User login
Reexamining the Role of Diet in Dermatology
Within the last decade, almost 3000 articles have been published on the role of diet in the prevention and management of dermatologic conditions. Patients are increasingly interested in—and employing—dietary modifications that may influence skin appearance and aid in the treatment of cutaneous disease.1 It is essential that dermatologists are familiar with existing evidence on the role of diet in dermatology to counsel patients appropriately. Herein, we discuss the compositions of several popular diets and their proposed utility for dermatologic purposes. We highlight the limited literature that exists surrounding this topic and emphasize the need for future, well-designed clinical trials that study the impact of diet on skin disease.
Ketogenic Diet
The ketogenic diet has a macronutrient profile composed of high fat, low to moderate protein, and very low carbohydrates. Nutritional ketosis occurs as the body begins to use free fatty acids (via beta oxidation) as the primary metabolite driving cellular metabolism. It has been suggested that the ketogenic diet may impart beneficial effects on skin disease; however, limited literature exists on the role of nutritional ketosis in the treatment of dermatologic conditions.
Mechanistically, the ketogenic diet decreases the secretion of insulin and insulinlike growth factor 1, resulting in a reduction of circulating androgens and increased activity of the retinoid X receptor.2 In acne vulgaris, it has been suggested that the ketogenic diet may be beneficial in decreasing androgen-induced sebum production and the overproliferation of keratinocytes.2-7 The ketogenic diet is one of the most rapidly effective dietary strategies for normalizing both insulin and androgens, thus it may theoretically be useful for other metabolic and hormone-dependent skin diseases, such as hidradenitis suppurativa.8,9
The cutaneous manifestations associated with chronic hyperinsulinemia and hyperglycemia are numerous and include acanthosis nigricans, acrochordons, diabetic dermopathy, scleredema diabeticorum, bullosis diabeticorum, keratosis pilaris, and generalized granuloma annulare. There also is an increased risk for bacterial and fungal skin infections associated with hyperglycemic states.10 The ketogenic diet is an effective nonpharmacologic tool for normalizing serum insulin and glucose levels in most patients and may have utility in the aforementioned conditions.11,12 In addition to improving insulin sensitivity, it has been used as a dietary strategy for weight loss.11-15 Because obesity and metabolic syndrome are highly correlated with common skin conditions such as psoriasis, hidradenitis suppurativa, and androgenetic alopecia, there may be a role for employing the ketogenic diet in these patient populations.16,17
Although robust clinical studies on ketogenic diets in skin disease are lacking, a recent single-arm, open-label clinical trial observed benefit in all 37 drug-naïve, overweight patients with chronic plaque psoriasis who underwent a ketogenic weight loss protocol. Significant reductions in psoriasis area and severity index (PASI) score and dermatology life quality index score were reported (P<.001).18 Another study of 30 patients with psoriasis found that a 4-week, low-calorie, ketogenic diet resulted in 50% improvement of PASI scores, 10% weight loss, and a reduction in the proinflammatory cytokines IL-1β and IL-2.19 Despite these results, it is a challenge to tease out if the specific dietary intervention or its associated weight loss was the main driver in these reported improvements in skin disease.
There is mixed evidence on the anti-inflammatory nature of the ketogenic diet, likely due to wide variation in the composition of foods included in individual diets. In many instances, the ketogenic diet is thought to possess considerable antioxidant and anti-inflammatory capabilities. Ketones are known activators of the nuclear factor erythroid 2–related factor 2 pathway, which upregulates the production of glutathione, a major endogenous intracellular antioxidant.20 Additionally, dietary compounds from foods that are encouraged while on the ketogenic diet, such as sulforaphane from broccoli, also are independent activators of nuclear factor erythroid 2–related factor 2.21 Ketones are efficiently utilized by mitochondria, which also may result in the decreased production of reactive oxygen species and lower oxidative stress.22 Moreover, the ketone body β-hydroxybutyrate has demonstrated the ability to reduce proinflammatory IL-1β levels via suppression of nucleotide-binding domain-like receptor protein 3 inflammasome activity.23,24 The activity of IL-1β is known to be elevated in many dermatologic conditions, including juvenile idiopathic arthritis, relapsing polychondritis, Schnitzler syndrome, hidradenitis suppurativa, Behçet disease, and other autoinflammatory syndromes.25 Ketones also have been shown to inhibit the nuclear factor–κB proinflammatory signaling pathway.22,26,27 Overexpression of IL-1β and aberrant activation of nuclear factor–κB are implicated in a variety of inflammatory, autoimmune, and oncologic cutaneous pathologies. The ketogenic diet may prove to be an effective adjunctive treatment for dermatologists to consider in select patient populations.23,24,28-30
For patients with keratinocyte carcinomas, the ketogenic diet may offer the aforementioned anti-inflammatory and antioxidant effects, as well as suppression of the mechanistic target of rapamycin, a major regulator of cell metabolism and proliferation.31,32 Inhibition of mechanistic target of rapamycin activity has been shown to slow tumor growth and reduce the development of squamous cell carcinoma.25,33,34 The ketogenic diet also may exploit the preferential utilization of glucose exhibited by many types of cancer cells, thereby “starving” the tumor of its primary fuel source.35,36 In vitro and animal studies in a variety of cancer types have demonstrated that a ketogenic metabolic state—achieved through the ketogenic diet or fasting—can sensitize tumor cells to chemotherapy and radiation while conferring a protective effect to normal cells.37-40 This recently described phenomenon is known as differential stress resistance, but it has not been studied in keratinocyte malignancies or melanoma to date. Importantly, some basal cell carcinomas and BRAF V600E–mutated melanomas have worsened while on the ketogenic diet, suggesting more data is needed before it can be recommended for all cancer patients.41,42 Furthermore, other skin conditions such as prurigo pigmentosa have been associated with initiation of the ketogenic diet.43
Low FODMAP Diet
Fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) are short-chain carbohydrates that are poorly absorbed, osmotically active, and rapidly fermented by intestinal bacteria.44 The low FODMAP diet has been shown to be efficacious for treatment of irritable bowel syndrome, small intestinal bacterial overgrowth (SIBO), and some cases of inflammatory bowel disease (IBD).44-49 A low FODMAP diet may have potential implications for several dermatologic conditions.
Rosacea has been associated with various gastrointestinal tract disorders including irritable bowel syndrome, SIBO, and IBD.50-54 A single study found that patients with rosacea had a 13-fold increased risk for SIBO.55,56 Treatment of 40 patients with SIBO using rifaximin resulted in complete resolution of rosacea in all patients, with no relapse after a 3-year follow-up period.55 Psoriasis also has been associated with SIBO and IBD.57,58 One small study found that eradication of SIBO in psoriatic patients resulted in improved PASI scores and colorimetric values.59
Although the long-term health consequences of the low FODMAP diet are unknown, further research on such dietary interventions for inflammatory skin conditions is warranted given the mounting evidence of a gut-skin connection and the role of the intestinal microbiome in skin health.50,51
Gluten-Free Diet
Gluten is a protein found in a variety of grains. Although the role of gluten in the pathogenesis of celiac disease and dermatitis herpetiformis is indisputable, the deleterious effects of gluten outside of the context of these diseases remain controversial. There may be a compelling case for eliminating gluten in psoriasis patients with seropositivity for celiac disease. A recent systematic review found a 2.2-fold increased risk for celiac disease in psoriasis patients.60 Antigliadin antibody titers also were found to be positively correlated with psoriatic disease severity.61 In addition, one open-label study found a reduction in PASI scores in 73% of patients with antigliadin antibodies after 3 months on a gluten-free diet compared to those without antibodies; however, the study only included 22 patients.62 Several other small studies have yielded similar results63,64; however, antigliadin antibodies are neither the most sensitive nor specific markers of celiac disease, and additional testing should be completed in any patient who may carry this diagnosis. A survey study by the National Psoriasis Foundation found that the dietary change associated with the greatest skin improvement was removal of gluten and nightshade vegetables in approximately 50% of the 1200 psoriasis patients that responded.65 Case reports of various dermatologic conditions including sarcoidosis, vitiligo, alopecia areata, lichen planus, dermatomyositis, pyoderma gangrenosum, erythema nodosum, leukocytoclastic vasculitis, linear IgA bullous dermatosis, and aphthous ulcerations have reportedly improved with a gluten-free diet; however, this should not be used as primary therapy in patients without celiac disease.66-71 Because gluten-free diets can be expensive and challenging to follow, a formal assessment for celiac disease should be considered before recommendation of this dietary intervention.
Low Histamine Diet
Histamine is a biogenic amine produced by the decarboxylation of the amino acid histidine.72 It is found in several foods in varying amounts. Because bacteria can convert histidine into histamine, many fermented and aged foods such as kimchi, sauerkraut, cheese, and red wine contain high levels of histamine. Individuals who have decreased activity of diamine oxidase (DAO), an enzyme that degrades histamine, may be more susceptible to histamine intolerance.72 The symptoms of histamine intolerance are numerous and include gastrointestinal tract distress, rhinorrhea and nasal congestion, headache, urticaria, flushing, and pruritus. Histamine intolerance can mimic an IgE-mediated food allergy; however, allergy testing is negative in these patients. Unfortunately, there is no laboratory test for histamine intolerance; a double-blind, placebo-controlled food challenge is considered the gold-standard test.72
As it pertains to dermatology, a low histamine diet may play a role in the treatment of certain patients with atopic dermatitis and chronic spontaneous urticaria. One study reported that 17 of 54 (31.5%) atopic patients had higher basal levels of serum histamine compared to controls.73 Another study found that a histamine-free diet led to improvement in both histamine intolerance symptoms and atopic dermatitis disease severity (SCORing atopic dermatitis) in patients with low DAO activity.74 In chronic spontaneous urticaria, a recent systematic review found that in 223 patients placed on a low histamine diet for 3 to 4 weeks, 12% and 44% achieved complete and partial remission, respectively.75 Although treatment response based on a patient’s DAO activity level has not been correlated, a diet low in histamine may prove useful for patients with persistent atopic dermatitis and chronic spontaneous urticaria who have negative food allergy tests and report exacerbation of symptoms after ingestion of histamine-rich foods.76,77
Mediterranean Diet
The Mediterranean diet has been touted as one of the healthiest diets to date, and large randomized clinical trials have demonstrated its effectiveness in weight loss, improving insulin sensitivity, and reducing inflammatory cytokine profiles.78,79 A major criticism of the Mediterranean diet is that it has considerable ambiguity and lacks a precise definition due to the variability of what is consumed in different Mediterranean regions. Generally, the diet emphasizes high consumption of colorful fruits and vegetables, aromatic herbs and spices, olive oil, nuts, and seafood, as well as modest amounts of dairy, eggs, and red meat.80 The anti-inflammatory effects of this diet largely have been attributed to its abundance of polyphenols, carotenoids, monounsaturated fatty acids, and omega-3 polyunsaturated fatty acids (PUFAs).80,81 Examples of polyphenols include resveratrol in red grapes, quercetin in apples and red onions, and curcumin in turmeric, while examples of carotenoids include lycopene in tomatoes and zeaxanthin in dark leafy greens. Oleic acid is a monounsaturated fatty acid present in high concentrations in olive oil, while eicosapentaenoic acid and docosahexaenoic acid are omega-3 PUFAs predominantly found in fish.82
Unfortunately, rigorous clinical trials regarding the Mediterranean diet as it pertains to dermatology have not been undertaken. Numerous observational studies in patients with psoriasis have suggested that close adherence to the Mediterranean diet was associated with improvement in PASI scores.83-86 The National Psoriasis Foundation now recommends a trial of the Mediterranean diet in some patients with psoriasis, emphasizing increased dietary intake of olive oil, fish, and vegetables.87 Adherence to a Mediterranean diet also has been inversely correlated to the severity of acne vulgaris and hidradenitis suppurativa88,89; however, these studies failed to account for the multifactorial risk factors associated with these conditions. Mediterranean diets also may impart a chemopreventive effect, supported by a number of in vivo and in vitro studies demonstrating the inhibition and/or reversal of cutaneous DNA damage induced by UV radiation through supplementation with various phytonutrients and omega-3 PUFAs.81,90-92 Although small case-control studies have found a decreased risk of basal cell carcinoma in those who closely adhered to a Mediterranean diet, more rigorous clinical research is needed.93
Whole-Food, Plant-Based Diet
A whole-food, plant-based (WFPB) diet is another popular dietary approach that consists of eating fruits, vegetables, legumes, nuts, seeds, and grains in their whole natural form.94 This diet discourages all animal products, including red meat, seafood, dairy, and eggs. It is similar to a vegan diet except that it eliminates all highly refined carbohydrates, vegetable oils, and other processed foods.94 Randomized clinical studies have demonstrated the WFPB diet to be effective in the treatment of obesity and metabolic syndrome.95,96
A WFPB diet has been shown to increase the antioxidant capacity of cells, lengthen telomeres, and reduce formation of advanced glycation end products.94,97,98 These benefits may help combat accelerated skin aging, including increased skin permeability, reduced elasticity and hydration, decreased angiogenesis, impaired immune function, and decreased vitamin D synthesis. Accelerated skin aging can result in delayed wound healing and susceptibility to skin tears and ecchymoses and also may promote the development of cutaneous malignancies.99 There remains a lack of clinical data studying a properly formulated WFPB diet in the dermatologic setting.
Paleolithic Diet
The paleolithic (Paleo) diet is an increasingly popular way of eating that attempts to mirror what our ancestors may have consumed between 10,000 and 2.5 million years ago.100 It is similar to the Mediterranean diet but excludes grains, dairy, legumes, and nightshade vegetables. It also calls for elimination of highly processed sugars and oils as well as chemical food additives and preservatives. There is a strict variation of the diet for individuals with autoimmune disease that also excludes eggs, nuts, and seeds, as these can be inflammatory or immunogenic in some patients.100-106 Other variations of the diet exist, including the ketogenic Paleo diet, pegan (Paleo vegan) diet, and lacto-Paleo diet.100 An often cited criticism of the Paleo diet is the low intake of calcium and risk for osteoporosis; however, consumption of calcium-rich foods or a calcium supplement can address this concern.107
Although small clinical studies have found the Paleo diet to be beneficial for various autoimmune diseases, clinical data evaluating the utility of the diet for cutaneous disease is lacking.108,109 Numerous randomized trials have demonstrated the Paleo diet to be effective for weight loss and improving insulin sensitivity and lipid levels.110-116 Thus, the Paleo diet may theoretically serve as a viable adjunct dietary approach to the treatment of cutaneous diseases associated with obesity and metabolic derangement.117
Carnivore Diet
Arguably the most controversial and radical diet is the carnivore diet. As the name implies, the carnivore diet is based on consuming solely animal products. A properly structured carnivore diet emphasizes a “nose-to-tail” eating approach where all parts of the animal including the muscle meats, organs, and fat are consumed. Proponents of the diet cite anthropologic evidence from fossil-stable carbon-13/carbon-12 isotope analyses, craniodental features, and numerous other adaptations that indicate increased consumption of meat during human evolution.118-122 Notably, many early humans ate a carnivore diet, but life span was very short at this time, suggesting the diet may not be as beneficial as has been suggested.
Despite the abundance of anecdotal evidence supporting its use for a variety of chronic conditions, including cutaneous autoimmune disease, there is a virtual absence of high-quality research on the carnivore diet.123-125
The purported benefits of the carnivore diet may be attributed to the consumption of organ meats that contain highly bioavailable essential vitamins and minerals, such as iron, zinc, copper, selenium, thiamine, niacin, folate, vitamin B6, vitamin B12, vitamin A, vitamin D, vitamin K, and choline.126-128 Other dietary compounds that have demonstrated benefit for skin health and are predominantly found in animal foods include carnosine, carnitine, creatine, taurine, coenzyme Q10, and collagen.129-134 Nevertheless, there is no data to recommend the elimination of antioxidant- and micronutrient-dense plant-based foods. Rigorous clinical research evaluating the efficacy and safety of the carnivore diet in dermatologic patients is needed. A carnivore diet should not be undertaken without the assistance of a dietician who can ensure adequate micronutrient and macronutrient support.
Final Thoughts
The adjunctive role of diet in the treatment of skin disease is expanding and becoming more widely accepted among dermatologists. Unfortunately, there remains a lack of randomized controlled trials confirming the efficacy of various dietary interventions in the dermatologic setting. Although evidence-based dietary recommendations currently are limited, it is important for dermatologists to be aware of the varied and nuanced dietary interventions employed by patients.
Ultimately, dietary recommendations must be personalized, considering a patient’s comorbidities, personal beliefs and preferences, and nutrigenetics. The emerging field of dermatonutrigenomics—the study of how dietary compounds interact with one’s genes to influence skin health—may allow for precise dietary recommendations to be made in dermatologic practice. Direct-to-consumer genetic tests targeted toward dermatology patients are already on the market, but their clinical utility awaits validation.1 Because nutritional science is a constantly evolving field, becoming familiar with these popular diets will serve both dermatologists and their patients well.
- Jaros J, Katta R, Shi VY. Dermatonutrigenomics: past, present, and future. Dermatology. 2019;235:164-166.
- Paoli A, Grimaldi K, Toniolo L, et al. Nutrition and acne: therapeutic potential of ketogenic diets. Skin Pharmacol Physiol. 2012;25:111-117.
- Melnik BC, Schmitz G. Role of insulin, insulin-like growth factor-1, hyperglycaemic food and milk consumption in the pathogenesis of acne vulgaris. Exp Dermatol. 2009;18:833-841.
- Smith RN, Mann NJ, Braue A, et al. The effect of a high-protein, low glycemic-load diet versus a conventional, high glycemic-load diet on biochemical parameters associated with acne vulgaris: a randomized, investigator-masked, controlled trial. J Am Acad Dermatol. 2007;57:247-256.
- Smith R, Mann N, Mäkeläinen H, et al. A pilot study to determine the short-term effects of a low glycemic load diet on hormonal markers of acne: a nonrandomized, parallel, controlled feeding trial. Mol Nutr Food Res. 2008;52:718-726.
- Smith RN, Braue A, Varigos GA, et al. The effect of a low glycemic load diet on acne vulgaris and the fatty acid composition of skin surface triglycerides. J Dermatol Sci. 2008;50:41-52.
- Kwon HH, Yoon JY, Hong JS, et al. Clinical and histological effect of a low glycaemic load diet in treatment of acne vulgaris in Korean patients: a randomized, controlled trial. Acta Derm Venereol. 2012;92:241-246.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a "male" therapy for a predominantly "female" disease. J Clin Aesthet Dermatol. 2016;9:44.
- Nikolakis G, Karagiannidis I, Vaiopoulos AG, et al. Endocrinological mechanisms in the pathophysiology of hidradenitis suppurativa [in German]. Hautarzt. 2020;71:762-771.
- Karadag AS, Ozlu E, Lavery MJ. Cutaneous manifestations of diabetes mellitus and the metabolic syndrome. Clin Dermatology. 2018;36:89-93.
- Gardner CD, Kiazand A, Alhassan S, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969-977.
- Anton SD, Hida A, Heekin K, et al. Effects of popular diets without specific calorie targets on weight loss outcomes: systematic review of findings from clinical trials. Nutrients. 2017;9:822.
- Castellana M, Conte E, Cignarelli A, et al. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: a systematic review and meta-analysis. Rev Endocr Metab Disord. 2020;21:5-16.
- Paoli A, Mancin L, Giacona MC, et al. Effects of a ketogenic diet in overweight women with polycystic ovary syndrome. J Transl Med. 2020;18:104.
- Dashti HM, Mathew TC, Hussein T, et al. Long-term effects of a ketogenic diet in obese patients. Exp Clin Cardiol. 2004;9:200-205.
- Lian N, Chen M. Metabolic syndrome and skin disease: potential connection and risk. Int J Dermatol Venereol. 2019;2:89-93.
- Hu Y, Zhu Y, Lian N, et al. Metabolic syndrome and skin diseases. Front Endocrinol (Lausanne). 2019;10:788.
- Castaldo G, Rastrelli L, Galdo G, et al. Aggressive weight-loss program with a ketogenic induction phase for the treatment of chronic plaque psoriasis: a proof-of-concept, single-arm, open-label clinical trial. Nutrition. 2020;74:110757.
- Castaldo G, Pagano I, Grimaldi M, et al. Effect of very-low-calorie ketogenic diet on psoriasis patients: a nuclear magnetic resonance-based metabolomic study. J Proteome Res. 2021;20:1509-1521.
- Milder J, Liang L-P, Patel M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis. 2010;40:238-244.
- Kubo E, Chhunchha B, Singh P, et al. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci Rep. 2017;7:14130.
- Pinto A, Bonucci A, Maggi E, et al. Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in Alzheimer's disease. Antioxidants (Basel). 2018;7:63.
- Youm Y-H, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263-269.
- Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328.
- Fomin DA, McDaniel B, Crane J. The promising potential role of ketones in inflammatory dermatologic disease: a new frontier in treatment research. J Dermatol Treat. 2017;28:484-487.
- Rahman M, Muhammad S, Khan MA, et al. The β-hydroxybutyrate receptor HCA 2 activates a neuroprotective subset of macrophages. Nat Commun. 2014;5:1-11.
- Lu Y, Yang YY, Zhou MW, et al. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-κB signaling pathways. Neurosci Lett. 2018;683:13-18.
- Hamarsheh S, Zeiser R. NLRP3 inflammasome activation in cancer: a double-edged sword. Front Immunol. 2020;11:1444.
- Bell S, Degitz K, Quirling M, et al. Involvement of NF-κB signalling in skin physiology and disease. Cell Signal. 2003;15:1-7.
- Goldminz AM, Au SC, Kim N, et al. NF-κB: an essential transcription factor in psoriasis. J Dermatol Sci. 2013;69:89-94.
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589.
- McDaniel S, Rensing N, Yamada K, et al. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:E7-E11.
- Alter M, Satzger I, Schrem H, et al. Non-melanoma skin cancer is reduced after switch of immunosuppression to mTOR-inhibitors in organ transplant recipients. J Dtsch Dermatol Ges. 2014;12:480-488.
- Feldmeyer L, Hofbauer GF, Böni T, et al. Mammalian target of rapamycin (mTOR) inhibitors slow skin carcinogenesis, but impair wound healing. Br J Dermatol. 2012;166:422-424.
- Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211-218.
- Li W. "Warburg effect" and mitochondrial metabolism in skin cancer.J Carcinogene Mutagene. 2012:S4.
- Naveed S, Aslam M, Ahmad A. Starvation based differential chemotherapy: a novel approach for cancer treatment. Oman Med J. 2014;29:391-398.
- Raffaghello L, Lee C, Safdie FM, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. 2008;105:8215-8220.
- Buono R, Longo VD. Starvation, stress resistance, and cancer. Trends Endocrinol Metab. 2018;29:271-280.
- de Groot S, Pijl H, van der Hoeven JJM, et al. Effects of short-term fasting on cancer treatment. J Exp Clin Cancer Res. 2019;38:209.
- Hosseini M, Kasraian Z, Rezvani HR. Energy metabolism in skin cancers: a therapeutic perspective. Biochim Biophys Acta Bioenerg. 2017;1858:712-722.
- Feichtinger RG, Lang R, Geilberger R, et al. Melanoma tumors exhibit a variable but distinct metabolic signature. Exp Dermatol. 2018;27:204-207.
- Alshaya MA, Turkmani MG, Alissa AM. Prurigo pigmentosa following ketogenic diet and bariatric surgery: a growing association. JAAD Case Rep. 2019;5:504-507.
- Bellini M, Tonarelli S, Nagy AG, et al. Low FODMAP diet: evidence, doubts, and hopes. Nutrients. 2020;12:148.
- Kwiatkowski L, Rice E, Langland J. Integrative treatment of chronic abdominal bloating and pain associated with overgrowth of small intestinal bacteria: a case report. Altern Ther Health Med. 2017;23:56-61.
- Hubkova T. No more pain in the gut: lifestyle medicine approach to irritable bowel syndrome. Am J Lifestyle Med. 2017;11:223-226.
- Schumann D, Klose P, Lauche R, et al. Low fermentable, oligo-, di-, mono-saccharides and polyol diet in the treatment of irritable bowel syndrome: a systematic review and meta-analysis. Nutrition. 2018;45:24-31.
- Cox SR, Prince AC, Myers CE, et al. Fermentable carbohydrates [FODMAPs] exacerbate functional gastrointestinal symptoms in patients with inflammatory bowel disease: a randomised, double-blind, placebo-controlled, cross-over, re-challenge trial. J Crohns Colitis. 2017;11:1420-1429.
- Damas OM, Garces L, Abreu MT. Diet as adjunctive treatment for inflammatory bowel disease: review and update of the latest literature. Curr Treat Options Gastroenterol. 2019;17:313-325.
- Wang FY, Chi CC. Rosacea, germs, and bowels: a review on gastrointestinal comorbidities and gut-skin axis of rosacea. Adv Ther. 2021;38:1415-1424.
- Daou H, Paradiso M, Hennessy K, et al. Rosacea and the microbiome: a systematic review. Dermatol Ther (Heidelb). 2021;11:1-12.
- Weinstock LB, Steinhoff M. Rosacea and small intestinal bacterial overgrowth: prevalence and response to rifaximin. J Am Acad Dermatol. 2013;68:875-876.
- Wu CY, Chang YT, Juan CK, et al. Risk of inflammatory bowel disease in patients with rosacea: results from a nationwide cohort study in Taiwan. J Am Acad Dermatol. 2017;76:911-917.
- Egeberg A, Weinstock LB, Thyssen EP, et al. Rosacea and gastrointestinal disorders: a population-based cohort study. Br J Dermatol. 2017;176:100-106.
- Drago F, De Col E, Agnoletti AF, et al. The role of small intestinal bacterial overgrowth in rosacea: a 3-year follow-up. J Am Acad Dermatol. 2016;75:E113-E115.
- Parodi A, Paolino S, Greco A, et al. Small intestinal bacterial overgrowth in rosacea: clinical effectiveness of its eradication. Clin Gastroenterol Hepatol. 2008;6:759-764.
- Ojetti V, De Simone C, Aguilar Sanchez J, et al. Malabsorption in psoriatic patients: cause or consequence? Scand J Gastroenterol. 2006;41:1267-1271.
- Kim M, Choi KH, Hwang SW, et al. Inflammatory bowel disease is associated with an increased risk of inflammatory skin diseases: a population-based cross-sectional study. J Am Acad Dermatol. 2017;76:40-48.
- Drago F, Ciccarese G, Indemini E, et al. Psoriasis and small intestine bacterial overgrowth. Int J Dermatol. 2018;57:112-113.
- Acharya P, Mathur M. Association between psoriasis and celiac disease: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:1376-1385.
- Bhatia BK, Millsop JW, Debbaneh M, et al. Diet and psoriasis, part II: celiac disease and role of a gluten-free diet. J Am Acad Dermatol. 2014;71:350-358.
- Michaëlsson G, Gerdén B, Hagforsen E, et al. Psoriasis patients with antibodies to gliadin can be improved by a gluten-free diet. Br J Dermatol. 2000;142:44-51.
- Kolchak NA, Tetarnikova MK, Theodoropoulou MS, et al. Prevalence of antigliadin IgA antibodies in psoriasis vulgaris and response of seropositive patients to a gluten-free diet. J Multidiscip Healthc. 2018;11:13-19.
- De Bastiani R, Gabrielli M, Lora L, et al. Association between coeliac disease and psoriasis: Italian primary care multicentre study. Dermatology. 2015;230:156-160.
- Afifi L, Danesh MJ, Lee KM, et al. Dietary behaviors in psoriasis: patient-reported outcomes from a U.S. national survey. Dermatol Ther (Heidelb). 2017;7:227-242.
- Loche F, Bazex J. Celiac disease associated with cutaneous sarcoidosic granuloma [in French]. Rev Med Interne. 1997;18:975-978.
- Rodríguez-García C, González-Hernández S, Pérez-Robayna N, et al. Repigmentation of vitiligo lesions in a child with celiac disease after a gluten-free diet. Pediatr Dermatol. 2011;28:209-210.
- Wijarnpreecha K, Panjawatanan P, Corral JE, et al. Celiac disease and risk of sarcoidosis: a systematic review and meta-analysis. J Evid Based Med. 2019;12:194-199.
- Rodrigo L, Beteta-Gorriti V, Alvarez N, et al. Cutaneous and mucosal manifestations associated with celiac disease. Nutrients. 2018;10:800.
- Song MS, Farber D, Bitton A, et al. Dermatomyositis associated with celiac disease: response to a gluten-free diet. Can J Gastroenterol. 2006;20:433-435.
