk but without adverse effects to the infant. Rifampin also is safe in pregnancy but should be used on a case-by-case basis in pregnant or nursing women because it is a cytochrome P450 inducer.
Dapsone—Dapsone has no increased risk for congenital anomalies. However, it is associated with hemolytic anemia and neonatal hyperbilirubinemia, especially in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency.12 Newborns exposed to dapsone are at an increased risk for methemoglobinemia owing to increased sensitivity of fetal erythrocytes to oxidizing agents.13 If dapsone use is necessary, stopping dapsone treatment in the last month of gestation is recommended to minimize risk for kernicterus.9 Dapsone can be found in high concentrations in breast milk at 14.3% of the maternal dose. It is still safe to use during breastfeeding, but there is a risk of the infant developing hyperbilirubinemia/G6PD deficiency.1,8 Thus, physicians may consider performing a G6PD screen on infants to determine if breastfeeding is safe.12
Fluoroquinolones—Quinolones are not contraindicated during pregnancy, but they can damage fetal cartilage and thus should be reserved for use in complicated infections when the benefits outweigh the risks.12 Quinolones are believed to increase risk for arthropathy but are safe for use in lactation. When quinolones are digested with milk, exposure decreases below pediatric doses because of the ionized property of calcium in milk.8
Tumor Necrosis Factor α Inhibitors—The safety of anti–tumor necrosis factor (TNF) α biologics in pregnancy is less certain when compared with antibiotics.1 Anti–TNF-α inhibitors such as etanercept, adalimumab, and infliximab are all labeled as FDA category B, meaning there are no well-controlled human studies of the drugs.9 There are limited data that support safe use of TNF-α inhibitors prior to the third trimester before maternal IgG antibodies are transferred to the fetus via the placenta.1,13 Anti–TNF-α inhibitors may be safe when breastfeeding because the drugs have large molecular weights that prevent them from entering breast milk in large amounts. Absorption also is limited due to the infant’s digestive acids and enzymes breaking down the protein structure of the medication.8 Overall, TNF-α inhibitor use is still controversial and only used if the benefits outweigh the risks during pregnancy or if there is no alternative treatment.1,3,9
Ustekinumab and Anakinra—Ustekinumab (an IL-12/IL-23 inhibitor) and anakinra (an IL-1α and IL-1β inhibitor) also are FDA category B drugs and have limited data supporting their use as HS treatment in pregnancy. Anakinra may have evidence of compatibility with breastfeeding, as endogenous IL-1α inhibitor is found in colostrum and mature breast milk.1
Immunosuppressants
Immunosuppressants that are used to treat HS include corticosteroids and cyclosporine.
Corticosteroids—Topical corticosteroids can be used safely in lactation if they are not applied directly to the nipple or any area that makes direct contact with the infant’s mouth. Intralesional corticosteroid injections are safe for use during both pregnancy and breastfeeding to decrease inflammation of acutely flaring lesions and can be considered first-line treatment.1 Oral glucocorticoids also can be safely used for acute flares during pregnancy; however, prolonged use is associated with pregnancy complications such as preeclampsia, eclampsia, premature delivery, and gestational diabetes.12 There also is a small risk of oral cleft deformity in the infant; thus, potent corticosteroids are recommended in short durations during pregnancy, and there are no adverse effects if the maternal dose is less than 10 mg daily.8,12 Systemic steroids are safe to use with breastfeeding, but patients should be advised to wait 4 hours after ingesting medication before breastfeeding.1,8
Cyclosporine—Topical and oral calcineurin inhibitors such as cyclosporine have low risk for transmission into breast milk; however, potential effects of exposure through breast milk are unknown. For that reason, manufacturers state that cyclosporine use is contraindicated during lactation.8 If cyclosporine is to be used by a breastfeeding woman, monitoring cyclosporine concentrations in the infant is suggested to ensure that the exposure is less than 5% to 10% of the therapeutic dose.13 The use of cyclosporine has been extensively studied in pregnant transplant patients and is considered relatively safe for use in pregnancy.14 Cyclosporine is lipid soluble and thus is quickly metabolized and spread throughout the body; it can easily cross the placenta.9,13 Blood concentration in the fetus is 30% to 64% that of the maternal circulation. However, cyclosporine is only toxic to the fetus at maternally toxic doses, which can result in low birth weight and increased prenatal and postnatal mortality.13
Isotretinoin, Oral Contraceptive Pills, and Spironolactone
Isotretinoin and hormonal treatments such as oral contraceptive pills and spironolactone (an androgen receptor blocker) commonly are used to treat HS, but all are contraindicated in pregnancy and lactation. Isotretinoin is a well-established teratogen, but adverse effects on nursing babies have not been described. However, the manufacturer of isotretinoin advises against its use in lactation. Oral contraceptive pill use in early pregnancy is associated with increased risk for Down syndrome. Oral contraceptive pill use also is contraindicated in lactation for 2 reasons: decreased milk production and risk for fetal feminization. Antiandrogenic agents such as spironolactone have been shown to be associated with hypospadias and feminization of the male fetus.7
COMMENT
Women with HS usually require ongoing medical treatment during pregnancy and immediately postpartum; thus, it is important that treatments are proven to be safe for use in this specific population. Current management guidelines are not entirely suitable for pregnant and breastfeeding women given that many HS drugs have teratogenic effects and/or can be excreted into breast milk.1 Several treatments have uncertain safety profiles in pregnancy and breastfeeding, which calls for dermatologists to change or create new regimens for their patients. Close management also is necessary to prevent excess inflammation of breast tissue and milk fistula formation, which would hinder normal breastfeeding.
The eTable lists medications used to treat HS. The FDA category is listed next to each drug. However, it should be noted that these FDA letter categories were replaced with the Pregnancy and Lactation Labeling Rule in 2015. The letter ratings were deemed overly simplistic and replaced with narrative-based labeling that provides more detailed adverse effects and clinical considerations.9

Risk Factors of HS—Predisposing risk factors for HS flares that are controllable include obesity and smoking.2 Pregnancy weight gain may cause increased skin maceration at intertriginous sites, which can contribute to worsening HS symptoms.1,5 Adipocytes play a role in HS exacerbation by promoting secretion of TNF-α, leading to increased inflammation.5 Dermatologists can help prevent postpartum HS flares by monitoring weight gain during pregnancy, encouraging smoking cessation, and promoting weight and nutrition goals as set by an obstetrician.1 In addition to medications, management of HS should include emotional support and education on wearing loose-fitting clothing to avoid irritation of the affected areas.3 An emphasis on dermatologist counseling for all patients with HS, even for those with milder disease, can reduce exacerbations during pregnancy.5

CONCLUSION
The selection of dermatologic drugs for the treatment of HS in the setting of pregnancy involves complex decision-making. Dermatologists need more guidelines and proven safety data in human trials, especially regarding use of biologics and immunosuppressants to better treat HS in pregnancy. With more data, they can create more evidence-based treatment regimens to help prevent postpartum exacerbations of HS. Thus, patients can breastfeed their infants comfortably and without any risks of impaired child development. In the meantime, dermatologists can continue to work together with obstetricians and psychiatrists to decrease disease flares through counseling patients on nutrition and weight gain and providing emotional support.
- Perng P, Zampella JG, Okoye GA. Management of hidradenitis suppurativa in pregnancy. J Am Acad Dermatol. 2017;76:979-989. doi:10.1016/j.jaad.2016.10.032
- Samuel S, Tremelling A, Murray M. Presentation and surgical management of hidradenitis suppurativa of the breast during pregnancy: a case report. Int J Surg Case Rep. 2018;51:21-24. doi:10.1016/j.ijscr.2018.08.013
- Yang CS, Teeple M, Muglia J, et al. Inflammatory and glandular skin disease in pregnancy. Clin Dermatol. 2016;34:335-343. doi:10.1016/j.clindermatol.2016.02.005
- Vossen AR, van Straalen KR, Prens EP, et al. Menses and pregnancy affect symptoms in hidradenitis suppurativa: a cross-sectional study. J Am Acad Dermatol. 2017;76:155-156. doi:10.1016/j.jaad.2016.07.024
- Lyons AB, Peacock A, McKenzie SA, et al. Evaluation of hidradenitis suppurativa disease course during pregnancy and postpartum. JAMA Dermatol. 2020;156:681-685. doi:10.1001/jamadermatol.2020.0777
- Riis PT, Ring HC, Themstrup L, et al. The role of androgens and estrogens in hidradenitis suppurativa—a systematic review. Acta Dermatovenerol Croat. 2016;24:239-249.
- Kong YL, Tey HL. Treatment of acne vulgaris during pregnancy and lactation. Drugs. 2013;73:779-787. doi:10.1007/s40265-013-0060-0
- Butler DC, Heller MM, Murase JE. Safety of dermatologic medications in pregnancy and lactation: part II. lactation. J Am Acad Dermatol. 2014;70:417:E1-E10. doi:10.1016/j.jaad.2013.09.009
- Wilmer E, Chai S, Kroumpouzos G. Drug safety: pregnancy rating classifications and controversies. Clin Dermatol. 2016;34:401-409. doi:10.1016/j.clindermatol.2016.02.013
- Inman WH, Rawson NS. Erythromycin estolate and jaundice. Br Med J (Clin Res Ed). 1983;286:1954-1955. doi:10.1136/bmj.286.6382.1954
- Workowski KA, Berman SM. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
- Murase JE, Heller MM, Butler DC. Safety of dermatologic medications in pregnancy and lactation: part I. pregnancy. J Am Acad Dermatol. 2014;70:401.e1-14; quiz 415. doi:10.1016/j.jaad.2013.09.010
- Brown SM, Aljefri K, Waas R, et al. Systemic medications used in treatment of common dermatological conditions: safety profile with respect to pregnancy, breast feeding and content in seminal fluid. J Dermatolog Treat. 2019;30:2-18. doi:10.1080/09546634.2016.1202402
- Kamarajah SK, Arntdz K, Bundred J, et al. Outcomes of pregnancy in recipients of liver transplants. Clin Gastroenterol Hepatol. 2019;17:1398-1404.e1. doi:10.1016/j.cgh.2018.11.055
Hidradenitis suppurativa (HS) is a chronic inflammatory skin disease associated with hyperandrogenism and is caused by occlusion or rupture of follicular units and inflammation of the apocrine glands.1-3 The disease most commonly affects women (female to male ratio of 3:1) of childbearing age.1,2,4,5 Body areas affected include the axillae and groin, and less commonly the perineum; perianal region; and skin folds, such as gluteal, inframammary, and infraumbilical folds.1,2 Symptoms manifest as painful subcutaneous nodules with possible accompanying purulent drainage, sinus tracts, and/or dermal contractures. Although the pathophysiology is unclear, androgens affect the course of HS during pregnancy by stimulating the affected glands and altering cytokines.1,2,6
During pregnancy, maternal immune function switches from cell-mediated T helper cell (TH1) to humoral TH2 cytokine production. The activity of sebaceous and eccrine glands increases while the activity of apocrine glands decreases, thus changing the inflammatory course of HS during pregnancy.3 Approximately 20% of women with HS experience improvement of symptoms during pregnancy, while the remainder either experience no relief or deterioration of symptoms.1 Improvement in symptoms during pregnancy was found to occur more frequently in those who had worsening symptoms during menses owing to the possible hormonal effect estrogen has on inhibiting TH1 and TH17 proinflammatory cytokines, which promotes an immunosuppressive environment.4
Lactation and breastfeeding abilities may be hindered if a woman has HS affecting the apocrine glands of breast tissue and a symptom flare in the postpartum period. If HS causes notable inflammation in the nipple-areolar complex during pregnancy, the patient may experience difficulties with lactation and milk fistula formation, leading to inability to breastfeed.2 Another reason why mothers with HS may not be able to breastfeed is that the medications required to treat the disease are unsafe if passed to the infant via breast milk. In addition, the teratogenic effects of HS medications may necessitate therapy adjustments in pregnancy.1 Here, we provide a brief overview of the medical management considerations of HS in the setting of pregnancy and the impact on breastfeeding.
MEDICAL MANAGEMENT AND DRUG SAFETY
Dermatologists prescribe a myriad of topical and systemic medications to ameliorate symptoms of HS. Therapy regimens often are multimodal and include antibiotics, biologics, and immunosuppressants.1,3
Antibiotics
First-line antibiotics include clindamycin, metronidazole, tetracyclines, erythromycin, rifampin, dapsone, and fluoroquinolones. Topical clindamycin 1%, metronidazole 0.75%, and erythromycin 2% are used for open or active HS lesions and are all safe to use in pregnancy since there is minimal systemic absorption and minimal excretion into breast milk.1 Topical antimicrobial washes such as benzoyl peroxide and chlorhexidine often are used in combination with systemic medications to treat HS. These washes are safe during pregnancy and lactation, as they have minimal systemic absorption.7
Of these first-line antibiotics, only tetracyclines are contraindicated during pregnancy and lactation, as they are deemed to be in category D by the US Food and Drug Administration (FDA).1 Aside from tetracyclines, these antibiotics do not cause birth defects and are safe for nursing infants.1,8 Systemic clindamycin is safe during pregnancy and breastfeeding. Systemic metronidazole also is safe for use in pregnant patients but needs to be discontinued 12 to 24 hours prior to breastfeeding, which often prohibits appropriate dosing.1
Systemic Erythromycin—There are several forms of systemic erythromycin, including erythromycin base, erythromycin estolate, erythromycin ethylsuccinate (EES), and erythromycin stearate. Erythromycin estolate is contraindicated in pregnancy because it is associated with reversible maternal hepatoxicity and jaundice.9-11 Erythromycin ethylsuccinate is the preferred form for pregnant patients. Providers should exercise caution when prescribing EES to lactating mothers, as small amounts are still secreted through breast milk.11 Some studies have shown an increased risk for development of infantile hypertrophic pyloric stenosis with systemic erythromycin use, especially if a neonate is exposed in the first 14 days of life. Thus, we recommend withholding EES for 2 weeks after delivery if the patient is breastfeeding. A follow-up study did not find any association between erythromycin and infantile hypertrophic pyloric stenosis; however, the American Academy of Pediatrics still recommends short-term use only of erythromycin if it is to be used in the systemic form.8
Rifampin—Rifampin is excreted into breast milk but without adverse effects to the infant. Rifampin also is safe in pregnancy but should be used on a case-by-case basis in pregnant or nursing women because it is a cytochrome P450 inducer.
