User login
Study: Pediatric cancer patients have high rate of germline mutations in predisposition genes
Children and adolescents with cancer were found to have a significantly higher rate of germline mutations in cancer predisposing genes compared with individuals with no known cancer; however, family history of cancer did not predict the presence of a predisposition syndrome for most patients.
Of the 1,120 children with cancer, 8.5% had mutations in predisposition genes, compared with 1.1% in the control group. Mutations in TP53 were most common (50 patients), followed by APC (6), BRCA2 (6), NF1 (4), PMS2 (4), RB1 (3), and RUNX1 (3). Germline TP53 mutations were present in 27 of 39 patients (69%) with adrenocortical tumors, 9 of 47 (19%) with hypodiploid acute lymphoblastic leukemia, and 1 of 4 (25%) with choroid plexus carcinoma (N Engl J Med. 2015 Nov. 18. doi: 10.1056/NEJMoa1508054).
Only 40% of the pediatric patients with pathogenic germline mutations had a family history of cancer, and in just half of those cases the history was consistent with a known cancer-predisposition syndrome. Among patients without predisposition germline mutations, a similar proportion (42%) had a family history of cancer.
“On the basis of these observations, family history cannot be the sole indication used to guide the provision of genetic testing,” wrote Jinghui Zhang, Ph.D., of the department of computational biology, St. Jude’s Research Hospital, Memphis, Tennessee, and her colleagues.
Unexpected germline mutations were found in several cases. Six patients with Ewing’s sarcoma had unexpected pathogenic germline mutations (TP53 in four patients, PMS2 in one and RET in one). Eight patents had heterozygous mutations in BRCA1, BRCA2, or PALB2, supporting the notion that mutations in these genes may play a role in pediatric as well as adult cancer. Other new associations included germline APC and SDHB mutations with neuroblastoma, and APC, VHL, CDH1, PTCH1, and SDHA germline mutations with leukemia.
The St. Jude–Washington University Pediatric Cancer Genome Project (PCGP) included 1,120 patients representing the major types of pediatric cancer, including 53% with leukemia, 22% with CNS tumors, and 26% with non-CNS tumors. Whole genomes were sequenced from 595 patients, whole exomes (coding regions only) from 456 patients, and both whole genomes and exomes from 69 patients. Whole exomes were sequenced from two control cohorts of individuals with no known cancer: 966 from the 1,000 Genome project and 723 from the National Database for Autism Research (cancer predisposition genes known to be associated with autism, NF1 and PTEN, were excluded from analysis with this control set).
The study sequenced whole genomes and exomes, but focused most of the analysis on 60 autosomal dominant cancer predisposition genes. Tumor types with the highest prevalence of germline mutations in these genes were non-CNS solid tumors (48 of 287 patients, 17%) and CNS tumors (21 of 245, 9%). Among patients with adrenocortical tumors, 69% had germline mutations. Despite inclusion of hypodiploid acute lymphoblastic leukemia, the lowest germline mutation prevalence was found in leukemia (26 of 588, 4%).
Dr. Zhang reported having no disclosures. One coauthor reported financial ties to an industry source.
The sequencing study by Zhang et al. found that 8.5% of 1,120 participants with pediatric cancer had pathogenic mutations in an autosomal dominant cancer-predisposition gene, and four of the mutations were mosaic (i.e., present in only a subset of normal cells, probably indicating the defect was not inherited). The four mosaic mutations probably would not be detected by standard genetic testing strategies.
Not surprisingly, more than 50% of the mutations were in the tumor suppressor gene TP53. A long list of other genes identified as potentially disease causing each occurred at a prevalence of less than 6%.
Although the study’s inclusion of certain high-risk childhood cancers could bias the results toward overestimating the proportion of germline cancer predisposition mutations, more likely the results are an underestimate. By evaluating mutations in only a small subset of candidate autosomal dominant genes, the findings to not reflect a thorough assessment of most genes in the genome. In addition, focusing only on the exome ignores DNA mutations in noncoding regions, especially in tissue-specific enhancers, which may have a role in cancer susceptibility. So-called epimutations may affect cancer susceptibility in a nonmendelian fashion. Finally, the ability to study the interaction of several of these events may contribute to our understanding of tumor initiation.
The study raises several important questions. Are children with mutations in APC, BRCA1, or BRCA2 at risk for childhood cancers? How does germline mosaicism influence disease penetrance, and how many of the mosaic mutations were inherited? How do the mutations identified interact with less well known gene mutations elsewhere in the genome to influence malignant transformation? How can the findings translate to the clinic?
The study highlights the fact that family history is insufficient to assess the likelihood of a cancer-predisposition syndrome in any patient with a newly diagnosed cancer.
Dr. John Maris is a pediatric oncologist in the division of oncology and Center for Childhood Cancer Research, Children’s Hospital of Philadelphia, and in the department of pediatrics at the Perelman School of Medicine, University of Pennsylvania. These remarks were part of an editorial accompanying the report (N Engl J Med. 2015 Nov. 18 doi: 10.1056/NEJMoa1508054). Dr. Maris reported having no disclosures.
The sequencing study by Zhang et al. found that 8.5% of 1,120 participants with pediatric cancer had pathogenic mutations in an autosomal dominant cancer-predisposition gene, and four of the mutations were mosaic (i.e., present in only a subset of normal cells, probably indicating the defect was not inherited). The four mosaic mutations probably would not be detected by standard genetic testing strategies.
Not surprisingly, more than 50% of the mutations were in the tumor suppressor gene TP53. A long list of other genes identified as potentially disease causing each occurred at a prevalence of less than 6%.
Although the study’s inclusion of certain high-risk childhood cancers could bias the results toward overestimating the proportion of germline cancer predisposition mutations, more likely the results are an underestimate. By evaluating mutations in only a small subset of candidate autosomal dominant genes, the findings to not reflect a thorough assessment of most genes in the genome. In addition, focusing only on the exome ignores DNA mutations in noncoding regions, especially in tissue-specific enhancers, which may have a role in cancer susceptibility. So-called epimutations may affect cancer susceptibility in a nonmendelian fashion. Finally, the ability to study the interaction of several of these events may contribute to our understanding of tumor initiation.
The study raises several important questions. Are children with mutations in APC, BRCA1, or BRCA2 at risk for childhood cancers? How does germline mosaicism influence disease penetrance, and how many of the mosaic mutations were inherited? How do the mutations identified interact with less well known gene mutations elsewhere in the genome to influence malignant transformation? How can the findings translate to the clinic?
The study highlights the fact that family history is insufficient to assess the likelihood of a cancer-predisposition syndrome in any patient with a newly diagnosed cancer.
Dr. John Maris is a pediatric oncologist in the division of oncology and Center for Childhood Cancer Research, Children’s Hospital of Philadelphia, and in the department of pediatrics at the Perelman School of Medicine, University of Pennsylvania. These remarks were part of an editorial accompanying the report (N Engl J Med. 2015 Nov. 18 doi: 10.1056/NEJMoa1508054). Dr. Maris reported having no disclosures.
The sequencing study by Zhang et al. found that 8.5% of 1,120 participants with pediatric cancer had pathogenic mutations in an autosomal dominant cancer-predisposition gene, and four of the mutations were mosaic (i.e., present in only a subset of normal cells, probably indicating the defect was not inherited). The four mosaic mutations probably would not be detected by standard genetic testing strategies.
Not surprisingly, more than 50% of the mutations were in the tumor suppressor gene TP53. A long list of other genes identified as potentially disease causing each occurred at a prevalence of less than 6%.
Although the study’s inclusion of certain high-risk childhood cancers could bias the results toward overestimating the proportion of germline cancer predisposition mutations, more likely the results are an underestimate. By evaluating mutations in only a small subset of candidate autosomal dominant genes, the findings to not reflect a thorough assessment of most genes in the genome. In addition, focusing only on the exome ignores DNA mutations in noncoding regions, especially in tissue-specific enhancers, which may have a role in cancer susceptibility. So-called epimutations may affect cancer susceptibility in a nonmendelian fashion. Finally, the ability to study the interaction of several of these events may contribute to our understanding of tumor initiation.
The study raises several important questions. Are children with mutations in APC, BRCA1, or BRCA2 at risk for childhood cancers? How does germline mosaicism influence disease penetrance, and how many of the mosaic mutations were inherited? How do the mutations identified interact with less well known gene mutations elsewhere in the genome to influence malignant transformation? How can the findings translate to the clinic?
The study highlights the fact that family history is insufficient to assess the likelihood of a cancer-predisposition syndrome in any patient with a newly diagnosed cancer.
Dr. John Maris is a pediatric oncologist in the division of oncology and Center for Childhood Cancer Research, Children’s Hospital of Philadelphia, and in the department of pediatrics at the Perelman School of Medicine, University of Pennsylvania. These remarks were part of an editorial accompanying the report (N Engl J Med. 2015 Nov. 18 doi: 10.1056/NEJMoa1508054). Dr. Maris reported having no disclosures.
Children and adolescents with cancer were found to have a significantly higher rate of germline mutations in cancer predisposing genes compared with individuals with no known cancer; however, family history of cancer did not predict the presence of a predisposition syndrome for most patients.
Of the 1,120 children with cancer, 8.5% had mutations in predisposition genes, compared with 1.1% in the control group. Mutations in TP53 were most common (50 patients), followed by APC (6), BRCA2 (6), NF1 (4), PMS2 (4), RB1 (3), and RUNX1 (3). Germline TP53 mutations were present in 27 of 39 patients (69%) with adrenocortical tumors, 9 of 47 (19%) with hypodiploid acute lymphoblastic leukemia, and 1 of 4 (25%) with choroid plexus carcinoma (N Engl J Med. 2015 Nov. 18. doi: 10.1056/NEJMoa1508054).
Only 40% of the pediatric patients with pathogenic germline mutations had a family history of cancer, and in just half of those cases the history was consistent with a known cancer-predisposition syndrome. Among patients without predisposition germline mutations, a similar proportion (42%) had a family history of cancer.
“On the basis of these observations, family history cannot be the sole indication used to guide the provision of genetic testing,” wrote Jinghui Zhang, Ph.D., of the department of computational biology, St. Jude’s Research Hospital, Memphis, Tennessee, and her colleagues.
Unexpected germline mutations were found in several cases. Six patients with Ewing’s sarcoma had unexpected pathogenic germline mutations (TP53 in four patients, PMS2 in one and RET in one). Eight patents had heterozygous mutations in BRCA1, BRCA2, or PALB2, supporting the notion that mutations in these genes may play a role in pediatric as well as adult cancer. Other new associations included germline APC and SDHB mutations with neuroblastoma, and APC, VHL, CDH1, PTCH1, and SDHA germline mutations with leukemia.
The St. Jude–Washington University Pediatric Cancer Genome Project (PCGP) included 1,120 patients representing the major types of pediatric cancer, including 53% with leukemia, 22% with CNS tumors, and 26% with non-CNS tumors. Whole genomes were sequenced from 595 patients, whole exomes (coding regions only) from 456 patients, and both whole genomes and exomes from 69 patients. Whole exomes were sequenced from two control cohorts of individuals with no known cancer: 966 from the 1,000 Genome project and 723 from the National Database for Autism Research (cancer predisposition genes known to be associated with autism, NF1 and PTEN, were excluded from analysis with this control set).
The study sequenced whole genomes and exomes, but focused most of the analysis on 60 autosomal dominant cancer predisposition genes. Tumor types with the highest prevalence of germline mutations in these genes were non-CNS solid tumors (48 of 287 patients, 17%) and CNS tumors (21 of 245, 9%). Among patients with adrenocortical tumors, 69% had germline mutations. Despite inclusion of hypodiploid acute lymphoblastic leukemia, the lowest germline mutation prevalence was found in leukemia (26 of 588, 4%).
Dr. Zhang reported having no disclosures. One coauthor reported financial ties to an industry source.
Children and adolescents with cancer were found to have a significantly higher rate of germline mutations in cancer predisposing genes compared with individuals with no known cancer; however, family history of cancer did not predict the presence of a predisposition syndrome for most patients.
Of the 1,120 children with cancer, 8.5% had mutations in predisposition genes, compared with 1.1% in the control group. Mutations in TP53 were most common (50 patients), followed by APC (6), BRCA2 (6), NF1 (4), PMS2 (4), RB1 (3), and RUNX1 (3). Germline TP53 mutations were present in 27 of 39 patients (69%) with adrenocortical tumors, 9 of 47 (19%) with hypodiploid acute lymphoblastic leukemia, and 1 of 4 (25%) with choroid plexus carcinoma (N Engl J Med. 2015 Nov. 18. doi: 10.1056/NEJMoa1508054).