- Egan CA, Smith EP, Taylor TB, et al. Linear IgA bullous dermatosis responsive to a gluten-free diet. Am J Gastroenterol. 2001;96:1927-1929.
- Comas-Basté O, Sánchez-Pérez S, Veciana-Nogués MT, et al. Histamine intolerance: the current state of the art. Biomolecules. 2020;10:1181.
- Ring J. Plasma histamine concentrations in atopic eczema. Clin Allergy. 1983;13:545-552.
- Maintz L, Benfadal S, Allam JP, et al. Evidence for a reduced histamine degradation capacity in a subgroup of patients with atopic eczema. J Allergy Clin Immunol. 2006;117:1106-1112.
- Cornillier H, Giraudeau B, Samimi M, et al. Effect of diet in chronic spontaneous urticaria: a systematic review. Acta Derm Venereol. 2019;99:127-132.
- Son JH, Chung BY, Kim HO, et al. A histamine-free diet is helpful for treatment of adult patients with chronic spontaneous urticaria. Ann Dermatol. 2018;30:164-172.
- Wagner N, Dirk D, Peveling-Oberhag A, et al. A popular myth - low-histamine diet improves chronic spontaneous urticaria - fact or fiction? J Eur Acad Dermatol Venereol. 2017;31:650-655.
- Esposito K, Marfella R, Ciotola M, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA. 2004;292:1440-1446.
- Steffen LM, Van Horn L, Daviglus ML, et al. A modified Mediterranean diet score is associated with a lower risk of incident metabolic syndrome over 25 years among young adults: the CARDIA (coronary artery risk development in young adults) study. Br J Nutr. 2014;112:1654-1661.
- Bower A, Marquez S, de Mejia EG. The health benefits of selected culinary herbs and spices found in the traditional Mediterranean diet. Crit Rev Food Sci Nutr. 2016;56:2728-2746.
- Bosch R, Philips N, Suárez-Pérez JA, et al. Mechanisms of photoaging and cutaneous photocarcinogenesis, and photoprotective strategies with phytochemicals. Antioxidants (Basel). 2015;4:248-268.
- Katsimbri P, Korakas E, Kountouri A, et al. The effect of antioxidant and anti-inflammatory capacity of diet on psoriasis and psoriatic arthritis phenotype: nutrition as therapeutic tool? Antioxidants. 2021;10:157.
- Molina-Leyva A, Cuenca-Barrales C, Vega-Castillo JJ, et al. Adherence to Mediterranean diet in Spanish patients with psoriasis: cardiovascular benefits? Dermatol Ther. 2019;32:E12810.
- Barrea L, Balato N, Di Somma C, et al. Nutrition and psoriasis: is there any association between the severity of the disease and adherence to the Mediterranean diet? J Transl Med. 2015;13:1-10.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort. JAMA Dermatol. 2018;154:1017-1024.
- Korovesi A, Dalamaga M, Kotopouli M, et al. Adherence to the Mediterranean diet is independently associated with psoriasis risk, severity, and quality of life: a cross-sectional observational study. Int J Dermatol. 2019;58:E164-E165.
- Ford AR, Siegel M, Bagel J, et al. Dietary recommendations for adults with psoriasis or psoriatic arthritis from the medical board of the National Psoriasis Foundation: a systematic review. JAMA Dermatol. 2018;154:934-950.
- Skroza N, Tolino E, Semyonov L, et al. Mediterranean diet and familial dysmetabolism as factors influencing the development of acne. Scand J Public Health. 2012;40:466-474.
- Barrea L, Fabbrocini G, Annunziata G, et al. Role of nutrition and adherence to the Mediterranean diet in the multidisciplinary approach of hidradenitis suppurativa: evaluation of nutritional status and its association with severity of disease. Nutrients. 2018;11:57.
- Nichols JA, Katiyar SK. Skin photoprotection by natural polyphenols: anti-inflammatory, antioxidant and DNA repair mechanisms. Arch Dermatol Res. 2010;302:71-83.
- Huang T-H, Wang P-W, Yang S-C, et al. Cosmetic and therapeutic applications of fish oil's fatty acids on the skin. Mar Drugs. 2018;16:256.
- Rizwan M, Rodriguez-Blanco I, Harbottle A, et al. Tomato paste rich in lycopene protects against cutaneous photodamage in humans in vivo: a randomized controlled trial. Br J Dermatol. 2011;164:154-162.
- Leone A, Martínez-González M, Martin-Gorgojo A, et al. Mediterranean diet, dietary approaches to stop hypertension, and pro-vegetarian dietary pattern in relation to the risk of basal cell carcinoma: a nested case-control study within the Seguimiento Universidad de Navarra (SUN) cohort. Am J Clin Nutr. 2020;112:364-372.
- Solway J, McBride M, Haq F, et al. Diet and dermatology: the role of a whole-food, plant-based diet in preventing and reversing skin aging--a review. J Clin Aesthet Dermatol. 2020;13:38-43.
- Greger M. A whole food plant-based diet is effective for weight loss: the evidence. Am J Lifestyle Med. 2020;14:500-510.
- Wright N, Wilson L, Smith M, et al. The BROAD study: a randomised controlled trial using a whole food plant-based diet in the community for obesity, ischaemic heart disease or diabetes. Nutr Diabetes. 2017;7:E256.
- Ornish D, Lin J, Chan JM, et al. Effect of comprehensive lifestyle changes on telomerase activity and telomere length in men with biopsy-proven low-risk prostate cancer: 5-year follow-up of a descriptive pilot study. Lancet Oncol. 2013;14:1112-1120.
- Ornish D, Lin J, Daubenmier J, et al. Increased telomerase activity and comprehensive lifestyle changes: a pilot study. Lancet Oncol. 2008;9:1048-1057.
- Zouboulis CC, Makrantonaki E. Clinical aspects and molecular diagnostics of skin aging. Clin Dermatol. 2011;29:3-14.
- Gupta L, Khandelwal D, Lal PR, et al. Palaeolithic diet in diabesity and endocrinopathies--a vegan's perspective. Eur Endocrinol. 2019;15:77-82.
- Chassaing B, Van de Wiele T, De Bodt J, et al. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut. 2017;66:1414-1427.
- Thorburn Alison N, Macia L, Mackay Charles R. Diet, metabolites, and "Western lifestyle" inflammatory diseases. Immunity. 2014;40:833-842.
- Katta R, Schlichte M. Diet and dermatitis: food triggers. J Clin Aesthet Dermatol. 2014;7:30-36.
- Dhar S, Srinivas SM. Food allergy in atopic dermatitis. Indian J Dermatol. 2016;61:645-648.
- Birmingham N, Thanesvorakul S, Gangur V. Relative immunogenicity of commonly allergenic foods versus rarely allergenic and nonallergenic foods in mice. J Food Prot. 2002;65:1988-1991.
- Yu W, Freeland DMH, Nadeau KC. Food allergy: immune mechanisms, diagnosis and immunotherapy. Nat Rev Immunol. 2016;16:751-765.
- Kowalski LM, Bujko J. Evaluation of biological and clinical potential of paleolithic diet [in Polish]. Rocz Panstw Zakl Hig. 2012;63:9-15.
- Lee JE, Titcomb TJ, Bisht B, et al. A modified MCT-based ketogenic diet increases plasma β-hydroxybutyrate but has less effect on fatigue and quality of life in people with multiple sclerosis compared to a modified paleolithic diet: a waitlist-controlled, randomized pilot study. J Am Coll Nutr. 2021;40:13-25.
- Abbott RD, Sadowski A, Alt AG. Efficacy of the autoimmune protocol diet as part of a multi-disciplinary, supported lifestyle intervention for Hashimoto's thyroiditis. Cureus. 2019;11:E4556.
- Lindeberg S, Jönsson T, Granfeldt Y, et al. A palaeolithic diet improves glucose tolerance more than a Mediterranean-like diet in individuals with ischaemic heart disease. Diabetologia. 2007;50:1795-1807.
- Jönsson T, Granfeldt Y, Ahrén B, et al. Beneficial effects of a paleolithic diet on cardiovascular risk factors in type 2 diabetes: a randomized cross-over pilot study. Cardiovasc Diabetol. 2009;8:35.
- Boers I, Muskiet FAJ, Berkelaar E, et al. Favourable effects of consuming a palaeolithic-type diet on characteristics of the metabolic syndrome: a randomized controlled pilot-study. Lipids Health Dis. 2014;13:160.
- Ghaedi E, Mohammadi M, Mohammadi H, et al. Effects of a paleolithic diet on cardiovascular disease risk factors: a systematic review and meta-analysis of randomized controlled trials. Adv Nutr. 2019;10:634-646.
- Mellberg C, Sandberg S, Ryberg M, et al. Long-term effects of a palaeolithic-type diet in obese postmenopausal women: a 2-year randomized trial. Eur J Clin Nutr. 2014;68:350-357.
- Pastore RL, Brooks JT, Carbone JW. Paleolithic nutrition improves plasma lipid concentrations of hypercholesterolemic adults to a greater extent than traditional heart-healthy dietary recommendations. Nutr Res. 2015;35:474-479.
- Otten J, Stomby A, Waling M, et al. Benefits of a paleolithic diet with and without supervised exercise on fat mass, insulin sensitivity, and glycemic control: a randomized controlled trial in individuals with type 2 diabetes. Diabetes Metab Res Rev. 2017;33:E2828.
- Stefanadi EC, Dimitrakakis G, Antoniou C-K, et al. Metabolic syndrome and the skin: a more than superficial association. reviewing the association between skin diseases and metabolic syndrome and a clinical decision algorithm for high risk patients. Diabetol Metab Syndr. 2018;10:9.
- Mann N. Meat in the human diet: an anthropological perspective. Nutr Dietetics. 2007;64(suppl 4):S102-S107.
- Bramble DM, Lieberman DE. Endurance running and the evolution of Homo. Nature. 2004;432:345-352.
- Kuhn JE. Throwing, the shoulder, and human evolution. Am J Orthop (Belle Mead NJ). 2016;45:110-114.
- Kobayashi H, Kohshima S. Unique morphology of the human eye and its adaptive meaning: comparative studies on external morphology of the primate eye. J Hum Evol. 2001;40:419-435.
- Cordain L, Eaton SB, Miller JB, et al. The paradoxical nature of hunter-gatherer diets: meat-based, yet non-atherogenic. Eur J Clin Nutr. 2002;56(suppl 1):S42-S52.
- McClellan WS, Du Bois EF. Clinical calorimetry: XLV. prolonged meat diets with a study of kidney function and ketosis. J Biol Chem. 1930;87:651-668.
- O'Hearn A. Can a carnivore diet provide all essential nutrients? Curr Opin Endocrinol Diabetes Obes. 2020;27:312-316.
- O'Hearn LA. A survey of improvements experienced on a carnivore diet compared to only carbohydrate restriction. Open Science Forum website. Published February 12, 2019. Accessed May 17, 2021. doi:10.17605/OSF.IO/5FU4D
- Williams P. Nutritional composition of red meat. Nutrition & Dietetics. 2007;64(suppl 4):S113-S119.
- Biel W, Czerniawska-Piątkowska E, Kowalczyk A. Offal chemical composition from veal, beef, and lamb maintained in organic production systems. Animals (Basel). 2019;9:489.
- Elmadfa I, Meyer AL. The role of the status of selected micronutrients in shaping the immune function. Endocr Metab Immune Disord Drug Targets. 2019;19:1100-1115.
- Babizhayev M. Treatment of skin aging and photoaging with innovative oral dosage forms of nonhydrolized carnosine and carcinine. Int J Clin Derm Res. 2017;5:116-143.
- Danby FW. Nutrition and aging skin: sugar and glycation. Clin Dermatol. 2010;28:409-411.
- Siefken W, Carstensen S, Springmann G, et al. Role of taurine accumulation in keratinocyte hydration. J Invest Dermatol. 2003;121:354-361.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19:3059.
- Fischer F, Achterberg V, März A, et al. Folic acid and creatineimprove the firmness of human skin in vivo. J Cosmet Dermatol. 2011;10:15-23.
- Blatt T, Lenz H, Weber T. Topical application of creatine is multibeneficial for human skin. J Am Acad Dermatol. 2005;52:P32.
Within the last decade, almost 3000 articles have been published on the role of diet in the prevention and management of dermatologic conditions. Patients are increasingly interested in—and employing—dietary modifications that may influence skin appearance and aid in the treatment of cutaneous disease.1 It is essential that dermatologists are familiar with existing evidence on the role of diet in dermatology to counsel patients appropriately. Herein, we discuss the compositions of several popular diets and their proposed utility for dermatologic purposes. We highlight the limited literature that exists surrounding this topic and emphasize the need for future, well-designed clinical trials that study the impact of diet on skin disease.
Ketogenic Diet
The ketogenic diet has a macronutrient profile composed of high fat, low to moderate protein, and very low carbohydrates. Nutritional ketosis occurs as the body begins to use free fatty acids (via beta oxidation) as the primary metabolite driving cellular metabolism. It has been suggested that the ketogenic diet may impart beneficial effects on skin disease; however, limited literature exists on the role of nutritional ketosis in the treatment of dermatologic conditions.
Mechanistically, the ketogenic diet decreases the secretion of insulin and insulinlike growth factor 1, resulting in a reduction of circulating androgens and increased activity of the retinoid X receptor.2 In acne vulgaris, it has been suggested that the ketogenic diet may be beneficial in decreasing androgen-induced sebum production and the overproliferation of keratinocytes.2-7 The ketogenic diet is one of the most rapidly effective dietary strategies for normalizing both insulin and androgens, thus it may theoretically be useful for other metabolic and hormone-dependent skin diseases, such as hidradenitis suppurativa.8,9
The cutaneous manifestations associated with chronic hyperinsulinemia and hyperglycemia are numerous and include acanthosis nigricans, acrochordons, diabetic dermopathy, scleredema diabeticorum, bullosis diabeticorum, keratosis pilaris, and generalized granuloma annulare. There also is an increased risk for bacterial and fungal skin infections associated with hyperglycemic states.10 The ketogenic diet is an effective nonpharmacologic tool for normalizing serum insulin and glucose levels in most patients and may have utility in the aforementioned conditions.11,12 In addition to improving insulin sensitivity, it has been used as a dietary strategy for weight loss.11-15 Because obesity and metabolic syndrome are highly correlated with common skin conditions such as psoriasis, hidradenitis suppurativa, and androgenetic alopecia, there may be a role for employing the ketogenic diet in these patient populations.16,17
Although robust clinical studies on ketogenic diets in skin disease are lacking, a recent single-arm, open-label clinical trial observed benefit in all 37 drug-naïve, overweight patients with chronic plaque psoriasis who underwent a ketogenic weight loss protocol. Significant reductions in psoriasis area and severity index (PASI) score and dermatology life quality index score were reported (P<.001).18 Another study of 30 patients with psoriasis found that a 4-week, low-calorie, ketogenic diet resulted in 50% improvement of PASI scores, 10% weight loss, and a reduction in the proinflammatory cytokines IL-1β and IL-2.19 Despite these results, it is a challenge to tease out if the specific dietary intervention or its associated weight loss was the main driver in these reported improvements in skin disease.
There is mixed evidence on the anti-inflammatory nature of the ketogenic diet, likely due to wide variation in the composition of foods included in individual diets. In many instances, the ketogenic diet is thought to possess considerable antioxidant and anti-inflammatory capabilities. Ketones are known activators of the nuclear factor erythroid 2–related factor 2 pathway, which upregulates the production of glutathione, a major endogenous intracellular antioxidant.20 Additionally, dietary compounds from foods that are encouraged while on the ketogenic diet, such as sulforaphane from broccoli, also are independent activators of nuclear factor erythroid 2–related factor 2.21 Ketones are efficiently utilized by mitochondria, which also may result in the decreased production of reactive oxygen species and lower oxidative stress.22 Moreover, the ketone body β-hydroxybutyrate has demonstrated the ability to reduce proinflammatory IL-1β levels via suppression of nucleotide-binding domain-like receptor protein 3 inflammasome activity.23,24 The activity of IL-1β is known to be elevated in many dermatologic conditions, including juvenile idiopathic arthritis, relapsing polychondritis, Schnitzler syndrome, hidradenitis suppurativa, Behçet disease, and other autoinflammatory syndromes.25 Ketones also have been shown to inhibit the nuclear factor–κB proinflammatory signaling pathway.22,26,27 Overexpression of IL-1β and aberrant activation of nuclear factor–κB are implicated in a variety of inflammatory, autoimmune, and oncologic cutaneous pathologies. The ketogenic diet may prove to be an effective adjunctive treatment for dermatologists to consider in select patient populations.23,24,28-30
For patients with keratinocyte carcinomas, the ketogenic diet may offer the aforementioned anti-inflammatory and antioxidant effects, as well as suppression of the mechanistic target of rapamycin, a major regulator of cell metabolism and proliferation.31,32 Inhibition of mechanistic target of rapamycin activity has been shown to slow tumor growth and reduce the development of squamous cell carcinoma.25,33,34 The ketogenic diet also may exploit the preferential utilization of glucose exhibited by many types of cancer cells, thereby “starving” the tumor of its primary fuel source.35,36 In vitro and animal studies in a variety of cancer types have demonstrated that a ketogenic metabolic state—achieved through the ketogenic diet or fasting—can sensitize tumor cells to chemotherapy and radiation while conferring a protective effect to normal cells.37-40 This recently described phenomenon is known as differential stress resistance, but it has not been studied in keratinocyte malignancies or melanoma to date. Importantly, some basal cell carcinomas and BRAF V600E–mutated melanomas have worsened while on the ketogenic diet, suggesting more data is needed before it can be recommended for all cancer patients.41,42 Furthermore, other skin conditions such as prurigo pigmentosa have been associated with initiation of the ketogenic diet.43
Low FODMAP Diet
Fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) are short-chain carbohydrates that are poorly absorbed, osmotically active, and rapidly fermented by intestinal bacteria.44 The low FODMAP diet has been shown to be efficacious for treatment of irritable bowel syndrome, small intestinal bacterial overgrowth (SIBO), and some cases of inflammatory bowel disease (IBD).44-49 A low FODMAP diet may have potential implications for several dermatologic conditions.
Rosacea has been associated with various gastrointestinal tract disorders including irritable bowel syndrome, SIBO, and IBD.50-54 A single study found that patients with rosacea had a 13-fold increased risk for SIBO.55,56 Treatment of 40 patients with SIBO using rifaximin resulted in complete resolution of rosacea in all patients, with no relapse after a 3-year follow-up period.55 Psoriasis also has been associated with SIBO and IBD.57,58 One small study found that eradication of SIBO in psoriatic patients resulted in improved PASI scores and colorimetric values.59
Although the long-term health consequences of the low FODMAP diet are unknown, further research on such dietary interventions for inflammatory skin conditions is warranted given the mounting evidence of a gut-skin connection and the role of the intestinal microbiome in skin health.50,51
Gluten-Free Diet
Gluten is a protein found in a variety of grains. Although the role of gluten in the pathogenesis of celiac disease and dermatitis herpetiformis is indisputable, the deleterious effects of gluten outside of the context of these diseases remain controversial. There may be a compelling case for eliminating gluten in psoriasis patients with seropositivity for celiac disease. A recent systematic review found a 2.2-fold increased risk for celiac disease in psoriasis patients.60 Antigliadin antibody titers also were found to be positively correlated with psoriatic disease severity.61 In addition, one open-label study found a reduction in PASI scores in 73% of patients with antigliadin antibodies after 3 months on a gluten-free diet compared to those without antibodies; however, the study only included 22 patients.62 Several other small studies have yielded similar results63,64; however, antigliadin antibodies are neither the most sensitive nor specific markers of celiac disease, and additional testing should be completed in any patient who may carry this diagnosis. A survey study by the National Psoriasis Foundation found that the dietary change associated with the greatest skin improvement was removal of gluten and nightshade vegetables in approximately 50% of the 1200 psoriasis patients that responded.65 Case reports of various dermatologic conditions including sarcoidosis, vitiligo, alopecia areata, lichen planus, dermatomyositis, pyoderma gangrenosum, erythema nodosum, leukocytoclastic vasculitis, linear IgA bullous dermatosis, and aphthous ulcerations have reportedly improved with a gluten-free diet; however, this should not be used as primary therapy in patients without celiac disease.66-71 Because gluten-free diets can be expensive and challenging to follow, a formal assessment for celiac disease should be considered before recommendation of this dietary intervention.
Low Histamine Diet
Histamine is a biogenic amine produced by the decarboxylation of the amino acid histidine.72 It is found in several foods in varying amounts. Because bacteria can convert histidine into histamine, many fermented and aged foods such as kimchi, sauerkraut, cheese, and red wine contain high levels of histamine. Individuals who have decreased activity of diamine oxidase (DAO), an enzyme that degrades histamine, may be more susceptible to histamine intolerance.72 The symptoms of histamine intolerance are numerous and include gastrointestinal tract distress, rhinorrhea and nasal congestion, headache, urticaria, flushing, and pruritus. Histamine intolerance can mimic an IgE-mediated food allergy; however, allergy testing is negative in these patients. Unfortunately, there is no laboratory test for histamine intolerance; a double-blind, placebo-controlled food challenge is considered the gold-standard test.72
As it pertains to dermatology, a low histamine diet may play a role in the treatment of certain patients with atopic dermatitis and chronic spontaneous urticaria. One study reported that 17 of 54 (31.5%) atopic patients had higher basal levels of serum histamine compared to controls.73 Another study found that a histamine-free diet led to improvement in both histamine intolerance symptoms and atopic dermatitis disease severity (SCORing atopic dermatitis) in patients with low DAO activity.74 In chronic spontaneous urticaria, a recent systematic review found that in 223 patients placed on a low histamine diet for 3 to 4 weeks, 12% and 44% achieved complete and partial remission, respectively.75 Although treatment response based on a patient’s DAO activity level has not been correlated, a diet low in histamine may prove useful for patients with persistent atopic dermatitis and chronic spontaneous urticaria who have negative food allergy tests and report exacerbation of symptoms after ingestion of histamine-rich foods.76,77
Mediterranean Diet
The Mediterranean diet has been touted as one of the healthiest diets to date, and large randomized clinical trials have demonstrated its effectiveness in weight loss, improving insulin sensitivity, and reducing inflammatory cytokine profiles.78,79 A major criticism of the Mediterranean diet is that it has considerable ambiguity and lacks a precise definition due to the variability of what is consumed in different Mediterranean regions. Generally, the diet emphasizes high consumption of colorful fruits and vegetables, aromatic herbs and spices, olive oil, nuts, and seafood, as well as modest amounts of dairy, eggs, and red meat.80 The anti-inflammatory effects of this diet largely have been attributed to its abundance of polyphenols, carotenoids, monounsaturated fatty acids, and omega-3 polyunsaturated fatty acids (PUFAs).80,81 Examples of polyphenols include resveratrol in red grapes, quercetin in apples and red onions, and curcumin in turmeric, while examples of carotenoids include lycopene in tomatoes and zeaxanthin in dark leafy greens. Oleic acid is a monounsaturated fatty acid present in high concentrations in olive oil, while eicosapentaenoic acid and docosahexaenoic acid are omega-3 PUFAs predominantly found in fish.82
Unfortunately, rigorous clinical trials regarding the Mediterranean diet as it pertains to dermatology have not been undertaken. Numerous observational studies in patients with psoriasis have suggested that close adherence to the Mediterranean diet was associated with improvement in PASI scores.83-86 The National Psoriasis Foundation now recommends a trial of the Mediterranean diet in some patients with psoriasis, emphasizing increased dietary intake of olive oil, fish, and vegetables.87 Adherence to a Mediterranean diet also has been inversely correlated to the severity of acne vulgaris and hidradenitis suppurativa88,89; however, these studies failed to account for the multifactorial risk factors associated with these conditions. Mediterranean diets also may impart a chemopreventive effect, supported by a number of in vivo and in vitro studies demonstrating the inhibition and/or reversal of cutaneous DNA damage induced by UV radiation through supplementation with various phytonutrients and omega-3 PUFAs.81,90-92 Although small case-control studies have found a decreased risk of basal cell carcinoma in those who closely adhered to a Mediterranean diet, more rigorous clinical research is needed.93
Whole-Food, Plant-Based Diet
A whole-food, plant-based (WFPB) diet is another popular dietary approach that consists of eating fruits, vegetables, legumes, nuts, seeds, and grains in their whole natural form.94 This diet discourages all animal products, including red meat, seafood, dairy, and eggs. It is similar to a vegan diet except that it eliminates all highly refined carbohydrates, vegetable oils, and other processed foods.94 Randomized clinical studies have demonstrated the WFPB diet to be effective in the treatment of obesity and metabolic syndrome.95,96
A WFPB diet has been shown to increase the antioxidant capacity of cells, lengthen telomeres, and reduce formation of advanced glycation end products.94,97,98 These benefits may help combat accelerated skin aging, including increased skin permeability, reduced elasticity and hydration, decreased angiogenesis, impaired immune function, and decreased vitamin D synthesis. Accelerated skin aging can result in delayed wound healing and susceptibility to skin tears and ecchymoses and also may promote the development of cutaneous malignancies.99 There remains a lack of clinical data studying a properly formulated WFPB diet in the dermatologic setting.
Paleolithic Diet
The paleolithic (Paleo) diet is an increasingly popular way of eating that attempts to mirror what our ancestors may have consumed between 10,000 and 2.5 million years ago.100 It is similar to the Mediterranean diet but excludes grains, dairy, legumes, and nightshade vegetables. It also calls for elimination of highly processed sugars and oils as well as chemical food additives and preservatives. There is a strict variation of the diet for individuals with autoimmune disease that also excludes eggs, nuts, and seeds, as these can be inflammatory or immunogenic in some patients.100-106 Other variations of the diet exist, including the ketogenic Paleo diet, pegan (Paleo vegan) diet, and lacto-Paleo diet.100 An often cited criticism of the Paleo diet is the low intake of calcium and risk for osteoporosis; however, consumption of calcium-rich foods or a calcium supplement can address this concern.107
Although small clinical studies have found the Paleo diet to be beneficial for various autoimmune diseases, clinical data evaluating the utility of the diet for cutaneous disease is lacking.108,109 Numerous randomized trials have demonstrated the Paleo diet to be effective for weight loss and improving insulin sensitivity and lipid levels.110-116 Thus, the Paleo diet may theoretically serve as a viable adjunct dietary approach to the treatment of cutaneous diseases associated with obesity and metabolic derangement.117
Carnivore Diet
Arguably the most controversial and radical diet is the carnivore diet. As the name implies, the carnivore diet is based on consuming solely animal products. A properly structured carnivore diet emphasizes a “nose-to-tail” eating approach where all parts of the animal including the muscle meats, organs, and fat are consumed. Proponents of the diet cite anthropologic evidence from fossil-stable carbon-13/carbon-12 isotope analyses, craniodental features, and numerous other adaptations that indicate increased consumption of meat during human evolution.118-122 Notably, many early humans ate a carnivore diet, but life span was very short at this time, suggesting the diet may not be as beneficial as has been suggested.
Despite the abundance of anecdotal evidence supporting its use for a variety of chronic conditions, including cutaneous autoimmune disease, there is a virtual absence of high-quality research on the carnivore diet.123-125
The purported benefits of the carnivore diet may be attributed to the consumption of organ meats that contain highly bioavailable essential vitamins and minerals, such as iron, zinc, copper, selenium, thiamine, niacin, folate, vitamin B6, vitamin B12, vitamin A, vitamin D, vitamin K, and choline.126-128 Other dietary compounds that have demonstrated benefit for skin health and are predominantly found in animal foods include carnosine, carnitine, creatine, taurine, coenzyme Q10, and collagen.129-134 Nevertheless, there is no data to recommend the elimination of antioxidant- and micronutrient-dense plant-based foods. Rigorous clinical research evaluating the efficacy and safety of the carnivore diet in dermatologic patients is needed. A carnivore diet should not be undertaken without the assistance of a dietician who can ensure adequate micronutrient and macronutrient support.
Final Thoughts
The adjunctive role of diet in the treatment of skin disease is expanding and becoming more widely accepted among dermatologists. Unfortunately, there remains a lack of randomized controlled trials confirming the efficacy of various dietary interventions in the dermatologic setting. Although evidence-based dietary recommendations currently are limited, it is important for dermatologists to be aware of the varied and nuanced dietary interventions employed by patients.
