User login
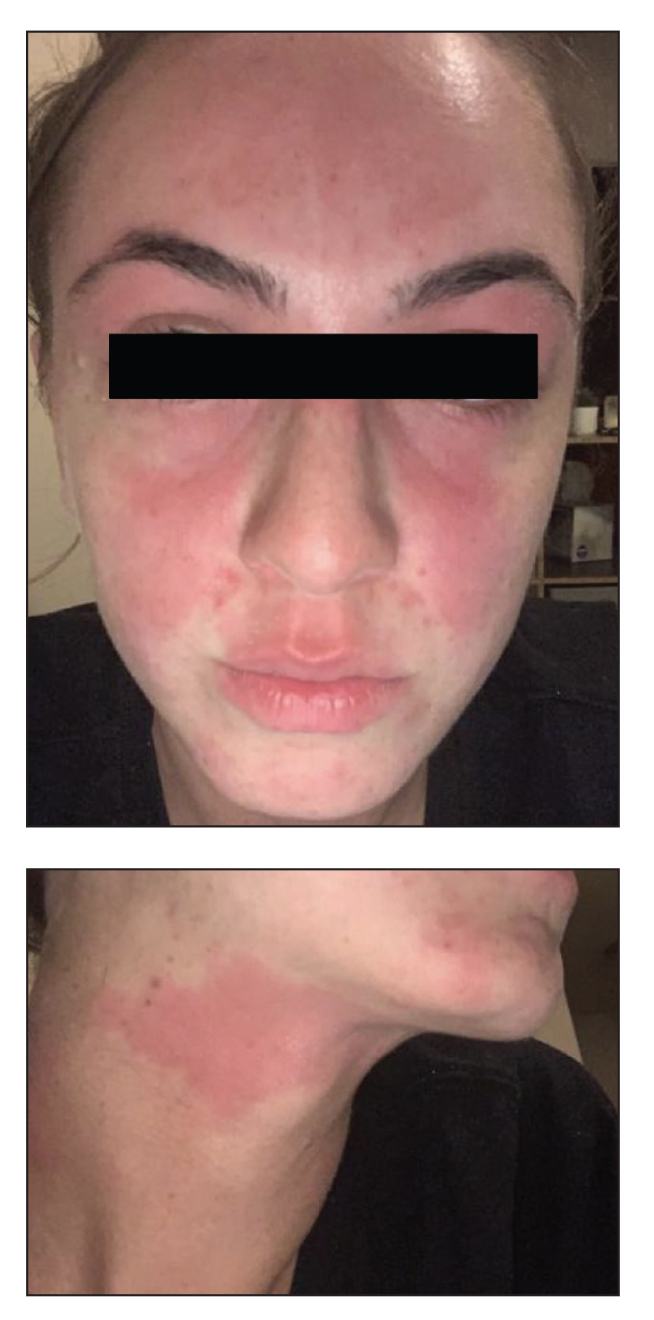
Erythematous Rash on the Face and Neck
The Diagnosis: Allergic Contact Dermatitis
In our patient, the erythematous pruritic rash on the face and neck, the lack of systemic symptoms, and her history of atopic dermatitis suggested a diagnosis of allergic contact dermatitis (ACD). She underwent patch testing with standard, fragrance, and cosmetic panels in addition to 6 of her personal care products. Her first patch test, which was read on day 2, showed a positive reaction to isopropyl myristate (IPM), a penetration enhancer used in cosmetics, topical medications (eg, tretinoin), and cosmeceuticals. The reading on day 5 showed a 2+ reaction to IPM, which was found in several of her personal care products, including her shampoo, leave-in conditioner, and eczema-calming cream. Isopropyl myristate is used in these products because of its ability to enhance their penetration into the skin and also can be found in commercially used products such as hand sanitizers. The patient was given information on this allergen and how to identify and avoid triggers. At follow-up, the ACD had resolved with avoidance of IPM.
Contact dermatitis is an inflammatory skin condition that is triggered by contact with a specific causative agent. There are 2 types of contact dermatitis: irritant and allergic; the irritant type is more common (approximately 80% of cases worldwide).1 Allergic contact dermatitis is a type IV (delayed-type) hypersensitivity reaction; common causative agents include shampoos, moisturizers, makeup, certain metals (eg, nickel), fragrances, latex, and certain plants (eg, poison ivy).2 In cases of ACD, a new reaction can develop from exposure to a product that the patient has used for years. It manifests clinically as erythema, pruritus, scaling, and vesicle formation.1 Certain populations, such as those with atopic dermatitis, are more prone to developing ACD due to a breakdown of the skin barrier, frequent use of topical products, and immune dysregulation.1,2 Patch testing performed by dermatologists and allergists is the gold standard for diagnosing ACD.1,3
Annually, allergists, dermatologists, and primary care physicians see thousands of cases of contact dermatitis.1 Early recognition and appropriate treatment can help reduce the severity and duration of symptoms and improve patient outcomes. The main treatment for ACD is identification of the causative agent followed by patient education on how to identify and avoid triggers.2 Once patch testing has been completed, patients can be given access to the American Contact Dermatitis Society’s Contact Allergen Management Program (CAMP) database (https://www.contactderm.org/resources/acds-camp) to help them identify and avoid products that contain triggering allergens.
Topical corticosteroids are the first-line pharmacologic treatments for atopic dermatitis.4 When our patient presented with the facial rash, her atopic dermatitis had been well controlled with both dupilumab and topical triamcinolone. The lack of response to previously successful therapies in a new area of involvement made a flare of atopic dermatitis less likely. For flares of ACD after exposure, topical corticosteroids and topical calcineurin inhibitors can help. If needed due to severity, oral corticosteroids also can be used.1
Dermatomyositis is an inflammatory myopathy that has several skin manifestations, including a heliotrope rash and poikiloderma.5 While our patient’s rash covered the periorbital area, she did not have other classic skin findings of dermatomyositis, such as nail-fold capillary changes or poikiloderma in a shawl or holster distribution.6 She also lacked signs of systemic involvement including myositis and elevated C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and creatine kinase levels.5
Erythematotelangiectatic rosacea is characterized by telangiectasias and transient flushing and erythema on the central face.5 Rosacea typically is triggered by temperature changes, alcohol consumption, sun exposure, spicy foods, and stress5 and would be expected to involve the nose, which was not observed in our patient. The fixed nature of our patient’s patches and the absence of telangiectasias also argued against this diagnosis.
The classic cutaneous finding of systemic lupus erythematosus is a malar rash, which appears as erythematous patches or thin plaques across the bridge of the nose and over the cheeks, sparing the nasolabial folds.5 Systemic lupus erythematosus is associated with laboratory abnormalities, such as positive antinuclear antibodies and elevated CRP and ESR levels.5 Our patient had notable sparing of the nose, negative antinuclear antibodies, and normal CRP and ESR levels, making systemic lupus erythematosus unlikely. Systemic lupus erythematosus also can manifest with photosensitivity,7 and involvement of the submental skin in our patient argued against a photosensitive eruption.
- Nassau S, Fonacier L. Allergic contact dermatitis. Med Clin North Am. 2020;104:61-76. doi:10.1016/j.mcna.2019.08.012
- Fonacier LS, Sher JM. Allergic contact dermatitis. Ann Allergy Asthma Immunol. 2014;113:9-12. doi:10.1016/j.anai.2014.03.018
- Uyesugi BA, Sheehan MP. Patch testing pearls. Clin Rev Allergy Immunol. 2019;56:110-118. doi:10.1007/s12016-018-8715-y
- Kapur S, Watson W, Carr S. Atopic dermatitis. Allergy Asthma Clin Immunol. 2018;14(suppl 2):52. doi:10.1186/s13223-018-0281-6
- Naji S. Malar rash. StatPearls. Updated September 4, 2023. Accessed June 30, 2025. https://www.statpearls.com/point-of-care/24661
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302. doi:10.1007 /s12016-015-8496-5
- Hannon CW, McCourt C, Lima HC, et al. Interventions for cutaneous disease in systemic lupus erythematosus. Cochrane Database Syst Rev. 2021;3(3):CD007478. doi:10.1002/14651858.CD007478.pub2
The Diagnosis: Allergic Contact Dermatitis
In our patient, the erythematous pruritic rash on the face and neck, the lack of systemic symptoms, and her history of atopic dermatitis suggested a diagnosis of allergic contact dermatitis (ACD). She underwent patch testing with standard, fragrance, and cosmetic panels in addition to 6 of her personal care products. Her first patch test, which was read on day 2, showed a positive reaction to isopropyl myristate (IPM), a penetration enhancer used in cosmetics, topical medications (eg, tretinoin), and cosmeceuticals. The reading on day 5 showed a 2+ reaction to IPM, which was found in several of her personal care products, including her shampoo, leave-in conditioner, and eczema-calming cream. Isopropyl myristate is used in these products because of its ability to enhance their penetration into the skin and also can be found in commercially used products such as hand sanitizers. The patient was given information on this allergen and how to identify and avoid triggers. At follow-up, the ACD had resolved with avoidance of IPM.
Contact dermatitis is an inflammatory skin condition that is triggered by contact with a specific causative agent. There are 2 types of contact dermatitis: irritant and allergic; the irritant type is more common (approximately 80% of cases worldwide).1 Allergic contact dermatitis is a type IV (delayed-type) hypersensitivity reaction; common causative agents include shampoos, moisturizers, makeup, certain metals (eg, nickel), fragrances, latex, and certain plants (eg, poison ivy).2 In cases of ACD, a new reaction can develop from exposure to a product that the patient has used for years. It manifests clinically as erythema, pruritus, scaling, and vesicle formation.1 Certain populations, such as those with atopic dermatitis, are more prone to developing ACD due to a breakdown of the skin barrier, frequent use of topical products, and immune dysregulation.1,2 Patch testing performed by dermatologists and allergists is the gold standard for diagnosing ACD.1,3
Annually, allergists, dermatologists, and primary care physicians see thousands of cases of contact dermatitis.1 Early recognition and appropriate treatment can help reduce the severity and duration of symptoms and improve patient outcomes. The main treatment for ACD is identification of the causative agent followed by patient education on how to identify and avoid triggers.2 Once patch testing has been completed, patients can be given access to the American Contact Dermatitis Society’s Contact Allergen Management Program (CAMP) database (https://www.contactderm.org/resources/acds-camp) to help them identify and avoid products that contain triggering allergens.
Topical corticosteroids are the first-line pharmacologic treatments for atopic dermatitis.4 When our patient presented with the facial rash, her atopic dermatitis had been well controlled with both dupilumab and topical triamcinolone. The lack of response to previously successful therapies in a new area of involvement made a flare of atopic dermatitis less likely. For flares of ACD after exposure, topical corticosteroids and topical calcineurin inhibitors can help. If needed due to severity, oral corticosteroids also can be used.1
Dermatomyositis is an inflammatory myopathy that has several skin manifestations, including a heliotrope rash and poikiloderma.5 While our patient’s rash covered the periorbital area, she did not have other classic skin findings of dermatomyositis, such as nail-fold capillary changes or poikiloderma in a shawl or holster distribution.6 She also lacked signs of systemic involvement including myositis and elevated C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and creatine kinase levels.5
Erythematotelangiectatic rosacea is characterized by telangiectasias and transient flushing and erythema on the central face.5 Rosacea typically is triggered by temperature changes, alcohol consumption, sun exposure, spicy foods, and stress5 and would be expected to involve the nose, which was not observed in our patient. The fixed nature of our patient’s patches and the absence of telangiectasias also argued against this diagnosis.
The classic cutaneous finding of systemic lupus erythematosus is a malar rash, which appears as erythematous patches or thin plaques across the bridge of the nose and over the cheeks, sparing the nasolabial folds.5 Systemic lupus erythematosus is associated with laboratory abnormalities, such as positive antinuclear antibodies and elevated CRP and ESR levels.5 Our patient had notable sparing of the nose, negative antinuclear antibodies, and normal CRP and ESR levels, making systemic lupus erythematosus unlikely. Systemic lupus erythematosus also can manifest with photosensitivity,7 and involvement of the submental skin in our patient argued against a photosensitive eruption.
The Diagnosis: Allergic Contact Dermatitis
In our patient, the erythematous pruritic rash on the face and neck, the lack of systemic symptoms, and her history of atopic dermatitis suggested a diagnosis of allergic contact dermatitis (ACD). She underwent patch testing with standard, fragrance, and cosmetic panels in addition to 6 of her personal care products. Her first patch test, which was read on day 2, showed a positive reaction to isopropyl myristate (IPM), a penetration enhancer used in cosmetics, topical medications (eg, tretinoin), and cosmeceuticals. The reading on day 5 showed a 2+ reaction to IPM, which was found in several of her personal care products, including her shampoo, leave-in conditioner, and eczema-calming cream. Isopropyl myristate is used in these products because of its ability to enhance their penetration into the skin and also can be found in commercially used products such as hand sanitizers. The patient was given information on this allergen and how to identify and avoid triggers. At follow-up, the ACD had resolved with avoidance of IPM.
Contact dermatitis is an inflammatory skin condition that is triggered by contact with a specific causative agent. There are 2 types of contact dermatitis: irritant and allergic; the irritant type is more common (approximately 80% of cases worldwide).1 Allergic contact dermatitis is a type IV (delayed-type) hypersensitivity reaction; common causative agents include shampoos, moisturizers, makeup, certain metals (eg, nickel), fragrances, latex, and certain plants (eg, poison ivy).2 In cases of ACD, a new reaction can develop from exposure to a product that the patient has used for years. It manifests clinically as erythema, pruritus, scaling, and vesicle formation.1 Certain populations, such as those with atopic dermatitis, are more prone to developing ACD due to a breakdown of the skin barrier, frequent use of topical products, and immune dysregulation.1,2 Patch testing performed by dermatologists and allergists is the gold standard for diagnosing ACD.1,3
Annually, allergists, dermatologists, and primary care physicians see thousands of cases of contact dermatitis.1 Early recognition and appropriate treatment can help reduce the severity and duration of symptoms and improve patient outcomes. The main treatment for ACD is identification of the causative agent followed by patient education on how to identify and avoid triggers.2 Once patch testing has been completed, patients can be given access to the American Contact Dermatitis Society’s Contact Allergen Management Program (CAMP) database (https://www.contactderm.org/resources/acds-camp) to help them identify and avoid products that contain triggering allergens.
Topical corticosteroids are the first-line pharmacologic treatments for atopic dermatitis.4 When our patient presented with the facial rash, her atopic dermatitis had been well controlled with both dupilumab and topical triamcinolone. The lack of response to previously successful therapies in a new area of involvement made a flare of atopic dermatitis less likely. For flares of ACD after exposure, topical corticosteroids and topical calcineurin inhibitors can help. If needed due to severity, oral corticosteroids also can be used.1
Dermatomyositis is an inflammatory myopathy that has several skin manifestations, including a heliotrope rash and poikiloderma.5 While our patient’s rash covered the periorbital area, she did not have other classic skin findings of dermatomyositis, such as nail-fold capillary changes or poikiloderma in a shawl or holster distribution.6 She also lacked signs of systemic involvement including myositis and elevated C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and creatine kinase levels.5
Erythematotelangiectatic rosacea is characterized by telangiectasias and transient flushing and erythema on the central face.5 Rosacea typically is triggered by temperature changes, alcohol consumption, sun exposure, spicy foods, and stress5 and would be expected to involve the nose, which was not observed in our patient. The fixed nature of our patient’s patches and the absence of telangiectasias also argued against this diagnosis.
The classic cutaneous finding of systemic lupus erythematosus is a malar rash, which appears as erythematous patches or thin plaques across the bridge of the nose and over the cheeks, sparing the nasolabial folds.5 Systemic lupus erythematosus is associated with laboratory abnormalities, such as positive antinuclear antibodies and elevated CRP and ESR levels.5 Our patient had notable sparing of the nose, negative antinuclear antibodies, and normal CRP and ESR levels, making systemic lupus erythematosus unlikely. Systemic lupus erythematosus also can manifest with photosensitivity,7 and involvement of the submental skin in our patient argued against a photosensitive eruption.
- Nassau S, Fonacier L. Allergic contact dermatitis. Med Clin North Am. 2020;104:61-76. doi:10.1016/j.mcna.2019.08.012
- Fonacier LS, Sher JM. Allergic contact dermatitis. Ann Allergy Asthma Immunol. 2014;113:9-12. doi:10.1016/j.anai.2014.03.018
- Uyesugi BA, Sheehan MP. Patch testing pearls. Clin Rev Allergy Immunol. 2019;56:110-118. doi:10.1007/s12016-018-8715-y
- Kapur S, Watson W, Carr S. Atopic dermatitis. Allergy Asthma Clin Immunol. 2018;14(suppl 2):52. doi:10.1186/s13223-018-0281-6
- Naji S. Malar rash. StatPearls. Updated September 4, 2023. Accessed June 30, 2025. https://www.statpearls.com/point-of-care/24661
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302. doi:10.1007 /s12016-015-8496-5
- Hannon CW, McCourt C, Lima HC, et al. Interventions for cutaneous disease in systemic lupus erythematosus. Cochrane Database Syst Rev. 2021;3(3):CD007478. doi:10.1002/14651858.CD007478.pub2
- Nassau S, Fonacier L. Allergic contact dermatitis. Med Clin North Am. 2020;104:61-76. doi:10.1016/j.mcna.2019.08.012
- Fonacier LS, Sher JM. Allergic contact dermatitis. Ann Allergy Asthma Immunol. 2014;113:9-12. doi:10.1016/j.anai.2014.03.018
- Uyesugi BA, Sheehan MP. Patch testing pearls. Clin Rev Allergy Immunol. 2019;56:110-118. doi:10.1007/s12016-018-8715-y
- Kapur S, Watson W, Carr S. Atopic dermatitis. Allergy Asthma Clin Immunol. 2018;14(suppl 2):52. doi:10.1186/s13223-018-0281-6
- Naji S. Malar rash. StatPearls. Updated September 4, 2023. Accessed June 30, 2025. https://www.statpearls.com/point-of-care/24661
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302. doi:10.1007 /s12016-015-8496-5
- Hannon CW, McCourt C, Lima HC, et al. Interventions for cutaneous disease in systemic lupus erythematosus. Cochrane Database Syst Rev. 2021;3(3):CD007478. doi:10.1002/14651858.CD007478.pub2
A 23-year-old woman with atopic dermatitis and seasonal allergic rhinitis presented to the dermatology department with an erythematous pruritic rash of 1 year’s duration involving the forehead, periorbital and submental skin, and neck. The patient’s atopic dermatitis was stable and had been well controlled with dupilumab and topical triamcinolone as needed for flares. The patient denied any other symptoms including fever, fatigue, and muscle weakness. Physical examination of the hands and nails revealed no abnormalities. She was treated with topical triamcinolone acetonide 0.1% without improvement. Short-term prednisone tapers fully resolved the rash, but it recurred within 5 days after discontinuation of prednisone. Results of testing for rheumatoid factor, antinuclear antibodies, complete blood count, comprehensive metabolic panel, C-reactive protein, erythrocyte sedimentation rate, and antistreptolysin O antibodies were unremarkable.
Multiple Fungating Plaques on the Face, Arms, and Legs
Multiple Fungating Plaques on the Face, Arms, and Legs
THE DIAGNOSIS: Mpox
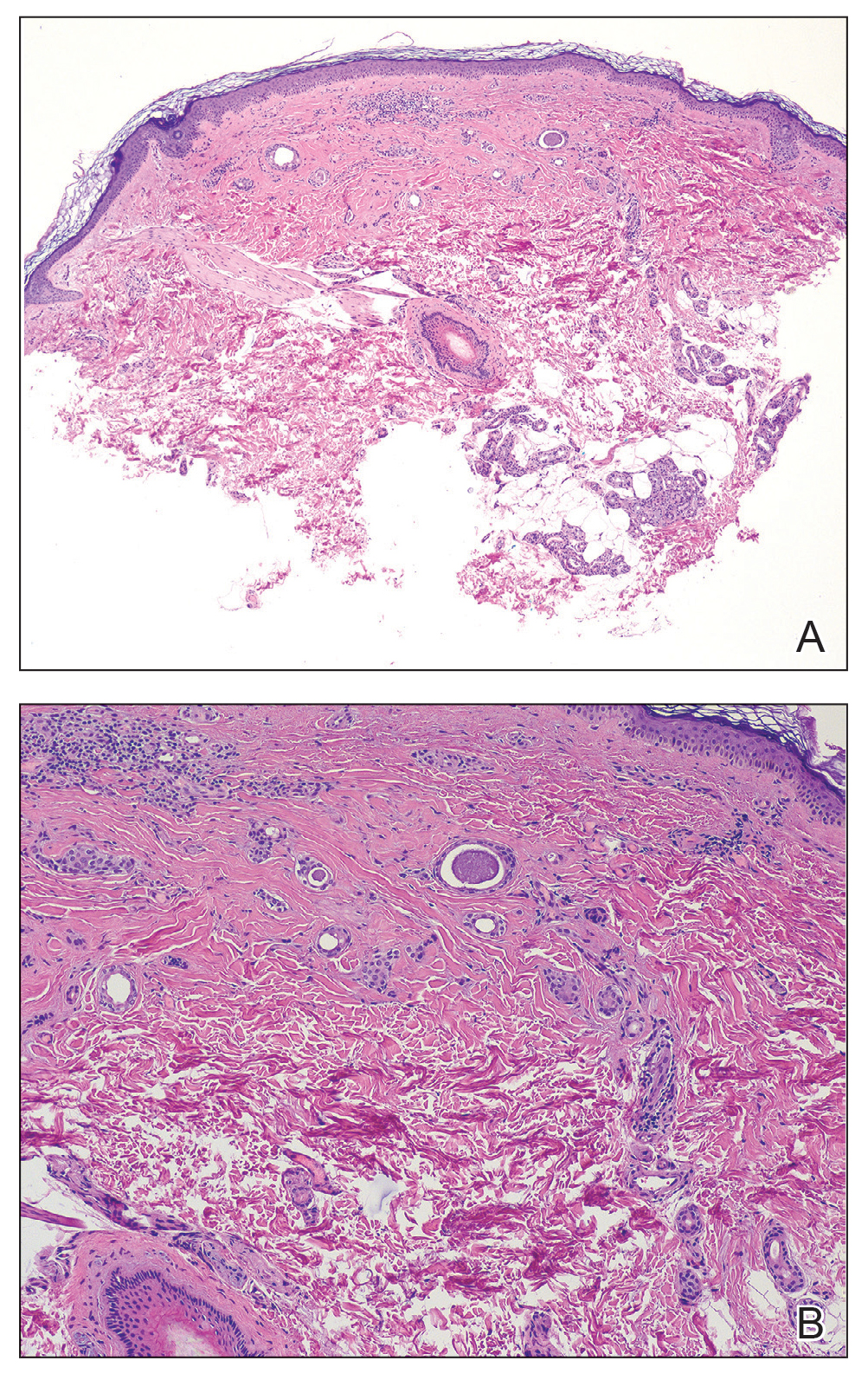
Histologic examination demonstrated dense aggregates of necrotic cellular debris composed of karyorrhectic nuclear fragments intermixed with neutrophils, lymphocytes, and histiocytes. Eosinophilic intracytoplasmic inclusions also were observed (Figure 1). The bacterial, fungal, and mycobacterial histologic special stains and cultures were negative. Three weeks after the initial visit with dermatology, the patient was admitted to the hospital for worsening symptoms of fever, chills, and painful erythema surrounding the skin lesions. Serology and viral workup revealed a positive mpox polymerase chain reaction test, suggesting a diagnosis of mpox. Following the Centers for Disease Control and Prevention protocol, the patient was started on oral tecovirimat 200 mg twice daily for 3 weeks and intravenous infusions of cidofovir 345 mg once weekly for 2 weeks. After treatment was initiated, the skin lesions showed rapid improvement (Figure 2), and he was discharged from the hospital after finishing the second dose of cidofovir. Four months after the initial dermatology consultation, the lesions had resolved completely with residual scarring. At that time, the patient had full movement of the right eye.
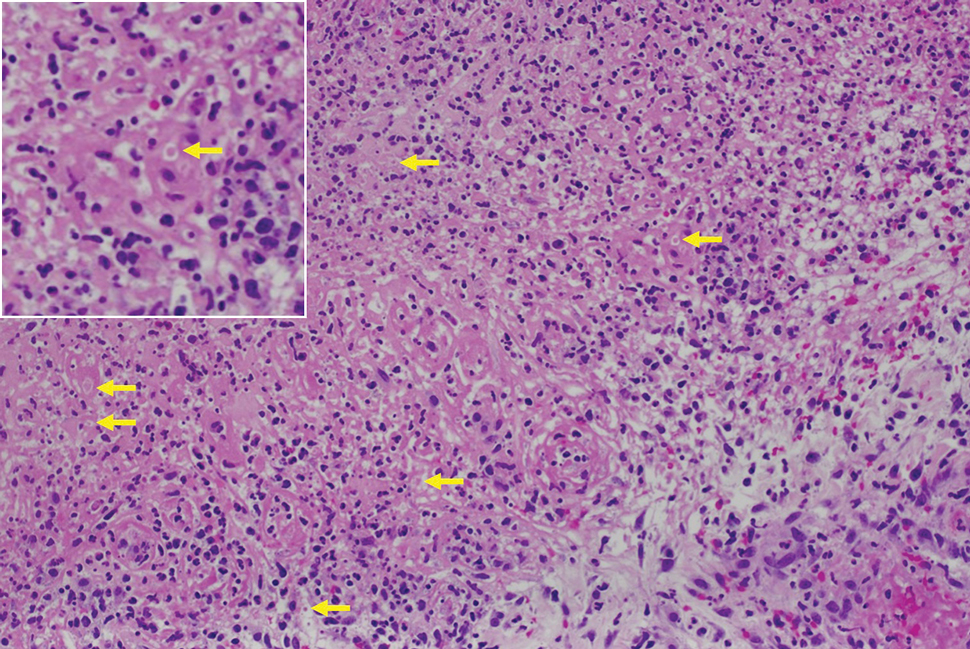
shows higher digital magnification of eosinophilic inclusions observed throughout the biopsy specimen (original magnification ×400).
Mpox virus is a member of the Poxviridae family of zoonotic viruses, which are transmitted from animals to humans. The mpox virus is brick-shaped (rectangular) and has a genome of linear double-stranded DNA encoding 180 proteins.1 Primates and rodents are the typical host reservoirs for viral circulation of mpox.2 Animal-to-human transmission occurs through direct contact with mucous membranes, bodily fluids, or tissues of an infected animal. Human-to-human transmission occurs through direct contact with infected mucous membranes, bodily fluids, respiratory droplets, and contaminated fomites.2
Symptoms typically occur within 1 week of exposure to the mpox virus. Prodromal symptoms of fever, sore throat, body aches, and headaches last for 3 days.1 Many patients experience a facial rash that spreads to the arms and legs over a period of 2 to 4 weeks. The rash initially manifests as small papules that progress to painful pustules and vesicles measuring 0.5 to 1.0 cm in diameter.3 The mpox virus is transmitted through these skin lesions until they crust over and re-epithelialize.1 The case fatality rate for mpox infection remains low (0.18%).4
Mpox outbreaks mainly were limited to central and western Africa prior to 2022. From May 17, 2022, through October 6, 2022, 26,384 cases of mpox were reported in the United States.5 During this outbreak, immunocompromised patients diagnosed with HIV and men who have sex with men were disproportionately affected.5
Due to the similarities between the smallpox virus and other orthopoxviruses, certain smallpox vaccines have been indicated for pre-exposure prophylaxis.6 The efficacy of prophylactic vaccination is believed to stem from the production of neutralizing antibodies that are cross-protective against other orthopoxviruses, including mpox.7 The 2 vaccines approved in the United States for mpox prophylaxis are JYNNEOS and ACAM2000, which are both live attenuated vaccines. Pre-exposure prophylaxis is indicated for patients at risk for severe disease, including men who have sex with men, individuals diagnosed with HIV or other immunosuppressive disorders, and individuals with recent diagnoses of one or more sexually transmitted diseases.8
Most mpox cases resolve within 2 to 4 weeks and only require supportive care (eg, nonsteroidal anti-inflammatory drugs, topical steroids, topical anesthetics) to treat pain.8 For patients at risk for severe disease, antiviral medications are warranted. Tecovirimat, brincidofovir, and cidofovir are antiviral medications used to treat smallpox that are thought to be effective against mpox.8,9 Tecovirimat and cidofovir have been shown to be effective against mpox in animal trials, but randomized or nonrandomized trials have not been performed in humans.9-11 Tecovirimat currently is available for the treatment of severe mpox in patients who meet the Centers for Disease Control and Prevention’s Investigational New Drug protocol; for these patients, a 200-mg course is administered orally or intravenously every 12 hours for 2 weeks.8
- Lu J, Xing H, Wang C, et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct Target Ther. 2023;8:458. doi:10.1038/s41392-023-01675-
- Lim CK, Roberts J, Moso M, et al. Mpox diagnostics: review of current and emerging technologies. J Med Virol. 2023;95:e28429. doi:10.1002/jmv.28429
- Brown K, Leggat PA. Human monkeypox: current state of knowledge and implications for the future. Trop Med Infect Dis. 2016;1:8. doi:10.3390/tropicalmed1010008
- World Health Organization. Mpox (monkeypox) World Health Organization. Published April 18, 2023. Accessed May 28, 2025. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- Kava CM, Rohraff DM, Wallace B, et al. Epidemiologic features of the monkeypox outbreak and the public health response—United States, May 17–October 6, 2022. 2022:1449-1456. https://www.cdc.gov/mmwr/volumes/71/wr/mm7145a4.htm?s_cid=mm7145a4_w
- Rizk JG, Lippi G, Henry BM, et al. Prevention and treatment of monkeypox. Drugs. 2022;82:957-963. doi:10.1007/s40265-022-01742-y
- Edghill-Smith Y, Golding H, Manischewitz J, et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740-747. doi:10.1038 /nm1261
- Centers for Disease Control and Prevention. Mpox treatment information for healthcare professionals. Updated June 18, 2024. Accessed May 28, 2025. https://www.cdc.gov/mpox/hcp/clinical-care/?CDC_AAref_Val=https://www.cdc.gov/poxvirus/mpox/clinicians/treatment.html
- Mitja O, Ogoina D, Titanji BK, et al. Monkeypox. Lancet. 2023;401:60-74. doi:10.1016/S0140-6736(22)02075-X
- Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53:2620-2625. doi:10.1128/aac.00021-09
- Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379:44-53. doi:10.1056 /nejmoa1705688
THE DIAGNOSIS: Mpox
Histologic examination demonstrated dense aggregates of necrotic cellular debris composed of karyorrhectic nuclear fragments intermixed with neutrophils, lymphocytes, and histiocytes. Eosinophilic intracytoplasmic inclusions also were observed (Figure 1). The bacterial, fungal, and mycobacterial histologic special stains and cultures were negative. Three weeks after the initial visit with dermatology, the patient was admitted to the hospital for worsening symptoms of fever, chills, and painful erythema surrounding the skin lesions. Serology and viral workup revealed a positive mpox polymerase chain reaction test, suggesting a diagnosis of mpox. Following the Centers for Disease Control and Prevention protocol, the patient was started on oral tecovirimat 200 mg twice daily for 3 weeks and intravenous infusions of cidofovir 345 mg once weekly for 2 weeks. After treatment was initiated, the skin lesions showed rapid improvement (Figure 2), and he was discharged from the hospital after finishing the second dose of cidofovir. Four months after the initial dermatology consultation, the lesions had resolved completely with residual scarring. At that time, the patient had full movement of the right eye.
shows higher digital magnification of eosinophilic inclusions observed throughout the biopsy specimen (original magnification ×400).