Only 40% of the pediatric patients with pathogenic germline mutations had a family history of cancer, and in just half of those cases the history was consistent with a known cancer-predisposition syndrome. Among patients without predisposition germline mutations, a similar proportion (42%) had a family history of cancer.
“On the basis of these observations, family history cannot be the sole indication used to guide the provision of genetic testing,” wrote Jinghui Zhang, Ph.D., of the department of computational biology, St. Jude’s Research Hospital, Memphis, Tennessee, and her colleagues.
Unexpected germline mutations were found in several cases. Six patients with Ewing’s sarcoma had unexpected pathogenic germline mutations (TP53 in four patients, PMS2 in one and RET in one). Eight patents had heterozygous mutations in BRCA1, BRCA2, or PALB2, supporting the notion that mutations in these genes may play a role in pediatric as well as adult cancer. Other new associations included germline APC and SDHB mutations with neuroblastoma, and APC, VHL, CDH1, PTCH1, and SDHA germline mutations with leukemia.
The St. Jude–Washington University Pediatric Cancer Genome Project (PCGP) included 1,120 patients representing the major types of pediatric cancer, including 53% with leukemia, 22% with CNS tumors, and 26% with non-CNS tumors. Whole genomes were sequenced from 595 patients, whole exomes (coding regions only) from 456 patients, and both whole genomes and exomes from 69 patients. Whole exomes were sequenced from two control cohorts of individuals with no known cancer: 966 from the 1,000 Genome project and 723 from the National Database for Autism Research (cancer predisposition genes known to be associated with autism, NF1 and PTEN, were excluded from analysis with this control set).
The study sequenced whole genomes and exomes, but focused most of the analysis on 60 autosomal dominant cancer predisposition genes. Tumor types with the highest prevalence of germline mutations in these genes were non-CNS solid tumors (48 of 287 patients, 17%) and CNS tumors (21 of 245, 9%). Among patients with adrenocortical tumors, 69% had germline mutations. Despite inclusion of hypodiploid acute lymphoblastic leukemia, the lowest germline mutation prevalence was found in leukemia (26 of 588, 4%).
Dr. Zhang reported having no disclosures. One coauthor reported financial ties to an industry source.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Children and adolescents with cancer have a significantly higher rate of germline mutations in cancer predisposition genes compared with individuals with no known cancer.
Major finding: Of the 1,120 children with cancer, 8.5% had mutations in predisposition genes, compared with 1.1% in the control group.
Data source: The St. Jude–Washington University Pediatric Cancer Genome Project (PCGP) sequenced whole genomes of 595 patients, whole exomes of 456 patients, and both whole genomes and exomes of 69 patients; whole exomes were sequenced from two control cohorts of 966 and 723 individuals.
Disclosures: Dr. Zhang reported having no disclosures. One coauthor reported financial ties to an industry source.
What’s on tap at ASH 2015
FROM A TELECONFERENCE – The American Society of Hematology’s (ASH) 57th annual meeting in Orlando is chock-full of much-anticipated results in cancer immunotherapies such as CAR T cell therapies and checkpoint inhibitors, advances in sickle cell disease, and practical advice on managing the latest drugs in the clinic, ASH officials said in a teleconference. Here are some of the day-by-day picks selected by ASH president Dr. David Williams and ASH secretary Dr. Stephanie J. Lee, who gave their recommendations during a conference call for the press. Meeting abstracts are now available online.
Saturday, Dec. 5
Clinical applications of newly approved drugs
The popular special education session on clinical applications of newly approved drugs returns on Saturday, Dec. 5 at 9:30 a.m., with didactic presentations that address issues clinicians may face such as drug-drug interactions, side effects, and adverse events. The three drugs to be discussed this year are: idarucizumab (Praxbind), the first specific reversal agent approved for dabigatran reversal; blinatumomab (Blincyto), approved for second-line treatment of Philadelphia chromosomenegative acute lymphoblastic leukemia; and the histone deacetylase (HDAC) inhibitor panobinostat (Farydak), approved for the treatment of multiple myeloma.
Adoptive immunotherapy
One presentation to look out for next month is abstract 99at 12:30 p.m. on Saturday, Dec. 5 in the adoptive immunotherapy session, Dr. Williams told reporters. The chimeric antigen receptor (CAR)-T-cell approach has relied on genetically engineering the patient’s own T cells to rev up the immune system. This group’s approach is to treat B-cell malignancies after allogeneic hematopoietic stem cell transplantation using a single infusion of anti-CD19 CAR-T cells from the patient’s transplant donor.
Eight of 20 patients treated with this strategy achieved remission, including six complete remissions and two partial remissions. Importantly, none of these patients developed acute graft-versus-host disease, a potential consequence of using allogeneic rather than autologous T cells, he said. The authors also noted that patients who responded and went into remission were marked by higher numbers of these infused CAR-T cells in their circulation, suggesting a biomarker of response.
Checkpoint, please?
Immunotherapy is a “very hot area,” so ASH has put together a special session at 4 p.m. Saturday called “Checkpoint, Please?” Dr. Williams said. Topics include the role of programmed death (PD)-1 and PD-ligand 1 in acute and chronic graft-versus-host disease, checkpoint blockade with neoantigen cancer vaccines, and insights into the mechanisms of action of anti-CTLA-4 (cytotoxic T-lymphocyte–associated protein 4) antibody therapy.
Sunday, Dec. 6
Precision medicine
Sunday’s plenary scientific session will include several noteworthy personalized medicine abstracts featuring emerging therapies targeted to specific genetic subtypes, Dr. Lee, from the University of Washington, Seattle, said.
Plenary abstract 6 is a large, multinational study looking at whether adding the multikinase inhibitor midostaurin to standard induction therapy and carried through 1 year of maintenance would improve outcomes in newly diagnosed acute myeloid leukemia with FLT3 mutations. Patients with these deleterious mutations do enter remission with chemotherapy, but often relapse.
Overall and event-free survival were better at 5 years by about 7% to 8% in the experimental arm using midostaurin, she said. Caveats are that complete response rates were similar in both arms and lower than reported in other trials.
“Because we know that patients with this FLT3 mutation have a very poor prognosis with standard chemotherapy, more than half of the patients in this trial received an allogeneic transplant,” Dr. Lee noted. “But the abstract does say that the results are similar if you censor at the time of the transplant.”
In this same vein of precision medicine is plenary abstract 1, testing whether adding rituximab to standard chemotherapy improves outcomes in adults with CD-20–positive, Philadelphia chromosome–negative, B-cell precursor acute lymphoblastic leukemia (ALL). Rituximab (Rituxan) binds to CD-20, which is found in about 30% to 50% of adult B-cell ALL, she said.
At 2 years, patients treated with rituximab had longer event-free survival than controls (65% vs. 52%; P = .038), but similar overall survival (71% vs. 64%; P = .09), according to the abstract. The rituximab arm also received more allogeneic transplants, but again, after censoring the data, the abstract states that both event-free and overall survival were longer with rituximab, Dr. Lee said.
Sickle cell anemia
Sunday’s plenary session will also feature the very important TWiTCH (TCD with Transfusions Changing to Hydroxyurea) study evaluating hydroxyurea therapy as an alternative to chronic blood transfusions to prevent stroke. Stroke is one of the most dreaded complications of sickle cell disease, occurring in up to 10% of children, Dr. Williams said. Though transfusions are effective, they have to be continued indefinitely and lead to iron overload. Hydroxyurea increases the amount of fetal hemoglobin and fetal red blood cells and has become a standard therapy to attenuate the complications of sickle cell.
The phase III noninferiority study, which used Transcranial Doppler (TCD) screening to identify children at elevated risk for stroke, showed that hydroxyurea “was as good as current therapy with red cell transfusions and there was some indication, although not significant, that it might even be superior in lowering the TCD levels,” Dr. Williams said. An added benefit of the hydroxyurea was that it improved the patients’ iron overload status. There were no strokes in either group.
Sunday’s abstract 202 is another presentation “that I’m sure will get a lot of attention,” Dr. Williams said. It offers updated details on outcomes from patients with sickle cell disease (SCD) treated with a novel gene therapy transduced with the LentiGlobin BB305 (Bluebird Bio) lentiviral vector. Patients with beta thalassemia major have remained transfusion-independent for more than a year after this treatment, with results now available from four patients with SCD. One patient with a severe phenotype has had no sickle cell complications and has been able to stop his transfusion therapy, while two of the other four patients are also transfusion-independent.
“This is an early study showing what appears to be efficacy of the gene therapy approach not in thalassemia, but in sickle cell disease,” Dr. Williams said, noting that abstract 3233 will also feature results using LentiGlobin gene therapy in severe SCD.
ASH/EHA joint symposium
Also noteworthy is a special joint ASH/European Hematology Association symposium looking at how well genomic data are being incorporated into practice in the U.S. and Europe.
Monday, Dec. 7
ASH/FDA joint symposium
A joint ASH/FDA symposium on late-breaking drug approvals is new this year and features drugs that gained approval in November 2015. FDA product-reviewers will discuss safety and efficacy issues in the clinical approval trials and toxicity studies, while clinicians will share their experiences in the real-world use of these drugs.
“This is information that is really going to be very hot off the press and presented in conjunction with the FDA,” Dr. Lee said.
Dr. Williams reported research funding from Bluebird Bio. Dr. Lee reported having no conflicts of interest.
FROM A TELECONFERENCE – The American Society of Hematology’s (ASH) 57th annual meeting in Orlando is chock-full of much-anticipated results in cancer immunotherapies such as CAR T cell therapies and checkpoint inhibitors, advances in sickle cell disease, and practical advice on managing the latest drugs in the clinic, ASH officials said in a teleconference. Here are some of the day-by-day picks selected by ASH president Dr. David Williams and ASH secretary Dr. Stephanie J. Lee, who gave their recommendations during a conference call for the press. Meeting abstracts are now available online.
Saturday, Dec. 5
Clinical applications of newly approved drugs
The popular special education session on clinical applications of newly approved drugs returns on Saturday, Dec. 5 at 9:30 a.m., with didactic presentations that address issues clinicians may face such as drug-drug interactions, side effects, and adverse events. The three drugs to be discussed this year are: idarucizumab (Praxbind), the first specific reversal agent approved for dabigatran reversal; blinatumomab (Blincyto), approved for second-line treatment of Philadelphia chromosomenegative acute lymphoblastic leukemia; and the histone deacetylase (HDAC) inhibitor panobinostat (Farydak), approved for the treatment of multiple myeloma.
Adoptive immunotherapy
One presentation to look out for next month is abstract 99at 12:30 p.m. on Saturday, Dec. 5 in the adoptive immunotherapy session, Dr. Williams told reporters. The chimeric antigen receptor (CAR)-T-cell approach has relied on genetically engineering the patient’s own T cells to rev up the immune system. This group’s approach is to treat B-cell malignancies after allogeneic hematopoietic stem cell transplantation using a single infusion of anti-CD19 CAR-T cells from the patient’s transplant donor.
Eight of 20 patients treated with this strategy achieved remission, including six complete remissions and two partial remissions. Importantly, none of these patients developed acute graft-versus-host disease, a potential consequence of using allogeneic rather than autologous T cells, he said. The authors also noted that patients who responded and went into remission were marked by higher numbers of these infused CAR-T cells in their circulation, suggesting a biomarker of response.
Checkpoint, please?
Immunotherapy is a “very hot area,” so ASH has put together a special session at 4 p.m. Saturday called “Checkpoint, Please?” Dr. Williams said. Topics include the role of programmed death (PD)-1 and PD-ligand 1 in acute and chronic graft-versus-host disease, checkpoint blockade with neoantigen cancer vaccines, and insights into the mechanisms of action of anti-CTLA-4 (cytotoxic T-lymphocyte–associated protein 4) antibody therapy.
Sunday, Dec. 6
Precision medicine
Sunday’s plenary scientific session will include several noteworthy personalized medicine abstracts featuring emerging therapies targeted to specific genetic subtypes, Dr. Lee, from the University of Washington, Seattle, said.
Plenary abstract 6 is a large, multinational study looking at whether adding the multikinase inhibitor midostaurin to standard induction therapy and carried through 1 year of maintenance would improve outcomes in newly diagnosed acute myeloid leukemia with FLT3 mutations. Patients with these deleterious mutations do enter remission with chemotherapy, but often relapse.