Ultimately, dietary recommendations must be personalized, considering a patient’s comorbidities, personal beliefs and preferences, and nutrigenetics. The emerging field of dermatonutrigenomics—the study of how dietary compounds interact with one’s genes to influence skin health—may allow for precise dietary recommendations to be made in dermatologic practice. Direct-to-consumer genetic tests targeted toward dermatology patients are already on the market, but their clinical utility awaits validation.1 Because nutritional science is a constantly evolving field, becoming familiar with these popular diets will serve both dermatologists and their patients well.
Within the last decade, almost 3000 articles have been published on the role of diet in the prevention and management of dermatologic conditions. Patients are increasingly interested in—and employing—dietary modifications that may influence skin appearance and aid in the treatment of cutaneous disease.1 It is essential that dermatologists are familiar with existing evidence on the role of diet in dermatology to counsel patients appropriately. Herein, we discuss the compositions of several popular diets and their proposed utility for dermatologic purposes. We highlight the limited literature that exists surrounding this topic and emphasize the need for future, well-designed clinical trials that study the impact of diet on skin disease.
Ketogenic Diet
The ketogenic diet has a macronutrient profile composed of high fat, low to moderate protein, and very low carbohydrates. Nutritional ketosis occurs as the body begins to use free fatty acids (via beta oxidation) as the primary metabolite driving cellular metabolism. It has been suggested that the ketogenic diet may impart beneficial effects on skin disease; however, limited literature exists on the role of nutritional ketosis in the treatment of dermatologic conditions.
Mechanistically, the ketogenic diet decreases the secretion of insulin and insulinlike growth factor 1, resulting in a reduction of circulating androgens and increased activity of the retinoid X receptor.2 In acne vulgaris, it has been suggested that the ketogenic diet may be beneficial in decreasing androgen-induced sebum production and the overproliferation of keratinocytes.2-7 The ketogenic diet is one of the most rapidly effective dietary strategies for normalizing both insulin and androgens, thus it may theoretically be useful for other metabolic and hormone-dependent skin diseases, such as hidradenitis suppurativa.8,9
The cutaneous manifestations associated with chronic hyperinsulinemia and hyperglycemia are numerous and include acanthosis nigricans, acrochordons, diabetic dermopathy, scleredema diabeticorum, bullosis diabeticorum, keratosis pilaris, and generalized granuloma annulare. There also is an increased risk for bacterial and fungal skin infections associated with hyperglycemic states.10 The ketogenic diet is an effective nonpharmacologic tool for normalizing serum insulin and glucose levels in most patients and may have utility in the aforementioned conditions.11,12 In addition to improving insulin sensitivity, it has been used as a dietary strategy for weight loss.11-15 Because obesity and metabolic syndrome are highly correlated with common skin conditions such as psoriasis, hidradenitis suppurativa, and androgenetic alopecia, there may be a role for employing the ketogenic diet in these patient populations.16,17
Although robust clinical studies on ketogenic diets in skin disease are lacking, a recent single-arm, open-label clinical trial observed benefit in all 37 drug-naïve, overweight patients with chronic plaque psoriasis who underwent a ketogenic weight loss protocol. Significant reductions in psoriasis area and severity index (PASI) score and dermatology life quality index score were reported (P<.001).18 Another study of 30 patients with psoriasis found that a 4-week, low-calorie, ketogenic diet resulted in 50% improvement of PASI scores, 10% weight loss, and a reduction in the proinflammatory cytokines IL-1β and IL-2.19 Despite these results, it is a challenge to tease out if the specific dietary intervention or its associated weight loss was the main driver in these reported improvements in skin disease.
There is mixed evidence on the anti-inflammatory nature of the ketogenic diet, likely due to wide variation in the composition of foods included in individual diets. In many instances, the ketogenic diet is thought to possess considerable antioxidant and anti-inflammatory capabilities. Ketones are known activators of the nuclear factor erythroid 2–related factor 2 pathway, which upregulates the production of glutathione, a major endogenous intracellular antioxidant.20 Additionally, dietary compounds from foods that are encouraged while on the ketogenic diet, such as sulforaphane from broccoli, also are independent activators of nuclear factor erythroid 2–related factor 2.21 Ketones are efficiently utilized by mitochondria, which also may result in the decreased production of reactive oxygen species and lower oxidative stress.22 Moreover, the ketone body β-hydroxybutyrate has demonstrated the ability to reduce proinflammatory IL-1β levels via suppression of nucleotide-binding domain-like receptor protein 3 inflammasome activity.23,24 The activity of IL-1β is known to be elevated in many dermatologic conditions, including juvenile idiopathic arthritis, relapsing polychondritis, Schnitzler syndrome, hidradenitis suppurativa, Behçet disease, and other autoinflammatory syndromes.25 Ketones also have been shown to inhibit the nuclear factor–κB proinflammatory signaling pathway.22,26,27 Overexpression of IL-1β and aberrant activation of nuclear factor–κB are implicated in a variety of inflammatory, autoimmune, and oncologic cutaneous pathologies. The ketogenic diet may prove to be an effective adjunctive treatment for dermatologists to consider in select patient populations.23,24,28-30
For patients with keratinocyte carcinomas, the ketogenic diet may offer the aforementioned anti-inflammatory and antioxidant effects, as well as suppression of the mechanistic target of rapamycin, a major regulator of cell metabolism and proliferation.31,32 Inhibition of mechanistic target of rapamycin activity has been shown to slow tumor growth and reduce the development of squamous cell carcinoma.25,33,34 The ketogenic diet also may exploit the preferential utilization of glucose exhibited by many types of cancer cells, thereby “starving” the tumor of its primary fuel source.35,36 In vitro and animal studies in a variety of cancer types have demonstrated that a ketogenic metabolic state—achieved through the ketogenic diet or fasting—can sensitize tumor cells to chemotherapy and radiation while conferring a protective effect to normal cells.37-40 This recently described phenomenon is known as differential stress resistance, but it has not been studied in keratinocyte malignancies or melanoma to date. Importantly, some basal cell carcinomas and BRAF V600E–mutated melanomas have worsened while on the ketogenic diet, suggesting more data is needed before it can be recommended for all cancer patients.41,42 Furthermore, other skin conditions such as prurigo pigmentosa have been associated with initiation of the ketogenic diet.43
Low FODMAP Diet
Fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) are short-chain carbohydrates that are poorly absorbed, osmotically active, and rapidly fermented by intestinal bacteria.44 The low FODMAP diet has been shown to be efficacious for treatment of irritable bowel syndrome, small intestinal bacterial overgrowth (SIBO), and some cases of inflammatory bowel disease (IBD).44-49 A low FODMAP diet may have potential implications for several dermatologic conditions.
Rosacea has been associated with various gastrointestinal tract disorders including irritable bowel syndrome, SIBO, and IBD.50-54 A single study found that patients with rosacea had a 13-fold increased risk for SIBO.55,56 Treatment of 40 patients with SIBO using rifaximin resulted in complete resolution of rosacea in all patients, with no relapse after a 3-year follow-up period.55 Psoriasis also has been associated with SIBO and IBD.57,58 One small study found that eradication of SIBO in psoriatic patients resulted in improved PASI scores and colorimetric values.59
Although the long-term health consequences of the low FODMAP diet are unknown, further research on such dietary interventions for inflammatory skin conditions is warranted given the mounting evidence of a gut-skin connection and the role of the intestinal microbiome in skin health.50,51
Gluten-Free Diet
Gluten is a protein found in a variety of grains. Although the role of gluten in the pathogenesis of celiac disease and dermatitis herpetiformis is indisputable, the deleterious effects of gluten outside of the context of these diseases remain controversial. There may be a compelling case for eliminating gluten in psoriasis patients with seropositivity for celiac disease. A recent systematic review found a 2.2-fold increased risk for celiac disease in psoriasis patients.60 Antigliadin antibody titers also were found to be positively correlated with psoriatic disease severity.61 In addition, one open-label study found a reduction in PASI scores in 73% of patients with antigliadin antibodies after 3 months on a gluten-free diet compared to those without antibodies; however, the study only included 22 patients.62 Several other small studies have yielded similar results63,64; however, antigliadin antibodies are neither the most sensitive nor specific markers of celiac disease, and additional testing should be completed in any patient who may carry this diagnosis. A survey study by the National Psoriasis Foundation found that the dietary change associated with the greatest skin improvement was removal of gluten and nightshade vegetables in approximately 50% of the 1200 psoriasis patients that responded.65 Case reports of various dermatologic conditions including sarcoidosis, vitiligo, alopecia areata, lichen planus, dermatomyositis, pyoderma gangrenosum, erythema nodosum, leukocytoclastic vasculitis, linear IgA bullous dermatosis, and aphthous ulcerations have reportedly improved with a gluten-free diet; however, this should not be used as primary therapy in patients without celiac disease.66-71 Because gluten-free diets can be expensive and challenging to follow, a formal assessment for celiac disease should be considered before recommendation of this dietary intervention.
Low Histamine Diet
Histamine is a biogenic amine produced by the decarboxylation of the amino acid histidine.72 It is found in several foods in varying amounts. Because bacteria can convert histidine into histamine, many fermented and aged foods such as kimchi, sauerkraut, cheese, and red wine contain high levels of histamine. Individuals who have decreased activity of diamine oxidase (DAO), an enzyme that degrades histamine, may be more susceptible to histamine intolerance.72 The symptoms of histamine intolerance are numerous and include gastrointestinal tract distress, rhinorrhea and nasal congestion, headache, urticaria, flushing, and pruritus. Histamine intolerance can mimic an IgE-mediated food allergy; however, allergy testing is negative in these patients. Unfortunately, there is no laboratory test for histamine intolerance; a double-blind, placebo-controlled food challenge is considered the gold-standard test.72
As it pertains to dermatology, a low histamine diet may play a role in the treatment of certain patients with atopic dermatitis and chronic spontaneous urticaria. One study reported that 17 of 54 (31.5%) atopic patients had higher basal levels of serum histamine compared to controls.73 Another study found that a histamine-free diet led to improvement in both histamine intolerance symptoms and atopic dermatitis disease severity (SCORing atopic dermatitis) in patients with low DAO activity.74 In chronic spontaneous urticaria, a recent systematic review found that in 223 patients placed on a low histamine diet for 3 to 4 weeks, 12% and 44% achieved complete and partial remission, respectively.75 Although treatment response based on a patient’s DAO activity level has not been correlated, a diet low in histamine may prove useful for patients with persistent atopic dermatitis and chronic spontaneous urticaria who have negative food allergy tests and report exacerbation of symptoms after ingestion of histamine-rich foods.76,77
Mediterranean Diet
The Mediterranean diet has been touted as one of the healthiest diets to date, and large randomized clinical trials have demonstrated its effectiveness in weight loss, improving insulin sensitivity, and reducing inflammatory cytokine profiles.78,79 A major criticism of the Mediterranean diet is that it has considerable ambiguity and lacks a precise definition due to the variability of what is consumed in different Mediterranean regions. Generally, the diet emphasizes high consumption of colorful fruits and vegetables, aromatic herbs and spices, olive oil, nuts, and seafood, as well as modest amounts of dairy, eggs, and red meat.80 The anti-inflammatory effects of this diet largely have been attributed to its abundance of polyphenols, carotenoids, monounsaturated fatty acids, and omega-3 polyunsaturated fatty acids (PUFAs).80,81 Examples of polyphenols include resveratrol in red grapes, quercetin in apples and red onions, and curcumin in turmeric, while examples of carotenoids include lycopene in tomatoes and zeaxanthin in dark leafy greens. Oleic acid is a monounsaturated fatty acid present in high concentrations in olive oil, while eicosapentaenoic acid and docosahexaenoic acid are omega-3 PUFAs predominantly found in fish.82
Unfortunately, rigorous clinical trials regarding the Mediterranean diet as it pertains to dermatology have not been undertaken. Numerous observational studies in patients with psoriasis have suggested that close adherence to the Mediterranean diet was associated with improvement in PASI scores.83-86 The National Psoriasis Foundation now recommends a trial of the Mediterranean diet in some patients with psoriasis, emphasizing increased dietary intake of olive oil, fish, and vegetables.87 Adherence to a Mediterranean diet also has been inversely correlated to the severity of acne vulgaris and hidradenitis suppurativa88,89; however, these studies failed to account for the multifactorial risk factors associated with these conditions. Mediterranean diets also may impart a chemopreventive effect, supported by a number of in vivo and in vitro studies demonstrating the inhibition and/or reversal of cutaneous DNA damage induced by UV radiation through supplementation with various phytonutrients and omega-3 PUFAs.81,90-92 Although small case-control studies have found a decreased risk of basal cell carcinoma in those who closely adhered to a Mediterranean diet, more rigorous clinical research is needed.93
Whole-Food, Plant-Based Diet
A whole-food, plant-based (WFPB) diet is another popular dietary approach that consists of eating fruits, vegetables, legumes, nuts, seeds, and grains in their whole natural form.94 This diet discourages all animal products, including red meat, seafood, dairy, and eggs. It is similar to a vegan diet except that it eliminates all highly refined carbohydrates, vegetable oils, and other processed foods.94 Randomized clinical studies have demonstrated the WFPB diet to be effective in the treatment of obesity and metabolic syndrome.95,96
A WFPB diet has been shown to increase the antioxidant capacity of cells, lengthen telomeres, and reduce formation of advanced glycation end products.94,97,98 These benefits may help combat accelerated skin aging, including increased skin permeability, reduced elasticity and hydration, decreased angiogenesis, impaired immune function, and decreased vitamin D synthesis. Accelerated skin aging can result in delayed wound healing and susceptibility to skin tears and ecchymoses and also may promote the development of cutaneous malignancies.99 There remains a lack of clinical data studying a properly formulated WFPB diet in the dermatologic setting.
Paleolithic Diet
The paleolithic (Paleo) diet is an increasingly popular way of eating that attempts to mirror what our ancestors may have consumed between 10,000 and 2.5 million years ago.100 It is similar to the Mediterranean diet but excludes grains, dairy, legumes, and nightshade vegetables. It also calls for elimination of highly processed sugars and oils as well as chemical food additives and preservatives. There is a strict variation of the diet for individuals with autoimmune disease that also excludes eggs, nuts, and seeds, as these can be inflammatory or immunogenic in some patients.100-106 Other variations of the diet exist, including the ketogenic Paleo diet, pegan (Paleo vegan) diet, and lacto-Paleo diet.100 An often cited criticism of the Paleo diet is the low intake of calcium and risk for osteoporosis; however, consumption of calcium-rich foods or a calcium supplement can address this concern.107
Although small clinical studies have found the Paleo diet to be beneficial for various autoimmune diseases, clinical data evaluating the utility of the diet for cutaneous disease is lacking.108,109 Numerous randomized trials have demonstrated the Paleo diet to be effective for weight loss and improving insulin sensitivity and lipid levels.110-116 Thus, the Paleo diet may theoretically serve as a viable adjunct dietary approach to the treatment of cutaneous diseases associated with obesity and metabolic derangement.117
Carnivore Diet
Arguably the most controversial and radical diet is the carnivore diet. As the name implies, the carnivore diet is based on consuming solely animal products. A properly structured carnivore diet emphasizes a “nose-to-tail” eating approach where all parts of the animal including the muscle meats, organs, and fat are consumed. Proponents of the diet cite anthropologic evidence from fossil-stable carbon-13/carbon-12 isotope analyses, craniodental features, and numerous other adaptations that indicate increased consumption of meat during human evolution.118-122 Notably, many early humans ate a carnivore diet, but life span was very short at this time, suggesting the diet may not be as beneficial as has been suggested.
Despite the abundance of anecdotal evidence supporting its use for a variety of chronic conditions, including cutaneous autoimmune disease, there is a virtual absence of high-quality research on the carnivore diet.123-125
The purported benefits of the carnivore diet may be attributed to the consumption of organ meats that contain highly bioavailable essential vitamins and minerals, such as iron, zinc, copper, selenium, thiamine, niacin, folate, vitamin B6, vitamin B12, vitamin A, vitamin D, vitamin K, and choline.126-128 Other dietary compounds that have demonstrated benefit for skin health and are predominantly found in animal foods include carnosine, carnitine, creatine, taurine, coenzyme Q10, and collagen.129-134 Nevertheless, there is no data to recommend the elimination of antioxidant- and micronutrient-dense plant-based foods. Rigorous clinical research evaluating the efficacy and safety of the carnivore diet in dermatologic patients is needed. A carnivore diet should not be undertaken without the assistance of a dietician who can ensure adequate micronutrient and macronutrient support.
Final Thoughts
The adjunctive role of diet in the treatment of skin disease is expanding and becoming more widely accepted among dermatologists. Unfortunately, there remains a lack of randomized controlled trials confirming the efficacy of various dietary interventions in the dermatologic setting. Although evidence-based dietary recommendations currently are limited, it is important for dermatologists to be aware of the varied and nuanced dietary interventions employed by patients.
Ultimately, dietary recommendations must be personalized, considering a patient’s comorbidities, personal beliefs and preferences, and nutrigenetics. The emerging field of dermatonutrigenomics—the study of how dietary compounds interact with one’s genes to influence skin health—may allow for precise dietary recommendations to be made in dermatologic practice. Direct-to-consumer genetic tests targeted toward dermatology patients are already on the market, but their clinical utility awaits validation.1 Because nutritional science is a constantly evolving field, becoming familiar with these popular diets will serve both dermatologists and their patients well.
- Jaros J, Katta R, Shi VY. Dermatonutrigenomics: past, present, and future. Dermatology. 2019;235:164-166.
- Paoli A, Grimaldi K, Toniolo L, et al. Nutrition and acne: therapeutic potential of ketogenic diets. Skin Pharmacol Physiol. 2012;25:111-117.
- Melnik BC, Schmitz G. Role of insulin, insulin-like growth factor-1, hyperglycaemic food and milk consumption in the pathogenesis of acne vulgaris. Exp Dermatol. 2009;18:833-841.
- Smith RN, Mann NJ, Braue A, et al. The effect of a high-protein, low glycemic-load diet versus a conventional, high glycemic-load diet on biochemical parameters associated with acne vulgaris: a randomized, investigator-masked, controlled trial. J Am Acad Dermatol. 2007;57:247-256.
- Smith R, Mann N, Mäkeläinen H, et al. A pilot study to determine the short-term effects of a low glycemic load diet on hormonal markers of acne: a nonrandomized, parallel, controlled feeding trial. Mol Nutr Food Res. 2008;52:718-726.
- Smith RN, Braue A, Varigos GA, et al. The effect of a low glycemic load diet on acne vulgaris and the fatty acid composition of skin surface triglycerides. J Dermatol Sci. 2008;50:41-52.
- Kwon HH, Yoon JY, Hong JS, et al. Clinical and histological effect of a low glycaemic load diet in treatment of acne vulgaris in Korean patients: a randomized, controlled trial. Acta Derm Venereol. 2012;92:241-246.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a "male" therapy for a predominantly "female" disease. J Clin Aesthet Dermatol. 2016;9:44.
- Nikolakis G, Karagiannidis I, Vaiopoulos AG, et al. Endocrinological mechanisms in the pathophysiology of hidradenitis suppurativa [in German]. Hautarzt. 2020;71:762-771.
- Karadag AS, Ozlu E, Lavery MJ. Cutaneous manifestations of diabetes mellitus and the metabolic syndrome. Clin Dermatology. 2018;36:89-93.
- Gardner CD, Kiazand A, Alhassan S, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969-977.
- Anton SD, Hida A, Heekin K, et al. Effects of popular diets without specific calorie targets on weight loss outcomes: systematic review of findings from clinical trials. Nutrients. 2017;9:822.
- Castellana M, Conte E, Cignarelli A, et al. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: a systematic review and meta-analysis. Rev Endocr Metab Disord. 2020;21:5-16.
- Paoli A, Mancin L, Giacona MC, et al. Effects of a ketogenic diet in overweight women with polycystic ovary syndrome. J Transl Med. 2020;18:104.
- Dashti HM, Mathew TC, Hussein T, et al. Long-term effects of a ketogenic diet in obese patients. Exp Clin Cardiol. 2004;9:200-205.
- Lian N, Chen M. Metabolic syndrome and skin disease: potential connection and risk. Int J Dermatol Venereol. 2019;2:89-93.
- Hu Y, Zhu Y, Lian N, et al. Metabolic syndrome and skin diseases. Front Endocrinol (Lausanne). 2019;10:788.
- Castaldo G, Rastrelli L, Galdo G, et al. Aggressive weight-loss program with a ketogenic induction phase for the treatment of chronic plaque psoriasis: a proof-of-concept, single-arm, open-label clinical trial. Nutrition. 2020;74:110757.
- Castaldo G, Pagano I, Grimaldi M, et al. Effect of very-low-calorie ketogenic diet on psoriasis patients: a nuclear magnetic resonance-based metabolomic study. J Proteome Res. 2021;20:1509-1521.
- Milder J, Liang L-P, Patel M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis. 2010;40:238-244.
- Kubo E, Chhunchha B, Singh P, et al. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci Rep. 2017;7:14130.
- Pinto A, Bonucci A, Maggi E, et al. Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in Alzheimer's disease. Antioxidants (Basel). 2018;7:63.
- Youm Y-H, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263-269.
- Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328.
- Fomin DA, McDaniel B, Crane J. The promising potential role of ketones in inflammatory dermatologic disease: a new frontier in treatment research. J Dermatol Treat. 2017;28:484-487.
- Rahman M, Muhammad S, Khan MA, et al. The β-hydroxybutyrate receptor HCA 2 activates a neuroprotective subset of macrophages. Nat Commun. 2014;5:1-11.
- Lu Y, Yang YY, Zhou MW, et al. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-κB signaling pathways. Neurosci Lett. 2018;683:13-18.
- Hamarsheh S, Zeiser R. NLRP3 inflammasome activation in cancer: a double-edged sword. Front Immunol. 2020;11:1444.
- Bell S, Degitz K, Quirling M, et al. Involvement of NF-κB signalling in skin physiology and disease. Cell Signal. 2003;15:1-7.
- Goldminz AM, Au SC, Kim N, et al. NF-κB: an essential transcription factor in psoriasis. J Dermatol Sci. 2013;69:89-94.
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589.
- McDaniel S, Rensing N, Yamada K, et al. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:E7-E11.
- Alter M, Satzger I, Schrem H, et al. Non-melanoma skin cancer is reduced after switch of immunosuppression to mTOR-inhibitors in organ transplant recipients. J Dtsch Dermatol Ges. 2014;12:480-488.
- Feldmeyer L, Hofbauer GF, Böni T, et al. Mammalian target of rapamycin (mTOR) inhibitors slow skin carcinogenesis, but impair wound healing. Br J Dermatol. 2012;166:422-424.
- Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211-218.
- Li W. "Warburg effect" and mitochondrial metabolism in skin cancer.J Carcinogene Mutagene. 2012:S4.
- Naveed S, Aslam M, Ahmad A. Starvation based differential chemotherapy: a novel approach for cancer treatment. Oman Med J. 2014;29:391-398.
- Raffaghello L, Lee C, Safdie FM, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. 2008;105:8215-8220.
- Buono R, Longo VD. Starvation, stress resistance, and cancer. Trends Endocrinol Metab. 2018;29:271-280.
- de Groot S, Pijl H, van der Hoeven JJM, et al. Effects of short-term fasting on cancer treatment. J Exp Clin Cancer Res. 2019;38:209.
- Hosseini M, Kasraian Z, Rezvani HR. Energy metabolism in skin cancers: a therapeutic perspective. Biochim Biophys Acta Bioenerg. 2017;1858:712-722.
- Feichtinger RG, Lang R, Geilberger R, et al. Melanoma tumors exhibit a variable but distinct metabolic signature. Exp Dermatol. 2018;27:204-207.
- Alshaya MA, Turkmani MG, Alissa AM. Prurigo pigmentosa following ketogenic diet and bariatric surgery: a growing association. JAAD Case Rep. 2019;5:504-507.
- Bellini M, Tonarelli S, Nagy AG, et al. Low FODMAP diet: evidence, doubts, and hopes. Nutrients. 2020;12:148.
- Kwiatkowski L, Rice E, Langland J. Integrative treatment of chronic abdominal bloating and pain associated with overgrowth of small intestinal bacteria: a case report. Altern Ther Health Med. 2017;23:56-61.
- Hubkova T. No more pain in the gut: lifestyle medicine approach to irritable bowel syndrome. Am J Lifestyle Med. 2017;11:223-226.
- Schumann D, Klose P, Lauche R, et al. Low fermentable, oligo-, di-, mono-saccharides and polyol diet in the treatment of irritable bowel syndrome: a systematic review and meta-analysis. Nutrition. 2018;45:24-31.
- Cox SR, Prince AC, Myers CE, et al. Fermentable carbohydrates [FODMAPs] exacerbate functional gastrointestinal symptoms in patients with inflammatory bowel disease: a randomised, double-blind, placebo-controlled, cross-over, re-challenge trial. J Crohns Colitis. 2017;11:1420-1429.
- Damas OM, Garces L, Abreu MT. Diet as adjunctive treatment for inflammatory bowel disease: review and update of the latest literature. Curr Treat Options Gastroenterol. 2019;17:313-325.
- Wang FY, Chi CC. Rosacea, germs, and bowels: a review on gastrointestinal comorbidities and gut-skin axis of rosacea. Adv Ther. 2021;38:1415-1424.
- Daou H, Paradiso M, Hennessy K, et al. Rosacea and the microbiome: a systematic review. Dermatol Ther (Heidelb). 2021;11:1-12.
- Weinstock LB, Steinhoff M. Rosacea and small intestinal bacterial overgrowth: prevalence and response to rifaximin. J Am Acad Dermatol. 2013;68:875-876.
- Wu CY, Chang YT, Juan CK, et al. Risk of inflammatory bowel disease in patients with rosacea: results from a nationwide cohort study in Taiwan. J Am Acad Dermatol. 2017;76:911-917.
- Egeberg A, Weinstock LB, Thyssen EP, et al. Rosacea and gastrointestinal disorders: a population-based cohort study. Br J Dermatol. 2017;176:100-106.
- Drago F, De Col E, Agnoletti AF, et al. The role of small intestinal bacterial overgrowth in rosacea: a 3-year follow-up. J Am Acad Dermatol. 2016;75:E113-E115.
- Parodi A, Paolino S, Greco A, et al. Small intestinal bacterial overgrowth in rosacea: clinical effectiveness of its eradication. Clin Gastroenterol Hepatol. 2008;6:759-764.
- Ojetti V, De Simone C, Aguilar Sanchez J, et al. Malabsorption in psoriatic patients: cause or consequence? Scand J Gastroenterol. 2006;41:1267-1271.
- Kim M, Choi KH, Hwang SW, et al. Inflammatory bowel disease is associated with an increased risk of inflammatory skin diseases: a population-based cross-sectional study. J Am Acad Dermatol. 2017;76:40-48.
- Drago F, Ciccarese G, Indemini E, et al. Psoriasis and small intestine bacterial overgrowth. Int J Dermatol. 2018;57:112-113.
- Acharya P, Mathur M. Association between psoriasis and celiac disease: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:1376-1385.
- Bhatia BK, Millsop JW, Debbaneh M, et al. Diet and psoriasis, part II: celiac disease and role of a gluten-free diet. J Am Acad Dermatol. 2014;71:350-358.
- Michaëlsson G, Gerdén B, Hagforsen E, et al. Psoriasis patients with antibodies to gliadin can be improved by a gluten-free diet. Br J Dermatol. 2000;142:44-51.
- Kolchak NA, Tetarnikova MK, Theodoropoulou MS, et al. Prevalence of antigliadin IgA antibodies in psoriasis vulgaris and response of seropositive patients to a gluten-free diet. J Multidiscip Healthc. 2018;11:13-19.
- De Bastiani R, Gabrielli M, Lora L, et al. Association between coeliac disease and psoriasis: Italian primary care multicentre study. Dermatology. 2015;230:156-160.
- Afifi L, Danesh MJ, Lee KM, et al. Dietary behaviors in psoriasis: patient-reported outcomes from a U.S. national survey. Dermatol Ther (Heidelb). 2017;7:227-242.
- Loche F, Bazex J. Celiac disease associated with cutaneous sarcoidosic granuloma [in French]. Rev Med Interne. 1997;18:975-978.
- Rodríguez-García C, González-Hernández S, Pérez-Robayna N, et al. Repigmentation of vitiligo lesions in a child with celiac disease after a gluten-free diet. Pediatr Dermatol. 2011;28:209-210.