Mpox virus is a member of the Poxviridae family of zoonotic viruses, which are transmitted from animals to humans. The mpox virus is brick-shaped (rectangular) and has a genome of linear double-stranded DNA encoding 180 proteins.1 Primates and rodents are the typical host reservoirs for viral circulation of mpox.2 Animal-to-human transmission occurs through direct contact with mucous membranes, bodily fluids, or tissues of an infected animal. Human-to-human transmission occurs through direct contact with infected mucous membranes, bodily fluids, respiratory droplets, and contaminated fomites.2
Symptoms typically occur within 1 week of exposure to the mpox virus. Prodromal symptoms of fever, sore throat, body aches, and headaches last for 3 days.1 Many patients experience a facial rash that spreads to the arms and legs over a period of 2 to 4 weeks. The rash initially manifests as small papules that progress to painful pustules and vesicles measuring 0.5 to 1.0 cm in diameter.3 The mpox virus is transmitted through these skin lesions until they crust over and re-epithelialize.1 The case fatality rate for mpox infection remains low (0.18%).4
Mpox outbreaks mainly were limited to central and western Africa prior to 2022. From May 17, 2022, through October 6, 2022, 26,384 cases of mpox were reported in the United States.5 During this outbreak, immunocompromised patients diagnosed with HIV and men who have sex with men were disproportionately affected.5
Due to the similarities between the smallpox virus and other orthopoxviruses, certain smallpox vaccines have been indicated for pre-exposure prophylaxis.6 The efficacy of prophylactic vaccination is believed to stem from the production of neutralizing antibodies that are cross-protective against other orthopoxviruses, including mpox.7 The 2 vaccines approved in the United States for mpox prophylaxis are JYNNEOS and ACAM2000, which are both live attenuated vaccines. Pre-exposure prophylaxis is indicated for patients at risk for severe disease, including men who have sex with men, individuals diagnosed with HIV or other immunosuppressive disorders, and individuals with recent diagnoses of one or more sexually transmitted diseases.8
Most mpox cases resolve within 2 to 4 weeks and only require supportive care (eg, nonsteroidal anti-inflammatory drugs, topical steroids, topical anesthetics) to treat pain.8 For patients at risk for severe disease, antiviral medications are warranted. Tecovirimat, brincidofovir, and cidofovir are antiviral medications used to treat smallpox that are thought to be effective against mpox.8,9 Tecovirimat and cidofovir have been shown to be effective against mpox in animal trials, but randomized or nonrandomized trials have not been performed in humans.9-11 Tecovirimat currently is available for the treatment of severe mpox in patients who meet the Centers for Disease Control and Prevention’s Investigational New Drug protocol; for these patients, a 200-mg course is administered orally or intravenously every 12 hours for 2 weeks.8
THE DIAGNOSIS: Mpox
Histologic examination demonstrated dense aggregates of necrotic cellular debris composed of karyorrhectic nuclear fragments intermixed with neutrophils, lymphocytes, and histiocytes. Eosinophilic intracytoplasmic inclusions also were observed (Figure 1). The bacterial, fungal, and mycobacterial histologic special stains and cultures were negative. Three weeks after the initial visit with dermatology, the patient was admitted to the hospital for worsening symptoms of fever, chills, and painful erythema surrounding the skin lesions. Serology and viral workup revealed a positive mpox polymerase chain reaction test, suggesting a diagnosis of mpox. Following the Centers for Disease Control and Prevention protocol, the patient was started on oral tecovirimat 200 mg twice daily for 3 weeks and intravenous infusions of cidofovir 345 mg once weekly for 2 weeks. After treatment was initiated, the skin lesions showed rapid improvement (Figure 2), and he was discharged from the hospital after finishing the second dose of cidofovir. Four months after the initial dermatology consultation, the lesions had resolved completely with residual scarring. At that time, the patient had full movement of the right eye.
shows higher digital magnification of eosinophilic inclusions observed throughout the biopsy specimen (original magnification ×400).
Mpox virus is a member of the Poxviridae family of zoonotic viruses, which are transmitted from animals to humans. The mpox virus is brick-shaped (rectangular) and has a genome of linear double-stranded DNA encoding 180 proteins.1 Primates and rodents are the typical host reservoirs for viral circulation of mpox.2 Animal-to-human transmission occurs through direct contact with mucous membranes, bodily fluids, or tissues of an infected animal. Human-to-human transmission occurs through direct contact with infected mucous membranes, bodily fluids, respiratory droplets, and contaminated fomites.2
Symptoms typically occur within 1 week of exposure to the mpox virus. Prodromal symptoms of fever, sore throat, body aches, and headaches last for 3 days.1 Many patients experience a facial rash that spreads to the arms and legs over a period of 2 to 4 weeks. The rash initially manifests as small papules that progress to painful pustules and vesicles measuring 0.5 to 1.0 cm in diameter.3 The mpox virus is transmitted through these skin lesions until they crust over and re-epithelialize.1 The case fatality rate for mpox infection remains low (0.18%).4
Mpox outbreaks mainly were limited to central and western Africa prior to 2022. From May 17, 2022, through October 6, 2022, 26,384 cases of mpox were reported in the United States.5 During this outbreak, immunocompromised patients diagnosed with HIV and men who have sex with men were disproportionately affected.5
Due to the similarities between the smallpox virus and other orthopoxviruses, certain smallpox vaccines have been indicated for pre-exposure prophylaxis.6 The efficacy of prophylactic vaccination is believed to stem from the production of neutralizing antibodies that are cross-protective against other orthopoxviruses, including mpox.7 The 2 vaccines approved in the United States for mpox prophylaxis are JYNNEOS and ACAM2000, which are both live attenuated vaccines. Pre-exposure prophylaxis is indicated for patients at risk for severe disease, including men who have sex with men, individuals diagnosed with HIV or other immunosuppressive disorders, and individuals with recent diagnoses of one or more sexually transmitted diseases.8
Most mpox cases resolve within 2 to 4 weeks and only require supportive care (eg, nonsteroidal anti-inflammatory drugs, topical steroids, topical anesthetics) to treat pain.8 For patients at risk for severe disease, antiviral medications are warranted. Tecovirimat, brincidofovir, and cidofovir are antiviral medications used to treat smallpox that are thought to be effective against mpox.8,9 Tecovirimat and cidofovir have been shown to be effective against mpox in animal trials, but randomized or nonrandomized trials have not been performed in humans.9-11 Tecovirimat currently is available for the treatment of severe mpox in patients who meet the Centers for Disease Control and Prevention’s Investigational New Drug protocol; for these patients, a 200-mg course is administered orally or intravenously every 12 hours for 2 weeks.8
- Lu J, Xing H, Wang C, et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct Target Ther. 2023;8:458. doi:10.1038/s41392-023-01675-
- Lim CK, Roberts J, Moso M, et al. Mpox diagnostics: review of current and emerging technologies. J Med Virol. 2023;95:e28429. doi:10.1002/jmv.28429
- Brown K, Leggat PA. Human monkeypox: current state of knowledge and implications for the future. Trop Med Infect Dis. 2016;1:8. doi:10.3390/tropicalmed1010008
- World Health Organization. Mpox (monkeypox) World Health Organization. Published April 18, 2023. Accessed May 28, 2025. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- Kava CM, Rohraff DM, Wallace B, et al. Epidemiologic features of the monkeypox outbreak and the public health response—United States, May 17–October 6, 2022. 2022:1449-1456. https://www.cdc.gov/mmwr/volumes/71/wr/mm7145a4.htm?s_cid=mm7145a4_w
- Rizk JG, Lippi G, Henry BM, et al. Prevention and treatment of monkeypox. Drugs. 2022;82:957-963. doi:10.1007/s40265-022-01742-y
- Edghill-Smith Y, Golding H, Manischewitz J, et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740-747. doi:10.1038 /nm1261
- Centers for Disease Control and Prevention. Mpox treatment information for healthcare professionals. Updated June 18, 2024. Accessed May 28, 2025. https://www.cdc.gov/mpox/hcp/clinical-care/?CDC_AAref_Val=https://www.cdc.gov/poxvirus/mpox/clinicians/treatment.html
- Mitja O, Ogoina D, Titanji BK, et al. Monkeypox. Lancet. 2023;401:60-74. doi:10.1016/S0140-6736(22)02075-X
- Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53:2620-2625. doi:10.1128/aac.00021-09
- Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379:44-53. doi:10.1056 /nejmoa1705688
- Lu J, Xing H, Wang C, et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct Target Ther. 2023;8:458. doi:10.1038/s41392-023-01675-
- Lim CK, Roberts J, Moso M, et al. Mpox diagnostics: review of current and emerging technologies. J Med Virol. 2023;95:e28429. doi:10.1002/jmv.28429
- Brown K, Leggat PA. Human monkeypox: current state of knowledge and implications for the future. Trop Med Infect Dis. 2016;1:8. doi:10.3390/tropicalmed1010008
- World Health Organization. Mpox (monkeypox) World Health Organization. Published April 18, 2023. Accessed May 28, 2025. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- Kava CM, Rohraff DM, Wallace B, et al. Epidemiologic features of the monkeypox outbreak and the public health response—United States, May 17–October 6, 2022. 2022:1449-1456. https://www.cdc.gov/mmwr/volumes/71/wr/mm7145a4.htm?s_cid=mm7145a4_w
- Rizk JG, Lippi G, Henry BM, et al. Prevention and treatment of monkeypox. Drugs. 2022;82:957-963. doi:10.1007/s40265-022-01742-y
- Edghill-Smith Y, Golding H, Manischewitz J, et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740-747. doi:10.1038 /nm1261
- Centers for Disease Control and Prevention. Mpox treatment information for healthcare professionals. Updated June 18, 2024. Accessed May 28, 2025. https://www.cdc.gov/mpox/hcp/clinical-care/?CDC_AAref_Val=https://www.cdc.gov/poxvirus/mpox/clinicians/treatment.html
- Mitja O, Ogoina D, Titanji BK, et al. Monkeypox. Lancet. 2023;401:60-74. doi:10.1016/S0140-6736(22)02075-X
- Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53:2620-2625. doi:10.1128/aac.00021-09
- Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379:44-53. doi:10.1056 /nejmoa1705688
Multiple Fungating Plaques on the Face, Arms, and Legs
Multiple Fungating Plaques on the Face, Arms, and Legs
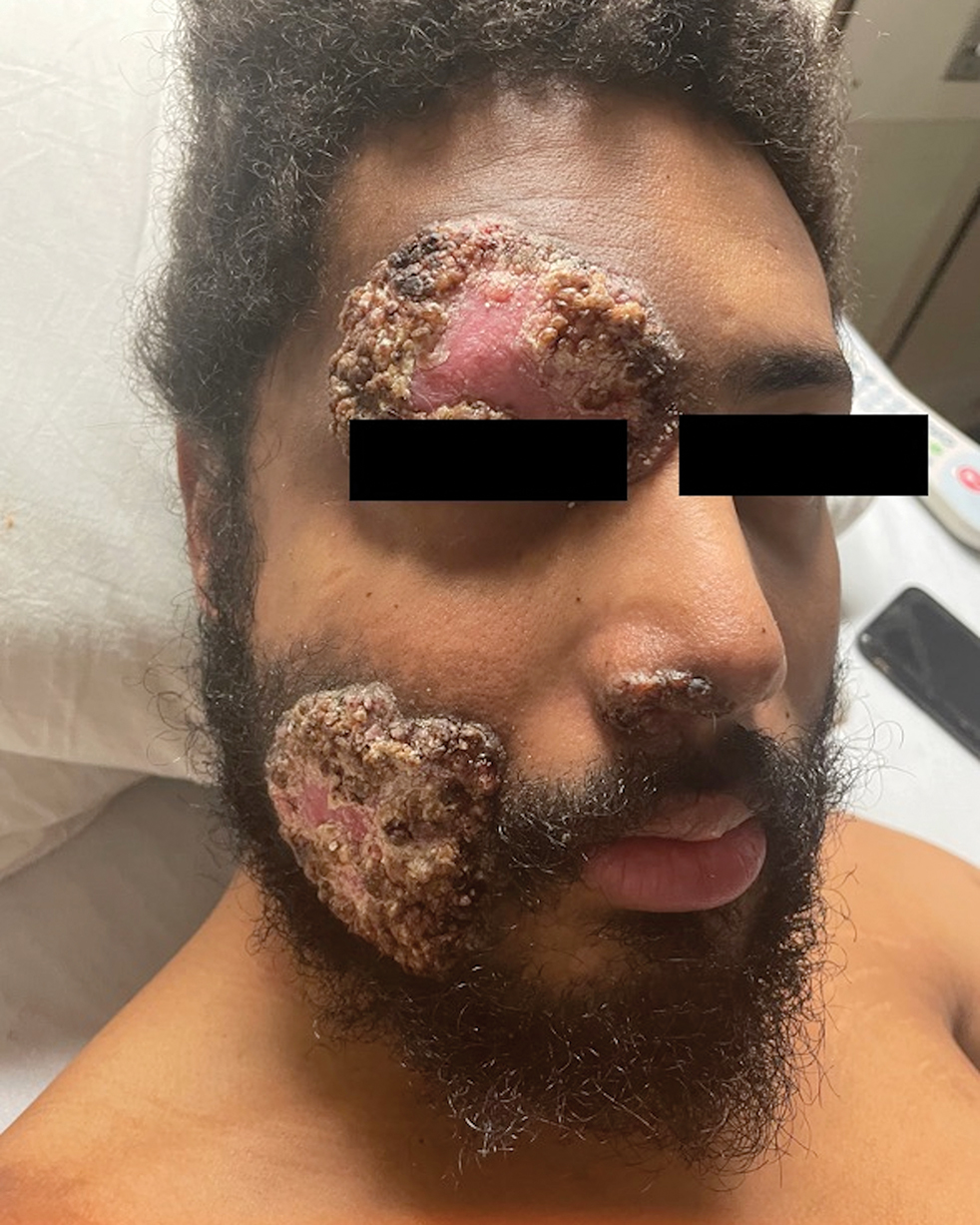
A 27-year-old man presented to his primary care physician after he was struck in the head by a tree branch while working outside. The next day, ulcerating lesions emerged on the right supraorbital ridge, along with subjective fevers, chills, fatigue, and shortness of breath. The patient reported a history of unprotected sexual intercourse with a male partner who was HIV positive. His medical history included syphilis status posttreatment with a course of 5 penicillin injections, hepatitis C, and HIV diagnosed one month prior to presentation (CD4 count, 169 cells/mm3 [reference range, 500-1500 cells/mm3]). A punch biopsy performed by the primary care physician revealed suppurative granulomatous inflammation, and the patient was prescribed antibiotics with mild improvement. He then was referred to dermatology for further evaluation of the ulcerating lesions.
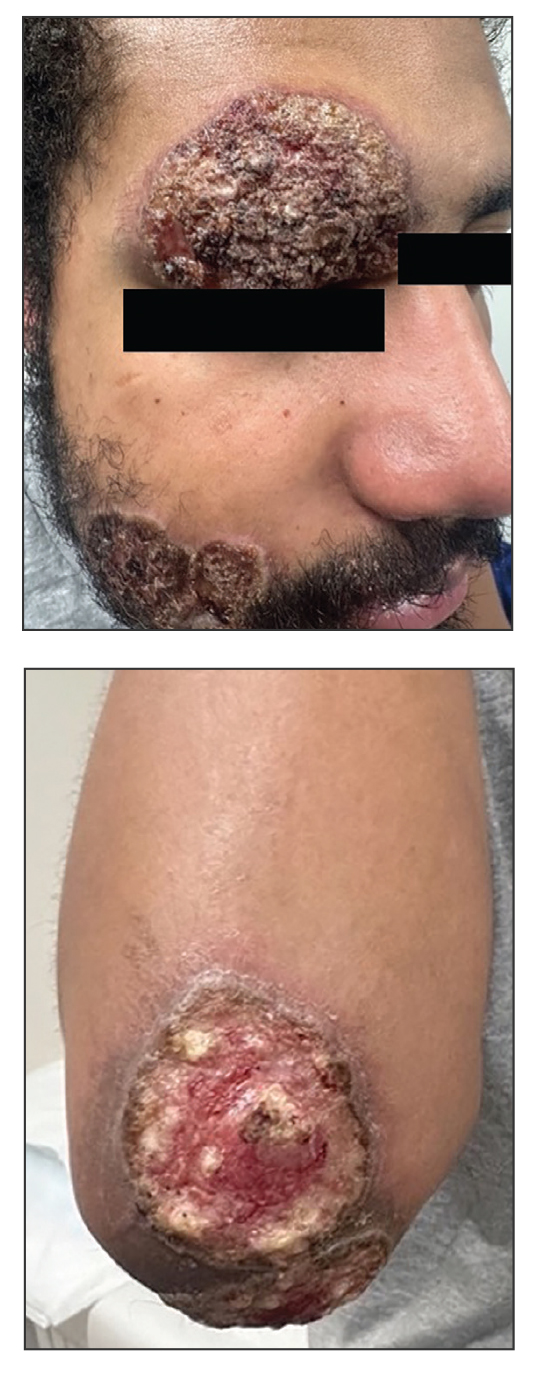
Three months after the initial trauma, the patient presented to the dermatology clinic for evaluation of multiple large fungating plaques affecting multiple sites on the face (top), arms (bottom), and legs. Physical examination revealed large circinate verrucous plaques involving the right supraorbital ridge and eyelid. The patient was unable to fully open the right eye. Similar plaques also were observed on the right malar cheek, arms, and feet. Four 5-mm punch biopsies from lesions on the right elbow and left ankle were obtained with fungal and bacterial cultures.

Eruptive Erythematous Papules on the Forearms
Eruptive Erythematous Papules on the Forearms
THE DIAGNOSIS: Acral Eruptive Syringoma
Syringomas are small, benign, often asymptomatic eccrine tumors that originate in the intraepidermal portion of eccrine sweat ducts.1 Clinically, they present as multiple symmetric white-to-yellow or discrete flesh-colored papules measuring 1 to 3 mm in diameter, often located on the face (most commonly on the eyelids), with a greater prevalence in middle-aged women. Occasionally, they manifest in other locations such as the cheeks, chest, axillae, abdomen, and groin.2
In 1987, Friedman and Butler3 developed a classification system categorizing syringomas into 4 clinical subtypes: familial syringoma, localized syringoma, Down syndrome–related syringoma, and generalized syringoma. The fourth subtype includes the variant of eruptive syringoma,3 a rare clinical manifestation that often develops before or during puberty with several flesh-colored or lightly pigmented papules on the neck, anterior chest, upper abdomen, axillae, periumbilical region, and/or genital region.1,4,5 The etiology of eruptive syringomas is unclear, although it has been linked to abnormal proliferation of sweat glands due to an underlying local inflammatory process.6
Acral distribution of syringomas is a rare variant that can manifest as part of generalized eruptive syringoma with consequent involvement of the arms and other areas.5,7 There are limited case reports on eruptive syringomas with predominant acral distribution.8 Compared to classic syringomas, the acral variant is associated with an older age of onset as well as a similar prevalence between men and women.9 Acral eruptive syringoma (AES) usually is isolated to the distal arms and legs. The most commonly affected region is the anterior surface of the forearms, although involvement of the dorsal hands, wrists, and feet also has been reported.10-16
The first known case of AES, which was reported in 1977, described eruptive syringomas on the dorsal hands of a healthy 31-year-old man.17 Several cases have been reported since then, mostly in patients aged 30 to 60 years, with predominant involvement of the dorsal hands and forearms.18-24 A review of Embase as well as PubMed articles indexed for MEDLINE using the search terms syringoma OR eccrine ductal tumor and eruptive OR acral OR arms OR forearms OR extremities identified 19 reported cases of AES between 1977 and 2023. For the reported AES cases, the mean (SD) age at diagnosis was 45.1 years (15.96 years), with patient ages ranging from 19 to 76 years. Notably, most cases occurred in individuals aged between 30 and 60 years, which deviates from the typical age of onset of localized syringomas, commonly seen during puberty or early adulthood.
Currently, AES is categorized within the clinical presentation of eruptive syringoma. Nevertheless, some authors have proposed classifying it as a distinct fifth clinical group due to specific features that distinguish it from generalized eruptive syringoma.9 This reclassification has considerable implications for the differential diagnosis, particularly because exclusive acral involvement poses a substantial diagnostic challenge and often requires histologic confirmation.
As shown in the Figure, histopathologic examination revealed tubular structures in the upper dermis with characteristic comma-shaped extensions. Some of these structures were lined with cuboidal cells and contained eosinophilic material within the lumen. There was no involvement of the epidermis or deeper dermis. The histologic features were consistent with syringoma, which is distinguished by its predominant involvement of the upper dermis and the presence of enlarged, dilated eccrine ducts, as observed in our case.
Treatment of syringomas often is challenging due to the high rate of recurrence and the risk for postinflammatory hyperpigmentation. Since the condition is benign, treatment typically is pursued for aesthetic reasons. Various therapeutic approaches have been reported, each with diverse response rates. The most common method involves surgical intervention, either with electrodesiccation or CO2 laser—both of which have shown satisfactory resolution of lesions without recurrence at 1-year follow-up, with no major scarring reported.25,26 Alternatively, topical management with retinoids daily over a 4-month period leads to flattening of the tumors with no further appearance of new lesions.27 Despite the availability of numerous management options, establishing a first-line treatment remains controversial due to the high risk for recurrence and the variability in the number and location of lesions among individual patients. In our case, given the benign nature of syringomas, the asymptomatic nature of the lesions, the involvement of noncritical aesthetic areas, and the limited response to noninvasive therapeutic options, the patient was informed of the diagnosis, and no further pharmacologic or surgical intervention was pursued.
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.E9. doi:10.1016 /j.jaad.2015.12.006
- Resende C, Araújo C, Santos R, et al. Late-onset of eruptive syringomas: a diagnostic challenge. An Bras Dermatol. 2015;90(3 suppl 1):239-241. doi:10.1590/abd1806-4841.20153899
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Avhad G, Ghuge P, Jerajani HR. Generalized eruptive syringoma. Indian J Dermatol. 2015;60:214. doi:10.4103/0019-5154.152586
- Ning WV, Bashey S, Cole C, et al. Multiple eruptive syringomas on the penis. Cutis. 2019;103:E15-E16.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Jamalipour M, Heidarpour M, Rajabi P. Generalized eruptive syringomas. Indian J Dermatol. 2009;54:65-67. doi:10.4103/0019-5154.48992
- Mohaghegh F, Amiri A, Fatemi Naeini F, et al. Acral eruptive syringoma: an unusual presentation with misdiagnosis. Case Rep Dermatol Med. 2020;2020:5416285. doi:10.1155/2020/5416285
- Valdivielso-Ramos M, de la Cueva P, Gimeno M, et al. Acral syringomas. Actas Dermosifiliogr. 2010;101:458-460.
- Patel K, Lundgren AD, Ahmed AM, et al. Disseminated syringomas of the upper extremities in a young woman. Cureus. 2018;10:E3619. doi:10.7759/cureus.3619
- Balci DD, Atik E, Altintas S. Coexistence of acral syringomas and multiple trichoepitheliomas on the face. J Cutan Med Surg. 2009;13:169-171. doi:10.2310/7750.2008.08011
- Martín-García RF, Muñoz CM. Acral syringomas presenting as a photosensitive papular eruption. Cutis. 2006;77:33-36.
- Varas-Meis E, Prada-García C, Samaniego-González E, et al. Acral syringomas associated with hematological neoplasm. Indian J Dermatol Venereol Leprol. 2017;83:136. doi:10.4103/0378-6323.192961
- Berbis P, Fabre JF, Jancovici E, et al. Late-onset syringomas of the upper extremities associated with a carcinoid tumor. Arch Dermatol. 1989;125:848-849.
- Metze D, Jurecka W, Gebhart W. Disseminated syringomas of the upper extremities. case history and immunohistochemical and ultrastructural study. Dermatologica. 1990;180:228-235. doi:10.1159/000248036
- Gómez-de Castro C, Vivanco Allende B, García-García B. Multiple acral syringomas. siringomas acrales múltiples. Actas Dermosifiliogr (Engl Ed). 2018;109:834-836. doi:10.1016/j.ad.2017.10.014
- Hughes PS, Apisarnthanarax P. Acral syringoma. Arch Dermatol. 1977;113:1435-1436.
- Asai Y, Ishii M, Hamada T. Acral syringoma: electron microscopic studies on its origin. Acta Derm Venereol. 1982;62:64-68.
- van den Broek H, Lundquist CD. Syringomas of the upper extremities with onset in the sixth decade. J Am Acad Dermatol. 1982,6:534-536. doi:10.1016/S0190-9622(82)80368-X
- Garcia C, Krunic AL, Grichnik J, et al. Multiple acral syringomata with uniform involvement of the hands and feet. Cutis. 1997;59:213-214, 216.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Iglesias Sancho M, Serra Llobet J, Salleras Redonnet M, et al. Siringomas disem- inados de inicio acral, aparecidos en la octava década. Actas Dermosifiliofr. 1999;90:253-257.
- Muniesa C, Fortuño Y, Moreno A, et al. Papules on the dorsum of the fingers. Actas Dermosifiliogr. 2008;99:812-813. doi:10.1016 /S1578-2190(08)70371-8
- Koh MJ. Multiple acral syringomas involving the hands. Clin Exp Dermatol. 2009;34:E438. doi:10.1111/j.1365-2230.2009.03462.x
- Karam P, Benedetto AV. Syringomas: new approach to an old technique. Int J Dermatol. 1996;35:219-220. doi:10.1111/j.1365-4362 .1996.tb01647.x
- Wang JI, Roenigk HH. Treatment of multiple facial syringomas with the carbon dioxide (CO2) laser. Dermatol Surg. 1999;25:136-139. doi:10.1046/j.1524-4725.1999.08111.x
- Gómez MI, Pérez B, Azaña JM, et al. Eruptive syringoma: treatment with topical tretinoin. Dermatology. 2009;189:105-106. doi:10.1159/000246803
THE DIAGNOSIS: Acral Eruptive Syringoma
Syringomas are small, benign, often asymptomatic eccrine tumors that originate in the intraepidermal portion of eccrine sweat ducts.1 Clinically, they present as multiple symmetric white-to-yellow or discrete flesh-colored papules measuring 1 to 3 mm in diameter, often located on the face (most commonly on the eyelids), with a greater prevalence in middle-aged women. Occasionally, they manifest in other locations such as the cheeks, chest, axillae, abdomen, and groin.2
In 1987, Friedman and Butler3 developed a classification system categorizing syringomas into 4 clinical subtypes: familial syringoma, localized syringoma, Down syndrome–related syringoma, and generalized syringoma. The fourth subtype includes the variant of eruptive syringoma,3 a rare clinical manifestation that often develops before or during puberty with several flesh-colored or lightly pigmented papules on the neck, anterior chest, upper abdomen, axillae, periumbilical region, and/or genital region.1,4,5 The etiology of eruptive syringomas is unclear, although it has been linked to abnormal proliferation of sweat glands due to an underlying local inflammatory process.6
Acral distribution of syringomas is a rare variant that can manifest as part of generalized eruptive syringoma with consequent involvement of the arms and other areas.5,7 There are limited case reports on eruptive syringomas with predominant acral distribution.8 Compared to classic syringomas, the acral variant is associated with an older age of onset as well as a similar prevalence between men and women.9 Acral eruptive syringoma (AES) usually is isolated to the distal arms and legs. The most commonly affected region is the anterior surface of the forearms, although involvement of the dorsal hands, wrists, and feet also has been reported.10-16
The first known case of AES, which was reported in 1977, described eruptive syringomas on the dorsal hands of a healthy 31-year-old man.17 Several cases have been reported since then, mostly in patients aged 30 to 60 years, with predominant involvement of the dorsal hands and forearms.18-24 A review of Embase as well as PubMed articles indexed for MEDLINE using the search terms syringoma OR eccrine ductal tumor and eruptive OR acral OR arms OR forearms OR extremities identified 19 reported cases of AES between 1977 and 2023. For the reported AES cases, the mean (SD) age at diagnosis was 45.1 years (15.96 years), with patient ages ranging from 19 to 76 years. Notably, most cases occurred in individuals aged between 30 and 60 years, which deviates from the typical age of onset of localized syringomas, commonly seen during puberty or early adulthood.
Currently, AES is categorized within the clinical presentation of eruptive syringoma. Nevertheless, some authors have proposed classifying it as a distinct fifth clinical group due to specific features that distinguish it from generalized eruptive syringoma.9 This reclassification has considerable implications for the differential diagnosis, particularly because exclusive acral involvement poses a substantial diagnostic challenge and often requires histologic confirmation.
As shown in the Figure, histopathologic examination revealed tubular structures in the upper dermis with characteristic comma-shaped extensions. Some of these structures were lined with cuboidal cells and contained eosinophilic material within the lumen. There was no involvement of the epidermis or deeper dermis. The histologic features were consistent with syringoma, which is distinguished by its predominant involvement of the upper dermis and the presence of enlarged, dilated eccrine ducts, as observed in our case.
Treatment of syringomas often is challenging due to the high rate of recurrence and the risk for postinflammatory hyperpigmentation. Since the condition is benign, treatment typically is pursued for aesthetic reasons. Various therapeutic approaches have been reported, each with diverse response rates. The most common method involves surgical intervention, either with electrodesiccation or CO2 laser—both of which have shown satisfactory resolution of lesions without recurrence at 1-year follow-up, with no major scarring reported.25,26 Alternatively, topical management with retinoids daily over a 4-month period leads to flattening of the tumors with no further appearance of new lesions.27 Despite the availability of numerous management options, establishing a first-line treatment remains controversial due to the high risk for recurrence and the variability in the number and location of lesions among individual patients. In our case, given the benign nature of syringomas, the asymptomatic nature of the lesions, the involvement of noncritical aesthetic areas, and the limited response to noninvasive therapeutic options, the patient was informed of the diagnosis, and no further pharmacologic or surgical intervention was pursued.
THE DIAGNOSIS: Acral Eruptive Syringoma
Syringomas are small, benign, often asymptomatic eccrine tumors that originate in the intraepidermal portion of eccrine sweat ducts.1 Clinically, they present as multiple symmetric white-to-yellow or discrete flesh-colored papules measuring 1 to 3 mm in diameter, often located on the face (most commonly on the eyelids), with a greater prevalence in middle-aged women. Occasionally, they manifest in other locations such as the cheeks, chest, axillae, abdomen, and groin.2
In 1987, Friedman and Butler3 developed a classification system categorizing syringomas into 4 clinical subtypes: familial syringoma, localized syringoma, Down syndrome–related syringoma, and generalized syringoma. The fourth subtype includes the variant of eruptive syringoma,3 a rare clinical manifestation that often develops before or during puberty with several flesh-colored or lightly pigmented papules on the neck, anterior chest, upper abdomen, axillae, periumbilical region, and/or genital region.1,4,5 The etiology of eruptive syringomas is unclear, although it has been linked to abnormal proliferation of sweat glands due to an underlying local inflammatory process.6
Acral distribution of syringomas is a rare variant that can manifest as part of generalized eruptive syringoma with consequent involvement of the arms and other areas.5,7 There are limited case reports on eruptive syringomas with predominant acral distribution.8 Compared to classic syringomas, the acral variant is associated with an older age of onset as well as a similar prevalence between men and women.9 Acral eruptive syringoma (AES) usually is isolated to the distal arms and legs. The most commonly affected region is the anterior surface of the forearms, although involvement of the dorsal hands, wrists, and feet also has been reported.10-16
The first known case of AES, which was reported in 1977, described eruptive syringomas on the dorsal hands of a healthy 31-year-old man.17 Several cases have been reported since then, mostly in patients aged 30 to 60 years, with predominant involvement of the dorsal hands and forearms.18-24 A review of Embase as well as PubMed articles indexed for MEDLINE using the search terms syringoma OR eccrine ductal tumor and eruptive OR acral OR arms OR forearms OR extremities identified 19 reported cases of AES between 1977 and 2023. For the reported AES cases, the mean (SD) age at diagnosis was 45.1 years (15.96 years), with patient ages ranging from 19 to 76 years. Notably, most cases occurred in individuals aged between 30 and 60 years, which deviates from the typical age of onset of localized syringomas, commonly seen during puberty or early adulthood.