Dapsone—Dapsone has no increased risk for congenital anomalies. However, it is associated with hemolytic anemia and neonatal hyperbilirubinemia, especially in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency.12 Newborns exposed to dapsone are at an increased risk for methemoglobinemia owing to increased sensitivity of fetal erythrocytes to oxidizing agents.13 If dapsone use is necessary, stopping dapsone treatment in the last month of gestation is recommended to minimize risk for kernicterus.9 Dapsone can be found in high concentrations in breast milk at 14.3% of the maternal dose. It is still safe to use during breastfeeding, but there is a risk of the infant developing hyperbilirubinemia/G6PD deficiency.1,8 Thus, physicians may consider performing a G6PD screen on infants to determine if breastfeeding is safe.12
Fluoroquinolones—Quinolones are not contraindicated during pregnancy, but they can damage fetal cartilage and thus should be reserved for use in complicated infections when the benefits outweigh the risks.12 Quinolones are believed to increase risk for arthropathy but are safe for use in lactation. When quinolones are digested with milk, exposure decreases below pediatric doses because of the ionized property of calcium in milk.8
Tumor Necrosis Factor α Inhibitors—The safety of anti–tumor necrosis factor (TNF) α biologics in pregnancy is less certain when compared with antibiotics.1 Anti–TNF-α inhibitors such as etanercept, adalimumab, and infliximab are all labeled as FDA category B, meaning there are no well-controlled human studies of the drugs.9 There are limited data that support safe use of TNF-α inhibitors prior to the third trimester before maternal IgG antibodies are transferred to the fetus via the placenta.1,13 Anti–TNF-α inhibitors may be safe when breastfeeding because the drugs have large molecular weights that prevent them from entering breast milk in large amounts. Absorption also is limited due to the infant’s digestive acids and enzymes breaking down the protein structure of the medication.8 Overall, TNF-α inhibitor use is still controversial and only used if the benefits outweigh the risks during pregnancy or if there is no alternative treatment.1,3,9
Ustekinumab and Anakinra—Ustekinumab (an IL-12/IL-23 inhibitor) and anakinra (an IL-1α and IL-1β inhibitor) also are FDA category B drugs and have limited data supporting their use as HS treatment in pregnancy. Anakinra may have evidence of compatibility with breastfeeding, as endogenous IL-1α inhibitor is found in colostrum and mature breast milk.1
Immunosuppressants
Immunosuppressants that are used to treat HS include corticosteroids and cyclosporine.
Corticosteroids—Topical corticosteroids can be used safely in lactation if they are not applied directly to the nipple or any area that makes direct contact with the infant’s mouth. Intralesional corticosteroid injections are safe for use during both pregnancy and breastfeeding to decrease inflammation of acutely flaring lesions and can be considered first-line treatment.1 Oral glucocorticoids also can be safely used for acute flares during pregnancy; however, prolonged use is associated with pregnancy complications such as preeclampsia, eclampsia, premature delivery, and gestational diabetes.12 There also is a small risk of oral cleft deformity in the infant; thus, potent corticosteroids are recommended in short durations during pregnancy, and there are no adverse effects if the maternal dose is less than 10 mg daily.8,12 Systemic steroids are safe to use with breastfeeding, but patients should be advised to wait 4 hours after ingesting medication before breastfeeding.1,8
Cyclosporine—Topical and oral calcineurin inhibitors such as cyclosporine have low risk for transmission into breast milk; however, potential effects of exposure through breast milk are unknown. For that reason, manufacturers state that cyclosporine use is contraindicated during lactation.8 If cyclosporine is to be used by a breastfeeding woman, monitoring cyclosporine concentrations in the infant is suggested to ensure that the exposure is less than 5% to 10% of the therapeutic dose.13 The use of cyclosporine has been extensively studied in pregnant transplant patients and is considered relatively safe for use in pregnancy.14 Cyclosporine is lipid soluble and thus is quickly metabolized and spread throughout the body; it can easily cross the placenta.9,13 Blood concentration in the fetus is 30% to 64% that of the maternal circulation. However, cyclosporine is only toxic to the fetus at maternally toxic doses, which can result in low birth weight and increased prenatal and postnatal mortality.13
Isotretinoin, Oral Contraceptive Pills, and Spironolactone
Isotretinoin and hormonal treatments such as oral contraceptive pills and spironolactone (an androgen receptor blocker) commonly are used to treat HS, but all are contraindicated in pregnancy and lactation. Isotretinoin is a well-established teratogen, but adverse effects on nursing babies have not been described. However, the manufacturer of isotretinoin advises against its use in lactation. Oral contraceptive pill use in early pregnancy is associated with increased risk for Down syndrome. Oral contraceptive pill use also is contraindicated in lactation for 2 reasons: decreased milk production and risk for fetal feminization. Antiandrogenic agents such as spironolactone have been shown to be associated with hypospadias and feminization of the male fetus.7
COMMENT
Women with HS usually require ongoing medical treatment during pregnancy and immediately postpartum; thus, it is important that treatments are proven to be safe for use in this specific population. Current management guidelines are not entirely suitable for pregnant and breastfeeding women given that many HS drugs have teratogenic effects and/or can be excreted into breast milk.1 Several treatments have uncertain safety profiles in pregnancy and breastfeeding, which calls for dermatologists to change or create new regimens for their patients. Close management also is necessary to prevent excess inflammation of breast tissue and milk fistula formation, which would hinder normal breastfeeding.
The eTable lists medications used to treat HS. The FDA category is listed next to each drug. However, it should be noted that these FDA letter categories were replaced with the Pregnancy and Lactation Labeling Rule in 2015. The letter ratings were deemed overly simplistic and replaced with narrative-based labeling that provides more detailed adverse effects and clinical considerations.9
Risk Factors of HS—Predisposing risk factors for HS flares that are controllable include obesity and smoking.2 Pregnancy weight gain may cause increased skin maceration at intertriginous sites, which can contribute to worsening HS symptoms.1,5 Adipocytes play a role in HS exacerbation by promoting secretion of TNF-α, leading to increased inflammation.5 Dermatologists can help prevent postpartum HS flares by monitoring weight gain during pregnancy, encouraging smoking cessation, and promoting weight and nutrition goals as set by an obstetrician.1 In addition to medications, management of HS should include emotional support and education on wearing loose-fitting clothing to avoid irritation of the affected areas.3 An emphasis on dermatologist counseling for all patients with HS, even for those with milder disease, can reduce exacerbations during pregnancy.5
CONCLUSION
The selection of dermatologic drugs for the treatment of HS in the setting of pregnancy involves complex decision-making. Dermatologists need more guidelines and proven safety data in human trials, especially regarding use of biologics and immunosuppressants to better treat HS in pregnancy. With more data, they can create more evidence-based treatment regimens to help prevent postpartum exacerbations of HS. Thus, patients can breastfeed their infants comfortably and without any risks of impaired child development. In the meantime, dermatologists can continue to work together with obstetricians and psychiatrists to decrease disease flares through counseling patients on nutrition and weight gain and providing emotional support.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin disease associated with hyperandrogenism and is caused by occlusion or rupture of follicular units and inflammation of the apocrine glands.1-3 The disease most commonly affects women (female to male ratio of 3:1) of childbearing age.1,2,4,5 Body areas affected include the axillae and groin, and less commonly the perineum; perianal region; and skin folds, such as gluteal, inframammary, and infraumbilical folds.1,2 Symptoms manifest as painful subcutaneous nodules with possible accompanying purulent drainage, sinus tracts, and/or dermal contractures. Although the pathophysiology is unclear, androgens affect the course of HS during pregnancy by stimulating the affected glands and altering cytokines.1,2,6
During pregnancy, maternal immune function switches from cell-mediated T helper cell (TH1) to humoral TH2 cytokine production. The activity of sebaceous and eccrine glands increases while the activity of apocrine glands decreases, thus changing the inflammatory course of HS during pregnancy.3 Approximately 20% of women with HS experience improvement of symptoms during pregnancy, while the remainder either experience no relief or deterioration of symptoms.1 Improvement in symptoms during pregnancy was found to occur more frequently in those who had worsening symptoms during menses owing to the possible hormonal effect estrogen has on inhibiting TH1 and TH17 proinflammatory cytokines, which promotes an immunosuppressive environment.4
Lactation and breastfeeding abilities may be hindered if a woman has HS affecting the apocrine glands of breast tissue and a symptom flare in the postpartum period. If HS causes notable inflammation in the nipple-areolar complex during pregnancy, the patient may experience difficulties with lactation and milk fistula formation, leading to inability to breastfeed.2 Another reason why mothers with HS may not be able to breastfeed is that the medications required to treat the disease are unsafe if passed to the infant via breast milk. In addition, the teratogenic effects of HS medications may necessitate therapy adjustments in pregnancy.1 Here, we provide a brief overview of the medical management considerations of HS in the setting of pregnancy and the impact on breastfeeding.
MEDICAL MANAGEMENT AND DRUG SAFETY
Dermatologists prescribe a myriad of topical and systemic medications to ameliorate symptoms of HS. Therapy regimens often are multimodal and include antibiotics, biologics, and immunosuppressants.1,3
Antibiotics
First-line antibiotics include clindamycin, metronidazole, tetracyclines, erythromycin, rifampin, dapsone, and fluoroquinolones. Topical clindamycin 1%, metronidazole 0.75%, and erythromycin 2% are used for open or active HS lesions and are all safe to use in pregnancy since there is minimal systemic absorption and minimal excretion into breast milk.1 Topical antimicrobial washes such as benzoyl peroxide and chlorhexidine often are used in combination with systemic medications to treat HS. These washes are safe during pregnancy and lactation, as they have minimal systemic absorption.7
Of these first-line antibiotics, only tetracyclines are contraindicated during pregnancy and lactation, as they are deemed to be in category D by the US Food and Drug Administration (FDA).1 Aside from tetracyclines, these antibiotics do not cause birth defects and are safe for nursing infants.1,8 Systemic clindamycin is safe during pregnancy and breastfeeding. Systemic metronidazole also is safe for use in pregnant patients but needs to be discontinued 12 to 24 hours prior to breastfeeding, which often prohibits appropriate dosing.1
Systemic Erythromycin—There are several forms of systemic erythromycin, including erythromycin base, erythromycin estolate, erythromycin ethylsuccinate (EES), and erythromycin stearate. Erythromycin estolate is contraindicated in pregnancy because it is associated with reversible maternal hepatoxicity and jaundice.9-11 Erythromycin ethylsuccinate is the preferred form for pregnant patients. Providers should exercise caution when prescribing EES to lactating mothers, as small amounts are still secreted through breast milk.11 Some studies have shown an increased risk for development of infantile hypertrophic pyloric stenosis with systemic erythromycin use, especially if a neonate is exposed in the first 14 days of life. Thus, we recommend withholding EES for 2 weeks after delivery if the patient is breastfeeding. A follow-up study did not find any association between erythromycin and infantile hypertrophic pyloric stenosis; however, the American Academy of Pediatrics still recommends short-term use only of erythromycin if it is to be used in the systemic form.8
Rifampin—Rifampin is excreted into breast milk but without adverse effects to the infant. Rifampin also is safe in pregnancy but should be used on a case-by-case basis in pregnant or nursing women because it is a cytochrome P450 inducer.