Overall and event-free survival were better at 5 years by about 7% to 8% in the experimental arm using midostaurin, she said. Caveats are that complete response rates were similar in both arms and lower than reported in other trials.
“Because we know that patients with this FLT3 mutation have a very poor prognosis with standard chemotherapy, more than half of the patients in this trial received an allogeneic transplant,” Dr. Lee noted. “But the abstract does say that the results are similar if you censor at the time of the transplant.”
In this same vein of precision medicine is plenary abstract 1, testing whether adding rituximab to standard chemotherapy improves outcomes in adults with CD-20–positive, Philadelphia chromosome–negative, B-cell precursor acute lymphoblastic leukemia (ALL). Rituximab (Rituxan) binds to CD-20, which is found in about 30% to 50% of adult B-cell ALL, she said.
At 2 years, patients treated with rituximab had longer event-free survival than controls (65% vs. 52%; P = .038), but similar overall survival (71% vs. 64%; P = .09), according to the abstract. The rituximab arm also received more allogeneic transplants, but again, after censoring the data, the abstract states that both event-free and overall survival were longer with rituximab, Dr. Lee said.
Sickle cell anemia
Sunday’s plenary session will also feature the very important TWiTCH (TCD with Transfusions Changing to Hydroxyurea) study evaluating hydroxyurea therapy as an alternative to chronic blood transfusions to prevent stroke. Stroke is one of the most dreaded complications of sickle cell disease, occurring in up to 10% of children, Dr. Williams said. Though transfusions are effective, they have to be continued indefinitely and lead to iron overload. Hydroxyurea increases the amount of fetal hemoglobin and fetal red blood cells and has become a standard therapy to attenuate the complications of sickle cell.
The phase III noninferiority study, which used Transcranial Doppler (TCD) screening to identify children at elevated risk for stroke, showed that hydroxyurea “was as good as current therapy with red cell transfusions and there was some indication, although not significant, that it might even be superior in lowering the TCD levels,” Dr. Williams said. An added benefit of the hydroxyurea was that it improved the patients’ iron overload status. There were no strokes in either group.
Sunday’s abstract 202 is another presentation “that I’m sure will get a lot of attention,” Dr. Williams said. It offers updated details on outcomes from patients with sickle cell disease (SCD) treated with a novel gene therapy transduced with the LentiGlobin BB305 (Bluebird Bio) lentiviral vector. Patients with beta thalassemia major have remained transfusion-independent for more than a year after this treatment, with results now available from four patients with SCD. One patient with a severe phenotype has had no sickle cell complications and has been able to stop his transfusion therapy, while two of the other four patients are also transfusion-independent.
“This is an early study showing what appears to be efficacy of the gene therapy approach not in thalassemia, but in sickle cell disease,” Dr. Williams said, noting that abstract 3233 will also feature results using LentiGlobin gene therapy in severe SCD.
ASH/EHA joint symposium
Also noteworthy is a special joint ASH/European Hematology Association symposium looking at how well genomic data are being incorporated into practice in the U.S. and Europe.
Monday, Dec. 7
ASH/FDA joint symposium
A joint ASH/FDA symposium on late-breaking drug approvals is new this year and features drugs that gained approval in November 2015. FDA product-reviewers will discuss safety and efficacy issues in the clinical approval trials and toxicity studies, while clinicians will share their experiences in the real-world use of these drugs.
“This is information that is really going to be very hot off the press and presented in conjunction with the FDA,” Dr. Lee said.
Dr. Williams reported research funding from Bluebird Bio. Dr. Lee reported having no conflicts of interest.
FROM A TELECONFERENCE – The American Society of Hematology’s (ASH) 57th annual meeting in Orlando is chock-full of much-anticipated results in cancer immunotherapies such as CAR T cell therapies and checkpoint inhibitors, advances in sickle cell disease, and practical advice on managing the latest drugs in the clinic, ASH officials said in a teleconference. Here are some of the day-by-day picks selected by ASH president Dr. David Williams and ASH secretary Dr. Stephanie J. Lee, who gave their recommendations during a conference call for the press. Meeting abstracts are now available online.
Saturday, Dec. 5
Clinical applications of newly approved drugs
The popular special education session on clinical applications of newly approved drugs returns on Saturday, Dec. 5 at 9:30 a.m., with didactic presentations that address issues clinicians may face such as drug-drug interactions, side effects, and adverse events. The three drugs to be discussed this year are: idarucizumab (Praxbind), the first specific reversal agent approved for dabigatran reversal; blinatumomab (Blincyto), approved for second-line treatment of Philadelphia chromosomenegative acute lymphoblastic leukemia; and the histone deacetylase (HDAC) inhibitor panobinostat (Farydak), approved for the treatment of multiple myeloma.
Adoptive immunotherapy
One presentation to look out for next month is abstract 99at 12:30 p.m. on Saturday, Dec. 5 in the adoptive immunotherapy session, Dr. Williams told reporters. The chimeric antigen receptor (CAR)-T-cell approach has relied on genetically engineering the patient’s own T cells to rev up the immune system. This group’s approach is to treat B-cell malignancies after allogeneic hematopoietic stem cell transplantation using a single infusion of anti-CD19 CAR-T cells from the patient’s transplant donor.
Eight of 20 patients treated with this strategy achieved remission, including six complete remissions and two partial remissions. Importantly, none of these patients developed acute graft-versus-host disease, a potential consequence of using allogeneic rather than autologous T cells, he said. The authors also noted that patients who responded and went into remission were marked by higher numbers of these infused CAR-T cells in their circulation, suggesting a biomarker of response.
Checkpoint, please?
Immunotherapy is a “very hot area,” so ASH has put together a special session at 4 p.m. Saturday called “Checkpoint, Please?” Dr. Williams said. Topics include the role of programmed death (PD)-1 and PD-ligand 1 in acute and chronic graft-versus-host disease, checkpoint blockade with neoantigen cancer vaccines, and insights into the mechanisms of action of anti-CTLA-4 (cytotoxic T-lymphocyte–associated protein 4) antibody therapy.
Sunday, Dec. 6
Precision medicine
Sunday’s plenary scientific session will include several noteworthy personalized medicine abstracts featuring emerging therapies targeted to specific genetic subtypes, Dr. Lee, from the University of Washington, Seattle, said.
Plenary abstract 6 is a large, multinational study looking at whether adding the multikinase inhibitor midostaurin to standard induction therapy and carried through 1 year of maintenance would improve outcomes in newly diagnosed acute myeloid leukemia with FLT3 mutations. Patients with these deleterious mutations do enter remission with chemotherapy, but often relapse.
Overall and event-free survival were better at 5 years by about 7% to 8% in the experimental arm using midostaurin, she said. Caveats are that complete response rates were similar in both arms and lower than reported in other trials.
“Because we know that patients with this FLT3 mutation have a very poor prognosis with standard chemotherapy, more than half of the patients in this trial received an allogeneic transplant,” Dr. Lee noted. “But the abstract does say that the results are similar if you censor at the time of the transplant.”
In this same vein of precision medicine is plenary abstract 1, testing whether adding rituximab to standard chemotherapy improves outcomes in adults with CD-20–positive, Philadelphia chromosome–negative, B-cell precursor acute lymphoblastic leukemia (ALL). Rituximab (Rituxan) binds to CD-20, which is found in about 30% to 50% of adult B-cell ALL, she said.
At 2 years, patients treated with rituximab had longer event-free survival than controls (65% vs. 52%; P = .038), but similar overall survival (71% vs. 64%; P = .09), according to the abstract. The rituximab arm also received more allogeneic transplants, but again, after censoring the data, the abstract states that both event-free and overall survival were longer with rituximab, Dr. Lee said.
Sickle cell anemia
Sunday’s plenary session will also feature the very important TWiTCH (TCD with Transfusions Changing to Hydroxyurea) study evaluating hydroxyurea therapy as an alternative to chronic blood transfusions to prevent stroke. Stroke is one of the most dreaded complications of sickle cell disease, occurring in up to 10% of children, Dr. Williams said. Though transfusions are effective, they have to be continued indefinitely and lead to iron overload. Hydroxyurea increases the amount of fetal hemoglobin and fetal red blood cells and has become a standard therapy to attenuate the complications of sickle cell.
The phase III noninferiority study, which used Transcranial Doppler (TCD) screening to identify children at elevated risk for stroke, showed that hydroxyurea “was as good as current therapy with red cell transfusions and there was some indication, although not significant, that it might even be superior in lowering the TCD levels,” Dr. Williams said. An added benefit of the hydroxyurea was that it improved the patients’ iron overload status. There were no strokes in either group.
Sunday’s abstract 202 is another presentation “that I’m sure will get a lot of attention,” Dr. Williams said. It offers updated details on outcomes from patients with sickle cell disease (SCD) treated with a novel gene therapy transduced with the LentiGlobin BB305 (Bluebird Bio) lentiviral vector. Patients with beta thalassemia major have remained transfusion-independent for more than a year after this treatment, with results now available from four patients with SCD. One patient with a severe phenotype has had no sickle cell complications and has been able to stop his transfusion therapy, while two of the other four patients are also transfusion-independent.
“This is an early study showing what appears to be efficacy of the gene therapy approach not in thalassemia, but in sickle cell disease,” Dr. Williams said, noting that abstract 3233 will also feature results using LentiGlobin gene therapy in severe SCD.
ASH/EHA joint symposium
Also noteworthy is a special joint ASH/European Hematology Association symposium looking at how well genomic data are being incorporated into practice in the U.S. and Europe.
Monday, Dec. 7
ASH/FDA joint symposium
A joint ASH/FDA symposium on late-breaking drug approvals is new this year and features drugs that gained approval in November 2015. FDA product-reviewers will discuss safety and efficacy issues in the clinical approval trials and toxicity studies, while clinicians will share their experiences in the real-world use of these drugs.
“This is information that is really going to be very hot off the press and presented in conjunction with the FDA,” Dr. Lee said.
Dr. Williams reported research funding from Bluebird Bio. Dr. Lee reported having no conflicts of interest.
Donor CAR T cells treat aggressive ALL in infant
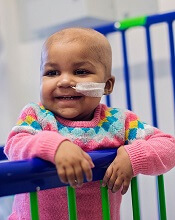
person treated with UCART19
Photo courtesy of GOSH
A hospital in the UK has reported success with the first-in-human use of an allogeneic chimeric antigen receptor (CAR) T-cell treatment.
The therapy, UCART19, helped treat an aggressive case of acute lymphoblastic leukemia (ALL) in an infant named Layla Richards.
Chemotherapy, a first transplant, and blinatumomab all failed to treat Layla’s disease. But UCART19 provided a bridge to a second transplant, and Layla
is now free of leukemia.
“We have only used this treatment on one very strong little girl, and we have to be cautious about claiming that this will be a suitable treatment option for all children,” said Waseem Qasim, MBBS, PhD, a professor at University College London’s Institute of Child Health and a consultant immunologist at Great Ormond Street Hospital (GOSH) in London, where Layla was treated.
“But this is a landmark in the use of new gene-engineering technology, and the effects for this child have been staggering. If replicated, it could represent a huge step forward in treating leukemia and other cancers.”
Layla’s history
Layla was born in June 2014 and, at 14 weeks old, was diagnosed with CD19+ ALL (t[11;19] rearrangement). Doctors at GOSH described her leukemia as “one of the most aggressive forms of the disease we have ever seen.”
Layla underwent several rounds of chemotherapy at GOSH and then received a transplant from a mismatched, unrelated donor. Seven weeks later, her leukemia had returned.
A second round of chemotherapy wasn’t an option, so Layla went on to receive blinatumomab. This, too, ultimately failed.
Layla’s family was unwilling to accept palliative care, so her doctors mentioned the possibility of using UCART19.
“The approach was looking incredibly successful in laboratory studies, and so when I heard there were no options left for treating this child’s disease, I thought, ‘Why don’t we use the new UCART19 cells?’” Dr Qasim said.
“The treatment was highly experimental, and we had to get special permissions, but she appeared ideally suited for this type of approach.”
UCART19 treatment
Before Layla received UCART19, investigators from GOSH, University College London, and the biotech company Cellectis had been developing off-the-shelf banks of the cells for use in clinical trials.
UCART19 consists of donor T cells modified using transcription activator-like effector nucleases (TALEN). Like some other CAR T-cell products, UCART19 is designed to target CD19+ cancer cells.