Currently, AES is categorized within the clinical presentation of eruptive syringoma. Nevertheless, some authors have proposed classifying it as a distinct fifth clinical group due to specific features that distinguish it from generalized eruptive syringoma.9 This reclassification has considerable implications for the differential diagnosis, particularly because exclusive acral involvement poses a substantial diagnostic challenge and often requires histologic confirmation.
As shown in the Figure, histopathologic examination revealed tubular structures in the upper dermis with characteristic comma-shaped extensions. Some of these structures were lined with cuboidal cells and contained eosinophilic material within the lumen. There was no involvement of the epidermis or deeper dermis. The histologic features were consistent with syringoma, which is distinguished by its predominant involvement of the upper dermis and the presence of enlarged, dilated eccrine ducts, as observed in our case.
Treatment of syringomas often is challenging due to the high rate of recurrence and the risk for postinflammatory hyperpigmentation. Since the condition is benign, treatment typically is pursued for aesthetic reasons. Various therapeutic approaches have been reported, each with diverse response rates. The most common method involves surgical intervention, either with electrodesiccation or CO2 laser—both of which have shown satisfactory resolution of lesions without recurrence at 1-year follow-up, with no major scarring reported.25,26 Alternatively, topical management with retinoids daily over a 4-month period leads to flattening of the tumors with no further appearance of new lesions.27 Despite the availability of numerous management options, establishing a first-line treatment remains controversial due to the high risk for recurrence and the variability in the number and location of lesions among individual patients. In our case, given the benign nature of syringomas, the asymptomatic nature of the lesions, the involvement of noncritical aesthetic areas, and the limited response to noninvasive therapeutic options, the patient was informed of the diagnosis, and no further pharmacologic or surgical intervention was pursued.
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.E9. doi:10.1016 /j.jaad.2015.12.006
- Resende C, Araújo C, Santos R, et al. Late-onset of eruptive syringomas: a diagnostic challenge. An Bras Dermatol. 2015;90(3 suppl 1):239-241. doi:10.1590/abd1806-4841.20153899
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Avhad G, Ghuge P, Jerajani HR. Generalized eruptive syringoma. Indian J Dermatol. 2015;60:214. doi:10.4103/0019-5154.152586
- Ning WV, Bashey S, Cole C, et al. Multiple eruptive syringomas on the penis. Cutis. 2019;103:E15-E16.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Jamalipour M, Heidarpour M, Rajabi P. Generalized eruptive syringomas. Indian J Dermatol. 2009;54:65-67. doi:10.4103/0019-5154.48992
- Mohaghegh F, Amiri A, Fatemi Naeini F, et al. Acral eruptive syringoma: an unusual presentation with misdiagnosis. Case Rep Dermatol Med. 2020;2020:5416285. doi:10.1155/2020/5416285
- Valdivielso-Ramos M, de la Cueva P, Gimeno M, et al. Acral syringomas. Actas Dermosifiliogr. 2010;101:458-460.
- Patel K, Lundgren AD, Ahmed AM, et al. Disseminated syringomas of the upper extremities in a young woman. Cureus. 2018;10:E3619. doi:10.7759/cureus.3619
- Balci DD, Atik E, Altintas S. Coexistence of acral syringomas and multiple trichoepitheliomas on the face. J Cutan Med Surg. 2009;13:169-171. doi:10.2310/7750.2008.08011
- Martín-García RF, Muñoz CM. Acral syringomas presenting as a photosensitive papular eruption. Cutis. 2006;77:33-36.
- Varas-Meis E, Prada-García C, Samaniego-González E, et al. Acral syringomas associated with hematological neoplasm. Indian J Dermatol Venereol Leprol. 2017;83:136. doi:10.4103/0378-6323.192961
- Berbis P, Fabre JF, Jancovici E, et al. Late-onset syringomas of the upper extremities associated with a carcinoid tumor. Arch Dermatol. 1989;125:848-849.
- Metze D, Jurecka W, Gebhart W. Disseminated syringomas of the upper extremities. case history and immunohistochemical and ultrastructural study. Dermatologica. 1990;180:228-235. doi:10.1159/000248036
- Gómez-de Castro C, Vivanco Allende B, García-García B. Multiple acral syringomas. siringomas acrales múltiples. Actas Dermosifiliogr (Engl Ed). 2018;109:834-836. doi:10.1016/j.ad.2017.10.014
- Hughes PS, Apisarnthanarax P. Acral syringoma. Arch Dermatol. 1977;113:1435-1436.
- Asai Y, Ishii M, Hamada T. Acral syringoma: electron microscopic studies on its origin. Acta Derm Venereol. 1982;62:64-68.
- van den Broek H, Lundquist CD. Syringomas of the upper extremities with onset in the sixth decade. J Am Acad Dermatol. 1982,6:534-536. doi:10.1016/S0190-9622(82)80368-X
- Garcia C, Krunic AL, Grichnik J, et al. Multiple acral syringomata with uniform involvement of the hands and feet. Cutis. 1997;59:213-214, 216.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Iglesias Sancho M, Serra Llobet J, Salleras Redonnet M, et al. Siringomas disem- inados de inicio acral, aparecidos en la octava década. Actas Dermosifiliofr. 1999;90:253-257.
- Muniesa C, Fortuño Y, Moreno A, et al. Papules on the dorsum of the fingers. Actas Dermosifiliogr. 2008;99:812-813. doi:10.1016 /S1578-2190(08)70371-8
- Koh MJ. Multiple acral syringomas involving the hands. Clin Exp Dermatol. 2009;34:E438. doi:10.1111/j.1365-2230.2009.03462.x
- Karam P, Benedetto AV. Syringomas: new approach to an old technique. Int J Dermatol. 1996;35:219-220. doi:10.1111/j.1365-4362 .1996.tb01647.x
- Wang JI, Roenigk HH. Treatment of multiple facial syringomas with the carbon dioxide (CO2) laser. Dermatol Surg. 1999;25:136-139. doi:10.1046/j.1524-4725.1999.08111.x
- Gómez MI, Pérez B, Azaña JM, et al. Eruptive syringoma: treatment with topical tretinoin. Dermatology. 2009;189:105-106. doi:10.1159/000246803
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.E9. doi:10.1016 /j.jaad.2015.12.006
- Resende C, Araújo C, Santos R, et al. Late-onset of eruptive syringomas: a diagnostic challenge. An Bras Dermatol. 2015;90(3 suppl 1):239-241. doi:10.1590/abd1806-4841.20153899
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Avhad G, Ghuge P, Jerajani HR. Generalized eruptive syringoma. Indian J Dermatol. 2015;60:214. doi:10.4103/0019-5154.152586
- Ning WV, Bashey S, Cole C, et al. Multiple eruptive syringomas on the penis. Cutis. 2019;103:E15-E16.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Jamalipour M, Heidarpour M, Rajabi P. Generalized eruptive syringomas. Indian J Dermatol. 2009;54:65-67. doi:10.4103/0019-5154.48992
- Mohaghegh F, Amiri A, Fatemi Naeini F, et al. Acral eruptive syringoma: an unusual presentation with misdiagnosis. Case Rep Dermatol Med. 2020;2020:5416285. doi:10.1155/2020/5416285
- Valdivielso-Ramos M, de la Cueva P, Gimeno M, et al. Acral syringomas. Actas Dermosifiliogr. 2010;101:458-460.
- Patel K, Lundgren AD, Ahmed AM, et al. Disseminated syringomas of the upper extremities in a young woman. Cureus. 2018;10:E3619. doi:10.7759/cureus.3619
- Balci DD, Atik E, Altintas S. Coexistence of acral syringomas and multiple trichoepitheliomas on the face. J Cutan Med Surg. 2009;13:169-171. doi:10.2310/7750.2008.08011
- Martín-García RF, Muñoz CM. Acral syringomas presenting as a photosensitive papular eruption. Cutis. 2006;77:33-36.
- Varas-Meis E, Prada-García C, Samaniego-González E, et al. Acral syringomas associated with hematological neoplasm. Indian J Dermatol Venereol Leprol. 2017;83:136. doi:10.4103/0378-6323.192961
- Berbis P, Fabre JF, Jancovici E, et al. Late-onset syringomas of the upper extremities associated with a carcinoid tumor. Arch Dermatol. 1989;125:848-849.
- Metze D, Jurecka W, Gebhart W. Disseminated syringomas of the upper extremities. case history and immunohistochemical and ultrastructural study. Dermatologica. 1990;180:228-235. doi:10.1159/000248036
- Gómez-de Castro C, Vivanco Allende B, García-García B. Multiple acral syringomas. siringomas acrales múltiples. Actas Dermosifiliogr (Engl Ed). 2018;109:834-836. doi:10.1016/j.ad.2017.10.014
- Hughes PS, Apisarnthanarax P. Acral syringoma. Arch Dermatol. 1977;113:1435-1436.
- Asai Y, Ishii M, Hamada T. Acral syringoma: electron microscopic studies on its origin. Acta Derm Venereol. 1982;62:64-68.
- van den Broek H, Lundquist CD. Syringomas of the upper extremities with onset in the sixth decade. J Am Acad Dermatol. 1982,6:534-536. doi:10.1016/S0190-9622(82)80368-X
- Garcia C, Krunic AL, Grichnik J, et al. Multiple acral syringomata with uniform involvement of the hands and feet. Cutis. 1997;59:213-214, 216.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Iglesias Sancho M, Serra Llobet J, Salleras Redonnet M, et al. Siringomas disem- inados de inicio acral, aparecidos en la octava década. Actas Dermosifiliofr. 1999;90:253-257.
- Muniesa C, Fortuño Y, Moreno A, et al. Papules on the dorsum of the fingers. Actas Dermosifiliogr. 2008;99:812-813. doi:10.1016 /S1578-2190(08)70371-8
- Koh MJ. Multiple acral syringomas involving the hands. Clin Exp Dermatol. 2009;34:E438. doi:10.1111/j.1365-2230.2009.03462.x
- Karam P, Benedetto AV. Syringomas: new approach to an old technique. Int J Dermatol. 1996;35:219-220. doi:10.1111/j.1365-4362 .1996.tb01647.x
- Wang JI, Roenigk HH. Treatment of multiple facial syringomas with the carbon dioxide (CO2) laser. Dermatol Surg. 1999;25:136-139. doi:10.1046/j.1524-4725.1999.08111.x
- Gómez MI, Pérez B, Azaña JM, et al. Eruptive syringoma: treatment with topical tretinoin. Dermatology. 2009;189:105-106. doi:10.1159/000246803
Eruptive Erythematous Papules on the Forearms
Eruptive Erythematous Papules on the Forearms
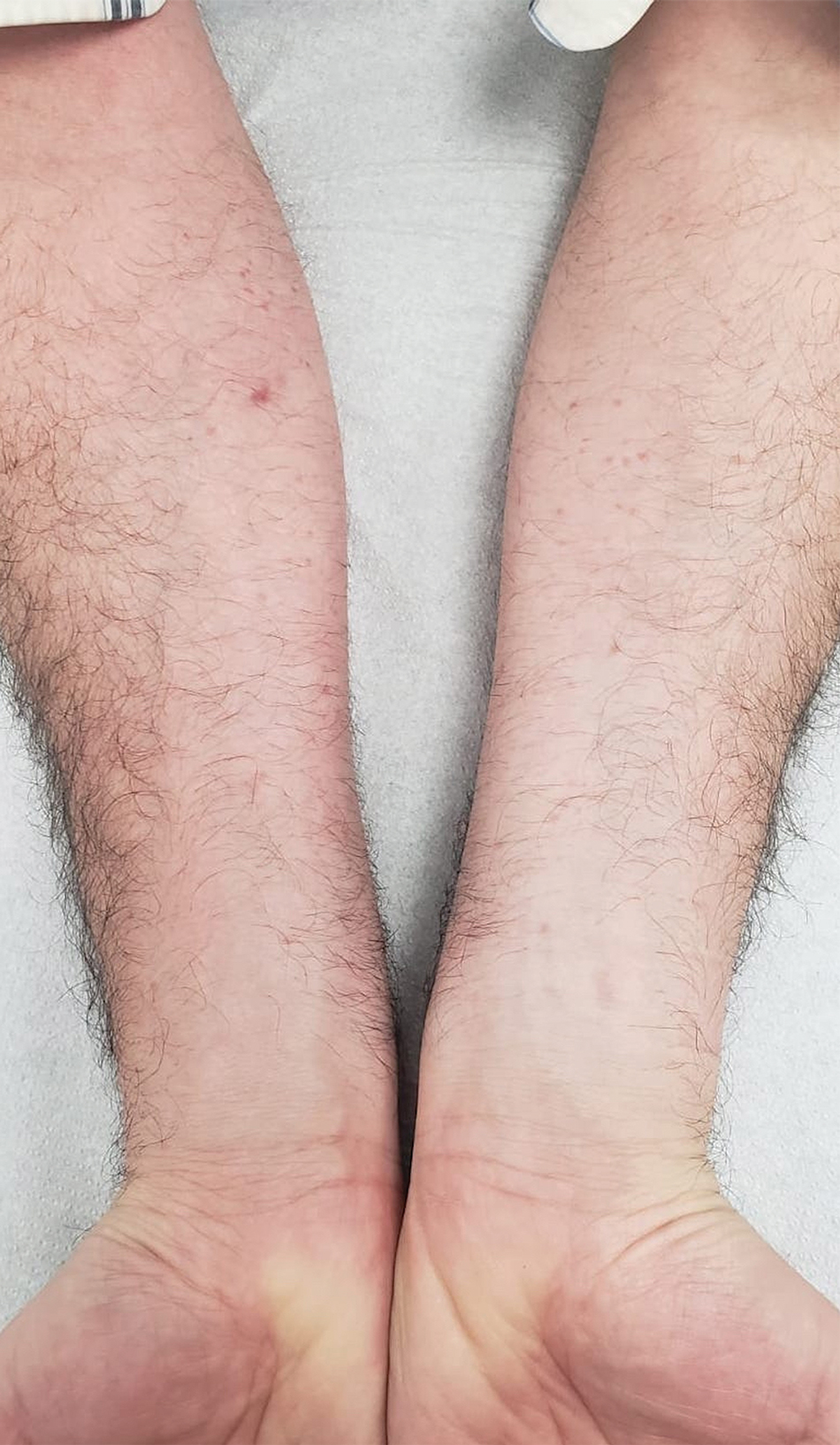
A 44-year-old man presented to the dermatology department with multiple eruptive, nonconfluent, erythematous papules on the anterior forearms of 2 years’ duration. The patient’s medical history was notable for right-sided testicular cancer diagnosed in childhood and 3 excised basal cell carcinomas, the most recent of which was concurrent with the present case. The patient denied any recent pruritus, exposure to irritants, or use of over-the-counter medications. Physical examination was remarkable for numerous monomorphic, symmetric, nonconfluent, flesh-colored to slightly pigmented papules on the dorsal aspect of the forearms. No involvement of the fingers or lower extremities was observed. Two punch biopsies of representative lesions on the right and left forearms were taken. Histopathologic examination revealed eccrine ductal proliferations lined by cuboidal cells embedded within bundles of sclerotic collagen.

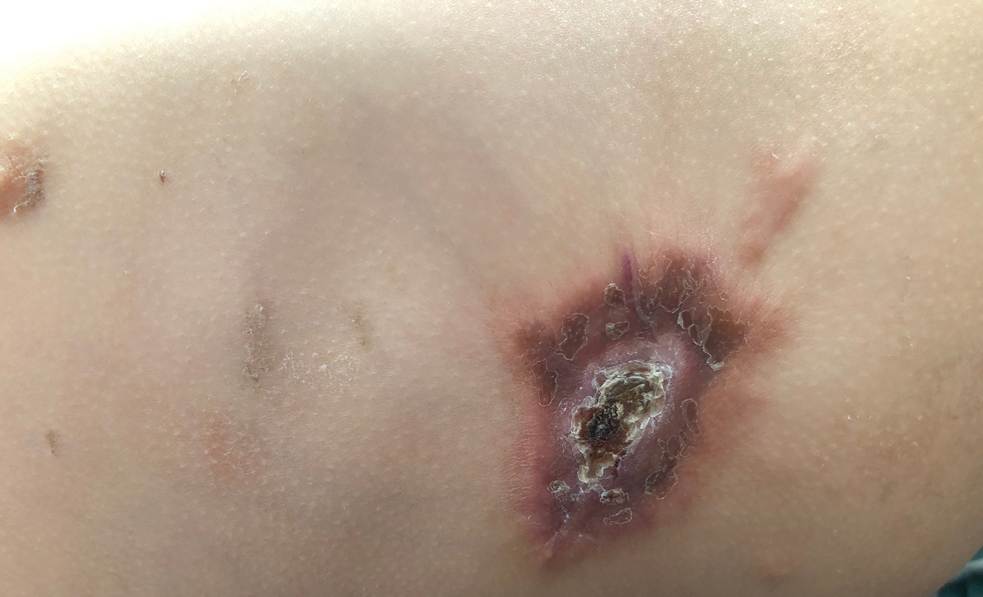
Nonhealing Ulcer on the Lower Lip
Nonhealing Ulcer on the Lower Lip
THE DIAGNOSIS: Syphilis
The differential diagnosis of oral lesions can be complex; in our patient, we considered conditions such as pyogenic granuloma, herpes simplex virus, and syphilis, despite the presence of pain. Immunohistochemical staining for spirochete antigens was positive, and serologic confirmation through a positive rapid plasma reagin (RPR) test confirmed the diagnosis of primary syphilis. The patient was promptly referred back to the primary care physician for treatment with intramuscular penicillin, leading to resolution of the lesion. At 3 months’ follow-up in our clinic, the lesion was fully resolved.
A primary syphilitic chancre is the initial lesion caused by Treponema pallidum, typically manifesting as a painless ulcer at the infection site, usually in the genital area; however, chancres also may manifest in other locations (eg, the anus or oral cavity) due to direct contact with infectious lesions on another individual. Our case represents an atypical presentation of an oral syphilitic chancre.
Syphilis is a sexually transmitted infection with various clinical manifestations. It is crucial to consider syphilis in the differential diagnosis of ulcerative lesions even when pain is present, especially in high-risk individuals such as those who engage in unprotected sex.1,2 Oral syphilitic chancres have been documented in the medical literature for more than a century, underscoring the importance of maintaining a high index of suspicion for diagnosis and a low threshold for obtaining an RPR test to facilitate early detection and treatment.2,3 Notably, the prevalence of syphilis is higher in men who have sex with men, particularly among those who engage in unprotected oral and anal sex. Increased screening and early treatment are essential to control the spread of disease within all populations. Doxycycline postexposure prophylaxis (doxyPEP) is used as a preventive measure for syphilis, chlamydia, and gonorrhea.4 This regimen consists of 200 mg of doxycycline taken within 24 hours but no later than 72 hours after unprotected anal, vaginal, or oral sex.
Our case highlights the importance of considering the differential diagnosis of oral ulcers, particularly in high-risk populations such as men who have sex with men. Prompt diagnosis, effective treatment, and preventive strategies such as doxyPEP are essential for controlling syphilis. Comprehensive patient education and regular follow-up appointments are critical components of successful management.
The United States has experienced a considerable rise in primary and congenital syphilis cases, with an 80% increase between 2018 and 2022.6 Serologic testing is the primary method for diagnosing, staging, and managing syphilis. Sexually active patients with suspected syphilis or unexplained symptoms should undergo testing. Prompt diagnosis and treatment can prevent systemic complications, including ocular involvement and permanent blindness.
Syphilis is transmitted through direct contact with a syphilitic ulcer or saliva or blood from an infected individual. Oral syphilitic ulcers can develop on the lips, tongue, oral mucosa, and tonsils. Chancres can range from a few millimeters to several centimeters, with an incubation period of 10 to 90 days (average, 21 days). The chancre lasts 3 to 6 weeks and heals spontaneously. Without treatment, primary syphilis can progress to secondary syphilis, characterized by a papulosquamous eruption and mucosal involvement, and potentially tertiary syphilis, which can affect the central nervous system, heart, bones, and skin.7
Immunocompromised patients, especially those diagnosed with HIV, face increased risks including altered clinical presentations (eg, multiple or deep chancres), delayed healing, overlapping stages of disease, and increased severity of organ involvement. All sexually active individuals should be screened for syphilis every 3 to 6 months, particularly those with unexplained oral ulcers.
Serologic testing is fundamental for syphilis diagnosis and management. Nontreponemal tests such as RPR and treponemal tests such as the fluorescent treponemal antibody absorption test provide comprehensive diagnostic information. Early diagnosis and empiric treatment are crucial in suspected cases. Ocular screening is recommended for suspected or confirmed syphilis cases.7
Management of syphilis includes treating all sexual partners and providing thorough patient education on the disease. Monitoring for the Jarisch-Herxheimer reaction—an acute febrile reaction following penicillin therapy—is important, especially in pregnant patients.5 Serologic evaluation at 6 and 12 months posttreatment is recommended, with more frequent evaluations if follow-up is uncertain, particularly for those with inconsistent access to health care or in whom reinfection is suspected. Guidelines from the Centers for Disease Control and Prevention advocate for intramuscular penicillin G benzathine as the preferred treatment, with specific dosing for adults and children.7 Due to the ongoing bicillin shortage, alternatives such as extencilline have temporarily been allowed for use in the United States.8
The rising incidence of syphilis in the United States underscores the critical need for enhanced public health initiatives focusing on education, screening, and early intervention. Comprehensive sexual education that includes information about syphilis and other sexually transmitted infections, proper use of prophylactic measures such as condoms, and the benefits of doxyPEP can considerably reduce transmission rates. Health care providers should routinely discuss these preventive measures with their patients, especially those in high-risk groups.
Our case highlights the importance of considering syphilis in the differential diagnosis of oral ulcers, particularly in high-risk populations. Timely diagnosis, effective treatment, and preventive measures such as doxyPEP are essential for managing and controlling syphilis. The rising incidence of syphilis in the United States warrants increased screening, patient education, and public health interventions to address this notable health challenge. The syphilis crisis calls for coordinated efforts from health care providers, public health officials, and community leaders to curb the spread of this infection and protect public health.
- Mayer KH, Traeger M, Marcus JL. Doxycycline postexposure prophylaxis and sexually transmitted infections. JAMA. 2023;330:1381-1382. doi:10.1001/jama.2023.16416
- Cossman JP, Fournier JB. Frequency of syphilis diagnoses by dermatologists. JAMA Dermatol. 2017;153:718-719. doi:10.1001 /jamadermatol.2017.0460
- Porterfield C, Brodell D, Dolohanty L, et al. Primary syphilis presenting as a chronic lip ulcer. Cureus. 2020;12:E7086. doi:10.7759 /cureus.7086
- Schamberg JF. An epidemic of chancres of the lip from kissing. JAMA. 1911;LVII:783-784. doi:10.1001/jama.1911.04260090005002
- Farmer TW. Jarisch-Herxheimer reaction in early syphilis. JAMA. 1948;138:480–485. doi:10.1001/jama.1948.02900070012003
- Winney A. Why is syphilis spiking in the U.S.? Johns Hopkins Bloomberg School of Public Health. Johns Hopkins Bloomberg School of Public Health. Published March 13, 2024. Accessed April 30, 2025. https://publichealth.jhu.edu/why-is-syphilis-spiking-in-the-us
- Koundanya VV, Tripathy K. Syphilis ocular manifestations. StatPearls Publishing; 2021. Updated August 25, 2023. Accessed May 6, 2025. https://www.ncbi.nlm.nih.gov/books/NBK558957/
- CDC. FDA announcement on availability of extencilline. National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention. Published July 19, 2024. Accessed April 30, 2025. https://www.cdc.gov/nchhstp/director-letters/extencilline-during-bicillin-l-a-shortage.html
THE DIAGNOSIS: Syphilis
The differential diagnosis of oral lesions can be complex; in our patient, we considered conditions such as pyogenic granuloma, herpes simplex virus, and syphilis, despite the presence of pain. Immunohistochemical staining for spirochete antigens was positive, and serologic confirmation through a positive rapid plasma reagin (RPR) test confirmed the diagnosis of primary syphilis. The patient was promptly referred back to the primary care physician for treatment with intramuscular penicillin, leading to resolution of the lesion. At 3 months’ follow-up in our clinic, the lesion was fully resolved.
A primary syphilitic chancre is the initial lesion caused by Treponema pallidum, typically manifesting as a painless ulcer at the infection site, usually in the genital area; however, chancres also may manifest in other locations (eg, the anus or oral cavity) due to direct contact with infectious lesions on another individual. Our case represents an atypical presentation of an oral syphilitic chancre.
Syphilis is a sexually transmitted infection with various clinical manifestations. It is crucial to consider syphilis in the differential diagnosis of ulcerative lesions even when pain is present, especially in high-risk individuals such as those who engage in unprotected sex.1,2 Oral syphilitic chancres have been documented in the medical literature for more than a century, underscoring the importance of maintaining a high index of suspicion for diagnosis and a low threshold for obtaining an RPR test to facilitate early detection and treatment.2,3 Notably, the prevalence of syphilis is higher in men who have sex with men, particularly among those who engage in unprotected oral and anal sex. Increased screening and early treatment are essential to control the spread of disease within all populations. Doxycycline postexposure prophylaxis (doxyPEP) is used as a preventive measure for syphilis, chlamydia, and gonorrhea.4 This regimen consists of 200 mg of doxycycline taken within 24 hours but no later than 72 hours after unprotected anal, vaginal, or oral sex.
Our case highlights the importance of considering the differential diagnosis of oral ulcers, particularly in high-risk populations such as men who have sex with men. Prompt diagnosis, effective treatment, and preventive strategies such as doxyPEP are essential for controlling syphilis. Comprehensive patient education and regular follow-up appointments are critical components of successful management.
The United States has experienced a considerable rise in primary and congenital syphilis cases, with an 80% increase between 2018 and 2022.6 Serologic testing is the primary method for diagnosing, staging, and managing syphilis. Sexually active patients with suspected syphilis or unexplained symptoms should undergo testing. Prompt diagnosis and treatment can prevent systemic complications, including ocular involvement and permanent blindness.
Syphilis is transmitted through direct contact with a syphilitic ulcer or saliva or blood from an infected individual. Oral syphilitic ulcers can develop on the lips, tongue, oral mucosa, and tonsils. Chancres can range from a few millimeters to several centimeters, with an incubation period of 10 to 90 days (average, 21 days). The chancre lasts 3 to 6 weeks and heals spontaneously. Without treatment, primary syphilis can progress to secondary syphilis, characterized by a papulosquamous eruption and mucosal involvement, and potentially tertiary syphilis, which can affect the central nervous system, heart, bones, and skin.7
Immunocompromised patients, especially those diagnosed with HIV, face increased risks including altered clinical presentations (eg, multiple or deep chancres), delayed healing, overlapping stages of disease, and increased severity of organ involvement. All sexually active individuals should be screened for syphilis every 3 to 6 months, particularly those with unexplained oral ulcers.
Serologic testing is fundamental for syphilis diagnosis and management. Nontreponemal tests such as RPR and treponemal tests such as the fluorescent treponemal antibody absorption test provide comprehensive diagnostic information. Early diagnosis and empiric treatment are crucial in suspected cases. Ocular screening is recommended for suspected or confirmed syphilis cases.7
Management of syphilis includes treating all sexual partners and providing thorough patient education on the disease. Monitoring for the Jarisch-Herxheimer reaction—an acute febrile reaction following penicillin therapy—is important, especially in pregnant patients.5 Serologic evaluation at 6 and 12 months posttreatment is recommended, with more frequent evaluations if follow-up is uncertain, particularly for those with inconsistent access to health care or in whom reinfection is suspected. Guidelines from the Centers for Disease Control and Prevention advocate for intramuscular penicillin G benzathine as the preferred treatment, with specific dosing for adults and children.7 Due to the ongoing bicillin shortage, alternatives such as extencilline have temporarily been allowed for use in the United States.8
The rising incidence of syphilis in the United States underscores the critical need for enhanced public health initiatives focusing on education, screening, and early intervention. Comprehensive sexual education that includes information about syphilis and other sexually transmitted infections, proper use of prophylactic measures such as condoms, and the benefits of doxyPEP can considerably reduce transmission rates. Health care providers should routinely discuss these preventive measures with their patients, especially those in high-risk groups.
Our case highlights the importance of considering syphilis in the differential diagnosis of oral ulcers, particularly in high-risk populations. Timely diagnosis, effective treatment, and preventive measures such as doxyPEP are essential for managing and controlling syphilis. The rising incidence of syphilis in the United States warrants increased screening, patient education, and public health interventions to address this notable health challenge. The syphilis crisis calls for coordinated efforts from health care providers, public health officials, and community leaders to curb the spread of this infection and protect public health.
THE DIAGNOSIS: Syphilis
The differential diagnosis of oral lesions can be complex; in our patient, we considered conditions such as pyogenic granuloma, herpes simplex virus, and syphilis, despite the presence of pain. Immunohistochemical staining for spirochete antigens was positive, and serologic confirmation through a positive rapid plasma reagin (RPR) test confirmed the diagnosis of primary syphilis. The patient was promptly referred back to the primary care physician for treatment with intramuscular penicillin, leading to resolution of the lesion. At 3 months’ follow-up in our clinic, the lesion was fully resolved.
A primary syphilitic chancre is the initial lesion caused by Treponema pallidum, typically manifesting as a painless ulcer at the infection site, usually in the genital area; however, chancres also may manifest in other locations (eg, the anus or oral cavity) due to direct contact with infectious lesions on another individual. Our case represents an atypical presentation of an oral syphilitic chancre.
Syphilis is a sexually transmitted infection with various clinical manifestations. It is crucial to consider syphilis in the differential diagnosis of ulcerative lesions even when pain is present, especially in high-risk individuals such as those who engage in unprotected sex.1,2 Oral syphilitic chancres have been documented in the medical literature for more than a century, underscoring the importance of maintaining a high index of suspicion for diagnosis and a low threshold for obtaining an RPR test to facilitate early detection and treatment.2,3 Notably, the prevalence of syphilis is higher in men who have sex with men, particularly among those who engage in unprotected oral and anal sex. Increased screening and early treatment are essential to control the spread of disease within all populations. Doxycycline postexposure prophylaxis (doxyPEP) is used as a preventive measure for syphilis, chlamydia, and gonorrhea.4 This regimen consists of 200 mg of doxycycline taken within 24 hours but no later than 72 hours after unprotected anal, vaginal, or oral sex.
Our case highlights the importance of considering the differential diagnosis of oral ulcers, particularly in high-risk populations such as men who have sex with men. Prompt diagnosis, effective treatment, and preventive strategies such as doxyPEP are essential for controlling syphilis. Comprehensive patient education and regular follow-up appointments are critical components of successful management.
The United States has experienced a considerable rise in primary and congenital syphilis cases, with an 80% increase between 2018 and 2022.6 Serologic testing is the primary method for diagnosing, staging, and managing syphilis. Sexually active patients with suspected syphilis or unexplained symptoms should undergo testing. Prompt diagnosis and treatment can prevent systemic complications, including ocular involvement and permanent blindness.
Syphilis is transmitted through direct contact with a syphilitic ulcer or saliva or blood from an infected individual. Oral syphilitic ulcers can develop on the lips, tongue, oral mucosa, and tonsils. Chancres can range from a few millimeters to several centimeters, with an incubation period of 10 to 90 days (average, 21 days). The chancre lasts 3 to 6 weeks and heals spontaneously. Without treatment, primary syphilis can progress to secondary syphilis, characterized by a papulosquamous eruption and mucosal involvement, and potentially tertiary syphilis, which can affect the central nervous system, heart, bones, and skin.7
Immunocompromised patients, especially those diagnosed with HIV, face increased risks including altered clinical presentations (eg, multiple or deep chancres), delayed healing, overlapping stages of disease, and increased severity of organ involvement. All sexually active individuals should be screened for syphilis every 3 to 6 months, particularly those with unexplained oral ulcers.
