User login
Nonhealing Ulcer in a Patient With Crohn Disease
The Diagnosis: Mycobacterium abscessus Infection
Upon further testing, cultures were positive for Mycobacterium abscessus. Our patient was referred to infectious disease for co-management, and his treatment plan consisted of intravenous amikacin 885 mg 3 times weekly, intravenous imipenem 1 g twice daily, azithromycin 500 mg/d, and omadacycline 150 mg/d for at least 3 months. Magnetic resonance imaging findings were consistent with a combination of cellulitis and osteomyelitis, and our patient was referred to plastic surgery for debridement. He subsequently was lost to follow-up.
Mycobacterium abscessus is classified as both a nontuberculous and rapidly growing mycobacterium. Mycobacterium abscessus recently has emerged as a pathogen of increasing public health concern, especially due to its high rate of antibiotic resistance.1-5 It is highly prevalent in the environment, and infection has been reported from a wide variety of environmental sources.6-8 Immunocompromised individuals, such as our patient, undergoing anti–tumor necrosis factor therapy are at increased risk for infection from all Mycobacterium species.9-11 Recognizing these infections quickly is a priority for patient care, as M abscessus can lead to disseminated infection and high mortality rates.1
Histopathology of M abscessus consists of granulomatous inflammation with mixed granulomas12; however, these findings are not always appreciable, and staining does not always reveal visible organisms. In our patient, histopathology revealed patchy plasmalymphocytic infiltrates of the dermis and subcutaneous tissue, which are signs of generalized inflammation (Figure). Therefore, cultures positive for M abscessus are the gold standard for diagnosis and established the diagnosis in this case.
.")
The differential diagnoses for our patient’s ulceration included squamous cell carcinoma, pyoderma gangrenosum, aseptic abscess ulcer, and pyodermatitispyostomatitis vegetans. Immunosuppressive therapy is a risk factor for squamous cell carcinoma13,14; however, ulcerated squamous cell carcinoma typically presents with prominent everted edges with a necrotic tumor base.15 Biopsy reveals cells with abundant eosinophilic cytoplasm, large nuclei, and variable keratin pearls.16 Pyoderma gangrenosum is an inflammatory skin condition associated with Crohn disease and often is a diagnosis of exclusion characterized by neutrophilic infiltrates on biopsy.17-19 Aseptic abscess ulcers are characterized by neutrophil-filled lesions that respond to corticosteroids but not antibiotics.20 Pyodermatitis-pyostomatitis vegetans is a rare skin manifestation of inflammatory bowel disease associated with a pustular eruption of the skin and/or mouth. Histopathology reveals pustules within or below the epidermis with many eosinophils or neutrophils. Granulomas do not occur as in M abscessus.21
Treatment of M abscessus infection requires the coadministration of several antibiotics across multiple classes to ensure complete disease resolution. High rates of antibiotic resistance are characterized by at least partial resistance to almost every antibiotic; clarithromycin has near-complete efficacy, but resistant strains have started to emerge. Amikacin and cefoxitin are other antibiotics that have reported a resistance rate of less than 50%, but they are only effective 90% and 70% of the time, respectively.1,22 The antibiotic omadacycline, which is approved by the US Food and Drug Administration to treat acute bacterial skin and soft-tissue infections, also may have utility in treating M abscessus infections.23,24 Finally, phage therapy may offer a potential mode of treatment for this bacterium and was used to treat pulmonary infection in a patient with cystic fibrosis.25 Despite these newer innovations, the current standard of care involves clarithromycin or azithromycin in combination with a parenteral antibiotic such as cefoxitin, amikacin, or imipenem for at least 4 months.1
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus complex. Semin Respir Crit Care Med. 2018;39:362-376.
- Cardenas DD, Yasmin T, Ahmed S. A rare insidious case of skin and soft tissue infection due to Mycobacterium abscessus: a case report. Cureus. 2022;14:E25725.
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-577.
- Dickison P, Howard V, O’Kane G, et al. Mycobacterium abscessus infection following penetrations through wetsuits. Australas J Dermatol. 2019;60:57-59.
- Choi H, Kim YI, Na CH, et al. Mycobacterium abscessus skin infection associated with shaving activity in a 75-year-old man. Ann Geriatr Med Res. 2018;22:204.
- Costa-Silva M, Cesar A, Gomes NP, et al. Mycobacterium abscessus infection in a spa worker. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:159-161.
- Besada E. Rapid growing mycobacteria and TNF-α blockers: case report of a fatal lung infection with Mycobacterium abscessus. Clin Exp Rheumatol. 2011;29:705-707.
- Mufti AH, Toye BW, Mckendry RR, et al. Mycobacterium abscessus infection after use of tumor necrosis factor α inhibitor therapy: case report and review of infectious complications associated with tumor necrosis factor α inhibitor use. Diagn Microbiol Infect Dis. 2005;53:233-238.
- Lee SK, Kim SY, Kim EY, et al. Mycobacterial infections in patients treated with tumor necrosis factor antagonists in South Korea. Lung. 2013;191:565-571.
- Rodríguez G, Ortegón M, Camargo D, et al. Iatrogenic Mycobacterium abscessus infection: histopathology of 71 patients. Br J Dermatol. 1997;137:214-218.
- Firnhaber JM. Diagnosis and treatment of basal cell and squamous cell carcinoma. Am Fam Physician. 2012;86:161-168.
- Walker HS, Hardwicke J. Non-melanoma skin cancer. Surgery (Oxford). 2022;40:39-45.
- Browse NL. The skin. In: Browse NL, ed. An Introduction to the Symptoms and Signs of Surgical Disease. 3rd ed. London Arnold Publications; 2001:66-69.
- Weedon D. Squamous cell carcinoma. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010;691-700.
- Powell F, Schroeter A, Su W, et al. Pyoderma gangrenosum: a review of 86 patients. QJM Int J Med. 1985;55:173-186.
- Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118:743-768.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- André MFJ, Piette JC, Kémény JL, et al. Aseptic abscesses: a study of 30 patients with or without inflammatory bowel disease and review of the literature. Medicine (Baltimore). 2007;86:145. doi:10.1097/md.0b013e18064f9f3
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015;36:67-78.
- Duah M, Beshay M. Omadacycline in first-line combination therapy for pulmonary Mycobacterium abscessus infection: a case series. Int J Infect Dis. 2022;122:953-956.
- Minhas R, Sharma S, Kundu S. Utilizing the promise of omadacycline in a resistant, non-tubercular mycobacterial pulmonary infection. Cureus. 2019;11:E5112.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-733.
The Diagnosis: Mycobacterium abscessus Infection
Upon further testing, cultures were positive for Mycobacterium abscessus. Our patient was referred to infectious disease for co-management, and his treatment plan consisted of intravenous amikacin 885 mg 3 times weekly, intravenous imipenem 1 g twice daily, azithromycin 500 mg/d, and omadacycline 150 mg/d for at least 3 months. Magnetic resonance imaging findings were consistent with a combination of cellulitis and osteomyelitis, and our patient was referred to plastic surgery for debridement. He subsequently was lost to follow-up.
Mycobacterium abscessus is classified as both a nontuberculous and rapidly growing mycobacterium. Mycobacterium abscessus recently has emerged as a pathogen of increasing public health concern, especially due to its high rate of antibiotic resistance.1-5 It is highly prevalent in the environment, and infection has been reported from a wide variety of environmental sources.6-8 Immunocompromised individuals, such as our patient, undergoing anti–tumor necrosis factor therapy are at increased risk for infection from all Mycobacterium species.9-11 Recognizing these infections quickly is a priority for patient care, as M abscessus can lead to disseminated infection and high mortality rates.1
Histopathology of M abscessus consists of granulomatous inflammation with mixed granulomas12; however, these findings are not always appreciable, and staining does not always reveal visible organisms. In our patient, histopathology revealed patchy plasmalymphocytic infiltrates of the dermis and subcutaneous tissue, which are signs of generalized inflammation (Figure). Therefore, cultures positive for M abscessus are the gold standard for diagnosis and established the diagnosis in this case.
The differential diagnoses for our patient’s ulceration included squamous cell carcinoma, pyoderma gangrenosum, aseptic abscess ulcer, and pyodermatitispyostomatitis vegetans. Immunosuppressive therapy is a risk factor for squamous cell carcinoma13,14; however, ulcerated squamous cell carcinoma typically presents with prominent everted edges with a necrotic tumor base.15 Biopsy reveals cells with abundant eosinophilic cytoplasm, large nuclei, and variable keratin pearls.16 Pyoderma gangrenosum is an inflammatory skin condition associated with Crohn disease and often is a diagnosis of exclusion characterized by neutrophilic infiltrates on biopsy.17-19 Aseptic abscess ulcers are characterized by neutrophil-filled lesions that respond to corticosteroids but not antibiotics.20 Pyodermatitis-pyostomatitis vegetans is a rare skin manifestation of inflammatory bowel disease associated with a pustular eruption of the skin and/or mouth. Histopathology reveals pustules within or below the epidermis with many eosinophils or neutrophils. Granulomas do not occur as in M abscessus.21
Treatment of M abscessus infection requires the coadministration of several antibiotics across multiple classes to ensure complete disease resolution. High rates of antibiotic resistance are characterized by at least partial resistance to almost every antibiotic; clarithromycin has near-complete efficacy, but resistant strains have started to emerge. Amikacin and cefoxitin are other antibiotics that have reported a resistance rate of less than 50%, but they are only effective 90% and 70% of the time, respectively.1,22 The antibiotic omadacycline, which is approved by the US Food and Drug Administration to treat acute bacterial skin and soft-tissue infections, also may have utility in treating M abscessus infections.23,24 Finally, phage therapy may offer a potential mode of treatment for this bacterium and was used to treat pulmonary infection in a patient with cystic fibrosis.25 Despite these newer innovations, the current standard of care involves clarithromycin or azithromycin in combination with a parenteral antibiotic such as cefoxitin, amikacin, or imipenem for at least 4 months.1
The Diagnosis: Mycobacterium abscessus Infection
Upon further testing, cultures were positive for Mycobacterium abscessus. Our patient was referred to infectious disease for co-management, and his treatment plan consisted of intravenous amikacin 885 mg 3 times weekly, intravenous imipenem 1 g twice daily, azithromycin 500 mg/d, and omadacycline 150 mg/d for at least 3 months. Magnetic resonance imaging findings were consistent with a combination of cellulitis and osteomyelitis, and our patient was referred to plastic surgery for debridement. He subsequently was lost to follow-up.
Mycobacterium abscessus is classified as both a nontuberculous and rapidly growing mycobacterium. Mycobacterium abscessus recently has emerged as a pathogen of increasing public health concern, especially due to its high rate of antibiotic resistance.1-5 It is highly prevalent in the environment, and infection has been reported from a wide variety of environmental sources.6-8 Immunocompromised individuals, such as our patient, undergoing anti–tumor necrosis factor therapy are at increased risk for infection from all Mycobacterium species.9-11 Recognizing these infections quickly is a priority for patient care, as M abscessus can lead to disseminated infection and high mortality rates.1
Histopathology of M abscessus consists of granulomatous inflammation with mixed granulomas12; however, these findings are not always appreciable, and staining does not always reveal visible organisms. In our patient, histopathology revealed patchy plasmalymphocytic infiltrates of the dermis and subcutaneous tissue, which are signs of generalized inflammation (Figure). Therefore, cultures positive for M abscessus are the gold standard for diagnosis and established the diagnosis in this case.
The differential diagnoses for our patient’s ulceration included squamous cell carcinoma, pyoderma gangrenosum, aseptic abscess ulcer, and pyodermatitispyostomatitis vegetans. Immunosuppressive therapy is a risk factor for squamous cell carcinoma13,14; however, ulcerated squamous cell carcinoma typically presents with prominent everted edges with a necrotic tumor base.15 Biopsy reveals cells with abundant eosinophilic cytoplasm, large nuclei, and variable keratin pearls.16 Pyoderma gangrenosum is an inflammatory skin condition associated with Crohn disease and often is a diagnosis of exclusion characterized by neutrophilic infiltrates on biopsy.17-19 Aseptic abscess ulcers are characterized by neutrophil-filled lesions that respond to corticosteroids but not antibiotics.20 Pyodermatitis-pyostomatitis vegetans is a rare skin manifestation of inflammatory bowel disease associated with a pustular eruption of the skin and/or mouth. Histopathology reveals pustules within or below the epidermis with many eosinophils or neutrophils. Granulomas do not occur as in M abscessus.21
Treatment of M abscessus infection requires the coadministration of several antibiotics across multiple classes to ensure complete disease resolution. High rates of antibiotic resistance are characterized by at least partial resistance to almost every antibiotic; clarithromycin has near-complete efficacy, but resistant strains have started to emerge. Amikacin and cefoxitin are other antibiotics that have reported a resistance rate of less than 50%, but they are only effective 90% and 70% of the time, respectively.1,22 The antibiotic omadacycline, which is approved by the US Food and Drug Administration to treat acute bacterial skin and soft-tissue infections, also may have utility in treating M abscessus infections.23,24 Finally, phage therapy may offer a potential mode of treatment for this bacterium and was used to treat pulmonary infection in a patient with cystic fibrosis.25 Despite these newer innovations, the current standard of care involves clarithromycin or azithromycin in combination with a parenteral antibiotic such as cefoxitin, amikacin, or imipenem for at least 4 months.1
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus complex. Semin Respir Crit Care Med. 2018;39:362-376.
- Cardenas DD, Yasmin T, Ahmed S. A rare insidious case of skin and soft tissue infection due to Mycobacterium abscessus: a case report. Cureus. 2022;14:E25725.