Dapsone—Dapsone has no increased risk for congenital anomalies. However, it is associated with hemolytic anemia and neonatal hyperbilirubinemia, especially in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency.12 Newborns exposed to dapsone are at an increased risk for methemoglobinemia owing to increased sensitivity of fetal erythrocytes to oxidizing agents.13 If dapsone use is necessary, stopping dapsone treatment in the last month of gestation is recommended to minimize risk for kernicterus.9 Dapsone can be found in high concentrations in breast milk at 14.3% of the maternal dose. It is still safe to use during breastfeeding, but there is a risk of the infant developing hyperbilirubinemia/G6PD deficiency.1,8 Thus, physicians may consider performing a G6PD screen on infants to determine if breastfeeding is safe.12
Fluoroquinolones—Quinolones are not contraindicated during pregnancy, but they can damage fetal cartilage and thus should be reserved for use in complicated infections when the benefits outweigh the risks.12 Quinolones are believed to increase risk for arthropathy but are safe for use in lactation. When quinolones are digested with milk, exposure decreases below pediatric doses because of the ionized property of calcium in milk.8
Tumor Necrosis Factor α Inhibitors—The safety of anti–tumor necrosis factor (TNF) α biologics in pregnancy is less certain when compared with antibiotics.1 Anti–TNF-α inhibitors such as etanercept, adalimumab, and infliximab are all labeled as FDA category B, meaning there are no well-controlled human studies of the drugs.9 There are limited data that support safe use of TNF-α inhibitors prior to the third trimester before maternal IgG antibodies are transferred to the fetus via the placenta.1,13 Anti–TNF-α inhibitors may be safe when breastfeeding because the drugs have large molecular weights that prevent them from entering breast milk in large amounts. Absorption also is limited due to the infant’s digestive acids and enzymes breaking down the protein structure of the medication.8 Overall, TNF-α inhibitor use is still controversial and only used if the benefits outweigh the risks during pregnancy or if there is no alternative treatment.1,3,9
Ustekinumab and Anakinra—Ustekinumab (an IL-12/IL-23 inhibitor) and anakinra (an IL-1α and IL-1β inhibitor) also are FDA category B drugs and have limited data supporting their use as HS treatment in pregnancy. Anakinra may have evidence of compatibility with breastfeeding, as endogenous IL-1α inhibitor is found in colostrum and mature breast milk.1
Immunosuppressants
Immunosuppressants that are used to treat HS include corticosteroids and cyclosporine.
Corticosteroids—Topical corticosteroids can be used safely in lactation if they are not applied directly to the nipple or any area that makes direct contact with the infant’s mouth. Intralesional corticosteroid injections are safe for use during both pregnancy and breastfeeding to decrease inflammation of acutely flaring lesions and can be considered first-line treatment.1 Oral glucocorticoids also can be safely used for acute flares during pregnancy; however, prolonged use is associated with pregnancy complications such as preeclampsia, eclampsia, premature delivery, and gestational diabetes.12 There also is a small risk of oral cleft deformity in the infant; thus, potent corticosteroids are recommended in short durations during pregnancy, and there are no adverse effects if the maternal dose is less than 10 mg daily.8,12 Systemic steroids are safe to use with breastfeeding, but patients should be advised to wait 4 hours after ingesting medication before breastfeeding.1,8
Cyclosporine—Topical and oral calcineurin inhibitors such as cyclosporine have low risk for transmission into breast milk; however, potential effects of exposure through breast milk are unknown. For that reason, manufacturers state that cyclosporine use is contraindicated during lactation.8 If cyclosporine is to be used by a breastfeeding woman, monitoring cyclosporine concentrations in the infant is suggested to ensure that the exposure is less than 5% to 10% of the therapeutic dose.13 The use of cyclosporine has been extensively studied in pregnant transplant patients and is considered relatively safe for use in pregnancy.14 Cyclosporine is lipid soluble and thus is quickly metabolized and spread throughout the body; it can easily cross the placenta.9,13 Blood concentration in the fetus is 30% to 64% that of the maternal circulation. However, cyclosporine is only toxic to the fetus at maternally toxic doses, which can result in low birth weight and increased prenatal and postnatal mortality.13
Isotretinoin, Oral Contraceptive Pills, and Spironolactone
Isotretinoin and hormonal treatments such as oral contraceptive pills and spironolactone (an androgen receptor blocker) commonly are used to treat HS, but all are contraindicated in pregnancy and lactation. Isotretinoin is a well-established teratogen, but adverse effects on nursing babies have not been described. However, the manufacturer of isotretinoin advises against its use in lactation. Oral contraceptive pill use in early pregnancy is associated with increased risk for Down syndrome. Oral contraceptive pill use also is contraindicated in lactation for 2 reasons: decreased milk production and risk for fetal feminization. Antiandrogenic agents such as spironolactone have been shown to be associated with hypospadias and feminization of the male fetus.7
COMMENT
Women with HS usually require ongoing medical treatment during pregnancy and immediately postpartum; thus, it is important that treatments are proven to be safe for use in this specific population. Current management guidelines are not entirely suitable for pregnant and breastfeeding women given that many HS drugs have teratogenic effects and/or can be excreted into breast milk.1 Several treatments have uncertain safety profiles in pregnancy and breastfeeding, which calls for dermatologists to change or create new regimens for their patients. Close management also is necessary to prevent excess inflammation of breast tissue and milk fistula formation, which would hinder normal breastfeeding.
The eTable lists medications used to treat HS. The FDA category is listed next to each drug. However, it should be noted that these FDA letter categories were replaced with the Pregnancy and Lactation Labeling Rule in 2015. The letter ratings were deemed overly simplistic and replaced with narrative-based labeling that provides more detailed adverse effects and clinical considerations.9
Risk Factors of HS—Predisposing risk factors for HS flares that are controllable include obesity and smoking.2 Pregnancy weight gain may cause increased skin maceration at intertriginous sites, which can contribute to worsening HS symptoms.1,5 Adipocytes play a role in HS exacerbation by promoting secretion of TNF-α, leading to increased inflammation.5 Dermatologists can help prevent postpartum HS flares by monitoring weight gain during pregnancy, encouraging smoking cessation, and promoting weight and nutrition goals as set by an obstetrician.1 In addition to medications, management of HS should include emotional support and education on wearing loose-fitting clothing to avoid irritation of the affected areas.3 An emphasis on dermatologist counseling for all patients with HS, even for those with milder disease, can reduce exacerbations during pregnancy.5
CONCLUSION
The selection of dermatologic drugs for the treatment of HS in the setting of pregnancy involves complex decision-making. Dermatologists need more guidelines and proven safety data in human trials, especially regarding use of biologics and immunosuppressants to better treat HS in pregnancy. With more data, they can create more evidence-based treatment regimens to help prevent postpartum exacerbations of HS. Thus, patients can breastfeed their infants comfortably and without any risks of impaired child development. In the meantime, dermatologists can continue to work together with obstetricians and psychiatrists to decrease disease flares through counseling patients on nutrition and weight gain and providing emotional support.
- Perng P, Zampella JG, Okoye GA. Management of hidradenitis suppurativa in pregnancy. J Am Acad Dermatol. 2017;76:979-989. doi:10.1016/j.jaad.2016.10.032
- Samuel S, Tremelling A, Murray M. Presentation and surgical management of hidradenitis suppurativa of the breast during pregnancy: a case report. Int J Surg Case Rep. 2018;51:21-24. doi:10.1016/j.ijscr.2018.08.013
- Yang CS, Teeple M, Muglia J, et al. Inflammatory and glandular skin disease in pregnancy. Clin Dermatol. 2016;34:335-343. doi:10.1016/j.clindermatol.2016.02.005
- Vossen AR, van Straalen KR, Prens EP, et al. Menses and pregnancy affect symptoms in hidradenitis suppurativa: a cross-sectional study. J Am Acad Dermatol. 2017;76:155-156. doi:10.1016/j.jaad.2016.07.024
- Lyons AB, Peacock A, McKenzie SA, et al. Evaluation of hidradenitis suppurativa disease course during pregnancy and postpartum. JAMA Dermatol. 2020;156:681-685. doi:10.1001/jamadermatol.2020.0777
- Riis PT, Ring HC, Themstrup L, et al. The role of androgens and estrogens in hidradenitis suppurativa—a systematic review. Acta Dermatovenerol Croat. 2016;24:239-249.
- Kong YL, Tey HL. Treatment of acne vulgaris during pregnancy and lactation. Drugs. 2013;73:779-787. doi:10.1007/s40265-013-0060-0
- Butler DC, Heller MM, Murase JE. Safety of dermatologic medications in pregnancy and lactation: part II. lactation. J Am Acad Dermatol. 2014;70:417:E1-E10. doi:10.1016/j.jaad.2013.09.009
- Wilmer E, Chai S, Kroumpouzos G. Drug safety: pregnancy rating classifications and controversies. Clin Dermatol. 2016;34:401-409. doi:10.1016/j.clindermatol.2016.02.013
- Inman WH, Rawson NS. Erythromycin estolate and jaundice. Br Med J (Clin Res Ed). 1983;286:1954-1955. doi:10.1136/bmj.286.6382.1954
- Workowski KA, Berman SM. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
- Murase JE, Heller MM, Butler DC. Safety of dermatologic medications in pregnancy and lactation: part I. pregnancy. J Am Acad Dermatol. 2014;70:401.e1-14; quiz 415. doi:10.1016/j.jaad.2013.09.010
- Brown SM, Aljefri K, Waas R, et al. Systemic medications used in treatment of common dermatological conditions: safety profile with respect to pregnancy, breast feeding and content in seminal fluid. J Dermatolog Treat. 2019;30:2-18. doi:10.1080/09546634.2016.1202402
- Kamarajah SK, Arntdz K, Bundred J, et al. Outcomes of pregnancy in recipients of liver transplants. Clin Gastroenterol Hepatol. 2019;17:1398-1404.e1. doi:10.1016/j.cgh.2018.11.055
- Perng P, Zampella JG, Okoye GA. Management of hidradenitis suppurativa in pregnancy. J Am Acad Dermatol. 2017;76:979-989. doi:10.1016/j.jaad.2016.10.032
- Samuel S, Tremelling A, Murray M. Presentation and surgical management of hidradenitis suppurativa of the breast during pregnancy: a case report. Int J Surg Case Rep. 2018;51:21-24. doi:10.1016/j.ijscr.2018.08.013
- Yang CS, Teeple M, Muglia J, et al. Inflammatory and glandular skin disease in pregnancy. Clin Dermatol. 2016;34:335-343. doi:10.1016/j.clindermatol.2016.02.005
- Vossen AR, van Straalen KR, Prens EP, et al. Menses and pregnancy affect symptoms in hidradenitis suppurativa: a cross-sectional study. J Am Acad Dermatol. 2017;76:155-156. doi:10.1016/j.jaad.2016.07.024
- Lyons AB, Peacock A, McKenzie SA, et al. Evaluation of hidradenitis suppurativa disease course during pregnancy and postpartum. JAMA Dermatol. 2020;156:681-685. doi:10.1001/jamadermatol.2020.0777
- Riis PT, Ring HC, Themstrup L, et al. The role of androgens and estrogens in hidradenitis suppurativa—a systematic review. Acta Dermatovenerol Croat. 2016;24:239-249.
- Kong YL, Tey HL. Treatment of acne vulgaris during pregnancy and lactation. Drugs. 2013;73:779-787. doi:10.1007/s40265-013-0060-0
- Butler DC, Heller MM, Murase JE. Safety of dermatologic medications in pregnancy and lactation: part II. lactation. J Am Acad Dermatol. 2014;70:417:E1-E10. doi:10.1016/j.jaad.2013.09.009
- Wilmer E, Chai S, Kroumpouzos G. Drug safety: pregnancy rating classifications and controversies. Clin Dermatol. 2016;34:401-409. doi:10.1016/j.clindermatol.2016.02.013
- Inman WH, Rawson NS. Erythromycin estolate and jaundice. Br Med J (Clin Res Ed). 1983;286:1954-1955. doi:10.1136/bmj.286.6382.1954
- Workowski KA, Berman SM. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
- Murase JE, Heller MM, Butler DC. Safety of dermatologic medications in pregnancy and lactation: part I. pregnancy. J Am Acad Dermatol. 2014;70:401.e1-14; quiz 415. doi:10.1016/j.jaad.2013.09.010
- Brown SM, Aljefri K, Waas R, et al. Systemic medications used in treatment of common dermatological conditions: safety profile with respect to pregnancy, breast feeding and content in seminal fluid. J Dermatolog Treat. 2019;30:2-18. doi:10.1080/09546634.2016.1202402
- Kamarajah SK, Arntdz K, Bundred J, et al. Outcomes of pregnancy in recipients of liver transplants. Clin Gastroenterol Hepatol. 2019;17:1398-1404.e1. doi:10.1016/j.cgh.2018.11.055
Practice Points
- Some medications used to treat hidradenitis suppurativa (HS) may have teratogenic effects and be contraindicated during breastfeeding.
- We summarize what treatments are proven to be safe in pregnancy and breastfeeding and highlight the need for more guidelines and safety data for dermatologists to manage their pregnant patients with HS.
Rapidly Enlarging Bullous Plaque
The Diagnosis: Bullous Pyoderma Gangrenosum
A bone marrow biopsy revealed 60% myeloblasts, leading to a diagnosis of acute myeloid leukemia (AML). A biopsy obtained from the edge of the bullous plaque demonstrated a dense dermal neutrophilic infiltrate with extravasated erythrocytes (Figure). Fite, Gram, and Grocott-Gomori methenamine-silver staining failed to reveal infectious organisms. Tissue and blood cultures were negative. Given the pathologic findings, clinical presentation including recent diagnosis of AML, and exclusion of other underlying disease processes including infection, the diagnosis of bullous pyoderma gangrenosum (PG) was made. The lesion improved with systemic steroids and treatment of the underlying AML with fludarabine and venetoclax chemotherapy.
.")