Serologic testing is fundamental for syphilis diagnosis and management. Nontreponemal tests such as RPR and treponemal tests such as the fluorescent treponemal antibody absorption test provide comprehensive diagnostic information. Early diagnosis and empiric treatment are crucial in suspected cases. Ocular screening is recommended for suspected or confirmed syphilis cases.7
Management of syphilis includes treating all sexual partners and providing thorough patient education on the disease. Monitoring for the Jarisch-Herxheimer reaction—an acute febrile reaction following penicillin therapy—is important, especially in pregnant patients.5 Serologic evaluation at 6 and 12 months posttreatment is recommended, with more frequent evaluations if follow-up is uncertain, particularly for those with inconsistent access to health care or in whom reinfection is suspected. Guidelines from the Centers for Disease Control and Prevention advocate for intramuscular penicillin G benzathine as the preferred treatment, with specific dosing for adults and children.7 Due to the ongoing bicillin shortage, alternatives such as extencilline have temporarily been allowed for use in the United States.8
The rising incidence of syphilis in the United States underscores the critical need for enhanced public health initiatives focusing on education, screening, and early intervention. Comprehensive sexual education that includes information about syphilis and other sexually transmitted infections, proper use of prophylactic measures such as condoms, and the benefits of doxyPEP can considerably reduce transmission rates. Health care providers should routinely discuss these preventive measures with their patients, especially those in high-risk groups.
Our case highlights the importance of considering syphilis in the differential diagnosis of oral ulcers, particularly in high-risk populations. Timely diagnosis, effective treatment, and preventive measures such as doxyPEP are essential for managing and controlling syphilis. The rising incidence of syphilis in the United States warrants increased screening, patient education, and public health interventions to address this notable health challenge. The syphilis crisis calls for coordinated efforts from health care providers, public health officials, and community leaders to curb the spread of this infection and protect public health.
- Mayer KH, Traeger M, Marcus JL. Doxycycline postexposure prophylaxis and sexually transmitted infections. JAMA. 2023;330:1381-1382. doi:10.1001/jama.2023.16416
- Cossman JP, Fournier JB. Frequency of syphilis diagnoses by dermatologists. JAMA Dermatol. 2017;153:718-719. doi:10.1001 /jamadermatol.2017.0460
- Porterfield C, Brodell D, Dolohanty L, et al. Primary syphilis presenting as a chronic lip ulcer. Cureus. 2020;12:E7086. doi:10.7759 /cureus.7086
- Schamberg JF. An epidemic of chancres of the lip from kissing. JAMA. 1911;LVII:783-784. doi:10.1001/jama.1911.04260090005002
- Farmer TW. Jarisch-Herxheimer reaction in early syphilis. JAMA. 1948;138:480–485. doi:10.1001/jama.1948.02900070012003
- Winney A. Why is syphilis spiking in the U.S.? Johns Hopkins Bloomberg School of Public Health. Johns Hopkins Bloomberg School of Public Health. Published March 13, 2024. Accessed April 30, 2025. https://publichealth.jhu.edu/why-is-syphilis-spiking-in-the-us
- Koundanya VV, Tripathy K. Syphilis ocular manifestations. StatPearls Publishing; 2021. Updated August 25, 2023. Accessed May 6, 2025. https://www.ncbi.nlm.nih.gov/books/NBK558957/
- CDC. FDA announcement on availability of extencilline. National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention. Published July 19, 2024. Accessed April 30, 2025. https://www.cdc.gov/nchhstp/director-letters/extencilline-during-bicillin-l-a-shortage.html
- Mayer KH, Traeger M, Marcus JL. Doxycycline postexposure prophylaxis and sexually transmitted infections. JAMA. 2023;330:1381-1382. doi:10.1001/jama.2023.16416
- Cossman JP, Fournier JB. Frequency of syphilis diagnoses by dermatologists. JAMA Dermatol. 2017;153:718-719. doi:10.1001 /jamadermatol.2017.0460
- Porterfield C, Brodell D, Dolohanty L, et al. Primary syphilis presenting as a chronic lip ulcer. Cureus. 2020;12:E7086. doi:10.7759 /cureus.7086
- Schamberg JF. An epidemic of chancres of the lip from kissing. JAMA. 1911;LVII:783-784. doi:10.1001/jama.1911.04260090005002
- Farmer TW. Jarisch-Herxheimer reaction in early syphilis. JAMA. 1948;138:480–485. doi:10.1001/jama.1948.02900070012003
- Winney A. Why is syphilis spiking in the U.S.? Johns Hopkins Bloomberg School of Public Health. Johns Hopkins Bloomberg School of Public Health. Published March 13, 2024. Accessed April 30, 2025. https://publichealth.jhu.edu/why-is-syphilis-spiking-in-the-us
- Koundanya VV, Tripathy K. Syphilis ocular manifestations. StatPearls Publishing; 2021. Updated August 25, 2023. Accessed May 6, 2025. https://www.ncbi.nlm.nih.gov/books/NBK558957/
- CDC. FDA announcement on availability of extencilline. National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention. Published July 19, 2024. Accessed April 30, 2025. https://www.cdc.gov/nchhstp/director-letters/extencilline-during-bicillin-l-a-shortage.html
Nonhealing Ulcer on the Lower Lip
Nonhealing Ulcer on the Lower Lip
A 54-year-old HIV-negative man with a history of having sex with men presented to his primary care physician with an ulcer on the lower lip of 3 weeks’ duration. The patient reported that the lesion had appeared as a typical cold sore with pain in the area. A 9-day course of oral valacyclovir prescribed by the primary care physician provided no relief or improvement. A 2-mm punch biopsy was performed.
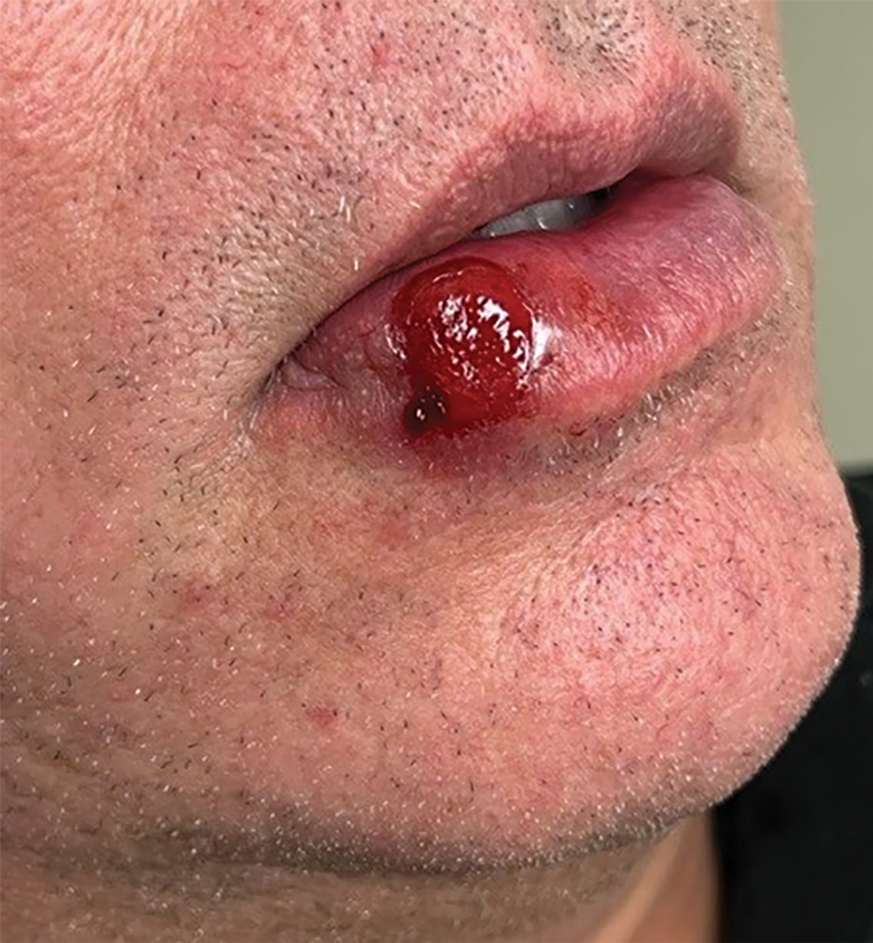

Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
THE DIAGNOSIS: Bullous Hemorrhagic Dermatosis
Biopsy results showed an intraepidermal blister with a floor composed of maturing epidermis. The roof of the blister was composed of necrotic keratinocytes with overlying orthokeratosis, and the cavity was filled with a moderate amount of fibrin and dead cells with neutrophils. Direct immunofluorescence (DIF) using specific antihuman IgG, IgM, IgA, C3, and fibrin was negative. Aerobic, anaerobic, and fungal cultures also were negative. With these histopathologic findings, medication exposure, and timing of bullae onset, our patient was diagnosed with bullous hemorrhagic dermatosis (BHD) secondary to enoxaparin administration. Enoxaparin was continued due to increased risk for coagulopathy, and there was complete resolution of the bullae after 5 weeks with no residual symptoms.
Bullous hemorrhagic dermatosis is a rare eruption that can occur after administration of heparin and low-molecular-weight heparin, with enoxaparin being the most commonly implicated drug.1 The lesions typically are seen in elderly men in the seventh decade of life and appear within a median of 7 days after drug exposure. The time course for the postexposure eruption can vary from 2 to 21 days, with reports of skin lesions appearing up to 4 months after exposure.1,2 hemorrhagic bullae (Figure) typically on the arms and legs, though lesions also can develop on the trunk. The lesions can occur in distant areas from the injection site, suggesting BHD may be a systemic reaction, although the etiology is poorly understood.1
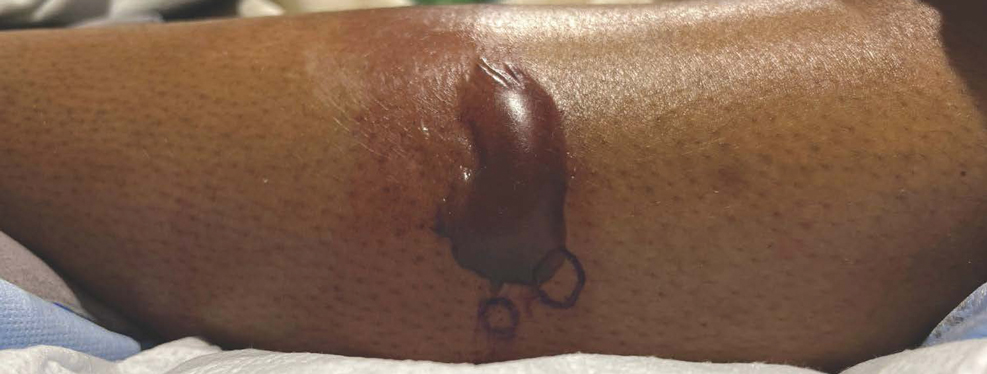
Another heparin reaction that can manifest similarly to BHD is heparin-induced skin necrosis.3 Patients with this condition also may have associated heparin-induced thrombocytopenia upon laboratory investigation and have a more aggressive clinical course than BHD. Biopsy can help differentiate BHD and early heparin-induced skin necrosis if the clinical manifestation is unclear. Histopathologically, BHD typically has intraepidermal bullae filled with blood, whereas heparin-induced skin necrosis has dermal thrombi.1,4 Treatment of both conditions differs in whether to discontinue anticoagulants: heparin-induced skin necrosis requires discontinuation of the medication, while BHD does not.2,3
In patients with BHD, the lesions are self-resolving, and treatment is supportive, although whether enoxaparin is discontinued varies among physicians.2 Lesions typically resolve within 2 weeks of onset, although it is unclear whether continuing anticoagulants delays resolution.1 Discontinuing anticoagulants in certain patients can be life-threatening due to complex comorbidities (eg, risk for venous thromboembolism or pulmonary embolism from prolonged hospitalization or severe trauma) and is not necessary for the resolution of BHD.
In addition to BHD and heparin-induced skin necrosis, our differential diagnosis included bullous pemphigoid, coma blisters, and Vibrio vulnificus infection. Although bullous pemphigoid can manifest with tense bullae that are pauci-inflammatory on histology, DIF would show linear IgG and C3 deposition at the dermal-epidermal junction. In our patient, DIF was negative and favored another etiology for the lesions. Coma blisters can occur in areas of sustained pressure and typically develop in patients with a prolonged hospitalization or those who are sedentary for long periods of time. The distribution of bullae on our patient’s bilateral pretibial shins made this diagnosis unlikely. Vibrio vulnificus infection can manifest as hemorrhagic bullae, though typically after a break in the skin exposed to brackish water. Vibrio vulnificus infection can be life-threatening, resulting in septicemia and increased mortality, and a thorough patient history is important for diagnosis.5
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose lowmolecular weight heparin: a case report and review of the literature. Exp Hematol Oncol. 2018;7:15. doi:10.1186/s40164-018-0108-7
- Dhattarwal N, Gurjar R. Bullous hemorrhagic dermatosis: a rare cutaneous reaction of heparin. J Postgrad Med. 2023;69:97-98. doi:10.4103/jpgm.jpgm_282_22
- Maldonado Cid P, Alonso de Celada RM, Noguera Morel L, et al. Cutaneous adverse events associated with heparin. Clin Exp Dermatol. 2012;37:707-711. doi:10.1111/j.1365-2230.2012.04395.x
- Handschin AE, Trentz O, Kock HJ, et al. Low molecular weight heparininduced skin necrosis-a systematic review. Langenbecks Arch Surg. 2005;390:249-254. doi:10.1007/s00423-004-0522-7
- Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723-1733. doi:10.1128/IAI.01046-08
THE DIAGNOSIS: Bullous Hemorrhagic Dermatosis
Biopsy results showed an intraepidermal blister with a floor composed of maturing epidermis. The roof of the blister was composed of necrotic keratinocytes with overlying orthokeratosis, and the cavity was filled with a moderate amount of fibrin and dead cells with neutrophils. Direct immunofluorescence (DIF) using specific antihuman IgG, IgM, IgA, C3, and fibrin was negative. Aerobic, anaerobic, and fungal cultures also were negative. With these histopathologic findings, medication exposure, and timing of bullae onset, our patient was diagnosed with bullous hemorrhagic dermatosis (BHD) secondary to enoxaparin administration. Enoxaparin was continued due to increased risk for coagulopathy, and there was complete resolution of the bullae after 5 weeks with no residual symptoms.
Bullous hemorrhagic dermatosis is a rare eruption that can occur after administration of heparin and low-molecular-weight heparin, with enoxaparin being the most commonly implicated drug.1 The lesions typically are seen in elderly men in the seventh decade of life and appear within a median of 7 days after drug exposure. The time course for the postexposure eruption can vary from 2 to 21 days, with reports of skin lesions appearing up to 4 months after exposure.1,2 hemorrhagic bullae (Figure) typically on the arms and legs, though lesions also can develop on the trunk. The lesions can occur in distant areas from the injection site, suggesting BHD may be a systemic reaction, although the etiology is poorly understood.1
Another heparin reaction that can manifest similarly to BHD is heparin-induced skin necrosis.3 Patients with this condition also may have associated heparin-induced thrombocytopenia upon laboratory investigation and have a more aggressive clinical course than BHD. Biopsy can help differentiate BHD and early heparin-induced skin necrosis if the clinical manifestation is unclear. Histopathologically, BHD typically has intraepidermal bullae filled with blood, whereas heparin-induced skin necrosis has dermal thrombi.1,4 Treatment of both conditions differs in whether to discontinue anticoagulants: heparin-induced skin necrosis requires discontinuation of the medication, while BHD does not.2,3
In patients with BHD, the lesions are self-resolving, and treatment is supportive, although whether enoxaparin is discontinued varies among physicians.2 Lesions typically resolve within 2 weeks of onset, although it is unclear whether continuing anticoagulants delays resolution.1 Discontinuing anticoagulants in certain patients can be life-threatening due to complex comorbidities (eg, risk for venous thromboembolism or pulmonary embolism from prolonged hospitalization or severe trauma) and is not necessary for the resolution of BHD.
In addition to BHD and heparin-induced skin necrosis, our differential diagnosis included bullous pemphigoid, coma blisters, and Vibrio vulnificus infection. Although bullous pemphigoid can manifest with tense bullae that are pauci-inflammatory on histology, DIF would show linear IgG and C3 deposition at the dermal-epidermal junction. In our patient, DIF was negative and favored another etiology for the lesions. Coma blisters can occur in areas of sustained pressure and typically develop in patients with a prolonged hospitalization or those who are sedentary for long periods of time. The distribution of bullae on our patient’s bilateral pretibial shins made this diagnosis unlikely. Vibrio vulnificus infection can manifest as hemorrhagic bullae, though typically after a break in the skin exposed to brackish water. Vibrio vulnificus infection can be life-threatening, resulting in septicemia and increased mortality, and a thorough patient history is important for diagnosis.5
THE DIAGNOSIS: Bullous Hemorrhagic Dermatosis
Biopsy results showed an intraepidermal blister with a floor composed of maturing epidermis. The roof of the blister was composed of necrotic keratinocytes with overlying orthokeratosis, and the cavity was filled with a moderate amount of fibrin and dead cells with neutrophils. Direct immunofluorescence (DIF) using specific antihuman IgG, IgM, IgA, C3, and fibrin was negative. Aerobic, anaerobic, and fungal cultures also were negative. With these histopathologic findings, medication exposure, and timing of bullae onset, our patient was diagnosed with bullous hemorrhagic dermatosis (BHD) secondary to enoxaparin administration. Enoxaparin was continued due to increased risk for coagulopathy, and there was complete resolution of the bullae after 5 weeks with no residual symptoms.
Bullous hemorrhagic dermatosis is a rare eruption that can occur after administration of heparin and low-molecular-weight heparin, with enoxaparin being the most commonly implicated drug.1 The lesions typically are seen in elderly men in the seventh decade of life and appear within a median of 7 days after drug exposure. The time course for the postexposure eruption can vary from 2 to 21 days, with reports of skin lesions appearing up to 4 months after exposure.1,2 hemorrhagic bullae (Figure) typically on the arms and legs, though lesions also can develop on the trunk. The lesions can occur in distant areas from the injection site, suggesting BHD may be a systemic reaction, although the etiology is poorly understood.1
Another heparin reaction that can manifest similarly to BHD is heparin-induced skin necrosis.3 Patients with this condition also may have associated heparin-induced thrombocytopenia upon laboratory investigation and have a more aggressive clinical course than BHD. Biopsy can help differentiate BHD and early heparin-induced skin necrosis if the clinical manifestation is unclear. Histopathologically, BHD typically has intraepidermal bullae filled with blood, whereas heparin-induced skin necrosis has dermal thrombi.1,4 Treatment of both conditions differs in whether to discontinue anticoagulants: heparin-induced skin necrosis requires discontinuation of the medication, while BHD does not.2,3
In patients with BHD, the lesions are self-resolving, and treatment is supportive, although whether enoxaparin is discontinued varies among physicians.2 Lesions typically resolve within 2 weeks of onset, although it is unclear whether continuing anticoagulants delays resolution.1 Discontinuing anticoagulants in certain patients can be life-threatening due to complex comorbidities (eg, risk for venous thromboembolism or pulmonary embolism from prolonged hospitalization or severe trauma) and is not necessary for the resolution of BHD.
In addition to BHD and heparin-induced skin necrosis, our differential diagnosis included bullous pemphigoid, coma blisters, and Vibrio vulnificus infection. Although bullous pemphigoid can manifest with tense bullae that are pauci-inflammatory on histology, DIF would show linear IgG and C3 deposition at the dermal-epidermal junction. In our patient, DIF was negative and favored another etiology for the lesions. Coma blisters can occur in areas of sustained pressure and typically develop in patients with a prolonged hospitalization or those who are sedentary for long periods of time. The distribution of bullae on our patient’s bilateral pretibial shins made this diagnosis unlikely. Vibrio vulnificus infection can manifest as hemorrhagic bullae, though typically after a break in the skin exposed to brackish water. Vibrio vulnificus infection can be life-threatening, resulting in septicemia and increased mortality, and a thorough patient history is important for diagnosis.5
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose lowmolecular weight heparin: a case report and review of the literature. Exp Hematol Oncol. 2018;7:15. doi:10.1186/s40164-018-0108-7
- Dhattarwal N, Gurjar R. Bullous hemorrhagic dermatosis: a rare cutaneous reaction of heparin. J Postgrad Med. 2023;69:97-98. doi:10.4103/jpgm.jpgm_282_22
- Maldonado Cid P, Alonso de Celada RM, Noguera Morel L, et al. Cutaneous adverse events associated with heparin. Clin Exp Dermatol. 2012;37:707-711. doi:10.1111/j.1365-2230.2012.04395.x
- Handschin AE, Trentz O, Kock HJ, et al. Low molecular weight heparininduced skin necrosis-a systematic review. Langenbecks Arch Surg. 2005;390:249-254. doi:10.1007/s00423-004-0522-7
- Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723-1733. doi:10.1128/IAI.01046-08
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose lowmolecular weight heparin: a case report and review of the literature. Exp Hematol Oncol. 2018;7:15. doi:10.1186/s40164-018-0108-7
- Dhattarwal N, Gurjar R. Bullous hemorrhagic dermatosis: a rare cutaneous reaction of heparin. J Postgrad Med. 2023;69:97-98. doi:10.4103/jpgm.jpgm_282_22
- Maldonado Cid P, Alonso de Celada RM, Noguera Morel L, et al. Cutaneous adverse events associated with heparin. Clin Exp Dermatol. 2012;37:707-711. doi:10.1111/j.1365-2230.2012.04395.x
- Handschin AE, Trentz O, Kock HJ, et al. Low molecular weight heparininduced skin necrosis-a systematic review. Langenbecks Arch Surg. 2005;390:249-254. doi:10.1007/s00423-004-0522-7
- Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723-1733. doi:10.1128/IAI.01046-08
Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
A 19-year-old man developed fluid-filled blisters on both legs within 1 month of a prolonged hospitalization following a gunshot wound that resulted in complete paralysis of the legs. His medical history was otherwise unremarkable. Medications started during hospitalization included moxifloxacin, levetiracetam, and prophylactic subcutaneous enoxaparin. Physical examination by dermatology revealed tense blood-filled bullae measuring several centimeters with well-demarcated, pink to red, irregularly shaped patches on both legs. A biopsy of a blister was taken.
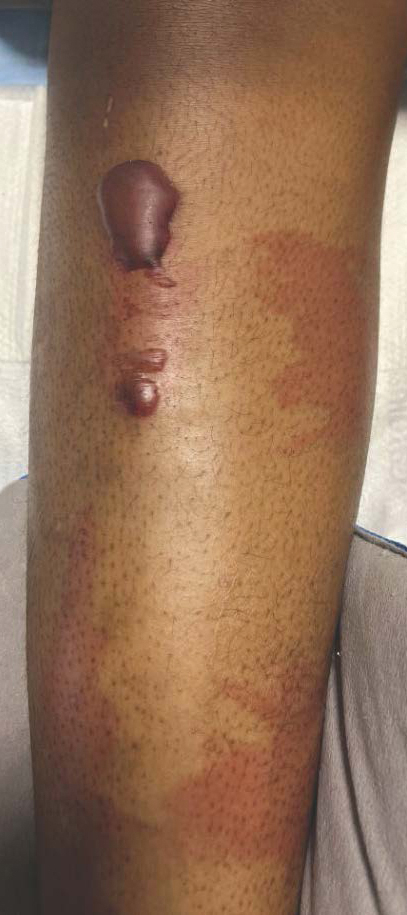

Pigmented Cystic Masses on the Scalp
THE DIAGNOSIS: Apocrine Hidrocystoma
Histology for all 3 lesions demonstrated similar cystic structures lined by a dual layer of epithelial cells, with the outermost layer composed of flattened myoepithelial cells and the inner layer composed of cells with apocrine features (Figure 1). Based on these findings, a diagnosis of apocrine hidrocystoma was made. The patient underwent successful surgical excision shortly thereafter without recurrence at follow-up 1 year later.
Apocrine hidrocystomas are rare benign cystic lesions that are considered to be adenomatous proliferations of apocrine glands. They typically manifest as solitary asymptomatic lesions measuring 3 to 15 mm.1 They tend to appear on the face, usually in the periorbital region, but also have been described on the neck, scalp, trunk, arms, and legs.2-4 Multiple apocrine hidrocystomas can be a marker of 2 rare inherited disorders: Gorlin-Goltz syndrome and Schopf-Schulz-Passarge syndrome.5 Apocrine hidrocystomas may be flesh colored or may have a blue, black, or brown appearance due to the Tyndall effect, in which light with shorter wavelengths is scattered by the contents of the lesions.2 Histologically, apocrine hidrocystomas are cysts lined by a dual layer of epithelial cells. The inner layer is composed of cells with apocrine features, and the outer layer is composed of flattened myoepithelial cells. Due to their range of colors and predilection for sun-exposed surfaces, apocrine hidrocystomas may be mistaken for various malignant neoplasms, including melanoma.6,7
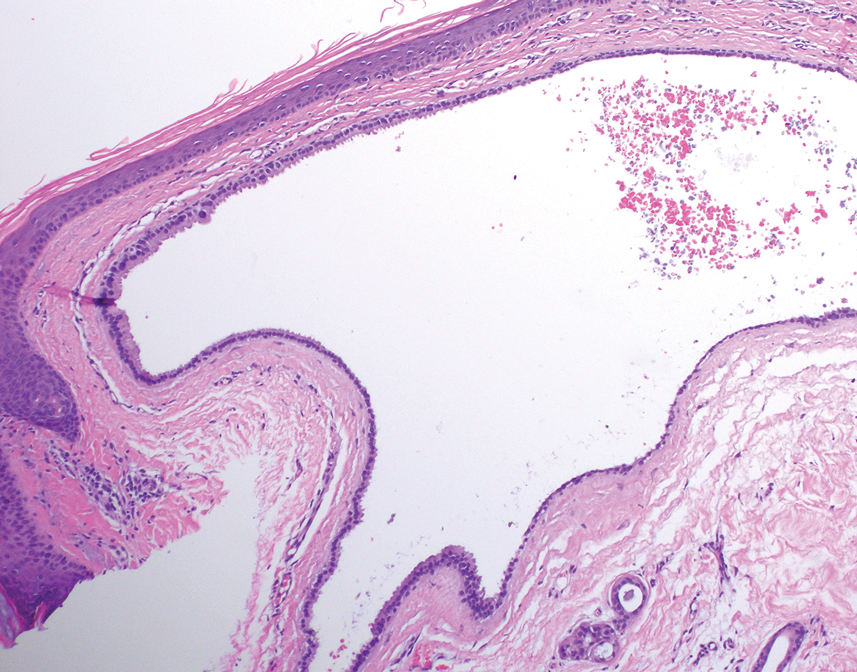
The differential diagnosis for our patient included agminated blue nevi, melanoma, pigmented basal cell carcinoma (BCC), and seborrheic keratosis. A blue nevus is a dermal melanocytic lesion that manifests as a well-demarcated, blue to blue-black papule that typically appears on the face, scalp, arms, legs, lower back, and buttocks. Although there are several histologic subtypes, the common blue nevus usually manifests as a solitary lesion measuring less than 1 cm, often developing during childhood to young adulthood.8 Histologically, common blue nevi are characterized by a dermal proliferation of deeply pigmented bipolar spindled melanocytes embedded in thickened collagen bundles, often with scattered epithelioid melanophages, and no conspicuous mitotic activity (Figure 2).9 There are other types of blue nevi, including cellular blue nevi, which tend to be larger and manifest commonly on the buttocks and sacrococcygeal region in early adulthood.9 Histologically, cellular blue nevi contain oval to spindled melanocytes with scattered melanophages forming a well-demarcated nodule typically in the reticular dermis. There may be bulbous extension into the subcutaneous adipose tissue. Occasional mitoses may be seen.9,10 Melanoma can arise from common or cellular blue nevi, though it more frequently occurs with cellular blue nevi. Other subtypes of blue nevi have been described, including the sclerosing, plaque-type, combined, hypomelanotic/amelanotic, and pigmented epithelioid melanocytoma.11 However, they typically have features of the common blue nevus or cellular blue nevus, such as oval/spindle cell morphology, some degree of melanin, and biphasic architecture, but are classified according to their dominant histologic characteristics.
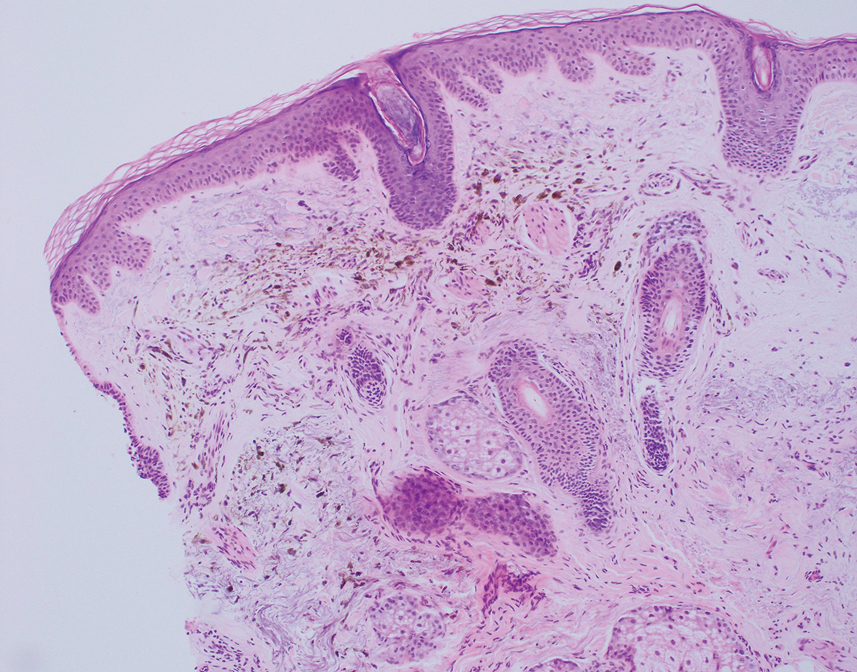
Given the location of our patient’s lesions on the scalp and his extensive history of sun exposure, malignancy was high in the differential. Multiple synchronous primary melanomas including nodular melanoma, blue nevus–like metastatic melanoma, and metastatic melanoma were considered. The leg and the scalp have the highest reported incidence of cutaneous metastases of melanoma, with many cases presenting as dermal or subcutaneous nodules and eruptive blue nevus–like papules, similar to our patient’s clinical presentation.12,13 Nodular melanoma (NM) is one of 4 major types of melanoma, accounting for approximately 15% to 30% of cases in the United States.14 Nodular melanoma typically manifests as a smooth, raised, symmetric, well-circumscribed lesion with variable pigmentation, from very dark to amelanotic. Histologically, NM is defined as a dermal mass, either in isolation or with an epidermal component, not to exceed 3 rete ridges beyond the dermal component.15 Tumor cells have a high cell density with pleomorphism, usually with atypical epithelioid cells with vesicular nuclei and irregular cytoplasm, and occasionally spindle cells (Figure 2).16 Mitoses and necrosis are frequent. Scalp location independently is responsible for worse survival, both overall and melanoma specific.17 Nodular melanoma tends to have greater Breslow thickness at diagnosis than other melanoma subtypes and often carries a worse prognosis.