- Wijarnpreecha K, Panjawatanan P, Corral JE, et al. Celiac disease and risk of sarcoidosis: a systematic review and meta-analysis. J Evid Based Med. 2019;12:194-199.
- Rodrigo L, Beteta-Gorriti V, Alvarez N, et al. Cutaneous and mucosal manifestations associated with celiac disease. Nutrients. 2018;10:800.
- Song MS, Farber D, Bitton A, et al. Dermatomyositis associated with celiac disease: response to a gluten-free diet. Can J Gastroenterol. 2006;20:433-435.
- Egan CA, Smith EP, Taylor TB, et al. Linear IgA bullous dermatosis responsive to a gluten-free diet. Am J Gastroenterol. 2001;96:1927-1929.
- Comas-Basté O, Sánchez-Pérez S, Veciana-Nogués MT, et al. Histamine intolerance: the current state of the art. Biomolecules. 2020;10:1181.
- Ring J. Plasma histamine concentrations in atopic eczema. Clin Allergy. 1983;13:545-552.
- Maintz L, Benfadal S, Allam JP, et al. Evidence for a reduced histamine degradation capacity in a subgroup of patients with atopic eczema. J Allergy Clin Immunol. 2006;117:1106-1112.
- Cornillier H, Giraudeau B, Samimi M, et al. Effect of diet in chronic spontaneous urticaria: a systematic review. Acta Derm Venereol. 2019;99:127-132.
- Son JH, Chung BY, Kim HO, et al. A histamine-free diet is helpful for treatment of adult patients with chronic spontaneous urticaria. Ann Dermatol. 2018;30:164-172.
- Wagner N, Dirk D, Peveling-Oberhag A, et al. A popular myth - low-histamine diet improves chronic spontaneous urticaria - fact or fiction? J Eur Acad Dermatol Venereol. 2017;31:650-655.
- Esposito K, Marfella R, Ciotola M, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA. 2004;292:1440-1446.
- Steffen LM, Van Horn L, Daviglus ML, et al. A modified Mediterranean diet score is associated with a lower risk of incident metabolic syndrome over 25 years among young adults: the CARDIA (coronary artery risk development in young adults) study. Br J Nutr. 2014;112:1654-1661.
- Bower A, Marquez S, de Mejia EG. The health benefits of selected culinary herbs and spices found in the traditional Mediterranean diet. Crit Rev Food Sci Nutr. 2016;56:2728-2746.
- Bosch R, Philips N, Suárez-Pérez JA, et al. Mechanisms of photoaging and cutaneous photocarcinogenesis, and photoprotective strategies with phytochemicals. Antioxidants (Basel). 2015;4:248-268.
- Katsimbri P, Korakas E, Kountouri A, et al. The effect of antioxidant and anti-inflammatory capacity of diet on psoriasis and psoriatic arthritis phenotype: nutrition as therapeutic tool? Antioxidants. 2021;10:157.
- Molina-Leyva A, Cuenca-Barrales C, Vega-Castillo JJ, et al. Adherence to Mediterranean diet in Spanish patients with psoriasis: cardiovascular benefits? Dermatol Ther. 2019;32:E12810.
- Barrea L, Balato N, Di Somma C, et al. Nutrition and psoriasis: is there any association between the severity of the disease and adherence to the Mediterranean diet? J Transl Med. 2015;13:1-10.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort. JAMA Dermatol. 2018;154:1017-1024.
- Korovesi A, Dalamaga M, Kotopouli M, et al. Adherence to the Mediterranean diet is independently associated with psoriasis risk, severity, and quality of life: a cross-sectional observational study. Int J Dermatol. 2019;58:E164-E165.
- Ford AR, Siegel M, Bagel J, et al. Dietary recommendations for adults with psoriasis or psoriatic arthritis from the medical board of the National Psoriasis Foundation: a systematic review. JAMA Dermatol. 2018;154:934-950.
- Skroza N, Tolino E, Semyonov L, et al. Mediterranean diet and familial dysmetabolism as factors influencing the development of acne. Scand J Public Health. 2012;40:466-474.
- Barrea L, Fabbrocini G, Annunziata G, et al. Role of nutrition and adherence to the Mediterranean diet in the multidisciplinary approach of hidradenitis suppurativa: evaluation of nutritional status and its association with severity of disease. Nutrients. 2018;11:57.
- Nichols JA, Katiyar SK. Skin photoprotection by natural polyphenols: anti-inflammatory, antioxidant and DNA repair mechanisms. Arch Dermatol Res. 2010;302:71-83.
- Huang T-H, Wang P-W, Yang S-C, et al. Cosmetic and therapeutic applications of fish oil's fatty acids on the skin. Mar Drugs. 2018;16:256.
- Rizwan M, Rodriguez-Blanco I, Harbottle A, et al. Tomato paste rich in lycopene protects against cutaneous photodamage in humans in vivo: a randomized controlled trial. Br J Dermatol. 2011;164:154-162.
- Leone A, Martínez-González M, Martin-Gorgojo A, et al. Mediterranean diet, dietary approaches to stop hypertension, and pro-vegetarian dietary pattern in relation to the risk of basal cell carcinoma: a nested case-control study within the Seguimiento Universidad de Navarra (SUN) cohort. Am J Clin Nutr. 2020;112:364-372.
- Solway J, McBride M, Haq F, et al. Diet and dermatology: the role of a whole-food, plant-based diet in preventing and reversing skin aging--a review. J Clin Aesthet Dermatol. 2020;13:38-43.
- Greger M. A whole food plant-based diet is effective for weight loss: the evidence. Am J Lifestyle Med. 2020;14:500-510.
- Wright N, Wilson L, Smith M, et al. The BROAD study: a randomised controlled trial using a whole food plant-based diet in the community for obesity, ischaemic heart disease or diabetes. Nutr Diabetes. 2017;7:E256.
- Ornish D, Lin J, Chan JM, et al. Effect of comprehensive lifestyle changes on telomerase activity and telomere length in men with biopsy-proven low-risk prostate cancer: 5-year follow-up of a descriptive pilot study. Lancet Oncol. 2013;14:1112-1120.
- Ornish D, Lin J, Daubenmier J, et al. Increased telomerase activity and comprehensive lifestyle changes: a pilot study. Lancet Oncol. 2008;9:1048-1057.
- Zouboulis CC, Makrantonaki E. Clinical aspects and molecular diagnostics of skin aging. Clin Dermatol. 2011;29:3-14.
- Gupta L, Khandelwal D, Lal PR, et al. Palaeolithic diet in diabesity and endocrinopathies--a vegan's perspective. Eur Endocrinol. 2019;15:77-82.
- Chassaing B, Van de Wiele T, De Bodt J, et al. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut. 2017;66:1414-1427.
- Thorburn Alison N, Macia L, Mackay Charles R. Diet, metabolites, and "Western lifestyle" inflammatory diseases. Immunity. 2014;40:833-842.
- Katta R, Schlichte M. Diet and dermatitis: food triggers. J Clin Aesthet Dermatol. 2014;7:30-36.
- Dhar S, Srinivas SM. Food allergy in atopic dermatitis. Indian J Dermatol. 2016;61:645-648.
- Birmingham N, Thanesvorakul S, Gangur V. Relative immunogenicity of commonly allergenic foods versus rarely allergenic and nonallergenic foods in mice. J Food Prot. 2002;65:1988-1991.
- Yu W, Freeland DMH, Nadeau KC. Food allergy: immune mechanisms, diagnosis and immunotherapy. Nat Rev Immunol. 2016;16:751-765.
- Kowalski LM, Bujko J. Evaluation of biological and clinical potential of paleolithic diet [in Polish]. Rocz Panstw Zakl Hig. 2012;63:9-15.
- Lee JE, Titcomb TJ, Bisht B, et al. A modified MCT-based ketogenic diet increases plasma β-hydroxybutyrate but has less effect on fatigue and quality of life in people with multiple sclerosis compared to a modified paleolithic diet: a waitlist-controlled, randomized pilot study. J Am Coll Nutr. 2021;40:13-25.
- Abbott RD, Sadowski A, Alt AG. Efficacy of the autoimmune protocol diet as part of a multi-disciplinary, supported lifestyle intervention for Hashimoto's thyroiditis. Cureus. 2019;11:E4556.
- Lindeberg S, Jönsson T, Granfeldt Y, et al. A palaeolithic diet improves glucose tolerance more than a Mediterranean-like diet in individuals with ischaemic heart disease. Diabetologia. 2007;50:1795-1807.
- Jönsson T, Granfeldt Y, Ahrén B, et al. Beneficial effects of a paleolithic diet on cardiovascular risk factors in type 2 diabetes: a randomized cross-over pilot study. Cardiovasc Diabetol. 2009;8:35.
- Boers I, Muskiet FAJ, Berkelaar E, et al. Favourable effects of consuming a palaeolithic-type diet on characteristics of the metabolic syndrome: a randomized controlled pilot-study. Lipids Health Dis. 2014;13:160.
- Ghaedi E, Mohammadi M, Mohammadi H, et al. Effects of a paleolithic diet on cardiovascular disease risk factors: a systematic review and meta-analysis of randomized controlled trials. Adv Nutr. 2019;10:634-646.
- Mellberg C, Sandberg S, Ryberg M, et al. Long-term effects of a palaeolithic-type diet in obese postmenopausal women: a 2-year randomized trial. Eur J Clin Nutr. 2014;68:350-357.
- Pastore RL, Brooks JT, Carbone JW. Paleolithic nutrition improves plasma lipid concentrations of hypercholesterolemic adults to a greater extent than traditional heart-healthy dietary recommendations. Nutr Res. 2015;35:474-479.
- Otten J, Stomby A, Waling M, et al. Benefits of a paleolithic diet with and without supervised exercise on fat mass, insulin sensitivity, and glycemic control: a randomized controlled trial in individuals with type 2 diabetes. Diabetes Metab Res Rev. 2017;33:E2828.
- Stefanadi EC, Dimitrakakis G, Antoniou C-K, et al. Metabolic syndrome and the skin: a more than superficial association. reviewing the association between skin diseases and metabolic syndrome and a clinical decision algorithm for high risk patients. Diabetol Metab Syndr. 2018;10:9.
- Mann N. Meat in the human diet: an anthropological perspective. Nutr Dietetics. 2007;64(suppl 4):S102-S107.
- Bramble DM, Lieberman DE. Endurance running and the evolution of Homo. Nature. 2004;432:345-352.
- Kuhn JE. Throwing, the shoulder, and human evolution. Am J Orthop (Belle Mead NJ). 2016;45:110-114.
- Kobayashi H, Kohshima S. Unique morphology of the human eye and its adaptive meaning: comparative studies on external morphology of the primate eye. J Hum Evol. 2001;40:419-435.
- Cordain L, Eaton SB, Miller JB, et al. The paradoxical nature of hunter-gatherer diets: meat-based, yet non-atherogenic. Eur J Clin Nutr. 2002;56(suppl 1):S42-S52.
- McClellan WS, Du Bois EF. Clinical calorimetry: XLV. prolonged meat diets with a study of kidney function and ketosis. J Biol Chem. 1930;87:651-668.
- O'Hearn A. Can a carnivore diet provide all essential nutrients? Curr Opin Endocrinol Diabetes Obes. 2020;27:312-316.
- O'Hearn LA. A survey of improvements experienced on a carnivore diet compared to only carbohydrate restriction. Open Science Forum website. Published February 12, 2019. Accessed May 17, 2021. doi:10.17605/OSF.IO/5FU4D
- Williams P. Nutritional composition of red meat. Nutrition & Dietetics. 2007;64(suppl 4):S113-S119.
- Biel W, Czerniawska-Piątkowska E, Kowalczyk A. Offal chemical composition from veal, beef, and lamb maintained in organic production systems. Animals (Basel). 2019;9:489.
- Elmadfa I, Meyer AL. The role of the status of selected micronutrients in shaping the immune function. Endocr Metab Immune Disord Drug Targets. 2019;19:1100-1115.
- Babizhayev M. Treatment of skin aging and photoaging with innovative oral dosage forms of nonhydrolized carnosine and carcinine. Int J Clin Derm Res. 2017;5:116-143.
- Danby FW. Nutrition and aging skin: sugar and glycation. Clin Dermatol. 2010;28:409-411.
- Siefken W, Carstensen S, Springmann G, et al. Role of taurine accumulation in keratinocyte hydration. J Invest Dermatol. 2003;121:354-361.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19:3059.
- Fischer F, Achterberg V, März A, et al. Folic acid and creatineimprove the firmness of human skin in vivo. J Cosmet Dermatol. 2011;10:15-23.
- Blatt T, Lenz H, Weber T. Topical application of creatine is multibeneficial for human skin. J Am Acad Dermatol. 2005;52:P32.
- Jaros J, Katta R, Shi VY. Dermatonutrigenomics: past, present, and future. Dermatology. 2019;235:164-166.
- Paoli A, Grimaldi K, Toniolo L, et al. Nutrition and acne: therapeutic potential of ketogenic diets. Skin Pharmacol Physiol. 2012;25:111-117.
- Melnik BC, Schmitz G. Role of insulin, insulin-like growth factor-1, hyperglycaemic food and milk consumption in the pathogenesis of acne vulgaris. Exp Dermatol. 2009;18:833-841.
- Smith RN, Mann NJ, Braue A, et al. The effect of a high-protein, low glycemic-load diet versus a conventional, high glycemic-load diet on biochemical parameters associated with acne vulgaris: a randomized, investigator-masked, controlled trial. J Am Acad Dermatol. 2007;57:247-256.
- Smith R, Mann N, Mäkeläinen H, et al. A pilot study to determine the short-term effects of a low glycemic load diet on hormonal markers of acne: a nonrandomized, parallel, controlled feeding trial. Mol Nutr Food Res. 2008;52:718-726.
- Smith RN, Braue A, Varigos GA, et al. The effect of a low glycemic load diet on acne vulgaris and the fatty acid composition of skin surface triglycerides. J Dermatol Sci. 2008;50:41-52.
- Kwon HH, Yoon JY, Hong JS, et al. Clinical and histological effect of a low glycaemic load diet in treatment of acne vulgaris in Korean patients: a randomized, controlled trial. Acta Derm Venereol. 2012;92:241-246.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a "male" therapy for a predominantly "female" disease. J Clin Aesthet Dermatol. 2016;9:44.
- Nikolakis G, Karagiannidis I, Vaiopoulos AG, et al. Endocrinological mechanisms in the pathophysiology of hidradenitis suppurativa [in German]. Hautarzt. 2020;71:762-771.
- Karadag AS, Ozlu E, Lavery MJ. Cutaneous manifestations of diabetes mellitus and the metabolic syndrome. Clin Dermatology. 2018;36:89-93.
- Gardner CD, Kiazand A, Alhassan S, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969-977.
- Anton SD, Hida A, Heekin K, et al. Effects of popular diets without specific calorie targets on weight loss outcomes: systematic review of findings from clinical trials. Nutrients. 2017;9:822.
- Castellana M, Conte E, Cignarelli A, et al. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: a systematic review and meta-analysis. Rev Endocr Metab Disord. 2020;21:5-16.
- Paoli A, Mancin L, Giacona MC, et al. Effects of a ketogenic diet in overweight women with polycystic ovary syndrome. J Transl Med. 2020;18:104.
- Dashti HM, Mathew TC, Hussein T, et al. Long-term effects of a ketogenic diet in obese patients. Exp Clin Cardiol. 2004;9:200-205.
- Lian N, Chen M. Metabolic syndrome and skin disease: potential connection and risk. Int J Dermatol Venereol. 2019;2:89-93.
- Hu Y, Zhu Y, Lian N, et al. Metabolic syndrome and skin diseases. Front Endocrinol (Lausanne). 2019;10:788.
- Castaldo G, Rastrelli L, Galdo G, et al. Aggressive weight-loss program with a ketogenic induction phase for the treatment of chronic plaque psoriasis: a proof-of-concept, single-arm, open-label clinical trial. Nutrition. 2020;74:110757.
- Castaldo G, Pagano I, Grimaldi M, et al. Effect of very-low-calorie ketogenic diet on psoriasis patients: a nuclear magnetic resonance-based metabolomic study. J Proteome Res. 2021;20:1509-1521.
- Milder J, Liang L-P, Patel M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis. 2010;40:238-244.
- Kubo E, Chhunchha B, Singh P, et al. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci Rep. 2017;7:14130.
- Pinto A, Bonucci A, Maggi E, et al. Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in Alzheimer's disease. Antioxidants (Basel). 2018;7:63.
- Youm Y-H, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263-269.
- Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328.
- Fomin DA, McDaniel B, Crane J. The promising potential role of ketones in inflammatory dermatologic disease: a new frontier in treatment research. J Dermatol Treat. 2017;28:484-487.
- Rahman M, Muhammad S, Khan MA, et al. The β-hydroxybutyrate receptor HCA 2 activates a neuroprotective subset of macrophages. Nat Commun. 2014;5:1-11.
- Lu Y, Yang YY, Zhou MW, et al. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-κB signaling pathways. Neurosci Lett. 2018;683:13-18.
- Hamarsheh S, Zeiser R. NLRP3 inflammasome activation in cancer: a double-edged sword. Front Immunol. 2020;11:1444.
- Bell S, Degitz K, Quirling M, et al. Involvement of NF-κB signalling in skin physiology and disease. Cell Signal. 2003;15:1-7.
- Goldminz AM, Au SC, Kim N, et al. NF-κB: an essential transcription factor in psoriasis. J Dermatol Sci. 2013;69:89-94.
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589.
- McDaniel S, Rensing N, Yamada K, et al. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:E7-E11.
- Alter M, Satzger I, Schrem H, et al. Non-melanoma skin cancer is reduced after switch of immunosuppression to mTOR-inhibitors in organ transplant recipients. J Dtsch Dermatol Ges. 2014;12:480-488.
- Feldmeyer L, Hofbauer GF, Böni T, et al. Mammalian target of rapamycin (mTOR) inhibitors slow skin carcinogenesis, but impair wound healing. Br J Dermatol. 2012;166:422-424.
- Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211-218.
- Li W. "Warburg effect" and mitochondrial metabolism in skin cancer.J Carcinogene Mutagene. 2012:S4.
- Naveed S, Aslam M, Ahmad A. Starvation based differential chemotherapy: a novel approach for cancer treatment. Oman Med J. 2014;29:391-398.
- Raffaghello L, Lee C, Safdie FM, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. 2008;105:8215-8220.
- Buono R, Longo VD. Starvation, stress resistance, and cancer. Trends Endocrinol Metab. 2018;29:271-280.
- de Groot S, Pijl H, van der Hoeven JJM, et al. Effects of short-term fasting on cancer treatment. J Exp Clin Cancer Res. 2019;38:209.
- Hosseini M, Kasraian Z, Rezvani HR. Energy metabolism in skin cancers: a therapeutic perspective. Biochim Biophys Acta Bioenerg. 2017;1858:712-722.
- Feichtinger RG, Lang R, Geilberger R, et al. Melanoma tumors exhibit a variable but distinct metabolic signature. Exp Dermatol. 2018;27:204-207.
- Alshaya MA, Turkmani MG, Alissa AM. Prurigo pigmentosa following ketogenic diet and bariatric surgery: a growing association. JAAD Case Rep. 2019;5:504-507.
- Bellini M, Tonarelli S, Nagy AG, et al. Low FODMAP diet: evidence, doubts, and hopes. Nutrients. 2020;12:148.
- Kwiatkowski L, Rice E, Langland J. Integrative treatment of chronic abdominal bloating and pain associated with overgrowth of small intestinal bacteria: a case report. Altern Ther Health Med. 2017;23:56-61.
- Hubkova T. No more pain in the gut: lifestyle medicine approach to irritable bowel syndrome. Am J Lifestyle Med. 2017;11:223-226.
- Schumann D, Klose P, Lauche R, et al. Low fermentable, oligo-, di-, mono-saccharides and polyol diet in the treatment of irritable bowel syndrome: a systematic review and meta-analysis. Nutrition. 2018;45:24-31.
- Cox SR, Prince AC, Myers CE, et al. Fermentable carbohydrates [FODMAPs] exacerbate functional gastrointestinal symptoms in patients with inflammatory bowel disease: a randomised, double-blind, placebo-controlled, cross-over, re-challenge trial. J Crohns Colitis. 2017;11:1420-1429.
- Damas OM, Garces L, Abreu MT. Diet as adjunctive treatment for inflammatory bowel disease: review and update of the latest literature. Curr Treat Options Gastroenterol. 2019;17:313-325.
- Wang FY, Chi CC. Rosacea, germs, and bowels: a review on gastrointestinal comorbidities and gut-skin axis of rosacea. Adv Ther. 2021;38:1415-1424.
- Daou H, Paradiso M, Hennessy K, et al. Rosacea and the microbiome: a systematic review. Dermatol Ther (Heidelb). 2021;11:1-12.
- Weinstock LB, Steinhoff M. Rosacea and small intestinal bacterial overgrowth: prevalence and response to rifaximin. J Am Acad Dermatol. 2013;68:875-876.
- Wu CY, Chang YT, Juan CK, et al. Risk of inflammatory bowel disease in patients with rosacea: results from a nationwide cohort study in Taiwan. J Am Acad Dermatol. 2017;76:911-917.
- Egeberg A, Weinstock LB, Thyssen EP, et al. Rosacea and gastrointestinal disorders: a population-based cohort study. Br J Dermatol. 2017;176:100-106.
- Drago F, De Col E, Agnoletti AF, et al. The role of small intestinal bacterial overgrowth in rosacea: a 3-year follow-up. J Am Acad Dermatol. 2016;75:E113-E115.
- Parodi A, Paolino S, Greco A, et al. Small intestinal bacterial overgrowth in rosacea: clinical effectiveness of its eradication. Clin Gastroenterol Hepatol. 2008;6:759-764.
- Ojetti V, De Simone C, Aguilar Sanchez J, et al. Malabsorption in psoriatic patients: cause or consequence? Scand J Gastroenterol. 2006;41:1267-1271.
- Kim M, Choi KH, Hwang SW, et al. Inflammatory bowel disease is associated with an increased risk of inflammatory skin diseases: a population-based cross-sectional study. J Am Acad Dermatol. 2017;76:40-48.
- Drago F, Ciccarese G, Indemini E, et al. Psoriasis and small intestine bacterial overgrowth. Int J Dermatol. 2018;57:112-113.
- Acharya P, Mathur M. Association between psoriasis and celiac disease: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:1376-1385.
- Bhatia BK, Millsop JW, Debbaneh M, et al. Diet and psoriasis, part II: celiac disease and role of a gluten-free diet. J Am Acad Dermatol. 2014;71:350-358.
- Michaëlsson G, Gerdén B, Hagforsen E, et al. Psoriasis patients with antibodies to gliadin can be improved by a gluten-free diet. Br J Dermatol. 2000;142:44-51.
- Kolchak NA, Tetarnikova MK, Theodoropoulou MS, et al. Prevalence of antigliadin IgA antibodies in psoriasis vulgaris and response of seropositive patients to a gluten-free diet. J Multidiscip Healthc. 2018;11:13-19.
- De Bastiani R, Gabrielli M, Lora L, et al. Association between coeliac disease and psoriasis: Italian primary care multicentre study. Dermatology. 2015;230:156-160.
- Afifi L, Danesh MJ, Lee KM, et al. Dietary behaviors in psoriasis: patient-reported outcomes from a U.S. national survey. Dermatol Ther (Heidelb). 2017;7:227-242.
- Loche F, Bazex J. Celiac disease associated with cutaneous sarcoidosic granuloma [in French]. Rev Med Interne. 1997;18:975-978.
- Rodríguez-García C, González-Hernández S, Pérez-Robayna N, et al. Repigmentation of vitiligo lesions in a child with celiac disease after a gluten-free diet. Pediatr Dermatol. 2011;28:209-210.
- Wijarnpreecha K, Panjawatanan P, Corral JE, et al. Celiac disease and risk of sarcoidosis: a systematic review and meta-analysis. J Evid Based Med. 2019;12:194-199.
- Rodrigo L, Beteta-Gorriti V, Alvarez N, et al. Cutaneous and mucosal manifestations associated with celiac disease. Nutrients. 2018;10:800.
- Song MS, Farber D, Bitton A, et al. Dermatomyositis associated with celiac disease: response to a gluten-free diet. Can J Gastroenterol. 2006;20:433-435.
- Egan CA, Smith EP, Taylor TB, et al. Linear IgA bullous dermatosis responsive to a gluten-free diet. Am J Gastroenterol. 2001;96:1927-1929.
- Comas-Basté O, Sánchez-Pérez S, Veciana-Nogués MT, et al. Histamine intolerance: the current state of the art. Biomolecules. 2020;10:1181.
- Ring J. Plasma histamine concentrations in atopic eczema. Clin Allergy. 1983;13:545-552.
- Maintz L, Benfadal S, Allam JP, et al. Evidence for a reduced histamine degradation capacity in a subgroup of patients with atopic eczema. J Allergy Clin Immunol. 2006;117:1106-1112.
- Cornillier H, Giraudeau B, Samimi M, et al. Effect of diet in chronic spontaneous urticaria: a systematic review. Acta Derm Venereol. 2019;99:127-132.
- Son JH, Chung BY, Kim HO, et al. A histamine-free diet is helpful for treatment of adult patients with chronic spontaneous urticaria. Ann Dermatol. 2018;30:164-172.
- Wagner N, Dirk D, Peveling-Oberhag A, et al. A popular myth - low-histamine diet improves chronic spontaneous urticaria - fact or fiction? J Eur Acad Dermatol Venereol. 2017;31:650-655.
- Esposito K, Marfella R, Ciotola M, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA. 2004;292:1440-1446.
- Steffen LM, Van Horn L, Daviglus ML, et al. A modified Mediterranean diet score is associated with a lower risk of incident metabolic syndrome over 25 years among young adults: the CARDIA (coronary artery risk development in young adults) study. Br J Nutr. 2014;112:1654-1661.
- Bower A, Marquez S, de Mejia EG. The health benefits of selected culinary herbs and spices found in the traditional Mediterranean diet. Crit Rev Food Sci Nutr. 2016;56:2728-2746.
- Bosch R, Philips N, Suárez-Pérez JA, et al. Mechanisms of photoaging and cutaneous photocarcinogenesis, and photoprotective strategies with phytochemicals. Antioxidants (Basel). 2015;4:248-268.
- Katsimbri P, Korakas E, Kountouri A, et al. The effect of antioxidant and anti-inflammatory capacity of diet on psoriasis and psoriatic arthritis phenotype: nutrition as therapeutic tool? Antioxidants. 2021;10:157.
- Molina-Leyva A, Cuenca-Barrales C, Vega-Castillo JJ, et al. Adherence to Mediterranean diet in Spanish patients with psoriasis: cardiovascular benefits? Dermatol Ther. 2019;32:E12810.
- Barrea L, Balato N, Di Somma C, et al. Nutrition and psoriasis: is there any association between the severity of the disease and adherence to the Mediterranean diet? J Transl Med. 2015;13:1-10.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort. JAMA Dermatol. 2018;154:1017-1024.
- Korovesi A, Dalamaga M, Kotopouli M, et al. Adherence to the Mediterranean diet is independently associated with psoriasis risk, severity, and quality of life: a cross-sectional observational study. Int J Dermatol. 2019;58:E164-E165.
- Ford AR, Siegel M, Bagel J, et al. Dietary recommendations for adults with psoriasis or psoriatic arthritis from the medical board of the National Psoriasis Foundation: a systematic review. JAMA Dermatol. 2018;154:934-950.
- Skroza N, Tolino E, Semyonov L, et al. Mediterranean diet and familial dysmetabolism as factors influencing the development of acne. Scand J Public Health. 2012;40:466-474.
- Barrea L, Fabbrocini G, Annunziata G, et al. Role of nutrition and adherence to the Mediterranean diet in the multidisciplinary approach of hidradenitis suppurativa: evaluation of nutritional status and its association with severity of disease. Nutrients. 2018;11:57.
- Nichols JA, Katiyar SK. Skin photoprotection by natural polyphenols: anti-inflammatory, antioxidant and DNA repair mechanisms. Arch Dermatol Res. 2010;302:71-83.
- Huang T-H, Wang P-W, Yang S-C, et al. Cosmetic and therapeutic applications of fish oil's fatty acids on the skin. Mar Drugs. 2018;16:256.
- Rizwan M, Rodriguez-Blanco I, Harbottle A, et al. Tomato paste rich in lycopene protects against cutaneous photodamage in humans in vivo: a randomized controlled trial. Br J Dermatol. 2011;164:154-162.