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-577.
- Dickison P, Howard V, O’Kane G, et al. Mycobacterium abscessus infection following penetrations through wetsuits. Australas J Dermatol. 2019;60:57-59.
- Choi H, Kim YI, Na CH, et al. Mycobacterium abscessus skin infection associated with shaving activity in a 75-year-old man. Ann Geriatr Med Res. 2018;22:204.
- Costa-Silva M, Cesar A, Gomes NP, et al. Mycobacterium abscessus infection in a spa worker. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:159-161.
- Besada E. Rapid growing mycobacteria and TNF-α blockers: case report of a fatal lung infection with Mycobacterium abscessus. Clin Exp Rheumatol. 2011;29:705-707.
- Mufti AH, Toye BW, Mckendry RR, et al. Mycobacterium abscessus infection after use of tumor necrosis factor α inhibitor therapy: case report and review of infectious complications associated with tumor necrosis factor α inhibitor use. Diagn Microbiol Infect Dis. 2005;53:233-238.
- Lee SK, Kim SY, Kim EY, et al. Mycobacterial infections in patients treated with tumor necrosis factor antagonists in South Korea. Lung. 2013;191:565-571.
- Rodríguez G, Ortegón M, Camargo D, et al. Iatrogenic Mycobacterium abscessus infection: histopathology of 71 patients. Br J Dermatol. 1997;137:214-218.
- Firnhaber JM. Diagnosis and treatment of basal cell and squamous cell carcinoma. Am Fam Physician. 2012;86:161-168.
- Walker HS, Hardwicke J. Non-melanoma skin cancer. Surgery (Oxford). 2022;40:39-45.
- Browse NL. The skin. In: Browse NL, ed. An Introduction to the Symptoms and Signs of Surgical Disease. 3rd ed. London Arnold Publications; 2001:66-69.
- Weedon D. Squamous cell carcinoma. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010;691-700.
- Powell F, Schroeter A, Su W, et al. Pyoderma gangrenosum: a review of 86 patients. QJM Int J Med. 1985;55:173-186.
- Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118:743-768.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- André MFJ, Piette JC, Kémény JL, et al. Aseptic abscesses: a study of 30 patients with or without inflammatory bowel disease and review of the literature. Medicine (Baltimore). 2007;86:145. doi:10.1097/md.0b013e18064f9f3
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015;36:67-78.
- Duah M, Beshay M. Omadacycline in first-line combination therapy for pulmonary Mycobacterium abscessus infection: a case series. Int J Infect Dis. 2022;122:953-956.
- Minhas R, Sharma S, Kundu S. Utilizing the promise of omadacycline in a resistant, non-tubercular mycobacterial pulmonary infection. Cureus. 2019;11:E5112.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-733.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus complex. Semin Respir Crit Care Med. 2018;39:362-376.
- Cardenas DD, Yasmin T, Ahmed S. A rare insidious case of skin and soft tissue infection due to Mycobacterium abscessus: a case report. Cureus. 2022;14:E25725.
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-577.
- Dickison P, Howard V, O’Kane G, et al. Mycobacterium abscessus infection following penetrations through wetsuits. Australas J Dermatol. 2019;60:57-59.
- Choi H, Kim YI, Na CH, et al. Mycobacterium abscessus skin infection associated with shaving activity in a 75-year-old man. Ann Geriatr Med Res. 2018;22:204.
- Costa-Silva M, Cesar A, Gomes NP, et al. Mycobacterium abscessus infection in a spa worker. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:159-161.
- Besada E. Rapid growing mycobacteria and TNF-α blockers: case report of a fatal lung infection with Mycobacterium abscessus. Clin Exp Rheumatol. 2011;29:705-707.
- Mufti AH, Toye BW, Mckendry RR, et al. Mycobacterium abscessus infection after use of tumor necrosis factor α inhibitor therapy: case report and review of infectious complications associated with tumor necrosis factor α inhibitor use. Diagn Microbiol Infect Dis. 2005;53:233-238.
- Lee SK, Kim SY, Kim EY, et al. Mycobacterial infections in patients treated with tumor necrosis factor antagonists in South Korea. Lung. 2013;191:565-571.
- Rodríguez G, Ortegón M, Camargo D, et al. Iatrogenic Mycobacterium abscessus infection: histopathology of 71 patients. Br J Dermatol. 1997;137:214-218.
- Firnhaber JM. Diagnosis and treatment of basal cell and squamous cell carcinoma. Am Fam Physician. 2012;86:161-168.
- Walker HS, Hardwicke J. Non-melanoma skin cancer. Surgery (Oxford). 2022;40:39-45.
- Browse NL. The skin. In: Browse NL, ed. An Introduction to the Symptoms and Signs of Surgical Disease. 3rd ed. London Arnold Publications; 2001:66-69.
- Weedon D. Squamous cell carcinoma. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010;691-700.
- Powell F, Schroeter A, Su W, et al. Pyoderma gangrenosum: a review of 86 patients. QJM Int J Med. 1985;55:173-186.
- Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118:743-768.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- André MFJ, Piette JC, Kémény JL, et al. Aseptic abscesses: a study of 30 patients with or without inflammatory bowel disease and review of the literature. Medicine (Baltimore). 2007;86:145. doi:10.1097/md.0b013e18064f9f3
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015;36:67-78.
- Duah M, Beshay M. Omadacycline in first-line combination therapy for pulmonary Mycobacterium abscessus infection: a case series. Int J Infect Dis. 2022;122:953-956.
- Minhas R, Sharma S, Kundu S. Utilizing the promise of omadacycline in a resistant, non-tubercular mycobacterial pulmonary infection. Cureus. 2019;11:E5112.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-733.
A 24-year-old man presented to our dermatology clinic with a painful lesion on the right buccal cheek of 4 months’ duration that had not changed in size or appearance. He had a history of Crohn disease that was being treated with 6-mercaptopurine and infliximab. He underwent jaw surgery 7 years prior for correction of an underbite, followed by subsequent surgery to remove the hardware 1 year after the initial procedure. He experienced recurring skin abscesses following the initial jaw surgery roughly once a year that were treated with bedside incision and drainage procedures in the emergency department followed by trimethoprim-sulfamethoxazole with complete resolution; however, treatment with mupirocin ointment 2%, trimethoprim-sulfamethoxazole, and azithromycin did not provide symptomatic relief or resolution for the current lesion. Physical examination revealed a 4-cm ulceration with actively draining serosanguineous discharge. Two punch biopsies were performed; 48-hour bacterial and fungal cultures, as well as Giemsa, acid-fast bacilli, and periodic acid–Schiff staining were negative.


Verrucous Plaque on the Foot
The Diagnosis: Eccrine Poroma
Histopathology demonstrated epidermal thickening, epidermal protrusions, a well-defined mass of tumor cells that extended from the epidermis down to the dermis, and luminal structures. Poroid cells and ovoid nuclei with basophilic cytoplasm also were evident (Figure 1). Dermoscopy showed papillomatous growth, milky-red areas, and dotted vessels (Figure 2). Reflectance confocal microscopy (RCM) at the spinous layer showed hyporefractile, dark, roundish lumina surrounded by keratinocytes (Figure 3). Based on the histologic, dermoscopic, and RCM findings, our patient was diagnosed with eccrine poroma.

Goldman et al1 first described poroma in 1956. Poromas, which include eccrine poroma, are a group of benign cutaneous neoplasms arising from the terminal eccrine or apocrine sweat gland ducts.2 Histologically, poroid cells appear as cuboidal keratinocytes with monomorphous ovoid nuclei and discrete nucleoli.3 They usually appear as nodules or plaques with colors varying from flesh colored to red, brown, or bluish, and they clinically mimic several benign and malignant skin tumors. The differential diagnosis may include keratoacanthoma, plantar wart, verrucous carcinoma, basal cell carcinoma, and squamous cell carcinoma. Poromas can be of eccrine or apocrine origin.4 They also belong to a broad group of neoplasms, including nodular hidradenomas, clear cell hidradenomas, hidroacanthoma simplex, dermal duct tumors, and hidradenomas.5 Four subtypes—poroma, poroid hidradenoma, hidroacanthoma simplex, and dermal duct tumor—have been documented.6 Because poromas have nonspecific and variable clinical presentations, they often are misdiagnosed as other skin neoplasms, and differentiation may be difficult. For example, some cases of poroma present with follicular, sebaceous, and/or apocrine differentiation, leading to difficulty in diagnosis.
.")
Characteristic features of eccrine poroma seen on dermoscopy and RCM have the potential to aid in the diagnosis compared to histopathology. Marchetti et al7 proposed 4 patterns of characteristic dermoscopic findings. Pattern 1 refers to the classic description with bleeding spots, a structureless yellow appearance, milkyred globules, and branched vessels. Patterns 2 and 3 simulate basal cell carcinoma, dermal nevus, or vascular tumors. Pattern 4 refers to tumors that are large in size and resemble keratinizing neoplasms.7 Brugués et al8 described poromas with the following RCM findings: an atypical honeycomb shape that was well separated from the normal epithelium, hyporefractile nests with atypical cells, lack of palisading, and dark holes. One study described RCM parameters as cords without palisading, dark holes, prominent vascularization, and abundant stroma—findings that were positively associated with poroma in a univariate analysis. These findings assist in distinguishing poromas from other conditions in the differential diagnosis.9
.")
There is a substantial overlap in clinical appearance with malignant conditions, including basal cell carcinoma, squamous cell carcinoma, cutaneous metastases, and Paget disease; therefore, the use of dermoscopy and RCM may be helpful in the diagnosis and recognition of specific features, as well as the corresponding patterns of poroma. Poromas commonly display vascularized features due to the variability of dermoscopic patterns of eccrine poroma, and further studies are required to establish the specificity of vascularized features.
Acral lesions are more likely to show the classic clinical features of erythema and exophytic growth. A case of a collision tumor with the verrucous changes of poroma, seborrheic keratosis, and viral wart has been described.10 The verrucous changes may lead to misdiagnosis as plantar warts or other neoplasms. Clinicians also should consider conditions that are induced by friction or trauma. In our patient, dermoscopy and RCM aided in the diagnosis of eccrine poroma due to the interference of prominent overlying verrucous changes.
Treatment of poroma is optional. Deeper lesions can be treated with surgical excision, and superficial lesions may be treated with electrosurgical destruction. Our patient was treated with surgical excision followed by repair of the surgical defect with a double V-Y flap.
- Goldman P, Pinkus H, Rogin JR. Eccrine poroma; tumors exhibiting features of the epidermal sweat duct unit. AMA Arch Derm. 1956; 74:511-521.
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma [published online January 28, 2022]. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Ahmed Jan N, Masood S. Poroma. StatPearls [Internet]. StatPearls Publishing; 2022. https://www.ncbi.nlm.nih.gov/books/NBK560909/
- Casper DJ, Glass LF, Shenefelt PD. An unusually large eccrine poroma: a case report and review of the literature. Cutis. 2011; 88:227-229.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms. Int J Dermatol. 2014;53:1053-1061.
- Betti R, Bombonato C, Cerri A, et al. Unusual sites for poromas are not very unusual: a survey of 101 cases. Clin Exp Dermatol. 2014; 39:119-122.
- Marchetti MA, Marino ML, Virmani P, et al. Dermoscopic features and patterns of poromas: a multicenter observational case-control study conducted by the International Dermoscopy Society (IDS). J Eur Acad Dermatol Venereol. 2018;32:1263-1271.
- Brugués A, Gamboa M, Alós L, et al. The challenging diagnosis of eccrine poromas. J Am Acad Dermatol. 2016;74:E113-E115.
- Di Tullio F, Mandel VD, Ignazio S, et al. The role of reflectance confocal microscopy in the diagnosis of eccrine poroma: a retrospective casecontrol study. Exp Dermatol. 2022;31:1779-1790.
- Bloom BS, Kamino H, Hale CS, et al. Collision tumor of eccrine poroma, seborrheic keratosis, and a viral wart. Dermatol Online J. 2014;20:13030/qt8tm0r9b9.
The Diagnosis: Eccrine Poroma
Histopathology demonstrated epidermal thickening, epidermal protrusions, a well-defined mass of tumor cells that extended from the epidermis down to the dermis, and luminal structures. Poroid cells and ovoid nuclei with basophilic cytoplasm also were evident (Figure 1). Dermoscopy showed papillomatous growth, milky-red areas, and dotted vessels (Figure 2). Reflectance confocal microscopy (RCM) at the spinous layer showed hyporefractile, dark, roundish lumina surrounded by keratinocytes (Figure 3). Based on the histologic, dermoscopic, and RCM findings, our patient was diagnosed with eccrine poroma.
Goldman et al1 first described poroma in 1956. Poromas, which include eccrine poroma, are a group of benign cutaneous neoplasms arising from the terminal eccrine or apocrine sweat gland ducts.2 Histologically, poroid cells appear as cuboidal keratinocytes with monomorphous ovoid nuclei and discrete nucleoli.3 They usually appear as nodules or plaques with colors varying from flesh colored to red, brown, or bluish, and they clinically mimic several benign and malignant skin tumors. The differential diagnosis may include keratoacanthoma, plantar wart, verrucous carcinoma, basal cell carcinoma, and squamous cell carcinoma. Poromas can be of eccrine or apocrine origin.4 They also belong to a broad group of neoplasms, including nodular hidradenomas, clear cell hidradenomas, hidroacanthoma simplex, dermal duct tumors, and hidradenomas.5 Four subtypes—poroma, poroid hidradenoma, hidroacanthoma simplex, and dermal duct tumor—have been documented.6 Because poromas have nonspecific and variable clinical presentations, they often are misdiagnosed as other skin neoplasms, and differentiation may be difficult. For example, some cases of poroma present with follicular, sebaceous, and/or apocrine differentiation, leading to difficulty in diagnosis.