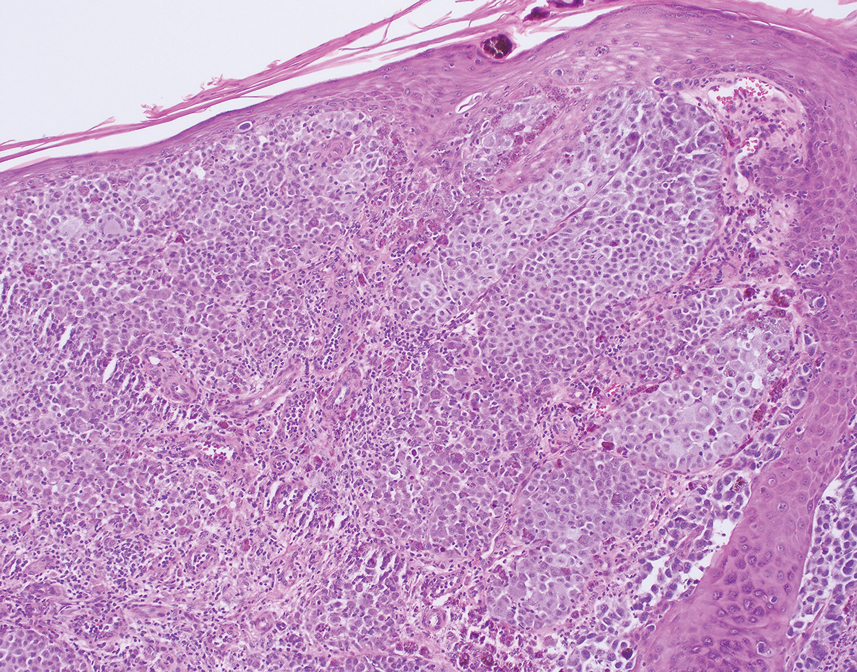
Malignant melanomas that develop from or in conjunction with or bear histologic resemblance to blue nevi are termed blue nevus–like melanoma or blue nevus–associated melanoma. These malignancies are exceedingly rare, accounting for only 0.3% of melanomas in one Turkey-based multicenter study.18 The histologic criteria for diagnosing blue nevus–like melanoma are poorly defined, and terminology of these lesions has led to some debate in naming conventions.19 Nevertheless, unlike blue nevus, blue nevus–like melanoma demonstrates histologic features of malignancy, including pleomorphism, prominent nucleoli, mitotic activity, vascular invasion, and potential necrosis.10 The lack of an inflammatory infiltrate, surrounding fibrosis, junctional activity, and pre-existing nevus can help distinguish cutaneous melanoma metastases from primary nodular melanoma. Immunohistochemical stains such as S100, Melan-A/MART1, or SOX-10 can help confirm melanocytic lineage.12
Pigmented BCC is a clinical and histologic variant of BCC characterized by increased melanin pigmentation due to melanocytes admixed with tumor cells. Dermoscopically, the pigment can have a maple leaf–like appearance with spoke-wheel areas, in-focus dots, and concentric structures at the dermoepidermal junction, which is more characteristic of superficial and infiltrating BCC.20 In nodular BCC, the pigment occurs as blue-gray ovoid nests and globules in deeper layers of the dermis.20
Seborrheic keratoses (SKs) can vary widely in clinical appearance, with pigmentation ranging from flesh colored to yellow to brown to black. Melanoacanthomas are acanthotic SKs that are highly pigmented due to intermixed epidermal melanocytes and subepidermal melanophages.21 Dermoscopy can help distinguish cutaneous malignancies from SKs, which often demonstrate fissures and ridges, comedolike openings, and milialike cysts. Biopsy sometimes is required to assess for malignancy, as was the case in our patient. The classic histologic features of SKs include acanthosis, papillomatosis, and hyperkeratosis.22
This case highlights the need to consider apocrine hidrocystoma, along with malignancy, in the differential diagnosis of pigmented cystic masses of the face and scalp. Because apocrine hidrocystomas are benign, they do not need to be treated but often are surgically excised for cosmesis or complete histopathologic examination. Destruction via electrodessication, carbon dioxide ablation, trichloroacetic acid chemical ablation, botulinum toxin injection, and anticholinergic creams sometimes is used, especially for cosmetic treatment of multiple small lesions.5 Our patient was treated with surgical excision with no evidence of recurrence on follow-up 1 year later.
- Ioannidis DG, Drivas EI, Papadakis CE, et al. Hidrocystoma of the external auditory canal: a case report. Cases J. 2009;2:79. doi:10.1186/1757- 1626-2-79
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp. Dermatol Online J. 2020;26. doi:10.5070/D3268049895
- Mendoza-Cembranos MD, Haro R, Requena L, et al. Digital apocrine hidrocystoma: the exception confirms the rule. Am J Dermatopathol. 2019;41:79. doi:10.1097/DAD.0000000000001044
- May C, Chang O, Compton N. A giant apocrine hidrocystoma of the trunk. Dermatol Online J. 2017;23. doi:10.5070/D3239036497
- Sarabi K, Khachemoune A. Hidrocystomas—a brief review. Medscape Gen Med. 2006;8:57.
- Kruse ALD, Zwahlen R, Bredell MG, et al. Apocrine hidrocystoma of the cheek. J Craniofac Surg. 2010;21:594-596. doi:10.1097 /SCS.0b013e3181d08c77
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405. doi:10.1002 /1097-0142(196803)21:3<393::aid-cncr2820210309>3.0.co;2-k
- Murali R, McCarthy SW, Scolyer RA. Blue nevi and related lesions: a review highlighting atypical and newly described variants, distinguishing features and diagnostic pitfalls. Adv Anat Pathol. 2009;16:365. doi:10.1097/PAP.0b013e3181bb6b53
- Borgenvik TL, Karlsvik TM, Ray S, et al. Blue nevus-like and blue nevusassociated melanoma: a comprehensive review of the literature. ANZ J Surg. 2017;87:345-349. doi:10.1111/ans.13946
- de la Fouchardiere A. Blue naevi and the blue tumour spectrum. Pathology. 2023;55:187-195. doi:10.1016/j.pathol.2022.12.342
- Lowe L. Metastatic melanoma and rare melanoma variants: a review. Pathology (Phila). 2023;55:236-244. doi:10.1016/j.pathol.2022.11.006
- Plaza JA, Torres-Cabala C, Evans H, et al. Cutaneous metastases of malignant melanoma: a clinicopathologic study of 192 cases with emphasis on the morphologic spectrum. Am J Dermatopathol. 2010;32:129-136. doi:10.1097/DAD.0b013e3181b34a19
- Shaikh WR, Xiong M, Weinstock MA. The contribution of nodular subtype to melanoma mortality in the United States, 1978 to 2007. Archives of Dermatology. 2012;148:30-36. doi:10.1001/archdermatol.2011.264
- Clark WH, From L, Bernardino EA, et al. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969;29:705-727.
- Bobos M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital J Dermatol Venereol. 2021;156:300-321. doi:10.23736/S2784-8671.21.06958-3
- Ozao-Choy J, Nelson DW, Hiles J, et al. The prognostic importance of scalp location in primary head and neck melanoma. J Surg Oncol. 2017;116:337-343. doi:10.1002/jso.24679
- Gamsizkan M, Yilmaz I, Buyukbabani N, et al. A retrospective multicenter evaluation of cutaneous melanomas in Turkey. Asian Pac J Cancer Prev APJCP. 2014;15:10451-10456. doi:10.7314 /apjcp.2014.15.23.10451
- Mones JM, Ackerman AB. “Atypical” blue nevus, “malignant” blue nevus, and “metastasizing” blue nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol. 2004;26:407-430. doi:10.1097/00000372-200410000-00012
- Tanese K. Diagnosis and management of basal cell carcinoma Curr Treat Options Oncol. 2019;20:13. doi:10.1007/s11864 -019-0610-0
- Barthelmann S, Butsch F, Lang BM, et al. Seborrheic keratosis. JDDG J Dtsch Dermatol Ges. 2023;21:265-277. doi:10.1111/ddg.14984
- Taylor S. Advancing the understanding of seborrheic keratosis. J Drugs Dermatol. 2017;16:419-424.
THE DIAGNOSIS: Apocrine Hidrocystoma
Histology for all 3 lesions demonstrated similar cystic structures lined by a dual layer of epithelial cells, with the outermost layer composed of flattened myoepithelial cells and the inner layer composed of cells with apocrine features (Figure 1). Based on these findings, a diagnosis of apocrine hidrocystoma was made. The patient underwent successful surgical excision shortly thereafter without recurrence at follow-up 1 year later.
Apocrine hidrocystomas are rare benign cystic lesions that are considered to be adenomatous proliferations of apocrine glands. They typically manifest as solitary asymptomatic lesions measuring 3 to 15 mm.1 They tend to appear on the face, usually in the periorbital region, but also have been described on the neck, scalp, trunk, arms, and legs.2-4 Multiple apocrine hidrocystomas can be a marker of 2 rare inherited disorders: Gorlin-Goltz syndrome and Schopf-Schulz-Passarge syndrome.5 Apocrine hidrocystomas may be flesh colored or may have a blue, black, or brown appearance due to the Tyndall effect, in which light with shorter wavelengths is scattered by the contents of the lesions.2 Histologically, apocrine hidrocystomas are cysts lined by a dual layer of epithelial cells. The inner layer is composed of cells with apocrine features, and the outer layer is composed of flattened myoepithelial cells. Due to their range of colors and predilection for sun-exposed surfaces, apocrine hidrocystomas may be mistaken for various malignant neoplasms, including melanoma.6,7
The differential diagnosis for our patient included agminated blue nevi, melanoma, pigmented basal cell carcinoma (BCC), and seborrheic keratosis. A blue nevus is a dermal melanocytic lesion that manifests as a well-demarcated, blue to blue-black papule that typically appears on the face, scalp, arms, legs, lower back, and buttocks. Although there are several histologic subtypes, the common blue nevus usually manifests as a solitary lesion measuring less than 1 cm, often developing during childhood to young adulthood.8 Histologically, common blue nevi are characterized by a dermal proliferation of deeply pigmented bipolar spindled melanocytes embedded in thickened collagen bundles, often with scattered epithelioid melanophages, and no conspicuous mitotic activity (Figure 2).9 There are other types of blue nevi, including cellular blue nevi, which tend to be larger and manifest commonly on the buttocks and sacrococcygeal region in early adulthood.9 Histologically, cellular blue nevi contain oval to spindled melanocytes with scattered melanophages forming a well-demarcated nodule typically in the reticular dermis. There may be bulbous extension into the subcutaneous adipose tissue. Occasional mitoses may be seen.9,10 Melanoma can arise from common or cellular blue nevi, though it more frequently occurs with cellular blue nevi. Other subtypes of blue nevi have been described, including the sclerosing, plaque-type, combined, hypomelanotic/amelanotic, and pigmented epithelioid melanocytoma.11 However, they typically have features of the common blue nevus or cellular blue nevus, such as oval/spindle cell morphology, some degree of melanin, and biphasic architecture, but are classified according to their dominant histologic characteristics.
Given the location of our patient’s lesions on the scalp and his extensive history of sun exposure, malignancy was high in the differential. Multiple synchronous primary melanomas including nodular melanoma, blue nevus–like metastatic melanoma, and metastatic melanoma were considered. The leg and the scalp have the highest reported incidence of cutaneous metastases of melanoma, with many cases presenting as dermal or subcutaneous nodules and eruptive blue nevus–like papules, similar to our patient’s clinical presentation.12,13 Nodular melanoma (NM) is one of 4 major types of melanoma, accounting for approximately 15% to 30% of cases in the United States.14 Nodular melanoma typically manifests as a smooth, raised, symmetric, well-circumscribed lesion with variable pigmentation, from very dark to amelanotic. Histologically, NM is defined as a dermal mass, either in isolation or with an epidermal component, not to exceed 3 rete ridges beyond the dermal component.15 Tumor cells have a high cell density with pleomorphism, usually with atypical epithelioid cells with vesicular nuclei and irregular cytoplasm, and occasionally spindle cells (Figure 2).16 Mitoses and necrosis are frequent. Scalp location independently is responsible for worse survival, both overall and melanoma specific.17 Nodular melanoma tends to have greater Breslow thickness at diagnosis than other melanoma subtypes and often carries a worse prognosis.
Malignant melanomas that develop from or in conjunction with or bear histologic resemblance to blue nevi are termed blue nevus–like melanoma or blue nevus–associated melanoma. These malignancies are exceedingly rare, accounting for only 0.3% of melanomas in one Turkey-based multicenter study.18 The histologic criteria for diagnosing blue nevus–like melanoma are poorly defined, and terminology of these lesions has led to some debate in naming conventions.19 Nevertheless, unlike blue nevus, blue nevus–like melanoma demonstrates histologic features of malignancy, including pleomorphism, prominent nucleoli, mitotic activity, vascular invasion, and potential necrosis.10 The lack of an inflammatory infiltrate, surrounding fibrosis, junctional activity, and pre-existing nevus can help distinguish cutaneous melanoma metastases from primary nodular melanoma. Immunohistochemical stains such as S100, Melan-A/MART1, or SOX-10 can help confirm melanocytic lineage.12
Pigmented BCC is a clinical and histologic variant of BCC characterized by increased melanin pigmentation due to melanocytes admixed with tumor cells. Dermoscopically, the pigment can have a maple leaf–like appearance with spoke-wheel areas, in-focus dots, and concentric structures at the dermoepidermal junction, which is more characteristic of superficial and infiltrating BCC.20 In nodular BCC, the pigment occurs as blue-gray ovoid nests and globules in deeper layers of the dermis.20
Seborrheic keratoses (SKs) can vary widely in clinical appearance, with pigmentation ranging from flesh colored to yellow to brown to black. Melanoacanthomas are acanthotic SKs that are highly pigmented due to intermixed epidermal melanocytes and subepidermal melanophages.21 Dermoscopy can help distinguish cutaneous malignancies from SKs, which often demonstrate fissures and ridges, comedolike openings, and milialike cysts. Biopsy sometimes is required to assess for malignancy, as was the case in our patient. The classic histologic features of SKs include acanthosis, papillomatosis, and hyperkeratosis.22
This case highlights the need to consider apocrine hidrocystoma, along with malignancy, in the differential diagnosis of pigmented cystic masses of the face and scalp. Because apocrine hidrocystomas are benign, they do not need to be treated but often are surgically excised for cosmesis or complete histopathologic examination. Destruction via electrodessication, carbon dioxide ablation, trichloroacetic acid chemical ablation, botulinum toxin injection, and anticholinergic creams sometimes is used, especially for cosmetic treatment of multiple small lesions.5 Our patient was treated with surgical excision with no evidence of recurrence on follow-up 1 year later.
THE DIAGNOSIS: Apocrine Hidrocystoma
Histology for all 3 lesions demonstrated similar cystic structures lined by a dual layer of epithelial cells, with the outermost layer composed of flattened myoepithelial cells and the inner layer composed of cells with apocrine features (Figure 1). Based on these findings, a diagnosis of apocrine hidrocystoma was made. The patient underwent successful surgical excision shortly thereafter without recurrence at follow-up 1 year later.
Apocrine hidrocystomas are rare benign cystic lesions that are considered to be adenomatous proliferations of apocrine glands. They typically manifest as solitary asymptomatic lesions measuring 3 to 15 mm.1 They tend to appear on the face, usually in the periorbital region, but also have been described on the neck, scalp, trunk, arms, and legs.2-4 Multiple apocrine hidrocystomas can be a marker of 2 rare inherited disorders: Gorlin-Goltz syndrome and Schopf-Schulz-Passarge syndrome.5 Apocrine hidrocystomas may be flesh colored or may have a blue, black, or brown appearance due to the Tyndall effect, in which light with shorter wavelengths is scattered by the contents of the lesions.2 Histologically, apocrine hidrocystomas are cysts lined by a dual layer of epithelial cells. The inner layer is composed of cells with apocrine features, and the outer layer is composed of flattened myoepithelial cells. Due to their range of colors and predilection for sun-exposed surfaces, apocrine hidrocystomas may be mistaken for various malignant neoplasms, including melanoma.6,7
The differential diagnosis for our patient included agminated blue nevi, melanoma, pigmented basal cell carcinoma (BCC), and seborrheic keratosis. A blue nevus is a dermal melanocytic lesion that manifests as a well-demarcated, blue to blue-black papule that typically appears on the face, scalp, arms, legs, lower back, and buttocks. Although there are several histologic subtypes, the common blue nevus usually manifests as a solitary lesion measuring less than 1 cm, often developing during childhood to young adulthood.8 Histologically, common blue nevi are characterized by a dermal proliferation of deeply pigmented bipolar spindled melanocytes embedded in thickened collagen bundles, often with scattered epithelioid melanophages, and no conspicuous mitotic activity (Figure 2).9 There are other types of blue nevi, including cellular blue nevi, which tend to be larger and manifest commonly on the buttocks and sacrococcygeal region in early adulthood.9 Histologically, cellular blue nevi contain oval to spindled melanocytes with scattered melanophages forming a well-demarcated nodule typically in the reticular dermis. There may be bulbous extension into the subcutaneous adipose tissue. Occasional mitoses may be seen.9,10 Melanoma can arise from common or cellular blue nevi, though it more frequently occurs with cellular blue nevi. Other subtypes of blue nevi have been described, including the sclerosing, plaque-type, combined, hypomelanotic/amelanotic, and pigmented epithelioid melanocytoma.11 However, they typically have features of the common blue nevus or cellular blue nevus, such as oval/spindle cell morphology, some degree of melanin, and biphasic architecture, but are classified according to their dominant histologic characteristics.
Given the location of our patient’s lesions on the scalp and his extensive history of sun exposure, malignancy was high in the differential. Multiple synchronous primary melanomas including nodular melanoma, blue nevus–like metastatic melanoma, and metastatic melanoma were considered. The leg and the scalp have the highest reported incidence of cutaneous metastases of melanoma, with many cases presenting as dermal or subcutaneous nodules and eruptive blue nevus–like papules, similar to our patient’s clinical presentation.12,13 Nodular melanoma (NM) is one of 4 major types of melanoma, accounting for approximately 15% to 30% of cases in the United States.14 Nodular melanoma typically manifests as a smooth, raised, symmetric, well-circumscribed lesion with variable pigmentation, from very dark to amelanotic. Histologically, NM is defined as a dermal mass, either in isolation or with an epidermal component, not to exceed 3 rete ridges beyond the dermal component.15 Tumor cells have a high cell density with pleomorphism, usually with atypical epithelioid cells with vesicular nuclei and irregular cytoplasm, and occasionally spindle cells (Figure 2).16 Mitoses and necrosis are frequent. Scalp location independently is responsible for worse survival, both overall and melanoma specific.17 Nodular melanoma tends to have greater Breslow thickness at diagnosis than other melanoma subtypes and often carries a worse prognosis.
Malignant melanomas that develop from or in conjunction with or bear histologic resemblance to blue nevi are termed blue nevus–like melanoma or blue nevus–associated melanoma. These malignancies are exceedingly rare, accounting for only 0.3% of melanomas in one Turkey-based multicenter study.18 The histologic criteria for diagnosing blue nevus–like melanoma are poorly defined, and terminology of these lesions has led to some debate in naming conventions.19 Nevertheless, unlike blue nevus, blue nevus–like melanoma demonstrates histologic features of malignancy, including pleomorphism, prominent nucleoli, mitotic activity, vascular invasion, and potential necrosis.10 The lack of an inflammatory infiltrate, surrounding fibrosis, junctional activity, and pre-existing nevus can help distinguish cutaneous melanoma metastases from primary nodular melanoma. Immunohistochemical stains such as S100, Melan-A/MART1, or SOX-10 can help confirm melanocytic lineage.12
Pigmented BCC is a clinical and histologic variant of BCC characterized by increased melanin pigmentation due to melanocytes admixed with tumor cells. Dermoscopically, the pigment can have a maple leaf–like appearance with spoke-wheel areas, in-focus dots, and concentric structures at the dermoepidermal junction, which is more characteristic of superficial and infiltrating BCC.20 In nodular BCC, the pigment occurs as blue-gray ovoid nests and globules in deeper layers of the dermis.20
Seborrheic keratoses (SKs) can vary widely in clinical appearance, with pigmentation ranging from flesh colored to yellow to brown to black. Melanoacanthomas are acanthotic SKs that are highly pigmented due to intermixed epidermal melanocytes and subepidermal melanophages.21 Dermoscopy can help distinguish cutaneous malignancies from SKs, which often demonstrate fissures and ridges, comedolike openings, and milialike cysts. Biopsy sometimes is required to assess for malignancy, as was the case in our patient. The classic histologic features of SKs include acanthosis, papillomatosis, and hyperkeratosis.22
This case highlights the need to consider apocrine hidrocystoma, along with malignancy, in the differential diagnosis of pigmented cystic masses of the face and scalp. Because apocrine hidrocystomas are benign, they do not need to be treated but often are surgically excised for cosmesis or complete histopathologic examination. Destruction via electrodessication, carbon dioxide ablation, trichloroacetic acid chemical ablation, botulinum toxin injection, and anticholinergic creams sometimes is used, especially for cosmetic treatment of multiple small lesions.5 Our patient was treated with surgical excision with no evidence of recurrence on follow-up 1 year later.
- Ioannidis DG, Drivas EI, Papadakis CE, et al. Hidrocystoma of the external auditory canal: a case report. Cases J. 2009;2:79. doi:10.1186/1757- 1626-2-79
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp. Dermatol Online J. 2020;26. doi:10.5070/D3268049895
- Mendoza-Cembranos MD, Haro R, Requena L, et al. Digital apocrine hidrocystoma: the exception confirms the rule. Am J Dermatopathol. 2019;41:79. doi:10.1097/DAD.0000000000001044
- May C, Chang O, Compton N. A giant apocrine hidrocystoma of the trunk. Dermatol Online J. 2017;23. doi:10.5070/D3239036497
- Sarabi K, Khachemoune A. Hidrocystomas—a brief review. Medscape Gen Med. 2006;8:57.
- Kruse ALD, Zwahlen R, Bredell MG, et al. Apocrine hidrocystoma of the cheek. J Craniofac Surg. 2010;21:594-596. doi:10.1097 /SCS.0b013e3181d08c77
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405. doi:10.1002 /1097-0142(196803)21:3<393::aid-cncr2820210309>3.0.co;2-k
- Murali R, McCarthy SW, Scolyer RA. Blue nevi and related lesions: a review highlighting atypical and newly described variants, distinguishing features and diagnostic pitfalls. Adv Anat Pathol. 2009;16:365. doi:10.1097/PAP.0b013e3181bb6b53
- Borgenvik TL, Karlsvik TM, Ray S, et al. Blue nevus-like and blue nevusassociated melanoma: a comprehensive review of the literature. ANZ J Surg. 2017;87:345-349. doi:10.1111/ans.13946
- de la Fouchardiere A. Blue naevi and the blue tumour spectrum. Pathology. 2023;55:187-195. doi:10.1016/j.pathol.2022.12.342
- Lowe L. Metastatic melanoma and rare melanoma variants: a review. Pathology (Phila). 2023;55:236-244. doi:10.1016/j.pathol.2022.11.006
- Plaza JA, Torres-Cabala C, Evans H, et al. Cutaneous metastases of malignant melanoma: a clinicopathologic study of 192 cases with emphasis on the morphologic spectrum. Am J Dermatopathol. 2010;32:129-136. doi:10.1097/DAD.0b013e3181b34a19
- Shaikh WR, Xiong M, Weinstock MA. The contribution of nodular subtype to melanoma mortality in the United States, 1978 to 2007. Archives of Dermatology. 2012;148:30-36. doi:10.1001/archdermatol.2011.264
- Clark WH, From L, Bernardino EA, et al. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969;29:705-727.
- Bobos M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital J Dermatol Venereol. 2021;156:300-321. doi:10.23736/S2784-8671.21.06958-3
- Ozao-Choy J, Nelson DW, Hiles J, et al. The prognostic importance of scalp location in primary head and neck melanoma. J Surg Oncol. 2017;116:337-343. doi:10.1002/jso.24679
- Gamsizkan M, Yilmaz I, Buyukbabani N, et al. A retrospective multicenter evaluation of cutaneous melanomas in Turkey. Asian Pac J Cancer Prev APJCP. 2014;15:10451-10456. doi:10.7314 /apjcp.2014.15.23.10451
- Mones JM, Ackerman AB. “Atypical” blue nevus, “malignant” blue nevus, and “metastasizing” blue nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol. 2004;26:407-430. doi:10.1097/00000372-200410000-00012
- Tanese K. Diagnosis and management of basal cell carcinoma Curr Treat Options Oncol. 2019;20:13. doi:10.1007/s11864 -019-0610-0
- Barthelmann S, Butsch F, Lang BM, et al. Seborrheic keratosis. JDDG J Dtsch Dermatol Ges. 2023;21:265-277. doi:10.1111/ddg.14984
- Taylor S. Advancing the understanding of seborrheic keratosis. J Drugs Dermatol. 2017;16:419-424.
- Ioannidis DG, Drivas EI, Papadakis CE, et al. Hidrocystoma of the external auditory canal: a case report. Cases J. 2009;2:79. doi:10.1186/1757- 1626-2-79
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp. Dermatol Online J. 2020;26. doi:10.5070/D3268049895
- Mendoza-Cembranos MD, Haro R, Requena L, et al. Digital apocrine hidrocystoma: the exception confirms the rule. Am J Dermatopathol. 2019;41:79. doi:10.1097/DAD.0000000000001044
- May C, Chang O, Compton N. A giant apocrine hidrocystoma of the trunk. Dermatol Online J. 2017;23. doi:10.5070/D3239036497
- Sarabi K, Khachemoune A. Hidrocystomas—a brief review. Medscape Gen Med. 2006;8:57.
- Kruse ALD, Zwahlen R, Bredell MG, et al. Apocrine hidrocystoma of the cheek. J Craniofac Surg. 2010;21:594-596. doi:10.1097 /SCS.0b013e3181d08c77
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405. doi:10.1002 /1097-0142(196803)21:3<393::aid-cncr2820210309>3.0.co;2-k
- Murali R, McCarthy SW, Scolyer RA. Blue nevi and related lesions: a review highlighting atypical and newly described variants, distinguishing features and diagnostic pitfalls. Adv Anat Pathol. 2009;16:365. doi:10.1097/PAP.0b013e3181bb6b53
- Borgenvik TL, Karlsvik TM, Ray S, et al. Blue nevus-like and blue nevusassociated melanoma: a comprehensive review of the literature. ANZ J Surg. 2017;87:345-349. doi:10.1111/ans.13946
- de la Fouchardiere A. Blue naevi and the blue tumour spectrum. Pathology. 2023;55:187-195. doi:10.1016/j.pathol.2022.12.342
- Lowe L. Metastatic melanoma and rare melanoma variants: a review. Pathology (Phila). 2023;55:236-244. doi:10.1016/j.pathol.2022.11.006
- Plaza JA, Torres-Cabala C, Evans H, et al. Cutaneous metastases of malignant melanoma: a clinicopathologic study of 192 cases with emphasis on the morphologic spectrum. Am J Dermatopathol. 2010;32:129-136. doi:10.1097/DAD.0b013e3181b34a19
- Shaikh WR, Xiong M, Weinstock MA. The contribution of nodular subtype to melanoma mortality in the United States, 1978 to 2007. Archives of Dermatology. 2012;148:30-36. doi:10.1001/archdermatol.2011.264
- Clark WH, From L, Bernardino EA, et al. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969;29:705-727.
- Bobos M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital J Dermatol Venereol. 2021;156:300-321. doi:10.23736/S2784-8671.21.06958-3
- Ozao-Choy J, Nelson DW, Hiles J, et al. The prognostic importance of scalp location in primary head and neck melanoma. J Surg Oncol. 2017;116:337-343. doi:10.1002/jso.24679
- Gamsizkan M, Yilmaz I, Buyukbabani N, et al. A retrospective multicenter evaluation of cutaneous melanomas in Turkey. Asian Pac J Cancer Prev APJCP. 2014;15:10451-10456. doi:10.7314 /apjcp.2014.15.23.10451
- Mones JM, Ackerman AB. “Atypical” blue nevus, “malignant” blue nevus, and “metastasizing” blue nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol. 2004;26:407-430. doi:10.1097/00000372-200410000-00012
- Tanese K. Diagnosis and management of basal cell carcinoma Curr Treat Options Oncol. 2019;20:13. doi:10.1007/s11864 -019-0610-0
- Barthelmann S, Butsch F, Lang BM, et al. Seborrheic keratosis. JDDG J Dtsch Dermatol Ges. 2023;21:265-277. doi:10.1111/ddg.14984
- Taylor S. Advancing the understanding of seborrheic keratosis. J Drugs Dermatol. 2017;16:419-424.
A 67-year-old man presented to the dermatology clinic with 3 asymptomatic pigmented papules on the scalp. The patient reported that he was unaware of the lesions until they were pointed out weeks earlier by his primary care physician during a routine visit. He then was referred to dermatology for follow-up. Physical examination at the current presentation revealed clustered firm, smooth, well-circumscribed, pigmented papules on the scalp measuring 5 to 8 mm. The patient reported no personal or family history of skin cancer but stated that he spent a lot of time outdoors and had a history of 6 blistering sunburns in his life. A punch biopsy of each lesion was performed.