- Leone A, Martínez-González M, Martin-Gorgojo A, et al. Mediterranean diet, dietary approaches to stop hypertension, and pro-vegetarian dietary pattern in relation to the risk of basal cell carcinoma: a nested case-control study within the Seguimiento Universidad de Navarra (SUN) cohort. Am J Clin Nutr. 2020;112:364-372.
- Solway J, McBride M, Haq F, et al. Diet and dermatology: the role of a whole-food, plant-based diet in preventing and reversing skin aging--a review. J Clin Aesthet Dermatol. 2020;13:38-43.
- Greger M. A whole food plant-based diet is effective for weight loss: the evidence. Am J Lifestyle Med. 2020;14:500-510.
- Wright N, Wilson L, Smith M, et al. The BROAD study: a randomised controlled trial using a whole food plant-based diet in the community for obesity, ischaemic heart disease or diabetes. Nutr Diabetes. 2017;7:E256.
- Ornish D, Lin J, Chan JM, et al. Effect of comprehensive lifestyle changes on telomerase activity and telomere length in men with biopsy-proven low-risk prostate cancer: 5-year follow-up of a descriptive pilot study. Lancet Oncol. 2013;14:1112-1120.
- Ornish D, Lin J, Daubenmier J, et al. Increased telomerase activity and comprehensive lifestyle changes: a pilot study. Lancet Oncol. 2008;9:1048-1057.
- Zouboulis CC, Makrantonaki E. Clinical aspects and molecular diagnostics of skin aging. Clin Dermatol. 2011;29:3-14.
- Gupta L, Khandelwal D, Lal PR, et al. Palaeolithic diet in diabesity and endocrinopathies--a vegan's perspective. Eur Endocrinol. 2019;15:77-82.
- Chassaing B, Van de Wiele T, De Bodt J, et al. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut. 2017;66:1414-1427.
- Thorburn Alison N, Macia L, Mackay Charles R. Diet, metabolites, and "Western lifestyle" inflammatory diseases. Immunity. 2014;40:833-842.
- Katta R, Schlichte M. Diet and dermatitis: food triggers. J Clin Aesthet Dermatol. 2014;7:30-36.
- Dhar S, Srinivas SM. Food allergy in atopic dermatitis. Indian J Dermatol. 2016;61:645-648.
- Birmingham N, Thanesvorakul S, Gangur V. Relative immunogenicity of commonly allergenic foods versus rarely allergenic and nonallergenic foods in mice. J Food Prot. 2002;65:1988-1991.
- Yu W, Freeland DMH, Nadeau KC. Food allergy: immune mechanisms, diagnosis and immunotherapy. Nat Rev Immunol. 2016;16:751-765.
- Kowalski LM, Bujko J. Evaluation of biological and clinical potential of paleolithic diet [in Polish]. Rocz Panstw Zakl Hig. 2012;63:9-15.
- Lee JE, Titcomb TJ, Bisht B, et al. A modified MCT-based ketogenic diet increases plasma β-hydroxybutyrate but has less effect on fatigue and quality of life in people with multiple sclerosis compared to a modified paleolithic diet: a waitlist-controlled, randomized pilot study. J Am Coll Nutr. 2021;40:13-25.
- Abbott RD, Sadowski A, Alt AG. Efficacy of the autoimmune protocol diet as part of a multi-disciplinary, supported lifestyle intervention for Hashimoto's thyroiditis. Cureus. 2019;11:E4556.
- Lindeberg S, Jönsson T, Granfeldt Y, et al. A palaeolithic diet improves glucose tolerance more than a Mediterranean-like diet in individuals with ischaemic heart disease. Diabetologia. 2007;50:1795-1807.
- Jönsson T, Granfeldt Y, Ahrén B, et al. Beneficial effects of a paleolithic diet on cardiovascular risk factors in type 2 diabetes: a randomized cross-over pilot study. Cardiovasc Diabetol. 2009;8:35.
- Boers I, Muskiet FAJ, Berkelaar E, et al. Favourable effects of consuming a palaeolithic-type diet on characteristics of the metabolic syndrome: a randomized controlled pilot-study. Lipids Health Dis. 2014;13:160.
- Ghaedi E, Mohammadi M, Mohammadi H, et al. Effects of a paleolithic diet on cardiovascular disease risk factors: a systematic review and meta-analysis of randomized controlled trials. Adv Nutr. 2019;10:634-646.
- Mellberg C, Sandberg S, Ryberg M, et al. Long-term effects of a palaeolithic-type diet in obese postmenopausal women: a 2-year randomized trial. Eur J Clin Nutr. 2014;68:350-357.
- Pastore RL, Brooks JT, Carbone JW. Paleolithic nutrition improves plasma lipid concentrations of hypercholesterolemic adults to a greater extent than traditional heart-healthy dietary recommendations. Nutr Res. 2015;35:474-479.
- Otten J, Stomby A, Waling M, et al. Benefits of a paleolithic diet with and without supervised exercise on fat mass, insulin sensitivity, and glycemic control: a randomized controlled trial in individuals with type 2 diabetes. Diabetes Metab Res Rev. 2017;33:E2828.
- Stefanadi EC, Dimitrakakis G, Antoniou C-K, et al. Metabolic syndrome and the skin: a more than superficial association. reviewing the association between skin diseases and metabolic syndrome and a clinical decision algorithm for high risk patients. Diabetol Metab Syndr. 2018;10:9.
- Mann N. Meat in the human diet: an anthropological perspective. Nutr Dietetics. 2007;64(suppl 4):S102-S107.
- Bramble DM, Lieberman DE. Endurance running and the evolution of Homo. Nature. 2004;432:345-352.
- Kuhn JE. Throwing, the shoulder, and human evolution. Am J Orthop (Belle Mead NJ). 2016;45:110-114.
- Kobayashi H, Kohshima S. Unique morphology of the human eye and its adaptive meaning: comparative studies on external morphology of the primate eye. J Hum Evol. 2001;40:419-435.
- Cordain L, Eaton SB, Miller JB, et al. The paradoxical nature of hunter-gatherer diets: meat-based, yet non-atherogenic. Eur J Clin Nutr. 2002;56(suppl 1):S42-S52.
- McClellan WS, Du Bois EF. Clinical calorimetry: XLV. prolonged meat diets with a study of kidney function and ketosis. J Biol Chem. 1930;87:651-668.
- O'Hearn A. Can a carnivore diet provide all essential nutrients? Curr Opin Endocrinol Diabetes Obes. 2020;27:312-316.
- O'Hearn LA. A survey of improvements experienced on a carnivore diet compared to only carbohydrate restriction. Open Science Forum website. Published February 12, 2019. Accessed May 17, 2021. doi:10.17605/OSF.IO/5FU4D
- Williams P. Nutritional composition of red meat. Nutrition & Dietetics. 2007;64(suppl 4):S113-S119.
- Biel W, Czerniawska-Piątkowska E, Kowalczyk A. Offal chemical composition from veal, beef, and lamb maintained in organic production systems. Animals (Basel). 2019;9:489.
- Elmadfa I, Meyer AL. The role of the status of selected micronutrients in shaping the immune function. Endocr Metab Immune Disord Drug Targets. 2019;19:1100-1115.
- Babizhayev M. Treatment of skin aging and photoaging with innovative oral dosage forms of nonhydrolized carnosine and carcinine. Int J Clin Derm Res. 2017;5:116-143.
- Danby FW. Nutrition and aging skin: sugar and glycation. Clin Dermatol. 2010;28:409-411.
- Siefken W, Carstensen S, Springmann G, et al. Role of taurine accumulation in keratinocyte hydration. J Invest Dermatol. 2003;121:354-361.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19:3059.
- Fischer F, Achterberg V, März A, et al. Folic acid and creatineimprove the firmness of human skin in vivo. J Cosmet Dermatol. 2011;10:15-23.
- Blatt T, Lenz H, Weber T. Topical application of creatine is multibeneficial for human skin. J Am Acad Dermatol. 2005;52:P32.
Practice Points
- Patients are increasingly interested in dietary modifications that may influence skin appearance and aid in the treatment of cutaneous disease.
- Although evidence-based dietary recommendations currently are limited, it is important for dermatologists to be aware of the varied and nuanced dietary interventions employed by patients.
- There remains a lack of randomized controlled trials assessing the efficacy of various dietary interventions in the dermatologic setting.
BEAT-LUPUS: Belimumab after rituximab delays severe flares
Using belimumab after rituximab to treat patients with systemic lupus erythematosus (SLE) refractory to conventional therapy not only significantly decreased levels of serum IgG anti-dsDNA antibody levels but also prolonged the time before severe flares of disease occurred in the phase 2b BEAT-LUPUS (Belimumab after B cell depletion in SLE) study.

The trial’s primary outcome of serum IgG anti-dsDNA antibody levels showed a decline from a geometric mean of 162 IU/mL at baseline to 69 IU/mL at 24 weeks and 47 IU/mL at 1 year in patients treated with belimumab (Benlysta) after rituximab (Rituxan and biosimilars). These reductions were significantly lower than the values seen in the placebo after rituximab arm (a respective 121 IU/mL, 99 IU/mL, and 103 IU/mL; P < .001).
Just 3 patients who received belimumab versus 10 who received placebo after rituximab experienced a severe BILAG (British Isles Lupus Assessment Group) index A flare by the end of the study at 52 weeks. The hazard ratio (HR) for the flare reduction was 0.27 (P = .03), indicating a 73% reduction.
The use of belimumab rather than a placebo also led to a small reduction in total serum IgG, and significantly suppressed B-cell repopulation (P = .03).
These results need confirming in a larger, phase 3 trial, the trial’s principal investigator, Michael Ehrenstein, PhD, said at the annual European Congress of Rheumatology. They are “clearly encouraging” and “support the hypothesis that BAFF [B-cell–activating factor] can drive flares after rituximab,” he said.
Although B-cell depletion with rituximab is recommended by national and international guidelines to treat some patients with SLE who are refractory to conventional therapy, its use is not licensed.
“Certainly, rituximab is a controversial drug in lupus,” Dr. Ehrenstein, a consultant rheumatologist based at University College London, said in an interview. Although there is real-world evidence from registries and open-label studies suggesting that it is widely used and effective in some patients, the randomized, controlled trials conducted with rituximab about 10 years ago failed to meet their primary endpoints.
“A lot has been written about why that was, but probably the biggest reason was the high dose of steroids in both groups,” Dr. Ehrenstein said. To try to avoid muddying the waters of the BEAT-LUPUS trial findings, the maximum dose of prednisolone allowed to be used as background therapy was 20 mg/day. The trial’s investigators were also encouraged to reduce the baseline steroid dose to at least 50% by the trial’s 6-month halfway point.
“We tried to reflect what was going on in the U.K.,” Dr. Ehrenstein said, noting that the inspiration for the trial was a patient who had received sequential rituximab treatment. Although she got better with each cycle of rituximab, when her disease flared it would be worse than the time before, with increasingly higher anti-dsDNA levels recorded. The reason for this seemed to be because of increasing BAFF levels, and so the hypothesis was that if rituximab was associated with increased BAFF levels, then co-targeting BAFF with belimumab should be able to prevent those flares from happening.
The BEAT-LUPUS trial has been a huge collaborative effort and was conducted across 16 U.K. centers. From initial funding to the data analysis, it has taken 6 years to complete and was made possible by a unique partnership between Versus Arthritis, University College London Hospitals Biomedical Research Center, the National Institute for Health Research UK Musculoskeletal Translational Research Collaboration, and GlaxoSmithKline (GSK). GSK provided belimumab free of charge, as well as additional funding, but had no role in the design of the study and will not have any role going forward.
From an initial 172 patients assessed for eligibility, 52 patients were finally enrolled into the trial and received rituximab as two infusions given 2 weeks apart. Patients were then randomized in a double-blind manner to receive either belimumab (n = 26) or placebo (n = 26) 4-8 weeks after their first dose of rituximab. The intention-to-treat population consisted of 43 patients.
The use of belimumab after rituximab did not increase the risk for infection – serious or otherwise – or adverse effects, Dr. Ehrenstein reported. Serious adverse events were reported in six (23%) patients in each arm, and serious infections were seen in two (8%) of the belimumab- and four (15%) of the placebo-treated patients.
“I think the take-home message is that it seems that belimumab can reduce the number of severe flares that occur after rituximab therapy,” Dr. Ehrenstein said. “It’s promising, but not definitive,” he added. The next step is of course to publish these data and to perform a phase 3 trial.

In the discussion time following the presentation, session moderator Xavier Mariette, MD, PhD, of Bicêtre Hospital, Paris-Saclay University, asked why not give belimumab first before rituximab if using belimumab afterward works?
“Our strategy was driven by the observation that BAFF levels surged after rituximab, and therefore it’s logical to give the belimumab to block that BAFF surge,” he answered.
“Certainly, there are ideas that belimumab releases mature B cells into the circulation and rituximab can target that,” he added. That strategy is under investigation in the BLISS-BELIEVE trial, which should also report by the end of this year. It’s a much larger, phase 3 trial, involving nearly 300 patients and is sponsored by GSK.
“Clearly, this is a combination treatment [but] whether you give one before the other is uncertain,” Dr. Ehrenstein observed.
Another member of the viewing audience asked whether it would have been a fairer comparison if another dose of rituximab had been given to patients at week 24 instead of no treatment. Dr. Ehrenstein noted that it was a “good point” to make, but the investigators mainly wanted to answer whether giving belimumab after rituximab would target BAFF and thereby drop serum anti-dsDNA antibody levels. He said that a full trial of rituximab for patients with SLE, perhaps adding this extra dose, needs to be conducted.
Dr. Ehrenstein disclosed receiving research funding and educational grants from GSK and participating in advisory panels for the company.
Using belimumab after rituximab to treat patients with systemic lupus erythematosus (SLE) refractory to conventional therapy not only significantly decreased levels of serum IgG anti-dsDNA antibody levels but also prolonged the time before severe flares of disease occurred in the phase 2b BEAT-LUPUS (Belimumab after B cell depletion in SLE) study.
The trial’s primary outcome of serum IgG anti-dsDNA antibody levels showed a decline from a geometric mean of 162 IU/mL at baseline to 69 IU/mL at 24 weeks and 47 IU/mL at 1 year in patients treated with belimumab (Benlysta) after rituximab (Rituxan and biosimilars). These reductions were significantly lower than the values seen in the placebo after rituximab arm (a respective 121 IU/mL, 99 IU/mL, and 103 IU/mL; P < .001).
Just 3 patients who received belimumab versus 10 who received placebo after rituximab experienced a severe BILAG (British Isles Lupus Assessment Group) index A flare by the end of the study at 52 weeks. The hazard ratio (HR) for the flare reduction was 0.27 (P = .03), indicating a 73% reduction.
The use of belimumab rather than a placebo also led to a small reduction in total serum IgG, and significantly suppressed B-cell repopulation (P = .03).
These results need confirming in a larger, phase 3 trial, the trial’s principal investigator, Michael Ehrenstein, PhD, said at the annual European Congress of Rheumatology. They are “clearly encouraging” and “support the hypothesis that BAFF [B-cell–activating factor] can drive flares after rituximab,” he said.
Although B-cell depletion with rituximab is recommended by national and international guidelines to treat some patients with SLE who are refractory to conventional therapy, its use is not licensed.
“Certainly, rituximab is a controversial drug in lupus,” Dr. Ehrenstein, a consultant rheumatologist based at University College London, said in an interview. Although there is real-world evidence from registries and open-label studies suggesting that it is widely used and effective in some patients, the randomized, controlled trials conducted with rituximab about 10 years ago failed to meet their primary endpoints.
“A lot has been written about why that was, but probably the biggest reason was the high dose of steroids in both groups,” Dr. Ehrenstein said. To try to avoid muddying the waters of the BEAT-LUPUS trial findings, the maximum dose of prednisolone allowed to be used as background therapy was 20 mg/day. The trial’s investigators were also encouraged to reduce the baseline steroid dose to at least 50% by the trial’s 6-month halfway point.
“We tried to reflect what was going on in the U.K.,” Dr. Ehrenstein said, noting that the inspiration for the trial was a patient who had received sequential rituximab treatment. Although she got better with each cycle of rituximab, when her disease flared it would be worse than the time before, with increasingly higher anti-dsDNA levels recorded. The reason for this seemed to be because of increasing BAFF levels, and so the hypothesis was that if rituximab was associated with increased BAFF levels, then co-targeting BAFF with belimumab should be able to prevent those flares from happening.
The BEAT-LUPUS trial has been a huge collaborative effort and was conducted across 16 U.K. centers. From initial funding to the data analysis, it has taken 6 years to complete and was made possible by a unique partnership between Versus Arthritis, University College London Hospitals Biomedical Research Center, the National Institute for Health Research UK Musculoskeletal Translational Research Collaboration, and GlaxoSmithKline (GSK). GSK provided belimumab free of charge, as well as additional funding, but had no role in the design of the study and will not have any role going forward.
From an initial 172 patients assessed for eligibility, 52 patients were finally enrolled into the trial and received rituximab as two infusions given 2 weeks apart. Patients were then randomized in a double-blind manner to receive either belimumab (n = 26) or placebo (n = 26) 4-8 weeks after their first dose of rituximab. The intention-to-treat population consisted of 43 patients.
The use of belimumab after rituximab did not increase the risk for infection – serious or otherwise – or adverse effects, Dr. Ehrenstein reported. Serious adverse events were reported in six (23%) patients in each arm, and serious infections were seen in two (8%) of the belimumab- and four (15%) of the placebo-treated patients.
“I think the take-home message is that it seems that belimumab can reduce the number of severe flares that occur after rituximab therapy,” Dr. Ehrenstein said. “It’s promising, but not definitive,” he added. The next step is of course to publish these data and to perform a phase 3 trial.
In the discussion time following the presentation, session moderator Xavier Mariette, MD, PhD, of Bicêtre Hospital, Paris-Saclay University, asked why not give belimumab first before rituximab if using belimumab afterward works?
“Our strategy was driven by the observation that BAFF levels surged after rituximab, and therefore it’s logical to give the belimumab to block that BAFF surge,” he answered.
“Certainly, there are ideas that belimumab releases mature B cells into the circulation and rituximab can target that,” he added. That strategy is under investigation in the BLISS-BELIEVE trial, which should also report by the end of this year. It’s a much larger, phase 3 trial, involving nearly 300 patients and is sponsored by GSK.
“Clearly, this is a combination treatment [but] whether you give one before the other is uncertain,” Dr. Ehrenstein observed.
Another member of the viewing audience asked whether it would have been a fairer comparison if another dose of rituximab had been given to patients at week 24 instead of no treatment. Dr. Ehrenstein noted that it was a “good point” to make, but the investigators mainly wanted to answer whether giving belimumab after rituximab would target BAFF and thereby drop serum anti-dsDNA antibody levels. He said that a full trial of rituximab for patients with SLE, perhaps adding this extra dose, needs to be conducted.
Dr. Ehrenstein disclosed receiving research funding and educational grants from GSK and participating in advisory panels for the company.
Using belimumab after rituximab to treat patients with systemic lupus erythematosus (SLE) refractory to conventional therapy not only significantly decreased levels of serum IgG anti-dsDNA antibody levels but also prolonged the time before severe flares of disease occurred in the phase 2b BEAT-LUPUS (Belimumab after B cell depletion in SLE) study.
The trial’s primary outcome of serum IgG anti-dsDNA antibody levels showed a decline from a geometric mean of 162 IU/mL at baseline to 69 IU/mL at 24 weeks and 47 IU/mL at 1 year in patients treated with belimumab (Benlysta) after rituximab (Rituxan and biosimilars). These reductions were significantly lower than the values seen in the placebo after rituximab arm (a respective 121 IU/mL, 99 IU/mL, and 103 IU/mL; P < .001).
Just 3 patients who received belimumab versus 10 who received placebo after rituximab experienced a severe BILAG (British Isles Lupus Assessment Group) index A flare by the end of the study at 52 weeks. The hazard ratio (HR) for the flare reduction was 0.27 (P = .03), indicating a 73% reduction.
The use of belimumab rather than a placebo also led to a small reduction in total serum IgG, and significantly suppressed B-cell repopulation (P = .03).
These results need confirming in a larger, phase 3 trial, the trial’s principal investigator, Michael Ehrenstein, PhD, said at the annual European Congress of Rheumatology. They are “clearly encouraging” and “support the hypothesis that BAFF [B-cell–activating factor] can drive flares after rituximab,” he said.
Although B-cell depletion with rituximab is recommended by national and international guidelines to treat some patients with SLE who are refractory to conventional therapy, its use is not licensed.
“Certainly, rituximab is a controversial drug in lupus,” Dr. Ehrenstein, a consultant rheumatologist based at University College London, said in an interview. Although there is real-world evidence from registries and open-label studies suggesting that it is widely used and effective in some patients, the randomized, controlled trials conducted with rituximab about 10 years ago failed to meet their primary endpoints.
“A lot has been written about why that was, but probably the biggest reason was the high dose of steroids in both groups,” Dr. Ehrenstein said. To try to avoid muddying the waters of the BEAT-LUPUS trial findings, the maximum dose of prednisolone allowed to be used as background therapy was 20 mg/day. The trial’s investigators were also encouraged to reduce the baseline steroid dose to at least 50% by the trial’s 6-month halfway point.
“We tried to reflect what was going on in the U.K.,” Dr. Ehrenstein said, noting that the inspiration for the trial was a patient who had received sequential rituximab treatment. Although she got better with each cycle of rituximab, when her disease flared it would be worse than the time before, with increasingly higher anti-dsDNA levels recorded. The reason for this seemed to be because of increasing BAFF levels, and so the hypothesis was that if rituximab was associated with increased BAFF levels, then co-targeting BAFF with belimumab should be able to prevent those flares from happening.
The BEAT-LUPUS trial has been a huge collaborative effort and was conducted across 16 U.K. centers. From initial funding to the data analysis, it has taken 6 years to complete and was made possible by a unique partnership between Versus Arthritis, University College London Hospitals Biomedical Research Center, the National Institute for Health Research UK Musculoskeletal Translational Research Collaboration, and GlaxoSmithKline (GSK). GSK provided belimumab free of charge, as well as additional funding, but had no role in the design of the study and will not have any role going forward.
From an initial 172 patients assessed for eligibility, 52 patients were finally enrolled into the trial and received rituximab as two infusions given 2 weeks apart. Patients were then randomized in a double-blind manner to receive either belimumab (n = 26) or placebo (n = 26) 4-8 weeks after their first dose of rituximab. The intention-to-treat population consisted of 43 patients.
The use of belimumab after rituximab did not increase the risk for infection – serious or otherwise – or adverse effects, Dr. Ehrenstein reported. Serious adverse events were reported in six (23%) patients in each arm, and serious infections were seen in two (8%) of the belimumab- and four (15%) of the placebo-treated patients.
“I think the take-home message is that it seems that belimumab can reduce the number of severe flares that occur after rituximab therapy,” Dr. Ehrenstein said. “It’s promising, but not definitive,” he added. The next step is of course to publish these data and to perform a phase 3 trial.
In the discussion time following the presentation, session moderator Xavier Mariette, MD, PhD, of Bicêtre Hospital, Paris-Saclay University, asked why not give belimumab first before rituximab if using belimumab afterward works?
“Our strategy was driven by the observation that BAFF levels surged after rituximab, and therefore it’s logical to give the belimumab to block that BAFF surge,” he answered.
“Certainly, there are ideas that belimumab releases mature B cells into the circulation and rituximab can target that,” he added. That strategy is under investigation in the BLISS-BELIEVE trial, which should also report by the end of this year. It’s a much larger, phase 3 trial, involving nearly 300 patients and is sponsored by GSK.
“Clearly, this is a combination treatment [but] whether you give one before the other is uncertain,” Dr. Ehrenstein observed.
Another member of the viewing audience asked whether it would have been a fairer comparison if another dose of rituximab had been given to patients at week 24 instead of no treatment. Dr. Ehrenstein noted that it was a “good point” to make, but the investigators mainly wanted to answer whether giving belimumab after rituximab would target BAFF and thereby drop serum anti-dsDNA antibody levels. He said that a full trial of rituximab for patients with SLE, perhaps adding this extra dose, needs to be conducted.
Dr. Ehrenstein disclosed receiving research funding and educational grants from GSK and participating in advisory panels for the company.
FROM THE EULAR 2021 CONGRESS
Intravenous immunoglobulin controls dermatomyositis in phase 3 trial
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
Nearly 50% achieve moderate improvement or better
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS
Ulcerative Heliotrope Rash in Antimelanoma Differentiation–Associated Gene 5 Dermatomyositis
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
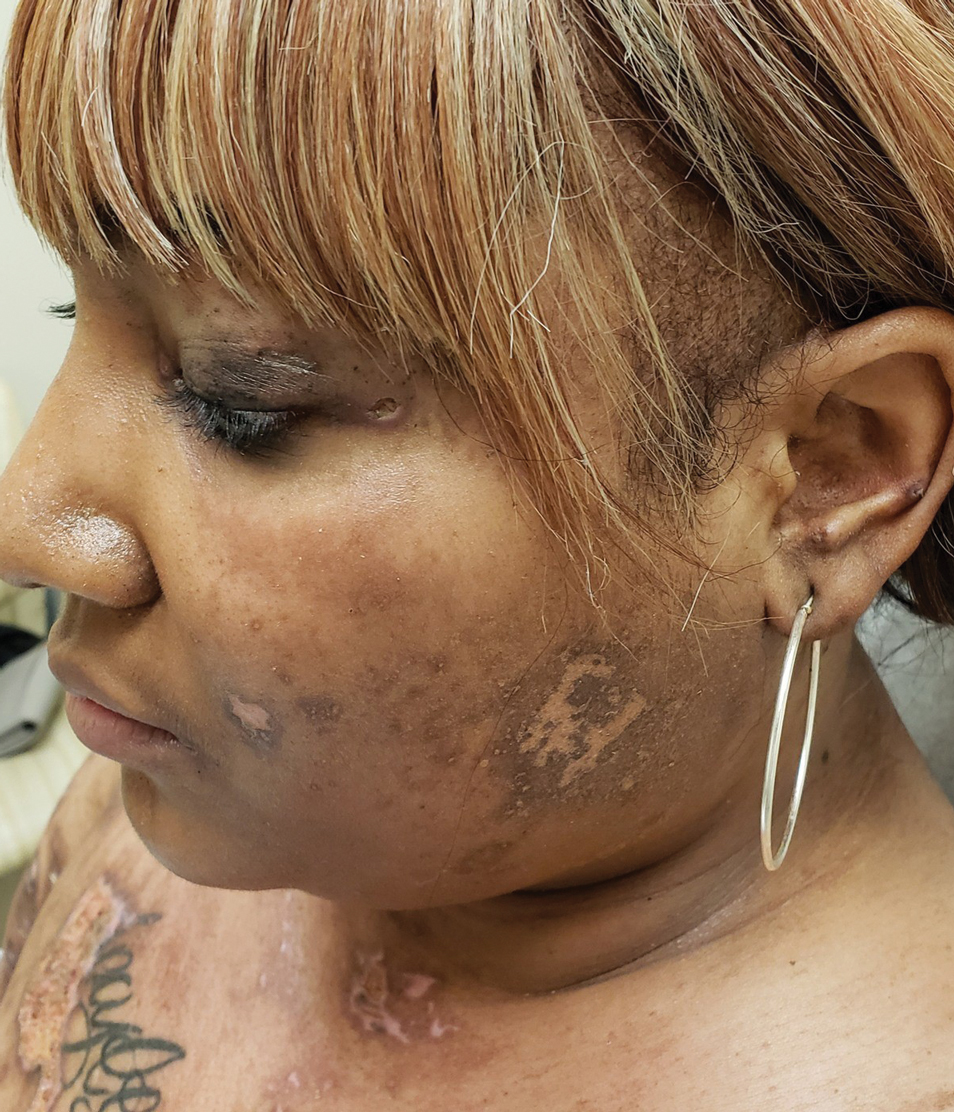
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
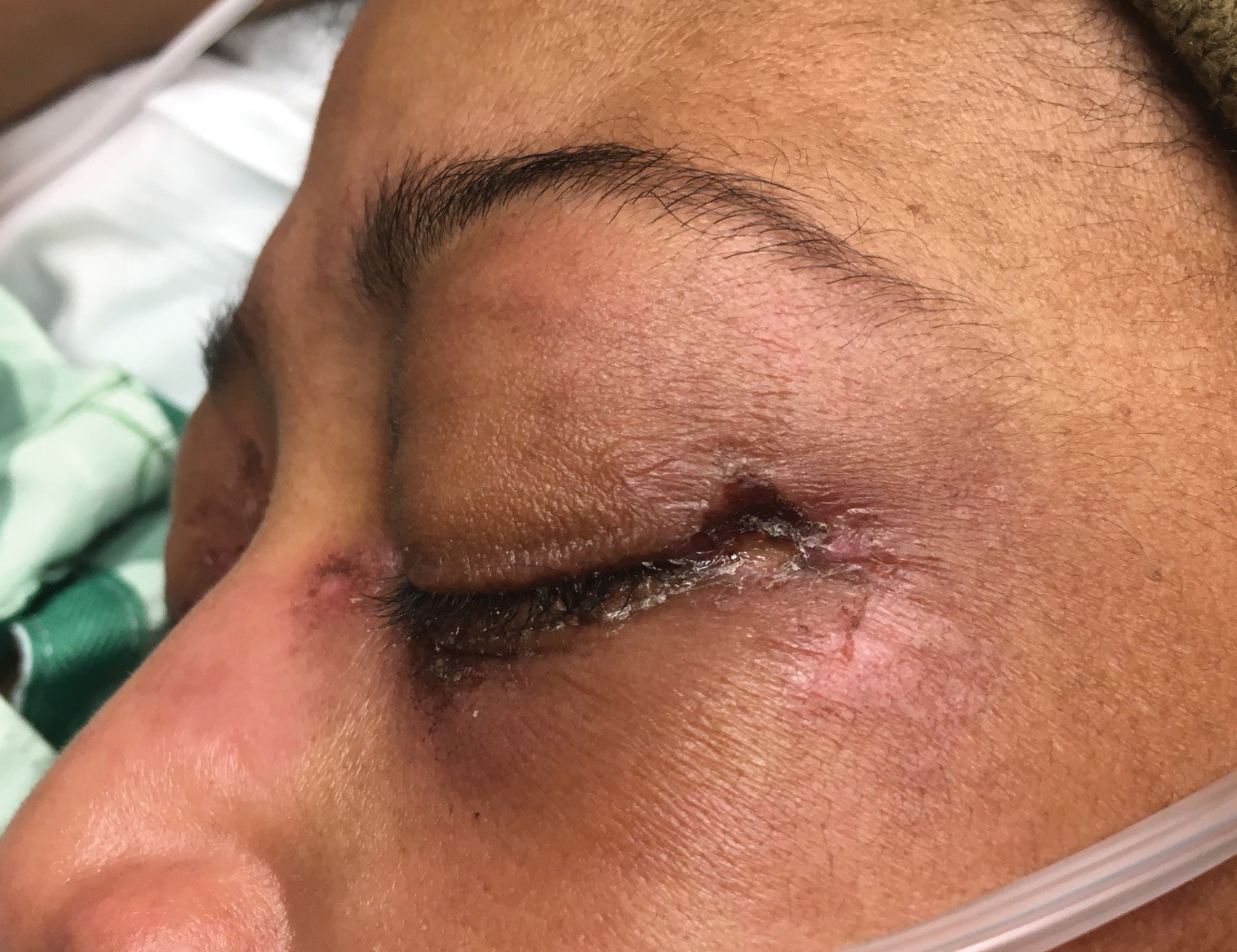
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
Practice Points
- Antimelanoma differentiation–associated gene 5 dermatomyositis (anti-MDA5 DM) can present with an ulcerative heliotrope rash.