Characteristic features of eccrine poroma seen on dermoscopy and RCM have the potential to aid in the diagnosis compared to histopathology. Marchetti et al7 proposed 4 patterns of characteristic dermoscopic findings. Pattern 1 refers to the classic description with bleeding spots, a structureless yellow appearance, milkyred globules, and branched vessels. Patterns 2 and 3 simulate basal cell carcinoma, dermal nevus, or vascular tumors. Pattern 4 refers to tumors that are large in size and resemble keratinizing neoplasms.7 Brugués et al8 described poromas with the following RCM findings: an atypical honeycomb shape that was well separated from the normal epithelium, hyporefractile nests with atypical cells, lack of palisading, and dark holes. One study described RCM parameters as cords without palisading, dark holes, prominent vascularization, and abundant stroma—findings that were positively associated with poroma in a univariate analysis. These findings assist in distinguishing poromas from other conditions in the differential diagnosis.9
There is a substantial overlap in clinical appearance with malignant conditions, including basal cell carcinoma, squamous cell carcinoma, cutaneous metastases, and Paget disease; therefore, the use of dermoscopy and RCM may be helpful in the diagnosis and recognition of specific features, as well as the corresponding patterns of poroma. Poromas commonly display vascularized features due to the variability of dermoscopic patterns of eccrine poroma, and further studies are required to establish the specificity of vascularized features.
Acral lesions are more likely to show the classic clinical features of erythema and exophytic growth. A case of a collision tumor with the verrucous changes of poroma, seborrheic keratosis, and viral wart has been described.10 The verrucous changes may lead to misdiagnosis as plantar warts or other neoplasms. Clinicians also should consider conditions that are induced by friction or trauma. In our patient, dermoscopy and RCM aided in the diagnosis of eccrine poroma due to the interference of prominent overlying verrucous changes.
Treatment of poroma is optional. Deeper lesions can be treated with surgical excision, and superficial lesions may be treated with electrosurgical destruction. Our patient was treated with surgical excision followed by repair of the surgical defect with a double V-Y flap.
The Diagnosis: Eccrine Poroma
Histopathology demonstrated epidermal thickening, epidermal protrusions, a well-defined mass of tumor cells that extended from the epidermis down to the dermis, and luminal structures. Poroid cells and ovoid nuclei with basophilic cytoplasm also were evident (Figure 1). Dermoscopy showed papillomatous growth, milky-red areas, and dotted vessels (Figure 2). Reflectance confocal microscopy (RCM) at the spinous layer showed hyporefractile, dark, roundish lumina surrounded by keratinocytes (Figure 3). Based on the histologic, dermoscopic, and RCM findings, our patient was diagnosed with eccrine poroma.
Goldman et al1 first described poroma in 1956. Poromas, which include eccrine poroma, are a group of benign cutaneous neoplasms arising from the terminal eccrine or apocrine sweat gland ducts.2 Histologically, poroid cells appear as cuboidal keratinocytes with monomorphous ovoid nuclei and discrete nucleoli.3 They usually appear as nodules or plaques with colors varying from flesh colored to red, brown, or bluish, and they clinically mimic several benign and malignant skin tumors. The differential diagnosis may include keratoacanthoma, plantar wart, verrucous carcinoma, basal cell carcinoma, and squamous cell carcinoma. Poromas can be of eccrine or apocrine origin.4 They also belong to a broad group of neoplasms, including nodular hidradenomas, clear cell hidradenomas, hidroacanthoma simplex, dermal duct tumors, and hidradenomas.5 Four subtypes—poroma, poroid hidradenoma, hidroacanthoma simplex, and dermal duct tumor—have been documented.6 Because poromas have nonspecific and variable clinical presentations, they often are misdiagnosed as other skin neoplasms, and differentiation may be difficult. For example, some cases of poroma present with follicular, sebaceous, and/or apocrine differentiation, leading to difficulty in diagnosis.
Characteristic features of eccrine poroma seen on dermoscopy and RCM have the potential to aid in the diagnosis compared to histopathology. Marchetti et al7 proposed 4 patterns of characteristic dermoscopic findings. Pattern 1 refers to the classic description with bleeding spots, a structureless yellow appearance, milkyred globules, and branched vessels. Patterns 2 and 3 simulate basal cell carcinoma, dermal nevus, or vascular tumors. Pattern 4 refers to tumors that are large in size and resemble keratinizing neoplasms.7 Brugués et al8 described poromas with the following RCM findings: an atypical honeycomb shape that was well separated from the normal epithelium, hyporefractile nests with atypical cells, lack of palisading, and dark holes. One study described RCM parameters as cords without palisading, dark holes, prominent vascularization, and abundant stroma—findings that were positively associated with poroma in a univariate analysis. These findings assist in distinguishing poromas from other conditions in the differential diagnosis.9
There is a substantial overlap in clinical appearance with malignant conditions, including basal cell carcinoma, squamous cell carcinoma, cutaneous metastases, and Paget disease; therefore, the use of dermoscopy and RCM may be helpful in the diagnosis and recognition of specific features, as well as the corresponding patterns of poroma. Poromas commonly display vascularized features due to the variability of dermoscopic patterns of eccrine poroma, and further studies are required to establish the specificity of vascularized features.
Acral lesions are more likely to show the classic clinical features of erythema and exophytic growth. A case of a collision tumor with the verrucous changes of poroma, seborrheic keratosis, and viral wart has been described.10 The verrucous changes may lead to misdiagnosis as plantar warts or other neoplasms. Clinicians also should consider conditions that are induced by friction or trauma. In our patient, dermoscopy and RCM aided in the diagnosis of eccrine poroma due to the interference of prominent overlying verrucous changes.
Treatment of poroma is optional. Deeper lesions can be treated with surgical excision, and superficial lesions may be treated with electrosurgical destruction. Our patient was treated with surgical excision followed by repair of the surgical defect with a double V-Y flap.
- Goldman P, Pinkus H, Rogin JR. Eccrine poroma; tumors exhibiting features of the epidermal sweat duct unit. AMA Arch Derm. 1956; 74:511-521.
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma [published online January 28, 2022]. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Ahmed Jan N, Masood S. Poroma. StatPearls [Internet]. StatPearls Publishing; 2022. https://www.ncbi.nlm.nih.gov/books/NBK560909/
- Casper DJ, Glass LF, Shenefelt PD. An unusually large eccrine poroma: a case report and review of the literature. Cutis. 2011; 88:227-229.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms. Int J Dermatol. 2014;53:1053-1061.
- Betti R, Bombonato C, Cerri A, et al. Unusual sites for poromas are not very unusual: a survey of 101 cases. Clin Exp Dermatol. 2014; 39:119-122.
- Marchetti MA, Marino ML, Virmani P, et al. Dermoscopic features and patterns of poromas: a multicenter observational case-control study conducted by the International Dermoscopy Society (IDS). J Eur Acad Dermatol Venereol. 2018;32:1263-1271.
- Brugués A, Gamboa M, Alós L, et al. The challenging diagnosis of eccrine poromas. J Am Acad Dermatol. 2016;74:E113-E115.
- Di Tullio F, Mandel VD, Ignazio S, et al. The role of reflectance confocal microscopy in the diagnosis of eccrine poroma: a retrospective casecontrol study. Exp Dermatol. 2022;31:1779-1790.
- Bloom BS, Kamino H, Hale CS, et al. Collision tumor of eccrine poroma, seborrheic keratosis, and a viral wart. Dermatol Online J. 2014;20:13030/qt8tm0r9b9.
- Goldman P, Pinkus H, Rogin JR. Eccrine poroma; tumors exhibiting features of the epidermal sweat duct unit. AMA Arch Derm. 1956; 74:511-521.
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma [published online January 28, 2022]. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Ahmed Jan N, Masood S. Poroma. StatPearls [Internet]. StatPearls Publishing; 2022. https://www.ncbi.nlm.nih.gov/books/NBK560909/
- Casper DJ, Glass LF, Shenefelt PD. An unusually large eccrine poroma: a case report and review of the literature. Cutis. 2011; 88:227-229.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms. Int J Dermatol. 2014;53:1053-1061.
- Betti R, Bombonato C, Cerri A, et al. Unusual sites for poromas are not very unusual: a survey of 101 cases. Clin Exp Dermatol. 2014; 39:119-122.
- Marchetti MA, Marino ML, Virmani P, et al. Dermoscopic features and patterns of poromas: a multicenter observational case-control study conducted by the International Dermoscopy Society (IDS). J Eur Acad Dermatol Venereol. 2018;32:1263-1271.
- Brugués A, Gamboa M, Alós L, et al. The challenging diagnosis of eccrine poromas. J Am Acad Dermatol. 2016;74:E113-E115.
- Di Tullio F, Mandel VD, Ignazio S, et al. The role of reflectance confocal microscopy in the diagnosis of eccrine poroma: a retrospective casecontrol study. Exp Dermatol. 2022;31:1779-1790.
- Bloom BS, Kamino H, Hale CS, et al. Collision tumor of eccrine poroma, seborrheic keratosis, and a viral wart. Dermatol Online J. 2014;20:13030/qt8tm0r9b9.
A 62-year-old man presented with an enlarging plaque on the foot of 3 years’ duration. He experienced minor pain while walking but reported no other symptoms. His family history was negative for similar anomalies, and his medical history was negative for the presence of malignant tumors. Physical examination revealed a 2-mm erythematous plaque on the plantar aspect of the right foot with prominent overlying verrucous changes and no ulceration or regional lymphadenopathy. Dermoscopy and reflectance confocal microscopy of the lesion were performed along with a histopathologic examination after complete surgical excision.


Hypotrichosis and Hair Loss on the Occipital Scalp
The Diagnosis: Monilethrix
A diagnosis of monilethrix was rendered based on the clinical and trichoscopic findings. Simple surveillance of the patient’s condition and prevention of further hair trauma were proposed as management options.
Monilethrix is a hair shaft disorder that is inherited in a predominantly autosomal-dominant pattern with variable expressiveness and penetrance resulting from heterozygous mutations in hair keratin genes KRT81, KRT83, and KRT86 in a region of chromosome 12q13.13.1,2 An autosomalrecessive form has been described with mutation in desmoglein 4, but it differs from the classical form by the variable periodicity of the region between the nodules.3,4
The morphologic alteration consists of the formation of fusiform nodules of normal structure alternated with narrow and dystrophic constrictions (Figure). These internodes are fragile areas that cause breakage at constricted points.5 Clinically, monilethrix presents as areas of focal or diffuse alopecia with frequent involvement of the terminal follicles, mainly in areas of friction. The hair is normal at birth due to the predominance of lanugo in the neonatal period, but it subsequently is replaced by abnormal hairs in the first months of life.6 Initial clinical signs begin to appear when the terminal hairs begin to form.7 Although rarer, the eyebrows and eyelashes, as well as the axillary, pubic, and body hair, may be involved.5
.")
Other hair shaft anomalies merit consideration in the differential diagnosis of monilethrix, including pseudomonilethrix, pressure alopecia, trichorrhexis invaginata, ectodermal dysplasia, tinea capitis, and trichothiodystrophy.6 The diagnosis is reached by clinical history and physical examination. Trichoscopy and light microscopy are used to confirm the diagnosis. Trichoscopic examination shows markedly higher rates of anagen hair. The shafts examined in our patient revealed 0.7- to 1-mm intervals between nodes. Hair can be better visualized under a polarized microscope, and the condition can be distinguished from pseudomonilethrix using this approach.5,6 In our patient, the diagnosis was made based on light microscopy and trichoscopic findings with no genetic testing; however, genetic testing for the classic mutations of the keratin genes would be desirable to confirm the diagnosis but was not done in our patient.6 The prognosis of monilethrix is variable; most cases persist into adulthood, though spontaneous improvement may occur with advancing age, during summer, and during pregnancy.8
There is no definitive therapy for monilethrix. Although there have been reports of cases treated with systemic corticosteroids, oral retinoids, topical minoxidil, vitamins, and peeling ointments (desquamative oil), the cornerstone of management is protecting the hair against traumatic procedures such as excessive combing, brushing, and friction, as well as parent and patient education about the benign nature of the condition.9 Additionally, some cases have shown improvement with minoxidil solution at 2% and 5% concentrations, oral minoxidil, or acitretin.7-9
- Fontenelle de Oliveira E, Cotta de Alencar Araripe AL. Monilethrix: a typical case report with microscopic and dermatoscopic findings. An Bras Dermatol. 2015;90:126-127.
- de Cruz R, Horev L, Green J, et al. A novel monilethrix mutation in coil 2A of KRT86 causing autosomal dominant monilethrix with incomplete penetrance. Br J Dermatol. 2012;166(suppl 2):20-26.
- Baltazard T, Dhaille F, Chaby G, et al. Value of dermoscopy for the diagnosis of monilethrix. Dermatol Online J. 2017;23:13030 /qt9hf1p3xm.
- Kato M, Shimizu A, Yokoyama Y, et al. An autosomal recessive mutation of DSG4 causes monilethrix through the ER stress response. J Invest Dermatol. 2015;135:1253-1260.
- Gummer CL, Dawber RP, Swift JA. Monilethrix: an electron microscopic and electron histochemical study. Br J Dermatol. 1981;105:529-541.
- Sharma VK, Chiramel MJ, Rao A. Dermoscopy: a rapid bedside tool to assess monilethrix. Indian J Dermatol Venereol Leprol. 2016;82:73-74.