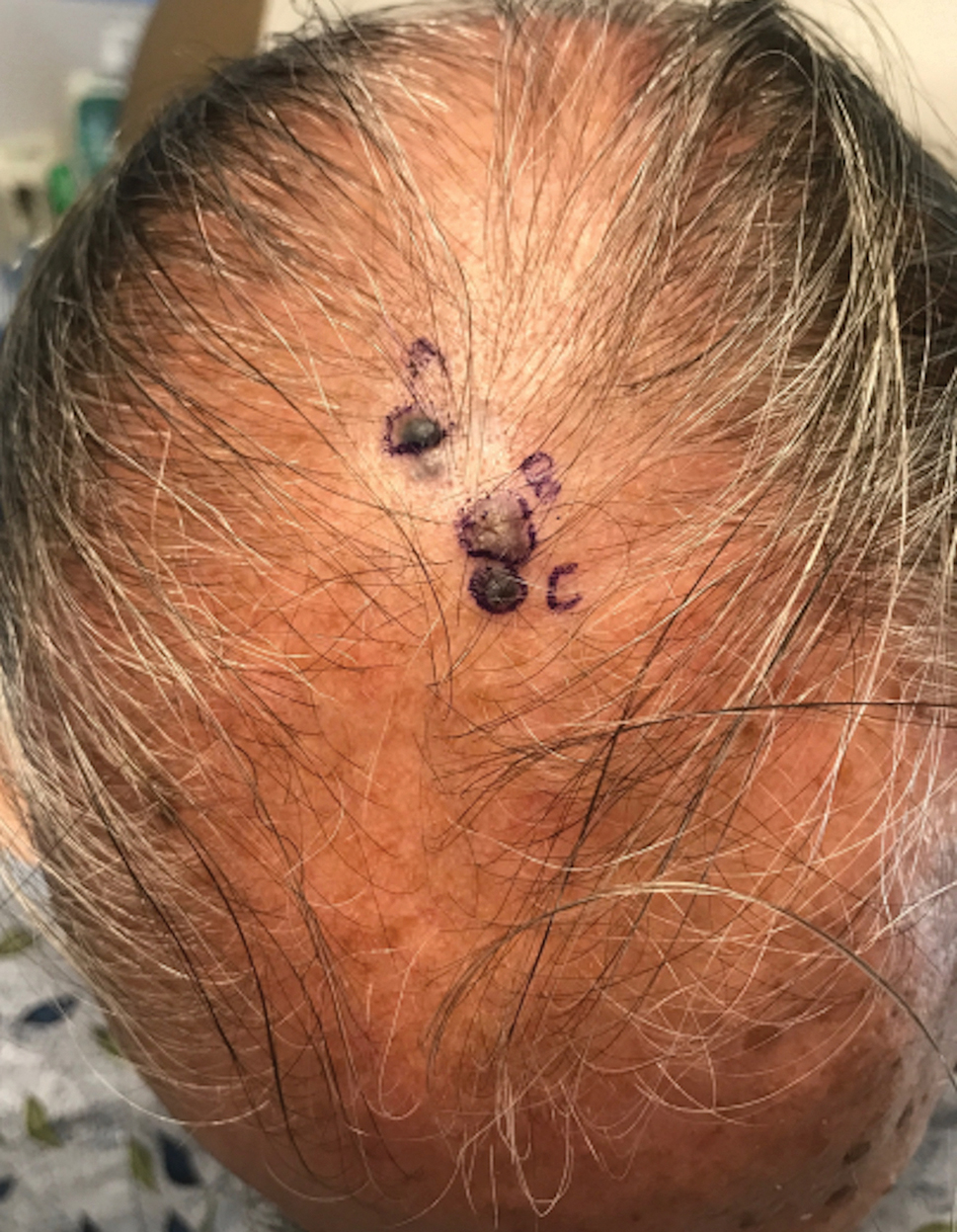
Papulonodules on the Ankle in a Patient with Lung Cancer
Papulonodules on the Ankle in a Patient with Lung Cancer
THE DIAGNOSIS: Pembrolizumab-Induced Eruptive Squamous Proliferation
Histopathology showed a broad squamous proliferation with acanthosis of the epidermis. Large glassy keratinocytes were seen with scattered necrotic keratinocytes (Figure), and a dense lichenoid band of inflammation was present subjacent to the proliferation. Notably, no hypergranulosis, remarkable keratinocyte atypia, or increased mitotic figures were seen. Based on the patient’s medical history and biopsy results, a diagnosis of pembrolizumab-induced eruptive squamous proliferation was made. The diagnosis was supported by a growing body of evidence of this type of reaction in patients taking programmed death 1 (PD-1) inhibitors.1,2 Conservative treatment with clobetasol ointment 0.05% was initiated with complete resolution of the lesions at the 2-month follow-up appointment. Other common treatments include topical steroids, injected corticosteroids, or cryosurgery to locally control the inflammation and atypical proliferation of cells.3
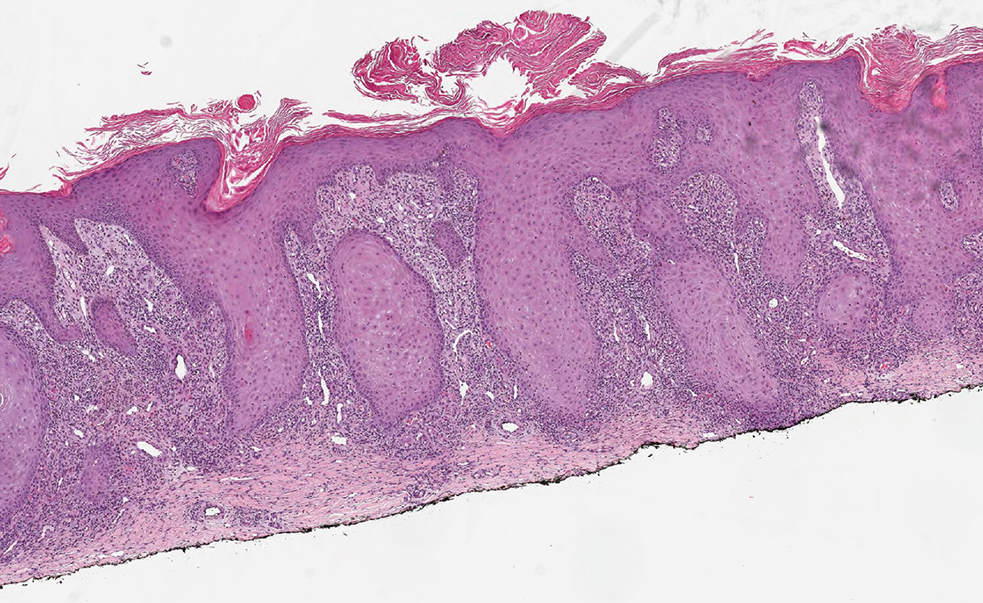
Pembrolizumab is a humanized IgG4 monoclonal antibody targeting the PD-1 receptor that has been utilized for its antitumor activity against various cancers, including unresectable and metastatic melanoma, head and neck cancers, and non–small cell lung cancer (NSCLC).1,4,5 While this drug has extended the lives of many patients with cancer, there are adverse reactions associated with PD-1 inhibitors (eg, pembrolizumab, nivolumab). Skin toxicity to PD-1 inhibitors is the one of most common immune-mediated reactions worldwide, occurring in approximately 30% of patients.6,7 Reactions can occur while a patient is taking the inciting drug and can continue up to 2 months after treatment discontinuation.8 Skin reactions associated with PD-1 inhibitors vary from lichenoid reactions and vitiligolike patches to psoriasis or eczema flares and are organized into 4 categories: inflammatory, immunobullous, alteration of keratinocytes, and alteration of melanocytes.9 Our patient demonstrated alteration of keratinocytes, which is characterized by overlapping features of hypertrophic lichen planus and early keratoacanthoma.
The differential diagnoses for pembrolizumab-induced eruptive squamous proliferation include squamous cell carcinoma (SCC), psoriasis, hypertrophic lichen planus, and cutaneous metastasis of NSCLC. Hypertrophic lupus erythematosus also is a well-documented reaction to use of immune-checkpoint inhibitors.10 Direct immunofluorescence could have helped differentiate hypertrophic lupus erythematosus from an eruptive squamous proliferation in our patient; however, due to her response to treatment, no additional workup was done.
Squamous cell carcinoma, which is the most common type of skin cancer in Black patients in the United States,11 has been shown to manifest after a PD-1 inhibitor is taken.12 Although it typically has a more chronic persistent course, the clinical appearance of SCC can be similar to the findings seen in our patient. Histologically, SCC may demonstrate necrosis, but the atypical proliferations will invade the dermis—a feature not seen in our patient’s histopathology.13
Lichen planus (LP) is an eruptive immune reaction of violaceous polygonal papules and plaques commonly seen on the ankles14 that has been shown to be an adverse effect of pembrolizumab.15 There are several subtypes of LP including hypertrophic versions, which can appear clinically similar to the findings seen in our patient. On dermoscopy, the classic finding of white lines, known as Wickham striae, is seen in all subtypes and can help diagnose this pathologic process. Under the microscope, LP can manifest with hyperkeratosis without parakeratosis, irregular thickening of the stratum granulosum, sawtooth rete ridges, and destruction of the basal layer.14
Psoriasis also has been shown to be exacerbated by anti–PD-1 therapy, although the majority of patients diagnosed with PD-1–induced psoriasis have a personal or family history of the disease.6 Clinically, psoriasis can have a hyperpigmented or violaceous appearance in patients with skin of color.16 The histopathology of psoriasis typically reveals confluent parakeratosis, neutrophils in the stratum corneum, regular acanthosis, thinning of the suprapapillary plates, and vessels in the dermal papillae.17
Although cutaneous metastasis of NSCLC may appear clinically similar to the current case, it is one of the rarer organ sites of metastasis for lung cancer.18 In our patient, biopsy quickly ruled out this diagnosis. If it had been a site of metastasis, histopathology would have shown a dermal-based proliferation of dysplastic cells without epidermal connection.19
It is important for dermatologists to recognize eruptive squamous proliferations associated with pembrolizumab, as they often respond to conservative treatment and typically do not require dose reduction or discontinuation of the inciting drug.
- Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. doi:10.1186/s40425-017-0242-5
- Preti BTB, Pencz A, Cowger JJM, et al. Skin deep: a fascinating case report of immunotherapy-triggered, treatment-refractory autoimmune lichen planus and keratoacanthoma. Case Rep Oncol. 2021;14: 1189-1193. doi:10.1159/000518313
- Fradet M, Sibaud V, Tournier E, et al. Multiple keratoacanthoma-like lesions in a patient treated with pembrolizumab. Acta Derm Venereol. 2019;99:1301-1302. doi:10.2340/00015555-3301
- Flynn JP, Gerriets V. Pembrolizumab. StatPearls [Internet]. StatPearls Publishing; 2023. Updated June 26, 2023. Accessed April 2, 2025.
- Antonov NK, Nair KG, Halasz CL. Transient eruptive keratoacanthomas associated with Nivolumab. JAAD Case Rep. 2019;5:342-345. doi:10.1016/j.jdcr.2019.01.025
- Voudouri D, Nikolaou V, Laschos K, et al. Anti-Pd1/Pdl1 induced psoriasis. Curr Probl Cancer. 2017;41:407-412. doi:10.1016 /j.currproblcancer.2017.10.003
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Coscarart A, Martel J, Lee MP, et al. Pembrolizumab-induced pseudoepitheliomatous eruption consistent with hypertrophic lichen planus. J Cutan Pathol. 2020;47:275-279. doi:10.1111/cup.13587
- Curry JL, Tetzlaff MT, Nagarajan P, et al. Diverse types of dermatologic toxicities from immune checkpoint blockade therapy. J Cutan Pathol. 2017;44:158-176. doi:10.1111/cup.12858
- Vitzthum von Eckstaedt H, Singh A, Reid P, et al. Immune checkpoint inhibitors and lupus erythematosus. Pharmaceuticals (Basel). 2024;2:15;17. doi:10.3390/ph17020252
- Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. 1995;75:667-673.
- Vu M, Chapman S, Lenz B, et al. Squamous cell carcinoma or squamous proliferation associated with nivolumab treatment for metastatic melanoma. Dermatol Online J. 2022;6:28. doi:10.5070/d328357786
- Howell JY, Ramsey ML. Squamous cell skin cancer. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 2, 2024. Accessed April 2, 2025.
- Arnold DL, Krishnamurthy K. Lichen planus. StatPearls [Internet]. StatPearls Publishing; 2024. Updated October 29, 2024. Accessed April 2, 2025.
- Yamashita A, Akasaka E, Nakano H, et al. Pembrolizumab-induced lichen planus on the scalp of a patient with non-small-cell lung carcinoma. Case Rep Dermatol. 2021;13:487-491. doi:10.1159/000519486
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25:524-528. doi:10.1016 /j.clindermatol.2007.08.005.
- Hidaka T, Ishii Y, Kitamura S. Clinical features of skin metastasis from lung cancer. Intern Med. 1996;35:459-462. doi:10.2169 /internalmedicine.35.459.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613–620. doi:10.1001/archderm.143.5.613
THE DIAGNOSIS: Pembrolizumab-Induced Eruptive Squamous Proliferation
Histopathology showed a broad squamous proliferation with acanthosis of the epidermis. Large glassy keratinocytes were seen with scattered necrotic keratinocytes (Figure), and a dense lichenoid band of inflammation was present subjacent to the proliferation. Notably, no hypergranulosis, remarkable keratinocyte atypia, or increased mitotic figures were seen. Based on the patient’s medical history and biopsy results, a diagnosis of pembrolizumab-induced eruptive squamous proliferation was made. The diagnosis was supported by a growing body of evidence of this type of reaction in patients taking programmed death 1 (PD-1) inhibitors.1,2 Conservative treatment with clobetasol ointment 0.05% was initiated with complete resolution of the lesions at the 2-month follow-up appointment. Other common treatments include topical steroids, injected corticosteroids, or cryosurgery to locally control the inflammation and atypical proliferation of cells.3
Pembrolizumab is a humanized IgG4 monoclonal antibody targeting the PD-1 receptor that has been utilized for its antitumor activity against various cancers, including unresectable and metastatic melanoma, head and neck cancers, and non–small cell lung cancer (NSCLC).1,4,5 While this drug has extended the lives of many patients with cancer, there are adverse reactions associated with PD-1 inhibitors (eg, pembrolizumab, nivolumab). Skin toxicity to PD-1 inhibitors is the one of most common immune-mediated reactions worldwide, occurring in approximately 30% of patients.6,7 Reactions can occur while a patient is taking the inciting drug and can continue up to 2 months after treatment discontinuation.8 Skin reactions associated with PD-1 inhibitors vary from lichenoid reactions and vitiligolike patches to psoriasis or eczema flares and are organized into 4 categories: inflammatory, immunobullous, alteration of keratinocytes, and alteration of melanocytes.9 Our patient demonstrated alteration of keratinocytes, which is characterized by overlapping features of hypertrophic lichen planus and early keratoacanthoma.
The differential diagnoses for pembrolizumab-induced eruptive squamous proliferation include squamous cell carcinoma (SCC), psoriasis, hypertrophic lichen planus, and cutaneous metastasis of NSCLC. Hypertrophic lupus erythematosus also is a well-documented reaction to use of immune-checkpoint inhibitors.10 Direct immunofluorescence could have helped differentiate hypertrophic lupus erythematosus from an eruptive squamous proliferation in our patient; however, due to her response to treatment, no additional workup was done.
Squamous cell carcinoma, which is the most common type of skin cancer in Black patients in the United States,11 has been shown to manifest after a PD-1 inhibitor is taken.12 Although it typically has a more chronic persistent course, the clinical appearance of SCC can be similar to the findings seen in our patient. Histologically, SCC may demonstrate necrosis, but the atypical proliferations will invade the dermis—a feature not seen in our patient’s histopathology.13
Lichen planus (LP) is an eruptive immune reaction of violaceous polygonal papules and plaques commonly seen on the ankles14 that has been shown to be an adverse effect of pembrolizumab.15 There are several subtypes of LP including hypertrophic versions, which can appear clinically similar to the findings seen in our patient. On dermoscopy, the classic finding of white lines, known as Wickham striae, is seen in all subtypes and can help diagnose this pathologic process. Under the microscope, LP can manifest with hyperkeratosis without parakeratosis, irregular thickening of the stratum granulosum, sawtooth rete ridges, and destruction of the basal layer.14
Psoriasis also has been shown to be exacerbated by anti–PD-1 therapy, although the majority of patients diagnosed with PD-1–induced psoriasis have a personal or family history of the disease.6 Clinically, psoriasis can have a hyperpigmented or violaceous appearance in patients with skin of color.16 The histopathology of psoriasis typically reveals confluent parakeratosis, neutrophils in the stratum corneum, regular acanthosis, thinning of the suprapapillary plates, and vessels in the dermal papillae.17
Although cutaneous metastasis of NSCLC may appear clinically similar to the current case, it is one of the rarer organ sites of metastasis for lung cancer.18 In our patient, biopsy quickly ruled out this diagnosis. If it had been a site of metastasis, histopathology would have shown a dermal-based proliferation of dysplastic cells without epidermal connection.19
It is important for dermatologists to recognize eruptive squamous proliferations associated with pembrolizumab, as they often respond to conservative treatment and typically do not require dose reduction or discontinuation of the inciting drug.
THE DIAGNOSIS: Pembrolizumab-Induced Eruptive Squamous Proliferation
Histopathology showed a broad squamous proliferation with acanthosis of the epidermis. Large glassy keratinocytes were seen with scattered necrotic keratinocytes (Figure), and a dense lichenoid band of inflammation was present subjacent to the proliferation. Notably, no hypergranulosis, remarkable keratinocyte atypia, or increased mitotic figures were seen. Based on the patient’s medical history and biopsy results, a diagnosis of pembrolizumab-induced eruptive squamous proliferation was made. The diagnosis was supported by a growing body of evidence of this type of reaction in patients taking programmed death 1 (PD-1) inhibitors.1,2 Conservative treatment with clobetasol ointment 0.05% was initiated with complete resolution of the lesions at the 2-month follow-up appointment. Other common treatments include topical steroids, injected corticosteroids, or cryosurgery to locally control the inflammation and atypical proliferation of cells.3
Pembrolizumab is a humanized IgG4 monoclonal antibody targeting the PD-1 receptor that has been utilized for its antitumor activity against various cancers, including unresectable and metastatic melanoma, head and neck cancers, and non–small cell lung cancer (NSCLC).1,4,5 While this drug has extended the lives of many patients with cancer, there are adverse reactions associated with PD-1 inhibitors (eg, pembrolizumab, nivolumab). Skin toxicity to PD-1 inhibitors is the one of most common immune-mediated reactions worldwide, occurring in approximately 30% of patients.6,7 Reactions can occur while a patient is taking the inciting drug and can continue up to 2 months after treatment discontinuation.8 Skin reactions associated with PD-1 inhibitors vary from lichenoid reactions and vitiligolike patches to psoriasis or eczema flares and are organized into 4 categories: inflammatory, immunobullous, alteration of keratinocytes, and alteration of melanocytes.9 Our patient demonstrated alteration of keratinocytes, which is characterized by overlapping features of hypertrophic lichen planus and early keratoacanthoma.
The differential diagnoses for pembrolizumab-induced eruptive squamous proliferation include squamous cell carcinoma (SCC), psoriasis, hypertrophic lichen planus, and cutaneous metastasis of NSCLC. Hypertrophic lupus erythematosus also is a well-documented reaction to use of immune-checkpoint inhibitors.10 Direct immunofluorescence could have helped differentiate hypertrophic lupus erythematosus from an eruptive squamous proliferation in our patient; however, due to her response to treatment, no additional workup was done.
Squamous cell carcinoma, which is the most common type of skin cancer in Black patients in the United States,11 has been shown to manifest after a PD-1 inhibitor is taken.12 Although it typically has a more chronic persistent course, the clinical appearance of SCC can be similar to the findings seen in our patient. Histologically, SCC may demonstrate necrosis, but the atypical proliferations will invade the dermis—a feature not seen in our patient’s histopathology.13
Lichen planus (LP) is an eruptive immune reaction of violaceous polygonal papules and plaques commonly seen on the ankles14 that has been shown to be an adverse effect of pembrolizumab.15 There are several subtypes of LP including hypertrophic versions, which can appear clinically similar to the findings seen in our patient. On dermoscopy, the classic finding of white lines, known as Wickham striae, is seen in all subtypes and can help diagnose this pathologic process. Under the microscope, LP can manifest with hyperkeratosis without parakeratosis, irregular thickening of the stratum granulosum, sawtooth rete ridges, and destruction of the basal layer.14
Psoriasis also has been shown to be exacerbated by anti–PD-1 therapy, although the majority of patients diagnosed with PD-1–induced psoriasis have a personal or family history of the disease.6 Clinically, psoriasis can have a hyperpigmented or violaceous appearance in patients with skin of color.16 The histopathology of psoriasis typically reveals confluent parakeratosis, neutrophils in the stratum corneum, regular acanthosis, thinning of the suprapapillary plates, and vessels in the dermal papillae.17
Although cutaneous metastasis of NSCLC may appear clinically similar to the current case, it is one of the rarer organ sites of metastasis for lung cancer.18 In our patient, biopsy quickly ruled out this diagnosis. If it had been a site of metastasis, histopathology would have shown a dermal-based proliferation of dysplastic cells without epidermal connection.19
It is important for dermatologists to recognize eruptive squamous proliferations associated with pembrolizumab, as they often respond to conservative treatment and typically do not require dose reduction or discontinuation of the inciting drug.
- Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. doi:10.1186/s40425-017-0242-5
- Preti BTB, Pencz A, Cowger JJM, et al. Skin deep: a fascinating case report of immunotherapy-triggered, treatment-refractory autoimmune lichen planus and keratoacanthoma. Case Rep Oncol. 2021;14: 1189-1193. doi:10.1159/000518313
- Fradet M, Sibaud V, Tournier E, et al. Multiple keratoacanthoma-like lesions in a patient treated with pembrolizumab. Acta Derm Venereol. 2019;99:1301-1302. doi:10.2340/00015555-3301
- Flynn JP, Gerriets V. Pembrolizumab. StatPearls [Internet]. StatPearls Publishing; 2023. Updated June 26, 2023. Accessed April 2, 2025.
- Antonov NK, Nair KG, Halasz CL. Transient eruptive keratoacanthomas associated with Nivolumab. JAAD Case Rep. 2019;5:342-345. doi:10.1016/j.jdcr.2019.01.025
- Voudouri D, Nikolaou V, Laschos K, et al. Anti-Pd1/Pdl1 induced psoriasis. Curr Probl Cancer. 2017;41:407-412. doi:10.1016 /j.currproblcancer.2017.10.003
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Coscarart A, Martel J, Lee MP, et al. Pembrolizumab-induced pseudoepitheliomatous eruption consistent with hypertrophic lichen planus. J Cutan Pathol. 2020;47:275-279. doi:10.1111/cup.13587
- Curry JL, Tetzlaff MT, Nagarajan P, et al. Diverse types of dermatologic toxicities from immune checkpoint blockade therapy. J Cutan Pathol. 2017;44:158-176. doi:10.1111/cup.12858
- Vitzthum von Eckstaedt H, Singh A, Reid P, et al. Immune checkpoint inhibitors and lupus erythematosus. Pharmaceuticals (Basel). 2024;2:15;17. doi:10.3390/ph17020252
- Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. 1995;75:667-673.
- Vu M, Chapman S, Lenz B, et al. Squamous cell carcinoma or squamous proliferation associated with nivolumab treatment for metastatic melanoma. Dermatol Online J. 2022;6:28. doi:10.5070/d328357786
- Howell JY, Ramsey ML. Squamous cell skin cancer. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 2, 2024. Accessed April 2, 2025.
- Arnold DL, Krishnamurthy K. Lichen planus. StatPearls [Internet]. StatPearls Publishing; 2024. Updated October 29, 2024. Accessed April 2, 2025.
- Yamashita A, Akasaka E, Nakano H, et al. Pembrolizumab-induced lichen planus on the scalp of a patient with non-small-cell lung carcinoma. Case Rep Dermatol. 2021;13:487-491. doi:10.1159/000519486
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25:524-528. doi:10.1016 /j.clindermatol.2007.08.005.
- Hidaka T, Ishii Y, Kitamura S. Clinical features of skin metastasis from lung cancer. Intern Med. 1996;35:459-462. doi:10.2169 /internalmedicine.35.459.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613–620. doi:10.1001/archderm.143.5.613
- Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. doi:10.1186/s40425-017-0242-5
- Preti BTB, Pencz A, Cowger JJM, et al. Skin deep: a fascinating case report of immunotherapy-triggered, treatment-refractory autoimmune lichen planus and keratoacanthoma. Case Rep Oncol. 2021;14: 1189-1193. doi:10.1159/000518313
- Fradet M, Sibaud V, Tournier E, et al. Multiple keratoacanthoma-like lesions in a patient treated with pembrolizumab. Acta Derm Venereol. 2019;99:1301-1302. doi:10.2340/00015555-3301
- Flynn JP, Gerriets V. Pembrolizumab. StatPearls [Internet]. StatPearls Publishing; 2023. Updated June 26, 2023. Accessed April 2, 2025.
- Antonov NK, Nair KG, Halasz CL. Transient eruptive keratoacanthomas associated with Nivolumab. JAAD Case Rep. 2019;5:342-345. doi:10.1016/j.jdcr.2019.01.025
- Voudouri D, Nikolaou V, Laschos K, et al. Anti-Pd1/Pdl1 induced psoriasis. Curr Probl Cancer. 2017;41:407-412. doi:10.1016 /j.currproblcancer.2017.10.003
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Coscarart A, Martel J, Lee MP, et al. Pembrolizumab-induced pseudoepitheliomatous eruption consistent with hypertrophic lichen planus. J Cutan Pathol. 2020;47:275-279. doi:10.1111/cup.13587
- Curry JL, Tetzlaff MT, Nagarajan P, et al. Diverse types of dermatologic toxicities from immune checkpoint blockade therapy. J Cutan Pathol. 2017;44:158-176. doi:10.1111/cup.12858
- Vitzthum von Eckstaedt H, Singh A, Reid P, et al. Immune checkpoint inhibitors and lupus erythematosus. Pharmaceuticals (Basel). 2024;2:15;17. doi:10.3390/ph17020252
- Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. 1995;75:667-673.
- Vu M, Chapman S, Lenz B, et al. Squamous cell carcinoma or squamous proliferation associated with nivolumab treatment for metastatic melanoma. Dermatol Online J. 2022;6:28. doi:10.5070/d328357786
- Howell JY, Ramsey ML. Squamous cell skin cancer. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 2, 2024. Accessed April 2, 2025.
- Arnold DL, Krishnamurthy K. Lichen planus. StatPearls [Internet]. StatPearls Publishing; 2024. Updated October 29, 2024. Accessed April 2, 2025.
- Yamashita A, Akasaka E, Nakano H, et al. Pembrolizumab-induced lichen planus on the scalp of a patient with non-small-cell lung carcinoma. Case Rep Dermatol. 2021;13:487-491. doi:10.1159/000519486
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25:524-528. doi:10.1016 /j.clindermatol.2007.08.005.
- Hidaka T, Ishii Y, Kitamura S. Clinical features of skin metastasis from lung cancer. Intern Med. 1996;35:459-462. doi:10.2169 /internalmedicine.35.459.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613–620. doi:10.1001/archderm.143.5.613
Papulonodules on the Ankle in a Patient with Lung Cancer
Papulonodules on the Ankle in a Patient with Lung Cancer
A 75-year-old woman presented to the dermatology department with well-circumscribed, round, hyperkeratotic papulonodules on the ankle of 3 months’ duration (top). The papulonodules also were evaluated by dermoscopy, which highlighted in greater detail the hyperkeratosis seen grossly (bottom). The patient had a history of chronic obstructive pulmonary disease and metastatic lung cancer and had been taking pembrolizumab for the past 2 years. The lesions initially appeared on the medial right foot and slowly spread proximally. Most of the lesions resolved spontaneously except for 2 on the right ankle. At the current presentation, one lesion was slightly tender to palpation, but both were otherwise asymptomatic. A lesion was biopsied and sent for dermatopathologic evaluation.
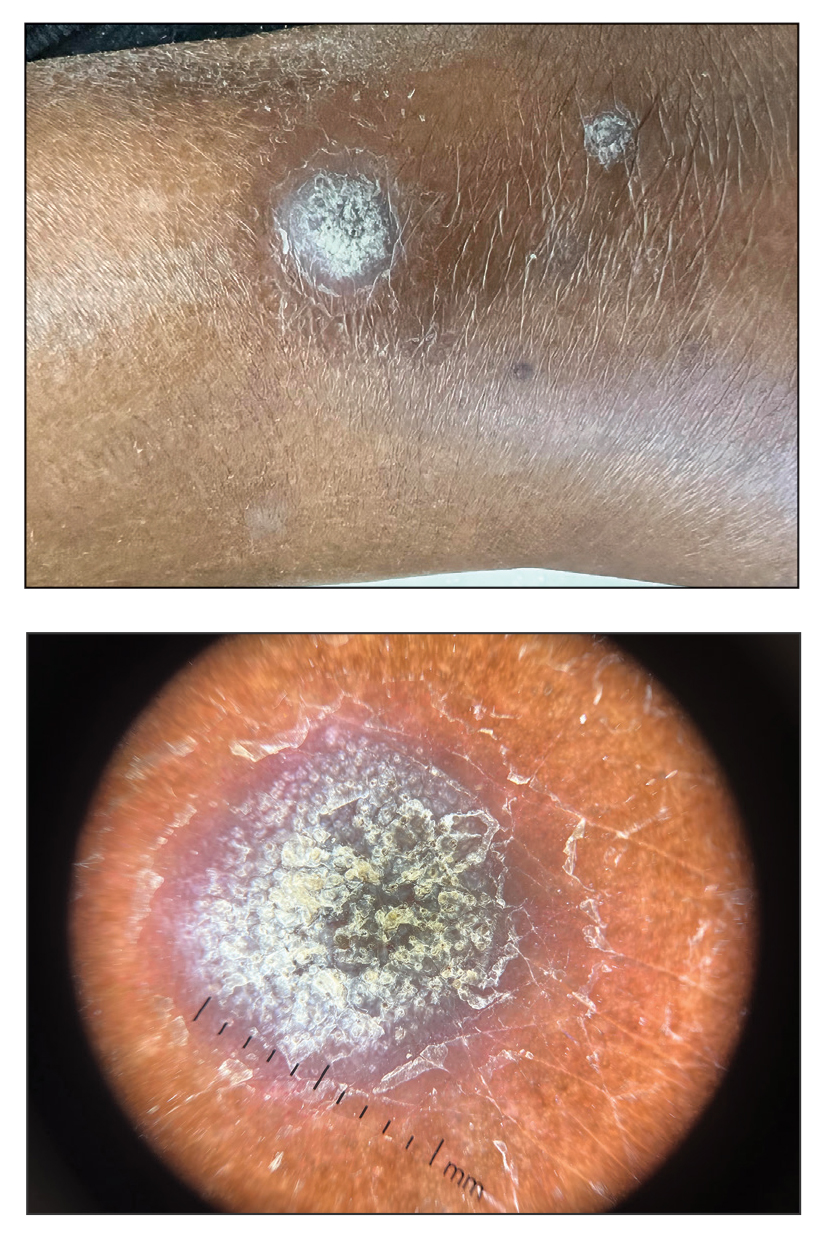

Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
THE DIAGNOSIS: Borderline-Borderline Leprosy With Type 1 Lepra Reaction
Punch biopsies from plaques on the right elbow and right shin revealed diffuse granulomatous dermatitis (Figure 1) with a narrow Grenz zone in the superficial dermis. The upper dermis contained a dense bandlike infiltrate of histiocytes with abundant foamy-gray cytoplasm and a moderate admixture of lymphocytes. The mid and deep dermis contained a nodular, perivascular, periadnexal, and perineural infiltrate of histiocytes and a dense admixture of lymphocytes. Periodic acid-Schiff and Gram stains were negative for microorganisms. Fite stain was positive for numerous organisms in histiocytes and small dermal nerves (Figure 2). These findings and the clinical examination confirmed a diagnosis of borderline-borderline leprosy with type 1 lepra reaction. The patient was started on dapsone 100 mg, rifampin 600 mg, and clofazimine 100 mg once daily and experienced clinical improvement within 6 months.