But UCART19 cells are also programmed to be insensitive to alemtuzumab. That way, a patient can receive the drug to prevent rejection of HLA-mismatched cells.
Before receiving UCART19, Layla was given vincristine, dexamethasone, and asparaginase, followed by fludarabine, cyclophosphamide, and alemtuzumab. She then received a single dose (4.5 x 106/kg) of UCART19.
After this, Layla spent several months in isolation to protect her from infections while her immune system was extremely weak. Within weeks of receiving UCART19, Layla demonstrated an immune response in the form of a rash.
The rash worsened, but, aside from that, Layla appeared to be well. To date, she has shown no signs of cytokine release syndrome.
She did, however, show signs of molecular and cytogenetic remission. Once she was deemed leukemia-free, Layla underwent a second transplant. One month later, she was well enough to go home. Today, Layla is still free of ALL.
More details on Layla’s story and UCART19 are scheduled to be presented at the 2015 ASH Annual Meeting (abstract 2046). Clinical trials of UCART19 (funded by Cellectis) are set to begin early next year. ![]()
person treated with UCART19
Photo courtesy of GOSH
A hospital in the UK has reported success with the first-in-human use of an allogeneic chimeric antigen receptor (CAR) T-cell treatment.
The therapy, UCART19, helped treat an aggressive case of acute lymphoblastic leukemia (ALL) in an infant named Layla Richards.
Chemotherapy, a first transplant, and blinatumomab all failed to treat Layla’s disease. But UCART19 provided a bridge to a second transplant, and Layla
is now free of leukemia.
“We have only used this treatment on one very strong little girl, and we have to be cautious about claiming that this will be a suitable treatment option for all children,” said Waseem Qasim, MBBS, PhD, a professor at University College London’s Institute of Child Health and a consultant immunologist at Great Ormond Street Hospital (GOSH) in London, where Layla was treated.
“But this is a landmark in the use of new gene-engineering technology, and the effects for this child have been staggering. If replicated, it could represent a huge step forward in treating leukemia and other cancers.”
Layla’s history
Layla was born in June 2014 and, at 14 weeks old, was diagnosed with CD19+ ALL (t[11;19] rearrangement). Doctors at GOSH described her leukemia as “one of the most aggressive forms of the disease we have ever seen.”
Layla underwent several rounds of chemotherapy at GOSH and then received a transplant from a mismatched, unrelated donor. Seven weeks later, her leukemia had returned.
A second round of chemotherapy wasn’t an option, so Layla went on to receive blinatumomab. This, too, ultimately failed.
Layla’s family was unwilling to accept palliative care, so her doctors mentioned the possibility of using UCART19.
“The approach was looking incredibly successful in laboratory studies, and so when I heard there were no options left for treating this child’s disease, I thought, ‘Why don’t we use the new UCART19 cells?’” Dr Qasim said.
“The treatment was highly experimental, and we had to get special permissions, but she appeared ideally suited for this type of approach.”
UCART19 treatment
Before Layla received UCART19, investigators from GOSH, University College London, and the biotech company Cellectis had been developing off-the-shelf banks of the cells for use in clinical trials.
UCART19 consists of donor T cells modified using transcription activator-like effector nucleases (TALEN). Like some other CAR T-cell products, UCART19 is designed to target CD19+ cancer cells.
But UCART19 cells are also programmed to be insensitive to alemtuzumab. That way, a patient can receive the drug to prevent rejection of HLA-mismatched cells.
Before receiving UCART19, Layla was given vincristine, dexamethasone, and asparaginase, followed by fludarabine, cyclophosphamide, and alemtuzumab. She then received a single dose (4.5 x 106/kg) of UCART19.
After this, Layla spent several months in isolation to protect her from infections while her immune system was extremely weak. Within weeks of receiving UCART19, Layla demonstrated an immune response in the form of a rash.
The rash worsened, but, aside from that, Layla appeared to be well. To date, she has shown no signs of cytokine release syndrome.
She did, however, show signs of molecular and cytogenetic remission. Once she was deemed leukemia-free, Layla underwent a second transplant. One month later, she was well enough to go home. Today, Layla is still free of ALL.
More details on Layla’s story and UCART19 are scheduled to be presented at the 2015 ASH Annual Meeting (abstract 2046). Clinical trials of UCART19 (funded by Cellectis) are set to begin early next year. ![]()
person treated with UCART19
Photo courtesy of GOSH
A hospital in the UK has reported success with the first-in-human use of an allogeneic chimeric antigen receptor (CAR) T-cell treatment.
The therapy, UCART19, helped treat an aggressive case of acute lymphoblastic leukemia (ALL) in an infant named Layla Richards.
Chemotherapy, a first transplant, and blinatumomab all failed to treat Layla’s disease. But UCART19 provided a bridge to a second transplant, and Layla
is now free of leukemia.
“We have only used this treatment on one very strong little girl, and we have to be cautious about claiming that this will be a suitable treatment option for all children,” said Waseem Qasim, MBBS, PhD, a professor at University College London’s Institute of Child Health and a consultant immunologist at Great Ormond Street Hospital (GOSH) in London, where Layla was treated.
“But this is a landmark in the use of new gene-engineering technology, and the effects for this child have been staggering. If replicated, it could represent a huge step forward in treating leukemia and other cancers.”
Layla’s history
Layla was born in June 2014 and, at 14 weeks old, was diagnosed with CD19+ ALL (t[11;19] rearrangement). Doctors at GOSH described her leukemia as “one of the most aggressive forms of the disease we have ever seen.”
Layla underwent several rounds of chemotherapy at GOSH and then received a transplant from a mismatched, unrelated donor. Seven weeks later, her leukemia had returned.
A second round of chemotherapy wasn’t an option, so Layla went on to receive blinatumomab. This, too, ultimately failed.
Layla’s family was unwilling to accept palliative care, so her doctors mentioned the possibility of using UCART19.
“The approach was looking incredibly successful in laboratory studies, and so when I heard there were no options left for treating this child’s disease, I thought, ‘Why don’t we use the new UCART19 cells?’” Dr Qasim said.
“The treatment was highly experimental, and we had to get special permissions, but she appeared ideally suited for this type of approach.”
UCART19 treatment
Before Layla received UCART19, investigators from GOSH, University College London, and the biotech company Cellectis had been developing off-the-shelf banks of the cells for use in clinical trials.
UCART19 consists of donor T cells modified using transcription activator-like effector nucleases (TALEN). Like some other CAR T-cell products, UCART19 is designed to target CD19+ cancer cells.
But UCART19 cells are also programmed to be insensitive to alemtuzumab. That way, a patient can receive the drug to prevent rejection of HLA-mismatched cells.
Before receiving UCART19, Layla was given vincristine, dexamethasone, and asparaginase, followed by fludarabine, cyclophosphamide, and alemtuzumab. She then received a single dose (4.5 x 106/kg) of UCART19.
After this, Layla spent several months in isolation to protect her from infections while her immune system was extremely weak. Within weeks of receiving UCART19, Layla demonstrated an immune response in the form of a rash.
The rash worsened, but, aside from that, Layla appeared to be well. To date, she has shown no signs of cytokine release syndrome.
She did, however, show signs of molecular and cytogenetic remission. Once she was deemed leukemia-free, Layla underwent a second transplant. One month later, she was well enough to go home. Today, Layla is still free of ALL.
More details on Layla’s story and UCART19 are scheduled to be presented at the 2015 ASH Annual Meeting (abstract 2046). Clinical trials of UCART19 (funded by Cellectis) are set to begin early next year. ![]()
Alternative splicing enables resistance to CTL019

Photo from Penn Medicine
New research has provided an explanation for resistance to CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy.
Investigators analyzed samples from children with B-cell acute lymphoblastic leukemia (B-ALL) and found evidence to suggest that CTL019 resistance can be caused by CD19 splicing alterations.
These alterations prompt the loss of certain parts of the CD19 protein that are recognized by the CAR T cells.
The team described this work in Cancer Discovery.
They noted that 10% to 20% of B-ALL patients treated with CD19-directed immunotherapy may experience relapse.
“Some of them can be successfully retreated, but, in others, a more pernicious kind of leukemia may emerge, which no longer responds to CTL019,” said study author Andrei Thomas-Tikhonenko, PhD, of the University of Pennsylvania in Philadelphia.
“In some cases, resistance is accompanied by the disappearance of the target CD19 protein from the cell surface . . . . Our goal was to figure out how the CD19 protein manages to vanish and whether it is gone for good or whether it could, under certain circumstances, be coaxed back.”
“Our initial finding from this study was that, in most cases, the CD19 genetic code was not irretrievably lost. We also discovered that the CD19 protein was still being made, but as a shorter version, which escapes detection by the immune system.”
To understand the mechanism of CTL019 resistance, Dr Thomas-Tikhonenko and his colleagues studied multiple tumor samples from 4 children with B-ALL. The samples were collected before the patients were treated with CTL019 and/or after they developed resistance to the therapy.
The investigators found that, in some cases, 1 copy of the gene coding for CD19 (located on chromosome 16) was deleted, and the other copy was damaged as a result of mutations in coding areas of the CD19 gene, most frequently in exon 2.
However, the team also discovered alternatively spliced CD19 messenger RNA species in which exons 2, 5, and 6 were frequently skipped, making mutations in exon 2 largely irrelevant.
Subsequent investigation revealed that deletion of exons 5 and 6 resulted in premature termination of CD19.
Deletion of exon 2 resulted in the production of a modified version of CD19, which was more stable than its standard version. The shortened protein was functional and could perform many of the tasks that CD19 is known to handle, but it cannot be targeted by CTL019.
The importance of exon skipping in CTL019 resistance cannot be overstated, Dr Thomas-Tikhonenko said.
“Without exons 5 and 6, the CD19 protein has no way of being retained on the cell surface,” he explained. “The case of missing exon 2 is more complex. Although the resultant protein can make it to the cell surface, albeit not very efficiently, it can no longer be recognized by CTL019.”
He and his colleagues believe this research can inform future use of CTL019 and immunotherapy in general.
“[A]lternative splicing could be a potent, built-in mechanism of resistance, and it might be better to target proteins that, unlike CD19, are not prone to exon skipping,” Dr Thomas-Tikhonenko said.
“[In addition,] it might be important to preselect patients for CTL019 and similar therapies and make sure that the alternatively spliced CD19 variants are not already present in their leukemias. If they are, resistance could develop very quickly.”
Designing new immunotherapeutics that can recognize the shortened version of CD19 is another approach to overcoming CTL019 resistance, he added.
He and his colleagues noted that this study was limited by the relatively small number of samples analyzed, which might have prevented the investigators from identifying additional mechanisms of resistance. ![]()
Photo from Penn Medicine
New research has provided an explanation for resistance to CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy.
Investigators analyzed samples from children with B-cell acute lymphoblastic leukemia (B-ALL) and found evidence to suggest that CTL019 resistance can be caused by CD19 splicing alterations.
These alterations prompt the loss of certain parts of the CD19 protein that are recognized by the CAR T cells.
The team described this work in Cancer Discovery.
They noted that 10% to 20% of B-ALL patients treated with CD19-directed immunotherapy may experience relapse.
“Some of them can be successfully retreated, but, in others, a more pernicious kind of leukemia may emerge, which no longer responds to CTL019,” said study author Andrei Thomas-Tikhonenko, PhD, of the University of Pennsylvania in Philadelphia.
“In some cases, resistance is accompanied by the disappearance of the target CD19 protein from the cell surface . . . . Our goal was to figure out how the CD19 protein manages to vanish and whether it is gone for good or whether it could, under certain circumstances, be coaxed back.”
“Our initial finding from this study was that, in most cases, the CD19 genetic code was not irretrievably lost. We also discovered that the CD19 protein was still being made, but as a shorter version, which escapes detection by the immune system.”
To understand the mechanism of CTL019 resistance, Dr Thomas-Tikhonenko and his colleagues studied multiple tumor samples from 4 children with B-ALL. The samples were collected before the patients were treated with CTL019 and/or after they developed resistance to the therapy.
The investigators found that, in some cases, 1 copy of the gene coding for CD19 (located on chromosome 16) was deleted, and the other copy was damaged as a result of mutations in coding areas of the CD19 gene, most frequently in exon 2.
However, the team also discovered alternatively spliced CD19 messenger RNA species in which exons 2, 5, and 6 were frequently skipped, making mutations in exon 2 largely irrelevant.