- Sinclair R. Treatment of monilethrix with oral minoxidil. JAAD Case Rep. 2016;2:212-215.
- Rakowska A, Slowinska M, Czuwara J, et al. Dermoscopy as a tool for rapid diagnosis of monilethrix. J Drugs Dermatol. 2007;6:222-224.
- Karincaoglu Y, Coskun BK, Seyhan ME, et al. Monilethrix. Am J Clin Dermatol. 2005;6:407-410.
The Diagnosis: Monilethrix
A diagnosis of monilethrix was rendered based on the clinical and trichoscopic findings. Simple surveillance of the patient’s condition and prevention of further hair trauma were proposed as management options.
Monilethrix is a hair shaft disorder that is inherited in a predominantly autosomal-dominant pattern with variable expressiveness and penetrance resulting from heterozygous mutations in hair keratin genes KRT81, KRT83, and KRT86 in a region of chromosome 12q13.13.1,2 An autosomalrecessive form has been described with mutation in desmoglein 4, but it differs from the classical form by the variable periodicity of the region between the nodules.3,4
The morphologic alteration consists of the formation of fusiform nodules of normal structure alternated with narrow and dystrophic constrictions (Figure). These internodes are fragile areas that cause breakage at constricted points.5 Clinically, monilethrix presents as areas of focal or diffuse alopecia with frequent involvement of the terminal follicles, mainly in areas of friction. The hair is normal at birth due to the predominance of lanugo in the neonatal period, but it subsequently is replaced by abnormal hairs in the first months of life.6 Initial clinical signs begin to appear when the terminal hairs begin to form.7 Although rarer, the eyebrows and eyelashes, as well as the axillary, pubic, and body hair, may be involved.5
Other hair shaft anomalies merit consideration in the differential diagnosis of monilethrix, including pseudomonilethrix, pressure alopecia, trichorrhexis invaginata, ectodermal dysplasia, tinea capitis, and trichothiodystrophy.6 The diagnosis is reached by clinical history and physical examination. Trichoscopy and light microscopy are used to confirm the diagnosis. Trichoscopic examination shows markedly higher rates of anagen hair. The shafts examined in our patient revealed 0.7- to 1-mm intervals between nodes. Hair can be better visualized under a polarized microscope, and the condition can be distinguished from pseudomonilethrix using this approach.5,6 In our patient, the diagnosis was made based on light microscopy and trichoscopic findings with no genetic testing; however, genetic testing for the classic mutations of the keratin genes would be desirable to confirm the diagnosis but was not done in our patient.6 The prognosis of monilethrix is variable; most cases persist into adulthood, though spontaneous improvement may occur with advancing age, during summer, and during pregnancy.8
There is no definitive therapy for monilethrix. Although there have been reports of cases treated with systemic corticosteroids, oral retinoids, topical minoxidil, vitamins, and peeling ointments (desquamative oil), the cornerstone of management is protecting the hair against traumatic procedures such as excessive combing, brushing, and friction, as well as parent and patient education about the benign nature of the condition.9 Additionally, some cases have shown improvement with minoxidil solution at 2% and 5% concentrations, oral minoxidil, or acitretin.7-9
The Diagnosis: Monilethrix
A diagnosis of monilethrix was rendered based on the clinical and trichoscopic findings. Simple surveillance of the patient’s condition and prevention of further hair trauma were proposed as management options.
Monilethrix is a hair shaft disorder that is inherited in a predominantly autosomal-dominant pattern with variable expressiveness and penetrance resulting from heterozygous mutations in hair keratin genes KRT81, KRT83, and KRT86 in a region of chromosome 12q13.13.1,2 An autosomalrecessive form has been described with mutation in desmoglein 4, but it differs from the classical form by the variable periodicity of the region between the nodules.3,4
The morphologic alteration consists of the formation of fusiform nodules of normal structure alternated with narrow and dystrophic constrictions (Figure). These internodes are fragile areas that cause breakage at constricted points.5 Clinically, monilethrix presents as areas of focal or diffuse alopecia with frequent involvement of the terminal follicles, mainly in areas of friction. The hair is normal at birth due to the predominance of lanugo in the neonatal period, but it subsequently is replaced by abnormal hairs in the first months of life.6 Initial clinical signs begin to appear when the terminal hairs begin to form.7 Although rarer, the eyebrows and eyelashes, as well as the axillary, pubic, and body hair, may be involved.5
Other hair shaft anomalies merit consideration in the differential diagnosis of monilethrix, including pseudomonilethrix, pressure alopecia, trichorrhexis invaginata, ectodermal dysplasia, tinea capitis, and trichothiodystrophy.6 The diagnosis is reached by clinical history and physical examination. Trichoscopy and light microscopy are used to confirm the diagnosis. Trichoscopic examination shows markedly higher rates of anagen hair. The shafts examined in our patient revealed 0.7- to 1-mm intervals between nodes. Hair can be better visualized under a polarized microscope, and the condition can be distinguished from pseudomonilethrix using this approach.5,6 In our patient, the diagnosis was made based on light microscopy and trichoscopic findings with no genetic testing; however, genetic testing for the classic mutations of the keratin genes would be desirable to confirm the diagnosis but was not done in our patient.6 The prognosis of monilethrix is variable; most cases persist into adulthood, though spontaneous improvement may occur with advancing age, during summer, and during pregnancy.8
There is no definitive therapy for monilethrix. Although there have been reports of cases treated with systemic corticosteroids, oral retinoids, topical minoxidil, vitamins, and peeling ointments (desquamative oil), the cornerstone of management is protecting the hair against traumatic procedures such as excessive combing, brushing, and friction, as well as parent and patient education about the benign nature of the condition.9 Additionally, some cases have shown improvement with minoxidil solution at 2% and 5% concentrations, oral minoxidil, or acitretin.7-9
- Fontenelle de Oliveira E, Cotta de Alencar Araripe AL. Monilethrix: a typical case report with microscopic and dermatoscopic findings. An Bras Dermatol. 2015;90:126-127.
- de Cruz R, Horev L, Green J, et al. A novel monilethrix mutation in coil 2A of KRT86 causing autosomal dominant monilethrix with incomplete penetrance. Br J Dermatol. 2012;166(suppl 2):20-26.
- Baltazard T, Dhaille F, Chaby G, et al. Value of dermoscopy for the diagnosis of monilethrix. Dermatol Online J. 2017;23:13030 /qt9hf1p3xm.
- Kato M, Shimizu A, Yokoyama Y, et al. An autosomal recessive mutation of DSG4 causes monilethrix through the ER stress response. J Invest Dermatol. 2015;135:1253-1260.
- Gummer CL, Dawber RP, Swift JA. Monilethrix: an electron microscopic and electron histochemical study. Br J Dermatol. 1981;105:529-541.
- Sharma VK, Chiramel MJ, Rao A. Dermoscopy: a rapid bedside tool to assess monilethrix. Indian J Dermatol Venereol Leprol. 2016;82:73-74.
- Sinclair R. Treatment of monilethrix with oral minoxidil. JAAD Case Rep. 2016;2:212-215.
- Rakowska A, Slowinska M, Czuwara J, et al. Dermoscopy as a tool for rapid diagnosis of monilethrix. J Drugs Dermatol. 2007;6:222-224.
- Karincaoglu Y, Coskun BK, Seyhan ME, et al. Monilethrix. Am J Clin Dermatol. 2005;6:407-410.
- Fontenelle de Oliveira E, Cotta de Alencar Araripe AL. Monilethrix: a typical case report with microscopic and dermatoscopic findings. An Bras Dermatol. 2015;90:126-127.
- de Cruz R, Horev L, Green J, et al. A novel monilethrix mutation in coil 2A of KRT86 causing autosomal dominant monilethrix with incomplete penetrance. Br J Dermatol. 2012;166(suppl 2):20-26.
- Baltazard T, Dhaille F, Chaby G, et al. Value of dermoscopy for the diagnosis of monilethrix. Dermatol Online J. 2017;23:13030 /qt9hf1p3xm.
- Kato M, Shimizu A, Yokoyama Y, et al. An autosomal recessive mutation of DSG4 causes monilethrix through the ER stress response. J Invest Dermatol. 2015;135:1253-1260.
- Gummer CL, Dawber RP, Swift JA. Monilethrix: an electron microscopic and electron histochemical study. Br J Dermatol. 1981;105:529-541.
- Sharma VK, Chiramel MJ, Rao A. Dermoscopy: a rapid bedside tool to assess monilethrix. Indian J Dermatol Venereol Leprol. 2016;82:73-74.
- Sinclair R. Treatment of monilethrix with oral minoxidil. JAAD Case Rep. 2016;2:212-215.
- Rakowska A, Slowinska M, Czuwara J, et al. Dermoscopy as a tool for rapid diagnosis of monilethrix. J Drugs Dermatol. 2007;6:222-224.
- Karincaoglu Y, Coskun BK, Seyhan ME, et al. Monilethrix. Am J Clin Dermatol. 2005;6:407-410.
A 6-month-old infant girl was referred to the dermatology service with hypotrichosis and hair loss on the occipital region of the scalp of 4 months’ duration (top). The patient was born at full term by cesarean delivery without complications. There were no comorbidities or family history of alopecia. Clinical examination revealed an alopecic plaque in the occipital region with broken hairs and some dystrophic hairs associated with follicular papules and perifollicular hyperkeratosis. A hair pull test was positive for telogen hairs. Trichoscopy revealed black dots and broken hairs resembling Morse code (bottom). Hair microscopy showed regular alternation of constriction zones separated by intervals of normal thickness.


Diffuse Pruritic Eruption in an Immunocompromised Patient
The Diagnosis: Scabies Infestation
Direct microscopy revealed the presence of a live scabies mite and numerous eggs (Figure), confirming the diagnosis of a scabies infestation. Scabies, caused by the Sarcoptes scabiei var hominis mite, characteristically presents in adults as pruritic hyperkeratotic plaques of the interdigital web spaces of the hands, flexor surfaces of the wrists and elbows, axillae, male genitalia, and breasts; however, an atypical presentation is common in immunocompromised or immunosuppressed individuals, such as our patient. In children, the palms, soles, and head (ie, face, scalp, neck) are common sites of involvement. Although dermatologists generally are familiar with severe atypical presentations such as Norwegian crusted scabies or bullous scabies, it is important that they are aware of other atypical presentations, such as the diffuse papulonodular variant observed in our patient.1 As such, a low threshold of suspicion for scabies infestations should be employed in immunocompromised patients with new-onset pruritic eruptions.
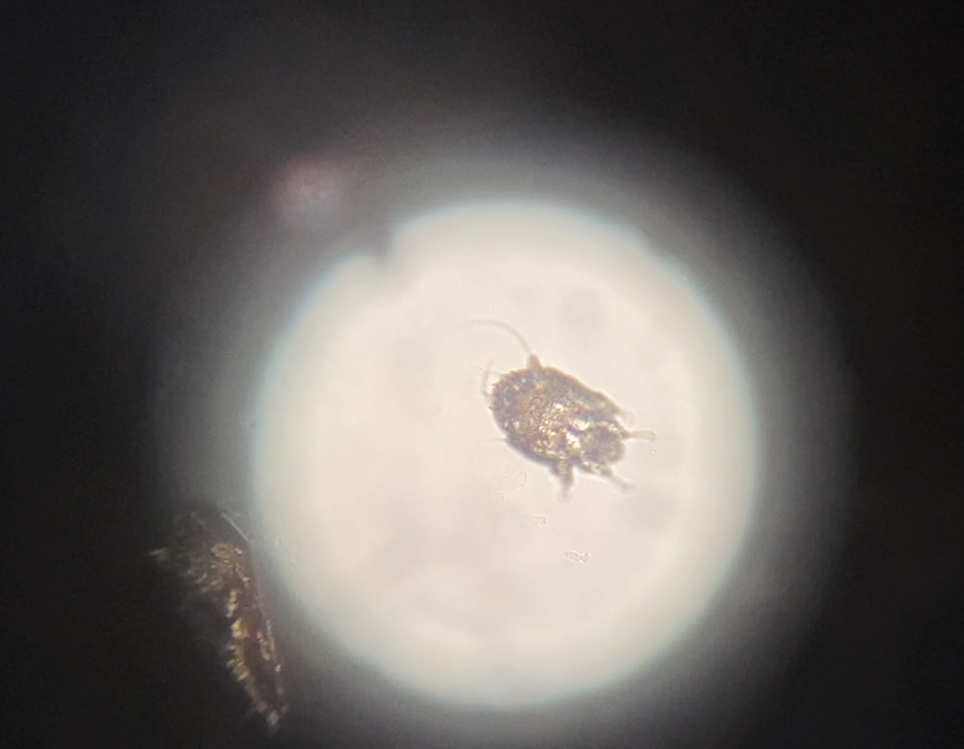
Direct microscopy is widely accepted as the gold standard for the diagnosis of scabies infestations; it is a fast and low-cost diagnostic tool. However, this technique displays variable sensitivity in clinical practice, requiring experience and a skilled hand.1,2 Other more sensitive diagnostic options for suspected scabies infestations include histopathology, serology, and molecular-based techniques such as DNA isolation and polymerase chain reaction. Although these tests do demonstrate greater sensitivity, they also are more invasive, time intensive, and costly.2 Therefore, they typically are not the first choice for a suspected scabies infestation. Dermoscopy has emerged as another tool to aid in the diagnosis of a suspected scabies infestation, enabling visualization of scaly burrows, eggs, and live mites. Classically, findings resembling a delta wing with contrail are seen on dermoscopic examination. The delta wing represents the brown triangular structure of the pigmented scabies mite head and anterior legs; the contrail is the lighter linear structures streaming behind the scabies mite (similar to visible vapor streams occurring behind flying jets), representing the burrow of the mite.