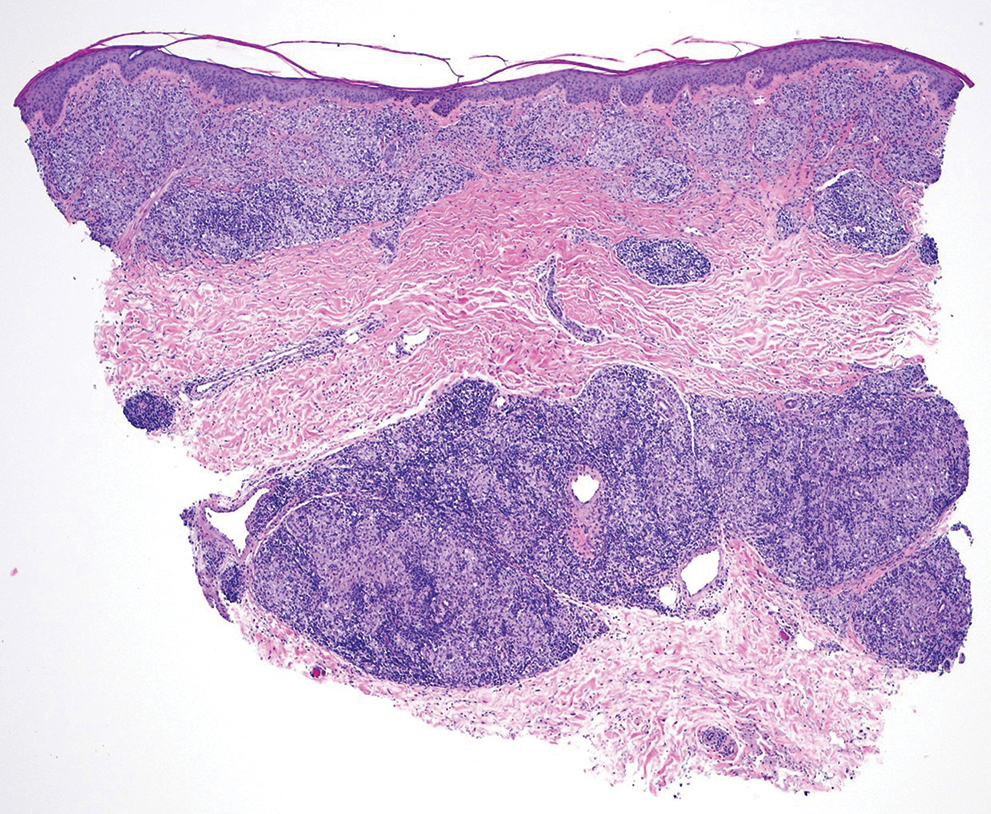
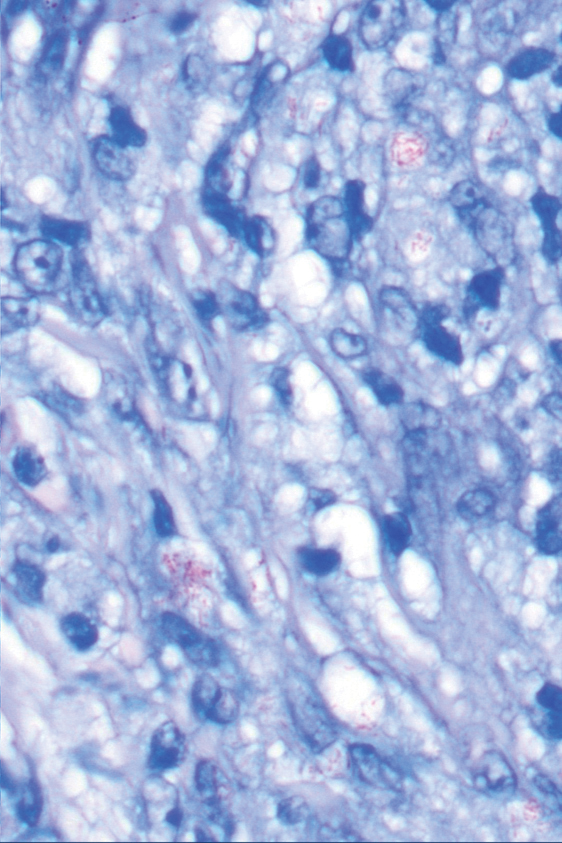
The World Health Organization reported more than 200,000 new leprosy cases globally in 2019, with most occurring in India, Brazil, and Indonesia.1 About 150 to 250 new cases are detected in the United States annually.1 The Ridley-Jopling classification of leprosy divides the condition into 5 categories: tuberculoid, borderline tuberculoid, borderline-borderline (BB), borderline lepromatous, and lepromatous. At one end of the spectrum, tuberculoid leprosy—a predominant Th1 immune response mediated by CD4 lymphocytes, interleukin (IL) 2, and interferon gamma2—is characterized by sharply demarcated erythematous and hypopigmented plaques with raised borders and an annular appearance.2,3 Lesions typically have atrophic and hypopigmented centers that often appear in an asymmetric distribution on the arms and legs.2,3 Histologic features include dermal tuberculoid granulomas with epithelioid cells—some located directly beneath the epidermis and others around deep vessels and nerves3—multinucleated Langerhans giant cells, thickened peripheral nerves with intraneural lymphocytic infiltrates, and granulomas with central necrosis. Fite-Faraco staining exhibits few bacteria.2
Lepromatous leprosy occurs in individuals with impaired T-cell immunity, leading to multiple red-brown nodular infiltrates in the skin and mucous membranes.2,3 Lesions typically are symmetric and favor the face and auricle of the ear.2,3 Histologically, there are bluish-gray foamy macrophages that form diffuse or nodular infiltrates with few lymphocytes,2 with a Grenz zone between the epidermis and dermis. Nerves may show lamination of the perineurium resembling an onion skin.2,3 Immunohistochemistry shows predominant CD8-positive infiltrates with a Th2 response and positive IL-4 and IL-10. Fite-Faraco stain shows numerous mycobacteria arranged in clusters and in histiocytes.2
Tuberculoid leprosy is treated with dapsone 100 mg and rifampin 600 mg once daily for 6 months,4 and lepromatous leprosy is treated with dapsone 100 mg, rifampin 600 mg, and clofazimine 50 mg once daily for 12 months.4 The prognosis for both is good with treatment; erythema and induration of skin lesions may improve within a few months, but residual nerve damage is common, especially in those with advanced disease prior to treatment.2 For direct contacts, a single dose of rifampin may be given.4
Borderline-borderline leprosy manifests with numerous asymmetric annular plaques, as seen in our patient (Figure 3). Histology findings can be variable and often overlap with other forms of leprosy. There can be epithelioid granulomas and only a few acid-fast bacilli (AFB) or diffuse histiocytic aggregates with foamy histiocytes containing large numbers of AFB.3 Nerve involvement is variable but can be severe in the setting of type 1 lepra reaction, which was present in our patient. Type 1 lepra reaction—a type IV cell-mediated allergic hypersensitivity reaction to Mycobacterium leprae antigens—manifests clinically with hyperesthesia, erythema, edema, and subsequent scaling.2 It occurs in up to 30% of patients with borderline leprosy, usually within 12 months of treatment initiation.2 Our patient had considerable edema and erythema of the hands and feet (Figure 4) along with extensive polyneuropathy prior to starting therapy.
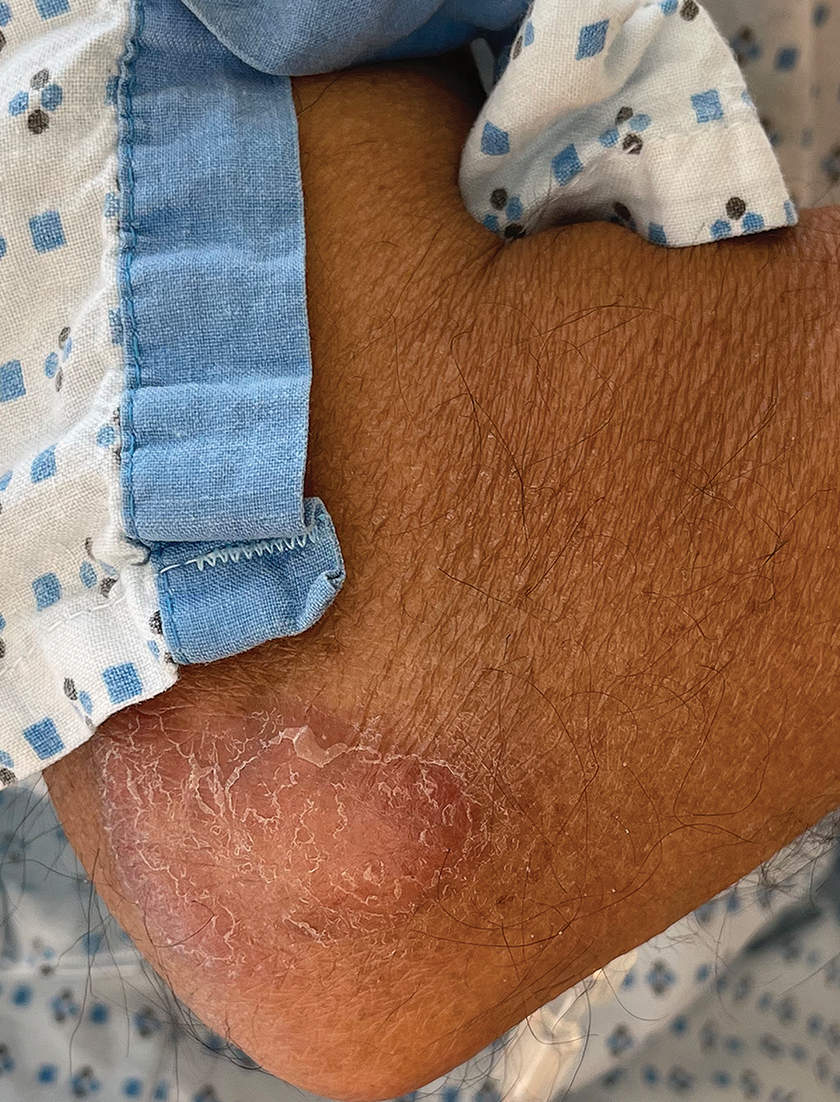
Lucio phenomenon is a rare leprosy reaction found in patients with untreated lepromatous leprosy characterized by erythematous to violaceous macules that lead to ulceronecrotic lesions.5 Histologically, there are many AFB in the vascular endothelium, leukocytoclastic vasculitis, and ischemic epidermal necrosis.5 Our patient did not have ulcerative or necrotic lesions.
The classic skin lesions of psoriasis vulgaris can be described as well-demarcated pink plaques with white or silvery scales that usually are distributed symmetrically and often are found on extensor surfaces.6 Rapidly progressive lesions can be annular with normal skin in the center, mimicking the lesions seen in tuberculoid leprosy. Clinically, both psoriasis and tuberculoid forms of leprosy are sharply demarcated; however, psoriatic lesions often have micaceous overlying scale that is not present in leprosy. Characteristic histologic findings of psoriasis are hyperkeratosis, parakeratosis, and acanthosis of the epidermis with dilated blood vessels and a lymphocytic infiltrate, predominantly into the dermis.7 Psoriatic arthritis has a variable clinical course but tends to emerge 5 to 12 years after initial skin manifestation.8 Classic clinical symptoms include swelling, tenderness, stiffness, and pain in joints and surrounding tissues.8 Other than edema, our patient did not exhibit signs of psoriatic arthritis.
Sarcoidosis is a systemic autoimmune disease characterized by noncaseating epithelioid granulomas affecting various organs, with cutaneous manifestations present in approximately 30% of all cases. Cutaneous manifestations can be variable, including maculopapular lesions, plaques, and nodules.9 Differentiating between cutaneous sarcoidosis and tuberculoid leprosy can be challenging, as both are granulomatous processes; however, histology of sarcoidosis demonstrates noncaseating granulomas in the dermis and/or subcutaneous tissues without AFB9 compared to granulomas with necrotic centers in tuberculoid leprosy.
Cutaneous tuberculosis has variable morphologies. One subtype, lupus vulgaris, can manifest with violaceous, scaly, eroded plaques that could be confused for leprosy. Lupus vulgaris usually results from hematogenous or lymphatic seeding in individuals with high or moderate immunity to M tuberculosis.10
Histologically, the dermis has tuberculoid granulomas containing multinucleated giant cells,10 which can mimic those seen in BB leprosy. Tuberculin skin test results often are positive10; while this test was not performed in our patient, chest radiography was unremarkable, making this diagnosis less likely.
Mycobacterium leprae infections should be considered in a patient with a worsening rash and progressive polyneuropathy. Clinical diagnosis can be challenging due to similarities with other diseases; however, histopathologic findings can help differentiate M leprae from other conditions. This infection is treatable, and early detection can minimize long-term patient morbidity.
- CDC. Hansen’s disease (leprosy). Accessed April 23, 2025. https://www.cdc.gov/leprosy/about/index.html
- Fischer M. Leprosy—an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827.
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020; 83:1-14.
- World Health Organization. Guidelines for the diagnosis, treatment and prevention of leprosy. October 6, 2018. Accessed April 2, 2025. https://www.who.int/publications/i/item/9789290226383
- Frade MAC, Coltro PS, Filho FB, et al. Lucio’s phenomenon: a systematic literature review of definition, clinical features, histopathogenesis and management. Indian J Dermatol Venereol Leprol. 2022;88:464-477.
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Evidence-Based Psoriasis. Springer International Publishing; 2018:1-16.
- Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271.
- Menter A. Psoriasis and psoriatic arthritis overview. Am J Manag Care. 2016;22(8 suppl):S216-S224.
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514.
- Hill MK, Sanders CV. Cutaneous tuberculosis. Microbiol Spectr. 2017;5:1-6.
THE DIAGNOSIS: Borderline-Borderline Leprosy With Type 1 Lepra Reaction
Punch biopsies from plaques on the right elbow and right shin revealed diffuse granulomatous dermatitis (Figure 1) with a narrow Grenz zone in the superficial dermis. The upper dermis contained a dense bandlike infiltrate of histiocytes with abundant foamy-gray cytoplasm and a moderate admixture of lymphocytes. The mid and deep dermis contained a nodular, perivascular, periadnexal, and perineural infiltrate of histiocytes and a dense admixture of lymphocytes. Periodic acid-Schiff and Gram stains were negative for microorganisms. Fite stain was positive for numerous organisms in histiocytes and small dermal nerves (Figure 2). These findings and the clinical examination confirmed a diagnosis of borderline-borderline leprosy with type 1 lepra reaction. The patient was started on dapsone 100 mg, rifampin 600 mg, and clofazimine 100 mg once daily and experienced clinical improvement within 6 months.
The World Health Organization reported more than 200,000 new leprosy cases globally in 2019, with most occurring in India, Brazil, and Indonesia.1 About 150 to 250 new cases are detected in the United States annually.1 The Ridley-Jopling classification of leprosy divides the condition into 5 categories: tuberculoid, borderline tuberculoid, borderline-borderline (BB), borderline lepromatous, and lepromatous. At one end of the spectrum, tuberculoid leprosy—a predominant Th1 immune response mediated by CD4 lymphocytes, interleukin (IL) 2, and interferon gamma2—is characterized by sharply demarcated erythematous and hypopigmented plaques with raised borders and an annular appearance.2,3 Lesions typically have atrophic and hypopigmented centers that often appear in an asymmetric distribution on the arms and legs.2,3 Histologic features include dermal tuberculoid granulomas with epithelioid cells—some located directly beneath the epidermis and others around deep vessels and nerves3—multinucleated Langerhans giant cells, thickened peripheral nerves with intraneural lymphocytic infiltrates, and granulomas with central necrosis. Fite-Faraco staining exhibits few bacteria.2
Lepromatous leprosy occurs in individuals with impaired T-cell immunity, leading to multiple red-brown nodular infiltrates in the skin and mucous membranes.2,3 Lesions typically are symmetric and favor the face and auricle of the ear.2,3 Histologically, there are bluish-gray foamy macrophages that form diffuse or nodular infiltrates with few lymphocytes,2 with a Grenz zone between the epidermis and dermis. Nerves may show lamination of the perineurium resembling an onion skin.2,3 Immunohistochemistry shows predominant CD8-positive infiltrates with a Th2 response and positive IL-4 and IL-10. Fite-Faraco stain shows numerous mycobacteria arranged in clusters and in histiocytes.2
Tuberculoid leprosy is treated with dapsone 100 mg and rifampin 600 mg once daily for 6 months,4 and lepromatous leprosy is treated with dapsone 100 mg, rifampin 600 mg, and clofazimine 50 mg once daily for 12 months.4 The prognosis for both is good with treatment; erythema and induration of skin lesions may improve within a few months, but residual nerve damage is common, especially in those with advanced disease prior to treatment.2 For direct contacts, a single dose of rifampin may be given.4
Borderline-borderline leprosy manifests with numerous asymmetric annular plaques, as seen in our patient (Figure 3). Histology findings can be variable and often overlap with other forms of leprosy. There can be epithelioid granulomas and only a few acid-fast bacilli (AFB) or diffuse histiocytic aggregates with foamy histiocytes containing large numbers of AFB.3 Nerve involvement is variable but can be severe in the setting of type 1 lepra reaction, which was present in our patient. Type 1 lepra reaction—a type IV cell-mediated allergic hypersensitivity reaction to Mycobacterium leprae antigens—manifests clinically with hyperesthesia, erythema, edema, and subsequent scaling.2 It occurs in up to 30% of patients with borderline leprosy, usually within 12 months of treatment initiation.2 Our patient had considerable edema and erythema of the hands and feet (Figure 4) along with extensive polyneuropathy prior to starting therapy.
Lucio phenomenon is a rare leprosy reaction found in patients with untreated lepromatous leprosy characterized by erythematous to violaceous macules that lead to ulceronecrotic lesions.5 Histologically, there are many AFB in the vascular endothelium, leukocytoclastic vasculitis, and ischemic epidermal necrosis.5 Our patient did not have ulcerative or necrotic lesions.
The classic skin lesions of psoriasis vulgaris can be described as well-demarcated pink plaques with white or silvery scales that usually are distributed symmetrically and often are found on extensor surfaces.6 Rapidly progressive lesions can be annular with normal skin in the center, mimicking the lesions seen in tuberculoid leprosy. Clinically, both psoriasis and tuberculoid forms of leprosy are sharply demarcated; however, psoriatic lesions often have micaceous overlying scale that is not present in leprosy. Characteristic histologic findings of psoriasis are hyperkeratosis, parakeratosis, and acanthosis of the epidermis with dilated blood vessels and a lymphocytic infiltrate, predominantly into the dermis.7 Psoriatic arthritis has a variable clinical course but tends to emerge 5 to 12 years after initial skin manifestation.8 Classic clinical symptoms include swelling, tenderness, stiffness, and pain in joints and surrounding tissues.8 Other than edema, our patient did not exhibit signs of psoriatic arthritis.
Sarcoidosis is a systemic autoimmune disease characterized by noncaseating epithelioid granulomas affecting various organs, with cutaneous manifestations present in approximately 30% of all cases. Cutaneous manifestations can be variable, including maculopapular lesions, plaques, and nodules.9 Differentiating between cutaneous sarcoidosis and tuberculoid leprosy can be challenging, as both are granulomatous processes; however, histology of sarcoidosis demonstrates noncaseating granulomas in the dermis and/or subcutaneous tissues without AFB9 compared to granulomas with necrotic centers in tuberculoid leprosy.
Cutaneous tuberculosis has variable morphologies. One subtype, lupus vulgaris, can manifest with violaceous, scaly, eroded plaques that could be confused for leprosy. Lupus vulgaris usually results from hematogenous or lymphatic seeding in individuals with high or moderate immunity to M tuberculosis.10
Histologically, the dermis has tuberculoid granulomas containing multinucleated giant cells,10 which can mimic those seen in BB leprosy. Tuberculin skin test results often are positive10; while this test was not performed in our patient, chest radiography was unremarkable, making this diagnosis less likely.
Mycobacterium leprae infections should be considered in a patient with a worsening rash and progressive polyneuropathy. Clinical diagnosis can be challenging due to similarities with other diseases; however, histopathologic findings can help differentiate M leprae from other conditions. This infection is treatable, and early detection can minimize long-term patient morbidity.
THE DIAGNOSIS: Borderline-Borderline Leprosy With Type 1 Lepra Reaction
Punch biopsies from plaques on the right elbow and right shin revealed diffuse granulomatous dermatitis (Figure 1) with a narrow Grenz zone in the superficial dermis. The upper dermis contained a dense bandlike infiltrate of histiocytes with abundant foamy-gray cytoplasm and a moderate admixture of lymphocytes. The mid and deep dermis contained a nodular, perivascular, periadnexal, and perineural infiltrate of histiocytes and a dense admixture of lymphocytes. Periodic acid-Schiff and Gram stains were negative for microorganisms. Fite stain was positive for numerous organisms in histiocytes and small dermal nerves (Figure 2). These findings and the clinical examination confirmed a diagnosis of borderline-borderline leprosy with type 1 lepra reaction. The patient was started on dapsone 100 mg, rifampin 600 mg, and clofazimine 100 mg once daily and experienced clinical improvement within 6 months.
The World Health Organization reported more than 200,000 new leprosy cases globally in 2019, with most occurring in India, Brazil, and Indonesia.1 About 150 to 250 new cases are detected in the United States annually.1 The Ridley-Jopling classification of leprosy divides the condition into 5 categories: tuberculoid, borderline tuberculoid, borderline-borderline (BB), borderline lepromatous, and lepromatous. At one end of the spectrum, tuberculoid leprosy—a predominant Th1 immune response mediated by CD4 lymphocytes, interleukin (IL) 2, and interferon gamma2—is characterized by sharply demarcated erythematous and hypopigmented plaques with raised borders and an annular appearance.2,3 Lesions typically have atrophic and hypopigmented centers that often appear in an asymmetric distribution on the arms and legs.2,3 Histologic features include dermal tuberculoid granulomas with epithelioid cells—some located directly beneath the epidermis and others around deep vessels and nerves3—multinucleated Langerhans giant cells, thickened peripheral nerves with intraneural lymphocytic infiltrates, and granulomas with central necrosis. Fite-Faraco staining exhibits few bacteria.2
Lepromatous leprosy occurs in individuals with impaired T-cell immunity, leading to multiple red-brown nodular infiltrates in the skin and mucous membranes.2,3 Lesions typically are symmetric and favor the face and auricle of the ear.2,3 Histologically, there are bluish-gray foamy macrophages that form diffuse or nodular infiltrates with few lymphocytes,2 with a Grenz zone between the epidermis and dermis. Nerves may show lamination of the perineurium resembling an onion skin.2,3 Immunohistochemistry shows predominant CD8-positive infiltrates with a Th2 response and positive IL-4 and IL-10. Fite-Faraco stain shows numerous mycobacteria arranged in clusters and in histiocytes.2
Tuberculoid leprosy is treated with dapsone 100 mg and rifampin 600 mg once daily for 6 months,4 and lepromatous leprosy is treated with dapsone 100 mg, rifampin 600 mg, and clofazimine 50 mg once daily for 12 months.4 The prognosis for both is good with treatment; erythema and induration of skin lesions may improve within a few months, but residual nerve damage is common, especially in those with advanced disease prior to treatment.2 For direct contacts, a single dose of rifampin may be given.4
Borderline-borderline leprosy manifests with numerous asymmetric annular plaques, as seen in our patient (Figure 3). Histology findings can be variable and often overlap with other forms of leprosy. There can be epithelioid granulomas and only a few acid-fast bacilli (AFB) or diffuse histiocytic aggregates with foamy histiocytes containing large numbers of AFB.3 Nerve involvement is variable but can be severe in the setting of type 1 lepra reaction, which was present in our patient. Type 1 lepra reaction—a type IV cell-mediated allergic hypersensitivity reaction to Mycobacterium leprae antigens—manifests clinically with hyperesthesia, erythema, edema, and subsequent scaling.2 It occurs in up to 30% of patients with borderline leprosy, usually within 12 months of treatment initiation.2 Our patient had considerable edema and erythema of the hands and feet (Figure 4) along with extensive polyneuropathy prior to starting therapy.
Lucio phenomenon is a rare leprosy reaction found in patients with untreated lepromatous leprosy characterized by erythematous to violaceous macules that lead to ulceronecrotic lesions.5 Histologically, there are many AFB in the vascular endothelium, leukocytoclastic vasculitis, and ischemic epidermal necrosis.5 Our patient did not have ulcerative or necrotic lesions.
The classic skin lesions of psoriasis vulgaris can be described as well-demarcated pink plaques with white or silvery scales that usually are distributed symmetrically and often are found on extensor surfaces.6 Rapidly progressive lesions can be annular with normal skin in the center, mimicking the lesions seen in tuberculoid leprosy. Clinically, both psoriasis and tuberculoid forms of leprosy are sharply demarcated; however, psoriatic lesions often have micaceous overlying scale that is not present in leprosy. Characteristic histologic findings of psoriasis are hyperkeratosis, parakeratosis, and acanthosis of the epidermis with dilated blood vessels and a lymphocytic infiltrate, predominantly into the dermis.7 Psoriatic arthritis has a variable clinical course but tends to emerge 5 to 12 years after initial skin manifestation.8 Classic clinical symptoms include swelling, tenderness, stiffness, and pain in joints and surrounding tissues.8 Other than edema, our patient did not exhibit signs of psoriatic arthritis.
Sarcoidosis is a systemic autoimmune disease characterized by noncaseating epithelioid granulomas affecting various organs, with cutaneous manifestations present in approximately 30% of all cases. Cutaneous manifestations can be variable, including maculopapular lesions, plaques, and nodules.9 Differentiating between cutaneous sarcoidosis and tuberculoid leprosy can be challenging, as both are granulomatous processes; however, histology of sarcoidosis demonstrates noncaseating granulomas in the dermis and/or subcutaneous tissues without AFB9 compared to granulomas with necrotic centers in tuberculoid leprosy.
Cutaneous tuberculosis has variable morphologies. One subtype, lupus vulgaris, can manifest with violaceous, scaly, eroded plaques that could be confused for leprosy. Lupus vulgaris usually results from hematogenous or lymphatic seeding in individuals with high or moderate immunity to M tuberculosis.10
Histologically, the dermis has tuberculoid granulomas containing multinucleated giant cells,10 which can mimic those seen in BB leprosy. Tuberculin skin test results often are positive10; while this test was not performed in our patient, chest radiography was unremarkable, making this diagnosis less likely.
Mycobacterium leprae infections should be considered in a patient with a worsening rash and progressive polyneuropathy. Clinical diagnosis can be challenging due to similarities with other diseases; however, histopathologic findings can help differentiate M leprae from other conditions. This infection is treatable, and early detection can minimize long-term patient morbidity.
- CDC. Hansen’s disease (leprosy). Accessed April 23, 2025. https://www.cdc.gov/leprosy/about/index.html
- Fischer M. Leprosy—an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827.
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020; 83:1-14.
- World Health Organization. Guidelines for the diagnosis, treatment and prevention of leprosy. October 6, 2018. Accessed April 2, 2025. https://www.who.int/publications/i/item/9789290226383
- Frade MAC, Coltro PS, Filho FB, et al. Lucio’s phenomenon: a systematic literature review of definition, clinical features, histopathogenesis and management. Indian J Dermatol Venereol Leprol. 2022;88:464-477.
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Evidence-Based Psoriasis. Springer International Publishing; 2018:1-16.
- Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271.
- Menter A. Psoriasis and psoriatic arthritis overview. Am J Manag Care. 2016;22(8 suppl):S216-S224.
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514.
- Hill MK, Sanders CV. Cutaneous tuberculosis. Microbiol Spectr. 2017;5:1-6.
- CDC. Hansen’s disease (leprosy). Accessed April 23, 2025. https://www.cdc.gov/leprosy/about/index.html
- Fischer M. Leprosy—an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827.
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020; 83:1-14.
- World Health Organization. Guidelines for the diagnosis, treatment and prevention of leprosy. October 6, 2018. Accessed April 2, 2025. https://www.who.int/publications/i/item/9789290226383
- Frade MAC, Coltro PS, Filho FB, et al. Lucio’s phenomenon: a systematic literature review of definition, clinical features, histopathogenesis and management. Indian J Dermatol Venereol Leprol. 2022;88:464-477.
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Evidence-Based Psoriasis. Springer International Publishing; 2018:1-16.
- Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271.
- Menter A. Psoriasis and psoriatic arthritis overview. Am J Manag Care. 2016;22(8 suppl):S216-S224.
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514.
- Hill MK, Sanders CV. Cutaneous tuberculosis. Microbiol Spectr. 2017;5:1-6.
Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
A 67-year-old man presented to his primary care physician with scaly plaques on the extensor surfaces along with distal neuropathy that had been slowly worsening over the past 6 months. The patient was prescribed triamcinolone cream 0.1% twice daily for presumed atopic dermatitis. Three months later, his symptoms rapidly worsened and he developed edema of the hands and feet. He was seen by neurology, and electromyography revealed severe distal sensorimotor neuropathy, prompting hospital admission for further evaluation of a potential rapidly progressive autoimmune disease. Laboratory workup and imaging were ordered, and the patient began an intravenous course of methylprednisolone. Minimal improvement in his symptoms was noted after 1 day, at which time dermatology was consulted.
Physical examination by dermatology revealed well-defined plaques with annular scale on extensor surfaces of the arms and legs, and edema on the hands and feet as well as distal sensorimotor neuropathy. The patient reported associated unspecified weight loss but denied any chest pain, shortness of breath, fevers, chills, cough, night sweats, exposure to chemicals, or recent travel. He reported that he had immigrated from India 37 years prior; his last visit to India was 6 years ago. He currently was taking famotidine for gastrointestinal reflux disease and losartan for hypertension. There was no personal or family history of autoimmune diseases. A complete workup for hematologic, thyroid, liver, and renal function was unremarkable. Initial autoimmune workup was negative for antinuclear antibodies and rheumatoid factor. Serum protein electrophoresis was normal. Results of testing for HIV, hepatitis B, and hepatitis C were negative. Chest radiography was unremarkable. Erythrocyte sedimentation rate and C-reactive protein level were elevated.
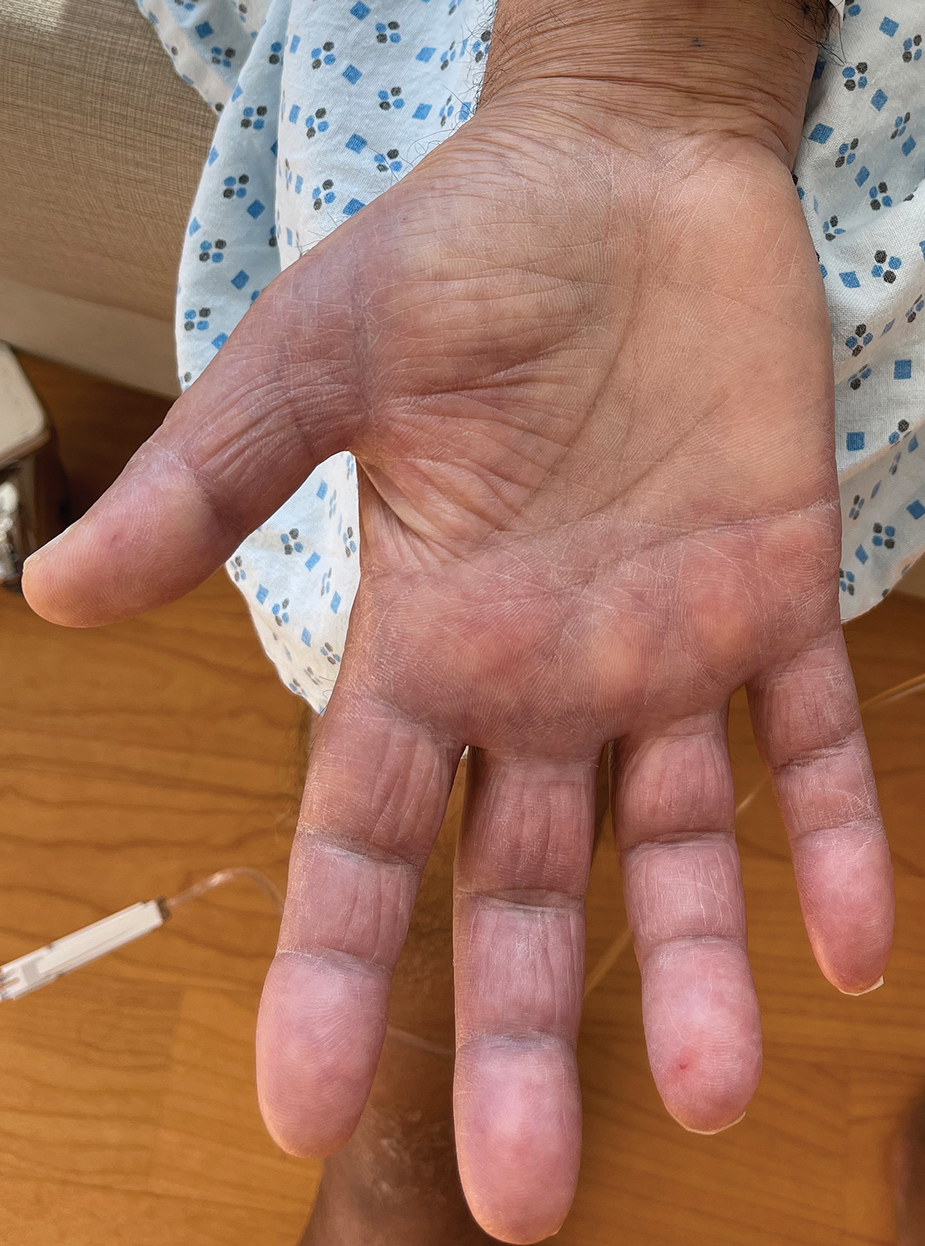
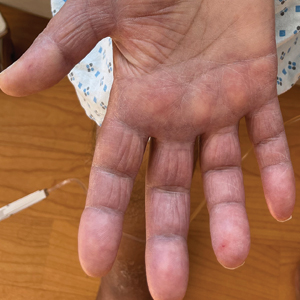
White Atrophic Plaques on the Thighs
White Atrophic Plaques on the Thighs
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
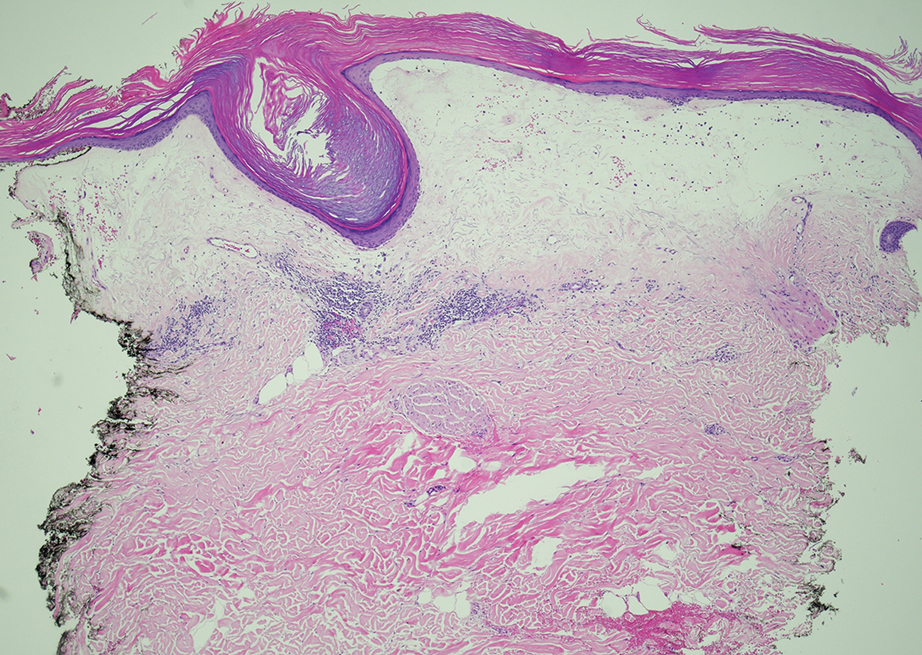
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
White Atrophic Plaques on the Thighs
White Atrophic Plaques on the Thighs
A 71-year-old woman presented to the dermatology clinic for evaluation of intense pruritus of the vaginal region and a nonpruritic rash on the inner thighs of 7 months’ duration. Physical examination revealed white atrophic plaques with scaling and a wrinkled appearance on the inner thighs. White atrophic patches also were noted on the vulva. The patient reported that she had tried over-the-counter antifungals with no improvement. A punch biopsy was performed.