Subsequent investigation revealed that deletion of exons 5 and 6 resulted in premature termination of CD19.
Deletion of exon 2 resulted in the production of a modified version of CD19, which was more stable than its standard version. The shortened protein was functional and could perform many of the tasks that CD19 is known to handle, but it cannot be targeted by CTL019.
The importance of exon skipping in CTL019 resistance cannot be overstated, Dr Thomas-Tikhonenko said.
“Without exons 5 and 6, the CD19 protein has no way of being retained on the cell surface,” he explained. “The case of missing exon 2 is more complex. Although the resultant protein can make it to the cell surface, albeit not very efficiently, it can no longer be recognized by CTL019.”
He and his colleagues believe this research can inform future use of CTL019 and immunotherapy in general.
“[A]lternative splicing could be a potent, built-in mechanism of resistance, and it might be better to target proteins that, unlike CD19, are not prone to exon skipping,” Dr Thomas-Tikhonenko said.
“[In addition,] it might be important to preselect patients for CTL019 and similar therapies and make sure that the alternatively spliced CD19 variants are not already present in their leukemias. If they are, resistance could develop very quickly.”
Designing new immunotherapeutics that can recognize the shortened version of CD19 is another approach to overcoming CTL019 resistance, he added.
He and his colleagues noted that this study was limited by the relatively small number of samples analyzed, which might have prevented the investigators from identifying additional mechanisms of resistance. ![]()
Photo from Penn Medicine
New research has provided an explanation for resistance to CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy.
Investigators analyzed samples from children with B-cell acute lymphoblastic leukemia (B-ALL) and found evidence to suggest that CTL019 resistance can be caused by CD19 splicing alterations.
These alterations prompt the loss of certain parts of the CD19 protein that are recognized by the CAR T cells.
The team described this work in Cancer Discovery.
They noted that 10% to 20% of B-ALL patients treated with CD19-directed immunotherapy may experience relapse.
“Some of them can be successfully retreated, but, in others, a more pernicious kind of leukemia may emerge, which no longer responds to CTL019,” said study author Andrei Thomas-Tikhonenko, PhD, of the University of Pennsylvania in Philadelphia.
“In some cases, resistance is accompanied by the disappearance of the target CD19 protein from the cell surface . . . . Our goal was to figure out how the CD19 protein manages to vanish and whether it is gone for good or whether it could, under certain circumstances, be coaxed back.”
“Our initial finding from this study was that, in most cases, the CD19 genetic code was not irretrievably lost. We also discovered that the CD19 protein was still being made, but as a shorter version, which escapes detection by the immune system.”
To understand the mechanism of CTL019 resistance, Dr Thomas-Tikhonenko and his colleagues studied multiple tumor samples from 4 children with B-ALL. The samples were collected before the patients were treated with CTL019 and/or after they developed resistance to the therapy.
The investigators found that, in some cases, 1 copy of the gene coding for CD19 (located on chromosome 16) was deleted, and the other copy was damaged as a result of mutations in coding areas of the CD19 gene, most frequently in exon 2.
However, the team also discovered alternatively spliced CD19 messenger RNA species in which exons 2, 5, and 6 were frequently skipped, making mutations in exon 2 largely irrelevant.
Subsequent investigation revealed that deletion of exons 5 and 6 resulted in premature termination of CD19.
Deletion of exon 2 resulted in the production of a modified version of CD19, which was more stable than its standard version. The shortened protein was functional and could perform many of the tasks that CD19 is known to handle, but it cannot be targeted by CTL019.
The importance of exon skipping in CTL019 resistance cannot be overstated, Dr Thomas-Tikhonenko said.
“Without exons 5 and 6, the CD19 protein has no way of being retained on the cell surface,” he explained. “The case of missing exon 2 is more complex. Although the resultant protein can make it to the cell surface, albeit not very efficiently, it can no longer be recognized by CTL019.”
He and his colleagues believe this research can inform future use of CTL019 and immunotherapy in general.
“[A]lternative splicing could be a potent, built-in mechanism of resistance, and it might be better to target proteins that, unlike CD19, are not prone to exon skipping,” Dr Thomas-Tikhonenko said.
“[In addition,] it might be important to preselect patients for CTL019 and similar therapies and make sure that the alternatively spliced CD19 variants are not already present in their leukemias. If they are, resistance could develop very quickly.”
Designing new immunotherapeutics that can recognize the shortened version of CD19 is another approach to overcoming CTL019 resistance, he added.
He and his colleagues noted that this study was limited by the relatively small number of samples analyzed, which might have prevented the investigators from identifying additional mechanisms of resistance. ![]()
Germline genetic variation linked to pediatric ALL

Photo courtesy of
St. Jude/Seth Dixon
Germline variations in the ETV6 gene are associated with an increased risk of developing pediatric acute lymphoblastic leukemia (ALL), according to research published in The Lancet Oncology.
Researchers said the magnitude of the risk must still be determined, as well as how the variants identified may promote ALL.
The evidence suggests that ETV6 variation alone is not sufficient to cause ALL but may play a significant role in inherited predisposition to childhood ALL.
The researchers discovered the association between the ETV6 variants and childhood ALL by sequencing the whole exome of a family in which the mother and 2 of the 3 children have a history of pediatric ALL.
All were treated at St. Jude Children’s Research Hospital in Memphis, Tennessee, and are now cancer-free.
The researchers identified a novel non-sense ETV6 variant (p.Arg359X) in this mother and her 3 children, including a daughter who has not been diagnosed with cancer. The father does not have the variant.
This variant is predicted to result in the production of a shortened ETV6 protein that cannot fulfill its normal function of binding to DNA and regulating the expression of other genes.
The researchers screened an additional 4405 children with ALL and found 31 ETV6 exonic variants—21 missense, 5 frameshift, 4 non-sense, and 1 splice site—that are potentially related to leukemia risk in 35 patients, or almost 1% of the patients screened.
The variants identified were unique to ALL patients or extremely rare in the general population, the researchers said.
Patients with the variants tended to be older when diagnosed with ALL (10.2 years vs 4.7 years; P=0.017) and were more likely to have hyperdiploid leukemia. Sixty-four percent of ALL cases with germline ETV6 variants were hyperdiploid, compared to 27% of ALL cases without the variants (P=0.0050).
The variants were not associated with a particular ethnicity or with the outcome of ALL therapy.
The researchers also noted that almost half of the ETV6 variants identified (n=15) clustered in the erythroblast transformation specific domain.
“That suggests the loss or alteration of this DNA-binding function of ETV6 may be critical to cancer promotion,” said study author Jun J. Yang, PhD, of St. Jude.
“This is the latest example of the important role that genetic variation and inheritance plays in ALL risk. That has clear clinical implications and will help us understand the biology driving this cancer.”
These findings build on previous work that revealed an association between inherited ETV6 variations and thrombocytopenia in families with a susceptibility to hematologic malignancies. The researchers said this new study further solidifies the association between ETV6 and pediatric ALL. ![]()
Photo courtesy of
St. Jude/Seth Dixon
Germline variations in the ETV6 gene are associated with an increased risk of developing pediatric acute lymphoblastic leukemia (ALL), according to research published in The Lancet Oncology.
Researchers said the magnitude of the risk must still be determined, as well as how the variants identified may promote ALL.
The evidence suggests that ETV6 variation alone is not sufficient to cause ALL but may play a significant role in inherited predisposition to childhood ALL.
The researchers discovered the association between the ETV6 variants and childhood ALL by sequencing the whole exome of a family in which the mother and 2 of the 3 children have a history of pediatric ALL.
All were treated at St. Jude Children’s Research Hospital in Memphis, Tennessee, and are now cancer-free.
The researchers identified a novel non-sense ETV6 variant (p.Arg359X) in this mother and her 3 children, including a daughter who has not been diagnosed with cancer. The father does not have the variant.
This variant is predicted to result in the production of a shortened ETV6 protein that cannot fulfill its normal function of binding to DNA and regulating the expression of other genes.
The researchers screened an additional 4405 children with ALL and found 31 ETV6 exonic variants—21 missense, 5 frameshift, 4 non-sense, and 1 splice site—that are potentially related to leukemia risk in 35 patients, or almost 1% of the patients screened.
The variants identified were unique to ALL patients or extremely rare in the general population, the researchers said.
Patients with the variants tended to be older when diagnosed with ALL (10.2 years vs 4.7 years; P=0.017) and were more likely to have hyperdiploid leukemia. Sixty-four percent of ALL cases with germline ETV6 variants were hyperdiploid, compared to 27% of ALL cases without the variants (P=0.0050).
The variants were not associated with a particular ethnicity or with the outcome of ALL therapy.
The researchers also noted that almost half of the ETV6 variants identified (n=15) clustered in the erythroblast transformation specific domain.
“That suggests the loss or alteration of this DNA-binding function of ETV6 may be critical to cancer promotion,” said study author Jun J. Yang, PhD, of St. Jude.
“This is the latest example of the important role that genetic variation and inheritance plays in ALL risk. That has clear clinical implications and will help us understand the biology driving this cancer.”
These findings build on previous work that revealed an association between inherited ETV6 variations and thrombocytopenia in families with a susceptibility to hematologic malignancies. The researchers said this new study further solidifies the association between ETV6 and pediatric ALL. ![]()
Photo courtesy of
St. Jude/Seth Dixon
Germline variations in the ETV6 gene are associated with an increased risk of developing pediatric acute lymphoblastic leukemia (ALL), according to research published in The Lancet Oncology.
Researchers said the magnitude of the risk must still be determined, as well as how the variants identified may promote ALL.
The evidence suggests that ETV6 variation alone is not sufficient to cause ALL but may play a significant role in inherited predisposition to childhood ALL.
The researchers discovered the association between the ETV6 variants and childhood ALL by sequencing the whole exome of a family in which the mother and 2 of the 3 children have a history of pediatric ALL.
All were treated at St. Jude Children’s Research Hospital in Memphis, Tennessee, and are now cancer-free.
The researchers identified a novel non-sense ETV6 variant (p.Arg359X) in this mother and her 3 children, including a daughter who has not been diagnosed with cancer. The father does not have the variant.
This variant is predicted to result in the production of a shortened ETV6 protein that cannot fulfill its normal function of binding to DNA and regulating the expression of other genes.
The researchers screened an additional 4405 children with ALL and found 31 ETV6 exonic variants—21 missense, 5 frameshift, 4 non-sense, and 1 splice site—that are potentially related to leukemia risk in 35 patients, or almost 1% of the patients screened.
The variants identified were unique to ALL patients or extremely rare in the general population, the researchers said.
Patients with the variants tended to be older when diagnosed with ALL (10.2 years vs 4.7 years; P=0.017) and were more likely to have hyperdiploid leukemia. Sixty-four percent of ALL cases with germline ETV6 variants were hyperdiploid, compared to 27% of ALL cases without the variants (P=0.0050).
The variants were not associated with a particular ethnicity or with the outcome of ALL therapy.
The researchers also noted that almost half of the ETV6 variants identified (n=15) clustered in the erythroblast transformation specific domain.
“That suggests the loss or alteration of this DNA-binding function of ETV6 may be critical to cancer promotion,” said study author Jun J. Yang, PhD, of St. Jude.
“This is the latest example of the important role that genetic variation and inheritance plays in ALL risk. That has clear clinical implications and will help us understand the biology driving this cancer.”
These findings build on previous work that revealed an association between inherited ETV6 variations and thrombocytopenia in families with a susceptibility to hematologic malignancies. The researchers said this new study further solidifies the association between ETV6 and pediatric ALL. ![]()
New test could help fight leukemia

Image courtesy of NIAID
Researchers say they have developed a test that can reveal how the immune system would respond to vaccines for leukemia.
To conduct this test, cancer-specific-proteins are spotted onto a microscope slide.
They are then incubated with a patient blood sample to show whether the immune system can recognize the proteins.
The researchers believe this test could inform immunotherapy trial development and eventually direct the treatment of leukemia.
They described the test in PLOS ONE.
The team explained that cellular arrays using peptide-MHC (pMHC) tetramers allow the simultaneous detection of different antigen-specific T-cell populations that are naturally circulating in leukemia patients and healthy individuals.
The researchers developed a pMHC array to detect CD8+ T-cell populations in leukemia patients that recognize epitopes within viral antigens and leukemia antigens.
Experiments showed this test was at least as sensitive as flow cytometry.