First recognized in 1916 by French dermatologist Louis Brocq, MD, PG is a sterile neutrophilic dermatosis that predominantly affects women older than 50 years.1,2 This disorder can develop idiopathically; secondary to trauma; or in association with systemic diseases such as inflammatory bowel disease, rheumatoid arthritis, and hematologic malignancies. The pathogenesis of PG remains unclear; however, overexpression of inflammatory cytokines may mediate its development by stimulating T cells and promoting neutrophilic chemotaxis.3
Pyoderma gangrenosum classically presents as a rapidly enlarging ulcer with cribriform scarring but manifests variably. Four variants of the disorder exist: classic ulcerative, pustular, bullous, and vegetative PG. Ulcerative PG is the most common variant. Bullous PG is associated with hematologic malignancies such as primary myelofibrosis, myelodysplastic disease, and AML. In these patients, hematologic malignancy often exists prior to the development of PG and portends a poorer prognosis. This association underscores the importance of timely diagnosis and thorough hematologic evaluation by obtaining a complete blood cell count with differential, peripheral smear, serum protein electrophoresis with immunofixation, and quantitative immunoglobulins (IgA, IgG, IgM). If any of the results are positive, prompt referral to a hematologist and bone marrow biopsy are paramount.3
The diagnosis of PG remains elusive, as no validated clinical or pathological criteria exist. Histopathologic evaluation may be nonspecific and variable depending on the subtype. Biopsy results for classic ulcerative PG may reveal a neutrophilic infiltrate with leukocytoclasia. Bullous PG may include subepidermal hemorrhagic bullae. Notably, bullous PG appears histologically similar to the superficial bullous variant of Sweet syndrome.
Sweet syndrome (also known as acute febrile neutrophilic dermatosis) is a type of neutrophilic dermatosis characterized by fever, neutrophilia, and the sudden onset of tender erythematous lesions. Variations include idiopathic, subcutaneous, and bullous Sweet syndrome, which present as plaques, nodules, or bullae, respectively.4 Similar to PG, Sweet syndrome can manifest in patients with hematologic malignancies. Both PG and Sweet syndrome are thought to exist along a continuum and can be considered intersecting diagnoses in the setting of leukemia or other hematologic malignancies.5 There have been reports of the coexistence of distinct PG and Sweet syndrome lesions on a single patient, further supporting the belief that these entities share a common pathologic mechanism.6 Sweet syndrome also commonly can be associated with upper respiratory infections; pregnancy; and medications, with culprits including granulocyte colony-stimulating factor, azathioprine, vemurafenib, and isotretinoin.7
Other differential diagnoses include brown recluse spider bite, bullous fixed drug eruption (FDE), and necrotizing fasciitis (NF). Venom from the brown recluse spider (Loxosceles reclusa) can trigger toxin-mediated hemolysis, complement-mediated erythrocyte destruction, and basement membrane zone degradation due to the synergistic effects of the toxin’s sphingomyelinase D and protease content.8 The inciting bite is painless. After 8 hours, the site becomes painful and pruritic and presents with peripheral erythema and central pallor. After 24 hours, the lesion blisters. The blister ruptures within 3 to 4 days, resulting in eschar formation with the subsequent development of an indurated blue ulcer with a stellate center. Ulcers can take months to heal.9 Based on the clinical findings in our patient, this diagnosis was less likely.
Fixed drug eruption is a localized cutaneous reaction that manifests in fixed locations minutes to days after exposure to medications such as trimethoprimsulfamethoxazole, nonsteroidal anti-inflammatory drugs, salicylates, and oral contraceptives. Commonly affected areas include the hands, legs, genitals, and trunk. Lesions initially present as well-demarcated, erythematous to violaceous, round plaques. A rarer variant manifesting as bullae also has been described. Careful consideration of the patient’s history and physical examination findings is sufficient for establishing this diagnosis; however, a punch biopsy can provide clarity. Histopathology reveals a lichenoid tissue reaction with dyskeratosis, broad epidermal necrosis, and damage to the stratum basalis. A lymphocytic perivascular infiltrate also may appear in the dermis.10 Both the clinical findings and histopathology of our case were not characteristic of FDE.
Necrotizing fasciitis is a fulminant, life-threatening, soft-tissue infection precipitated by polymicrobial flora. Early recognition of NF is difficult, as in its early stages it can mimic cellulitis. As the infection takes its course, necrosis can extend from the skin and into the subcutaneous tissue. Patients also develop fever, leukocytosis, and signs of sepsis. Histopathology demonstrates neutrophilic infiltration with bacterial invasion as well as necrosis of the superficial fascia and subepidermal edema.11 Pyoderma gangrenosum previously has been reported to mimic NF; however, lack of responsiveness to antibiotic therapy would favor a diagnosis of PG over NF.12
Treatment of PG is driven by the extent of cutaneous involvement. In mild cases, wound care and topical therapy with corticosteroids and tacrolimus may suffice. Severe cases necessitate systemic therapy with oral corticosteroids or cyclosporine; biologic therapy also may play a role in treatment.4 In patients with hematologic malignancy, chemotherapy alone may partially or completely resolve the lesion; however, systemic corticosteroids commonly are included in management.3
- Brocq L. A new contribution to the study of geometric phagedenism. Ann Dermatol Syphiligr. 1916;9:1-39.
- Xu A, Balgobind A, Strunk A, et al. Prevalence estimates for pyoderma gangrenosum in the United States: an age- and sexadjusted population analysis. J Am Acad Dermatol. 2020;83:425-429. doi:10.1016/j.jaad.2019.08.001
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359. doi:10.1016/j.jaad.2019.09.032
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi:10.1186/1750-1172-2-34
- George C, Deroide F, Rustin M. Pyoderma gangrenosum—a guide to diagnosis and management. Clin Med (Lond). 2019;19:224‐228. doi:10.7861/clinmedicine.19-3-224
- Caughman W, Stern R, Haynes H. Neutrophilic dermatosis of myeloproliferative disorders. atypical forms of pyoderma gangrenosum and Sweet’s syndrome associated with myeloproliferative disorders. J Am Acad Dermatol. 1983;9:751-758. doi:10.1016/s0190-9622(83)70191-x
- Wallach D, Vignon-Pennamen M. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Manzoni-de-Almeida D, Squaiella-Baptistão CC, Lopes PH, et al. Loxosceles venom sphingomyelinase D activates human blood leukocytes: role of the complement system. Mol Immunol. 2018;94:45-53.
- Wilson JR, Hagood CO Jr, Prather ID. Brown recluse spider bites: a complex problem wound. a brief review and case study. Ostomy Wound Manage. 2005;51:59-66.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727. doi:10.14423/SMJ.0000000000000195
- Bakleh M, Wold LE, Mandrekar JN, et al. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40:410-414. doi:10.1086/427286
- de Souza EF, da Silva GA, Dos Santos GR, et al. Pyoderma gangrenosum simulating necrotizing fasciitis. Case Rep Med. 2015;2015:504970. doi:10.1155/2015/504970
The Diagnosis: Bullous Pyoderma Gangrenosum
A bone marrow biopsy revealed 60% myeloblasts, leading to a diagnosis of acute myeloid leukemia (AML). A biopsy obtained from the edge of the bullous plaque demonstrated a dense dermal neutrophilic infiltrate with extravasated erythrocytes (Figure). Fite, Gram, and Grocott-Gomori methenamine-silver staining failed to reveal infectious organisms. Tissue and blood cultures were negative. Given the pathologic findings, clinical presentation including recent diagnosis of AML, and exclusion of other underlying disease processes including infection, the diagnosis of bullous pyoderma gangrenosum (PG) was made. The lesion improved with systemic steroids and treatment of the underlying AML with fludarabine and venetoclax chemotherapy.
First recognized in 1916 by French dermatologist Louis Brocq, MD, PG is a sterile neutrophilic dermatosis that predominantly affects women older than 50 years.1,2 This disorder can develop idiopathically; secondary to trauma; or in association with systemic diseases such as inflammatory bowel disease, rheumatoid arthritis, and hematologic malignancies. The pathogenesis of PG remains unclear; however, overexpression of inflammatory cytokines may mediate its development by stimulating T cells and promoting neutrophilic chemotaxis.3
Pyoderma gangrenosum classically presents as a rapidly enlarging ulcer with cribriform scarring but manifests variably. Four variants of the disorder exist: classic ulcerative, pustular, bullous, and vegetative PG. Ulcerative PG is the most common variant. Bullous PG is associated with hematologic malignancies such as primary myelofibrosis, myelodysplastic disease, and AML. In these patients, hematologic malignancy often exists prior to the development of PG and portends a poorer prognosis. This association underscores the importance of timely diagnosis and thorough hematologic evaluation by obtaining a complete blood cell count with differential, peripheral smear, serum protein electrophoresis with immunofixation, and quantitative immunoglobulins (IgA, IgG, IgM). If any of the results are positive, prompt referral to a hematologist and bone marrow biopsy are paramount.3
The diagnosis of PG remains elusive, as no validated clinical or pathological criteria exist. Histopathologic evaluation may be nonspecific and variable depending on the subtype. Biopsy results for classic ulcerative PG may reveal a neutrophilic infiltrate with leukocytoclasia. Bullous PG may include subepidermal hemorrhagic bullae. Notably, bullous PG appears histologically similar to the superficial bullous variant of Sweet syndrome.
Sweet syndrome (also known as acute febrile neutrophilic dermatosis) is a type of neutrophilic dermatosis characterized by fever, neutrophilia, and the sudden onset of tender erythematous lesions. Variations include idiopathic, subcutaneous, and bullous Sweet syndrome, which present as plaques, nodules, or bullae, respectively.4 Similar to PG, Sweet syndrome can manifest in patients with hematologic malignancies. Both PG and Sweet syndrome are thought to exist along a continuum and can be considered intersecting diagnoses in the setting of leukemia or other hematologic malignancies.5 There have been reports of the coexistence of distinct PG and Sweet syndrome lesions on a single patient, further supporting the belief that these entities share a common pathologic mechanism.6 Sweet syndrome also commonly can be associated with upper respiratory infections; pregnancy; and medications, with culprits including granulocyte colony-stimulating factor, azathioprine, vemurafenib, and isotretinoin.7
Other differential diagnoses include brown recluse spider bite, bullous fixed drug eruption (FDE), and necrotizing fasciitis (NF). Venom from the brown recluse spider (Loxosceles reclusa) can trigger toxin-mediated hemolysis, complement-mediated erythrocyte destruction, and basement membrane zone degradation due to the synergistic effects of the toxin’s sphingomyelinase D and protease content.8 The inciting bite is painless. After 8 hours, the site becomes painful and pruritic and presents with peripheral erythema and central pallor. After 24 hours, the lesion blisters. The blister ruptures within 3 to 4 days, resulting in eschar formation with the subsequent development of an indurated blue ulcer with a stellate center. Ulcers can take months to heal.9 Based on the clinical findings in our patient, this diagnosis was less likely.
Fixed drug eruption is a localized cutaneous reaction that manifests in fixed locations minutes to days after exposure to medications such as trimethoprimsulfamethoxazole, nonsteroidal anti-inflammatory drugs, salicylates, and oral contraceptives. Commonly affected areas include the hands, legs, genitals, and trunk. Lesions initially present as well-demarcated, erythematous to violaceous, round plaques. A rarer variant manifesting as bullae also has been described. Careful consideration of the patient’s history and physical examination findings is sufficient for establishing this diagnosis; however, a punch biopsy can provide clarity. Histopathology reveals a lichenoid tissue reaction with dyskeratosis, broad epidermal necrosis, and damage to the stratum basalis. A lymphocytic perivascular infiltrate also may appear in the dermis.10 Both the clinical findings and histopathology of our case were not characteristic of FDE.
Necrotizing fasciitis is a fulminant, life-threatening, soft-tissue infection precipitated by polymicrobial flora. Early recognition of NF is difficult, as in its early stages it can mimic cellulitis. As the infection takes its course, necrosis can extend from the skin and into the subcutaneous tissue. Patients also develop fever, leukocytosis, and signs of sepsis. Histopathology demonstrates neutrophilic infiltration with bacterial invasion as well as necrosis of the superficial fascia and subepidermal edema.11 Pyoderma gangrenosum previously has been reported to mimic NF; however, lack of responsiveness to antibiotic therapy would favor a diagnosis of PG over NF.12
Treatment of PG is driven by the extent of cutaneous involvement. In mild cases, wound care and topical therapy with corticosteroids and tacrolimus may suffice. Severe cases necessitate systemic therapy with oral corticosteroids or cyclosporine; biologic therapy also may play a role in treatment.4 In patients with hematologic malignancy, chemotherapy alone may partially or completely resolve the lesion; however, systemic corticosteroids commonly are included in management.3
The Diagnosis: Bullous Pyoderma Gangrenosum
A bone marrow biopsy revealed 60% myeloblasts, leading to a diagnosis of acute myeloid leukemia (AML). A biopsy obtained from the edge of the bullous plaque demonstrated a dense dermal neutrophilic infiltrate with extravasated erythrocytes (Figure). Fite, Gram, and Grocott-Gomori methenamine-silver staining failed to reveal infectious organisms. Tissue and blood cultures were negative. Given the pathologic findings, clinical presentation including recent diagnosis of AML, and exclusion of other underlying disease processes including infection, the diagnosis of bullous pyoderma gangrenosum (PG) was made. The lesion improved with systemic steroids and treatment of the underlying AML with fludarabine and venetoclax chemotherapy.