- Ulceration of the heliotrope rash in anti-MDA5 DM may indicate disease progression.
- Rapidly progressive interstitial lung disease is highly associated with anti-MDA5 DM.
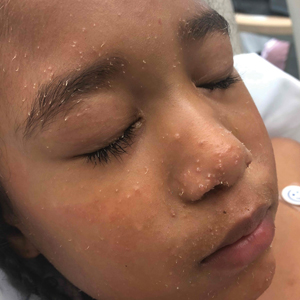
Psoriatic Alopecia in a Patient With Crohn Disease: An Uncommon Manifestation of Tumor Necrosis Factor α Inhibitors
Tumor necrosis factor α (TNF-α) inhibitor–induced psoriasis is a known paradoxical adverse effect of this family of medications, which includes infliximab, adalimumab, etanercept, golimumab, and certolizumab. In the pediatric population, these therapies recently gained approval for nondermatologic conditions—meaning that this phenomenon is encountered more frequently.1 In a systematic review of TNF-α inhibitor–induced psoriasis, severe scalp involvement was associated with alopecia in 7.5% of cases.2 Onset of scalp psoriasis with alopecia in patients being treated with a TNF-α inhibitor should lead to consideration of this condition.
Psoriatic alopecia is an uncommon presentation of psoriasis. Although well described, alopecia as a clinical manifestation of scalp psoriasis is not a well-known concept among clinicians and has never been widely accepted. Adding to the diagnostic challenge is that psoriatic alopecia secondary to TNF-α inhibitor–induced psoriasis rarely has been reported in adults or children.3-5 Including our case, our review of the literature yielded 7 pediatric cases (≤18 years) of TNF-α inhibitor–induced psoriatic alopecia.6,7 A primary literature search of PubMed articles indexed for MEDLINE was conducted using the terms psoriatic alopecia, psoriasiform alopecia, TNF-α inhibitors, infliximab, adalimumab, etanercept, golimumab, and certolizumab.
We present the case of a pediatric patient with psoriatic alopecia secondary to treatment with adalimumab for Crohn disease (CD). We also provide a review of reported cases of psoriatic alopecia induced by a TNF-α inhibitor in the literature.
Case Report
A 12-year-old girl presented to our dermatology clinic with erythematous scaly plaques on the trunk, scalp, arms, and legs of 2 months’ duration. The lesions involved approximately 15% of the body surface area. The patient’s medical history was remarkable for CD diagnosed 4 years prior to presentation of the skin lesions. She had been treated for the past 2 years with adalimumab 40 mg once every 2 weeks and azathioprine 100 mg once daily. Because her CD was poorly controlled, the dosage of adalimumab was increased to 40 mg once weekly 6 months prior to the current presentation.
Our diagnosis was TNF-α inhibitor-induced psoriasis secondary to treatment with adalimumab.
The patient was treated with mometasone lotion 0.1% for the scalp lesions and triamcinolone cream 0.1% for the body lesions. Because of the extent of the psoriasis, we recommended changing adalimumab to ustekinumab, which is approved for CD in adults but is off label in children.
At 1-month follow-up, after receiving the induction dose of ustekinumab, the patient presented with partial improvement of the skin lesions but had developed a large, alopecic, erythematous plaque with thick yellowish scales on the scalp (Figure 1). She also had a positive hair pull test. The presumptive initial diagnosis of the alopecic scalp lesion was tinea capitis, for which multiple potassium hydroxide preparations of scales were performed, all yielding negative results. In addition, histopathologic examination with hematoxylin and eosin staining was performed (Figures 2A and 2B). Sterile tissue cultures for bacteria, fungi, and acid-fast bacilli were obtained and showed no growth. Periodic acid–Schiff staining was negative for fungal structures.
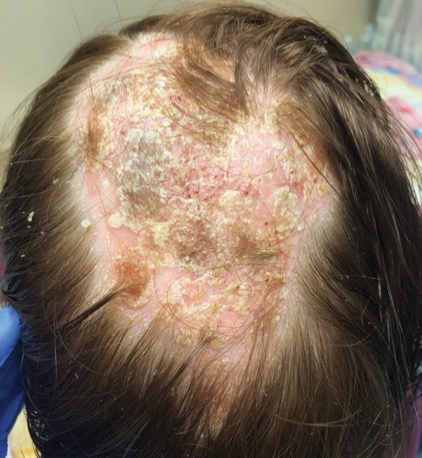
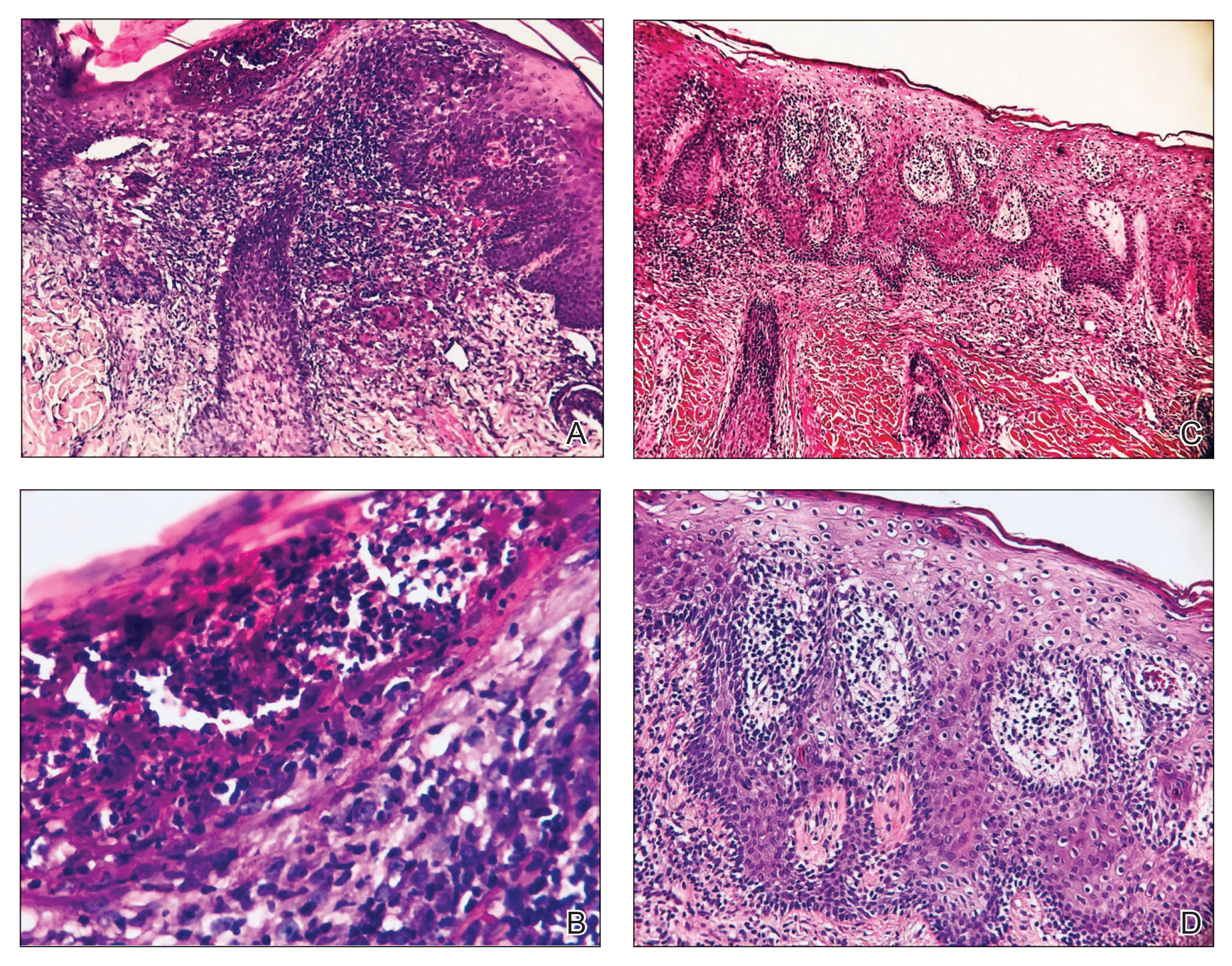
A second biopsy showed a psoriasiform pattern, parakeratosis, and hypogranulosis, highly suggestive of psoriasis (Figure 2C and 2D). Based on those findings, a diagnosis of psoriatic alopecia was made. The mometasone was switched to clobetasol lotion 0.05%. The patient continued treatment with ustekinumab. At 6-month follow-up, her CD was well controlled and she showed hair regrowth in previously alopecic areas (Figure 3).

Comment
Psoriatic alopecia induced by a TNF-α inhibitor was first reported in 2007 in a 30-year-old woman with ankylosing spondylitis who was being treated with adalimumab.8 She had erythematous, scaly, alopecic plaques on the scalp and palmoplantar pustulosis. Findings on skin biopsy were compatible with psoriasis. The patient’s severe scalp psoriasis failed to respond to topical steroid treatment and adalimumab cessation. The extensive hair loss responded to cyclosporine 3 mg/kg daily.8
After conducting an extensive literature review, we found 26 cases of TNF-α–induced psoriatic alopecia, including the current case (Table).6-16 The mean age at diagnosis was 27.8 years (SD, 13.6 years; range, 7–60 years). The female-to-male ratio was 3.3:1. The most common underlying condition for which TNF-α inhibitors were prescribed was CD (77% [20/26]). Psoriatic alopecia most commonly was reported secondary to treatment with infliximab (54% [14/26]), followed by adalimumab (42% [11/26]). Golimumab was the causative drug in 1 (4%) case. We did not find reports of etanercept or certolizumab having induced this manifestation. The onset of the scalp lesions occurred 2 to 46 months after starting treatment with the causative medication.
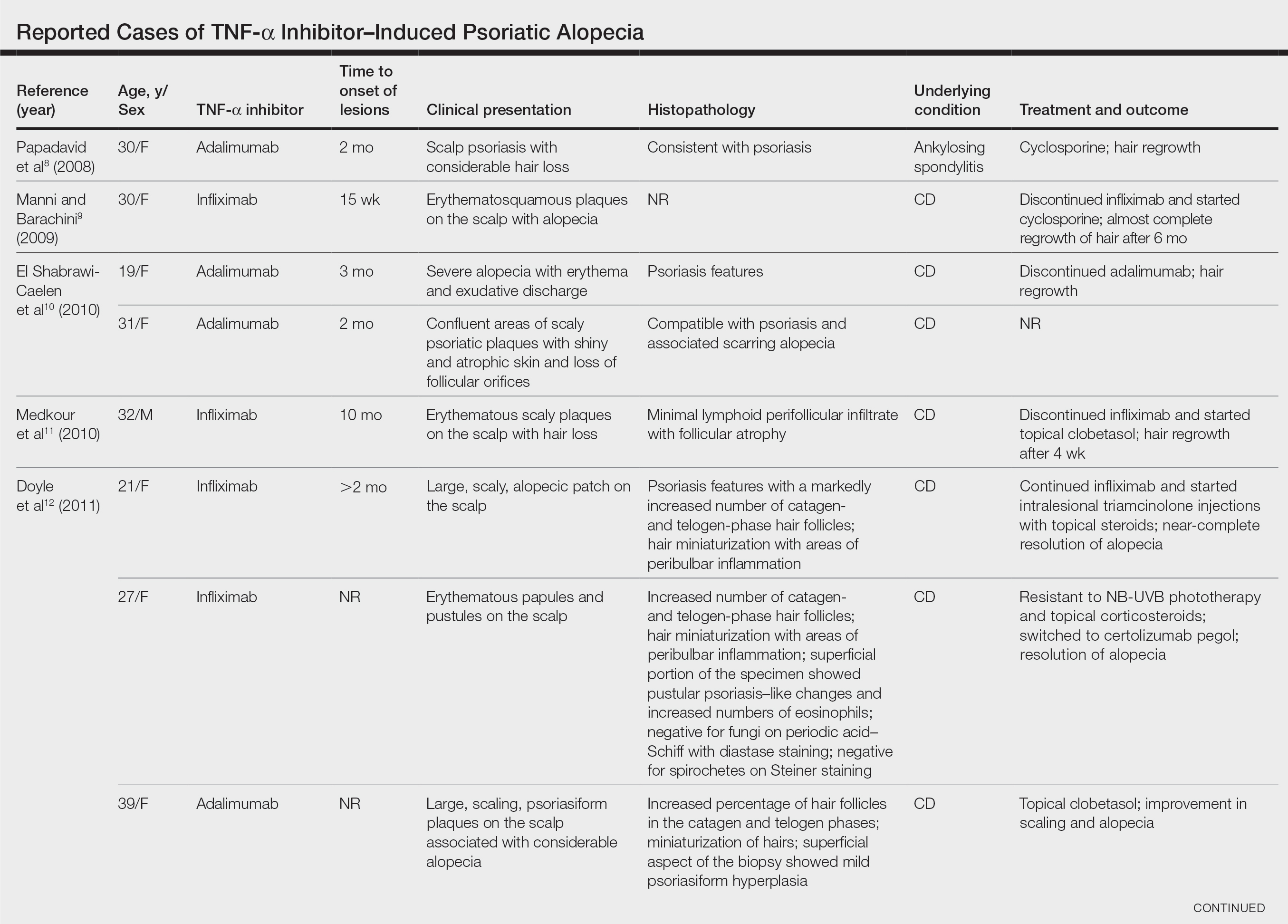
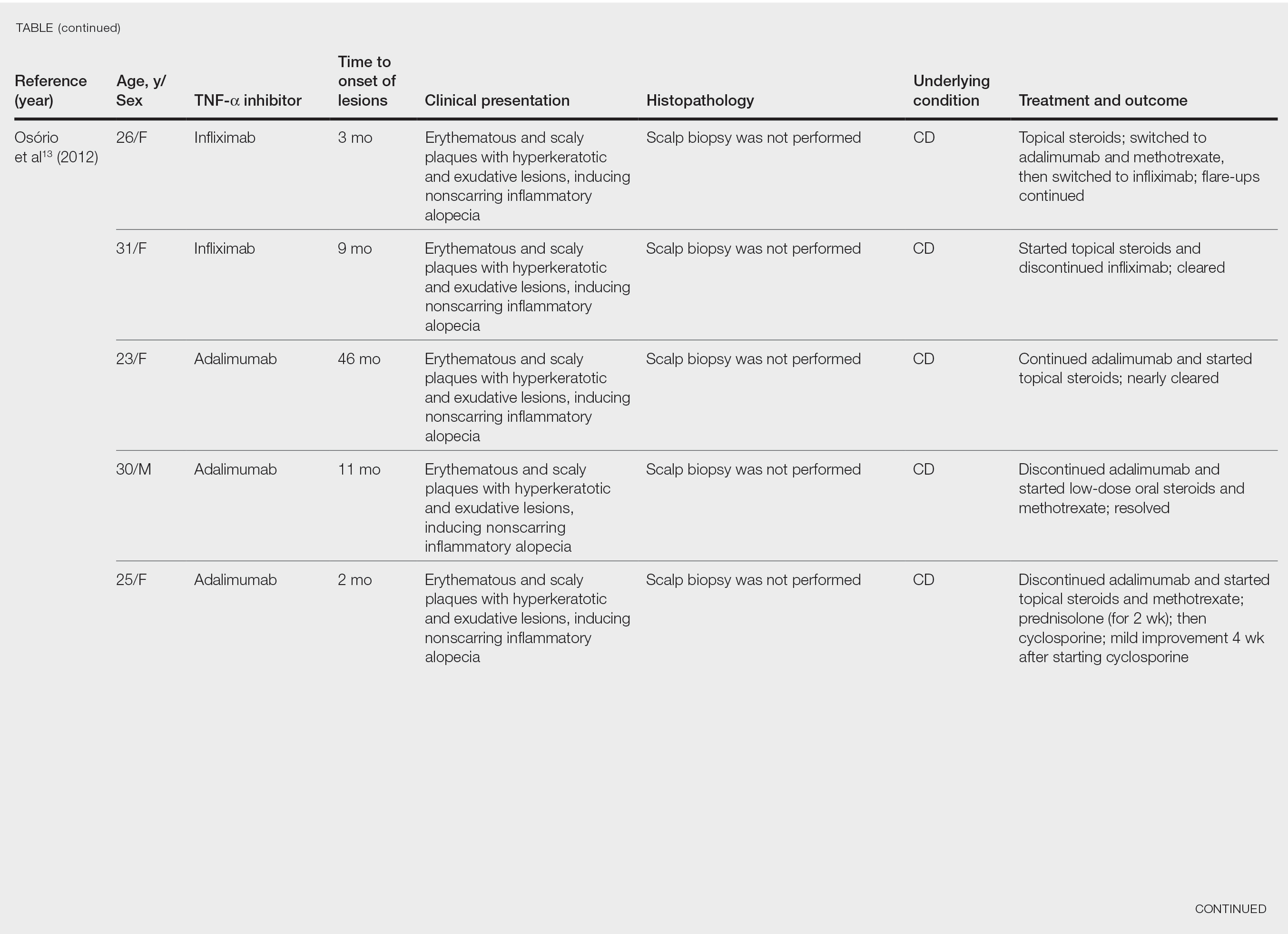
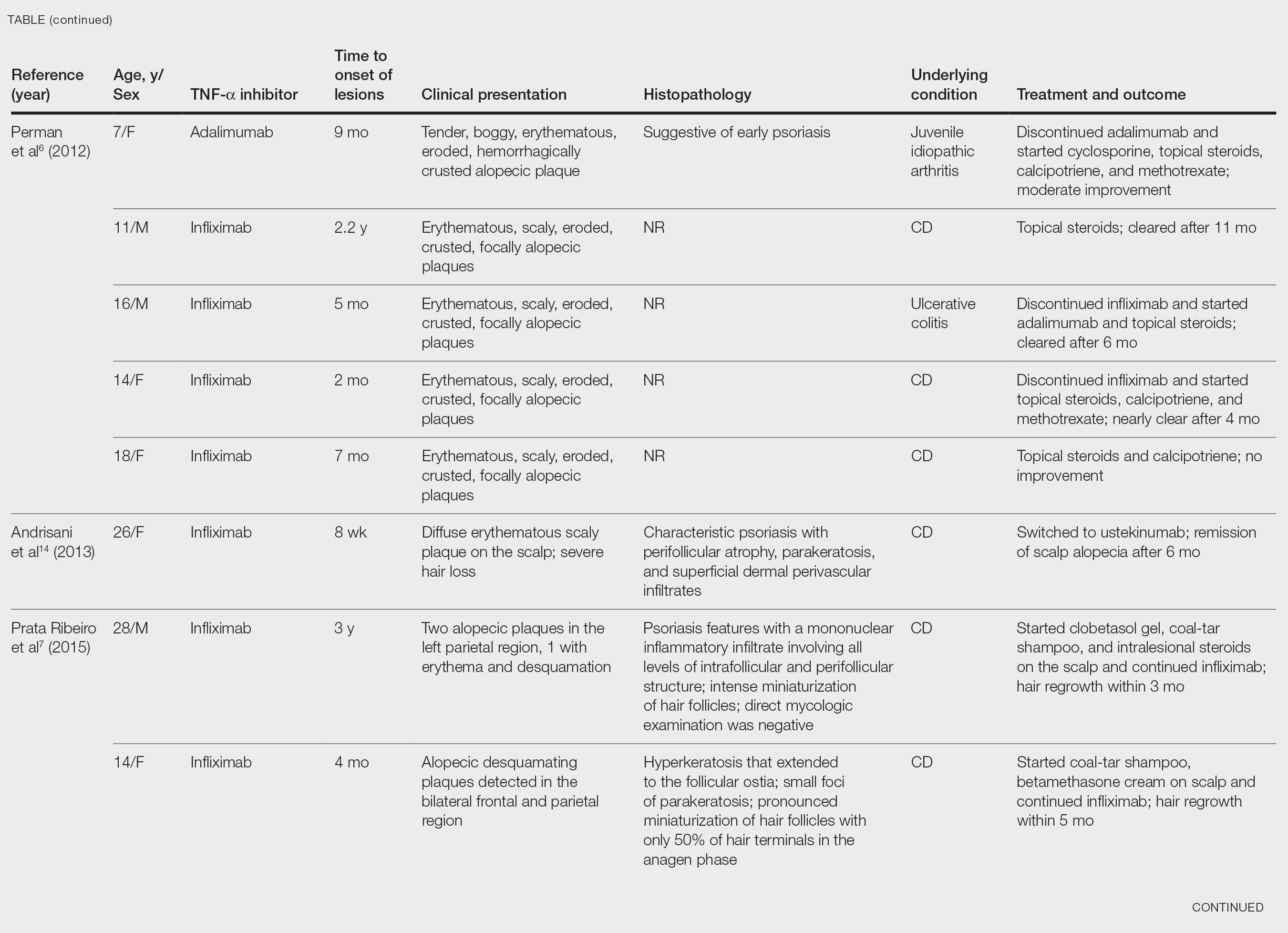
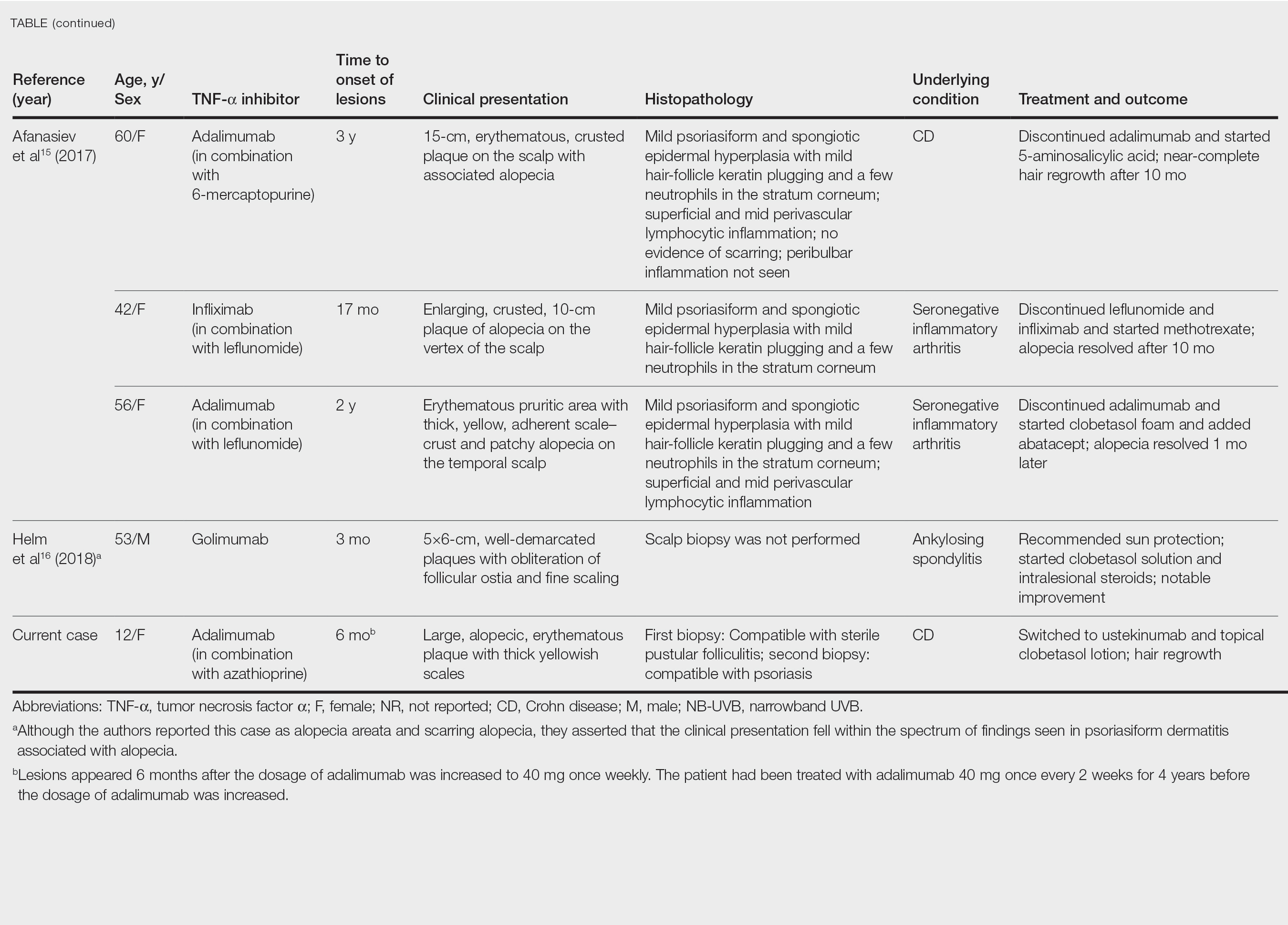
Laga et al17 reported that TNF-α inhibitor–induced psoriasis can have a variety of histopathologic findings, including typical findings of various stages of psoriasis, a lichenoid pattern mimicking remnants of lichen planus, and sterile pustular folliculitis. Our patient’s 2 scalp biopsies demonstrated results consistent with findings reported by Laga et al.17 In the first biopsy, findings were consistent with a dense neutrophilic infiltrate with negative sterile cultures and negative periodic acid–Schiff stain (sterile folliculitis), with crust and areas of parakeratosis. The second biopsy demonstrated psoriasiform hyperplasia, parakeratosis, and an absent granular layer, all typical features of psoriasis (Figure 2).