The pMHC array successfully identified more than 40 T-cell populations. It identified T cells that recognized various tumor antigen epitopes in patients with acute myeloid leukemia and acute lymphoblastic leukemia.
“This [test] would allow us to know how good a patients’ immune system is and potentially which proteins their immune system will react to, allowing us to prioritize which proteins we use to develop anticancer vaccines,” said study author Barbara Guinn, PhD, of the University of Southampton in the UK.
“In the future, we may be able to monitor patient immune responses as they are treated in clinical trials, helping us to direct the immune system more efficiently against cancer cells.”
Dr Guinn has spent a large part of her career investigating the differences between cancer cells and normal cells in terms of the proteins they make. She has been able to identify a number of proteins that are overexpressed in tumor cells but not healthy cells.
“Some of these proteins act as biomarkers for patient survival,” she said, “and some of them have helped us understand more about how cancer develops in subgroups of patients with leukemia.” ![]()
Image courtesy of NIAID
Researchers say they have developed a test that can reveal how the immune system would respond to vaccines for leukemia.
To conduct this test, cancer-specific-proteins are spotted onto a microscope slide.
They are then incubated with a patient blood sample to show whether the immune system can recognize the proteins.
The researchers believe this test could inform immunotherapy trial development and eventually direct the treatment of leukemia.
They described the test in PLOS ONE.
The team explained that cellular arrays using peptide-MHC (pMHC) tetramers allow the simultaneous detection of different antigen-specific T-cell populations that are naturally circulating in leukemia patients and healthy individuals.
The researchers developed a pMHC array to detect CD8+ T-cell populations in leukemia patients that recognize epitopes within viral antigens and leukemia antigens.
Experiments showed this test was at least as sensitive as flow cytometry.
The pMHC array successfully identified more than 40 T-cell populations. It identified T cells that recognized various tumor antigen epitopes in patients with acute myeloid leukemia and acute lymphoblastic leukemia.
“This [test] would allow us to know how good a patients’ immune system is and potentially which proteins their immune system will react to, allowing us to prioritize which proteins we use to develop anticancer vaccines,” said study author Barbara Guinn, PhD, of the University of Southampton in the UK.
“In the future, we may be able to monitor patient immune responses as they are treated in clinical trials, helping us to direct the immune system more efficiently against cancer cells.”
Dr Guinn has spent a large part of her career investigating the differences between cancer cells and normal cells in terms of the proteins they make. She has been able to identify a number of proteins that are overexpressed in tumor cells but not healthy cells.
“Some of these proteins act as biomarkers for patient survival,” she said, “and some of them have helped us understand more about how cancer develops in subgroups of patients with leukemia.” ![]()
Image courtesy of NIAID
Researchers say they have developed a test that can reveal how the immune system would respond to vaccines for leukemia.
To conduct this test, cancer-specific-proteins are spotted onto a microscope slide.
They are then incubated with a patient blood sample to show whether the immune system can recognize the proteins.
The researchers believe this test could inform immunotherapy trial development and eventually direct the treatment of leukemia.
They described the test in PLOS ONE.
The team explained that cellular arrays using peptide-MHC (pMHC) tetramers allow the simultaneous detection of different antigen-specific T-cell populations that are naturally circulating in leukemia patients and healthy individuals.
The researchers developed a pMHC array to detect CD8+ T-cell populations in leukemia patients that recognize epitopes within viral antigens and leukemia antigens.
Experiments showed this test was at least as sensitive as flow cytometry.
The pMHC array successfully identified more than 40 T-cell populations. It identified T cells that recognized various tumor antigen epitopes in patients with acute myeloid leukemia and acute lymphoblastic leukemia.
“This [test] would allow us to know how good a patients’ immune system is and potentially which proteins their immune system will react to, allowing us to prioritize which proteins we use to develop anticancer vaccines,” said study author Barbara Guinn, PhD, of the University of Southampton in the UK.
“In the future, we may be able to monitor patient immune responses as they are treated in clinical trials, helping us to direct the immune system more efficiently against cancer cells.”
Dr Guinn has spent a large part of her career investigating the differences between cancer cells and normal cells in terms of the proteins they make. She has been able to identify a number of proteins that are overexpressed in tumor cells but not healthy cells.
“Some of these proteins act as biomarkers for patient survival,” she said, “and some of them have helped us understand more about how cancer develops in subgroups of patients with leukemia.” ![]()
Team aims to inhibit Notch safely

Photo courtesy of the
University of Michigan
A new study suggests a potential way to block one of the most common cancer-causing genes without causing severe side effects.
The Notch gene plays a role in many cancers, and it’s the most common cancer-causing gene in T-cell acute lymphoblastic leukemia (T-ALL).
About 60% of children and adults with T-ALL harbor a Notch mutation.
Unfortunately, drugs that inhibit Notch can cause serious side effects, such as skin cancers.
Now, investigators have discovered a potential new target to inhibit Notch without the toxic effects.
They found that a protein called Zmiz1 sticks to Notch, prompting the gene to turn on its cancer function. But Zmiz1 does not impact normal, healthy Notch functions.
“Notch controls the genes that cause cancer, but it’s also important for normal health,” said Mark Chiang, MD, PhD, of the University of Michigan in Ann Arbor.
“The challenge is to knock out the cancer function of Notch but preserve its normal function. If you unstick Zmiz1 from Notch, the cancer cells die. And Zmiz1 seems to be selective in turning on the cancer functions of Notch.”
Dr Chiang and his colleagues found that mice lived longer when Zmiz1 was deleted. The mice had normal body weight and no severe side effects from Zmiz1 deletion.
The investigators reported these results in Immunity.
“Our goal is to develop a drug to sit right between Notch and Zmiz1 that could break apart the bond,” Dr Chiang said. “We think this would block the Notch cancer pathway without causing toxic side effects, like we see with current Notch inhibitors.”
He noted that a majority of children with T-ALL are cured, but about 20% will relapse. Those children face a grim prognosis.
“We need to develop therapies against Notch to help kids with relapsed cancer and to cure kids with fewer toxicities or long-term effects,” Dr Chiang said. “Our current treatments may often be curative, but there can be a huge price to pay in late effects.”
To further this research, Dr Chiang and his colleagues plan to use X-ray crystallography to create a 3-dimensional image of Notch and Zmiz1 in an effort to understand how they are sticking together. This could help the team to design a drug to separate the proteins. ![]()
Photo courtesy of the
University of Michigan
A new study suggests a potential way to block one of the most common cancer-causing genes without causing severe side effects.
The Notch gene plays a role in many cancers, and it’s the most common cancer-causing gene in T-cell acute lymphoblastic leukemia (T-ALL).
About 60% of children and adults with T-ALL harbor a Notch mutation.
Unfortunately, drugs that inhibit Notch can cause serious side effects, such as skin cancers.
Now, investigators have discovered a potential new target to inhibit Notch without the toxic effects.
They found that a protein called Zmiz1 sticks to Notch, prompting the gene to turn on its cancer function. But Zmiz1 does not impact normal, healthy Notch functions.
“Notch controls the genes that cause cancer, but it’s also important for normal health,” said Mark Chiang, MD, PhD, of the University of Michigan in Ann Arbor.
“The challenge is to knock out the cancer function of Notch but preserve its normal function. If you unstick Zmiz1 from Notch, the cancer cells die. And Zmiz1 seems to be selective in turning on the cancer functions of Notch.”
Dr Chiang and his colleagues found that mice lived longer when Zmiz1 was deleted. The mice had normal body weight and no severe side effects from Zmiz1 deletion.
The investigators reported these results in Immunity.
“Our goal is to develop a drug to sit right between Notch and Zmiz1 that could break apart the bond,” Dr Chiang said. “We think this would block the Notch cancer pathway without causing toxic side effects, like we see with current Notch inhibitors.”
He noted that a majority of children with T-ALL are cured, but about 20% will relapse. Those children face a grim prognosis.
“We need to develop therapies against Notch to help kids with relapsed cancer and to cure kids with fewer toxicities or long-term effects,” Dr Chiang said. “Our current treatments may often be curative, but there can be a huge price to pay in late effects.”
To further this research, Dr Chiang and his colleagues plan to use X-ray crystallography to create a 3-dimensional image of Notch and Zmiz1 in an effort to understand how they are sticking together. This could help the team to design a drug to separate the proteins. ![]()
Photo courtesy of the
University of Michigan
A new study suggests a potential way to block one of the most common cancer-causing genes without causing severe side effects.
The Notch gene plays a role in many cancers, and it’s the most common cancer-causing gene in T-cell acute lymphoblastic leukemia (T-ALL).
About 60% of children and adults with T-ALL harbor a Notch mutation.
Unfortunately, drugs that inhibit Notch can cause serious side effects, such as skin cancers.
Now, investigators have discovered a potential new target to inhibit Notch without the toxic effects.
They found that a protein called Zmiz1 sticks to Notch, prompting the gene to turn on its cancer function. But Zmiz1 does not impact normal, healthy Notch functions.
“Notch controls the genes that cause cancer, but it’s also important for normal health,” said Mark Chiang, MD, PhD, of the University of Michigan in Ann Arbor.
“The challenge is to knock out the cancer function of Notch but preserve its normal function. If you unstick Zmiz1 from Notch, the cancer cells die. And Zmiz1 seems to be selective in turning on the cancer functions of Notch.”
Dr Chiang and his colleagues found that mice lived longer when Zmiz1 was deleted. The mice had normal body weight and no severe side effects from Zmiz1 deletion.
The investigators reported these results in Immunity.
“Our goal is to develop a drug to sit right between Notch and Zmiz1 that could break apart the bond,” Dr Chiang said. “We think this would block the Notch cancer pathway without causing toxic side effects, like we see with current Notch inhibitors.”
He noted that a majority of children with T-ALL are cured, but about 20% will relapse. Those children face a grim prognosis.
“We need to develop therapies against Notch to help kids with relapsed cancer and to cure kids with fewer toxicities or long-term effects,” Dr Chiang said. “Our current treatments may often be curative, but there can be a huge price to pay in late effects.”
To further this research, Dr Chiang and his colleagues plan to use X-ray crystallography to create a 3-dimensional image of Notch and Zmiz1 in an effort to understand how they are sticking together. This could help the team to design a drug to separate the proteins. ![]()
Novel compound could treat leukemia
A small-molecule compound that has previously shown activity against Ewing sarcoma and prostate cancer may fight leukemia as well, according to preclinical research published in Oncotarget.
The compound, YK-4-279, inhibits the oncogenic activity of the fusion protein EWS-FLI1.
“EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma,” said study author Aykut Üren, MD, of Georgetown University Medical Center in Washington, DC.
“It also appears to drive cancer cell growth in some prostate cancers.”
ETS family fusion proteins are found in patients with acute myeloid leukemia and acute lymphoblastic leukemia as well.
So Dr Üren and his colleagues decided to create a mouse model of EWS-FLI1-induced leukemia and assess the activity of YK-4-279 in this model.
Mice with EWS-FLI1-induced leukemia presented with severe hepatomegaly, splenomegaly, and anemia, followed by rapid death.
The investigators treated these mice with injections of YK-4-279 five days a week for 2 weeks or vehicle intraperitoneal injections on the same schedule.
The team said treatment with YK-4-279 significantly reduced white blood cell counts, nucleated erythroblasts in the peripheral blood, splenomegaly, and hepatomegaly.
They noted that mice experienced reductions in the weight of their spleens and livers without experiencing reductions in total body weight.
In addition, mice that received YK-4-279 had significantly better overall survival than control mice. The median survival times were 60.5 days and 21 days, respectively.
The investigators also noted that treated mice did not exhibit overt toxicity in the liver, spleen, or bone marrow.
“The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can’t be answered until we conduct clinical trials,” Dr Üren said.
Nevertheless, he and his colleagues believe these results support the continued preclinical development of YK-4-279 for Ewing sarcoma, prostate cancers, and leukemias with highly homologous translocation products or with a clear ETS-driven gene signature. ![]()
A small-molecule compound that has previously shown activity against Ewing sarcoma and prostate cancer may fight leukemia as well, according to preclinical research published in Oncotarget.
The compound, YK-4-279, inhibits the oncogenic activity of the fusion protein EWS-FLI1.
“EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma,” said study author Aykut Üren, MD, of Georgetown University Medical Center in Washington, DC.
“It also appears to drive cancer cell growth in some prostate cancers.”