First recognized in 1916 by French dermatologist Louis Brocq, MD, PG is a sterile neutrophilic dermatosis that predominantly affects women older than 50 years.1,2 This disorder can develop idiopathically; secondary to trauma; or in association with systemic diseases such as inflammatory bowel disease, rheumatoid arthritis, and hematologic malignancies. The pathogenesis of PG remains unclear; however, overexpression of inflammatory cytokines may mediate its development by stimulating T cells and promoting neutrophilic chemotaxis.3
Pyoderma gangrenosum classically presents as a rapidly enlarging ulcer with cribriform scarring but manifests variably. Four variants of the disorder exist: classic ulcerative, pustular, bullous, and vegetative PG. Ulcerative PG is the most common variant. Bullous PG is associated with hematologic malignancies such as primary myelofibrosis, myelodysplastic disease, and AML. In these patients, hematologic malignancy often exists prior to the development of PG and portends a poorer prognosis. This association underscores the importance of timely diagnosis and thorough hematologic evaluation by obtaining a complete blood cell count with differential, peripheral smear, serum protein electrophoresis with immunofixation, and quantitative immunoglobulins (IgA, IgG, IgM). If any of the results are positive, prompt referral to a hematologist and bone marrow biopsy are paramount.3
The diagnosis of PG remains elusive, as no validated clinical or pathological criteria exist. Histopathologic evaluation may be nonspecific and variable depending on the subtype. Biopsy results for classic ulcerative PG may reveal a neutrophilic infiltrate with leukocytoclasia. Bullous PG may include subepidermal hemorrhagic bullae. Notably, bullous PG appears histologically similar to the superficial bullous variant of Sweet syndrome.
Sweet syndrome (also known as acute febrile neutrophilic dermatosis) is a type of neutrophilic dermatosis characterized by fever, neutrophilia, and the sudden onset of tender erythematous lesions. Variations include idiopathic, subcutaneous, and bullous Sweet syndrome, which present as plaques, nodules, or bullae, respectively.4 Similar to PG, Sweet syndrome can manifest in patients with hematologic malignancies. Both PG and Sweet syndrome are thought to exist along a continuum and can be considered intersecting diagnoses in the setting of leukemia or other hematologic malignancies.5 There have been reports of the coexistence of distinct PG and Sweet syndrome lesions on a single patient, further supporting the belief that these entities share a common pathologic mechanism.6 Sweet syndrome also commonly can be associated with upper respiratory infections; pregnancy; and medications, with culprits including granulocyte colony-stimulating factor, azathioprine, vemurafenib, and isotretinoin.7
Other differential diagnoses include brown recluse spider bite, bullous fixed drug eruption (FDE), and necrotizing fasciitis (NF). Venom from the brown recluse spider (Loxosceles reclusa) can trigger toxin-mediated hemolysis, complement-mediated erythrocyte destruction, and basement membrane zone degradation due to the synergistic effects of the toxin’s sphingomyelinase D and protease content.8 The inciting bite is painless. After 8 hours, the site becomes painful and pruritic and presents with peripheral erythema and central pallor. After 24 hours, the lesion blisters. The blister ruptures within 3 to 4 days, resulting in eschar formation with the subsequent development of an indurated blue ulcer with a stellate center. Ulcers can take months to heal.9 Based on the clinical findings in our patient, this diagnosis was less likely.
Fixed drug eruption is a localized cutaneous reaction that manifests in fixed locations minutes to days after exposure to medications such as trimethoprimsulfamethoxazole, nonsteroidal anti-inflammatory drugs, salicylates, and oral contraceptives. Commonly affected areas include the hands, legs, genitals, and trunk. Lesions initially present as well-demarcated, erythematous to violaceous, round plaques. A rarer variant manifesting as bullae also has been described. Careful consideration of the patient’s history and physical examination findings is sufficient for establishing this diagnosis; however, a punch biopsy can provide clarity. Histopathology reveals a lichenoid tissue reaction with dyskeratosis, broad epidermal necrosis, and damage to the stratum basalis. A lymphocytic perivascular infiltrate also may appear in the dermis.10 Both the clinical findings and histopathology of our case were not characteristic of FDE.
Necrotizing fasciitis is a fulminant, life-threatening, soft-tissue infection precipitated by polymicrobial flora. Early recognition of NF is difficult, as in its early stages it can mimic cellulitis. As the infection takes its course, necrosis can extend from the skin and into the subcutaneous tissue. Patients also develop fever, leukocytosis, and signs of sepsis. Histopathology demonstrates neutrophilic infiltration with bacterial invasion as well as necrosis of the superficial fascia and subepidermal edema.11 Pyoderma gangrenosum previously has been reported to mimic NF; however, lack of responsiveness to antibiotic therapy would favor a diagnosis of PG over NF.12
Treatment of PG is driven by the extent of cutaneous involvement. In mild cases, wound care and topical therapy with corticosteroids and tacrolimus may suffice. Severe cases necessitate systemic therapy with oral corticosteroids or cyclosporine; biologic therapy also may play a role in treatment.4 In patients with hematologic malignancy, chemotherapy alone may partially or completely resolve the lesion; however, systemic corticosteroids commonly are included in management.3
- Brocq L. A new contribution to the study of geometric phagedenism. Ann Dermatol Syphiligr. 1916;9:1-39.
- Xu A, Balgobind A, Strunk A, et al. Prevalence estimates for pyoderma gangrenosum in the United States: an age- and sexadjusted population analysis. J Am Acad Dermatol. 2020;83:425-429. doi:10.1016/j.jaad.2019.08.001
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359. doi:10.1016/j.jaad.2019.09.032
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi:10.1186/1750-1172-2-34
- George C, Deroide F, Rustin M. Pyoderma gangrenosum—a guide to diagnosis and management. Clin Med (Lond). 2019;19:224‐228. doi:10.7861/clinmedicine.19-3-224
- Caughman W, Stern R, Haynes H. Neutrophilic dermatosis of myeloproliferative disorders. atypical forms of pyoderma gangrenosum and Sweet’s syndrome associated with myeloproliferative disorders. J Am Acad Dermatol. 1983;9:751-758. doi:10.1016/s0190-9622(83)70191-x
- Wallach D, Vignon-Pennamen M. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Manzoni-de-Almeida D, Squaiella-Baptistão CC, Lopes PH, et al. Loxosceles venom sphingomyelinase D activates human blood leukocytes: role of the complement system. Mol Immunol. 2018;94:45-53.
- Wilson JR, Hagood CO Jr, Prather ID. Brown recluse spider bites: a complex problem wound. a brief review and case study. Ostomy Wound Manage. 2005;51:59-66.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727. doi:10.14423/SMJ.0000000000000195
- Bakleh M, Wold LE, Mandrekar JN, et al. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40:410-414. doi:10.1086/427286
- de Souza EF, da Silva GA, Dos Santos GR, et al. Pyoderma gangrenosum simulating necrotizing fasciitis. Case Rep Med. 2015;2015:504970. doi:10.1155/2015/504970
- Brocq L. A new contribution to the study of geometric phagedenism. Ann Dermatol Syphiligr. 1916;9:1-39.
- Xu A, Balgobind A, Strunk A, et al. Prevalence estimates for pyoderma gangrenosum in the United States: an age- and sexadjusted population analysis. J Am Acad Dermatol. 2020;83:425-429. doi:10.1016/j.jaad.2019.08.001
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359. doi:10.1016/j.jaad.2019.09.032
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi:10.1186/1750-1172-2-34
- George C, Deroide F, Rustin M. Pyoderma gangrenosum—a guide to diagnosis and management. Clin Med (Lond). 2019;19:224‐228. doi:10.7861/clinmedicine.19-3-224
- Caughman W, Stern R, Haynes H. Neutrophilic dermatosis of myeloproliferative disorders. atypical forms of pyoderma gangrenosum and Sweet’s syndrome associated with myeloproliferative disorders. J Am Acad Dermatol. 1983;9:751-758. doi:10.1016/s0190-9622(83)70191-x
- Wallach D, Vignon-Pennamen M. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Manzoni-de-Almeida D, Squaiella-Baptistão CC, Lopes PH, et al. Loxosceles venom sphingomyelinase D activates human blood leukocytes: role of the complement system. Mol Immunol. 2018;94:45-53.
- Wilson JR, Hagood CO Jr, Prather ID. Brown recluse spider bites: a complex problem wound. a brief review and case study. Ostomy Wound Manage. 2005;51:59-66.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727. doi:10.14423/SMJ.0000000000000195
- Bakleh M, Wold LE, Mandrekar JN, et al. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40:410-414. doi:10.1086/427286
- de Souza EF, da Silva GA, Dos Santos GR, et al. Pyoderma gangrenosum simulating necrotizing fasciitis. Case Rep Med. 2015;2015:504970. doi:10.1155/2015/504970

A 26-year-old previously healthy man presented to the emergency department with a new asymptomatic enlarging lesion on the lower leg that had appeared 4 days prior as a self-described “pimple” and rapidly evolved. The patient also reported chills, fatigue, and decreased appetite during that time. Physical examination revealed a red to violaceous, well-demarcated, bullous plaque involving much of the left lower leg. Laboratory studies demonstrated a hemoglobin level of 8.1 g/dL (reference range, 14.0–17.5 g/dL), hematocrit level of 23.7% (reference range, 41%–50%), platelet count of 26×103 /μL (reference range, 150–350×103 /μL), and a population of circulating blast cells and metamyelocytes.

Iododerma Following Exposure to Iodine: A Case of Explosive Acneform Eruption Overnight
To the Editor:
Iododerma is a rare dermatologic condition caused by exposure to iodinated contrast media, oral iodine suspensions, or topical povidone-iodine that can manifest as eruptive acneform lesions.1-3
A 27-year-old woman in septic shock presented for worsening facial lesions that showed no improvement on broad-spectrum antibiotics, antifungals, and antivirals. She initially presented to an outside hospital with abdominal pain and underwent computed tomography (CT) with intravenous (IV) iodinated contrast; 24 hours after this imaging study, the family reported the appearance of “explosive acne overnight.” The lesions first appeared as vegetative and acneform ulcerations on the face. A second abdominal CT scan with IV contrast was performed 4 days after the initial scan, given the concern for spontaneous bacterial peritonitis. Hours after the second study, the lesions progressed to involve the buccal mucosae, tongue, mucosal airway, and distal arms and legs. She became progressively disoriented and developed an altered mentation over the course of the following week. Due to progressive facial edema, she required intubation 5 days after the second CT scan.

The patient had a medical history of end-stage renal disease secondary to crescenteric glomerulonephritis on peritoneal dialysis. Physical examination revealed numerous beefy-red, heaped-up, weepy, crusted nodules clustered on the face (Figure 1) and a few newer bullous-appearing lesions on the hands and feet. She had similar lesions involving the buccal mucosae and tongue with substantial facial edema. Infectious workup was notable for a positive skin culture growing methicillin-susceptible Staphylococcus aureus. All blood and tissue cultures as well as serologies for fungal and viral etiologies were negative. A tissue biopsy revealed necrosis with a neutrophilic infiltrate with mixed cell inflammation (Figure 2), and direct immunofluorescence was negative.

The patient initially was thought to be septic due to viral or bacterial infection. She was transferred from an outside hospital 7 days after the initial appearance of the acneform lesions, having already received IV contrast on 2 occasions within the first 48 hours of illness. Infectious disease was consulted and initiated broad-spectrum antiviral, antimicrobial, and antifungal therapy with acyclovir, linezolid, meropenem, and later micafungin without improvement. The diagnosis of iododerma ultimately was established based on the patient’s elevated urinary iodine levels with preceding iodine exposure in the context of renal failure. The preferential involvement of sebaceous areas and pathology findings were supportive of this diagnosis. Aggressive supportive measures including respiratory support, IV fluids, and dialysis were initiated. Topical iodine solutions, iodine-containing medications, and additional contrast subsequently were avoided. Despite these supportive measures, the patient died within 48 hours of admission from acute respiratory failure. Her autopsy attributed “septic complications of multifocal ulcerative cutaneous disease” as the anatomic cause of death.
Iododerma is an extremely rare neutrophilic dermatosis. The proposed mechanism of action involves a cell-mediated hypersensitivity reaction to iodine with induction of neutrophil degranulation.2 There have been documented cases with exposure to oral potassium iodide supplements, amiodarone, topical povidone-iodine, and IV iodinated contrast material.1-3 Iododerma typically presents 1 to 3 days after exposure to iodine. The most common source is IV radiocontrast. Diagnosis is based on the clinical presentation including acneform to vegetative nodular or bullous eruptions involving sebaceous areas in the context of recent iodine exposure. Elevated urinary iodine levels and histologic findings of neutrophilic infiltrate of the dermis support the diagnosis.3,4
Although there have been reported cases of iododerma in patients with normal renal function, patients with renal failure are much more susceptible due to the decreased clearance of iodine.5 The plasma half-life of radiocontrast is 23 hours in patients with end-stage renal disease vs 2 hours in patients with normal kidney function.3 Dosage adjustments for renal impairment have not been well studied, and no specific guidelines exist for the prevention of iododerma in patients with renal failure.
The first step in treating iododerma is to remove the offending iodine-containing agent. In most cases, cutaneous lesions resolve in 4 to 6 weeks after discontinuation of the source of iodine; however, there have been reported fatalities in the literature secondary to pulmonary edema in patients with iododerma.6,7 Despite the rarity and diagnostically challenging nature of iododerma, early recognition of this disease is crucial. Although our patient showed symptoms of iododerma after 1 dose of radiocontrast, she was not diagnosed at that time and received a second imaging study with contrast less than 48 hours later. These 2 consecutive exposures to iodine as well as the delayed diagnosis unfortunately resulted in rapid clinical deterioration.