Including the current case, our review of the literature yielded 7 pediatric (ie, 0–18 years of age) cases of TNF-α inhibitor–induced psoriatic alopecia. Of the 6 previously reported pediatric cases, 5 occurred after administration of infliximab.6,7
Similar to our case, TNF-α inhibitor–induced psoriatic alopecia was reported in a 7-year-old girl who was treated with adalimumab for juvenile idiopathic arthritis.6 Nine months after starting treatment, that patient presented with a tender, erythematous, eroded, and crusted alopecic plaque along with scaly plaques on the scalp. Adalimumab was discontinued, and cyclosporine and topical steroids were started. Cyclosporine was then discontinued due to partial resolution of the psoriasis; the patient was started on abatacept, with persistence of the psoriasis and alopecia. The patient was then started on oral methotrexate 12.5 mg once weekly with moderate improvement and mild to moderate exacerbations.
Tumor necrosis factor α inhibitor–induced psoriasis may occur as a result of a cytokine imbalance. A TNF-α blockade leads to upregulation of interferon α (IFN-α) and TNF-α production by plasmacytoid dendritic cells (pDCs), usually in genetically susceptible people.6,7,9-15 The IFN-α induces maturation of myeloid dendritic cells (mDCs) responsible for increasing proinflammatory cytokines that contribute to psoriasis.11 Generation of TNF-α by pDCs leads to mature or activated dendritic cells derived from pDCs through autocrine TNF-α production and paracrine IFN-α production from immature mDCs.9 Once pDCs mature, they are incapable of producing IFN-α; TNF-α then inhibits IFN-α production by inducing pDC maturation.11 Overproduction of IFN-α during TNF-α inhibition induces expression of the chemokine receptor CXCR3 on T cells, which recruits T cells to the dermis. The T cells then produce TNF-α, causing psoriatic skin lesions.10,11,13,14
Although TNF-α inhibitor–induced psoriatic alopecia is uncommon, the condition should be considered in female patients with underlying proinflammatory disease—CD in particular. Perman et al6 reported 5 cases of psoriatic alopecia in which 3 patients initially were treated with griseofulvin because of suspected tinea capitis.
Conditions with similar clinical findings should be ruled out before making a diagnosis of TNF-α inhibitor–induced psoriatic alopecia. Although clinicopathologic correlation is essential for making the diagnosis, it is possible that the histologic findings will not be specific for psoriasis.17 It is important to be aware of this condition in patients being treated with a TNF-α inhibitor as early as 2 months to 4 years or longer after starting treatment.
Previously reported cases have demonstrated various treatment options that yielded improvement or resolution of TNF-α inhibitor–induced psoriatic alopecia. These include either continuation or discontinuation of the TNF-α inhibitor combined with topical or intralesional steroids, methotrexate, or cyclosporine. Another option is to switch the TNF-α inhibitor to another biologic. Outcomes vary from patient to patient, making the physician’s clinical judgment crucial in deciding which treatment route to take. Our patient showed notable improvement when she was switched from adalimumab to ustekinumab as well as the combination of ustekinumab and clobetasol lotion 0.05%.
Conclusion
We recommend an individualized approach that provides patients with the safest and least invasive treatment option for TNF-α inhibitor–induced psoriatic alopecia. In most reported cases, the problem resolved with treatment, thereby classifying this form of alopecia as noncicatricial alopecia.
- Horneff G, Seyger MMB, Arikan D, et al. Safety of adalimumab in pediatric patients with polyarticular juvenile idiopathic arthritis, enthesitis-related arthritis, psoriasis, and Crohn’s disease. J Pediatr. 2018;201:166-175.e3. doi:10.1016/j.jpeds.2018.05.042
- Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
- George SMC, Taylor MR, Farrant PBJ. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77. doi:10.1111/j.1365-2133.1972.tb05103.x
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? a clinicohistopathologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Perman MJ, Lovell DJ, Denson LA, et al. Five cases of anti-tumor necrosis factor alpha-induced psoriasis presenting with severe scalp involvement in children. Pediatr Dermatol. 2012;29:454-459. doi:10.1111/j.1525-1470.2011.01521.x
- Prata Ribeiro LB, Gonçalves Rego JC, Duque Estrada B, et al. Alopecia secondary to anti-tumor necrosis factor-alpha therapy. An Bras Dermatol. 2015;90:232–235. doi:10.1590/abd1806-4841.20153084
- Papadavid E, Gazi S, Dalamaga M, et al. Palmoplantar and scalp psoriasis occurring during anti-tumour necrosis factor-alpha therapy: a case series of four patients and guidelines for management. J Eur Acad Dermatol Venereol. 2008;22:380-382. doi:10.1111/j.1468-3083.2007.02335.x
- Manni E, Barachini P. Psoriasis induced by infliximab in a patient suffering from Crohn’s disease. Int J Immunopathol Pharmacol. 2009;22:841-844. doi:10.1177/039463200902200331
- El Shabrawi-Caelen L, La Placa M, Vincenzi C, et al. Adalimumab-induced psoriasis of the scalp with diffuse alopecia: a severe potentially irreversible cutaneous side effect of TNF-alpha blockers. Inflamm Bowel Dis. 2010;16:182-183. doi:10.1002/ibd.20954
- Medkour F, Babai S, Chanteloup E, et al. Development of diffuse psoriasis with alopecia during treatment of Crohn’s disease with infliximab. Gastroenterol Clin Biol. 2010;34:140-141. doi:10.1016/j.gcb.2009.10.021
- Doyle LA, Sperling LC, Baksh S, et al. Psoriatic alopecia/alopecia areata-like reactions secondary to anti-tumor necrosis factor-α therapy: a novel cause of noncicatricial alopecia. Am J Dermatopathol. 2011;33:161-166. doi:10.1097/DAD.0b013e3181ef7403
- Osório F, Magro F, Lisboa C, et al. Anti-TNF-alpha induced psoriasiform eruptions with severe scalp involvement and alopecia: report of five cases and review of the literature. Dermatology. 2012;225:163-167. doi:10.1159/000342503
- Andrisani G, Marzo M, Celleno L, et al. Development of psoriasis scalp with alopecia during treatment of Crohn’s disease with infliximab and rapid response to both diseases to ustekinumab. Eur Rev Med Pharmacol Sci. 2013;17:2831-2836.
- Afanasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-569. doi:10.1111/cup.12932
- Helm MM, Haddad S. Alopecia areata and scarring alopecia presenting during golimumab therapy for ankylosing spondylitis. N Am J Med Sci. 2018;11:22-24. doi:10.7156/najms.2018.110122
- Laga AC, Vleugels RA, Qureshi AA, et al. Histopathologic spectrum of psoriasiform skin reactions associated with tumor necrosis factor-a inhibitor therapy. a study of 16 biopsies. Am J Dermatopathol. 2010;32:568-573. doi:10.1097/DAD.0b013e3181cb3ff7
Tumor necrosis factor α (TNF-α) inhibitor–induced psoriasis is a known paradoxical adverse effect of this family of medications, which includes infliximab, adalimumab, etanercept, golimumab, and certolizumab. In the pediatric population, these therapies recently gained approval for nondermatologic conditions—meaning that this phenomenon is encountered more frequently.1 In a systematic review of TNF-α inhibitor–induced psoriasis, severe scalp involvement was associated with alopecia in 7.5% of cases.2 Onset of scalp psoriasis with alopecia in patients being treated with a TNF-α inhibitor should lead to consideration of this condition.
Psoriatic alopecia is an uncommon presentation of psoriasis. Although well described, alopecia as a clinical manifestation of scalp psoriasis is not a well-known concept among clinicians and has never been widely accepted. Adding to the diagnostic challenge is that psoriatic alopecia secondary to TNF-α inhibitor–induced psoriasis rarely has been reported in adults or children.3-5 Including our case, our review of the literature yielded 7 pediatric cases (≤18 years) of TNF-α inhibitor–induced psoriatic alopecia.6,7 A primary literature search of PubMed articles indexed for MEDLINE was conducted using the terms psoriatic alopecia, psoriasiform alopecia, TNF-α inhibitors, infliximab, adalimumab, etanercept, golimumab, and certolizumab.
We present the case of a pediatric patient with psoriatic alopecia secondary to treatment with adalimumab for Crohn disease (CD). We also provide a review of reported cases of psoriatic alopecia induced by a TNF-α inhibitor in the literature.
Case Report
A 12-year-old girl presented to our dermatology clinic with erythematous scaly plaques on the trunk, scalp, arms, and legs of 2 months’ duration. The lesions involved approximately 15% of the body surface area. The patient’s medical history was remarkable for CD diagnosed 4 years prior to presentation of the skin lesions. She had been treated for the past 2 years with adalimumab 40 mg once every 2 weeks and azathioprine 100 mg once daily. Because her CD was poorly controlled, the dosage of adalimumab was increased to 40 mg once weekly 6 months prior to the current presentation.
Our diagnosis was TNF-α inhibitor-induced psoriasis secondary to treatment with adalimumab.
The patient was treated with mometasone lotion 0.1% for the scalp lesions and triamcinolone cream 0.1% for the body lesions. Because of the extent of the psoriasis, we recommended changing adalimumab to ustekinumab, which is approved for CD in adults but is off label in children.
At 1-month follow-up, after receiving the induction dose of ustekinumab, the patient presented with partial improvement of the skin lesions but had developed a large, alopecic, erythematous plaque with thick yellowish scales on the scalp (Figure 1). She also had a positive hair pull test. The presumptive initial diagnosis of the alopecic scalp lesion was tinea capitis, for which multiple potassium hydroxide preparations of scales were performed, all yielding negative results. In addition, histopathologic examination with hematoxylin and eosin staining was performed (Figures 2A and 2B). Sterile tissue cultures for bacteria, fungi, and acid-fast bacilli were obtained and showed no growth. Periodic acid–Schiff staining was negative for fungal structures.
A second biopsy showed a psoriasiform pattern, parakeratosis, and hypogranulosis, highly suggestive of psoriasis (Figure 2C and 2D). Based on those findings, a diagnosis of psoriatic alopecia was made. The mometasone was switched to clobetasol lotion 0.05%. The patient continued treatment with ustekinumab. At 6-month follow-up, her CD was well controlled and she showed hair regrowth in previously alopecic areas (Figure 3).
Comment
Psoriatic alopecia induced by a TNF-α inhibitor was first reported in 2007 in a 30-year-old woman with ankylosing spondylitis who was being treated with adalimumab.8 She had erythematous, scaly, alopecic plaques on the scalp and palmoplantar pustulosis. Findings on skin biopsy were compatible with psoriasis. The patient’s severe scalp psoriasis failed to respond to topical steroid treatment and adalimumab cessation. The extensive hair loss responded to cyclosporine 3 mg/kg daily.8
After conducting an extensive literature review, we found 26 cases of TNF-α–induced psoriatic alopecia, including the current case (Table).6-16 The mean age at diagnosis was 27.8 years (SD, 13.6 years; range, 7–60 years). The female-to-male ratio was 3.3:1. The most common underlying condition for which TNF-α inhibitors were prescribed was CD (77% [20/26]). Psoriatic alopecia most commonly was reported secondary to treatment with infliximab (54% [14/26]), followed by adalimumab (42% [11/26]). Golimumab was the causative drug in 1 (4%) case. We did not find reports of etanercept or certolizumab having induced this manifestation. The onset of the scalp lesions occurred 2 to 46 months after starting treatment with the causative medication.
Laga et al17 reported that TNF-α inhibitor–induced psoriasis can have a variety of histopathologic findings, including typical findings of various stages of psoriasis, a lichenoid pattern mimicking remnants of lichen planus, and sterile pustular folliculitis. Our patient’s 2 scalp biopsies demonstrated results consistent with findings reported by Laga et al.17 In the first biopsy, findings were consistent with a dense neutrophilic infiltrate with negative sterile cultures and negative periodic acid–Schiff stain (sterile folliculitis), with crust and areas of parakeratosis. The second biopsy demonstrated psoriasiform hyperplasia, parakeratosis, and an absent granular layer, all typical features of psoriasis (Figure 2).
Including the current case, our review of the literature yielded 7 pediatric (ie, 0–18 years of age) cases of TNF-α inhibitor–induced psoriatic alopecia. Of the 6 previously reported pediatric cases, 5 occurred after administration of infliximab.6,7
Similar to our case, TNF-α inhibitor–induced psoriatic alopecia was reported in a 7-year-old girl who was treated with adalimumab for juvenile idiopathic arthritis.6 Nine months after starting treatment, that patient presented with a tender, erythematous, eroded, and crusted alopecic plaque along with scaly plaques on the scalp. Adalimumab was discontinued, and cyclosporine and topical steroids were started. Cyclosporine was then discontinued due to partial resolution of the psoriasis; the patient was started on abatacept, with persistence of the psoriasis and alopecia. The patient was then started on oral methotrexate 12.5 mg once weekly with moderate improvement and mild to moderate exacerbations.
Tumor necrosis factor α inhibitor–induced psoriasis may occur as a result of a cytokine imbalance. A TNF-α blockade leads to upregulation of interferon α (IFN-α) and TNF-α production by plasmacytoid dendritic cells (pDCs), usually in genetically susceptible people.6,7,9-15 The IFN-α induces maturation of myeloid dendritic cells (mDCs) responsible for increasing proinflammatory cytokines that contribute to psoriasis.11 Generation of TNF-α by pDCs leads to mature or activated dendritic cells derived from pDCs through autocrine TNF-α production and paracrine IFN-α production from immature mDCs.9 Once pDCs mature, they are incapable of producing IFN-α; TNF-α then inhibits IFN-α production by inducing pDC maturation.11 Overproduction of IFN-α during TNF-α inhibition induces expression of the chemokine receptor CXCR3 on T cells, which recruits T cells to the dermis. The T cells then produce TNF-α, causing psoriatic skin lesions.10,11,13,14
Although TNF-α inhibitor–induced psoriatic alopecia is uncommon, the condition should be considered in female patients with underlying proinflammatory disease—CD in particular. Perman et al6 reported 5 cases of psoriatic alopecia in which 3 patients initially were treated with griseofulvin because of suspected tinea capitis.
Conditions with similar clinical findings should be ruled out before making a diagnosis of TNF-α inhibitor–induced psoriatic alopecia. Although clinicopathologic correlation is essential for making the diagnosis, it is possible that the histologic findings will not be specific for psoriasis.17 It is important to be aware of this condition in patients being treated with a TNF-α inhibitor as early as 2 months to 4 years or longer after starting treatment.
Previously reported cases have demonstrated various treatment options that yielded improvement or resolution of TNF-α inhibitor–induced psoriatic alopecia. These include either continuation or discontinuation of the TNF-α inhibitor combined with topical or intralesional steroids, methotrexate, or cyclosporine. Another option is to switch the TNF-α inhibitor to another biologic. Outcomes vary from patient to patient, making the physician’s clinical judgment crucial in deciding which treatment route to take. Our patient showed notable improvement when she was switched from adalimumab to ustekinumab as well as the combination of ustekinumab and clobetasol lotion 0.05%.
Conclusion
We recommend an individualized approach that provides patients with the safest and least invasive treatment option for TNF-α inhibitor–induced psoriatic alopecia. In most reported cases, the problem resolved with treatment, thereby classifying this form of alopecia as noncicatricial alopecia.
Tumor necrosis factor α (TNF-α) inhibitor–induced psoriasis is a known paradoxical adverse effect of this family of medications, which includes infliximab, adalimumab, etanercept, golimumab, and certolizumab. In the pediatric population, these therapies recently gained approval for nondermatologic conditions—meaning that this phenomenon is encountered more frequently.1 In a systematic review of TNF-α inhibitor–induced psoriasis, severe scalp involvement was associated with alopecia in 7.5% of cases.2 Onset of scalp psoriasis with alopecia in patients being treated with a TNF-α inhibitor should lead to consideration of this condition.
Psoriatic alopecia is an uncommon presentation of psoriasis. Although well described, alopecia as a clinical manifestation of scalp psoriasis is not a well-known concept among clinicians and has never been widely accepted. Adding to the diagnostic challenge is that psoriatic alopecia secondary to TNF-α inhibitor–induced psoriasis rarely has been reported in adults or children.3-5 Including our case, our review of the literature yielded 7 pediatric cases (≤18 years) of TNF-α inhibitor–induced psoriatic alopecia.6,7 A primary literature search of PubMed articles indexed for MEDLINE was conducted using the terms psoriatic alopecia, psoriasiform alopecia, TNF-α inhibitors, infliximab, adalimumab, etanercept, golimumab, and certolizumab.
We present the case of a pediatric patient with psoriatic alopecia secondary to treatment with adalimumab for Crohn disease (CD). We also provide a review of reported cases of psoriatic alopecia induced by a TNF-α inhibitor in the literature.
Case Report
A 12-year-old girl presented to our dermatology clinic with erythematous scaly plaques on the trunk, scalp, arms, and legs of 2 months’ duration. The lesions involved approximately 15% of the body surface area. The patient’s medical history was remarkable for CD diagnosed 4 years prior to presentation of the skin lesions. She had been treated for the past 2 years with adalimumab 40 mg once every 2 weeks and azathioprine 100 mg once daily. Because her CD was poorly controlled, the dosage of adalimumab was increased to 40 mg once weekly 6 months prior to the current presentation.
Our diagnosis was TNF-α inhibitor-induced psoriasis secondary to treatment with adalimumab.
The patient was treated with mometasone lotion 0.1% for the scalp lesions and triamcinolone cream 0.1% for the body lesions. Because of the extent of the psoriasis, we recommended changing adalimumab to ustekinumab, which is approved for CD in adults but is off label in children.
At 1-month follow-up, after receiving the induction dose of ustekinumab, the patient presented with partial improvement of the skin lesions but had developed a large, alopecic, erythematous plaque with thick yellowish scales on the scalp (Figure 1). She also had a positive hair pull test. The presumptive initial diagnosis of the alopecic scalp lesion was tinea capitis, for which multiple potassium hydroxide preparations of scales were performed, all yielding negative results. In addition, histopathologic examination with hematoxylin and eosin staining was performed (Figures 2A and 2B). Sterile tissue cultures for bacteria, fungi, and acid-fast bacilli were obtained and showed no growth. Periodic acid–Schiff staining was negative for fungal structures.
A second biopsy showed a psoriasiform pattern, parakeratosis, and hypogranulosis, highly suggestive of psoriasis (Figure 2C and 2D). Based on those findings, a diagnosis of psoriatic alopecia was made. The mometasone was switched to clobetasol lotion 0.05%. The patient continued treatment with ustekinumab. At 6-month follow-up, her CD was well controlled and she showed hair regrowth in previously alopecic areas (Figure 3).
Comment
Psoriatic alopecia induced by a TNF-α inhibitor was first reported in 2007 in a 30-year-old woman with ankylosing spondylitis who was being treated with adalimumab.8 She had erythematous, scaly, alopecic plaques on the scalp and palmoplantar pustulosis. Findings on skin biopsy were compatible with psoriasis. The patient’s severe scalp psoriasis failed to respond to topical steroid treatment and adalimumab cessation. The extensive hair loss responded to cyclosporine 3 mg/kg daily.8
After conducting an extensive literature review, we found 26 cases of TNF-α–induced psoriatic alopecia, including the current case (Table).6-16 The mean age at diagnosis was 27.8 years (SD, 13.6 years; range, 7–60 years). The female-to-male ratio was 3.3:1. The most common underlying condition for which TNF-α inhibitors were prescribed was CD (77% [20/26]). Psoriatic alopecia most commonly was reported secondary to treatment with infliximab (54% [14/26]), followed by adalimumab (42% [11/26]). Golimumab was the causative drug in 1 (4%) case. We did not find reports of etanercept or certolizumab having induced this manifestation. The onset of the scalp lesions occurred 2 to 46 months after starting treatment with the causative medication.
Laga et al17 reported that TNF-α inhibitor–induced psoriasis can have a variety of histopathologic findings, including typical findings of various stages of psoriasis, a lichenoid pattern mimicking remnants of lichen planus, and sterile pustular folliculitis. Our patient’s 2 scalp biopsies demonstrated results consistent with findings reported by Laga et al.17 In the first biopsy, findings were consistent with a dense neutrophilic infiltrate with negative sterile cultures and negative periodic acid–Schiff stain (sterile folliculitis), with crust and areas of parakeratosis. The second biopsy demonstrated psoriasiform hyperplasia, parakeratosis, and an absent granular layer, all typical features of psoriasis (Figure 2).
Including the current case, our review of the literature yielded 7 pediatric (ie, 0–18 years of age) cases of TNF-α inhibitor–induced psoriatic alopecia. Of the 6 previously reported pediatric cases, 5 occurred after administration of infliximab.6,7
Similar to our case, TNF-α inhibitor–induced psoriatic alopecia was reported in a 7-year-old girl who was treated with adalimumab for juvenile idiopathic arthritis.6 Nine months after starting treatment, that patient presented with a tender, erythematous, eroded, and crusted alopecic plaque along with scaly plaques on the scalp. Adalimumab was discontinued, and cyclosporine and topical steroids were started. Cyclosporine was then discontinued due to partial resolution of the psoriasis; the patient was started on abatacept, with persistence of the psoriasis and alopecia. The patient was then started on oral methotrexate 12.5 mg once weekly with moderate improvement and mild to moderate exacerbations.
Tumor necrosis factor α inhibitor–induced psoriasis may occur as a result of a cytokine imbalance. A TNF-α blockade leads to upregulation of interferon α (IFN-α) and TNF-α production by plasmacytoid dendritic cells (pDCs), usually in genetically susceptible people.6,7,9-15 The IFN-α induces maturation of myeloid dendritic cells (mDCs) responsible for increasing proinflammatory cytokines that contribute to psoriasis.11 Generation of TNF-α by pDCs leads to mature or activated dendritic cells derived from pDCs through autocrine TNF-α production and paracrine IFN-α production from immature mDCs.9 Once pDCs mature, they are incapable of producing IFN-α; TNF-α then inhibits IFN-α production by inducing pDC maturation.11 Overproduction of IFN-α during TNF-α inhibition induces expression of the chemokine receptor CXCR3 on T cells, which recruits T cells to the dermis. The T cells then produce TNF-α, causing psoriatic skin lesions.10,11,13,14
Although TNF-α inhibitor–induced psoriatic alopecia is uncommon, the condition should be considered in female patients with underlying proinflammatory disease—CD in particular. Perman et al6 reported 5 cases of psoriatic alopecia in which 3 patients initially were treated with griseofulvin because of suspected tinea capitis.
Conditions with similar clinical findings should be ruled out before making a diagnosis of TNF-α inhibitor–induced psoriatic alopecia. Although clinicopathologic correlation is essential for making the diagnosis, it is possible that the histologic findings will not be specific for psoriasis.17 It is important to be aware of this condition in patients being treated with a TNF-α inhibitor as early as 2 months to 4 years or longer after starting treatment.
Previously reported cases have demonstrated various treatment options that yielded improvement or resolution of TNF-α inhibitor–induced psoriatic alopecia. These include either continuation or discontinuation of the TNF-α inhibitor combined with topical or intralesional steroids, methotrexate, or cyclosporine. Another option is to switch the TNF-α inhibitor to another biologic. Outcomes vary from patient to patient, making the physician’s clinical judgment crucial in deciding which treatment route to take. Our patient showed notable improvement when she was switched from adalimumab to ustekinumab as well as the combination of ustekinumab and clobetasol lotion 0.05%.
Conclusion
We recommend an individualized approach that provides patients with the safest and least invasive treatment option for TNF-α inhibitor–induced psoriatic alopecia. In most reported cases, the problem resolved with treatment, thereby classifying this form of alopecia as noncicatricial alopecia.
- Horneff G, Seyger MMB, Arikan D, et al. Safety of adalimumab in pediatric patients with polyarticular juvenile idiopathic arthritis, enthesitis-related arthritis, psoriasis, and Crohn’s disease. J Pediatr. 2018;201:166-175.e3. doi:10.1016/j.jpeds.2018.05.042
- Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
- George SMC, Taylor MR, Farrant PBJ. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77. doi:10.1111/j.1365-2133.1972.tb05103.x
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? a clinicohistopathologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Perman MJ, Lovell DJ, Denson LA, et al. Five cases of anti-tumor necrosis factor alpha-induced psoriasis presenting with severe scalp involvement in children. Pediatr Dermatol. 2012;29:454-459. doi:10.1111/j.1525-1470.2011.01521.x
- Prata Ribeiro LB, Gonçalves Rego JC, Duque Estrada B, et al. Alopecia secondary to anti-tumor necrosis factor-alpha therapy. An Bras Dermatol. 2015;90:232–235. doi:10.1590/abd1806-4841.20153084
- Papadavid E, Gazi S, Dalamaga M, et al. Palmoplantar and scalp psoriasis occurring during anti-tumour necrosis factor-alpha therapy: a case series of four patients and guidelines for management. J Eur Acad Dermatol Venereol. 2008;22:380-382. doi:10.1111/j.1468-3083.2007.02335.x
- Manni E, Barachini P. Psoriasis induced by infliximab in a patient suffering from Crohn’s disease. Int J Immunopathol Pharmacol. 2009;22:841-844. doi:10.1177/039463200902200331
- El Shabrawi-Caelen L, La Placa M, Vincenzi C, et al. Adalimumab-induced psoriasis of the scalp with diffuse alopecia: a severe potentially irreversible cutaneous side effect of TNF-alpha blockers. Inflamm Bowel Dis. 2010;16:182-183. doi:10.1002/ibd.20954
- Medkour F, Babai S, Chanteloup E, et al. Development of diffuse psoriasis with alopecia during treatment of Crohn’s disease with infliximab. Gastroenterol Clin Biol. 2010;34:140-141. doi:10.1016/j.gcb.2009.10.021
- Doyle LA, Sperling LC, Baksh S, et al. Psoriatic alopecia/alopecia areata-like reactions secondary to anti-tumor necrosis factor-α therapy: a novel cause of noncicatricial alopecia. Am J Dermatopathol. 2011;33:161-166. doi:10.1097/DAD.0b013e3181ef7403
- Osório F, Magro F, Lisboa C, et al. Anti-TNF-alpha induced psoriasiform eruptions with severe scalp involvement and alopecia: report of five cases and review of the literature. Dermatology. 2012;225:163-167. doi:10.1159/000342503
- Andrisani G, Marzo M, Celleno L, et al. Development of psoriasis scalp with alopecia during treatment of Crohn’s disease with infliximab and rapid response to both diseases to ustekinumab. Eur Rev Med Pharmacol Sci. 2013;17:2831-2836.
- Afanasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-569. doi:10.1111/cup.12932
- Helm MM, Haddad S. Alopecia areata and scarring alopecia presenting during golimumab therapy for ankylosing spondylitis. N Am J Med Sci. 2018;11:22-24. doi:10.7156/najms.2018.110122
- Laga AC, Vleugels RA, Qureshi AA, et al. Histopathologic spectrum of psoriasiform skin reactions associated with tumor necrosis factor-a inhibitor therapy. a study of 16 biopsies. Am J Dermatopathol. 2010;32:568-573. doi:10.1097/DAD.0b013e3181cb3ff7
- Horneff G, Seyger MMB, Arikan D, et al. Safety of adalimumab in pediatric patients with polyarticular juvenile idiopathic arthritis, enthesitis-related arthritis, psoriasis, and Crohn’s disease. J Pediatr. 2018;201:166-175.e3. doi:10.1016/j.jpeds.2018.05.042
- Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
- George SMC, Taylor MR, Farrant PBJ. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77. doi:10.1111/j.1365-2133.1972.tb05103.x
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? a clinicohistopathologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Perman MJ, Lovell DJ, Denson LA, et al. Five cases of anti-tumor necrosis factor alpha-induced psoriasis presenting with severe scalp involvement in children. Pediatr Dermatol. 2012;29:454-459. doi:10.1111/j.1525-1470.2011.01521.x
- Prata Ribeiro LB, Gonçalves Rego JC, Duque Estrada B, et al. Alopecia secondary to anti-tumor necrosis factor-alpha therapy. An Bras Dermatol. 2015;90:232–235. doi:10.1590/abd1806-4841.20153084
- Papadavid E, Gazi S, Dalamaga M, et al. Palmoplantar and scalp psoriasis occurring during anti-tumour necrosis factor-alpha therapy: a case series of four patients and guidelines for management. J Eur Acad Dermatol Venereol. 2008;22:380-382. doi:10.1111/j.1468-3083.2007.02335.x
- Manni E, Barachini P. Psoriasis induced by infliximab in a patient suffering from Crohn’s disease. Int J Immunopathol Pharmacol. 2009;22:841-844. doi:10.1177/039463200902200331
- El Shabrawi-Caelen L, La Placa M, Vincenzi C, et al. Adalimumab-induced psoriasis of the scalp with diffuse alopecia: a severe potentially irreversible cutaneous side effect of TNF-alpha blockers. Inflamm Bowel Dis. 2010;16:182-183. doi:10.1002/ibd.20954
- Medkour F, Babai S, Chanteloup E, et al. Development of diffuse psoriasis with alopecia during treatment of Crohn’s disease with infliximab. Gastroenterol Clin Biol. 2010;34:140-141. doi:10.1016/j.gcb.2009.10.021
- Doyle LA, Sperling LC, Baksh S, et al. Psoriatic alopecia/alopecia areata-like reactions secondary to anti-tumor necrosis factor-α therapy: a novel cause of noncicatricial alopecia. Am J Dermatopathol. 2011;33:161-166. doi:10.1097/DAD.0b013e3181ef7403
- Osório F, Magro F, Lisboa C, et al. Anti-TNF-alpha induced psoriasiform eruptions with severe scalp involvement and alopecia: report of five cases and review of the literature. Dermatology. 2012;225:163-167. doi:10.1159/000342503
- Andrisani G, Marzo M, Celleno L, et al. Development of psoriasis scalp with alopecia during treatment of Crohn’s disease with infliximab and rapid response to both diseases to ustekinumab. Eur Rev Med Pharmacol Sci. 2013;17:2831-2836.
- Afanasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-569. doi:10.1111/cup.12932
- Helm MM, Haddad S. Alopecia areata and scarring alopecia presenting during golimumab therapy for ankylosing spondylitis. N Am J Med Sci. 2018;11:22-24. doi:10.7156/najms.2018.110122
- Laga AC, Vleugels RA, Qureshi AA, et al. Histopathologic spectrum of psoriasiform skin reactions associated with tumor necrosis factor-a inhibitor therapy. a study of 16 biopsies. Am J Dermatopathol. 2010;32:568-573. doi:10.1097/DAD.0b013e3181cb3ff7
Practice Points
- Psoriatic alopecia is a rare nonscarring alopecia that can present as a complication of treatment with tumor necrosis factor α inhibitors.
- This finding commonly is seen in females undergoing treatment with infliximab or adalimumab, usually for Crohn disease.
- Histopathologic findings can show a psoriasiform-pattern, neutrophil-rich, inflammatory infiltrate involving hair follicles or a lichenoid pattern.
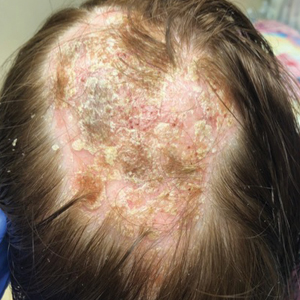
FDA panel narrowly backs avacopan approval
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.

At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.

“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”

Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.

During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.
At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.
“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”
Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.
During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.
At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.
“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”
Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.
During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
Black patients with cutaneous sarcoidosis may have more systemic and CV disease
according to a retrospective chart review of patients seen at Massachusetts General Hospital and Brigham and Women’s Hospital, both in Boston.
Black patients were also significantly more likely to have two or more organs involved and have higher rates of cardiac involvement, the latter of which is associated with worse prognosis. “Our data suggest there may be substantial variations in organ involvement between racial groups of patients presenting with cutaneous sarcoidosis,” said medical student Kylee Kus, a medical student at Oakland University, Auburn Hills, Mich., who presented the findings with Bina Kassamali, a medical student at Harvard University, Boston, at the annual Skin of Color Society scientific symposium.
Sotonye Imadojemu, MD, MBE; Avery LeChance, MD, MPH; and Ruth Anne Vleugels, MD, MPH, MBA; of Brigham and Women’s Hospital, are cosenior authors of the abstract.
The researchers identified 111 patients who were diagnosed with cutaneous sarcoidosis over a 20-year period (January 2000–December 2019), 50 of whom presented without established extracutaneous disease. They examined the charts of these 50 patients for whether subsequent work-up revealed systemic disease.
Of the 50 patients, 9 were Black. Seven of these nine patients (77.8%), were found to have systemic involvement, compared with 14 of 41 (46.3%) non-Black patients – a 31.5% higher probability (P < .05). One-third of the nine Black patients were found to have disease in one organ, and 44.4% in two or more organs. In non-Black patients, these rates were 12.2% and 34.1%, respectively.
Cardiovascular involvement was not found in any of the non-Black patients who had extracutaneous disease, but was found in 29% of the Black patients with extracutaneous disease, a statistically significant difference.
Black patients are known to be at higher risk for sarcoidosis than non-Black patients, and because “there is an association between cardiac sarcoid involvement and poor prognosis largely due to manifestations such as heart block, arrhythmias, and heart failure ... the study helps demonstrate how this organ involvement can disproportionately affect the Black population,” Ms. Kassamali said in an interview after the meeting.
A separate, recently published analysis of data from the same patient population examined the work-ups that patients received after a dermatologist’s diagnosis of sarcoidosis and found that patients with no previous systemic work-up were subsequently assessed for cardiac involvement in only 58.3% of cases. Assessment for pulmonary and ocular disease was completed more than 90% of the time.
“Crucial testing for cardiac involvement fell short,” Dr. Imadojemu, of the department of dermatology, Brigham and Women’s Hospital, and coinvestigators wrote in the research letter.
“Because the cutaneous manifestations of sarcoidosis often present at disease onset, dermatologists may be the first physicians to diagnose a patient with sarcoidosis,” they wrote. “As such, dermatologists are often responsible for initiating the appropriate evaluation of patients with sarcoidosis.”
Pulmonary involvement occurs in nearly all cases of sarcoidosis, while ocular and cardiac disease develop in approximately 25% and 10% of patients, respectively. Cardiac sarcoidosis is usually asymptomatic and accounts for 13%-25% of sarcoidosis-related deaths in the United States, they wrote.
An electrocardiogram is the appropriate initial screening tool and “is warranted in all patients with sarcoidosis,” they advised.
according to a retrospective chart review of patients seen at Massachusetts General Hospital and Brigham and Women’s Hospital, both in Boston.
Black patients were also significantly more likely to have two or more organs involved and have higher rates of cardiac involvement, the latter of which is associated with worse prognosis. “Our data suggest there may be substantial variations in organ involvement between racial groups of patients presenting with cutaneous sarcoidosis,” said medical student Kylee Kus, a medical student at Oakland University, Auburn Hills, Mich., who presented the findings with Bina Kassamali, a medical student at Harvard University, Boston, at the annual Skin of Color Society scientific symposium.
Sotonye Imadojemu, MD, MBE; Avery LeChance, MD, MPH; and Ruth Anne Vleugels, MD, MPH, MBA; of Brigham and Women’s Hospital, are cosenior authors of the abstract.
The researchers identified 111 patients who were diagnosed with cutaneous sarcoidosis over a 20-year period (January 2000–December 2019), 50 of whom presented without established extracutaneous disease. They examined the charts of these 50 patients for whether subsequent work-up revealed systemic disease.
Of the 50 patients, 9 were Black. Seven of these nine patients (77.8%), were found to have systemic involvement, compared with 14 of 41 (46.3%) non-Black patients – a 31.5% higher probability (P < .05). One-third of the nine Black patients were found to have disease in one organ, and 44.4% in two or more organs. In non-Black patients, these rates were 12.2% and 34.1%, respectively.
Cardiovascular involvement was not found in any of the non-Black patients who had extracutaneous disease, but was found in 29% of the Black patients with extracutaneous disease, a statistically significant difference.
Black patients are known to be at higher risk for sarcoidosis than non-Black patients, and because “there is an association between cardiac sarcoid involvement and poor prognosis largely due to manifestations such as heart block, arrhythmias, and heart failure ... the study helps demonstrate how this organ involvement can disproportionately affect the Black population,” Ms. Kassamali said in an interview after the meeting.
A separate, recently published analysis of data from the same patient population examined the work-ups that patients received after a dermatologist’s diagnosis of sarcoidosis and found that patients with no previous systemic work-up were subsequently assessed for cardiac involvement in only 58.3% of cases. Assessment for pulmonary and ocular disease was completed more than 90% of the time.
“Crucial testing for cardiac involvement fell short,” Dr. Imadojemu, of the department of dermatology, Brigham and Women’s Hospital, and coinvestigators wrote in the research letter.
“Because the cutaneous manifestations of sarcoidosis often present at disease onset, dermatologists may be the first physicians to diagnose a patient with sarcoidosis,” they wrote. “As such, dermatologists are often responsible for initiating the appropriate evaluation of patients with sarcoidosis.”
Pulmonary involvement occurs in nearly all cases of sarcoidosis, while ocular and cardiac disease develop in approximately 25% and 10% of patients, respectively. Cardiac sarcoidosis is usually asymptomatic and accounts for 13%-25% of sarcoidosis-related deaths in the United States, they wrote.
An electrocardiogram is the appropriate initial screening tool and “is warranted in all patients with sarcoidosis,” they advised.
according to a retrospective chart review of patients seen at Massachusetts General Hospital and Brigham and Women’s Hospital, both in Boston.
Black patients were also significantly more likely to have two or more organs involved and have higher rates of cardiac involvement, the latter of which is associated with worse prognosis. “Our data suggest there may be substantial variations in organ involvement between racial groups of patients presenting with cutaneous sarcoidosis,” said medical student Kylee Kus, a medical student at Oakland University, Auburn Hills, Mich., who presented the findings with Bina Kassamali, a medical student at Harvard University, Boston, at the annual Skin of Color Society scientific symposium.
Sotonye Imadojemu, MD, MBE; Avery LeChance, MD, MPH; and Ruth Anne Vleugels, MD, MPH, MBA; of Brigham and Women’s Hospital, are cosenior authors of the abstract.
The researchers identified 111 patients who were diagnosed with cutaneous sarcoidosis over a 20-year period (January 2000–December 2019), 50 of whom presented without established extracutaneous disease. They examined the charts of these 50 patients for whether subsequent work-up revealed systemic disease.
Of the 50 patients, 9 were Black. Seven of these nine patients (77.8%), were found to have systemic involvement, compared with 14 of 41 (46.3%) non-Black patients – a 31.5% higher probability (P < .05). One-third of the nine Black patients were found to have disease in one organ, and 44.4% in two or more organs. In non-Black patients, these rates were 12.2% and 34.1%, respectively.
Cardiovascular involvement was not found in any of the non-Black patients who had extracutaneous disease, but was found in 29% of the Black patients with extracutaneous disease, a statistically significant difference.
Black patients are known to be at higher risk for sarcoidosis than non-Black patients, and because “there is an association between cardiac sarcoid involvement and poor prognosis largely due to manifestations such as heart block, arrhythmias, and heart failure ... the study helps demonstrate how this organ involvement can disproportionately affect the Black population,” Ms. Kassamali said in an interview after the meeting.
A separate, recently published analysis of data from the same patient population examined the work-ups that patients received after a dermatologist’s diagnosis of sarcoidosis and found that patients with no previous systemic work-up were subsequently assessed for cardiac involvement in only 58.3% of cases. Assessment for pulmonary and ocular disease was completed more than 90% of the time.
“Crucial testing for cardiac involvement fell short,” Dr. Imadojemu, of the department of dermatology, Brigham and Women’s Hospital, and coinvestigators wrote in the research letter.
“Because the cutaneous manifestations of sarcoidosis often present at disease onset, dermatologists may be the first physicians to diagnose a patient with sarcoidosis,” they wrote. “As such, dermatologists are often responsible for initiating the appropriate evaluation of patients with sarcoidosis.”
Pulmonary involvement occurs in nearly all cases of sarcoidosis, while ocular and cardiac disease develop in approximately 25% and 10% of patients, respectively. Cardiac sarcoidosis is usually asymptomatic and accounts for 13%-25% of sarcoidosis-related deaths in the United States, they wrote.
An electrocardiogram is the appropriate initial screening tool and “is warranted in all patients with sarcoidosis,” they advised.
FROM SOC SOCIETY 2021
Squamous Cell Carcinoma in Hidradenitis Suppurativa Lesions Following Tumor Necrosis Factor α Inhibitors
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
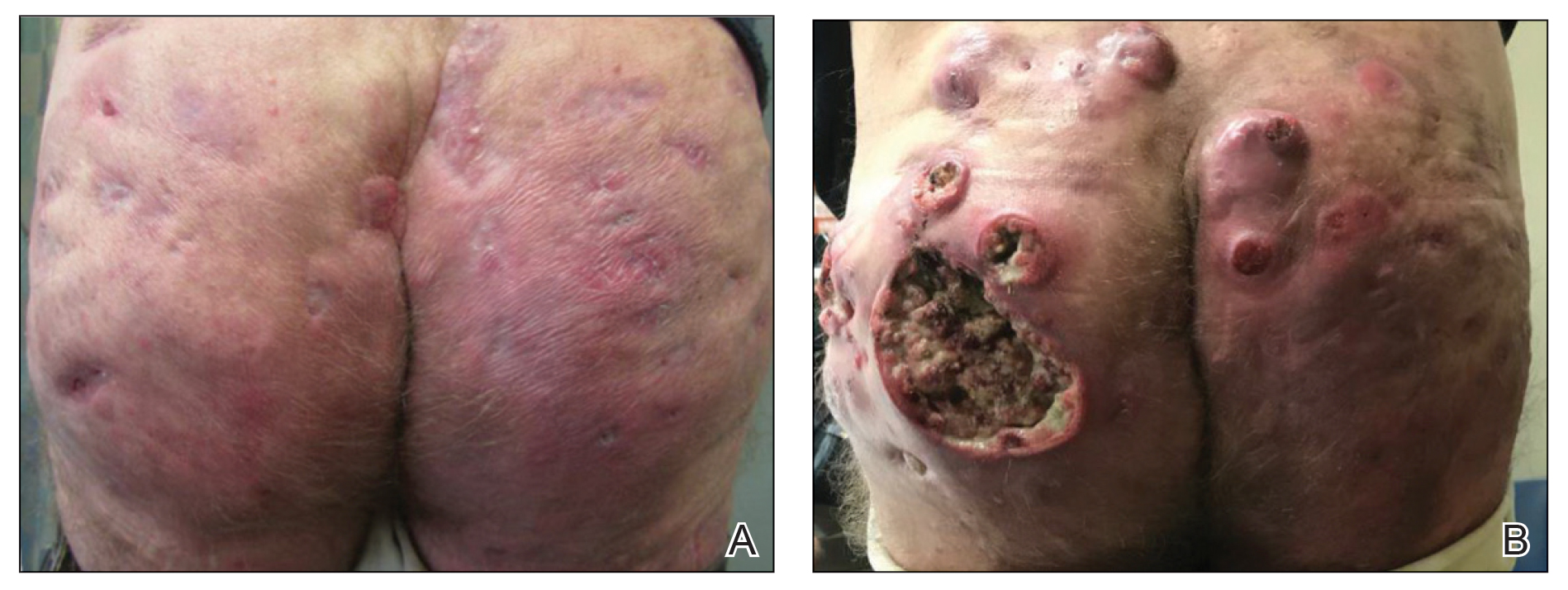
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
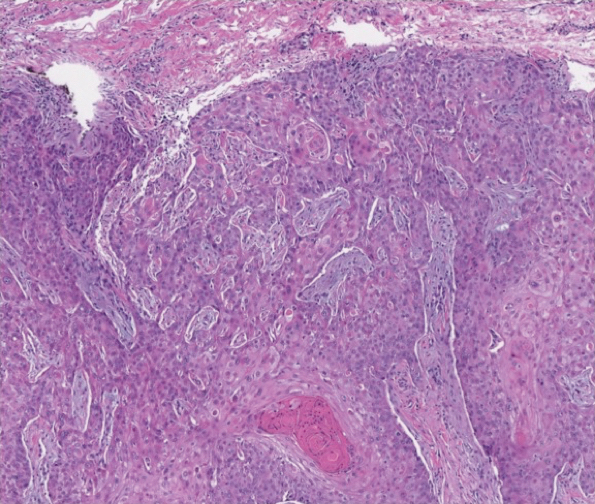
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
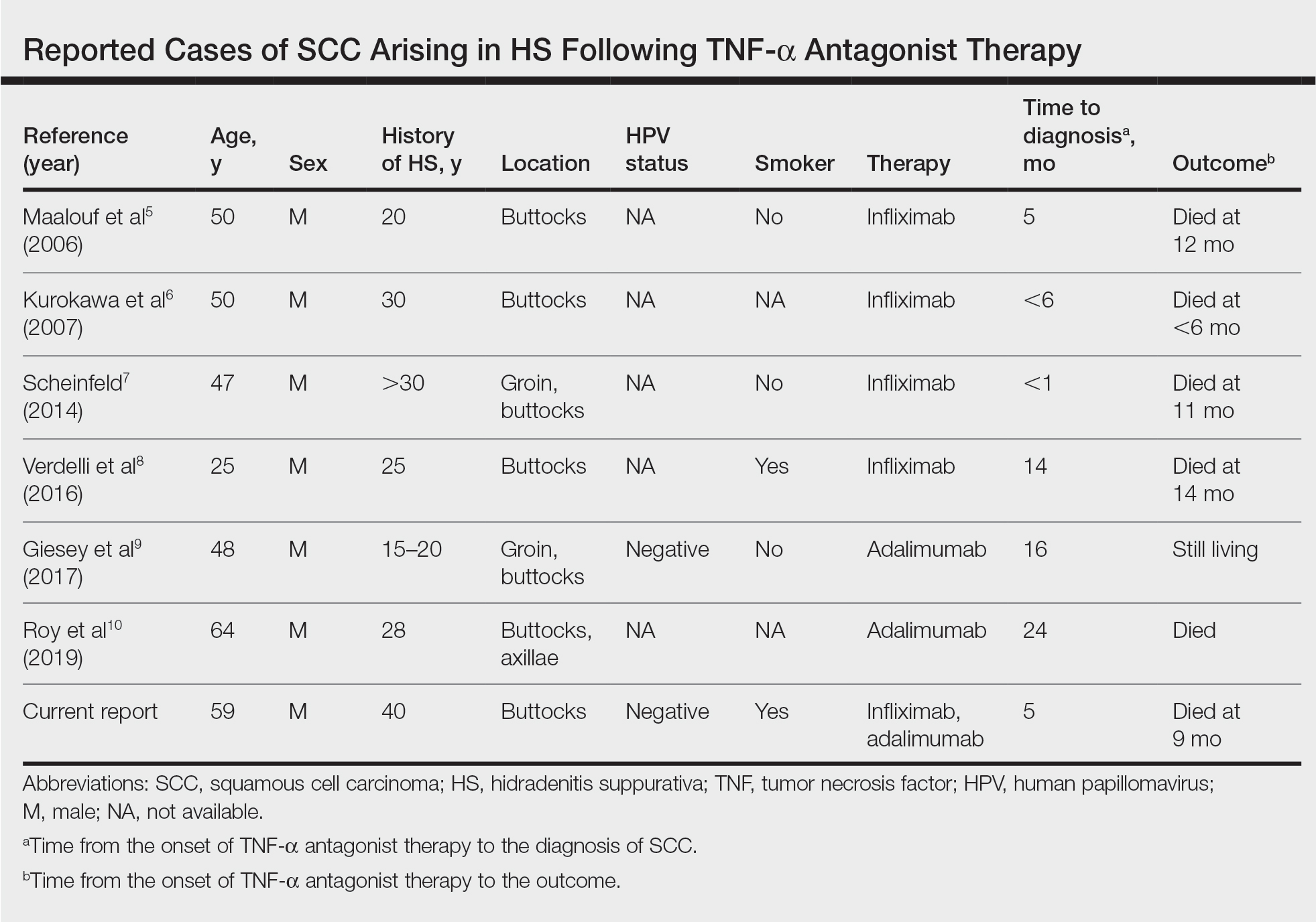
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
Practice Points
- Consider biopsy of representative lesions in men older than 20 years with moderate to severe disease of the groin and/or buttocks prior to initiation of tumor necrosis factor inhibitors.
- Consider more frequent clinical monitoring with a decrease in threshold to perform biopsy of any new or ulcerating lesions.
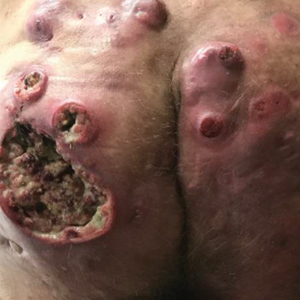
COVID-19’s impact on lupus inpatients examined in study
Severe COVID-19 infection was more likely in hospitalized patients with systemic lupus erythematosus (SLE) who had comorbidities and risk factors associated with severe infection in the general population, notably older age, male gender, and hypertension, based on data from a nationwide epidemiologic study of inpatients in France.
“Recently, anti-interferon antibodies have been implicated in severe SARS-CoV-2 infection while it has been known for decades that patients with SLE may produce such autoantibodies,” but large-scale data on the risk of severe COVID-19 infection in SLE patients are limited, Arthur Mageau, MD, of Bichat–Claude Bernard Hospital in Paris, and colleagues wrote.
In a research letter published in Annals of the Rheumatic Diseases, the researchers used the French health care database Programme de Médicalisation des Systèmes d’Information to identify 11,055 adult SLE patients who had at least one hospital stay between March 1, 2020, and Oct.31, 2020. Of these, 1,411 (12.8%) also were diagnosed with COVID-19, and these patients had a total of 1,721 hospital stays.
Overall, in-hospital mortality was approximately four times higher among SLE patients with COVID-19 infection, compared with SLE patients without COVID-19 infection (9.5% vs. 2.4%, P < .001), and 293 (17%) of the COVID-19 hospital stays involved an intensive care unit. In the ICU, 78 (26.7%) of the COVID-19 patients required invasive ventilation, and 71 (24.7%) required noninvasive mechanical ventilation.
The SLE patients with COVID-19 who died were significantly more likely than the SLE patients with COVID-19 who recovered to be older and male, and to have conditions including chronic kidney disease, high blood pressure, chronic pulmonary disease, and a history of cardiovascular events or lupus nephritis. The study findings were limited by the focus on hospitalized patients only, so the results cannot be generalized to all lupus patients, the researchers said.
“Interestingly, while the overall mortality rate was lower in SLE/COVID-19–positive inpatients as compared with the total population admitted for SARS-CoV-2 infection in France during the same period (9.5% vs 15.7%, P < .0001), the mortality rate at a younger age tended to be higher in patients with SLE,” the researchers wrote, but the difference for these younger patients was not statistically significant. This disparity may be caused by the reduced need for immunosuppressive drugs in SLE patients as they age, and the observed increased mortality in younger SLE patients, compared with the general population, suggests that SLE may promote poor outcomes from COVID-19 infection.
Dr. Mageau received PhD fellowship support from the Agence Nationale pour la recherche. He and the other researchers had no financial conflicts to disclose. The study received no outside funding.
Severe COVID-19 infection was more likely in hospitalized patients with systemic lupus erythematosus (SLE) who had comorbidities and risk factors associated with severe infection in the general population, notably older age, male gender, and hypertension, based on data from a nationwide epidemiologic study of inpatients in France.
“Recently, anti-interferon antibodies have been implicated in severe SARS-CoV-2 infection while it has been known for decades that patients with SLE may produce such autoantibodies,” but large-scale data on the risk of severe COVID-19 infection in SLE patients are limited, Arthur Mageau, MD, of Bichat–Claude Bernard Hospital in Paris, and colleagues wrote.
In a research letter published in Annals of the Rheumatic Diseases, the researchers used the French health care database Programme de Médicalisation des Systèmes d’Information to identify 11,055 adult SLE patients who had at least one hospital stay between March 1, 2020, and Oct.31, 2020. Of these, 1,411 (12.8%) also were diagnosed with COVID-19, and these patients had a total of 1,721 hospital stays.
Overall, in-hospital mortality was approximately four times higher among SLE patients with COVID-19 infection, compared with SLE patients without COVID-19 infection (9.5% vs. 2.4%, P < .001), and 293 (17%) of the COVID-19 hospital stays involved an intensive care unit. In the ICU, 78 (26.7%) of the COVID-19 patients required invasive ventilation, and 71 (24.7%) required noninvasive mechanical ventilation.
The SLE patients with COVID-19 who died were significantly more likely than the SLE patients with COVID-19 who recovered to be older and male, and to have conditions including chronic kidney disease, high blood pressure, chronic pulmonary disease, and a history of cardiovascular events or lupus nephritis. The study findings were limited by the focus on hospitalized patients only, so the results cannot be generalized to all lupus patients, the researchers said.
“Interestingly, while the overall mortality rate was lower in SLE/COVID-19–positive inpatients as compared with the total population admitted for SARS-CoV-2 infection in France during the same period (9.5% vs 15.7%, P < .0001), the mortality rate at a younger age tended to be higher in patients with SLE,” the researchers wrote, but the difference for these younger patients was not statistically significant. This disparity may be caused by the reduced need for immunosuppressive drugs in SLE patients as they age, and the observed increased mortality in younger SLE patients, compared with the general population, suggests that SLE may promote poor outcomes from COVID-19 infection.
Dr. Mageau received PhD fellowship support from the Agence Nationale pour la recherche. He and the other researchers had no financial conflicts to disclose. The study received no outside funding.
Severe COVID-19 infection was more likely in hospitalized patients with systemic lupus erythematosus (SLE) who had comorbidities and risk factors associated with severe infection in the general population, notably older age, male gender, and hypertension, based on data from a nationwide epidemiologic study of inpatients in France.
“Recently, anti-interferon antibodies have been implicated in severe SARS-CoV-2 infection while it has been known for decades that patients with SLE may produce such autoantibodies,” but large-scale data on the risk of severe COVID-19 infection in SLE patients are limited, Arthur Mageau, MD, of Bichat–Claude Bernard Hospital in Paris, and colleagues wrote.
In a research letter published in Annals of the Rheumatic Diseases, the researchers used the French health care database Programme de Médicalisation des Systèmes d’Information to identify 11,055 adult SLE patients who had at least one hospital stay between March 1, 2020, and Oct.31, 2020. Of these, 1,411 (12.8%) also were diagnosed with COVID-19, and these patients had a total of 1,721 hospital stays.
Overall, in-hospital mortality was approximately four times higher among SLE patients with COVID-19 infection, compared with SLE patients without COVID-19 infection (9.5% vs. 2.4%, P < .001), and 293 (17%) of the COVID-19 hospital stays involved an intensive care unit. In the ICU, 78 (26.7%) of the COVID-19 patients required invasive ventilation, and 71 (24.7%) required noninvasive mechanical ventilation.
The SLE patients with COVID-19 who died were significantly more likely than the SLE patients with COVID-19 who recovered to be older and male, and to have conditions including chronic kidney disease, high blood pressure, chronic pulmonary disease, and a history of cardiovascular events or lupus nephritis. The study findings were limited by the focus on hospitalized patients only, so the results cannot be generalized to all lupus patients, the researchers said.
“Interestingly, while the overall mortality rate was lower in SLE/COVID-19–positive inpatients as compared with the total population admitted for SARS-CoV-2 infection in France during the same period (9.5% vs 15.7%, P < .0001), the mortality rate at a younger age tended to be higher in patients with SLE,” the researchers wrote, but the difference for these younger patients was not statistically significant. This disparity may be caused by the reduced need for immunosuppressive drugs in SLE patients as they age, and the observed increased mortality in younger SLE patients, compared with the general population, suggests that SLE may promote poor outcomes from COVID-19 infection.
Dr. Mageau received PhD fellowship support from the Agence Nationale pour la recherche. He and the other researchers had no financial conflicts to disclose. The study received no outside funding.
FROM ANNALS OF THE RHEUMATIC DISEASES
Candida Esophagitis Associated With Adalimumab for Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
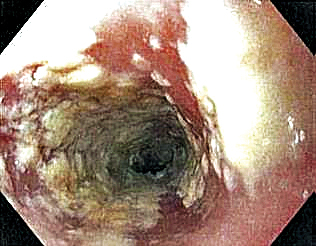
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
Practice Points
- Adalimumab is an effective treatment for patients with hidradenitis suppurativa.
- There is risk for opportunistic infections with adalimumab, and patients should be monitored closely.