Although treatment of scabies infestations typically can be accomplished with permethrin cream 5%, the diffuse nature of our patient’s lesions in combination with his immunocompromised state made oral therapy a more appropriate choice. Based on Centers for Disease Control and Prevention recommendations, the patient received 2 doses of oral weight-based ivermectin (200 μg/kg per dose) administered 1 week apart.1,3 The initial dose at day 1 serves to eliminate any scabies mites that are present, while the second dose 1 week later eliminates any residual eggs. Our patient experienced complete resolution of the symptoms following this treatment regimen.
It was important to differentiate our patient’s scabies infestation from other intensely pruritic conditions and morphologic mimics including papular urticaria, lichenoid drug eruptions, tinea corporis, and prurigo nodularis. Papular urticaria is an intensely pruritic hypersensitivity reaction to insect bites that commonly affects the extremities or other exposed areas. Visible puncta may be present.4 Our patient’s lesion distribution involved areas covered by clothing, no puncta were present, and he had no history of a recent arthropod assault, making the diagnosis of papular urticaria less likely.
Lichenoid drug eruptions classically present with symmetric, diffuse, pruritic, violaceous, scaling papules and plaques that present 2 to 3 months after exposure to an offending agent.5 Our patient’s eruption was papulonodular with no violaceous plaques, and he did not report changes to his medications, making a lichenoid drug eruption less likely.
Tinea corporis is another intensely pruritic condition that should be considered, especially in immunocompromised patients. It is caused by dermatophytes and classically presents as erythematous pruritic plaques with an annular, advancing, scaling border.6 Although immunocompromised patients may display extensive involvement, our patient’s lesions were papulonodular with no annular morphology or scale, rendering tinea corporis less likely.
Prurigo nodularis is a chronic condition characterized by pruritic, violaceous, dome-shaped, smooth or crusted nodules secondary to repeated scratching or pressure. Although prurigo nodules can develop as a secondary change due to chronic excoriations in scabies infestations, prurigo nodules usually do not develop in areas such as the midline of the back that are not easily reached by the fingernails,7 which made prurigo nodularis less likely in our patient.
This case describes a unique papulonodular variant of scabies presenting in an immunocompromised cancer patient. Timely recognition and diagnosis of atypical scabies infestations can decrease morbidity and improve the quality of life of these patients.
- Chandler DJ, Fuller LC. A review of scabies: an infestation more than skin deep. Dermatology. 2019;235:79-90. doi:10.1159/000495290
- Siddig EE, Hay R. Laboratory-based diagnosis of scabies: a review of the current status. Trans R Soc Trop Med Hyg. 2022;116:4-9. doi:10.1093/trstmh/trab049
- Centers for Disease Control and Prevention. Parasites—scabies. medications. Accessed September 19, 2023. https://www.cdc.gov/parasites/ scabies/health_professionals/meds.html
- Örnek S, Zuberbier T, Kocatürk E. Annular urticarial lesions. Clin Dermatol. 2022;40:480-504. doi:10.1016/j.clindermatol .2021.12.010
- Cheraghlou S, Levy LL. Fixed drug eruptions, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6. doi:10.7573/dic.2020-5-6
- Kwon CD, Khanna R, Williams KA, et al. Diagnostic workup and evaluation of patients with prurigo nodularis. Medicines (Basel). 2019;6:97. doi:10.3390/medicines6040097
The Diagnosis: Scabies Infestation
Direct microscopy revealed the presence of a live scabies mite and numerous eggs (Figure), confirming the diagnosis of a scabies infestation. Scabies, caused by the Sarcoptes scabiei var hominis mite, characteristically presents in adults as pruritic hyperkeratotic plaques of the interdigital web spaces of the hands, flexor surfaces of the wrists and elbows, axillae, male genitalia, and breasts; however, an atypical presentation is common in immunocompromised or immunosuppressed individuals, such as our patient. In children, the palms, soles, and head (ie, face, scalp, neck) are common sites of involvement. Although dermatologists generally are familiar with severe atypical presentations such as Norwegian crusted scabies or bullous scabies, it is important that they are aware of other atypical presentations, such as the diffuse papulonodular variant observed in our patient.1 As such, a low threshold of suspicion for scabies infestations should be employed in immunocompromised patients with new-onset pruritic eruptions.
Direct microscopy is widely accepted as the gold standard for the diagnosis of scabies infestations; it is a fast and low-cost diagnostic tool. However, this technique displays variable sensitivity in clinical practice, requiring experience and a skilled hand.1,2 Other more sensitive diagnostic options for suspected scabies infestations include histopathology, serology, and molecular-based techniques such as DNA isolation and polymerase chain reaction. Although these tests do demonstrate greater sensitivity, they also are more invasive, time intensive, and costly.2 Therefore, they typically are not the first choice for a suspected scabies infestation. Dermoscopy has emerged as another tool to aid in the diagnosis of a suspected scabies infestation, enabling visualization of scaly burrows, eggs, and live mites. Classically, findings resembling a delta wing with contrail are seen on dermoscopic examination. The delta wing represents the brown triangular structure of the pigmented scabies mite head and anterior legs; the contrail is the lighter linear structures streaming behind the scabies mite (similar to visible vapor streams occurring behind flying jets), representing the burrow of the mite.
Although treatment of scabies infestations typically can be accomplished with permethrin cream 5%, the diffuse nature of our patient’s lesions in combination with his immunocompromised state made oral therapy a more appropriate choice. Based on Centers for Disease Control and Prevention recommendations, the patient received 2 doses of oral weight-based ivermectin (200 μg/kg per dose) administered 1 week apart.1,3 The initial dose at day 1 serves to eliminate any scabies mites that are present, while the second dose 1 week later eliminates any residual eggs. Our patient experienced complete resolution of the symptoms following this treatment regimen.
It was important to differentiate our patient’s scabies infestation from other intensely pruritic conditions and morphologic mimics including papular urticaria, lichenoid drug eruptions, tinea corporis, and prurigo nodularis. Papular urticaria is an intensely pruritic hypersensitivity reaction to insect bites that commonly affects the extremities or other exposed areas. Visible puncta may be present.4 Our patient’s lesion distribution involved areas covered by clothing, no puncta were present, and he had no history of a recent arthropod assault, making the diagnosis of papular urticaria less likely.
Lichenoid drug eruptions classically present with symmetric, diffuse, pruritic, violaceous, scaling papules and plaques that present 2 to 3 months after exposure to an offending agent.5 Our patient’s eruption was papulonodular with no violaceous plaques, and he did not report changes to his medications, making a lichenoid drug eruption less likely.
Tinea corporis is another intensely pruritic condition that should be considered, especially in immunocompromised patients. It is caused by dermatophytes and classically presents as erythematous pruritic plaques with an annular, advancing, scaling border.6 Although immunocompromised patients may display extensive involvement, our patient’s lesions were papulonodular with no annular morphology or scale, rendering tinea corporis less likely.
Prurigo nodularis is a chronic condition characterized by pruritic, violaceous, dome-shaped, smooth or crusted nodules secondary to repeated scratching or pressure. Although prurigo nodules can develop as a secondary change due to chronic excoriations in scabies infestations, prurigo nodules usually do not develop in areas such as the midline of the back that are not easily reached by the fingernails,7 which made prurigo nodularis less likely in our patient.
This case describes a unique papulonodular variant of scabies presenting in an immunocompromised cancer patient. Timely recognition and diagnosis of atypical scabies infestations can decrease morbidity and improve the quality of life of these patients.
The Diagnosis: Scabies Infestation
Direct microscopy revealed the presence of a live scabies mite and numerous eggs (Figure), confirming the diagnosis of a scabies infestation. Scabies, caused by the Sarcoptes scabiei var hominis mite, characteristically presents in adults as pruritic hyperkeratotic plaques of the interdigital web spaces of the hands, flexor surfaces of the wrists and elbows, axillae, male genitalia, and breasts; however, an atypical presentation is common in immunocompromised or immunosuppressed individuals, such as our patient. In children, the palms, soles, and head (ie, face, scalp, neck) are common sites of involvement. Although dermatologists generally are familiar with severe atypical presentations such as Norwegian crusted scabies or bullous scabies, it is important that they are aware of other atypical presentations, such as the diffuse papulonodular variant observed in our patient.1 As such, a low threshold of suspicion for scabies infestations should be employed in immunocompromised patients with new-onset pruritic eruptions.
Direct microscopy is widely accepted as the gold standard for the diagnosis of scabies infestations; it is a fast and low-cost diagnostic tool. However, this technique displays variable sensitivity in clinical practice, requiring experience and a skilled hand.1,2 Other more sensitive diagnostic options for suspected scabies infestations include histopathology, serology, and molecular-based techniques such as DNA isolation and polymerase chain reaction. Although these tests do demonstrate greater sensitivity, they also are more invasive, time intensive, and costly.2 Therefore, they typically are not the first choice for a suspected scabies infestation. Dermoscopy has emerged as another tool to aid in the diagnosis of a suspected scabies infestation, enabling visualization of scaly burrows, eggs, and live mites. Classically, findings resembling a delta wing with contrail are seen on dermoscopic examination. The delta wing represents the brown triangular structure of the pigmented scabies mite head and anterior legs; the contrail is the lighter linear structures streaming behind the scabies mite (similar to visible vapor streams occurring behind flying jets), representing the burrow of the mite.
Although treatment of scabies infestations typically can be accomplished with permethrin cream 5%, the diffuse nature of our patient’s lesions in combination with his immunocompromised state made oral therapy a more appropriate choice. Based on Centers for Disease Control and Prevention recommendations, the patient received 2 doses of oral weight-based ivermectin (200 μg/kg per dose) administered 1 week apart.1,3 The initial dose at day 1 serves to eliminate any scabies mites that are present, while the second dose 1 week later eliminates any residual eggs. Our patient experienced complete resolution of the symptoms following this treatment regimen.
It was important to differentiate our patient’s scabies infestation from other intensely pruritic conditions and morphologic mimics including papular urticaria, lichenoid drug eruptions, tinea corporis, and prurigo nodularis. Papular urticaria is an intensely pruritic hypersensitivity reaction to insect bites that commonly affects the extremities or other exposed areas. Visible puncta may be present.4 Our patient’s lesion distribution involved areas covered by clothing, no puncta were present, and he had no history of a recent arthropod assault, making the diagnosis of papular urticaria less likely.
Lichenoid drug eruptions classically present with symmetric, diffuse, pruritic, violaceous, scaling papules and plaques that present 2 to 3 months after exposure to an offending agent.5 Our patient’s eruption was papulonodular with no violaceous plaques, and he did not report changes to his medications, making a lichenoid drug eruption less likely.
Tinea corporis is another intensely pruritic condition that should be considered, especially in immunocompromised patients. It is caused by dermatophytes and classically presents as erythematous pruritic plaques with an annular, advancing, scaling border.6 Although immunocompromised patients may display extensive involvement, our patient’s lesions were papulonodular with no annular morphology or scale, rendering tinea corporis less likely.
Prurigo nodularis is a chronic condition characterized by pruritic, violaceous, dome-shaped, smooth or crusted nodules secondary to repeated scratching or pressure. Although prurigo nodules can develop as a secondary change due to chronic excoriations in scabies infestations, prurigo nodules usually do not develop in areas such as the midline of the back that are not easily reached by the fingernails,7 which made prurigo nodularis less likely in our patient.
This case describes a unique papulonodular variant of scabies presenting in an immunocompromised cancer patient. Timely recognition and diagnosis of atypical scabies infestations can decrease morbidity and improve the quality of life of these patients.
- Chandler DJ, Fuller LC. A review of scabies: an infestation more than skin deep. Dermatology. 2019;235:79-90. doi:10.1159/000495290
- Siddig EE, Hay R. Laboratory-based diagnosis of scabies: a review of the current status. Trans R Soc Trop Med Hyg. 2022;116:4-9. doi:10.1093/trstmh/trab049
- Centers for Disease Control and Prevention. Parasites—scabies. medications. Accessed September 19, 2023. https://www.cdc.gov/parasites/ scabies/health_professionals/meds.html
- Örnek S, Zuberbier T, Kocatürk E. Annular urticarial lesions. Clin Dermatol. 2022;40:480-504. doi:10.1016/j.clindermatol .2021.12.010
- Cheraghlou S, Levy LL. Fixed drug eruptions, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6. doi:10.7573/dic.2020-5-6
- Kwon CD, Khanna R, Williams KA, et al. Diagnostic workup and evaluation of patients with prurigo nodularis. Medicines (Basel). 2019;6:97. doi:10.3390/medicines6040097
- Chandler DJ, Fuller LC. A review of scabies: an infestation more than skin deep. Dermatology. 2019;235:79-90. doi:10.1159/000495290
- Siddig EE, Hay R. Laboratory-based diagnosis of scabies: a review of the current status. Trans R Soc Trop Med Hyg. 2022;116:4-9. doi:10.1093/trstmh/trab049
- Centers for Disease Control and Prevention. Parasites—scabies. medications. Accessed September 19, 2023. https://www.cdc.gov/parasites/ scabies/health_professionals/meds.html
- Örnek S, Zuberbier T, Kocatürk E. Annular urticarial lesions. Clin Dermatol. 2022;40:480-504. doi:10.1016/j.clindermatol .2021.12.010
- Cheraghlou S, Levy LL. Fixed drug eruptions, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6. doi:10.7573/dic.2020-5-6
- Kwon CD, Khanna R, Williams KA, et al. Diagnostic workup and evaluation of patients with prurigo nodularis. Medicines (Basel). 2019;6:97. doi:10.3390/medicines6040097
A 54-year-old man presented to our dermatology clinic for evaluation of a widespread intensely pruritic rash of 4 weeks’ duration. Calamine lotion and oral hydroxyzine provided minimal relief. He was being treated for a myeloproliferative disorder with immunosuppressive therapy consisting of a combination of cladribine, low-dose cytarabine, and fedratinib. Physical examination revealed multiple excoriated papules and indurated nodules on the extensor and flexor surfaces of the arms and legs (top), chest, midline of the back (bottom), and groin. No lesions were noted on the volar aspect of the patient’s wrists or interdigital spaces, and no central puncta or scales were present. He denied any preceding arthropod bites, trauma, new environmental exposures, or changes to his medications. Scrapings from several representative lesions were obtained for mineral oil preparation and microscopic evaluation.