ETS family fusion proteins are found in patients with acute myeloid leukemia and acute lymphoblastic leukemia as well.
So Dr Üren and his colleagues decided to create a mouse model of EWS-FLI1-induced leukemia and assess the activity of YK-4-279 in this model.
Mice with EWS-FLI1-induced leukemia presented with severe hepatomegaly, splenomegaly, and anemia, followed by rapid death.
The investigators treated these mice with injections of YK-4-279 five days a week for 2 weeks or vehicle intraperitoneal injections on the same schedule.
The team said treatment with YK-4-279 significantly reduced white blood cell counts, nucleated erythroblasts in the peripheral blood, splenomegaly, and hepatomegaly.
They noted that mice experienced reductions in the weight of their spleens and livers without experiencing reductions in total body weight.
In addition, mice that received YK-4-279 had significantly better overall survival than control mice. The median survival times were 60.5 days and 21 days, respectively.
The investigators also noted that treated mice did not exhibit overt toxicity in the liver, spleen, or bone marrow.
“The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can’t be answered until we conduct clinical trials,” Dr Üren said.
Nevertheless, he and his colleagues believe these results support the continued preclinical development of YK-4-279 for Ewing sarcoma, prostate cancers, and leukemias with highly homologous translocation products or with a clear ETS-driven gene signature. ![]()
A small-molecule compound that has previously shown activity against Ewing sarcoma and prostate cancer may fight leukemia as well, according to preclinical research published in Oncotarget.
The compound, YK-4-279, inhibits the oncogenic activity of the fusion protein EWS-FLI1.
“EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma,” said study author Aykut Üren, MD, of Georgetown University Medical Center in Washington, DC.
“It also appears to drive cancer cell growth in some prostate cancers.”
ETS family fusion proteins are found in patients with acute myeloid leukemia and acute lymphoblastic leukemia as well.
So Dr Üren and his colleagues decided to create a mouse model of EWS-FLI1-induced leukemia and assess the activity of YK-4-279 in this model.
Mice with EWS-FLI1-induced leukemia presented with severe hepatomegaly, splenomegaly, and anemia, followed by rapid death.
The investigators treated these mice with injections of YK-4-279 five days a week for 2 weeks or vehicle intraperitoneal injections on the same schedule.
The team said treatment with YK-4-279 significantly reduced white blood cell counts, nucleated erythroblasts in the peripheral blood, splenomegaly, and hepatomegaly.
They noted that mice experienced reductions in the weight of their spleens and livers without experiencing reductions in total body weight.
In addition, mice that received YK-4-279 had significantly better overall survival than control mice. The median survival times were 60.5 days and 21 days, respectively.
The investigators also noted that treated mice did not exhibit overt toxicity in the liver, spleen, or bone marrow.
“The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can’t be answered until we conduct clinical trials,” Dr Üren said.
Nevertheless, he and his colleagues believe these results support the continued preclinical development of YK-4-279 for Ewing sarcoma, prostate cancers, and leukemias with highly homologous translocation products or with a clear ETS-driven gene signature. ![]()
Drug granted breakthrough designation for ALL

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant).
*Information in the abstract differs from the presentation.
In ALL, early treatment decisions have “irrevocable” implications
SAN FRANCISCO – “The best opportunity to improve acute lymphoblastic leukemia (ALL) outcomes is to make the best evidence-based choices early at the time of diagnosis or early at the time of relapse. This is a disease where early choices are irrevocable, and if you make the wrong choices, patients suffer,” Dr. Joseph C. Alvarnas asserted at the National Comprehensive Cancer Network 10th Annual Congress: Hematologic Malignancies.
Hematologists must also stay up on novel agents being added to the ALL treatment armamentarium, he stressed. “The state of the art is one that evolves over the course of months, not over the course of years. So maintaining current [knowledge] in this is essential. And many of these patients benefit from being referred quickly to an expert institution,” he said.

Cytogenetics and genomics help risk-adapt therapy
“Cytogenetic, molecular, and genomic data are essential to making great early choices,” maintained Dr. Alvarnas, who is an associate clinical professor in the department of hematology & hematopoietic cell transplantation, and director of Value Based Analytics, at the City of Hope Comprehensive Cancer Center in Duarte, California.
Patients with Philadelphia chromosome (Ph)-positive ALL should receive tyrosine kinase inhibitors (TKIs) concomitantly with age-adapted induction and consolidation therapy, he recommended. In those with a poor response, a mutational analysis is key to guiding next steps.
“While in the young pediatric population – we are talking ages 5-10 years – there is a trend away from offering transplant to patients with Ph-positive disease because some of them are actually cured through the combination of induction pediatric regimens followed by TKI-based therapy, for adults, the standard of care until demonstrated otherwise is prompt referral for transplant,” he said.
Indeed, long-term survival is nearly doubled for Ph-positive patients if they have a transplant in a first complete remission versus later (54% vs 29%) (Blood. 2008;112;903-9).
Patients with the high-risk MLL rearrangement are likely to fare poorly and should also be considered for early transplant in first complete remission, according to Dr. Alvarnas.
A novel genetic subtype of ALL identified by looking at networks of genes – Ph-like ALL – has a poor prognosis, especially when affected patients are young adults as compared with children or adolescents (N Engl J Med. 2014;371:1005-15). Analyses have identified the presence of a cluster of genetic abnormalities involving ABL, JAK2, and RAS, among others.
“If you think strategically about how we might be able to better treat these patients … targeted agents like ruxolitinib (Jakafi), dasatinib (Sprycel), and crizotinib (Xalkori) may all play a role,” he said. “Now this is not ready for prime time yet – I’m not ready to advocate that you begin treating patients with targeted therapies. I think in fact this patient population should be referred to an academic cancer center for treatment on protocol. But as we look at what’s likely to change over the next year to 5 years, genomic alterations may make these patients better candidates for treatment with TKIs.”
Demographics can guide treatment choices as well
Patient demographics, especially age, should also be used to risk-adapt ALL therapy, according to Dr. Alvarnas. The adolescent and young adult (AYA) subset of patients – aged 15-39 – tend to be fitter and can therefore benefit from pediatric or pediatric-inspired regimens.
“These regimens don’t use novel therapeutics, for the most part; they increase the dose density or dose intensity of existing agents, particularly L-asparaginase. And a lot of adult doctors used to treating older patients don’t like L-asparaginase because of the significant morbidities, particularly pancreatitis, that can arise with this agent,” he said. “But when you get a younger, fitter group of patients, you can use very intensive doses of L-asparaginase not only with impunity, but with greater cure rates.”
AYA patients have superior event-free and overall survival when treated with a pediatric or pediatric-inspired regimen than when treated with an adult regimen (Blood. 2008;112:1646-54).
“So think of it this way: patients 15-39 years of age are receiving inferior therapy if they are receiving adult regimens,” Dr. Alvarnas said. “Now the caveat there is they have to be … physiologically fit, and there may be specific contraindications to these pediatric regimens. But this should be an opt out, not an opt in. The pediatric-inspired regimens are, I would say, the standard care for this population.”
At the other end of the age spectrum, patients 65 years and older with ALL have poorer outcomes, which may be due to both biology of disease and physiology. “We need to be very mindful and think carefully of how best to treat these patients in a patient-centric fashion,” he said.
He recommended consideration of comorbidities and use of a comprehensive geriatric assessment when contemplating care options for this age group. “We want to make sure that the therapy used matches the patient before us,” he added, pointing to an algorithm that is helpful in this setting (Blood. 2013;122:1366-75).
“Where it’s possible, I would encourage the use of clinical trials, particularly geared to the older age population. And that said, in the older, fitter patient with a good initial response to therapy, do not discount the appropriateness of allogeneic stem cell transplant,” Dr. Alvarnas advised.
At the same time, hematologists should have a frank discussion with these older patients about the goals of care and advanced directives, and should involve supportive care early.
“NCCN has an absolutely beautiful document on the care of older oncology patients as well as a beautiful set of guidelines regarding supportive care,” he added. “Please look at those. I think are an invaluable resource.”
Immunotherapy shows promise in the salvage setting
“At the time of salvage, immunotherapy-based approaches are very powerful, so don’t overtreat the patient with modalities that aren’t going to work,” Dr. Alvarnas recommended.
“Immunotherapeutic approaches are going to play an increasingly important role in patients with ALL, and we see these novel therapeutics completely upending what we knew about this disease even a year or two ago,” he said. A variety of monoclonal antibodies against CD20, CD19, CD22, and CD52 have shown promise when tested in various patient populations (Blood. 2015;125:4010-6).
This concept has been taken a step further with blinatumomab (Blincyto), an antibody having two antigen recognition sites that brings CD19-positive tumor cells in contact with T lymphocytes. It is the first such agent to be approved by the FDA for an ALL indication (currently for refractory or relapsed Ph-negative B-cell ALL). “It has nonoverlapping activity with cytotoxic chemotherapy, which makes it an ideal agent,” Dr. Alvarnas noted.
The main risk with blinatumomab is a cytokine release syndrome, which is most common in patients with a high disease burden and requires drug discontinuation and treatment with high-dose dexamethasone. Neurotoxicity is also noteworthy as it can be fatal.
Responses to blinatumomab tend to be dramatic and deep, but brief, according to Dr. Alvarnas. “Even though it’s profoundly powerful, it’s not a curative agent. It really provides a bridge towards cure, with that cure coming through the use of allogeneic stem cell transplant,” he elaborated. “So if someone relapses and you begin blinatumomab, get them referred very quickly to a transplant center.”
Another promising immunotherapy is inotuzumab ozagamicin, an antibody-drug conjugate that targets CD22-expressing cells. It has been associated with a complete response rate of 19%, although veno-occlusive toxicity has been problematic (Cancer. 2013;119:2728-36). “This agent has not yet received FDA approval, but it’s one that we are awaiting expectantly,” he said.
Finally, phase 1 trials from various academic centers have shown that chimeric antigen receptor (CAR) T-cell therapy achieves complete response rates of 67%-90% in patients with high-risk refractory disease, according to Dr. Alvarnas, who disclosed that he had no relevant financial relationships.
“This has lead to culmination in phase 2 trials, and I really do see this as an important component in the management of patients with relapsed and refractory ALL,” he concluded. “It’s not something that is available at every center. Right now it’s restricted largely to academic centers capable of producing these therapeutics in their own GLP [Good Laboratory Practice] facility.”
SAN FRANCISCO – “The best opportunity to improve acute lymphoblastic leukemia (ALL) outcomes is to make the best evidence-based choices early at the time of diagnosis or early at the time of relapse. This is a disease where early choices are irrevocable, and if you make the wrong choices, patients suffer,” Dr. Joseph C. Alvarnas asserted at the National Comprehensive Cancer Network 10th Annual Congress: Hematologic Malignancies.
Hematologists must also stay up on novel agents being added to the ALL treatment armamentarium, he stressed. “The state of the art is one that evolves over the course of months, not over the course of years. So maintaining current [knowledge] in this is essential. And many of these patients benefit from being referred quickly to an expert institution,” he said.
Cytogenetics and genomics help risk-adapt therapy
“Cytogenetic, molecular, and genomic data are essential to making great early choices,” maintained Dr. Alvarnas, who is an associate clinical professor in the department of hematology & hematopoietic cell transplantation, and director of Value Based Analytics, at the City of Hope Comprehensive Cancer Center in Duarte, California.
Patients with Philadelphia chromosome (Ph)-positive ALL should receive tyrosine kinase inhibitors (TKIs) concomitantly with age-adapted induction and consolidation therapy, he recommended. In those with a poor response, a mutational analysis is key to guiding next steps.
“While in the young pediatric population – we are talking ages 5-10 years – there is a trend away from offering transplant to patients with Ph-positive disease because some of them are actually cured through the combination of induction pediatric regimens followed by TKI-based therapy, for adults, the standard of care until demonstrated otherwise is prompt referral for transplant,” he said.
Indeed, long-term survival is nearly doubled for Ph-positive patients if they have a transplant in a first complete remission versus later (54% vs 29%) (Blood. 2008;112;903-9).
Patients with the high-risk MLL rearrangement are likely to fare poorly and should also be considered for early transplant in first complete remission, according to Dr. Alvarnas.
A novel genetic subtype of ALL identified by looking at networks of genes – Ph-like ALL – has a poor prognosis, especially when affected patients are young adults as compared with children or adolescents (N Engl J Med. 2014;371:1005-15). Analyses have identified the presence of a cluster of genetic abnormalities involving ABL, JAK2, and RAS, among others.