The mainstay of therapy for iododerma includes avoidance of iodine-containing materials as soon as the diagnosis is suspected as well as supportive care. Patients have been successfully treated with systemic corticosteroids, with the addition of cyclosporine and hemodialysis in severe cases.3 Patients with a history of iododerma are advised to avoid iodine in their diet, in topical preparations, and in future imaging studies.8
- Aliagaoglu C, Turan H, Uslu E, et al. Iododerma following topical povidone-iodine application. Cutan Ocul Toxicol. 2013;32:339-340.
- Torkamani, N, Sinclair R. Iododerma in pregnancy secondary to iodinated multivitamins. Australas J Dermatol. 2015;56:235-236.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast material. Br J Dermatol. 2014;170:1377-1379.
- Stavert R, Bunick CG, Modi B, et al. Vegetative plaques and hemorrhagic pustules. JAMA Dermatol. 2013;149:1231-1232.
- Rothman LR, Levender MM, Scharf MD, et al. Iododerma following serial computed tomography scans in a lung cancer patient. J Drugs Dermatol. 2013;12:574-576.
- Miranda-Romero A, Sánchez-Sambucety P, Gómez JE, et al. Vegetating iododerma with fatal outcome. Dermatology. 1999;198:295-297.
- Vailant L, Pengloan J, Blanchier D, et al. Iododerma and acute respiratory distress with leucocytoclastic vasculitis following the intravenous injection of contrast medium. Clin Exp Dermatol. 1990;15:232-233.
- Massé M, Flanaga V, Zhou LH. Use of topical povidone iodine resulting in an iododerma-like eruption. J Dermatol. 2008;35:744-747.
To the Editor:
Iododerma is a rare dermatologic condition caused by exposure to iodinated contrast media, oral iodine suspensions, or topical povidone-iodine that can manifest as eruptive acneform lesions.1-3
A 27-year-old woman in septic shock presented for worsening facial lesions that showed no improvement on broad-spectrum antibiotics, antifungals, and antivirals. She initially presented to an outside hospital with abdominal pain and underwent computed tomography (CT) with intravenous (IV) iodinated contrast; 24 hours after this imaging study, the family reported the appearance of “explosive acne overnight.” The lesions first appeared as vegetative and acneform ulcerations on the face. A second abdominal CT scan with IV contrast was performed 4 days after the initial scan, given the concern for spontaneous bacterial peritonitis. Hours after the second study, the lesions progressed to involve the buccal mucosae, tongue, mucosal airway, and distal arms and legs. She became progressively disoriented and developed an altered mentation over the course of the following week. Due to progressive facial edema, she required intubation 5 days after the second CT scan.
The patient had a medical history of end-stage renal disease secondary to crescenteric glomerulonephritis on peritoneal dialysis. Physical examination revealed numerous beefy-red, heaped-up, weepy, crusted nodules clustered on the face (Figure 1) and a few newer bullous-appearing lesions on the hands and feet. She had similar lesions involving the buccal mucosae and tongue with substantial facial edema. Infectious workup was notable for a positive skin culture growing methicillin-susceptible Staphylococcus aureus. All blood and tissue cultures as well as serologies for fungal and viral etiologies were negative. A tissue biopsy revealed necrosis with a neutrophilic infiltrate with mixed cell inflammation (Figure 2), and direct immunofluorescence was negative.
The patient initially was thought to be septic due to viral or bacterial infection. She was transferred from an outside hospital 7 days after the initial appearance of the acneform lesions, having already received IV contrast on 2 occasions within the first 48 hours of illness. Infectious disease was consulted and initiated broad-spectrum antiviral, antimicrobial, and antifungal therapy with acyclovir, linezolid, meropenem, and later micafungin without improvement. The diagnosis of iododerma ultimately was established based on the patient’s elevated urinary iodine levels with preceding iodine exposure in the context of renal failure. The preferential involvement of sebaceous areas and pathology findings were supportive of this diagnosis. Aggressive supportive measures including respiratory support, IV fluids, and dialysis were initiated. Topical iodine solutions, iodine-containing medications, and additional contrast subsequently were avoided. Despite these supportive measures, the patient died within 48 hours of admission from acute respiratory failure. Her autopsy attributed “septic complications of multifocal ulcerative cutaneous disease” as the anatomic cause of death.
Iododerma is an extremely rare neutrophilic dermatosis. The proposed mechanism of action involves a cell-mediated hypersensitivity reaction to iodine with induction of neutrophil degranulation.2 There have been documented cases with exposure to oral potassium iodide supplements, amiodarone, topical povidone-iodine, and IV iodinated contrast material.1-3 Iododerma typically presents 1 to 3 days after exposure to iodine. The most common source is IV radiocontrast. Diagnosis is based on the clinical presentation including acneform to vegetative nodular or bullous eruptions involving sebaceous areas in the context of recent iodine exposure. Elevated urinary iodine levels and histologic findings of neutrophilic infiltrate of the dermis support the diagnosis.3,4
Although there have been reported cases of iododerma in patients with normal renal function, patients with renal failure are much more susceptible due to the decreased clearance of iodine.5 The plasma half-life of radiocontrast is 23 hours in patients with end-stage renal disease vs 2 hours in patients with normal kidney function.3 Dosage adjustments for renal impairment have not been well studied, and no specific guidelines exist for the prevention of iododerma in patients with renal failure.
The first step in treating iododerma is to remove the offending iodine-containing agent. In most cases, cutaneous lesions resolve in 4 to 6 weeks after discontinuation of the source of iodine; however, there have been reported fatalities in the literature secondary to pulmonary edema in patients with iododerma.6,7 Despite the rarity and diagnostically challenging nature of iododerma, early recognition of this disease is crucial. Although our patient showed symptoms of iododerma after 1 dose of radiocontrast, she was not diagnosed at that time and received a second imaging study with contrast less than 48 hours later. These 2 consecutive exposures to iodine as well as the delayed diagnosis unfortunately resulted in rapid clinical deterioration.
The mainstay of therapy for iododerma includes avoidance of iodine-containing materials as soon as the diagnosis is suspected as well as supportive care. Patients have been successfully treated with systemic corticosteroids, with the addition of cyclosporine and hemodialysis in severe cases.3 Patients with a history of iododerma are advised to avoid iodine in their diet, in topical preparations, and in future imaging studies.8
To the Editor:
Iododerma is a rare dermatologic condition caused by exposure to iodinated contrast media, oral iodine suspensions, or topical povidone-iodine that can manifest as eruptive acneform lesions.1-3
A 27-year-old woman in septic shock presented for worsening facial lesions that showed no improvement on broad-spectrum antibiotics, antifungals, and antivirals. She initially presented to an outside hospital with abdominal pain and underwent computed tomography (CT) with intravenous (IV) iodinated contrast; 24 hours after this imaging study, the family reported the appearance of “explosive acne overnight.” The lesions first appeared as vegetative and acneform ulcerations on the face. A second abdominal CT scan with IV contrast was performed 4 days after the initial scan, given the concern for spontaneous bacterial peritonitis. Hours after the second study, the lesions progressed to involve the buccal mucosae, tongue, mucosal airway, and distal arms and legs. She became progressively disoriented and developed an altered mentation over the course of the following week. Due to progressive facial edema, she required intubation 5 days after the second CT scan.
The patient had a medical history of end-stage renal disease secondary to crescenteric glomerulonephritis on peritoneal dialysis. Physical examination revealed numerous beefy-red, heaped-up, weepy, crusted nodules clustered on the face (Figure 1) and a few newer bullous-appearing lesions on the hands and feet. She had similar lesions involving the buccal mucosae and tongue with substantial facial edema. Infectious workup was notable for a positive skin culture growing methicillin-susceptible Staphylococcus aureus. All blood and tissue cultures as well as serologies for fungal and viral etiologies were negative. A tissue biopsy revealed necrosis with a neutrophilic infiltrate with mixed cell inflammation (Figure 2), and direct immunofluorescence was negative.
The patient initially was thought to be septic due to viral or bacterial infection. She was transferred from an outside hospital 7 days after the initial appearance of the acneform lesions, having already received IV contrast on 2 occasions within the first 48 hours of illness. Infectious disease was consulted and initiated broad-spectrum antiviral, antimicrobial, and antifungal therapy with acyclovir, linezolid, meropenem, and later micafungin without improvement. The diagnosis of iododerma ultimately was established based on the patient’s elevated urinary iodine levels with preceding iodine exposure in the context of renal failure. The preferential involvement of sebaceous areas and pathology findings were supportive of this diagnosis. Aggressive supportive measures including respiratory support, IV fluids, and dialysis were initiated. Topical iodine solutions, iodine-containing medications, and additional contrast subsequently were avoided. Despite these supportive measures, the patient died within 48 hours of admission from acute respiratory failure. Her autopsy attributed “septic complications of multifocal ulcerative cutaneous disease” as the anatomic cause of death.
Iododerma is an extremely rare neutrophilic dermatosis. The proposed mechanism of action involves a cell-mediated hypersensitivity reaction to iodine with induction of neutrophil degranulation.2 There have been documented cases with exposure to oral potassium iodide supplements, amiodarone, topical povidone-iodine, and IV iodinated contrast material.1-3 Iododerma typically presents 1 to 3 days after exposure to iodine. The most common source is IV radiocontrast. Diagnosis is based on the clinical presentation including acneform to vegetative nodular or bullous eruptions involving sebaceous areas in the context of recent iodine exposure. Elevated urinary iodine levels and histologic findings of neutrophilic infiltrate of the dermis support the diagnosis.3,4
Although there have been reported cases of iododerma in patients with normal renal function, patients with renal failure are much more susceptible due to the decreased clearance of iodine.5 The plasma half-life of radiocontrast is 23 hours in patients with end-stage renal disease vs 2 hours in patients with normal kidney function.3 Dosage adjustments for renal impairment have not been well studied, and no specific guidelines exist for the prevention of iododerma in patients with renal failure.
The first step in treating iododerma is to remove the offending iodine-containing agent. In most cases, cutaneous lesions resolve in 4 to 6 weeks after discontinuation of the source of iodine; however, there have been reported fatalities in the literature secondary to pulmonary edema in patients with iododerma.6,7 Despite the rarity and diagnostically challenging nature of iododerma, early recognition of this disease is crucial. Although our patient showed symptoms of iododerma after 1 dose of radiocontrast, she was not diagnosed at that time and received a second imaging study with contrast less than 48 hours later. These 2 consecutive exposures to iodine as well as the delayed diagnosis unfortunately resulted in rapid clinical deterioration.
The mainstay of therapy for iododerma includes avoidance of iodine-containing materials as soon as the diagnosis is suspected as well as supportive care. Patients have been successfully treated with systemic corticosteroids, with the addition of cyclosporine and hemodialysis in severe cases.3 Patients with a history of iododerma are advised to avoid iodine in their diet, in topical preparations, and in future imaging studies.8
- Aliagaoglu C, Turan H, Uslu E, et al. Iododerma following topical povidone-iodine application. Cutan Ocul Toxicol. 2013;32:339-340.
- Torkamani, N, Sinclair R. Iododerma in pregnancy secondary to iodinated multivitamins. Australas J Dermatol. 2015;56:235-236.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast material. Br J Dermatol. 2014;170:1377-1379.
- Stavert R, Bunick CG, Modi B, et al. Vegetative plaques and hemorrhagic pustules. JAMA Dermatol. 2013;149:1231-1232.
- Rothman LR, Levender MM, Scharf MD, et al. Iododerma following serial computed tomography scans in a lung cancer patient. J Drugs Dermatol. 2013;12:574-576.
- Miranda-Romero A, Sánchez-Sambucety P, Gómez JE, et al. Vegetating iododerma with fatal outcome. Dermatology. 1999;198:295-297.
- Vailant L, Pengloan J, Blanchier D, et al. Iododerma and acute respiratory distress with leucocytoclastic vasculitis following the intravenous injection of contrast medium. Clin Exp Dermatol. 1990;15:232-233.
- Massé M, Flanaga V, Zhou LH. Use of topical povidone iodine resulting in an iododerma-like eruption. J Dermatol. 2008;35:744-747.
- Aliagaoglu C, Turan H, Uslu E, et al. Iododerma following topical povidone-iodine application. Cutan Ocul Toxicol. 2013;32:339-340.
- Torkamani, N, Sinclair R. Iododerma in pregnancy secondary to iodinated multivitamins. Australas J Dermatol. 2015;56:235-236.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast material. Br J Dermatol. 2014;170:1377-1379.
- Stavert R, Bunick CG, Modi B, et al. Vegetative plaques and hemorrhagic pustules. JAMA Dermatol. 2013;149:1231-1232.
- Rothman LR, Levender MM, Scharf MD, et al. Iododerma following serial computed tomography scans in a lung cancer patient. J Drugs Dermatol. 2013;12:574-576.
- Miranda-Romero A, Sánchez-Sambucety P, Gómez JE, et al. Vegetating iododerma with fatal outcome. Dermatology. 1999;198:295-297.
- Vailant L, Pengloan J, Blanchier D, et al. Iododerma and acute respiratory distress with leucocytoclastic vasculitis following the intravenous injection of contrast medium. Clin Exp Dermatol. 1990;15:232-233.
- Massé M, Flanaga V, Zhou LH. Use of topical povidone iodine resulting in an iododerma-like eruption. J Dermatol. 2008;35:744-747.
Practice Points
- Iododerma should be considered for patients who develop rapidly progressive, vegetative lesions, especially in those with renal failure. A thorough history should be obtained in these cases, focusing on medications and recent studies involving iodinated contrast.
- The most important first step in treating iododerma is to remove the iodine-containing agent to avoid continued exposure.