Transient Skin Rippling in an Infant
The Diagnosis: Infantile Transient Smooth Muscle Contraction of the Skin
A diagnosis of infantile transient smooth muscle contraction of the skin (ITSMC) was made based on our patient’s clinical presentation and eliminating the diagnoses in the differential. No treatment ultimately was indicated, as episodes became less frequent over time.
The term infantile transient smooth muscle contraction of the skin was first proposed in 2013 by Torrelo et al,1 who described 9 newborns with episodic skin rippling occasionally associated with exposure to cold or friction. The authors postulated that ITSMC was the result of a transient contraction of the arrector pili smooth muscle fibers of the skin, secondary to autonomic immaturity, primitive reflexes, or smooth muscle hypersensitivity.1 Since this first description, ITSMC has remained a rarely reported and poorly understood phenomenon with rare identified cases in the literature.2,3 Clinical history and examination of infants with intermittent transient skin rippling help to distinguish ITSMC from other diagnoses without the need for biopsy, which is particularly undesirable in the pediatric population.
Congenital smooth muscle hamartoma is a benign proliferation of mature smooth muscle that also can arise from the arrector pili muscles.4 In contrast to ITSMC, a hamartoma does not clear; rather, it persists and grows proportionally with the child and is associated with overlying hyperpigmentation and hypertrichosis. The transient nature of ITSMC may be worrisome for mastocytoma; however, this condition presents as erythematous, yellow, red, or brown macules, papules, plaques, or nodules with a positive Darier sign.5 Although the differential diagnosis includes the shagreen patch characteristic of tuberous sclerosis, this irregular plaque typically is located on the lower back with overlying peau d’orange skin changes, and our patient lacked other features indicative of this condition.6 Becker nevus also remains a consideration in patients with rippled skin, but this entity typically becomes more notable at puberty and is associated with hyperpigmentation and hypertrichosis and is a type of smooth muscle hamartoma.4
Our case highlighted the unusual presentation of ITSMC, a condition that can easily go unrecognized, leading to unnecessary referrals and concern. Familiarity with this benign diagnosis is essential to inform prognosis and guide management.
- Torrelo A, Moreno S, Castro C, et al. Infantile transient smooth muscle contraction of the skin. J Am Acad Dermatol. 2013;69:498-500. doi:10.1016/j.jaad.2013.04.029
- Theodosiou G, Belfrage E, Berggård K, et al. Infantile transient smooth muscle contraction of the skin: a case report and literature review. Eur J Dermatol. 2021;31:260-261. doi:10.1684/ejd.2021.3996
- Topham C, Deacon DC, Bowen A, et al. More than goosebumps: a case of marked skin dimpling in an infant. Pediatr Dermatol. 2019;36:E71-E72. doi:10.1111/pde.13791
- Raboudi A, Litaiem N. Congenital smooth muscle hamartoma. StatPearls. StatPearls Publishing; 2022.
- Leung AKC, Lam JM, Leong KF. Childhood solitary cutaneous mastocytoma: clinical manifestations, diagnosis, evaluation, and management. Curr Pediatr Rev. 2019;15:42-46. doi:10.2174/1573396315666 181120163952
- Bongiorno MA, Nathan N, Oyerinde O, et al. Clinical characteristics of connective tissue nevi in tuberous sclerosis complex with special emphasis on shagreen patches. JAMA Dermatol. 2017;153:660-665. doi:10.1001/jamadermatol.2017.0298
The Diagnosis: Infantile Transient Smooth Muscle Contraction of the Skin
A diagnosis of infantile transient smooth muscle contraction of the skin (ITSMC) was made based on our patient’s clinical presentation and eliminating the diagnoses in the differential. No treatment ultimately was indicated, as episodes became less frequent over time.
The term infantile transient smooth muscle contraction of the skin was first proposed in 2013 by Torrelo et al,1 who described 9 newborns with episodic skin rippling occasionally associated with exposure to cold or friction. The authors postulated that ITSMC was the result of a transient contraction of the arrector pili smooth muscle fibers of the skin, secondary to autonomic immaturity, primitive reflexes, or smooth muscle hypersensitivity.1 Since this first description, ITSMC has remained a rarely reported and poorly understood phenomenon with rare identified cases in the literature.2,3 Clinical history and examination of infants with intermittent transient skin rippling help to distinguish ITSMC from other diagnoses without the need for biopsy, which is particularly undesirable in the pediatric population.
Congenital smooth muscle hamartoma is a benign proliferation of mature smooth muscle that also can arise from the arrector pili muscles.4 In contrast to ITSMC, a hamartoma does not clear; rather, it persists and grows proportionally with the child and is associated with overlying hyperpigmentation and hypertrichosis. The transient nature of ITSMC may be worrisome for mastocytoma; however, this condition presents as erythematous, yellow, red, or brown macules, papules, plaques, or nodules with a positive Darier sign.5 Although the differential diagnosis includes the shagreen patch characteristic of tuberous sclerosis, this irregular plaque typically is located on the lower back with overlying peau d’orange skin changes, and our patient lacked other features indicative of this condition.6 Becker nevus also remains a consideration in patients with rippled skin, but this entity typically becomes more notable at puberty and is associated with hyperpigmentation and hypertrichosis and is a type of smooth muscle hamartoma.4
Our case highlighted the unusual presentation of ITSMC, a condition that can easily go unrecognized, leading to unnecessary referrals and concern. Familiarity with this benign diagnosis is essential to inform prognosis and guide management.
The Diagnosis: Infantile Transient Smooth Muscle Contraction of the Skin
A diagnosis of infantile transient smooth muscle contraction of the skin (ITSMC) was made based on our patient’s clinical presentation and eliminating the diagnoses in the differential. No treatment ultimately was indicated, as episodes became less frequent over time.
The term infantile transient smooth muscle contraction of the skin was first proposed in 2013 by Torrelo et al,1 who described 9 newborns with episodic skin rippling occasionally associated with exposure to cold or friction. The authors postulated that ITSMC was the result of a transient contraction of the arrector pili smooth muscle fibers of the skin, secondary to autonomic immaturity, primitive reflexes, or smooth muscle hypersensitivity.1 Since this first description, ITSMC has remained a rarely reported and poorly understood phenomenon with rare identified cases in the literature.2,3 Clinical history and examination of infants with intermittent transient skin rippling help to distinguish ITSMC from other diagnoses without the need for biopsy, which is particularly undesirable in the pediatric population.
Congenital smooth muscle hamartoma is a benign proliferation of mature smooth muscle that also can arise from the arrector pili muscles.4 In contrast to ITSMC, a hamartoma does not clear; rather, it persists and grows proportionally with the child and is associated with overlying hyperpigmentation and hypertrichosis. The transient nature of ITSMC may be worrisome for mastocytoma; however, this condition presents as erythematous, yellow, red, or brown macules, papules, plaques, or nodules with a positive Darier sign.5 Although the differential diagnosis includes the shagreen patch characteristic of tuberous sclerosis, this irregular plaque typically is located on the lower back with overlying peau d’orange skin changes, and our patient lacked other features indicative of this condition.6 Becker nevus also remains a consideration in patients with rippled skin, but this entity typically becomes more notable at puberty and is associated with hyperpigmentation and hypertrichosis and is a type of smooth muscle hamartoma.4
Our case highlighted the unusual presentation of ITSMC, a condition that can easily go unrecognized, leading to unnecessary referrals and concern. Familiarity with this benign diagnosis is essential to inform prognosis and guide management.
- Torrelo A, Moreno S, Castro C, et al. Infantile transient smooth muscle contraction of the skin. J Am Acad Dermatol. 2013;69:498-500. doi:10.1016/j.jaad.2013.04.029
- Theodosiou G, Belfrage E, Berggård K, et al. Infantile transient smooth muscle contraction of the skin: a case report and literature review. Eur J Dermatol. 2021;31:260-261. doi:10.1684/ejd.2021.3996
- Topham C, Deacon DC, Bowen A, et al. More than goosebumps: a case of marked skin dimpling in an infant. Pediatr Dermatol. 2019;36:E71-E72. doi:10.1111/pde.13791
- Raboudi A, Litaiem N. Congenital smooth muscle hamartoma. StatPearls. StatPearls Publishing; 2022.
- Leung AKC, Lam JM, Leong KF. Childhood solitary cutaneous mastocytoma: clinical manifestations, diagnosis, evaluation, and management. Curr Pediatr Rev. 2019;15:42-46. doi:10.2174/1573396315666 181120163952
- Bongiorno MA, Nathan N, Oyerinde O, et al. Clinical characteristics of connective tissue nevi in tuberous sclerosis complex with special emphasis on shagreen patches. JAMA Dermatol. 2017;153:660-665. doi:10.1001/jamadermatol.2017.0298
- Torrelo A, Moreno S, Castro C, et al. Infantile transient smooth muscle contraction of the skin. J Am Acad Dermatol. 2013;69:498-500. doi:10.1016/j.jaad.2013.04.029
- Theodosiou G, Belfrage E, Berggård K, et al. Infantile transient smooth muscle contraction of the skin: a case report and literature review. Eur J Dermatol. 2021;31:260-261. doi:10.1684/ejd.2021.3996
- Topham C, Deacon DC, Bowen A, et al. More than goosebumps: a case of marked skin dimpling in an infant. Pediatr Dermatol. 2019;36:E71-E72. doi:10.1111/pde.13791
- Raboudi A, Litaiem N. Congenital smooth muscle hamartoma. StatPearls. StatPearls Publishing; 2022.
- Leung AKC, Lam JM, Leong KF. Childhood solitary cutaneous mastocytoma: clinical manifestations, diagnosis, evaluation, and management. Curr Pediatr Rev. 2019;15:42-46. doi:10.2174/1573396315666 181120163952
- Bongiorno MA, Nathan N, Oyerinde O, et al. Clinical characteristics of connective tissue nevi in tuberous sclerosis complex with special emphasis on shagreen patches. JAMA Dermatol. 2017;153:660-665. doi:10.1001/jamadermatol.2017.0298
A healthy, full-term, 5-month-old infant boy presented to dermatology for evaluation of an intermittent, asymptomatic, rippled skin texture of the left thigh that resolved completely between flares. The parents noted fewer than 10 intermittent flares prior to the initial presentation at 5 months. Physical examination of the patient’s skin revealed no epidermal abnormalities, dermatographism, or subcutaneous nodules, and there was no positive Darier sign. A subsequent flare at 9 months of age occurred concurrently with fevers up to 39.4 °C (103 °F), and a corresponding photograph (quiz image) provided by the parents due to the intermittent and transient nature of the condition demonstrated an ill-defined, raised, rippled plaque on the left lateral thigh.


Disseminated Papules and Nodules on the Skin and Oral Mucosa in an Infant
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
.")
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.

Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
A 38-week-old infant boy presented at birth with disseminated, nonblanching, purple to dark red papules and nodules on the skin and oral mucosa. He was born spontaneously after an uncomplicated pregnancy. The mother experienced an episode of oral herpes simplex virus during pregnancy. The infant was otherwise healthy. Laboratory tests including a complete blood cell count and routine serum biochemical analyses were within reference range; however, an infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies. Ophthalmologic and auditory screenings were normal.


Raised Linear Plaques on the Back
The Diagnosis: Flagellate Dermatitis
Upon further questioning by dermatology, the patient noted recent ingestion of shiitake mushrooms, which were not a part of his typical diet. Based on the appearance of the rash in the context of ingesting shiitake mushrooms, our patient was diagnosed with flagellate dermatitis. At 6-week followup, the patient’s rash had resolved spontaneously without further intervention.
Flagellate dermatitis usually appears on the torso as linear whiplike streaks.1 The eruption often is pruritic and may be preceded by severe pruritus. Flagellate dermatitis also is a well-documented complication of bleomycin sulfate therapy with an incidence rate of 8% to 66%.2
Other chemotherapeutic causes include peplomycin, bendamustine, docetaxel, cisplatin, and trastuzumab.3 Flagellate dermatitis also is seen in some patients with dermatomyositis.4 A thorough patient history, including medications and dietary habits, is necessary to differentiate flagellate dermatitis from dermatomyositis.