“If you think strategically about how we might be able to better treat these patients … targeted agents like ruxolitinib (Jakafi), dasatinib (Sprycel), and crizotinib (Xalkori) may all play a role,” he said. “Now this is not ready for prime time yet – I’m not ready to advocate that you begin treating patients with targeted therapies. I think in fact this patient population should be referred to an academic cancer center for treatment on protocol. But as we look at what’s likely to change over the next year to 5 years, genomic alterations may make these patients better candidates for treatment with TKIs.”
Demographics can guide treatment choices as well
Patient demographics, especially age, should also be used to risk-adapt ALL therapy, according to Dr. Alvarnas. The adolescent and young adult (AYA) subset of patients – aged 15-39 – tend to be fitter and can therefore benefit from pediatric or pediatric-inspired regimens.
“These regimens don’t use novel therapeutics, for the most part; they increase the dose density or dose intensity of existing agents, particularly L-asparaginase. And a lot of adult doctors used to treating older patients don’t like L-asparaginase because of the significant morbidities, particularly pancreatitis, that can arise with this agent,” he said. “But when you get a younger, fitter group of patients, you can use very intensive doses of L-asparaginase not only with impunity, but with greater cure rates.”
AYA patients have superior event-free and overall survival when treated with a pediatric or pediatric-inspired regimen than when treated with an adult regimen (Blood. 2008;112:1646-54).
“So think of it this way: patients 15-39 years of age are receiving inferior therapy if they are receiving adult regimens,” Dr. Alvarnas said. “Now the caveat there is they have to be … physiologically fit, and there may be specific contraindications to these pediatric regimens. But this should be an opt out, not an opt in. The pediatric-inspired regimens are, I would say, the standard care for this population.”
At the other end of the age spectrum, patients 65 years and older with ALL have poorer outcomes, which may be due to both biology of disease and physiology. “We need to be very mindful and think carefully of how best to treat these patients in a patient-centric fashion,” he said.
He recommended consideration of comorbidities and use of a comprehensive geriatric assessment when contemplating care options for this age group. “We want to make sure that the therapy used matches the patient before us,” he added, pointing to an algorithm that is helpful in this setting (Blood. 2013;122:1366-75).
“Where it’s possible, I would encourage the use of clinical trials, particularly geared to the older age population. And that said, in the older, fitter patient with a good initial response to therapy, do not discount the appropriateness of allogeneic stem cell transplant,” Dr. Alvarnas advised.
At the same time, hematologists should have a frank discussion with these older patients about the goals of care and advanced directives, and should involve supportive care early.
“NCCN has an absolutely beautiful document on the care of older oncology patients as well as a beautiful set of guidelines regarding supportive care,” he added. “Please look at those. I think are an invaluable resource.”
Immunotherapy shows promise in the salvage setting
“At the time of salvage, immunotherapy-based approaches are very powerful, so don’t overtreat the patient with modalities that aren’t going to work,” Dr. Alvarnas recommended.
“Immunotherapeutic approaches are going to play an increasingly important role in patients with ALL, and we see these novel therapeutics completely upending what we knew about this disease even a year or two ago,” he said. A variety of monoclonal antibodies against CD20, CD19, CD22, and CD52 have shown promise when tested in various patient populations (Blood. 2015;125:4010-6).
This concept has been taken a step further with blinatumomab (Blincyto), an antibody having two antigen recognition sites that brings CD19-positive tumor cells in contact with T lymphocytes. It is the first such agent to be approved by the FDA for an ALL indication (currently for refractory or relapsed Ph-negative B-cell ALL). “It has nonoverlapping activity with cytotoxic chemotherapy, which makes it an ideal agent,” Dr. Alvarnas noted.
The main risk with blinatumomab is a cytokine release syndrome, which is most common in patients with a high disease burden and requires drug discontinuation and treatment with high-dose dexamethasone. Neurotoxicity is also noteworthy as it can be fatal.
Responses to blinatumomab tend to be dramatic and deep, but brief, according to Dr. Alvarnas. “Even though it’s profoundly powerful, it’s not a curative agent. It really provides a bridge towards cure, with that cure coming through the use of allogeneic stem cell transplant,” he elaborated. “So if someone relapses and you begin blinatumomab, get them referred very quickly to a transplant center.”
Another promising immunotherapy is inotuzumab ozagamicin, an antibody-drug conjugate that targets CD22-expressing cells. It has been associated with a complete response rate of 19%, although veno-occlusive toxicity has been problematic (Cancer. 2013;119:2728-36). “This agent has not yet received FDA approval, but it’s one that we are awaiting expectantly,” he said.
Finally, phase 1 trials from various academic centers have shown that chimeric antigen receptor (CAR) T-cell therapy achieves complete response rates of 67%-90% in patients with high-risk refractory disease, according to Dr. Alvarnas, who disclosed that he had no relevant financial relationships.
“This has lead to culmination in phase 2 trials, and I really do see this as an important component in the management of patients with relapsed and refractory ALL,” he concluded. “It’s not something that is available at every center. Right now it’s restricted largely to academic centers capable of producing these therapeutics in their own GLP [Good Laboratory Practice] facility.”
SAN FRANCISCO – “The best opportunity to improve acute lymphoblastic leukemia (ALL) outcomes is to make the best evidence-based choices early at the time of diagnosis or early at the time of relapse. This is a disease where early choices are irrevocable, and if you make the wrong choices, patients suffer,” Dr. Joseph C. Alvarnas asserted at the National Comprehensive Cancer Network 10th Annual Congress: Hematologic Malignancies.
Hematologists must also stay up on novel agents being added to the ALL treatment armamentarium, he stressed. “The state of the art is one that evolves over the course of months, not over the course of years. So maintaining current [knowledge] in this is essential. And many of these patients benefit from being referred quickly to an expert institution,” he said.
Cytogenetics and genomics help risk-adapt therapy
“Cytogenetic, molecular, and genomic data are essential to making great early choices,” maintained Dr. Alvarnas, who is an associate clinical professor in the department of hematology & hematopoietic cell transplantation, and director of Value Based Analytics, at the City of Hope Comprehensive Cancer Center in Duarte, California.
Patients with Philadelphia chromosome (Ph)-positive ALL should receive tyrosine kinase inhibitors (TKIs) concomitantly with age-adapted induction and consolidation therapy, he recommended. In those with a poor response, a mutational analysis is key to guiding next steps.
“While in the young pediatric population – we are talking ages 5-10 years – there is a trend away from offering transplant to patients with Ph-positive disease because some of them are actually cured through the combination of induction pediatric regimens followed by TKI-based therapy, for adults, the standard of care until demonstrated otherwise is prompt referral for transplant,” he said.
Indeed, long-term survival is nearly doubled for Ph-positive patients if they have a transplant in a first complete remission versus later (54% vs 29%) (Blood. 2008;112;903-9).
Patients with the high-risk MLL rearrangement are likely to fare poorly and should also be considered for early transplant in first complete remission, according to Dr. Alvarnas.
A novel genetic subtype of ALL identified by looking at networks of genes – Ph-like ALL – has a poor prognosis, especially when affected patients are young adults as compared with children or adolescents (N Engl J Med. 2014;371:1005-15). Analyses have identified the presence of a cluster of genetic abnormalities involving ABL, JAK2, and RAS, among others.
“If you think strategically about how we might be able to better treat these patients … targeted agents like ruxolitinib (Jakafi), dasatinib (Sprycel), and crizotinib (Xalkori) may all play a role,” he said. “Now this is not ready for prime time yet – I’m not ready to advocate that you begin treating patients with targeted therapies. I think in fact this patient population should be referred to an academic cancer center for treatment on protocol. But as we look at what’s likely to change over the next year to 5 years, genomic alterations may make these patients better candidates for treatment with TKIs.”
Demographics can guide treatment choices as well
Patient demographics, especially age, should also be used to risk-adapt ALL therapy, according to Dr. Alvarnas. The adolescent and young adult (AYA) subset of patients – aged 15-39 – tend to be fitter and can therefore benefit from pediatric or pediatric-inspired regimens.
“These regimens don’t use novel therapeutics, for the most part; they increase the dose density or dose intensity of existing agents, particularly L-asparaginase. And a lot of adult doctors used to treating older patients don’t like L-asparaginase because of the significant morbidities, particularly pancreatitis, that can arise with this agent,” he said. “But when you get a younger, fitter group of patients, you can use very intensive doses of L-asparaginase not only with impunity, but with greater cure rates.”
AYA patients have superior event-free and overall survival when treated with a pediatric or pediatric-inspired regimen than when treated with an adult regimen (Blood. 2008;112:1646-54).
“So think of it this way: patients 15-39 years of age are receiving inferior therapy if they are receiving adult regimens,” Dr. Alvarnas said. “Now the caveat there is they have to be … physiologically fit, and there may be specific contraindications to these pediatric regimens. But this should be an opt out, not an opt in. The pediatric-inspired regimens are, I would say, the standard care for this population.”
At the other end of the age spectrum, patients 65 years and older with ALL have poorer outcomes, which may be due to both biology of disease and physiology. “We need to be very mindful and think carefully of how best to treat these patients in a patient-centric fashion,” he said.
He recommended consideration of comorbidities and use of a comprehensive geriatric assessment when contemplating care options for this age group. “We want to make sure that the therapy used matches the patient before us,” he added, pointing to an algorithm that is helpful in this setting (Blood. 2013;122:1366-75).
“Where it’s possible, I would encourage the use of clinical trials, particularly geared to the older age population. And that said, in the older, fitter patient with a good initial response to therapy, do not discount the appropriateness of allogeneic stem cell transplant,” Dr. Alvarnas advised.
At the same time, hematologists should have a frank discussion with these older patients about the goals of care and advanced directives, and should involve supportive care early.
“NCCN has an absolutely beautiful document on the care of older oncology patients as well as a beautiful set of guidelines regarding supportive care,” he added. “Please look at those. I think are an invaluable resource.”
Immunotherapy shows promise in the salvage setting
“At the time of salvage, immunotherapy-based approaches are very powerful, so don’t overtreat the patient with modalities that aren’t going to work,” Dr. Alvarnas recommended.
“Immunotherapeutic approaches are going to play an increasingly important role in patients with ALL, and we see these novel therapeutics completely upending what we knew about this disease even a year or two ago,” he said. A variety of monoclonal antibodies against CD20, CD19, CD22, and CD52 have shown promise when tested in various patient populations (Blood. 2015;125:4010-6).
This concept has been taken a step further with blinatumomab (Blincyto), an antibody having two antigen recognition sites that brings CD19-positive tumor cells in contact with T lymphocytes. It is the first such agent to be approved by the FDA for an ALL indication (currently for refractory or relapsed Ph-negative B-cell ALL). “It has nonoverlapping activity with cytotoxic chemotherapy, which makes it an ideal agent,” Dr. Alvarnas noted.
The main risk with blinatumomab is a cytokine release syndrome, which is most common in patients with a high disease burden and requires drug discontinuation and treatment with high-dose dexamethasone. Neurotoxicity is also noteworthy as it can be fatal.
Responses to blinatumomab tend to be dramatic and deep, but brief, according to Dr. Alvarnas. “Even though it’s profoundly powerful, it’s not a curative agent. It really provides a bridge towards cure, with that cure coming through the use of allogeneic stem cell transplant,” he elaborated. “So if someone relapses and you begin blinatumomab, get them referred very quickly to a transplant center.”
Another promising immunotherapy is inotuzumab ozagamicin, an antibody-drug conjugate that targets CD22-expressing cells. It has been associated with a complete response rate of 19%, although veno-occlusive toxicity has been problematic (Cancer. 2013;119:2728-36). “This agent has not yet received FDA approval, but it’s one that we are awaiting expectantly,” he said.
Finally, phase 1 trials from various academic centers have shown that chimeric antigen receptor (CAR) T-cell therapy achieves complete response rates of 67%-90% in patients with high-risk refractory disease, according to Dr. Alvarnas, who disclosed that he had no relevant financial relationships.
“This has lead to culmination in phase 2 trials, and I really do see this as an important component in the management of patients with relapsed and refractory ALL,” he concluded. “It’s not something that is available at every center. Right now it’s restricted largely to academic centers capable of producing these therapeutics in their own GLP [Good Laboratory Practice] facility.”
EXPERT ANALYSIS AT NCCN ANNUAL CONGRESS: HEMATOLOGIC MALIGNANCIES