- Therapies for iododerma include supportive care, cyclosporine, systemic corticosteroids, and hemodialysis in severe cases.

Patch Testing on Dupilumab: Reliable or Not?
In patients with persistent atopic dermatitis (AD) who are taking dupilumab, is there benefit of patch testing to determine if allergic contact dermatitis (ACD) also is contributing to their disease? Results of patch testing are likely be influenced by the immunomodulatory effects of dupilumab. Similar to the recommendation for patients to refrain from using topical or systemic corticosteroids for 1 week or more prior to patch testing to eliminate false negatives, we reviewed the literature to create practice guidelines for dermatologists regarding patch testing while a patient is taking dupilumab.
Pathophysiology and Pathomechanism
Dupilumab functions through the blockade of T helper 2 (TH2) cells; ACD is propagated through the T helper 1 (TH1) cellular pathway. However, patients with ACD that is unresponsive to allergen avoidance and traditional therapies, such as topical and oral corticosteroids, have responded to dupilumab. The more common reports of this responsiveness are with fragrances; multiple case series described patients with ACD to fragrance mix I1 and balsam of Peru1,2 who improved on dupilumab when other treatments failed. There also are reports of response when ACD was secondary to nickel,2,3p-phenylenediamine,1 Compositae,4 and non–formaldehyde-releasing preservatives (non-FRPs).5 Therefore, not all ACD is propagated through the TH1 cellular pathway.
As noted in these cases, ACD can be a response to an allergen whose pathogenesis involves the TH2 pathway or when patient characteristics favor a TH2 response. It has been suggested that AD patients are more susceptible to TH2-mediated contact sensitization to less-potent allergens, such as fragrances.6
Patch Test Results
Positive patch test results for allergens have been reported while patients are on dupilumab therapy, including a few studies in which results prior to starting dupilumab were compared with those while patients were on dupilumab therapy. In a retrospective chart review of 48 patients on dupilumab for AD with persistent disease, 23 patients were patch tested before and during dupilumab therapy. In these patients, the majority of contact allergies were persistent and only 10% (13/125) of patch test–positive results resolved on dupilumab therapy.7 Contact allergies that resolved included those to emulsifiers (propylene glycol, Amerchol L101 [lanolin-containing products found in cosmetics and other goods], dimethylaminopropylamine), fragrances (fragrance mix I, balsam of Peru), sunscreens (sulisobenzone, phenylbenzimidazole-5-sulfonic acid), and metals (vanadium chloride, phenylmercuric acetate).7 The following results observed in individual cases demonstrated conflicting findings: persistence of allergy to non-FRPs (methylisothiazolinone [MI]) but resolution of allergy to formaldehyde8; persistence of allergy to corticosteroids (budesonide and alclometasone)9; persistence of allergy to an antibiotic (neomycin sulfate) but resolution of allergies to a different antibiotic (bacitracin), glues (ethyl acrylate), bleach, and glutaraldehyde9; persistence of nickel allergy but resolution of allergies to fragrances (cinnamic aldehyde, balsam of Peru) and non-FRPs (methylchloroisothiazolinone or MI)10; and persistence of allergies to non-FRPs (MI) and FRPs (bronopol) but resolution of allergies to nickel, fragrances (hydroperoxides of linalool), and Compositae.11 Additional case reports of positive patch test results while on dupilumab but with no pretreatment results for comparison include allergies to rubber additives,12-14 nickel,14 textile dyes,14 cosmetic and hair care additives,12,14,15 corticosteroids,15 FRPs,15 fragrances,15,16 emulsifiers,16 and non-FRPs.17
An evident theme in the dupilumab patch-testing literature has been that results are variable and case specific: a given patient with ACD to an allergen will respond to dupilumab treatment and have subsequent negative patch testing, while another patient will not respond to dupilumab treatment and have persistent positive patch testing. This is likely because, in certain individuals, the allergen-immune system combination shifts ACD pathogenesis from a purely TH1 response to at least a partial TH2 response, thus allowing for benefit from dupilumab therapy. T helper 1 cell–mediated ACD should not be affected by dupilumab; therefore, reliable results can be elucidated from patch testing despite the drug.
Final Thoughts
We propose that AD patients with residual disease after taking dupilumab undergo patch testing. Positive results indicate allergens that are not inhibited by the drug. Patients will need to follow strict allergen avoidance to resolve this component of their disease; failure to improve might suggest the result was a nonrelevant positive.
If patch testing is negative, an alternative cause for residual disease must be sought. We do not recommend stopping dupilumab prior to patch testing to avoid a disease flare from AD or possible TH2-mediated ACD.
- Chipalkatti N, Lee N, Zancanaro P, et al. Dupilumab as a treatment for allergic contact dermatitis. Dermatitis. 2018;29:347-348. doi:10.1097/DER.0000000000000414
- Jacob SE, Sung CT, Machler BC. Dupilumab for systemic allergy syndrome with dermatitis. Dermatitis. 2019;30:164-167. doi:10.1097/DER.0000000000000446
- Joshi SR, Khan DA. Effective use of dupilumab in managing systemic allergic contact dermatitis. Dermatitis. 2018;29:282-284. doi:10.1097/DER.0000000000000409
- Ruge IF, Skov L, Zachariae C, et al. Dupilumab treatment in two patients with severe allergic contact dermatitis caused by sesquiterpene lactones. Contact Dermatitis. 2020:83;137-139. doi:10.1111/cod.13545
- Goldminz AM, Scheinman PL. A case series of dupilumab-treated allergic contact dermatitis patients. Dermatol Ther. 2018;31:e12701. doi:10.1111/dth.12701
- Kohli N, Nedorost S. Inflamed skin predisposes to sensitization to less potent allergens. J Am Acad Dermatol. 2016;75:312-317. doi:10.1016/j.jaad.2016.03.010
- Raffi J, Suresh R, Botto N, et al. The impact of dupilumab on patch testing and the prevalence of comorbid allergic contact dermatitis in recalcitrant atopic dermatitis: a retrospective chart review. J Am Acad Dermatol. 2020;82:132-138. doi:10.1016/j.jaad.2019.09.028
- Puza CJ, Atwater AR. Positive patch test reaction in a patient taking dupilumab. Dermatitis. 2018;29:89. doi:10.1097/DER.0000000000000346
- Suresh R, Murase JE. The role of expanded series patch testing in identifying causality of residual facial dermatitis following initiation of dupilumab therapy. JAAD Case Rep. 2018;4:899-904. doi:10.1016/j.jdcr.2018.08.027
- Stout M, Silverberg JI. Variable impact of dupilumab on patch testing results and allergic contact dermatitis in adults with atopic dermatitis. J Am Acad Dermatol. 2019;81:157-162. doi:10.1016/j.jaad.2019.03.020
- Raffi J, Botto N. Patch testing and allergen-specific inhibition in a patient taking dupilumab. JAMA Dermatol. 2019;155:120-121. doi:10.1001/jamadermatol.2018.4098
- Hoot JW, Douglas JD, Falo LD Jr. Patch testing in a patient on dupilumab. Dermatitis. 2018;29:164. doi:10.1097/DER.0000000000000357
- Crepy M-N, Nosbaum A, Bensefa-Colas L. Blocking type 2 inflammation by dupilumab does not control classic (type 1-driven) allergic contact dermatitis in chronic hand eczema. Contact Dermatitis. 2019;81:145-147. doi:10.1111/cod.13266
- Raffi J, Chen R, Botto N. Wide dye reactors. JAAD Case Rep. 2019;5:877-879. doi:10.1016/j.jdcr.2019.08.005
- Koblinski JE, Hamann D. Mixed occupational and iatrogenic allergic contact dermatitis in a hairdresser. Occup Med (Lond). 2020;70:523-526. doi:10.1093/occmed/kqaa152
- Raffi J, Suresh R, Fishman H, et al. Investigating the role of allergic contact dermatitis in residual ocular surface disease on dupilumab (ROSDD). Int J Womens Dermatol. 2019;5:308-313. doi:10.1016/j.ijwd.2019.10.001
- Zhu GA, Chen JK, Chiou A, et al. Repeat patch testing in a patient with allergic contact dermatitis improved on dupilumab. JAAD Case Rep. 2019;5:336-338. doi:10.1016/j.jdcr.2019.01.023
In patients with persistent atopic dermatitis (AD) who are taking dupilumab, is there benefit of patch testing to determine if allergic contact dermatitis (ACD) also is contributing to their disease? Results of patch testing are likely be influenced by the immunomodulatory effects of dupilumab. Similar to the recommendation for patients to refrain from using topical or systemic corticosteroids for 1 week or more prior to patch testing to eliminate false negatives, we reviewed the literature to create practice guidelines for dermatologists regarding patch testing while a patient is taking dupilumab.
Pathophysiology and Pathomechanism
Dupilumab functions through the blockade of T helper 2 (TH2) cells; ACD is propagated through the T helper 1 (TH1) cellular pathway. However, patients with ACD that is unresponsive to allergen avoidance and traditional therapies, such as topical and oral corticosteroids, have responded to dupilumab. The more common reports of this responsiveness are with fragrances; multiple case series described patients with ACD to fragrance mix I1 and balsam of Peru1,2 who improved on dupilumab when other treatments failed. There also are reports of response when ACD was secondary to nickel,2,3p-phenylenediamine,1 Compositae,4 and non–formaldehyde-releasing preservatives (non-FRPs).5 Therefore, not all ACD is propagated through the TH1 cellular pathway.
As noted in these cases, ACD can be a response to an allergen whose pathogenesis involves the TH2 pathway or when patient characteristics favor a TH2 response. It has been suggested that AD patients are more susceptible to TH2-mediated contact sensitization to less-potent allergens, such as fragrances.6
Patch Test Results
Positive patch test results for allergens have been reported while patients are on dupilumab therapy, including a few studies in which results prior to starting dupilumab were compared with those while patients were on dupilumab therapy. In a retrospective chart review of 48 patients on dupilumab for AD with persistent disease, 23 patients were patch tested before and during dupilumab therapy. In these patients, the majority of contact allergies were persistent and only 10% (13/125) of patch test–positive results resolved on dupilumab therapy.7 Contact allergies that resolved included those to emulsifiers (propylene glycol, Amerchol L101 [lanolin-containing products found in cosmetics and other goods], dimethylaminopropylamine), fragrances (fragrance mix I, balsam of Peru), sunscreens (sulisobenzone, phenylbenzimidazole-5-sulfonic acid), and metals (vanadium chloride, phenylmercuric acetate).7 The following results observed in individual cases demonstrated conflicting findings: persistence of allergy to non-FRPs (methylisothiazolinone [MI]) but resolution of allergy to formaldehyde8; persistence of allergy to corticosteroids (budesonide and alclometasone)9; persistence of allergy to an antibiotic (neomycin sulfate) but resolution of allergies to a different antibiotic (bacitracin), glues (ethyl acrylate), bleach, and glutaraldehyde9; persistence of nickel allergy but resolution of allergies to fragrances (cinnamic aldehyde, balsam of Peru) and non-FRPs (methylchloroisothiazolinone or MI)10; and persistence of allergies to non-FRPs (MI) and FRPs (bronopol) but resolution of allergies to nickel, fragrances (hydroperoxides of linalool), and Compositae.11 Additional case reports of positive patch test results while on dupilumab but with no pretreatment results for comparison include allergies to rubber additives,12-14 nickel,14 textile dyes,14 cosmetic and hair care additives,12,14,15 corticosteroids,15 FRPs,15 fragrances,15,16 emulsifiers,16 and non-FRPs.17
An evident theme in the dupilumab patch-testing literature has been that results are variable and case specific: a given patient with ACD to an allergen will respond to dupilumab treatment and have subsequent negative patch testing, while another patient will not respond to dupilumab treatment and have persistent positive patch testing. This is likely because, in certain individuals, the allergen-immune system combination shifts ACD pathogenesis from a purely TH1 response to at least a partial TH2 response, thus allowing for benefit from dupilumab therapy. T helper 1 cell–mediated ACD should not be affected by dupilumab; therefore, reliable results can be elucidated from patch testing despite the drug.
Final Thoughts
We propose that AD patients with residual disease after taking dupilumab undergo patch testing. Positive results indicate allergens that are not inhibited by the drug. Patients will need to follow strict allergen avoidance to resolve this component of their disease; failure to improve might suggest the result was a nonrelevant positive.
If patch testing is negative, an alternative cause for residual disease must be sought. We do not recommend stopping dupilumab prior to patch testing to avoid a disease flare from AD or possible TH2-mediated ACD.
In patients with persistent atopic dermatitis (AD) who are taking dupilumab, is there benefit of patch testing to determine if allergic contact dermatitis (ACD) also is contributing to their disease? Results of patch testing are likely be influenced by the immunomodulatory effects of dupilumab. Similar to the recommendation for patients to refrain from using topical or systemic corticosteroids for 1 week or more prior to patch testing to eliminate false negatives, we reviewed the literature to create practice guidelines for dermatologists regarding patch testing while a patient is taking dupilumab.
Pathophysiology and Pathomechanism
Dupilumab functions through the blockade of T helper 2 (TH2) cells; ACD is propagated through the T helper 1 (TH1) cellular pathway. However, patients with ACD that is unresponsive to allergen avoidance and traditional therapies, such as topical and oral corticosteroids, have responded to dupilumab. The more common reports of this responsiveness are with fragrances; multiple case series described patients with ACD to fragrance mix I1 and balsam of Peru1,2 who improved on dupilumab when other treatments failed. There also are reports of response when ACD was secondary to nickel,2,3p-phenylenediamine,1 Compositae,4 and non–formaldehyde-releasing preservatives (non-FRPs).5 Therefore, not all ACD is propagated through the TH1 cellular pathway.