Flagellate dermatitis, also known as shiitake dermatitis, is observed as erythematous flagellate eruptions involving the trunk or extremities that present within 2 hours to 5 days of handling or consuming undercooked or raw shiitake mushrooms (Lentinula edodes),5,6 as was observed in our patient. Lentinan is the polysaccharide component of the shiitake species and is destabilized by heat.6 Ingestion of polysaccharide is associated with dermatitis, particularly in Japan, China, and Korea; however, the consumption of shiitake mushrooms has increased worldwide, and cases increasingly are reported outside of these typical regions. The rash typically resolves spontaneously; therefore, treatment is supportive. However, more severe symptomatic cases may require courses of topical corticosteroids and antihistamines.6
In our case, the differential diagnosis consisted of acute urticaria, cutaneous dermatomyositis, dermatographism, and maculopapular cutaneous mastocytosis. Acute urticaria displays well-circumscribed edematous papules or plaques, and individual lesions last less than 24 hours. Cutaneous dermatomyositis includes additional systemic manifestations such as fatigue, malaise, and myalgia, as well as involvement of the gastrointestinal, respiratory, or cardiac organs. Dermatographism is evoked by stroking or rubbing of the skin, which results in asymptomatic lesions that persist for 15 to 30 minutes. Cases of maculopapular cutaneous mastocytosis more often are seen in children, and the histamine release most often causes gastrointestinal tract symptoms such as nausea, vomiting, and diarrhea, as well as flushing, blushing, pruritus, respiratory difficulty, and malaise.
- Biswas A, Chaudhari PB, Sharma P, et al. Bleomycin induced flagellate erythema: revisiting a unique complication. J Cancer Res Ther. 2013;9:500-503.
- Yagoda A, Mukherji B, Young C, et al. Bleomycin, an anti-tumor antibiotic: clinical experience in 274 patients. Ann Intern Med. 1972;77:861-870.
- Cohen PR. Trastuzumab-associated flagellate erythema: report in a woman with metastatic breast cancer and review of antineoplastic therapy-induced flagellate dermatoses. Dermatol Ther (Heidelb). 2015;5:253-264. doi:10.1007/s13555-015-0085-2
- Grynszpan R, Niemeyer-Corbellini JP, Lopes MS, et al. Bleomycininduced flagellate dermatitis. BMJ Case Rep. 2013;2013:bcr2013009764. doi:10.1136/bcr-2013-009764
- Stephany MP, Chung S, Handler MZ, et al. Shiitake mushroom dermatitis: a review. Am J Clin Dermatol. 2016;17:485-489.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
The Diagnosis: Flagellate Dermatitis
Upon further questioning by dermatology, the patient noted recent ingestion of shiitake mushrooms, which were not a part of his typical diet. Based on the appearance of the rash in the context of ingesting shiitake mushrooms, our patient was diagnosed with flagellate dermatitis. At 6-week followup, the patient’s rash had resolved spontaneously without further intervention.
Flagellate dermatitis usually appears on the torso as linear whiplike streaks.1 The eruption often is pruritic and may be preceded by severe pruritus. Flagellate dermatitis also is a well-documented complication of bleomycin sulfate therapy with an incidence rate of 8% to 66%.2
Other chemotherapeutic causes include peplomycin, bendamustine, docetaxel, cisplatin, and trastuzumab.3 Flagellate dermatitis also is seen in some patients with dermatomyositis.4 A thorough patient history, including medications and dietary habits, is necessary to differentiate flagellate dermatitis from dermatomyositis.
Flagellate dermatitis, also known as shiitake dermatitis, is observed as erythematous flagellate eruptions involving the trunk or extremities that present within 2 hours to 5 days of handling or consuming undercooked or raw shiitake mushrooms (Lentinula edodes),5,6 as was observed in our patient. Lentinan is the polysaccharide component of the shiitake species and is destabilized by heat.6 Ingestion of polysaccharide is associated with dermatitis, particularly in Japan, China, and Korea; however, the consumption of shiitake mushrooms has increased worldwide, and cases increasingly are reported outside of these typical regions. The rash typically resolves spontaneously; therefore, treatment is supportive. However, more severe symptomatic cases may require courses of topical corticosteroids and antihistamines.6
In our case, the differential diagnosis consisted of acute urticaria, cutaneous dermatomyositis, dermatographism, and maculopapular cutaneous mastocytosis. Acute urticaria displays well-circumscribed edematous papules or plaques, and individual lesions last less than 24 hours. Cutaneous dermatomyositis includes additional systemic manifestations such as fatigue, malaise, and myalgia, as well as involvement of the gastrointestinal, respiratory, or cardiac organs. Dermatographism is evoked by stroking or rubbing of the skin, which results in asymptomatic lesions that persist for 15 to 30 minutes. Cases of maculopapular cutaneous mastocytosis more often are seen in children, and the histamine release most often causes gastrointestinal tract symptoms such as nausea, vomiting, and diarrhea, as well as flushing, blushing, pruritus, respiratory difficulty, and malaise.
The Diagnosis: Flagellate Dermatitis
Upon further questioning by dermatology, the patient noted recent ingestion of shiitake mushrooms, which were not a part of his typical diet. Based on the appearance of the rash in the context of ingesting shiitake mushrooms, our patient was diagnosed with flagellate dermatitis. At 6-week followup, the patient’s rash had resolved spontaneously without further intervention.
Flagellate dermatitis usually appears on the torso as linear whiplike streaks.1 The eruption often is pruritic and may be preceded by severe pruritus. Flagellate dermatitis also is a well-documented complication of bleomycin sulfate therapy with an incidence rate of 8% to 66%.2
Other chemotherapeutic causes include peplomycin, bendamustine, docetaxel, cisplatin, and trastuzumab.3 Flagellate dermatitis also is seen in some patients with dermatomyositis.4 A thorough patient history, including medications and dietary habits, is necessary to differentiate flagellate dermatitis from dermatomyositis.
Flagellate dermatitis, also known as shiitake dermatitis, is observed as erythematous flagellate eruptions involving the trunk or extremities that present within 2 hours to 5 days of handling or consuming undercooked or raw shiitake mushrooms (Lentinula edodes),5,6 as was observed in our patient. Lentinan is the polysaccharide component of the shiitake species and is destabilized by heat.6 Ingestion of polysaccharide is associated with dermatitis, particularly in Japan, China, and Korea; however, the consumption of shiitake mushrooms has increased worldwide, and cases increasingly are reported outside of these typical regions. The rash typically resolves spontaneously; therefore, treatment is supportive. However, more severe symptomatic cases may require courses of topical corticosteroids and antihistamines.6
In our case, the differential diagnosis consisted of acute urticaria, cutaneous dermatomyositis, dermatographism, and maculopapular cutaneous mastocytosis. Acute urticaria displays well-circumscribed edematous papules or plaques, and individual lesions last less than 24 hours. Cutaneous dermatomyositis includes additional systemic manifestations such as fatigue, malaise, and myalgia, as well as involvement of the gastrointestinal, respiratory, or cardiac organs. Dermatographism is evoked by stroking or rubbing of the skin, which results in asymptomatic lesions that persist for 15 to 30 minutes. Cases of maculopapular cutaneous mastocytosis more often are seen in children, and the histamine release most often causes gastrointestinal tract symptoms such as nausea, vomiting, and diarrhea, as well as flushing, blushing, pruritus, respiratory difficulty, and malaise.
- Biswas A, Chaudhari PB, Sharma P, et al. Bleomycin induced flagellate erythema: revisiting a unique complication. J Cancer Res Ther. 2013;9:500-503.
- Yagoda A, Mukherji B, Young C, et al. Bleomycin, an anti-tumor antibiotic: clinical experience in 274 patients. Ann Intern Med. 1972;77:861-870.
- Cohen PR. Trastuzumab-associated flagellate erythema: report in a woman with metastatic breast cancer and review of antineoplastic therapy-induced flagellate dermatoses. Dermatol Ther (Heidelb). 2015;5:253-264. doi:10.1007/s13555-015-0085-2
- Grynszpan R, Niemeyer-Corbellini JP, Lopes MS, et al. Bleomycininduced flagellate dermatitis. BMJ Case Rep. 2013;2013:bcr2013009764. doi:10.1136/bcr-2013-009764
- Stephany MP, Chung S, Handler MZ, et al. Shiitake mushroom dermatitis: a review. Am J Clin Dermatol. 2016;17:485-489.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Biswas A, Chaudhari PB, Sharma P, et al. Bleomycin induced flagellate erythema: revisiting a unique complication. J Cancer Res Ther. 2013;9:500-503.
- Yagoda A, Mukherji B, Young C, et al. Bleomycin, an anti-tumor antibiotic: clinical experience in 274 patients. Ann Intern Med. 1972;77:861-870.
- Cohen PR. Trastuzumab-associated flagellate erythema: report in a woman with metastatic breast cancer and review of antineoplastic therapy-induced flagellate dermatoses. Dermatol Ther (Heidelb). 2015;5:253-264. doi:10.1007/s13555-015-0085-2
- Grynszpan R, Niemeyer-Corbellini JP, Lopes MS, et al. Bleomycininduced flagellate dermatitis. BMJ Case Rep. 2013;2013:bcr2013009764. doi:10.1136/bcr-2013-009764
- Stephany MP, Chung S, Handler MZ, et al. Shiitake mushroom dermatitis: a review. Am J Clin Dermatol. 2016;17:485-489.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
A 77-year-old man with a history of hypertension, hyperlipidemia, and nonmelanoma skin cancer presented to the dermatology clinic for evaluation of a new rash of 2 days’ duration. He trialed a previously prescribed triamcinolone cream 0.1% without improvement. The patient denied any recent travel, as well as fever, nausea, vomiting, or changes in bowel habits. Physical examination revealed diffuse, erythematous, raised, linear plaques on the mid to lower back.


Diffuse Annular Plaques in an Infant
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
A 5-week-old infant boy presented with a rash at birth (left). The pregnancy was full term without complications, and he was otherwise healthy. A family history revealed that his older brother developed a similar rash 2 weeks after birth (right). Physical examination revealed polycyclic annular patches with an erythematous border and central clearing diffusely located on the trunk, extremities, scalp, and face with periorbital edema.


Shiny Indurated Plaques on the Legs
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
A 70-year-old woman presented with pain and swelling in both legs of many years’ duration. She had no history of skin disease. Physical examination revealed shiny indurated plaques on the legs, ankles, and toes with limited range of motion in the ankles (top). Marked thickening of the hands and index fingers also was noted (bottom). A punch biopsy of the distal pretibial region was performed.


Violaceous Plaque on the Metacarpophalangeal Joints
The Diagnosis: Mycobacterial Infection
Mycobacterium marinum is a waterborne nontuberculous mycobacterium prevailing in salt water, brackish water, and still or streaming fresh water that infects fish and amphibians worldwide.1,2 Although first described in 1926 as the organism responsible for the demise of fish in an aquarium in Philadelphia, Pennsylvania, it was not until 1954 that the organism was linked to the cause of infection in humans after it was identified in 80 individuals who had utilized the same swimming pool.1 Due to its ability to secondarily contaminate aquariums, swimming pools, and rivers, this species can give rise to infection in humans, likely though an impaired skin barrier or points of trauma. It commonly is known as swimming pool or fish tank granuloma.3,4
Infection by M marinum commonly presents with lesions on the upper extremities, particularly the hands, that appear approximately 2 to 3 weeks following exposure to the organism.2 Lesions are categorized as superficial (type 1), granulomatous (type 2), or deep (type 3).1 Superficial lesions usually are solitary and painless; may exhibit purulent secretions; and consist of papulonodular, verrucose, or ulcerated granulomatous inflammation.1 These lesions may spread in a sporotrichoidlike pattern or in a linear fashion along lymphatic channels, similar to sporotrichosis. Granulomatous lesions present as solitary or numerous granulomas that typically are swollen, tender, and purulent. Deep lesions are the rarest form and primarily are seen in immunocompromised patients, particularly transplant recipients. Infection can lead to arthritis, tenosynovitis, or osteomyelitis.1
Mycobacterium marinum infection is diagnosed via tissue biopsy for concomitant histopathologic examination and culture from a nonulcerated area close to the lesion.1,2 If cultures do not grow, polymerase chain reaction (PCR) or PCR restriction fragment length polymorphism analysis can be conducted. These techniques can exclude other potential diagnoses; however, PCR is unable to provide information on antibiotic susceptibility.1 Biopsy of lesions reveals a nonspecific inflammatory type of reaction within the dermis consisting of lymphocytes, polymorphonuclear cells, and histiocytes.1,4 Additionally, a granulomatous inflammatory infiltrate resembling tuberculoid granuloma, sarcoidlike granuloma, or rheumatoidlike nodules also may be observed.1 With staining, the acid-fast organisms can be viewed within histiocytes, sometimes demonstrating transverse bands.4
The preferred treatment of M marinum infection is antibiotic therapy.2 It generally is not recommended to obtain in vitro drug sensitivity testing, as mutational resistance to the commonly utilized drugs is minimal. Microbiologic investigation may be warranted in cases of treatment failure or persistently positive cultures over a period of several months.1,2 Due to its rarity, no clinical trials exist to guide optimal management of M marinum infection, according to a search of ClinicalTrials.gov. Nonetheless, anecdotal evidence of prior cases can direct the selection of antibiotics. Mycobacterium marinum appears to respond to certain tetracyclines, including minocycline followed by doxycycline. Other options include clarithromycin, clarithromycin in combination with rifampin, rifampin in combination with ethambutol, trimethoprimsulfamethoxazole, and ciprofloxacin.1,2 Surgical debridement or excision may be indicated, especially in an infection involving deep structures, though recurrences have been reported in some individuals following surgery.2,4 Nonspecific treatment such as hyperthermic or liquid nitrogen local treatment have been used experimentally with positive outcomes; however, experience with this treatment modality is limited.2
Sarcoidosis is an immune-mediated systemic disorder that most commonly affects the lungs and skin. Histopathology shows sarcoidal granulomas with features similar to M marinum infection. The clinical presentation often is described as red-brown macules or papules affecting the face, rarely with overlying scale or ulceration.5 Majocchi granuloma is a dermatophyte fungal infection involving the hair follicles. Although application of topical steroids can worsen the involvement, it commonly displays perifollicular pustules,6 which were not seen in our patient. Granuloma annulare is a benign granulomatous disorder that will spontaneously resolve, typically within 2 years of onset. It presents as an annular or arcuate red-brown papule or plaque without overlying scale or ulceration,7 unlike the lesion seen in our patient. Cutaneous lymphoma is a malignant lymphoproliferative disease most commonly affecting middle-aged White men. The presentation is variable and may include an ulcerated plaque8; the lack of systemic symptoms and notable progression over several years in our patient made this a less likely diagnosis.