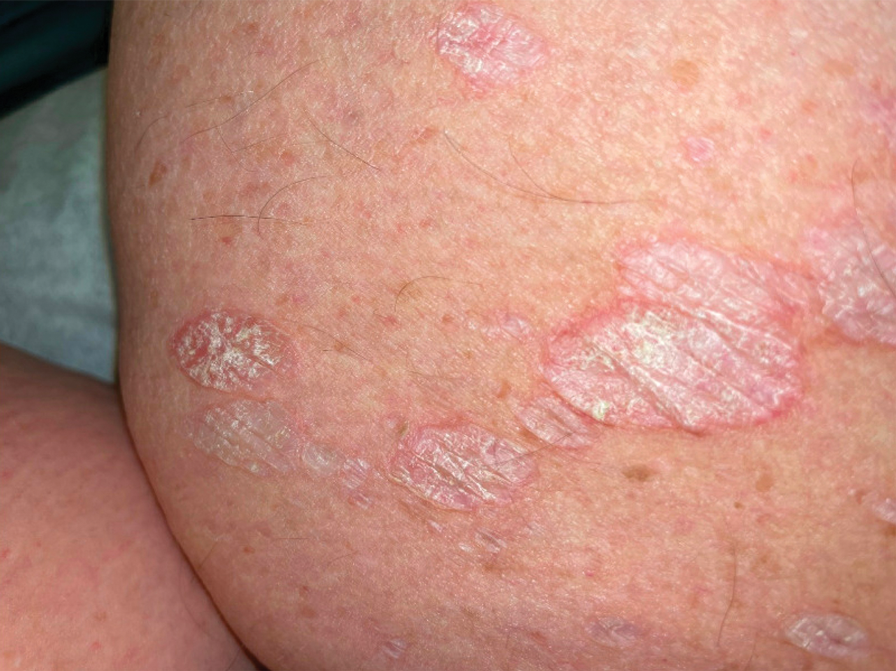

Plaque With Central Ulceration on the Abdomen
Plaque With Central Ulceration on the Abdomen
THE DIAGNOSIS: Plaquelike Myofibroblastic Tumor
An incisional biopsy of the plaque demonstrated a hypercellular proliferation of bland spindle cells in the dermis that infiltrated the subcutis. The overlying epidermis was mildly acanthotic with both ulceration and follicular induction. There was trapping of individual adipocytes in a honeycomb pattern with foci of erythrocyte extravasation, microvesiculation, and widened fibrous septa (Figure 1). Immunohistochemistry was positive for vimentin, actin, and smooth muscle actin (SMA)(Figure 2A). Variable positivity for Factor XIIIa antibodies was noted. CD68 staining was focal positive, suggesting fibrohistiocytic lineage. Expression of CD31, CD34, S100, and anaplastic lymphoma kinase was negative, and Ki-67 was present in less than 10% of cells (Figure 2B).
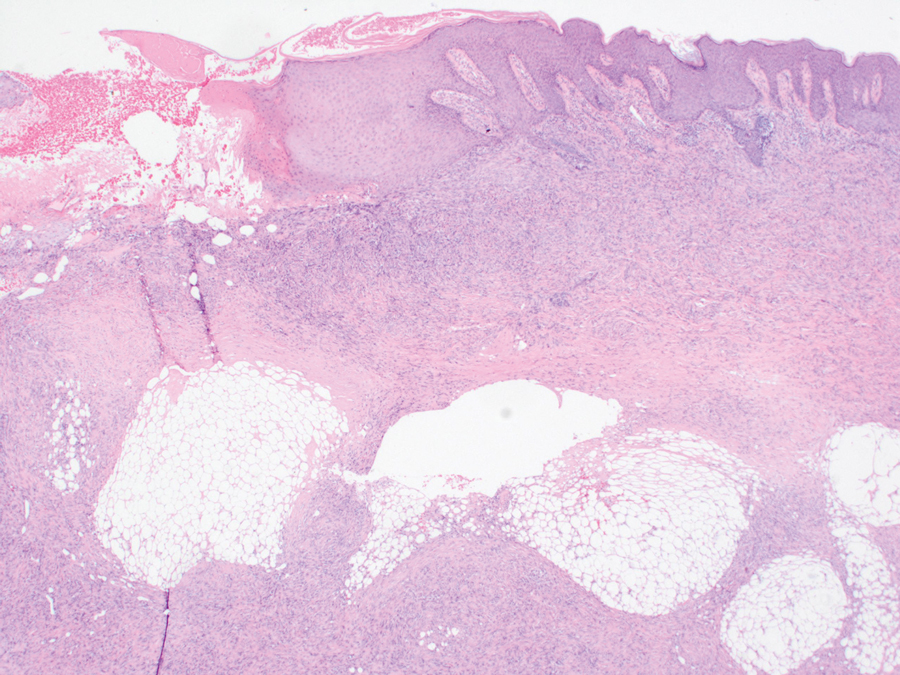
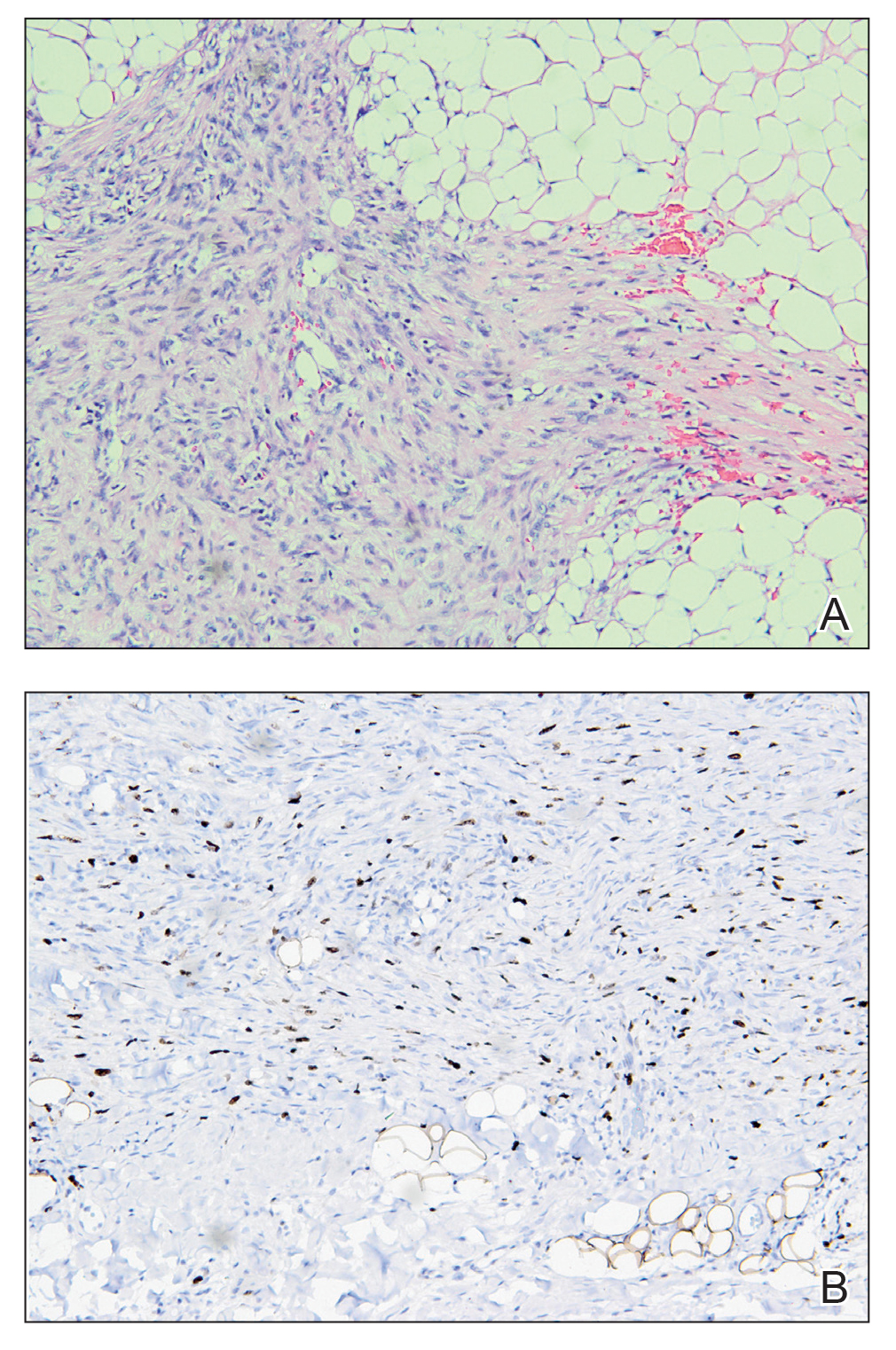
We reviewed the case in conjunction with a soft-tissue pathologist (Y.L.), and based on the clinical and immunophenotypic features, a diagnosis of plaquelike myofibroblastic tumor (PLMT) was made. The patient’s parents refused further treatment, and there was no sign of disease progression at 6-month follow-up.
Plaquelike myofibroblastic tumor is an unusual pediatric dermal tumor that was first described by Clarke et al1 in 2007. Clinical manifestation of PLMT on the right abdomen was unique in our patient, as the lesions typically present as indurated plaques on the lower back, but the central ulceration in our case resembled a report by Marqueling et al.2 Ulceration and induration of PLMT developing at 8 months of age can suggest an aggressive disease course corresponding with deep infiltration and is seen mostly in children.
The histopathologic features of PLMT include an acanthotic epidermis and follicular induction, which also are characteristic of dermatofibroma (DF). The proliferation of spindle cells extended deep into the fat with foci of erythrocyte extravasation and microvesiculation of the stroma similar to nodular fasciitis and proliferative fasciitis. The presentation of infiltrating and expanding fibrous septae and trapping of individual adipocytes in a honeycomb pattern is similar to dermatofibrosarcoma protuberans (DFSP). Most cases of PLMT are positive for SMA. Factor XIIIa typically is variably positive, and in one report, 31% (4/13) of cases showed positive staining for calponin.3 Rapid growth, ulceration, and recurrence emphasize that PLMT can be locally aggressive, similar to DFSP.4
The main differential diagnoses include DF and its variants, dermatomyofibroma, DFSP, and proliferative fasciitis.3,5 In the cases mentioned above, microscopic features were similar with a relatively well-circumscribed proliferation of spindle cells arranged in short fascicles through the entire reticular dermis, and the overlying epidermis was acanthotic.
Dermatofibroma commonly manifests in adults as a minor nodular lesion (commonly <1 cm), and usually is located on the legs. It has several clinical and histologic variants, including multiple clustered DF (MCDF)—a rare condition that has been reported in children and young adults and generally appears in the first and second decades of life. Of the reported cases of MCDF, immunohistochemical staining for SMA was performed in 8 cases. All these cases showed negative or minimal staining.3-5 Smooth muscle actin staining in DFs is negative, or weak and patchy, unlike in PLMT where it is diffuse, uniform, and strong.
Dermatofibrosarcoma protuberans typically occurs in young adults and manifests as dermal and subcutaneous nodular/multinodular or plaquelike masses, with rare congenital cases. Immunohistochemical staining for CD34, which typically is firmly and diffusely positive, is the most reliable marker of DFSP.6 Factor XIIIA in DFSP typically is negative for focal staining, mainly at periphery or in scattered dendritic cells. The prognosis of DFSP generally is excellent, with local recurrences in up to 30% of cases and extremely low metastatic potential (essentially only in cases with fibrosarcomatous transformation).6 Dermatomyofibroma is another rare benign dermal myofibroblastic tumor that typically manifests with indurated hyperpigmented or erythematous plaques or nodules on the shoulders and torso.6 This condition occurs mainly in adolescents and young adults, unlike PLMT. The most striking features of dermatomyofibroma are the horizontal orientation of the spindle cell nuclei and the pattern of the proliferation concerning the adnexal structures, especially hair follicles. The hair follicles have a normal appearance, and the proliferation extends up to each follicle, then continues to the other side without any displacement of the follicle. Tumor cells are variably positive for SMA in dermatomyofibromas and are negative for muscle-specific actin, desmin, S100, CD34, and Factor XIIIA.6
Immunohistochemistry can be very useful in differentiating PLMT from other conditions. Neoplastic cells stain positively for CD34 but not for Factor XIIIa and SMA in cases of DFSP. Dermatofibroma and its variants always present with collagen trapping at the periphery of the lesions and may demonstrate foamy macrophages, hemosiderin, or plasma cells FXIIIA(+), CD34(-), and variable SMA reactivity. This positivity usually is less prominent in DF than in PLMT. Neoplastic cells in dermatomyofibroma often stain positive for calponin, but only focally for SMA. The clinical features of dermatomyofibroma include early onset, large size, multiple nodules, and plaquelike morphology. Moulonguet et al4 hypothesized that, although MCDF and PLMT appear to show some distinctive clinical and histologic features, they also show similarities that could suggest they form part of the myofibroblastic spectrum. Furthermore, Moradi et al7 also considered them as part of the same disease spectrum because of their overlapping clinical, histologic, and immunohistochemical features.
The microscopic features in our case are notable, as the lesion demonstrated overlying acanthosis and follicular induction, resembling DF. The stroma contained microvesicular changes and erythrocyte extravasation, characteristic of nodular or proliferative fasciitis. Additionally, densely packed spindle cells infiltrated deep into the subcutaneous adipose tissue, similar to DFSP.2,3 Our findings expand on the reported histopathologic spectrum of this tumor to date.
- Clarke JT, Clarke LE, Miller C, et al. Plaque-like myofibroblastic tumor of infancy. Pediatr Dermatol. 2007;24:E83-E87. doi:10.1111 /j.1525-1470.2007.00449.x
- Marqueling AL, Dasher D, Friedlander SF, et al. Plaque-like myofibroblastic tumor: report of three cases. Pediatr Dermatol. 2013;30:600-607. doi:10.1111/pde.12185
- Sekar T, Mushtaq J, AlBadry W, et al. Plaque-like myofibroblastic tumor: a series of 2 cases of this unusual dermal tumor which occurs in infancy and early childhood. Pediatr Dev Pathol. 2018;21:444-448. doi: 10.1177/1093526617746807
- Moulonguet I, Biaggi A, Eschard C, et al. Plaque-like myofibroblastic tumor: report of 4 cases. Am J Dermatopathol. 2017;39:767-772. doi: 10.1097/DAD.0000000000000869
- Virdi A, Baraldi C, Barisani A, et al. Plaque-like myofibroblastic tumor, a rare entity of childhood: possible pitfalls in differential diagnosis. J Cutan Pathol. 2019;46:389-392. doi:10.1111/cup.13441
- Cassarino DS. Diagnostic Pathology: Neoplastic Dermatopathology. 2nd ed. Elsevier; 2021.
- Moradi S, Mnayer L, Earle J, et al. Plaque-like dermatofibroma: case report of a rare entity. Dermatopathology (Basel). 2021;8:337-341. doi:10.3390/dermatopathology8030038
THE DIAGNOSIS: Plaquelike Myofibroblastic Tumor
An incisional biopsy of the plaque demonstrated a hypercellular proliferation of bland spindle cells in the dermis that infiltrated the subcutis. The overlying epidermis was mildly acanthotic with both ulceration and follicular induction. There was trapping of individual adipocytes in a honeycomb pattern with foci of erythrocyte extravasation, microvesiculation, and widened fibrous septa (Figure 1). Immunohistochemistry was positive for vimentin, actin, and smooth muscle actin (SMA)(Figure 2A). Variable positivity for Factor XIIIa antibodies was noted. CD68 staining was focal positive, suggesting fibrohistiocytic lineage. Expression of CD31, CD34, S100, and anaplastic lymphoma kinase was negative, and Ki-67 was present in less than 10% of cells (Figure 2B).
We reviewed the case in conjunction with a soft-tissue pathologist (Y.L.), and based on the clinical and immunophenotypic features, a diagnosis of plaquelike myofibroblastic tumor (PLMT) was made. The patient’s parents refused further treatment, and there was no sign of disease progression at 6-month follow-up.
Plaquelike myofibroblastic tumor is an unusual pediatric dermal tumor that was first described by Clarke et al1 in 2007. Clinical manifestation of PLMT on the right abdomen was unique in our patient, as the lesions typically present as indurated plaques on the lower back, but the central ulceration in our case resembled a report by Marqueling et al.2 Ulceration and induration of PLMT developing at 8 months of age can suggest an aggressive disease course corresponding with deep infiltration and is seen mostly in children.
The histopathologic features of PLMT include an acanthotic epidermis and follicular induction, which also are characteristic of dermatofibroma (DF). The proliferation of spindle cells extended deep into the fat with foci of erythrocyte extravasation and microvesiculation of the stroma similar to nodular fasciitis and proliferative fasciitis. The presentation of infiltrating and expanding fibrous septae and trapping of individual adipocytes in a honeycomb pattern is similar to dermatofibrosarcoma protuberans (DFSP). Most cases of PLMT are positive for SMA. Factor XIIIa typically is variably positive, and in one report, 31% (4/13) of cases showed positive staining for calponin.3 Rapid growth, ulceration, and recurrence emphasize that PLMT can be locally aggressive, similar to DFSP.4
The main differential diagnoses include DF and its variants, dermatomyofibroma, DFSP, and proliferative fasciitis.3,5 In the cases mentioned above, microscopic features were similar with a relatively well-circumscribed proliferation of spindle cells arranged in short fascicles through the entire reticular dermis, and the overlying epidermis was acanthotic.
Dermatofibroma commonly manifests in adults as a minor nodular lesion (commonly <1 cm), and usually is located on the legs. It has several clinical and histologic variants, including multiple clustered DF (MCDF)—a rare condition that has been reported in children and young adults and generally appears in the first and second decades of life. Of the reported cases of MCDF, immunohistochemical staining for SMA was performed in 8 cases. All these cases showed negative or minimal staining.3-5 Smooth muscle actin staining in DFs is negative, or weak and patchy, unlike in PLMT where it is diffuse, uniform, and strong.
Dermatofibrosarcoma protuberans typically occurs in young adults and manifests as dermal and subcutaneous nodular/multinodular or plaquelike masses, with rare congenital cases. Immunohistochemical staining for CD34, which typically is firmly and diffusely positive, is the most reliable marker of DFSP.6 Factor XIIIA in DFSP typically is negative for focal staining, mainly at periphery or in scattered dendritic cells. The prognosis of DFSP generally is excellent, with local recurrences in up to 30% of cases and extremely low metastatic potential (essentially only in cases with fibrosarcomatous transformation).6 Dermatomyofibroma is another rare benign dermal myofibroblastic tumor that typically manifests with indurated hyperpigmented or erythematous plaques or nodules on the shoulders and torso.6 This condition occurs mainly in adolescents and young adults, unlike PLMT. The most striking features of dermatomyofibroma are the horizontal orientation of the spindle cell nuclei and the pattern of the proliferation concerning the adnexal structures, especially hair follicles. The hair follicles have a normal appearance, and the proliferation extends up to each follicle, then continues to the other side without any displacement of the follicle. Tumor cells are variably positive for SMA in dermatomyofibromas and are negative for muscle-specific actin, desmin, S100, CD34, and Factor XIIIA.6
Immunohistochemistry can be very useful in differentiating PLMT from other conditions. Neoplastic cells stain positively for CD34 but not for Factor XIIIa and SMA in cases of DFSP. Dermatofibroma and its variants always present with collagen trapping at the periphery of the lesions and may demonstrate foamy macrophages, hemosiderin, or plasma cells FXIIIA(+), CD34(-), and variable SMA reactivity. This positivity usually is less prominent in DF than in PLMT. Neoplastic cells in dermatomyofibroma often stain positive for calponin, but only focally for SMA. The clinical features of dermatomyofibroma include early onset, large size, multiple nodules, and plaquelike morphology. Moulonguet et al4 hypothesized that, although MCDF and PLMT appear to show some distinctive clinical and histologic features, they also show similarities that could suggest they form part of the myofibroblastic spectrum. Furthermore, Moradi et al7 also considered them as part of the same disease spectrum because of their overlapping clinical, histologic, and immunohistochemical features.
The microscopic features in our case are notable, as the lesion demonstrated overlying acanthosis and follicular induction, resembling DF. The stroma contained microvesicular changes and erythrocyte extravasation, characteristic of nodular or proliferative fasciitis. Additionally, densely packed spindle cells infiltrated deep into the subcutaneous adipose tissue, similar to DFSP.2,3 Our findings expand on the reported histopathologic spectrum of this tumor to date.
THE DIAGNOSIS: Plaquelike Myofibroblastic Tumor
An incisional biopsy of the plaque demonstrated a hypercellular proliferation of bland spindle cells in the dermis that infiltrated the subcutis. The overlying epidermis was mildly acanthotic with both ulceration and follicular induction. There was trapping of individual adipocytes in a honeycomb pattern with foci of erythrocyte extravasation, microvesiculation, and widened fibrous septa (Figure 1). Immunohistochemistry was positive for vimentin, actin, and smooth muscle actin (SMA)(Figure 2A). Variable positivity for Factor XIIIa antibodies was noted. CD68 staining was focal positive, suggesting fibrohistiocytic lineage. Expression of CD31, CD34, S100, and anaplastic lymphoma kinase was negative, and Ki-67 was present in less than 10% of cells (Figure 2B).
We reviewed the case in conjunction with a soft-tissue pathologist (Y.L.), and based on the clinical and immunophenotypic features, a diagnosis of plaquelike myofibroblastic tumor (PLMT) was made. The patient’s parents refused further treatment, and there was no sign of disease progression at 6-month follow-up.
Plaquelike myofibroblastic tumor is an unusual pediatric dermal tumor that was first described by Clarke et al1 in 2007. Clinical manifestation of PLMT on the right abdomen was unique in our patient, as the lesions typically present as indurated plaques on the lower back, but the central ulceration in our case resembled a report by Marqueling et al.2 Ulceration and induration of PLMT developing at 8 months of age can suggest an aggressive disease course corresponding with deep infiltration and is seen mostly in children.
The histopathologic features of PLMT include an acanthotic epidermis and follicular induction, which also are characteristic of dermatofibroma (DF). The proliferation of spindle cells extended deep into the fat with foci of erythrocyte extravasation and microvesiculation of the stroma similar to nodular fasciitis and proliferative fasciitis. The presentation of infiltrating and expanding fibrous septae and trapping of individual adipocytes in a honeycomb pattern is similar to dermatofibrosarcoma protuberans (DFSP). Most cases of PLMT are positive for SMA. Factor XIIIa typically is variably positive, and in one report, 31% (4/13) of cases showed positive staining for calponin.3 Rapid growth, ulceration, and recurrence emphasize that PLMT can be locally aggressive, similar to DFSP.4
The main differential diagnoses include DF and its variants, dermatomyofibroma, DFSP, and proliferative fasciitis.3,5 In the cases mentioned above, microscopic features were similar with a relatively well-circumscribed proliferation of spindle cells arranged in short fascicles through the entire reticular dermis, and the overlying epidermis was acanthotic.
Dermatofibroma commonly manifests in adults as a minor nodular lesion (commonly <1 cm), and usually is located on the legs. It has several clinical and histologic variants, including multiple clustered DF (MCDF)—a rare condition that has been reported in children and young adults and generally appears in the first and second decades of life. Of the reported cases of MCDF, immunohistochemical staining for SMA was performed in 8 cases. All these cases showed negative or minimal staining.3-5 Smooth muscle actin staining in DFs is negative, or weak and patchy, unlike in PLMT where it is diffuse, uniform, and strong.
Dermatofibrosarcoma protuberans typically occurs in young adults and manifests as dermal and subcutaneous nodular/multinodular or plaquelike masses, with rare congenital cases. Immunohistochemical staining for CD34, which typically is firmly and diffusely positive, is the most reliable marker of DFSP.6 Factor XIIIA in DFSP typically is negative for focal staining, mainly at periphery or in scattered dendritic cells. The prognosis of DFSP generally is excellent, with local recurrences in up to 30% of cases and extremely low metastatic potential (essentially only in cases with fibrosarcomatous transformation).6 Dermatomyofibroma is another rare benign dermal myofibroblastic tumor that typically manifests with indurated hyperpigmented or erythematous plaques or nodules on the shoulders and torso.6 This condition occurs mainly in adolescents and young adults, unlike PLMT. The most striking features of dermatomyofibroma are the horizontal orientation of the spindle cell nuclei and the pattern of the proliferation concerning the adnexal structures, especially hair follicles. The hair follicles have a normal appearance, and the proliferation extends up to each follicle, then continues to the other side without any displacement of the follicle. Tumor cells are variably positive for SMA in dermatomyofibromas and are negative for muscle-specific actin, desmin, S100, CD34, and Factor XIIIA.6
Immunohistochemistry can be very useful in differentiating PLMT from other conditions. Neoplastic cells stain positively for CD34 but not for Factor XIIIa and SMA in cases of DFSP. Dermatofibroma and its variants always present with collagen trapping at the periphery of the lesions and may demonstrate foamy macrophages, hemosiderin, or plasma cells FXIIIA(+), CD34(-), and variable SMA reactivity. This positivity usually is less prominent in DF than in PLMT. Neoplastic cells in dermatomyofibroma often stain positive for calponin, but only focally for SMA. The clinical features of dermatomyofibroma include early onset, large size, multiple nodules, and plaquelike morphology. Moulonguet et al4 hypothesized that, although MCDF and PLMT appear to show some distinctive clinical and histologic features, they also show similarities that could suggest they form part of the myofibroblastic spectrum. Furthermore, Moradi et al7 also considered them as part of the same disease spectrum because of their overlapping clinical, histologic, and immunohistochemical features.
The microscopic features in our case are notable, as the lesion demonstrated overlying acanthosis and follicular induction, resembling DF. The stroma contained microvesicular changes and erythrocyte extravasation, characteristic of nodular or proliferative fasciitis. Additionally, densely packed spindle cells infiltrated deep into the subcutaneous adipose tissue, similar to DFSP.2,3 Our findings expand on the reported histopathologic spectrum of this tumor to date.
- Clarke JT, Clarke LE, Miller C, et al. Plaque-like myofibroblastic tumor of infancy. Pediatr Dermatol. 2007;24:E83-E87. doi:10.1111 /j.1525-1470.2007.00449.x
- Marqueling AL, Dasher D, Friedlander SF, et al. Plaque-like myofibroblastic tumor: report of three cases. Pediatr Dermatol. 2013;30:600-607. doi:10.1111/pde.12185
- Sekar T, Mushtaq J, AlBadry W, et al. Plaque-like myofibroblastic tumor: a series of 2 cases of this unusual dermal tumor which occurs in infancy and early childhood. Pediatr Dev Pathol. 2018;21:444-448. doi: 10.1177/1093526617746807
- Moulonguet I, Biaggi A, Eschard C, et al. Plaque-like myofibroblastic tumor: report of 4 cases. Am J Dermatopathol. 2017;39:767-772. doi: 10.1097/DAD.0000000000000869
- Virdi A, Baraldi C, Barisani A, et al. Plaque-like myofibroblastic tumor, a rare entity of childhood: possible pitfalls in differential diagnosis. J Cutan Pathol. 2019;46:389-392. doi:10.1111/cup.13441
- Cassarino DS. Diagnostic Pathology: Neoplastic Dermatopathology. 2nd ed. Elsevier; 2021.
- Moradi S, Mnayer L, Earle J, et al. Plaque-like dermatofibroma: case report of a rare entity. Dermatopathology (Basel). 2021;8:337-341. doi:10.3390/dermatopathology8030038
- Clarke JT, Clarke LE, Miller C, et al. Plaque-like myofibroblastic tumor of infancy. Pediatr Dermatol. 2007;24:E83-E87. doi:10.1111 /j.1525-1470.2007.00449.x
- Marqueling AL, Dasher D, Friedlander SF, et al. Plaque-like myofibroblastic tumor: report of three cases. Pediatr Dermatol. 2013;30:600-607. doi:10.1111/pde.12185
- Sekar T, Mushtaq J, AlBadry W, et al. Plaque-like myofibroblastic tumor: a series of 2 cases of this unusual dermal tumor which occurs in infancy and early childhood. Pediatr Dev Pathol. 2018;21:444-448. doi: 10.1177/1093526617746807
- Moulonguet I, Biaggi A, Eschard C, et al. Plaque-like myofibroblastic tumor: report of 4 cases. Am J Dermatopathol. 2017;39:767-772. doi: 10.1097/DAD.0000000000000869
- Virdi A, Baraldi C, Barisani A, et al. Plaque-like myofibroblastic tumor, a rare entity of childhood: possible pitfalls in differential diagnosis. J Cutan Pathol. 2019;46:389-392. doi:10.1111/cup.13441
- Cassarino DS. Diagnostic Pathology: Neoplastic Dermatopathology. 2nd ed. Elsevier; 2021.
- Moradi S, Mnayer L, Earle J, et al. Plaque-like dermatofibroma: case report of a rare entity. Dermatopathology (Basel). 2021;8:337-341. doi:10.3390/dermatopathology8030038
Plaque With Central Ulceration on the Abdomen
Plaque With Central Ulceration on the Abdomen
A 14-month-old girl presented to the dermatology department with a firm asymptomatic lesion on the abdomen of 6 months’ duration. The lesion started as a flesh-colored papule and developed slowly into an indurated plaque that darkened in color. The patient had no history of trauma to the area. Physical examination revealed a dark reddish–brown, indurated, irregularly shaped plaque with central ulceration and elevated borders on the right abdomen. The plaque measured 2×3 cm with a few smaller satellite nodules distributed along the periphery. Abdominal ultrasonography revealed a multinodular proliferation in the dermis and subcutis of the right abdomen.