As noted in these cases, ACD can be a response to an allergen whose pathogenesis involves the TH2 pathway or when patient characteristics favor a TH2 response. It has been suggested that AD patients are more susceptible to TH2-mediated contact sensitization to less-potent allergens, such as fragrances.6
Patch Test Results
Positive patch test results for allergens have been reported while patients are on dupilumab therapy, including a few studies in which results prior to starting dupilumab were compared with those while patients were on dupilumab therapy. In a retrospective chart review of 48 patients on dupilumab for AD with persistent disease, 23 patients were patch tested before and during dupilumab therapy. In these patients, the majority of contact allergies were persistent and only 10% (13/125) of patch test–positive results resolved on dupilumab therapy.7 Contact allergies that resolved included those to emulsifiers (propylene glycol, Amerchol L101 [lanolin-containing products found in cosmetics and other goods], dimethylaminopropylamine), fragrances (fragrance mix I, balsam of Peru), sunscreens (sulisobenzone, phenylbenzimidazole-5-sulfonic acid), and metals (vanadium chloride, phenylmercuric acetate).7 The following results observed in individual cases demonstrated conflicting findings: persistence of allergy to non-FRPs (methylisothiazolinone [MI]) but resolution of allergy to formaldehyde8; persistence of allergy to corticosteroids (budesonide and alclometasone)9; persistence of allergy to an antibiotic (neomycin sulfate) but resolution of allergies to a different antibiotic (bacitracin), glues (ethyl acrylate), bleach, and glutaraldehyde9; persistence of nickel allergy but resolution of allergies to fragrances (cinnamic aldehyde, balsam of Peru) and non-FRPs (methylchloroisothiazolinone or MI)10; and persistence of allergies to non-FRPs (MI) and FRPs (bronopol) but resolution of allergies to nickel, fragrances (hydroperoxides of linalool), and Compositae.11 Additional case reports of positive patch test results while on dupilumab but with no pretreatment results for comparison include allergies to rubber additives,12-14 nickel,14 textile dyes,14 cosmetic and hair care additives,12,14,15 corticosteroids,15 FRPs,15 fragrances,15,16 emulsifiers,16 and non-FRPs.17
An evident theme in the dupilumab patch-testing literature has been that results are variable and case specific: a given patient with ACD to an allergen will respond to dupilumab treatment and have subsequent negative patch testing, while another patient will not respond to dupilumab treatment and have persistent positive patch testing. This is likely because, in certain individuals, the allergen-immune system combination shifts ACD pathogenesis from a purely TH1 response to at least a partial TH2 response, thus allowing for benefit from dupilumab therapy. T helper 1 cell–mediated ACD should not be affected by dupilumab; therefore, reliable results can be elucidated from patch testing despite the drug.
Final Thoughts
We propose that AD patients with residual disease after taking dupilumab undergo patch testing. Positive results indicate allergens that are not inhibited by the drug. Patients will need to follow strict allergen avoidance to resolve this component of their disease; failure to improve might suggest the result was a nonrelevant positive.
If patch testing is negative, an alternative cause for residual disease must be sought. We do not recommend stopping dupilumab prior to patch testing to avoid a disease flare from AD or possible TH2-mediated ACD.
- Chipalkatti N, Lee N, Zancanaro P, et al. Dupilumab as a treatment for allergic contact dermatitis. Dermatitis. 2018;29:347-348. doi:10.1097/DER.0000000000000414
- Jacob SE, Sung CT, Machler BC. Dupilumab for systemic allergy syndrome with dermatitis. Dermatitis. 2019;30:164-167. doi:10.1097/DER.0000000000000446
- Joshi SR, Khan DA. Effective use of dupilumab in managing systemic allergic contact dermatitis. Dermatitis. 2018;29:282-284. doi:10.1097/DER.0000000000000409
- Ruge IF, Skov L, Zachariae C, et al. Dupilumab treatment in two patients with severe allergic contact dermatitis caused by sesquiterpene lactones. Contact Dermatitis. 2020:83;137-139. doi:10.1111/cod.13545
- Goldminz AM, Scheinman PL. A case series of dupilumab-treated allergic contact dermatitis patients. Dermatol Ther. 2018;31:e12701. doi:10.1111/dth.12701
- Kohli N, Nedorost S. Inflamed skin predisposes to sensitization to less potent allergens. J Am Acad Dermatol. 2016;75:312-317. doi:10.1016/j.jaad.2016.03.010
- Raffi J, Suresh R, Botto N, et al. The impact of dupilumab on patch testing and the prevalence of comorbid allergic contact dermatitis in recalcitrant atopic dermatitis: a retrospective chart review. J Am Acad Dermatol. 2020;82:132-138. doi:10.1016/j.jaad.2019.09.028
- Puza CJ, Atwater AR. Positive patch test reaction in a patient taking dupilumab. Dermatitis. 2018;29:89. doi:10.1097/DER.0000000000000346
- Suresh R, Murase JE. The role of expanded series patch testing in identifying causality of residual facial dermatitis following initiation of dupilumab therapy. JAAD Case Rep. 2018;4:899-904. doi:10.1016/j.jdcr.2018.08.027
- Stout M, Silverberg JI. Variable impact of dupilumab on patch testing results and allergic contact dermatitis in adults with atopic dermatitis. J Am Acad Dermatol. 2019;81:157-162. doi:10.1016/j.jaad.2019.03.020
- Raffi J, Botto N. Patch testing and allergen-specific inhibition in a patient taking dupilumab. JAMA Dermatol. 2019;155:120-121. doi:10.1001/jamadermatol.2018.4098
- Hoot JW, Douglas JD, Falo LD Jr. Patch testing in a patient on dupilumab. Dermatitis. 2018;29:164. doi:10.1097/DER.0000000000000357
- Crepy M-N, Nosbaum A, Bensefa-Colas L. Blocking type 2 inflammation by dupilumab does not control classic (type 1-driven) allergic contact dermatitis in chronic hand eczema. Contact Dermatitis. 2019;81:145-147. doi:10.1111/cod.13266
- Raffi J, Chen R, Botto N. Wide dye reactors. JAAD Case Rep. 2019;5:877-879. doi:10.1016/j.jdcr.2019.08.005
- Koblinski JE, Hamann D. Mixed occupational and iatrogenic allergic contact dermatitis in a hairdresser. Occup Med (Lond). 2020;70:523-526. doi:10.1093/occmed/kqaa152
- Raffi J, Suresh R, Fishman H, et al. Investigating the role of allergic contact dermatitis in residual ocular surface disease on dupilumab (ROSDD). Int J Womens Dermatol. 2019;5:308-313. doi:10.1016/j.ijwd.2019.10.001
- Zhu GA, Chen JK, Chiou A, et al. Repeat patch testing in a patient with allergic contact dermatitis improved on dupilumab. JAAD Case Rep. 2019;5:336-338. doi:10.1016/j.jdcr.2019.01.023
- Chipalkatti N, Lee N, Zancanaro P, et al. Dupilumab as a treatment for allergic contact dermatitis. Dermatitis. 2018;29:347-348. doi:10.1097/DER.0000000000000414
- Jacob SE, Sung CT, Machler BC. Dupilumab for systemic allergy syndrome with dermatitis. Dermatitis. 2019;30:164-167. doi:10.1097/DER.0000000000000446
- Joshi SR, Khan DA. Effective use of dupilumab in managing systemic allergic contact dermatitis. Dermatitis. 2018;29:282-284. doi:10.1097/DER.0000000000000409
- Ruge IF, Skov L, Zachariae C, et al. Dupilumab treatment in two patients with severe allergic contact dermatitis caused by sesquiterpene lactones. Contact Dermatitis. 2020:83;137-139. doi:10.1111/cod.13545
- Goldminz AM, Scheinman PL. A case series of dupilumab-treated allergic contact dermatitis patients. Dermatol Ther. 2018;31:e12701. doi:10.1111/dth.12701
- Kohli N, Nedorost S. Inflamed skin predisposes to sensitization to less potent allergens. J Am Acad Dermatol. 2016;75:312-317. doi:10.1016/j.jaad.2016.03.010
- Raffi J, Suresh R, Botto N, et al. The impact of dupilumab on patch testing and the prevalence of comorbid allergic contact dermatitis in recalcitrant atopic dermatitis: a retrospective chart review. J Am Acad Dermatol. 2020;82:132-138. doi:10.1016/j.jaad.2019.09.028
- Puza CJ, Atwater AR. Positive patch test reaction in a patient taking dupilumab. Dermatitis. 2018;29:89. doi:10.1097/DER.0000000000000346
- Suresh R, Murase JE. The role of expanded series patch testing in identifying causality of residual facial dermatitis following initiation of dupilumab therapy. JAAD Case Rep. 2018;4:899-904. doi:10.1016/j.jdcr.2018.08.027
- Stout M, Silverberg JI. Variable impact of dupilumab on patch testing results and allergic contact dermatitis in adults with atopic dermatitis. J Am Acad Dermatol. 2019;81:157-162. doi:10.1016/j.jaad.2019.03.020
- Raffi J, Botto N. Patch testing and allergen-specific inhibition in a patient taking dupilumab. JAMA Dermatol. 2019;155:120-121. doi:10.1001/jamadermatol.2018.4098
- Hoot JW, Douglas JD, Falo LD Jr. Patch testing in a patient on dupilumab. Dermatitis. 2018;29:164. doi:10.1097/DER.0000000000000357
- Crepy M-N, Nosbaum A, Bensefa-Colas L. Blocking type 2 inflammation by dupilumab does not control classic (type 1-driven) allergic contact dermatitis in chronic hand eczema. Contact Dermatitis. 2019;81:145-147. doi:10.1111/cod.13266
- Raffi J, Chen R, Botto N. Wide dye reactors. JAAD Case Rep. 2019;5:877-879. doi:10.1016/j.jdcr.2019.08.005
- Koblinski JE, Hamann D. Mixed occupational and iatrogenic allergic contact dermatitis in a hairdresser. Occup Med (Lond). 2020;70:523-526. doi:10.1093/occmed/kqaa152
- Raffi J, Suresh R, Fishman H, et al. Investigating the role of allergic contact dermatitis in residual ocular surface disease on dupilumab (ROSDD). Int J Womens Dermatol. 2019;5:308-313. doi:10.1016/j.ijwd.2019.10.001
- Zhu GA, Chen JK, Chiou A, et al. Repeat patch testing in a patient with allergic contact dermatitis improved on dupilumab. JAAD Case Rep. 2019;5:336-338. doi:10.1016/j.jdcr.2019.01.023
Practice Points
- Allergic contact dermatitis is an important diagnostic consideration in patients with refractory or persistent dermatitis.
- Patch testing is important to help determine a possible allergic contactant, but there is confusion about its accuracy in patients taking dupilumab.
- Patients with residual dermatitis while on dupilumab are likely to benefit from patch testing.
Leukemia Cutis Manifesting as Nonpalpable Purpura
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.

The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).

Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
 and numerous notched nuclei")
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.

Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
Practice Points
- Leukemia cutis complicates 5% to 15% of all cases of acute myeloid leukemia (AML) in adults.
- The appearance of leukemia cutis may be highly variable. Therefore, it should be included in the differential diagnosis for any cutaneous presentation in patients with an existing diagnosis or high likelihood of AML.
- Leukemic infiltrates are associated with sites of inflammation.

Treatment of Elephantiasic Pretibial Myxedema With Rituximab Therapy
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.

The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
. B, Colloidal iron staining highlighted the prominent mucin within the dermis")
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
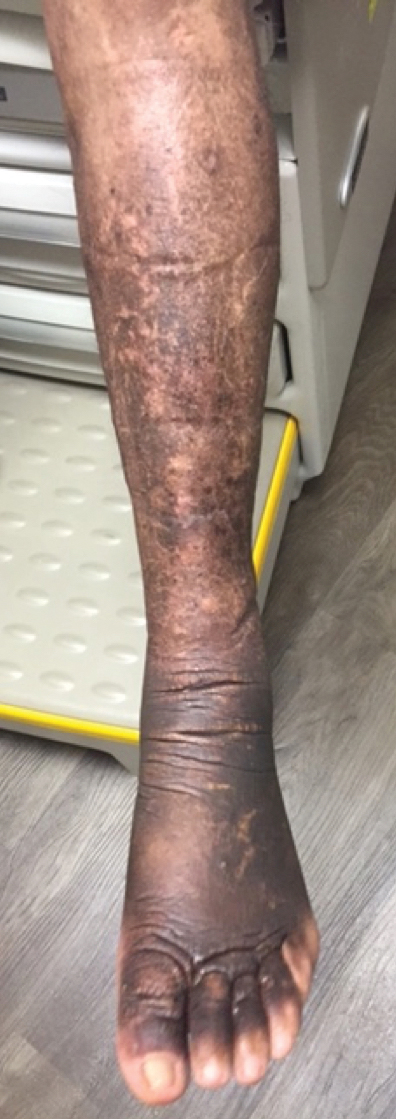
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
Practice Points
- Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet.
- Although many therapeutic modalities have been described for the management of the elephantiasis variant of PTM, few treatments have shown notable efficacy.
- Rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.