- Karim S, Devani A, Brassard A. Dermacase. can you identify this condition? Mycobacterium marinum infection. Can Fam Physician. 2013;59:53-54.
- Petrini B. Mycobacterium marinum: ubiquitous agent of waterborne granulomatous skin infections. Eur J Clin Microbiol Infect Dis. 2006; 25:609-613. doi:10.1007/s10096-006-0201-4
- Gray SF, Smith RS, Reynolds NJ, et al. Fish tank granuloma. BMJ. 1990;300:1069-1070. doi:10.1136/bmj.300.6731.1069
- Philpott JA Jr, Woodburne AR, Philpott OS, et al. Swimming pool granuloma: a study of 290 cases. Arch Dermatol. 1963;88:158-162. doi:10.1001/archderm.1963.01590200046008
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- Boral H, Durdu M, Ilkit M. Majocchi’s granuloma: current perspectives [published online May 22, 2018]. Infect Drug Resist. 2018;11:751-760. doi:10.2147/IDR.S145027
- Joshi TP, Duvic M. Granuloma annulare: an updated review of epidemiology, pathogenesis, and treatment options. Am J Clin Dermatol. 2022;23:37-50. doi:10.1007/s40257-021-00636-1
- Charli-Joseph YV, Gatica-Torres M, Pincus LB. Approach to cutaneous lymphoid infiltrates: when to consider lymphoma? Indian J Dermatol. 2016;61:351-374. doi:10.4103/0019-5154.185698
The Diagnosis: Mycobacterial Infection
Mycobacterium marinum is a waterborne nontuberculous mycobacterium prevailing in salt water, brackish water, and still or streaming fresh water that infects fish and amphibians worldwide.1,2 Although first described in 1926 as the organism responsible for the demise of fish in an aquarium in Philadelphia, Pennsylvania, it was not until 1954 that the organism was linked to the cause of infection in humans after it was identified in 80 individuals who had utilized the same swimming pool.1 Due to its ability to secondarily contaminate aquariums, swimming pools, and rivers, this species can give rise to infection in humans, likely though an impaired skin barrier or points of trauma. It commonly is known as swimming pool or fish tank granuloma.3,4
Infection by M marinum commonly presents with lesions on the upper extremities, particularly the hands, that appear approximately 2 to 3 weeks following exposure to the organism.2 Lesions are categorized as superficial (type 1), granulomatous (type 2), or deep (type 3).1 Superficial lesions usually are solitary and painless; may exhibit purulent secretions; and consist of papulonodular, verrucose, or ulcerated granulomatous inflammation.1 These lesions may spread in a sporotrichoidlike pattern or in a linear fashion along lymphatic channels, similar to sporotrichosis. Granulomatous lesions present as solitary or numerous granulomas that typically are swollen, tender, and purulent. Deep lesions are the rarest form and primarily are seen in immunocompromised patients, particularly transplant recipients. Infection can lead to arthritis, tenosynovitis, or osteomyelitis.1
Mycobacterium marinum infection is diagnosed via tissue biopsy for concomitant histopathologic examination and culture from a nonulcerated area close to the lesion.1,2 If cultures do not grow, polymerase chain reaction (PCR) or PCR restriction fragment length polymorphism analysis can be conducted. These techniques can exclude other potential diagnoses; however, PCR is unable to provide information on antibiotic susceptibility.1 Biopsy of lesions reveals a nonspecific inflammatory type of reaction within the dermis consisting of lymphocytes, polymorphonuclear cells, and histiocytes.1,4 Additionally, a granulomatous inflammatory infiltrate resembling tuberculoid granuloma, sarcoidlike granuloma, or rheumatoidlike nodules also may be observed.1 With staining, the acid-fast organisms can be viewed within histiocytes, sometimes demonstrating transverse bands.4
The preferred treatment of M marinum infection is antibiotic therapy.2 It generally is not recommended to obtain in vitro drug sensitivity testing, as mutational resistance to the commonly utilized drugs is minimal. Microbiologic investigation may be warranted in cases of treatment failure or persistently positive cultures over a period of several months.1,2 Due to its rarity, no clinical trials exist to guide optimal management of M marinum infection, according to a search of ClinicalTrials.gov. Nonetheless, anecdotal evidence of prior cases can direct the selection of antibiotics. Mycobacterium marinum appears to respond to certain tetracyclines, including minocycline followed by doxycycline. Other options include clarithromycin, clarithromycin in combination with rifampin, rifampin in combination with ethambutol, trimethoprimsulfamethoxazole, and ciprofloxacin.1,2 Surgical debridement or excision may be indicated, especially in an infection involving deep structures, though recurrences have been reported in some individuals following surgery.2,4 Nonspecific treatment such as hyperthermic or liquid nitrogen local treatment have been used experimentally with positive outcomes; however, experience with this treatment modality is limited.2
Sarcoidosis is an immune-mediated systemic disorder that most commonly affects the lungs and skin. Histopathology shows sarcoidal granulomas with features similar to M marinum infection. The clinical presentation often is described as red-brown macules or papules affecting the face, rarely with overlying scale or ulceration.5 Majocchi granuloma is a dermatophyte fungal infection involving the hair follicles. Although application of topical steroids can worsen the involvement, it commonly displays perifollicular pustules,6 which were not seen in our patient. Granuloma annulare is a benign granulomatous disorder that will spontaneously resolve, typically within 2 years of onset. It presents as an annular or arcuate red-brown papule or plaque without overlying scale or ulceration,7 unlike the lesion seen in our patient. Cutaneous lymphoma is a malignant lymphoproliferative disease most commonly affecting middle-aged White men. The presentation is variable and may include an ulcerated plaque8; the lack of systemic symptoms and notable progression over several years in our patient made this a less likely diagnosis.
The Diagnosis: Mycobacterial Infection
Mycobacterium marinum is a waterborne nontuberculous mycobacterium prevailing in salt water, brackish water, and still or streaming fresh water that infects fish and amphibians worldwide.1,2 Although first described in 1926 as the organism responsible for the demise of fish in an aquarium in Philadelphia, Pennsylvania, it was not until 1954 that the organism was linked to the cause of infection in humans after it was identified in 80 individuals who had utilized the same swimming pool.1 Due to its ability to secondarily contaminate aquariums, swimming pools, and rivers, this species can give rise to infection in humans, likely though an impaired skin barrier or points of trauma. It commonly is known as swimming pool or fish tank granuloma.3,4
Infection by M marinum commonly presents with lesions on the upper extremities, particularly the hands, that appear approximately 2 to 3 weeks following exposure to the organism.2 Lesions are categorized as superficial (type 1), granulomatous (type 2), or deep (type 3).1 Superficial lesions usually are solitary and painless; may exhibit purulent secretions; and consist of papulonodular, verrucose, or ulcerated granulomatous inflammation.1 These lesions may spread in a sporotrichoidlike pattern or in a linear fashion along lymphatic channels, similar to sporotrichosis. Granulomatous lesions present as solitary or numerous granulomas that typically are swollen, tender, and purulent. Deep lesions are the rarest form and primarily are seen in immunocompromised patients, particularly transplant recipients. Infection can lead to arthritis, tenosynovitis, or osteomyelitis.1
Mycobacterium marinum infection is diagnosed via tissue biopsy for concomitant histopathologic examination and culture from a nonulcerated area close to the lesion.1,2 If cultures do not grow, polymerase chain reaction (PCR) or PCR restriction fragment length polymorphism analysis can be conducted. These techniques can exclude other potential diagnoses; however, PCR is unable to provide information on antibiotic susceptibility.1 Biopsy of lesions reveals a nonspecific inflammatory type of reaction within the dermis consisting of lymphocytes, polymorphonuclear cells, and histiocytes.1,4 Additionally, a granulomatous inflammatory infiltrate resembling tuberculoid granuloma, sarcoidlike granuloma, or rheumatoidlike nodules also may be observed.1 With staining, the acid-fast organisms can be viewed within histiocytes, sometimes demonstrating transverse bands.4
The preferred treatment of M marinum infection is antibiotic therapy.2 It generally is not recommended to obtain in vitro drug sensitivity testing, as mutational resistance to the commonly utilized drugs is minimal. Microbiologic investigation may be warranted in cases of treatment failure or persistently positive cultures over a period of several months.1,2 Due to its rarity, no clinical trials exist to guide optimal management of M marinum infection, according to a search of ClinicalTrials.gov. Nonetheless, anecdotal evidence of prior cases can direct the selection of antibiotics. Mycobacterium marinum appears to respond to certain tetracyclines, including minocycline followed by doxycycline. Other options include clarithromycin, clarithromycin in combination with rifampin, rifampin in combination with ethambutol, trimethoprimsulfamethoxazole, and ciprofloxacin.1,2 Surgical debridement or excision may be indicated, especially in an infection involving deep structures, though recurrences have been reported in some individuals following surgery.2,4 Nonspecific treatment such as hyperthermic or liquid nitrogen local treatment have been used experimentally with positive outcomes; however, experience with this treatment modality is limited.2
Sarcoidosis is an immune-mediated systemic disorder that most commonly affects the lungs and skin. Histopathology shows sarcoidal granulomas with features similar to M marinum infection. The clinical presentation often is described as red-brown macules or papules affecting the face, rarely with overlying scale or ulceration.5 Majocchi granuloma is a dermatophyte fungal infection involving the hair follicles. Although application of topical steroids can worsen the involvement, it commonly displays perifollicular pustules,6 which were not seen in our patient. Granuloma annulare is a benign granulomatous disorder that will spontaneously resolve, typically within 2 years of onset. It presents as an annular or arcuate red-brown papule or plaque without overlying scale or ulceration,7 unlike the lesion seen in our patient. Cutaneous lymphoma is a malignant lymphoproliferative disease most commonly affecting middle-aged White men. The presentation is variable and may include an ulcerated plaque8; the lack of systemic symptoms and notable progression over several years in our patient made this a less likely diagnosis.
- Karim S, Devani A, Brassard A. Dermacase. can you identify this condition? Mycobacterium marinum infection. Can Fam Physician. 2013;59:53-54.
- Petrini B. Mycobacterium marinum: ubiquitous agent of waterborne granulomatous skin infections. Eur J Clin Microbiol Infect Dis. 2006; 25:609-613. doi:10.1007/s10096-006-0201-4
- Gray SF, Smith RS, Reynolds NJ, et al. Fish tank granuloma. BMJ. 1990;300:1069-1070. doi:10.1136/bmj.300.6731.1069
- Philpott JA Jr, Woodburne AR, Philpott OS, et al. Swimming pool granuloma: a study of 290 cases. Arch Dermatol. 1963;88:158-162. doi:10.1001/archderm.1963.01590200046008
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- Boral H, Durdu M, Ilkit M. Majocchi’s granuloma: current perspectives [published online May 22, 2018]. Infect Drug Resist. 2018;11:751-760. doi:10.2147/IDR.S145027
- Joshi TP, Duvic M. Granuloma annulare: an updated review of epidemiology, pathogenesis, and treatment options. Am J Clin Dermatol. 2022;23:37-50. doi:10.1007/s40257-021-00636-1
- Charli-Joseph YV, Gatica-Torres M, Pincus LB. Approach to cutaneous lymphoid infiltrates: when to consider lymphoma? Indian J Dermatol. 2016;61:351-374. doi:10.4103/0019-5154.185698
- Karim S, Devani A, Brassard A. Dermacase. can you identify this condition? Mycobacterium marinum infection. Can Fam Physician. 2013;59:53-54.
- Petrini B. Mycobacterium marinum: ubiquitous agent of waterborne granulomatous skin infections. Eur J Clin Microbiol Infect Dis. 2006; 25:609-613. doi:10.1007/s10096-006-0201-4
- Gray SF, Smith RS, Reynolds NJ, et al. Fish tank granuloma. BMJ. 1990;300:1069-1070. doi:10.1136/bmj.300.6731.1069
- Philpott JA Jr, Woodburne AR, Philpott OS, et al. Swimming pool granuloma: a study of 290 cases. Arch Dermatol. 1963;88:158-162. doi:10.1001/archderm.1963.01590200046008
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- Boral H, Durdu M, Ilkit M. Majocchi’s granuloma: current perspectives [published online May 22, 2018]. Infect Drug Resist. 2018;11:751-760. doi:10.2147/IDR.S145027
- Joshi TP, Duvic M. Granuloma annulare: an updated review of epidemiology, pathogenesis, and treatment options. Am J Clin Dermatol. 2022;23:37-50. doi:10.1007/s40257-021-00636-1
- Charli-Joseph YV, Gatica-Torres M, Pincus LB. Approach to cutaneous lymphoid infiltrates: when to consider lymphoma? Indian J Dermatol. 2016;61:351-374. doi:10.4103/0019-5154.185698
A 24-year-old man presented with a slowly growing, asymptomatic lesion on the left dorsal fourth and fifth metacarpophalangeal joints of 5 years’ duration that was recalcitrant to potent topical corticosteroids. Physical examination revealed an L-shaped, violaceous, firm plaque with focal areas of serous crust. There was no regional lymphadenopathy or lymphangitic spread. The patient had no history of recent travel, and he reported no associated pain or signs of systemic infection.

